 Alberto Granzotto
Alberto Granzotto Lorella M. T. Canzoniero
Lorella M. T. Canzoniero Stefano L. Sensi
Stefano L. Sensi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci., 26 November 2020
Sec. Molecular Signalling and Pathways
Volume 13 - 2020 | https://doi.org/10.3389/fnmol.2020.600089
This article is part of the Research TopicExcitotoxicity Turns 50View all 7 articles
Fifty years ago, the seminal work by John Olney provided the first evidence of the neurotoxic properties of the excitatory neurotransmitter glutamate. A process hereafter termed excitotoxicity. Since then, glutamate-driven neuronal death has been linked to several acute and chronic neurological conditions, like stroke, traumatic brain injury, Alzheimer’s, Parkinson’s, and Huntington’s diseases, and Amyotrophic Lateral Sclerosis. Mechanisms linked to the overactivation of glutamatergic receptors involve an aberrant cation influx, which produces the failure of the ionic neuronal milieu. In this context, zinc, the second most abundant metal ion in the brain, is a key but still somehow underappreciated player of the excitotoxic cascade. Zinc is an essential element for neuronal functioning, but when dysregulated acts as a potent neurotoxin. In this review, we discuss the ionic changes and downstream effects involved in the glutamate-driven neuronal loss, with a focus on the role exerted by zinc. Finally, we summarize our work on the fascinating distinct properties of NADPH-diaphorase neurons. This neuronal subpopulation is spared from excitotoxic insults and represents a powerful tool to understand mechanisms of resilience against excitotoxic processes.
Excitotoxicity is a form of neuronal death triggered by excessive and/or sustained exposure to the amino acid glutamate, the primary excitatory neurotransmitter in the brain. Evidence accumulated in the past four decades indicates that excitotoxicity is a critical contributor to the neuronal demise occurring upon acute and chronic neurological conditions, like stroke, Alzheimer’s disease (AD), Huntington’s disease (HD), Amyotrophic Lateral Sclerosis (ALS), and Parkinson’s disease (PD) (Mehta et al., 2013).
Although, 50 years have passed since the first description of glutamate’s neurotoxic activity (Olney, 1969), therapeutic strategies set at counteracting these processes have been only partially exploited. In that regard, the targeting of upstream mechanisms of glutamate-driven neurotoxicity has produced, in the late 80s, an early wave of enthusiasm and fueled a level of optimism that has not been corroborated in the following years. These approaches have been found promising in preclinical models (Lee et al., 1999) but failed in clinical trials (Lee et al., 1999; Ikonomidou and Turski, 2002; Chamorro et al., 2016; Choi, 2020). Only riluzole and memantine, two drugs that target glutamate-driven neuronal death, have been approved for the treatment of ALS and AD, respectively.
Although, most of the preclinical findings failed “the bench to bed” translation, this experimental evidence has significantly helped dissect the molecular underpinnings of excitotoxicity. These studies have also helped provide support for the excitotoxic cascade hypothesis (Zivin and Choi, 1991; Choi, 2020). The construct posits that excitotoxic neuronal death is primarily mediated by the glutamate-driven activation of N-methyl-D-aspartate receptors (NMDARs) and the subsequent toxic intraneuronal accumulation of calcium (Ca2+). The NMDAR-driven Ca2+ overload is, in fact, a mandatory step in the process as most of the downstream mechanisms of the cascade, like the generation of reactive oxygen species (ROS; of mitochondrial and non-mitochondrial origin), or reactive nitrogen species (RNS), the concurrent mitochondrial dysfunction, metabolic impairment, as well as the activation of necrotic/apoptotic pathways, are all Ca2+-dependent processes (Lee et al., 1999; Lai et al., 2014; Bano and Ankarcrona, 2018; Choi, 2020; Swanson and Wang, 2020). However, Ca2+ is not alone, and other cations find a way to participate in the death banquet. Zinc (Zn2+) is, for instance, a VIP guest.
In the review, we provide a brief overview of the role of Zn2+ in the brain and discuss its neurotoxic properties and how they intertwine with the excitotoxic cascade. Finally, we focus on the distinct features of the NADPH-diaphorase neurons, a subpopulation spared from excitotoxic insults offering an intriguing model to further our understanding of neuroprotective mechanisms.
After iron, Zn2+ is the most abundant metal ion in the brain. The cation can be found in either structural or labile forms (Sensi et al., 2009). Structural Zn2+ is tightly bound to proteins/peptides and acts as a critical component for proper protein folding or as the catalytic/co-catalytic element required for several enzymes (McCall et al., 2000).
Labile, free Zn2+ is either stored in the lumen of intracellular organelles, like synaptic “zinkergic” vesicles, mitochondria, lysosomes, the endoplasmic reticulum (ER), and the Golgi apparatus, or bound to metallothioneins (MTs), a class of metal-binding redox-sensitive proteins (Maret, 1994). Under physiological conditions, cytosolic Zn2+ concentrations are kept in a picomolar to a low nanomolar range (Outten and O’Halloran, 2001) through the carefully orchestrated activity of Zn2+ transporters (ZnTs), Zrt-, Irt-related proteins (ZIPs), Zn2+-stores and binding proteins (Sekler et al., 2007; Sensi et al., 2009).
Zn2+ within synaptic vesicles is released, along with glutamate, during excitatory neurotransmission (Sensi et al., 2009). Once released in the synaptic cleft, the cation shapes the post-synaptic glutamate responses by modulating the activity of glutamatergic receptors, like NMDARs and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) (Paoletti et al., 1997; Kalappa et al., 2015). Zn2+ exerts an inhibitory effect on NMDARs by acting on a high (nanomolar) and a low (micromolar) affinity site that is located on the GluN2A and GluN2B subunits, respectively (Rachline et al., 2005). As for AMPARs, the cation has been proposed to exert an inhibitory activity by acting on the histidine residues of the receptor ligand-binding domain (Kalappa et al., 2015). However, direct structural evidence for this interaction is still missing. Of note, recent findings indicate that ZnT1-dependent cation extrusion in the proximity of synaptic NMDARs is required for the Zn2+-dependent inhibition of the receptor (Mellone et al., 2015; Krall et al., 2020). A novel post-synaptic mechanism that may put under a new light the cation’s modulating activities as the metal has been so far thought to only act via its presynaptic release. Extracellular Zn2+ is also implicated in the modulation of neurotrophic signaling as the cation is critical for the activity of the matrix metalloproteinases (MMPs), a class of enzymes involved in matrix remodeling and the maturation of the brain-derived neurotrophic factor (BDNF) from its precursor form proBDNF (Hwang et al., 2005), a process activated by Zn2+ supplementation and impaired by metal chelation (Corona et al., 2010; Frazzini et al., 2018). The proBDNF/BDNF balance is critical for neuronal functioning as the two peptides exert opposite effects. BDNF affects long-term memory processes and neuronal survival. On the contrary, proBDNF inhibits GABAergic neurotransmission (Riffault et al., 2014), facilitates long-term depression (Woo et al., 2005), and activates neuronal death-related pathways (Teng et al., 2005; Mizui et al., 2016). Zn2+ has also been shown to activate the BDNF receptor TrkB directly. This process, called transactivation, is triggered by synaptically-released Zn2+ and/or ROS-driven intracellular Zn2+ elevations (Huang et al., 2008; Huang and McNamara, 2012). However, the mechanisms described “in vitro” settings do not entirely translate in “in vivo” conditions as, surprisingly, TrkB activation has been found to be unaffected in transgenic mice devoid of vesicular Zn2+ (Helgager et al., 2014).
Unlike what is known about vesicular Zn2+, the intracellular labile pools’ physiological significance has been only partially unraveled. Along with its role as a metal reservoir, compelling evidence indicates that releasable Zn2+ can affect mitochondria and lysosomal functioning and, in close analogy with Ca2+, act as a signaling molecule (Yamasaki et al., 2007).
Most importantly, like Ca2+, when dysregulated, Zn2+ can turn into a potent neurotoxin (Sensi et al., 2009).
The contribution of Zn2+ in neurodegenerative processes has been extensively investigated. In conditions characterized by the overactivation of excitatory signaling, synaptically released Zn2+ can flux into post-synaptic neurons through Zn2+ permeable channels (Sensi et al., 2009). Zn2+ entry occurs primarily through voltage-gated Ca2+ channels (VGCCs) and Ca2+ permeable AMPARs (CP-AMPARs) (Sensi et al., 1997, 1999b; McDonald et al., 1998; Colvin et al., 2000; Sheline et al., 2002). AMPAR permeability to Ca2+ and Zn2+ ions is restricted to certain neuronal populations or occurs upon disease associated challenges. The distinct expression pattern and the high permeability to Ca2+ and Zn2+ contribute to the unique role of CP-AMPAR in selective neurodegeneration (see Weiss, 2011 for an extensive review on the topic). NMDARs are poorly permeable to Zn2+ ions (Sensi et al., 1997).
Additional routes of entry are the Na+-Zn2+ exchanger and transient receptor potential channels (TRP); however, their contribution to the cation’s toxic accumulation is still mostly unexplored (Bouron and Oberwinkler, 2014). The exact amount of Zn2+ released from presynaptic terminals and the degree of its contribution to cation accumulation in the post-synaptic neurons are also not completely clear and have been matters of lively debates (Kay and Toth, 2008; Vergnano et al., 2014).
Zn2+ released from intracellular pools also participates in the cation’s cytosolic build-up (McCord and Aizenman, 2014). In this regard, MTs are a significant source of intracellular Zn2+ (Maret, 1994; Aizenman et al., 2000). MTs mobilize a large amount of Zn2+ (ranging 10–100 nM) in response to Ca2+-driven generation of ROS/RNS as well as in conditions of mild acidosis, a combination found in excitotoxic settings and several neurodegenerative conditions. The critical role played by Zn2+ released from MTs in the production of neuronal death is supported by the ability of oxidative agents [i.e., 2,2′-dithiodipyridine (DTDP) or N-ethylmaleimide (NEM)] to trigger widespread apoptotic neuronal death (Aizenman et al., 2000; Gibon et al., 2010). This process is mostly reduced by Zn2+chelators and independent of Ca2+ load. Intraneuronal Zn2+ rises are not the final step of the toxic cascade but are critical to trigger mitochondrial and lysosomal dysfunction, as well as the activation of neurotoxic pathways in the cytosol (Sensi et al., 2009; Ji et al., 2019; Koh et al., 2019).
Mitochondria are a primary target of intracellular Zn2+ as the cation accumulates in the organelles thanks to their steep electrochemical gradient (Δp). Once sequestered, Zn2+, along with Ca2+, contributes to Δp loss and promotes ROS generation (Sensi et al., 1999a; Ji and Weiss, 2018). Zn2+ mobilization is an essential prerequisite to trigger irreversible mitochondrial dysfunction as the cation, by acting in close synergy with Ca2+ damaging effects, promote the full demise of the organelles and, eventually, cell death (Jiang et al., 2001; Granzotto and Sensi, 2015). Within mitochondria, Zn2+ acts by inhibiting complexes of the electron transport chain (ETC) and α-ketoglutarate dehydrogenase (αKGDH) of the Krebs cycle, thereby promoting aberrant ROS production and metabolic failure (Sensi et al., 2009; Ji et al., 2019). Zn2+ interactions with αKGDH and the matrix-facing complexes of the ETC support the presence of the cation in the mitochondrial matrix. Moreover, recent findings indicate that mitochondrial Zn2+ uptake through the activation of the mitochondrial Ca2+ uniporter (MCU) participates in producing the neuronal death found in preclinical models of brain ischemia (Ji et al., 2019, 2020). Zn2+ also triggers the permeabilization of the mitochondrial membrane through the activation of the mitochondrial permeability transition pore (MPTP; a key promoter of cell death; Bernardi et al., 2015), thereby generating the release/production of pro-apoptotic factors [like cytochrome c, apoptosis-inducing factor (AIF), and ΔN-Bcl-XL] (Jiang et al., 2001; Bossy-Wetzel et al., 2004; Bonanni et al., 2006; Ji et al., 2019).
In addition, Zn2+ elevations target lysosomes (Koh et al., 2019). Lysosomal Zn2+ rises, coupled with the accumulation of lipid peroxidation byproducts (4-hydroxinonenal), are instrumental for organelle membrane permeabilization (LMP). LMP results in cation release in the cytosol, along with the activation of lysosomal degrading enzymes. These events are critical for neuronal and astrocyte death when exposed to oxidative challenges (Lee and Koh, 2010; Koh et al., 2019).
Zn2+ also affects many cytosolic pathways to promote demise in the CNS cells, including activation of apoptotic/necrotic pathways, modulation of plasma membrane channels, depletion of metabolic substrates, and the induction of cytosolic oxidative enzymes. In neurons and astrocytes, the metal contributes to NADPH oxidase activation, resulting in aberrant O2– generation (Noh and Koh, 2000; Brennan et al., 2009; Swanson and Wang, 2020). Similarly, Zn2+ activates the neuronal isoform of the nitric oxide synthase (nNOS), thereby promoting increased production of nitric oxide (NO) (Kim and Koh, 2002). These two pathways converge in a process in which O2– + NO generate ONOO– (peroxynitrite), a potently neurotoxic RNS (Bossy-Wetzel et al., 2004). Of note, the Zn2+-driven ROS/RNS production promotes further metal release from intracellular redox-sensitive stores (like MTs), thereby exacerbating a vicious feed-forward loop of cation dyshomeostasis (Corona et al., 2011; Slepchenko et al., 2017). At the cytosolic level, Zn2+ promotes NAD+ depletion, thereby resulting in glyceraldehyde-3-phosphate dehydrogenase (GAPDH, a critical enzyme in the glycolytic pathway) inhibition, ATP breakdown, and eventually energetic neuronal failures (Sheline et al., 2000). This NAD+ depletion may critically impinge on mechanisms that are relevant to aging (Lautrup et al., 2019). Zn2+ also targets and promotes activation of PARP (Kim and Koh, 2002), cyclin-dependent kinase 5 (Cdk5; Tuo et al., 2018), and AMPK (Kim et al., 2020), three molecules involved in cell death pathways.
Finally, Zn2+ mobilization, by contributing to the activation of the CamKII/p38/syntaxin/calcineurin axis, promotes outward potassium (K+) currents, a critical step in the production of neuronal apoptosis (Yu et al., 1997; McCord and Aizenman, 2013; Shah and Aizenman, 2014; Aizenman et al., 2020).
Intriguingly, some neuronal subpopulations are mostly insensitive to excitotoxicity. The phenomenon is present in oculomotor neurons, Onuf’s nucleus neurons, and NADPH-diaphorase neurons (Koh et al., 1986; Brockington et al., 2013). NADPH-diaphorase neurons are a subset of medium-sized aspiny interneurons that are largely spared following excitotoxic hits (Koh et al., 1986; Koh and Choi, 1988; Uemura et al., 1990; Weiss et al., 1994; Granzotto and Sensi, 2015). The subpopulation is characterized by the overexpression of nNOS [also known as NOS1; hereafter termed nNOS (+) neurons (Dawson et al., 1991; Hope et al., 1991)]. nNOS (+) neurons are present with a relatively more significant percentage in the striatum but are also expressed in good numbers in the hippocampus and the cerebral cortex. The subpopulation encompasses various cellular subtypes characterized by distinct morphological, transcriptomic, and functional features (Tricoire and Vitalis, 2012). Early studies have shown that these neurons survive instead of the widespread neuronal loss documented by brain autopsy of AD, HD, and PD patients, three conditions characterized by a robust glutamatergic overdrive (Ferrante et al., 1985; Graveland et al., 1985; Mufson and Brandabur, 1994).
Our group has recently exploited this neuronal subpopulation’s unique features to evaluate the mechanisms that promote resilience to excitotoxicity. Employing an array of single-cell imaging and biochemical approaches, we have demonstrated that nNOS (+) neurons fail to generate ROS in response to excitotoxic stimuli (Canzoniero et al., 2013; Granzotto and Sensi, 2015), a critical step that contributes to their resilience and enhanced survival upon glutamate-driven neurodegeneration.
The investigation of these processes has indicated an intriguing scenario in which the ROS-dependent release of intracellular Zn2+ acts as a critical intermediate step of the excitotoxic process (Granzotto and Sensi, 2015). Thus, experimental data support the notion that Zn2+ participates, with glutamate and Ca2+, in a neurotoxic ménage-à-trois.
Overactivation of NMDARs is the first mandatory step in the excitotoxic cascade; compelling evidence indicates that the receptor triggers the activation of early signaling pathways involving PSD95 and nNOS recruitment as well as aberrant Ca2+-driven induction of nNOS (Szydlowska and Tymianski, 2010; Fricker et al., 2018; Wu and Tymianski, 2018). Disruption of the NMDAR/PSD95/nNOS axis prevents excitotoxic damage in in vitro and in vivo preclinical models of cerebral ischemia (Aarts et al., 2002). Functional, transcriptomic, and biochemical analysis, however, indicate that nNOS (+) neurons express fully operational NMDARs that do not differ from the ones present in the general population of nNOS (−) neurons (Price et al., 1993; Landwehrmeyer et al., 1995; Canzoniero et al., 2013; Granzotto and Sensi, 2015; Granzotto and Sensi, 2015, observations). Interestingly, additional studies have also indicated that nNOS (+) neurons are positive to cobalt staining, a maneuver employed to identify CP-AMPARs, thereby suggesting that these cells possess a significant number of these glutamate receptor subtypes (Weiss et al., 1994). This set of findings supports the notion that NMDAR-driven Ca2+ overload and nNOS activation are necessary but not sufficient steps for the initiation and development of the excitotoxic cascade. Additional downstream processes are required, and Zn2+ participates in these mechanisms with a leading role.
Early studies indicated that mitochondria are critical hubs for the development of the excitotoxic cascade (Ankarcrona et al., 1995). The organelles participate in the clearance of NMDAR-driven cytosolic Ca2+ raises and are instrumental for the activation of apoptotic and necrotic processes (Ankarcrona et al., 1995; Schinder et al., 1996). Mitochondrial Ca2+ overload results in organelle dysfunction, aberrant ROS generation, and, ultimately, neuronal loss (Dugan et al., 1995; Stout et al., 1998; Duchen, 2012; Rizzuto et al., 2012).
Mitochondria of nNOS (+) cells are insensitive to excitotoxicity and have emerged as a critical switch to turn off the injurious process (Canzoniero et al., 2013; Granzotto and Sensi, 2015; Figure 1). Although, mitochondria of these neurons take up large amounts of Ca2+, the organelles respond with minimal Δp losses and negligible generation of ROS (Canzoniero et al., 2013; Granzotto and Sensi, 2015). Early studies have shown that, to counteract the detrimental effects linked to peroxynitrite generation, nNOS (+) neurons express high levels of SOD2, the ROS quenching enzyme that is strategically localized inside of mitochondria (Gonzalez-Zulueta et al., 1998). Therefore, it is conceivable that this constitutive overexpression of SOD2 makes the subpopulation better equipped to cope with the oxidative surge produced by the excitotoxic challenges.
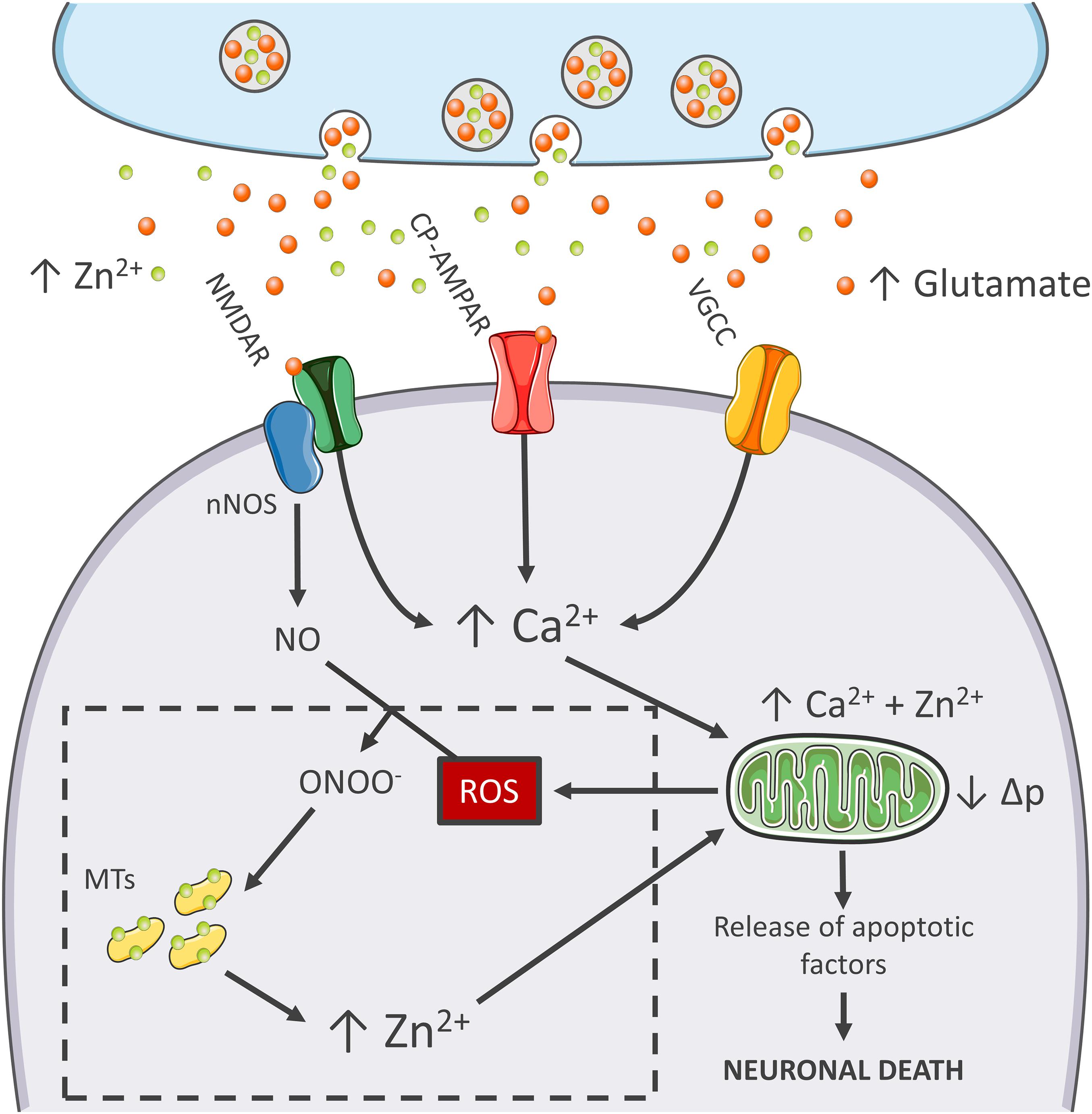
Figure 1. Zn2+ in the excitotoxic cascade. Aberrant release of glutamate from presynaptic terminals triggers NMDAR activation, which, in turn, promotes Ca2+ entry and generation of RNS and ROS of mitochondrial and extramitochondrial origin. The surge of ROS and RNS is required for intraneuronal Zn2+ mobilization from metallothioneins (MTs; Zn2+ buffering redox-sensitive proteins prone to release Zn2+ following oxidative stimuli). Intraneuronal Zn2+ rises target mitochondria and, along with Ca2+, contribute to the organelle impairment. Dysfunctional mitochondria fail to cope with Ca2+ clearance and further exacerbate Ca2+ dysregulation and ROS production. The lack of ROS generation in nNOS (+) neurons is a critical point of divergence in the excitotoxic cascade. By missing the injurious interaction between ROS and RNS, the subpopulation fails to mobilize neurotoxic Zn2+, prevents mitochondrial failure, and eventually neuronal death (dashed line box). In the general population of nNOS (−) neurons, pharmacological Zn2+ chelation prevents the full development of the excitotoxic cascade and mimics nNOS (+) cells’ behavior. These findings lend support to the idea that intraneuronal Zn2+ release is a critical regulator of excitotoxicity.
The idea that mitochondrial dysfunction and oxidative stress are prerequisites for NMDA-driven neuronal loss is in line with the “source-specific” hypothesis of excitotoxicity. The construct posits that the neurotoxic cascade depends on the route of Ca2+ entry, mainly NMDARs, rather than the magnitude of cation load (Wu and Tymianski, 2018). In agreement with this view, abundant Ca2+ entry through VGCCs, a maneuver devoid of neurotoxic effects, fails to trigger ROS and Δp changes (Table 1). This phenomenon shows great analogies with the effects of Ca2+ rises observed in nNOS (+) neurons following NMDAR activation (Granzotto and Sensi, 2015). Although, NMDAR and VGCC activation produces large Ca2+ rises, differences can be found when dissecting the temporal progression of the two stimuli. Unlike VGCC-driven Ca2+ entry, NMDAR overactivation promotes a prolonged and sustained build-up of Ca2+, a phenomenon likely due to impaired cation handling. Conceivably, the NMDAR-driven generation of RNS and ROS can severely affect the mitochondrial Ca2+ buffering as well as the defective extrusion of the cation.

Table 1. The functional hallmarks of excitotoxicity.
RNS/ROS can per se contribute to mitochondrial damage (Murphy, 2009). However, an alternative angle is offered by the mitotoxic properties of Zn2+. The cation represents a critical point of convergence between Ca2+, ROS, and mitochondrial failure. By missing ROS generation, nNOS (+) neurons fail to mobilize intracellular Zn2+ upon NMDAR overactivation (Granzotto and Sensi, 2015). Chelation experiments support the hypothesis that NMDAR-triggered Zn2+ rises are required for the full development of the excitotoxic cascade. In that respect, chelation prevents Zn2+ rises without affecting the upstream mechanisms of the cascade (i.e., Ca2+ entry or the Ca2+-driven generation of RNS/ROS). In nNOS (−) neurons, blockade of excitotoxic-driven Zn2+ elevations results in reduced mitochondrial dysfunction and improved intracellular Ca2+ cycling; two functional changes that closely match the ones observed in the nNOS (+) subpopulation (Granzotto and Sensi, 2015) and provide neuroprotection in excitotoxic settings (Wang and Thayer, 2002). On the contrary, nNOS (+) neurons are extremely vulnerable to pharmacological maneuvers that promote Zn2+ elevations, thereby suggesting that the damaging effects of the cation can override the protective machinery of nNOS (+) neurons (Granzotto and Sensi, 2015, and unpublished observations).
These results are in line with several studies showing that Zn2+ chelation is highly neuroprotective as the maneuver prevents mitochondrial failure, irreversible dysregulation of Ca2+ homeostasis, and eventually neuronal demise (Jiang et al., 2001; Bossy-Wetzel et al., 2004; Medvedeva et al., 2009; Vander Jagt et al., 2009; Clausen et al., 2013; Medvedeva and Weiss, 2014; Ji and Weiss, 2018; Ji et al., 2020; Table 1).
The peculiar milieu offered by nNOS (+) neurons replicates these mechanisms in a naïve, patho-physiologically relevant setting and allows inference on the central role played by Zn2+ in the excitotoxic process (Figure 1). Zn2+ is, therefore, not an accomplice or an amplifier of Ca2+-driven toxicity but rather the downstream executioner. Zn2+ actively promotes mitochondrial dysfunction, Ca2+ dysregulation and, eventually, neuronal death.
The nNOS (+) neurons’ intriguing behavior provides critical insights into the molecular mechanisms involved in excitotoxicity and fosters a critical re-evaluation of the role played by the metal in the modulation of age-related neurodegenerative diseases.
Aging is the primary risk factor for most neurodegenerative conditions, and evidence accumulated over the past 30 years has shown that brain aging is strongly associated with the production of Ca2+ dyshomeostasis and oxidative stress (Beckman and Ames, 1998; Alzheimer’s Association Calcium Hypothesis Workgroup, 2017). Zn2+ can concur in these processes.
In an aging-dependent condition like AD, for example, Zn2+ affects β-amyloid (Aβ) metabolism and the resultant oxidative stress as well as promote tau pathology, the two hallmarks of the disease (Sensi et al., 2018; Figure 2). As for amyloid, Zn2+ is avidly sequestered by Aβ and found highly enriched in senile plaques of AD patients and AD transgenic mice (Bush, 2003; Granzotto et al., 2011). In that regard, a critical role is played by the metal released from presynaptic terminals. Crossing APP mutant mice with ZnT3 KO mice that lack pools of releasable Zn2+ dramatically decreases the Aβ burden (Lee et al., 2002). Of note, the ZnT3 KO mouse model has been proposed to phenocopy AD pathogenesis as the mice develop age-dependent cognitive deficits that overlap with the ones found in AD, thereby supporting the idea that the synaptic deficiency of the cation may participate in shaping disease progression (Deshpande et al., 2009; Adlard et al., 2010). Zn2+ released from presynaptic terminals by inhibiting AMPARs- and NMDARs is also critical for modulating excitatory neurotransmission (Paoletti et al., 2009; Figure 2). Disruption of this process has been associated with seizure-like activity in the hippocampus (Cole et al., 2000), a feature also observed in AD patients and preclinical AD models (Busche and Konnerth, 2015). In agreement, a substantial body of evidence also indicates that, in the early stages, AD is characterized by an aberrant glutamatergic activation (Busche and Konnerth, 2015), a process that leads to Ca2+ and Zn2+ dysregulation (Corona et al., 2011). In this context, the two cations can cooperate to promote the initial steps in the pathogenic cascade that generates the AD-related neuronal loss (Figure 2).
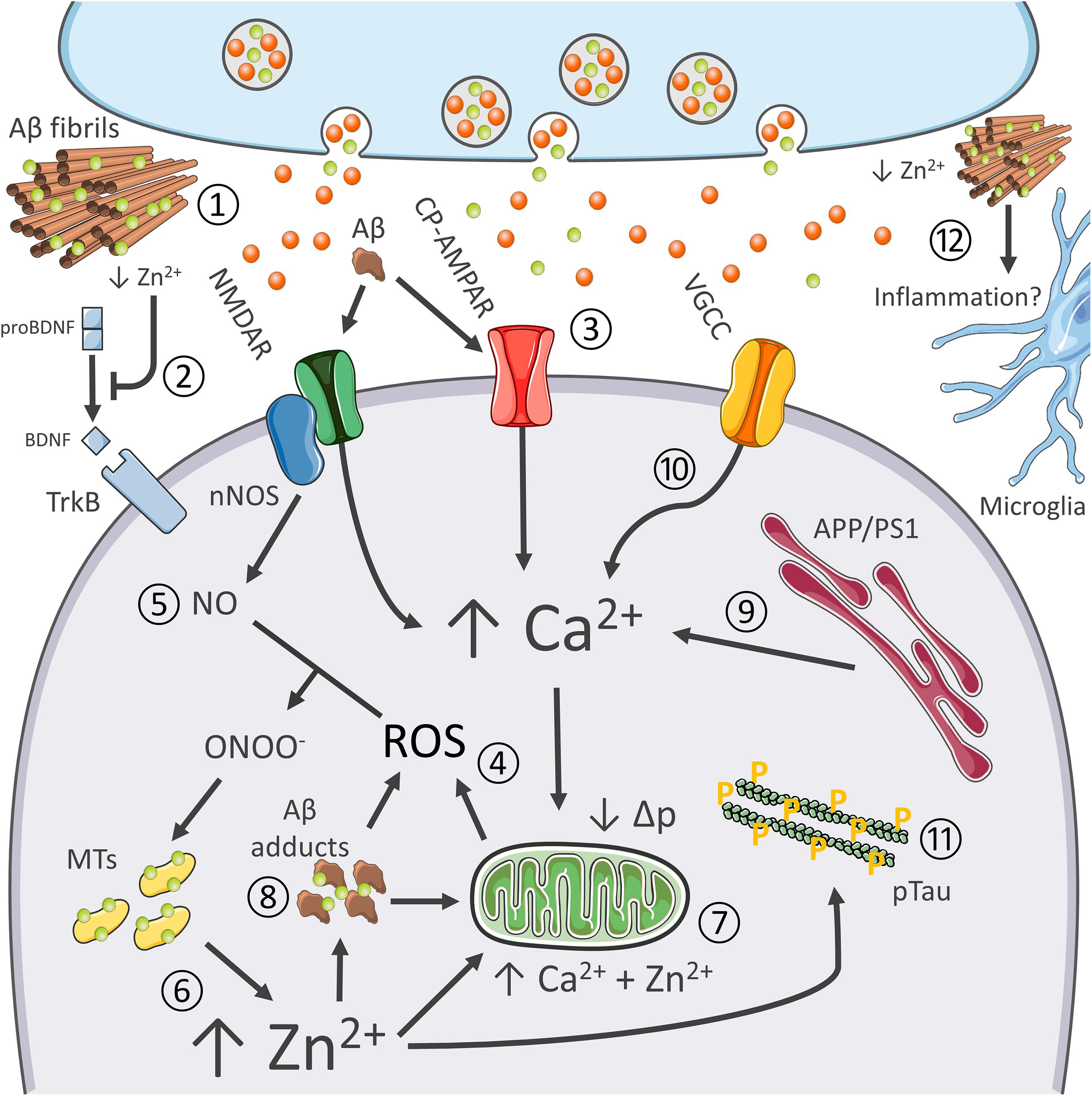
Figure 2. Synergistic contribution of Aβ and tau pathology, oxidative stress, excitotoxicity, Ca2+ dysregulation, and inflammation in AD. Role of Zn2+ in the process. The pictogram summarizes the synergistic interaction between AD-related molecular changes, and the cation dysregulation triggered by altered glutamatergic neurotransmission. Aberrant glutamatergic signaling represents a critical point of convergence of many of the molecular changes observed in AD. Aβ avidly sequesters synaptically released Zn2+ into senile plaques (1). Cation removal from the cleft negatively affects BDNF maturation and, therefore, impinges on neurotrophic signaling (2) as well as Zn2+-dependent NMDAR blockade. Aβ adducts can also directly activate NMDARs and CP-AMPARs, thereby further promoting the glutamatergic overdrive (3). NMDAR and CP-AMPAR overactivation promotes Ca2+ accumulation, increase the intracellular production of ROS (4), as well as the generation of nitric oxide (NO) from nNOS (5). ROS and RNS species are instrumental for Zn2+ release from MTs (6). Zn2+ build-up is potently neurotoxic as the cation, in synergy with Ca2+, further promotes mitochondrial dysfunction, ROS generation, and the release of apoptotic molecules (7). Zn2+ rises may also trigger intraneuronal Aβ aggregation (8). Aβ adducts may further contribute to mitochondrial impairment and generation of ROS. AD-related mutations on PS1 and APP and oxidative stress enhance Ca2+ dyshomeostasis by altering cation handling by the ER (9) and influx through VGCC (10). Ca2+ and Zn2+ rises, along with oxidative stress, also promote tau hyperphosphorylation (11). Finally, recent findings pinpoint at Zn2+ as a critical modulator of neuroinflammatory processes (12).
In the context of AD, multiple factors promote Ca2+ dysregulation. Altered glutamatergic neurotransmission represents a critical point of convergence of many of the molecular changes observed in AD, like vascular dysfunctions, metabolic deficits, and the aggregation of misfolded neurotoxic proteins (Sensi et al., 2018). AD-linked PS1 and APP mutation and accumulation of Aβ adducts can significantly interfere with both Ca2+ homeostasis and glutamatergic neurotransmission (Zhang et al., 2010; Kipanyula et al., 2012); Aβ-driven overactivation of glutamatergic signaling further exacerbate the process (Mattson et al., 1992; Figure 2). Of note, Aβ can also promote the endocytosis of NMDA-type glutamate receptors, a mechanism that, later on, may contribute to the synaptic failure observed in AD (Snyder et al., 2005). In parallel, Ca2+ dyshomeostasis alters APP processing and promotes neurofibrillary tangles (Figure 2).
Changes in cytosolic Ca2+ levels result in mitochondrial dysfunction, nitro/oxidative stress, and eventually neuronal death (Bezprozvanny and Mattson, 2008). Aβ further contributes to ROS’s toxic build-up elicited by Ca2+ dysregulation (Behl et al., 1994). In a self-feeding vicious cycle, oxidative stress can increase Ca2+ influx through VGCC (Todorovic and Jevtovic-Todorovic, 2014), Ca2+ release from intracellular stores through the redox-sensitive Ryanodine receptors (RyRs; Sanmartín et al., 2017), and impair mitochondrial Ca2+ buffering (Görlach et al., 2015).
The milieu triggered by Ca2+ dyshomeostasis and oxidative stress is therefore instrumental in setting in motion the neurotoxic activities of Zn2+ (Figure 2). The hypothesis that oxidative stress and Zn2+ dysregulation concur to promote damage in the aging AD brain is supported by evidence indicating age-dependent changes of molecules that control the brain metal homeostasis (Smith et al., 2006; Lyubartseva et al., 2010). For instance, the gene encoding expression for the neuronal isoform of MT3 is more abundant in aging hippocampal neurons (Giacconi et al., 2003; Scudiero et al., 2017) and AD brains (Hidalgo et al., 2006). While the abundance of MTs may reflect an endogenous protective response to a sub-chronic state of oxidative stress, the proteins can also offer increased availability of releasable Zn2+.
Although, still poorly investigated, Zn2+ dyshomeostasis may also participate in the functional and structural impairment of AD synapses. Evidence indicates that the metal modulates the assembly of critical components of the post-synaptic density (PSD; Grabrucker, 2014). The cation also coordinates the assembly of Shank scaffold proteins within the PSD (Tao-Cheng et al., 2016), complexes that, in turn, modulate synaptic stability and the recruitment of functional AMPARs (Ha et al., 2018). A key phenomenon as these receptors are critical modulators of long-term potentiation and depression processes that underlie memory and learning. Dietary Zn2+ deficiency, or cation sequestration by Aβ, negatively impinges the process, thereby potentially contributing to the synaptic dysfunctions observed in AD (Gong et al., 2009; Grabrucker et al., 2011).
Moreover, neuroinflammation is an emerging contributor factor in AD, and compelling evidence indicates that Zn2+ contributes to microglial activation (Kauppinen et al., 2008). Intriguingly, aspecific Zn2+-related transcriptomic changes have been recently described in xenotransplanted human microglia challenged with pro-inflammatory stimuli (Hasselmann et al., 2019).
Finally, as mentioned above, Zn2+ is also an essential component of MMPs (Page-McCaw et al., 2007). The enzymes, along with their activity on Aβ degradation, participate in the activation of BDNF (Hwang et al., 2005). Zn2+ can, therefore, modulate the neurotrophic axis and affect structural synaptic remodeling, two critical processes associated with learning and memory performances, and found impaired in AD (Weinstein et al., 2014). These findings support the notion of a causal link between Aβ dysmetabolism, Zn2+ dyshomeostasis, and the dysregulation of crucial brain signaling molecules (Figure 2).
Zn2+ dysregulation may also contribute to HD, a condition characterized by the occurrence of a chronic glutamatergic overdrive, mitochondrial dysfunction, and oxidative stress and in which nNOS (+) neurons are spared (Ferrante et al., 1985; Leavitt et al., 2020). Although, still mostly unexplored, similar processes may take place in the motor neuron (MN) loss observed in ALS. MNs show selective expression of CP-AMPARs, thereby making this subpopulation particularly vulnerable to the glutamate-driven oxidative stress and Ca2+ dysregulation (Boillee et al., 2006; Weiss, 2011). The glutamate-Ca2+-Zn2+ cascade described here offers the rationale for exploring mechanisms involved in the degeneration of MN that occurs in ALS.
The selective vulnerability of dopaminergic neurons has been observed in PD, a phenomenon that, in analogy with HD and ALS, is associated with altered glutamatergic signaling. Furthermore, the neuronal loss in PD is also complemented by signs of mitochondrial dysfunctions and oxidative damage, processes that are also modulated by glutamate-driven Zn2+ dysregulation (Iovino et al., 2020).
Glutamate-driven Zn2+ dysregulation plays a significant role in ischemic and traumatic brain injury. Converging evidence accrued from preclinical stroke models indicates that the pharmacological chelation of the cation, as well as the reduction of mitochondrial Zn2+ uptake, prevent ischemic neuronal damage and the development of post-ischemic neurological sequelae (Koh et al., 1996; Frederickson et al., 2004; Zhao et al., 2018; Ji et al., 2019).
While a wealth of evidence has consolidated the notion that Zn2+ dysregulation plays an essential role in the glutamate-driven neuronal demise that occurs in stroke, brain trauma, and AD, less evidence has been gathered in other excitotoxic conditions like ALS, HD, or PD. This lack of information on the role of Zn2+ dyshomeostasis in these conditions constitutes the basis for a call to arms for the neurobiology community. This is becoming particularly relevant as a growing body of evidence is now demonstrating that the damaging processes set in motion in AD, PD, and ALS result from the synergistic activities of many polygenic, epigenetic, environmental, vascular, and metabolic factors, but most importantly make use of a final common neurodegenerative pathway (Sensi et al., 2018).
Furthermore, it is now clear that nosographic distinctions between different neurodegenerative conditions are becoming blurrier and blurrier. Most of the patients, if carefully investigated, exhibit a neuropathology mix characterized by different arrays of neurotoxic proteins like Aβ, tau, prion proteins (PrP), α-synuclein, and TAR DNA binding protein-43 (TDP-43) (Boyle et al., 2018; Karanth et al., 2020). This neuropathological mix is the rule and not the exception in AD and PD- and ALS-related neurodegeneration (Boyle et al., 2018; Karanth et al., 2020). Of great therapeutic and clinical importance, the molecular switches that act up or downstream of the “common neurodegenerative pathway” to turn the process toward the expression of specific clinical phenotypes are still not entirely known. We believe, and that is, in a nutshell, the main goal of this brief review paper, that further research is needed to fully disclose the role of Zn2+ in these processes and the generalizability of its damaging activities across several neurological conditions.
Targeting Zn2+ dysregulation appears, in fact, as a therapeutically exploitable venue (Lynes et al., 2007). Early studies employing clioquinol and its derivative PBT2, two Zn2+ (and copper) chelators, have shown promising effects in preclinical AD models (Cherny et al., 2001; Adlard et al., 2008), as well as in AD and HD clinical trials (Lannfelt et al., 2008; Faux et al., 2010; Angus et al., 2015). However, Zn2+ chelation therapy is not devoid of potential drawbacks. As indicated above, reducing brain Zn2+ levels could also negatively impinge on glutamate neurotransmission (Vergnano et al., 2014; Krall et al., 2020), neurotrophic signaling (Frazzini et al., 2018), and inflammation (Kauppinen et al., 2008; Olechnowicz et al., 2018). Thus, preclinical and clinical studies are needed to evaluate when, where, and how much Zn2+ dysregulation should be therapeutically addressed.
In summary, by bridging Ca2+ dysregulation and oxidative stress, Zn2+ dyshomeostasis may represent a critical common event in many age-related neurodegenerative processes. Dissecting the complex liaisones between Ca2+, ROS, and Zn2+, as well as deciphering their net contribution to specific disease-related pathological changes, may provide new clues for the development of novel therapeutic strategies. Finally, in the complex scenario of neurodegenerative conditions, considerable attention has been put on the selective vulnerability of specific neuronal subtypes (Saxena and Caroni, 2011; Surmeier et al., 2017; Fu et al., 2018). Therefore, the unique features of nNOS (+) neurons may lay the ground for the investigation of the molecular signature of “selective neuronal resilience.”
AG, LC, and SS conceived and designed the study and wrote the manuscript. All authors contributed to the article and approved the submitted version.
SS was supported by research fundings from the Italian Department of Health (RF-2013–02358785 and NET-2011-02346784-1), from the AIRAlzh Onlus (ANCC-COOP), from the Alzheimer’s Association–Part the Cloud: Translational Research Funding for Alzheimer’s Disease (18PTC-19-602325) and the Alzheimer’s Association–GAAIN Exploration to Evaluate Novel Alzheimer’s Queries (GEENA-Q-19-596282). AG was supported by the European Union’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement iMIND—No. 841665.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all the members of the Molecular and Behavioral Neurology Unit for helpful discussions. AG is grateful to Prof. Matt Blurton-Jones, Hayk Davtyan, Ian Smith, and all the members of the Blurton-Jones Lab for the support provided during these difficult times.
Aarts, M., Liu, Y., Liu, L., Besshoh, S., Arundine, M., Gurd, J. W., et al. (2002). Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 298, 846–850. doi: 10.1126/science.1072873
Adlard, P. A., Cherny, R. A., Finkelstein, D. I., Gautier, E., Robb, E., Cortes, M., et al. (2008). Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Aβ. Neuron 59, 43–55. doi: 10.1016/J.NEURON.2008.06.018
Adlard, P. A., Parncutt, J. M., Finkelstein, D. I., and Bush, A. I. (2010). Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 30, 1631–1636. doi: 10.1523/JNEUROSCI.5255-09.2010
Aizenman, E., Loring, R. H., Reynolds, I. J., and Rosenberg, P. A. (2020). The redox biology of excitotoxic processes: the NMDA receptor, TOPA quinone, and the oxidative liberation of intracellular zinc. Front. Neurosci. 14:778. doi: 10.3389/fnins.2020.00778
Aizenman, E., Stout, A. K., Hartnett, K. A., Dineley, K. E., McLaughlin, B., and Reynolds, I. J. (2000). Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J. Neurochem. 75, 1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x
Alzheimer’s Association Calcium Hypothesis Workgroup (2017). Calcium hypothesis of Alzheimer’s disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 13, 178–182.e17. doi: 10.1016/j.jalz.2016.12.006
Angus, D., Herd, C., Stone, C., Stout, J., Wieler, M., Reilmann, R., et al. (2015). Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 14, 39–47. doi: 10.1016/S1474-4422(14)70262-5
Ankarcrona, M., Dypbukt, J. M., Bonfoco, E., Zhivotovsky, B., Orrenius, S., Lipton, S. A., et al. (1995). Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron 15, 961–973. doi: 10.1016/0896-6273(95)90186-8
Bano, D., and Ankarcrona, M. (2018). Beyond the critical point: an overview of excitotoxicity, calcium overload and the downstream consequences. Neurosci. Lett. 663, 79–85. doi: 10.1016/j.neulet.2017.08.048
Beckman, K. B., and Ames, B. N. (1998). The free radical theory of aging matures. Physiol. Rev. 78, 547–581. doi: 10.1152/physrev.1998.78.2.547
Behl, C., Davis, J. B., Lesley, R., and Schubert, D. (1994). Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 77, 817–827. doi: 10.1016/0092-8674(94)90131-7
Bernardi, P., Rasola, A., Forte, M., and Lippe, G. (2015). The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 95, 1111–1155. doi: 10.1152/physrev.00001.2015
Bezprozvanny, I., and Mattson, M. P. (2008). Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 31, 454–463. doi: 10.1016/j.tins.2008.06.005
Boillee, S., Vande Velde, C., and Cleveland, D. W. (2006). ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59. doi: 10.1016/j.neuron.2006.09.018
Bonanni, L., Chachar, M., Jover-Mengual, T., Li, H., Jones, A., Yokota, H., et al. (2006). Zinc-dependent multi-conductance channel activity in mitochondria isolated from ischemic brain. J. Neurosci. 26, 6851–6862. doi: 10.1523/JNEUROSCI.5444-05.2006
Bossy-Wetzel, E., Talantova, M. V., Lee, W. D., Scholzke, M. N., Harrop, A., Mathews, E., et al. (2004). Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron 41, 351–365. doi: 10.1016/s0896-6273(04)00015-7
Bouron, A., and Oberwinkler, J. (2014). Contribution of calcium-conducting channels to the transport of zinc ions. Pflügers Arch. Eur. J. Physiol. 466, 381–387. doi: 10.1007/s00424-013-1295-z
Boyle, P. A., Yu, L., Wilson, R. S., Leurgans, S. E., Schneider, J. A., and Bennett, D. A. (2018). Person-specific contribution of neuropathologies to cognitive loss in old age. Ann. Neurol. 83, 74–83. doi: 10.1002/ana.25123
Brennan, A. M., Suh, S. W., Won, S. J., Narasimhan, P., Kauppinen, T. M., Lee, H., et al. (2009). NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 12, 857–863. doi: 10.1038/nn.2334
Brockington, A., Ning, K., Heath, P. R., Wood, E., Kirby, J., Fusi, N., et al. (2013). Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 125, 95–109. doi: 10.1007/s00401-012-1058-5
Busche, M. A., and Konnerth, A. (2015). Neuronal hyperactivity–A key defect in Alzheimer’s disease? Bioessays 37, 624–632. doi: 10.1002/bies.201500004
Bush, A. I. (2003). The metallobiology of Alzheimer’s disease. Trends Neurosci. 26, 207–214. doi: 10.1016/S0166-2236(03)00067-5
Canzoniero, L. M. T., Granzotto, A., Turetsky, D. M., Choi, D. W., Dugan, L. L., and Sensi, S. L. (2013). nNOS(+) striatal neurons, a subpopulation spared in Huntington’s Disease, possess functional NMDA receptors but fail to generate mitochondrial ROS in response to an excitotoxic challenge. Front. Physiol. 4:112. doi: 10.3389/fphys.2013.00112
Chamorro, Á, Dirnagl, U., Urra, X., and Planas, A. M. (2016). Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 15, 869–881. doi: 10.1016/S1474-4422(16)00114-9
Cherny, R. A., Atwood, C. S., Xilinas, M. E., Gray, D. N., Jones, W. D., McLean, C. A., et al. (2001). Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 30, 665–676. doi: 10.1016/S0896-6273(01)00317-8
Choi, D. W. (2020). Excitotoxicity: still hammering the ischemic brain in 2020. Front. Neurosci. 14:1104. doi: 10.3389/FNINS.2020.579953
Clausen, A., McClanahan, T., Ji, S. G., and Weiss, J. H. (2013). Mechanisms of rapid reactive oxygen species generation in response to cytosolic Ca2+ or Zn2+ loads in cortical neurons. PLoS One 8:e83347. doi: 10.1371/journal.pone.0083347
Cole, T. B., Robbins, C. A., Wenzel, H. J., Schwartzkroin, P. A., and Palmiter, R. D. (2000). Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res. 39, 153–169. doi: 10.1016/s0920-1211(99)00121-7
Colvin, R. A., Davis, N., Nipper, R. W., and Carter, P. A. (2000). Zinc transport in the brain: routes of zinc influx and efflux in neurons. J. Nutr. 130, 1484S–1487S.
Corona, C., Masciopinto, F., Silvestri, E., Del Viscovo, A., Lattanzio, R., La Sorda, R., et al. (2010). Dietary zinc supplementation of 3xTg-AD mice increases BDNF levels and prevents cognitive deficits as well as mitochondrial dysfunction. Cell Death Dis. 1:e91. doi: 10.1038/cddis.2010.73
Corona, C., Pensalfini, A., Frazzini, V., and Sensi, S. L. (2011). New therapeutic targets in Alzheimer’s disease: brain deregulation of calcium and zinc. Cell Death Dis. 2:e176. doi: 10.1038/cddis.2011.57
Dawson, T. M., Bredt, D. S., Fotuhi, M., Hwang, P. M., and Snyder, S. H. (1991). Nitric oxide synthase and neuronal NADPH diaphorase are identical in brain and peripheral tissues. Proc. Natl. Acad. Sci. U.S.A. 88, 7797–7801. doi: 10.1073/pnas.88.17.7797
Deshpande, A., Kawai, H., Metherate, R., Glabe, C. G., and Busciglio, J. (2009). A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J. Neurosci. 29, 4004–4015. doi: 10.1523/JNEUROSCI.5980-08.2009
Duchen, M. R. (2012). Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflügers Arch. Eur. J. Physiol. 464, 111–121. doi: 10.1007/s00424-012-1112-0
Dugan, L. L., Sensi, S. L., Canzoniero, L. M., Handran, S. D., Rothman, S. M., Lin, T. S., et al. (1995). Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-D-aspartate. J. Neurosci. 15, 6377–6388. doi: 10.1523/jneurosci.15-10-06377.1995
Faux, N. G., Ritchie, C. W., Gunn, A., Rembach, A., Tsatsanis, A., Bedo, J., et al. (2010). PBT2 rapidly improves cognition in Alzheimer’s disease: additional phase II analyses. J. Alzheimers Dis. 20, 509–516. doi: 10.3233/JAD-2010-1390
Ferrante, R. J., Kowall, N. W., Beal, M. F., Richardson, E. P. Jr., Bird, E. D., and Martin, J. B. (1985). Selective sparing of a class of striatal neurons in Huntington’s disease. Science 230, 561–563. doi: 10.1126/science.2931802
Frazzini, V., Granzotto, A., Bomba, M., Massetti, N., Castelli, V., D’Aurora, M., et al. (2018). The pharmacological perturbation of brain zinc impairs BDNF-related signaling and the cognitive performances of young mice. Sci. Rep. 8:9768. doi: 10.1038/s41598-018-28083-9
Frederickson, C. J., Maret, W., and Cuajungco, M. P. (2004). Zinc and excitotoxic brain injury: a new model. Neuroscientist 10, 18–25. doi: 10.1177/1073858403255840
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880. doi: 10.1152/physrev.00011.2017
Fu, H., Hardy, J., and Duff, K. E. (2018). Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 21, 1350–1358. doi: 10.1038/s41593-018-0221-2
Giacconi, R., Cipriano, C., Muzzioli, M., Gasparini, N., Orlando, F., and Mocchegiani, E. (2003). Interrelationships among brain, endocrine and immune response in ageing and successful ageing: role of metallothionein III isoform. Mech. Ageing Dev. 124, 371–378. doi: 10.1016/S0047-6374(03)00011-3
Gibon, J., Tu, P., Frazzini, V., Sensi, S. L., and Bouron, A. (2010). The thiol-modifying agent N-ethylmaleimide elevates the cytosolic concentration of free Zn(2+) but not of Ca(2+) in murine cortical neurons. Cell Calcium 48, 37–43. doi: 10.1016/j.ceca.2010.06.004
Gong, Y., Lippa, C. F., Zhu, J., Lin, Q., and Rosso, A. L. (2009). Disruption of glutamate receptors at Shank-postsynaptic platform in Alzheimer’s disease. Brain Res. 1292, 191–198. doi: 10.1016/j.brainres.2009.07.056
Gonzalez-Zulueta, M., Ensz, L. M., Mukhina, G., Lebovitz, R. M., Zwacka, R. M., Engelhardt, J. F., et al. (1998). Manganese superoxide dismutase protects nNOS neurons from NMDA and nitric oxide-mediated neurotoxicity. J. Neurosci. 18, 2040–2055. doi: 10.1523/jneurosci.18-06-02040.1998
Görlach, A., Bertram, K., Hudecova, S., and Krizanova, O. (2015). Calcium and ROS: a mutual interplay. Redox Biol. 6, 260–271. doi: 10.1016/j.redox.2015.08.010
Grabrucker, A. M. (2014). A role for synaptic zinc in ProSAP/Shank PSD scaffold malformation in autism spectrum disorders. Dev. Neurobiol. 74, 136–146. doi: 10.1002/dneu.22089
Grabrucker, A. M., Schmeisser, M. J., Udvardi, P. T., Arons, M., Schoen, M., Woodling, N. S., et al. (2011). Amyloid beta protein-induced zinc sequestration leads to synaptic loss via dysregulation of the ProSAP2/Shank3 scaffold. Mol. Neurodegener. 6, 65. doi: 10.1186/1750-1326-6-65
Granzotto, A., Bolognin, S., Scancar, J., Milacic, R., and Zatta, P. (2011). Beta-amyloid toxicity increases with hydrophobicity in the presence of metal ions. Monatshefte Fur Chem. 142, 421–430. doi: 10.1007/s00706-011-0470-1
Granzotto, A., and Sensi, S. L. (2015). Intracellular zinc is a critical intermediate in the excitotoxic cascade. Neurobiol. Dis. 81, 25–37. doi: 10.1016/j.nbd.2015.04.010
Graveland, G. A., Williams, R. S., and DiFiglia, M. (1985). Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 227, 770–773. doi: 10.1126/science.3155875
Ha, H. T. T., Leal-Ortiz, S., Lalwani, K., Kiyonaka, S., Hamachi, I., Mysore, S. P., et al. (2018). Shank and zinc mediate an AMPA receptor subunit switch in developing neurons. Front. Mol. Neurosci. 11:405. doi: 10.3389/fnmol.2018.00405
Hasselmann, J., Coburn, M. A., England, W., Figueroa Velez, D. X., Kiani Shabestari, S., Tu, C. H., et al. (2019). Development of a chimeric model to study and manipulate human microglia in vivo. Neuron 103, 1016–1033.e10. doi: 10.1016/j.neuron.2019.07.002
Helgager, J., Huang, Y. Z., and Mcnamara, J. O. (2014). Brain-derived neurotrophic factor but not vesicular zinc promotes TrkB activation within mossy fibers of mouse hippocampus in vivo. J. Comp. Neurol. 522, 3885–3899. doi: 10.1002/cne.23647
Hidalgo, J., Penkowa, M., Espejo, C., Martínez-Cáceres, E. M., Carrasco, J., Quintana, A., et al. (2006). Expression of metallothionein-I, -II, and -III in Alzheimer disease and animal models of neuroinflammation. Exp. Biol. Med. 231, 1450–1458. doi: 10.1177/153537020623100902
Hope, B. T., Michael, G. J., Knigge, K. M., and Vincent, S. R. (1991). Neuronal NADPH diaphorase is a nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 88, 2811–2814.
Huang, Y. Z., and McNamara, J. O. (2012). Neuroprotective effects of reactive oxygen species mediated by BDNF-independent activation of TrkB. J. Neurosci. 32, 15521–15532. doi: 10.1523/JNEUROSCI.0755-12.2012
Huang, Y. Z., Pan, E., Xiong, Z. Q., and McNamara, J. O. (2008). Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron 57, 546–558. doi: 10.1016/j.neuron.2007.11.026
Hwang, J. J., Park, M.-H., Choi, S.-Y., and Koh, J.-Y. (2005). Activation of the Trk signaling pathway by extracellular zinc. Role of metalloproteinases. J. Biol. Chem. 280, 11995–12001. doi: 10.1074/jbc.M403172200
Ikonomidou, C., and Turski, L. (2002). Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 1, 383–386. doi: 10.1016/s1474-4422(02)00164-3
Iovino, L., Tremblay, M. E., and Civiero, L. (2020). Glutamate-induced excitotoxicity in Parkinson’s disease: the role of glial cells. J. Pharmacol. Sci. 144, 151–164. doi: 10.1016/j.jphs.2020.07.011
Ji, S. G., Medvedeva, Y. V., Wang, H.-L., Yin, H. Z., and Weiss, J. H. (2019). Mitochondrial Zn2+ accumulation: a potential trigger of hippocampal ischemic injury. Neuroscience 25, 126–138. doi: 10.1177/1073858418772548
Ji, S. G., Medvedeva, Y. V., and Weiss, J. H. (2020). Zn2+ entry through the mitochondrial calcium uniporter is a critical contributor to mitochondrial dysfunction and neurodegeneration. Exp. Neurol. 325:113161. doi: 10.1016/j.expneurol.2019.113161
Ji, S. G., and Weiss, J. H. (2018). Zn2+-induced disruption of neuronal mitochondrial function: synergism with Ca2+, critical dependence upon cytosolic Zn2+ buffering, and contributions to neuronal injury. Exp. Neurol. 302, 181–195. doi: 10.1016/j.expneurol.2018.01.012
Jiang, D., Sullivan, P. G., Sensi, S. L., Steward, O., and Weiss, J. H. (2001). Zn(2+) induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J. Biol. Chem. 276, 47524–47529. doi: 10.1074/jbc.M108834200
Kalappa, B. I., Anderson, C. T., Goldberg, J. M., Lippard, S. J., and Tzounopoulos, T. (2015). AMPA receptor inhibition by synaptically released zinc. Proc. Natl. Acad. Sci. U.S.A. 112, 15749–15754. doi: 10.1073/pnas.1512296112
Karanth, S., Karanth, S., Nelson, P. T., Nelson, P. T., Katsumata, Y., Katsumata, Y., et al. (2020). Prevalence and clinical phenotype of quadruple misfolded proteins in older adults. JAMA Neurol. doi: 10.1001/jamaneurol.2020.1741 [Epub ahead of print].
Kauppinen, T. M., Higashi, Y., Suh, S. W., Escartin, C., Nagasawa, K., and Swanson, R. A. (2008). Zinc triggers microglial activation. J. Neurosci. 28, 5827–5835. doi: 10.1523/JNEUROSCI.1236-08.2008
Kay, A. R., and Toth, K. (2008). Is Zinc a Neuromodulator? Sci. Signal. 1:re3. doi: 10.1126/stke.119re3
Kim, Y. H., Eom, J. W., and Koh, J. Y. (2020). Mechanism of zinc excitotoxicity: a focus on AMPK. Front. Neurosci. 14:958. doi: 10.3389/fnins.2020.577958
Kim, Y.-H., and Koh, J.-Y. (2002). The role of NADPH oxidase and neuronal nitric oxide synthase in zinc-induced poly(ADP-ribose) polymerase activation and cell death in cortical culture. Exp. Neurol. 177, 407–418. doi: 10.1006/exnr.2002.7990
Kipanyula, M. J., Contreras, L., Zampese, E., Lazzari, C., Wong, A. K. C., Pizzo, P., et al. (2012). Ca 2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 11, 885–893. doi: 10.1111/j.1474-9726.2012.00858.x
Koh, J. Y., and Choi, D. W. (1988). Vulnerability of cultured cortical neurons to damage by excitotoxins: differential susceptibility of neurons containing NADPH-diaphorase. J. Neurosci. 8, 2153–2163. doi: 10.1523/jneurosci.08-06-02153.1988
Koh, J. Y., Kim, H. N., Hwang, J. J., Kim, Y. H., and Park, S. E. (2019). Lysosomal dysfunction in proteinopathic neurodegenerative disorders: possible therapeutic roles of cAMP and zinc. Mol. Brain 12, 18. doi: 10.1186/s13041-019-0439-2
Koh, J. Y., Peters, S., and Choi, D. W. (1986). Neurons containing NADPH-diaphorase are selectively resistant to quinolinate toxicity. Science 234, 73–76. doi: 10.1126/science.2875522
Koh, J. Y., Suh, S. W., Gwag, B. J., He, Y. Y., Hsu, C. Y., and Choi, D. W. (1996). The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 272, 1013–1016. doi: 10.1126/science.272.5264.1013
Krall, R. F., Moutal, A., Phillips, M. B., Asraf, H., Johnson, J. W., Khanna, R., et al. (2020). Synaptic zinc inhibition of NMDA receptors depends on the association of GluN2A with the zinc transporter ZnT1. Sci. Adv. 6:eabb1515. doi: 10.1126/sciadv.abb1515
Lai, T. W., Zhang, S., and Wang, Y. T. (2014). Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog. Neurobiol. 115C, 157–188. doi: 10.1016/j.pneurobio.2013.11.006
Landwehrmeyer, G. B., Standaert, D. G., Testa, C. M., Penney, J. B. Jr., and Young, A. B. (1995). NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J. Neurosci. 15, 5297–5307. doi: 10.1523/jneurosci.15-07-05297.1995
Lannfelt, L., Blennow, K., Zetterberg, H., Batsman, S., Ames, D., Harrison, J., et al. (2008). Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 7, 779–786. doi: 10.1016/S1474-4422(08)70167-4
Lautrup, S., Sinclair, D. A., Mattson, M. P., and Fang, E. F. (2019). NAD+ in brain aging and neurodegenerative disorders. Cell Metab. 30, 630–655. doi: 10.1016/j.cmet.2019.09.001
Leavitt, B. R., Kordasiewicz, H. B., and Schobel, S. A. (2020). Huntingtin-lowering therapies for Huntington disease: a review of the evidence of potential benefits and risks. JAMA Neurol. 77, 764–772. doi: 10.1001/jamaneurol.2020.0299
Lee, J.-M. M., Zipfel, G. J., and Choi, D. W. (1999). The changing landscape of ischaemic brain injury mechanisms. Nature 399, A7–A14. doi: 10.1038/399a007
Lee, J.-Y., Cole, T. B., Palmiter, R. D., Suh, S. W., and Koh, J.-Y. (2002). Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 99, 7705–7710. doi: 10.1073/pnas.092034699
Lee, S.-J., and Koh, J.-Y. (2010). Roles of zinc and metallothionein-3 in oxidative stress-induced lysosomal dysfunction, cell death, and autophagy in neurons and astrocytes. Mol. Brain 3:30. doi: 10.1186/1756-6606-3-30
Lynes, M. A., Kang, Y. J., Sensi, S. L., Perdrizet, G. A., and Hightower, L. E. (2007). Heavy metal ions in normal physiology, toxic stress, and cytoprotection. Ann. N. Y. Acad. Sci. 1113, 159–172. doi: 10.1196/annals.1391.010
Lyubartseva, G., Smith, J. L., Markesbery, W. R., and Lovell, M. A. (2010). Alterations of zinc transporter proteins ZnT-1, ZnT-4 and ZnT-6 in preclinical Alzheimer’s disease brain. Brain Pathol. 20, 343–350. doi: 10.1111/j.1750-3639.2009.00283.x
Maret, W. (1994). Oxidative metal release from metallothionein via zinc-thiol/disulfide interchange. Proc. Natl. Acad. Sci. U.S.A. 91, 237–241. doi: 10.1073/pnas.91.1.237
Mattson, M. P., Cheng, B., Davis, D., Bryant, K., Lieberburg, I., and Rydel, R. E. (1992). beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 12, 376–389. doi: 10.1523/jneurosci.12-02-00376.1992
McCall, K. A., Huang, C., and Fierke, C. A. (2000). Function and mechanism of zinc metalloenzymes. J. Nutr. 130, 1437S–1446S.
McCord, M. C., and Aizenman, E. (2013). Convergent Ca2+ and Zn2+ signaling regulates apoptotic Kv2.1 K+ currents. Proc. Natl. Acad. Sci. U.S.A. 110, 13988–13993. doi: 10.1073/pnas.1306238110
McCord, M. C., and Aizenman, E. (2014). The role of intracellular zinc release in aging, oxidative stress, and Alzheimer’s disease. Front. Aging Neurosci. 6:77. doi: 10.3389/fnagi.2014.00077
McDonald, J. W., Bhattacharyya, T., Sensi, S. L., Lobner, D., Ying, H. S., Canzoniero, L. M., et al. (1998). Extracellular acidity potentiates AMPA receptor-mediated cortical neuronal death. J. Neurosci. 18, 6290–6299. doi: 10.1523/jneurosci.18-16-06290.1998
Medvedeva, Y. V., Lin, B., Shuttleworth, C. W., and Weiss, J. H. (2009). Intracellular Zn2+ accumulation contributes to synaptic failure, mitochondrial depolarization, and cell death in an acute slice oxygen-glucose deprivation model of ischemia. J. Neurosci. 29, 1105–1114. doi: 10.1523/JNEUROSCI.4604-08.2009
Medvedeva, Y. V., and Weiss, J. H. (2014). Intramitochondrial Zn2+ accumulation via the Ca2+ uniporter contributes to acute ischemic neurodegeneration. Neurobiol. Dis. 68, 137–144. doi: 10.1016/j.nbd.2014.04.011
Mehta, A., Prabhakar, M., Kumar, P., Deshmukh, R., and Sharma, P. L. (2013). Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 698, 6–18. doi: 10.1016/j.ejphar.2012.10.032
Mellone, M., Pelucchi, S., Alberti, L., Genazzani, A. A., Di Luca, M., and Gardoni, F. (2015). Zinc transporter-1: a novel NMDA receptor-binding protein at the postsynaptic density. J. Neurochem. 132, 159–168. doi: 10.1111/jnc.12968
Mizui, T., Ishikawa, Y., Kumanogoh, H., and Kojima, M. (2016). Neurobiological actions by three distinct subtypes of brain-derived neurotrophic factor: multi-ligand model of growth factor signaling. Pharmacol. Res. 105, 93–98. doi: 10.1016/j.phrs.2015.12.019
Mufson, E. J., and Brandabur, M. M. (1994). Sparing of NADPH-diaphorase striatal neurons in Parkinson’s and Alzheimer’s diseases. Neuroreport 5, 705–708. doi: 10.1097/00001756-199402000-00011
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Noh, K. M., and Koh, J. Y. (2000). Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J. Neurosci. 20:RC111.
Olechnowicz, J., Tinkov, A., Skalny, A., and Suliburska, J. (2018). Zinc status is associated with inflammation, oxidative stress, lipid, and glucose metabolism. J. Physiol. Sci. 68, 19–31. doi: 10.1007/s12576-017-0571-7
Olney, J. W. (1969). Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 164, 719–721. doi: 10.1126/science.164.3880.719
Outten, C. E., and O’Halloran, T. V. (2001). Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science 292, 2488–2492. doi: 10.1126/science.1060331
Page-McCaw, A., Ewald, A. J., and Werb, Z. (2007). Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 8, 221–233. doi: 10.1038/nrm2125
Paoletti, P., Ascher, P., and Neyton, J. (1997). High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J. Neurosci. 17, 5711–5725. doi: 10.1523/jneurosci.17-15-05711.1997
Paoletti, P., Vergnano, A. M., Barbour, B., and Casado, M. (2009). Zinc at glutamatergic synapses. Neuroscience 158, 126–136. doi: 10.1016/j.neuroscience.2008.01.061
Price, R. H. Jr., Mayer, B., and Beitz, A. J. (1993). Nitric oxide synthase neurons in rat brain express more NMDA receptor mRNA than non-NOS neurons. Neuroreport 4, 807–810. doi: 10.1097/00001756-199306000-00053
Rachline, J., Perin-Dureau, F., Le Goff, A., Neyton, J., and Paoletti, P. (2005). The micromolar zinc-binding domain on the NMDA receptor subunit NR2B. J. Neurosci. 25, 308–317. doi: 10.1523/JNEUROSCI.3967-04.2005
Riffault, B., Medina, I., Dumon, C., Thalman, C., Ferrand, N., Friedel, P., et al. (2014). Pro-brain-derived neurotrophic factor inhibits gabaergic neurotransmission by activating endocytosis and repression of GABAA receptors. J. Neurosci. 34, 13516–13534. doi: 10.1523/JNEUROSCI.2069-14.2014
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578. doi: 10.1038/nrm3412
Sanmartín, C. D., Veloso, P., Adasme, T., Lobos, P., Bruna, B., Galaz, J., et al. (2017). RyR2-mediated Ca2+ release and mitochondrial ROS generation partake in the synaptic dysfunction caused by amyloid β peptide oligomers. Front. Mol. Neurosci. 10:115. doi: 10.3389/fnmol.2017.00115
Saxena, S., and Caroni, P. (2011). Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 71, 35–48. doi: 10.1016/j.neuron.2011.06.031
Schinder, A. F., Olson, E. C., Spitzer, N. C., and Montal, M. (1996). Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J. Neurosci. 16, 6125–6133. doi: 10.1523/jneurosci.16-19-06125.1996
Scudiero, R., Cigliano, L., and Verderame, M. (2017). Age-related changes of metallothionein 1/2 and metallothionein 3 expression in rat brain. Comptes Rendus Biol. 340, 13–17. doi: 10.1016/j.crvi.2016.11.003
Sekler, I., Sensi, S. L., Hershfinkel, M., and Silverman, W. F. (2007). Mechanism and regulation of cellular zinc transport. Mol. Med. 13, 337–343. doi: 10.2119/2007-00037.sekler
Sensi, S. L., Canzoniero, L. M., Yu, S. P., Ying, H. S., Koh, J. Y., Kerchner, G. A., et al. (1997). Measurement of intracellular free zinc in living cortical neurons: routes of entry. J. Neurosci. 17, 9554–9564. doi: 10.1523/jneurosci.17-24-09554.1997
Sensi, S. L., Paoletti, P., Bush, A. I., and Sekler, I. (2009). Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 10, 780–791. doi: 10.1038/nrn2734
Sensi, S. L., Yin, H. Z., Carriedo, S. G., Rao, S. S., and Weiss, J. H. (1999a). Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc. Natl. Acad. Sci. U.S.A. 96, 2414–2419. doi: 10.1073/PNAS.96.5.2414
Sensi, S. L., Yin, H. Z., and Weiss, J. H. (1999b). Glutamate triggers preferential Zn2+ flux through Ca2+ permeable AMPA channels and consequent ROS production. Neuroreport 10, 1723–1727. doi: 10.1097/00001756-199906030-00018
Sensi, S. L. S. L., Granzotto, A., Siotto, M., and Squitti, R. (2018). Copper and zinc dysregulation in Alzheimer’s disease. Trends Pharmacol. Sci. 39, 1049–1063. doi: 10.1016/j.tips.2018.10.001
Shah, N. H., and Aizenman, E. (2014). Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Transl. Stroke Res. 5, 38–58. doi: 10.1007/s12975-013-0297-7
Sheline, C. T., Behrens, M. M., and Choi, D. W. (2000). Zinc-induced cortical neuronal death: contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis. J. Neurosci. 20, 3139–3146. doi: 10.1523/jneurosci.20-09-03139.2000
Sheline, C. T., Ying, H. S., Ling, C. S., Canzoniero, L. M. T., and Choi, D. W. (2002). Depolarization-induced 65zinc influx into cultured cortical neurons. Neurobiol. Dis. 10, 41–53. doi: 10.1006/nbdi.2002.0497
Slepchenko, K. G., Lu, Q., and Li, Y. V. (2017). Cross talk between increased intracellular zinc (Zn2+) and accumulation of reactive oxygen species in chemical ischemia. Am. J. Physiol. Cell Physiol. 313, C48–C459. doi: 10.1152/ajpcell.00048.2017
Smith, J. L., Xiong, S., Markesbery, W. R., and Lovell, M. A. (2006). Altered expression of zinc transporters-4 and -6 in mild cognitive impairment, early and late Alzheimer’s disease brain. Neuroscience 140, 879–888. doi: 10.1016/j.neuroscience.2006.02.049
Snyder, E. M., Nong, Y., Almeida, C. G., Paul, S., Moran, T., Choi, E. Y., et al. (2005). Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 8, 1051–1058. doi: 10.1038/nn1503
Stout, A. K., Raphael, H. M., Kanterewicz, B. I., Klann, E., and Reynolds, I. J. (1998). Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat. Neurosci. 1, 366–373. doi: 10.1038/1577
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017). Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 18, 101–113. doi: 10.1038/nrn.2016.178
Swanson, R. A., and Wang, J. (2020). Superoxide and non-ionotropic signaling in neuronal excitotoxicity. Front. Neurosci. 14:861. doi: 10.3389/FNINS.2020.00861
Szydlowska, K., and Tymianski, M. (2010). Calcium, ischemia and excitotoxicity. Cell Calcium 47, 122–129. doi: 10.1016/j.ceca.2010.01.003
Tao-Cheng, J.-H., Toy, D., Winters, C. A., Reese, T. S., and Dosemeci, A. (2016). Zinc stabilizes Shank3 at the postsynaptic density of hippocampal synapses. PLoS One 11:e0153979. doi: 10.1371/journal.pone.0153979
Teng, H. K., Teng, K. K., Lee, R., Wright, S., Tevar, S., Almeida, R. D., et al. (2005). ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 25, 5455–5463. doi: 10.1523/JNEUROSCI.5123-04.2005
Todorovic, S. M., and Jevtovic-Todorovic, V. (2014). Redox regulation of neuronal voltage-gated calcium channels. Antioxid. Redox Signal. 21, 880–891. doi: 10.1089/ars.2013.5610
Tricoire, L., and Vitalis, T. (2012). Neuronal nitric oxide synthase expressing neurons: a journey from birth to neuronal circuits. Front. Neural Circuits 6:82. doi: 10.3389/fncir.2012.00082
Tuo, Q. Z., Liuyang, Z. Y., Lei, P., Yan, X., Shentu, Y. P., Liang, J. W., et al. (2018). Zinc induces CDK5 activation and neuronal death through CDK5-Tyr15 phosphorylation in ischemic stroke. Cell Death Dis. 9:870. doi: 10.1038/s41419-018-0929-7
Uemura, Y., Kowall, N. W., and Beal, M. F. (1990). Selective sparing of NADPH-diaphorase-somatostatin-neuropeptide Y neurons in ischemic gerbil striatum. Ann. Neurol. 27, 620–625. doi: 10.1002/ana.410270606
Vander Jagt, T. A., Connor, J. A., Weiss, J. H., and Shuttleworth, C. W. (2009). Intracellular Zn2+ increases contribute to the progression of excitotoxic Ca2+ increases in apical dendrites of CA1 pyramidal neurons. Neuroscience 159, 104–114. doi: 10.1016/j.neuroscience.2008.11.052
Vergnano, A. M., Rebola, N., Savtchenko, L. P., Pinheiro, P. S., Casado, M., Kieffer, B. L., et al. (2014). Zinc dynamics and action at excitatory synapses. Neuron 82, 1101–1114. doi: 10.1016/j.neuron.2014.04.034
Wang, G. J., and Thayer, S. A. (2002). NMDA-induced calcium loads recycle across the mitochondrial inner membrane of hippocampal neurons in culture. J. Neurophysiol. 87, 740–749. doi: 10.1152/jn.00345.2001
Weinstein, G., Beiser, A. S., Choi, S. H., Preis, S. R., Chen, T. C., Vorgas, D., et al. (2014). Serum brain-derived neurotrophic factor and the risk for dementia: the Framingham Heart Study. JAMA Neurol. 71, 55–61. doi: 10.1001/jamaneurol.2013.4781
Weiss, J. H. (2011). Ca2+ permeable AMPA channels in diseases of the nervous system. Front. Mol. Neurosci. 4:42. doi: 10.3389/fnmol.2011.00042
Weiss, J. H., Turetsky, D., Wilke, G., and Choi, D. W. (1994). AMPA/kainate receptor-mediated damage to NADPH-diaphorase-containing neurons is Ca2+ dependent. Neurosci. Lett. 167, 93–96. doi: 10.1016/0304-3940(94)91035-9
Woo, N. H., Teng, H. K., Siao, C.-J., Chiaruttini, C., Pang, P. T., Milner, T. A., et al. (2005). Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat. Neurosci. 8, 1069–1077. doi: 10.1038/nn1510
Wu, Q. J., and Tymianski, M. (2018). Targeting nmda receptors in stroke: new hope in neuroprotection Tim Bliss. Mol. Brain 11, 15. doi: 10.1186/s13041-018-0357-8
Yamasaki, S., Sakata-Sogawa, K., Hasegawa, A., Suzuki, T., Kabu, K., Sato, E., et al. (2007). Zinc is a novel intracellular second messenger. J. Cell Biol. 177, 637–645. doi: 10.1083/jcb.200702081
Yu, S. P., Yeh, C. H., Sensi, S. L., Gwag, B. J., Canzoniero, L. M., Farhangrazi, Z. S., et al. (1997). Mediation of neuronal apoptosis by enhancement of outward potassium current. Science 278, 114–117. doi: 10.1126/science.278.5335.114
Zhang, H., Sun, S., Herreman, A., De Strooper, B., and Bezprozvanny, I. (2010). Role of presenilins in neuronal calcium homeostasis. J. Neurosci. 30, 8566–8580. doi: 10.1523/JNEUROSCI.1554-10.2010
Zhao, Y., Yan, F., Yin, J., Pan, R., Shi, W., Qi, Z., et al. (2018). Synergistic interaction between zinc and reactive oxygen species amplifies ischemic brain injury in rats. Stroke 49, 2200–2210. doi: 10.1161/STROKEAHA.118.021179
Keywords: Alzheimer’s disease, Amyotrophic lateral sclerosis, Huntington’s disease, Parkinson’s disease, mitochondria, reactive oxygen species, reactive nitrogen species, NADPH-diaphorase
Citation: Granzotto A, Canzoniero LMT and Sensi SL (2020) A Neurotoxic Ménage-à-trois: Glutamate, Calcium, and Zinc in the Excitotoxic Cascade. Front. Mol. Neurosci. 13:600089. doi: 10.3389/fnmol.2020.600089
Received: 28 August 2020; Accepted: 30 October 2020;
Published: 26 November 2020.
Edited by:
Fiorenzo Conti, Marche Polytechnic University, ItalyReviewed by:
Carlos B. Duarte, University of Coimbra, PortugalCopyright © 2020 Granzotto, Canzoniero and Sensi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alberto Granzotto, YWxiZXJ0by5ncmFuem90dG9AdW5pY2guaXQ=; YWxiZXJ0by5ncmFuem90dG9AdWNpLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.