 Rithvik R. Nalamalapu
Rithvik R. Nalamalapu Michelle Yue
Michelle Yue Aaron R. Stone
Aaron R. Stone Samantha Murphy
Samantha Murphy Margaret S. Saha
Margaret S. Saha
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 28 June 2021
Sec. Methods and Model Organisms
Volume 14 - 2021 | https://doi.org/10.3389/fnmol.2021.672511
This article is part of the Research Topic Developmental Genes and Molecular Approaches: From Embryo to Human Diseases View all 9 articles
The tweety genes encode gated chloride channels that are found in animals, plants, and even simple eukaryotes, signifying their deep evolutionary origin. In vertebrates, the tweety gene family is highly conserved and consists of three members—ttyh1, ttyh2, and ttyh3—that are important for the regulation of cell volume. While research has elucidated potential physiological functions of ttyh1 in neural stem cell maintenance, proliferation, and filopodia formation during neural development, the roles of ttyh2 and ttyh3 are less characterized, though their expression patterns during embryonic and fetal development suggest potential roles in the development of a wide range of tissues including a role in the immune system in response to pathogen-associated molecules. Additionally, members of the tweety gene family have been implicated in various pathologies including cancers, particularly pediatric brain tumors, and neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease. Here, we review the current state of research using information from published articles and open-source databases on the tweety gene family with regard to its structure, evolution, expression during development and adulthood, biochemical and cellular functions, and role in human disease. We also identify promising areas for further research to advance our understanding of this important, yet still understudied, family of genes.
The first member of the tweety gene family was initially identified in Drosophila melanogaster as a transcriptional unit in the flightless locus and was therefore named after a cartoon character that lacked the ability to fly (Campbell et al., 1993). Present in most invertebrates as a single gene or one with paralogs, the tweety genes encode chloride channels that are also highly conserved in most vertebrates where duplication events resulted in three distinct members, ttyh1–3, each with unique expression patterns (Campbell et al., 2000; Suzuki and Mizuno, 2004; Matthews et al., 2007; Kumada et al., 2010). The role of chloride channels in general, and the tweety family in particular, have been implicated in a wide variety of cellular and physiological processes in the mature organism, including cell volume regulation, muscle and neuron excitability, neurite growth, nociception, immune cell activation, stem cell migration, and wound healing (Chen et al., 2010; Duran et al., 2010; Stefaniuk et al., 2010; Guo et al., 2016; Kim et al., 2018; Han et al., 2019, 2021; Okada, 2019; Yasko et al., 2019). However, tweety genes are also prominently expressed throughout embryonic development, particularly in the nervous system, suggesting an important role in embryogenesis (Chen et al., 2010; Brown et al., 2015; Halleran et al., 2015; Poroca et al., 2017; Jentsch and Pusch, 2018; Karimi et al., 2018; Kim et al., 2018; Han et al., 2019). Additionally, recent research has implicated dysfunction of tweety family genes in a variety of human diseases (Rae et al., 2001; Xu et al., 2006; Toiyama et al., 2007; van Dijk et al., 2012; Wiernasz et al., 2014; Taylor et al., 2016; Jung et al., 2017). While a growing body of research has begun to reveal the critical importance of tweety genes in embryonic development, adult physiology, and pathological conditions, many intriguing questions remain unanswered regarding their function. In this review, we discuss our current knowledge of the tweety gene family and also identify current gaps in our knowledge of tweety genes and the proteins they encode as well as promising areas for further research that will extend our knowledge of this important and ancient gene family.
While various organisms have different numbers of tweety genes based on the number of gene/genome duplication events, all Tweety proteins are anion channels that exhibit a common structure consisting of multiple transmembrane domains (Campbell et al., 2000; Rae et al., 2001; Suzuki and Mizuno, 2004; He et al., 2008b). The structure of several of the Tweety proteins, however, has historically been contested. It was initially proposed that both human TTYH1 and TTYH2 have five transmembrane domains in a 2–2–1 arrangement, also referred to as the Tweety domain, consisting of a pair of transmembrane domains near the N-terminus, another pair of transmembrane domains separated from the first pair by a hydrophilic region, and a fifth transmembrane domain at the C-terminus (Campbell et al., 2000; Rae et al., 2001; Lu et al., 2020). However, Han et al. (2019), using the topology prediction software TMHMM, proposed that the previously predicted transmembrane domain at the C-terminus of Ttyh1 is instead an intramembrane domain. They verified this by expressing a murine Ttyh1-GFP fusion protein in HEK293T cells and conducting immunocytochemistry with two separate antibodies, one that binds specifically to the N-terminus of Ttyh1 and another that binds specifically to the C-terminus GFP. They demonstrated that both the N- and C-termini were located intracellularly for Ttyh1, confirming that, topologically, this last membrane domain must be intramembrane (Han et al., 2019). Interestingly, their TMHMM results placed the N- and C-termini of Ttyh1 extracellularly, which contradicted their experimental results. However, given that the TMHMM program only determines probabilities, Han et al. concluded that the N- and C-termini were located intracellularly for Ttyh1 (Sonnhammer et al., 1998; Krogh et al., 2001; Han et al., 2019). The structure of TTYH3 has also been disputed. Using transmembrane domain prediction tools and empirical studies based on the location of glycosylated residues, it was initially proposed that TTYH3 has six transmembrane domains (Suzuki and Mizuno, 2004). However, this was questioned in a subsequent article that also used hydropathy analysis and glycosylation assays and concluded that TTYH3 has five predicted transmembrane domains (He et al., 2008b).
Among orthologs and paralogs, the transmembrane domains of Tweety proteins are relatively conserved, although significant variation exists at the proteins’ C-terminal ends. For example, the original Tweety protein discovered in D. melanogaster and its paralog CG3638 have an extended C-terminus consisting of 520 and 205 additional hydrophilic residues repeated in short 4–6 amino acid sequences (Campbell et al., 2000). In humans, the C-terminal region of TTYH1 is shorter than those of the two previously mentioned homologs (Suzuki and Mizuno, 2004). Interestingly, this C-terminus seems to only occur for D. melanogaster Tty and not for other invertebrate Tweety orthologs such as those from Branchiostoma floridae (amphioxus) and Ciona intestinalis (tunicate; NCBI Resource Coordinators, 2018). The functional importance of this C-terminal extension is not known.
Recent work has also identified additional domains required for Tweety function. In mice, Han et al. (2019) identified amino acids that are necessary for channel-pore formation in Ttyh1 and Ttyh2 through systematic 20 amino acid truncations of the proteins and subsequent measurement of anion current for each deletion mutant. After identifying potential candidates in loop 2 of the proteins, further analysis identified Arginine 165 in mouse Ttyh1 and Arginine 164 in mouse Ttyh2 as being necessary for channel-pore formation, residues also present in other mammalian Ttyh1 proteins (Han et al., 2019; Uniprot Consortium, 2019). The amino acids required for the formation of the channel pore in invertebrate and non-mammalian vertebrate Tweety proteins remain unknown.
In summary, Tweety proteins all contain the Tweety domain, the amino acids forming the characteristic 2–2–1 of hydrophobic regions. The channel pore-forming residue was found to be Arginine 165 in mouse Ttyh1 and Arginine 164 in mouse Ttyh2, but the channel pore-forming residues for non-mammalian Tweety proteins are still not known. While the transmembrane regions have been identified, the locations of the N- and C-termini for mammalian Ttyh2 and Ttyh3 and non-mammalian Tweety proteins have not been experimentally determined.
Although the original Drosophila Tweety protein was initially hypothesized to be an iron transporter due to the similarity of its transmembrane domains to those of iron transporters, later research contradicted this finding (Campbell et al., 2000; Suzuki and Mizuno, 2004). There is now agreement that tweety genes encode chloride channels, but the ongoing discussion still exists over the precise subtype of chloride channel (Table 1).
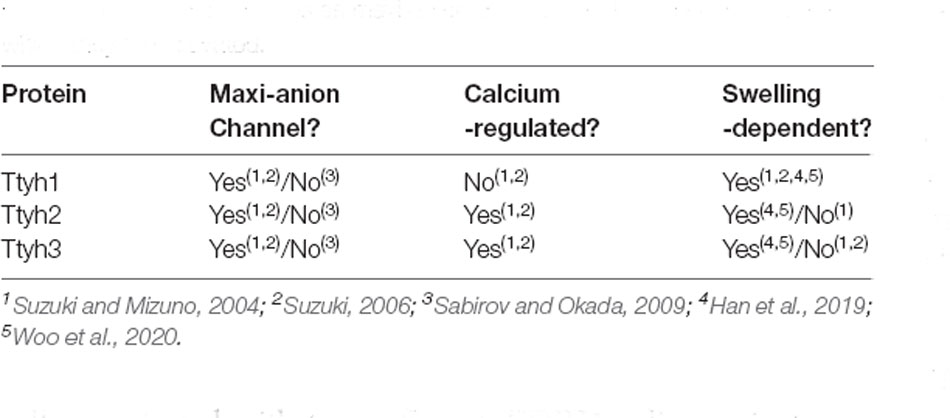
Table 1. Summary table describing what is and is not agreed upon about the roles of the Tweety homologs as maxi-anion channels and the mechanisms by which they are activated.
Using patch-clamp recording, Suzuki and Mizuno (2004) maintained that the Tweety family of proteins are gated chloride channels with maxi-anion channel properties, that is, wide-pore channels with large conductance that allow the passage of larger anions such as ATP and glutamate. However, because HEK293T cells transfected with two different TTYH1 splice variants did not show typical maxi-anion phenotypes, the Tweety proteins themselves may not serve as maxi-anion channels (as reported in Sabirov and Okada, 2009; Han et al., 2019).
While additional experimental data may be required to fully assess the identity of the Tweety homologs as maxi-anion channels, there is general agreement that TTYH2 and TTYH3 are calcium-dependent channels while TTYH1 is calcium-independent. However, all three act in a swelling-dependent manner and function as volume-regulated anion channels (VRAC; Suzuki and Mizuno, 2004; Suzuki, 2006; Han et al., 2019). These channels display ICl, Swell chloride currents that occur in response to an influx of water molecules causing cell swelling which in turn triggers an efflux of chloride ions through a VRACswell channel bringing intracellular volume to normal levels. All three murine Tweety paralogs display activity of swelling-dependent volume-regulated anion channels (VRACswell) in mouse astrocyte primary culture when assessed by whole-cell patch-clamp recording and shRNA-mediated knockdown diminished current (Han et al., 2019; Supplementary Table 1). Additionally, expression of each TTYH in HEK293T and CHO-K1 cells resulted in an ICl, Swell similar to that of native astrocytes, and all three Tweety paralogs were shown to be involved in the regulated volume decrease through a VRAC current in hippocampal astrocytes (Han et al., 2019; Woo et al., 2020). Bae et al. (2019) found that TTYH1 and TTYH2 are both responsible for generating VRAC currents in the SNU-601 gastric cancer cell line after using CRISPR-Cas9 technology to delete exon 7 of the TTYH1 gene and exons 2 and 3 of the TTYH2 gene, which reduced VRAC currents in the resulting knockout cells. They also found that TTYH1 and TTYH2 can generate VRAC currents independently in cancer cells after observing VRAC currents in the HepG2 cell line—which expresses only TTYH1—and in the LoVo cancer cell line—which expresses only TTYH2 (Bae et al., 2019).
Experiments to determine the subcellular location of Tweety proteins are consistent with its identification as a transmembrane channel. Matthews et al. (2007) localized expression to the Golgi apparatus or endoplasmic reticulum (ER) with the localization shifting to the plasma membrane and inducing filopodia formation. Another study using fractionation techniques localized Tyh1 expression to the smooth ER (Kumada et al., 2010). However, Stefaniuk et al. (2010) suggested that the localization to the ER could simply be a result of normal protein processing to the plasma membrane. Consistent with earlier work, Wiernasz et al. (2014), using double immunoflourescent staining with subcellular markers in rat hippocampal neurons, observed expression in the Golgi and ER as well as clathrin-coated vesicles, late endosomes, and lysosomes. Thus, the expression of these proteins in these organelles are consistent with post-translational processing and trafficking to the cell membrane and support its function as a transmembrane chloride channel involved in the regulation of cell volume through the creation of ICl, Swell (Table 1).
While there are only two studies focused specifically on proteins that directly interact with individual Tweety proteins, there are several large-scale interactome studies that shed light on putative interacting partners. In terms of Tweety-focused studies, TTYH2 has also been shown through yeast two-hybrid assay to interact with β-COP, a subunit of Coat Protein Complex I, a protein required for transport from the ER to the Golgi (Ryu et al., 2019). This was further supported through Co-IP experiments. Additionally, β-COP was shown to play a role in trafficking TTYH2 to the plasma membrane as coexpression of TTYH2 and β-COP in COS-7 cells reduced TTYH2 expression at the cell surface, likely due to its role in both retrograde and anterograde transport (Ryu et al., 2019). Additionally, TTYH2 and TTYH3 expression was shown to be regulated through ubiquitination via Nedd4-2, a ubiquitin ligase in HEK293 cells (He et al., 2008a).
There is additional experimental evidence suggesting interactions between TTYH3 and CDKAL1, LMAN2, DHRS9 P2RY12, LPAR1, CD70, FAM189A2, FAM134C, GPR141, TMEM206, ASGR2, CD27, PTGIR, S1PR1, SLCD6A1, TNFRSF1A, GYPB, TMEM171, IPPK, and FTR2; TTYH1 and CPSF4; and TTYH2 and MANSC1 in two separate interactome studies through high-throughput affinity purification mass-spectrometry in HEK293T cells (Huttlin et al., 2015, 2017; Szklarczyk et al., 2019). TTYH2 may also interact with CCND2, KAT2A, CDK2NB, GRM1, KDELR2, NF2, PDGFRA, ERBB2, FGFR4, and IGF1R based on a study using time-resolved fluorescence resonance energy transfer in multiple lung cancer cell lines (Ivanov et al., 2017; Li et al., 2017; Szklarczyk et al., 2019). Another study showed that TTYH2 may interact with GRB2 by using affinity purification-selected reaction monitoring mass-spectrometry in HEK293T cells (Bisson et al., 2011; Szklarczyk et al., 2019). A human protein microarray suggested that TTYH2 also interacts with IKBKG (Fenner et al., 2010; Szklarczyk et al., 2019). While the Tweety proteins have many potential interactants, the precise nature of these interactions is not fully understood; rigorous analysis of these putative binding partners of the Tweety proteins may provide additional insights into their structure and biochemical functions.
Current work concurs that the tweety gene family represents an ancient and phylogenetically diverse group of genes, which are summarized in the tree diagram depicting gene duplication events that resulted in three paralogs found in most vertebrates and varying numbers of homologs found in other eukaryotes (Figure 1). In the first phylogenetic analysis of Tweety proteins, Campbell et al. (2000) compared Tweety coding regions in Drosophila melanogaster (fruit fly), Caenorhabditis elegans (roundworm), Mus musculus (house mouse), and Homo sapiens. They identified two tweety paralogs in D. melanogaster: tty and CG3638. PANTHER, a program that annotates proteins based on homologous sequences, also identified the uncharacterized gene CG14540 as a potential third paralog in D. melanogaster (Thomas et al., 2003). The human TTYH1 shares 27% identity with the D. melanogaster Tty and 29% with the second D. melanogaster Tweety paralog at the amino acid level. Campbell et al. (2000) constructed an unrooted distance neighbor-joining tree, revealing a relatively distant relationship between the two Tweety proteins in D. melanogaster compared to the Tweety homologs found in mice and humans, suggesting an early divergence of these genes from a duplication event (Campbell et al., 2000).

Figure 1. Tree diagram depicting the Tweety protein homologs identified across multiple lineages based on phylogenetic analyses by past literature. The positions of potential orthologs identified by OrthoDB are assumed based on a revised eukaryotic tree incorporating multiple phylogenomic studies, while the positions of potential amphioxus and tunicate Tweety homologs identified through BLAST searches are assumed based on the current understanding of chordate evolution (Donoghue, 2017; Kriventseva et al., 2019; Burki et al., 2020). These have been labeled with a “(?)” in front of the species name. Multiple duplication events lead to the three Tweety paralogs found in most vertebrate species, while other eukaryotes have varying numbers of homologs (Campbell et al., 2000; Matthews et al., 2007; Han et al., 2019). The diagram does not attempt any phylogenetic analyses between the homologs and is only a summary of the findings by past literature and potential homologs identified through database searches. Protein accession numbers not included in the diagram are given in the Supplementary Material (Supplementary Accession Numbers; NCBI Resource Coordinators, 2018; https://www.ncbi.nlm.nih.gov/protein).
Extending the phylogenetic analysis of Tweety proteins to a broader range of eukaryotes, Matthews et al. (2007) found that the conserved Tweety domain has a deep evolutionary history. BLAST searches conducted by Matthews et al. (2007) using the D. melanogaster Tty protein identified three homologs in Dictyostelium discoideum (slime mold), one homolog in Entamoeba histolytica (parasitic protozoan), and four homologs in Arabidopsis thaliana (flowering plant; Altschul et al., 19901). The identified amino acid sequences have low homology but contain a similar arrangement of transmembrane segments to that proposed by Campbell et al. (2000).
In the alignment, Matthews et al. (2007) trimmed the N-terminal region before the first predicted transmembrane region and the C-terminal region after the last predicted transmembrane region to construct a neighbor-joining tree rooted on E. histolytica. The constructed tree reveals the origin of the tweety genes as early as in the common ancestor of animals and plants, signifying its functional importance among eukaryotes (Matthews et al., 2007). Of the three D. discoideum homologs identified, one is closely related to animal Tweety proteins, while the other two are closer to Tweety in plants (A. thaliana). This is consistent with other phylogenetic analyses of proteins in the genome of D. discoideum, which diverged from the ancestor of animals and fungi after the divergence of plants (Williams, 2010). One homolog was identified in Caenorhabditis briggsae (roundworm), which has close relatedness to the C. elegans tweety homolog. Anopheles gambiae (mosquito) was also identified to have two homologs related to tweety, each closely related to the paralogs found in D. melanogaster. Matthews et al. (2007) constructed a second tree focusing on the evolution of tweety in vertebrates. They note a duplication event resulting in a divergence of ttyh3 from the ancestral gene for ttyh1 and ttyh2 as well as a second duplication event leading to the emergence of ttyh1 and ttyh2, resulting in three tweety genes found in most vertebrates. The relationship between vertebrate Tweety proteins was also analyzed by Han et al. (2019) who constructed a maximum-likelihood tree that was overall consistent with the findings of Matthews et al. (Matthews et al., 2007; Han et al., 2019).
Recent advances in genomic sequencing and the publication of a diverse set of non-model organism genomes allow for a more detailed phylogenetic analysis of tweety-like genes and proteins. BLAST searches on publicly available genomes reveal potential Tweety proteins in the following simple organisms: five homologs in Salpingoeca rosetta (choanoflagellate), one homolog in Ostreococcus tauri (chlorophyte), and two homologs in Sphaeroforma arctica (unicellular eukaryote). BLAST searches also reveal potential Tweety proteins in animals: one homolog in Amphimedon queenslandica (sponge), Ciona intestinalis (tunicate), Branchiostoma floridae (amphioxus), and four homologs in Petromyzon marinus (sea lamprey; Altschul et al., 1990; NCBI Resource Coordinators, 2018). The potential amphioxus and tunicate Tweety homologs have been included in the tree diagram (Figure 1) with their positions assumed based on the current understanding of vertebrate evolution (Donoghue, 2017). According to NCBI’s conserved domain database, these Tweety proteins contain a conserved domain called the Tweety_N superfamily, which is based on their similarities with the N-terminal domain of the D. melanogaster Tty protein (domain accession: cl12141). A member within the superfamily is a conserved domain also named Tweety_N (cd07912), which includes a putative pore region as its feature (Lu et al., 20202). OrthoDB, a database that collects genomes from public databases such as NCBI and assesses gene homology, identified potential tweety orthologs in specific fungi and protists: one ortholog in Thalassiosira oceanica (centric diatom), Thalassiosira pseudonana (centric diatom), Metarhizium album (entomopathogenic fungi), and Scleroderma citrinum (common earthball), and eight orthologs in Sphaerobolus stellatus (shotgun fungi; Kriventseva et al., 20193). Most of these orthologs identified by OrthoDB are included in the tree diagram (Figure 1) with their positions assumed based on a revised eukaryotic tree incorporating multiple phylogenomic studies (Burki et al., 2020). Unlike the Tweety proteins in vertebrates, most Tweety homologs in other eukaryotes have not been characterized, including the three homologs in the plant A. thaliana, which are annotated in NCBI as plasma membrane fusion protein (protein accession: NP_178935.2), envelope glycoprotein B (NP_178169.4), and transmembrane protein (NP_177267.2; NCBI Resource Coordinators, 20184).
Current research reveals multiple independent gene duplication events in vertebrates giving rise to numerous paralogous tweety genes. Xenopus laevis (African clawed frog), an allotetraploid, has homeologous pairs for ttyh1 (ttyh1.s, encoding Ttyh1b, and ttyh1.l, encoding Ttyh1a) and ttyh3 (ttyh3.s and ttyh3.l), unlike its closely related species, Xenopus tropicalis (western clawed frog), which is a diploid and lacks these pairs. The homeologous pair for ttyh2.s (encoding Ttyh2) could not be identified and is most likely lost in X. laevis. The ttyh3.l gene in X. laevis encodes a notably shorter protein compared to ttyh3.s and other vertebrate Tweety proteins, suggesting the possibility of a specialized function (Karimi et al., 2018). Danio rerio (zebrafish), Tetraodon nigroviridis (green spotted puffer), and Takifugu rubripes (tiger puffer) have paralogous pairs for ttyh2 (ttyh2 and ttyh2-like, also referred to as ttyh2l) and ttyh3 (ttyh3a and ttyh3b), which is consistent with the findings of Matthews et al. (2007) and with duplication events noted in fish. Specific subfamilies within teleost fish have considerably more tweety paralogs correlating to their polyploidization events, which were identified through the gene gain/loss tree of the tweety family by ENSEMBL (Leggatt and Iwama, 2003; Yates et al., 20205). The subfamily Cyprininae has paralogs ranging from 8 in Cyprinus carpio (common carp) to 15 in Carassius auratus (goldfish), while in the subfamily Salmoninae, Hucho hucho (huchen) contains 13 paralogs (Yates et al., 2020). Interestingly, only ttyh2 and ttyh3 could be identified in birds Taeniopygia guttata (zebra finch) and Gallus gallus (red junglefowl), suggesting the possibility that ttyh1 was lost (Altschul et al., 1990; NCBI Resource Coordinators, 2018).
An interesting finding from BLAST searches with the D. melanogaster Tty protein is a 100% alignment with a portion of a hypothetical protein in Acinetobacter baumannii, a gram-negative bacterium (protein accession: WP_143046262.1). While only 96 amino acids in length, this bacterium’s protein sequence is identical to the D. melanogaster Tty sequence from position 215–310 (Altschul et al., 1990; NCBI Resource Coordinators, 2018). According to SwissProt, these positions in the Tty protein contain two transmembrane segments with a cytoplasmic domain between them followed by an extracellular domain (Uniprot Consortium, 20196). These positions also have high Tweety protein alignments in both vertebrates and other eukaryotes. Additionally, the A. baumannii sequence aligns with the Tweety conserved domain Tweety_N superfamily. No sequences with significant similarities were found in any other bacterial species using the BLAST search tool, suggesting the possibility of a horizontal gene transfer event in which A. baumannii acquired the sequence from the D. melanogaster genome. While the sequence could be a potential Tweety homolog, no conclusions could be made on the sequence’s origin due to the lack of experimental evidence.
In summary, the tweety gene family has a deep evolutionary history, extending as early as the common ancestor of animals and plants. Multiple studies have shown that tweety genes in vertebrates evolved through two separate gene duplication events resulting in three paralogs (with the possibility of ttyh1 being lost in birds). They have also demonstrated that the tweety paralogs underwent further duplication events in X. laevis and fish. While a relatively clear view of the evolutionary history of tweety genes exists for vertebrates, the tweety homologs have not been extensively studied in other eukaryotes, which vary in the number of homologs. This includes non-vertebrate chordates such as tunicates and amphioxi within the Deuterostomia superphylum; the Nematoda and Arthropoda phyla within the Ecdysozoa superphylum; and other clades of Eukarya including plants, fungi, and choanoflagellates. Further analysis of the expression patterns and functions of Tweety proteins across all lineages will provide additional insight into the evolution and role of the ancestral tweety genes.
A complete understanding of the functional role of tweety family genes requires a thorough analysis of mRNA and protein expression patterns throughout development and beyond. While RNA-Seq analysis has provided a wealth of data, somewhat surprisingly, there are relatively few analyses of tweety gene expression using detailed histological analysis to identify cell/tissue type specificity. Here we review current tweety expression data throughout development (Supplementary Table 2) and in the mature organism (Supplementary Table 3) using all available sources of mRNA and protein expression data including published articles as well as gene expression and organism-specific expression databases.
RNA-Seq data available on FlyBase indicates expression of both tty and its paralog CG3638 across embryonic, larval, and pupal development, although not in identical patterns (Thurmond et al., 2019). Tissue-specific RNA-Seq data shows both genes being consistently expressed in the CNS and imaginal discs during the larval wandering stage and the fat bodies during the pupal stage. CG3638 is generally expressed at higher levels and, unlike tty, is also expressed within the digestive system during the larval wandering stage (Thurmond et al., 2019). Proteomics confirms CG3638’s—but not Tty’s—expression throughout development (Thurmond et al., 2019). It should be noted that the Fly-FISH in situ hybridization database did not detect tty expression at any developmental stage (Lécuyer et al., 2007; Wilk et al., 2016). C. elegans’s only tweety homolog ttyh1 is expressed throughout early embryonic stage into larval development and adulthood, with demonstrated expression in the embryonic pharynx and several adult tissues (Levin et al., 2012; Hashimshony et al., 2015; Harris et al., 2020). In echinoderms, RNA-Seq data on S. pupuratus reports that ttyh2 expression is absent until 18 h after fertilization, when levels increase through early gastrulation (Cameron et al., 2009; Tu et al., 2014; Kudtarkar and Cameron, 2017; Cary et al., 2018).
In adults, both tty and CG3638 are expressed across several tissues in D. melanogaster, particularly the head; however, expression of CG3638 is stronger than that of tty across most sampled adult organs and is present within the digestive tract while tty is not (Thurmond et al., 2019).
Although expression studies (primarily RNA-Seq) are largely restricted to model organisms, tweety family genes in invertebrates generally display broad temporal and spatial tissue distribution. However, given that ISH was performed only in flies, there is a need for additional tissue-specific analysis at the RNA and protein level.
Gamete expression data derives primarily from the tetraploid X. laevis and M. musculus. qRT-PCR on X. laevis eggs revealed low levels of expression of all three tweety genes in unfertilized eggs (Halleran et al., 2015). Mouse RNA-Seq experiments have also detected very low to moderate levels of ttyh2 and tthy3 expression in oocytes; however, only one study detected significant expression of ttyh1 (Bult et al., 2019; Papatheodorou et al., 2020).
Data on tweety gene family expression during cleavage and blastula stages also stem largely from amphibians, with some data available from mice. In mice, at the single-cell stage, ttyh1 expression was essentially undetectable while tthy2 was expressed at low levels and ttyh3 at moderate levels. By the two-cell stage, all three genes were expressed at detectable levels (Bult et al., 2019).
More extensive-expression data is available from non-mammalian vertebrates. In X. tropicalis, RNA-Seq data shows very low levels of ttyh1 and ttyh2 during cleavage stages while ttyh3 displays a steady increase in expression, peaking at the late blastula stages (Karimi et al., 2018). In X. laevis, pre-gastrulation expression varies between genes. While no signal was detectable using in situ hybridization (ISH), qRT-PCR revealed low levels of tweety family gene expression through blastula stages with slightly elevated levels near the mid-blastula transition for ttyh1 and ttyh3, before a subsequent drop back to basal levels (Halleran et al., 2015). According to RNA-Seq data, although ttyh2 expression remains absent throughout blastula stages, ttyh1.s is expressed in fertilized eggs, before decreasing during the cleavage through blastula stages. Both homeologs of ttyh3 increase in expression at the mid-blastula transition (Karimi et al., 2018). Similar to Xenopus, D. rerio tweety gene expression during the blastula and cleavage stages vary between genes. While ttyh1 expression is absent during cleavage and blastula stages, ttyh2, ttyh2l, ttyh3a, and ttyh3b are expressed at low to moderate levels depending on the gene (White et al., 2017).
Although limited to a few model organisms, all studied species express one or more of the tweety genes in oocytes through the blastula stages, with specific temporal patterns varying among genes and species. These data, which rely almost exclusively on RNA-Seq and RT-PCR results, could benefit from histological analysis.
From gastrulation onwards, mRNA levels of all three tweety family genes increase significantly and show distinct expression patterns.
During gastrulation and organogenesis, ttyh1 expression is localized to neural tissues in most vertebrate organisms. In mice, immunohisotochemistry revealed broad expression starting at late gastrula stages with elevated levels in M-phase cells (Kumada et al., 2010). By E14, ISH and immunohistochemistry experiments detected prominent expression localized to the brain and spinal cord with continuing pronounced expression in dividing cells, presumably neural progenitors (Kumada et al., 2010). ISH experiments also detected expression in the ventricular zone of the mouse brain and forebrain during organogenesis (Abramova et al., 2005; Kawaguchi et al., 2008) in addition to strong expression throughout the nervous system (Blackshaw et al., 2004; Visel et al., 2007; Allen Institute for Brain Science, 2008a; Diez-Roux et al., 2011; Thompson et al., 2014; Bult et al., 2019). Expression was also observed outside of the nervous system in the pancreas and ear and less prominently in several other organ systems (Visel et al., 2007; Bult et al., 2019). RNA-Seq experiments in embryonic mice have corroborated the localization of ttyh1 to the CNS through the fetal stages (Brown et al., 2015; Papatheodorou et al., 2020).
Rat RNA-seq data also localizes the highest levels of expression to the developing brain, with much lower levels of expression present in the developing testes (Papatheodorou et al., 2020). Experiments conducted in hippocampus embryonic neuronal cell cultures further confirm neuronal expression (Stefaniuk et al., 2010; Wiernasz et al., 2014). Experiments in rat pup cell cultures detect glial (Wiernasz et al., 2014) and neuronal (Matthews et al., 2007) expression, with neuronal expression being preferentially localized to the axons. Human mass-spectrometry and RNA-Seq datasets show inconsistent expression in other tissues but consistent expression in the CNS (Kim et al., 2014; Brown et al., 2015; Papatheodorou et al., 2020).
In non-mammalian vertebrates, ttyh1 also localizes to the developing CNS during gastrulation and organogenesis. In X. laevis, ttyh1 mRNA ISH signal was detectable at neurula stages, with strong expression in the midbrain and eyes by the late neurula stages. By the tailbud stages, expression extended along most of the anterior-posterior neural axis (Halleran et al., 2015). As in mice, ttyh1 is localized to the ventricular area, a region of actively dividing cells. Both qRT-PCR and RNA-Seq data show significant increases in transcript levels throughout neurulation (Halleran et al., 2015; Karimi et al., 2018). In D. rerio, previously absent or low ttyh1 expression also appears during gastrulation (White et al., 2017).
Mouse ISH data show weak to moderate expression throughout the CNS, in a single cell, punctate distribution pattern, in addition to expression in the adrenal gland, brain vasculature, and parts of the retina (Blackshaw et al., 2004; Diez-Roux et al., 2011; Hupe et al., 2017; Bult et al., 2019). RNA-Seq mouse data shows at least low levels of ttyh2 expression in all sampled embryonic and fetal tissues, with highest expression being in the brain or testis/ovaries depending on study and stage (Brown et al., 2015; Papatheodorou et al., 2020).
The most detailed studies of ttyh2 expression are from non-mammalian vertebrates. In Xenopus, a strong ISH ttyh2 signal was present within the ganglia V, VII, IX, and X, with signal first detectable at the hatching stage (Halleran et al., 2015). In zebrafish, ISH data detected both ttyh2 and ttyh2l at segmentation onwards, although only signals for ttyh2l were present in early to mid-somitogenesis, with signals being detected consistently in the adaxial (muscle precursor) cells but also in the neural tube, neurons, and trigeminal placode. By late somitogenesis, ttyh2l expression is restricted to the cranial ganglion and optic vesicle while expression of ttyh2 appears in the CNS before extending to the retina as development progresses. In the mid-pharyngeal stages, ttyh2l expression appears in the retina, where it will remain in the hatching stages, and spinal cord, where it will diminish (Thisse and Thisse, 2004; Ruzicka et al., 2019). These expression patterns are consistent with RNA-Seq data showing increased expression following gastrulation (White et al., 2017).
Mice RNA-Seq datasets identify ttyh3 expression from somitogenesis into fetal stages. Two of these datasets also characterize tissue localization, with both reporting significant expression in the CNS and in several other tissues (Brown et al., 2015; Papatheodorou et al., 2020). Consistent with mouse expression, rat RNA-Seq data shows expression across multiple different tissue types, particularly the brain, ovary, and testis (Papatheodorou et al., 2020). In Xenopus laevis, qRT-PCR data identifies peak expression during neurula and early tailbud stages followed by a subsequent decline (Halleran et al., 2015). RNA-Seq data, however, identifies an earlier peak (Karimi et al., 2018). ISH experiments detected strong ttyh3 signals throughout the anterior nervous system by the neurula stage with signals extending to the spinal cord by tailbud stages. Transient signals in the somites were also detected. Unlike ttyh1, ttyh3 was detected primarily in postmitotic neural cells (Halleran et al., 2015). In zebrafish, RNA-Seq data also reveals expression levels increasing after gastrulation (White et al., 2017).
In summary, expression analyses point to consistent patterns of tweety gene expression across all vertebrate taxa analyzed, with ttyh2 and ttyh3 typically demonstrating expression in a broader array of tissues compared to ttyh1 which is localized to the central nervous system. RNA-Seq analysis remains the primary mode of study for ttyh3 while ttyh2 and ttyh1 patterns incorporate more detailed histological data, creating an opportunity for future study of ttyh3.
Although detailed histological analysis of tweety gene expression in the mature organism is lacking, RNA-Seq data shows that tweety genes are still expressed in the fully developed organism with expression patterns similar to those observed in later organogenesis stages.
Similar to embryonic and fetal development, ttyh1 is prominently expressed in the CNS. RNA-Seq studies in rats show a consistent pattern of maximal expression in the CNS, particularly in the visual cortex, and to a lesser extent in the testes and pituitary gland (Steen et al., 1999). RT-PCR in adult rat brains detected high levels of expression in the brain stem, cerebellum, and cerebral cortex (Morciano et al., 2009). ISH experiments confirm brain expression (Matthews et al., 2007; Morciano et al., 2009), with one finding strong mRNA signals in the olfactory bulb, cerebral cortex, and cerebellum, with less intense expression in other regions of the brain (Morciano et al., 2009). A combination of western blots and immunohistochemistry experiments have shown clear expression in rat neurons (Stefaniuk and Lukasiuk, 2010; Stefaniuk et al., 2010). One study further co-localized ttyh1 expression to the presynaptic active zone; however, two subsequent studies have only detected “negligible” (Wiernasz et al., 2014) or “rare” (Stefaniuk et al., 2010) colocalization with synaptic/synaptic vesicle markers.
Data for murine ttyh1 mRNA and Ttyh1 protein is consistent with rat data as the highest levels of expression were found within the nervous system and the testis (Brown et al., 2015; Schmidt et al., 2018; Papatheodorou et al., 2020; Samaras et al., 2020). ISH further confirms the presence of ttyh1 expression throughout the brain (Lein et al., 2007). RT-PCR analysis specifically identified ttyh1 expression in the dorsal root ganglion (Al-Jumaily et al., 2007). RNA-Seq and mass-spectrometry datasets in other mammals align with these trends, with human RNA-Seq and mass-spectrometry datasets localizing the highest expression to the CNS across multiple isoforms (Supplementary Table 3). Adult expression in non-mammalian vertebrates also continues to be restricted to the CNS, specifically the brain in both Xenopus laevis and tropicalis (Karimi et al., 2018; Papatheodorou et al., 2020).
Expression data on ttyh2 in mature organisms almost exclusively derives from RNA-Seq data and shows broad tissue distribution. In mice, RNA-Seq studies report prominent expression in the CNS, adrenal gland, digestive system, liver, and testes, with ISH data confirming expression in the brain and spinal cord (Lein et al., 2007; Allen Institute for Brain Science, 2008b; Brown et al., 2015; Papatheodorou et al., 2020). Mass-spectrometry data further supports the expression of multiple Ttyh2 isoforms within the brain (Schmidt et al., 2018; Samaras et al., 2020). Like ttyh1, RT-PCR experiments identified ttyh2 expression in adult dorsal root ganglia (Al-Jumaily et al., 2007). A single microarray experiment notes upregulated expression of ttyh2 within adult mice myelinating oligodendrocyte cells (Edgar et al., 2013). In rat RNA-Seq datasets, ttyh2 expression was typically highest in the brain, adrenal gland, or liver depending on the strain and study, with an additional study recording notably higher ttyh2 expression in the superior olivary complex relative to the hippocampus or corpus striatum (Steen et al., 1999; Nothwang et al., 2006). Human RNA-Seq datasets typically identify the highest levels of expression in the CNS (Brown et al., 2015; Papatheodorou et al., 2020). Earlier northern blot experiments also detected expression in the brain and testis, with comparatively lower levels of expression in the heart and ovary and even lower levels in leukocytes, skeletal muscle, and spleen (Rae et al., 2001). Proteomics consistently notes expression in the brain; however, depending on the experiment, expression can be comparatively higher in places such as the breast and colon muscle (Stelzer et al., 2016; Schmidt et al., 2018; Samaras et al., 2020) or bone marrow (Papatheodorou et al., 2020). RNA-Seq datasets from other mammalian species are largely consistent with rat, human, and mice datasets (Supplementary Table 3).
Non-mammalian vertebrate expression is similar to that observed in mammals. In X. laevis, RNA-Seq analysis identified maximal adult expression within the eyes then brain and lower levels in the intestine, spleen, kidney, liver, lung, and testes, a result consistent with that in X. tropicalis (Karimi et al., 2018; Papatheodorou et al., 2020). In bird species G. gallus and reptile species A. carolinesis, ttyh2 expression was the highest in the brain, with similar levels of expression being detected in the spleen in one G. gallus experiment (Papatheodorou et al., 2020).
Like ttyh2, ttyh3 is expressed in a wide range of tissues in the mature organism. In mice, most RNA-Seq studies localized maximal expression to the CNS, which mass-spectrometry data corroborates (Schmidt et al., 2018; Papatheodorou et al., 2020; Samaras et al., 2020); however, one study localized highest expression to the adrenal. Nonetheless, all RNA-Seq studies recorded significant expression across most sampled tissues (Brown et al., 2015; Papatheodorou et al., 2020). Like ttyh1 and ttyh2, ISH work identified expression in the brain, and RT-PCR identified ttyh3 expression in the dorsal root ganglia (Al-Jumaily et al., 2007; Lein et al., 2007). Rat RNA-Seq experiments identified significant expression in the spleen, lung, and brain and detectable expression in many organs/tissues (Steen et al., 1999).
Across human RNA-Seq and mass-spectrometry datasets, significant levels of adult expression were found in the CNS, kidney, spleen, bone, platelets, or immune cells depending upon the specific study, with some mass-spectrometry datasets reporting notably narrower expression ranges than RNA-Seq datasets (Kim et al., 2014; Brown et al., 2015; Stelzer et al., 2016; Schmidt et al., 2018; Papatheodorou et al., 2020; Samaras et al., 2020). Whereas mass-spectrometry data showed adult expression of all identified TTYH3 isoforms as maximal in bone, RNA-Seq data on splicing variants localized the highest expression to the brain, although only this dataset does not include values for the bone (GTex Consortium, 2015; Schmidt et al., 2018; Samaras et al., 2020). In other primate species, RNA-Seq analysis reports a wide tissue distribution of expression that varies among species (Thierry-Mieg and Thierry-Mieg, 2006). In non-primate mammals, expression levels varied with the organism being assayed, but the highest expression was typically reported in the lung, brain, or spleen (Steen et al., 1999; Papatheodorou et al., 2020).
In non-mammalian vertebrate species G. gallus and X. laevis, expression was broad, with the highest levels of expression usually localized to either the CNS and/or spleen depending on the study (Karimi et al., 2018; Papatheodorou et al., 2020). In summary, similar to embryonic expression patterns in vertebrates, ttyh1 expression remains restricted to the CNS while ttyh2 and ttyh3 are expressed in a broader array of tissues including the liver and adrenal gland for ttyh2 and spleen, lungs, and immune cells for ttyh3. These data are based almost exclusively on RNA-Seq and mass spectrometry data; there is a significant need for more corroborative histological analysis.
While the expression patterns of the various tweety genes provide critical information suggestive of their possible roles in the developing and mature organism, functional experiments are required to confirm these putative roles. Despite relatively few experimental studies that attempt to assess the function of the tweety genes, particularly for ttyh2 and ttyh3, the current body of work points to a wide range of functions for the tweety gene family in both embryonic development and the mature organism (Supplementary Table 1).
An early study attempting to determine the role of ttyh1 suggested its involvement in mitosis and its critical role in development at the morula and blastula stages in mice. ttyh1 was observed to be strongly upregulated in some mouse embryo mitotic cells, suggesting that ttyh1 is involved in progenitor cell and stem cell mitosis (Kumada et al., 2010). Immunostaining against Ttyh1 showed elevated expression of Ttyh1 in the ER of cells during metaphase and anaphase in parasagittal sections of E7.5, E11.5, and E14.5 mouse embryos (Kumada et al., 2010). Furthermore, an engineered germline loss of function mutation in ttyh1 in which exons 5–10 were replaced with a neomycin resistance gene cassette showed early embryonic lethality in mice embryos (Kumada et al., 2010). Localization of Ttyh1 to the ER led to the hypothesis that Ttyh1 was essential for embryonic development through a possible role in maintaining calcium homeostasis during mitosis (Kumada et al., 2010). However, there is no additional evidence supporting this claim. Another study observed the localization of Ttyh1 to the ER but hypothesized that this observation was likely due to being a site of post-translational processing rather than suggesting a novel function of the Ttyh1 protein (Stefaniuk et al., 2010). Additionally, a later article contested the observation that knocking out ttyh1 causes embryonic lethality; they proposed that the observed phenotypes were due to the knockout of a long noncoding RNA gene NR_033548.1 which is located within the ttyh1 gene (Wu et al., 2019). This hypothesis was further supported after mouse embryos from a knockout mouse line, created by using CRISPR-Cas9 technology to delete exon 4 of the ttyh1 allele, developed normally. The deletion of exon 4 in ttyh1 resulted in a truncated protein that left the NR-033548.1 gene intact (Wu et al., 2019). However, disruption of ttyh1 did result in a reduction of neural stem cell stemness in the embryo; neurospheres derived from ttyh1-deficient mice were smaller in size and number (Wu et al., 2019).
While Ttyh1 does not appear necessary for embryonic development at the blastula stage, consistent with its very low levels of expression at this stage, there is general agreement that it plays a role in maintaining neural stem cell properties in the embryo based on its expression patterns and functional analyses. This was demonstrated by neurosphere frequency increasing after overexpressing Ttyh1 in mouse E14.5 primary neural progenitors using a retroviral vector containing the murine stem cell virus long terminal repeat which drives the overexpression of ttyh1 (Kim et al., 2018).
Ttyh1 was shown to maintain neural stem cell properties by increasing the expression of the direct downstream targets of Notch following overexpression in neural progenitor cells. ttyh1 increased Notch IntraCellular Domain (NICD) production through the degradation of RER1 and subsequently increased maturation of γ-secretase complexes (Kim et al., 2018). The increase in NICD production was supported by western blot data using an α-NICD antibody (Kim et al., 2018). Thus, ttyh1 appears to maintain neural stem cell properties by increasing Notch activity (Kim et al., 2018). Using microarray analysis, another study did find, however, that ttyh1 expression is significantly upregulated in late neurula stage Xenopus laevis embryos after over-expressing NICD when compared to GFP-treated embryos (Vasiliu et al., 2015). Furthermore, a second study supported the idea that ttyh1 acts downstream of Notch as treatment of mouse neurospheres with a γ-secretase inhibitor, which downregulates Notch activity, caused a downregulation of ttyh1 at the mRNA and protein levels as assessed by qRT-PCR and western blot data (Wu et al., 2019). Where ttyh1 is positioned in the Notch pathway signaling—whether it is upstream or downstream of NICD activation, or whether there is an auto-regulatory loop—remains an open question.
Consistent with its role in stem cell proliferation, ttyh1 was also strongly upregulated in newly postmitotic Müller glia, the ventricular zone of the E13 and E14 mouse brain, apical progenitor cells, the presynaptic active zone of 3- to 4-week old Wistar rats, and the ventricular zone of the human hypothalamus—all highly proliferative regions (Blackshaw et al., 2004; Abramova et al., 2005; Kawaguchi et al., 2008; Morciano et al., 2009; Okamoto et al., 2016; Zhou et al., 2020).
In the developing nervous system, ttyh1 has also been implicated in neurite growth. An siRNA-mediated knockdown of ttyh1 in hippocampal neurons cultured from E18-E19 Wistar rats showed increased MAP2 aggregation (Stefaniuk et al., 2010). TTYH1 also appears to play a role in filopodia formation and cell adhesion as deconvolution microscopy showed colocalization of the engineered TTYH1-GFP fusion protein with filopodia and integrin on HEK293 cells (Matthews et al., 2007). Enhanced filopodia formation was also observed in neurons that overexpressed TTYH1 further supporting this idea (Stefaniuk et al., 2010). Additionally, overexpression of ttyh1 in a hippocampal neuron cell culture showed increased neuritogenesis (Stefaniuk et al., 2010).
In the mature nervous system, several studies suggest that ttyh1 is involved in nociception. An RNA-Seq experiment analyzed nociceptor sensory neurons in mice with and without the production of a spinal cord injury using a vessel clip (which applies a force to the wound). RNA-Seq analysis showed a downregulation of ttyh1 in mice with a spinal cord injury when compared to sham mice, which underwent the same procedure without the vessel clip (Yasko et al., 2019). Another study found that the knockout of ttyh1 in mice via CRISPR-Cas9 led to a reduced pain response and reduced nociceptor excitability in mice. Knocking down ttyh1 in nociceptors resulted in reduced nociception and pain hypersensitivity in mice, further supporting the idea that ttyh1 plays a role in nociception (Han et al., 2021).
Finally, regarding a completely different role, ttyh1 was shown to be downregulated (among other genes) in human epithelial Caco-2 cells when treated with vitamin D3 (Claro da Silva et al., 2016). However, additional work is required to determine the role of ttyh1 in the vitamin D3 pathway. In summary, ttyh1 appears to play a central role in neural development and maintaining neural stem cell-like properties during early neurogenesis, and it may do so by interacting with the Notch signaling pathway. ttyh1 also appears to play a role in mitosis and nociception, although these two potential roles of ttyh1 still need more definitive evidence to flesh out mechanistic details.
Compared to ttyh1, ttyh2 is not as extensively studied. While expression studies suggest that ttyh2 may play a role in neural development given its strong expression in both Xenopus and zebrafish cranial nerves during neural embryonic development, this putative role requires functional experimental analysis (Halleran et al., 2015; Ruzicka et al., 2019). Likewise, single-cell RNA-Seq analysis revealed relatively high expression of ttyh2 in most of the radial glial cells and outer radial glia that were analyzed, indicating a possible role in the development of this cell type (Johnson et al., 2015).
Ttyh2 has also been shown to be involved in the formation of cell membrane extensions as Hori et al. (2019) found that overexpressing ttyh2 in retinal pigment epithelium cells caused an increase in membrane extensions in those cells.
Other investigations have focused on the role of ttyh2 in immune function. One study found that infection of mice with lymphocytic choriomeningitis mammarenavirus caused ttyh2 downregulation in splenic natural killer cells when compared to those of uninfected mice (Papatheodorou et al., 2020). Another study found that treatment of H1N1-infected human monocyte-derived dendritic cells with Poly I:C, an immunostimulant used to simulate viral infection, upregulated ttyh2 (Papatheodorou et al., 2020). Likewise, treatment of primary human macrophages from patients with ulcerative colitis cultured with heat-killed E. coli reported ttyh2 upregulation (Papatheodorou et al., 2020). These studies suggest a role for ttyh2 in the immune response, although additional functional studies are needed to confirm this. This role is, however, consistent with the strong expression of ttyh2 in organs rich with immune cells, like the spleen. Overall, ttyh2 may be involved in the immune response; ttyh2 may also play a role in neural development given its strong expression in the cranial nerves and glial cells of developing embryos.
ttyh3, like ttyh2, is relatively understudied compared to ttyh1. ttyh3 also likely plays a role in neural development given its strong expression in neural tissue in early embryonic neural development, but its precise role remains unknown in the absence of functional studies (Brown et al., 2015; Halleran et al., 2015; Ruzicka et al., 2019).
ttyh3 may be involved in Schwann cell repair after nerve injury as RNA-Seq analysis of Schwann cells following injury to the sciatic nerve of mice showed an upregulation of ttyh3 when compared to mice without nerve injury (Papatheodorou et al., 2020). However, more definitive experiments are needed to determine if ttyh3 is involved in this process.
Like ttyh2, ttyh3 also seems to play a role in the adult immune system, regulating immune cell activation in response to pathogens, specifically in response to pathogen-associated molecular patterns (PAMPs). Ttyh3 was shown to facilitate ATP release in response to PAMPs (Chen et al., 2010). Additionally, neutrophils treated with lipoteichoic acid (an agonist for Toll-like receptor 2) were shown to upregulate ttyh3 mRNA, possibly through downregulation of has-miR-1271-5p (Yen et al., 2019). TargetScan analysis showed that ttyh3 was a downstream target of this miRNA, however, miRTarBase analysis did not corroborate this (Yen et al., 2019). It is hypothesized that this miRNA may interact with ttyh3 among other genes in neutrophil activation (Yen et al., 2019). However, another study found that infection of mice with lymphocytic choriomeningitis mammarenavirus caused downregulation of ttyh3 when compared to those of uninfected mice (Papatheodorou et al., 2020).
While this could be a pathogen-specific response, the downregulation of ttyh3 in response to a pathogen in splenic killer cells is inconsistent with the results of the other studies that have shown ttyh3 upregulation in response to pathogen and MAMP exposure, results that call for further investigation to determine the role of ttyh3 in the immune system. In general, ttyh3 may be involved in regulating immune cell activation in response to PAMPs. ttyh3 may also play a role in neural development given its strong expression in neural tissue in embryonic neural development.
Drosophila tty was initially discovered in the flightless locus in Drosophila along with fli, dodo, and penguin (Campbell et al., 1993, 2000). A study that crossed a UAS-driven inverted repeat of tty with a Gal4 fly strain to induce RNAi knockdown of tty in F1 offspring found that the resulting embryos were viable and had a wild-type phenotype that was capable of flying (Mummery-Widmer et al., 2009). Another study, using the same UAS with the muscle-specific Mef2-Gal4 driver, found that the resulting offspring were viable (Schnorrer et al., 2010). To assess which genes are involved in heat nociception, Neely et al. (2010) used the pan-neuronal elav-Gal4 driver and found that resulting offspring were viable and did not exhibit decreased avoidance to noxious heat, indicating that tty does not play a role in heat nociception.
A paralog of Drosophila tty, CG3638, was shown to be embryonic lethal in Drosophila embryos in a P-element mutagenesis assay (Bourbon et al., 2002). This was further corroborated by a study showing that flies with UAS-driven inverted repeats of the CG3638 gene crossed with flies expressing Gal4 produced F1 offspring that were embryonic lethal prior to the pupal stage (Mummery-Widmer et al., 2009). However, another study, using the muscle specific Mef2-Gal4 driver, found that the resulting offspring were viable (Schnorrer et al., 2010). In the study assessing the roles of certain genes in heat nociception, Neely et al. (2010) used the pan-neuronal elav-Gal4 driver and found that the resulting offspring were viable and did not exhibit decreased avoidance to noxious heat, indicating that CG3638 does not play a role in heat nociception. Another study created a transgenic Drosophila line in which a YFP-RAB1 fusion gene was inserted into the CG3638 gene, and this insertion proved to be viable in embryos (Zhang et al., 2007). CG3638 may also play a role in the stress response, as the creation of heterozygous Drosophila containing only one functional CG3638 allele led to decreased starvation resistance (Harbison et al., 2004). The gene may also play a role in Drosophila immune response as RNAi knockdown of CG3638 resulted in reduced phagocytosis of C. albicans cells in plasmatocytes (Stroschein-Stevenson et al., 2006). It is also possible that CG3638 somehow influences organismal behavior as a homozygous knockout of CG3638 through a P-element insertion assay resulting in male Drosophila displaying significantly increased levels of aggressive behavior (Edwards et al., 2009).
Even though the tweety gene family was identified over two decades ago, much remains unknown regarding its functions within the developing and mature organism, both in Drosophila and vertebrates. Despite being in the flightless locus in Drosophila, tty is not necessary for Drosophila flight. Additionally, there is some experimental evidence suggesting that the paralog of Drosophila tty, CG3638, may produce an embryonic lethal phenotype in developing embryos when knocked down. CG3638 may also be involved in the Drosophila immune response. These two potential roles of CG3638 still need more definitive evidence to understand the role of CG3638 during development and the immune response.
While the role of the tweety gene family in cancer (particularly brain and colon cancer) is well established (Supplementary Table 4), a growing number of studies are documenting its role in neurological diseases (e.g., status epilepticus, Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis; Supplementary Table 5) and other disorders (e.g., hypertension, cystic fibrosis, and Turner syndrome; Supplementary Table 6). Much of our knowledge about the role of tweety genes in disease derives from differential gene expression experiments comparing normal and pathological tissues; however, as described below, studies are increasingly attempting to investigate the role of this gene family in disease on a more mechanistic level.
A strong association exists between TTYH1 and brain cancers, particularly glioblastoma. Notably, TTYH1 shRNA knockdowns in glioblastoma cell lines caused reduced glioma invasiveness, abnormal neurite membrane protrusion morphology, decreased tumor size, and increased survival in mice (Jung et al., 2017). Additionally, linear regression revealed that Ttyh1 levels quantified and analyzed using western blot positively correlated with glioblastoma invasiveness in vivo (Jung et al., 2017). Furthermore, an oncogenic fusion between TTYH3 and BRAF intron 1 was identified in a glioblastoma multiforme patient (Weinberg et al., 2019). This fusion protein is associated with increased MEK/ERK phosphorylation and pathway signal transduction, which is important in cell proliferation and transformation (Khokhlatchev et al., 1998; Chang and Karin, 2001; Kolch, 2005; Kyriakis and Avruch, 2012; Weinberg et al., 2019).
Additional evidence for the role of tweety genes in gliomas comes from the association between TTYH1 and tumor microtube formation in glioblastoma cells (Osswald et al., 2015; Jung et al., 2017). Prominent in gliomas, tumor microtubes are cell membrane protrusions extending ten to hundreds of microns long and are likely involved in intercellular communication and glioma propagation (Osswald et al., 2015; Jung et al., 2017). Immunocytochemistry showed human glioblastoma stem cells in vitro having punctate TTYH1 localization concentrated at the membrane and growth cone-like tips of tumor microtubes, suggesting TTYH1’s possible involvement in tumor microtube growth (Jung et al., 2017). Furthermore, glioblastomas with 1p/19q chromosome arms codeletions (associated with diminished glioma propagation and impaired tumor microtube formation and function) show TTYH1 and TTYH2 downregulation and TTYH3 upregulation compared to 1p/19q non-codeleted gliomas (Osswald et al., 2015; Jung et al., 2017).
Transplant studies also corroborate this association between tweety and microtube formation. When glioblastoma cells grown under differentiating, serum-containing conditions were implanted into mice brains, tumor microtube formation became impaired and RNA microarray showed corresponding ttyh1 downregulation (Jung et al., 2017). Additionally, in vivo two-photon microscopy in mice implanted with human glioma cells showed that TTYH1-positivity was tumor microtube count-dependent as higher TTYH1-positivity was found in glioma cells with one to two tumor microtubes compared to those with more than four (Jung et al., 2017). Cells with one to two tumor microtubes were more motile than those with more than four, suggesting TTYH1’s role in facilitating glioma propagation, potentially through tumor microtube-associated mechanisms (Jung et al., 2017). Overall, reasonably consistent correlations exist between tweety 1 downregulation, decreased tumor microtube function, and decreased glioma propagation, suggesting tweety 1 may facilitate glioma invasiveness, potentially through tumor microtube-associated mechanisms.
A caveat when comparing pathological to normal tissues and cells is that RNA-Seq and microarray experiments demonstrate how TTYH1 and TTYH2 expression patterns and cancers may be cell and tissue-type-dependent. For example, TTYH1 and TTYH2 were upregulated in infiltrating compared to core neoplastic cells in glioblastoma patients (Darmanis et al., 2017). tweety 2 differential expression assays also demonstrated cell and tissue type-dependent expression—RNA microarray of mouse brain glioma-associated macrophages revealed ttyh2 upregulation while RNA-Seq of human glioma biopsies showed TTYH2 downregulation (Huang et al., 2014; Papatheodorou et al., 2020). However, TTYH3 consistently demonstrated upregulation in glioma tissues (Papatheodorou et al., 2020).
TTYH1 is also implicated in other brain cancer types. In particular, RNA-Seq of embryonal tumors with multilayered rosettes demonstrated that C19MC (the chr19q13.41 miRNA cluster) amplification was due to a TTYH1-C19MC fusion (Archer and Pomeroy, 2014; Kleinman et al., 2014). In embryonal brain tumors associated with C19MC, this fusion dysregulated and increased expression of a DNMT3B isoform—a DNA methyltransferase that is found specifically in embryonic brains and is associated with significant overexpression in pediatric brain tumors (Sin-Chan and Huang, 2014; Gowher and Jeltsch, 2018). In medulloblastoma tumors, microarrays demonstrate consistent TTYH1 downregulation and one microarray demonstrated TTYH3 upregulation (Papatheodorou et al., 2020). Other microarrays demonstrate TTYH1 and TTYH2 downregulation in subependymal giant cell astrocytoma samples as well as TTYH2 downregulation and TTYH3 upregulation in atypical teratoid rhabdoid tumors (Papatheodorou et al., 2020). Interestingly, across gliomas and brain cancers, TTYH3 shows consistent upregulation in pathological cells and tissue. Overall, RNA-Seq and microarray experiments provide correlations between brain cancers and one or more tweety genes while functional gene knockdowns and microscopic image analyses suggest functional associations including tweety 1’s facilitation of glioma invasiveness through tumor microtube-associated mechanisms, TTYH1-C19MC’s role in embryonal brain tumors, and TTYH3-BRAF’s role in glioma.
TTYH2 has been studied in association with colon cancer through gene expression, functional gene knockdowns, and binding assays. RT-PCR found TTYH2 upregulation in colon cancer cell lines (Caco-2, LoVo, and DLD-1) and human colon cancer tissue, although RNA-Seq of human blood platelets from colorectal cancer patients showed TTYH2 downregulation (Toiyama et al., 2007; Papatheodorou et al., 2020). Similar to gliomas, differential TTYH2 expression patterns between pathological and normal cells and tissue are seemingly cell and tissue type-dependent in colon cancer. Additionally, siRNA knockdown of TTYH2 in colon cancer-derived cell lines Caco-2 and DLD-1 showed increased cell aggregation and impaired cell proliferation, suggesting a role of TTYH2 in tumor growth and metastasis (Toiyama et al., 2007). The studies attempting to characterize TTYH2 in colon cancer consistently suggest TTYH2 facilitates tumor growth and/or metastasis.
In contrast with the functional studies conducted on TTYH2 in colon cancer, TTYH1 and TTYH3 have only been studied in colon cancer through gene expression assays. RNA-Seq of human blood platelets from colorectal cancer patients demonstrated TTYH1 downregulation (Papatheodorou et al., 2020). TTYH3 demonstrated upregulation in association with colon cancer, consistent with the TTYH3 upregulation pattern in brain cancers (Rhodes et al., 2004; Papatheodorou et al., 2020).
In addition to central nervous system-associated cancers and colon cancers, tweety is implicated in other cancers including breast, clear cell sarcoma, gastric, histiocytic sarcoma, kidney, osteosarcoma, and thyroid cancers. Expression from immunohistochemistry images and clinical outcome data in patients with gastric cancer analyzed using various databases (e.g., Human Protein Atlas, Oncomine, UALCAN, and Gene Expression Omnibus) revealed upregulated TTYH3 in gastric cancer compared to normal tissue (Rhodes et al., 2004; Saha et al., 20197). Furthermore, Kaplan-Meier analysis (modeling patient survival probability over time) demonstrated a correlation between TTYH3 upregulation and decreased patient survival (Saha et al., 2019). These findings suggest TTYH3 facilitates gastric cancer pathology.
Although Darweesh et al. (2014) could not validate this pattern, overall, TTYH1 downregulation in breast cancer-associated cells and tissue is consistent across RNA-Seq and microarray experiments (Rhodes et al., 2004; Darweesh et al., 2014; Papatheodorou et al., 2020 ). A Kaplan-Meier analysis also demonstrated a positive correlation between TTYH1 expression and life expectancy in triple-negative breast cancer patients, conferring TTYH1 as a potential disease biomarker and suggesting that TTYH1 may hinder breast cancer progression (Zhong et al., 2020). In contrast with TTYH1, TTYH2 and TTYH3 have only been studied in breast cancer through RNA-Seq and microarray analyses with TTYH2 showing downregulation and TTYH3 showing upregulation in breast cancer-associated cells and tissues (Papatheodorou et al., 2020). The upregulation of TTYH3 in breast cancer-associated cells is consistent with the upregulation reported in brain and colon cancers.
In addition to breast cancer, TTYH2’s role has been investigated in osteosarcoma where RNA-Seq showed TTYH2 upregulation in mouse osteosarcoma samples (Papatheodorou et al., 2020). Additionally, an siRNA knockdown of TTYH2 performed on osteosarcoma cell line U2OS (which showed high TTYH2 expression) attempted to identify possible associations between TTYH2 expression and cancerous properties of U2OS cells (Moon et al., 2019). This siRNA knockdown resulted in impaired U2OS invasion and migration but not cell proliferation (Moon et al., 2019). It also resulted in downregulation of transcription factors SLUG and ZEB1, which regulate epithelial-to-mesenchymal transition (EMT), a phenomenon characteristic of many osteosarcoma-associated signaling pathways (Moon et al., 2019). In contrast with TTYH2, TTYH1 and TTYH3 have only been studied in osteosarcoma through microarray and RNA-Seq experiments showing TTYH1 downregulation and TTYH3 upregulation (Papatheodorou et al., 2020). Again, TTYH3 upregulation in osteosarcoma tissues and cells is consistent with TTYH3 upregulation patterns seen in other cancers. In clear cell sarcomas, RNA-Seq demonstrated ttyh1, ttyh2, and ttyh3 upregulation suggesting tweety may encourage clear cell sarcoma pathology (Papatheodorou et al., 2020). An intra-chromosomal fusion between exon 12 of TTYH3 and exon 8 of BRAF on chromosome 7 was also identified in a case of histiocytic sarcoma without any other known cancer driver mutation (Egan et al., 2020).
In kidney cancers, DD-PCR and RT-PCR of human renal cell carcinoma samples demonstrated TTYH2 upregulation (Rae et al., 2001). Additionally, microarrays demonstrated ttyh1 upregulation, ttyh2 downregulation, and ttyh3 upregulation in mouse secondary tumors (Papatheodorou et al., 2020). Again, ttyh3 upregulation in kidney tumor cells is consistent with ttyh3 expression trends in other cancers (Papatheodorou et al., 2020). Additionally, a fusion transcript of TTYH3 and BRAF has been identified in kinase fusion-positive thyroid carcinomas as one of many kinase fusions (Chu et al., 2020).
Finally, microarray and RNA-Seq have demonstrated abnormal tweety gene expression in various other cancers. For example, tweety 1 has shown upregulation in lung cancer (in mouse lung epithelial cells) and tongue squamous cell carcinoma as well as downregulation in esophageal squamous cell carcinoma, esophageal adenocarcinoma, hepatobiliary carcinoma (in human blood platelets), lung cancer (in human blood platelets), pancreatic cancer, and skin squamous cell carcinoma (Papatheodorou et al., 2020). Overall, tweety 1 is more commonly downregulated than upregulated in association with cancer, but this is not consistent among all tissues.
Microarray and RNA-Seq analyses have also demonstrated tweety 2 upregulation in esophageal squamous cell carcinoma, esophageal adenocarcinoma, lung cancer (in mouse lung cells), lymphoma, and skin squamous cell carcinoma as well as downregulation in lung cancer (in human blood platelets), ovarian cancer, and pancreatic adenocarcinoma (Rhodes et al., 2004; Papatheodorou et al., 2020). Overall, tweety 2 seems to be more commonly downregulated than upregulated in association with cancer, although not consistently in all tissues.
Through microarray and RNA-Seq analyses, tweety 3 has demonstrated upregulation in esophageal squamous cell carcinoma, esophageal adenocarcinoma, hepatobiliary carcinoma (in human blood platelets), liver cancer, lung cancer, melanoma, pancreatic cancer, synovial sarcoma, squamous cell carcinoma, and uterine leiomyosarcoma as well as downregulation in lung cancer (in human blood platelets) and myxosarcoma (Rhodes et al., 2004; Papatheodorou et al., 2020). Overall, this suggests tweety 3 is more commonly upregulated than downregulated in association with cancer, consistent with tweety 3 upregulation exhibited in previously described cancers such as glioma, brain cancers, and colon cancer.
Overall, expression and functional studies demonstrate associations between tweety genes and cancers and suggest that they may serve as promising biomarker candidates for disease diagnosis or prognosis. As cancers are characterized by uncontrolled proliferation of pathological cells, the pattern of tweety 1 downregulation, tweety 2 upregulation, and tweety 3 upregulation may suggest differential functions among the tweety genes with tweety 1 regulating cell proliferation or impeding cancerous phenotypes and tweety 2 and tweety 3 facilitating cell proliferation or promoting cancerous phenotypes (Supplementary Table 4).
In addition to cancers, tweety has also been implicated in neurological disorders including status epilepticus, Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (Supplementary Table 5). Both microarrays and immunohistochemistry have demonstrated ttyh1 overexpression in rat neurons during epileptogenesis and 2 weeks after epileptogenic stimulus (Lukasiuk et al., 2003; Stefaniuk and Lukasiuk, 2010; Stefaniuk et al., 2010). Additionally, in vivo immunostaining revealed Ttyh1 immunoreactivity in reactive astrocytes after amygdala-induced status epilepticus in mice, further suggesting ttyh1’s role in epileptogenesis (Wiernasz et al., 2014).
Microarrays also demonstrate brain location-dependent TTYH1 expression patterns in Alzheimer’s disease patients as the hippocampus and entorhinal cortex samples showed TTYH1 downregulation while superior frontal gyrus and middle temporal gyrus samples revealed TTYH1 (and TTYH2) upregulation (Xu et al., 2006; Liang et al., 2007; Papatheodorou et al., 2020). Studies have not been conducted to demonstrate a functional link between tweety and Alzheimer’s disease; therefore, functional mutagenesis studies should be conducted to elucidate the functional relevance of tweety in Alzheimer’s disease.
Additionally, mass-spectrometry demonstrated TTYH1 downregulation in the locus coeruleus of Parkinson’s disease patients when compared to controls (van Dijk et al., 2012). TTYH1, along with various other proteins associated with calcium homeostasis, were identified as having differential expression in Parkinson’s disease patients, suggesting a role of calcium homeostasis (and TTYH1’s association with it) in Parkinson’s disease pathogenesis (van Dijk et al., 2012).
TTYH1 also showed differential expression in MN1 neurons expressing a TDP-43A315T mutant when compared to MN1 neurons expressing human TDP-43 WT protein, suggesting TTYH1 as a potential target of translation for the hTDP-43A315T protein. Dysfunctional regulation of RNA-binding protein TDP is implicated in amyotrophic lateral sclerosis development, and the TDP-43A315T mutation is found in a subset of amyotrophic lateral sclerosis patients, suggesting a functional association between TTYH1 and amyotrophic lateral sclerosis through mechanisms associated with TDP-43A315T (Neelagandan et al., 2019).
Additional RNA-Seq and microarray experiments have demonstrated TTYH3 downregulation in ataxia-telangiectasia, TTYH2 upregulation in cerebral small vessel disease, and TTYH2 downregulation in pick disease and progressive supranuclear palsy (Papatheodorou et al., 2020). Because of limited differential expression data, the TTYH3 downregulation pattern present in association with cancers is not easily identifiable (if present) for neurological disorders.
Although expression patterns of tweety genes under pathological conditions are more frequently studied in cancers and neurological disorders, their roles in other disorders such as hypertension, cystic fibrosis, and Turner syndrome have also been explored (Supplementary Table 6). For example, genome-wide association studies conducted on African Americans show an SNP in the intronic region of TTYH2 is associated with systolic blood pressure levels and may impact hypertension incidence (Taylor et al., 2016). Additionally, TTYH3’s involvement in cystic fibrosis is suggested by strong downregulation of mTTYH3 in intestines of cftrtumor microtubule1Cam mice, although mTTYH3 is not downregulated in cftrTgH(neoim)Hgu mice with a weaker cystic fibrosis phenotype (Braun et al., 2010). Furthermore, high-resolution array-based comparative genomic hybridization performed on 17 Turner Syndrome patients found TTYH3 (in addition to AMZ1 and GNA12) as copy number variations at 7p22.3 (Li et al., 2019). Finally, genomic microarrays identified an overlapping 0.47 Mb microdeletion containing TTYH3 at 7p22.3p22.2 in five patients exhibiting distinct facial features (e.g., broad nasal root and prominent forehead) and varying delays in development (Yu et al., 2017).
RNA-Seq and microarray experiments also demonstrate abnormal tweety expression in myriad other diseases. When comparing pathological to normal tissues and cells, TTYH1 was upregulated in human atopic dermatitis skin samples and downregulated in association with actinic keratosis, chronic pancreatitis, down syndrome, familial hemophagocytic lymphohistiocytosis, Klinefelter syndrome, nevus sebaceous of Jadassohn, and psoriasis (Papatheodorou et al., 2020).
Microarray and RNA-Seq analyses also demonstrate TTYH2 upregulation in actinic keratosis, non-dysplastic Barrett’s esophagus, juvenile idiopathic arthritis, metopic craniosynostosis, nevus sebaceous of Jadassohn, post-traumatic osteoarthritis, sagittal craniosynostosis, non-primary Sjogren syndrome, autism, and Streptococcus pneumoniae infection paired with chronic obstructive pulmonary disorder as well as downregulation in Klinefelter’s syndrome, Lyme disease, sepsis, and Streptococcus pneumoniae infection (Papatheodorou et al., 2020). Finally, microarray and RNA-Seq studies show TTYH3 upregulation in actinic keratosis, Barrett’s esophagus, Crohn’s disease, nevus sebaceous of Jadassohn, pulmonary sarcoidosis, Streptococcus pneumoniae infection paired with chronic obstructive pulmonary disorder, and ulcerative colitis as well as downregulation in influenza and Streptococcus pneumoniae infection (Rhodes et al., 2004; Papatheodorou et al., 2020).
Overall, the disease-specific tweety expression patterns confer tweety as promising biomarker candidates for disease diagnosis or prognosis. Additionally, similar to the differential tweety expression patterns identified in cancers, the patterns of tweety 1 downregulation, tweety 2 upregulation, and tweety 3 upregulation seem present among these diverse diseases and disorders. This may indicate tweety genes serve a fundamental function in maintaining physiological normal biological conditions. As more functional gene knockout studies, mutagenesis experiments, and binding assays elucidate the functions of tweety genes under physiological conditions, their role in pathological conditions may also become clearer.
Since the discovery of the first tweety gene family member several decades ago, ongoing research has deepened our understanding of not only the structure, function, phylogenetic distribution, and expression of this gene family, but also its importance in normal development and physiology as well as pathological conditions. However, despite significant progress, many questions about this relatively understudied family of genes remain. This review of the current literature points to several particularly important avenues for further research on the tweety gene family: comprehensive and phylogenetically diverse analyses of expression patterns, additional perturbation experiments to assess function, protein structure-function analyses using physical methods, identification of the position of tweety genes in signaling and gene networks, and the investigation of how tweety gene expression is regulated in both wild type and pathological conditions.
Evident from published work, there are relatively few comprehensive in situ analyses of tweety gene expression patterns during embryogenesis or in tissues of the mature organism for virtually any species. More detailed histological analysis to document cell and tissue-type specificity is required to complement functional studies. Based on discrepancies between reported mRNA and protein expression in some instances (Rae et al., 2001; Kim et al., 2014; Brown et al., 2015; Stelzer et al., 2016; Schmidt et al., 2018; Papatheodorou et al., 2020; Samaras et al., 2020), expression analyses at both these levels are essential to document translational control. In particular, ttyh2 and ttyh3 have received little attention. In addition to standard ISH and immunohistochemistry, employing techniques such as CLARITY, multiplexed immunohistochemistry techniques such as Immuno-SABER, or single-molecule fluorescent in situ hybridization (smFISH) will provide enhanced quantitative spatial specificity and co-localization with putative regulators, an approach productively employed to examine cardiac sodium and potassium currents (Raj et al., 2008; Chung et al., 2013; Saka et al., 2019).
The use of these techniques is especially important in pathological tissues to determine the cell types in which tweety genes are differentially and/or inappropriately expressed. Given the demonstrated interactions of ttyh1 with the Notch signaling pathway to maintain stem cell-like properties, the interactions between ttyh1 and the Notch signaling pathway in neural cancers cancer stem cells could uncover how ttyh1 may regulate maintenance and metastasis of neural cancers (Vasiliu et al., 2015; Jung et al., 2017; Kim et al., 2018; Wu et al., 2019). Analysis of subcellular localization, particularly the dynamics of localization using in vivo imaging, could further shed light on the role of Tweety proteins in normal and pathological conditions.
While the detailed spatial and temporal investigation in a wide array of model and non-model organisms provides essential baseline information for functional and evolutionary analysis, a full complement of knockout and overexpression experiments—at the very least in select model organisms—is essential for determining the function of the tweety genes. These experiments are especially important given that even in mice, there is still conflicting evidence as to whether ttyh1 is embryonic lethal (Kumada et al., 2010; Wu et al., 2019). Particularly low-hanging fruit for further investigation is the set of mutants already available for all three tweety genes in zebrafish (Ruzicka et al., 2019). Combining comparative genomics with CRISPR-based perturbation studies in a larger array of non-model organisms will uncover the level of functional conservation among tweety orthologs. A particularly intriguing avenue for future research is the role of ttyh2 and ttyh3 in immune cell activation, a function that may be conserved in non-mammalian vertebrates (Chen et al., 2010; Papatheodorou et al., 2020).
With respect to structure-function analysis, questions remain regarding the basic structure number of Tweety protein transmembrane domains. While bioinformatics techniques have been the primary source for Tweety protein structure prediction and analysis, additional technologies such as NMR, X-ray crystallography, and cryo-electron microscopy could corroborate current predicted structures, particularly when combined with targeted point mutation analysis to assess functional outcomes of mutations. For example, studies employing X-ray crystallography with cryo-EM have been extremely useful in determining structure-function relationships for other ion channels (Eichel et al., 2019). While evidence refutes the role of tweety genes as maxi anion channels (Suzuki and Mizuno, 2004; Suzuki, 2006; Sabirov and Okada, 2009; Han et al., 2019), questions still remain about the respective roles of the three Tweety proteins in generating volume regulated currents (VSOR and VRAC), particularly regarding whether they can activate VSOR current in a swelling-independent manner via reactive oxygen species or GPCR signaling. Integrating structural analysis with RNAi or CRISPR knockouts and electrophysiology data will further elucidate their biochemical function. The past decade has seen an exponential increase in ‘omics’ data for model and non-model organisms. Employing these data for comparative phylogenetic analysis will also provide a deeper understanding of structure-function relationships of the tweety gene family from an evolutionary perspective.
The position of the tweety family in gene and signaling networks remains an open but important question. ttyh1 has already been shown to be involved in the Notch signaling pathway; however, whether ttyh1 acts upstream or downstream (or both) of Notch ICD activation is unclear given that ICD overexpression upregulates ttyh1 expression and that ttyh1 activates ICD (Vasiliu et al., 2015; Kim et al., 2018; Wu et al., 2019). In a Drosophila study attempting to find novel regulators of the Notch signaling pathway through an RNAi screen, tty was not identified; this, however, does not preclude it being a downstream target of Notch signaling, or its association with Notch being a vertebrate-specific phenomenon (Mummery-Widmer et al., 2009). When, and precisely how, a tweety gene began interacting with the Notch pathway in evolutionary history is not fully understood. Nor is it known whether ttyh2 and ttyh3 also play a role in the Notch signaling pathway. Searching for differential expression of tweety genes in existing RNA-Seq datasets in which various genes were perturbed combined with performing RNA-Seq on embryos, tissues, or cell lines or performing RNA-Seq where tweety genes were knocked out or overexpressed will allow investigators to determine if tweety genes are associated with other signaling pathways and will provide the raw material for positioning tweety homologs in gene networks.
Finally, how tweety gene expression is regulated remains largely unknown. At the transcriptional level, the regulatory elements and associated binding proteins that regulate transcription have not been empirically identified in any organism, despite intriguing data from oncogenic gene fusions suggesting that tweety promoters are implicated in driving tumor development. Comparative genomics of putative upstream and intronic regulatory regions and systematic mining of databases such as ENCODE combined with functional analysis using transgenic approaches can be employed to identify regions that govern the appropriate spatial and temporal expression of the tweety genes in phylogenetically diverse organisms (Andersson and Sandelin, 2020). On the RNA processing level, the tweety genes, like many ion channels, have many exons (~15) and are known to have between two and five alternatively spliced variants; however, whether these variants are differentially expressed or functionally relevant is not known (Matthews et al., 2007; Stelzer et al., 2016). Finally, at the post-translational level, while all three Tweety proteins in humans undergo post-translational processing (Stelzer et al., 2016), comprehensive studies of interacting proteins are lacking. Pull-down assays and two-hybrid screens in combination with the bioinformatic information on putative protein interactors will allow investigators to flesh out this important level of regulation.
Overall, ongoing research has opened promising avenues of investigation for this intriguing but understudied gene family. More comprehensive characterization of expression, structure-function relationships, regulatory mechanisms, and perturbation experiments will elucidate the role of tweety genes in development and disease. Furthermore, the observed differential expression patterns between the three tweety genes demonstrated across diseases can inform functional assays that attempt to characterize potentially different biological roles possessed by each particular tweety gene. Finally, as more differential tweety expression data associated with diseases become increasingly available, evaluating the tweety genes for their capacity to act as biomarkers or even therapeutic targets becomes an increasingly promising avenue for future investigation.
Conceptualization: MS. Review of literature: AS, MS, MY, RN, and SM. Writing: AS, MY, RN, and SM. Review and editing: AS, MS, MY, RN, and SM. Creation of Tables and Figures: AS, MY, RN, and SM. Supervision and Funding: MS. All authors contributed to the article and approved the submitted version.
This work was supported by NIH grant 1R15HD096415-01 and NSF IOS 1257895 to MS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge the following databases for the important information they provided for this review: Allen Brain Atlas, LifeMap Discovery, Echinobase, ENSEMBL, Expression Atlas, FlyBase, Fly-FISH, NCBI, NCBI AceView, NCBI Gene, GeneCards, GTEx Portal, Human Proteome Map, MGI, OncoPPi, Oncomine, OrthoDB, ProteomicsDB, RGD, SwissProt, UniProt, WormBase, and Xenbase.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2021.672511/full#supplementary-material.
Allen Institute for Brain Science. (2008a). Allen Developing Mouse Brain Atlas. Available online at: developingmouse.brain-map.org
Allen Institute for Brain Science. (2008b). Allen Spinal Cord Atlas. Available online at: mousespinal.brain-map.org.
Abramova, N., Charniga, C., Goderie, S. K., and Temple, S. (2005). Stage-specific changes in gene expression in acutely isolated mouse CNS progenitor cells. Dev. Biol. 283, 269–281. doi: 10.1016/j.ydbio.2005.03.040
Al-Jumaily, M., Kozlenkov, A., Mechaly, I., Fichard, A., Matha, V., Scamps, F., et al. (2007). Expression of three distinct families of calcium-activated chloride channel genes in the mouse dorsal root ganglion. Neurosci. Bull. 23, 293–299. doi: 10.1007/s12264-007-0044-8
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andersson, R., and Sandelin, A. (2020). Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 21, 71–87. doi: 10.1038/s41576-019-0173-8
Archer, T. C., and Pomeroy, S. L. (2014). A developmental program drives aggressive embryonal brain tumors. Nat. Genet. 46, 2–3. doi: 10.1038/ng.2857
Bae, Y., Kim, A., Cho, C. H., Kim, D., Jung, H. G., Kim, S. S., et al. (2019). TTYH1 and TTYH2 serve as LRRC8A-independent volume-regulated anion channels in cancer cells. Cells 8:562. doi: 10.3390/cells8060562
Barbosa-Morais, N. L., Irimia, M., Pan, Q., Xiong, H. Y., Gueroussov, S., Lee, L. J., et al. (2012). The evolutionary landscape of alternative splicing in vertebrate species. Science 338, 1587–1593. doi: 10.1126/science.1230612
Best, M. G., Sol, N., Kooi, I., Tannous, J., Westerman, B. A., Rustenburg, F., et al. (2015). RNA-seq of tumor-educated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell 28, 666–676. doi: 10.1016/j.ccell.2015.09.018
Birks, D. K., Donson, A. M., Patel, P. R., Sufit, A., Algar, E. M., Dunham, C., et al. (2013). Pediatric rhabdoid tumors of kidney and brain show many differences in gene expression but share dysregulation of cell cycle and epigenetic effector genes. Pediatr. Blood Cancer 60, 1095–1102. doi: 10.1002/pbc.24481
Bisson, N., James, D. A., Ivosev, G., Tate, S. A., Bonner, R., Taylor, L., et al. (2011). Selected reaction monitoring mass spectrometry reveals the dynamics of signaling through the GRB2 adaptor. Nat. Biotechnol. 29, 653–658. doi: 10.1038/nbt.1905
Blackshaw, S., Harpavat, S., Trimarchi, J., Cai, L., Huang, H., Kuo, W. P., et al. (2004). Genomic analysis of mouse retinal development. PLoS Biol. 2:E247. doi: 10.1371/journal.pbio.0020247
Bourbon, H.-M., Gonzy-Treboul, G., Peronnet, F., Alin, M.-F., Ardourel, C., Benassayag, C., et al. (2002). A P-insertion screen identifying novel X-linked essential genes in Drosophila. Mech. Dev. 110, 71–83. doi: 10.1016/s0925-4773(01)00566-4
Braun, J., Mundhenk, L., Range, F., and Gruber, A. D. (2010). Quantitative expression analyses of candidates for alternative anion conductance in cystic fibrosis mouse models. J. Cyst. Fibros. 9, 351–364. doi: 10.1016/j.jcf.2010.06.003
Brawand, D., Soumillon, M., Necsulea, A., Julien, P., Csárdi, G., Harrigan, P., et al. (2011). The evolution of gene expression levels in mammalian organs. Nature 478, 343–348. doi: 10.1038/nature10532
Bronner, I. F., Bochdanovits, Z., Rizzu, P., Kamphorst, W., Ravid, R., van Swieten, J. C., et al. (2009). Comprehensive mRNA expression profiling distinguishes tauopathies and identifies shared molecular pathways. PLoS One 4:e6826. doi: 10.1371/journal.pone.0006826
Brown, J. B., Boley, N., Eisman, R., May, G. E., Stoiber, M. H., Duff, M. O., et al. (2014). Diversity and dynamics of the Drosophila transcriptome. Nature 512, 393–399. doi: 10.1038/nature12962
Brown, G. R., Hem, V., Katz, K. S., Ovetsky, M., Wallin, C., Ermolaeva, O., et al. (2015). Gene: a gene-centered information resource at NCBI. Nucleic Acids Res. 43, D36–D42. doi: 10.1093/nar/gku1055
Brune, V., Tiacci, E., Pfeil, I., Döring, C., Eckerle, S., van Noesel, C. J., et al. (2008). Origin and pathogenesis of nodular lymphocyte-predominant Hodgkin lymphoma as revealed by global gene expression analysis. J. Exp. Med. 205, 2251–2268. doi: 10.1084/jem.20080809
Bult, C. J., Blake, J. A., Smith, C. L., Kadin, J. A., Richardson, J. E., and Mouse Genome Database Group. (2019). Mouse genome database (MGD) 2019. Nucleic Acids Res. 47, D801–D806. doi: 10.1093/nar/gky1056
Burki, F., Roger, A. J., Brown, M. W., and Simpson, A. G. B. (2020). The new tree of eukaryotes. Trends Ecol. Evol. 35, 43–55. doi: 10.1016/j.tree.2019.08.008
Cameron, R. A., Samanta, M., Yuan, A., He, D., and Davidson, E. (2009). SpBase: the sea urchin genome database and web site. Nucleic Acids Res. 37, D750–D754. doi: 10.1093/nar/gkn887
Campbell, H. D., Kamei, M., Claudianos, C., Woollatt, E., Sutherland, G. R., Suzuki, Y., et al. (2000). Human and mouse homologues of the Drosophila melanogaster tweety (tty) gene: a novel gene family encoding predicted transmembrane proteins. Genomics 68, 89–92. doi: 10.1006/geno.2000.6259
Campbell, H. D., Schimansky, T., Claudianos, C., Ozsarac, N., Kasprzak, A. B., Cotsell, J. N., et al. (1993). The Drosophila melanogaster flightless-I gene involved in gastrulation and muscle degeneration encodes gelsolin-like and leucine-rich repeat domains and is conserved in Caenorhabditis elegans and humans. Proc. Natl. Acad. Sci. U S A 90, 11386–11390. doi: 10.1073/pnas.90.23.11386
Cary, G. A., Cameron, R. A., and Hinman, V. F. (2018). EchinoBase: tools for echinoderm genome analyses. Methods Mol. Biol. 1757, 349–369. doi: 10.1007/978-1-4939-7737-6_12
Chang, L., and Karin, M. (2001). Mammalian MAP kinase signalling cascades. Nature 410, 37–40. doi: 10.1038/35065000
Chang, W. L. W., Coro, E. S., Rau, F. C., Xiao, Y., Erle, D. J., and Baumgarth, N. (2007). Influenza virus infection causes global respiratory tract B cell response modulation via innate immune signals. J. Immunol. 178, 1457–1467. doi: 10.4049/jimmunol.178.3.1457
Chen, X., Leung, S. Y., Yuen, S. T., Chu, K.-M., Ji, J., Li, R., et al. (2003). Variation in gene expression patterns in human gastric cancers. Mol. Biol. Cell 14, 3208–3215. doi: 10.1091/mbc.e02-12-0833
Chen, Y., Yao, Y., Sumi, Y., Li, A., Kim To, U., Abdallah, E., et al. (2010). Purinergic signaling: a fundamental mechanism in neutrophil activation. Sci. Signal. 3:ra45. doi: 10.1126/scisignal.2000549
Cho, J. Y., Lim, J. Y., Cheong, J. H., Park, Y.-Y., Yoon, S.-L., Kim, S. M., et al. (2011). Gene expression signature-based prognostic risk score in gastric cancer. Clin. Cancer Res. 17, 1850–1857. doi: 10.1158/1078-0432.CCR-10-2180
Choi, H., Sheng, J., Gao, D., Li, F., Durrans, A., Ryu, S., et al. (2015). Transcriptome analysis of individual stromal cell populations identifies stroma-tumor crosstalk in mouse lung cancer model. Cell Rep. 10, 1187–1201. doi: 10.1016/j.celrep.2015.01.040
Chu, Y.-H., Wirth, L. J., Farahani, A. A., Nosé, V., Faquin, W. C., Dias-Santagata, D., et al. (2020). Clinicopathologic features of kinase fusion-related thyroid carcinomas: an integrative analysis with molecular characterization. Mod. Pathol. 33, 2458–2472. doi: 10.1038/s41379-020-0638-5
Chung, K., Wallace, J., Kim, S.-Y., Kalyanasundaram, S., Andalman, A. S., Davidson, T. J., et al. (2013). Structural and molecular interrogation of intact biological systems. Nature 497, 332–337. doi: 10.1038/nature12107
Claro da Silva, T., Hiller, C., Gai, Z., and Kullak-Ublick, G. A. (2016). Vitamin D3 transactivates the zinc and manganese transporter SLC30A10 via the Vitamin D receptor. J. Steroid Biochem. Mol. Biol. 163, 77–87. doi: 10.1016/j.jsbmb.2016.04.006
Coleman, S. J., Zeng, Z., Hestand, M. S., Liu, J., and Macleod, J. N. (2013). Analysis of unannotated equine transcripts identified by mRNA sequencing. PLoS One 8:e70125. doi: 10.1371/journal.pone.0070125
Compagno, M., Lim, W. K., Grunn, A., Nandula, S. V., Brahmachary, M., Shen, Q., et al. (2009). Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B- cell lymphoma. Nature 459, 717–721. doi: 10.1038/nature07968
Crnogorac-Jurcevic, T., Chelala, C., Barry, S., Harada, T., Bhakta, V., Lattimore, S., et al. (2013). Molecular analysis of precursor lesions in familial pancreatic cancer. PLoS One 8:e54830. doi: 10.1371/journal.pone.0054830
Curtis, C., Shah, S. P., Chin, S.-F., Turashvili, G., Rueda, O. M., Dunning, M. J., et al. (2012). The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352. doi: 10.1038/nature10983
D’Errico, M., de Rinaldis, E., Blasi, M. F., Viti, V., Falchetti, M., Calcagnile, A., et al. (2009). Genome-wide expression profile of sporadic gastric cancers with microsatellite instability. Eur. J. Cancer 45, 461–469. doi: 10.1016/j.ejca.2008.10.032
Darmanis, S., Sloan, S. A., Croote, D., Mignardi, M., Chernikova, S., Samghababi, P., et al. (2017). Single-cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 21, 1399–1410. doi: 10.1016/j.celrep.2017.10.030
Darweesh, A. S., Louka, M. L., Hana, M., Rashad, S., El-Shinawi, M., Sharaf-Eldin, A., et al. (2014). Validation of analytical breast cancer microarray analysis in medical laboratory. Med. Oncol. 31:201. doi: 10.1007/s12032-014-0201-7
Diez-Roux, G., Banfi, S., Sultan, M., Geffers, L., Anand, S., Rozado, D., et al. (2011). A high- resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. 9:e1000582. doi: 10.1371/journal.pbio.1000582
Donoghue, P. (2017). Evolution: divining the nature of the ancestral vertebrate. Curr. Biol. 27, R277–R279. doi: 10.1016/j.cub.2017.02.029
Duran, C., Thompson, C. H., Xiao, Q., and Hartzell, H. C. (2010). Chloride channels: often enigmatic, rarely predictable. Annu. Rev. Physiol. 72, 95–121. doi: 10.1146/annurev-physiol-021909-135811
Edgar, R., Mazor, Y., Rinon, A., Blumenthal, J., Golan, Y., Buzhor, E., et al. (2013). LifeMap DiscoveryTM: the embryonic development, stem cells, and regenerative medicine research portal. PLoS One 8:e66629. doi: 10.1371/journal.pone.0066629
Edwards, A. C., Zwarts, L., Yamamoto, A., Callaerts, P., and Mackay, T. F. (2009). Mutations in many genes affect aggressive behavior in Drosophila melanogaster. BMC Biol. 7:29. doi: 10.1186/1741-7007-7-29
Egan, C., Nicolae, A., Lack, J., Chung, H.-J., Skarshaug, S., Pham, T. A., et al. (2020). Genomic profiling of primary histiocytic sarcoma reveals two molecular subgroups. Haematologica 105, 951–960. doi: 10.3324/haematol.2019.230375
Eichel, C. A., Ríos-Pérez, E. B., Liu, F., Jameson, M. B., Jones, D. K., Knickelbine, J. J., et al. (2019). A microtranslatome coordinately regulates sodium and potassium currents in the human heart. eLife 8:e52654. doi: 10.7554/eLife.52654
Eswaran, J., Cyanam, D., Mudvari, P., Reddy, S. D., Pakala, S. B., Nair, S. S., et al. (2012). Transcriptomic landscape of breast cancers through mRNA sequencing. Sci. Rep. 2:264. doi: 10.1038/srep00264
Fagerberg, L., Hallström, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J., et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteomics 13, 397–406. doi: 10.1074/mcp.M113.035600
Fenner, B. J., Scannell, M., and Prehn, J. H. (2010). Expanding the substantial interactome of NEMO using protein microarrays. PLoS One 5:e8799. doi: 10.1371/journal.pone.0008799
Funke, B., Lasitschka, F., Roth, W., Penzel, R., Meuer, S., Saile, M., et al. (2011). Selective downregulation of retinoic acid-inducible gene I within the intestinal epithelial compartment in Crohn’s disease. Inflamm. Bowel Dis. 17, 1943–1954. doi: 10.1002/ibd.21572
Gaedcke, J., Grade, M., Jung, K., Camps, J., Jo, P., Emons, G., et al. (2010). Mutated KRAS results in overexpression of DUSP4, a MAP-kinase phosphatase and SMYD3, a histone methyltransferase, in rectal carcinomas. Genes Chromosomes Cancer 49, 1024–1034. doi: 10.1002/gcc.20811
Galamb, O., Györffy, B., Sipos, F., Spisák, S., Németh, A. M., Miheller, P., et al. (2008). Inflammation, adenoma and cancer: objective classification of colon biopsy specimens with gene expression signature. Dis. Markers 25, 1–16. doi: 10.1155/2008/586721
Gill, B. J., Pisapia, D. J., Malone, H. R., Goldstein, H., Lei, L., Sonabend, A., et al. (2014). MRI- localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc. Natl. Acad. Sci. U S A 111, 12550–12555. doi: 10.1073/pnas.1405839111
Gittler, J. K., Shemer, A., Suárez-Fariñas, M., Fuentes-Duculan, J., Gulewicz, K. J., Wang, C. Q., et al. (2012). Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 130, 1344–1354. doi: 10.1016/j.jaci.2012.07.012
Gowher, H., and Jeltsch, A. (2018). Mammalian DNA methyltransferases: new discoveries and open questions. Biochem. Soc. Trans. 46, 1191–1202. doi: 10.1042/BST20170574
Graveley, B. R., Brooks, A. N., Carlson, J. W., Duff, M. O., Landolin, J. M., Yang, L., et al. (2011). The developmental transcriptome of Drosophila melanogaster. Nature 471, 473–479. doi: 10.1038/nature09715
Griesinger, A. M., Birks, D. K., Donson, A. M., Amani, V., Hoffman, L. M., Waziri, A., et al. (2013). Characterization of distinct immunophenotypes across pediatric brain tumor types. J. Immunol. 191, 4880–4888. doi: 10.4049/jimmunol.1301966
Griesinger, A. M., Josephson, R. J., Donson, A. M., Mulcahy Levy, J. M., Amani, V., Birks, D. K., et al. (2015). Interleukin-6/STAT3 pathway signaling drives an inflammatory phenotype in group a ependymoma. Cancer Immunol. Res. 3, 1165–1174. doi: 10.1158/2326-6066.CIR-15-0061
GTex Consortium. (2015). Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. doi: 10.1126/science.1262110
Guo, R., Pan, F., Tian, Y., Li, H., Li, S., and Cao, C. (2016). Down-regulation of ClC-3 expression reduces epidermal stem cell migration by inhibiting volume-activated chloride currents. J. Membr. Biol. 249, 281–292. doi: 10.1007/s00232-015-9867-9
Haldar, M., Hancock, J. D., Coffin, C. M., Lessnick, S. L., and Capecchi, M. R. (2007). A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer Cell 11, 375–388. doi: 10.1016/j.ccr.2007.01.016
Halleran, A. D., Sehdev, M., Rabe, B. A., Huyck, R. W., Williams, C. C., and Saha, M. S. (2015). Characterization of tweety gene (ttyh1–3) expression in Xenopus laevis during embryonic development. Gene Expr. Patterns 17, 38–44. doi: 10.1016/j.gep.2014.12.002
Han, Y.-E., Kwon, J., Won, J., An, H., Jang, M. W., Woo, J., et al. (2019). Tweety-homolog (Ttyh) family encodes the pore-forming subunits of the swelling-dependent volume- regulated anion channel (VRACswell) in the brain. Exp. Neurobiol. 28, 183–215. doi: 10.5607/en.2019.28.2.183
Han, W.-J., Ma, S.-B., Wu, W.-B., Wang, F.-D., Cao, X.-L., Wang, D.-H., et al. (2021). Tweety- homolog 1 facilitates pain via enhancement of nociceptor excitability and spinal synaptic transmission. Neurosci. Bull. 37, 478–496. doi: 10.1007/s12264-020-00617-0
Hanley, M. P., Hahn, M. A., Li, A. X., Wu, X., Lin, J., Wang, J., et al. (2017). Genome-wide DNA methylation profiling reveals cancer-associated changes within early colonic neoplasia. Oncogene 36, 5035–5044. doi: 10.1038/onc.2017.130
Haqq, C., Nosrati, M., Sudilovsky, D., Crothers, J., Khodabakhsh, D., Pulliam, B. L., et al. (2005). The gene expression signatures of melanoma progression. Proc. Natl. Acad. Sci. U S A 102, 6092–6097. doi: 10.1073/pnas.0501564102
Harbison, S. T., Yamamoto, A. H., Fanara, J. J., Norga, K. K., and Mackay, T. F. (2004). Quantitative trait loci affecting starvation resistance in Drosophila melanogaster. Genetics 166, 1807–1823. doi: 10.1534/genetics.166.4.1807
Harris, T. W., Arnaboldi, V., Cain, S., Chan, J., Chen, W. J., Cho, J., et al. (2020). WormBase: a modern model organism information resource. Nucleic Acids Res. 48, D762–D767. doi: 10.1093/nar/gkz920
Hashimshony, T., Feder, M., Levin, M., Hall, B. K., and Yanai, I. (2015). Spatiotemporal transcriptomics reveals the evolutionary history of the endoderm germ layer. Nature 519, 219–222. doi: 10.1038/nature13996
He, Y., Hryciw, D. H., Carroll, M. L., Myers, S. A., Whitbread, A. K., Kumar, S., et al. (2008a). The ubiquitin-protein ligase Nedd4–2 differentially interacts with and regulates members of the Tweety family of chloride ion channels. J. Biol. Chem. 283, 24000–24010. doi: 10.1074/jbc.M803361200
He, Y., Ramsay, A. J., Hunt, M. L., Whitbread, A. K., Myers, S. A., and Hooper, J. D. (2008b). N-glycosylation analysis of the human Tweety family of putative chloride ion channels supports a penta-spanning membrane arrangement: impact of N-glycosylation on cellular processing of Tweety homologue 2 (TTYH2). Biochem. J. 412, 45–55. doi: 10.1042/BJ20071722
Hibaoui, Y., Grad, I., Letourneau, A., Sailani, M. R., Dahoun, S., Santoni, F. A., et al. (2014). Modelling and rescuing neurodevelopmental defect of Down syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. EMBO Mol. Med. 6, 259–277. doi: 10.1002/emmm.201302848
Hiraoka, N., Yamazaki-Itoh, R., Ino, Y., Mizuguchi, Y., Yamada, T., Hirohashi, S., et al. (2011). CXCL17 and ICAM2 are associated with a potential anti-tumor immune response in early intraepithelial stages of human pancreatic carcinogenesis. Gastroenterology 140, 310–321. doi: 10.1053/j.gastro.2010.10.009
Hori, A., Nishide, K., Yasukuni, Y., Haga, K., Kakuta, W., Ishikawa, Y., et al. (2019). Prominin- 1 modulates Rho/ROCK-mediated membrane morphology and calcium-dependent intracellular chloride flux. Sci. Rep. 9:15911. doi: 10.1038/s41598-019-52040-9
Horvath, S., Nazmul-Hossain, A. N., Pollard, R. P., Kroese, F. G., Vissink, A., Kallenberg, C. G., et al. (2012). Systems analysis of primary Sjögren’s syndrome pathogenesis in salivary glands identifies shared pathways in human and a mouse model. Arthritis Res. Ther. 14:R238. doi: 10.1186/ar4081
Howell, K. J., Kraiczy, J., Nayak, K. M., Gasparetto, M., Ross, A., Lee, C., et al. (2018). DNA methylation and transcription patterns in intestinal epithelial cells from pediatric patients with inflammatory bowel diseases differentiate disease subtypes and associate with outcome. Gastroenterology 154, 585–598. doi: 10.1053/j.gastro.2017.10.007
Huang, Y., Hoffman, C., Rajappa, P., Kim, J. H., Hu, W., Huse, J., et al. (2014). Oligodendrocyte progenitor cells promote neovascularization in glioma by disrupting the blood-brain barrier. Cancer Res. 74, 1011–1021. doi: 10.1158/0008-5472.CAN-13-1072
Huang, Y., Kim, J. K., Do, D. V., Lee, C., Penfold, C. A., Zylicz, J. J., et al. (2017). Stella modulates transcriptional and endogenous retrovirus programs during maternal-to- zygotic transition. eLife 6:e22345. doi: 10.7554/eLife.22345
Hunt-Newbury, R., Viveiros, R., Johnsen, R., Mah, A., Anastas, D., Fang, L., et al. (2007). High- throughput in vivo analysis of gene expression in Caenorhabditis elegans. PLoS Biol. 5:e237. doi: 10.1371/journal.pbio.0050237
Hupe, M., Li, M. X., Kneitz, S., Davydova, D., Yokota, C., Kele, J., et al. (2017). Gene expression profiles of brain endothelial cells during embryonic development at bulk and single-cell levels. Sci. Signal. 10:eaag2476. doi: 10.1126/scisignal.aag2476
Huttlin, E. L., Bruckner, R. J., Paulo, J. A., Cannon, J. R., Ting, L., Baltier, K., et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. doi: 10.1038/nature22366
Huttlin, E. L., Jedrychowski, M. P., Elias, J. E., Goswami, T., Rad, R., Beausoleil, S. A., et al. (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. doi: 10.1016/j.cell.2010.12.001
Huttlin, E. L., Ting, L., Bruckner, R. J., Gebreab, F., Gygi, M. P., Szpyt, J., et al. (2015). The BioPlex network: a systematic exploration of the human interactome. Cell 162, 425–440. doi: 10.1016/j.cell.2015.06.043
Ivanov, A. A., Gonzalez-Pecchi, V., Khuri, L. F., Niu, Q., Wang, Y., Xu, Y., et al. (2017). OncoPPi-informed discovery of mitogen-activated protein kinase kinase 3 as a novel binding partner of c-Myc. Oncogene 36, 5852–5860. doi: 10.1038/onc.2017.180
Jabbari, A., Suárez-Fariñas, M., Fuentes-Duculan, J., Gonzalez, J., Cueto, I., Franks, A. G., et al. (2014). Dominant Th1 and minimal Th17 skewing in discoid lupus revealed by transcriptomic comparison with psoriasis. J. Invest. Dermatol. 134, 87–95. doi: 10.1038/jid.2013.269
Jentsch, T. J., and Pusch, M. (2018). CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol. Rev. 98, 1493–1590. doi: 10.1152/physrev.00047.2017
Jiang, Y., Xie, M., Chen, W., Talbot, R., Maddox, J. F., Faraut, T., et al. (2014). The sheep genome illuminates biology of the rumen and lipid metabolism. Science 344, 1168–1173. doi: 10.1126/science.1252806
Johnson, M. B., Wang, P. P., Atabay, K. D., Murphy, E. A., Doan, R. N., Hecht, J. L., et al. (2015). Single-cell analysis reveals transcriptional heterogeneity of neural progenitors in human cortex. Nat. Neurosci. 18, 637–646. doi: 10.1038/nn.3980
Jones, K. B., Salah, Z., Del Mare, S., Galasso, M., Gaudio, E., Nuovo, G. J., et al. (2012). miRNA signatures associate with pathogenesis and progression of osteosarcoma. Cancer Res. 72, 1865–1877. doi: 10.1158/0008-5472.CAN-11-2663
Jung, E., Osswald, M., Blaes, J., Wiestler, B., Sahm, F., Schmenger, T., et al. (2017). Tweety- homolog 1 drives brain colonization of gliomas. J. Neurosci. 37, 6837–6850. doi: 10.1523/JNEUROSCI.3532-16.2017
Kanth, P., Bronner, M. P., Boucher, K. M., Burt, R. W., Neklason, D. W., Hagedorn, C. H., et al. (2016). Gene signature in sessile serrated polyps identifies colon cancer subtype. Cancer Prev. Res. 9, 456–465. doi: 10.1158/1940-6207.CAPR-15-0363
Karimi, K., Fortriede, J. D., Lotay, V. S., Burns, K. A., Wang, D. Z., Fisher, M. E., et al. (2018). Xenbase: a genomic, epigenomic and transcriptomic model organism database. Nucleic Acids Res. 46, D861–D868. doi: 10.1093/nar/gkx936
Kawaguchi, A., Ikawa, T., Kasukawa, T., Ueda, H. R., Kurimoto, K., Saitou, M., et al. (2008). Single-cell gene profiling defines differential progenitor subclasses in mammalian neurogenesis. Development 135, 3113–3124. doi: 10.1242/dev.022616
Keane, T. M., Goodstadt, L., Danecek, P., White, M. A., Wong, K., Yalcin, B., et al. (2011). Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294. doi: 10.1038/nature10413
Khokhlatchev, A. V., Canagarajah, B., Wilsbacher, J., Robinson, M., Atkinson, M., Goldsmith, E., et al. (1998). Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell 93, 605–615. doi: 10.1016/s0092-8674(00)81189-7
Kim, J., Han, D., Byun, S. H., Kwon, M., Cho, J. Y., Pleasure, S. J., et al. (2018). Ttyh1 regulates embryonic neural stem cell properties by enhancing the Notch signaling pathway. EMBO Rep. 19:e45472. doi: 10.15252/embr.201745472
Kim, S. M., Park, Y.-Y., Park, E. S., Cho, J. Y., Izzo, J. G., Zhang, D., et al. (2010). Prognostic biomarkers for esophageal adenocarcinoma identified by analysis of tumor transcriptome. PLoS One 5:e15074. doi: 10.1371/journal.pone.0015074
Kim, M.-S., Pinto, S. M., Getnet, D., Nirujogi, R. S., Manda, S. S., Chaerkady, R., et al. (2014). A draft map of the human proteome. Nature 509, 575–581. doi: 10.1038/nature13302
Kleinman, C. L., Gerges, N., Papillon-Cavanagh, S., Sin-Chan, P., Pramatarova, A., Quang, D. A., et al. (2014). Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat. Genet. 46, 39–44. doi: 10.1038/ng.2849
Kolch, W. (2005). Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 6, 827–837. doi: 10.1038/nrm1743
Komatsu, M., Yoshimaru, T., Matsuo, T., Kiyotani, K., Miyoshi, Y., Tanahashi, T., et al. (2013). Molecular features of triple negative breast cancer cells by genome-wide gene expression profiling analysis. Int. J. Oncol. 42, 478–506. doi: 10.3892/ijo.2012.1744
Kriventseva, E. V., Kuznetsov, D., Tegenfeldt, F., Manni, M., Dias, R., Simão, F. A., et al. (2019). OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47, D807–D811. doi: 10.1093/nar/gky1053
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kudtarkar, P., and Cameron, R. A. (2017). Echinobase: an expanding resource for echinoderm genomic information. Database 2017:bax074. doi: 10.1093/database/bax074
Kumada, T., Yamanaka, Y., Kitano, A., Shibata, M., Awaya, T., Kato, T., et al. (2010). Ttyh1, a Ca(2+)-binding protein localized to the endoplasmic reticulum, is required for early embryonic development. Dev. Dyn. 239, 2233–2245. doi: 10.1002/dvdy.22348
Kyriakis, J. M., and Avruch, J. (2012). Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol. Rev. 92, 689–737. doi: 10.1152/physrev.00028.2011
Lécuyer, E., Yoshida, H., Parthasarathy, N., Alm, C., Babak, T., Cerovina, T., et al. (2007). Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell 131, 174–187. doi: 10.1016/j.cell.2007.08.003
Lee, S., Medina, D., Tsimelzon, A., Mohsin, S. K., Mao, S., Wu, Y., et al. (2007). Alterations of gene expression in the development of early hyperplastic precursors of breast cancer. Am. J. Pathol. 171, 252–262. doi: 10.2353/ajpath.2007.061010
Leggatt, R. A., and Iwama, G. K. (2003). Occurrence of polyploidy in the fishes. Rev. Fish Biol. Fisheries 13, 237–246. doi: 10.1023/b:rfbf.0000033049.00668.fe
Lein, E. S., Hawrylycz, M. J., Ao, N., Ayres, M., Bensinger, A., Bernard, A., et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. doi: 10.1038/nature05453
Levin, M., Hashimshony, T., Wagner, F., and Yanai, I. (2012). Developmental milestones punctuate gene expression in the Caenorhabditis embryo. Dev. Cell 22, 1101–1108. doi: 10.1016/j.devcel.2012.04.004
Li, Z., Ivanov, A. A., Su, R., Gonzalez-Pecchi, V., Qi, Q., Liu, S., et al. (2017). The OncoPPi network of cancer-focused protein-protein interactions to inform biological insights and therapeutic strategies. Nat. Commun. 8:14356. doi: 10.1038/ncomms14356
Li, L., Li, Q., Wang, Q., Liu, L., Li, R., Liu, H., et al. (2019). Rare copy number variants in the genome of Chinese female children and adolescents with Turner syndrome. Biosci. Rep. 39:BSR20181305. doi: 10.1042/BSR20181305
Li, B., Tsoi, L. C., Swindell, W. R., Gudjonsson, J. E., Tejasvi, T., Johnston, A., et al. (2014). Transcriptome analysis of psoriasis in a large case-control sample: RNA-seq provides insights into disease mechanisms. J. Invest. Dermatol. 134, 1828–1838. doi: 10.1038/jid.2014.28
Liang, W. S., Dunckley, T., Beach, T. G., Grover, A., Mastroeni, D., Walker, D. G., et al. (2007). Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiol. Genomics 28, 311–322. doi: 10.1152/physiolgenomics.00208.2006
Liao, X., Bao, H., Meng, Y., Plastow, G., Moore, S., and Stothard, P. (2014). Sequence, structural and expression divergence of duplicate genes in the bovine genome. PLoS One 9:e102868. doi: 10.1371/journal.pone.0102868
Linsley, P. S., Speake, C., Whalen, E., and Chaussabel, D. (2014). Copy number loss of the interferon gene cluster in melanomas is linked to reduced T cell infiltrate and poor patient prognosis. PLoS One 9:e109760. doi: 10.1371/journal.pone.0109760
Logsdon, C. D., Simeone, D. M., Binkley, C., Arumugam, T., Greenson, J. K., Giordano, T. J., et al. (2003). Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Res. 63, 2649–2657. Available online at: https://cancerres.aacrjournals.org/content/63/10/2649.long.
Lu, S., Wang, J., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., et al. (2020). CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48, D265–D268. doi: 10.1093/nar/gkz991
Lukasiuk, K., Kontula, L., and Pitkänen, A. (2003). cDNA profiling of epileptogenesis in the rat brain. Eur. J. Neurosci. 17, 271–279. doi: 10.1046/j.1460-9568.2003.02461.x
Ma, Y., Li, C., Gu, J., Tang, F., Li, P., Ping, P., et al. (2012). Aberrant gene expression profiles in pluripotent stem cells induced from fibroblasts of a Klinefelter syndrome patient. J. Biol. Chem. 287, 38970–38979. doi: 10.1074/jbc.M112.380204
Maag, J. L. V., Fisher, O. M., Levert-Mignon, A., Kaczorowski, D. C., Thomas, M. L., Hussey, D. J., et al. (2017). Novel aberrations uncovered in barrett’s esophagus and esophageal adenocarcinoma using whole transcriptome sequencing. Mol. Cancer Res. 15, 1558–1569. doi: 10.1158/1541-7786.MCR-17-0332
Marcinkiewicz, K. M., and Gudas, L. J. (2014). Altered epigenetic regulation of homeobox genes in human oral squamous cell carcinoma cells. Exp. Cell Res. 320, 128–143. doi: 10.1016/j.yexcr.2013.09.011
Matthews, C. A., Shaw, J. E., Hooper, J. A., Young, I. G., Crouch, M. F., and Campbell, H. D. (2007). Expression and evolution of the mammalian brain gene Ttyh1. J. Neurochem. 100, 693–707. doi: 10.1111/j.1471-4159.2006.04237.x
McKay, S. J., Johnsen, R., Khattra, J., Asano, J., Baillie, D. L., Chan, S., et al. (2003). Gene expression profiling of cells, tissues and developmental stages of the nematode C. elegans. Cold Spring Harb. Symp. Quant. Biol. 68, 159–169. doi: 10.1101/sqb.2003.68.159
Merkin, J., Russell, C., Chen, P., and Burge, C. B. (2012). Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 338, 1593–1599. doi: 10.1126/science.1228186
Moon, D. K., Bae, Y. J., Jeong, G.-R., Cho, C.-H., and Hwang, S. C. (2019). Upregulated TTYH2 expression is critical for the invasion and migration of U2OS human osteosarcoma cell lines. Biochem. Biophys. Res. Commun. 516, 521–525. doi: 10.1016/j.bbrc.2019.06.047
Morciano, M., Beckhaus, T., Karas, M., Zimmermann, H., and Volknandt, W. (2009). The proteome of the presynaptic active zone: from docked synaptic vesicles to adhesion molecules and maxi-channels. J. Neurochem. 108, 662–675. doi: 10.1111/j.1471-4159.2008.05824.x
Mummery-Widmer, J. L., Yamazaki, M., Stoeger, T., Novatchkova, M., Bhalerao, S., Chen, D., et al. (2009). Genome-wide analysis of Notch signalling in Drosophila by transgenic RNAi. Nature 458, 987–992. doi: 10.1038/nature07936
Naqvi, S., Godfrey, A. K., Hughes, J. F., Goodheart, M. L., Mitchell, R. N., and Page, D. C. (2019). Conservation, acquisition, and functional impact of sex-biased gene expression in mammals. Science 365:eaaw7317. doi: 10.1126/science.aaw7317
NCBI Resource Coordinators. (2018). Database resources of the national center for biotechnology information. Nucleic Acids Res. 46, D8–D13. doi: 10.1093/nar/gkx1095
Neelagandan, N., Gonnella, G., Dang, S., Janiesch, P. C., Miller, K. K., Küchler, K., et al. (2019). TDP-43 enhances translation of specific mRNAs linked to neurodegenerative disease. Nucleic Acids Res. 47, 341–361. doi: 10.1093/nar/gky972
Neely, G. G., Hess, A., Costigan, M., Keene, A. C., Goulas, S., Langeslag, M., et al. (2010). A genome-wide Drosophila screen for heat nociception identifies α2δ3 as an evolutionarily conserved pain gene. Cell 143, 628–638. doi: 10.1016/j.cell.2010.09.047
Nothwang, H. G., Koehl, A., and Friauf, E. (2006). Comparative gene expression analysis reveals a characteristic molecular profile of the superior olivary complex. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 288, 409–423. doi: 10.1002/ar.a.20301
Ohnishi, K., Semi, K., Yamamoto, T., Shimizu, M., Tanaka, A., Mitsunaga, K., et al. (2014). Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 156, 663–677. doi: 10.1016/j.cell.2014.01.005
Okada, Y. (2019). Tweety homologs (TTYH) freshly join the journey of molecular identification of the VRAC/VSOR channel pore. Exp. Neurobiol. 28, 131–133. doi: 10.5607/en.2019.28.2.131
Okamoto, M., Miyata, T., Konno, D., Ueda, H. R., Kasukawa, T., Hashimoto, M., et al. (2016). Cell-cycle-independent transitions in temporal identity of mammalian neural progenitor cells. Nat. Commun. 7:11349. doi: 10.1038/ncomms11349
Osswald, M., Jung, E., Sahm, F., Solecki, G., Venkataramani, V., Blaes, J., et al. (2015). Brain tumour cells interconnect to a functional and resistant network. Nature 528, 93–98. doi: 10.1038/nature16071
Owens, N. D. L., Blitz, I. L., Lane, M. A., Patrushev, I., Overton, J. D., Gilchrist, M. J., et al. (2016). Measuring absolute RNA copy numbers at high temporal resolution reveals transcriptome kinetics in development. Cell Rep. 14, 632–647. doi: 10.1016/j.celrep.2015.12.050
Papatheodorou, I., Moreno, P., Manning, J., Fuentes, A. M., George, N., Fexova, S., et al. (2020). Expression atlas update: from tissues to single cells. Nucleic Acids Res. 48, D77–D83. doi: 10.1093/nar/gkz947
Peeters, J. G., Vervoort, S. J., Tan, S. C., Mijnheer, G., de Roock, S., Vastert, S. J., et al. (2015). Inhibition of super-enhancer activity in autoinflammatory site-derived T cells reduces disease-associated gene expression. Cell Rep. 12, 1986–1996. doi: 10.1016/j.celrep.2015.08.046
Pfeiffer, M. J., Taher, L., Drexler, H., Suzuki, Y., Makałowski, W., Schwarzer, C., et al. (2015). Differences in embryo quality are associated with differences in oocyte composition: a proteomic study in inbred mice. Proteomics 15, 675–687. doi: 10.1002/pmic.201400334
Pipes, L., Li, S., Bozinoski, M., Palermo, R., Peng, X., Blood, P., et al. (2013). The non- human primate reference transcriptome resource (NHPRTR) for comparative functional genomics. Nucleic Acids Res. 41, D906–D914. doi: 10.1093/nar/gks1268
Poroca, D. R., Pelis, R. M., and Chappe, V. M. (2017). ClC channels and transporters: structure, physiological functions, and implications in human chloride channelopathies. Front. Pharmacol. 8:151. doi: 10.3389/fphar.2017.00151
Rae, F. K., Hooper, J. D., Eyre, H. J., Sutherland, G. R., Nicol, D. L., and Clements, J. A. (2001). TTYH2, a human homologue of the Drosophila melanogaster gene tweety, is located on 17q24 and upregulated in renal cell carcinoma. Genomics 77, 200–207. doi: 10.1006/geno.2001.6629
Raj, A., van den Bogaard, P., Rifkin, S. A., van Oudenaarden, A., and Tyagi, S. (2008). Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5, 877–879. doi: 10.1038/nmeth.1253
Rhodes, D. R., Yu, J., Shanker, K., Deshpande, N., Varambally, R., Ghosh, D., et al. (2004). ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 6, 1–6. doi: 10.1016/s1476-5586(04)80047-2
Rojas-Peña, M. L., Olivares-Navarrete, R., Hyzy, S., Arafat, D., Schwartz, Z., Boyan, B. D., et al. (2014). Characterization of distinct classes of differential gene expression in osteoblast cultures from non-syndromic craniosynostosis bone. J. Genomics 2, 121–130. doi: 10.7150/jgen.8833
Ruzicka, L., Howe, D. G., Ramachandran, S., Toro, S., Van Slyke, C. E., Bradford, Y. M., et al. (2019). The Zebrafish Information Network: new support for non-coding genes, richer Gene Ontology annotations and the Alliance of Genome Resources. Nucleic Acids Res. 47, D867–D873. doi: 10.1093/nar/gky1090
Ryu, J., Kim, D. G., Lee, Y. S., Bae, Y., Kim, A., Park, N., et al. (2019). Surface expression of TTYH2 is attenuated by direct interaction with β-COP. BMB Rep. 52, 445–450. doi: 10.5483/BMBRep.2019.52.7.188
Sabates-Bellver, J., Van der Flier, L. G., de Palo, M., Cattaneo, E., Maake, C., Rehrauer, H., et al. (2007). Transcriptome profile of human colorectal adenomas. Mol. Cancer Res. 5, 1263–1275. doi: 10.1158/1541-7786.MCR-07-0267
Sabirov, R. Z., and Okada, Y. (2009). The maxi-anion channel: a classical channel playing novel roles through an unidentified molecular entity. J. Physiol. Sci. 59, 3–21. doi: 10.1007/s12576-008-0008-4
Saha, S. K., Biswas, P. K., Gil, M., and Cho, S. G. (2019). High expression of TTYH3 is related to poor outcomes in human gastric cancer. J. Clin. Med. 8:1762. doi: 10.3390/jcm8111762
Saka, S. K., Wang, Y., Kishi, J. Y., Zhu, A., Zeng, Y., Xie, W., et al. (2019). Immuno-SABER enables highly multiplexed and amplified protein imaging in tissues. Nat. Biotechnol. 37, 1080–1090. doi: 10.1038/s41587-019-0207-y
Samaras, P., Schmidt, T., Frejno, M., Gessulat, S., Reinecke, M., Jarzab, A., et al. (2020). ProteomicsDB: a multi-omics and multi-organism resource for life science research. Nucleic Acids Res. 48, D1153–D1163. doi: 10.1093/nar/gkz974
Scalise, M., Torella, M., Marino, F., Ravo, M., Giurato, G., Vicinanza, C., et al. (2020). Atrial myxomas arise from multipotent cardiac stem cells. Eur. Heart J. 41, 4332–4345. doi: 10.1093/eurheartj/ehaa156
Schmidt, T., Samaras, P., Frejno, M., Gessulat, S., Barnert, M., Kienegger, H., et al. (2018). ProteomicsDB. Nucleic Acids Res. 46, D1271–D1281. doi: 10.1093/nar/gkx1029
Schnorrer, F., Schönbauer, C., Langer, C. C., Dietzl, G., Novatchkova, M., Schernhuber, K., et al. (2010). Systematic genetic analysis of muscle morphogenesis and function in Drosophila. Nature 464, 287–291. doi: 10.1038/nature08799
Selamat, S. A., Chung, B. S., Girard, L., Zhang, W., Zhang, Y., Campan, M., et al. (2012). Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 22, 1197–1211. doi: 10.1101/gr.132662.111
Session, A. M., Uno, Y., Kwon, T., Chapman, J. A., Toyoda, A., Takahashi, S., et al. (2016). Genome evolution in the allotetraploid frog Xenopus laevis. Nature 538, 336–343. doi: 10.1038/nature19840
Sin-Chan, P., and Huang, A. (2014). DNMTs as potential therapeutic targets in high-risk pediatric embryonal brain tumors. Expert Opin. Ther. Targets 18, 1103–1107. doi: 10.1517/14728222.2014.938052
Sonnhammer, E. L., von Heijne, G., and Krogh, A. (1998). A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6, 175–182.
Steen, R. G., Kwitek-Black, A. E., Glenn, C., Gullings-Handley, J., Van Etten, W., Atkinson, O. S., et al. (1999). A high-density integrated genetic linkage and radiation hybrid map of the laboratory rat. Genome Res. 9, AP1–AP8.
Stefaniuk, M., and Lukasiuk, K. (2010). Cloning of expressed sequence tags (ESTs) representing putative epileptogenesis-related genes and the localization of their expression in the normal brain. Neurosci. Lett. 482, 230–234. doi: 10.1016/j.neulet.2010.07.045
Stefaniuk, M., Swiech, L., Dzwonek, J., and Lukasiuk, K. (2010). Expression of Ttyh1, a member of the Tweety family in neurons in vitro and in vivo and its potential role in brain pathology. J. Neurochem. 115, 1183–1194. doi: 10.1111/j.1471-4159.2010.07023.x
Stelzer, G., Rosen, N., Plaschkes, I., Zimmerman, S., Twik, M., Fishilevich, S., et al. (2016). The genecards suite: from gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 54, 1.30.1–1.30.33. doi: 10.1002/cpbi.5
Straessler, K. M., Jones, K. B., Hu, H., Jin, H., van de Rijn, M., and Capecchi, M. R. (2013). Modeling clear cell sarcomagenesis in the mouse: cell of origin differentiation state impacts tumor characteristics. Cancer Cell 23, 215–227. doi: 10.1016/j.ccr.2012.12.019
Stroschein-Stevenson, S. L., Foley, E., O’Farrell, P. H., and Johnson, A. D. (2006). Identification of Drosophila gene products required for phagocytosis of Candida albicans. PLoS Biol. 4:e4. doi: 10.1371/journal.pbio.0040004
Suzuki, M. (2006). The Drosophila tweety family: molecular candidates for large-conductance Ca2+-activated Cl− channels. Exp. Physiol. 91, 141–147. doi: 10.1113/expphysiol.2005.031773
Suzuki, M., and Mizuno, A. (2004). A novel human Cl− channel family related to Drosophila flightless locus. J. Biol. Chem. 279, 22461–22468. doi: 10.1074/jbc.M313813200
Szabo, L., Morey, R., Palpant, N. J., Wang, P. L., Afari, N., Jiang, C., et al. (2015). Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 16:126. doi: 10.1186/s13059-015-0690-5
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. doi: 10.1093/nar/gky1131
Tahiri, A., Leivonen, S. K., Lüders, T., Steinfeld, I., Ragle Aure, M., Geisler, J., et al. (2014). Deregulation of cancer-related miRNAs is a common event in both benign and malignant human breast tumors. Carcinogenesis 35, 76–85. doi: 10.1093/carcin/bgt333
Tan, T. Z., Miow, Q. H., Miki, Y., Noda, T., Mori, S., Huang, R. Y., et al. (2014). Epithelial- mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 6, 1279–1293. doi: 10.15252/emmm.201404208
Taylor, J. Y., Schwander, K., Kardia, S. L., Arnett, D., Liang, J., Hunt, S. C., et al. (2016). A Genome-wide study of blood pressure in African Americans accounting for gene- smoking interaction. Sci. Rep. 6:18812. doi: 10.1038/srep18812
Thierry-Mieg, D., and Thierry-Mieg, J. (2006). AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 7, S12.1–S12.14. doi: 10.1186/gb-2006-7-s1-s12
Thisse, B., and Thisse, C. (2004). Fast Release Clones: A High Throughput Expression Analysis. ZFIN Direct Data Submission. Available online at: http://zfin.org.
Thomas, P. D., Campbell, M. J., Kejariwal, A., Mi, H., Karlak, B., Daverman, R., et al. (2003). PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141. doi: 10.1101/gr.772403
Thompson, C. L., Ng, L., Menon, V., Martinez, S., Lee, C. K., Glattfelder, K., et al. (2014). A high-resolution spatiotemporal atlas of gene expression of the developing mouse brain. Neuron 83, 309–323. doi: 10.1016/j.neuron.2014.05.033
Thurmond, J., Goodman, J. L., Strelets, V. B., Attrill, H., Gramates, L. S., Marygold, S. J., et al. (2019). FlyBase 2.0: the next generation. Nucleic Acids Res. 47, D759–D765. doi: 10.1093/nar/gky1003
Toiyama, Y., Mizoguchi, A., Kimura, K., Hiro, J., Inoue, Y., Tutumi, T., et al. (2007). TTYH2, a human homologue of the Drosophila melanogaster gene tweety, is up-regulated in colon carcinoma and involved in cell proliferation and cell aggregation. World J. Gastroenterol. 13, 2717–2721. doi: 10.3748/wjg.v13.i19.2717
Tong, M., Chan, K. W., Bao, J. Y., Wong, K. Y., Chen, J. N., Kwan, P. S., et al. (2012). Rab25 is a tumor suppressor gene with antiangiogenic and anti-invasive activities in esophageal squamous cell carcinoma. Cancer Res. 72, 6024–6035. doi: 10.1158/0008-5472.CAN-12-1269
Tu, Q., Cameron, R. A., and Davidson, E. H. (2014). Quantitative developmental transcriptomes of the sea urchin Strongylocentrotus purpuratus. Dev. Biol. 385, 160–167. doi: 10.1016/j.ydbio.2013.11.019
Tyburczy, M. E., Kotulska, K., Pokarowski, P., Mieczkowski, J., Kucharska, J., Grajkowska, W., et al. (2010). Novel proteins regulated by mTOR in subependymal giant cell astrocytomas of patients with tuberous sclerosis complex and new therapeutic implications. Am. J. Pathol. 176, 1878–1890. doi: 10.2353/ajpath.2010.090950
Ulmasov, B., Oshima, K., Rodriguez, M. G., Cox, R. D., and Neuschwander-Tetri, B. A. (2013). Differences in the degree of cerulein-induced chronic pancreatitis in C57BL/6 mouse substrains lead to new insights in identification of potential risk factors in the development of chronic pancreatitis. Am. J. Pathol. 183, 692–708. doi: 10.1016/j.ajpath.2013.05.020
Uniprot Consortium. (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515. doi: 10.1093/nar/gky1049
van Dijk, K. D., Berendse, H. W., Drukarch, B., Fratantoni, S. A., Pham, T. V., Piersma, S. R., et al. (2012). The proteome of the locus ceruleus in Parkinson’s disease: relevance to pathogenesis. Brain Pathol. 22, 485–498. doi: 10.1111/j.1750-3639.2011.00540.x
Vasiliu, D., Clamons, S., McDonough, M., Rabe, B., and Saha, M. (2015). A regression-based differential expression detection algorithm for microarray studies with ultra-low sample size. PLoS One 10:e0118198. doi: 10.1371/journal.pone.0118198
Visel, A., Carson, J., Oldekamp, J., Warnecke, M., Jakubcakova, V., Zhou, X., et al. (2007). Regulatory pathway analysis by high-throughput in situ hybridization. PLoS Genet. 3, 1867–1883. doi: 10.1371/journal.pgen.0030178
Voineagu, I., Wang, X., Johnston, P., Lowe, J. K., Tian, Y., Horvath, S., et al. (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. doi: 10.1038/nature10110
Wang, D., Eraslan, B., Wieland, T., Hallström, B., Hopf, T., Zolg, D. P., et al. (2019). A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 15:e8503. doi: 10.15252/msb.20188503
Weinberg, F., Griffin, R., Fröhlich, M., Heining, C., Braun, S., Spohr, C., et al. (2019). Identification and characterization of a BRAF fusion oncoprotein with retained autoinhibitory domains. Oncogene 39, 814–832. doi: 10.1038/s41388-019-1021-1
White, R. J., Collins, J. E., Sealy, I. M., Wali, N., Dooley, C. M., Digby, Z., et al. (2017). A high-resolution mRNA expression time course of embryonic development in zebrafish. eLife 6:e30860. doi: 10.7554/eLife.30860
Wiernasz, E., Kaliszewska, A., Brutkowski, W., Bednarczyk, J., Gorniak, M., Kaza, B., et al. (2014). Ttyh1 protein is expressed in glia in vitro and shows elevated expression in activated astrocytes following status epilepticus. Neurochem. Res. 39, 2516–2526. doi: 10.1007/s11064-014-1455-3
Wilk, R., Hu, J., Blotsky, D., and Krause, H. M. (2016). Diverse and pervasive subcellular distributions for both coding and long noncoding RNAs. Genes Dev. 30, 594–609. doi: 10.1101/gad.276931.115
Williams, J. G. (2010). Dictyostelium finds new roles to model. Genetics 185, 717–726. doi: 10.1534/genetics.110.119297
Woo, J., Jang, M. W., Lee, J., Koh, W., Mikoshiba, K., and Lee, C. J. (2020). The molecular mechanism of synaptic activity-induced astrocytic volume transient. J. Physiol. 598, 4555–4572. doi: 10.1113/JP279741
Woodward, W. A., Krishnamurthy, S., Yamauchi, H., El-Zein, R., Ogura, D., Kitadai, E., et al. (2013). Genomic and expression analysis of microdissected inflammatory breast cancer. Breast Cancer Res. Treat. 138, 761–772. doi: 10.1007/s10549-013-2501-6
Wu, H.-N., Cao, X.-L., Fang, Z., Zhang, Y.-F., Han, W.-J., Yue, K.-Y., et al. (2019). Deficiency of Ttyh1 downstream to Notch signaling results in precocious differentiation of neural stem cells. Biochem. Biophys. Res. Commun. 514, 842–847. doi: 10.1016/j.bbrc.2019.04.181
Xu, P.-T., Li, Y.-J., Qin, X.-J., Scherzer, C. R., Xu, H., Schmechel, D. E., et al. (2006). Differences in apolipoprotein E3/3 and E4/4 allele-specific gene expression in hippocampus in Alzheimer disease. Neurobiol. Dis. 21, 256–275. doi: 10.1016/j.nbd.2005.07.004
Yasko, J. R., Moss, I. L., and Mains, R. E. (2019). Transcriptional profiling of non-injured nociceptors after spinal cord injury reveals diverse molecular changes. Front. Mol. Neurosci. 12:284. doi: 10.3389/fnmol.2019.00284
Yates, A. D., Achuthan, P., Akanni, W., Allen, J., Alvarez-Jarreta, J., Amode, M. R., et al. (2020). Ensembl 2020. Nucleic Acids Res. 48, D682–D688. doi: 10.1093/nar/gkz966
Yen, M.-C., Yeh, I.-J., Liu, K.-T., Jian, S.-F., Lin, C.-J., Tsai, M.-J., et al. (2019). Next-generation sequencing predicts interaction network between miRNA and target genes in lipoteichoic acid-stimulated human neutrophils. Int. J. Mol. Med. 44, 1436–1446. doi: 10.3892/ijmm.2019.4295
Yeung, T.-L., Leung, C. S., Wong, K.-K., Samimi, G., Thompson, M. S., Liu, J., et al. (2013). TGF-β modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 73, 5016–5028. doi: 10.1158/0008-5472.CAN-13-0023
Yu, Y., Fuscoe, J. C., Zhao, C., Guo, C., Jia, M., Qing, T., et al. (2014). A rat RNA-Seq transcriptomic BodyMap across 11 organs and 4 developmental stages. Nat. Commun. 5:3230. doi: 10.1038/ncomms4230
Yu, A. C., Zambrano, R. M., Cristian, I., Price, S., Bernhard, B., Zucker, M., et al. (2017). Variable developmental delays and characteristic facial features-A novel 7p22.3p22.2 microdeletion syndrome? Am. J. Med. Genet. A 173, 1593–1600. doi: 10.1002/ajmg.a.38241
Yue, F., Cheng, Y., Breschi, A., Vierstra, J., Wu, W., Ryba, T., et al. (2014). A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364. doi: 10.1038/nature13992
Zhang, J., Schulze, K. L., Hiesinger, P. R., Suyama, K., Wang, S., Fish, M., et al. (2007). Thirty- one flavors of Drosophila rab proteins. Genetics 176, 1307–1322. doi: 10.1534/genetics.106.066761
Zhong, G., Lou, W., Shen, Q., Yu, K., and Zheng, Y. (2020). Identification of key genes as potential biomarkers for triple-negative breast cancer using integrating genomics analysis. Mol. Med. Rep. 21, 557–566. doi: 10.3892/mmr.2019.10867
Keywords: tweety, ttyh1, ttyh2, ttyh3, embryo, neural development, TTY, chloride channel
Citation: Nalamalapu RR, Yue M, Stone AR, Murphy S and Saha MS (2021) The tweety Gene Family: From Embryo to Disease. Front. Mol. Neurosci. 14:672511. doi: 10.3389/fnmol.2021.672511
Received: 25 February 2021; Accepted: 18 May 2021;
Published: 28 June 2021.
Edited by:
Tania Cristina Leite de Sampaio e Spohr, Centogene GmbH, GermanyReviewed by:
Takashi Saito, Nagoya City University, JapanCopyright © 2021 Nalamalapu, Yue, Stone, Murphy and Saha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margaret S. Saha,bXNzYWhhQHdtLmVkdQ==
† These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.