 Andrea N. Suarez
Andrea N. Suarez Emily E. Noble
Emily E. Noble Scott E. Kanoski
Scott E. Kanoski
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 18 April 2019
Sec. Neuroplasticity and Development
Volume 12 - 2019 | https://doi.org/10.3389/fnmol.2019.00101
This article is part of the Research Topic Learning and Memory View all 59 articles
The hippocampus (HPC) controls fundamental learning and memory processes, including memory for visuospatial navigation (spatial memory) and flexible memory for facts and autobiographical events (declarative memory). Emerging evidence reveals that hippocampal-dependent memory function is regulated by various peripheral biological systems that are traditionally known for their roles in appetite and body weight regulation. Here, we argue that these effects are consistent with a framework that it is evolutionarily advantageous to encode and recall critical features surrounding feeding behavior, including the spatial location of a food source, social factors, post-absorptive processing, and other episodic elements of a meal. We review evidence that gut-to-brain communication from the vagus nerve and from feeding-relevant endocrine systems, including ghrelin, insulin, leptin, and glucagon-like peptide-1 (GLP-1), promote hippocampal-dependent spatial and declarative memory via neurotrophic and neurogenic mechanisms. The collective literature reviewed herein supports a model in which various stages of feeding behavior and hippocampal-dependent memory function are closely linked.
“Ponder well on this point: the pleasant hours of our life are all connected by a more or less tangible link with some memory of the table”
—Charles Monselet.
In most corners of the globe food is readily available with minimal effort required for foraging. This luxury of the modern environment was not enjoyed throughout human evolution, as food in nature was often irregularly available to our ancestors and required substantial labor and resources to obtain. A survival advantage common to early humans and lower-order mammals is to accurately remember the physical location of a food source, and to then efficiently navigate back to a shelter/dwelling. Thus, it follows that the biological systems that regulate feeding behavior may share common origins with those involved in learning about one’s external environment. Here, we present a framework that the ability to remember the location and features of environmental locations (visuospatial and contextual memory, respectively) is intimately linked with endocrine and neural pathways that also regulate appetite and feeding behavior.
In addition to external spatial and contextual cues, learning about other aspects of feeding behavior also provides a survival advantage. For example, in humans and in lower-order mammals (including rodents), social cues strongly influence both food choice and amount consumed, in part, by conveying that a particular food is safe and/or nutritive (de Castro et al., 1990; Levitsky, 2005; Herman and Higgs, 2015). Further, memory of the physical location of a food source is modulated by temporal factors, including longer-term seasonal changes for vegetation and shorter-term diurnal fluctuations for predator-prey dynamics. Remembering these and other aspects of the entire “feeding episode” are useful to more efficiently guide future foraging and feeding behavior. Thus, biological systems that fluctuate around and influence feeding behavior may also promote a hippocampal-dependent memory process known as declarative memory, which is the flexible memory for facts and episodic events (Eichenbaum and Cohen, 2014). We note, however, that declarative memory and visuospatial memory are not necessarily two distinct processes, but rather, may be two manifestations of the same (or similar) fundamental memory process that is mediated by hippocampal neurons (Buzsáki and Moser, 2013; Milivojevic and Doeller, 2013; Eichenbaum and Cohen, 2014).
During the preprandial (before a meal), prandial, and postprandial stages of feeding behavior, various endocrine, neuropeptidergic, and neural signals are released to regulate appetite, meal size, and the inter-meal interval. Emerging evidence reveals that in addition to regulating feeding behavior, these energy status-related biological systems also play a critical role in learning and memory function, particularly with regards to remembering feeding-relevant episodic experiences and visuospatial information such as recalling when, how, and where food was obtained and consumed. Here, we review evidence that these energy status-related biological signals converge with external sensory-related cues in the hippocampus (HPC), a brain region that is most famously linked with learning and memory and more recently with the higher-order controls of feeding behavior (Davidson et al., 2007; Parent et al., 2014; Kanoski and Grill, 2017). Indeed, HPC neurons play an important role in processing relative information about one’s external environment and spatial orientation (Morris et al., 1982; Burgess et al., 2002). The HPC is also required for flexible memory for autobiographical events (“episodic memory,” a component of declarative memory; Tulving and Markowitsch, 1998; Eichenbaum and Cohen, 2014; Parent, 2016).
Consistent with a role for HPC neurons in regulating spatial and declarative memory processes relevant to foraging and feeding, selective HPC lesions impair both meal-related spatial and episodic memory used to relocate previously stored food sources in scrub jays (Clayton and Dickinson, 1998). Similarly, intact rhesus monkeys preferentially return to a foraging site where food was previously obtained in a matching-to-location task, whereas monkeys with HPC lesions do not show this learned matching tendency (Hampton et al., 2004). While selective targeted lesions to the HPC is not a feasible experimental approach in humans for obvious reasons, amnesic humans with bilateral nonselective damage to the HPC and surrounding medial temporal lobe structures (including the amygdala) will eat a full second meal that is offered immediately after consuming a meal with minimal change in self-reported hunger/satiety ratings (Hebben et al., 1985). Analogous findings have been observed in rodents in which reversible postprandial inactivation of either dorsal (dHPC) or ventral (vHPC) hippocampus using either muscimol infusions (Henderson et al., 2013; Hannapel et al., 2017) or optogenetic inhibition (Hannapel et al., 2019) decreases latency to initiate the subsequent meal and increases subsequent intake, results that are hypothesized to be based on disrupted meal-related episodic memory consolidation (Parent, 2016). These findings further indicate that both the dorsal and ventral HPC subregions are involved in meal-related memory processing, which is notable in light data suggesting that these subregions may be functionally distinct with regards to spatial memory (dHPC) and emotional-related memory processing (vHPC; see Moser and Moser, 1998; Fanselow and Dong, 2010; Kanoski and Grill, 2017; for further review on dorsal vs. ventral dissociation of function). Also of note from Parent and colleagues’ work is that reversible optogenetic inactivation of either dHPC or vHPC neurons reduces the latency to initiate a subsequent meal following nonnutritive saccharine consumption (Hannapel et al., 2019), suggesting that HPC control of meal-related episodic memory may be based, in part, on hedonic orosensory properties independent of post-ingestive caloric consequences.
These findings indicate that a critical bridge between previous eating episodes, interoceptive energy status cues, and ongoing feeding behavior resides within the HPC. Consistent with this framework, selective lesions to the HPC in rats yields increased food intake and body weight gain (Davidson et al., 2010), as well as increased meal frequency (Clifton et al., 1998). These results were obtained in rats free feeding in the home cage, and therefore are unlikely to be based on deficits in visuospatial memory and foraging, but rather, may be based on impaired episodic memory of recent eating occasions (e.g., see Parent et al., 2014).
In this review article, we discuss the common biological mechanisms through which feeding behavior and HPC-dependent memory function are closely linked. More specifically, we focus primarily on how peripherally-derived feeding-relevant signals that are released before, during, and after a meal exert their action directly on their respective targets in HPC neurons to influence both feeding behavior and memory function, particularly mnemonic processes related to autobiographical events, foraging, and food location.
Ghrelin, often referred to as the “hunger hormone,” is a 28-amino acid peptide hormone produced and secreted by P/D1 cells in the fundus of the stomach (Kojima et al., 1999; Date et al., 2000; Dornonville de la Cour et al., 2001). Ghrelin binds to and activates its seven transmembrane G protein couple receptor, the type 1a growth hormone secretagogue receptor (GHSR1a, aka “ghrelin receptor”; Howard et al., 1996; Sun et al., 2004). The pre-pro ghrelin precursor protein is first cleaved into two peptides, obestatin and des-acyl ghrelin (Gualillo et al., 2006). Des-acyl ghrelin is present in the stomach and bloodstream and is the inactive form of ghrelin, as it does not engage GHSR1a signaling at physiological concentrations (Hosoda et al., 2000; Tong et al., 2013). During times of energy insufficiency, the ghrelin O-acyltransferase (GOAT) enzyme is upregulated and converts des-acyl into its active acyl form by adding an acyl chain on Ser3 residue of ghrelin (Gahete et al., 2010; Zhao et al., 2010). Following this post-translational modification, acyl ghrelin can then access the active GHSR1a binding sites to augment both appetite and food intake (Yang et al., 2008). Consistent with ghrelin’s orexigenic effects, ghrelin levels are elevated during energy restriction, peak preprandially (Wren et al., 2001a; Drazen et al., 2006; Blum et al., 2009; Davis et al., 2011), and rapidly decrease in response to eating (Ariyasu et al., 2001; Cummings et al., 2001; Nass et al., 2008).
Early research on ghrelin signaling in the brain largely focused on regions classically associated with energy homeostasis [e.g., arcuate nucleus of the hypothalamus (ARH), nucleus tractus solitarius (NTS) in the caudal brainstem] (Wren et al., 2001b; Faulconbridge et al., 2003). However, GHSR1as are also expressed in “higher-order” (limbic, cortical) brain regions involved in memory and cognition, including the HPC (Guan et al., 1997; Zigman et al., 2006). More specifically, GHSR1a is robustly expressed in the dentate gyrus (DG), CA1, CA2, and CA3 regions of the HPC, particularly in the vHPC (Guan et al., 1997; Diano et al., 2006; Zigman et al., 2006; Mani et al., 2014; Hsu et al., 2015b). While the HPC is protected by the blood-brain barrier (BBB), radiolabeled ghrelin is present in the mouse HPC following peripheral administration (Harrold et al., 2008). Bioactive ghrelin may reach HPC neurons either through acyl ghrelin BBB saturable transport, or through des-acyl ghrelin crossing the BBB from blood to brain uni-directionally and conversion to acyl ghrelin within the central nerve system (CNS) via GOAT transcripts expressed in the brain (for review, see Edwards and Abizaid, 2017), as minimal evidence supports the idea that ghrelin is produced in the brain (Ferrini et al., 2009).
Research on ghrelin has predominantly focused on its role in appetite and food intake. Ghrelin signaling also influences memory and cognition, and here we consider that these lesser studied effects are very much connected with ghrelin’s role in appetite. Early evidence for ghrelin’s role in modulating cognitive function comes from Carlini et al. (2002), who demonstrated that administration of acyl ghrelin in the cerebral ventricles (Carlini et al., 2002) or the HPC directly (Carlini et al., 2004) immediately after training improved memory retention in a step-down inhibitory avoidance assay in a dose-dependent manner in rats, indicating a stimulatory effect of ghrelin on memory consolidation for contextual episodic memory. Further work from this group revealed that intra-hippocampal ghrelin administration before training sessions improved memory for the contextual location of aversive reinforcement (Carlini et al., 2010). Diano et al. (2006) extended this work and demonstrated that subcutaneous ghrelin administration in rats improves performance in a spontaneous alternation plus-maze task, whereas intracerebroventricular (ICV) ghrelin administration following training enhanced retention performance in T-maze foot shock avoidance and step-down passive avoidance tasks in mice.
While these pharmacological findings suggest a role for ghrelin in memory consolidation, it is important to consider whether endogenous ghrelin plays a physiological role in memory. Indeed, ghrelin KO mice (Diano et al., 2006) are impaired in a novel object recognition (NOR) task, and these deficits are rescued following subcutaneous ghrelin replacement. Recently the endogenous ghrelin antagonist, liver-expressed antimicrobial peptide 2 (LEAP2), was identified. Not surprisingly, LEAP2 administration reduces food intake, blocks fasting-induced growth hormone secretion, and impairs the maintenance of glucose levels during chronic caloric restriction, however, to our knowledge its role in memory function and neuronal plasticity has yet to be examined (Ge et al., 2018).
The rodent model work described above uses memory tasks that involve learning about external cues and/or episodic memory based on either passive or aversive reinforcement. These fundamental learning processes may be advantageous to inform about future feeding behavior, however, more direct evidence that ghrelin promotes feeding-relevant memory comes from studies that utilize food reinforcement. Considering that ghrelin levels peak before a meal, ghrelin may facilitate food seeking by enhancing HPC-dependent spatial and contextual memory to remember the physical location of food sources, as well as other features (e.g., social factors) that comprise episodic memory relating to appetitive and consummatory behavior. Indeed, wild-type mice treated with a GHSR antagonist and GHSR-null mice fail to show conditioned place preference (CPP) to a high fat diet (HFD; Perello et al., 2010; Chuang et al., 2011; Disse et al., 2011), demonstrating that ghrelin plays a role in enhancing memory for the location of reward-based food intake. Consistent with this framework, wildtype mice exhibit Pavlovian cue-induced hyperphagia following extensive cue-food conditioning (i.e., “cue-potentiated feeding”), whereas GHSR1a-null mice do not (Walker et al., 2012). This deficit may be based, in part, on the loss of GHSR1a in the ventral subregion of the HPC (vHPC), as vHPC ghrelin administration increases food-motivated behaviors for sucrose reinforcement and increases initiation of meals in response to external food-related cues in rats (Kanoski et al., 2013). Moreover, vHPC GHSR1a blockade prior to chow access reduces food intake in meal-entrained rats that had previously learned to consume all of their daily calories in a 4 h period, yet has no effect on intake in rats that were equally food restricted but were not previously meal-entrained (Hsu et al., 2015b). Consistent with these pharmacological findings, GHSR1a-null mice lack food anticipatory activity to habituated feeding responses (Davis et al., 2011).
In addition to promoting appetitive-related memory based on external discrete cues, contextual cues, and temporal scheduled feeding cues, additional rodent model work reveals that ghrelin signaling in HPC promotes social-based memory related to feeding. We recently examined the effects of RNA interference-mediated knockdown (KD) of ventral CA1 GHSR1a [via an adeno-associated virus (AAV) expressing short hairpin RNAs targeting GHSR1a] on learning the olfactory-based social transmission of food preference task (STFP; Hsu et al., 2018). In this task, “observers” rats learn to prefer a flavor of chow based on a brief social interaction with another “demonstrator” rat that had recently consumed the flavored chow. The “transmission” of food preference in observer rats, demonstrated subsequently as a preference for the demonstrator-paired flavored chow vs. a novel flavored chow, is based on exposure to olfactory cues from the breath of the demonstrator rat during the social interaction (Countryman et al., 2005). Rats that received a control AAV in the vHPC (with scrambled sequence for the short hairpin RNAs) preferred the demonstrator-paired flavored chow vs. the novel flavored chow, whereas rats with vHPC GHSR-1a KD were impaired in learning the STFP task. Importantly, a non-social olfactory transmission of food preference control task was performed in this study to determine whether the deficits found in vHPC GHSR1a KD is specific to olfactory processing of social cues (Hsu et al., 2018). This task replicates the olfactory components of the STFP task but excludes social interaction with a demonstrator rat in the social interaction arena. Results from this control procedure show that both vHPC GHSR1a KD and control animals preferred the flavored chow that was paired with the bedding, indicating vHPC GHSR1a KD selectively impairs learned, social transmission of food preference without deficits in olfactory processing or habituation learning.
Evidence from humans also supports the idea that ghrelin signaling enhances attention to external food-related cues, and this enhanced attention may serve to promote the acquisition and consolidation of food-related memories via action in the HPC. For example, ghrelin administration to healthy volunteers during functional magnetic resonance imaging increases cerebral blood flow (CBF) response to food cues, and in the HPC and amygdala, this effect is specific to food cues with no change in CBF in response to scenery pictures (Malik et al., 2008). A more recent article expanded this work by showing that the ability of hyperghrelinemia (either endogenous or exogenous-based) to enhance food-cue induced increased CBF, including in the HPC, is not explicable by consistent changes in other metabolic markers that vary with energy status, including glucose, insulin, peptide YY, and glucagon-like peptide-1 (GLP-1; Goldstone et al., 2014). Moreover, intravenous ghrelin administration in healthy humans enhances the formation of food-related cue-reward associations by increasing HPC signaling to ventral striatum (Han et al., 2018), results consistent with a previously discussed study in rodents demonstrating an important role for vHPC ghrelin signaling in external social-based food cues (Hsu et al., 2018).
Ghrelin enhances memory function, in part, by promoting adult hippocampal neurogenesis and synaptic plasticity. Ghrelin administered peripherally induces proliferation and differentiation of adult progenitor cells in the DG (Moon et al., 2009; Zhao et al., 2014), whereas immunoneutralization of ghrelin in the DG reduces these effects (Moon et al., 2009). Furthermore, ghrelin regulates morphological changes in HPC neurons as ghrelin receptor KO rodent models are associated with reduced HPC spine density (Cahill et al., 2014), whereas reduced HPC spine density is rescued following peripheral ghrelin administration in ghrelin deficient rodents (Diano et al., 2006). In vitro ghrelin administration also enhances long-term potentiation (LTP) in HPC slices (Diano et al., 2006), whereas in vivo ghrelin administration directly in the DG enhances synaptic plasticity [e.g., long-lasting potentiation of excitatory postsynaptic potentials (EPSPs)] and improves spatial memory in the Morris water maze task via activation of the PI3K signaling pathway (Chen et al., 2011). Collectively these findings show that ghrelin enhances neurogenesis and neural plasticity in the HPC and that these processes are feasible neurobiological mechanisms through which ghrelin promotes memory processes.
Considering that plasma ghrelin concentrations increase during calorie restriction (Yang et al., 2007), and that calorie restriction increases synaptic plasticity (Fontán-Lozano et al., 2007) as well as ghrelin BBB transport (Harrold et al., 2008; Schaeffer et al., 2013), it is likely that ghrelin’s role in enhancing memory function is very much dependent on energy status. Consistent with this notion, overnight calorie restriction (or intraperitoneal acyl-ghrelin administration) increased levels of early growth response-1 (Egr-1; a neurogenic transcription factor) in the DG, and 2 weeks on a calorie restriction diet improved contextual fear memory and increased neurogenesis in the DG of wild type but not GHSR deficient mice (Hornsby et al., 2016). Additionally, when ghrelin KO and wild-type mice were maintained on either dietary restriction or ad libitum feeding for 3 months, the ghrelin KO mice on an ad lib diet demonstrated reduced neurogenesis, whereas dietary restriction increased the survival of newborn cells in wild type, but not ghrelin knock out mice (Kim et al., 2015). These studies indicate that endogenous GHSR and ghrelin signaling during calorie restriction play a critical role in enhancing HPC neurogenesis and memory function induced by energy deficits.
On the other side of the energy balance scale, obese humans (English et al., 2002; Yildiz et al., 2004) and diet-induced obese rodents (Perreault et al., 2004; Williams et al., 2006; Uchida et al., 2014) exhibit attenuated ghrelin secretion and reduced circulating plasma ghrelin levels. Obese mice also demonstrate deficits in ghrelin BBB permeability (Banks et al., 2008), as well as reduced hyperphagia (Perreault et al., 2004) and hypothalamic arcuate NPY/AgRP mRNA expression (Briggs et al., 2010) in response to peripheral administration of ghrelin. In humans, lean individuals homozygous for the obesity risk-associated fat mass and obesity-related (FTO) gene allele demonstrate attenuated postprandial reduction of circulating levels of acyl-ghrelin, as well as attenuated difference in BOLD responsiveness to high-calorie vs. low-calorie food images in homeostatic and hedonic brain regions in the fed vs. fasted state (Karra et al., 2013). Similarly, studies have identified CNS “resistance” to ghrelin-induced food reward-associated behaviors in DIO mice. For example, peripheral ghrelin administration enhances operant progressive ratio (PR) responding for sucrose reward (Finger et al., 2012) and induces CPP for palatable food availability in normal but not DIO mice (Lockie et al., 2015). Whether DIO impairs feeding-related memory function via vHPC GHSR1A signaling remains to be determined, but HFD-induced ghrelin resistance has been identified at the intracellular signaling level in HPC neurons, as ICV ghrelin administration increases vHPC PI3K and Akt signaling in chow-fed, but not HFD-rats (Kanoski et al., 2013).
Overall these findings reveal that ghrelin signaling in the HPC is physiologically relevant for conditioned aspects of appetitive behavior. In addition to promoting HPC-dependent memory based on external spatial and contextual cues in tasks that involve passive or aversive reinforcement, emerging research detailed above shows that ghrelin promotes HPC-dependent memory directly related to feeding behavior, including learning about discreet external cues, contextual external cues, temporal entrainment cues, and social cues that inform about feeding. When in a state of chronic or acute energy deficit, ghrelin signaling may be adaptive to memory formation and retrieval of a food source location and other episodic elements of feeding-related events (Figure 1). On the other hand, the capacity of ghrelin signaling to promote food intake, conditioned appetitive behaviors, and intracellular signaling cascades in HPC neurons may be blunted in obesity and with HFD maintenance. Given that ghrelin levels rapidly decrease following the onset of eating, it follows that additional feeding-relevant systems that mediate prandial, postprandial, and overall energy status are also likely to interact with HPC-dependent memory function. These systems are discussed below.
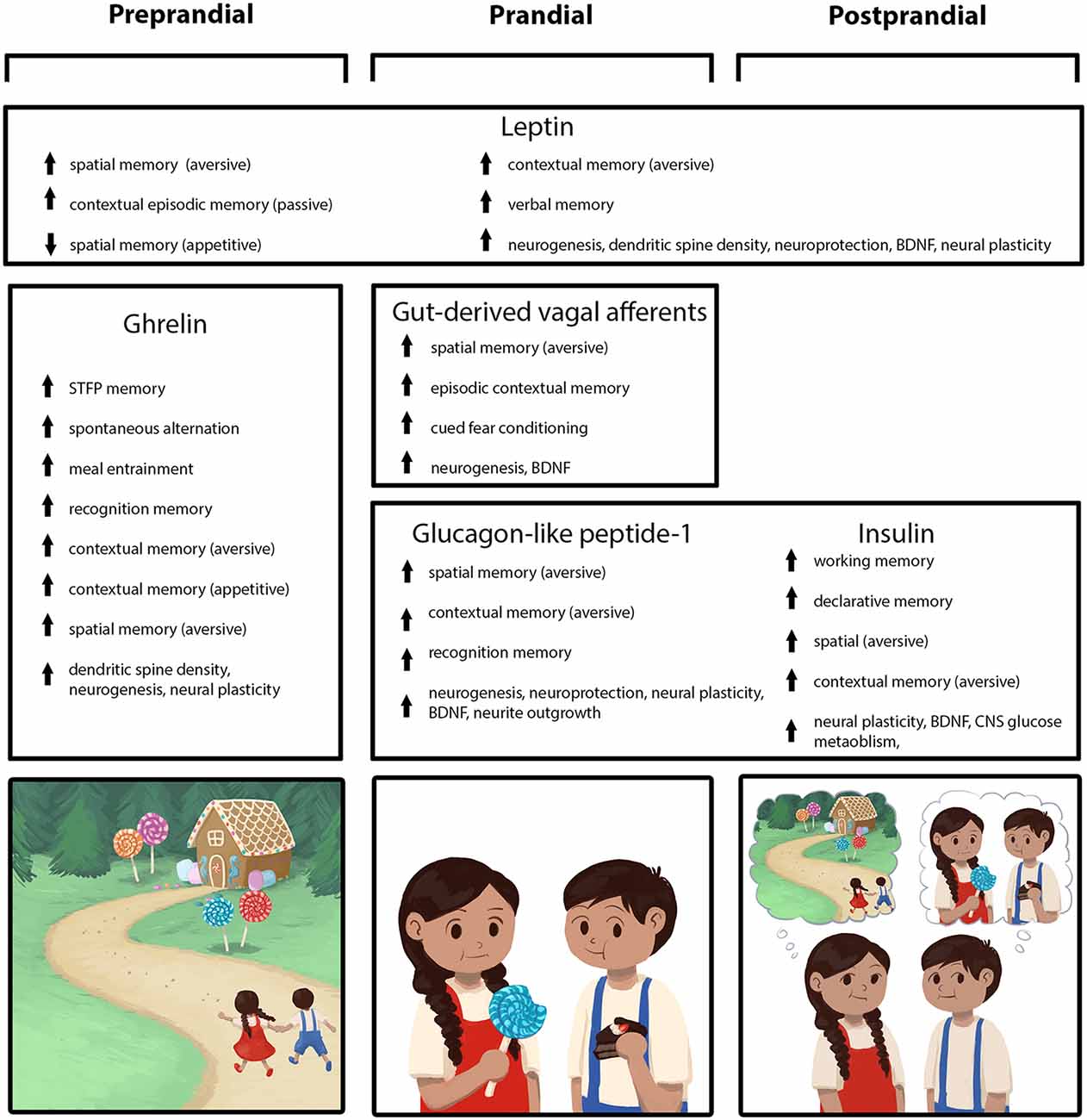
Figure 1. Peripherally-derived feeding-relevant signals occurring during the preprandial (left column), prandial (center column), and postprandial (right column) stages of feeding converge with neural processing in the hippocampus (HPC) to promote learning and memory for various elements of a feeding episode. These mnemonic episodic elements include the spatial location of a food source, as well as the temporal, nutritive, and social aspects of a meal (mnemonic elements depicted in the cartoon across the bottom row). During the preprandial/appetitive stage, elevated ghrelin signaling promotes HPC-dependent spatial and episodic memory formation related to food procurement via GHSR action in HPC neurons. In the prandial stage, within-meal gut-derived vagal sensory signaling enhances HPC-dependent memory for visuospatial and external contextual features related to a feeding episode via an ascending multisynaptic hindbrain-septal-HPC pathway. In addition, endocrine signals (glucagon-like peptide-1, GLP-1, insulin) are released prandially and immediately postprandially, which each independently contribute to meal-related episodic mnemonic elements (e.g., memory of the nutritive quality of a meal, food location) via direct action on HPC neurons, as well as indirectly through metabolic pathways. The adipokine leptin is presented as a signal that influences HPC-dependent memory (via direct action on HPC neurons) across all feeding stages, potentially via a modulatory mechanism such that optimal leptin levels associated with healthy energy status promote food-related memory.
Insulin is a peptide hormone produced by pancreatic β cells in the islets of Langerhans that plays an important role in nutrient metabolism and energy homeostasis. A major function of insulin in the periphery is to facilitate postprandial storage of nutrients, thereby maintaining nutrient homeostasis in the blood following a meal. The most well-characterized of the insulin secretion pathways is metabolic, whereby intracellular catabolism of nutrients by the β cell triggers a rise in intracellular calcium and insulin secretion (for review, see Skelin Klemen et al., 2017). While glucose is the nutrient most commonly associated with insulin secretion, foods that are low in glucose but high in proteins and fats also raise plasma insulin levels (Holt et al., 1997). In addition to this endocrine metabolic pathway, a neural pathway via vagal inputs to the pancreas mediates a cephalic insulin response, which elevates insulin secretion within the first few minutes of initiating a meal (triggered, in part, by oral nutrient detection; Berthoud et al., 1981; Powley, 2000; Ahrén and Holst, 2001). Finally, a humoral pathway for insulin secretion exists involving the incretin hormones GLP-1 and glucose-dependent insulinotropic polypeptide (GIP), which act in concert with rising glucose levels to stimulate insulin synthesis and secretion via an adenylate cyclase mediate pathway (Cernea and Raz, 2011).
Insulin acts on the insulin receptor (IR), which belongs to the receptor tyrosine kinase superfamily (De Meyts, 2000). Insulin binding to IR in muscle or adipose tissue promotes glucose uptake into the cell, as well as glycogen, fat, and protein synthesis (Tatulian, 2015). In addition to being expressed in peripheral tissues capable of storing nutrients, IR is expressed in the brain, where it plays a role in both peripheral energy metabolism and reducing food intake (Woods et al., 1996; Bruning et al., 2000; Schwartz et al., 2000; Obici et al., 2002a,b). High levels of IR expression in the brain are found in the olfactory bulb, cerebral cortex, hypothalamus, cerebellum, cortex, and HPC, with robust expression in the pyramidal region of the CA1, CA3, and granule layer of the DG (Unger et al., 1989; Marks et al., 1990; Schulingkamp et al., 2000). The major source of insulin in the brain is believed to come either predominantly or exclusively from the periphery via a BBB saturable transporter system (Baura et al., 1993; Banks, 2004; Ferrario and Reagan, 2018). The process through which insulin is transported across the BBB was recently elucidated, in part, by Gray et al. (2017), who showed that insulin enters the brain via IR-mediated brain endothelial cell transcytosis, a vesicle-mediated process that does not require downstream signaling of the classic IR intracellular PI3K signaling cascade. Insulin uptake across the BBB is not uniform across brain regions, however, and some regions appear to be more permeable for insulin transport than others. Notably, along with the pons, medulla, and the hypothalamus, the HPC is one of the regions with the highest uptake for insulin from the blood (Banks and Kastin, 1998). Thus, the HPC is a region containing both high levels of IR and for which the BBB is particularly permeable for insulin, suggesting that endogenous peripheral changes in insulin levels influence hippocampal function.
Early evidence for insulin’s involvement in memory was reported by Strong et al. (1990) who discovered that peripheral injections of insulin completely reversed deficits in working memory attributable to the ischemic stroke. Subsequently, Craft et al. (1996) showed that insulin alone, independent of the consequential lowering of blood glucose, is sufficient to significantly improve hippocampal-dependent declarative memory function in human subjects with mild symptoms of dementia. In healthy human subjects, 8 weeks of intranasal insulin treatment (4×/day) significantly improves hippocampal-dependent declarative memory (Benedict et al., 2004). Additional evidence for a role of insulin in supporting cognitive functioning comes from rodent models of diabetes, in which insulin secretion is reduced. For example, rats treated with intravenous injection of streptozotocin (STZ), which kills insulin-secreting pancreatic β cells, have impaired hippocampal-dependent place/location learning and reduced synaptic plasticity (Biessels et al., 1996). Rats treated similarly with STZ also have impaired short-term spatial memory, a deficit that is restored by peripheral treatment with insulin (Kumar et al., 2011).
In addition to impaired peripheral and central insulin signaling seen in diabetes models, impaired memory function in Alzheimer’s disease (AD) is associated with disrupted brain IR signaling. In fact, it has been suggested that AD be considered as “diabetes type 3” (Rivera et al., 2005; Steen et al., 2005; de la Monte and Wands, 2008). In humans, intranasal treatment with insulin has been shown to improve delayed memory recall associated with both mild cognitive impairment (MCI) and mild-to-moderate AD (Reger et al., 2008; Craft et al., 2012, 2017). Extracellular deposits of amyloid β and neuronal loss in the HPC are hallmarks of AD, as is impairment in hippocampal-dependent memory function. In rats, ICV injections of STZ, an established model for AD, impairs spatial and working memory and reduces IR signaling molecules, effects that are normalized by intranasal insulin treatment (Rajasekar et al., 2017; Rostami et al., 2017). There is also evidence to support a role for IR signaling in hippocampal-dependent learning and memory performance in the absence of cognitive deficit and/or dementia. For example, insulin injected into the dHPC enhances spatial working memory performance in rats in the spontaneous alternation task (McNay et al., 2010). Similarly, in healthy rats, bilateral dHPC injections of insulin transiently enhance long-term memory in contextual fear conditioning and inhibitory avoidance tasks (Stern et al., 2014) and insulin injected into the CA1 region of the dHPC immediately post-training enhances memory performance in an inhibitory avoidance task (Babri et al., 2007).
In addition to pharmacologically injected insulin, endogenous IR signaling in the HPC is critical for learning and memory function in healthy lab rats. For example, blockade of endogenous insulin in the HPC via microinjection of a small, anti-insulin, antibody-like protein, impairs spatial working memory performance in the spontaneous alternation task (McNay et al., 2010) and a 70% KD of dorsal HPC (dHPC) IR impairs HPC-dependent long-term spatial memory without affecting energy balance or peripheral glucose metabolism (Grillo et al., 2015). Consistent with these effects, learning per se appears to augment IR expression in the HPC. For example, spatial learning in the Morris water maze is associated with increased IR expression in the CA1 pyramidal region of HPC, as well as enrichment of the receptor in nuclear and dendritic compartments (Zhao et al., 1999). Together, the literature strongly supports a role for IR signaling in HPC-dependent learning and memory function, and further suggests that memory function can be enhanced with exogenous insulin treatment under both pathological and healthy conditions.
Whether mechanisms for central IR impairment are similar to peripheral IR resistance is unclear. One major difference between central and peripheral IR function is that a major function of insulin in the periphery is to enhance GLUT4 migration and glucose uptake, whereas glucose uptake in the brain is thought to be largely insulin independent (Tatulian, 2015). However, hippocampal neurons contain GLUT4 transporters and insulin enhances GLUT4 migration to the membrane of hippocampal neurons via a PI3K mediated pathway (Reagan, 2005; Grillo et al., 2009). In rats, intrahippocampal injections of insulin increase local glycolytic activity and enhances spatial working memory performance in the spontaneous alternation task via a PI3K-mediated pathway (McNay et al., 2010), whereas blocking GLUT4 mediated glucose uptake prevents performance improvements in the task by administration of memory-enhancing doses of insulin to the HPC (Pearson-Leary et al., 2018). Collectively these findings suggest that insulin’s memory-enhancing effects require increased glucose utilization. Indeed, spatial memory is limited by glucose availability and utilization in the HPC (McNay et al., 2000). Thus, the action of insulin in mediating neuronal glucose uptake may, in part, contribute to insulin’s memory enhancing effects.
In addition to improving glucose uptake, at the neuronal level, mechanisms for how central insulin may improve memory function include increasing synaptic plasticity. For example, IR signaling promotes synaptic plasticity (e.g., LTP) in hippocampal neurons, a physiological process widely considered to play a critical role in memory (Lee and Silva, 2009; Lisman et al., 2018). On the other hand, genetically knocking down dHPC IR impairs hippocampal-dependent memory and blocks LTP in the CA1 and DG (Grillo et al., 2015). Related to IR-mediated increases in neuronal plasticity, insulin increases the expression of the growth and plasticity factor brain-derived neurotrophic factor (BDNF) and its receptor TrkB in the HPC (Haas et al., 2016), an effect that is abolished in aged rats. The accumulation of amyloid β in the HPC reduces BDNF function, cell membrane IRs, and IR pathway mediated signaling (De Felice et al., 2009, 2014; Takach et al., 2015). Moreover, the function of BDNF is restored by activating the downstream mediators of IR signaling, suggesting that AB may reduce neuronal plasticity in part by interfering with IR function (Takach et al., 2015). Thus, overall the mechanisms through which central IR signaling may enhance HPC functioning include improved glucose utilization and enhanced neuronal plasticity via neurotrophic pathways.
Central IR function appears to be potently regulated by dietary and metabolic factors. For example, insulin resistance in the HPC is observed in the obese Zucker fa/fa rat model (Špolcová et al., 2014). Similarly, DIO rodents are less responsive to the memory enhancing effects of intrahippocampal insulin (McNay et al., 2010). In females, estrogen treatment normalizes both the peripheral and central IR sensitivity due to ovariectomy in rats fed standard chow, however, peripheral but not central IR sensitivity is restored by estrogen treatment in ovariectomized rats fed a HFD (Pratchayasakul et al., 2014). Even short-term (7 days) feeding of a diet high in saturated fat and fructose is associated with reduced hippocampal IR signaling (Calvo-Ochoa et al., 2014). Similarly, a diet high in fructose (without elevated fat) reduces IR phosphorylation in hippocampal neurons and activation of the downstream signaling molecules IR Substrate-1 (Agrawal et al., 2016), AKT, and PI3K (Wu et al., 2015), outcomes also associated with impaired spatial memory (Wu et al., 2015; Agrawal et al., 2016).
Given that insulin secretion is triggered by immediate nutrient availability, one can speculate that it would be advantageous from an evolutionary perspective to have enhanced spatial and contextual memory during the prandial state in order to remember the location of the food supply, as well as to mnemonically encode additional episodic information that facilitated feeding (Figure 1). Indeed, insulin is an ideal candidate to enhance such memories surrounding the prandial period, as its release occurs at the onset, during, and after a meal. One possible explanation for why memory function is enhanced by insulin and other endocrine factors that are triggered during an ongoing meal is that the memory of the meal-related episodic event may be important for influencing the timing of and amount consumed in a subsequent meal in order to maintain optimal energy balance. Similarly, it is advantageous for animals to be able to encode the memory of the nutritive quality of a meal to better guide future foraging behavior. These memory-promoting effects of insulin appear to mirror the peripheral metabolic effects of insulin in that HFD consumption and obesity are associated with dysregulated IR signaling pathways. Corroborating the idea that the evolutionary function for insulin’s memory enhancing effects might be related to the prandial timing of its secretion, additional prandial factors have been shown to similarly enhance hippocampal-dependent learning and memory (as discussed in the subsequent sections).
The vagus nerve (aka, the 10th cranial nerve) is the primary communicator between the gastrointestinal (GI) tract and the brain, delivering energy-status signals from the gut to the brain via vagal afferent (sensory) nerves. There are discrete classes of afferent fibers that innervate GI organs to either detect stomach volume or intestinal nutrient contents (Powley and Phillips, 2004; Brookes et al., 2013; Williams et al., 2016). These afferent fibers contain cell bodies in the nodose ganglia that synapse to the CNS. The medial NTS (mNTS) of the hindbrain is an important integrator in gut-to-brain communication, as it is the first brain region to receive vagal-mediated signaling from the GI organs (Grill and Hayes, 2012).
The HPC is a new player in the world of gut-to-brain communication. For example, increased GI signaling by within-meal satiation signals (e.g., gastric distension, intestinal nutrient infusion) activates CBF in hippocampal neurons in rodents (Xu et al., 2008; Min et al., 2011). Additionally, HPC CBF is robustly activated following gastric vagus nerve stimulation (VNS) in obese humans (Wang et al., 2006). However, the mNTS does not directly project to the HPC (Rinaman, 2010; Hsu et al., 2015a). This suggests that the connection between the mNTS and the HPC must be through multi-order neural projection pathways. The locus coeruleus (LC) and medial septum (MS) have been identified as potential brain region relay sites connecting the mNTS to the ventral CA1 HPC (Castle et al., 2005). Consistent with the MS as a potential relay between mNTS and CA1, recent studies demonstrate that stimulation of the vagus nerve induces dHPC theta rhythms via MS cholinergic signaling (Broncel et al., 2018). Further, we recently utilized monosynaptic and transsynaptic viral tracing techniques to identify the MS as a relay region connecting the mNTS to glutamatergic neurons in the dorsal CA3 and DG subfields of the dHPC (Suarez et al., 2018). These studies provide support for a multi-order neuroanatomical connection linking GI vagally-mediated signaling to higher-order HPC neural processing.
While traditionally linked with the mediation of GI-derived satiation/meal termination signaling, the role of the vagus nerve with regards to cognitive function has been a topic of recent interest. Studies from Clark et al. (1995) demonstrate that unilateral cervical VNS and stimulation of vagal afferents with inactivation of efferents (Clark et al., 1998) improves retention of inhibitory-avoidance memory in rats, whereas in humans, VNS enhances retention of recognition memory when stimulation occurred after learning (Clark et al., 1999). Moreover, VNS in rodents improves spatial memory in the Morris water maze as well as inhibitory avoidance learning, whereas elimination of norepinephrine levels in the HPC via ICV administration of a neurotoxin selective for noradrenergic neurons reverses these VNS-induced memory enhancements in rats with cerebral ischemia/reperfusion injury (Liu et al., 2016).
While VNS enhances memory retention in a variety of HPC-dependent memory tasks, it is important to consider the physiological role of vagal signaling to memory function. The subdiaphragmatic deafferentation (SDA) procedure, which eliminates all GI vagal sensory signaling to the brain, while leaving 50% of supradiaphragmatic sensory and 50% of vagal motor signaling intact (Norgren and Smith, 1994), has been shown to impair extinction of auditory-cued fear conditioning (Klarer et al., 2014), but did not affect NOR memory or working memory in a non-spatial alternation task (Klarer et al., 2017). Of more direct relevance to HPC-dependent memory, our group has recently shown that subdiaphragmatic vagotomy (SDV), which eliminates both GI sensory and motor signaling below the diaphragm, and nodose ganglia injections of CCK-saporin, a novel surgical method which selectively eliminates ~80% of GI-derived vagal sensory signaling below the diaphragm while preserving 100% of motor signaling, both impair HPC-dependent spatial working and contextual episodic memory function in a Barnes’ maze and novel object in context task. On the other hand, SDV does not affect appetitive learning based on internal energy-state cues and social transmission of food-related cues (Suarez et al., 2018). Thus, these findings suggest that GI-derived vagal sensory signaling, which is engaged during feeding, may promote learning about meal-related visuospatial external environmental cues to remember a food location to facilitate future foraging behavior, while having less impact on memory for nonspatial information surrounding a feeding episode (e.g., social cues and interoceptive circulating energy status cues).
Vagal signaling plays an important role in hippocampal neurogenesis and synaptic plasticity. VNS increases BDNF mRNA (Follesa et al., 2007) and activates BDNF receptor TrkB phosphorylation (Furmaga et al., 2012) in rat HPC. Moreover, VNS induces LTP (Zuo et al., 2007), enhances excitatory synaptic transmission (Ura et al., 2013), and increases BrdU (a thymidine analogue incorporated into the DNA of dividing cells; marker of cell proliferation) and doublecortin (a microtubule-associated protein; marker of cell differentiation) immunoreactivity (Biggio et al., 2009) within the DG subfield of the HPC. Importantly, other studies provide physiological relevance of vagus nerve signaling in neurogenesis and synaptic function. Capsaicin-induced sensory vagus nerve injury, which damages non-myelinated vagal afferents, reduced cell proliferation and differentiation in granule cell layer of the HPC in rats (Ronchi et al., 2012). In adult mice, SDV reduces BDNF mRNA in all subregions of the HPC (e.g., DG, CA1, CA3), the number of immature HPC neurons, proliferation in the dHPC, and survival of newly born cells in both dHPC and vHPC (O’Leary et al., 2018). Moreover, rats with either SDV or selective elimination of GI-derived vagal afferent signaling via nodose injections of CCK-saporin have significantly reduced BDNF and DCX levels in the dHPC relative to controls, and importantly, these reductions are correlated with the magnitude of HPC-dependent memory impairment (Suarez et al., 2018). Collectively these findings suggest that gut-derived vagal signaling promotes HPC-dependent memory by enhancing neurotrophic and neurogenic pathways, and that the results from O’Leary et al. (2018) discussed above are based on elimination of sensory, and not motor vagal signaling.
Diet and metabolic factors strongly regulate the sensitivity of vagal afferent neurons to peripheral GI-derived signals. For example, studies have demonstrated significantly reduced c-Fos (a marker of neuronal activation) in the mNTS following a meal in obese rats (Covasa et al., 2000a,b), reduced mechanosensitivity to stomach distension in obese mice (Daly et al., 2011; Kentish et al., 2012), and reduced chemosensitivity to GI hormones that induce satiation in mice maintained on a high-fat diet vs. low-fat-diet (Covasa and Ritter, 1998). This impaired sensitivity of vagal afferent signals that develops in diet-induced obesity may promote reduced satiation and overconsumption of food. In DIO minipigs, chronic VNS decreases food intake, and in a three-choice meal test, reduces sweet food consumption relative to DIO sham-treated animals (Val-Laillet et al., 2010). These findings suggest that VNS may rescue obesity-associated vagally-based deficits in satiation processing. Of relevance to memory function, obese humans with an implantable gastric stimulator (IGS), a device that expands the stomach through electrical stimulation of the vagus nerve, demonstrated 18% higher brain glucose metabolism in the right HPC, measured via positron emission tomography (PET), and this was associated with lower emotional eating scores with IGS on relative to off (Wang et al., 2006). These results suggest that obesity-induced blunting of vagal afferent signaling impairs the ability of HPC neural activity to modulate eating behaviors, however, this hypothesis requires further testing.
Within-meal gut-derived vagal sensory signaling may be part of the prandial mechanism that facilitates learning and remembering the physical location of a food source to inform future foraging behavior. These effects likely involve a multi-order neural pathway from the mNTS through the MS to the dHPC that promotes neurogenic and neurotrophic action in HPC neurons. Based on recent results from our group (Suarez et al., 2018), it appears that this pathway is more important for remembering external visuospatial and contextual information compared to social-related cues and circulating energy balance cues related to feeding behavior (Figure 1). Future work is needed to sort out the role of the reinforcer (appetitive vs. aversive vs. passive) in mediating these effects, as well as to examine whether obesity-associate deficits in GI-derived satiation signal processing extend to vagal modulation of memory and cognitive outcomes.
GLP-1 is produced from the preproglucagon gene. As an incretin hormone, GLP-1 is secreted from the distal intestines and has blood glucose lowering effects (Kreymann et al., 1987; Baggio and Drucker, 2007), attributed primarily to enhanced glucose-stimulated insulin secretion from pancreatic β cells (Mojsov et al., 1987). The primary impetus for GLP-1 secretion from the ileal endocrine L cells is the presence of nutrients in the gut (Baggio and Drucker, 2007). In addition to its incretin actions, GLP-1 signaling also reduces food intake, in part, via a reduction in gastric emptying rate (for review, see Shah and Vella, 2014; Drucker, 2018).
In addition to the intestinal GLP-1 secretion, GLP-1 is also released in the brain by the preproglucagon (PPG)-expressing neurons located in the hindbrain mNTS, the caudal medullary reticular formation, and the olfactory bulb (Merchenthaler et al., 1999). Given the short half-life of peripherally-released GLP-1 due to rapid enzymatic degradation by dipeptidyl peptidase-4 (DPP-4), the predominant source of GLP-1 in the brain is thought to be the PPG neurons (Holst, 2007), which project extensively throughout the brain (Gu et al., 2013; Trapp and Cork, 2015). Similarly, GLP-1R is widely expressed throughout the neuraxis (Cork et al., 2015), and several specific nuclei have been identified that are functionally relevant to GLP-1’s food intake reducing effects (for review, see Kanoski et al., 2016).
Of direct relevance to this review, GLP-1 receptor (GLP-1R) is robustly expressed in the HPC, particularly within the vHPC (Merchenthaler et al., 1999; Cork et al., 2015). GLP-1R activation in the vHPC reduces food intake by reducing meal size and conditioned motivated responding for palatable food via downstream connectivity with the medial prefrontal cortex (Hsu et al., 2015a, 2018). In addition to these hypophagic effects, GLP-1 also acts in hippocampal neurons to enhance learning and memory. Early evidence for GLP-1’s role in hippocampal memory function comes from During et al. (2003), who demonstrated that injections of GLP-1 (7–36) amide in the brain (ICV) enhances hippocampal-dependent spatial memory in rodents (During et al., 2003). This report further revealed that GLP1-R KD impairs contextual fear memory in mice and that these impairments are reversed by virogenetic re-expression of the GLP1-R in the HPC (During et al., 2003). These data suggest that GLP-1R signaling in the HPC improves learning and memory in normal healthy animals. However, while GLP-1 crosses the BBB (Kastin et al., 2002), the rapid enzymatic degradation in peripheral circulation by DPP-4 (Holst, 2007) raises the question as to what extent peripheral GLP-1 reaches the brain (and HPC) in meaningful concentrations. Related, GLP-1-releasing PPG neurons in the hindbrain, considered to be the predominant source of endogenous GLP-1 in signaling in the brain, do not project to the HPC, thereby reducing the likelihood of a direct GLP-1 synaptic connection between the NTS and the HPC. Given that GLP-1 immunoreactive terminals directly contact the ependymal cells contacting the cerebral spinal fluid (CSF) in the cerebral ventricles, and that active GLP-1 is present in both the CSF and in hippocampal tissue lysates under physiological conditions, we have previously proposed CSF ventricular “volume transmission” of GLP-1 as a mechanism through which GLP-1 released from PPG neurons reaches HPC neurons and glial cells (Hsu et al., 2015a). Additional support for this biological signaling pathway in the control of feeding behavior was recently provided for the orexigenic neuropeptides, melanin-concentrating hormone and orexin (Noble et al., 2018), thereby further supporting the feasibility of CSF transmission of GLP-1 as a mechanism for communication to hippocampal neurons.
Clinically-relevant evidence supporting a role for GLP-1 in promoting hippocampal-dependent memory comes from studies using peripheral administration of FDA-approved GLP-1-based diabetes and obesity drugs. For example, peripheral treatment with the DPP4 inhibitor sitagliptin, which delays the degradation of GLP-1 and therefore elevates its endogenous half-life, increases markers of hippocampal neurogenesis and improves recognition memory in mice fed a high-fat diet (Gault et al., 2015). Further, chronic peripheral treatment with the long-acting GLP-1 analog, exenatide, significantly improves performance in the spatial radial arm maze in healthy rats (Isacson et al., 2011). Similarly, chronic treatment with a different long-acting GLP-1 analog, liraglutide, improves deficits in spatial memory observed in a rat model of central streptozotocin (STZ)-induced neurotoxicity and diabetes (Palleria et al., 2017), an outcome also observed following exenatide treatment (Chen et al., 2012). In addition to improving hippocampal function in animals with central STZ-induced neurotoxicity, exenatide treatment improves hippocampal-dependent spatial memory in rats made insulin resistant with a high fructose diet (Gad et al., 2016) and in a mouse model of peripheral STZ-induced diabetes (Huang et al., 2012; Gumuslu et al., 2016). Unfortunately, it is not possible from these studies to distinguish the peripheral from the central effects of the GLP-1 analogs, as both exenatide and liraglutide cross the BBB following peripheral administration (Kastin and Akerstrom, 2003; Knudsen et al., 2016). However, ICV injections of GLP-1 attenuate spatial memory impairments in a juvenile model of STZ-induced diabetes (Iwai et al., 2009), suggesting that upregulating central GLP-1R signaling per se improves hippocampal-dependent learning and memory in animals with peripheral hyperglycemia and/or reduced insulin production.
It is noteworthy that the food intake-reducing effects of peripherally-delivered GLP-1 analogs (exendin-4 and liraglutide) require vagus nerve-independent BBB transport and direct action on central GLP-1Rs (Kanoski et al., 2011a), suggesting that these GLP-1 analogs may have clinical relevance for memory disorders. Indeed, in rodent models GLP1R agonists have shown promise for reducing the mnemonic deficits associated with AD. In rats, intra-hippocampal injections of GLP1R agonists prevent impairments in spatial memory induced by central injections of amyloid β (Wang et al., 2010; Qi et al., 2016). Similarly, in the amyloid precursor protein/presenilin (APP/PS1) double and APP/PS1/Tau triple transgenic mouse models of AD, liraglutide treatment attenuates spatial memory impairments (McClean et al., 2011; Chen et al., 2017). The memory improvements associated with GLP-1 receptor agonists are not limited to conditions in which amyloid β or hyperphosphorylated tau are present, as GLP-1 receptor agonists are neuroprotective and promote memory retention in senescence accelerated mouse prone 8 (SAMP8) mice, a model of age-related spontaneous AD not associated with amyloid plaques (Hansen et al., 2015).
The mechanisms through which GLP-1R activation affects hippocampal-dependent learning and memory are not completely understood, however, evidence suggests that GLP-1R activation increases synaptic plasticity (Gilman et al., 2003; Abbas et al., 2009; Gault et al., 2010; Han et al., 2013; Cai et al., 2014) and increases neurite outgrowth (Perry et al., 2002). GLP-1R agonism has also been shown to affect gene expression levels of the neurotrophic and neuroplasticity factor BDNF and its receptor TrkB (Lennox et al., 2014; Gumuslu et al., 2016). However, other studies found no effect of GLP1R agonist liraglutide treatment on expression levels of NTRK2, BDNF, or synaptophysin, but nevertheless observed an increase in hippocampal LTP in the CA1 region which was associated with elevated levels of the mammalian achaete-scute homologue 1 [Mash1; a marker for neurogenesis (Isacson et al., 2011; Porter et al., 2013)]. GLP-1R agonists also increase neurogenesis in the HPC, evidenced by increased expression of the neurogenic marker doublecortin, elevated BrdU incorporation, and an increase in neuronal number in the HPC (Isacson et al., 2011; Hansen et al., 2015). Thus, taken together GLP-1 may improve HPC-dependent learning and memory by increasing neuronal plasticity, neurogenesis, and/or both.
Unlike previous systems discussed above, results on whether dietary and metabolic factors regulate GLP-1 sensitivity are mixed. Postprandial GLP-1 release is attenuated in obese individuals (Näslund et al., 1998) and higher levels of GLP-1 appear to be required to produce anorectic effects and terminate a meal (Näslund et al., 1999), suggesting obesity-induced GLP-1 insensitivity, or “resistance” with regards to feeding outcomes. Consistent with these effects, rats maintained on a HFD demonstrate attenuated satiation effects following GLP-1 receptor activation (Williams et al., 2011). Of relevance to memory outcomes, peripheral administration of GLP-1 receptor agonists [e.g., Liraglutide (Val8)GLP-1(GluPAL), Exendin-4] in DIO mice rescues HPC LTP and normalizes object recognition memory to performance levels similar to lean control mice (Gault et al., 2010; Porter et al., 2010; Lennox et al., 2014). Additionally, oral administration of Sitagliptin (a DPP-4 inhibitor that elevates GLP-1 concentrations) reverses object recognition memory deficits and elevates HPC doublecortin expression in HFD-maintained mice (Gault et al., 2015).
In contrast with these findings, however, human studies demonstrate limited or no evidence of obesity-associated GLP-1 resistance following chronic pharmacological GLP1 analog treatment. For example, exenatide treatment produces significantly greater weight loss in extremely obese individuals relative to that observed in overweight or lean individuals (Blonde et al., 2006). Moreover, intravenous administration of exenatide reduces blood-oxygen-level dependent responses in a memory-related brain regions (including the right HPC) in response to high-calorie food pictures in obese subjects but not in lean subjects (Eldor et al., 2016). Overall the influence of dietary and metabolic factors on GLP-1-based effects on incretin signaling, feeding behavior, and memory outcomes is thus far poorly understood.
As highlighted throughout this review, there may be an evolutionary benefit in remembering the details surrounding a meal, such as remembering where the food source comes from, as well as whether the meal rewarding/nutritive or did it cause malaise. Given the role of the HPC in memory function related to spatial and temporal events, contextual elements associated with reward or aversive stimuli (Behrendt, 2013), or physiological energy status cues (reviewed in Kanoski and Grill, 2017), it seems logical that the time during and immediately following food consumption is a key time for the HPC contribute to encode memory. While the GLP-1 system has similar effects to insulin on hippocampal function, the fact that there are direct effects of GLP-1 in the brain independent of the incretin effects of GLP-1 in the periphery (During et al., 2003; Iwai et al., 2009) suggests that these two systems have independent effects on memory function. This redundancy supports a framework in which enhanced memory function during and surrounding meal consumption involves multiple energy balance relevant systems that have different temporal secretion dynamics, as well systems that have opposing effects on food intake (Figure 1). Consistent with data discussed above indicating that these systems are modulated by energy status, now we discuss a system, leptin, that is a key signal for chronic energy status.
Leptin is a 16 kD hormone synthesized and released into the blood primarily by white adipose tissue (Zhang et al., 1994). Circulating plasma leptin levels are positively correlated with fat mass and fluctuate with nutrient status (Frederich et al., 1995; Maffei et al., 1995), with low plasma levels during nutrient deficiency and high levels postprandially and in obese states (Dalamaga et al., 2013). Additionally, there is a stomach-derived source of leptin, which is secreted during a meal and absorbed from the small intestine into the blood stream (Cammisotto and Bendayan, 2012). Leptin exerts its main effect on long-term regulation of energy homeostasis by binding to the long isoform of leptin receptor (LepRb) expressed in peripheral tissues and the brain. Leptin acts as an energy status signal to reduce food intake and body weight during times of nutritional abundance, whereas mutation of leptin or LepRb results in severe hyperphagia, decreased energy expenditure, and early onset obesity in humans and rodents (Tartaglia et al., 1995; Chua et al., 1996; Montague et al., 1997; Farooqi et al., 1999; Kelesidis et al., 2010). Leptin crosses the BBB via a saturable transporter system (Banks et al., 1996) and binds to LepRbs expressed in the hypothalamus, caudal brainstem, and elsewhere to regulate energy balance control (Friedman and Halaas, 1998; Bates and Myers, 2003; Myers et al., 2009; Hayes et al., 2010; Kanoski et al., 2012). Recent findings have also highlighted the physiological relevance of LepRb signaling in higher-order (limbic, cortical) brain regions, including the lateral hypothalamus (Leinninger et al., 2009, 2011; Goforth et al., 2014; Laque et al., 2015), ventral tegmental area (Fulton et al., 2006; Hommel et al., 2006; Mietlicki-Baase et al., 2015), and of direct relevance to this review, the CA1, CA3, and DG subregions of the HPC (Huang et al., 1996; Couce et al., 1997; Shioda et al., 1998; Burguera et al., 2000b; Ur et al., 2002).
In addition to its more widely studied role in energy balance, leptin also promotes memory function, particularly through its action in hippocampal neurons. For example, intravenous and peripheral administration of leptin enhances performance in HPC-dependent spatial and contextual memory tasks in rodents (e.g., Morris water maze and passive avoidance task, respectively; Oomura et al., 2006), as well as a novel place recognition task that measures contextual episodic memory (Malekizadeh et al., 2017). In humans, leptin deficiency is associated with impaired verbal memory function, which is rescued following subcutaneous leptin administration (Paz-Filho et al., 2008). Within the brain, direct dHPC leptin administration improves HPC-dependent memory retention in mice in a T-maze foot shock avoidance and step-down avoidance tasks (Farr et al., 2006). Collectively, these behavioral studies indicate that leptin signaling improves HPC-dependent memory function associated with passive reinforcement tasks and memory tasks based on escape/aversive reinforcement.
Leptin’s pharmacological effects on memory may be particularly influenced by the nature of the reinforcement. For example, the studies mentioned above reveal that leptin, administered either peripherally or directly to the dHPC, improves memory function for spatial and contextual-based tasks that involve passive or aversive reinforcement. In contrast, leptin administration in the vHPC impairs memory consolidation for the spatial location of food, whereas dHPC administration had no effect (Kanoski et al., 2011b). Consistent with these findings, vHPC lesions, which eliminate leptin signaling in the vHPC, enhance memory for an association between environmental cues and food reward (Ferbinteanu and McDonald, 2001). It could be that increased leptin signaling in the vHPC, associated with energy surplus, reduces the ability of environmental cues to elicit memory for food-related information in favor of remembering non-food-related cues. Whether this putative reinforcement-dependent role for leptin in memory function is differentially mediated by dorsal vs. ventral hippocampal neurons requires further investigation.
At the neuronal level, leptin acts on HPC neurons to promote synaptic function, in part, by protecting HPC neurons from apoptosis (Guo et al., 2008), by promoting proliferation of HPC progenitor cells (Garza et al., 2008), and by increasing dendritic spine formation (O’Malley et al., 2007; Stranahan et al., 2009; Dhar et al., 2014a,b). LepRb deficient rodents (e.g., Zucker fa/fa rats and db/db mice) demonstrate impaired LTP and LDT in HPC CA1 synapses, as well as impaired spatial memory performance in the Morris water maze (Li et al., 2002). These changes in LTP and LTD are mediated, in part, by leptin-dependent regulation of N-Methyl-D-aspartate (NMDA) receptor activation (Shanley et al., 2001; Durakoglugil et al., 2005), as well as through activation of Akt/PI3K and ERK/MAPK intracellular signaling pathways (Niswender et al., 2001; Irving et al., 2006; Weng et al., 2007; Garza et al., 2008; Guo et al., 2008). PI3K signaling, in particular, is implicated in leptin-dependent induction and maintenance of LTP, LTD, and NMDA receptors in the HPC (Shanley et al., 2001; Durakoglugil et al., 2005) via stimulation of calcium/calmodulin-dependent protein kinase II (CaMKII) activity (Li et al., 2002; Oomura et al., 2006). In addition to enhancing synaptic plasticity, leptin induces morphological changes by forming dendritic protrusions through activation of TrpC signaling, which is important for BDNF-induced spine formation in the HPC (Li et al., 2010), via activation of CaMK signaling (Dhar et al., 2014a), and by increasing dendritic spine density of HPC synapses through activation of NMDA receptors that is mediated by MAPK/ERK signaling pathway (O’Malley et al., 2007).
While leptin plays an important role in enhancing neuronal plasticity and memory function, studies demonstrate that variations in nutritional status can alter these leptin-mediated effects. Low serum levels of leptin have been linked to severe malnutrition (Caro et al., 1996; Pinos et al., 2007; Amirkalali et al., 2010), acceleration of brain aging, and higher risk to develop AD (Power et al., 2001; Lieb et al., 2009; Fadel et al., 2013). Further, serum leptin levels are positively correlated with HPC gray matter volume (Lieb et al., 2009; Narita et al., 2009) and MCI patients demonstrate lower serum leptin levels independent of body fat, as well as reduced memory performance on an auditory verbal learning test and decreased volume and microstructural integrity of HPC subfields relative to healthy controls (Witte et al., 2016). Direct administration of leptin to HPC neurons, either in vitro or in vivo, facilitates LTP induction, whereas smaller and larger doses of leptin actually inhibit LTP (Wayner et al., 2004). These findings suggest that a specific concentration of leptin when optimal energy reserves are met may enhance synaptic plasticity, with opposite effects observed in the presence of either insufficient or excess energy reserves. Moreover, leptin rescues the increased neuroapoptosis and impaired cognitive function in the Morris water maze via regulation of 5′-AMP-activated protein kinase (AMPK) activity that occurs under severe dietary restriction (Dagon et al., 2005). Therefore, nutrient deficiency (associated with low circulating leptin levels) impairs HPC integrity and cognitive function, further supporting the notion that leptin’s role as an energy-status signal may extend to modulating HPC-dependent spatial and other memory processes.
Obese rodents and humans demonstrate leptin resistance and peripheral hyperleptinemia, in part, due to reduced leptin transport across the BBB and reduced ability of CNS leptin to engage intracellular signaling pathways downstream of LepRb binding (Montague et al., 1997; Strobel et al., 1998; Banks et al., 1999; Farooqi et al., 1999; Burguera et al., 2000a; El-Haschimi et al., 2000; Banks, 2001; Munzberg et al., 2005). Considering that obesity is associated with impaired cognitive function (Elias et al., 2005; Cournot et al., 2006; Wolf et al., 2007), leptin resistance may play a role in HPC-dependent memory impairment associated with obesity (see Van Doorn et al., 2017 for further review on this topic). Related, rats maintained on a high fructose and HFD develop hyperleptinemia, lower central leptin levels, and impaired performance on the Morris water maze task (Lin et al., 2017). These effects could be due to elevated stearoyl-CoA desaturase-1 (SCD-1) expression in the brain, an enzyme involved in fatty acid metabolism with expression correlated with obesity and suppressed by leptin (Biddinger et al., 2006). Moreover, mice maintained on a HFD demonstrate impaired spatial memory in a novel location recognition (NLR) task with desensitization of PI3K/Akt signaling pathway coupled to HPC LepRbs (Valladolid-Acebes et al., 2013).
Excessive caloric consumption may interfere with leptin’s capacity to promote memory function through interactions with BDNF, a neurotrophin that promotes plasticity and is reduced in the HPC following HFD maintenance in rodents (Molteni et al., 2002; Kanoski et al., 2007; Noble et al., 2014). For example, CNS leptin increases BDNF expression in the HPC, and this effect is diminished with HFD maintenance (Yamada et al., 2011; Kanoski et al., 2014). Consistent with this framework, obese rats (induced via lentivirus-mediated downregulation of hypothalamic IRs) demonstrate hyperleptinemia and impaired LTP in CA1 HPC (Grillo et al., 2011a), as well as deficits in HPC synaptic plasticity, reduction in HPC dendritic spine density, and reduced c-Fos expression in the CA1 subregion of the HPC following fear conditioning tasks (Grillo et al., 2011b). Rats on a high-calorie diet also demonstrate reduced HPC LTP at CA1 synapses and deficits in spatial memory function through BDNF-mediated effects on dendritic spine density (Stranahan et al., 2008).
Results discussed above indicate that the HPC receives and responds to leptin-induced energy status signaling to influence memory function. Interestingly, data support a U-shaped mechanism whereby low and high concentrations of leptin present in malnutrition and obesity, respectively, negatively impact HPC synaptic plasticity and cognitive function. Therefore, when energy reserves are met and leptin levels are optimal to maintain energy homeostasis, the HPC appears to be able to integrate information about healthy energy status to promote spatial and contextual memory, which may be relevant to efficiently learning the location of food and other episodic elements surrounding feeding (Figure 1). Future research is needed to determine whether HPC leptin signaling differentially promotes memory based on food vs. nonfood reinforcement, and related, whether these effects are dependent on HPC subregion (dorsal vs. ventral), as well as overall energy status and circulating leptin levels.
Eating is paramount in the hierarchy of mammalian physiological and psychological needs. The literature reviewed herein highlights that the neurobiological control of learning and memory is strongly linked with signaling from biological peripherally-derived systems that fluctuate around and strongly influence the consumption of a meal (Figure 1). Preprandial (ghrelin) and prandial/immediate postprandial signals, including insulin, GLP-1, and gut-derived vagal nerve activation, improve HPC-dependent contextual, episodic, and spatial memory. Similarly, the adipocyte-derived hormone leptin, an overall energy status marker, has similar beneficial effects on HPC neural and mnemonic outcomes. While beyond the scope of this review, it is also the case that various CNS-derived neuropeptide systems that potently influence appetite and feeding enhance hippocampal-dependent memory via similar neuronal mechanisms as the peripheral-derived systems discussed herein. For example, the hypothalamic-derived feeding-related neuropeptides, orexin (aka, hypocretin; Yang et al., 2013; Mavanji et al., 2017), oxytocin (Lee et al., 2015), melanin-concentrating hormone (Monzon et al., 1999; Adamantidis et al., 2005), and neurotensin (Ohinata et al., 2007; Zhang et al., 2016), have all been established to promote HPC memory function and plasticity in rodent models. Collectively, these peripheral- and central-derived biological systems represent a diverse array of neurobiological pathways that bi-directionally influence feeding behavior, yet generally, have common promoting/enhancing effects on HPC function, with some exceptions (e.g., based on pharmacological dose effects, nature of reinforcement, etc.).
The common neurobiological mechanisms through which these diverse systems improve memory function include (but are not limited to) enhanced neuroplasticity and neurogenesis in hippocampal neurons. Based on evidence that multiple signals elevate BDNF-TrkB signaling, which is important for both synaptic plasticity and hippocampal neurogenesis, it is possible that this neurotrophic pathway may be downstream of both preprandial and prandial/postprandial feeding-related biological signals, and upstream of neuroplasticity, neurogenic, and mnemonic outcomes. Though this hypothesis remains to be tested, it is possible that surrounding the event of a meal, energy balance-related signaling factors promote a temporary rise in BDNF/TrkB signaling which primes neuronal systems for learning about the critical elements of the meal. Importantly, dietary and metabolic factors appear to potently regulate the capacity of these systems to promote these neurobiological mechanisms and enhance memories of a meal, and generally-speaking, the memory-promoting capacity of these systems are reduced with obesity and HFD maintenance.
This review highlights functional/behavioral data from both humans and mechanistic experimental rodent model experiments revealing that hippocampal-dependent memory function is enhanced by biological systems that show sharp and dynamic fluctuations surrounding mealtime. Such mnemonic enhancement likely provides an important evolutionary advantage. For example, the ability to recall the details surrounding a nutritive meal, including the physical location of the food, how to procure and prepare it, and what external episodic factors modulate the availability and nutritive quality of the food, may serve to increase the success and efficiency of future foraging and feeding behavior. The reviewed literature suggests that these meal-related systems also enhance the capacity for memory for events not directly related to feeding behavior, including those that involve passive or aversive reinforcement. Whether these effects are a by-product of systems that are engineered to promote feeding-related memories, and related, whether the nature of the reinforcer (food vs. nonfood, hedonic vs. homeostatic, appetitive vs. aversive) is a critical factor in the ability of these systems to promote memory function, are important areas for future research.
All authors made comparable scholarship, creative, and writing contributions to this review article.
This work was supported by NIH (National Institute of Diabetes and Digestive and Kidney Diseases, NIDDK) grants: DK104897 and DK118402 (SK), and DK116558 (AS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Alyssa Cortella for her artistic and conceptual contributions to the figure.
Abbas, T., Faivre, E., and Hölscher, C. (2009). Impairment of synaptic plasticity and memory formation in GLP-1 receptor KO mice: interaction between type 2 diabetes and Alzheimer’s disease. Behav. Brain Res. 205, 265–271. doi: 10.1016/j.bbr.2009.06.035
Adamantidis, A., Thomas, E., Foidart, A., Tyhon, A., Coumans, B., Minet, A., et al. (2005). Disrupting the melanin-concentrating hormone receptor 1 in mice leads to cognitive deficits and alterations of NMDA receptor function. Eur. J. Neurosci. 21, 2837–2844. doi: 10.1111/j.1460-9568.2005.04100.x
Agrawal, R., Noble, E., Vergnes, L., Ying, Z., Reue, K., and Gomez-Pinilla, F. (2016). Dietary fructose aggravates the pathobiology of traumatic brain injury by influencing energy homeostasis and plasticity. J. Cereb. Blood Flow Metab. 36, 941–953. doi: 10.1177/0271678x15606719
Ahrén, B., and Holst, J. J. (2001). The cephalic insulin response to meal ingestion in humans is dependent on both cholinergic and noncholinergic mechanisms and is important for postprandial glycemia. Diabetes 50, 1030–1038. doi: 10.2337/diabetes.50.5.1030
Amirkalali, B., Sharifi, F., Fakhrzadeh, H., Mirarefein, M., Ghaderpanahi, M., Badamchizadeh, Z., et al. (2010). Low serum leptin serves as a biomarker of malnutrition in elderly patients. Nutr. Res. 30, 314–319. doi: 10.1016/j.nutres.2010.05.002
Ariyasu, H., Takaya, K., Tagami, T., Ogawa, Y., Hosoda, K., Akamizu, T., et al. (2001). Stomach is a major source of circulating ghrelin and feeding state determines plasma ghrelin-like immunoreactivity levels in humans. J. Clin. Endocrinol. Metab. 86, 4753–4758. doi: 10.1210/jc.86.10.4753
Babri, S., Badie, H. G., Khamenei, S., and Seyedlar, M. O. (2007). Intrahippocampal insulin improves memory in a passive-avoidance task in male wistar rats. Brain Cogn. 64, 86–91. doi: 10.1016/j.bandc.2007.01.002
Baggio, L. L., and Drucker, D. J. (2007). Biology of incretins: GLP-1 and GIP. Gastroenterology 132, 2131–2157. doi: 10.1053/j.gastro.2007.03.054
Banks, W. A. (2001). Leptin transport across the blood-brain barrier: implications for the cause and treatment of obesity. Curr. Pharm. Des. 7, 125–133. doi: 10.2174/1381612013398310
Banks, W. A. (2004). The source of cerebral insulin. Eur. J. Pharmacol. 490, 5–12. doi: 10.1016/j.ejphar.2004.02.040
Banks, W. A., Burney, B. O., and Robinson, S. M. (2008). Effects of triglycerides, obesity, and starvation on ghrelin transport across the blood-brain barrier. Peptides 29, 2061–2065. doi: 10.1016/j.peptides.2008.07.001
Banks, W. A., Dipalma, C. R., and Farrell, C. L. (1999). Impaired transport of leptin across the blood-brain barrier in obesity. Peptides 20, 1341–1345. doi: 10.1016/s0196-9781(99)00139-4
Banks, W. A., and Kastin, A. J. (1998). Differential permeability of the blood-brain barrier to two pancreatic peptides: insulin and amylin. Peptides 19, 883–889. doi: 10.1016/s0196-9781(98)00018-7
Banks, W. A., Kastin, A. J., Huang, W., Jaspan, J. B., and Maness, L. M. (1996). Leptin enters the brain by a saturable system independent of insulin. Peptides 17, 305–311. doi: 10.1016/0196-9781(96)00025-3
Bates, S. H., and Myers, M. G. Jr. (2003). The role of leptin receptor signaling in feeding and neuroendocrine function. Trends Endocrinol. Metab. 14, 447–452. doi: 10.1016/j.tem.2003.10.003
Baura, G. D., Foster, D. M., Porte, D. Jr., Kahn, S. E., Bergman, R. N., Cobelli, C., et al. (1993). Saturable transport of insulin from plasma into the central nervous system of dogs in vivo. A mechanism for regulated insulin delivery to the brain. J. Clin. Invest. 92, 1824–1830. doi: 10.1172/jci116773
Behrendt, R. P. (2013). Conscious experience and episodic memory: hippocampus at the crossroads. Front. Psychol. 4:304. doi: 10.3389/fpsyg.2013.00304
Benedict, C., Hallschmid, M., Hatke, A., Schultes, B., Fehm, H. L., Born, J., et al. (2004). Intranasal insulin improves memory in humans. Psychoneuroendocrinology 29, 1326–1334. doi: 10.1016/j.psyneuen.2004.04.003
Berthoud, H. R., Bereiter, D. A., Trimble, E. R., Siegel, E. G., and Jeanrenaud, B. (1981). Cephalic phase, reflex insulin secretion neuroanatomical and physiological characterization. Diabetologia 20, 393–401. doi: 10.1007/bf00254508
Biddinger, S. B., Miyazaki, M., Boucher, J., Ntambi, J. M., and Kahn, C. R. (2006). Leptin suppresses stearoyl-CoA desaturase 1 by mechanisms independent of insulin and sterol regulatory element-binding protein-1c. Diabetes 55, 2032–2041. doi: 10.2337/db05-0742
Biessels, G. J., Kamal, A., Ramakers, G. M., Urban, I. J., Spruijt, B. M., Erkelens, D. W., et al. (1996). Place learning and hippocampal synaptic plasticity in streptozotocin-induced diabetic rats. Diabetes 45, 1259–1266. doi: 10.2337/diab.45.9.1259
Biggio, F., Gorini, G., Utzeri, C., Olla, P., Marrosu, F., Mocchetti, I., et al. (2009). Chronic vagus nerve stimulation induces neuronal plasticity in the rat hippocampus. Int. J. Neuropsychopharmacol. 12, 1209–1221. doi: 10.1017/S1461145709000200
Blonde, L., Klein, E. J., Han, J., Zhang, B., Mac, S. M., Poon, T. H., et al. (2006). Interim analysis of the effects of exenatide treatment on A1C, weight and cardiovascular risk factors over 82 weeks in 314 overweight patients with type 2 diabetes. Diabetes Obes. Metab. 8, 436–447. doi: 10.1111/j.1463-1326.2006.00602.x
Blum, I. D., Patterson, Z., Khazall, R., Lamont, E. W., Sleeman, M. W., Horvath, T. L., et al. (2009). Reduced anticipatory locomotor responses to scheduled meals in ghrelin receptor deficient mice. Neuroscience 164, 351–359. doi: 10.1016/j.neuroscience.2009.08.009
Briggs, D. I., Enriori, P. J., Lemus, M. B., Cowley, M. A., and Andrews, Z. B. (2010). Diet-induced obesity causes ghrelin resistance in arcuate NPY/AgRP neurons. Endocrinology 151, 4745–4755. doi: 10.1210/en.2010-0556
Broncel, A., Bocian, R., Kłos-Wojtczak, P., and Konopacki, J. (2018). Medial septal cholinergic mediation of hippocampal theta rhythm induced by vagal nerve stimulation. PLoS One 13:e0206532. doi: 10.1371/journal.pone.0206532
Brookes, S. J., Spencer, N. J., Costa, M., and Zagorodnyuk, V. P. (2013). Extrinsic primary afferent signalling in the gut. Nat. Rev. Gastroenterol. Hepatol. 10, 286–296. doi: 10.1038/nrgastro.2013.29
Bruning, J. C., Gautam, D., Burks, D. J., Gillette, J., Schubert, M., Orban, P. C., et al. (2000). Role of brain insulin receptor in control of body weight and reproduction. Science 289, 2122–2125. doi: 10.1126/science.289.5487.2122
Burgess, N., Maguire, E. A., and O’Keefe, J. (2002). The human hippocampus and spatial and episodic memory. Neuron 35, 625–641. doi: 10.1016/s0896-6273(02)00830-9
Burguera, B., Couce, M. E., Curran, G. L., Jensen, M. D., Lloyd, R. V., Cleary, M. P., et al. (2000a). Obesity is associated with a decreased leptin transport across the blood-brain barrier in rats. Diabetes 49, 1219–1223. doi: 10.2337/diabetes.49.7.1219
Burguera, B., Couce, M. E., Long, J., Lamsam, J., Laakso, K., Jensen, M. D., et al. (2000b). The long form of the leptin receptor (OB-Rb) is widely expressed in the human brain. Neuroendocrinology 71, 187–195. doi: 10.1159/000054536
Buzsáki, G., and Moser, E. I. (2013). Memory, navigation and theta rhythm in the hippocampal-entorhinal system. Nat. Neurosci. 16, 130–138. doi: 10.1038/nn.3304
Cahill, S. P., Hatchard, T., Abizaid, A., and Holahan, M. R. (2014). An examination of early neural and cognitive alterations in hippocampal-spatial function of ghrelin receptor-deficient rats. Behav. Brain Res. 264, 105–115. doi: 10.1016/j.bbr.2014.02.004
Cai, H. Y., Hölscher, C., Yue, X. H., Zhang, S. X., Wang, X. H., Qiao, F., et al. (2014). Lixisenatide rescues spatial memory and synaptic plasticity from amyloid β protein-induced impairments in rats. Neuroscience 277, 6–13. doi: 10.1016/j.neuroscience.2014.02.022
Calvo-Ochoa, E., Hernández-Ortega, K., Ferrera, P., Morimoto, S., and Arias, C. (2014). Short-term high-fat-and-fructose feeding produces insulin signaling alterations accompanied by neurite and synaptic reduction and astroglial activation in the rat hippocampus. J. Cereb. Blood Flow Metab. 34, 1001–1008. doi: 10.1038/jcbfm.2014.48
Cammisotto, P., and Bendayan, M. (2012). A review on gastric leptin: the exocrine secretion of a gastric hormone. Anat. Cell Biol. 45, 1–16. doi: 10.5115/acb.2012.45.1.1
Carlini, V. P., Ghersi, M., Schiöth, H. B., and de Barioglio, S. R. (2010). Ghrelin and memory: differential effects on acquisition and retrieval. Peptides 31, 1190–1193. doi: 10.1016/j.peptides.2010.02.021
Carlini, V. P., Monzón, M. E., Varas, M. M., Cragnolini, A. B., Schióth, H. B., Scimonelli, T. N., et al. (2002). Ghrelin increases anxiety-like behavior and memory retention in rats. Biochem. Biophys. Res. Commun. 299, 739–743. doi: 10.1016/s0006-291x(02)02740-7
Carlini, V. P., Varas, M. M., Cragnolini, A. B., Schiöth, H. B., Scimonelli, T. N., and de Barioglio, S. R. (2004). Differential role of the hippocampus, amygdala and dorsal raphe nucleus in regulating feeding, memory, and anxiety-like behavioral responses to ghrelin. Biochem. Biophys. Res. Commun. 313, 635–641. doi: 10.1016/j.bbrc.2003.11.150
Caro, J. F., Sinha, M. K., Kolaczynski, J. W., Zhang, P. L., and Considine, R. V. (1996). Leptin: the tale of an obesity gene. Diabetes 45, 1455–1462. doi: 10.2337/diab.45.11.1455
Castle, M., Comoli, E., and Loewy, A. D. (2005). Autonomic brainstem nuclei are linked to the hippocampus. Neuroscience 134, 657–669. doi: 10.1016/j.neuroscience.2005.04.031
Cernea, S., and Raz, I. (2011). Therapy in the early stage: incretins. Diabetes Care 34, S264–S271. doi: 10.2337/dc11-s223
Chen, S., Liu, A. R., An, F. M., Yao, W. B., and Gao, X. D. (2012). Amelioration of neurodegenerative changes in cellular and rat models of diabetes-related Alzheimer’s disease by exendin-4. Age 34, 1211–1224. doi: 10.1007/s11357-011-9303-8
Chen, S., Sun, J., Zhao, G., Guo, A., Chen, Y., Fu, R., et al. (2017). Liraglutide improves water maze learning and memory performance while reduces hyperphosphorylation of tau and neurofilaments in APP/PS1/Tau triple transgenic mice. Neurochem. Res. 42, 2326–2335. doi: 10.1007/s11064-017-2250-8
Chen, L., Xing, T., Wang, M., Miao, Y., Tang, M., Chen, J., et al. (2011). Local infusion of ghrelin enhanced hippocampal synaptic plasticity and spatial memory through activation of phosphoinositide 3-kinase in the dentate gyrus of adult rats. Eur. J. Neurosci. 33, 266–275. doi: 10.1111/j.1460-9568.2010.07491.x
Chua, S. C. Jr., Chung, W. K., Wu-Peng, X. S., Zhang, Y., Liu, S. M., Tartaglia, L., et al. (1996). Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science 271, 994–996. doi: 10.1126/science.271.5251.994
Chuang, J. C., Perello, M., Sakata, I., Osborne-Lawrence, S., Savitt, J. M., Lutter, M., et al. (2011). Ghrelin mediates stress-induced food-reward behavior in mice. J. Clin. Invest. 121, 2684–2692. doi: 10.1172/jci57660
Clark, K. B., Krahl, S. E., Smith, D. C., and Jensen, R. A. (1995). Post-training unilateral vagal stimulation enhances retention performance in the rat. Neurobiol. Learn. Mem. 63, 213–216. doi: 10.1006/nlme.1995.1024
Clark, K. B., Naritoku, D. K., Smith, D. C., Browning, R. A., and Jensen, R. A. (1999). Enhanced recognition memory following vagus nerve stimulation in human subjects. Nat. Neurosci. 2, 94–98. doi: 10.1038/4600
Clark, K. B., Smith, D. C., Hassert, D. L., Browning, R. A., Naritoku, D. K., and Jensen, R. A. (1998). Posttraining electrical stimulation of vagal afferents with concomitant vagal efferent inactivation enhances memory storage processes in the rat. Neurobiol. Learn. Mem. 70, 364–373. doi: 10.1006/nlme.1998.3863
Clayton, N. S., and Dickinson, A. (1998). Episodic-like memory during cache recovery by scrub jays. Nature 395, 272–274. doi: 10.1038/26216
Clifton, P. G., Vickers, S. P., and Somerville, E. M. (1998). Little and often: ingestive behavior patterns following hippocampal lesions in rats. Behav. Neurosci. 112, 502–511. doi: 10.1037/0735-7044.112.3.502
Cork, S. C., Richards, J. E., Holt, M. K., Gribble, F. M., Reimann, F., and Trapp, S. (2015). Distribution and characterisation of Glucagon-like peptide-1 receptor expressing cells in the mouse brain. Mol. Metab. 4, 718–731. doi: 10.1016/j.molmet.2015.07.008
Couce, M. E., Burguera, B., Parisi, J. E., Jensen, M. D., and Lloyd, R. V. (1997). Localization of leptin receptor in the human brain. Neuroendocrinology 66, 145–150. doi: 10.1159/000127232
Countryman, R. A., Kaban, N. L., and Colombo, P. J. (2005). Hippocampal c-fos is necessary for long-term memory of a socially transmitted food preference. Neurobiol. Learn. Mem. 84, 175–183. doi: 10.1016/j.nlm.2005.07.005
Cournot, M., Marquié, J. C., Ansiau, D., Martinaud, C., Fonds, H., Ferrières, J., et al. (2006). Relation between body mass index and cognitive function in healthy middle-aged men and women. Neurology 67, 1208–1214. doi: 10.1212/01.wnl.0000238082.13860.50
Covasa, M., and Ritter, R. C. (1998). Rats maintained on high-fat diets exhibit reduced satiety in response to CCK and bombesin. Peptides 19, 1407–1415. doi: 10.1016/s0196-9781(98)00096-5
Covasa, M., Grahn, J., and Ritter, R. C. (2000a). High fat maintenance diet attenuates hindbrain neuronal response to CCK. Regul. Pept. 86, 83–88. doi: 10.1016/s0167-0115(99)00084-1
Covasa, M., Grahn, J., and Ritter, R. C. (2000b). Reduced hindbrain and enteric neuronal response to intestinal oleate in rats maintained on high-fat diet. Auton. Neurosci. 84, 8–18. doi: 10.1016/s1566-0702(00)00176-4
Craft, S., Baker, L. D., Montine, T. J., Minoshima, S., Watson, G. S., Claxton, A., et al. (2012). Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch. Neurol. 69, 29–38. doi: 10.1001/archneurol.2011.233
Craft, S., Claxton, A., Baker, L. D., Hanson, A. J., Cholerton, B., Trittschuh, E. H., et al. (2017). Effects of regular and long-acting insulin on cognition and Alzheimer’s disease biomarkers: a pilot clinical trial. J. Alzheimers Dis. 57, 1325–1334. doi: 10.3233/jad-161256
Craft, S., Newcomer, J., Kanne, S., Dagogo-Jack, S., Cryer, P., Sheline, Y., et al. (1996). Memory improvement following induced hyperinsulinemia in Alzheimer’s disease. Neurobiol. Aging 17, 123–130. doi: 10.1016/0197-4580(95)02002-0
Cummings, D. E., Purnell, J. Q., Frayo, R. S., Schmidova, K., Wisse, B. E., and Weigle, D. S. (2001). A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50, 1714–1719. doi: 10.2337/diabetes.50.8.1714
Dagon, Y., Avraham, Y., Magen, I., Gertler, A., Ben-Hur, T., and Berry, E. M. (2005). Nutritional status, cognition, and survival: a new role for leptin and AMP kinase. J. Biol. Chem. 280, 42142–42148. doi: 10.1074/jbc.m507607200
Dalamaga, M., Chou, S. H., Shields, K., Papageorgiou, P., Polyzos, S. A., and Mantzoros, C. S. (2013). Leptin at the intersection of neuroendocrinology and metabolism: current evidence and therapeutic perspectives. Cell Metab. 18, 29–42. doi: 10.1016/j.cmet.2013.05.010
Daly, D. M., Park, S. J., Valinsky, W. C., and Beyak, M. J. (2011). Impaired intestinal afferent nerve satiety signalling and vagal afferent excitability in diet induced obesity in the mouse. J. Physiol. 589, 2857–2870. doi: 10.1113/jphysiol.2010.204594
Date, Y., Kojima, M., Hosoda, H., Sawaguchi, A., Mondal, M. S., Suganuma, T., et al. (2000). Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 141, 4255–4261. doi: 10.1210/en.141.11.4255
Davidson, T. L., Kanoski, S. E., Chan, K., Clegg, D. J., Benoit, S. C., and Jarrard, L. E. (2010). Hippocampal lesions impair retention of discriminative responding based on energy state cues. Behav. Neurosci. 124, 97–105. doi: 10.1037/a0018402
Davidson, T. L., Kanoski, S. E., Schier, L. A., Clegg, D. J., and Benoit, S. C. (2007). A potential role for the hippocampus in energy intake and body weight regulation. Curr. Opin. Pharmacol. 7, 613–616. doi: 10.1016/j.coph.2007.10.008
Davis, J. F., Choi, D. L., Clegg, D. J., and Benoit, S. C. (2011). Signaling through the ghrelin receptor modulates hippocampal function and meal anticipation in mice. Physiol. Behav. 103, 39–43. doi: 10.1016/j.physbeh.2010.10.017
de Castro, J. M., Brewer, E. M., Elmore, D. K., and Orozco, S. (1990). Social facilitation of the spontaneous meal size of humans occurs regardless of time, place, alcohol or snacks. Appetite 15, 89–101. doi: 10.1016/0195-6663(90)90042-7
De Felice, F. G., Lourenco, M. V., and Ferreira, S. T. (2014). How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement. 10, S26–S32. doi: 10.1016/j.jalz.2013.12.004
De Felice, F. G., Vieira, M. N., Bomfim, T. R., Decker, H., Velasco, P. T., Lambert, M. P., et al. (2009). Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Aβ oligomers. Proc. Natl. Acad. Sci. U S A 106, 1971–1976. doi: 10.1073/pnas.0809158106
de la Monte, S. M., and Wands, J. R. (2008). Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2, 1101–1113. doi: 10.1177/193229680800200619
De Meyts, P. (2000). “The insulin receptor and its signal transduction network,” in Endotext,, eds L. J. De Groot, G. Chrousos, K. Dungan, K. R. Feingold, A. Grossman, J. M. Hershman, et al. (South Dartmouth, MA: MDText.com, Inc.).
Dhar, M., Wayman, G. A., Zhu, M., Lambert, T. J., Davare, M. A., and Appleyard, S. M. (2014a). Leptin-induced spine formation requires TrpC channels and the CaM kinase cascade in the hippocampus. J. Neurosci. 34, 10022–10033. doi: 10.1523/JNEUROSCI.2868-13.2014
Dhar, M., Zhu, M., Impey, S., Lambert, T. J., Bland, T., Karatsoreos, I. N., et al. (2014b). Leptin induces hippocampal synaptogenesis via CREB-regulated microRNA-132 suppression of p250GAP. Mol. Endocrinol. 28, 1073–1087. doi: 10.1210/me.2013-1332
Diano, S., Farr, S. A., Benoit, S. C., McNay, E. C., da Silva, I., Horvath, B., et al. (2006). Ghrelin controls hippocampal spine synapse density and memory performance. Nat. Neurosci. 9, 381–388. doi: 10.1038/nn1656
Disse, E., Bussier, A. L., Deblon, N., Pfluger, P. T., Tschöp, M. H., Laville, M., et al. (2011). Systemic ghrelin and reward: effect of cholinergic blockade. Physiol. Behav. 102, 481–484. doi: 10.1016/j.physbeh.2010.12.006
Dornonville de la Cour, C., Björkqvist, M., Sandvik, A. K., Bakke, I., Zhao, C. M., Chen, D., et al. (2001). A-like cells in the rat stomach contain ghrelin and do not operate under gastrin control. Regul. Pept. 99, 141–150. doi: 10.1016/s0167-0115(01)00243-9
Drazen, D. L., Vahl, T. P., D’Alessio, D. A., Seeley, R. J., and Woods, S. C. (2006). Effects of a fixed meal pattern on ghrelin secretion: evidence for a learned response independent of nutrient status. Endocrinology 147, 23–30. doi: 10.1210/en.2005-0973
Drucker, D. J. (2018). Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. 27, 740–756. doi: 10.1016/j.cmet.2018.03.001
Durakoglugil, M., Irving, A. J., and Harvey, J. (2005). Leptin induces a novel form of NMDA receptor-dependent long-term depression. J. Neurochem. 95, 396–405. doi: 10.1111/j.1471-4159.2005.03375.x
During, M. J., Cao, L., Zuzga, D. S., Francis, J. S., Fitzsimons, H. L., Jiao, X., et al. (2003). Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat. Med. 9, 1173–1179. doi: 10.1038/nm919
Edwards, A., and Abizaid, A. (2017). Clarifying the ghrelin system’s ability to regulate feeding behaviours despite enigmatic spatial separation of the GHSR and its endogenous ligand. Int. J. Mol. Sci. 18:E859. doi: 10.3390/ijms18040859
Eichenbaum, H., and Cohen, N. J. (2014). Can we reconcile the declarative memory and spatial navigation views on hippocampal function? Neuron 83, 764–770. doi: 10.1016/j.neuron.2014.07.032
Eldor, R., Daniele, G., Huerta, C., Al-Atrash, M., Adams, J., Defronzo, R., et al. (2016). Discordance between central (Brain) and pancreatic action of exenatide in lean and obese subjects. Diabetes Care 39, 1804–1810. doi: 10.2337/dc15-2706
El-Haschimi, K., Pierroz, D. D., Hileman, S. M., Bjørbaek, C., and Flier, J. S. (2000). Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J. Clin. Invest. 105, 1827–1832. doi: 10.1172/jci9842
Elias, M. F., Elias, P. K., Sullivan, L. M., Wolf, P. A., and D’Agostino, R. B. (2005). Obesity, diabetes and cognitive deficit: the Framingham heart study. Neurobiol. Aging 26, 11–16. doi: 10.1016/j.neurobiolaging.2005.08.019
English, P. J., Ghatei, M. A., Malik, I. A., Bloom, S. R., and Wilding, J. P. (2002). Food fails to suppress ghrelin levels in obese humans. J. Clin. Endocrinol. Metab. 87:2984. doi: 10.1210/jc.87.6.2984
Fadel, J. R., Jolivalt, C. G., and Reagan, L. P. (2013). Food for thought: the role of appetitive peptides in age-related cognitive decline. Ageing Res. Rev. 12, 764–776. doi: 10.1016/j.arr.2013.01.009
Fanselow, M. S., and Dong, H. W. (2010). Are the dorsal and ventral hippocampus functionally distinct structures. Neuron 65, 7–19. doi: 10.1016/j.neuron.2009.11.031
Farooqi, I. S., Jebb, S. A., Langmack, G., Lawrence, E., Cheetham, C. H., Prentice, A. M., et al. (1999). Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N. Engl. J. Med. 341, 879–884. doi: 10.1056/NEJM199909163411204
Farr, S. A., Banks, W. A., and Morley, J. E. (2006). Effects of leptin on memory processing. Peptides 27, 1420–1425. doi: 10.1016/j.peptides.2005.10.006
Faulconbridge, L. F., Cummings, D. E., Kaplan, J. M., and Grill, H. J. (2003). Hyperphagic effects of brainstem ghrelin administration. Diabetes 52, 2260–2265. doi: 10.2337/diabetes.52.9.2260
Ferbinteanu, J., and McDonald, R. J. (2001). Dorsal/ventral hippocampus, fornix, and conditioned place preference. Hippocampus 11, 187–200. doi: 10.1002/hipo.1036
Ferrario, C. R., and Reagan, L. P. (2018). Insulin-mediated synaptic plasticity in the CNS: anatomical, functional and temporal contexts. Neuropharmacology 136, 182–191. doi: 10.1016/j.neuropharm.2017.12.001
Ferrini, F., Salio, C., Lossi, L., and Merighi, A. (2009). Ghrelin in central neurons. Curr. Neuropharmacol. 7, 37–49. doi: 10.2174/157015909787602779
Finger, B. C., Dinan, T. G., and Cryan, J. F. (2012). Diet-induced obesity blunts the behavioural effects of ghrelin: studies in a mouse-progressive ratio task. Psychopharmacology 220, 173–181. doi: 10.1007/s00213-011-2468-0
Follesa, P., Biggio, F., Gorini, G., Caria, S., Talani, G., Dazzi, L., et al. (2007). Vagus nerve stimulation increases norepinephrine concentration and the gene expression of BDNF and bFGF in the rat brain. Brain Res. 1179, 28–34. doi: 10.1016/j.brainres.2007.08.045
Fontán-Lozano, A., Sáez-Cassanelli, J. L., Inda, M. C., de los Santos-Arteaga, M., Sierra-Domínguez, S. A., López-Lluch, G., et al. (2007). Caloric restriction increases learning consolidation and facilitates synaptic plasticity through mechanisms dependent on NR2B subunits of the NMDA receptor. J. Neurosci. 27, 10185–10195. doi: 10.1523/JNEUROSCI.2757-07.2007
Frederich, R. C., Hamann, A., Anderson, S., Löllmann, B., Lowell, B. B., and Flier, J. S. (1995). Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat. Med. 1, 1311–1314. doi: 10.1038/nm1295-1311
Friedman, J. M., and Halaas, J. L. (1998). Leptin and the regulation of body weight in mammals. Nature 395, 763–770. doi: 10.1038/27376
Fulton, S., Pissios, P., Manchon, R. P., Stiles, L., Frank, L., Pothos, E. N., et al. (2006). Leptin regulation of the mesoaccumbens dopamine pathway. Neuron 51, 811–822. doi: 10.1016/j.neuron.2006.09.006
Furmaga, H., Carreno, F. R., and Frazer, A. (2012). Vagal nerve stimulation rapidly activates brain-derived neurotrophic factor receptor TrkB in rat brain. PLoS One 7:e34844. doi: 10.1371/journal.pone.0034844
Gad, E. S., Zaitone, S. A., and Moustafa, Y. M. (2016). Pioglitazone and exenatide enhance cognition and downregulate hippocampal β amyloid oligomer and microglia expression in insulin-resistant rats. Can. J. Physiol. Pharmacol. 94, 819–828. doi: 10.1139/cjpp-2015-0242
Gahete, M. D., Córdoba-Chacón, J., Salvatori, R., Castaño, J. P., Kineman, R. D., and Luque, R. M. (2010). Metabolic regulation of ghrelin O-acyl transferase (GOAT) expression in the mouse hypothalamus, pituitary and stomach. Mol. Cell. Endocrinol. 317, 154–160. doi: 10.1016/j.mce.2009.12.023
Garza, J. C., Guo, M., Zhang, W., and Lu, X. Y. (2008). Leptin increases adult hippocampal neurogenesis in vivo and in vitro. J. Biol. Chem. 283, 18238–18247. doi: 10.1074/jbc.M800053200
Gault, V. A., Lennox, R., and Flatt, P. R. (2015). Sitagliptin, a dipeptidyl peptidase-4 inhibitor, improves recognition memory, oxidative stress and hippocampal neurogenesis and upregulates key genes involved in cognitive decline. Diabetes Obes. Metab. 17, 403–413. doi: 10.1111/dom.12432
Gault, V. A., Porter, W. D., Flatt, P. R., and Holscher, C. (2010). Actions of exendin-4 therapy on cognitive function and hippocampal synaptic plasticity in mice fed a high-fat diet. Int. J. Obes. 34, 1341–1344. doi: 10.1038/ijo.2010.59
Ge, X., Yang, H., Bednarek, M. A., Galon-Tilleman, H., Chen, P., Chen, M., et al. (2018). LEAP2 is an endogenous antagonist of the ghrelin receptor. Cell Metab. 27, 461.e6–469.e6. doi: 10.1016/j.cmet.2017.10.016
Gilman, C. P., Perry, T., Furukawa, K., Grieg, N. H., Egan, J. M., and Mattson, M. P. (2003). Glucagon-like peptide 1 modulates calcium responses to glutamate and membrane depolarization in hippocampal neurons. J. Neurochem. 87, 1137–1144. doi: 10.1046/j.1471-4159.2003.02073.x
Goforth, P. B., Leinninger, G. M., Patterson, C. M., Satin, L. S., and Myers, M. G. Jr. (2014). Leptin acts via lateral hypothalamic area neurotensin neurons to inhibit orexin neurons by multiple GABA-independent mechanisms. J. Neurosci. 34, 11405–11415. doi: 10.1523/JNEUROSCI.5167-13.2014
Goldstone, A. P., Prechtl, C. G., Scholtz, S., Miras, A. D., Chhina, N., Durighel, G., et al. (2014). Ghrelin mimics fasting to enhance human hedonic, orbitofrontal cortex, and hippocampal responses to food. Am. J. Clin. Nutr. 99, 1319–1330. doi: 10.3945/ajcn.113.075291
Gray, S. M., Aylor, K. W., and Barrett, E. J. (2017). Unravelling the regulation of insulin transport across the brain endothelial cell. Diabetologia 60, 1512–1521. doi: 10.1007/s00125-017-4285-4
Grill, H. J., and Hayes, M. R. (2012). Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab. 16, 296–309. doi: 10.1016/j.cmet.2012.06.015
Grillo, C. A., Piroli, G. G., Evans, A. N., Macht, V. A., Wilson, S. P., Scott, K. A., et al. (2011a). Obesity/hyperleptinemic phenotype adversely affects hippocampal plasticity: effects of dietary restriction. Physiol. Behav. 104, 235–241. doi: 10.1016/j.physbeh.2010.10.020
Grillo, C. A., Piroli, G. G., Junor, L., Wilson, S. P., Mott, D. D., Wilson, M. A., et al. (2011b). Obesity/hyperleptinemic phenotype impairs structural and functional plasticity in the rat hippocampus. Physiol. Behav. 105, 138–144. doi: 10.1016/j.physbeh.2011.02.028
Grillo, C. A., Piroli, G. G., Hendry, R. M., and Reagan, L. P. (2009). Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain Res. 1296, 35–45. doi: 10.1016/j.brainres.2009.08.005
Grillo, C. A., Piroli, G. G., Lawrence, R. C., Wrighten, S. A., Green, A. J., Wilson, S. P., et al. (2015). Hippocampal insulin resistance impairs spatial learning and synaptic plasticity. Diabetes 64, 3927–3936. doi: 10.2337/db15-0596
Gu, G., Roland, B., Tomaselli, K., Dolman, C. S., Lowe, C., and Heilig, J. S. (2013). Glucagon-like peptide-1 in the rat brain: distribution of expression and functional implication. J. Comp. Neurol. 521, 2235–2261. doi: 10.1002/cne.23282
Gualillo, O., Lago, F., Casanueva, F. F., and Dieguez, C. (2006). One ancestor, several peptides post-translational modifications of preproghrelin generate several peptides with antithetical effects. Mol. Cell. Endocrinol. 256, 1–8. doi: 10.1016/j.mce.2006.05.007
Guan, X. M., Yu, H., Palyha, O. C., McKee, K. K., Feighner, S. D., Sirinathsinghji, D. J., et al. (1997). Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Mol. Brain Res. 48, 23–29. doi: 10.1016/s0169-328x(97)00071-5
Gumuslu, E., Mutlu, O., Celikyurt, I. K., Ulak, G., Akar, F., Erden, F., et al. (2016). Exenatide enhances cognitive performance and upregulates neurotrophic factor gene expression levels in diabetic mice. Fundam. Clin. Pharmacol. 30, 376–384. doi: 10.1111/fcp.12192
Guo, Z., Jiang, H., Xu, X., Duan, W., and Mattson, M. P. (2008). Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J. Biol. Chem. 283, 1754–1763. doi: 10.1074/jbc.m703753200
Haas, C. B., Kalinine, E., Zimmer, E. R., Hansel, G., Brochier, A. W., Oses, J. P., et al. (2016). Brain insulin administration triggers distinct cognitive and neurotrophic responses in young and aged rats. Mol. Neurobiol. 53, 5807–5817. doi: 10.1007/s12035-015-9494-6
Hampton, R. R., Hampstead, B. M., and Murray, E. A. (2004). Selective hippocampal damage in rhesus monkeys impairs spatial memory in an open-field test. Hippocampus 14, 808–818. doi: 10.1002/hipo.10217
Han, J. E., Frasnelli, J., Zeighami, Y., Larcher, K., Boyle, J., McConnell, T., et al. (2018). Ghrelin enhances food odor conditioning in healthy humans: an fMRI study. Cell Rep. 25, 2643.e4–2652.e4. doi: 10.1016/j.celrep.2018.11.026
Han, W. N., Hölscher, C., Yuan, L., Yang, W., Wang, X. H., Wu, M. N., et al. (2013). Liraglutide protects against amyloid-β protein-induced impairment of spatial learning and memory in rats. Neurobiol. Aging 34, 576–588. doi: 10.1016/j.neurobiolaging.2012.04.009
Hannapel, R. C., Henderson, Y. H., Nalloor, R., Vazdarjanova, A., and Parent, M. B. (2017). Ventral hippocampal neurons inhibit postprandial energy intake. Hippocampus 27, 274–284. doi: 10.1002/hipo.22692
Hannapel, R., Ramesh, J., Ross, A., Lalumiere, R. T., Roseberry, A. G., and Parent, M. B. (2019). Postmeal optogenetic inhibition of dorsal or ventral hippocampal pyramidal neurons increases future intake. eNeuro 6:ENEURO.0457-18.2018. doi: 10.1523/eneuro.0457-18.2018
Hansen, H. H., Fabricius, K., Barkholt, P., Niehoff, M. L., Morley, J. E., Jelsing, J., et al. (2015). The GLP-1 receptor agonist liraglutide improves memory function and increases hippocampal CA1 neuronal numbers in a senescence-accelerated mouse model of Alzheimer’s disease. J. Alzheimers Dis. 46, 877–888. doi: 10.3233/jad-143090
Harrold, J. A., Dovey, T., Cai, X. J., Halford, J. C., and Pinkney, J. (2008). Autoradiographic analysis of ghrelin receptors in the rat hypothalamus. Brain Res. 1196, 59–64. doi: 10.1016/j.brainres.2007.12.055
Hayes, M. R., Skibicka, K. P., Leichner, T. M., Guarnieri, D. J., DiLeone, R. J., Bence, K. K., et al. (2010). Endogenous leptin signaling in the caudal nucleus tractus solitarius and area postrema is required for energy balance regulation. Cell Metab. 11, 77–83. doi: 10.1016/j.cmet.2009.10.009
Hebben, N., Corkin, S., Eichenbaum, H., and Shedlack, K. (1985). Diminished ability to interpret and report internal states after bilateral medial temporal resection: case H.M. Behav. Neurosci. 99, 1031–1039. doi: 10.1037//0735-7044.99.6.1031
Henderson, Y. O., Smith, G. P., and Parent, M. B. (2013). Hippocampal neurons inhibit meal onset. Hippocampus 23, 100–107. doi: 10.1002/hipo.22062
Herman, C. P., and Higgs, S. (2015). Social influences on eating. An introduction to the special issue. Appetite 86, 1–2. doi: 10.1016/j.appet.2014.10.027
Holst, J. J. (2007). The physiology of glucagon-like peptide 1. Physiol. Rev. 87, 1409–1439. doi: 10.1152/physrev.00034.2006
Holt, S. H., Miller, J. C., and Petocz, P. (1997). An insulin index of foods: the insulin demand generated by 1000-kJ portions of common foods. Am. J. Clin. Nutr. 66, 1264–1276. doi: 10.1093/ajcn/66.5.1264
Hommel, J. D., Trinko, R., Sears, R. M., Georgescu, D., Liu, Z. W., Gao, X. B., et al. (2006). Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron 51, 801–810. doi: 10.1016/j.neuron.2006.08.023
Hornsby, A. K., Redhead, Y. T., Rees, D. J., Ratcliff, M. S., Reichenbach, A., Wells, T., et al. (2016). Short-term calorie restriction enhances adult hippocampal neurogenesis and remote fear memory in a Ghsr-dependent manner. Psychoneuroendocrinology 63, 198–207. doi: 10.1016/j.psyneuen.2015.09.023
Hosoda, H., Kojima, M., Matsuo, H., and Kangawa, K. (2000). Ghrelin and des-acyl ghrelin: two major forms of rat ghrelin peptide in gastrointestinal tissue. Biochem. Biophys. Res. Commun. 279, 909–913. doi: 10.1006/bbrc.2000.4039
Howard, A. D., Feighner, S. D., Cully, D. F., Arena, J. P., Liberator, P. A., Rosenblum, C. I., et al. (1996). A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 273, 974–977. doi: 10.1126/science.273.5277.974
Hsu, T. M., Hahn, J. D., Konanur, V. R., Lam, A., and Kanoski, S. E. (2015a). Hippocampal GLP-1 receptors influence food intake, meal size, and effort-based responding for food through volume transmission. Neuropsychopharmacology 40, 327–337. doi: 10.1038/npp.2014.175
Hsu, T. M., Hahn, J. D., Konanur, V. R., Noble, E. E., Suarez, A. N., Thai, J., et al. (2015b). Hippocampus ghrelin signaling mediates appetite through lateral hypothalamic orexin pathways. Elife 4:e11190. doi: 10.7554/elife.11190
Hsu, T. M., Noble, E. E., Liu, C. M., Cortella, A. M., Konanur, V. R., Suarez, A. N., et al. (2018). A hippocampus to prefrontal cortex neural pathway inhibits food motivation through glucagon-like peptide-1 signaling. Mol. Psychiatry 23, 1555–1565. doi: 10.1038/mp.2017.91
Hsu, T. M., Noble, E. E., Reiner, D. J., Liu, C. M., Suarez, A. N., Konanur, V. R., et al. (2018). Hippocampus ghrelin receptor signaling promotes socially-mediated learned food preference. Neuropharmacology 131, 487–496. doi: 10.1016/j.neuropharm.2017.11.039
Huang, H. J., Chen, Y. H., Liang, K. C., Jheng, Y. S., Jhao, J. J., Su, M. T., et al. (2012). Exendin-4 protected against cognitive dysfunction in hyperglycemic mice receiving an intrahippocampal lipopolysaccharide injection. PLoS One 7:e39656. doi: 10.1371/journal.pone.0039656
Huang, X. F., Koutcherov, I., Lin, S., Wang, H. Q., and Storlien, L. (1996). Localization of leptin receptor mRNA expression in mouse brain. Neuroreport 7, 2635–2638. doi: 10.1097/00001756-199611040-00045
Irving, A. J., Wallace, L., Durakoglugil, D., and Harvey, J. (2006). Leptin enhances NR2B-mediated N-methyl-D-aspartate responses via a mitogen-activated protein kinase-dependent process in cerebellar granule cells. Neuroscience 138, 1137–1148. doi: 10.1016/j.neuroscience.2005.11.042
Isacson, R., Nielsen, E., Dannaeus, K., Bertilsson, G., Patrone, C., Zachrisson, O., et al. (2011). The glucagon-like peptide 1 receptor agonist exendin-4 improves reference memory performance and decreases immobility in the forced swim test. Eur. J. Pharmacol. 650, 249–255. doi: 10.1016/j.ejphar.2010.10.008
Iwai, T., Suzuki, M., Kobayashi, K., Mori, K., Mogi, Y., and Oka, J. (2009). The influences of juvenile diabetes on memory and hippocampal plasticity in rats: improving effects of glucagon-like peptide-1. Neurosci. Res. 64, 67–74. doi: 10.1016/j.neures.2009.01.013
Kanoski, S. E., Fortin, S. M., Arnold, M., Grill, H. J., and Hayes, M. R. (2011a). Peripheral and central GLP-1 receptor populations mediate the anorectic effects of peripherally administered GLP-1 receptor agonists, liraglutide and exendin-4. Endocrinology 152, 3103–3112. doi: 10.1210/en.2011-0174
Kanoski, S. E., Hayes, M. R., Greenwald, H. S., Fortin, S. M., Gianessi, C. A., Gilbert, J. R., et al. (2011b). Hippocampal leptin signaling reduces food intake and modulates food-related memory processing. Neuropsychopharmacology 36, 1859–1870. doi: 10.1038/npp.2011.70
Kanoski, S. E., Fortin, S. M., Ricks, K. M., and Grill, H. J. (2013). Ghrelin signaling in the ventral hippocampus stimulates learned and motivational aspects of feeding via PI3K-Akt signaling. Biol. Psychiatry 73, 915–923. doi: 10.1016/j.biopsych.2012.07.002
Kanoski, S. E., and Grill, H. J. (2017). Hippocampus contributions to food intake control: mnemonic, neuroanatomical, and endocrine mechanisms. Biol. Psychiatry 81, 748–756. doi: 10.1016/j.biopsych.2015.09.011
Kanoski, S. E., Hayes, M. R., and Skibicka, K. P. (2016). GLP-1 and weight loss: unraveling the diverse neural circuitry. Am. J. Physiol. Regul. Integr. Comp. Physiol. 310, R885–R895. doi: 10.1152/ajpregu.00520.2015
Kanoski, S. E., Hsu, T. H., and Pennell, S. (2014). “Obesity, western diet intake, and cognitive impairment,” in Omega-3 Fatty Acids in Brain and Neurological Health, eds R. R. Watson and W. D. Meester (New York, NY: Academic Press), 57–62.
Kanoski, S. E., Meisel, R. L., Mullins, A. J., and Davidson, T. L. (2007). The effects of energy-rich diets on discrimination reversal learning and on BDNF in the hippocampus and prefrontal cortex of the rat. Behav. Brain Res. 182, 57–66. doi: 10.1016/j.bbr.2007.05.004
Kanoski, S. E., Zhao, S., Guarnieri, D. J., DiLeone, R. J., Yan, J., De Jonghe, B. C., et al. (2012). Endogenous leptin receptor signaling in the medial nucleus tractus solitarius affects meal size and potentiates intestinal satiation signals. Am. J. Physiol. Endocrinol. Metab. 303, E496–E503. doi: 10.1152/ajpendo.00205.2012
Karra, E., O’Daly, O. G., Choudhury, A. I., Yousseif, A., Millership, S., Neary, M. T., et al. (2013). A link between FTO, ghrelin, and impaired brain food-cue responsivity. J. Clin. Invest. 123, 3539–3551. doi: 10.1172/jci44403
Kastin, A. J., and Akerstrom, V. (2003). Entry of exendin-4 into brain is rapid but may be limited at high doses. Int. J. Obes. Relat. Metab. Disord. 27, 313–318. doi: 10.1038/sj.ijo.0802206
Kastin, A. J., Akerstrom, V., and Pan, W. (2002). Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J. Mol. Neurosci. 18, 7–14. doi: 10.1385/jmn:18:1-2:07
Kelesidis, T., Kelesidis, I., Chou, S., and Mantzoros, C. S. (2010). Narrative review: the role of leptin in human physiology: emerging clinical applications. Ann. Intern. Med. 152, 93–100. doi: 10.7326/0003-4819-152-2-201001190-00008
Kentish, S., Li, H., Philp, L. K., O’donnell, T. A., Isaacs, N. J., Young, R. L., et al. (2012). Diet-induced adaptation of vagal afferent function. J. Physiol. 590, 209–221. doi: 10.1113/jphysiol.2011.222158
Kim, Y., Kim, S., Kim, C., Sato, T., Kojima, M., and Park, S. (2015). Ghrelin is required for dietary restriction-induced enhancement of hippocampal neurogenesis: lessons from ghrelin knockout mice. Endocr. J. 62, 269–275. doi: 10.1507/endocrj.ej14-0436
Klarer, M., Arnold, M., Gunther, L., Winter, C., Langhans, W., and Meyer, U. (2014). Gut vagal afferents differentially modulate innate anxiety and learned fear. J. Neurosci. 34, 7067–7076. doi: 10.1523/JNEUROSCI.0252-14.2014
Klarer, M., Weber-Stadlbauer, U., Arnold, M., Langhans, W., and Meyer, U. (2017). Cognitive effects of subdiaphragmatic vagal deafferentation in rats. Neurobiol. Learn. Mem. 142, 190–199. doi: 10.1016/j.nlm.2017.05.006
Knudsen, L. B., Secher, A., Hecksher-Sørensen, J., and Pyke, C. (2016). Long-acting glucagon-like peptide-1 receptor agonists have direct access to and effects on pro-opiomelanocortin/cocaine- and amphetamine-stimulated transcript neurons in the mouse hypothalamus. J. Diabetes Investig. 7, 56–63. doi: 10.1111/jdi.12463
Kojima, M., Hosoda, H., Date, Y., Nakazato, M., Matsuo, H., and Kangawa, K. (1999). Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402, 656–660. doi: 10.1038/45230
Kreymann, B., Williams, G., Ghatei, M. A., and Bloom, S. R. (1987). Glucagon-like peptide-1 7–36: a physiological incretin in man. Lancet 2, 1300–1304. doi: 10.1016/s0140-6736(87)91194-9
Kumar, P. T., Antony, S., Nandhu, M. S., Sadanandan, J., Naijil, G., and Paulose, C. S. (2011). Vitamin D3 restores altered cholinergic and insulin receptor expression in the cerebral cortex and muscarinic M3 receptor expression in pancreatic islets of streptozotocin induced diabetic rats. J. Nutr. Biochem. 22, 418–425. doi: 10.1016/j.jnutbio.2010.03.010
Laque, A., Yu, S., Qualls-Creekmore, E., Gettys, S., Schwartzenburg, C., Bui, K., et al. (2015). Leptin modulates nutrient reward via inhibitory galanin action on orexin neurons. Mol. Metab. 4, 706–717. doi: 10.1016/j.molmet.2015.07.002
Lee, S. Y., Park, S. H., Chung, C., Kim, J. J., Choi, S. Y., and Han, J. S. (2015). Oxytocin protects hippocampal memory and plasticity from uncontrollable stress. Sci. Rep. 5:18540. doi: 10.1038/srep18540
Lee, Y. S., and Silva, A. J. (2009). The molecular and cellular biology of enhanced cognition. Nat. Rev. Neurosci. 10, 126–140. doi: 10.1038/nrn2572
Leinninger, G. M., Jo, Y. H., Leshan, R. L., Louis, G. W., Yang, H., Barrera, J. G., et al. (2009). Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metab. 10, 89–98. doi: 10.1016/j.cmet.2009.06.011
Leinninger, G. M., Opland, D. M., Jo, Y. H., Faouzi, M., Christensen, L., Cappellucci, L. A., et al. (2011). Leptin action via neurotensin neurons controls orexin, the mesolimbic dopamine system and energy balance. Cell Metab. 14, 313–323. doi: 10.1016/j.cmet.2011.06.016
Lennox, R., Porter, D. W., Flatt, P. R., Holscher, C., Irwin, N., and Gault, V. A. (2014). Comparison of the independent and combined effects of sub-chronic therapy with metformin and a stable GLP-1 receptor agonist on cognitive function, hippocampal synaptic plasticity and metabolic control in high-fat fed mice. Neuropharmacology 86, 22–30. doi: 10.1016/j.neuropharm.2014.06.026
Levitsky, D. A. (2005). The non-regulation of food intake in humans: hope for reversing the epidemic of obesity. Physiol. Behav. 86, 623–632. doi: 10.1016/j.physbeh.2005.08.053
Li, X. L., Aou, S., Oomura, Y., Hori, N., Fukunaga, K., and Hori, T. (2002). Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience 113, 607–615. doi: 10.1016/s0306-4522(02)00162-8
Li, Y., Calfa, G., Inoue, T., Amaral, M. D., and Pozzo-Miller, L. (2010). Activity-dependent release of endogenous BDNF from mossy fibers evokes a TRPC3 current and Ca2+ elevations in CA3 pyramidal neurons. J. Neurophysiol. 103, 2846–2856. doi: 10.1152/jn.01140.2009
Lieb, W., Beiser, A. S., Vasan, R. S., Tan, Z. S., Au, R., Harris, T. B., et al. (2009). Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA 302, 2565–2572. doi: 10.1001/jama.2009.1836
Lin, C. I., Shen, C. F., Hsu, T. H., and Lin, S. H. (2017). A high-fructose-high-coconut oil diet induces dysregulating expressions of hippocampal leptin and stearoyl-CoA desaturase and spatial memory deficits in rats. Nutrients 9:E619. doi: 10.3390/nu9060619
Lisman, J., Cooper, K., Sehgal, M., and Silva, A. J. (2018). Memory formation depends on both synapse-specific modifications of synaptic strength and cell-specific increases in excitability. Nat. Neurosci. 21, 309–314. doi: 10.1038/s41593-018-0076-6
Liu, A. F., Zhao, F. B., Wang, J., Lu, Y. F., Tian, J., Zhao, Y., et al. (2016). Effects of vagus nerve stimulation on cognitive functioning in rats with cerebral ischemia reperfusion. J. Transl. Med. 14:101. doi: 10.1186/s12967-016-0858-0
Lockie, S. H., Dinan, T., Lawrence, A. J., Spencer, S. J., and Andrews, Z. B. (2015). Diet-induced obesity causes ghrelin resistance in reward processing tasks. Psychoneuroendocrinology 62, 114–120. doi: 10.1016/j.psyneuen.2015.08.004
Maffei, M., Halaas, J., Ravussin, E., Pratley, R. E., Lee, G. H., Zhang, Y., et al. (1995). Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1, 1155–1161. doi: 10.1038/nm1195-1155
Malekizadeh, Y., Holiday, A., Redfearn, D., Ainge, J. A., Doherty, G., and Harvey, J. (2017). A leptin fragment mirrors the cognitive enhancing and neuroprotective actions of leptin. Cereb. Cortex 27, 4769–4782. doi: 10.1093/cercor/bhw272
Malik, S., McGlone, F., Bedrossian, D., and Dagher, A. (2008). Ghrelin modulates brain activity in areas that control appetitive behavior. Cell Metab. 7, 400–409. doi: 10.1016/j.cmet.2008.03.007
Mani, B. K., Walker, A. K., Lopez Soto, E. J., Raingo, J., Lee, C. E., Perello, M., et al. (2014). Neuroanatomical characterization of a growth hormone secretagogue receptor-green fluorescent protein reporter mouse. J. Comp. Neurol. 522, 3644–3666. doi: 10.1002/cne.23627
Marks, J. L., Porte, D. Jr., Stahl, W. L., and Baskin, D. G. (1990). Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology 127, 3234–3236. doi: 10.1210/endo-127-6-3234
Mavanji, V., Butterick, T. A., Duffy, C. M., Nixon, J. P., Billington, C. J., and Kotz, C. M. (2017). Orexin/hypocretin treatment restores hippocampal-dependent memory in orexin-deficient mice. Neurobiol. Learn. Mem. 146, 21–30. doi: 10.1016/j.nlm.2017.10.014
McClean, P. L., Parthsarathy, V., Faivre, E., and Hölscher, C. (2011). The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. 31, 6587–6594. doi: 10.1523/JNEUROSCI.0529-11.2011
McNay, E. C., Fries, T. M., and Gold, P. E. (2000). Decreases in rat extracellular hippocampal glucose concentration associated with cognitive demand during a spatial task. Proc. Natl. Acad. Sci. U S A 97, 2881–2885. doi: 10.1073/pnas.050583697
McNay, E. C., Ong, C. T., McCrimmon, R. J., Cresswell, J., Bogan, J. S., and Sherwin, R. S. (2010). Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol. Learn. Mem. 93, 546–553. doi: 10.1016/j.nlm.2010.02.002
Merchenthaler, I., Lane, M., and Shughrue, P. (1999). Distribution of pre-pro-glucagon and glucagon-like peptide-1 receptor messenger RNAs in the rat central nervous system. J. Comp. Neurol. 403, 261–280. doi: 10.1002/(sici)1096-9861(19990111)403:2<261::aid-cne8>3.0.co;2-5
Mietlicki-Baase, E. G., Olivos, D. R., Jeffrey, B. A., and Hayes, M. R. (2015). Cooperative interaction between leptin and amylin signaling in the ventral tegmental area for the control of food intake. Am. J. Physiol. Endocrinol. Metab. 308, E1116–E1122. doi: 10.1152/ajpendo.00087.2015
Milivojevic, B., and Doeller, C. F. (2013). Mnemonic networks in the hippocampal formation: from spatial maps to temporal and conceptual codes. J. Exp. Psychol. Gen. 142, 1231–1241. doi: 10.1037/a0033746
Min, D. K., Tuor, U. I., and Chelikani, P. K. (2011). Gastric distention induced functional magnetic resonance signal changes in the rodent brain. Neuroscience 179, 151–158. doi: 10.1016/j.neuroscience.2011.01.051
Mojsov, S., Weir, G. C., and Habener, J. F. (1987). Insulinotropin: glucagon-like peptide I (7–37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J. Clin. Invest. 79, 616–619. doi: 10.1172/JCI112855
Molteni, R., Barnard, R. J., Ying, Z., Roberts, C. K., and Gómez-Pinilla, F. (2002). A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience 112, 803–814. doi: 10.1016/s0306-4522(02)00123-9
Montague, C. T., Farooqi, I. S., Whitehead, J. P., Soos, M. A., Rau, H., Wareham, N. J., et al. (1997). Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 387, 903–908. doi: 10.1038/43185
Monzon, M. E., de Souza, M. M., Izquierdo, L. A., Izquierdo, I., Barros, D. M., and de Barioglio, S. R. (1999). Melanin-concentrating hormone (MCH) modifies memory retention in rats. Peptides 20, 1517–1519. doi: 10.1016/s0196-9781(99)00164-3
Moon, M., Kim, S., Hwang, L., and Park, S. (2009). Ghrelin regulates hippocampal neurogenesis in adult mice. Endocr. J. 56, 525–531. doi: 10.1507/endocrj.k09e-089
Morris, R. G., Garrud, P., Rawlins, J. N., and O’Keefe, J. (1982). Place navigation impaired in rats with hippocampal lesions. Nature 297, 681–683. doi: 10.1038/297681a0
Moser, M. B., and Moser, E. I. (1998). Functional differentiation in the hippocampus. Hippocampus 8, 608–619. doi: 10.1002/(sici)1098-1063(1998)8:6<608::aid-hipo3>3.0.co;2-7
Munzberg, H., Björnholm, M., Bates, S. H., and Myers, M. G. Jr. (2005). Leptin receptor action and mechanisms of leptin resistance. Cell. Mol. Life Sci. 62, 642–652. doi: 10.1007/s00018-004-4432-1
Myers, M. G. Jr., Münzberg, H., Leinninger, G. M., and Leshan, R. L. (2009). The geometry of leptin action in the brain: more complicated than a simple ARC. Cell Metab. 9, 117–123. doi: 10.1016/j.cmet.2008.12.001
Narita, K., Kosaka, H., Okazawa, H., Murata, T., and Wada, Y. (2009). Relationship between plasma leptin level and brain structure in elderly: a voxel-based morphometric study. Biol. Psychiatry 65, 992–994. doi: 10.1016/j.biopsych.2008.10.006
Näslund, E., Barkeling, B., King, N., Gutniak, M., Blundell, J. E., Holst, J. J., et al. (1999). Energy intake and appetite are suppressed by glucagon-like peptide-1 (GLP-1) in obese men. Int. J. Obes. Relat. Metab. Disord. 23, 304–311. doi: 10.1038/sj.ijo.0800818
Näslund, E., Gutniak, M., Skogar, S., Rössner, S., and Hellström, P. M. (1998). Glucagon-like peptide 1 increases the period of postprandial satiety and slows gastric emptying in obese men. Am. J. Clin. Nutr. 68, 525–530. doi: 10.1093/ajcn/68.3.525
Nass, R., Farhy, L. S., Liu, J., Prudom, C. E., Johnson, M. L., Veldhuis, P., et al. (2008). Evidence for acyl-ghrelin modulation of growth hormone release in the fed state. J. Clin. Endocrinol. Metab. 93, 1988–1994. doi: 10.1210/jc.2007-2234
Niswender, K. D., Morton, G. J., Stearns, W. H., Rhodes, C. J., Myers, M. G. Jr., and Schwartz, M. W. (2001). Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature 413, 794–795. doi: 10.1038/35101657
Noble, E. E., Hahn, J. D., Konanur, V. R., Hsu, T. M., Page, S. J., Cortella, A. M., et al. (2018). Control of feeding behavior by cerebral ventricular volume transmission of melanin-concentrating hormone. Cell Metab. 28, 55.e7–68.e7. doi: 10.1016/j.cmet.2018.05.001
Noble, E. E., Mavanji, V., Little, M. R., Billington, C. J., Kotz, C. M., and Wang, C. (2014). Exercise reduces diet-induced cognitive decline and increases hippocampal brain-derived neurotrophic factor in CA3 neurons. Neurobiol. Learn. Mem. 114, 40–50. doi: 10.1016/j.nlm.2014.04.006
Norgren, R., and Smith, G. P. (1994). A method for selective section of vagal afferent or efferent axons in the rat. Am. J. Physiol. 267, R1136–R1141. doi: 10.1152/ajpregu.1994.267.4.R1136
Obici, S., Feng, Z., Karkanias, G., Baskin, D. G., and Rossetti, L. (2002a). Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat. Neurosci. 5, 566–572. doi: 10.1038/nn861
Obici, S., Zhang, B. B., Karkanias, G., and Rossetti, L. (2002b). Hypothalamic insulin signaling is required for inhibition of glucose production. Nat. Med. 8, 1376–1382. doi: 10.1038/nm798
Ohinata, K., Sonoda, S., Inoue, N., Yamauchi, R., Wada, K., and Yoshikawa, M. (2007). β-Lactotensin, a neurotensin agonist peptide derived from bovine β-lactoglobulin, enhances memory consolidation in mice. Peptides 28, 1470–1474. doi: 10.1016/j.peptides.2007.06.002
O’Leary, O. F., Ogbonnaya, E. S., Felice, D., Levone, B. R., C Conroy, L., Fitzgerald, P., et al. (2018). The vagus nerve modulates BDNF expression and neurogenesis in the hippocampus. Eur. Neuropsychopharmacol. 28, 307–316. doi: 10.1016/j.euroneuro.2017.12.004
O’Malley, D., MacDonald, N., Mizielinska, S., Connolly, C. N., Irving, A. J., and Harvey, J. (2007). Leptin promotes rapid dynamic changes in hippocampal dendritic morphology. Mol. Cell. Neurosci. 35, 559–572. doi: 10.1016/j.mcn.2007.05.001
Oomura, Y., Hori, N., Shiraishi, T., Fukunaga, K., Takeda, H., Tsuji, M., et al. (2006). Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides 27, 2738–2749. doi: 10.1016/j.peptides.2006.07.001
Palleria, C., Leo, A., Andreozzi, F., Citraro, R., Iannone, M., Spiga, R., et al. (2017). Liraglutide prevents cognitive decline in a rat model of streptozotocin-induced diabetes independently from its peripheral metabolic effects. Behav. Brain Res. 321, 157–169. doi: 10.1016/j.bbr.2017.01.004
Parent, M. B. (2016). Cognitive control of meal onset and meal size: role of dorsal hippocampal-dependent episodic memory. Physiol. Behav. 162, 112–119. doi: 10.1016/j.physbeh.2016.03.036
Parent, M. B., Darling, J. N., and Henderson, Y. O. (2014). Remembering to eat: hippocampal regulation of meal onset. Am. J. Physiol. Regul. Integr. Comp. Physiol. 306, R701–R713. doi: 10.1152/ajpregu.00496.2013
Paz-Filho, G. J., Babikian, T., Asarnow, R., Delibasi, T., Esposito, K., Erol, H. K., et al. (2008). Leptin replacement improves cognitive development. PLoS One 3:e3098. doi: 10.1371/journal.pone.0003098
Pearson-Leary, J., Jahagirdar, V., Sage, J., and McNay, E. C. (2018). Insulin modulates hippocampally-mediated spatial working memory via glucose transporter-4. Behav. Brain Res. 338, 32–39. doi: 10.1016/j.bbr.2017.09.033
Perello, M., Sakata, I., Birnbaum, S., Chuang, J. C., Osborne-Lawrence, S., Rovinsky, S. A., et al. (2010). Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner. Biol. Psychiatry 67, 880–886. doi: 10.1016/j.biopsych.2009.10.030
Perreault, M., Istrate, N., Wang, L., Nichols, A. J., Tozzo, E., and Stricker-Krongrad, A. (2004). Resistance to the orexigenic effect of ghrelin in dietary-induced obesity in mice: reversal upon weight loss. Int. J. Obes. Relat. Metab. Disord. 28, 879–885. doi: 10.1038/sj.ijo.0802640
Perry, T., Lahiri, D. K., Chen, D., Zhou, J., Shaw, K. T., Egan, J. M., et al. (2002). A novel neurotrophic property of glucagon-like peptide 1: a promoter of nerve growth factor-mediated differentiation in PC12 cells. J. Pharmacol. Exp. Ther. 300, 958–966. doi: 10.1124/jpet.300.3.958
Pinos, H., Ortega, E., Carrillo, B., Pérez-Izquierdo, M. A., and Collado, P. (2007). Differential effects of undernourishment and nutritional rehabilitation on serum leptin levels in male and female rats. Neurochem. Res. 32, 407–413. doi: 10.1007/s11064-006-9240-6
Porter, D. W., Kerr, B. D., Flatt, P. R., Holscher, C., and Gault, V. A. (2010). Four weeks administration of Liraglutide improves memory and learning as well as glycaemic control in mice with high fat dietary-induced obesity and insulin resistance. Diabetes Obes. Metab. 12, 891–899. doi: 10.1111/j.1463-1326.2010.01259.x
Porter, W. D., Flatt, P. R., Holscher, C., and Gault, V. A. (2013). Liraglutide improves hippocampal synaptic plasticity associated with increased expression of Mash1 in ob/ob mice. Int. J. Obes. 37, 678–684. doi: 10.1038/ijo.2012.91
Power, D. A., Noel, J., Collins, R., and O’neill, D. (2001). Circulating leptin levels and weight loss in Alzheimer’s disease patients. Dement. Geriatr. Cogn. Disord. 12, 167–170. doi: 10.1159/000051252
Powley, T. L. (2000). Vagal circuitry mediating cephalic-phase responses to food. Appetite 34, 184–188. doi: 10.1006/appe.1999.0279
Powley, T. L., and Phillips, R. J. (2004). Gastric satiation is volumetric, intestinal satiation is nutritive. Physiol. Behav. 82, 69–74. doi: 10.1016/j.physbeh.2004.04.037
Pratchayasakul, W., Chattipakorn, N., and Chattipakorn, S. C. (2014). Estrogen restores brain insulin sensitivity in ovariectomized non-obese rats, but not in ovariectomized obese rats. Metabolism 63, 851–859. doi: 10.1016/j.metabol.2014.03.009
Qi, L., Ke, L., Liu, X., Liao, L., Ke, S., Liu, X., et al. (2016). Subcutaneous administration of liraglutide ameliorates learning and memory impairment by modulating tau hyperphosphorylation via the glycogen synthase kinase-3β pathway in an amyloid β protein induced alzheimer disease mouse model. Eur. J. Pharmacol. 783, 23–32. doi: 10.1016/j.ejphar.2016.04.052
Rajasekar, N., Nath, C., Hanif, K., and Shukla, R. (2017). Intranasal insulin administration ameliorates streptozotocin (ICV)-induced insulin receptor dysfunction, neuroinflammation, amyloidogenesis, and memory impairment in rats. Mol. Neurobiol. 54, 6507–6522. doi: 10.1007/s12035-016-0169-8
Reagan, L. P. (2005). Neuronal insulin signal transduction mechanisms in diabetes phenotypes. Neurobiol. Aging 26, 56–59. doi: 10.1016/j.neurobiolaging.2005.09.001
Reger, M. A., Watson, G. S., Green, P. S., Wilkinson, C. W., Baker, L. D., Cholerton, B., et al. (2008). Intranasal insulin improves cognition and modulates β-amyloid in early AD. Neurology 70, 440–448. doi: 10.1212/01.WNL.0000265401.62434.36
Rinaman, L. (2010). Ascending projections from the caudal visceral nucleus of the solitary tract to brain regions involved in food intake and energy expenditure. Brain Res. 1350, 18–34. doi: 10.1016/j.brainres.2010.03.059
Rivera, E. J., Goldin, A., Fulmer, N., Tavares, R., Wands, J. R., and de la Monte, S. M. (2005). Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J. Alzheimers Dis. 8, 247–268. doi: 10.3233/jad-2005-8304
Ronchi, G., Ryu, V., Fornaro, M., and Czaja, K. (2012). Hippocampal plasticity after a vagus nerve injury in the rat. Neural Regen. Res. 7, 1055–1063. doi: 10.3969/j.issn.1673-5374.2012.14.003
Rostami, F., Javan, M., Moghimi, A., Haddad-Mashadrizeh, A., and Fereidoni, M. (2017). Streptozotocin-induced hippocampal astrogliosis and insulin signaling malfunction as experimental scales for subclinical sporadic Alzheimer model. Life Sci. 188, 172–185. doi: 10.1016/j.lfs.2017.08.025
Schaeffer, M., Langlet, F., Lafont, C., Molino, F., Hodson, D. J., Roux, T., et al. (2013). Rapid sensing of circulating ghrelin by hypothalamic appetite-modifying neurons. Proc. Natl. Acad. Sci. U S A 110, 1512–1517. doi: 10.1073/pnas.1212137110
Schulingkamp, R. J., Pagano, T. C., Hung, D., and Raffa, R. B. (2000). Insulin receptors and insulin action in the brain: review and clinical implications. Neurosci. Biobehav. Rev. 24, 855–872. doi: 10.1016/s0149-7634(00)00040-3
Schwartz, M. W., Woods, S. C., Porte, D. Jr., Seeley, R. J., and Baskin, D. G. (2000). Central nervous system control of food intake. Nature 404, 661–671. doi: 10.1038/35007534
Shah, M., and Vella, A. (2014). Effects of GLP-1 on appetite and weight. Rev. Endocr. Metab. Disord. 15, 181–187. doi: 10.1007/s11154-014-9289-5
Shanley, L. J., Irving, A. J., and Harvey, J. (2001). Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J. Neurosci. 21:RC186. doi: 10.1523/JNEUROSCI.21-24-j0001.2001
Shioda, S., Funahashi, H., Nakajo, S., Yada, T., Maruta, O., and Nakai, Y. (1998). Immunohistochemical localization of leptin receptor in the rat brain. Neurosci. Lett. 243, 41–44. doi: 10.1016/s0304-3940(98)00082-2
Skelin Klemen, M., Dolensek, J., Slak Rupnik, M., and Stozer, A. (2017). The triggering pathway to insulin secretion: functional similarities and differences between the human and the mouse β cells and their translational relevance. Islets 9, 109–139. doi: 10.1080/19382014.2017.1342022
Špolcová, A., Mikulášková, B., Kršková, K., Gajdošechová, L., Zórad, Š., Olszanecki, R., et al. (2014). Deficient hippocampal insulin signaling and augmented Tau phosphorylation is related to obesity- and age-induced peripheral insulin resistance: a study in Zucker rats. BMC Neurosci. 15:111. doi: 10.1186/1471-2202-15-111
Steen, E., Terry, B. M., Rivera, E. J., Cannon, J. L., Neely, T. R., Tavares, R., et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—is this type 3 diabetes? J. Alzheimers Dis. 7, 63–80. doi: 10.3233/jad-2005-7107
Stern, S. A., Chen, D. Y., and Alberini, C. M. (2014). The effect of insulin and insulin-like growth factors on hippocampus- and amygdala-dependent long-term memory formation. Learn. Mem. 21, 556–563. doi: 10.1101/lm.029348.112
Stranahan, A. M., Lee, K., Martin, B., Maudsley, S., Golden, E., Cutler, R. G., et al. (2009). Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus 19, 951–961. doi: 10.1002/hipo.20577
Stranahan, A. M., Norman, E. D., Lee, K., Cutler, R. G., Telljohann, R. S., Egan, J. M., et al. (2008). Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus 18, 1085–1088. doi: 10.1002/hipo.20470
Strobel, A., Issad, T., Camoin, L., Ozata, M., and Strosberg, A. D. (1998). A leptin missense mutation associated with hypogonadism and morbid obesity. Nat. Genet. 18, 213–215. doi: 10.1038/ng0398-213
Strong, A. J., Fairfield, J. E., Monteiro, E., Kirby, M., Hogg, A. R., Snape, M., et al. (1990). Insulin protects cognitive function in experimental stroke. J. Neurol. Neurosurg. Psychiatry 53, 847–853. doi: 10.1136/jnnp.53.10.847
Suarez, A. N., Hsu, T. M., Liu, C. M., Noble, E. E., Cortella, A. M., Nakamoto, E. M., et al. (2018). Gut vagal sensory signaling regulates hippocampus function through multi-order pathways. Nat. Commun. 9:2181. doi: 10.1038/s41467-018-04639-1
Sun, Y., Wang, P., Zheng, H., and Smith, R. G. (2004). Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc. Natl. Acad. Sci. U S A 101, 4679–4684. doi: 10.1073/pnas.0305930101
Takach, O., Gill, T. B., and Silverman, M. A. (2015). Modulation of insulin signaling rescues BDNF transport defects independent of tau in amyloid-β oligomer-treated hippocampal neurons. Neurobiol. Aging 36, 1378–1382. doi: 10.1016/j.neurobiolaging.2014.11.018
Tartaglia, L. A., Dembski, M., Weng, X., Deng, N., Culpepper, J., Devos, R., et al. (1995). Identification and expression cloning of a leptin receptor, OB-R. Cell 83, 1263–1271. doi: 10.1016/0092-8674(95)90151-5
Tatulian, S. A. (2015). Structural dynamics of insulin receptor and transmembrane signaling. Biochemistry 54, 5523–5532. doi: 10.1021/acs.biochem.5b00805
Tong, J., Dave, N., Mugundu, G. M., Davis, H. W., Gaylinn, B. D., Thorner, M. O., et al. (2013). The pharmacokinetics of acyl, des-acyl, and total ghrelin in healthy human subjects. Eur. J. Endocrinol. 168, 821–828. doi: 10.1530/eje-13-0072
Trapp, S., and Cork, S. C. (2015). PPG neurons of the lower brain stem and their role in brain GLP-1 receptor activation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 309, R795–R804. doi: 10.1152/ajpregu.00333.2015
Tulving, E., and Markowitsch, H. J. (1998). Episodic and declarative memory: role of the hippocampus. Hippocampus 8, 198–204. doi: 10.1002/(sici)1098-1063(1998)8:3<198::aid-hipo2>3.3.co;2-j
Uchida, A., Zechner, J. F., Mani, B. K., Park, W. M., Aguirre, V., and Zigman, J. M. (2014). Altered ghrelin secretion in mice in response to diet-induced obesity and Roux-en-Y gastric bypass. Mol. Metab. 3, 717–730. doi: 10.1016/j.molmet.2014.07.009
Unger, J., McNeill, T. H., Moxley, R. T. III., White, M., Moss, A., and Livingston, J. N. (1989). Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience 31, 143–157. doi: 10.1016/0306-4522(89)90036-5
Ur, E., Wilkinson, D. A., Morash, B. A., and Wilkinson, M. (2002). Leptin immunoreactivity is localized to neurons in rat brain. Neuroendocrinology 75, 264–272. doi: 10.1159/000054718
Ura, H., Sugaya, Y., Ohata, H., Takumi, I., Sadamoto, K., Shibasaki, T., et al. (2013). Vagus nerve stimulation induced long-lasting enhancement of synaptic transmission and decreased granule cell discharge in the hippocampal dentate gyrus of urethane-anesthetized rats. Brain Res. 1492, 63–71. doi: 10.1016/j.brainres.2012.11.024
Valladolid-Acebes, I., Fole, A., Martín, M., Morales, L., Cano, M. V., Ruiz-Gayo, M., et al. (2013). Spatial memory impairment and changes in hippocampal morphology are triggered by high-fat diets in adolescent mice. Is there a role of leptin? Neurobiol. Learn. Mem. 106, 18–25. doi: 10.1016/j.nlm.2013.06.012
Val-Laillet, D., Biraben, A., Randuineau, G., and Malbert, C. H. (2010). Chronic vagus nerve stimulation decreased weight gain, food consumption and sweet craving in adult obese minipigs. Appetite 55, 245–252. doi: 10.1016/j.appet.2010.06.008
Van Doorn, C., Macht, V. A., Grillo, C. A., and Reagan, L. P. (2017). Leptin resistance and hippocampal behavioral deficits. Physiol. Behav. 176, 207–213. doi: 10.1016/j.physbeh.2017.03.002
Walker, A. K., Ibia, I. E., and Zigman, J. M. (2012). Disruption of cue-potentiated feeding in mice with blocked ghrelin signaling. Physiol. Behav. 108, 34–43. doi: 10.1016/j.physbeh.2012.10.003
Wang, X. H., Li, L., Holscher, C., Pan, Y. F., Chen, X. R., and Qi, J. S. (2010). Val8-glucagon-like peptide-1 protects against Aβ1–40-induced impairment of hippocampal late-phase long-term potentiation and spatial learning in rats. Neuroscience 170, 1239–1248. doi: 10.1016/j.neuroscience.2010.08.028
Wang, G. J., Yang, J., Volkow, N. D., Telang, F., Ma, Y., Zhu, W., et al. (2006). Gastric stimulation in obese subjects activates the hippocampus and other regions involved in brain reward circuitry. Proc. Natl. Acad. Sci. U S A 103, 15641–15645. doi: 10.1073/pnas.0601977103
Wayner, M. J., Armstrong, D. L., Phelix, C. F., and Oomura, Y. (2004). Orexin-A (Hypocretin-1) and leptin enhance LTP in the dentate gyrus of rats in vivo. Peptides 25, 991–996. doi: 10.1016/j.peptides.2004.03.018
Weng, Z., Signore, A. P., Gao, Y., Wang, S., Zhang, F., Hastings, T., et al. (2007). Leptin protects against 6-hydroxydopamine-induced dopaminergic cell death via mitogen-activated protein kinase signaling. J. Biol. Chem. 282, 34479–34491. doi: 10.1074/jbc.M705426200
Williams, E. K., Chang, R. B., Strochlic, D. E., Umans, B. D., Lowell, B. B., and Liberles, S. D. (2016). Sensory neurons that detect stretch and nutrients in the digestive system. Cell 166, 209–221. doi: 10.1016/j.cell.2016.05.011
Williams, D. L., Grill, H. J., Cummings, D. E., and Kaplan, J. M. (2006). Overfeeding-induced weight gain suppresses plasma ghrelin levels in rats. J. Endocrinol. Invest. 29, 863–868. doi: 10.1007/bf03349188
Williams, D. L., Hyvarinen, N., Lilly, N., Kay, K., Dossat, A., Parise, E., et al. (2011). Maintenance on a high-fat diet impairs the anorexic response to glucagon-like-peptide-1 receptor activation. Physiol. Behav. 103, 557–564. doi: 10.1016/j.physbeh.2011.04.005
Witte, A. V., Kobe, T., Graunke, A., Schuchardt, J. P., Hahn, A., Tesky, V. A., et al. (2016). Impact of leptin on memory function and hippocampal structure in mild cognitive impairment. Hum. Brain Mapp. 37, 4539–4549. doi: 10.1002/hbm.23327
Wolf, P. A., Beiser, A., Elias, M. F., Au, R., Vasan, R. S., and Seshadri, S. (2007). Relation of obesity to cognitive function: importance of central obesity and synergistic influence of concomitant hypertension. The Framingham heart study. Curr. Alzheimer Res. 4, 111–116. doi: 10.2174/156720507780362263
Woods, S. C., Chavez, M., Park, C. R., Riedy, C., Kaiyala, K., Richardson, R. D., et al. (1996). The evaluation of insulin as a metabolic signal influencing behavior via the brain. Neurosci. Biobehav. Rev. 20, 139–144. doi: 10.1016/0149-7634(95)00044-f
Wren, A. M., Seal, L. J., Cohen, M. A., Brynes, A. E., Frost, G. S., Murphy, K. G., et al. (2001a). Ghrelin enhances appetite and increases food intake in humans. J. Clin. Endocrinol. Metab. 86:5992. doi: 10.1210/jc.86.12.5992
Wren, A. M., Small, C. J., Abbott, C. R., Dhillo, W. S., Seal, L. J., Cohen, M. A., et al. (2001b). Ghrelin causes hyperphagia and obesity in rats. Diabetes 50, 2540–2547. doi: 10.2337/diabetes.50.11.2540
Wu, H. W., Ren, L. F., Zhou, X., and Han, D. W. (2015). A high-fructose diet induces hippocampal insulin resistance and exacerbates memory deficits in male Sprague-Dawley rats. Nutr. Neurosci. 18, 323–328. doi: 10.1179/1476830514y.0000000133
Xu, L., Sun, X., Lu, J., Tang, M., and Chen, J. D. (2008). Effects of gastric electric stimulation on gastric distention responsive neurons and expressions of CCK in rodent hippocampus. Obesity 16, 951–957. doi: 10.1038/oby.2008.17
Yamada, N., Katsuura, G., Ochi, Y., Ebihara, K., Kusakabe, T., Hosoda, K., et al. (2011). Impaired CNS leptin action is implicated in depression associated with obesity. Endocrinology 152, 2634–2643. doi: 10.1210/en.2011-0004
Yang, J., Brown, M. S., Liang, G., Grishin, N. V., and Goldstein, J. L. (2008). Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 132, 387–396. doi: 10.1016/j.cell.2008.01.017
Yang, H., Youm, Y. H., Nakata, C., and Dixit, V. D. (2007). Chronic caloric restriction induces forestomach hypertrophy with enhanced ghrelin levels during aging. Peptides 28, 1931–1936. doi: 10.1016/j.peptides.2007.07.030
Yang, L., Zou, B., Xiong, X., Pascual, C., Xie, J., Malik, A., et al. (2013). Hypocretin/orexin neurons contribute to hippocampus-dependent social memory and synaptic plasticity in mice. J. Neurosci. 33, 5275–5284. doi: 10.1523/JNEUROSCI.3200-12.2013
Yildiz, B. O., Suchard, M. A., Wong, M. L., McCann, S. M., and Licinio, J. (2004). Alterations in the dynamics of circulating ghrelin, adiponectin and leptin in human obesity. Proc. Natl. Acad. Sci. U S A 101, 10434–10439. doi: 10.1073/pnas.0403465101
Zhang, H., Dong, H., Cilz, N. I., Kurada, L., Hu, B., Wada, E., et al. (2016). Neurotensinergic excitation of dentate gyrus granule cells via galphaq-coupled inhibition of TASK-3 channels. Cereb. Cortex 26, 977–990. doi: 10.1093/cercor/bhu267
Zhang, Y., Proenca, R., Maffei, M., Barone, M., Leopold, L., and Friedman, J. M. (1994). Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–432. doi: 10.1038/372425a0
Zhao, W., Chen, H., Xu, H., Moore, E., Meiri, N., Quon, M. J., et al. (1999). Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation and signaling molecules in the hippocampus of water maze trained rats. J. Biol. Chem. 274, 34893–34902. doi: 10.1074/jbc.274.49.34893
Zhao, T. J., Liang, G., Li, R. L., Xie, X., Sleeman, M. W., Murphy, A. J., et al. (2010). Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc. Natl. Acad. Sci. U S A 107, 7467–7472. doi: 10.1073/pnas.1002271107
Zhao, Z., Liu, H., Xiao, K., Yu, M., Cui, L., Zhu, Q., et al. (2014). Ghrelin administration enhances neurogenesis but impairs spatial learning and memory in adult mice. Neuroscience 257, 175–185. doi: 10.1016/j.neuroscience.2013.10.063
Zigman, J. M., Jones, J. E., Lee, C. E., Saper, C. B., and Elmquist, J. K. (2006). Expression of ghrelin receptor mRNA in the rat and the mouse brain. J. Comp. Neurol. 494, 528–548. doi: 10.1002/cne.21171
Keywords: hippocampus, memory, obesity, vagus nerve, GLP-1, learning, plasticity, neurogenesis
Citation: Suarez AN, Noble EE and Kanoski SE (2019) Regulation of Memory Function by Feeding-Relevant Biological Systems: Following the Breadcrumbs to the Hippocampus. Front. Mol. Neurosci. 12:101. doi: 10.3389/fnmol.2019.00101
Received: 14 January 2019; Accepted: 03 April 2019;
Published: 18 April 2019.
Edited by:
Victor Ramírez-Amaya, Medical Research Institute Mercedes and Martín Ferreyra (INIMEC), ArgentinaReviewed by:
Hélène Volkoff, Memorial University of Newfoundland, CanadaCopyright © 2019 Suarez, Noble and Kanoski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Scott E. Kanoski, a2Fub3NraUB1c2MuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.