 Jinxia Lu1†
Jinxia Lu1† Qin Cao2†
Qin Cao2† Chuchu Wang3,4†
Chuchu Wang3,4† Jing Zheng5
Jing Zheng5 Feng Luo3,4Jingfei Xie3,4Yichen Li1Xiaojuan Ma3,4
Feng Luo3,4Jingfei Xie3,4Yichen Li1Xiaojuan Ma3,4 Lin He1,5David Eisenberg2James Nowick6
Lin He1,5David Eisenberg2James Nowick6 Lin Jiang7*
Lin Jiang7* Dan Li1*
Dan Li1*- 1Key Laboratory for the Genetics of Developmental and Neuropsychiatric Disorders (Ministry of Education), Bio-X Institutes, Shanghai Jiao Tong University, Shanghai, China
- 2UCLA-DOE Institute for Genomics and Proteomics, University of California, Los Angeles, Los Angeles, CA, United States
- 3Interdisciplinary Research Center on Biology and Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, China
- 4University of Chinese Academy of Sciences, Beijing, China
- 5Shanghai Center for Women and Children’s Health, Shanghai, China
- 6Department of Chemistry, University of California, Irvine, Irvine, CA, United States
- 7Department of Neurology, Easton Center for Alzheimer’s Disease Research, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, United States
Many human neurodegenerative diseases are associated with amyloid fibril formation. Inhibition of amyloid formation is of importance for therapeutics of the related diseases. However, the development of selective potent amyloid inhibitors remains challenging. Here based on the structures of amyloid β (Aβ) fibrils and their amyloid-forming segments, we designed a series of peptide inhibitors using RosettaDesign. We further utilized a chemical scaffold to constrain the designed peptides into β-strand conformation, which significantly improves the potency of the inhibitors against Aβ aggregation and toxicity. Furthermore, we show that by targeting different Aβ segments, the designed peptide inhibitors can selectively recognize different species of Aβ. Our study developed an approach that combines the structure-based rational design with chemical modification for the development of amyloid inhibitors, which could be applied to the development of therapeutics for different amyloid-related diseases.
Introduction
Amyloid diseases, including many neurodegenerative diseases, are increasingly prevalent in aging societies (Eisenberg and Jucker, 2012; Dobson, 2017). The pathogenesis of these devastating diseases is closely associated with aberrant protein aggregation (Chiti and Dobson, 2006). In the progression of amyloid aggregation, soluble proteins undergo a series of conformational changes and self-assemble into insoluble amyloid fibrils (Riek and Eisenberg, 2016). Plaques containing amyloid fibrils are one of the histological hallmarks of Alzheimer’s and Parkinson’s diseases (Lee et al., 1991; Spillantini et al., 1997; Koo et al., 1999). Various strategies have been exploited to interfere with the process of amyloid aggregation by targeting different conformational species, including stabilizing monomers by antibodies (Ladiwala et al., 2012), redirecting monomers to nontoxic off-pathway oligomers by polyphenolic compounds (Ehrnhoefer et al., 2008), accelerating mature fibril formation by fibril binders (Bieschke et al., 2012; Jiang et al., 2013), inhibiting fibril growing by peptide blockers (Seidler et al., 2018), and disrupting amyloid assembly by nanomaterials (Hamley, 2012; Huang et al., 2014; Lee et al., 2014; Li et al., 2018; Han and He, 2018). Many of these strategies show promising inhibitory effects against toxic amyloid aggregation (Härd and Lendel, 2012; Arosio et al., 2014), but so far none has led to clinical drugs because of unsettled issues such as target selectivity, side effects, membrane permeability and penetration of the blood-brain barrier.
Amyloid β (Aβ) has long been targeted for drug development and therapeutic treatment of Alzheimer’s disease (Caputo and Salama, 1989; Haass and Selkoe, 2007; Sevigny et al., 2016). In addition to the common difficulties in targeting amyloid proteins, Aβ is especially challenging since it contains multiple species with various lengths generated by γ-secretases (Acx et al., 2014; Kummer and Heneka, 2014; Szaruga et al., 2017). Many studies have shown that Aβ42 rather than Aβ40 is more prone to form toxic aggregates, and the ratio of Aβ42/Aβ40 is better correlated with the pathology rather than the amount of each individual Aβ species (Lewczuk et al., 2004; Jan et al., 2008; Kuperstein et al., 2010). However, selective inhibition of Aβ42 is very difficult because it is only two residues longer than Aβ40 at the C-terminus. In this work, we targeted two key amyloid-forming segments of Aβ42 (16KLVFFA21 and 37GGVVIA42) based on the cryo-EM structure of Aβ42 fibrils reported recently (Gremer et al., 2017). We designed peptide binders of these two segments using RosettaDesign with the atomic structures of these two segments as templates (Sawaya et al., 2007; Colletier et al., 2011). The designed sequences showed inhibitory effect to Aβ42 fibril formation. We further utilized a macrocyclic β-sheet mimic scaffold (Zheng et al., 2011; Cheng et al., 2012, 2013) to constrain the designed peptide inhibitors in β-conformation, which significantly enhanced the inhibitory effect on Aβ42 aggregation. Furthermore, we show that the peptide inhibitor designed to target the C-terminus of Aβ42 can selectively inhibit Aβ42 aggregation, but not to that of Aβ40 or other amyloid proteins. Our work shed light on the application of structure-based rational design combined with chemical modification in the development of therapeutics for Alzheimer’s disease and other amyloid-related diseases.
Materials and Methods
Structure-Based Design by Rosetta Software Package
Initial Structure Model for Design
We chose two key amyloidogenic Aβ segments, 16KLVFFA21 and 37GGVVIA42, for our inhibitor design. The design templates were taken from the crystal structures of KLVFFA (PDB ID: 2Y2A) and GGVVIA (PDB ID: 2ONV). The backbone of the inhibiting pentapeptide was fully extended to mimic β-conformation. This extended peptide was aligned with the N, C, and O backbone atoms of the template.
Rosetta Design of Fibril-Inhibiting Peptides
The peptide inhibitors were subsequently designed to ensure maximal interaction, while keeping the template amino acid sequence fixed. Computational designs were carried out using the RosettaDesign software package1. This algorithm involves building side-chain rotamers of all L-amino acids onto a fixed peptide backbone. The optimal set of side-chain rotamers at each position with the best interaction energy is then identified, with the guidance of a full-atom energy function containing a Lennard-Jones potential, an orientation-dependent hydrogen bond potential, an implicit solvation term, amino acid-dependent reference energies, and a statistical torsional potential that depends on the backbone and side-chain dihedral angles. Finally, the entire structure was refined by simultaneously optimizing degrees of freedom on: (1) the rigid-body geometry between the inhibiting peptide and template; (2) backbone torsions of each peptide; and (3) side chain torsions of each peptide. The lowest-energy model was picked and the interaction energies of each final model from different peptide inhibitors are listed in Table 1.
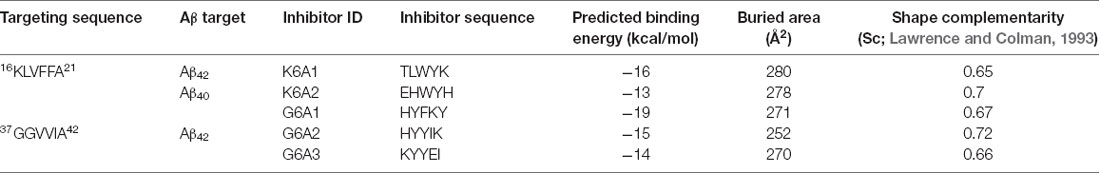
Table 1. Characteristics of designed peptide inhibitors.
Circular Dichroism Spectroscopy (CD)
Chirascan spectrometer (Applied Photophysics) equipped with a Peltier temperature controller (Quantum Northwest) is used to acquire the CD spectra. Far UV spectra (240–180 nm) are collected in 0.05 cm path-length quartz cells. Sample concentration is 600 μM. All measurements are conducted at 23°C. Water is used as blank for subtraction from corresponding samples. Secondary structure is predicted from CD using CDPro (Eisenberg and Jucker, 2012).
Preparation of Aβ42 and Aβ40
Both Aβ42 and Aβ40 were purified from E. coli expression system as reported previously (Dobson, 2017). The expression constructs contain an N-terminal His-tag, followed by 19 repeats of Asn-Ala-Asn-Pro, the Tobacco etch virus (TEV) protease site, and the sequence of Aβ42 or Aβ40. Purification of Aβ42 and Aβ40 follows the same experimental procedure. Briefly, the Aβ fusion protein was overexpressed into inclusion bodies in E. coli BL21(DE3) cells. The inclusion bodies were solubilized in 8 M urea, followed by washing in a high salt and detergent-containing solution. The Aβ fusion proteins were purified through HisTrapTM HP Columns, followed by reversed-phase high-performance liquid chromatography (RP-HPLC). After cleavage by TEV protease, Aβ was released from fusion protein, and purified through RP-HPLC followed by lyophilization. To disrupt preformed Aβ aggregates, lyophilized Aβ powder was resuspended in 100% HFIP and incubated at room temperature for 2 h. HFIP was fully removed by evaporation. Before used in ThT or MTT assay, Aβ was freshly dissolved in 10 mM NaOH, solubilized by sonication. Aβ is further diluted to 200 μM in phosphate buffer saline (PBS) as a stock solution.
Synthesis of Designed Macrocyclic Peptides
Designed macrocyclic peptides were synthesized by standard Fmoc solid-phase peptide synthesis. In brief, with Boc-Orn(Fmoc)-OH attached onto 2-chlorotrityl chloride resin, the linear peptide was elongated by standard automated Fmoc solid-phase peptide synthesis. Then, the peptide was cleaved from the resin under mildly acidic conditions, followed by being cyclized to the corresponding protected cyclic peptide by slow addition to HCTU and DIEA in dilute (ca. 0.5 mM) DMF solution. Since the C-terminus of the protected linear peptide comprises an amino acid carbamate (Boc-NH-CHR-COOH), the cyclization condition efficiently avoids problematic epimerization. The final deprotection with TFA solution followed by RP-HPLC purification yielded macrocyclic peptides in 18%–43% overall yield, based on the loading of Boc- Orn(Fmoc)-OH attached onto the resin.
1H NMR Spectroscopy
1H NMR experiments for the designed macrocyclic peptides were performed in D2O with the internal standard 4,4-Dimethyl-4-silapentane-1-ammonium trifluoroacetate (DSA) at 500 MHz (Brüker Avance) or 600 MHz (Brüker Avance). All peptides were studied at 2 mM in D2O at 298 K. Sample solutions were prepared gravimetrically by dissolving the macrocyclic peptides directly in solvent. All amino groups were assumed to be protonated as the TFA salts for molecular weight calculation. The data were processed with the Brüker XwinNMR software.
ThT Fluorescence Assay
Thioflavin T (ThT) fluorescence assays were performed to monitor the real-time aggregation of Aβ42 and Aβ40 in the absence or presence of designed peptides. ThT assays were conducted in 96-well plates (black with flat optical bottom) in a Varioskan fluorescence plate reader (Thermo Scientific, 444 nm excitation, 484 nm emission). Each experiment was run in triplicates. The reaction solution contained 30 μM pre-disaggregated Aβ42 or Aβ40, 10 μM ThT, and designed peptides at indicated concentrations in PBS. The ThT assay was conducted at 37°C, without shaking for the Aβ42 aggregation assay, and with shaking (300 rpm) for Aβ40 aggregation assay. The fluorescence readings were collected every 2 min.
Native Gel Electrophoresis
Purified Aβ42 powder was pre-treated by HFIP and dissolved in PBS buffer as described above. Aβ42 solution was diluted to a final concentration of 10 μM with or without the macrocyclic peptides mcG6A1, mcG6A2, and mcK6A1 (the final concentration of the inhibitors was 50 μM), and incubated at 37°C for 7.5 h. The samples were separated by a NativePAGE 4%–16% BisTris Gel (Novex, USA) and transferred to a nitrocellulose membrane pre-packed in iBlot 2 NC Mini Stacks (Novex, USA) by iBlot 2 Dry Blotting System (Life technologies, USA). The membrane was probed by β amyloid, 1–16 (6E10) Monoclonal Antibody (Covance, USA) and secondary anti-mouse IgG-HRP (MBL, USA), and detected with SuperSignal West Pico Chemiluminescent Substrate (Thermo, USA). The freshly made Aβ42 sample without inhibitors was loaded to a separated native gel and detected by the same method as a 0-h control. The molecular weight of the protein aggregates or monomer were accurately determined by the protein standard especially for native gel (Life technologies; cat. # LC0725).
Transmission Electron Microscopy (TEM)
For specimen preparation, 5 μl of each sample was deposited onto a glow-discharged carbon film on 400 mesh copper grids, followed by washing in water twice. The grids were then stained in 0.75% uranyl formate. A Tecnai G2 Spirit transmission electron microscope operating at an accelerating voltage of 120 kV was used to examine and visualize the samples. Images were collected by a 4k × 4k charge-coupled device camera (BM-Eagle, FEI).
Cell Viability Assay
We performed MTT-based cell viability assays to evaluate the toxicity of Aβ42 in the absence or presence of the designed peptides. We used a CellTiter 96 aqueous non-radioactive cell proliferation assay kit (Promega cat. # G4100). PC-12 cell lines (ATCC; cat. # CRL-1721) were used to test the cytotoxicity of Aβ42 under different conditions. PC-12 cells were cultured in ATCC-formulated RPMI 1640 medium (ATCC; cat. # 30-2001) with 5% fetal bovine serum and 10% heat-inactivated horse serum. Before the cell viability experiment, PC-12 cells were plated at 10,000 cells per well in 96-well plates (Costar; cat. # 3596), and cultured for 20 h at 37°C in 5% CO2. For the preparation of Aβ42 and peptide inhibitors mixture solutions, purified and pre-disaggregated Aβ42 samples were dissolved in PBS to a final concentration of 5 μM, followed by the addition of different peptide inhibitors at indicated concentrations. The mixture solution was filtered through a 0.22 μm filter, followed by incubation at 37°C without shaking for 16 h. To initiate the cell viability assay, 10 μl of pre-incubated mixture was added to each well containing 90 μl medium. After incubation at 37°C in 5% CO2 for 24 h. Fifteen microliter Dye solution (Promega; cat. # G4102) was applied into each well. After incubation for 4 h at 37°C, 100 μl solubilization Solution/Stop Mix (Promega; cat. # G4101) were added. After further incubation at room temperature for 12 h, the absorbance reading was collected at 570 nm with background reading at 700 nm. Four replicates were measured in parallel for each sample. The cell survival rate was normalized by using the PBS-treated cells as 100% and 0.02% SDS-treated cells as 0% viability.
Results
Structure-Based Design of Peptide Inhibitors
To effectively inhibit Aβ fibril formation, we targeted two key amyloid-forming segments of Aβ42: 16KLVFFA21 and 37GGVVIA42 (Figure 1A). The 16KLVFFA21 segment has been identified as a key segment accounting for both Aβ42 and Aβ40 nucleation and fibrillation (Ahmed et al., 2010; Colletier et al., 2011; Fawzi et al., 2011; Lu et al., 2013). In the known structures of Aβ fibrils including the recent cryo-EM structure of Aβ42 and the previous solid-state NMR structure of Aβ40 (Paravastu et al., 2008; Ahmed et al., 2010), this segment forms extended β-strands and stacks repetitively along the fibril axis to form the Aβ fibril core (Supplementary Figure S1). Thus, we selected 16KLVFFA21 as one of our design targets. In addition, the cryo-EM structure of Aβ42 fibril shows that the C-terminal segment 37GGVVIA42 plays an essential role in the fibril formation (Supplementary Figure S2). 37GGVVIA42 of one protofilament interdigitates via side chains with its counterpart of the neighboring protofilament forming a steric-zipper-like interaction to compose the mature fibril. Therefore, preventing the self-assembly of either 16KLVFFA21 or 37GGVVIA42 may potentially inhibit the assembly of Aβ42 fibrils.
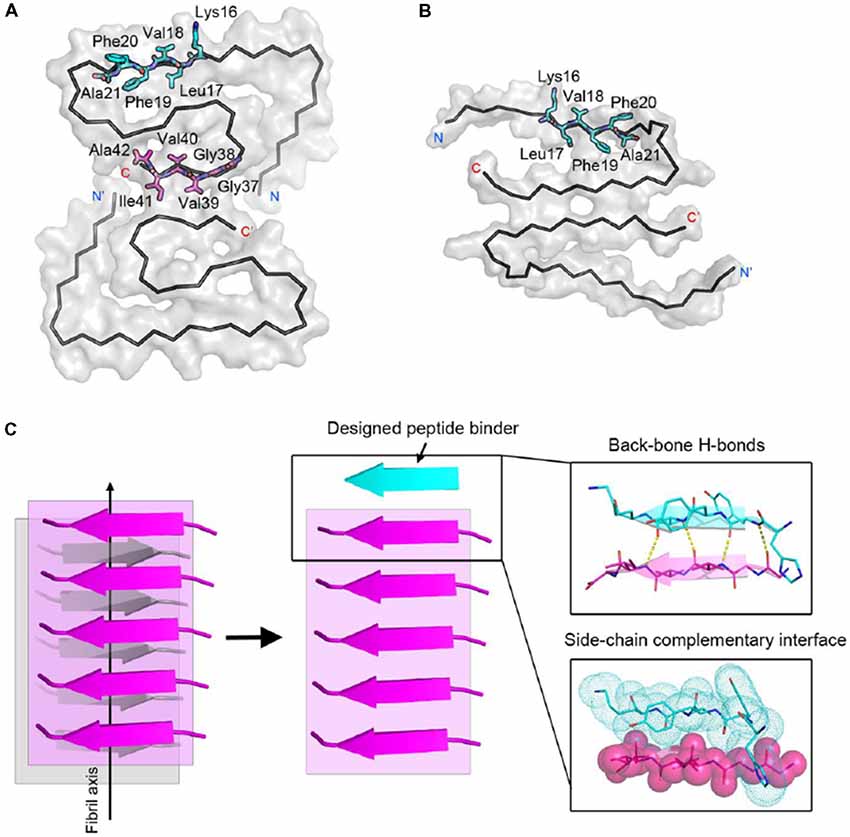
Figure 1. Structure-based peptide inhibitor design of amyloid β (Aβ) amyloid aggregation. (A) The fibril structure of full-length Aβ42 determined by cryo-EM (PDB ID: 5OQV) is shown as black ribbons. The atomic crystal structures of peptides KLVFFA (PDB ID: 2Y2A, cyan) and GGVVIA (PDB ID: 2ONV, magenta) are aligned on one of the two protofilaments of the full-length Aβ42 fibril structure and shown as sticks. (B) The fibril structure of full-length Aβ40 determined by solid-state NMR (PDB ID: 2LMN) is shown as black ribbons. The atomic crystal structure of peptides KLVFFA is aligned on one of the two protofilaments of the full-length Aβ40 fibril structure and shown as sticks. (C) Design strategy for peptide inhibitors of amyloid fibrils. The designing template is a five-stranded sheet extracted from the fibrillar structure of the targeting segment. Peptide inhibitors (in cyan) are designed to have the optimal interactions with the target via backbone hydrogen bonds (yellow dashed lines) and complementary side-chain interactions (shown as spheres and dots). Oxygen atoms are in red. Nitrogen atoms are in blue.
For structure-based computational design, we used the atomic structures of 16KLVFFA21 (PDB ID: 2Y2A) and 37GGVVIA42 (PDB ID: 2ONV) as templates. The atomic structures of these two segments represent their conformations in the context of the full-length Aβ fibrils (Figures 1A,B). Based on the structures of the two targeting templates, we designed pentapeptides that bind the targeting segments to block the stacking of Aβ molecules along the fibril axis, thus inhibiting fibril growth (Figure 1C). We extracted a five-stranded layer from the steric-zipper structure of each segment, and docked a fully extended pentapeptide backbone on one end of the β-sheet. Then, we maximized the backbone interaction with the template by forming a backbone H-bonding network. To further increase the binding affinity and selectivity, we searched for the canonical L-amino acids at each position of the pentapeptide, using RosettaDesign (Leaver-Fay et al., 2011) for the side chains and their conformations, that provide maximal interactions with the template.
Next, we calculated the binding energy, buried surface area and shape complementarity of the binding interfaces of the predicted binding models, and proceeded with experimental validation for the top-ranking designs. Using ThT fluorescence assay, we observed that the top-5 designs showed inhibitory effects on Aβ42 amyloid aggregation by significantly delaying the aggregation lag time (Xue et al., 2008; Knowles et al., 2009; Figure 2). Among them, two peptide inhibitors (K6A1 and K6A2) were designed for targeting 16KLVFFA21 and three (G6A1-G6A3) were for 37GGVVIA42 (Table 1). Furthermore, unlike their targeting segments, the five designed peptides do not form amyloid fibrils by themselves (Supplementary Figure S3).
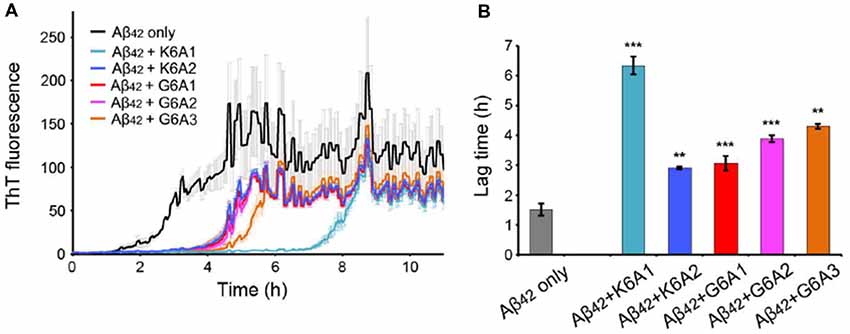
Figure 2. Inhibitory effects of designed peptides on Aβ42 amyloid aggregation measured by the thioflavin T (ThT) fluorescence assay. (A) The ThT fluorescence curves of Aβ42 in the presence of designed peptide inhibitors. The molar ratio of Aβ:peptide-inhibitor is 1:5. Three replicates were measured for each curve. The lag time of Aβ42 aggregation in the presence of peptide inhibitors is compared in (B). *p-value < 0.05; **p-value < 0.01; ***p-value < 0.001.
Constraining the Structures of Designed Peptides With a Chemical Scaffold
We next sought to enhance the potency of the peptide inhibitors. In our design, the peptide inhibitors were expected to adopt an extended β-strand conformation to maximize the interaction with the template (Figure 1C). However, in solution, the peptides are mainly unstructured (Supplementary Table S1). Thus, upon binding to the template, the peptides need to undergo conformational change to form extended β strands, which causes an entropy decrease and thus weakens the binding affinity of the peptides to the template. To overcome the entropy lost during the conformational change, we adopted a macrocyclic β-sheet mimic scaffold to fix the peptide binders into β strands (Figure 3A). The Nowick group has developed a series of macrocycles in different sizes as robust scaffolds for displaying peptides of interest in β-conformation (Liu et al., 2012; Cheng et al., 2013; Salveson et al., 2016; Kreutzer et al., 2017). According to the length of our designed peptides, we chose a 42-membered macrocyclic β-sheet mimic and grafted the designed sequence into the open strand of the macrocyclic scaffold with appropriate amino acids in the blocking strand for proper solubility and stability (Figure 3B). The β-strand conformation of the grafted sequence was validated by measuring the α-H shifts and δOrn anisotropy using 1H NMR experiments (Supplementary Figures S4, S5) in solution. Furthermore, we confirmed that the macrocycles carrying the designed peptides do not form amyloid aggregation in solution, while those carrying native amyloid-forming sequences may form amyloid fibrils with an out-of-register packing (Lu et al., 2013; Supplementary Figure S3).
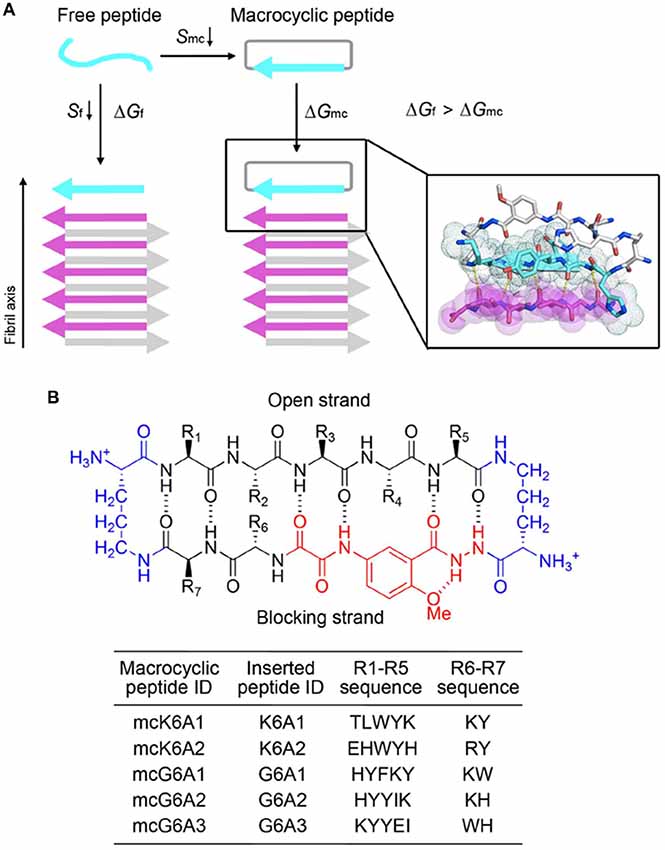
Figure 3. Design of macrocyclic peptide inhibitors. (A) The schematic shows that as the macrocyclic β-sheet mimic scaffold constrains the designed peptide sequence into a β-strand, the entropy loss is diminished during the process of target binding. “f” represents free peptide; “mc” represents macrocyclic peptide. The zoom-in view shows the structure model of a macrocyclic inhibitor binding to the targeting segment. The targeting segment is in magenta. The designed sequence is in cyan. The macrocyclic scaffold is in gray. H-bonds between the designed sequence and the targeting sequence are labeled by yellow dotted lines. (B) The 42-membered macrocyclic scaffold used in this study. The open strand (positions R1 to R5) accommodates the designed peptides in β-conformation. Two δ-linked ornithine turn units are in blue. The Hao unit in the blocking strand is in red. Sequences of R1-R7 are listed in the table below.
Next, we tested the inhibitory effects of the macrocyclic peptides on Aβ42 amyloid aggregation. The result showed that, in comparison with the free peptides, the macrocyclic peptides remarkably enhanced the inhibition on Aβ42 aggregation (Figures 2B, 4A,B, and Supplementary Figures S6–S10). For instance, the macrocycle carrying K6A1 (mcK6A1) is about 10 times more potent than free K6A1 in prolonging the lag time of Aβ aggregation. The macrocyclic peptides inhibited the amyloid aggregation of Aβ42 in a dose-dependent manner. McK6A1, mcG6A1 and mcG6A2 showed remarkably strong inhibition with a 7–10-fold increase of the lag time at sub-stoichiometric concentrations of 0.2 molar equivalence to Aβ42 monomer (Figure 4A).
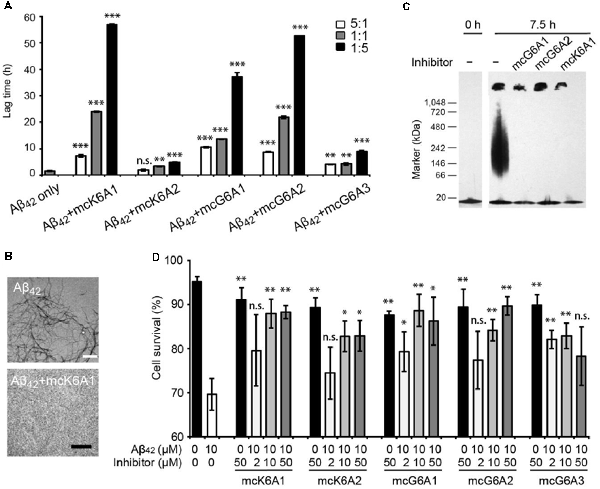
Figure 4. Inhibitory effects of designed macrocyclic peptides on Aβ42 amyloid aggregation and cytotoxicity. (A) The designed macrocyclic peptides, in particular mcK6A1, mcG6A1 and mcG6A2, significantly inhibit the amyloid fibril formation of Aβ42 in a dose-dependent manner. (B) Transmission electron microscopy (TEM) images of Aβ42 (20 μM) after incubation without inhibitors (top) and with 1.0 equivalent of mcK6A1 (bottom) to Aβ monome for 15 h. The scale bars are 200 nm. (C) Inhibition of Aβ42 oligomers. Aβ42 oligomers formed after 7.5 h of incubation at a concentration of 5 μM (by Aβ42 monomer equivalence) were invisible on the native gel with the addition of five molar excess of designed macrocyclic peptides. (D) The designed peptide inhibitors ameliorated Aβ42 cytotoxicity to PC-12 cells. The first column is the cells treated with 0.1 mM NaOH and phosphate buffer saline (PBS) as a positive control. Error bars correspond to standard deviations three replicates of each experiment. *p-value < 0.05; **p-value < 0.01; ***p-value < 0.001; n.s. represents “not significant”.
Moreover, we found that the designed macrocyclic peptides can inhibit the formation of Aβ42 oligomers, the toxic intermediates of Aβ aggregation, monitored by the native gel (Figure 4C). This result demonstrated that targeting 16KLVFFA21 and 37GGVVIA42 can prevent both oligomer and fibril formation, indicating the potential important role of these two segments in the early stage of Aβ42 aggregation. To further assess whether the designed peptides can reduce Aβ cytotoxicity, we performed the MTT-based cell viability assay. The result showed that the designed macrocyclic peptides can significantly reduce the cytotoxicity of Aβ42 to PC-12 cells even with a molar ratio of inhibitor to Aβ42 as low as 0.2:1 (Figure 4D). Also, the designed macrocyclic peptides showed little toxicity to the PC-12 cells (Figure 4D). In addition, the designed inhibitors of Aβ42 showed no inhibition of the amyloid aggregation of other amyloid proteins (e.g., α-synuclein and the K19 variant of Tau), indicating that the designed peptides are highly sequence-specific (Supplementary Figure S11).
Designed Peptides Selectively Inhibit the Aggregation of Aβ42 but Not Aβ40
Selective inhibition of Aβ42 aggregation over that of Aβ40 is challenging because Aβ42 is only two residues longer than Aβ40 at the C- terminus (Figure 5A). Since segment 37GGVVIA42 exists only in Aβ42, the designed peptides that target this segment may selectively inhibit the aggregation of Aβ42 but not that of Aβ40. As shown in the designed models, mcG6A1 that is designed to target 37GGVVIA42 forms extensive side-chain interactions with 37GGVVIA42 (Figure 5B). The aromatic residues Tyr and Phe of mcG6A1 interact with Ile41 of 37GGVVIA42 via van der Waals forces. The absence of Ile41 and Ala42 in Aβ40 diminishes the binding of mcG6A1 to Aβ40. Indeed, the experimental data showed that mcG6A1 and mcG6A2 that strongly inhibit the amyloid aggregation of Aβ42, cannot effectively inhibit the aggregation of Aβ40, as measured by ThT assay (Figure 5C, Supplementary Figures S12, S13). Note that a weak inhibitory effect of mcG6A1 and mcG6A2 to Aβ40 remains, which might come from non-specific backbone interactions between the inhibitors and Aβ40 (Figure 5B). In contrast, mcK6A1 that was designed to target the 16KLVFFA21 segment, a segment important for the amyloid aggregation of both Aβ42 and Aβ40, showed a dose-dependent inhibition of both Aβ42 and Aβ40 aggregation (Figures 4A, 5C,D, and Supplementary Figure S14). However, the inhibitory efficiency of mcK6A1 on Aβ40 is weaker than that on Aβ42, indicating that 16KLVFFA21 may play a more important role in Aβ42 aggregation than that of Aβ40. This implication is in agreement with the hypothesis that Aβ42 and Aβ40 may employ different amyloid nucleation and aggregation process (Sánchez et al., 2011; Meisl et al., 2014).
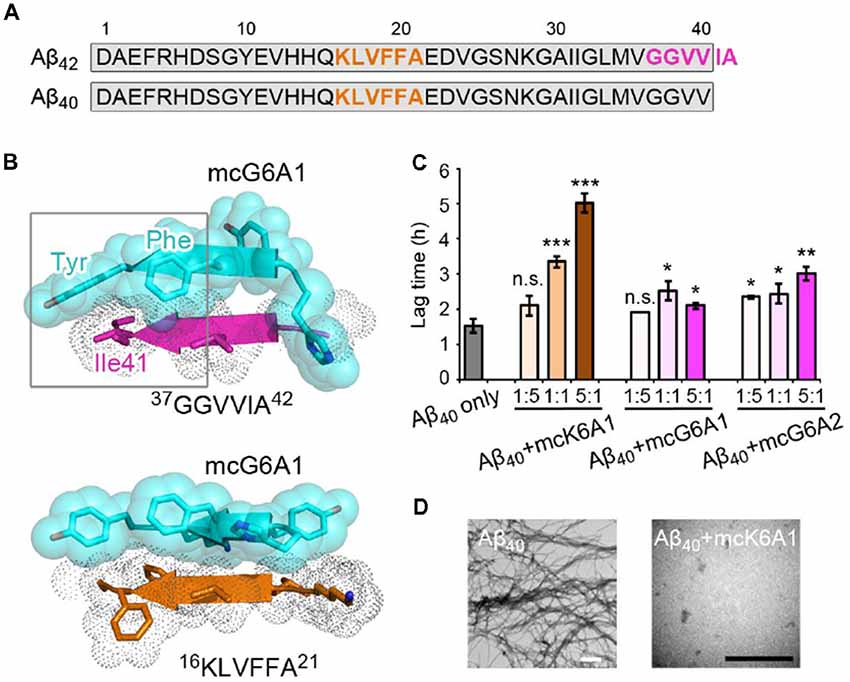
Figure 5. Specificity of designed macrocyclic peptides for the inhibition of Aβ42 and Aβ40 aggregation. (A) The sequences of Aβ42 and Aβ40. The amyloid-forming segment 16KLVFFA21 (highlighted in orange) is present in both Aβ42 and Aβ40, while segment 37GGVVIA42 (highlighted in magenta) is present only in Aβ42. The consensus sequence of Aβ42 and Aβ40 is highlighted in gray. (B) The structure models of mcG6A1 (cyan) in complex with 37GGVVIA42 (magenta) and 16KLVFFA21 (orange), respectively. McG6A1 was designed based on the structure of GGVVIA. Residues Tyr and Phe of mcG6A1, and Ile41 of GGVVIA (highlighted with a gray frame) engage in van der Waals interactions at the inhibitor-target interface. In contrast, mcG6A1 designed for GGVVIA has no specific side-chain interactions, but merely non-specific back-bone interactions with KLVFFA. (C) The effects of mcK6A, mcG6A1 and mcG6A2 on Aβ40 aggregation (30 μM by Aβ40 monomer equivalence), measured by ThT assay. Error bars correspond to standard deviations of three replicates of each experiment. *p-value < 0.05; **p-value < 0.01; ***p-value < 0.001; n.s. represents “not significant.” (D) TEM images of Aβ40 (30 μM) after incubation without inhibitors (left), and with 1.0 equivalent of mcK6A1 to Aβ monome (right). The scale bars are 200 nm.
Discussion
Development of peptide-based drugs is gaining greater attentions. In general, peptide-protein interactions have a high density of hydrogen bonds and highly complementary packing via hot-spot binding residues, leading to high binding affinity and exquisite selectivity with fewer off-target side effects (Kaspar and Reichert, 2013). Many attempts have been made to rationally design peptide inhibitors of amyloid protein aggregation, including modified internal segments of parent amyloid proteins, non-natural amino-acid inhibitors, proline substitutions, and other methods (Abedini et al., 2007; Sievers et al., 2011). Recently, RosettaDesign shows effectiveness for designing novel proteins and peptides with predicted structures having atomic accuracy (Bhardwaj et al., 2016; Huang et al., 2016). This technical advance has enabled the peptide inhibitor design of Tau aggregation (Abedini et al., 2007; Seidler et al., 2018). In this study, we designed peptides that can efficiently inhibit Aβ42 aggregation. Notably, the designed peptides show selectivity for the intended amyloid target, in contrast to small molecule inhibitors (e.g., EGCG and methylene blue) that broadly interfere amyloid aggregation of many proteins (Necula et al., 2007; Jiang et al., 2013; Palhano et al., 2013). Furthermore, the designed peptides can differentiate Aβ42 from Aβ40, demonstrating the accuracy and potency of structure-based rational design.
Short peptides composed of natural amino acids normally form unstructured ensembles in solution. If a defined conformation is required for target binding, conformational changes may occur upon binding, at a large entropic cost. This counteracts enthalpy gain from the favorable interaction of the designed peptide and its target, and consequently reduces the binding affinity of the peptide with its target. Therefore, constraining the designed peptide in the desired conformation (“pre-organization”) can minimize the entropic cost and increase the binding affinity. Chemical scaffolds provide a powerful toolbox for constraining peptides in defined secondary or tertiary structures in solution (Mowery et al., 2009; Azzarito et al., 2013; Cheng et al., 2013; Johnson and Gellman, 2013). In this work, we use a macrocyclic β-sheet mimic scaffold to constrain the designed peptides into β strands. Our results show significant enhancement of inhibition gained by the conformational constraint, which highlights the importance of conformation-constraint and the advantage of a chemical scaffold in the development of peptide binders. In addition, biopharmaceutical properties, such as degradation resistance and membrane permeability, may be achieved by modifying the chemical scaffold, rather than changing the inhibitor sequences.
Macrocyclic β-sheet mimics have been shown to be a useful model system to study the structural basis of amyloid-like oligomers and fibrils (Liu et al., 2012; Cheng et al., 2013; Zheng et al., 2013; Salveson et al., 2016). A variety of key amyloidogenic segments from different amyloid proteins (e.g., Aβ, α-synuclein and prion) were constructed into the macrocycles (Zheng et al., 2011; Cheng et al., 2012). However, the self-assembling and potential toxic properties of macrocyclic molecules that contain native amyloid-forming sequences hinder application of macrocycles in the development of amyloid inhibitors (Liu et al., 2012; Salveson et al., 2016). In this study, by using RosettaDesign approach, we developed novel sequences and incorporated them into macrocycles. These designed macrocyclic peptides resist self-assembly and exhibit little cytotoxicity. In additional to Aβ, the structures of many other pathogenic amyloid fibrils have been determined recently (Tuttle et al., 2006; Fitzpatrick et al., 2017; Murray et al., 2017). Thus, the strategy of combining RosettaDesign and chemical scaffolds may be useful for peptide inhibitor design of different amyloid proteins for a variety of amyloid-related diseases.
Data Availability
All datasets generated for this study are included in the manuscript and/or the supplementary files.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was sponsored by Shanghai Pujiang Program (18PJ1404300); the National Natural Science Foundation (NSF) of China (31872716, 91853113); National Institutes of Health (NIH; 5R01 GM097562 and R01 AG029430), the 1000 Talents Program, China; and Howard Hughes Medical Institute (HHMI), USA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2019.00054/full#supplementary-material
References
Abedini, A., Meng, F., and Raleigh, D. P. (2007). A single-point mutation converts the highly amyloidogenic human islet amyloid polypeptide into a potent fibrillization inhibitor. J. Am. Chem. Soc. 129, 11300–11301. doi: 10.1021/ja072157y
Acx, H., Chávez-Gutiérrez, L., Serneels, L., Lismont, S., Benurwar, M., Elad, N., et al. (2014). Signature amyloid β profiles are produced by different γ-secretase complexes. J. Biol. Chem. 289, 4346–4355. doi: 10.1074/jbc.M113.530907
Ahmed, M., Davis, J., Aucoin, D., Sato, T., Ahuja, S., Aimoto, S., et al. (2010). Structural conversion of neurotoxic amyloid-β1–42 oligomers to fibrils. Nat. Struct. Mol. Biol. 17, 561–567. doi: 10.1038/nsmb.1799
Arosio, P., Vendruscolo, M., Dobson, C. M., and Knowles, T. P. (2014). Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol. Sci. 35, 127–135. doi: 10.1016/j.tips.2013.12.005
Azzarito, V., Long, K., Murphy, N. S., and Wilson, A. J. (2013). Inhibition of α-helix-mediated protein-protein interactions using designed molecules. Nat. Chem. 5, 161–173. doi: 10.1038/nchem.1568
Bhardwaj, G., Mulligan, V. K., Bahl, C. D., Gilmore, J. M., Harvey, P. J., Cheneval, O., et al. (2016). Accurate de novo design of hyperstable constrained peptides. Nature 538, 329–335. doi: 10.1038/nature19791
Bieschke, J., Herbst, M., Wiglenda, T., Friedrich, R. P., Boeddrich, A., Schiele, F., et al. (2012). Small-molecule conversion of toxic oligomers to nontoxic β-sheet-rich amyloid fibrils. Nat. Chem. Biol. 8, 93–101. doi: 10.1038/nchembio.719
Caputo, C. B., and Salama, A. I. (1989). The amyloid proteins of Alzheimer’s disease as potential targets for drug therapy. Neurobiol. Aging 10, 451–461. doi: 10.1016/0197-4580(89)90096-1
Cheng, P. N., Liu, C., Zhao, M., Eisenberg, D., and Nowick, J. S. (2012). Amyloid β-sheet mimics that antagonize protein aggregation and reduce amyloid toxicity. Nat. Chem. 4, 927–933. doi: 10.1038/nchem.1433
Cheng, P. N., Pham, J. D., and Nowick, J. S. (2013). The supramolecular chemistry of β-sheets. J. Am. Chem. Soc. 135, 5477–5492. doi: 10.1021/ja3088407
Chiti, F., and Dobson, C. M. (2006). Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366. doi: 10.1146/annurev.biochem.75.101304.123901
Colletier, J. P., Laganowsky, A., Landau, M., Zhao, M., Soriaga, A. B., Goldschmidt, L., et al. (2011). Molecular basis for amyloid-β polymorphism. Proc. Natl. Acad. Sci. U S A 108, 16938–16943. doi: 10.1073/pnas.1112600108
Dobson, C. M. (2017). The amyloid phenomenon and its links with human disease. Cold Spring Harb. Perspect. Biol. 9:a023648. doi: 10.1101/cshperspect.a023648
Ehrnhoefer, D. E., Bieschke, J., Boeddrich, A., Herbst, M., Masino, L., Lurz, R., et al. (2008). EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 15, 558–566. doi: 10.1038/nsmb.1437
Eisenberg, D., and Jucker, M. (2012). The amyloid state of proteins in human diseases. Cell 148, 1188–1203. doi: 10.1016/j.cell.2012.02.022
Fawzi, N. L., Ying, J., Ghirlando, R., Torchia, D. A., and Clore, G. M. (2011). Atomic-resolution dynamics on the surface of amyloid-β protofibrils probed by solution NMR. Nature 480, 268–272. doi: 10.1038/nature10577
Fitzpatrick, A. W. P., Falcon, B., He, S., Murzin, A. G., Murshudov, G., Garringer, H. J., et al. (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. doi: 10.1038/nature23002
Gremer, L., Schölzel, D., Schenk, C., Reinartz, E., Labahn, J., Ravelli, R. B. G., et al. (2017). Fibril structure of amyloid-β1–42 by cryo-electron microscopy. Science 358, 116–119. doi: 10.1126/science.aao2825
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Hamley, I. W. (2012). The amyloid β peptide: a chemist’s perspective role in Alzheimer’s and fibrillization. Chem. Rev. 112, 5147–5192. doi: 10.1021/cr3000994
Han, X., and He, G. (2018). Toward a rational design to regulate β-amyloid fibrillation for alzheimer’s disease treatment. ACS Chem. Neurosci. 9, 198–210. doi: 10.1021/acschemneuro.7b00477
Härd, T., and Lendel, C. (2012). Inhibition of amyloid formation. J. Mol. Biol. 421, 441–465. doi: 10.1016/j.jmb.2011.12.062
Huang, P. S., Boyken, S. E., and Baker, D. (2016). The coming of age of de novo protein design. Nature 537, 320–327. doi: 10.1038/nature19946
Huang, F., Wang, J., Qu, A., Shen, L., Liu, J., Liu, J., et al. (2014). Maintenance of amyloid β peptide homeostasis by artificial chaperones based on mixed-shell polymeric micelles. Angew. Chem. Int. Ed. Engl. 53, 8985–8990. doi: 10.1002/anie.201400735
Jan, A., Gokce, O., Luthi-Carter, R., and Lashuel, H. A. (2008). The ratio of monomeric to aggregated forms of Aβ40 and Aβ42 is an important determinant of amyloid-β aggregation, fibrillogenesis, and toxicity. J. Biol. Chem. 283, 28176–28189. doi: 10.1074/jbc.m803159200
Jiang, L., Liu, C., Leibly, D., Landau, M., Zhao, M., Hughes, M. P., et al. (2013). Structure-based discovery of fiber-binding compounds that reduce the cytotoxicity of amyloid β. Elife 2:e00857. doi: 10.7554/eLife.00857
Johnson, L. M., and Gellman, S. H. (2013). α-Helix mimicry with α/β-peptides. Methods Enzymol. 523, 407–429. doi: 10.1016/B978-0-12-394292-0.00019-9
Kaspar, A. A., and Reichert, J. M. (2013). Future directions for peptide therapeutics development. Drug Discov. Today 18, 807–817. doi: 10.1016/j.drudis.2013.05.011
Knowles, T. P., Waudby, C. A., Devlin, G. L., Cohen, S. I., Aguzzi, A., Vendruscolo, M., et al. (2009). An analytical solution to the kinetics of breakable filament assembly. Science 326, 1533–1537. doi: 10.1126/science.1178250
Koo, E. H., Lansbury, P. T. Jr., and Kelly, J. W. (1999). Amyloid diseases: abnormal protein aggregation in neurodegeneration. Proc. Natl. Acad. Sci. U S A 96, 9989–9990. doi: 10.1073/pnas.96.18.9989
Kreutzer, A. G., Yoo, S., Spencer, R. K., and Nowick, J. S. (2017). Stabilization, assembly, and toxicity of trimers derived from Aβ. J. Am. Chem. Soc. 139, 966–975. doi: 10.1021/jacs.6b11748
Kummer, M. P., and Heneka, M. T. (2014). Truncated and modified amyloid-β species. Alzheimers. Res. Ther. 6:28. doi: 10.1186/alzrt258
Kuperstein, I., Broersen, K., Benilova, I., Rozenski, J., Jonckheere, W., Debulpaep, M., et al. (2010). Neurotoxicity of Alzheimer’s disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 29, 3408–3420. doi: 10.1038/emboj.2010.211
Ladiwala, A. R., Bhattacharya, M., Perchiacca, J. M., Cao, P., Raleigh, D. P., Abedini, A., et al. (2012). Rational design of potent domain antibody inhibitors of amyloid fibril assembly. Proc. Natl. Acad. Sci. U S A 109, 19965–19970. doi: 10.1073/pnas.1208797109
Lawrence, M. C., and Colman, P. M. (1993). Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234, 946–950. doi: 10.1006/jmbi.1993.1648
Leaver-Fay, A., Tyka, M., Lewis, S. M., Lange, O. F., Thompson, J., Jacak, R., et al. (2011). ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Meth. Enzymol. 487, 545–574. doi: 10.1016/B978-0-12-381270-4.00019-6
Lee, H. H., Choi, T. S., Lee, S. J., Lee, J. W., Park, J., Ko, Y. H., et al. (2014). Supramolecular inhibition of amyloid fibrillation by cucurbituril. Angew. Chem. Int. Ed. Engl. 53, 7461–7465. doi: 10.1002/anie.201402496
Lee, V., Balin, B., Otvos, L., and Trojanowski, J. (1991). A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science 251, 675–678. doi: 10.1126/science.1899488
Lewczuk, P., Esselmann, H., Otto, M., Maler, J. M., Henkel, A. W., Henkel, M. K., et al. (2004). Neurochemical diagnosis of Alzheimer’s dementia by CSF Aβ42, Aβ42/Aβ40 ratio and total tau. Neurobiol. Aging 25, 273–281. doi: 10.1016/S0197-4580(03)00086-1
Li, G., Yang, W.-Y., Zhao, Y.-F., Chen, Y.-X., Hong, L., and Li, Y.-M. (2018). Differentialmodulation of the aggregation of n-terminal truncated aβ via cucurbiturils. Chem. Eur. J. 24, 13647–13653. doi: 10.1002/chem.201802655
Liu, C., Zhao, M., Jiang, L., Cheng, P. N., Park, J., Sawaya, M. R., et al. (2012). Out-of-register β-sheets suggest a pathway to toxic amyloid aggregates. Proc. Natl. Acad. Sci. U S A 109, 20913–20918. doi: 10.1073/pnas.1218792109
Lu, J. X., Qiang, W., Yau, W. M., Schwieters, C. D., Meredith, S. C., and Tycko, R. (2013). Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 154, 1257–1268. doi: 10.1016/j.cell.2013.08.035
Meisl, G., Yang, X., Hellstrand, E., Frohm, B., Kirkegaard, J. B., Cohen, S. I., et al. (2014). Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc. Natl. Acad. Sci. U S A 111, 9384–9389. doi: 10.1073/pnas.1401564111
Mowery, B. P., Lindner, A. H., Weisblum, B., Stahl, S. S., and Gellman, S. H. (2009). Structure-activity relationships among random nylon-3 copolymers that mimic antibacterial host-defense peptides. J. Am. Chem. Soc. 131, 9735–9745. doi: 10.1021/ja901613g
Murray, D. T., Kato, M., Lin, Y., Thurber, K. R., Hung, I., McKnight, S. L., et al. (2017). Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell 171, 615.e16–627.e16. doi: 10.1016/j.cell.2017.08.048
Necula, M., Kayed, R., Milton, S., and Glabe, C. G. (2007). Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 282, 10311–10324. doi: 10.1074/jbc.m608207200
Palhano, F. L., Lee, J., Grimster, N. P., and Kelly, J. W. (2013). Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils. J. Am. Chem. Soc. 135, 7503–7510. doi: 10.1021/ja3115696
Paravastu, A. K., Leapman, R. D., Yau, W. M., and Tycko, R. (2008). Molecular structural basis for polymorphism in Alzheimer’s β-amyloid fibrils. Proc. Natl. Acad. Sci. U S A 105, 18349–18354. doi: 10.1073/pnas.0806270105
Riek, R., and Eisenberg, D. S. (2016). The activities of amyloids from a structural perspective. Nature 539, 227–235. doi: 10.1038/nature20416
Salveson, P. J., Spencer, R. K., and Nowick, J. S. (2016). X-ray crystallographic structure of oligomers formed by a toxic β-hairpin derived from α-synuclein: trimers and higher-order oligomers. J. Am. Chem. Soc. 138, 4458–4467. doi: 10.1021/jacs.5b13261
Sánchez, L., Madurga, S., Pukala, T., Vilaseca, M., López-Iglesias, C., Robinson, C. V., et al. (2011). Aβ40 and Aβ42 amyloid fibrils exhibit distinct molecular recycling properties. J. Am. Chem. Soc. 133, 6505–6508. doi: 10.1021/ja1117123
Sawaya, M. R., Sambashivan, S., Nelson, R., Ivanova, M. I., Sievers, S. A., Apostol, M. I., et al. (2007). Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447, 453–457. doi: 10.1038/nature05695
Seidler, P., Boyer, D. R., Rodriguez, J. A., Sawaya, M. R., Cascio, D., Murray, K., et al. (2018). Structure-based inhibitors of tau aggregation. Nat. Chem. 10, 170–176. doi: 10.1038/nchem.2889
Sevigny, J., Chiao, P., Bussière, T., Weinreb, P. H., Williams, L., Maier, M., et al. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56. doi: 10.1038/nature19323
Sievers, S. A., Karanicolas, J., Chang, H. W., Zhao, A., Jiang, L., Zirafi, O., et al. (2011). Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 475, 96–100. doi: 10.1038/nature10154
Spillantini, M. G., Schmidt, M. L., Lee, M. Y., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). α-synuclein in lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Szaruga, M., Munteanu, B., Lismont, S., Veugelen, S., Horré, K., Mercken, M., et al. (2017). Alzheimer’s-causing mutations shift Aβ length by destabilizing γ-secretase-Aβn interactions. Cell 170, 443.e14–456.e14. doi: 10.1016/j.cell.2017.07.004
Tuttle, M. D., Comellas, G., Nieuwkoop, A. J., Covell, D. J., Berthold, D. A., Kloepper, K. D., et al. (2006). Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 23, 409–415. doi: 10.1038/nsmb.3194
Xue, W. F., Homans, S. W., and Radford, S. E. (2008). Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc. Natl. Acad. Sci. U S A 105, 8926–8931. doi: 10.1073/pnas.0711664105
Zheng, J., Baghkhanian, A. M., and Nowick, J. S. (2013). A hydrophobic surface is essential to inhibit the aggregation of a tau-protein-derived hexapeptide. J. Am. Chem. Soc. 135, 6846–6852. doi: 10.1021/ja310817d
Keywords: neurodegenerative diseases, Alzheimer’s disease, Aβ fibril, protein misfolding, structure-based inhibitor design
Citation: Lu J, Cao Q, Wang C, Zheng J, Luo F, Xie J, Li Y, Ma X, He L, Eisenberg D, Nowick J, Jiang L and Li D (2019) Structure-Based Peptide Inhibitor Design of Amyloid-β Aggregation. Front. Mol. Neurosci. 12:54. doi: 10.3389/fnmol.2019.00054
Received: 12 January 2019; Accepted: 12 February 2019;
Published: 04 March 2019.
Edited by:
Jiajie Diao, University of Cincinnati, United StatesReviewed by:
Kailu Yang, Stanford University, United StatesYing Lai, Stanford University, United States
Yanmei Li, Tsinghua University, China
Copyright © 2019 Lu, Cao, Wang, Zheng, Luo, Xie, Li, Ma, He, Eisenberg, Nowick, Jiang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Jiang, amlhbmdsaW5AdWNsYS5lZHU=
Dan Li, bGlkYW4yMDE3QHNqdHUuZWR1LmNu
† These authors have contributed equally to this work