 Bindu D. Paul1*
Bindu D. Paul1* Solomon H. Snyder1,2,3
Solomon H. Snyder1,2,3- 1The Solomon H. Snyder Department of Neuroscience, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 2Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 3Department of Pharmacology and Molecular Sciences, Johns Hopkins University School of Medicine, Baltimore, MD, United States
Huntington’s disease (HD) is a neurodegenerative disease triggered by expansion of polyglutamine repeats in the protein huntingtin. Mutant huntingtin (mHtt) aggregates and elicits toxicity by multiple mechanisms which range from dysregulated transcription to disturbances in several metabolic pathways in both the brain and peripheral tissues. Hallmarks of HD include elevated oxidative stress and imbalanced redox signaling. Disruption of antioxidant defense mechanisms, involving antioxidant molecules and enzymes involved in scavenging or reversing oxidative damage, have been linked to the pathophysiology of HD. In addition, mitochondrial function is compromised in HD leading to impaired bioenergetics and elevated production of free radicals in cells. However, the exact mechanisms linking redox imbalance to neurodegeneration are still elusive. This review will focus on the current understanding of aberrant redox homeostasis in HD and potential therapeutic interventions.
Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease caused by expansion of CAG repeats in the gene huntingtin, htt, present on chromosome 4 (MacDonald et al., 1993). About 1 in 7500 individuals are affected by HD worldwide, and no satisfactory cure exists so far (Fisher and Hayden, 2014). HD primarily affects the corpus striatum of the brain and manifests as abnormal involuntary movements, motor and cognitive deficits (Bates et al., 2015; McColgan and Tabrizi, 2018). Once symptoms appear, median survival is about 18 years (Ross et al., 2014). The number of CAG repeats in mutant htt (mHtt) inversely correlates with the age of onset of disease (Illarioshkin et al., 1994; Brandt et al., 1996). Translation of the CAG repeats results in an abnormally long polyglutamine repeat at the N-terminal end of Htt. mHtt is proteolytically cleaved and the N-terminal fragments aggregate to form inclusion bodies, a characteristic feature of the disease, although its role in disease progression is debated (Arrasate and Finkbeiner, 2012). Immunostaining with antibodies against mHtt led to the discovery of intranuclear inclusions in mouse models and patient samples, a hallmark of the disease (Davies et al., 1997; DiFiglia et al., 1997). mHtt impacts multiple cellular processes ranging from transcriptional and translational regulation, mitochondrial function, DNA replication and repair to nucleocytoplasmic transport which leads to neurotoxicity (Gu et al., 1996; Luthi-Carter et al., 2002; Lu X.H. et al., 2014; Banez-Coronel et al., 2015; Grima et al., 2017; Maiuri et al., 2017).
A prominent feature of HD is elevated oxidative stress. Oxidative stress is usually defined as a balance of prooxidant-antioxidant tendencies favoring the former. However, with advances in the field, oxidative stress is more appropriately defined as a disruption of redox signaling (Jones, 2006). Physiological levels of oxidative stress may be important for cellular processes (eustress) but excess, uncontrolled oxidative stress may be deleterious (distress) (Sies et al., 2017). The brain is particularly vulnerable to damage induced by oxidative stress. The brain is amongst the most metabolically active organ in the body and accounts for about 20% of the oxygen consumed (Gadoth and Goebel, 2011). The lipid-rich composition of the brain with its suboptimal antioxidant defense mechanisms as compared to peripheral tissues makes it a target for free radical-induced damage (Floyd and Carney, 1992). Reactive oxygen, nitrogen and sulfur species (ROS, RNS, and RSS) can induce protein oxidation, lipid peroxidation and DNA damage (Sbodio et al., 2018b). As in HD, oxidative stress has been observed in other neurodegenerative disorders including Alzheimer’s disease (AD), Parkinson’s disease (PD), Amyotrophic lateral sclerosis (ALS) and Ataxias and is linked to pathogenesis (Sbodio et al., 2018b).
Redox Imbalance and HD
Several studies have reported oxidative damage in cells and tissues from HD models and patient samples. Elevated markers of damage such as protein oxidation, lipid peroxidation, and DNA damage have been linked to HD. The damage could stem from a variety of abnormalities arising due to the toxic effects of mHtt (summarized in Figure 1).
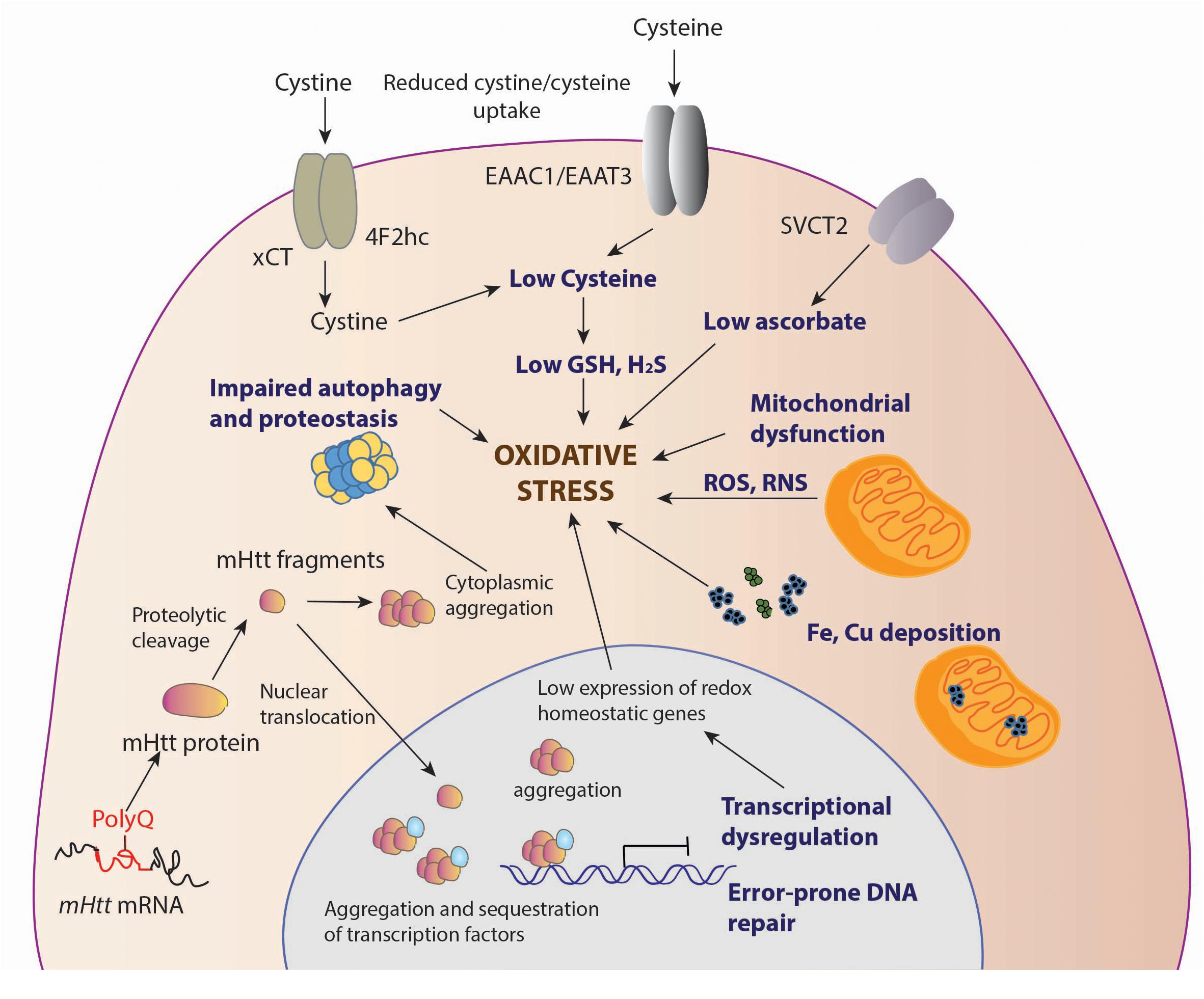
Figure 1. Sources of oxidative stress in HD. Decreased levels of antioxidants, cysteine and ascorbate is observed in HD. The cysteine transporter, excitatory amino acid transporter 3 (EAAT3/EAAC1) is dysregulated in HD leading to decreased intake of cysteine. The uptake of the oxidized form of cysteine, cystine, mediated by the system, composed of the light chain xCT, and the heavy chain 4F2hc, is also reduced, contributing to low cysteine levels. Ascorbate influx via the SVCT2 transporter is also limited in HD, which leads to reduced antioxidant defense in neurons. Deposition of transition metals such as iron (Fe) has been observed both in the cytoplasm and mitochondria, leading to elevated levels of free radicals which can damage cellular components. Mutant huntingtin (mHtt) aggregates both in the nucleus and cytoplasm affecting multiple cellular processes, which include mitochondrial function, autophagy and proteostasis, which leads to elevated oxidative stress. In the nucleus, mHtt sequesters or affects transcription factors, several of which are involved in regulation of antioxidant defense mechanisms, further contributing to redox imbalance in cells. mHtt also affects DNA repair processes, which results in error prone repair and damage.
Deposition of Metal Ions
Metal ions such as iron (Fe), copper (Cu), manganese (Mn), and Zinc (Zn) serve as cofactors for a variety of enzymes and participate in processes such as electron transport, redox regulation, and oxygen transport among others. These metals are beneficial in trace amounts, but excess accumulation leads to several pathological conditions. Iron is redox active, existing in the ferrous (Fe2+) and ferric (Fe3+) states. The Fe2+ form participates in the Fenton reaction reacting with hydrogen peroxide (H2O2) to generate the highly reactive hydroxyl radical (.OH) and HO2, which can cause oxidative damage to cellular components. Elevated iron content has been observed in the basal ganglia in symptomatic and late stage HD (Bartzokis et al., 1999, 2007; Bartzokis and Tishler, 2000). Iron accumulates in both neurons and glia, and treatment with deferoxamine, an iron chelator, affords neuroprotection in the R6/2 mouse model of HD (Simmons et al., 2007; Chen et al., 2013). Conversely, iron supplementation in the diet of neonatal R6/2 mice promotes neurodegeneration in the R6/2 mice (Berggren et al., 2015). Neonatal iron supplementation resulted in iron accumulation in mitochondria due to the increased expression of the mitochondrial iron transporter mitoferrin 2 (Agrawal et al., 2018). In addition to iron, excess copper deposition also mediates neurodegeneration in HD (Dexter et al., 1992; Fox et al., 2007). Copper binds the N-terminal region of mHtt, promotes its aggregation and delays its clearance (Fox et al., 2011). Accordingly, therapies preventing the accumulation of these redox active metals may prove beneficial.
Altered Levels of Antioxidant Molecules and Enzymes
Cells harbor an array of metabolites and molecules that counteract oxidative damage. These may be endogenously synthesized or obtained from the diet. Diminished levels of the antioxidants cysteine, glutathione (GSH), coenzyme Q10 (CoQ10) and ascorbate have been observed in HD and could potentiate disease progression (Andrich et al., 2004; Paul et al., 2014).
Vitamin C/Ascorbate
Vitamin C/ascorbate is a water soluble molecule and cofactor for several enzymatic processes, which regulates metabolism and protects neurons against oxidative stress (Padh, 1990; Castro et al., 2009). During neuronal activity, glutamate is taken up and ascorbate released by astrocytes, which is accumulated by neurons via a specific transporter, SVCT2 (Wilson et al., 2000; Castro et al., 2001). Neuronal ascorbate promotes utilization of lactate over glucose during synaptic activity and also modulates redox balance. The uptake of ascorbate was compromised in cell culture and R6/2 mouse models of HD due to impaired translocation of SVCT2 to the plasma membrane and these changes preceded mitochondrial dysfunction (Acuna et al., 2013). Supplementation of ascorbate reversed the deficits.
Cysteine
Cysteine is a semi-essential amino acid which is synthesized endogenously as well as obtained from the diet. The availability of cysteine is the rate limiting step for glutathione biosynthesis. We have shown previously that cysteine metabolism is compromised in HD (Paul et al., 2014, 2018). Expression of the biosynthetic enzyme for cysteine, cystathionine γ-lyase (CSE) is drastically decreased in HD due to the sequestration of its transcription factor, specificity protein1 (SP1) by mHtt. SP1 regulates transcription of CSE during basal conditions. During stress, expression of CSE is controlled by the stress-responsive activating transcription factor 4 (ATF4). In HD cells, induction of ATF4 is also suboptimal leading to decreased CSE expression and cysteine biosynthesis during stress (Sbodio et al., 2016). Both the biosynthesis and uptake of cysteine and its oxidized form are impaired in HD. Activity of the neuronal cysteine transporter, EAAT3/EAAC1 is decreased in HD due to inhibition of its trafficking to the plasma membrane (Li et al., 2010). Cysteine exists as its oxidized form, cystine, in extracellular fluids and is taken up by the cystine transport system, system, composed of the light-chain (xCT, encoded by the SLC7A11 gene and the heavy-chain subunit 4F2hc, encoded by the SLC3A2 gene) into cells, where the reducing atmosphere converts it to its monomeric, reduced form, cysteine. The activity of xCT is decreased in HD leading to suboptimal cystine metabolism (Frederick et al., 2014). This is not surprising, considering that ATF4 is one of the transcription factors for the transporter’s expression. Supplementing cysteine and its stable precursor, N-acetyl cysteine (NAC) in the diet of R6/2 mice alleviated some of the symptoms and delayed disease progression. Cysteine is also the substrate for the generation of hydrogen sulfide (H2S), a gaseous signaling molecule that participates in a myriad of physiological processes. Three enzymes, CSE, cystathionine β-synthase (CBS) and 3-mercaptopyruvate sulfur transferase (3-MST), utilize cysteine to generate H2S. H2S signals by sulfhydration also called persulfidation, wherein the reactive –SH group of a cysteine residue is converted to a persulfide or –SSH group (Mustafa et al., 2009). Sulfhydration regulates the activity of several proteins and thus modulates several signal transduction cascades (Paul and Snyder, 2012, 2015a,b, 2018). As cysteine metabolism and CSE expression are altered in HD, levels of H2S are diminished in HD. We demonstrated that upregulating the transsulfuration pathway to induce CSE expression and H2S production has therapeutic benefits (Sbodio et al., 2018a,c). Cysteine is utilized by cells to generate several sulfur containing molecules such as cystamine, taurine, lanthionine, homolanthionine, and coenzyme A. The metabolism of these molecules in HD remain to be investigated.
Glutathione
Glutathione, a tripeptide composed of glutamate, cysteine and glycine, is a major antioxidant involved in maintenance of redox homeostasis in cells (Meister and Anderson, 1983). GSH metabolism is dysregulated in HD, which could contribute to the redox imbalance observed in HD. Plasma GSH levels are inversely correlated to caudate atrophy in HD patients (Pena-Sanchez et al., 2015). Decrease in GSH in the postmortem cortex of HD patients has also been observed (Beal et al., 1992). However, other studies have detected increased intracellular glutathione levels in cell culture HD models and increased GSH in cortical and striatal mitochondria of the R6/2 mouse model of HD, although this increase was not sufficient to alleviate the oxidative stress associated with the disease (Choo et al., 2005; Ribeiro et al., 2012), which could reflect differences in the systems utilized or compensatory mechanisms.
Antioxidant Proteins and Pathways
In addition to small molecule antioxidants, cells are equipped with an array of proteins and enzymes which scavenge or neutralize reactive oxygen and nitrogen species. These include superoxide dismutases (SOD1), glutathione peroxidases (GPx), peroxiredoxins (PRDXs), and catalase (Sbodio et al., 2018b). The activities of GPx and SOD1, which act on lipid hydroperoxides and superoxide, were decreased in erythtocytes of HD patients (Chen et al., 2007). Cytosolic SOD1 activity was also decreased in the HD parietal cortex and putamen (Browne et al., 1997). Increasing the activity of GPx1, either by genetic or pharmacologic means in cell culture, yeast and Drosophila models of HD mitigated toxicity (Mason et al., 2013). In addition to effects on antioxidant enzymes, mHtt affects the activity of several transcription factors which regulate antioxidant defense and redox signaling pathways. For instance, as explained earlier, mHtt alters SP1 and ATF4 function, thereby affecting expression of CSE and cysteine metabolism (Paul and Snyder, 2014; Paul et al., 2014; Sbodio et al., 2016). Elevated oxidative stress perturbs the compensatory cytoprotective responses in HD. Our studies using a cell culture model of HD revealed that excessive oxidative stress perturbs signaling mediated by ATF4, a master regulator of amino acid homeostasis. Reduction of oxidative stress improves the response of ATF4 to stress stimuli (Sbodio et al., 2016). Nuclear factor erythroid 2-related factor (Nrf2), which orchestrates gene expression pathways which involved in redox balance and proteostasis (Kensler et al., 2007; Tsvetkov et al., 2013; Dinkova-Kostova et al., 2018) is another transcription factor influenced in HD. Blunted Nrf2 signaling was observed in cell culture models of HD (Jin et al., 2013; Rotblat et al., 2014). Thus, stimulation of Nrf2 signaling pathway may be beneficial in HD (Bresciani et al., 2017).
Mitochondrial Dysfunction
In addition to compromised antioxidant defense as described in the previous section, mitochondrial dysfunction plays a central role in redox imbalance. Mitochondria are the predominant source of ROS and free radicals. Mitochondria are also sites of oxidative phosphorylation (OXPHOS) which is carried out by five multisubunit electron transport complexes (ETCs), designated complexes I–V. Electrons are transported from NADH to NADH coenzyme Q reductase (complex I) to coenzyme Q, which also receives electrons from succinate dehydrogenase (SDH/complex II). Coenzyme Q then relays the electrons to cytochrome C oxidase (complex IV) via complex III (cytochrome bc1). Complex IV reduces molecular oxygen to water using these electrons. Mitochondrial malfunction can lead to oxidative and nitrosative stress through several pathways.
Suboptimal Functioning of the Electron Transport Chain (ETC)
Mitochondrial malfunction in HD was first implied by nuclear magnetic resonance spectroscopy, which revealed increased lactate levels in the striatum and cortex of HD patients (Jenkins et al., 1993). Activities of complex II, III and IV are decreased in HD brains (Gu et al., 1996; Browne et al., 1997). During the process of respiration, the highly reactive is generated, especially by complexes I and III (Raha and Robinson, 2000; Murphy, 2009). Electron transport through the complexes is coupled to proton translocation via the mitochondrial inter membrane space, forming an electrochemical proton gradient (electromotive force) and a mitochondrial transmembrane potential (Δψm), necessary for generation of ATP. In HD, the transmembrane potential is perturbed, impacting mitochondrial bioenergetics (Carmo et al., 2018). The close proximity of the ETC to the mitochondrial genome makes it highly susceptible to oxidative and nitrosative damage. Oxidative DNA damage, as measured by levels of 8-hydroxy-2-deoxyguanosine (OH8dG), to mitochondrial DNA (mtDNA) occurs predominantly in the parietal region of the human HD brain, as opposed to the cerebellum or the frontal cortex with the medium spiny neurons (MSNs) accumulating the greatest damage (Polidori et al., 1999). Consistent with these findings, inhibition of complex II by 3-NPA induced HD-like symptoms and neurotoxicity (Beal et al., 1993). mHtt can affect multiple aspects of mitochondrial function which contribute to elevated oxidative stress (Figure 2).
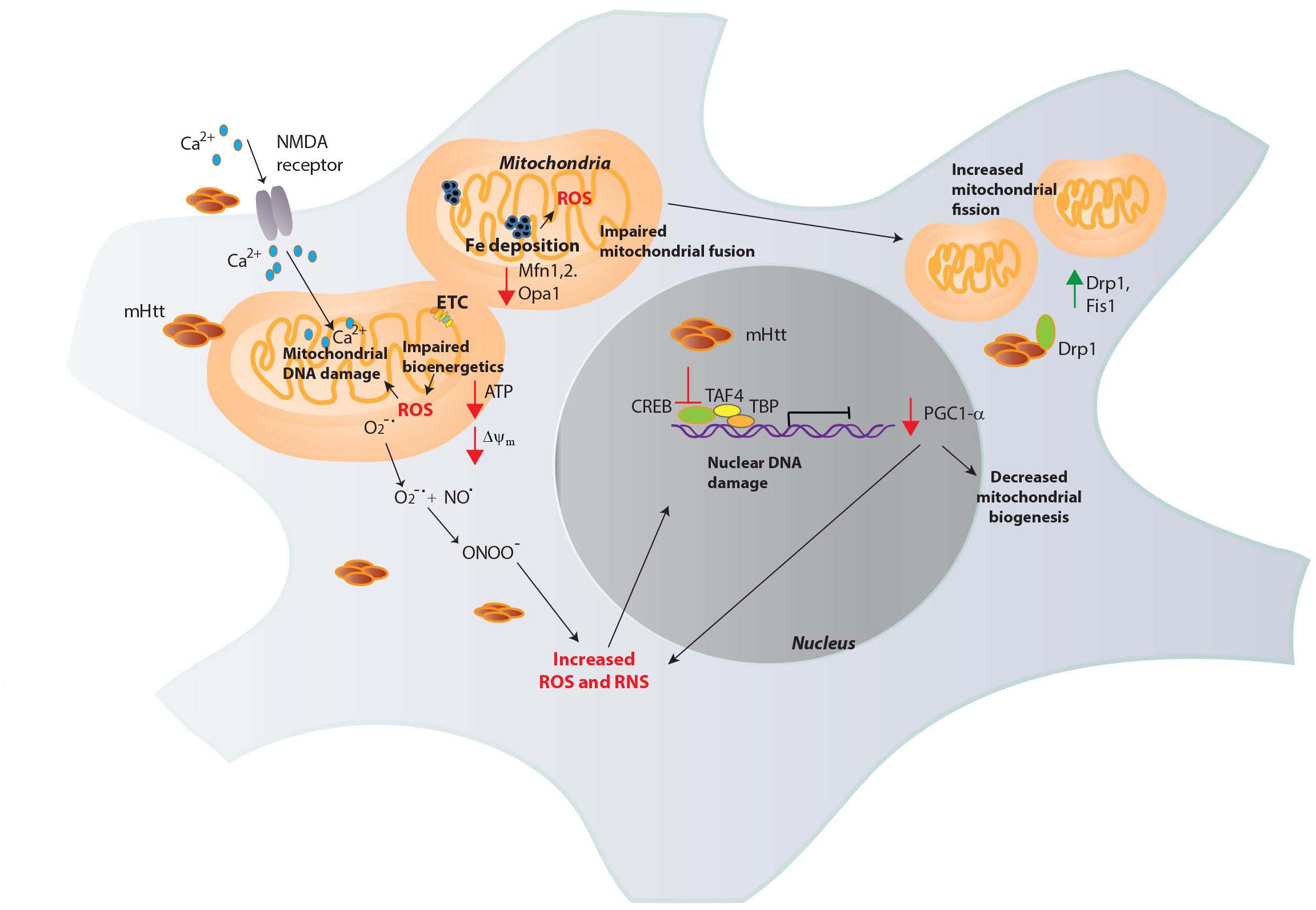
Figure 2. Mitochondrial dysfunction in HD. Mutant huntingtin (mHtt) elicits mitochondrial dysfunction through several mechanisms. mHtt interacts with the outer mitochondrial membrane and could be responsible for several of the observed deficits. mHtt impacts the electron transport chain (ETC), leading to decreased generation of ATP and increased production of ROS such as superoxide (O2-∙). Increased levels of oxidants damage mitochondrial DNA and nuclear DNA which further compromise mitochondrial function to generate more ROS in a vicious cycle. mHtt also induces defects in Ca2+ homeostasis, increasing Ca2+ influx via the N-methyl-D-aspartate (NMDA) receptors and excitotoxicity. Increased Fe deposition is observed in HD, which can participate in the Fenton reaction to produce the highly reactive OH∙, which causes DNA damage. OH∙ react with nitric oxide (NO) to generate peroxynitrite (ONOO-), which causes nitrosative damage to cellular components. mHtt impairs the transcriptional activity of several transcription factors involved in mitochondrial maintenance and function. The expression of the peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1-α) transcriptional coactivator, a regulator of mitochondrial biogenesis is diminished in HD by mHtt. mHtt affects the activity of the transcription factor cAMP response element binding protein (CREB) and other proteins such as TATA-box binding protein (TBP) and TBP-associated factor 4 (TAF4), comprising the transcription initiation complex to decrease PGC1-α expression. As PGC1-α has roles in redox homeostasis, its depletion results in oxidative distress. In addition to these effects, mHtt causes mitochondrial fragmentation, binding to the GTPase dynamin related protein 1 (Drp1, whose expression, along with Fis1 is increased in HD) and stimulating its activity to increase fission. Mitochondrial fusion is also impaired in HD due to decreased levels of the proteins involved in fusion, optic atrophy 1 (Opa1) and Mitofusins (Mfn1 and 2).
Abnormal Ca2+ Homeostasis
Mitochondrial Ca2+ is perturbed in HD, which impacts diverse mitochondrial dynamics (Kolobkova et al., 2017). mHtt interacts with the outer mitochondrial membrane and affects the mitochondrial permeability transition pore (mPTP), decreasing the Ca2+ threshold to trigger mPTP opening, and lowering ATP levels within the organelle (Choo et al., 2004). mHtt has also been shown to interact with Ca2+-binding proteins such as calmodulin; disruption of this interaction has proved beneficial (Bao et al., 1996; Dudek et al., 2010). Interaction of mHtt with type 1 inositol (1,4,5)-triphosphate receptor (InsP3R1), an intracellular Ca2+ release channel, has also been reported (Tang et al., 2003). Although Ca2+ has no direct effect on redox reactions, mitochondrial Ca2+ overload induces elevations in ROS levels which in turn cause mtDNA damage (Peng and Jou, 2010; Wang et al., 2013).
Impaired Function of Transcription Factors and Proteins Involved in Mitochondrial Function
Mutant huntingtin interacts with and affects the function of a number of transcription factors involved in maintenance of mitochondrial function. For instance, mHtt binds to PPARγ coactivator 1α (PGC-1α), a regulator of several metabolic processes including mitochondrial respiration and biogenesis (Cui et al., 2006). PGC-1α also regulates a battery of proteins involved in antioxidant defense and suppresses the formation of ROS in cells (St-Pierre et al., 2006). In addition, mHtt associates with the PGC-1α promoter and interferes with the transcriptional activation functions of promoter-bound transcription factors, CREB and TAF4 leading to diminished PGC-1α expression, mitochondrial abnormalities, and elevated oxidative stress. Besides these processes, mHtt affects mitochondrial trafficking, fission and fusion, resulting in low ATP levels and impaired bioenergetics (Reddy and Shirendeb, 2012; Carmo et al., 2018). The mitochondrial network in cells is dynamic, undergoing fission and fusion to maintain morphology and function. mHtt interferes with the balance of fission and fusion. mHtt binds to dynamin-related protein 1 (Drp1), whose expression is increased in HD, and enhances its GTPase activity to increase mitochondrial fragmentation and distribution (Costa et al., 2010; Song et al., 2011). Furthermore, nitrosylation of Drp1, which increases its activity, has been reported in HD (Nakamura et al., 2010). In addition, expression of the adaptor protein mitochondrial fission 1 (Fis1) is elevated in HD. Mitochondrial fusion mediated by the proteins mitofusin 1 and 2 (Mfn 1 and 2) and the optic atrophy 1 (OPA1) is also affected, as revealed by decreased levels of these proteins in the striatum and cortex of HD patients (Shirendeb et al., 2011). Accordingly, specifically targeting mitochondrial fission and fusion processes may be beneficial (Yin et al., 2016).
Autophagy
As discussed above, damaged mitochondria can further exacerbate oxidative stress hence their removal by autophagy can prevent further damage (Murphy, 2009). Autophagy is a conserved lysosomal degradation pathway which acts by clearance of damaged molecules and organelles and mobilizes cellular nutrients in response to several forms of stress (Klionsky and Emr, 2000; Kroemer et al., 2010; Bento et al., 2016; Leidal et al., 2018). Autophagy and oxidative stress have been linked in several studies. There are numerous reports on dysregulated autophagy in HD (Harding and Tong, 2018). Autophagy plays a vital role in maintaining healthy mitochondria in cells by mitochondrial-specific recycling termed mitophagy. Mitochondrial quality control in neurons is highly dependent on autophagy and plays a central role in neuronal bioenergetics (Batlevi and La Spada, 2011; Redmann et al., 2017). As mitochondrial metabolism generates free radicals, autophagic clearance of damaged mitochondria would limit the levels of oxidative and nitrosative damage (Giordano et al., 2014).
Endoplasmic Reticulum Stress (ER Stress)
Another pathway whose dysregulation leads to elevated oxidative stress is the ER stress signaling cascade. The ER is essential for several cellular functions including but not limited to protein synthesis, folding, maturation, quality control, calcium homeostasis, and glucose metabolism (Almanza et al., 2019). Accumulation of misfolded proteins in the ER triggers a stress response termed the ER stress response, which is a characteristic feature of several neurodegenerative diseases involving protein misfolding and aggregation, such as HD (Hetz and Saxena, 2017). The ER also generates ROS during its normal functions. Expression of N-terminal huntingtin proteins with expanded polyglutamine repeats has been reported to induce cell death in neuronal PC6.3 cell lines that involves endoplasmic reticulum (ER) stress and inhibition of ER stress improved viability and decreased protein aggregation caused by mHtt (Reijonen et al., 2008). One of the proteins involved in the ER stress response is the transcription factor, nuclear factor-κB (NF-κB), which mediates anti-oxidant and anti-apoptotic signaling (Pahl and Baeuerle, 1995; Deng et al., 2004; Sen et al., 2012; Han and Kaufman, 2017). Overexpression of mutant huntingtin proteins in the PC6.3 cell lines results in diminished NF-κB expression and concomitant increase in oxidative stress, decrease in antioxidant levels and enhanced cell death (Reijonen et al., 2010). Accordingly drugs that target the NF-κB pathway may be beneficial in HD. For instance, PRE084, an agonist of the Sigma-1 receptor, Sig-1R, an ER resident chaperone protein, increases NF-κB expression and improves cell viability and mitigates the toxicity mediated by mutant huntingtin proteins in neuronal PC6.3 cells (Hyrskyluoto et al., 2013). Dysregulated NF-κB has also been reported in other studies using RNA-seq and network analysis, where activation has been reported (Miller et al., 2016; Al-Ramahi et al., 2018). Thus, it appears that stage-specific analysis with additional cell types would be necessary to obtain a comprehensive picture of the immune signaling axis in HD.
Defective DNA Repair
Besides, the processes outlined above, defective DNA repair is linked to elevated oxidative stress and contributes to disease progression. Oxidative DNA damage as assessed by formation of 8-OHdG has been observed in both nuclear and mtDNA (Browne et al., 1997). The expanded CAG repeats in mHtt display both germline and somatic instability; the extent of oxidative damage positively correlates with the degree of expansion (Mangiarini et al., 1997; Kovtun et al., 2007). Expansion of the triplet repeats occurs during repair of DNA damage, especially the oxidized guanosine bases, a process relying on the DNA glycosylase OGG1. This base excision repair, BER, is error prone and leads to expansion of the polyglutamine repeats in a process involving single strand breaks and strand slippage. Expansion of the polyglutamine tract further induces oxidative damage and error-prone repair of the lesions, forming a vicious oxidative cycle (Kovtun et al., 2007). Another DNA repair pathway affected in HD involves the ataxia-telangectasia mutated (ATM) protein (Lu X.H. et al., 2014). ATM initiates repair and is activated during oxidative stress and upon DNA damage (Shiloh and Ziv, 2013). Excessive activation of ATM has been reported in HD; inhibiting its activity delays disease progression in mouse models of HD (Lu X.H. et al., 2014). The N terminal end of normal Htt acts as a sensor of ROS and has been shown to detach from the ER, get phosphorylated and translocate to the nucleus when a methionine residue (M8) is oxidized (DiGiovanni et al., 2016). Htt acts as a scaffolding protein for ATM at sites of DNA damage; the CAG repeats may affect the DNA repair process (Maiuri et al., 2017).
Exploring Therapeutic Options in HD
A central feature of complex neurodegenerative diseases such as HD is elevated redox stress, which is intimately linked to disease progression. The current understanding of redox signaling and the intricate interplay between the various pathways involved in protection against oxidative and nitrosative stress is not complete. Whether elevated levels of ROS/RNS are the cause or consequence of disease is still not clear, however increased redox stress exacerbates neurodegeneration and blunts cytoprotective responses. Aberrant redox signaling has been observed in other complex disease states such as cancer. While cancer is associated with uncontrolled cell proliferation, neurodegeneration is characterized by premature cell death. Tumorigenesis and neurodegeneration appear to be at two ends of a homeostatic regulatory breakdown. Antioxidant defense mechanisms such as cysteine and glutathione synthesis are bolstered in cancers leading to reduced sensitivity to radio and chemotherapy, whereas the converse appears to operate in most neurodegenerative diseases (Benfeitas et al., 2017; Vucetic et al., 2017; Sbodio et al., 2018b). Thus, targeting cancer cells by increasing ROS levels have proved beneficial in several cancers (Trachootham et al., 2009; Wondrak, 2009; Ishimoto et al., 2011). In neurodegeneration associated with compromised redox homeostasis, diminishing ROS levels may afford therapeutic benefit. Although antioxidant supplementation has been beneficial in mouse models of HD (Table 1), human clinical trials using antioxidants have been largely unsuccessful in HD and other neurodegenerative diseases (Kim et al., 2015). Several factors may underlie the inefficacy of these antioxidants. Most antioxidants used cannot completely intercept or neutralize oxidant species, leading to a slow buildup of oxidative damage. The antioxidants used target only certain species of free radicals and oxidants; a single antioxidant cannot target all oxidant species. Antioxidant therapy can damage normal cellular processes such as autophagy (Underwood et al., 2010). Accordingly, antioxidants that do not negatively interact with normal cellular processes or have off-target effects may be desirable. Reversing or preventing oxidative damage as well as targeting cytoprotective pathways as a whole, as opposed to simple scavenging of oxidants, may afford greater protection against neurodegeneration. Development of non-cytotoxic drugs that possess multitarget, combinatorial selectivity may be more effective (Varshavsky, 1998). Another aspect which has been largely unexplored in HD is the complex interaction between soluble and cellular immune system components in neurodegeneration. In HD, analysis of redox signaling pathways as a function of disease progression is crucial, as end-stage analysis of tissues likely lack the cells of interest which may have already died. Moreover, population studies by themselves may not be sufficient, as individual variations and heterogeneity in redox pathways and genes that control them may affect the treatment outcome. Comprehensive analyses coupling genomics, transcriptomics, proteomics, metabolomics, and clinical data may reveal central hubs for therapeutic intervention. Such approaches are being pursued for cancer therapy and may have applications in HD (Bidkhori et al., 2018). The timing of intervention, the site of delivery and recycling of the antioxidant should also be considered to arrive at effective redox-active molecules. Development of therapies specific for the various reactive oxygen, nitrogen species and other free radicals, would facilitate a greater understanding of disease progression. Combination therapy involving antioxidant supplementation in conjunction with strategies that strengthen cellular antioxidant capacity may also be considered.
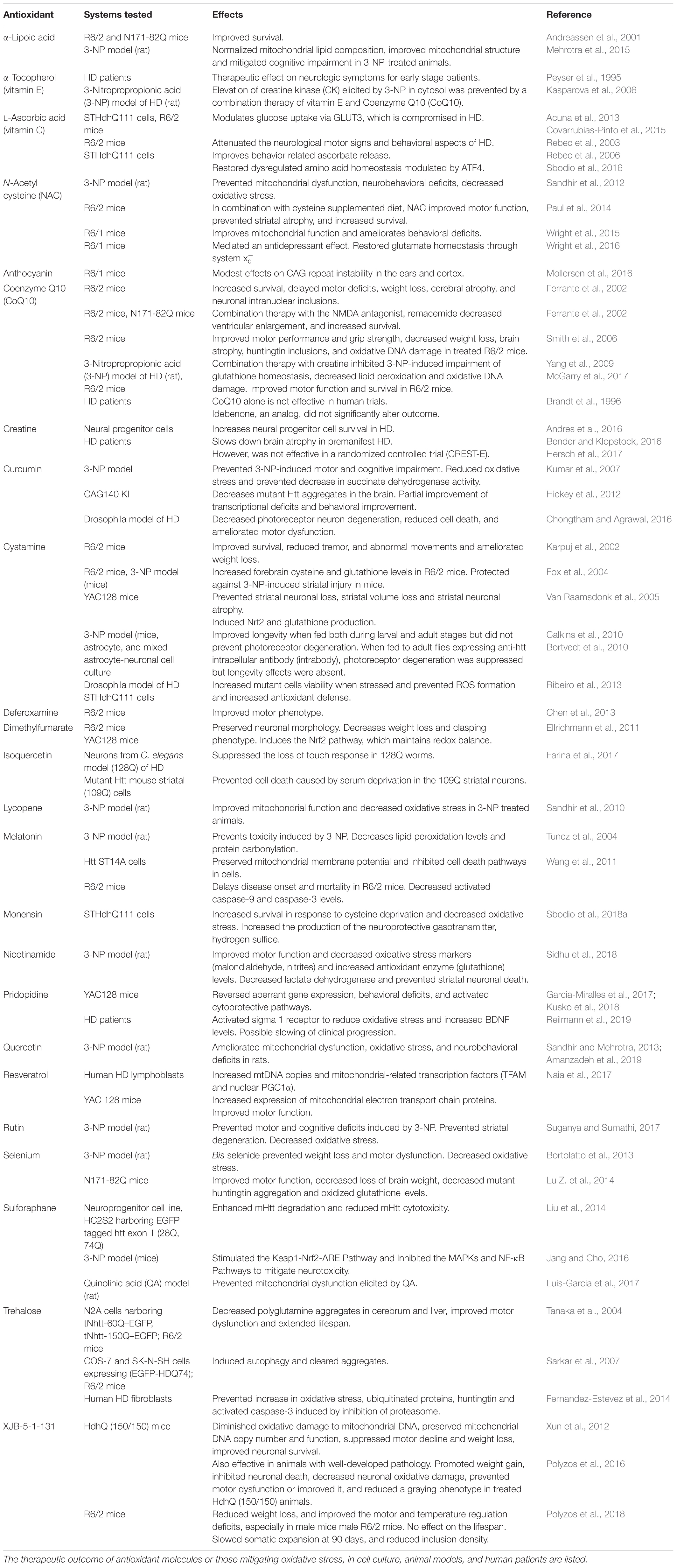
Table 1. Molecules with antioxidant and neuroprotective effects in HD.
Concluding Remarks
It is becoming increasingly evident that our knowledge of redox signaling and its bearing on the origin of these complex diseases is not complete. Repurposing/repositioning/recycling/reprofiling drugs, which have cytoprotective effects in the brain, may offer viable options to arrive at safe and effective drugs for these complex diseases. Such strategies are currently actively investigated for cancers and neurodegeneration (Duraes et al., 2018; Turanli et al., 2018). Studies encompassing diverse genetic, epigenetic and environmental factors may facilitate development of specific therapies targeting redox diseases.
Author Contributions
BP conceptualized the theme of the review. BP and SS wrote the manuscript.
Funding
This work was supported by grants from the US Public Health Service (MH18501 and DA000266 to SS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Acuna, A. I., Esparza, M., Kramm, C., Beltran, F. A., Parra, A. V., Cepeda, C., et al. (2013). A failure in energy metabolism and antioxidant uptake precede symptoms of Huntington’s disease in mice. Nat. Commun. 4:2917. doi: 10.1038/ncomms3917
Agrawal, S., Fox, J., Thyagarajan, B., and Fox, J. H. (2018). Brain mitochondrial iron accumulates in Huntington’s disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free Radic. Biol. Med. 120, 317–329. doi: 10.1016/j.freeradbiomed.2018.04.002
Almanza, A., Carlesso, A., Chintha, C., Creedican, S., Doultsinos, D., Leuzzi, B., et al. (2019). Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J. 286, 241–278. doi: 10.1111/febs.14608
Al-Ramahi, I., Lu, B., Di Paola, S., Pang, K., de Haro, M., Peluso, I., et al. (2018). High-throughput functional analysis distinguishes pathogenic, nonpathogenic, and compensatory transcriptional changes in neurodegeneration. Cell Syst. 7, 28.e4–40.e4. doi: 10.1016/j.cels.2018.05.010
Amanzadeh, E., Esmaeili, A., Rahgozar, S., and Nourbakhshnia, M. (2019). Application of quercetin in neurological disorders: from nutrition to nanomedicine. Rev. Neurosci. doi: 10.1515/revneuro-2018-0080 [Epub ahead of print].
Andreassen, O. A., Ferrante, R. J., Dedeoglu, A., and Beal, M. F. (2001). Lipoic acid improves survival in transgenic mouse models of Huntington’s disease. Neuroreport 12, 3371–3373. doi: 10.1097/00001756-200110290-00044
Andres, R. H., Wallimann, T., and Widmer, H. R. (2016). Creatine supplementation improves neural progenitor cell survival in Huntington’s disease. Brain Circ. 2, 133–137. doi: 10.4103/2394-8108.192519
Andrich, J., Saft, C., Gerlach, M., Schneider, B., Arz, A., Kuhn, W., et al. (2004). Coenzyme Q10 serum levels in Huntington’s disease. J. Neural. Trans. Suppl. 68, 111–116. doi: 10.1007/978-3-7091-0579-5_13
Arrasate, M., and Finkbeiner, S. (2012). Protein aggregates in Huntington’s disease. Exp. Neurol. 238, 1–11. doi: 10.1016/j.expneurol.2011.12.013
Banez-Coronel, M., Ayhan, F., Tarabochia, A. D., Zu, T., Perez, B. A., Tusi, S. K., et al. (2015). RAN translation in huntington disease. Neuron 88, 667–677. doi: 10.1016/j.neuron.2015.10.038
Bao, J., Sharp, A. H., Wagster, M. V., Becher, M., Schilling, G., Ross, C. A., et al. (1996). Expansion of polyglutamine repeat in huntingtin leads to abnormal protein interactions involving calmodulin. Proc. Natl. Acad. Sci. U.S.A. 93, 5037–5042. doi: 10.1073/pnas.93.10.5037
Bartzokis, G., Cummings, J., Perlman, S., Hance, D. B., and Mintz, J. (1999). Increased basal ganglia iron levels in Huntington disease. Arch. Neurol. 56, 569–574. doi: 10.1001/archneur.56.5.569
Bartzokis, G., Lu, P. H., Tishler, T. A., Fong, S. M., Oluwadara, B., Finn, J. P., et al. (2007). Myelin breakdown and iron changes in Huntington’s disease: pathogenesis and treatment implications. Neurochem. Res. 32, 1655–1664. doi: 10.1007/s11064-007-9352-7
Bartzokis, G., and Tishler, T. A. (2000). MRI evaluation of basal ganglia ferritin iron and neurotoxicity in Alzheimer’s and Huntingon’s disease. Cell Mol. Biol. 46, 821–833.
Bates, G. P., Dorsey, R., Gusella, J. F., Hayden, M. R., Kay, C., Leavitt, B. R., et al. (2015). Huntington disease. Nat. Rev. Dis. Prim. 1:15005. doi: 10.1038/nrdp.2015.5
Batlevi, Y., and La Spada, A. R. (2011). Mitochondrial autophagy in neural function, neurodegenerative disease, neuron cell death, and aging. Neurobiol. Dis. 43, 46–51. doi: 10.1016/j.nbd.2010.09.009
Beal, M. F., Brouillet, E., Jenkins, B. G., Ferrante, R. J., Kowall, N. W., Miller, J. M., et al. (1993). Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 13, 4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993
Beal, M. F., Matson, W. R., Storey, E., Milbury, P., Ryan, E. A., Ogawa, T., et al. (1992). Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex. J. Neurol. Sci. 108, 80–87. doi: 10.1016/0022-510X(92)90191-M
Bender, A., and Klopstock, T. (2016). Creatine for neuroprotection in neurodegenerative disease: end of story? Amino Acids 48, 1929–1940. doi: 10.1007/s00726-015-2165-0
Benfeitas, R., Uhlen, M., Nielsen, J., and Mardinoglu, A. (2017). New challenges to study heterogeneity in cancer redox metabolism. Front. Cell Dev. Biol. 5:65. doi: 10.3389/fcell.2017.00065
Bento, C. F., Renna, M., Ghislat, G., Puri, C., Ashkenazi, A., Vicinanza, M., et al. (2016). Mammalian autophagy: how does it work? Annu. Rev. Biochem. 85, 685–713. doi: 10.1146/annurev-biochem-060815-014556
Berggren, K. L., Chen, J., Fox, J., Miller, J., Dodds, L., Dugas, B., et al. (2015). Neonatal iron supplementation potentiates oxidative stress, energetic dysfunction and neurodegeneration in the R6/2 mouse model of Huntington’s disease. Redox. Biol. 4, 363–374. doi: 10.1016/j.redox.2015.02.002
Bidkhori, G., Benfeitas, R., Klevstig, M., Zhang, C., Nielsen, J., Uhlen, M., et al. (2018). Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes. Proc. Natl. Acad. Sci. U.S.A. 115, E11874–E11883. doi: 10.1073/pnas.1807305115
Bortolatto, C. F., Jesse, C. R., Wilhelm, E. A., Chagas, P. M., and Nogueira, C. W. (2013). Organoselenium bis selenide attenuates 3-nitropropionic acid-induced neurotoxicity in rats. Neurotox. Res. 23, 214–224. doi: 10.1007/s12640-012-9336-5
Bortvedt, S. F., McLear, J. A., Messer, A., Ahern-Rindell, A. J., and Wolfgang, W. J. (2010). Cystamine and intrabody co-treatment confers additional benefits in a fly model of Huntington’s disease. Neurobiol. Dis. 40, 130–134. doi: 10.1016/j.nbd.2010.04.007
Brandt, J., Bylsma, F. W., Gross, R., Stine, O. C., Ranen, N., and Ross, C. A. (1996). Trinucleotide repeat length and clinical progression in Huntington’s disease. Neurology 46, 527–531. doi: 10.1212/WNL.46.2.527
Bresciani, A., Missineo, A., Gallo, M., Cerretani, M., Fezzardi, P., Tomei, L., et al. (2017). Nuclear factor (erythroid-derived 2)-like 2 (NRF2) drug discovery: biochemical toolbox to develop NRF2 activators by reversible binding of Kelch-like ECH-associated protein 1 (KEAP1). Arch. Biochem. Biophys. 631, 31–41. doi: 10.1016/j.abb.2017.08.003
Browne, S. E., Bowling, A. C., MacGarvey, U., Baik, M. J., Berger, S. C., Muqit, M. M., et al. (1997). Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann. Neurol. 41, 646–653. doi: 10.1002/ana.410410514
Calkins, M. J., Townsend, J. A., Johnson, D. A., and Johnson, J. A. (2010). Cystamine protects from 3-nitropropionic acid lesioning via induction of nf-e2 related factor 2 mediated transcription. Exp. Neurol. 224, 307–317. doi: 10.1016/j.expneurol.2010.04.008
Carmo, C., Naia, L., Lopes, C., and Rego, A. C. (2018). Mitochondrial dysfunction in huntington’s disease. Adv. Exp. Med. Biol. 1049, 59–83. doi: 10.1007/978-3-319-71779-1_3
Castro, M., Caprile, T., Astuya, A., Millan, C., Reinicke, K., Vera, J. C., et al. (2001). High-affinity sodium-vitamin C co-transporters (SVCT) expression in embryonic mouse neurons. J. Neurochem. 78, 815–823. doi: 10.1046/j.1471-4159.2001.00461.x
Castro, M. A., Beltran, F. A., Brauchi, S., and Concha, I. I. (2009). A metabolic switch in brain: glucose and lactate metabolism modulation by ascorbic acid. J. Neurochem. 110, 423–440. doi: 10.1111/j.1471-4159.2009.06151.x
Chen, C. M., Wu, Y. R., Cheng, M. L., Liu, J. L., Lee, Y. M., Lee, P. W., et al. (2007). Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem. Biophys. Res. Commun. 359, 335–340. doi: 10.1016/j.bbrc.2007.05.093
Chen, J., Marks, E., Lai, B., Zhang, Z., Duce, J. A., Lam, L. Q., et al. (2013). Iron accumulates in huntington’s disease neurons: protection by deferoxamine. PLoS One 8:e77023. doi: 10.1371/journal.pone.0077023
Chongtham, A., and Agrawal, N. (2016). Curcumin modulates cell death and is protective in huntington’s disease model. Sci. Rep. 6:18736. doi: 10.1038/srep18736
Choo, Y. S., Johnson, G. V., MacDonald, M., Detloff, P. J., and Lesort, M. (2004). Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 13, 1407–1420. doi: 10.1093/hmg/ddh162
Choo, Y. S., Mao, Z., Johnson, G. V., and Lesort, M. (2005). Increased glutathione levels in cortical and striatal mitochondria of the R6/2 Huntington’s disease mouse model. Neurosci. Lett. 386, 63–68. doi: 10.1016/j.neulet.2005.05.065
Costa, V., Giacomello, M., Hudec, R., Lopreiato, R., Ermak, G., Lim, D., et al. (2010). Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol. Med. 2, 490–503. doi: 10.1002/emmm.201000102
Covarrubias-Pinto, A., Moll, P., Solis-Maldonado, M., Acuna, A. I., Riveros, A., Miro, M. P., et al. (2015). Beyond the redox imbalance: oxidative stress contributes to an impaired GLUT3 modulation in Huntington’s disease. Free Radic. Biol. Med. 89, 1085–1096. doi: 10.1016/j.freeradbiomed.2015.09.024
Cui, L., Jeong, H., Borovecki, F., Parkhurst, C. N., Tanese, N., and Krainc, D. (2006). Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127, 59–69. doi: 10.1016/j.cell.2006.09.015
Davies, S. W., Turmaine, M., Cozens, B. A., DiFiglia, M., Sharp, A. H., Ross, C. A., et al. (1997). Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548. doi: 10.1016/S0092-8674(00)80513-9
Deng, J., Lu, P. D., Zhang, Y., Scheuner, D., Kaufman, R. J., Sonenberg, N., et al. (2004). Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell Biol. 24, 10161–10168. doi: 10.1128/MCB.24.23.10161-10168.2004
Dexter, D. T., Jenner, P., Schapira, A. H., and Marsden, C. D. (1992). Alterations in levels of iron, ferritin, and other trace metals in neurodegenerative diseases affecting the basal ganglia. the royal kings and queens parkinson’s disease research group. Ann. Neurol. 32, S94–S100. doi: 10.1002/ana.410320716
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
DiGiovanni, L. F., Mocle, A. J., Xia, J., and Truant, R. (2016). Huntingtin N17 domain is a reactive oxygen species sensor regulating huntingtin phosphorylation and localization. Hum. Mol. Genet. 25, 3937–3945. doi: 10.1093/hmg/ddw234
Dinkova-Kostova, A. T., Kostov, R. V., and Kazantsev, A. G. (2018). The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 285, 3576–3590. doi: 10.1111/febs.14379
Dudek, N. L., Dai, Y., and Muma, N. A. (2010). Neuroprotective effects of calmodulin peptide 76-121aa: disruption of calmodulin binding to mutant huntingtin. Brain Pathol. 20, 176–189. doi: 10.1111/j.1750-3639.2008.00258.x
Duraes, F., Pinto, M., and Sousa, E. (2018). Old drugs as new treatments for neurodegenerative diseases. Pharmaceuticals 11:E44. doi: 10.3390/ph11020044
Ellrichmann, G., Petrasch-Parwez, E., Lee, D. H., Reick, C., Arning, L., Saft, C., et al. (2011). Efficacy of fumaric acid esters in the R6/2 and YAC128 models of huntington’s disease. PLoS One 6:e16172. doi: 10.1371/journal.pone.0016172
Farina, F., Lambert, E., Commeau, L., Lejeune, F. X., Roudier, N., Fonte, C., et al. (2017). The stress response factor daf-16/FOXO is required for multiple compound families to prolong the function of neurons with Huntington’s disease. Sci. Rep. 7:4014. doi: 10.1038/s41598-017-04256-w
Fernandez-Estevez, M. A., Casarejos, M. J., Lopez Sendon, J., Garcia Caldentey, J., Ruiz, C., Gomez, A., et al. (2014). Trehalose reverses cell malfunction in fibroblasts from normal and Huntington’s disease patients caused by proteosome inhibition. PLoS One 9:e90202. doi: 10.1371/journal.pone.0090202
Ferrante, R. J., Andreassen, O. A., Dedeoglu, A., Ferrante, K. L., Jenkins, B. G., Hersch, S. M., et al. (2002). Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington’s disease. J. Neurosci. 22, 1592–1599. doi: 10.1523/JNEUROSCI.22-05-01592.2002
Fisher, E. R., and Hayden, M. R. (2014). Multisource ascertainment of Huntington disease in Canada: prevalence and population at risk. Mov. Disord. 29, 105–114. doi: 10.1002/mds.25717
Floyd, R. A., and Carney, J. M. (1992). Free radical damage to protein and DNA: mechanisms involved and relevant observations on brain undergoing oxidative stress. Ann. Neurol. 32, S22–S27. doi: 10.1002/ana.410320706
Fox, J. H., Barber, D. S., Singh, B., Zucker, B., Swindell, M. K., Norflus, F., et al. (2004). Cystamine increases L-cysteine levels in Huntington’s disease transgenic mouse brain and in a PC12 model of polyglutamine aggregation. J. Neurochem. 91, 413–422. doi: 10.1111/j.1471-4159.2004.02726.x
Fox, J. H., Connor, T., Stiles, M., Kama, J., Lu, Z., Dorsey, K., et al. (2011). Cysteine oxidation within N-terminal mutant huntingtin promotes oligomerization and delays clearance of soluble protein. J. Biol. Chem. 286, 18320–18330. doi: 10.1074/jbc.M110.199448
Fox, J. H., Kama, J. A., Lieberman, G., Chopra, R., Dorsey, K., Chopra, V., et al. (2007). Mechanisms of copper ion mediated Huntington’s disease progression. PLoS One 2:e334. doi: 10.1371/journal.pone.0000334
Frederick, N. M., Bertho, J., Patel, K. K., Petr, G. T., Bakradze, E., Smith, S. B., et al. (2014). Dysregulation of system xc(-) expression induced by mutant huntingtin in a striatal neuronal cell line and in R6/2 mice. Neurochem. Int. 76, 59–69. doi: 10.1016/j.neuint.2014.06.017
Gadoth, N., and Goebel, H. H. (2011). Oxidative Stress and Free Radical Damage in Neurology. New York, NY: Humana Press. doi: 10.1007/978-1-60327-514-9
Garcia-Miralles, M., Geva, M., Tan, J. Y., Yusof, N., Cha, Y., Kusko, R., et al. (2017). Early pridopidine treatment improves behavioral and transcriptional deficits in YAC128 Huntington disease mice. JCI Insight 2:95665. doi: 10.1172/jci.insight.95665
Giordano, S., Darley-Usmar, V., and Zhang, J. (2014). Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2, 82–90. doi: 10.1016/j.redox.2013.12.013
Grima, J. C., Daigle, J. G., Arbez, N., Cunningham, K. C., Zhang, K., Ochaba, J., et al. (2017). Mutant huntingtin disrupts the nuclear pore complex. Neuron 94, 93.e6–107e6. doi: 10.1016/j.neuron.2017.03.023
Gu, M., Gash, M. T., Mann, V. M., Javoy-Agid, F., Cooper, J. M., and Schapira, A. H. (1996). Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol. 39, 385–389. doi: 10.1002/ana.410390317
Han, J., and Kaufman, R. J. (2017). Physiological/pathological ramifications of transcription factors in the unfolded protein response. Genes Dev. 31, 1417–1438. doi: 10.1101/gad.297374.117
Harding, R. J., and Tong, Y. F. (2018). Proteostasis in Huntington’s disease: disease mechanisms and therapeutic opportunities. Acta Pharmacol. Sin. 39, 754–769. doi: 10.1038/aps.2018.11
Hersch, S. M., Schifitto, G., Oakes, D., Bredlau, A. L., Meyers, C. M., Nahin, R., et al. (2017). The CREST-E study of creatine for huntington disease: a randomized controlled trial. Neurology 89, 594–601. doi: 10.1212/WNL.0000000000004209
Hetz, C., and Saxena, S. (2017). ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 13, 477–491. doi: 10.1038/nrneurol.2017.99
Hickey, M. A., Zhu, C., Medvedeva, V., Lerner, R. P., Patassini, S., Franich, N. R., et al. (2012). Improvement of neuropathology and transcriptional deficits in CAG 140 knock-in mice supports a beneficial effect of dietary curcumin in Huntington’s disease. Mol. Neurodegener. 7:12. doi: 10.1186/1750-1326-7-12
Hyrskyluoto, A., Pulli, I., Tornqvist, K., Ho, T. H., Korhonen, L., and Lindholm, D. (2013). Sigma-1 receptor agonist PRE084 is protective against mutant huntingtin-induced cell degeneration: involvement of calpastatin and the NF-kappaB pathway. Cell Death Dis. 4:e646. doi: 10.1038/cddis.2013.170
Illarioshkin, S. N., Igarashi, S., Onodera, O., Markova, E. D., Nikolskaya, N. N., Tanaka, H., et al. (1994). Trinucleotide repeat length and rate of progression of Huntington’s disease. Ann. Neurol. 36, 630–635. doi: 10.1002/ana.410360412
Ishimoto, T., Nagano, O., Yae, T., Tamada, M., Motohara, T., Oshima, H., et al. (2011). CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 19, 387–400. doi: 10.1016/j.ccr.2011.01.038
Jang, M., and Cho, I. H. (2016). Sulforaphane ameliorates 3-nitropropionic acid-induced striatal toxicity by activating the keap1-Nrf2-ARE pathway and inhibiting the MAPKs and NF-kappaB pathways. Mol. Neurobiol. 53, 2619–2635. doi: 10.1007/s12035-015-9230-2
Jenkins, B. G., Koroshetz, W. J., Beal, M. F., and Rosen, B. R. (1993). Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology 43, 2689–2695. doi: 10.1212/WNL.43.12.2689
Jin, Y. N., Yu, Y. V., Gundemir, S., Jo, C., Cui, M., Tieu, K., et al. (2013). Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full-length mutant huntingtin. PLoS One 8:e57932. doi: 10.1371/journal.pone.0057932
Jones, D. P. (2006). Redefining oxidative stress. Antioxid. Redox. Signal. 8, 1865–1879. doi: 10.1089/ars.2006.8.1865
Karpuj, M. V., Becher, M. W., Springer, J. E., Chabas, D., Youssef, S., Pedotti, R., et al. (2002). Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat. Med. 8, 143–149. doi: 10.1038/nm0202-143
Kasparova, S., Sumbalova, Z., Bystricky, P., Kucharska, J., Liptaj, T., Mlynarik, V., et al. (2006). Effect of coenzyme Q10 and vitamin E on brain energy metabolism in the animal model of Huntington’s disease. Neurochem. Int. 48, 93–99. doi: 10.1016/j.neuint.2005.09.002
Kensler, T. W., Wakabayashi, N., and Biswal, S. (2007). Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 47, 89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046
Kim, G. H., Kim, J. E., Rhie, S. J., and Yoon, S. (2015). The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 24, 325–340. doi: 10.5607/en.2015.24.4.325
Klionsky, D. J., and Emr, S. D. (2000). Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721. doi: 10.1126/science.290.5497.1717
Kolobkova, Y. A., Vigont, V. A., Shalygin, A. V., and Kaznacheyeva, E. V. (2017). Huntington’s disease: calcium dyshomeostasis and pathology models. Acta Nat. 9, 34–46.
Kovtun, I. V., Liu, Y., Bjoras, M., Klungland, A., Wilson, S. H., and McMurray, C. T. (2007). OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 447, 447–452. doi: 10.1038/nature05778
Kroemer, G., Marino, G., and Levine, B. (2010). Autophagy and the integrated stress response. Mol. Cell 40, 280–293. doi: 10.1016/j.molcel.2010.09.023
Kumar, P., Padi, S. S., Naidu, P. S., and Kumar, A. (2007). Possible neuroprotective mechanisms of curcumin in attenuating 3-nitropropionic acid-induced neurotoxicity. Methods Find. Exp. Clin. Pharmacol. 29, 19–25. doi: 10.1358/mf.2007.29.1.1063492
Kusko, R., Dreymann, J., Ross, J., Cha, Y., Escalante-Chong, R., Garcia-Miralles, M., et al. (2018). Large-scale transcriptomic analysis reveals that pridopidine reverses aberrant gene expression and activates neuroprotective pathways in the YAC128 HD mouse. Mol. Neurodegener. 13:25. doi: 10.1186/s13024-018-0259-3
Leidal, A. M., Levine, B., and Debnath, J. (2018). Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 20, 1338–1348. doi: 10.1038/s41556-018-0235-8
Li, X., Valencia, A., Sapp, E., Masso, N., Alexander, J., Reeves, P., et al. (2010). Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington’s disease. J. Neurosci. 30, 4552–4561. doi: 10.1523/JNEUROSCI.5865-09.2010
Liu, Y., Hettinger, C. L., Zhang, D., Rezvani, K., Wang, X., and Wang, H. (2014). Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington’s disease. J. Neurochem. 129, 539–547. doi: 10.1111/jnc.12647
Lu, X. H., Mattis, V. B., Wang, N., Al-Ramahi, I., van den Berg, N., Fratantoni, S. A., et al. (2014). Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci. Transl. Med. 6:268ra178. doi: 10.1126/scitranslmed.3010523
Lu, Z., Marks, E., Chen, J., Moline, J., Barrows, L., Raisbeck, M., et al. (2014). Altered selenium status in Huntington’s disease: neuroprotection by selenite in the N171-82Q mouse model. Neurobiol. Dis. 71, 34–42. doi: 10.1016/j.nbd.2014.06.022
Luis-Garcia, E. R., Limon-Pacheco, J. H., Serrano-Garcia, N., Hernandez-Perez, A. D., Pedraza-Chaverri, J., and Orozco-Ibarra, M. (2017). Sulforaphane prevents quinolinic acid-induced mitochondrial dysfunction in rat striatum. J. Biochem. Mol. Toxicol. 31:e21837. doi: 10.1002/jbt.21837
Luthi-Carter, R., Hanson, S. A., Strand, A. D., Bergstrom, D. A., Chun, W., Peters, N. L., et al. (2002). Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum. Mol. Genet. 11, 1911–1926. doi: 10.1093/hmg/11.17.1911
MacDonald, M. E., Ambrose, C. M., Duyao, M. P., Myers, R. H., Lin, C., Srinidhi, L., et al. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The huntington’s disease collaborative research group. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585-E
Maiuri, T., Mocle, A. J., Hung, C. L., Xia, J., van Roon-Mom, W. M., and Truant, R. (2017). Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum. Mol. Genet. 26, 395–406. doi: 10.1093/hmg/ddw395
Mangiarini, L., Sathasivam, K., Mahal, A., Mott, R., Seller, M., and Bates, G. P. (1997). Instability of highly expanded CAG repeats in mice transgenic for the Huntington’s disease mutation. Nat. Genet. 15, 197–200. doi: 10.1038/ng0297-197
Mason, R. P., Casu, M., Butler, N., Breda, C., Campesan, S., Clapp, J., et al. (2013). Glutathione peroxidase activity is neuroprotective in models of Huntington’s disease. Nat. Genet. 45, 1249–1254. doi: 10.1038/ng.2732
McColgan, P., and Tabrizi, S. J. (2018). Huntington’s disease: a clinical review. Eur. J. Neurol. 25, 24–34. doi: 10.1111/ene.13413
McGarry, A., McDermott, M., Kieburtz, K., de Blieck, E. A., Beal, F., Marder, K., et al. (2017). A randomized, double-blind, placebo-controlled trial of coenzyme Q10 in Huntington disease. Neurology 88, 152–159. doi: 10.1212/WNL.0000000000003478
Mehrotra, A., Sood, A., and Sandhir, R. (2015). Mitochondrial modulators improve lipid composition and attenuate memory deficits in experimental model of Huntington’s disease. Mol. Cell Biochem. 410, 281–292. doi: 10.1007/s11010-015-2561-5
Meister, A., and Anderson, M. E. (1983). Glutathione. Annu. Rev. Biochem. 52, 711–760. doi: 10.1146/annurev.bi.52.070183.003431
Miller, J. R., Lo, K. K., Andre, R., Hensman Moss, D. J., Trager, U., Stone, T. C., et al. (2016). RNA-Seq of Huntington’s disease patient myeloid cells reveals innate transcriptional dysregulation associated with proinflammatory pathway activation. Hum. Mol. Genet. 25, 2893–2904. doi: 10.1093/hmg/ddw142
Mollersen, L.,Rowe, A. D., Bjolgerud, A., Holm, I., Tveteras, L., et al. (2016). Effects of anthocyanins on CAG repeat instability and behaviour in huntington’s disease R6/1 Mice. PLoS Curr. 8. doi: 10.1371/currents.hd.58d04209ab6d5de0844db7ef5628ff67
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Mustafa, A. K., Gadalla, M. M., Sen, N., Kim, S., Mu, W., Gazi, S. K., et al. (2009). H2S signals through protein S-sulfhydration. Sci. Signal. 2:ra72. doi: 10.1126/scisignal.2000464
Naia, L., Rosenstock, T. R., Oliveira, A. M., Oliveira-Sousa, S. I., Caldeira, G. L., Carmo, C., et al. (2017). Comparative mitochondrial-based protective effects of resveratrol and nicotinamide in huntington’s disease models. Mol. Neurobiol. 54, 5385–5399. doi: 10.1007/s12035-016-0048-3
Nakamura, T., Cieplak, P., Cho, D. H., Godzik, A., and Lipton, S. A. (2010). S-nitrosylation of Drp1 links excessive mitochondrial fission to neuronal injury in neurodegeneration. Mitochondrion 10, 573–578. doi: 10.1016/j.mito.2010.04.007
Padh, H. (1990). Cellular functions of ascorbic acid. Biochem. Cell Biol. 68, 1166–1173. doi: 10.1139/o90-173
Pahl, H. L., and Baeuerle, P. A. (1995). A novel signal transduction pathway from the endoplasmic reticulum to the nucleus is mediated by transcription factor NF-kappa B. EMBO J. 14, 2580–2588. doi: 10.1002/j.1460-2075.1995.tb07256.x
Paul, B. D., Sbodio, J. I., and Snyder, S. H. (2018). Cysteine metabolism in neuronal redox homeostasis. Trends Pharmacol. Sci. 39, 513–524. doi: 10.1016/j.tips.2018.02.007
Paul, B. D., Sbodio, J. I., Xu, R., Vandiver, M. S., Cha, J. Y., Snowman, A. M., et al. (2014). Cystathionine gamma-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature 509, 96–100. doi: 10.1038/nature13136
Paul, B. D., and Snyder, S. H. (2012). H(2)S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 13, 499–507. doi: 10.1038/nrm3391
Paul, B. D., and Snyder, S. H. (2014). Neurodegeneration in Huntington’s disease involves loss of cystathionine gamma-lyase. Cell Cycle 13, 2491–2493. doi: 10.4161/15384101.2014.950538
Paul, B. D., and Snyder, S. H. (2015a). H2S: a novel gasotransmitter that signals by sulfhydration. Trends Biochem. Sci. 40, 687–700. doi: 10.1016/j.tibs.2015.08.007
Paul, B. D., and Snyder, S. H. (2015b). Modes of physiologic H2S signaling in the brain and peripheral tissues. Antioxid. Redox. Signal. 22, 411–423. doi: 10.1089/ars.2014.5917
Paul, B. D., and Snyder, S. H. (2018). Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 149, 101–109. doi: 10.1016/j.bcp.2017.11.019
Pena-Sanchez, M., Riveron-Forment, G., Zaldivar-Vaillant, T., Soto-Lavastida, A., Borrero-Sanchez, J., Lara-Fernandez, G., et al. (2015). Association of status redox with demographic, clinical and imaging parameters in patients with Huntington’s disease. Clin. Biochem. 48, 1258–1263. doi: 10.1016/j.clinbiochem.2015.06.014
Peng, T. I., and Jou, M. J. (2010). Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 1201, 183–188. doi: 10.1111/j.1749-6632.2010.05634.x
Peyser, C. E., Folstein, M., Chase, G. A., Starkstein, S., Brandt, J., Cockrell, J. R., et al. (1995). Trial of d-alpha-tocopherol in Huntington’s disease. Am. J. Psychiatry 152, 1771–1775. doi: 10.1176/ajp.152.12.1771
Polidori, M. C., Mecocci, P., Browne, S. E., Senin, U., and Beal, M. F. (1999). Oxidative damage to mitochondrial DNA in huntington’s disease parietal cortex. Neurosci. Lett. 272, 53–56. doi: 10.1016/S0304-3940(99)00578-9
Polyzos, A., Holt, A., Brown, C., Cosme, C., Wipf, P., Gomez-Marin, A., et al. (2016). Mitochondrial targeting of XJB-5-131 attenuates or improves pathophysiology in HdhQ150 animals with well-developed disease phenotypes. Hum. Mol. Genet. 25, 1792–1802. doi: 10.1093/hmg/ddw051
Polyzos, A. A., Wood, N. I., Williams, P., Wipf, P., Morton, A. J., and McMurray, C. T. (2018). XJB-5-131-mediated improvement in physiology and behaviour of the R6/2 mouse model of Huntington’s disease is age- and sex- dependent. PLoS One 13:e0194580. doi: 10.1371/journal.pone.0194580
Raha, S., and Robinson, B. H. (2000). Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 25, 502–508. doi: 10.1016/S0968-0004(00)01674-1
Rebec, G. V., Barton, S. J., Marseilles, A. M., and Collins, K. (2003). Ascorbate treatment attenuates the Huntington behavioral phenotype in mice. Neuroreport 14, 1263–1265. doi: 10.1097/01.wnr.0000081868.45938.12
Rebec, G. V., Conroy, S. K., and Barton, S. J. (2006). Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: variations with behavioral state and repeated ascorbate treatment. Neuroscience 137, 327–336. doi: 10.1016/j.neuroscience.2005.08.062
Reddy, P. H., and Shirendeb, U. P. (2012). Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington’s disease. Biochim. Biophys. Acta 1822, 101–110. doi: 10.1016/j.bbadis.2011.10.016
Redmann, M., Benavides, G. A., Berryhill, T. F., Wani, W. Y., Ouyang, X., Johnson, M. S., et al. (2017). Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox. Biol. 11, 73–81. doi: 10.1016/j.redox.2016.11.004
Reijonen, S., Kukkonen, J. P., Hyrskyluoto, A., Kivinen, J., Kairisalo, M., Takei, N., et al. (2010). Downregulation of NF-kappaB signaling by mutant huntingtin proteins induces oxidative stress and cell death. Cell Mol. Life Sci. 67, 1929–1941. doi: 10.1007/s00018-010-0305-y
Reijonen, S., Putkonen, N., Norremolle, A., Lindholm, D., and Korhonen, L. (2008). Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp. Cell Res. 314, 950–960. doi: 10.1016/j.yexcr.2007.12.025
Reilmann, R., McGarry, A., Grachev, I. D., Savola, J. M., Borowsky, B., Eyal, E., et al. (2019). Safety and efficacy of pridopidine in patients with Huntington’s disease (PRIDE-HD): a phase 2, randomised, placebo-controlled, multicentre, dose-ranging study. Lancet Neurol. 18, 165–176. doi: 10.1016/S1474-4422(18)30391-0
Ribeiro, M., Rosenstock, T. R., Cunha-Oliveira, T., Ferreira, I. L., Oliveira, C. R., and Rego, A. C. (2012). Glutathione redox cycle dysregulation in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 53, 1857–1867. doi: 10.1016/j.freeradbiomed.2012.09.004
Ribeiro, M., Silva, A. C., Rodrigues, J., Naia, L., and Rego, A. C. (2013). Oxidizing effects of exogenous stressors in Huntington’s disease knock-in striatal cells–protective effect of cystamine and creatine. Toxicol. Sci. 136, 487–499. doi: 10.1093/toxsci/kft199
Ross, C. A., Aylward, E. H., Wild, E. J., Langbehn, D. R., Long, J. D., Warner, J. H., et al. (2014). Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 10, 204–216. doi: 10.1038/nrneurol.2014.24
Rotblat, B., Southwell, A. L., Ehrnhoefer, D. E., Skotte, N. H., Metzler, M., Franciosi, S., et al. (2014). HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc. Natl. Acad. Sci. U.S.A. 111, 3032–3037. doi: 10.1073/pnas.1314421111
Sandhir, R., and Mehrotra, A. (2013). Quercetin supplementation is effective in improving mitochondrial dysfunctions induced by 3-nitropropionic acid: implications in Huntington’s disease. Biochim. Biophys. Acta 1832, 421–430. doi: 10.1016/j.bbadis.2012.11.018
Sandhir, R., Mehrotra, A., and Kamboj, S. S. (2010). Lycopene prevents 3-nitropropionic acid-induced mitochondrial oxidative stress and dysfunctions in nervous system. Neurochem. Int. 57, 579–587. doi: 10.1016/j.neuint.2010.07.005
Sandhir, R., Sood, A., Mehrotra, A., and Kamboj, S. S. (2012). N-Acetylcysteine reverses mitochondrial dysfunctions and behavioral abnormalities in 3-nitropropionic acid-induced Huntington’s disease. Neurodegener. Dis. 9, 145–157. doi: 10.1159/000334273
Sarkar, S., Davies, J. E., Huang, Z., Tunnacliffe, A., and Rubinsztein, D. C. (2007). Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 282, 5641–5652. doi: 10.1074/jbc.M609532200
Sbodio, J. I., Snyder, S. H., and Paul, B. D. (2016). Transcriptional control of amino acid homeostasis is disrupted in Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 113, 8843–8848. doi: 10.1073/pnas.1608264113
Sbodio, J. I., Snyder, S. H., and Paul, B. D. (2018a). Golgi stress response reprograms cysteine metabolism to confer cytoprotection in Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 115, 780–785. doi: 10.1073/pnas.1717877115
Sbodio, J. I., Snyder, S. H., and Paul, B. D. (2018b). Redox mechanisms in neurodegeneration: from disease outcomes to therapeutic opportunities. Antioxid. Redox. Signal. 30, 1450–1499. doi: 10.1089/ars.2017.7321
Sbodio, J. I., Snyder, S. H., and Paul, B. D. (2018c). Regulators of the transsulfuration pathway. Br. J. Pharmacol. 176, 583–593. doi: 10.1111/bph.14446
Sen, N., Paul, B. D., Gadalla, M. M., Mustafa, A. K., Sen, T., Xu, R., et al. (2012). Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol. Cell 45, 13–24. doi: 10.1016/j.molcel.2011.10.021
Shiloh, Y., and Ziv, Y. (2013). The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 14, 197–210. doi: 10.1038/nrm3546
Shirendeb, U., Reddy, A. P., Manczak, M., Calkins, M. J., Mao, P., Tagle, D. A., et al. (2011). Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum. Mol. Genet. 20, 1438–1455. doi: 10.1093/hmg/ddr024
Sidhu, A., Diwan, V., Kaur, H., Bhateja, D., Singh, C. K., Sharma, S., et al. (2018). Nicotinamide reverses behavioral impairments and provides neuroprotection in 3-nitropropionic acid induced animal model ofHuntington’s disease: implication of oxidative stress- poly(ADP- ribose) polymerase pathway. Metab. Brain Dis. 33, 1911–1921. doi: 10.1007/s11011-018-0297-0
Sies, H., Berndt, C., and Jones, D. P. (2017). Oxidative stress. Annu. Rev. Biochem. 86, 715–748. doi: 10.1146/annurev-biochem-061516-045037
Simmons, D. A., Casale, M., Alcon, B., Pham, N., Narayan, N., and Lynch, G. (2007). Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia 55, 1074–1084. doi: 10.1002/glia.20526
Smith, K. M., Matson, S., Matson, W. R., Cormier, K., Del Signore, S. J., Hagerty, S. W., et al. (2006). Dose ranging and efficacy study of high-dose coenzyme Q10 formulations in Huntington’s disease mice. Biochim. Biophys. Acta 1762, 616–626. doi: 10.1016/j.bbadis.2006.03.004
Song, W., Chen, J., Petrilli, A., Liot, G., Klinglmayr, E., Zhou, Y., et al. (2011). Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 17, 377–382. doi: 10.1038/nm.2313
St-Pierre, J., Drori, S., Uldry, M., Silvaggi, J. M., Rhee, J., Jager, S., et al. (2006). Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127, 397–408. doi: 10.1016/j.cell.2006.09.024
Suganya, S. N., and Sumathi, T. (2017). Effect of rutin against a mitochondrial toxin, 3-nitropropionicacid induced biochemical, behavioral and histological alterations-a pilot study on Huntington’s disease model in rats. Metab. Brain Dis. 32, 471–481. doi: 10.1007/s11011-016-9929-4
Tanaka, M., Machida, Y., Niu, S., Ikeda, T., Jana, N. R., Doi, H., et al. (2004). Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 10, 148–154. doi: 10.1038/nm985
Tang, T. S., Tu, H., Chan, E. Y., Maximov, A., Wang, Z., Wellington, C. L., et al. (2003). Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39, 227–239. doi: 10.1016/S0896-6273(03)00366-0
Trachootham, D., Alexandre, J., and Huang, P. (2009). Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discov. 8, 579–591. doi: 10.1038/nrd2803
Tsvetkov, A. S., Arrasate, M., Barmada, S., Ando, D. M., Sharma, P., Shaby, B. A., et al. (2013). Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat. Chem. Biol. 9, 586–592. doi: 10.1038/nchembio.1308
Tunez, I., Montilla, P., Del Carmen Munoz, M., Feijoo, M., and Salcedo, M. (2004). Protective effect of melatonin on 3-nitropropionic acid-induced oxidative stress in synaptosomes in an animal model of Huntington’s disease. J. Pineal. Res. 37, 252–256. doi: 10.1111/j.1600-079X.2004.00163.x
Turanli, B., Grotli, M., Boren, J., Nielsen, J., Uhlen, M., Arga, K. Y., et al. (2018). Drug repositioning for effective prostate cancer treatment. Front. Physiol. 9:500. doi: 10.3389/fphys.2018.00500
Underwood, B. R., Imarisio, S., Fleming, A., Rose, C., Krishna, G., Heard, P., et al. (2010). Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Hum. Mol. Genet. 19, 3413–3429. doi: 10.1093/hmg/ddq253
Van Raamsdonk, J. M., Pearson, J., Bailey, C. D., Rogers, D. A., Johnson, G. V., Hayden, M. R., et al. (2005). Cystamine treatment is neuroprotective in the YAC128 mouse model of Huntington disease. J. Neurochem. 95, 210–220. doi: 10.1111/j.1471-4159.2005.03357.x
Varshavsky, A. (1998). Codominant interference, antieffectors, and multitarget drugs. Proc. Natl. Acad. Sci. U.S.A. 95, 2094–2099. doi: 10.1073/pnas.95.5.2094
Vucetic, M., Cormerais, Y., Parks, S. K., and Pouyssegur, J. (2017). The central role of amino acids in cancer redox homeostasis: vulnerability points of the cancer redox code. Front. Oncol. 7:319. doi: 10.3389/fonc.2017.00319
Wang, J. Q., Chen, Q., Wang, X., Wang, Q. C., Wang, Y., Cheng, H. P., et al. (2013). Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. J. Biol. Chem. 288, 3070–3084. doi: 10.1074/jbc.M112.407726
Wang, X., Sirianni, A., Pei, Z., Cormier, K., Smith, K., Jiang, J., et al. (2011). The melatonin MT1 receptor axis modulates mutant Huntingtin-mediated toxicity. J. Neurosci. 31, 14496–14507. doi: 10.1523/JNEUROSCI.3059-11.2011
Wilson, J. X., Peters, C. E., Sitar, S. M., Daoust, P., and Gelb, A. W. (2000). Glutamate stimulates ascorbate transport by astrocytes. Brain Res. 858, 61–66. doi: 10.1016/S0006-8993(99)02433-6
Wondrak, G. T. (2009). Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxid. Redox. Signal. 11, 3013–3069. doi: 10.1089/ARS.2009.2541
Wright, D. J., Gray, L. J., Finkelstein, D. I., Crouch, P. J., Pow, D., Pang, T. Y., et al. (2016). N-acetylcysteine modulates glutamatergic dysfunction and depressive behavior in Huntington’s disease. Hum. Mol. Genet. 25, 2923–2933. doi: 10.1093/hmg/ddw144
Wright, D. J., Renoir, T., Smith, Z. M., Frazier, A. E., Francis, P. S., Thorburn, D. R., et al. (2015). N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl. Psychiatry 5:e492. doi: 10.1038/tp.2014.131
Xun, Z., Rivera-Sanchez, S., Ayala-Pena, S., Lim, J., Budworth, H., Skoda, E. M., et al. (2012). Targeting of XJB-5-131 to mitochondria suppresses oxidative DNA damage and motor decline in a mouse model of Huntington’s disease. Cell Rep. 2, 1137–1142. doi: 10.1016/j.celrep.2012.10.001
Yang, L., Calingasan, N. Y., Wille, E. J., Cormier, K., Smith, K., Ferrante, R. J., et al. (2009). Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J. Neurochem. 109, 1427–1439. doi: 10.1111/j.1471-4159.2009.06074.x
Keywords: Huntington’s disease, oxidative stress, redox signaling, cysteine, mitochondria
Citation: Paul BD and Snyder SH (2019) Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications. Front. Mol. Neurosci. 12:68. doi: 10.3389/fnmol.2019.00068
Received: 29 December 2018; Accepted: 04 March 2019;
Published: 19 March 2019.
Edited by:
Daniela De Zio, Danish Cancer Society, DenmarkReviewed by:
Dan Lindholm, University of Helsinki, FinlandMauro Cozzolino, Istituto di Farmacologia Traslazionale (IFT), Italy
Copyright © 2019 Paul and Snyder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bindu D. Paul, YnBhdWw4QGpobWkuZWR1