 Audrey Hector
Audrey Hector Jonathan Brouillette
Jonathan Brouillette
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 07 January 2021
Sec. Brain Disease Mechanisms
Volume 13 - 2020 | https://doi.org/10.3389/fnmol.2020.600084
This article is part of the Research Topic Beta Amyloid: From Physiology to Pathogenesis View all 15 articles
Soluble amyloid-beta oligomers (Aβo) start to accumulate in the human brain one to two decades before any clinical symptoms of Alzheimer's disease (AD) and are implicated in synapse loss, one of the best predictors of memory decline that characterize the illness. Cognitive impairment in AD was traditionally thought to result from a reduction in synaptic activity which ultimately induces neurodegeneration. More recent evidence indicates that in the early stages of AD synaptic failure is, at least partly, induced by neuronal hyperactivity rather than hypoactivity. Here, we review the growing body of evidence supporting the implication of soluble Aβo on the induction of neuronal hyperactivity in AD animal models, in vitro, and in humans. We then discuss the impact of Aβo-induced hyperactivity on memory performance, cell death, epileptiform activity, gamma oscillations, and slow wave activity. We provide an overview of the cellular and molecular mechanisms that are emerging to explain how Aβo induce neuronal hyperactivity. We conclude by providing an outlook on the impact of hyperactivity for the development of disease-modifying interventions at the onset of AD.
Synapse loss that precedes neuronal death is the strongest predictor of cognitive decline in Alzheimer's disease (AD) (Alzheimer's, 2020). Although we still need to uncover all the cellular and molecular events leading to neurodegeneration in AD, it is well-established that toxic soluble low-molecular-weight amyloid-beta oligomers (Aβo) play an essential role in synapse loss and strongly correlate with the clinical state of AD patients (Selkoe, 2002; Brouillette, 2014). Since soluble Aβo start to accumulate in the brain up to two decades before the appearance of clinical symptoms (Cline et al., 2018), understanding how Aβ pathology disturbs cell functioning and neuronal networks would be exceedingly beneficial to develop novel therapeutic approaches to prevent memory deficits at the onset of AD before neurodegeneration induces irreversible brain damages that drastically compromises the quality of life of the patient.
Aβ peptides are composed of 36–43 amino acids and are produced by the proteolytic cleavage of the transmembrane amyloid precursor protein (APP) by β- and γ-secretases (Haass et al., 2012). Given their hydrolytic properties, Aβ peptides (especially Aβ1−42) tend to oligomerize rapidly and dynamically until they form insoluble fibrils that aggregate into plaques. Many Aβ species have been shown to be neurotoxic such as dimers, trimers, tetramers, nonamers, dodecamers, protofibrils, and fibrils (Brouillette, 2014). Whereas some reports have highlighted the neurotoxic effects of particular Aβ intermediates with a defined size and structure, other studies have used mixtures of various Aβ species to measure the global impact of the different species that are found simultaneous in the brain. Although a large amount of studies have consistently reported the deleterious impact of soluble Aβo on synapse function and cognitive performance using different types of Aβ preparations in AD mouse models, in vitro, and in humans, the primary events disturbed by Aβo which drive the neurodegenerative process still need to be elucidated.
Cognitive impairment in AD was traditionally assumed to originate from lower synaptic activity that eventually lead to neurodegeneration. Multiple lines of evidence now indicate that, particularly in the early stages of AD, synapse dysfunction and loss are first induced by neuronal hyperactivity rather than hypoactivity (Busche et al., 2012, 2015a). Over the past few years, a growing body of evidence has highlighted the major role of soluble Aβo in the induction of neuronal hyperactivity at the onset of AD. Based on AD animal models, in vitro experiments and human studies, Aβo-induced neuronal hyperactivity has emerged as an early functional hallmark of AD which triggers synaptic failure, memory dysfunction, epileptiform activity, and neurodegeneration.
In human, brain activity can be investigated by functional magnetic resonance imaging (fMRI), positron emission tomography (PET), single-photon emission computed tomography (SPECT), and electroencephalogram (EEG) recordings at resting state or while executing a cognitive task. Hippocampal hyperactivation has been detected by fMRI during memory-encoding tasks in people with mild cognitive impairment (MCI), a prodromal stage of AD, as well as in pre-symptomatic individuals carrying the E280A presenilin-1 (PS1) mutation, the most common cause of early-onset familial AD (Dickerson et al., 2005; Celone et al., 2006; Quiroz et al., 2010; Bakker et al., 2012; Sepulveda-Falla et al., 2012) (Table 1). Higher hippocampal activation was also observed before any clinical symptoms in carriers of the APOE4 allele, the most important genetic risk factor for late-onset sporadic AD (Bookheimer et al., 2000; Trivedi et al., 2008; Filippini et al., 2009; Kunz et al., 2015).
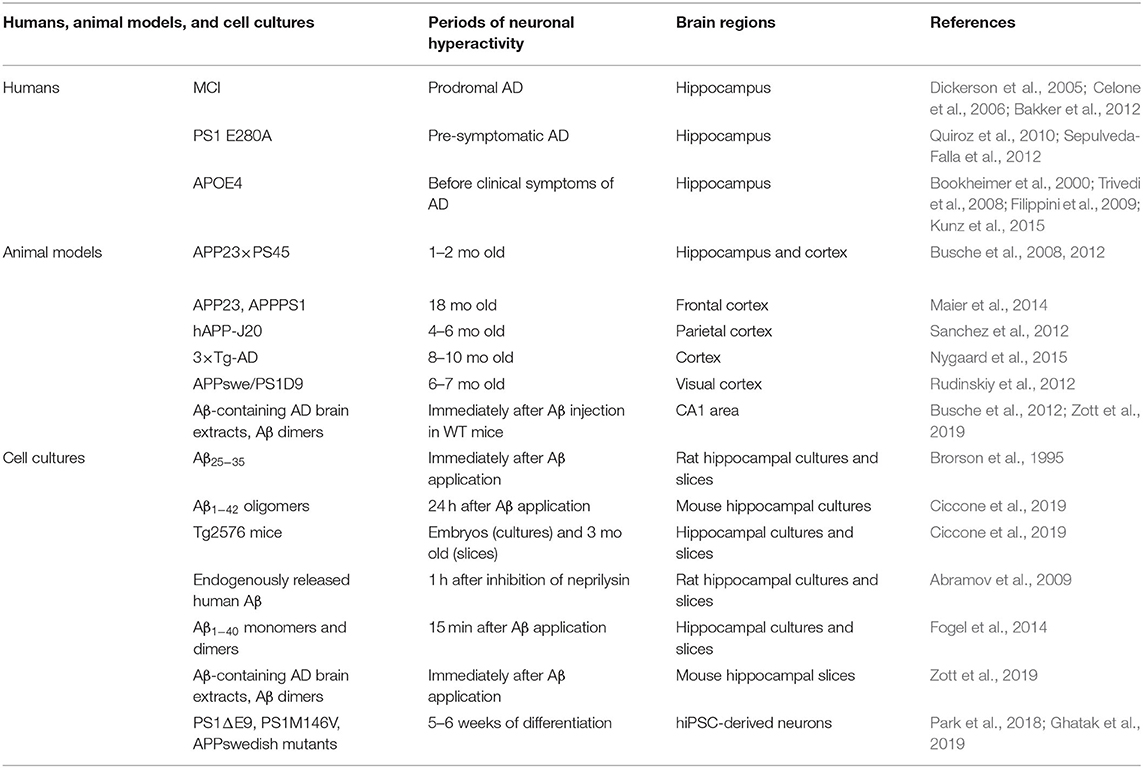
Table 1. Neuronal hyperactivity in humans, AD animal models, and cell cultures.
As the disease progresses, neuronal networks gradually switch to hypoactivity in AD during memory encoding (Celone et al., 2006; Persson et al., 2008; Reiman et al., 2012). Although there is currently many different compounds such as the Pittsburgh Compound B that can efficiently detect Aβ plaques in the brain using imaging techniques (Chetelat et al., 2020), the level of soluble Aβo cannot yet be directly measured in the brain of live patients. Although we know that the level of soluble Aβo begin to increase in the brain ~10–15 years before any clinical symptoms of AD (Cline et al., 2018), it still need to be established if the hyperactivity observed in early AD patients is induced, at least partly, by this progressive accumulation of soluble Aβo in the brain as shown in vitro and in animal models. A way to bypass this limitation would be to investigate the level of Aβ1−42 and Aβ1−40 in the CSF or plasma of AD patients while measuring hippocampal hyperactivity by imaging techniques, although this method would only allow to investigate the global impact of Aβo on specific brain area dysfunctions.
Neuronal hyperactivity has been detected in many transgenic AD mice such as the hAPP-J20, 3 × Tg-AD, APP23 × PS45, APP23, and APPswe/PS1D9 mice (Busche et al., 2008, 2012, 2015a; Rudinskiy et al., 2012; Sanchez et al., 2012; Maier et al., 2014; Nygaard et al., 2015) (Table 1). Using two-photon Ca2+ imaging, it was observed that 21% of cortical neurons displayed an increase of Ca2+ influx predominantly near the amyloid plaques in the APP23 × PS45 mouse model (Busche et al., 2008). A similar level of hyperactivity was also observed in the CA1 region of the hippocampus in young (1–2 months of age) APP23 × PS45 mice when Aβo begin to accumulate but no plaques are detected (Busche et al., 2012).
These results suggest that hyperactivity is an early pathological event that depends on the accumulation of Aβo rather than plaques per se, and that plaques might serve as a reservoir of toxic Aβo that amplify this excessive neuronal activity responsible, at least in part, for the marked synaptic and neuronal losses observed around plaques (Hefendehl et al., 2016). In parallel to this hyperactivity, another fraction (29%) of cortical neurons were also found to be hypoactive in 6–10 months old APP23 × PS45 mice when plaques are present (Busche et al., 2008). Since hypoactive neurones were only found after plaque formation, it is hypothesized that initial neuronal hyperactivity progressively switch to hypoactivity in AD (Busche and Konnerth, 2016), although the cellular and molecular mechanisms underpinning this shift still need to be determined.
To determine the direct implication of soluble Aβo on neuronal hyperactivation in vivo, exogenous Aβ species were also injected into the brain of wild-type mice. A single injection of Aβ-containing AD brain extracts and Aβ dimers were both found to induce a marked neuronal hyperactivity in CA1 neurons of wild-type mice (Busche et al., 2012; Zott et al., 2019). However, it should be noted that overexpression of additional APP fragments other than Aβo were also shown to induce hyperactivity and seizures in another mouse model (APP/TTA) (Born et al., 2014). Since it is difficult to tease apart the specific effects of each APP metabolites that are overexpressed in transgenic mouse models, the use of an animal model where fresh solutions of soluble Aβo are injected chronically into the hippocampus, such as the one we developed (Brouillette et al., 2012), could be advantageous to investigate the specific impact of Aβo on neuronal hyperactivity over time.
In line with these observations in AD mouse models, a myriad of studies performed in vitro also support the implication of Aβ on neuronal hyperexcitability using different types of Aβ solutions (Table 1). Indeed, application of the toxic Aβ peptide fragment consisting of amino acid residues 25 through 35 (Aβ25−35) to rat hippocampal cultures increased the intracellular levels of Ca2+ and the action potential activity in neurons (Brorson et al., 1995). Another study found that synthetic Aβ1−42 oligomers applied in primary neuronal cultures induced a dose-dependent decrease in neuronal viability that was cause, at least partly, by neuronal overexcitation (Sanchez-Mejia et al., 2008). Moreover, Aβ1−42 oligomers were found to induce aberrant neuronal activity in primary hippocampal neurons and in hippocampal slices from 3-month-old Tg2576 mice (Ciccone et al., 2019). Extracellular elevation of endogenously released human Aβ induced by inhibiting its degradation also rise up the synaptic vesicle release probability, and results in neuronal overexcitation in rat hippocampal cultures and in acute hippocampal slices (Abramov et al., 2009).
Similarly, higher levels of extracellular human Aβ1−40 monomers and dimers augmented synaptic vesicle release which in turn leads to hyperactivity of excitatory synapses in cultured hippocampal neurons and acute hippocampal slices (Fogel et al., 2014). More recently, it was shown that Aβ-containing AD brain extracts and purified cross-linked Aβ dimers were able to induce hyperactivity in active CA1 neurons treated with bicuculline in wild-type mouse hippocampal slices (Zott et al., 2019). Furthermore, increased Ca2+ transients and excessive neuronal excitability have been observed in neurons derived from human induced pluripotent stem cell (hiPSC) lines carrying familial AD mutations (Park et al., 2018; Ghatak et al., 2019).
Different studies have revealed various cellular and molecular mechanisms to explain how Aβo might induce neuronal hyperactivity (Figure 1). Several lines of evidence obtained in AD mouse models suggest that soluble Aβo alter the excitation/inhibition balance by decreasing the inhibitory GABAergic function, which in turn induced an excessive activation of the excitatory glutamatergic system in AD mice (Busche et al., 2008; Palop and Mucke, 2010; Busche and Konnerth, 2016; Styr and Slutsky, 2018). Indeed, hyperactivity of the cortical neurons in APP23 × PS45 mice was linked to lower GABAergic inhibition instead of higher glutamatergic transmission, and the activity of the hyperactive neurons was found to be decreased by diazepam, a benzodiazepine that increase the probability of opening the γ-aminobutyric acid type A (GABAA) receptor channels (Busche et al., 2008).
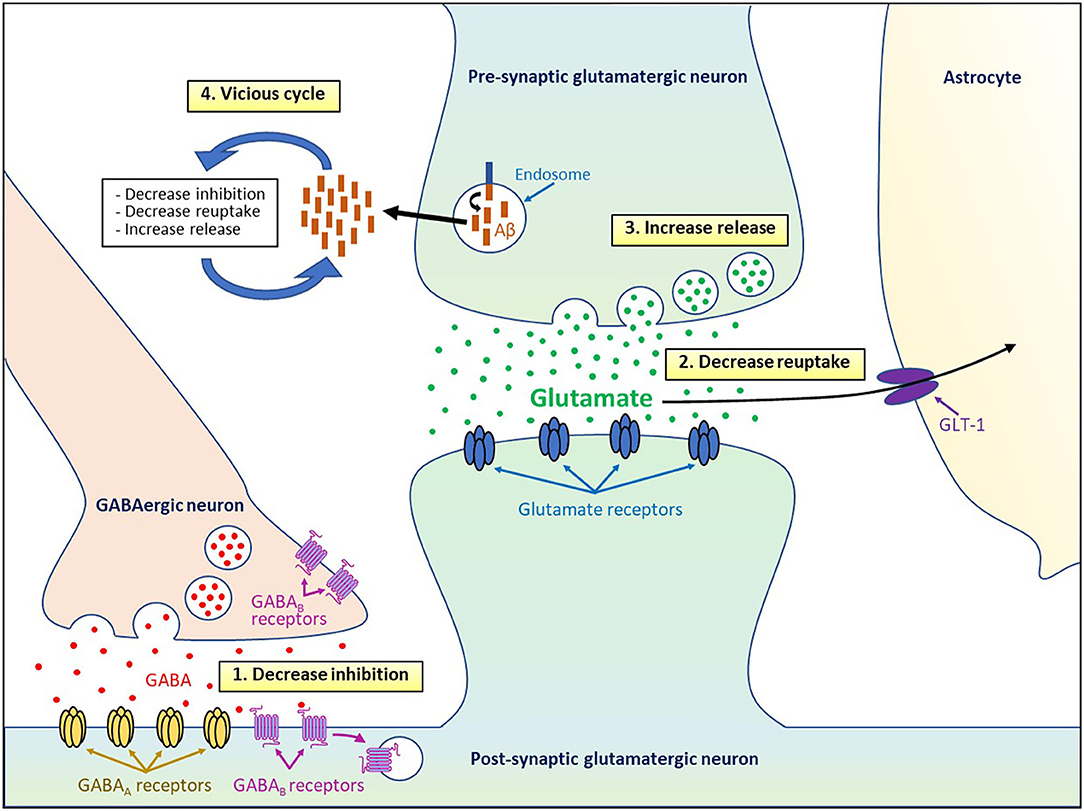
Figure 1. Cellular and molecular mechanisms underpinning Aβo-induced neuronal hyperactivity.
These results are consistent with another study showing that GABAA receptors localized in the temporal cortex of AD patients have a reduction of current, a higher rate of desensitization, and are less sensitive to GABA (Limon et al., 2012). Higher excitatory and lower inhibitory synaptic activities have also been reported in AD hiPSC-derived neurons (Ghatak et al., 2019). On the other hand, aberrant excitatory neuronal activity triggers by Aβ in the cortex and hippocampus of hAPP-J20 mice was found to induce subsequent maladaptive inhibitory mechanisms that reduce overexcitation (Palop et al., 2007), which could potentially be involved in the gradual switch to hypoactivity seen in animal models and AD patients (Celone et al., 2006; Persson et al., 2008; Sperling et al., 2009; Reiman et al., 2012; Busche and Konnerth, 2016).
Another mechanism that could explain hyperactivity generates by Aβo relies on the accumulation of glutamate at the synapse. Indeed, in vivo infusion of Aβ1−42 and Aβ25−35 into the rat cholinergic magnocellular nucleus basalis was shown to induce extracellular glutamate accumulation (Harkany et al., 2000). Fibrillar Aβ was also reported to decrease glutamate reuptake by both neuronal and glial cells (Harris et al., 1996; Parpura-Gill et al., 1997). More recently, it was found that Aβo-dependent hyperactivity in active CA1 neurons was triggered by impaired reuptake of synaptically released glutamate, which in turn potentiate excitatory glutamatergic transmission (Zott et al., 2019).
This reuptake suppression was shown to be induced by lower levels and membrane diffusion obstruction of the astroglial excitatory amino-acid transporter 2 (EAAT2; termed GLT-1 in mice) (Jacob et al., 2007; Hefendehl et al., 2016; Zott et al., 2019), a glutamate transporter that is predominant in the CA1 area and whose activity is reduced in the early stages of AD (Masliah et al., 1996; Hefendehl et al., 2016). In line with these observations, neuronal hyperexcitability observed in 5 × FAD mice was attenuated by increasing the expression of GLT-1 and by reducing changes in dendrite morphology, synaptic strength, and NMDA/AMPA receptors activity ratios after inhibiting the nuclear factor of activated T cells 4 (Sompol et al., 2017), a protein overactivated in the early stages of AD (Abdul et al., 2009).
An alternative mechanism by which Aβo may deregulate glutamate homeostasis implicates aberrant release of glutamate stored in pre-synaptic vesicles. Soluble Aβo have been shown to increase the release of pre-synaptic vesicles in hippocampal neuronal cultures, whereas the activation of inhibitory GABAA receptors by the agonist taurine was able to block the accumulation of glutamate at the synaptic cleft (Brito-Moreira et al., 2011). Moreover, application of Aβ1−42 oligomers on hippocampal cultures was reported to increase the amount of synaptic vesicles and their exocytosis by disrupting the synaptophysin/VAMP2 complex at the pre-synaptic terminals (Russell et al., 2012). Even a small elevation of endogenous Aβ40 and Aβ42 peptides of different lengths and molecular conformations was able to accelerate the vesicle exocytosis rate and increased release probability of active neurons in hippocampal cultures (Abramov et al., 2009). Given that both higher and lower levels of endogenous extracellular Aβ oligomers reduced short-term facilitation of vesicle release (Abramov et al., 2009), these results indicate that the level of Aβ peptides needs to be tightly control to keep the vesicle release probability in the optimal range. Application of Aβ40 monomers or dimers was also shown to induce hyperactivity by augmenting vesicle release probability at excitatory synapses after promoting pre-synaptic CA2+ influx via APP homodimerization in hippocampal cultures and slices (Fogel et al., 2014). Interestingly, various Aβ peptides such as Aβ1−42, Aβ1−40, Aβ1−28, and Aβ25−35 were all found to increase potassium-evoked glutamate release from hippocampal slices in a dose-dependent manner (Kabogo et al., 2010).
Lower reuptake and higher release of glutamate can also act synergistically to increase the load of glutamate in the synaptic cleft and lead to its “spillover” to activate extrasynaptic GluN2B-containing NMDA receptors that were found to promote neuronal death (Parsons and Raymond, 2014). Interestingly, prolonged activation of NMDA receptors has been shown to induce endocytosis and lysosomal degradation of the post-synaptic GABAB receptors (Terunuma et al., 2010), which could in turn amplify neuronal excitability by decreasing the inhibitory action of GABA in AD. Moreover, lower axonal trafficking and reduced expression of the pre-synaptic GABAB receptors in AD were reported to increase Aβ formation (Dinamarca et al., 2019). Since neuronal and synaptic activity were shown to increase the production and secretion of Aβ (Cirrito et al., 2005; Dolev et al., 2013; Yamamoto et al., 2015), the hyperactivity induced by Aβo can also favor an excessive release of Aβ and consequently causes a vicious cycle that amplifies and perpetuates the deleterious effects of Aβo on cell function. Using a chemogenetic approach, it was reported that chronic attenuation of aberrant neuronal activity was able to reduce amyloid plaque formation and synapse loss (Yuan and Grutzendler, 2016).
By blocking glutamate reuptake and facilitating its pre-synaptic release, soluble Aβo increased glutamate concentration at the synaptic cleft and subsequently affect post-synaptic neurons by overactivating glutamatergic NMDA and AMPA receptors. It was shown that higher pre-synaptic release of glutamate induced by soluble Aβ generated a massive entry of Ca2+ and Na+ through NMDA receptors, which in turn impaired intracellular signaling pathways involved in synaptic plasticity and produced deleterious effects on neurons leading ultimately to cell death (Calvo-Rodriguez and Bacskai, 2020) (Figure 2). In physiological condition, Ca2+ concentration is finely balanced to maintain a lower level in the cytosol than in the extracellular space or some cell organelles such as the endoplasmic reticulum (ER) and lysosome. When this balance is disturbed in AD, overactivation of Ca2+-dependent intracellular pathways impaired energy metabolism, produced reactive oxygen species (ROS), and oxidative stress that eventually lead to cell death (Belkacemi and Ramassamy, 2012; Calvo-Rodriguez and Bacskai, 2020).
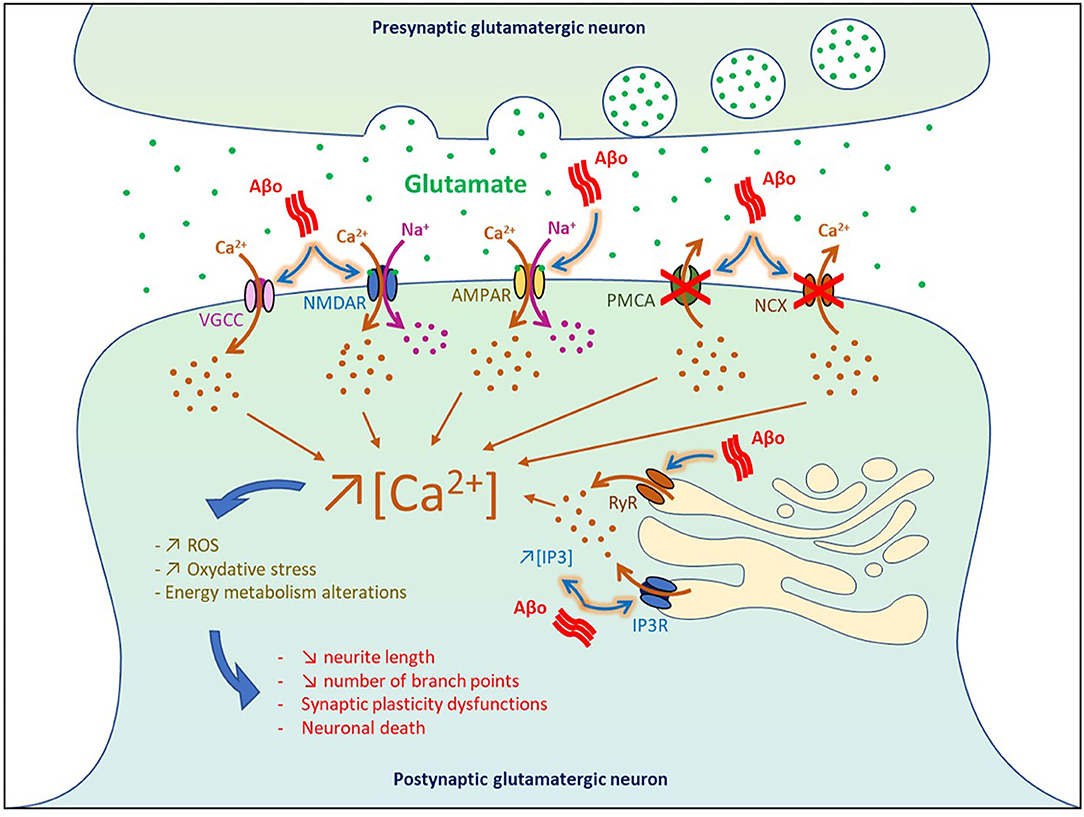
Figure 2. Impact of Aβo-induced hyperactivity on cell death. VGCC, voltage-gated Ca2+ channel; NMDAR, NMDA receptor; AMPAR, AMPA receptor; PMCA, plasma membrane calcium ATPase NCX, Na+/Ca2+ exchanger; IP3, inositol 1,4,5-trisphosphate; IP3R, IP3 receptor; RyR, ryanodine receptor.
Using neuronal culture and entorhinal–hippocampal organotypic slices, it was found that Aβ1−42 oligomers dysregulated Ca2+ homeostasis and triggered neuronal death through both NMDA and AMPA receptors by generating ROS that derived in part from mitochondrial sources (De Felice et al., 2007; Wang and Zheng, 2019). In vivo infusion of Aβ1−42 and Aβ25−35 in the rat cholinergic magnocellular nucleus basalis induced a rapid accumulation of intracellular Ca2+ in the vicinity of the injection site followed by cell death 3 days post-injection (Harkany et al., 2000). In human cortical cell cultures, Aβ1−38 and Aβ25−35 increased the intracellular basal level of Ca2+ and amplified Ca2+ influx induced by excitatory amino acid (EAA), thereby potentiating EAA-induced neuronal degeneration (Mattson et al., 1992). Injection of Aβ-containing AD extracts in the CA1 area was also reported to reduce neurite length and the number of branch points in wild-type mice (Zott et al., 2019).
At the level of the plasma membrane, Aβo can also increase the intracellular levels of Ca2+ by (1) inhibiting the Ca2+-efflux ATPase or exchangers (Wu et al., 1997; Kim et al., 1999; Mata, 2018), and by (2) intensifying Ca2+ influx through L-type, T-type, and N-type voltage-gated Ca2+ channels (Ueda et al., 1997; Ekinci et al., 1999; MacManus et al., 2000; Thibault et al., 2012; Min et al., 2013). Moreover, Aβo were found to increase Ca2+ release from the ER to the cytosol by enhancing the function of ryanodine receptors and by increasing inositol 1,4,5-trisphosphate receptor (IP3) production and binding to its receptors (Cowburn et al., 1995; Shtifman et al., 2010; Demuro and Parker, 2013; SanMartin et al., 2017). These results are in line with the beneficial effects observed in some AD patients with the memantine compound, an antagonist of the NMDA receptors that reduced Ca2+ influx into cells (Robinson and Keating, 2006).
It is well-established that in healthy individuals hippocampal activity increased when performing different types of memory tasks such as spatial navigation, episodic and associative memory tasks (Sperling et al., 2003; Zeineh et al., 2003; Moser et al., 2017). This higher neuronal activity is essential to induce synaptic plasticity to encode and consolidate new information learned while executing the task. But what happen when the hippocampus gets overactivated? Excessive neuronal activity in the hippocampus was first observed in animal models of aging and has been shown to induce age-related memory deficits (Koh et al., 2010; Thome et al., 2016; Haberman et al., 2017). In AD, hippocampal hyperactivity can be detected in the preclinical et prodromal stages of the disease when memory deficits are still very subtle and can hardly be perceived by neurocognitive exams (Mondadori et al., 2006; Filippini et al., 2009; Bateman et al., 2012; Reiman et al., 2012). Indeed, before clinical symptoms become apparent in APOE4 carriers, higher hippocampal activation was associated with lower grid-cell like representation in the entorhinal cortex when performing a virtual spatial-memory task (Bookheimer et al., 2000; Kunz et al., 2015).
In APP knock-in mice with human APP containing three mutations, grid cells were shown to degenerate when Aβ depositions are emerging, and started to lose connection with place cells in the hippocampus when mice were getting old, which prevented the hippocampus to recreate spatial maps to distinguish between different environments (Jun et al., 2020). Memory deficits were also observed in APOE4 knock-in mice, in which the APOE gene is replaced by knocking in the human ε4 allele (Andrews-Zwilling et al., 2010). Transplantation of interneuron precursor cells and treatment with pentobarbital to promote the inhibitory action of GABA were both able to attenuate these cognitive dysfunctions in APOE4 knock-in mice (Andrews-Zwilling et al., 2010).
In presymptomatic individuals carrying the AD-associated PS1 E280A mutation, increased activation of the right anterior hippocampus was observed when performing a face-name associative encoding task (Quiroz et al., 2010). Hippocampal hyperactivity was even detected in elderly with Aβ plaque deposition who doesn't showed episodic memory impairment (Mormino et al., 2012), suggesting that Aβ-dependent hyperactivation is an early event that might be present before memory deficits become apparent in some hippocampal-dependent memory tasks. In cognitively normal elderly, higher hippocampal activation at baseline has been shown to be correlated with increased longitudinal Aβ plaque deposition and progressive memory decline across time (Leal et al., 2017).
In another study, the presence of Aβ plaques in the neocortex was associated with impaired episodic memory deficits in both asymptomatic elderly and MCI individuals (Pike et al., 2007). Amnestic MCI and non-demented older adults also showed aberrant activity in the dentate gyrus and CA3 regions of the hippocampus during a pattern-separation task, which markedly depends on the hippocampus (Yassa et al., 2011; Bakker et al., 2012, 2015). Moreover, MCI patients with Aβ plaque depositions were found to have more pronounce hippocampal activation at baseline and faster clinical progression compared to Aβ negative MCI elderly (Huijbers et al., 2015).
Transcranial magnetic stimulation (TMS) is a non-invasive form of brain stimulation technique that not only allow to monitor variations in intracortical inhibition and excitation but might also serve as a diagnostic tool and a way to modulate cortex activity to ameliorate memory function in AD patients. Indeed, repetitive TMS (rTMS) applied to the dorsolateral prefrontal cortex (DLPFC) has been shown to improve performance on an action naming memory task in mild AD as well as object naming in moderate to severe AD patients (Cotelli et al., 2006, 2008). A longer treatment (five times a week for 4 weeks) with rTMS over the left DLPFC was even able to enhance language performance of AD patients that lasted for 8 weeks after ending the stimulations (Cotelli et al., 2011). Moreover, high-frequency rTMS over the DLPFC improved memory performance in the mini-mental state examination (MMSE) in patients with mild to moderate AD, whereas high-frequency rTMS over the right inferior frontal gyrus increased attention and psychomotor speed of MCI and mild AD patients in the trail making test (Eliasova et al., 2014). Another study has found that application of rTMS for 6 weeks over the parietal P3/P4 and posterior temporal T5/T6 areas improved cognitive function in mild to moderate AD patients in three different neuropsychological tests (Zhao et al., 2017). Although we still don't know if rTMS can impact Aβ accumulation, this technique holds great promise to tackle neuronal hyperactivity and acts on it to improve cognitive performance of AD patients.
Neuronal hyperactivity also affects memory performance in various animal models. All the transgenic AD mouse models showing network hyperexcitability such as the hAPP-J20, 3 × Tg-AD, APP23 × PS45, APP23, Tg2576, and APPswe/PS1D9 mice, were found to have memory deficits in various memory tasks (Busche et al., 2008, 2012, 2015a; Rudinskiy et al., 2012; Sanchez et al., 2012; Maier et al., 2014; Nygaard et al., 2015). Direct injections of soluble Aβ1−42 oligomers into the hippocampus also induced memory deficits that were reversed by sequestering Aβo with transthyretin (Brouillette et al., 2012). Cognitive functions were also improved in hAPP-J20 and 3 × Tg-AD mice by suppressing neuronal overactivation with levetiracetam, an anti-epileptic drug that facilitate inhibitory GABAergic neurotransmission (Sanchez et al., 2012; Nygaard et al., 2015).
Memory plasticity can be modeled by inducing long-term potentiation (LTP) or long-term depression (LTD) in cell cultures or animal models (Nabavi et al., 2014). Nanomolar and micromolar levels of Aβ dimers and trimers were shown to inhibit LTP, increase LTD and reduce dendritic spine density in organotypic hippocampal slices (Townsend et al., 2006; Shankar et al., 2007, 2008; Li et al., 2009). It was found that Aβo altered LTP and LTD by decreasing neuronal glutamate reuptake, thereby contributing to the diffusion of glutamate outside the post-synaptic density where it can activate extrasynaptic GluN2B-containing NMDA receptors and induced cell death (Li et al., 2009, 2011; Hardingham and Bading, 2010). On the contrary, smaller (picomolar) concentration of Aβ42 was shown to enhance LTP and memory formation (Puzzo et al., 2008), suggesting that the level of Aβ needs to be finely tuned to prevent synaptic failure and ensuing cognitive impairment.
As the disease progresses and cognitive decline worsens, hippocampal activation decreased gradually at the basal level and when AD patients performed a task-related hippocampal activity (Dickerson et al., 2005; Pariente et al., 2005; Celone et al., 2006). In a prospective study it was found that MCI individuals shifted from hippocampal hyperexcitability to hypoactivation at the baseline level over time, and that deterioration of memory performance was associated with the rate of decrease in hippocampal activity (O'Brien et al., 2010). Collectively, these studies suggest that high neuronal activity induced, at least in part, by Aβ accumulation is a very early phenomenon in AD pathogenesis that has a deleterious impact on memory abilities.
Since neuronal hyperactivation is characterized by an increase in frequency and amplitude of neuronal firing, it is not surprising to observe abnormal level of synchronization between excitatory glutamatergic neurons that fired together at the same time, which in turn increased the incidence of epileptiform activity and seizure observed in AD patients and AD animal models. Although the prevalence rates vary considerably between studies (1.5–64%) because of limitation and methodological issues to detect non-convulsive epileptiform activity, a rate of 64% has been observed in cohorts monitored carefully at all stages of AD (Friedman et al., 2012).
Interestingly, seizures have been shown to occur more frequently in younger AD patients (Vossel et al., 2013; Sherzai et al., 2014), when neuronal hyperactivity is more prominent. In patients with early-onset AD that developed the disease before 65 years old, seizures were detected in 45% of cases (Samson et al., 1996). A seizure rate of 28% was also observed in people with familial AD carrying mutations in APP, PS1, or PS2 genes (Shea et al., 2016). In a prospective study of 8 years, seizures were observed in 84% of patients with Down's syndrome who developed AD because of the duplication of chromosome 21 that contains the APP gene (Lai and Williams, 1989). The higher and earlier accumulation of Aβ in familial cases of AD and Down's syndrome supports the notion that neuronal hyperactivation induced by soluble Aβo is involved in epileptogenic activity seen at the onset of AD.
In the more common form of sporadic AD, epileptiform activity might be more prominent than previously recognized. Indeed, it was first found that only 2% of AD patients had subclinical non-convulsive epileptiform activity when recording EEGs for 30 min in awake patients (Liedorp et al., 2010). However, a more recent study detected subclinical epileptiform activity in 42% of the cases (four times more often than in healthy controls), using 24h EEGs in combination with 1 h magnetoencephalography (MEG) (Vossel et al., 2016). Interestingly, 90% of epileptiform activity occurred during sleep, and AD patients with subclinical epileptiform activity showed a faster rate of cognitive decline (Vossel et al., 2016). Using intracranial recording, clinically silent hippocampal seizures and epileptiform spikes were also observed during sleep in two AD patients without a history or EEG evidence of seizures (Lam et al., 2017).
These results are in line with the manifestation of non-convulsive seizure activity and epileptiform spike discharges observed using EEGs in various AD transgenic models. Like in humans, most of the epilepsies seen in AD mice are non-convulsive, with the exception of mice overexpressing human APPswe and PS1ΔE9 which have recurrent motor seizures (Minkeviciene et al., 2009; Palop and Mucke, 2010; Um et al., 2012). In hAPP-J20 and APPswe/PS1ΔE9 mice, pathological elevation of Aβo has been shown to elicit hyperexcitability and spontaneous non-convulsive epileptic activity, including spikes and sharp waves, in cortical and hippocampal networks (Palop et al., 2007; Minkeviciene et al., 2009).
As in humans, spontaneous epileptiform discharges were found to arise mainly during resting periods in hAPPJ20 mice (Verret et al., 2012). Enhancing inhibitory GABA current by restoring the level of voltage-gated sodium channels subunit Nav1.1 was shown to reduce network hypersynchrony, memory deficits, and premature mortality in hAPP-J20 mice (Verret et al., 2012). Aβ1−42 oligomers were also found to up-regulate the level of Nav1.6 subtype, which contribute to neuronal hyperexcitability observed in primary hippocampal neurons and in hippocampal slices from 3-month-old Tg2576 mice (Ciccone et al., 2019). In APPswe/PS1ΔE9 mice, electrographic and motor seizures were prevented by deleting the cellular prion protein, which was shown to interact with Aβ and triggered dendritic spine loss (Um et al., 2012).
Normal neuronal synchrony is critical to generate oscillatory rhythmic activities within a certain range that allow different brain regions to communicate efficiently together in function of the brain state. Brain rhythms are formed when neuronal ensembles depolarized (most often with firing) and hyperpolarized their membrane potentials together in synchronized repeating sequences (Buzsaki and Watson, 2012). Five widely recognized brain waves have been characterized in function of their frequencies; delta (1–4 Hz), theta (4–8 Hz), alpha (8–12 Hz), beta (12–30 Hz), and gamma (30–150 Hz) oscillations. Each brain waves have been associated with a particular brain state, where delta oscillations are more prominent during non-rapid eye movement (NREM) sleep whereas gamma oscillations are mostly detected when concentration is required, and tend to be localized to neuronal networks directly implicated in the task (Timofeev and Chauvette, 2017; Adaikkan and Tsai, 2020). For example, the amplitude (power) of gamma oscillatory activity was shown to be increased in the hippocampus during memory encoding and to predict effective memory formation in humans and mice (Jensen et al., 2007; Sederberg et al., 2007; Matsumoto et al., 2013; Yamamoto et al., 2014).
Given that Aβo-induced hyperactivity favors hypersynchrony, which in turn affects brain waves, one could expect that brain rhythms are altered at the onset of AD. In fact, gamma power has been shown to be reduced in MCI and AD patients (Herrmann and Demiralp, 2005; van Deursen et al., 2008), as well as in various AD mouse models (Verret et al., 2012; Goutagny et al., 2013; Iaccarino et al., 2016; Mably et al., 2017; Mondragon-Rodriguez et al., 2018). Interestingly, it was found recently that restoring slow gamma oscillation (40 Hz) in a non-invasive manner by simply exposing AD mice to 1 h of 40 Hz tons per day for a week was sufficient to reduce amyloid and tau pathologies not only in the auditory cortex but also in the hippocampus, to activate microglia, and to improve cognitive performance (Martorell et al., 2019). A stronger microglia response and a larger reduction of amyloid plaques were also found by combining auditory with visual stimulation to induce 40 Hz gamma waves (Martorell et al., 2019). Moreover, optogenetic stimulation of medial septal parvalbumin neurons at 40 Hz was reported to restore hippocampal slow gamma oscillations power and to ameliorate spatial memory in hAPP J20 mice (Etter et al., 2019).
Slow wave activity (SWA)—comprising slow oscillations (0.6–1 Hz) and delta waves—that is present during NREM sleep was also found to be disrupted in the early stages of AD (Lee et al., 2020). It is well-established that NREM sleep is particularly important to consolidate memories newly acquired during the awake state, and that SWA is critical to transfer novel information from the hippocampus to long-term memory storage across cortical areas (Steriade et al., 1993; Clemens et al., 2005; Diekelmann and Born, 2010). In individuals with MCI, lower delta and theta power during sleep was associated with declarative memory impairments, and more fragmentation of slow-wave sleep was observed relative to healthy elders (Hita-Yanez et al., 2012; Westerberg et al., 2012). It was shown that disruption of NREM SWA and deficits in hippocampus-dependent memory consolidation correlated with the level of Aβ plaque deposition in the medial prefrontal cortex of older adults (Mander et al., 2015). Cortical Aβ burden was also able to predict the lower amplitude of slow oscillations in elderly (Winer et al., 2019). Moreover, reduce slow-wave sleep was associated with higher level of Aβ in the plasma of MCI individuals (Sanchez-Espinosa et al., 2014). Interestingly, restoring slow oscillations by transcranial direct current stimulation was shown to improve memory performance in patients with early AD (Ladenbauer et al., 2017).
In line with these human studies showing the involvement of Aβ on SWA impairment at the onset of AD, disruption of SWA was also detected in mouse models of β-amyloidosis. SWA has been shown to be markedly disrupted in the hippocampus, neocortex and thalamus of APP23 × PS45 mice and in wild-type mice injected with synthetic Aβ (Busche et al., 2015b). Slow wave power was also decreased in young and older APPswe/PS1ΔE9 mice (Kastanenka et al., 2017, 2019). Moreover, both the APP/PS1 and Tg2576 mouse models exhibited an age-dependent decreased in delta and theta power (Kent et al., 2018), whereas 3 × Tg-AD mice showed slow waves at lower frequency (Castano-Prat et al., 2019). Remarkably, restoring slow oscillations using a GABA receptors agonist (benzodiazepine), a suppressor of Aβ production (β-secretase), or by optogenetic manipulation have all been shown to rescue memory deficits in various AD mouse models (Busche et al., 2015b; Kastanenka et al., 2017; Keskin et al., 2017).
Since Aβo-induced hyperactivity is an early pathological event that precedes plaque formation when soluble low-molecular-weight Aβo begin to accumulate in the human brain up to two decades before the symptomatic phase of the disease (Cline et al., 2018), acting on this detrimental phenomenon might prove beneficial to develop therapeutic approaches to prevent or at least slow down the disease progression. Since the excitation/inhibition balance has been shown to be compromised at the onset of AD primarily because of insufficient GABAergic inhibition (Busche et al., 2008; Palop and Mucke, 2010; Busche and Konnerth, 2016; Styr and Slutsky, 2018), using drugs that are capable of restoring the GABAergic system might potentially lower the hyperactivity triggers by Aβo and consequently AD pathogenesis.
The GABAA receptors agonist taurine was found to attenuate neuronal hyperactivity by decreasing glutamate level released at the synapse (Brito-Moreira et al., 2011) (Table 2). Hyperactivity was also reduced in cortical neurons of APP23 × PS45 mice by increasing the inhibitory effect of GABA with diazepam (Busche et al., 2008). Topical application of GABA directly on the somatosensory cortex was reported to restore slow oscillations in APP mice (Kastanenka et al., 2017, 2019), whereas the topical application of the GABAA agonist midazolam rescued the frequency and long-range coherence of slow waves in the frontal and occipital cortex of APP23 × PS45 mice and in wild-type mice infused with Aβo (Busche et al., 2015b). Moreover, intraperitoneal injection of the benzodiazepine clonazepam, which increase GABAergic function by acting on GABAA receptors, has been shown to rescue slow waves and sleep-dependent memory consolidation in APP23 × PS45 mice (Busche et al., 2015b).
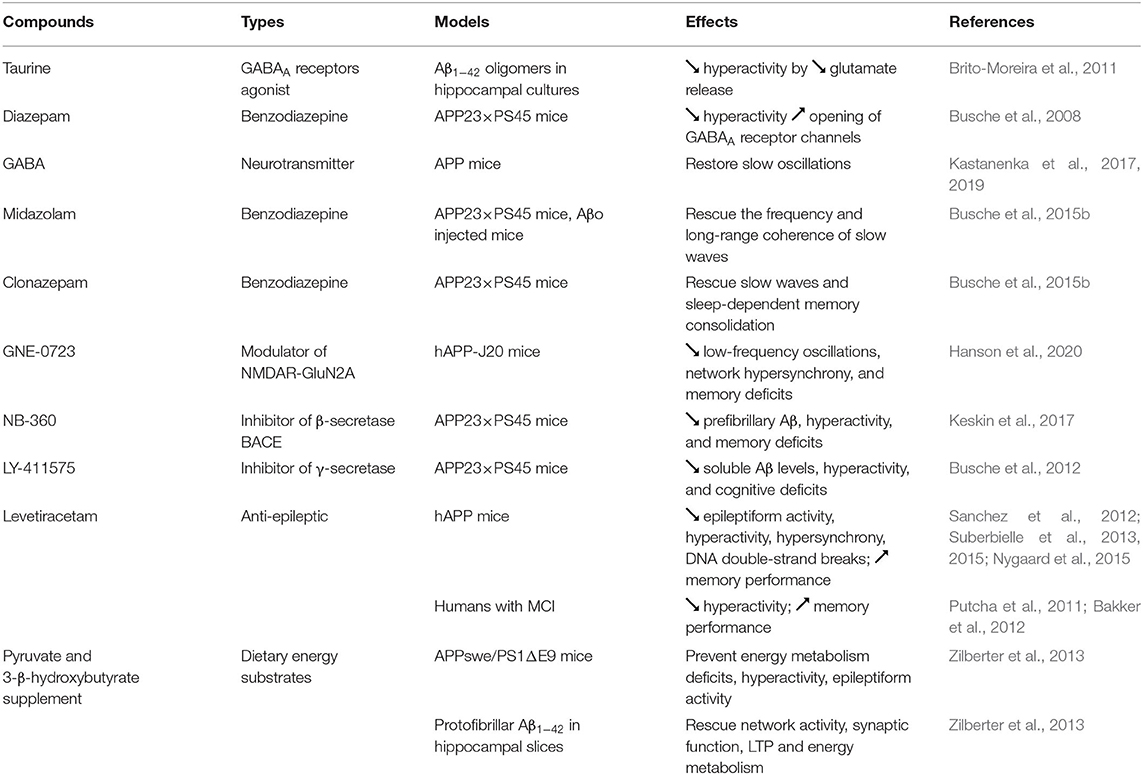
Table 2. Treatments to counteract neuronal hyperactivation in AD.
Recently, a positive allosteric modulator called GNE-0723 that can boost the activity of NMDAR containing GluN2A subunit contained in both excitatory pyramidal neurons and inhibitory interneurons has been tested in hAPP-J20 mice (Hanson et al., 2020). This compound was found to decrease aberrant low-frequency oscillations (12–20 Hz), network hypersynchrony, and memory deficits in hAPP-J20 mice, suggesting that this drug is able to reinstate the excitation/inhibition balance. Inhibitors of β-secretase BACE and γ-secretase, two enzymes involved in the production of Aβ, have also been shown to decrease Aβo-induced hyperactivity and cognitive impairments in APP23 × PS45 mice (Busche et al., 2012; Keskin et al., 2017). However, given the clinical trial failures obtained so far with these types of compounds, additional experiments are requested to develop more Aβ specific BACE and γ-secretase inhibitors and to find the appropriate doses and time of administration for an efficient therapeutic intervention.
Levetiracetam (Keppra) is an atypical anti-epileptic drug that is assumed to decrease impulse conduction across excitatory synapses by inhibiting pre-synaptic Ca2+ channels, and by acting on the synaptic vesicle protein SV2A (Lynch et al., 2004; Vogl et al., 2012). Interestingly, levetiracetam not only decreased epileptiform activity in hAPP mice, but also lower neuronal hyperactivation and hypersynchrony, improved memory performance, and reduced neuronal DNA double-strand breaks in AD mouse models (Sanchez et al., 2012; Suberbielle et al., 2013, 2015; Nygaard et al., 2015). In MCI individuals, treatment with a low dose of levetiracetam for two weeks was found to attenuate hippocampal hyperactivation and to ameliorate performance in a pattern-separation memory task (Putcha et al., 2011; Bakker et al., 2012).
Since higher level of glucose is required to provide the increase of energy associated with neuronal hyperexcitability, several cellular energy deficiencies have also been detected at the onset of AD (Velliquette et al., 2005; Guglielmotto et al., 2009; Avila et al., 2010). To compensate for this neuronal energy supply deficiency, an energy substrate-enriched diet (standard diet supplemented with pyruvate and 3-β-hydroxybutyrate) was administered for 5 weeks to APPswe/PS1ΔE9 mice (Zilberter et al., 2013). By restoring the level of glycogen in the brain of these AD mice, this treatment was able to prevent energy metabolism deficits, neuronal hyperexcitability, and epileptiform activity. Moreover, alterations in network activity, synaptic function, LTP, and energy metabolism induced by protofibrillar Aβ1−42 in hippocampal slices were rescued by using artificial cerebrospinal fluid supplemented with pyruvate and 3-β-hydroxybutyrate (Zilberter et al., 2013).
A myriad of studies performed in humans, cell cultures, hiPSC lines carrying familial AD mutations, AD mouse models, and wild-type mice injected with soluble Aβo indicate that neuronal hyperactivity is an early detrimental event in AD pathogenesis. Multiple lines of evidence strongly suggest that the accumulation of soluble low-molecular-weight Aβo plays a major role in neuronal hyperexcitability observed at the onset of AD, although other factors might also contribute, such as tau, other APP metabolites, APOE4, glial responses, neuroinflammation, and oxidative stress. Encouragingly, a growing body of evidence indicates that neuronal hyperactivity may be potentially reversed, which could prevent cell death, improve cognitive impairments, decrease epileptiform activity, restore gamma oscillations, and slow wave activity. Decreasing the abnormal accumulation of soluble Aβo to avoid an excess of glutamate at the synaptic cleft and re-establishing the balance between synaptic excitation and inhibition might prove useful to ameliorate memory performance in the early stages of AD and prevent, or at least slow down, the neurodegenerative process that progressively takes place in the course of AD.
AH and JB wrote and approved the final manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdul, H. M., Sama, M. A., Furman, J. L., Mathis, D. M., Beckett, T. L., Weidner, A. M., et al. (2009). Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J. Neurosci. 29, 12957–12969. doi: 10.1523/JNEUROSCI.1064-09.2009
Abramov, E., Dolev, I., Fogel, H., Ciccotosto, G. D., Ruff, E., and Slutsky, I. (2009). Amyloid-β as a positive endogenous regulator of release probability at hippocampal synapses. Nat. Neurosci. 12, 1567–1576. doi: 10.1038/nn.2433
Adaikkan, C., and Tsai, L. H. (2020). Gamma entrainment: impact on neurocircuits, glia, and therapeutic opportunities. Trends Neurosci. 43, 24–41. doi: 10.1016/j.tins.2019.11.001
Alzheimer's, A. (2020). 2020 Alzheimer's disease facts and figures. Alzheimers Dement. 16, 391–460. doi: 10.1002/alz.12068
Andrews-Zwilling, Y., Bien-Ly, N., Xu, Q., Li, G., Bernardo, A., Yoon, S. Y., et al. (2010). Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci. 30, 13707–13717. doi: 10.1523/JNEUROSCI.4040-10.2010
Avila, J., Wandosell, F., and Hernandez, F. (2010). Role of glycogen synthase kinase-3 in Alzheimer's disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev. Neurother. 10, 703–710. doi: 10.1586/ern.10.40
Bakker, A., Albert, M. S., Krauss, G., Speck, C. L., and Gallagher, M. (2015). Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. Neuroimage Clin. 7, 688–698. doi: 10.1016/j.nicl.2015.02.009
Bakker, A., Krauss, G. L., Albert, M. S., Speck, C. L., Jones, L. R., Stark, C. E., et al. (2012). Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 74, 467–474. doi: 10.1016/j.neuron.2012.03.023
Bateman, R. J., Xiong, C., Benzinger, T. L., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Belkacemi, A., and Ramassamy, C. (2012). Time sequence of oxidative stress in the brain from transgenic mouse models of Alzheimer's disease related to the amyloid-beta cascade. Free Radic. Biol. Med. 52, 593–600. doi: 10.1016/j.freeradbiomed.2011.11.020
Bookheimer, S. Y., Strojwas, M. H., Cohen, M. S., Saunders, A. M., Pericak-Vance, M. A., Mazziotta, J. C., et al. (2000). Patterns of brain activation in people at risk for Alzheimer's disease. N. Engl. J. Med. 343, 450–456. doi: 10.1056/NEJM200008173430701
Born, H. A., Kim, J. Y., Savjani, R. R., Das, P., Dabaghian, Y. A., Guo, Q., et al. (2014). Genetic suppression of transgenic APP rescues Hypersynchronous network activity in a mouse model of Alzeimer's disease. J. Neurosci. 34, 3826–3840. doi: 10.1523/JNEUROSCI.5171-13.2014
Brito-Moreira, J., Paula-Lima, A. C., Bomfim, T. R., Oliveira, F. B., Sepulveda, F. J., De Mello, F. G., et al. (2011). Abeta oligomers induce glutamate release from hippocampal neurons. Curr. Alzheimer Res. 8, 552–562. doi: 10.2174/156720511796391917
Brorson, J. R., Bindokas, V. P., Iwama, T., Marcuccilli, C. J., Chisholm, J. C., and Miller, R. J. (1995). The Ca2+ influx induced by beta-amyloid peptide 25-35 in cultured hippocampal neurons results from network excitation. J. Neurobiol. 26, 325–338. doi: 10.1002/neu.480260305
Brouillette, J. (2014). The effects of soluble Abeta oligomers on neurodegeneration in Alzheimer's disease. Curr. Pharm. Des. 20, 2506–2519. doi: 10.2174/13816128113199990498
Brouillette, J., Caillierez, R., Zommer, N., Alves-Pires, C., Benilova, I., Blum, D., et al. (2012). Neurotoxicity and memory deficits induced by soluble low-molecular-weight amyloid-β1–42 oligomers are revealed in vivo by using a novel animal model. J. Neurosci. 32, 7852–7861. doi: 10.1523/JNEUROSCI.5901-11.2012
Busche, M. A., Chen, X., Henning, H. A., Reichwald, J., Staufenbiel, M., Sakmann, B., et al. (2012). Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U. S. A. 109, 8740–8745. doi: 10.1073/pnas.1206171109
Busche, M. A., Eichhoff, G., Adelsberger, H., Abramowski, D., Wiederhold, K. H., Haass, C., et al. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science 321, 1686–1689. doi: 10.1126/science.1162844
Busche, M. A., Grienberger, C., Keskin, A. D., Song, B., Neumann, U., Staufenbiel, M., et al. (2015a). Decreased amyloid-beta and increased neuronal hyperactivity by immunotherapy in Alzheimer's models. Nat. Neurosci. 18, 1725–1727. doi: 10.1038/nn.4163
Busche, M. A., Kekus, M., Adelsberger, H., Noda, T., Forstl, H., Nelken, I., et al. (2015b). Rescue of long-range circuit dysfunction in Alzheimer's disease models. Nat. Neurosci. 18, 1623–1630. doi: 10.1038/nn.4137
Busche, M. A., and Konnerth, A. (2016). Impairments of neural circuit function in Alzheimer's disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371:20150429. doi: 10.1098/rstb.2015.0429
Buzsaki, G., and Watson, B. O. (2012). Brain rhythms and neural syntax: implications for efficient coding of cognitive content and neuropsychiatric disease. Dialogues Clin. Neurosci. 14, 345–367. doi: 10.31887/DCNS.2012.14.4/gbuzsaki
Calvo-Rodriguez, M., and Bacskai, B. J. (2020). Mitochondria and calcium in Alzheimer's disease: from cell signaling to neuronal cell death. Trends Neurosci. 4. doi: 10.1016/j.tins.2020.10.004
Castano-Prat, P., Perez-Mendez, L., Perez-Zabalza, M., Sanfeliu, C., Gimenez-Llort, L., and Sanchez-Vives, M. V. (2019). Altered slow (<1 Hz) and fast (beta and gamma) neocortical oscillations in the 3 × Tg-AD mouse model of Alzheimer's disease under anesthesia. Neurobiol. Aging 79, 142–151. doi: 10.1016/j.neurobiolaging.2019.02.009
Celone, K. A., Calhoun, V. D., Dickerson, B. C., Atri, A., Chua, E. F., Miller, S. L., et al. (2006). Alterations in memory networks in mild cognitive impairment and Alzheimer's disease: an independent component analysis. J. Neurosci. 26, 10222–10231. doi: 10.1523/JNEUROSCI.2250-06.2006
Chetelat, G., Arbizu, J., Barthel, H., Garibotto, V., Law, I., Morbelli, S., et al. (2020). Amyloid-PET and (18)F-FDG-PET in the diagnostic investigation of Alzheimer's disease and other dementias. Lancet Neurol. 19, 951–962. doi: 10.1016/S1474-4422(20)30314-8
Ciccone, R., Franco, C., Piccialli, I., Boscia, F., Casamassa, A., de Rosa, V., et al. (2019). Amyloid beta-induced upregulation of Nav1.6 underlies neuronal hyperactivity in Tg2576 Alzheimer's disease mouse model. Sci. Rep. 9, 13592. doi: 10.1038/s41598-019-50018-1
Cirrito, J. R., Yamada, K. A., Finn, M. B., Sloviter, R. S., Bales, K. R., May, P. C., et al. (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922. doi: 10.1016/j.neuron.2005.10.028
Clemens, Z., Fabo, D., and Halasz, P. (2005). Overnight verbal memory retention correlates with the number of sleep spindles. Neuroscience 132, 529–535. doi: 10.1016/j.neuroscience.2005.01.011
Cline, E. N., Bicca, M. A., Viola, K. L., and Klein, W. L. (2018). The amyloid-beta oligomer hypothesis: beginning of the third decade. J. Alzheimers Dis. 64, S567–S610. doi: 10.3233/JAD-179941
Cotelli, M., Calabria, M., Manenti, R., Rosini, S., Zanetti, O., Cappa, S. F., et al. (2011). Improved language performance in Alzheimer disease following brain stimulation. J. Neurol. Neurosurg. Psychiatry 82, 794–797. doi: 10.1136/jnnp.2009.197848
Cotelli, M., Manenti, R., Cappa, S. F., Geroldi, C., Zanetti, O., Rossini, P. M., et al. (2006). Effect of transcranial magnetic stimulation on action naming in patients with Alzheimer disease. Arch. Neurol. 63, 1602–1604. doi: 10.1001/archneur.63.11.1602
Cotelli, M., Manenti, R., Cappa, S. F., Zanetti, O., and Miniussi, C. (2008). Transcranial magnetic stimulation improves naming in Alzheimer disease patients at different stages of cognitive decline. Eur. J. Neurol. 15, 1286–1292. doi: 10.1111/j.1468-1331.2008.02202.x
Cowburn, R. F., Wiehager, B., and Sundstrom, E. (1995). β-Amyloid peptides enhance binding of the calcium mobilising second messengers, inositol(1,4,5)trisphosphate and inositol-(1,3,4,5)tetrakisphosphate to their receptor sites in rat cortical membranes. Neurosci. Lett. 191, 31–34. doi: 10.1016/0304-3940(94)11549-3
De Felice, F. G., Velasco, P. T., Lambert, M. P., Viola, K., Fernandez, S. J., Ferreira, S. T., et al. (2007). Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 282, 11590–11601. doi: 10.1074/jbc.M607483200
Demuro, A., and Parker, I. (2013). Cytotoxicity of intracellular aβ42 amyloid oligomers involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J Neurosci. 33, 3824–3833. doi: 10.1523/JNEUROSCI.4367-12.2013
Dickerson, B. C., Salat, D. H., Greve, D. N., Chua, E. F., Rand-Giovannetti, E., Rentz, D. M., et al. (2005). Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology 65, 404–411. doi: 10.1212/01.wnl.0000171450.97464.49
Diekelmann, S., and Born, J. (2010). The memory function of sleep. Nat. Rev. Neurosci. 11, 114–126. doi: 10.1038/nrn2762
Dinamarca, M. C., Raveh, A., Schneider, A., Fritzius, T., Fruh, S., Rem, P. D., et al. (2019). Complex formation of APP with GABAB receptors links axonal trafficking to amyloidogenic processing. Nat Commun. 10, 1331. doi: 10.1038/s41467-019-09164-3
Dolev, I., Fogel, H., Milshtein, H., Berdichevsky, Y., Lipstein, N., Brose, N., et al. (2013). Spike bursts increase amyloid-beta 40/42 ratio by inducing a presenilin-1 conformational change. Nat. Neurosci. 16, 587–595. doi: 10.1038/nn.3376
Ekinci, F. J., Malik, K. U., and Shea, T. B. (1999). Activation of the L voltage-sensitive calcium channel by mitogen-activated protein (MAP) kinase following exposure of neuronal cells to β-amyloid. MAP kinase mediates beta-amyloid-induced neurodegeneration. J. Biol. Chem. 274, 30322–30327. doi: 10.1074/jbc.274.42.30322
Eliasova, I., Anderkova, L., Marecek, R., and Rektorova, I. (2014). Non-invasive brain stimulation of the right inferior frontal gyrus may improve attention in early Alzheimer's disease: a pilot study. J. Neurol. Sci. 346, 318–322. doi: 10.1016/j.jns.2014.08.036
Etter, G., van der Veldt, S., Manseau, F., Zarrinkoub, I., Trillaud-Doppia, E., and Williams, S. (2019). Optogenetic gamma stimulation rescues memory impairments in an Alzheimer's disease mouse model. Nat. Commun. 10, 5322. doi: 10.1038/s41467-019-13260-9
Filippini, N., MacIntosh, B. J., Hough, M. G., Goodwin, G. M., Frisoni, G. B., Smith, S. M., et al. (2009). Distinct patterns of brain activity in young carriers of the APOE-ε4 allele. Proc. Natl. Acad. Sci. U. S. A. 106, 7209–7214. doi: 10.1073/pnas.0811879106
Fogel, H., Frere, S., Segev, O., Bharill, S., Shapira, I., Gazit, N., et al. (2014). APP homodimers transduce an amyloid-beta-mediated increase in release probability at excitatory synapses. Cell Rep. 7, 1560–1576. doi: 10.1016/j.celrep.2014.04.024
Friedman, D., Honig, L. S., and Scarmeas, N. (2012). Seizures and epilepsy in Alzheimer's disease. CNS Neurosci. Ther. 18, 285–294. doi: 10.1111/j.1755-5949.2011.00251.x
Ghatak, S., Dolatabadi, N., Trudler, D., Zhang, X., Wu, Y., Mohata, M., et al. (2019). Mechanisms of hyperexcitability in Alzheimer's disease hiPSC-derived neurons and cerebral organoids vs isogenic controls. Elife. 8:e50333. doi: 10.7554/eLife.50333.sa2
Goutagny, R., Gu, N., Cavanagh, C., Jackson, J., Chabot, J. G., Quirion, R., et al. (2013). Alterations in hippocampal network oscillations and theta-gamma coupling arise before Abeta overproduction in a mouse model of Alzheimer's disease. Eur. J. Neurosci. 37, 1896–1902. doi: 10.1111/ejn.12233
Guglielmotto, M., Tamagno, E., and Danni, O. (2009). Oxidative stress and hypoxia contribute to Alzheimer's disease pathogenesis: two sides of the same coin. ScientificWorldJournal 9, 781–791. doi: 10.1100/tsw.2009.93
Haass, C., Kaether, C., Thinakaran, G., and Sisodia, S. (2012). Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2, a006270. doi: 10.1101/cshperspect.a006270
Haberman, R. P., Koh, M. T., and Gallagher, M. (2017). Heightened cortical excitability in aged rodents with memory impairment. Neurobiol. Aging 54, 144–151. doi: 10.1016/j.neurobiolaging.2016.12.021
Hanson, J. E., Ma, K., Elstrott, J., Weber, M., Saillet, S., Khan, A. S., et al. (2020). GluN2A NMDA receptor enhancement improves brain oscillations, synchrony, and cognitive functions in dravet syndrome and Alzheimer's disease models. Cell Rep. 30, 381–396.e384. doi: 10.1016/j.celrep.2019.12.030
Hardingham, G. E., and Bading, H. (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696. doi: 10.1038/nrn2911
Harkany, T., Abraham, I., Timmerman, W., Laskay, G., Toth, B., Sasvari, M., et al. (2000). β-amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur. J. Neurosci. 12, 2735–2745. doi: 10.1046/j.1460-9568.2000.00164.x
Harris, M. E., Wang, Y., Pedigo, N. W. Jr., Hensley, K., Butterfield, D. A., and Carney, J. M. (1996). Amyloid beta peptide (25-35) inhibits Na+-dependent glutamate uptake in rat hippocampal astrocyte cultures. J. Neurochem. 67, 277–286. doi: 10.1046/j.1471-4159.1996.67010277.x
Hefendehl, J. K., LeDue, J., Ko, R. W., Mahler, J., Murphy, T. H., and MacVicar, B. A. (2016). Mapping synaptic glutamate transporter dysfunction in vivo to regions surrounding Abeta plaques by iGluSnFR two-photon imaging. Nat. Commun. 7, 13441. doi: 10.1038/ncomms13441
Herrmann, C. S., and Demiralp, T. (2005). Human EEG gamma oscillations in neuropsychiatric disorders. Clin. Neurophysiol. 116, 2719–2733. doi: 10.1016/j.clinph.2005.07.007
Hita-Yanez, E., Atienza, M., Gil-Neciga, E., and Cantero, J. L. (2012). Disturbed sleep patterns in elders with mild cognitive impairment: the role of memory decline and ApoE ε4 genotype. Curr. Alzheimer Res. 9, 290–297. doi: 10.2174/156720512800107609
Huijbers, W., Mormino, E. C., Schultz, A. P., Wigman, S., Ward, A. M., Larvie, M., et al. (2015). Amyloid-beta deposition in mild cognitive impairment is associated with increased hippocampal activity, atrophy and clinical progression. Brain 138, 1023–1035. doi: 10.1093/brain/awv007
Iaccarino, H. F., Singer, A. C., Martorell, A. J., Rudenko, A., Gao, F., Gillingham, T. Z., et al. (2016). Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 540, 230–235. doi: 10.1038/nature20587
Jacob, C. P., Koutsilieri, E., Bartl, J., Neuen-Jacob, E., Arzberger, T., Zander, N., et al. (2007). Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer's disease. J. Alzheimers Dis. 11, 97–116. doi: 10.3233/JAD-2007-11113
Jensen, O., Kaiser, J., and Lachaux, J. P. (2007). Human gamma-frequency oscillations associated with attention and memory. Trends Neurosci. 30, 317–324. doi: 10.1016/j.tins.2007.05.001
Jun, H., Bramian, A., Soma, S., Saito, T., Saido, T. C., and Igarashi, K. M. (2020). Disrupted place cell remapping and impaired grid cells in a knockin model of Alzheimer's disease. Neuron. 107, 1095–1112.e6. doi: 10.1016/j.neuron.2020.06.023
Kabogo, D., Rauw, G., Amritraj, A., Baker, G., and Kar, S. (2010). ss-amyloid-related peptides potentiate K+-evoked glutamate release from adult rat hippocampal slices. Neurobiol. Aging 31, 1164–1172. doi: 10.1016/j.neurobiolaging.2008.08.009
Kastanenka, K. V., Calvo-Rodriguez, M., Hou, S. S., Zhou, H., Takeda, S., Arbel-Ornath, M., et al. (2019). Frequency-dependent exacerbation of Alzheimer's disease neuropathophysiology. Sci. Rep. 9, 8964. doi: 10.1038/s41598-019-44964-z
Kastanenka, K. V., Hou, S. S., Shakerdge, N., Logan, R., Feng, D., Wegmann, S., et al. (2017). Optogenetic restoration of disrupted slow oscillations halts amyloid deposition and restores calcium homeostasis in an animal model of Alzheimer's disease. PLoS One 12, e0170275. doi: 10.1371/journal.pone.0170275
Kent, B. A., Strittmatter, S. M., and Nygaard, H. B. (2018). Sleep and EEG power spectral analysis in three transgenic mouse models of Alzheimer's disease: APP/PS1, 3 × TgAD, and Tg2576. J. Alzheimers Dis. 64, 1325–1336. doi: 10.3233/JAD-180260
Keskin, A. D., Kekus, M., Adelsberger, H., Neumann, U., Shimshek, D. R., Song, B., et al. (2017). BACE inhibition-dependent repair of Alzheimer's pathophysiology. Proc. Natl. Acad. Sci. U. S. A. 114, 8631–8636. doi: 10.1073/pnas.1708106114
Kim, H. S., Lee, J. H., and Suh, Y. H. (1999). C-terminal fragment of Alzheimer's amyloid precursor protein inhibits sodium/calcium exchanger activity in SK-N-SH cell. Neuroreport 10, 113–116. doi: 10.1097/00001756-199901180-00022
Koh, M. T., Haberman, R. P., Foti, S., McCown, T. J., and Gallagher, M. (2010). Treatment strategies targeting excess hippocampal activity benefit aged rats with cognitive impairment. Neuropsychopharmacology 35, 1016–1025. doi: 10.1038/npp.2009.207
Kunz, L., Schroder, T. N., Lee, H., Montag, C., Lachmann, B., Sariyska, R., et al. (2015). Reduced grid-cell-like representations in adults at genetic risk for Alzheimer's disease. Science 350, 430–433. doi: 10.1126/science.aac8128
Ladenbauer, J., Ladenbauer, J., Kulzow, N., de Boor, R., Avramova, E., Grittner, U., et al. (2017). Promoting sleep oscillations and their functional coupling by transcranial stimulation enhances memory consolidation in mild cognitive impairment. J. Neurosci. 37, 7111–7124. doi: 10.1523/JNEUROSCI.0260-17.2017
Lai, F., and Williams, R. S. (1989). A prospective study of Alzheimer disease in Down syndrome. Arch. Neurol. 46, 849–853. doi: 10.1001/archneur.1989.00520440031017
Lam, A. D., Deck, G., Goldman, A., Eskandar, E. N., Noebels, J., and Cole, A. J. (2017). Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer's disease. Nat. Med. 23, 678–680. doi: 10.1038/nm.4330
Leal, S. L., Landau, S. M., Bell, R. K., and Jagust, W. J. (2017). Hippocampal activation is associated with longitudinal amyloid accumulation and cognitive decline. Elife. 6:e22978. doi: 10.7554/eLife.22978.012
Lee, Y. F., Gerashchenko, D., Timofeev, I., Bacskai, B. J., and Kastanenka, K. V. (2020). Slow wave sleep is a promising intervention target for Alzheimer's disease. Front. Neurosci. 14, 705. doi: 10.3389/fnins.2020.00705
Li, S., Hong, S., Shepardson, N. E., Walsh, D. M., Shankar, G. M., and Selkoe, D. (2009). Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801. doi: 10.1016/j.neuron.2009.05.012
Li, S., Jin, M., Koeglsperger, T., Shepardson, N. E., Shankar, G. M., and Selkoe, D. J. (2011). Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 31, 6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011
Liedorp, M., Stam, C. J., van der Flier, W. M., Pijnenburg, Y. A., and Scheltens, P. (2010). Prevalence and clinical significance of epileptiform EEG discharges in a large memory clinic cohort. Dement. Geriatr. Cogn. Disord. 29, 432–437. doi: 10.1159/000278620
Limon, A., Reyes-Ruiz, J. M., and Miledi, R. (2012). Loss of functional GABA (A) receptors in the Alzheimer diseased brain. Proc. Natl. Acad. Sci. U. S. A. 109, 10071–10076. doi: 10.1073/pnas.1204606109
Lynch, B. A., Lambeng, N., Nocka, K., Kensel-Hammes, P., Bajjalieh, S. M., Matagne, A., et al. (2004). The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. U. S. A. 101, 9861–9866. doi: 10.1073/pnas.0308208101
Mably, A. J., Gereke, B. J., Jones, D. T., and Colgin, L. L. (2017). Impairments in spatial representations and rhythmic coordination of place cells in the 3 × Tg mouse model of Alzheimer's disease. Hippocampus 27, 378–392. doi: 10.1002/hipo.22697
MacManus, A., Ramsden, M., Murray, M., Henderson, Z., Pearson, H. A., and Campbell, V. A. (2000). Enhancement of (45)Ca(2+) influx and voltage-dependent Ca(2+) channel activity by beta-amyloid-(1-40) in rat cortical synaptosomes and cultured cortical neurons. Modulation by the proinflammatory cytokine interleukin-1β. J. Biol. Chem. 275, 4713–4718. doi: 10.1074/jbc.275.7.4713
Maier, F. C., Wehrl, H. F., Schmid, A. M., Mannheim, J. G., Wiehr, S., Lerdkrai, C., et al. (2014). Longitudinal PET-MRI reveals beta-amyloid deposition and rCBF dynamics and connects vascular amyloidosis to quantitative loss of perfusion. Nat. Med. 20, 1485–1492. doi: 10.1038/nm.3734
Mander, B. A., Marks, S. M., Vogel, J. W., Rao, V., Lu, B., Saletin, J. M., et al. (2015). β-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat. Neurosci. 18, 1051–1057. doi: 10.1038/nn.4035
Martorell, A. J., Paulson, A. L., Suk, H. J., Abdurrob, F., Drummond, G. T., Guan, W., et al. (2019). Multi-sensory gamma stimulation ameliorates Alzheimer's-associated pathology and improves cognition. Cell 177, 256–271.e222. doi: 10.1016/j.cell.2019.02.014
Masliah, E., Alford, M., DeTeresa, R., Mallory, M., and Hansen, L. (1996). Deficient glutamate transport is associated with neurodegeneration in Alzheimer's disease. Ann. Neurol. 40, 759–766. doi: 10.1002/ana.410400512
Mata, A. M. (2018). Functional interplay between plasma membrane Ca(2+)-ATPase, amyloid beta-peptide and tau. Neurosci. Lett. 663, 55–59. doi: 10.1016/j.neulet.2017.08.004
Matsumoto, J. Y., Stead, M., Kucewicz, M. T., Matsumoto, A. J., Peters, P. A., Brinkmann, B. H., et al. (2013). Network oscillations modulate interictal epileptiform spike rate during human memory. Brain 136, 2444–2456. doi: 10.1093/brain/awt159
Mattson, M. P., Cheng, B., Davis, D., Bryant, K., Lieberburg, I., and Rydel, R. E. (1992). β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 12, 376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992
Min, D., Guo, F., Zhu, S., Xu, X., Mao, X., Cao, Y., et al. (2013). The alterations of Ca2+/calmodulin/CaMKII/CaV1.2 signaling in experimental models of Alzheimer's disease and vascular dementia. Neurosci. Lett. 538, 60–65. doi: 10.1016/j.neulet.2013.02.001
Minkeviciene, R., Rheims, S., Dobszay, M. B., Zilberter, M., Hartikainen, J., Fulop, L., et al. (2009). Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci. 29, 3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009
Mondadori, C. R., Buchmann, A., Mustovic, H., Schmidt, C. F., Boesiger, P., Nitsch, R. M., et al. (2006). Enhanced brain activity may precede the diagnosis of Alzheimer's disease by 30 years. Brain 129, 2908–2922. doi: 10.1093/brain/awl266
Mondragon-Rodriguez, S., Gu, N., Manseau, F., and Williams, S. (2018). Alzheimer's transgenic model is characterized by very early brain network alterations and beta-CTF fragment accumulation: reversal by beta-secretase inhibition. Front. Cell Neurosci. 12, 121. doi: 10.3389/fncel.2018.00121
Mormino, E. C., Brandel, M. G., Madison, C. M., Marks, S., Baker, S. L., and Jagust, W. J. (2012). Abeta Deposition in aging is associated with increases in brain activation during successful memory encoding. Cereb. Cortex 22, 1813–1823. doi: 10.1093/cercor/bhr255
Moser, E. I., Moser, M. B., and McNaughton, B. L. (2017). Spatial representation in the hippocampal formation: a history. Nat. Neurosci. 20, 1448–1464. doi: 10.1038/nn.4653
Nabavi, S., Fox, R., Proulx, C. D., Lin, J. Y., Tsien, R. Y., and Malinow, R. (2014). Engineering a memory with LTD and LTP. Nature 511, 348–352. doi: 10.1038/nature13294
Nygaard, H. B., Kaufman, A. C., Sekine-Konno, T., Huh, L. L., Going, H., Feldman, S. J., et al. (2015). Brivaracetam, but not ethosuximide, reverses memory impairments in an Alzheimer's disease mouse model. Alzheimers Res. Ther. 7, 25. doi: 10.1186/s13195-015-0110-9
O'Brien, J. L., O'Keefe, K. M., LaViolette, P. S., DeLuca, A. N., Blacker, D., Dickerson, B. C., et al. (2010). Longitudinal fMRI in elderly reveals loss of hippocampal activation with clinical decline. Neurology 74, 1969–1976. doi: 10.1212/WNL.0b013e3181e3966e
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron 55, 697–711. doi: 10.1016/j.neuron.2007.07.025
Palop, J. J., and Mucke, L. (2010). Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat. Neurosci. 13, 812–818. doi: 10.1038/nn.2583
Pariente, J., Cole, S., Henson, R., Clare, L., Kennedy, A., Rossor, M., et al. (2005). Alzheimer's patients engage an alternative network during a memory task. Ann. Neurol. 58, 870–879. doi: 10.1002/ana.20653
Park, J., Wetzel, I., Marriott, I., Dreau, D., D'Avanzo, C., Kim, D. Y., et al. (2018). A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer's disease. Nat. Neurosci. 21, 941–951. doi: 10.1038/s41593-018-0175-4
Parpura-Gill, A., Beitz, D., and Uemura, E. (1997). The inhibitory effects of beta-amyloid on glutamate and glucose uptakes by cultured astrocytes. Brain Res. 754, 65–71. doi: 10.1016/S0006-8993(97)00043-7
Parsons, M. P., and Raymond, L. A. (2014). Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 82, 279–293. doi: 10.1016/j.neuron.2014.03.030
Persson, J., Lind, J., Larsson, A., Ingvar, M., Sleegers, K., Van Broeckhoven, C., et al. (2008). Altered deactivation in individuals with genetic risk for Alzheimer's disease. Neuropsychologia 46, 1679–1687. doi: 10.1016/j.neuropsychologia.2008.01.026
Pike, K. E., Savage, G., Villemagne, V. L., Ng, S., Moss, S. A., Maruff, P., et al. (2007). Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer's disease. Brain 130, 2837–2844. doi: 10.1093/brain/awm238
Putcha, D., Brickhouse, M., O'Keefe, K., Sullivan, C., Rentz, D., Marshall, G., et al. (2011). Hippocampal hyperactivation associated with cortical thinning in Alzheimer's disease signature regions in non-demented elderly adults. J. Neurosci. 31, 17680–17688. doi: 10.1523/JNEUROSCI.4740-11.2011
Puzzo, D., Privitera, L., Leznik, E., Fa, M., Staniszewski, A., Palmeri, A., et al. (2008). Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 28, 14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008
Quiroz, Y. T., Budson, A. E., Celone, K., Ruiz, A., Newmark, R., Castrillon, G., et al. (2010). Hippocampal hyperactivation in presymptomatic familial Alzheimer's disease. Ann. Neurol. 68, 865–875. doi: 10.1002/ana.22105
Reiman, E. M., Quiroz, Y. T., Fleisher, A. S., Chen, K., Velez-Pardo, C., Jimenez-Del-Rio, M., et al. (2012). Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer's disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 11, 1048–1056. doi: 10.1016/S1474-4422(12)70228-4
Robinson, D. M., and Keating, G. M. (2006). Memantine: a review of its use in Alzheimer's disease. Drugs 66, 1515–1534. doi: 10.2165/00003495-200666110-00015
Rudinskiy, N., Hawkes, J. M., Betensky, R. A., Eguchi, M., Yamaguchi, S., Spires-Jones, T. L., et al. (2012). Orchestrated experience-driven Arc responses are disrupted in a mouse model of Alzheimer's disease. Nat. Neurosci. 15, 1422–1429. doi: 10.1038/nn.3199
Russell, C. L., Semerdjieva, S., Empson, R. M., Austen, B. M., Beesley, P. W., and Alifragis, P. (2012). Amyloid-beta acts as a regulator of neurotransmitter release disrupting the interaction between synaptophysin and VAMP2. PLoS One 7, e43201. doi: 10.1371/journal.pone.0043201
Samson, W. N., van Duijn, C. M., Hop, W. C., and Hofman, A. (1996). Clinical features and mortality in patients with early-onset Alzheimer's disease. Eur. Neurol. 36, 103–106. doi: 10.1159/000117218
Sanchez, P. E., Zhu, L., Verret, L., Vossel, K. A., Orr, A. G., Cirrito, J. R., et al. (2012). Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc. Natl. Acad. Sci. U. S. A. 109, E2895–2903. doi: 10.1073/pnas.1121081109
Sanchez-Espinosa, M. P., Atienza, M., and Cantero, J. L. (2014). Sleep deficits in mild cognitive impairment are related to increased levels of plasma amyloid-beta and cortical thinning. Neuroimage 98, 395–404. doi: 10.1016/j.neuroimage.2014.05.027
Sanchez-Mejia, R. O., Newman, J. W., Toh, S., Yu, G. Q., Zhou, Y., Halabisky, B., et al. (2008). Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer's disease. Nat. Neurosci. 11, 1311–1318. doi: 10.1038/nn.2213
SanMartin, C. D., Veloso, P., Adasme, T., Lobos, P., Bruna, B., Galaz, J., et al. (2017). RyR2-Mediated Ca(2+) release and mitochondrial ROS generation partake in the synaptic dysfunction caused by amyloid beta peptide oligomers. Front. Mol. Neurosci. 10, 115. doi: 10.3389/fnmol.2017.00115
Sederberg, P. B., Schulze-Bonhage, A., Madsen, J. R., Bromfield, E. B., McCarthy, D. C., Brandt, A., et al. (2007). Hippocampal and neocortical gamma oscillations predict memory formation in humans. Cereb. Cortex 17, 1190–1196. doi: 10.1093/cercor/bhl030
Selkoe, D. J. (2002). Alzheimer's disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Sepulveda-Falla, D., Glatzel, M., and Lopera, F. (2012). Phenotypic profile of early-onset familial Alzheimer's disease caused by presenilin-1 E280A mutation. J. Alzheimers Dis. 32, 1–12. doi: 10.3233/JAD-2012-120907
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Shea, Y. F., Chu, L. W., Chan, A. O., Ha, J., Li, Y., and Song, Y. Q. (2016). A systematic review of familial Alzheimer's disease: Differences in presentation of clinical features among three mutated genes and potential ethnic differences. J. Formos. Med. Assoc. 115, 67–75. doi: 10.1016/j.jfma.2015.08.004
Sherzai, D., Losey, T., Vega, S., and Sherzai, A. (2014). Seizures and dementia in the elderly: Nationwide Inpatient Sample 1999-2008. Epilepsy Behav. 36, 53–56. doi: 10.1016/j.yebeh.2014.04.015
Shtifman, A., Ward, C. W., Laver, D. R., Bannister, M. L., Lopez, J. R., Kitazawa, M., et al. (2010). Amyloid-beta protein impairs Ca2+ release and contractility in skeletal muscle. Neurobiol. Aging 31, 2080–2090. doi: 10.1016/j.neurobiolaging.2008.11.003
Sompol, P., Furman, J. L., Pleiss, M. M., Kraner, S. D., Artiushin, I. A., Batten, S. R., et al. (2017). Calcineurin/NFAT signaling in activated astrocytes drives network hyperexcitability in abeta-bearing mice. J. Neurosci. 37, 6132–6148. doi: 10.1523/JNEUROSCI.0877-17.2017
Sperling, R., Chua, E., Cocchiarella, A., Rand-Giovannetti, E., Poldrack, R., Schacter, D. L., et al. (2003). Putting names to faces: successful encoding of associative memories activates the anterior hippocampal formation. Neuroimage 20, 1400–1410. doi: 10.1016/S1053-8119(03)00391-4
Sperling, R. A., Laviolette, P. S., O'Keefe, K., O'Brien, J., Rentz, D. M., Pihlajamaki, M., et al. (2009). Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63, 178–188. doi: 10.1016/j.neuron.2009.07.003
Steriade, M., Nunez, A., and Amzica, F. (1993). A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J. Neurosci. 13, 3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993
Styr, B., and Slutsky, I. (2018). Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer's disease. Nat. Neurosci. 21, 463–473. doi: 10.1038/s41593-018-0080-x
Suberbielle, E., Djukic, B., Evans, M., Kim, D. H., Taneja, P., Wang, X., et al. (2015). DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 6, 8897. doi: 10.1038/ncomms9897
Suberbielle, E., Sanchez, P. E., Kravitz, A. V., Wang, X., Ho, K., Eilertson, K., et al. (2013). Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nat. Neurosci. 16, 613–621. doi: 10.1038/nn.3356
Terunuma, M., Vargas, K. J., Wilkins, M. E., Ramirez, O. A., Jaureguiberry-Bravo, M., Pangalos, M. N., et al. (2010). Prolonged activation of NMDA receptors promotes dephosphorylation and alters postendocytic sorting of GABAB receptors. Proc. Natl. Acad. Sci. U. S. A. 107, 13918–13923. doi: 10.1073/pnas.1000853107
Thibault, O., Pancani, T., Landfield, P. W., and Norris, C. M. (2012). Reduction in neuronal L-type calcium channel activity in a double knock-in mouse model of Alzheimer's disease. Biochim. Biophys. Acta 1822, 546–549. doi: 10.1016/j.bbadis.2012.01.004
Thome, A., Gray, D. T., Erickson, C. A., Lipa, P., and Barnes, C. A. (2016). Memory impairment in aged primates is associated with region-specific network dysfunction. Mol. Psychiatry 21, 1257–1262. doi: 10.1038/mp.2015.160
Timofeev, I., and Chauvette, S. (2017). Sleep slow oscillation and plasticity. Curr. Opin. Neurobiol. 44, 116–126. doi: 10.1016/j.conb.2017.03.019
Townsend, M., Shankar, G. M., Mehta, T., Walsh, D. M., and Selkoe, D. J. (2006). Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J. Physiol. 572, 477–492. doi: 10.1113/jphysiol.2005.103754
Trivedi, M. A., Schmitz, T. W., Ries, M. L., Hess, T. M., Fitzgerald, M. E., Atwood, C. S., et al. (2008). fMRI activation during episodic encoding and metacognitive appraisal across the lifespan: risk factors for Alzheimer's disease. Neuropsychologia 46, 1667–1678. doi: 10.1016/j.neuropsychologia.2007.11.035
Ueda, K., Shinohara, S., Yagami, T., Asakura, K., and Kawasaki, K. (1997). Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J. Neurochem. 68, 265–271. doi: 10.1046/j.1471-4159.1997.68010265.x
Um, J. W., Nygaard, H. B., Heiss, J. K., Kostylev, M. A., Stagi, M., Vortmeyer, A., et al. (2012). Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235. doi: 10.1038/nn.3178
van Deursen, J. A., Vuurman, E. F., Verhey, F. R., van Kranen-Mastenbroek, V. H., and Riedel, W. J. (2008). Increased EEG gamma band activity in Alzheimer's disease and mild cognitive impairment. J. Neural Transm. (Vienna) 115, 1301–1311. doi: 10.1007/s00702-008-0083-y
Velliquette, R. A., O'Connor, T., and Vassar, R. (2005). Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer's disease pathogenesis. J. Neurosci. 25, 10874–10883. doi: 10.1523/JNEUROSCI.2350-05.2005
Verret, L., Mann, E. O., Hang, G. B., Barth, A. M., Cobos, I., Ho, K., et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721. doi: 10.1016/j.cell.2012.02.046
Vogl, C., Mochida, S., Wolff, C., Whalley, B. J., and Stephens, G. J. (2012). The synaptic vesicle glycoprotein 2A ligand levetiracetam inhibits presynaptic Ca2+ channels through an intracellular pathway. Mol. Pharmacol. 82, 199–208. doi: 10.1124/mol.111.076687
Vossel, K. A., Beagle, A. J., Rabinovici, G. D., Shu, H., Lee, S. E., Naasan, G., et al. (2013). Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 70, 1158–1166. doi: 10.1001/jamaneurol.2013.136
Vossel, K. A., Ranasinghe, K. G., Beagle, A. J., Mizuiri, D., Honma, S. M., Dowling, A. F., et al. (2016). Incidence and impact of subclinical epileptiform activity in Alzheimer's disease. Ann. Neurol. 80, 858–870. doi: 10.1002/ana.24794
Wang, X., and Zheng, W. (2019). Ca(2+) homeostasis dysregulation in Alzheimer's disease: a focus on plasma membrane and cell organelles. FASEB J. 33, 6697–6712. doi: 10.1096/fj.201801751R
Westerberg, C. E., Mander, B. A., Florczak, S. M., Weintraub, S., Mesulam, M. M., Zee, P. C., et al. (2012). Concurrent impairments in sleep and memory in amnestic mild cognitive impairment. J. Int. Neuropsychol. Soc. 18, 490–500. doi: 10.1017/S135561771200001X
Winer, J. R., Mander, B. A., Helfrich, R. F., Maass, A., Harrison, T. M., Baker, S. L., et al. (2019). Sleep as a potential biomarker of tau and β-amyloid burden in the human brain. J. Neurosci. 39, 6315–6324. doi: 10.1523/JNEUROSCI.0503-19.2019
Wu, A., Derrico, C. A., Hatem, L., and Colvin, R. A. (1997). Alzheimer's amyloid-beta peptide inhibits sodium/calcium exchange measured in rat and human brain plasma membrane vesicles. Neuroscience 80, 675–684. doi: 10.1016/S0306-4522(97)00053-5
Yamamoto, J., Suh, J., Takeuchi, D., and Tonegawa, S. (2014). Successful execution of working memory linked to synchronized high-frequency gamma oscillations. Cell 157, 845–857. doi: 10.1016/j.cell.2014.04.009
Yamamoto, K., Tanei, Z. I., Hashimoto, T., Wakabayashi, T., Okuno, H., Naka, Y., et al. (2015). Chronic optogenetic activation augments abeta pathology in a mouse model of Alzheimer disease. Cell Rep. 11, 859–865. doi: 10.1016/j.celrep.2015.04.017
Yassa, M. A., Lacy, J. W., Stark, S. M., Albert, M. S., Gallagher, M., and Stark, C. E. (2011). Pattern separation deficits associated with increased hippocampal CA3 and dentate gyrus activity in nondemented older adults. Hippocampus 21, 968–979. doi: 10.1002/hipo.20808
Yuan, P., and Grutzendler, J. (2016). Attenuation of β-amyloid deposition and neurotoxicity by chemogenetic modulation of neural activity. J. Neurosci. 36, 632–641. doi: 10.1523/JNEUROSCI.2531-15.2016
Zeineh, M. M., Engel, S. A., Thompson, P. M., and Bookheimer, S. Y. (2003). Dynamics of the hippocampus during encoding and retrieval of face-name pairs. Science 299, 577–580. doi: 10.1126/science.1077775
Zhao, J., Li, Z., Cong, Y., Zhang, J., Tan, M., Zhang, H., et al. (2017). Repetitive transcranial magnetic stimulation improves cognitive function of Alzheimer's disease patients. Oncotarget 8, 33864–33871. doi: 10.18632/oncotarget.13060
Zilberter, M., Ivanov, A., Ziyatdinova, S., Mukhtarov, M., Malkov, A., Alpar, A., et al. (2013). Dietary energy substrates reverse early neuronal hyperactivity in a mouse model of Alzheimer's disease. J. Neurochem. 125, 157–171. doi: 10.1111/jnc.12127
Keywords: amyloid-beta oligomers, hyperactivity, neurodegeneration, memory, epileptiform activity, gamma oscillations, slow wave
Citation: Hector A and Brouillette J (2021) Hyperactivity Induced by Soluble Amyloid-β Oligomers in the Early Stages of Alzheimer's Disease. Front. Mol. Neurosci. 13:600084. doi: 10.3389/fnmol.2020.600084
Received: 28 August 2020; Accepted: 01 December 2020;
Published: 07 January 2021.
Edited by:
Robert A. Nichols, University of Hawaii at Manoa, United StatesReviewed by:
Shaomin Li, Brigham and Women's Hospital and Harvard Medical School, United StatesCopyright © 2021 Hector and Brouillette. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Brouillette, am9uYXRoYW4uYnJvdWlsbGV0dGVAdW1vbnRyZWFsLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.