 Xiaolin Zhu
Xiaolin Zhu Yu Zhang1
Yu Zhang1 Xin Yang
Xin Yang Chunyan Hao
Chunyan Hao Hubin Duan
Hubin Duan- 1Department of Neurosurgery, First Hospital of Shanxi Medical University, Taiyuan, China
- 2Department of Geriatrics, First Hospital of Shanxi Medical University, Taiyuan, China
- 3Department of Neurosurgery, Lvliang People’s Hospital, Lvliang, China
The pathogenesis of neurodegenerative diseases (NDDs) is complex and diverse. Over the decades, our understanding of NDD has been limited to pathological features. However, recent advances in gene sequencing have facilitated elucidation of NDD at a deeper level. Gene editing techniques have uncovered new genetic links to phenotypes, promoted the development of novel treatment strategies and equipped researchers with further means to construct effective cell and animal models. The current review describes the history of evolution of gene editing tools, with the aim of improving overall understanding of this technology, and focuses on the four most common NDD disorders to demonstrate the potential future applications and research directions of gene editing.
Introduction
Neurodegenerative disease (NDD) refers to a group of chronic disorders characterized by progressive loss of neurons in the brain and spinal cord. Due to technical limitations, our initial understanding of NDD was initially restricted to the pathological manifestations of abnormal protein aggregation, such as Aβ protein in Alzheimer’s disease (AD), huntingtin (HTT) protein in Huntington’s disease, α-synuclein in Parkinson’s disease, and neurofilament in amyotrophic lateral sclerosis. However, treatments targeting abnormal protein levels have constantly faced setbacks in clinical trials. By the end of the 20th century, revolutionary advances in sequencing techniques offered a novel perspective to interpret the mechanisms underlying progression of NDD and gene mutations were identified as drivers of phenotype changes. Thereafter, several studies on NDD at the gene level were conducted. With progression of sequencing methods to third-generation technology, numerous NDD-related mutations and single nucleotide polymorphism (SNP) sites were progressively identified. However, gene mutations cannot explain 100% of NDD cases and sporadic cases exist, even for Huntington’s disease (HD) that is generally considered an autosomal dominant disorder. Thus, in recent years, research focus has expanded from direct gene expression to regulation of expression, which encompasses the fields of transcriptomics, proteomics, and epigenomics.
The concept of gene therapy, first proposed in 1972 (Friedmann and Roblin, 1972), refers to targeted changes in gene sequences through molecular means. In a narrow sense, gene editing is primarily achieved through inducing specific DNA double-strand breaks (DSB) to replace or modify target genes based on the donor sequence. From zinc finger endonuclease to the CRISPR/Cas9 system, the rapidly progressing geneediting field has significant therapeutic potential. The CRISPR/Cas system is particularly outstanding with the advantages of minimal molecular weight and natural existence. Free of artificial design, its convenience and effectiveness has greatly aided in providing insights into the mechanisms underlying NDD, reduced the cost of gene editing, and enabled construction of disease models in both cell and animal systems, in addition to facilitating multiple gene editing valuable for complex diseases such as NDD.
This report provides an overview of the history of gene editing and recent research focus on the four most common NDD disorders, specifically, AD, Parkinson’s disease (PD), HD, and amyotrophic lateral sclerosis (ALS).
Evolution of Gene Editing
Homologous Recombination
Homologous recombination is the earliest genome editing technique based on natural DNA damage and the cellular repair system. Chemicals, radiation, and by-products of cellular biological processes, such as reactive oxygen species produced during aerobic respiration, nitrogen compounds produced by inflammatory cells and free radicals generated in hydrolysis reactions, can lead to DNA damage (Jackson and Bartek, 2009). Cells in the human body experience tens of thousands of DNA lesions per day (Lindahl and Barnes, 2000). Among the multiple forms of damage, double-stranded breaks (DSB) are one of the most toxic and difficult to repair (Khanna and Jackson, 2001).
Cells have evolved various mechanisms to repair DSB, two of the most important being homologous recombination (HR) and non-homologous end-joining (NHEJ) (Lieber, 2010). Compared with template-free NHEJ, HR is confined to S and G2 phases (Hustedt and Durocher, 2016; Zhao et al., 2017; Murray and Carr, 2018) while NHEJ is prevalent over the entire cell cycle (Symington and Gautier, 2011). As an important method of allelic exchange, HR also plays a key role in meiosis and mitosis (San Filippo et al., 2008). NHEJ is the dominant repair pathway in mammals and HR competes with other mechanisms (Symington and Gautier, 2011; Kowalczykowski, 2015; Haber, 2016). The repair process of HR is based on multiple pathways, starting from Mre11-Rad50-Nbs1 (MRN) binding to the DSB site followed by 5′-to-3′ resection to generate single-stranded DNA (ssDNA), eventually initiating sub-pathway repair (Mehta and Haber, 2014; Skoneczna et al., 2015; Haber, 2016). HR uses homologous sequences of the sister chromatid as templates in the natural route (San Filippo et al., 2008; Heyer et al., 2010). In genetic engineering, plasmid or viral vector-mediated sequences homologous to DSB ends serve as the new template. However, due to competition with NHEJ, the natural frequency of HR repair alleles in eukaryotes is extremely low (Allen et al., 2002), greatly limiting the efficiency of gene editing, which could be augmented with human intervention in the future.
Evidence indicates that DSBs trigger HR (Rouet et al., 1994; Choulika et al., 1995) and an enzyme designated “meganuclease” or “homing endonuclease” that is naturally present in mitochondria and chloroplasts of microorganisms specifically recognizes 12–30 bp DNA sequences for cleavage without affecting the whole genome (Colleaux et al., 1986; Thierry and Dujon, 1992; Choulika et al., 1994). Owing to the advantages of long recognition sites and 3′ overhang production after DNA cleavage, meganucleases exhibit lower toxicity and better precision than other restriction enzymes (Kc and Steer, 2019). While hundreds of meganucleases have been identified to date, the likelihood of locating the enzyme required for a specific site remains low. On the other hand, since DNA binding and cleavage sites of meganuclease are interspersed in the same domain and are difficult to separate, tailoring the required meganuclease through engineered modifications remains a big challenge (Khan et al., 2018). Overall, the clinical application of meganuclease continues to face technical difficulties (Gaj et al., 2016).
Zinc Finger Nucleases
In the 1990s, the discovery of Flavobacterium okeanokoites (FokI) enzyme (Sugisaki and Kanazawa, 1981; Li et al., 1992; Kim and Chandrasegaran, 1994) and zinc finger structure (Fegan et al., 1985; Lee et al., 1989) promoted further development of the gene editing technique. Hydrolyzed FokI enzyme (a type IIS restriction endonuclease) in Flavobacterium contains N-terminal DNA-binding and C-terminal domains with non-specific DNA cleavage activity (Li et al., 1992, 1993) that can be easily separated (Waugh and Sauer, 1993). Owing to the modularity of FokI enzyme, engineering is relatively simple.
Zinc finger is an independently folded binding domain that coordinates zinc ions to stabilize the structure. Repeated zinc-binding motifs were first reported in Xenopus transcription factor IIIA (TFIIIA) (Miller et al., 1985). After the first single zinc finger was described in 1989 (Lee et al., 1989), vast complexes were successively identified. Considering Cys2-His2, the most common zinc finger domain as an example, a zinc finger is composed of about 30 amino acids in a conserved ββα configuration (Beerli and Barbas, 2002) and builds contacts with three base pairs in DNA sequences. Zinc finger proteins with conserved sequences are arranged in a certain order followed by attachment of FokI to the 3′ end of the protein, ultimately generating a zinc-finger nuclease (ZFN) consisting of both DNA binding and DNA cleavage domains that recognizes a 9–18 bp sequence (Liu et al., 1997). After binding of ZFN to DNA, the FokI nuclease induces cleavage as a dimer, resulting in DSB. HR and NHEJ are activated to complete gene editing via the intracellular DNA repair mechanism (Bitinaite et al., 1998; Smith et al., 2000). Theoretically, ZFNs recognize almost all 64 possible nucleotide triplets but several of these fail in terms of pairing, design and selection (Ramirez et al., 2008; Kim et al., 2010). The specificity and affinity of ZFN is also an issue although optimal fingers have a certain affinity for similar sequences. Increasing the number of zinc fingers can improve specificity and affinity but also raises the issue of inability to access sequences at certain sites, such as those with close chromatin structure and DNA modification (Carroll, 2011). Additionally, ZFNs are reported to exert a significant cytotoxic effect (Khalil, 2020).
Transcription Activator-Like Effector Nuclease
The discovery of transcription activator-like effector in the plant pathogen Xanthomonas (Boch et al., 2009; Moscou and Bogdanove, 2009) promoted the development of transcription activator-like effector nuclease (TALEN) (Miller et al., 2011), a second-generation nuclease editing technique. The central structure of TALEN protein is a highly conserved sequence of 33–35 amino acids with two variable residues at positions 12 and 13, referred to as repeat variable di-residues (RVD). Each motif relies on RVDs to recognize a single nucleotide (Deng et al., 2012). Similar to ZFN, construction of TALEN is based on the modularity and DNA cleavage function of FokI (Sun and Zhao, 2013). TALEN is easier to design and produce than ZFN but requires about three times as many coding genes. Moreover, its higher molecular weight makes transfection into mammalian cells difficult, especially using virus vectors with limited packaging capacity (Chandrasegaran and Carroll, 2016; Maeder and Gersbach, 2016).
CRISPR
Clustered regularly interspaced palindromic repeat (CRISPR) was first described in 1987 (Ishino et al., 1987) and its gene editing ability confirmed in human cells in 2013 (Cho et al., 2013). CRISPR exists in 40% bacteria and 90% archaeal genomes and functions as an adaptive immune defense system (Horvath and Barrangou, 2010). The CRISPR system can specifically capture gene sequences adjacent to protospacer adjacent motif (PAM) for cleavage using Cas nuclease into spacer segments derived from the exogenous genome. Spacers are subsequently incorporated into the CRISPR locus of host cells, separated by palindromic sequences, and eventually transcribed to CRISPR RNA (crRNA) with spacer characteristics (Barrangou et al., 2007; Garneau et al., 2010; Horvath and Barrangou, 2010). CrRNA pairs with invading foreign gene sequences in a complementary manner. Simultaneously, Cas nuclease destroys target DNA and completes the entire immune response. By capturing exogenous gene segments from invading phages, viruses and plasmids and incorporating them into host genomic loci, the CRISPR/Cas system sustains acquired immune function (Barrangou et al., 2007; Garneau et al., 2010; Horvath and Barrangou, 2010).
Type II CRISPR/Cas9 composed of Cas9 endonuclease, crRNA and trans-activating crRNA (tracrRNA) is the most commonly used system in genetic engineering (Jinek et al., 2012). Cas nuclease is the core functional element of the CRISPR system. TracrRNA and precursor crRNA (pre-crRNA) bind via base pairing, are trimmed by RNaseIII, self-folded into a partial double-stranded RNA structure, and interact with Cas9 to form a complex with DNA cleavage ability. The crRNA-tracrRNA duplex functions as a single guide RNA (sgRNA) that effectively pairs with the target sequence. After binding to the target site, Cas9 undergoes conformational changes and induces DSBs 3–4 nucleotides upstream of PAM (Jinek et al., 2012; Nishimasu et al., 2014). Domains in Cas9 not only interact with the PAM motif but also assist with sgRNA binding to the target sequence (Nishimasu et al., 2014; Barman et al., 2020).
Compared with ZFN and TALEN, the editing horizon of CRISPR is elevated from protein to RNA and technical difficulties from design to assembly are greatly simplified. In terms of target recognition, specificity is higher and binding is more stable. In addition, Cas9 acts as monomer in contrast to FokI, which only cleaves DNA in a dimeric form (Bitinaite et al., 1998; Smith et al., 2000). However, CRISPR does not alter the nature of HR induction through DSBs. In fact, NHEJ is a more prevalent pathway for DSB repair in the entire cell cycle (Chapman et al., 2012). Although NHEJ inhibitors (e.g., Scr7) (Maruyama et al., 2015) and HR promoters (e.g., Cas9-RecA fusion protein) (Cai et al., 2019) are expected to improve the efficiency of HR, CRISPR technology requires further improvement to improve the accuracy of gene editing.
Latest Developments in Genome Editing
In 2017, a study published in Nature reported a technique denoted “Base Editor” (BE) that achieved single base conversion independently of DSB and HD (Komor et al., 2016; Gaudelli et al., 2018). Based on a complex composed of dCas9 (inactive or dead Cas9) or Cas9n (Cas9 nickase with single-strand DNA incisional enzyme activity), cytosine deaminase (yCD), uracil DNA glycosylase inhibitor (UGI) and sgRNA, BE can achieve four types of accurate base substitution between C/T and G/A (Komor et al., 2016). A new technique known as “Prime Editor” (PE) was reported in 2019 (Anzalone et al., 2019) involving coupling of Cas9 protein with reverse transcriptase. Under guidance of prime editing guide RNA (pegRNA), the PE complex cuts a single strand of DNA at the target site and synthesizes new sequences with the aid of reverse transcriptase. Unpaired sequences in pegRNA are used as templates. The newly synthesized sequence is finally incorporated into the host genome for completion of the gene editing process. PE is free of DNA template and can achieve precise single nucleotide substitutions in sequences inaccessible for BE. While the advent of BE and PE has created new possibilities for gene editing, several concerns remain. For instance, dependence on the Cas9 enzyme limits their recognition window, since Cas9 can only act on sequences adjacent to PAM. BE converts all editable bases in the editing window in a non-specific manner. Moreover, BE can achieve transition of purine–purine and pyrimidine–pyrimidine but not transversion of purine-pyrimidine. In addition, BE and PE only perform edits on single nucleotides and are unable to achieve targeted integration of DNA. The off-target effects of BE and PE in practical applications remain to be established.
In 2019, a new technique using CRISPR-associated transposon (CAST) for DNA transposition was reported in Science (Strecker et al., 2019) and another similar report published in Nature (Klompe et al., 2019). CAST utilizes the ability of Tn7-like transposons to recruit the CRISPR/Cas system in bacteria (Peters et al., 2017). After instrumentalization, Tn7-like transposons can be used for targeted DNA insertion. Independent from DSBs, CAST can effectively carry cargo genes up to 10 kB, which is far superior to the current gene knock-in tool (Hou and Zhang, 2019).
Alzheimer’s Disease
Alzheimer’s disease is a clinical syndrome characterized by brain amyloid-beta (Aβ) protein deposition in senile plaques (SPs), downstream neuronal degeneration, and tau protein hyperphosphorylation (p-tau) forming neurofibrillary tangles (NFTs) (McKhann et al., 2011). Over the past 20 years, the amyloid cascade hypothesis has dominated research on the pathogenesis of AD. However, identification of mutations within three autosomal dominant genes, specifically, APP on chromosome 21 (Goate et al., 1991), PSEN1 on chromosome 14 (Sherrington et al., 1995), and PSEN2 on chromosome 1 (Levy-Lahad et al., 1995), has significantly changed research perspective. Subsequent genome-wide association studies (GWAS) have resulted in the identification of another risk gene, ApoE4, and further AD-related SNP sites. These genes encode proteins implicated in various biological processes of AD, which may serve as future editing targets.
Recent GWAS have led to the discovery of dozens of risk loci (Lambert et al., 2013; Kunkle et al., 2019; Vacher et al., 2019; Kunkle et al., 2021). Among these, clear evidence of function has been obtained for ApoE, ABCA7, BIN1, TREM2, SORL1, ADAM10, SPI1, and CR1. In particular, ApoE4 has been extensively characterized in different disease models (Burnham et al., 2020). A recent study showed that Klotho hormone in its biological form reduces risk of AD onset in individuals carrying ApoE4. Moreover, a heterozygous state of KL-VS (KL-VSHET+) genotype was suggested in association with reduced burden of AD and Aβ protein (Belloy et al., 2020). However, no association of KL-VS, the variant of Klotho, with cognitive decline of patients was observed in another clinical study (Porter et al., 2019). The potential contribution of ApoE4 to AD was further examined from multiple perspectives. REST, a central regulator of neural differentiation, is suggested to be related to the ApoE4-induced phenotype (Meyer et al., 2019). Moreover, considering the energy metabolism failure in patients with AD, the ApoE4 genotype may have a regulatory effect on metabolism. These metabolic changes have additionally been linked to gender differences (Arnold et al., 2020). One advantage is that ApoE4 has only one nucleotide difference from its allele ApoE3. Therefore, it is feasible to induce single nucleotide changes, especially with the PE technique. Triggering receptor expressed on myeloid cells 2 (TREM2) is a genetic locus shared by AD and PD. Aggravated neurodegeneration has been detected in TREM2-deleted mice, which may be related to microglial activation (Guo et al., 2019). Another in vivo study exhibited transformational value for clinical treatment. Researchers successfully improved Aβ pathology in APP transgenic mice (Nagata et al., 2018; see Table 1).
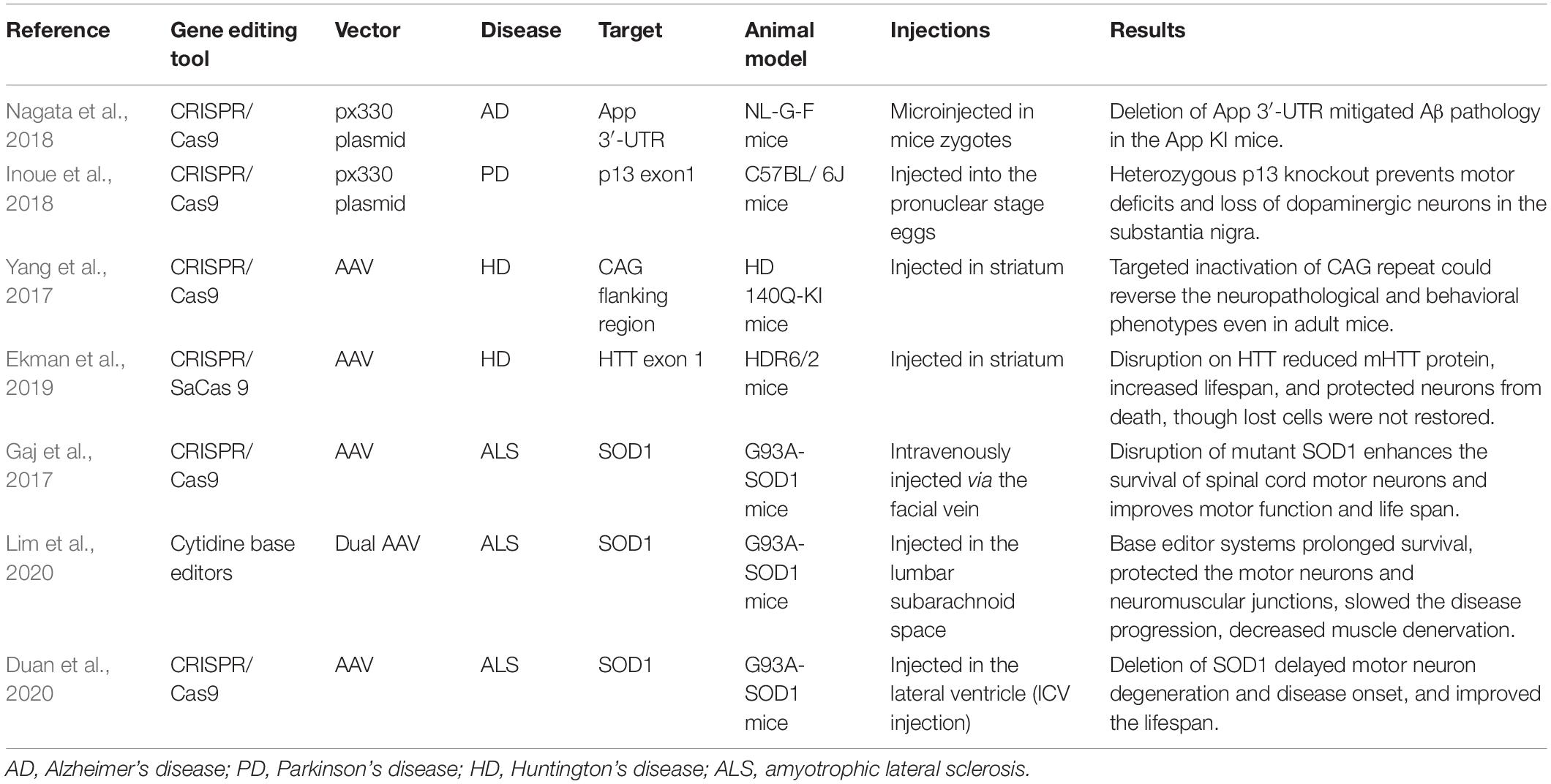
Table 1. Pre-clinical studies of gene therapy in neurodegenerative diseases.
A number of studies have also focused on the genetic background of AD pathological manifestations. For instance, CHRFAM7A exerts an antagonistic effect on cholinergic receptors in induced pluripotent stem cell (iPSCs) transfected with TALEN (Szigeti et al., 2020). PSENLIN2 is associated with greater amyloid β protein accumulation than PSENLIN1 (Lessard et al., 2019). However, editing the C-terminus of APP via CRISPR led to successful reduction of Aβ protein generation in iPSCs (Sun et al., 2019). The Aβ protein-related phenotype was also inhibited by phosphorylation of Threonine 205 (T205) in APP transgenic mice. The post-synaptic mitogen-activated protein kinase (MAPK), p38γ, is further proposed to be involved in regulation (Ittner et al., 2020). Other epigenetically dysregulated loci have been described in a genetic model of Caenorhabditis elegans. In another study, mimicking of phosphorylation of Threonine 231 (T231) and acetylation of Lysine 274 (K274) and Lysine 281 (K281) in C. elegans was associated with age-related reduction in touch sensation and neuronal morphological abnormalities (Guha et al., 2020).
In addition to the loci that have been extensively investigated, other risk loci identified by GWAS remain to be validated in cell/animal models. A number of studies have included peripheral tissues (such as skin tissue) for analysis. However, the pathogenic significance of these newly identified genes in AD remains to be confirmed (Gerring et al., 2020).
Parkinson’s Disease
Parkinson’s disease, another important age-related chronic progressive neurodegenerative disorder, is characterized by aggregation of α-synuclein protein. Numerous studies have focused on PD patients with family history-specific mutations in LRRK2, PARK2, DJ-1, PINK1, and SNCA (Sundal et al., 2012). SNCA is directly related to expression of α-synuclein and one of the most significant prediction sites for sporadic PD (Ferreira and Massano, 2017). Mutation and triplication of SNCA A53T affects nucleocytoplasmic transport mediated by α-synuclein (Chen V. et al., 2020). This regulation has been further confirmed in CRISPR-edited iPSCs (Barbuti et al., 2020). Another newly discovered α-syn SNP site, rs12411216, is reported to regulate the function of glucocerebrosidase, which promotes distribution of α-syn protein (Jiang et al., 2020). An improved SCNA-specific CRISPR technique has been applied to generate a PD cell model (Arias-Fuenzalida et al., 2017). A novel CRISPR-based lentiviral vector has additionally been designed to downregulate transcription and expression though targeted methylation of intron 1 of SNCA (Kantor et al., 2018). Another study showed that cell lines depleted of SNCA present resistance to Lewy pathology (Chen X. et al., 2020).
P13, PINK, and PARKIN are additionally highlighted as therapeutic targets on account of their involvement in regulation of mitochondrial function. Several groups have investigated the effect of PARKIN mutation on expression of PD-related proteins in iPSCs lines (Suda et al., 2018). Decreased expression of P13 is reported to exert neuroprotective effects on genetic PD and toxin-induced PD models. In contrast, overexpression of P13 has been shown to promote the emergence of phenotypes in toxin-induced PD mice (Inoue et al., 2018). Some researchers have proposed references for the construction of a LRRK2-related PD stem cell model through cytogenetic analysis (Vetchinova et al., 2018). Another LRRK2 iPSC model constructed with the TALEN technique could additionally serve as a reference (Ohta et al., 2020). In an interesting study, PARKIN, DJ-1, and ATP13A2 genes were deleted using the CRISPR/Cas system in nigral dopaminergic neurons (DN). Through integration of transcriptome and proteome data, oxidative stress was identified as the common dysregulation pathway of all the isogenic cell lines (Ahfeldt et al., 2020). With elucidation of the molecular mechanisms underlying PD, traditional clinical typing may no longer be applicable. More precise delineation of PD subtypes is required, whereby knowledge of molecular etiology could provide further therapeutic perspectives that may be applicable to all NDD disease types.
A novel mutation in DNAJC6 potentially contributes to early impairment of PD in human embryonic stem cells (hESC) (Wulansari et al., 2021). Moreover, PD-related behavioral deficits have been reported in LIN28A knockout mice (Chang et al., 2019). Similar to AD, several recent GWAS for PD have been conducted (Chang et al., 2017; Nabais et al., 2021). However, the issue of whether these disclosed mutations are valuable for clinical prediction requires further study in cell/animal models.
Huntington’s Disease
Huntington’s disease, a hereditary neurodegenerative disorder characterized by involuntary dance movements and continuous deterioration of behavior and cognition, is commonly associated with disability and early death. HD is distinguished by neuronal loss and astrocytosis in terms of pathology and progressive brain atrophy on imaging. Confirmation of diagnosis is mainly based on family history, clinical symptoms and genetic mutations. Duplication of CAG trinucleotides on exon 1 of Huntington’s gene (HTT) is associated with occurrence of HD (Horvath et al., 2016). Normal CAG repeats on HTT are less than 27 and complete penetration is accomplished when CAG repetition exceeds 39 (McColgan and Tabrizi, 2018). The proteins encoded by the mutated HTT gene (mHTT) cannot participate in physiological cellular mechanisms like their normal protein counterparts and additionally display cytotoxicity. As a disorder caused by single mutation and single abnormal protein, HD is an ideal environment for application of gene therapy.
The construction of HD cell models with gene editing techniques that can be used to validate the efficacy of therapeutic agents has been described in several articles (An et al., 2012, 2014; Xu et al., 2017; Dunbar et al., 2019; Ooi et al., 2019; Malankhanova et al., 2020a). Earlier studies have reported high calcium influx (Vigont et al., 2021) and ultrastructural synapse defects (Malankhanova et al., 2020b) in a HD cell model. Furthermore, the frequency of ultrastructural synapse defects is related to the number of CAG repeats (Morozova et al., 2018).
In another study, deletion of neuronal mHTT was induced via CRISPR/Cas9 in HD140Q-KI mice, which led to a significant reduction in reactive astrocytes and improvement of motor dysfunction in the experimental group (Yang et al., 2017). Targeting on the exon 1 of CAG repeat, another in vivo study successfully interfered HTT expression as well (Ekman et al., 2019; see Table 1). Based on current studies, although inactivation of CAG expression can effectively alleviate the HD phenotype, the apoptotic cells cannot be restored. Following the success of the non-allele-specific CRISPR system in the PD mouse model, allele-specific CRISPR was shown to be effective in two studies (Shin et al., 2016; Monteys et al., 2017). In addition to directly targeting HTT mutations, CITP2, which interacts with mutant huntingtin (Fjodorova et al., 2019), was edited. ZFN and TALEN were also applied to correct repeated expansion of CAG (Fink et al., 2016; Zeitler et al., 2019).
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis is an adult-onset, fatal neurodegenerative disorder. In this disease, apoptosis of the upper motor neurons (e.g., spinal cord, brain stem, and motor cortex) triggers progressive weakness and atrophy of muscles throughout the body, resulting in paralysis and death within 3–5 years after the onset of symptoms. Similar to many other NDDs, ALS is currently incurable. The major pathogenic genes in ALS have been identified as C9orf72, SOD1 (Rosen et al., 1993), FUS, TARDBP, and TBK1 (Müller et al., 2018). However, ALS does not have strong genetic background due to its most cases are sporadic.
Several cell and animal models targeted on SOD1 have been reported (Gaj et al., 2017; Kim et al., 2020). The adeno-associated virus (AAV)-mediated CRISPR system was applied to disrupt SOD1, leading to decreased expression of SOD1 protein in spinal cord and reduction of muscle atrophy in mice. With improvement of motor function, the average survival time of mice increased by 28–30 days (Gaj et al., 2017). Similar favorable results of SOD1 deletion were reported in other animal studies as well (Duan et al., 2020; Lim et al., 2020). Such in vivo studies were summarized in Table 1.
The G4C2 hexanucleotide repeat in C9orf72 is a newly described pathogenic factor (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Its pathogenic mechanism can be complex. Researchers found that the deletion of C9orf72 aggravated the axonal defects and thus increased cell apoptosis (Abo-Rady et al., 2020). While recent studies suggest that this pathogenic expansion can be fully corrected using the CRISPR/Cas system (Ababneh et al., 2020), expression of C9orf72 is also reported to affect efficacy of the gene editing process (Moore et al., 2019) and DSB repair (Andrade et al., 2020). The pathogenic effect may achieved through affecting the GluA Q/R site RNA editing (Konen et al., 2020; Moore et al., 2019) and mitochondrial Ca2+ uptake impairment (Dafinca et al., 2020). Many other risk sites have also been reported, such as KIF5A associating to the cytoskeletal defects in ALS (Nicolas et al., 2018). However, further in-depth studies are currently insufficient on these GWAS-identified sites.
Discussion
In recent decades, integration of the fields of computer science and biology has fueled the development of bioinformatics, with significant improvements in efficacy of analysis and utilization of sequencing data. For instance, genome-wide analysis facilitates prediction of potential risk loci at low cost, which is valuable for complex diseases. To an extent, computer science has revolutionized the paradigm and efficacy of research in traditional experimental biology. These changes have significant implications for gene editing techniques. Despite the fact that gene editing tools are rapidly evolving in terms of improved ease of use and accuracy, actual editing efficacy remains unpredictable, especially in vivo. As an auxiliary discipline, computer science is highly valuable in helping to improve the efficacy of editing tools. To this end, researchers have integrated cell-specific information based on gene expression profiles and biological networks to further develop CRISPR sgRNA design tools and predict the efficacy of the CRISPR/Cas system (Liu et al., 2019). Computer science has become an indispensable part of biological research. With increasingly comprehensive research at the molecular level, the accuracy of interpretation of DNA and RNA sequences depends on the advancement of natural language processing techniques. Based on review of the studies on gene editing tools in NDD, we propose the following transformation of future research patterns: clinical studies provide patient information, computers process the profile and make predictions, experimentalists verify the hypotheses, and the data are collectively used to obtain meaningful conclusions.
From the earliest anti-protein treatment strategy to the gene editing technique, one common feature is the translational gap between human and animal models. Often the performance of therapy in humans does not conform to predictions, which could be attributed to the complexity of the human body. In complex organisms, expression of genes is regulated on a multiple and not linear scale. Similarly, expressed products participate in multiple regulatory mechanisms that form a regulatory DNA-RNA-protein network in the human body. Therefore, the actual results of gene editing are inconsistent due to unknown compensation effects (Sun et al., 2019). However, different types of NDD share common pathways, including mitochondrial dysfunction, cytoskeletal integrity, and DNA repair defects (Chia et al., 2018), suggesting that patient stratification via molecular typing or genotyping is valuable for treatment. The next step in NDD analysis is to explain the association between clinical syndromes and molecular pathogenesis. Considering that changes in the neuronal phenotype can be directly detected through knockin/knockout, gene editing tools should significantly enrich our knowledge of regulation networks within neurons. In the foreseeable future, gene sequencing will become a routine procedure that directly impacts clinical practice (Chia et al., 2018).
In general, research on gene editing techniques in neurogenerative disease has primarily centered on cell/animal models to explore the underlying biological mechanisms and involves multiple disciplines including molecular biology, cell reprogramming, computer science, statistics, and multi-omics. The major current challenge for NDD is unknown pathogenesis. Researchers have attempted to explore the pathological changes of NDD at the molecular level, whereby gene editing tools play a significant role in clarifying the gene-phenotype relationships. While gene editing tools have been updated at a rapid pace, their clinical transformation may not be easily achievable in the near future. In addition, digitalization has been explored as a critical research direction, from designing of editing tools to construction of disease models. Further studies on NDD incorporating participants from diverse academic backgrounds with large-scale studies are warranted.
Author Contributions
XZ: manuscript drafting and review and editing. YZ and XY: conceptualization. CH and HD: supervision and review and editing. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Fund Program for the Scientific Activities of Selected Returned Overseas Professionals in Shanxi Province and Scientific Research Project of Shanxi Provincial Health Commission.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ababneh, N. A., Scaber, J., Flynn, R., Douglas, A., Barbagallo, P., Candalija, A., et al. (2020). Correction of amyotrophic lateral sclerosis related phenotypes in induced pluripotent stem cell-derived motor neurons carrying a hexanucleotide expansion mutation in C9orf72 by CRISPR/Cas9 genome editing using homology-directed repair. Hum. Mol. Genet. 29, 2200–2217. doi: 10.1093/hmg/ddaa106
Abo-Rady, M., Kalmbach, N., Pal, A., Schludi, C., Janosch, A., Richter, T., et al. (2020). Knocking out C9ORF72 exacerbates axonal trafficking defects associated with hexanucleotide repeat expansion and reduces levels of heat shock proteins. Stem Cell Rep. 14, 390–405. doi: 10.1016/j.stemcr.2020.01.010
Ahfeldt, T., Ordureau, A., Bell, C., Sarrafha, L., Sun, C., Piccinotti, S., et al. (2020). Pathogenic pathways in early-onset autosomal recessive Parkinson’s disease discovered using isogenic human dopaminergic neurons. Stem Cell Rep. 14, 75–90. doi: 10.1016/j.stemcr.2019.12.005
Allen, C., Kurimasa, A., Brenneman, M. A., Chen, D. J., and Nickoloff, J. A. (2002). DNA-dependent protein kinase suppresses double-strand break-induced and spontaneous homologous recombination. Proc. Natl. Acad. Sci. U S A. 99, 3758–3763. doi: 10.1073/pnas.052545899
An, M. C., O’Brien, R. N., Zhang, N., Patra, B. N., De La Cruz, M., Ray, A., et al. (2014). Polyglutamine disease modeling: epitope based screen for homologous recombination using CRISPR/Cas9 system. PLoS Curr. 6:ecurrents.hd.0242d2e7ad72225efa72f6964589369a. doi: 10.1371/currents.hd.0242d2e7ad72225efa72f6964589369a
An, M. C., Zhang, N., Scott, G., Montoro, D., Wittkop, T., Mooney, S., et al. (2012). Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell 11, 253–263. doi: 10.1016/j.stem.2012.04.026
Andrade, N. S., Ramic, M., Esanov, R., Liu, W., Rybin, M. J., Gaidosh, G., et al. (2020). Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol. Neurodegener. 15:13. doi: 10.1186/s13024-020-00365-369
Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W., Levy, J. M., et al. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157. doi: 10.1038/s41586-019-1711-1714
Arias-Fuenzalida, J., Jarazo, J., Qing, X., Walter, J., Gomez-Giro, G., Nickels, S. L., et al. (2017). FACS-Assisted CRISPR-Cas9 genome editing facilitates Parkinson’s disease modeling. Stem Cell Rep. 9, 1423–1431. doi: 10.1016/j.stemcr.2017.08.026
Arnold, M., Nho, K., Kueider-Paisley, A., Massaro, T., Huynh, K., Brauner, B., et al. (2020). Sex and APOE ε4 genotype modify the Alzheimer’s disease serum metabolome. Nat. Commun. 11:1148. doi: 10.1038/s41467-020-14959-w
Barbuti, P., Antony, P., Santos, B., Massart, F., Cruciani, G., Dording, C., et al. (2020). Using high-content screening to generate single-cell gene-corrected patient-derived iPS clones reveals excess alpha-synuclein with familial Parkinson’s disease point mutation A30P. Cells 9:2065. doi: 10.3390/cells9092065
Barman, N. C., Khan, N. M., Islam, M., Nain, Z., Roy, R. K., Haque, A., et al. (2020). CRISPR-Cas9: a promising genome editing therapeutic tool for Alzheimer’s Disease-A narrative review. Neurol. Ther. 9, 419–434. doi: 10.1007/s40120-020-00218-z
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. doi: 10.1126/science.1138140
Beerli, R. R., and Barbas, C. F. III (2002). Engineering polydactyl zinc-finger transcription factors. Nat. Biotechnol. 20, 135–141. doi: 10.1038/nbt0202-135
Belloy, M. E., Napolioni, V., Han, S. S., Le Guen, Y., and Greicius, M. D. (2020). Association of Klotho-VS heterozygosity with risk of alzheimer disease in individuals who carry APOE4. JAMA Neurol. 77, 849–862. doi: 10.1001/jamaneurol.2020.0414
Bitinaite, J., Wah, D. A., Aggarwal, A. K., and Schildkraut, I. (1998). FokI dimerization is required for DNA cleavage. Proc. Natl. Acad. Sci. U S A. 95, 10570–10575. doi: 10.1073/pnas.95.18.10570
Boch, J., Scholze, H., Schornack, S., Landgraf, A., Hahn, S., Kay, S., et al. (2009). Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509–1512. doi: 10.1126/science.1178811
Burnham, S. C., Laws, S. M., Budgeon, C. A., Doré, V., Porter, T., Bourgeat, P., et al. (2020). Impact of APOE-ε4 carriage on the onset and rates of neocortical Aβ-amyloid deposition. Neurobiol. Aging 95, 46–55. doi: 10.1016/j.neurobiolaging.2020.06.001
Cai, Y., Cheng, T., Yao, Y., Li, X., Ma, Y., Li, L., et al. (2019). In vivo genome editing rescues photoreceptor degeneration via a Cas9/RecA-mediated homology-directed repair pathway. Sci. Adv. 5:eaav3335. doi: 10.1126/sciadv.aav3335
Carroll, D. (2011). Genome engineering with zinc-finger nucleases. Genetics 188, 773–782. doi: 10.1534/genetics.111.131433
Chandrasegaran, S., and Carroll, D. (2016). Origins of programmable nucleases for genome engineering. J. Mol. Biol. 428, 963–989. doi: 10.1016/j.jmb.2015.10.014
Chang, D., Nalls, M. A., Hallgrímsdóttir, I. B., Hunkapiller, J., van der Brug, M., Cai, F., et al. (2017). A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 49, 1511–1516. doi: 10.1038/ng.3955
Chang, M. Y., Oh, B., Choi, J. E., Sulistio, Y. A., Woo, H. J., Jo, A., et al. (2019). LIN28A loss of function is associated with Parkinson’s disease pathogenesis. EMBO J. 38:e101196. doi: 10.15252/embj.2018101196
Chapman, J. R., Taylor, M. R., and Boulton, S. J. (2012). Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 47, 497–510. doi: 10.1016/j.molcel.2012.07.029
Chen, V., Moncalvo, M., Tringali, D., Tagliafierro, L., Shriskanda, A., Ilich, E., et al. (2020). The mechanistic role of alpha-synuclein in the nucleus: impaired nuclear function caused by familial Parkinson’s disease SNCA mutations. Hum. Mol. Genet. 29, 3107–3121. doi: 10.1093/hmg/ddaa183
Chen, X., Xie, C., Tian, W., Sun, L., Zheng, W., Hawes, S., et al. (2020). Parkinson’s disease-related Leucine-rich repeat kinase 2 modulates nuclear morphology and genomic stability in striatal projection neurons during aging. Mol. Neurodegener. 15:12. doi: 10.1186/s13024-020-00360-360
Chia, R., Chiò, A., and Traynor, B. J. (2018). Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 17, 94–102. doi: 10.1016/s1474-4422(17)30401-30405
Cho, S. W., Kim, S., Kim, J. M., and Kim, J. S. (2013). Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230–232. doi: 10.1038/nbt.2507
Choulika, A., Perrin, A., Dujon, B., and Nicolas, J. F. (1994). The yeast I-Sce I meganuclease induces site-directed chromosomal recombination in mammalian cells. C R Acad. Sci. III 317, 1013–1019.
Choulika, A., Perrin, A., Dujon, B., and Nicolas, J. F. (1995). Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Mol. Cell Biol. 15, 1968–1973. doi: 10.1128/mcb.15.4.1968
Colleaux, L., d’Auriol, L., Betermier, M., Cottarel, G., Jacquier, A., Galibert, F., et al. (1986). Universal code equivalent of a yeast mitochondrial intron reading frame is expressed into E. coli as a specific double strand endonuclease. Cell 44, 521–533. doi: 10.1016/0092-8674(86)90262-x
Dafinca, R., Barbagallo, P., Farrimond, L., Candalija, A., Scaber, J., Ababneh, N. A., et al. (2020). Impairment of mitochondrial calcium buffering links mutations in C9ORF72 and TARDBP in iPS-Derived motor neurons from patients with ALS/FTD. Stem Cell Rep. 14, 892–908. doi: 10.1016/j.stemcr.2020.03.023
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Deng, D., Yan, C., Pan, X., Mahfouz, M., Wang, J., Zhu, J. K., et al. (2012). Structural basis for sequence-specific recognition of DNA by TAL effectors. Science 335, 720–723. doi: 10.1126/science.1215670
Duan, W., Guo, M., Yi, L., Liu, Y., Li, Z., Ma, Y., et al. (2020). The deletion of mutant SOD1 via CRISPR/Cas9/sgRNA prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Ther. 27, 157–169. doi: 10.1038/s41434-019-0116-111
Dunbar, G. L., Koneru, S., Kolli, N., Sandstrom, M., Maiti, P., and Rossignol, J. (2019). Silencing of the mutant huntingtin gene through CRISPR-Cas9 improves the mitochondrial biomarkers in an in vitro model of Huntington’s disease. Cell Transplant 28, 460–463. doi: 10.1177/0963689719840662
Ekman, F. K., Ojala, D. S., Adil, M. M., Lopez, P. A., Schaffer, D. V., and Gaj, T. (2019). CRISPR-Cas9-Mediated genome editing increases lifespan and improves motor deficits in a Huntington’s disease mouse model. Mol. Ther. Nucleic Acids 17, 829–839. doi: 10.1016/j.omtn.2019.07.009
Fegan, C., Sunter, J. P., and Miller, I. A. (1985). Menetrier’s disease complicated by development of the Zollinger-Ellison syndrome. Br. J. Surg. 72, 929–930. doi: 10.1002/bjs.1800721131
Ferreira, M., and Massano, J. (2017). An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta Neurol. Scand. 135, 273–284. doi: 10.1111/ane.12616
Fink, K. D., Deng, P., Gutierrez, J., Anderson, J. S., Torrest, A., Komarla, A., et al. (2016). Allele-Specific reduction of the mutant huntingtin allele using transcription activator-like effectors in human Huntington’s disease fibroblasts. Cell Transplant 25, 677–686. doi: 10.3727/096368916x690863
Fjodorova, M., Louessard, M., Li, Z., De La Fuente, D. C., Dyke, E., Brooks, S. P., et al. (2019). CTIP2-Regulated reduction in PKA-Dependent DARPP32 phosphorylation in human medium spiny neurons: implications for Huntington disease. Stem Cell Rep. 13, 448–457. doi: 10.1016/j.stemcr.2019.07.015
Friedmann, T., and Roblin, R. (1972). Gene therapy for human genetic disease? Science 175, 949–955. doi: 10.1126/science.175.4025.949
Gaj, T., Ojala, D. S., Ekman, F. K., Byrne, L. C., Limsirichai, P., and Schaffer, D. V. (2017). In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci Adv. 3:eaar3952. doi: 10.1126/sciadv.aar3952
Gaj, T., Sirk, S. J., Shui, S. L., and Liu, J. (2016). Genome-Editing technologies: principles and applications. Cold Spring Harb. Perspect. Biol. 8:a023754. doi: 10.1101/cshperspect.a023754
Garneau, J. E., Dupuis, M., Villion, M., Romero, D. A., Barrangou, R., Boyaval, P., et al. (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71. doi: 10.1038/nature09523
Gaudelli, N. M., Komor, A. C., Rees, H. A., Packer, M. S., Badran, A. H., Bryson, D. I., et al. (2018). Publisher correction: programmable base editing of AT to GC in genomic DNA without DNA cleavage. Nature 559:E8. doi: 10.1038/s41586-018-0070-x
Gerring, Z. F., Lupton, M. K., Edey, D., Gamazon, E. R., and Derks, E. M. (2020). An analysis of genetically regulated gene expression across multiple tissues implicates novel gene candidates in Alzheimer’s disease. Alzheimers Res. Ther. 12:43. doi: 10.1186/s13195-020-00611-618
Goate, A., Chartier-Harlin, M. C., Mullan, M., Brown, J., Crawford, F., Fidani, L., et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706. doi: 10.1038/349704a0
Guha, S., Fischer, S., Johnson, G. V. W., and Nehrke, K. (2020). Tauopathy-associated tau modifications selectively impact neurodegeneration and mitophagy in a novel C. elegans single-copy transgenic model. Mol. Neurodegener. 15:65. doi: 10.1186/s13024-020-00410-417
Guo, Y., Wei, X., Yan, H., Qin, Y., Yan, S., Liu, J., et al. (2019). TREM2 deficiency aggravates α-synuclein-induced neurodegeneration and neuroinflammation in Parkinson’s disease models. FASEB J. 33, 12164–12174. doi: 10.1096/fj.201900992R
Haber, J. E. (2016). A life investigating pathways that repair broken chromosomes. Annu. Rev. Genet. 50, 1–28. doi: 10.1146/annurev-genet-120215-135043
Heyer, W. D., Ehmsen, K. T., and Liu, J. (2010). Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 44, 113–139. doi: 10.1146/annurev-genet-051710-150955
Horvath, P., and Barrangou, R. (2010). CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170. doi: 10.1126/science.1179555
Horvath, S., Langfelder, P., Kwak, S., Aaronson, J., Rosinski, J., Vogt, T. F., et al. (2016). Huntington’s disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging (Albany NY) 8, 1485–1512. doi: 10.18632/aging.101005
Hou, Z., and Zhang, Y. (2019). Inserting DNA with CRISPR. Science 365, 25–26. doi: 10.1126/science.aay2056
Hustedt, N., and Durocher, D. (2016). The control of DNA repair by the cell cycle. Nat. Cell Biol. 19, 1–9. doi: 10.1038/ncb3452
Inoue, N., Ogura, S., Kasai, A., Nakazawa, T., Ikeda, K., Higashi, S., et al. (2018). Knockdown of the mitochondria-localized protein p13 protects against experimental parkinsonism. EMBO Rep. 19:e44860. doi: 10.15252/embr.201744860
Ishino, S., Mizukami, T., Yamaguchi, K., Katsumata, R., and Araki, K. (1987). Nucleotide sequence of the meso-diaminopimelate D-dehydrogenase gene from Corynebacterium glutamicum. Nucleic Acids Res. 15:3917. doi: 10.1093/nar/15.9.3917
Ittner, A., Asih, P. R., Tan, A. R. P., Prikas, E., Bertz, J., Stefanoska, K., et al. (2020). Reduction of advanced tau-mediated memory deficits by the MAP kinase p38γ. Acta Neuropathol. 140, 279–294. doi: 10.1007/s00401-020-02191-2191
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. doi: 10.1038/nature08467
Jiang, Z., Huang, Y., Zhang, P., Han, C., Lu, Y., Mo, Z., et al. (2020). Characterization of a pathogenic variant in GBA for Parkinson’s disease with mild cognitive impairment patients. Mol. Brain 13:102. doi: 10.1186/s13041-020-00637-x
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Kantor, B., Tagliafierro, L., Gu, J., Zamora, M. E., Ilich, E., Grenier, C., et al. (2018). Downregulation of SNCA expression by targeted editing of DNA methylation: a potential strategy for precision therapy in PD. Mol. Ther. 26, 2638–2649. doi: 10.1016/j.ymthe.2018.08.019
Kc, M., and Steer, C. J. (2019). A new era of gene editing for the treatment of human diseases. Swiss Med Wkly 149:w20021. doi: 10.4414/smw.2019.20021
Khalil, A. M. (2020). The genome editing revolution: review. J. Genet. Eng. Biotechnol. 18:68. doi: 10.1186/s43141-020-00078-y
Khan, S., Mahmood, M. S., Rahman, S. U., Zafar, H., Habibullah, S., Khan, Z., et al. (2018). CRISPR/Cas9: the Jedi against the dark empire of diseases. J. Biomed. Sci. 25:29. doi: 10.1186/s12929-018-0425-425
Khanna, K. K., and Jackson, S. P. (2001). DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27, 247–254. doi: 10.1038/85798
Kim, B. W., Ryu, J., Jeong, Y. E., Kim, J., and Martin, L. J. (2020). Human motor neurons with SOD1-G93A mutation generated from CRISPR/Cas9 gene-Edited iPSCs develop pathological features of amyotrophic lateral sclerosis. Front. Cell Neurosci. 14:604171. doi: 10.3389/fncel.2020.604171
Kim, J. S., Lee, H. J., and Carroll, D. (2010). Genome editing with modularly assembled zinc-finger nucleases. Nat. Methods 7:91. doi: 10.1038/nmeth0210-91a
Kim, Y. G., and Chandrasegaran, S. (1994). Chimeric restriction endonuclease. Proc. Natl. Acad. Sci. U S A. 91, 883–887. doi: 10.1073/pnas.91.3.883
Klompe, S. E., Vo, P. L. H., Halpin-Healy, T. S., and Sternberg, S. H. (2019). Transposon-encoded CRISPR-Cas systems direct RNA-guided DNA integration. Nature 571, 219–225. doi: 10.1038/s41586-019-1323-z
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A., and Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. doi: 10.1038/nature17946
Konen, L. M., Wright, A. L., Royle, G. A., Morris, G. P., Lau, B. K., Seow, P. W., et al. (2020). A new mouse line with reduced GluA2 Q/R site RNA editing exhibits loss of dendritic spines, hippocampal CA1-neuron loss, learning and memory impairments and NMDA receptor-independent seizure vulnerability. Mol. Brain 13:27. doi: 10.1186/s13041-020-0545-541
Kowalczykowski, S. C. (2015). An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 7:a016410. doi: 10.1101/cshperspect.a016410
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J. C., Damotte, V., Naj, A. C., et al. (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430. doi: 10.1038/s41588-019-0358-352
Kunkle, B. W., Schmidt, M., Klein, H. U., Naj, A. C., Hamilton-Nelson, K. L., Larson, E. B., et al. (2021). Novel Alzheimer disease risk loci and pathways in african american individuals using the african genome resources panel: a meta-analysis. JAMA Neurol. 78, 102–113. doi: 10.1001/jamaneurol.2020.3536
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Lee, M. S., Gippert, G. P., Soman, K. V., Case, D. A., and Wright, P. E. (1989). Three-dimensional solution structure of a single zinc finger DNA-binding domain. Science 245, 635–637. doi: 10.1126/science.2503871
Lessard, C. B., Rodriguez, E., Ladd, T. B., Minter, L. M., Osborne, B. A., Miele, L., et al. (2019). Individual and combined presenilin 1 and 2 knockouts reveal that both have highly overlapping functions in HEK293T cells. J. Biol. Chem. 294, 11276–11285. doi: 10.1074/jbc.RA119.008041
Levy-Lahad, E., Wijsman, E. M., Nemens, E., Anderson, L., Goddard, K. A., Weber, J. L., et al. (1995). A familial Alzheimer’s disease locus on chromosome 1. Science 269, 970–973. doi: 10.1126/science.7638621
Li, L., Wu, L. P., and Chandrasegaran, S. (1992). Functional domains in Fok I restriction endonuclease. Proc. Natl. Acad. Sci. U S A. 89, 4275–4279. doi: 10.1073/pnas.89.10.4275
Li, L., Wu, L. P., Clarke, R., and Chandrasegaran, S. (1993). C-terminal deletion mutants of the FokI restriction endonuclease. Gene 133, 79–84. doi: 10.1016/0378-1119(93)90227-t
Lieber, M. R. (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211. doi: 10.1146/annurev.biochem.052308.093131
Lim, C. K. W., Gapinske, M., Brooks, A. K., Woods, W. S., Powell, J. E., Zeballos, C. M., et al. (2020). Treatment of a mouse model of ALS by in vivo base editing. Mol. Ther. 28, 1177–1189. doi: 10.1016/j.ymthe.2020.01.005
Lindahl, T., and Barnes, D. E. (2000). Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 65, 127–133. doi: 10.1101/sqb.2000.65.127
Liu, Q., He, D., and Xie, L. (2019). Prediction of off-target specificity and cell-specific fitness of CRISPR-Cas System using attention boosted deep learning and network-based gene feature. PLoS Comput. Biol. 15:e1007480. doi: 10.1371/journal.pcbi.1007480
Liu, Q., Segal, D. J., Ghiara, J. B., and Barbas, C. F. III (1997). Design of polydactyl zinc-finger proteins for unique addressing within complex genomes. Proc. Natl. Acad. Sci. U S A. 94, 5525–5530. doi: 10.1073/pnas.94.11.5525
Maeder, M. L., and Gersbach, C. A. (2016). Genome-editing technologies for gene and cell therapy. Mol. Ther. 24, 430–446. doi: 10.1038/mt.2016.10
Malankhanova, T., Sorokin, M., Medvedev, S., Zakian, S., and Malakhova, A. (2020a). Introducing an expanded trinucleotide repeat tract into the human genome for Huntington’s disease modeling in vitro. Curr. Protoc. Hum. Genet. 106:e100. doi: 10.1002/cphg.100
Malankhanova, T., Suldina, L., Grigor’eva, E., Medvedev, S., Minina, J., Morozova, K., et al. (2020b). A human induced pluripotent stem cell-derived isogenic model of Huntington’s disease based on neuronal cells has several relevant phenotypic abnormalities. J. Pers. Med. 10:215. doi: 10.3390/jpm10040215
Maruyama, T., Dougan, S. K., Truttmann, M. C., Bilate, A. M., Ingram, J. R., and Ploegh, H. L. (2015). Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 33, 538–542. doi: 10.1038/nbt.3190
McColgan, P., and Tabrizi, S. J. (2018). Huntington’s disease: a clinical review. Eur. J. Neurol. 25, 24–34. doi: 10.1111/ene.13413
McKhann, G. M., Knopman, D. S., Chertkow, H., Hyman, B. T., Jack, C. R. Jr., Kawas, C. H., et al. (2011). The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269. doi: 10.1016/j.jalz.2011.03.005
Mehta, A., and Haber, J. E. (2014). Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 6:a016428. doi: 10.1101/cshperspect.a016428
Meyer, K., Feldman, H. M., Lu, T., Drake, D., Lim, E. T., Ling, K. H., et al. (2019). REST and neural gene network dysregulation in iPSC Models of Alzheimer’s disease. Cell Rep. 26, 1112–1127.e9. doi: 10.1016/j.celrep.2019.01.023
Miller, J., McLachlan, A. D., and Klug, A. (1985). Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J. 4, 1609–1614. doi: 10.1002/j.1460-2075.1985.tb03825.x
Miller, J. C., Tan, S., Qiao, G., Barlow, K. A., Wang, J., Xia, D. F., et al. (2011). A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 29, 143–148. doi: 10.1038/nbt.1755
Monteys, A. M., Ebanks, S. A., Keiser, M. S., and Davidson, B. L. (2017). CRISPR/Cas9 editing of the mutant huntingtin allele in vitro and in vivo. Mol. Ther. 25, 12–23. doi: 10.1016/j.ymthe.2016.11.010
Moore, S., Alsop, E., Lorenzini, I., Starr, A., Rabichow, B. E., Mendez, E., et al. (2019). ADAR2 mislocalization and widespread RNA editing aberrations in C9orf72-mediated ALS/FTD. Acta Neuropathol. 138, 49–65. doi: 10.1007/s00401-019-01999-w
Morozova, K. N., Suldina, L. A., Malankhanova, T. B., Grigor’eva, E. V., Zakian, S. M., Kiseleva, E., et al. (2018). Introducing an expanded CAG tract into the huntingtin gene causes a wide spectrum of ultrastructural defects in cultured human cells. PLoS One 13:e0204735. doi: 10.1371/journal.pone.0204735
Moscou, M. J., and Bogdanove, A. J. (2009). A simple cipher governs DNA recognition by TAL effectors. Science 326:1501. doi: 10.1126/science.1178817
Müller, K., Brenner, D., Weydt, P., Meyer, T., Grehl, T., Petri, S., et al. (2018). Comprehensive analysis of the mutation spectrum in 301 German ALS families. J. Neurol. Neurosurg. Psychiatry 89, 817–827. doi: 10.1136/jnnp-2017-317611
Murray, J. M., and Carr, A. M. (2018). Integrating DNA damage repair with the cell cycle. Curr. Opin. Cell Biol. 52, 120–125. doi: 10.1016/j.ceb.2018.03.006
Nabais, M. F., Laws, S. M., Lin, T., Vallerga, C. L., Armstrong, N. J., Blair, I. P., et al. (2021). Meta-analysis of genome-wide DNA methylation identifies shared associations across neurodegenerative disorders. Genome Biol. 22:90. doi: 10.1186/s13059-021-02275-2275
Nagata, K., Takahashi, M., Matsuba, Y., Okuyama-Uchimura, F., Sato, K., Hashimoto, S., et al. (2018). Generation of App knock-in mice reveals deletion mutations protective against Alzheimer’s disease-like pathology. Nat. Commun. 9:1800. doi: 10.1038/s41467-018-04238-4230
Nicolas, A., Kenna, K. P., Renton, A. E., Ticozzi, N., Faghri, F., Chia, R., et al. (2018). Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268–1283.e6. doi: 10.1016/j.neuron.2018.02.027
Nishimasu, H., Ran, F. A., Hsu, P. D., Konermann, S., Shehata, S. I., Dohmae, N., et al. (2014). Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949. doi: 10.1016/j.cell.2014.02.001
Ohta, E., Sone, T., Ukai, H., Hisamatsu, T., Kitagawa, T., Ishikawa, M., et al. (2020). Generation of gene-corrected iPSCs line (KEIUi001-A) from a PARK8 patient iPSCs with familial Parkinson’s disease carrying the I2020T mutation in LRRK2. Stem Cell Res. 49:102073. doi: 10.1016/j.scr.2020.102073
Ooi, J., Langley, S. R., Xu, X., Utami, K. H., Sim, B., Huang, Y., et al. (2019). Unbiased profiling of isogenic Huntington disease hPSC-Derived CNS and peripheral cells reveals strong cell-type specificity of CAG length effects. Cell Rep. 26, 2494–2508.e7. doi: 10.1016/j.celrep.2019.02.008
Peters, J. E., Makarova, K. S., Shmakov, S., and Koonin, E. V. (2017). Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proc. Natl. Acad. Sci. U S A. 114, E7358–E7366. doi: 10.1073/pnas.1709035114
Porter, T., Burnham, S. C., Milicic, L., Savage, G., Maruff, P., Lim, Y. Y., et al. (2019). Klotho allele status is not associated with Aβ and APOE ε4-related cognitive decline in preclinical Alzheimer’s disease. Neurobiol. Aging 76, 162–165. doi: 10.1016/j.neurobiolaging.2018.12.014
Ramirez, C. L., Foley, J. E., Wright, D. A., Müller-Lerch, F., Rahman, S. H., Cornu, T. I., et al. (2008). Unexpected failure rates for modular assembly of engineered zinc fingers. Nat. Methods 5, 374–375. doi: 10.1038/nmeth0508-374
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rouet, P., Smih, F., and Jasin, M. (1994). Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl. Acad. Sci. U S A. 91, 6064–6068. doi: 10.1073/pnas.91.13.6064
San Filippo, J., Sung, P., and Klein, H. (2008). Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 77, 229–257. doi: 10.1146/annurev.biochem.77.061306.125255
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., et al. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760. doi: 10.1038/375754a0
Shin, J. W., Kim, K. H., Chao, M. J., Atwal, R. S., Gillis, T., MacDonald, M. E., et al. (2016). Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 25, 4566–4576. doi: 10.1093/hmg/ddw286
Skoneczna, A., Kaniak, A., and Skoneczny, M. (2015). Genetic instability in budding and fission yeast-sources and mechanisms. FEMS Microbiol. Rev. 39, 917–967. doi: 10.1093/femsre/fuv028
Smith, J., Bibikova, M., Whitby, F. G., Reddy, A. R., Chandrasegaran, S., and Carroll, D. (2000). Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res. 28, 3361–3369. doi: 10.1093/nar/28.17.3361
Strecker, J., Ladha, A., Gardner, Z., Schmid-Burgk, J. L., Makarova, K. S., Koonin, E. V., et al. (2019). RNA-guided DNA insertion with CRISPR-associated transposases. Science 365, 48–53. doi: 10.1126/science.aax9181
Suda, Y., Kuzumaki, N., Sone, T., Narita, M., Tanaka, K., Hamada, Y., et al. (2018). Down-regulation of ghrelin receptors on dopaminergic neurons in the substantia nigra contributes to Parkinson’s disease-like motor dysfunction. Mol. Brain 11:6. doi: 10.1186/s13041-018-0349-348
Sugisaki, H., and Kanazawa, S. (1981). New restriction endonucleases from Flavobacterium okeanokoites (FokI) and Micrococcus luteus (MluI). Gene 16, 73–78. doi: 10.1016/0378-1119(81)90062-90067
Sun, J., Carlson-Stevermer, J., Das, U., Shen, M., Delenclos, M., Snead, A. M., et al. (2019). CRISPR/Cas9 editing of APP C-terminus attenuates β-cleavage and promotes α-cleavage. Nat. Commun. 10:53. doi: 10.1038/s41467-018-07971-7978
Sun, N., and Zhao, H. (2013). Transcription activator-like effector nucleases (TALENs): a highly efficient and versatile tool for genome editing. Biotechnol. Bioeng. 110, 1811–1821. doi: 10.1002/bit.24890
Sundal, C., Fujioka, S., Uitti, R. J., and Wszolek, Z. K. (2012). Autosomal dominant Parkinson’s disease. Parkinsonism Relat. Disord. 18, (Suppl. 1), S7–S10. doi: 10.1016/s1353-8020(11)70005-70000
Symington, L. S., and Gautier, J. (2011). Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271. doi: 10.1146/annurev-genet-110410-132435
Szigeti, K., Ihnatovych, I., Birkaya, B., Chen, Z., Ouf, A., Indurthi, D. C., et al. (2020). CHRFAM7A: a human specific fusion gene, accounts for the translational gap for cholinergic strategies in Alzheimer’s disease. EBioMedicine 59:102892. doi: 10.1016/j.ebiom.2020.102892
Thierry, A., and Dujon, B. (1992). Nested chromosomal fragmentation in yeast using the meganuclease I-Sce I: a new method for physical mapping of eukaryotic genomes. Nucleic Acids Res. 20, 5625–5631. doi: 10.1093/nar/20.21.5625
Vacher, M., Porter, T., Villemagne, V. L., Milicic, L., Peretti, M., Fowler, C., et al. (2019). Validation of a priori candidate Alzheimer’s disease SNPs with brain amyloid-beta deposition. Sci. Rep. 9:17069. doi: 10.1038/s41598-019-53604-53605
Vetchinova, A. S., Simonova, V. V., Novosadova, E. V., Manuilova, E. S., Nenasheva, V. V., Tarantul, V. Z., et al. (2018). Cytogenetic analysis of the results of genome editing on the cell model of Parkinson’s disease. Bull. Exp. Biol. Med. 165, 378–381. doi: 10.1007/s10517-018-4174-y
Vigont, V. A., Grekhnev, D. A., Lebedeva, O. S., Gusev, K. O., Volovikov, E. A., Skopin, A. Y., et al. (2021). STIM2 mediates excessive store-operated calcium entry in patient-specific iPSC-Derived neurons modeling a juvenile form of Huntington’s disease. Front. Cell Dev. Biol. 9:625231. doi: 10.3389/fcell.2021.625231
Waugh, D. S., and Sauer, R. T. (1993). Single amino acid substitutions uncouple the DNA binding and strand scission activities of Fok I endonuclease. Proc. Natl. Acad. Sci. U S A. 90, 9596–9600. doi: 10.1073/pnas.90.20.9596
Wulansari, N., Darsono, W. H. W., Woo, H. J., Chang, M. Y., Kim, J., Bae, E. J., et al. (2021). Neurodevelopmental defects and neurodegenerative phenotypes in human brain organoids carrying Parkinson’s disease-linked DNAJC6 mutations. Sci. Adv. 7:eabb1540. doi: 10.1126/sciadv.abb1540
Xu, X., Tay, Y., Sim, B., Yoon, S. I., Huang, Y., Ooi, J., et al. (2017). Reversal of phenotypic abnormalities by CRISPR/Cas9-mediated gene correction in huntington disease patient-derived induced pluripotent stem cells. Stem Cell Rep. 8, 619–633. doi: 10.1016/j.stemcr.2017.01.022
Yang, S., Chang, R., Yang, H., Zhao, T., Hong, Y., Kong, H. E., et al. (2017). CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Invest. 127, 2719–2724. doi: 10.1172/jci92087
Zeitler, B., Froelich, S., Marlen, K., Shivak, D. A., Yu, Q., Li, D., et al. (2019). Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 25, 1131–1142. doi: 10.1038/s41591-019-0478-473
Keywords: gene editing, neurodegenerative disease, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis
Citation: Zhu X, Zhang Y, Yang X, Hao C and Duan H (2021) Gene Therapy for Neurodegenerative Disease: Clinical Potential and Directions. Front. Mol. Neurosci. 14:618171. doi: 10.3389/fnmol.2021.618171
Received: 16 October 2020; Accepted: 07 May 2021;
Published: 14 June 2021.
Edited by:
David Woldbye, University of Copenhagen, DenmarkReviewed by:
Jerome Mertens, Salk Institute for Biological Studies, United StatesShyam Gajavelli, University of Florida, United States
Copyright © 2021 Zhu, Zhang, Yang, Hao and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hubin Duan, aHViaW5kdWFuNjhAMTYzLmNvbQ==; Chunyan Hao, aGFvY2h1bnlhbjY4QDEyNi5jb20=