 Hannah K. Shorrock
Hannah K. Shorrock Thomas H. Gillingwater
Thomas H. Gillingwater Ewout J. N. Groen
Ewout J. N. Groen- 1Edinburgh Medical School: Biomedical Sciences, The University of Edinburgh, Edinburgh, United Kingdom
- 2Euan MacDonald Centre for Motor Neurone Disease Research, The University of Edinburgh, Edinburgh, United Kingdom
Activation of skeletal muscle in response to acetylcholine release from the neuromuscular junction triggered by motor neuron firing forms the basis of all mammalian locomotion. Intricate feedback and control mechanisms, both from within the central nervous system and from sensory organs in the periphery, provide essential inputs that regulate and finetune motor neuron activity. Interestingly, in motor neuron diseases, such as spinal muscular atrophy (SMA), pathological studies in patients have identified alterations in multiple parts of the sensory-motor system. This has stimulated significant research efforts across a range of different animal models of SMA in order to understand these defects and their contribution to disease pathogenesis. Several recent studies have demonstrated that defects in sensory components of the sensory-motor system contribute to dysfunction of motor neurons early in the pathogenic process. In this review, we provide an overview of these findings, with a specific focus on studies that have provided mechanistic insights into the molecular processes that underlie dysfunction of the sensory-motor system in SMA. These findings highlight the role that cell types other than motor neurons play in SMA pathogenesis, and reinforce the need for therapeutic interventions that target and rescue the wide array of defects that occur in SMA.
Introduction
Lower motor neurons, whose cell bodies are resident in the ventral gray horn of spinal cord, represent the “final common pathway” (Sherrington, 1906) through which neuronal activity has to pass in order to generate the contraction of skeletal muscle required for movement. Whilst a large proportion of the synaptic inputs controlling lower motor neuron function arise from descending pathways, including upper motor neurons, pioneering work begun by Sir John Eccles and colleagues in the 1950s has revealed the additional influence of synaptic inputs arising from proprioceptive sensory neurons (including group Ia afferents), whose cell bodies are located in the dorsal root ganglia (DRG) (Eccles et al., 1957; Brown, 1981; Mears and Frank, 1997). These sensory inputs have been shown to form monosynaptic connections with lower motor neurons (Figure 1), whereby they can regulate motor neuron firing patterns as part of the monosynaptic spinal stretch reflex arc as well as in other locomotor behaviors (Rossignol et al., 2006).
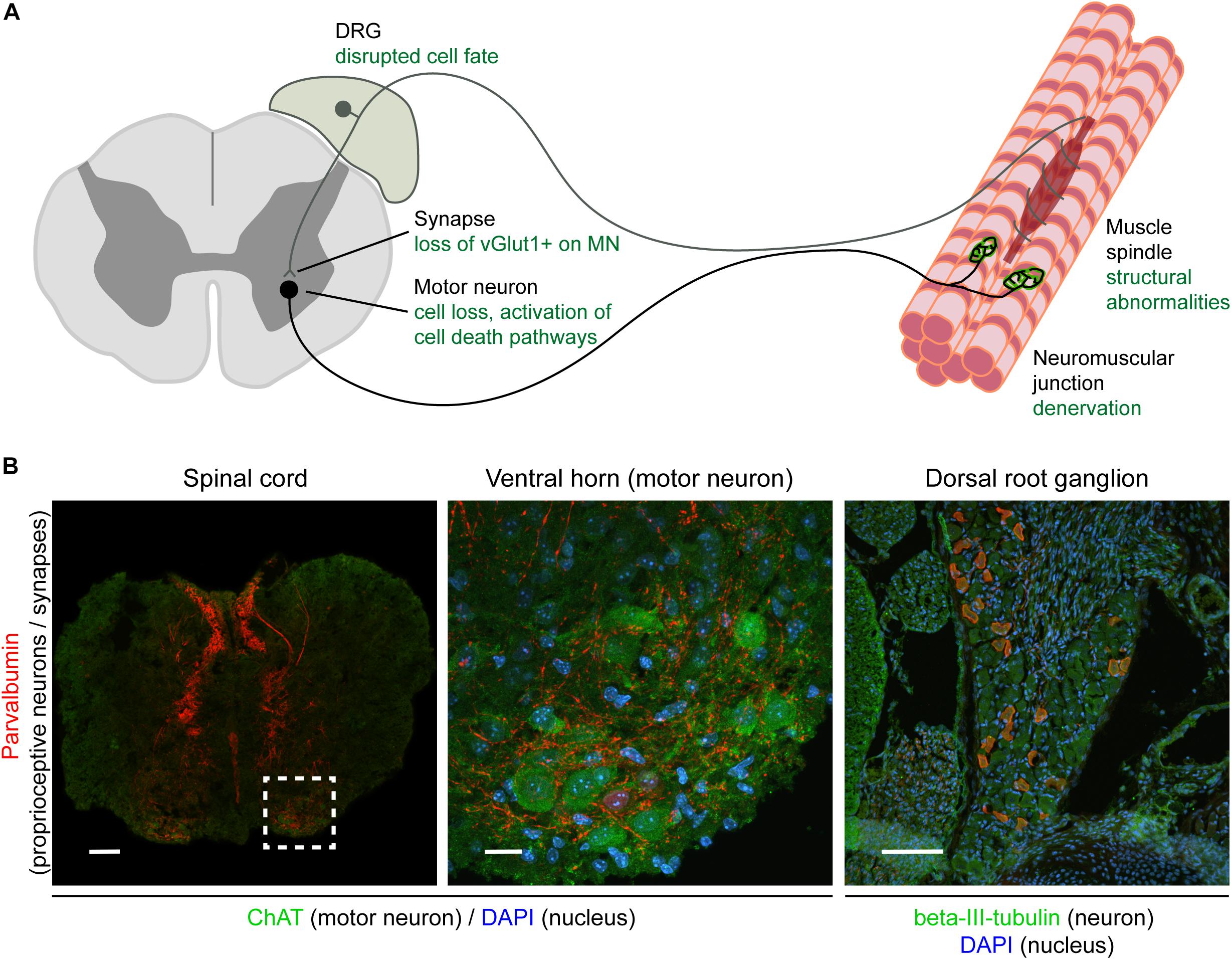
Figure 1. Overview of the sensory-motor system and pathologies observed in SMA. Schematic (A) and immunohistochemical (B) representation of the sensory-motor system, focusing on structures, cell types, and subcellular compartments that have been implicated in SMA pathogenesis. In (A), the text in green indicates the primary pathological changes occurring in specific cell types or in specific subcellular compartments. In (B), immunofluorescence was used to label parvalbumin-expressing cells and projections (proprioceptive neurons; red), neuronal cell bodies (ChAT for motor neurons, beta-III-tubulin for DRG cell bodies; green), and nuclei (DAPI; blue). Note that only a subgroup of DRG cell bodies express parvalbumin and that other markers such as NF200+ (mechano- and proprioceptive) and peripherin+ (nociceptive) can also be used to identify other types of sensory neurons. Scale bars: 100 μm (spinal cord and DRG); 20 μm (motor neuron). DRG, dorsal root ganglion; ChAT, acetylcholine transferase; DAPI, 4,6-diamidino-2-phenylindole; vGlut1, vesicular glutamate transporter 1; NF200, neurofilament heavy polypeptide (200 kDa).
Recent work has begun to uncover the complex molecular pathways regulating the formation and maintenance of such sensory-motor connectivity in the spinal cord, highlighting key roles for pathways incorporating Sema3e–Plxnd1 signaling (Pecho-Vrieseling et al., 2009) and the RNaseIII protein Dicer (Imai et al., 2016). Moreover, several studies have highlighted how disruption of sensory-motor connectivity can result from perturbations in genes and proteins underlying a diverse range of neurodegenerative conditions, including; amyotrophic lateral sclerosis (ALS) (Jiang et al., 2009), post-polio muscular atrophy (PPMA) (Soliven and Maselli, 1992), and spinal muscular atrophy (SMA) (see below). In this review we provide an overview of the contributions that sensory-motor connectivity defects make to the pathogenesis of SMA, incorporating new insights into underlying molecular pathways that suggest the existence of common mechanisms across diverse neuromuscular conditions.
Sensory-Motor Defects in Spinal Muscular Atrophy (SMA)
SMA is a hereditary form of motor neuron disease, characterized by degeneration and death of lower (alpha) motor neurons in the ventral horn of spinal cord (Lunn and Wang, 2008; Groen et al., 2018b). This leads to progressive proximal muscle weakness, atrophy and, in severe cases, paralysis and death. SMA is caused by homozygous deletion of, or other deleterious variants in, the SMN1 gene, leading to a significant reduction in the expression of full-length survival motor neuron protein (SMN). SMN is a ubiquitously expressed protein that has been implicated in numerous cellular processes, including snRNP biogenesis, cytoskeletal dynamics, protein homeostasis, and mitochondrial function and is required for cellular survival. A detailed discussion of the cellular roles of SMN can be found in a number of recent review papers [for example (Hosseinibarkooie et al., 2017; Singh et al., 2017; Chaytow et al., 2018)].
In line with the ubiquitous expression of SMN, SMA is not solely a disease of the lower motor neuron. For example, it is now known that multiple different organ systems and cell types are affected in SMA (Nash et al., 2016), including defects in the heart, vasculature and skeletal muscles (Hamilton and Gillingwater, 2013; Shababi et al., 2014). Alongside these systemic pathologies, defects in sensory neurons have been reported in SMA patients, including abnormal sensory conduction (Duman et al., 2013; Yonekawa et al., 2013) or complete absence of sensory nerve action potentials (Yuan and Jiang, 2015; Reid et al., 2016), along with axonal degeneration and loss of myelinated fibers within sensory nerves (Korinthenberg et al., 1997; Omran et al., 1998; Rudnik-Schoneborn et al., 2003). Depending on the type of SMA, varying structural abnormalities in muscle spindles have been described, although some of these findings are contradictory and would require further investigation (Marshall and Duchen, 1975; Bobele et al., 1996; Kararizou et al., 2006). Several studies conducted on presumed SMA cases (patients diagnosed with SMA before genetic testing was available for the disease) also identified degeneration of sensory nerves, glial bundles within dorsal roots, ballooned neurons and chromatolysis within the DRG (Marshall and Duchen, 1975; Carpenter et al., 1978; Shishikura et al., 1983; Murayama et al., 1991). Importantly, reduced synaptophysin expression has been observed adjacent to anterior horn neuron cell bodies in SMA patients, indicating a reduction in the number of or disconnection of afferent nerve fibers, including sensory synapses (Ikemoto et al., 1996). Not only do these studies demonstrate defects within the sensory neurons themselves but they also demonstrate the possibility of alterations and lack of connectivity at both extremities of the sensory neuron.
Sensory-Motor Defects in Mouse Models of SMA
The SMA research field has benefited greatly from the availability of a number of animal models that mimic key pathological features of SMA, such as motor neuron death, degeneration of the neuromuscular junction and organ pathology (Hamilton and Gillingwater, 2013; Edens et al., 2015). Indeed, model systems of SMA also recapitulate core defects of the sensory nervous system seen in SMA patients. For example, sensory neurons cultured from a severe mouse model of SMA show defects in neurite outgrowth and growth cone morphology, along with a reduction of beta-actin protein and mRNA in growth cones (Jablonka et al., 2006) – a phenotype that is similar to that observed in SMN deficient motor neurons (Rossoll et al., 2003). In mouse models of SMA, sensory neurons are smaller, consistent with an appearance of overall reduced size of DRGs (Shorrock et al., 2018b). Moreover, the ratio of different subtypes of sensory neurons in SMA DRG is altered (see Section “Molecular Mechanisms Associated With Sensory Neuron Dysfunction in SMA and Related Conditions”) (Shorrock et al., 2018b). It has also been shown that there is a reduction in myelinated dorsal root axons in SMA mice compared to controls (Ling et al., 2010) and a reduction of sensory fibers passing into the ventral horn in SMA mice (Mentis et al., 2011).
Importantly, in line with what has been observed in SMA patients, these structural defects occurring in sensory neurons have also been associated with defects at the level of synapses formed by sensory neurons in SMA mice (Fletcher and Mentis, 2016). The total number of synapses onto motor neurons in the lumbar region of the spinal cord is significantly reduced in mouse models of SMA (Ling et al., 2010). The largest contribution to this overall reduction of sensory synapses comes from a reduction in the number of vGlut1-positive synapses which primarily originate from proprioceptive neurons. This central feature of sensory-motor pathology has now been observed across multiple different SMA mouse models, including the commonly used Taiwanese and delta7 models, suggesting that it is a conserved feature of the disease (Ling et al., 2010; Mentis et al., 2011; Shorrock et al., 2018b). Within the lumbar region of spinal cord, the reduction in proprioceptive synapses onto motor neurons is most severe at the level of L1 motor neurons and medial L5 motor neurons (Mentis et al., 2011). Interestingly, this reduction is observed presymptomatically, (Mentis et al., 2011; Fletcher et al., 2017) and is associated with significant functional deficits (Fletcher et al., 2017). Thus, sensory-motor connectivity defects reported in SMA mice consistently affect DRGs and motor neurons related to the body regions primarily affected in SMA patients, suggesting that defects occurring in SMA animal models are comparable to sensory-motor pathology observed in patients (Figure 1).
SMN Expression Is Required Throughout the Sensory-Motor System
Whilst motor neuron death is central to the pathogenesis of SMA, in mouse models motor neuron death is preceded by dysfunction of motor neuron and neuromuscular transmission characterized by hyperexcitability, increased input resistance, and decreased synaptic efficacy (Ling et al., 2010; Mentis et al., 2011; Fletcher et al., 2017). These functional defects, in combination with pathological findings that show sensory neuron and synapse loss in SMA patients and mouse models, indicate a complex interplay between the various components of the sensory-motor system eventually leading to motor neuron defects and death.
As loss of SMN expression is central to SMA pathogenesis, recent research has aimed to determine the requirements for SMN expression in the sensory-motor system. In wild-type mice, SMN is expressed throughout all components of the sensory-motor system, including DRGs (Shorrock et al., 2018b), muscle, brain and spinal cord (Lee et al., 2012; Groen et al., 2018a), as well as peripheral nerves (Hunter et al., 2016). In Drosophila models of SMA, pan-neuronal restoration of SMN rescues locomotion and motor rhythm phenotypes (Imlach et al., 2012). In mouse models of SMA, pan-neuronal SMN restoration completely rescues motor neuron numbers and the number of vGlut1+ synapses onto motor neurons (Lee et al., 2012). To explore this phenomenon further, several research groups have investigated the requirements of SMN expression by restoring SMN in specific cell types of the sensory-motor system, using a range of genetic tools and animal lines (Gogliotti et al., 2012; Imlach et al., 2012; Martinez et al., 2012; Hao le et al., 2015; Simon et al., 2016; Fletcher et al., 2017). However, it remains difficult to generate a consensus from these studies, meaning that the exact cell-type and temporal requirements for SMN expression in the sensory-motor system remain to be fully determined (Table 1). Overall, however, these studies indicate that motor neuron death may be primarily due to cell-autonomous effects while the underlying cause of motor neuron dysfunction has a larger contribution from defects in proprioceptive neurons which provide a substantial non-cell autonomous component of motor neuron pathology. These findings highlight important issues when considering temporal and tissue specific requirements of SMN-targeted therapies for SMA as restoring SMN expression specifically in proprioceptive and motor neurons at the same time led to the biggest improvement in SMA-associated pathology compared to restoring SMN expression in either motor or proprioceptive neurons alone (Fletcher et al., 2017).
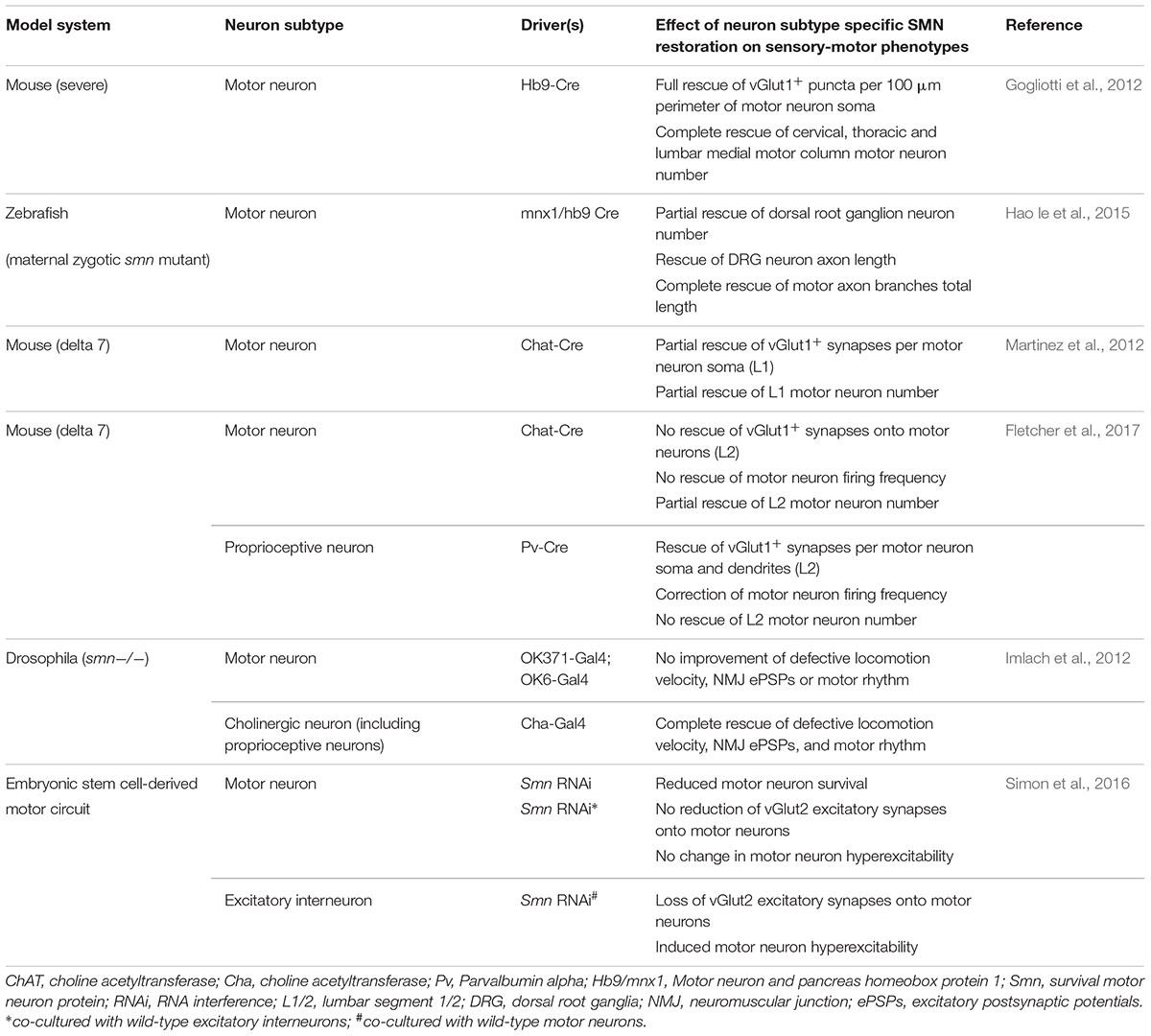
Table 1. Overview of studies investigating SMN expression requirements in various components of the sensory-motor system.
The importance of non-cell autonomous effects on motor neurons is further illustrated by several studies that have investigated the effect of defects intrinsic to specific types of sensory neurons on motor neuron function, in otherwise healthy animals. For example, blocking neurotransmission specifically in proprioceptive neurons in wild-type mice can cause severe motor defects, shortened lifespan and motor neuron dysfunction (Fletcher et al., 2017). Similarly, inhibiting cholinergic neuron activity in Drosophila causes reduced locomotion, increased spontaneous rhythmic motor activity and increased EPSPs at the neuromuscular junction by reducing excitatory cholinergic input to motor neurons (Imlach et al., 2012). Likewise, pharmacological inhibition of excitatory neurotransmission in an embryonic stem cell model of the motor circuit induced motor neuron hyperexcitability but not motor neuron death (Simon et al., 2016). Finally, mutations in the mechanosensitive ion channel responsible for mechanosensation of light touch and proprioception, PIEZO2, cause a neuromuscular disease characterized by muscle atrophy, aberrant muscle development and function, mild sensory involvement, delayed motor milestones, and scoliosis (Chesler et al., 2016; Delle Vedove et al., 2016). Together these studies indicate that defects within sensory neurons themselves can lead to both motor neuron and muscle dysfunction in the absence of defects intrinsic to motor neurons.
In summary, the above work illustrates that SMA mouse models reliably reflect pathological changes in the sensory-motor system of SMA patients. Moreover, it also illustrates the importance of homeostasis of the sensory-motor system: correct functioning of each of its parts is required for it to function correctly as a whole. Mechanistically, these studies have largely focused on motor neuron pathology and SMN requirements. Further molecular studies will be required to better understand the cellular pathways that are involved in regulating these pathological changes. In the next section of this review, we will focus on studies that have aimed to address this issue, also drawing on molecular insights obtained by studying other diseases of the sensory-motor system.
Molecular Mechanisms Underlying Sensory-Motor Connectivity Defects in SMA
The presence of sensory-motor connectivity defects in SMA (as illustrated by the loss of proprioceptive vGlut1+ synapses), alterations in sensory neuron development, and the requirement for SMN expression in different neuronal subpopulations have all been clearly established (Figure 1). In SMA, SMN depletion is at the basis of each of these pathological changes. However, the molecular mechanisms that modulate these changes downstream of SMN depletion are still unclear. It is by further studying the changes downstream of SMN depletion that we will be able to better understand why certain cell types are more sensitive to SMN depletion than others, why defects in certain cellular pathways affect certain cell types more than others, and, ultimately, how SMN depletion leads to SMA, including disruption of sensory-motor connectivity.
Molecular Mechanisms Linked to Motor Neuron Death and Pathology in SMA
As illustrated by the cell-type specific SMN requirements discussed above, pathways leading to motor neuron death appear to be largely cell autonomous. A better understanding of the molecular mechanisms associated with motor neuron death was gained from studies that compared gene expression profiles of vulnerable and resistant pools of motor neurons. These studies identified differences in basal bioenergetics profiles between vulnerable and resistant motor neuron pools (Boyd et al., 2017), as well as differences in activation of the cell death-regulating protein p53 and differential regulation of its downstream targets (Murray et al., 2015; Simon et al., 2017). Mechanistically, it has been shown that the alternative splicing of Mdm2 and Mdm4 downstream of and concurrently with disrupted snRNP biogenesis acts synergistically to induce p53 accumulation in SMA (Van Alstyne et al., 2018). However, despite rescuing the reduction of MN numbers seen in SMA mice, neither the inhibition of p53 signaling nor virus-mediated overexpression of full-length Mdm2 and Mdm4 is able to rescue vGlut1+-synapse loss (Simon et al., 2017; Van Alstyne et al., 2018).
Other factors have also been linked to motor circuit function in SMA, including stasimon (Lotti et al., 2012). Decreasing stasimon expression in zebrafish phenocopied motor axon branching defects seen in smn morpholino zebrafish, which in turn could be rescued by overexpressing stasimon. Similarly, in a Drosophila model of SMA, expressing stasimon in cholinergic (proprioceptive) neurons, but not motor neurons, rescued neurotransmitter release at the NMJ and muscle size defects (Lotti et al., 2012). All of these studies indicate that, while motor neuron death itself can be rescued by targeting the pathways that directly lead to motor neuron death (such as p53 activation, and Mdm2 and Mdm4 missplicing), this does not rescue motor neuron dysfunction or other motor neuron-associated phenotypes in models of SMA (Lotti et al., 2012; Van Alstyne et al., 2018). Other molecular factors such as agrin and plastin3 that are linked to and important for NMJ organization and AMPA receptor functioning have also been implicated in SMA (Zhang et al., 2013; Hosseinibarkooie et al., 2016; Kim et al., 2017). These factors could be important for the reduced synaptic transmission seen in SMA. Moreover, complement C1q, an important factor in postsynaptic pruning, is upregulated in SMA motor neurons (Zhang et al., 2013); a event which, along with an increased association of microglia with SMA motor neurons (Ling et al., 2010), might contribute to the impaired sensory-motor connectivity observed in SMA.
In summary, the above studies have provided important insights into the molecular mechanisms that underlie motor neuron death in SMA. However, in line with previous pathological and SMN expression studies (see Section “SMN Expression Is Required Throughout the Sensory-Motor System”), these studies also illustrate the importance of non cell-autonomous processes in sensory-motor dysfunction in SMA.
Molecular Mechanisms Associated With Sensory Neuron Dysfunction in SMA and Related Conditions
The number of studies investigating the molecular mechanisms that underlie sensory neuron dysfunction in SMA is limited. Therefore, adapting biological findings generated from studies of other, related neuromuscular diseases that are characterized by sensory neuron dysfunction has been of benefit to the SMA research field. For example, Charcot-Marie-Tooth (CMT) disease is a hereditary motor and sensory neuropathy that, depending on its genetic cause, has a variable clinical presentation. CMT type 2D, caused by mutations in the tRNA synthetase GARS (glycine tRNA-ligase or GlyRS), has a predominant motor phenotype, although sensory defects are also present (Motley et al., 2010). In CMT2D, sensory defects have been shown to be linked to a disruption in sensory neuron fate, downstream of increased expression of mutant GARS (Sleigh et al., 2017). Specifically, when determining sensory neuron subtypes in DRGs from two CMT2D mouse models, fewer large cell body NF200-positive (neurofilament heavy) cell bodies were present in favor of an increased number of peripherin-positive, smaller neurons (Sleigh et al., 2017). Taking this observation into the SMA context provides a possible explanation for the loss of sensory synapses onto lower motor neurons that occurs in the SMA spinal cord, as a shift in sensory neuron cell fate would also lead to changes at the level of their synaptic contacts. Indeed, when investigating sensory neuron development in the Taiwanese mouse model of SMA, it was shown that a similar change in sensory neuron fate occurs when SMN expression levels are reduced: a decrease in the number of NF200-positive mechano- and proprioceptive sensory neurons is accompanied by an increase in the number of peripherin-positive nociceptive sensory neurons (Shorrock et al., 2018b). Correspondingly, protein levels of GARS were also found to be changed in the spinal cord of SMA mice, as well as in DRGs, in a way that is comparable to changes in GARS observed in mouse models of CMT2D.
The upregulation of GARS in SMA was shown to be regulated by the E1 ubiquitin-activating enzyme, UBA1. UBA1 expression has previously been shown to be significantly decreased in SMA from very early in the pathogenic process and is therapeutically targetable (Wishart et al., 2014; Powis et al., 2016). When investigating protein changes that occur downstream of UBA1, GARS expression was found to depend on UBA1 expression (Shorrock et al., 2018b). In line with this, GARS-dependent pathological features of SMA, including altered sensory neuron fate and loss of proprioceptive synapses in the spinal cord, were rescued when UBA1 levels were elevated using a gene therapy approach (Shorrock et al., 2018b). These findings provide an interesting pathological link between SMA and CMT2D, but also indicate possible molecular pathways that may underlie sensory-motor pathology in SMA and provide starting points for further research. For example, possible mechanistic links related to disruption of GARS-mediated pathways have been shown to involve dysfunctional Nrp1 / VEGF and Trk signaling (He et al., 2015; Sleigh et al., 2017). Mutant GARS can aberrantly bind these proteins and this aberrant binding is thought to disrupt their function. However, to what extent these pathways are relevant for sensory defects associated with SMA remains to be determined.
In addition to defects in Trk and Nrp1/VEGF signaling, mutations in GARS are known to lead to increased levels of alpha-tubulin acetylation (d’Ydewalle et al., 2011). Alpha-tubulin acetylation depends on HDAC activity (Zhang et al., 2003), and, interestingly, both a broad spectrum HDAC inhibitor (trichostatin A) as well as a specific HDAC6 inhibitor (tubastatin A) reversed increases in alpha-tubulin acetylation in a range of cellular and animal models of CMT2D (d’Ydewalle et al., 2011; Benoy et al., 2018; Mo et al., 2018). Intriguingly, the broad spectrum HDAC inhibitor trichostatin A was previously found to rescue vGlut1+-synapse loss on spinal motor neurons in the delta7 model of SMA (Mentis et al., 2011). To what extent this process is also related to alpha-tubulin acetylation or is due to other effects of HDAC inhibition in SMA remains to be determined. However, this provides an intriguing possible link between HDAC inhibition, GARS function, DRG pathology and sensory synapse loss.
In summary, these findings illustrate how molecular pathways that were previously identified in other disorders, such as CMT2D, can assist discovery of novel mechanisms and therapeutic targets that underlie sensory-motor connectivity defects in SMA.
Concluding Remarks
Motor neurons rely on a range of synaptic inputs, both from within the central nervous system and from sensory neurons projecting from the periphery, to regulate and finetune their activity. In SMA, multiple components of this sensory-motor system have been shown to be disrupted, contributing to motor neuron dysfunction and death. Several molecular pathways have now been identified that regulate sensory neuron dysfunction and these pathways form interesting novel targets for further study and potential therapy development.
The SMA research field is currently at a defining moment. The first disease-modifying therapy for SMA was recently approved, while other therapies are currently in advanced stages of clinical development (Shorrock et al., 2018a). However, the fact that current therapies are targeted to the nervous system specifically and do not benefit all patients equally, illustrates that fundamental SMA research is still needed, perhaps more than ever (Groen et al., 2018b). Furthering our understanding of the molecular mechanisms that underlie pathological changes occurring in specific cell types, such as sensory neurons, will aid the discovery of novel therapeutic strategies. Moreover, this process may be sped up by scrutinizing the existing knowledge from other, related disorders, as is illustrated by the recent identification of shared molecular mechanisms between SMA and CMT2D. Combining advances in these areas of research will be necessary to be able to eventually provide therapies that will benefit and support all SMA patients equally and efficiently.
Author Contributions
HS and EG performed the literature search and generated the table and figure. HS, TG, and EG prepared and wrote the manuscript.
Funding
The authors gratefully acknowledge funding from the Wellcome Trust (to EG and TG, grant 106098/Z/14/Z), the Euan MacDonald Centre for MND Research (to HS and TG), SMA Europe (to TG and EG), MND Scotland (to TG), and the UK SMA Research Consortium (by the SMA Trust, to TG). Open access publication fees are covered by the Charity Open Access Fund, administered through the University of Edinburgh.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Benoy, V., Van Helleputte, L., Prior, R., d’Ydewalle, C., Haeck, W., Geens, N., et al. (2018). HDAC6 is a therapeutic target in mutant GARS-induced Charcot-Marie-Tooth disease. Brain 141, 673–687. doi: 10.1093/brain/awx375
Bobele, G. B., Feeback, D. L., Leech, R. W., and Brumback, R. A. (1996). Hypertrophic intrafusal muscle fibers in infantile spinal muscular atrophy. J. Child. Neurol. 11, 246–248. doi: 10.1177/088307389601100318
Boyd, P. J., Tu, W. Y., Shorrock, H. K., Groen, E. J. N., Carter, R. N., Powis, R. A., et al. (2017). Bioenergetic status modulates motor neuron vulnerability and pathogenesis in a zebrafish model of spinal muscular atrophy. PLoS Genet. 13:e1006744. doi: 10.1371/journal.pgen.1006744
Brown, A. G. (1981). Organization in the Spinal Cord : The Anatomy and Physiology of Identified Neurones. New York, NY: Springer-Verlag. doi: 10.1007/978-1-4471-1305-8
Carpenter, S., Karpati, G., Rothman, S., Watters, G., and Andermann, F. (1978). Pathological involvement of primary sensory neurons in Werdnig-Hoffmann disease. Acta Neuropathol. 42, 91–97. doi: 10.1007/BF00690973
Chaytow, H., Huang, Y. T., Gillingwater, T. H., and Faller, K. M. E. (2018). The role of survival motor neuron protein (SMN) in protein homeostasis. Cell Mol. Life Sci. 75, 3877–3894. doi: 10.1007/s00018-018-2849-1
Chesler, A. T., Szczot, M., Bharucha-Goebel, D., Ceko, M., Donkervoort, S., Laubacher, C., et al. (2016). The Role of PIEZO2 in Human Mechanosensation. N. Engl. J. Med. 375, 1355–1364. doi: 10.1056/NEJMoa1602812
Delle Vedove, A., Storbeck, M., Heller, R., Holker, I., Hebbar, M., Shukla, A., et al. (2016). Biallelic loss of proprioception-related PIEZO2 causes muscular atrophy with perinatal respiratory distress, arthrogryposis, and scoliosis. Am. J. Hum. Genet. 99, 1206–1216. doi: 10.1016/j.ajhg.2016.09.019
Duman, O., Uysal, H., Skjei, K. L., Kizilay, F., Karauzum, S., and Haspolat, S. (2013). Sensorimotor polyneuropathy in patients with SMA type-1: electroneuromyographic findings. Muscle Nerve 48, 117–121. doi: 10.1002/mus.23722
d’Ydewalle, C., Krishnan, J., Chiheb, D. M., Van Damme, P., Irobi, J., Kozikowski, A. P., et al. (2011). HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced charcot-marie-tooth disease. Nat. Med. 17, 968–974. doi: 10.1038/nm.2396
Eccles, J. C., Eccles, R. M., and Lundberg, A. (1957). The convergence of monosynaptic excitatory afferents on to many different species of alpha motoneurones. J. Physiol. 137, 22–50. doi: 10.1113/jphysiol.1957.sp005794
Edens, B. M., Ajroud-Driss, S., Ma, L., and Ma, Y. C. (2015). Molecular mechanisms and animal models of spinal muscular atrophy. Biochim. Biophys. Acta 1852, 685–692. doi: 10.1016/j.bbadis.2014.07.024
Fletcher, E. V., and Mentis, G. Z. (2016). “Motor circuit dysfunction in spinal muscular atrophy,” in Spinal Muscular Atrophy, eds C. J. Sumner, S. Paushkin, and C. P. Ko (London: Elsevier), 153–165.
Fletcher, E. V., Simon, C. M., Pagiazitis, J. G., Chalif, J. I., Vukojicic, A., Drobac, E., et al. (2017). Reduced sensory synaptic excitation impairs motor neuron function via Kv2.1 in spinal muscular atrophy. Nat. Neurosci. 20, 905–916. doi: 10.1038/nn.4561
Gogliotti, R. G., Quinlan, K. A., Barlow, C. B., Heier, C. R., Heckman, C. J., and Didonato, C. J. (2012). Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J. Neurosci. 32, 3818–3829. doi: 10.1523/JNEUROSCI.5775-11.2012
Groen, E. J. N., Perenthaler, E., Courtney, N. L., Jordan, C. Y., Shorrock, H. K., van der Hoorn, D., et al. (2018a). Temporal and tissue-specific variability of SMN protein levels in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 27, 2851–2862. doi: 10.1093/hmg/ddy195
Groen, E. J. N., Talbot, K., and Gillingwater, T. H. (2018b). Advances in therapy for spinal muscular atrophy: promises and challenges. Nat. Rev. Neurol. 14, 214–224. doi: 10.1038/nrneurol.2018.4
Hamilton, G., and Gillingwater, T. H. (2013). Spinal muscular atrophy: going beyond the motor neuron. Trends Mol. Med. 19, 40–50. doi: 10.1016/j.molmed.2012.11.002
Hao le, T., Duy, P. Q., Jontes, J. D., and Beattie, C. E. (2015). Motoneuron development influences dorsal root ganglia survival and Schwann cell development in a vertebrate model of spinal muscular atrophy. Hum. Mol. Genet. 24, 346–360. doi: 10.1093/hmg/ddu447
He, W., Bai, G., Zhou, H., Wei, N., White, N. M., Lauer, J., et al. (2015). CMT2D neuropathy is linked to the neomorphic binding activity of glycyl-tRNA synthetase. Nature 526, 710–714. doi: 10.1038/nature15510
Hosseinibarkooie, S., Peters, M., Torres-Benito, L., Rastetter, R. H., Hupperich, K., Hoffmann, A., et al. (2016). The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue sma phenotype. Am. J. Hum. Genet. 99, 647–665. doi: 10.1016/j.ajhg.2016.07.014
Hosseinibarkooie, S., Schneider, S., and Wirth, B. (2017). Advances in understanding the role of disease-associated proteins in spinal muscular atrophy. Expert Rev. Proteomics 14, 581–592. doi: 10.1080/14789450.2017.1345631
Hunter, G., Powis, R. A., Jones, R. A., Groen, E. J., Shorrock, H. K., Lane, F. M., et al. (2016). Restoration of SMN in Schwann cells reverses myelination defects and improves neuromuscular function in spinal muscular atrophy. Hum. Mol. Genet. 25, 2853–2861. doi: 10.1093/hmg/ddw141
Ikemoto, A., Hirano, A., Matsumoto, S., Akiguchi, I., and Kimura, J. (1996). Synaptophysin expression in the anterior horn of werdnig-hoffmann disease. J. Neurol. Sci. 136, 94–100. doi: 10.1016/0022-510X(95)00297-F
Imai, F., Chen, X., Weirauch, M. T., and Yoshida, Y. (2016). Requirement for dicer in maintenance of monosynaptic sensory-motor circuits in the spinal cord. Cell Rep. 17, 2163–2172. doi: 10.1016/j.celrep.2016.10.083
Imlach, W. L., Beck, E. S., Choi, B. J., Lotti, F., Pellizzoni, L., and McCabe, B. D. (2012). SMN is required for sensory-motor circuit function in Drosophila. Cell 151, 427–439. doi: 10.1016/j.cell.2012.09.011
Jablonka, S., Karle, K., Sandner, B., Andreassi, C., von Au, K., and Sendtner, M. (2006). Distinct and overlapping alterations in motor and sensory neurons in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 15, 511–518. doi: 10.1093/hmg/ddi467
Jiang, M., Schuster, J. E., Fu, R., Siddique, T., and Heckman, C. J. (2009). Progressive changes in synaptic inputs to motoneurons in adult sacral spinal cord of a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 29, 15031–15038. doi: 10.1523/JNEUROSCI.0574-09.2009
Kararizou, E., Manta, P., Kalfakis, N., Gkiatas, K., and Vassilopoulos, D. (2006). Morphological and morphometrical study of human muscle spindles in Werdnig-Hoffmann disease (infantile spinal muscular atrophy type I). Acta Histochem. 108, 265–269. doi: 10.1016/j.acthis.2006.03.020
Kim, J. K., Caine, C., Awano, T., Herbst, R., and Monani, U. R. (2017). Motor neuronal repletion of the NMJ organizer, agrin, modulates the severity of the spinal muscular atrophy disease phenotype in model mice. Hum. Mol. Genet. 26, 2377–2385. doi: 10.1093/hmg/ddx124
Korinthenberg, R., Sauer, M., Ketelsen, U. P., Hanemann, C. O., Stoll, G., Graf, M., et al. (1997). Congenital axonal neuropathy caused by deletions in the spinal muscular atrophy region. Ann. Neurol. 42, 364–368. doi: 10.1002/ana.410420314
Lee, A. J., Awano, T., Park, G. H., and Monani, U. R. (2012). Limited phenotypic effects of selectively augmenting the SMN protein in the neurons of a mouse model of severe spinal muscular atrophy. PLoS One 7:e46353. doi: 10.1371/journal.pone.0046353
Ling, K. K., Lin, M. Y., Zingg, B., Feng, Z., and Ko, C. P. (2010). Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS One 5:e15457. doi: 10.1371/journal.pone.0015457
Lotti, F., Imlach, W. L., Saieva, L., Beck, E. S., Hao le, T., Li, D. K., et al. (2012). An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151, 440–454. doi: 10.1016/j.cell.2012.09.012
Lunn, M. R., and Wang, C. H. (2008). Spinal muscular atrophy. Lancet 371, 2120–2133. doi: 10.1016/S0140-6736(08)60921-6
Marshall, A., and Duchen, L. W. (1975). Sensory system involvement in infantile spinal muscular atrophy. J. Neurol. Sci. 26, 349–359. doi: 10.1016/0022-510X(75)90207-5
Martinez, T. L., Kong, L., Wang, X., Osborne, M. A., Crowder, M. E., Van Meerbeke, J. P., et al. (2012). Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J. Neurosci. 32, 8703–8715. doi: 10.1523/JNEUROSCI.0204-12.2012
Mears, S. C., and Frank, E. (1997). Formation of specific monosynaptic connections between muscle spindle afferents and motoneurons in the mouse. J. Neurosci. 17, 3128–3135. doi: 10.1523/JNEUROSCI.17-09-03128.1997
Mentis, G. Z., Blivis, D., Liu, W., Drobac, E., Crowder, M. E., Kong, L., et al. (2011). Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 69, 453–467. doi: 10.1016/j.neuron.2010.12.032
Mo, Z., Zhao, X., Liu, H., Hu, Q., Chen, X. Q., Pham, J., et al. (2018). Aberrant GlyRS-HDAC6 interaction linked to axonal transport deficits in Charcot-Marie-Tooth neuropathy. Nat. Commun. 9:1007. doi: 10.1038/s41467-018-03461-z
Motley, W. W., Talbot, K., and Fischbeck, K. H. (2010). GARS axonopathy: not every neuron’s cup of tRNA. Trends Neurosci. 33, 59–66. doi: 10.1016/j.tins.2009.11.001
Murayama, S., Bouldin, T. W., and Suzuki, K. (1991). Immunocytochemical and ultrastructural studies of werdnig-hoffmann disease. Acta Neuropathol. 81, 408–417. doi: 10.1007/BF00293462
Murray, L. M., Beauvais, A., Gibeault, S., Courtney, N. L., and Kothary, R. (2015). Transcriptional profiling of differentially vulnerable motor neurons at pre-symptomatic stage in the Smn (2b/-) mouse model of spinal muscular atrophy. Acta Neuropathol. Commun. 3:55. doi: 10.1186/s40478-015-0231-1
Nash, L. A., Burns, J. K., Chardon, J. W., Kothary, R., and Parks, R. J. (2016). Spinal muscular atrophy: more than a disease of motor neurons? Curr. Mol. Med. 16, 779–792. doi: 10.2174/1566524016666161128113338
Omran, H., Ketelsen, U. P., Heinen, F., Sauer, M., Rudnik-Schoneborn, S., Wirth, B., et al. (1998). Axonal neuropathy and predominance of type II myofibers in infantile spinal muscular atrophy. J. Child. Neurol. 13, 327–331. doi: 10.1177/088307389801300704
Pecho-Vrieseling, E., Sigrist, M., Yoshida, Y., Jessell, T. M., and Arber, S. (2009). Specificity of sensory-motor connections encoded by Sema3e-Plxnd1 recognition. Nature 459, 842–846. doi: 10.1038/nature08000
Powis, R. A., Karyka, E., Boyd, P., Come, J., Jones, R. A., Zheng, Y., et al. (2016). Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight 1:e87908. doi: 10.1172/jci.insight.87908
Reid, D., Zinger, Y., and Raheja, D. (2016). Sensory neuronopathy in spinal muscular atrophy: a case presentation. J. Clin. Neuromuscul. Dis. 18, 44–46. doi: 10.1097/CND.0000000000000124
Rossignol, S., Dubuc, R., and Gossard, J. P. (2006). Dynamic sensorimotor interactions in locomotion. Physiol. Rev. 86, 89–154. doi: 10.1152/physrev.00028.2005
Rossoll, W., Jablonka, S., Andreassi, C., Kroning, A. K., Karle, K., Monani, U. R., et al. (2003). Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J. Cell Biol. 163, 801–812. doi: 10.1083/jcb.200304128
Rudnik-Schoneborn, S., Goebel, H. H., Schlote, W., Molaian, S., Omran, H., Ketelsen, U., et al. (2003). Classical infantile spinal muscular atrophy with SMN deficiency causes sensory neuronopathy. Neurology 60, 983–987. doi: 10.1212/01.WNL.0000052788.39340.45
Shababi, M., Lorson, C. L., and Rudnik-Schoneborn, S. S. (2014). Spinal muscular atrophy: a motor neuron disorder or a multi-organ disease? J. Anat. 224, 15–28. doi: 10.1111/joa.12083
Sherrington, C. S. (1906). The Integrative Action of the Nervous System. New York, NY: C. Scribner’s sons.
Shishikura, K., Hara, M., Sasaki, Y., and Misugi, K. (1983). A neuropathologic study of Werdnig-Hoffmann disease with special reference to the thalamus and posterior roots. Acta Neuropathol. 60, 99–106. doi: 10.1007/BF00685353
Shorrock, H. K., Gillingwater, T. H., and Groen, E. J. N. (2018a). Overview of current drugs and molecules in development for spinal muscular atrophy therapy. Drugs 78, 293–305. doi: 10.1007/s40265-018-0868-8
Shorrock, H. K., van der Hoorn, D., Boyd, P. J., Llavero Hurtado, M., Lamont, D. J., Wirth, B., et al. (2018b). UBA1/GARS-dependent pathways drive sensory-motor connectivity defects in spinal muscular atrophy. Brain 141, 2878–2894. doi: 10.1093/brain/awy237
Simon, C. M., Dai, Y., Van Alstyne, M., Koutsioumpa, C., Pagiazitis, J. G., Chalif, J. I., et al. (2017). Converging mechanisms of p53 activation drive motor neuron degeneration in spinal muscular atrophy. Cell Rep. 21, 3767–3780. doi: 10.1016/j.celrep.2017.12.003
Simon, C. M., Janas, A. M., Lotti, F., Tapia, J. C., Pellizzoni, L., and Mentis, G. Z. (2016). A stem cell model of the motor circuit uncouples motor neuron death from hyperexcitability induced by SMN deficiency. Cell Rep. 16, 1416–1430. doi: 10.1016/j.celrep.2016.06.087
Singh, R. N., Howell, M. D., Ottesen, E. W., and Singh, N. N. (2017). Diverse role of survival motor neuron protein. Biochim. Biophys. Acta Gene. Regul. Mech. 1860, 299–315. doi: 10.1016/j.bbagrm.2016.12.008
Sleigh, J. N., Dawes, J. M., West, S. J., Wei, N., Spaulding, E. L., Gomez-Martin, A., et al. (2017). Trk receptor signaling and sensory neuron fate are perturbed in human neuropathy caused by Gars mutations. Proc. Natl. Acad. Sci. U.S.A. 114, E3324–E3333. doi: 10.1073/pnas.1614557114
Soliven, B., and Maselli, R. A. (1992). Single motor unit H-reflex in motor neuron disorders. Muscle Nerve 15, 656–660. doi: 10.1002/mus.880150604
Van Alstyne, M., Simon, C. M., Sardi, S. P., Shihabuddin, L. S., Mentis, G. Z., and Pellizzoni, L. (2018). Dysregulation of Mdm2 and Mdm4 alternative splicing underlies motor neuron death in spinal muscular atrophy. Genes Dev. 32, 1045–1059. doi: 10.1101/gad.316059.118
Wishart, T. M., Mutsaers, C. A., Riessland, M., Reimer, M. M., Hunter, G., Hannam, M. L., et al. (2014). Dysregulation of ubiquitin homeostasis and beta-catenin signaling promote spinal muscular atrophy. J. Clin. Invest. 124, 1821–1834. doi: 10.1172/JCI71318
Yonekawa, T., Komaki, H., Saito, Y., Sugai, K., and Sasaki, M. (2013). Peripheral nerve abnormalities in pediatric patients with spinal muscular atrophy. Brain Dev. 35, 165–171. doi: 10.1016/j.braindev.2012.03.009
Yuan, P., and Jiang, L. (2015). Clinical characteristics of three subtypes of spinal muscular atrophy in children. Brain Dev. 37, 537–541. doi: 10.1016/j.braindev.2014.08.007
Zhang, Y., Li, N., Caron, C., Matthias, G., Hess, D., Khochbin, S., et al. (2003). HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 22, 1168–1179. doi: 10.1093/emboj/cdg115
Keywords: spinal muscular atrophy, SMN, sensory-motor circuit, proprioception, motor neuron, neurodegenaration
Citation: Shorrock HK, Gillingwater TH and Groen EJN (2019) Molecular Mechanisms Underlying Sensory-Motor Circuit Dysfunction in SMA. Front. Mol. Neurosci. 12:59. doi: 10.3389/fnmol.2019.00059
Received: 14 December 2018; Accepted: 15 February 2019;
Published: 04 March 2019.
Edited by:
Andrew Paul Tosolini, University College London, United KingdomReviewed by:
Chien-Ping Ko, University of Southern California, United StatesGeorge Mentis, Columbia University, United States
Copyright © 2019 Shorrock, Gillingwater and Groen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ewout J. N. Groen, ZS5ncm9lbkBlZC5hYy51aw==
†Present address: Hannah K. Shorrock, Center for NeuroGenetics, Department of Molecular Genetics and Microbiology, College of Medicine, University of Florida, Gainesville, FL, United States