 Jennifer Mundhenk
Jennifer Mundhenk Camilla Fusi
Camilla Fusi Michael R. Kreutz
Michael R. Kreutz- 1RG Neuroplasticity, Leibniz Institute for Neurobiology, Magdeburg, Germany
- 2Leibniz Group “Dendritic Organelles and Synaptic Function”, Center for Molecular Neurobiology, ZMNH, Hamburg, Germany
The calmodulin (CaM)-like Ca2+-sensor proteins caldendrin, calneuron-1 and -2 are members of the neuronal calcium-binding protein (nCaBP)-family, a family that evolved relatively late during vertebrate evolution. All three proteins are abundant in brain but show a strikingly different subcellular localization. Whereas caldendrin is enriched in the postsynaptic density (PSD), calneuron-1 and -2 accumulate at the trans-Golgi-network (TGN). Caldendrin exhibit a unique bipartite structure with a basic and proline-rich N-terminus while calneurons are the only EF-Hand CaM-like transmembrane proteins. These uncommon structural features come along with highly specialized functions of calneurons in Golgi-to-plasma-membrane trafficking and for caldendrin in actin-remodeling in dendritic spine synapses. In this review article, we will provide a synthesis of available data on the structure and biophysical properties of all three proteins. We will then discuss their cellular function with special emphasis on synaptic neurotransmission. Finally, we will summarize the evidence for a role of these proteins in neuropsychiatric disorders.
Introduction
Neuronal Ca2+-signaling is based on spatio-temporal gradients called Ca2+ waves, spikes, transients and puffs. The strict segmentation of such gradients allows for complex signaling events at the micro- and even nanoscale (Berridge et al., 2003). Consequently, the huge variety of Ca2+-evoked processes require a highly specialized machinery leading to alterations in cellular functions and Ca2+ sensor proteins from the calmodulin (CaM) family are instrumental in this regard.
The nCaBP-Family of EF-Hand CaM-Like Calcium Sensor Proteins
The predominant neuronally expressed Ca2+-binding proteins of the neuronal calcium sensor (NCS)- and neuronal calcium-binding protein (nCaBP)-family share the same EF-hand organization as their ancestor CaM (Figure 1). In contrast to CaM, however, these proteins exhibit a more restricted and cell type specific expression. They are frequently targeted to subcellular compartments, contain at least one cryptic EF-hand incapable of Ca2+-binding, while many of them exhibit a higher global Ca2+-binding affinity and a different conformational response upon Ca2+-binding than CaM. It is nowadays widely believed that NCS and nCaBP family members are involved in distinct signaling processes and thereby increase the versatility of the Ca2+-signaling tool kit (Mikhaylova et al., 2011). NCS proteins are evolutionarily related to Frequenin and, on the basis of their sequences, they have been grouped into five subfamilies (Burgoyne, 2007; Mikhaylova et al., 2011), comprising Frequenin/NCS-1, Recoverin, VILIPs 1–3/Hippocalcin/Neurocalcin delta, GCAP1–3 and the voltage-gated K+ (Kv) channel-interacting proteins (KChiPs) 1–4/DREAM (Figure 1).
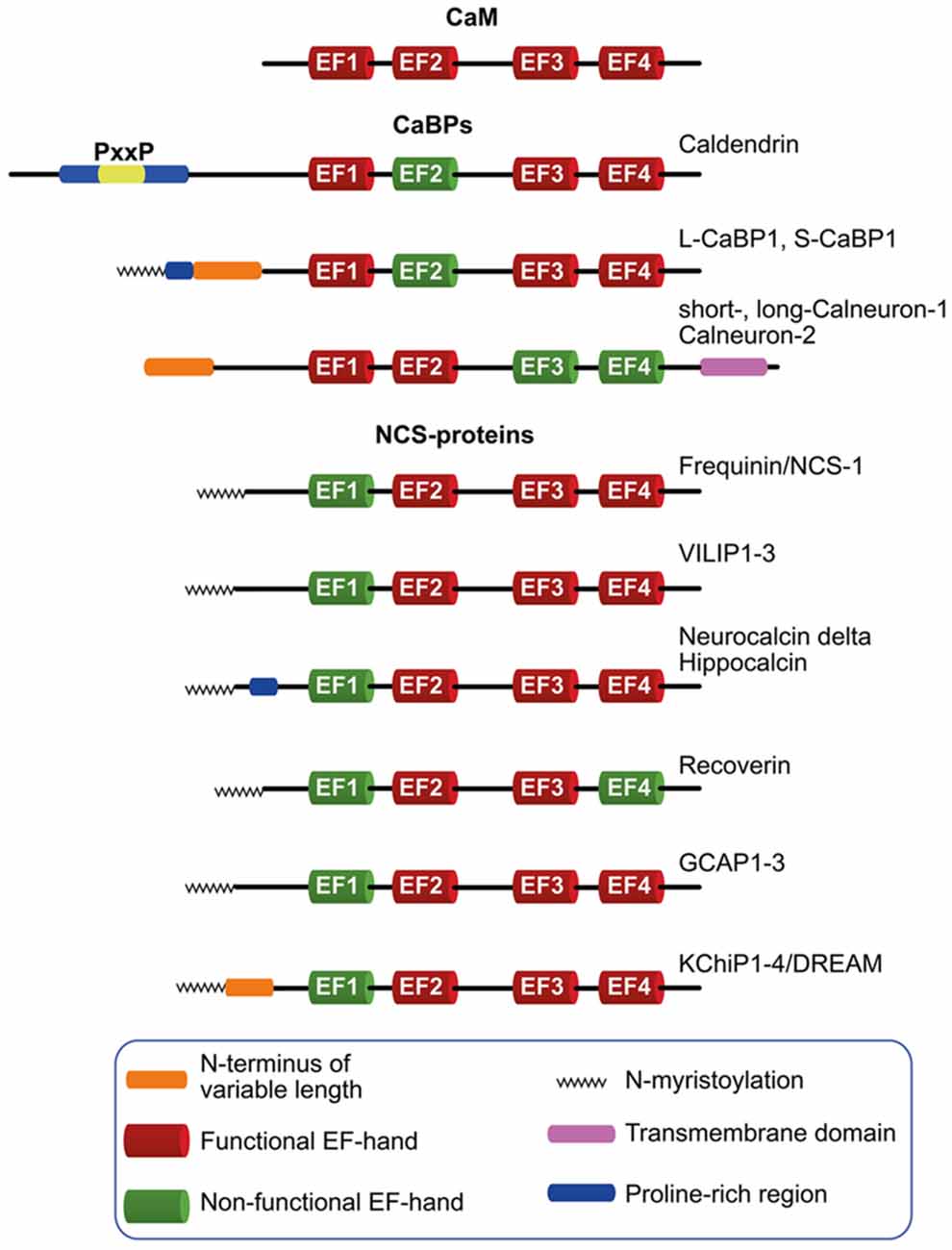
Figure 1. Schematic diagram depicting the neuronal calcium sensor (NCS) and neuronal calcium-binding proteins (nCaBPs) families. Analogous to their ancestral protein calmodulin (CaM), all nCaBPs and NCS-proteins harbor four EF-hands out of which at least one is not capable of binding Ca2+ and is therefore referred to as non-functional (green). All NCS family members carry a N-myristoylation (zigzag line) which can be masked in a Ca2+-dependent manner in VILIP-proteins, Hippocalcin and Recoverin. Caldendrin, L-CaBP1 and S-CaBP1 share the same EF-hand organization with different N-termini which might define the subcellular distribution of the proteins. The extended N-terminus of caldendrin is characterized by the presence of tandem PxxP motifs. Compared to other nCaBPs and NCS-proteins, calneurons harbor an extension of the C-terminus which is crucial for their targeting to the Golgi-membrane, where they are involved in the regulation of Golgi to plasma membrane trafficking.
The nCaBP family consists of six family members, caldendrin, L-CaBP1, S-CaBP1, short- and long-calneuron-1, calneuron-2 (Figure 1). Caldendrin harbors a cryptic EF-hand 2, whereas in calneuron-1 and -2 EF-hands 3 and 4 are non-functional (Figure 1; Mikhaylova et al., 2006; McCue et al., 2010). This excludes interdomain cooperativity in Ca2+-binding like it was reported for other CaM-like EF-hand Ca2+-sensor proteins (Fefeu et al., 2000; Heidarsson et al., 2013; Ranaghan et al., 2013) including caldendrin (Kiran et al., 2017). Two shorter isoforms arise from alternative splicing of the caldendrin/CaBP1 gene that are denoted L-CaBP1 and S-CaBP1, respectively. Similar to caldendrin, L-CaBP1 and S-CaBP1 harbor a non-functional EF-hand 2 (Haeseleer et al., 2000; Laube et al., 2002). Caldendrin is prominently expressed in brain, whereas S- and L-CaBP1 like all other CaBPs are mainly abundant in retina and the cochlea (Seidenbecher et al., 1998; Haeseleer et al., 2000; Laube et al., 2002; Landwehr et al., 2003; Kim et al., 2014). Thus, in addition to caldendrin, from the nCaBP family only calneuron-1 and -2 are abundant in brain (Hradsky et al., 2015).
Structural Features and Biophysical Properties of Caldendrin
Caldendrin exhibits a unique bipartite structure with a N-terminal half that contains seven phosphorylation sites with an unusual high proline content (13%) and multiple PxxP motifs (Figure 1). The N-terminus shows no sequence homology to other family members. The shorter splice isoforms share the C-terminus with caldendrin (Figure 1) that resembles the structure of CaM (Figure 1). In contrast to CaM, however, the first EF-hand motif will bind both Mg2+ and Ca2+ while the second EF-hand is not capable of Ca2+ binding. The global affinity of Ca2+ and Mg2+ binding was found to be in the low μM range (Wingard et al., 2005; Reddy et al., 2014). Since the Mg2+-binding affinity is clearly higher than in S-CaBP1 it is likely that the extended N-terminus will impact Mg2+-binding to the first EF-hand (Wingard et al., 2005; Reddy et al., 2014). Several lines of evidence suggest complex intra- and intermolecular interactions of caldendrin (Reddy et al., 2014). However, the protein shows very little change conformational change as evidenced by alterations in surface hydrophobicity and secondary as well as tertiary structure upon Ca2+-binding to the Mg2+-saturated protein (Reddy et al., 2014). Caldendrin is in the C-terminal part the closest homolog of CaM in brain and shares with its ancestor a flexible linker region between both EF-hand domains. But in case of caldendrin insertion of four additional amino acids will allow for a more flexible orientation of both domains that might allow for binding to sterically more complex target interactions (Seidenbecher et al., 1998; Haeseleer et al., 2000; Laube et al., 2002). It was shown that the folding of the EF-hand domains can occur independently and in any order in CaM and probably also all other nCaBP. Unfortunately, still very little is known about the structure-function relationships in relation to ion binding properties of this sensor. Although NMR and crystal structures have been reported for the shorter isoforms (Li et al., 2009; Findeisen and Minor, 2010), the unique N-terminal region and reports pointing towards functional differences between caldendrin and the shorter splice isoforms asks for an independent analysis of their function (Tippens and Lee, 2007; Findeisen and Minor, 2010). Along these lines the rather complex inter- and intramolecular interactions in Caldendrin that are regulated by Ca2+-binding, the exceptionally high Mg2+- and the relatively low Ca2+-binding affinity, the rigid first EF-hand domain and the rather modest changes in confirmation upon binding of Ca2+ make it plausible that the association with binding partners might follow different molecular principles in comparison to other neuronal Ca2+-sensors.
The Caldendrin Interactome and Cellular Functions of Caldendrin
Caldendrin is enriched at synapses and in the somatodendritic compartment of pyramidal neurons in cortex and hippocampus (Seidenbecher et al., 1998; Bernstein et al., 2007). Synaptic [Ca2+]i levels can be increased by activation of L-type voltage activated Ca2+ channels like CaV1.2, N-methyl-D-aspartate receptors (NMDARs) or Inositol trisphosphate receptors (InsP3Rs; Sala and Segal, 2014). It is well established that the activity of InsP3Rs is regulated by CaM (Kasri et al., 2002), however, several reports also indicate that CaBP1, can modulate the activity of InsP3Rs (Haynes et al., 2004; Kasri et al., 2004; Li et al., 2009, 2013). Of note, two binding sites for CaBP1 were identified in InsP3Rs and a high affinity interaction requires self-association and a close proximity of both binding domains (Li et al., 2009). Although binding was originally described in contrast to those of CaM to be Ca2+-independent, it turned out that Ca2+-binding to EF-hands-3 and -4 clearly enhances binding affinity and strengthens the interaction (Li et al., 2009). In addition, it was reported that phosphorylation of CaBP1 by casein kinase II promotes the association (Kasri et al., 2004). In functional terms CaBP1 binding reduces InsP3-triggered [Ca2+]i release (Haynes et al., 2004; Kasri et al., 2004). Based on NMR studies, it was postulated that a cluster of hydrophobic residues in the C-terminal domain of CaBP1 interact with a complementary cluster of hydrophobic residues in the β-trefold domain of InsP3R, trapping the channel in a closed conformation (Li et al., 2013). Upon binding of Ca2+ the affinity of CaBP1 was found to be increased for InsP3R, locking the channel in the closed state (Li et al., 2009, 2013). Importantly, Li et al. (2009) could show that binding of CaBP1 to InsP3R is much stronger than those of CaM to InsP3Rs, indicating that the former interaction will easily dominate and is more significant for InsP3R function and given the abundance of Caldendrin in spines it will clearly outcompete CaM. Furthermore, the low expression of CaBP1 in the hippocampus makes it likely that caldendrin is the Ca2+ sensor for regulating hippocampal InsP3Rs and might thereby play an important role in synapto-dendritic Ca2+ signaling as a prominent inhibitor of InsP3Rs in the hippocampus, a prediction that can be easily tested now due to the existence of caldendrin/CaBP1 knockout mice models (Kim et al., 2014; Mikhaylova et al., 2018; Yang et al., 2018).
Several reports indicate that the association the association of caldendrin/CaBP1 with the α1-C subunit of L-type voltage-dependent Ca2+ channels (CaV1.2) is important for its physiological function (Zhou et al., 2004, 2005; Tippens and Lee, 2007; Findeisen and Minor, 2010; Oz et al., 2011, 2013). Caldendrin/CaBP1 reduce the Ca2+ dependent inactivation (CDI) of CaV1.2 L-type voltage gated Ca2+ channels and thereby facilitate Ca2+ currents (Zhou et al., 2004, 2005; Tippens and Lee, 2007; Oz et al., 2011, 2013). In contrast, binding of CaM reportedly results in an inactivation of Ca2+ influx and competes with caldendrin/CaBP1 for the same interaction site (Zhou et al., 2004, 2005; Tippens and Lee, 2007). It is plausible that the interaction takes place in the postsynaptic membrane since co-immunoprecipitation of caldendrin and CaV1.2 Ca2+ channels was shown with synaptic protein preparations (Zhou et al., 2004, 2005; Tippens and Lee, 2007). The emerging scenario from several studies shows that the regulation of CaV1.2 Ca2+ channels by caldendrin, CaBP1 and CaM is complex and that differential binding will allow for a high degree of fine-tuning of synapto-dendritic Ca2+ signaling. All three sensors interact with an IQ-domain that is part of the C-terminal cytoplasmic domain of the α1-subunit of CaV1.2. In addition, CaBP1 binds to a second region in the N-terminus and this interaction is in contrast to those of CaM Ca2+-independent (Zhou et al., 2005; Dick et al., 2008). Of note, the presence of this domain is essential for inactivation of CaV1.2 Ca2+ currents by Caldendrin, but not the CDI mediated by CaM (Zhou et al., 2004). Interestingly, CaBP1 seems to more efficiently inhibit inactivation of CaV1.2 channels than caldendrin (Zhou et al., 2005; Tippens and Lee, 2007). Whereas caldendrin displaces CaM and CaBP1 from the C-terminal IQ-domain it does not interact with the N-terminus of CaV1.2 (Tippens and Lee, 2007). It is therefore likely that the functionally different roles of CaM, CaBP1 and caldendrin are mediated by association to two different domains with different Ca2+ requirements.
Other interactions have been studied in much less detail. In functional terms a role in synapse-to-nucleus communication has been ascribed to caldendrin by preventing the nuclear import of the synapto-nuclear messenger Jacob (Dieterich et al., 2008). Caldendrin/CaBP1 knockout mice show a rapid depression at inhibitory presynaptic sites that is related to binding and inactivation of CaV2.1 calcium channels in control of short-term synaptic plasticity (Lee et al., 2002; Few et al., 2011; Leal et al., 2012; Nanou et al., 2018). Other potential binding partners where a cellular and in particular synaptic function is less well investigated include light chain 3 (Seidenbecher et al., 2004), myo1c (Tang et al., 2007), recoverin (Fries et al., 2010), metabotropic glutamate receptors (Nakajima, 2011), AKAP79/150 (Seeger et al., 2012) and CaV1.3 calcium channels (Findeisen et al., 2013; Yang et al., 2018).
A Specific Synaptic Function of Caldendrin
Interestingly, caldendrin is highly enriched in the postsynaptic density (PSD) of spine synapses, which is very uncommon for a CaM-like Ca2+-sensor protein (Seidenbecher et al., 1998; Laube et al., 2002). Dendritic spines are considered as microcompartments of Ca2+ signaling (Raghuram et al., 2012) with exceptionally fast Ca2+ decay times, much faster than in dendrites (Cornelisse et al., 2007). The presence of “fast” Ca2+ buffers are thought to mediate the fast decay of Ca2+-transients in spines immediately following influx through Ca2+ channels or release from Ca2+ stores (Keller et al., 2008). Fast buffers are the first to intercept free Ca2+ in the spine. Surprisingly, it was found utilizing UV flash photolysis of DM-nitrophen caged Ca2+ that CaM binds Ca2+ with a very high on-rate at the N-terminal lobe, even faster than those of classical known calcium buffers like calbindin (Faas et al., 2011). In conclusion, CaM, due to high abundance, its ubiquitous expression and the fast Ca2+-binding capacity, has to be considered as the principal buffer in pyramidal neurons. In addition numerous Ca2+-dependent targets have been identified which in regulate different aspects of cellular function. In light of these arguments the simple question arises how members of the NCS and nCaBP family operate as calcium sensors in the presence of CaM? Or in other terms will CaM-like sensors even have a chance to compete with CaM for Ca2+ binding in spines?
In a recently published study we addressed this question systematically (Mikhaylova et al., 2018). We first found that caldendrin has a similar on rate like CaM for Ca2+-binding and can therefore by abundance and properties easily compete with CaM in spines (Mikhaylova et al., 2018). Alterations in synaptic strength require an intimate link between functional and structural plasticity. The latter one is based on the unique cytoskeletal organization of differentially arranged actin filaments. Compartmentalization of calcium-dependent plasticity allows for rapid actin remodeling and in a landmark study, Bosch et al. (2014) addressed the question of structural and molecular remodeling of dendritic spines following the induction of long-term potentiation (LTP). LTP induction results in rapid cofilin-dependent severing of filamentous actin and a massive increase in actin remodeling proteins, which is then followed by a stabilization phase where different F-actin stabilizing and capping proteins gradually increase and only at this later stage reorganization of the PSD comes into play. What is still unclear is how a minimal stable pool of branched actin that is essential for remodeling of F-actin is maintained in spines undergoing plasticity. In fact, molecular machineries coupling local and rapid synaptic elevation of [Ca2+]i to actin remodeling in the initial reorganization phase are still unknown. Finally, the role of spatially segregated spinous F-actin pools in transition from short-term to long-term synaptic potentiation was unclear.
We could show that caldendrin activates the actin-binding protein cortactin in dendritic spines and thereby stabilizes a synaptic pool of branched F-actin that is essential for the maintenance of LTP (Mikhaylova et al., 2018). We found that steep elevations in spinous [Ca2+]i disrupt an intramolecular interaction of caldendrin that hinders access to a series of PxxP-motifs. Opening of the intramolecular interaction results in a rapid association with the SH3 domain of cortactin (Figure 2). A fast on and a slow off rate of binding keep cortactin in an active, F-actin-stabilizing conformation. In this conformation the protein protects a minimal stable spinous F-actin pool against cofilin-induced severing and primes cortactin for sequential binding to N-WASP/Arp2/3 complex in vicinity of the PSD. Caldendrin gene knockout or protein knockdown result in higher actin turnover caused by loss of stable pool of actin filaments and a disordered topology of spinous F-actin which lead to a loss of spatial segregation of F-actin nanodomains, defects in structural spine plasticity, LTP and hippocampus dependent learning (Mikhaylova et al., 2018). We think that caldendrin directly couples elevation of [Ca2+]i to the stabilization of actin branches in a very early step of temporary gating for F-actin remodeling in dendritic spines and thus controls the life time of functionally different F-actin pools during the reorganization phase (Figure 2). This mechanism is an essential component of structural plasticity. Accordingly, it was reported in a recent study (Yang et al., 2018) that caldendrin knock-out mice exhibit deficits in spatial learning and memory and also fear-related memories. Surprisingly, it was also found that adult neurogenesis in the hippocampus is severely impaired in knock-out animals by yet unknown mechanisms (Yang et al., 2018). It is likely that this impairment will also contribute to learning and memory deficits in these mice.
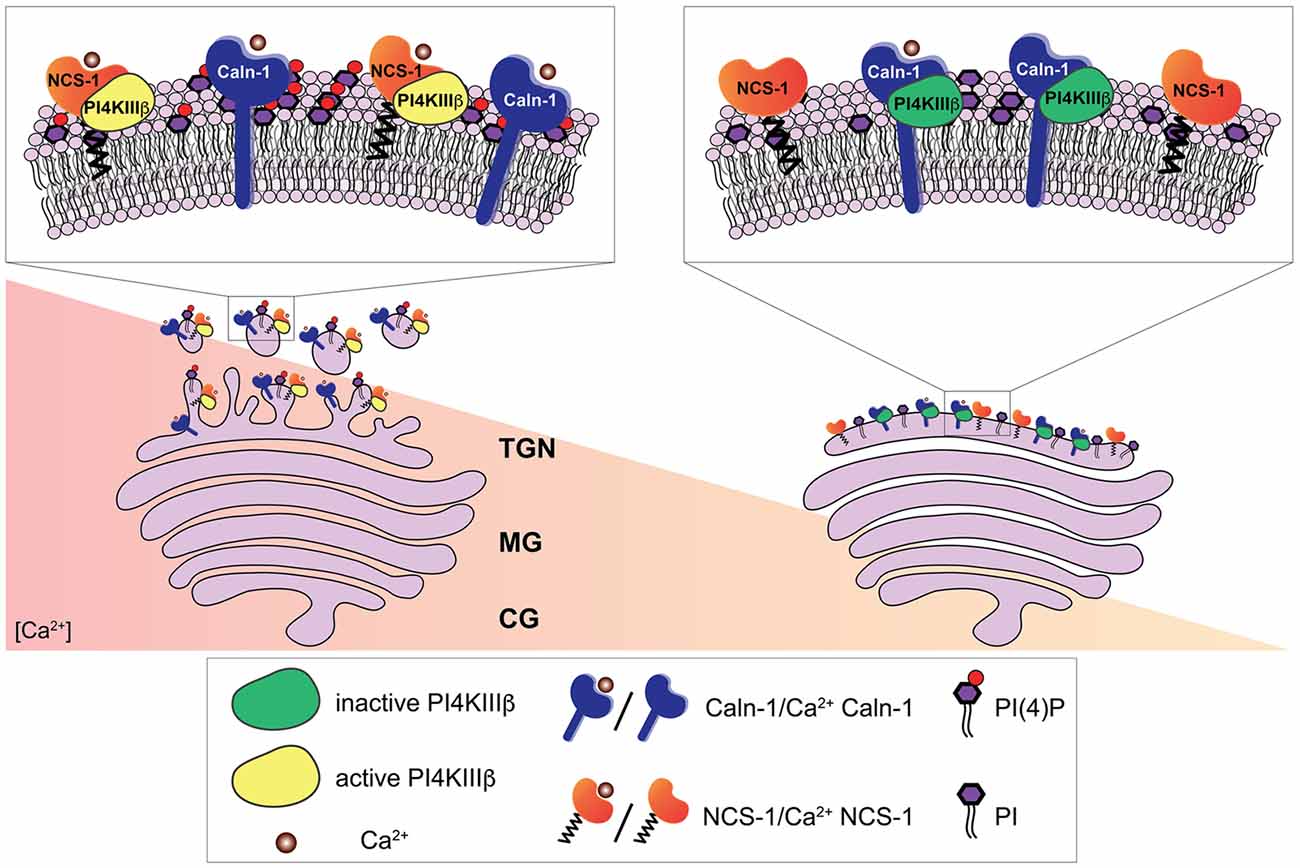
Figure 2. Ca2+-dependent regulation of the phosphatidylinositol 4-kinase IIIβ (PI4KIIIβ) by calneuron-1 (Caln-1) and NCS-1 at the trans-Golgi network (TGN). The PI4KIIIβ-catalyzed synthesis of phosphatidylinositol 4-phosphate (PI(4)P) from phosphatidylinositol (PI) at the TGN membrane is a key step in the process of vesicle budding for TGN to plasma membrane trafficking. Ca2+-bound calneuron-1 inhibits the activity of PI4KIIIβ at basal intracellular Ca2+ concentrations, [Ca2+], (right). Middle and high Ca2+ concentration (left) can lead to a fully Ca2+-bound NCS-1 protein, which then can replace calneuron-1 from PI4KIIIβ thus leading to a strong increase in the production of PI(4)P and vesicle membrane budding.
The Transmembrane nCaBP Family Members Calneuron-1 and -2
Calneuron-1 and -2 (also called CaBP8 and CaBP7; McCue et al., 2010) configure a separate subfamily within the nCaBP family (Mikhaylova et al., 2006, 2011). In contrast to other NCS- and nCaBP proteins they harbor a non-functional C-terminal EF-hand domain (Figure 1; Wu et al., 2001; Mikhaylova et al., 2006; McCue et al., 2010). Both calneurons show 64% identity at the amino acid level when compared to each other whereas less than 30% identity exists in comparison to CaBPs and caldendrin (Mikhaylova et al., 2006; McCue et al., 2010). The loops of the functional EF-hands 1 and 2 are almost identical (Mikhaylova et al., 2006) and also the loops of EF-hand 3 exhibit high similarity. Calneurons have the highest Ca2+-affinity of all identified CaM-like EF-hand Ca2+-sensors with a global dissociation constant of 230 and 180 nM for calneuron-1 and -2, respectively (Mikhaylova et al., 2009). However, these affinities were measured for proteins without membrane insertion, which might have obscured the outcome of these measurements. Apart from differences within the EF-hand domains calneurons harbor in addition an extension of the C-terminus compared to other nCaBPs and NCS-proteins, which is crucial for the targeting of these proteins to the Golgi-membrane (McCue et al., 2010; Hradsky et al., 2011), where they are involved in the regulation of Golgi to plasma membrane trafficking (Mikhaylova et al., 2009). Being almost identical in both proteins the C-terminus contains a highly hydrophobic stretch, which configures a transmembrane domain (TMD; Hradsky et al., 2011; McCue et al., 2011). Since both calneurons do not harbor a N-terminal myristoylation motif, transmembrane insertion of these proteins would explain their strong membranal association as compared to other neuronal Ca2+-sensors like caldendrin. Of note they are the only transmembrane CaM-like calcium sensors and this points to a probably unique role in the neuronal Ca2+-sensing toolkit.
Calneuron-1 and -2 Are Tail Anchored Proteins That Regulate Golgi-to-Plasma Membrane Trafficking
The C-terminal position of the transmembrane segment indicates that calneurons belong to the group of tail-anchored proteins, which have to undergo post-translational insertion (Borgese et al., 2003, 2007). It turned out that calneurons are indeed non-classical type II tail-anchored proteins and their posttranslational insertion into the ER membrane via an association of the TMD with the TRC40/Asna1 chaperone complex was demonstrated (Hradsky et al., 2011). Their tight association with the trans-Golgi-network (TGN) might be explained by the length of the TMD and phosphatidylinositol 4-phosphate (PI(4)P) lipid binding (Hradsky et al., 2011). Self-association in vitro and in vivo occurs via the TMD and EF-hand containing N-terminus. Despite the fact that dimerization will hinder TRC40/Asna1 binding and in consequence membrane insertion, evidence for the existence of a cytosolic non-membrane associated pool of calneurons is currently lacking and dimerization was only found for membrane inserted protein (Hradsky et al., 2011). This almost exclusive and unique association with membranes of the secretory pathway indicates a probably highly specific function with a limited number of target interactions.
In functional terms calneurons play an important role at the Golgi-apparatus where they control TGN to plasma membrane trafficking by regulating the activity of phosphatidylinositol 4-OH kinase IIIβ (PI-4KIIIβ; Mikhaylova et al., 2009). PI-4KIIIβ catalyzes local synthesis of phosphoinositides necessary for vesicle budding at the TGN. Calneurons directly bind to PI-4KIIIβ and inhibit the enzyme at low [Ca2+] levels (Figure 3; Mikhaylova et al., 2009). With increased [Ca2+] levels the inhibition is released and PI4KIIIβ is activated via a preferential association with NCS-1. Taken together that data suggest that calneurons establish a [Ca2+] threshold for activation of the enzyme (Figure 3; Mikhaylova et al., 2009). Of note, the tight association of calneurons to the Golgi can be even used to target proteins to the TGN (Bera et al., 2016). The TMD of calneuron-2 was employed to develop a plasmid-based expression system called pGolt that has the advantage to fuse other proteins to the extraluminal part. This in turn makes it possible to study protein-protein interactions outside of the Golgi lumen (Bera et al., 2016). An obvious and particularly promising application in neuroscience is to use this Golgi-tracker system for the visualization of Golgi outposts (GOs). In non-neuronal cells the organelles of the secretory pathway have a highly restricted spatial organization. In stark contrast in neuronal cells along with the localization of secretory organelles in the cell soma, numerous discrete and discontinuous structures resembling Golgi cisternae are present along dendrites, which are known as GOs. We next showed with pGolt the presence of Golgi-related organelles in all dendrites of pyramidal neurons in close proximity to endoplasmic reticulum-Golgi intermediate compartment and retromer (Mikhaylova et al., 2016). We found that this Golgi-Satellite secretory system (GS) in dendrites is much more widespread than previously described GOs. Most importantly, this GS contains at least part of the cellular glycosylation machinery but as opposed to GOs lacks many protein components for sorting and organization of Golgi cisternae. Moreover, we realized that a broad spectrum of synaptic transmembrane proteins (including GluA1, GluN1, GluN2B, NCAM and Neuroligin-1) might pass and even recycle through these organelles and that also calneuron-1 is present at GS (Mikhaylova et al., 2016). Collectively the study suggest that GS will enable local glycosylation of proteins, and that these proteins will be subsequently recruited to membranes in spatially confined dendritic segments. It will be interesting to investigate in the future whether calneuron-1 has a similar role at GS like at the TGN. Of note, another report (Rajamanoharan et al., 2015) indicated that calneuron-2 mediated inhibition of PI4KIIIβ is instrumental for cytokinesis in HeLa cells. In this study, it was reported that calneuron-2 associates with lysosomes and regulates their clustering and that PI4KIIIβ plays an important role for normal cytokinesis (Rajamanoharan et al., 2015).
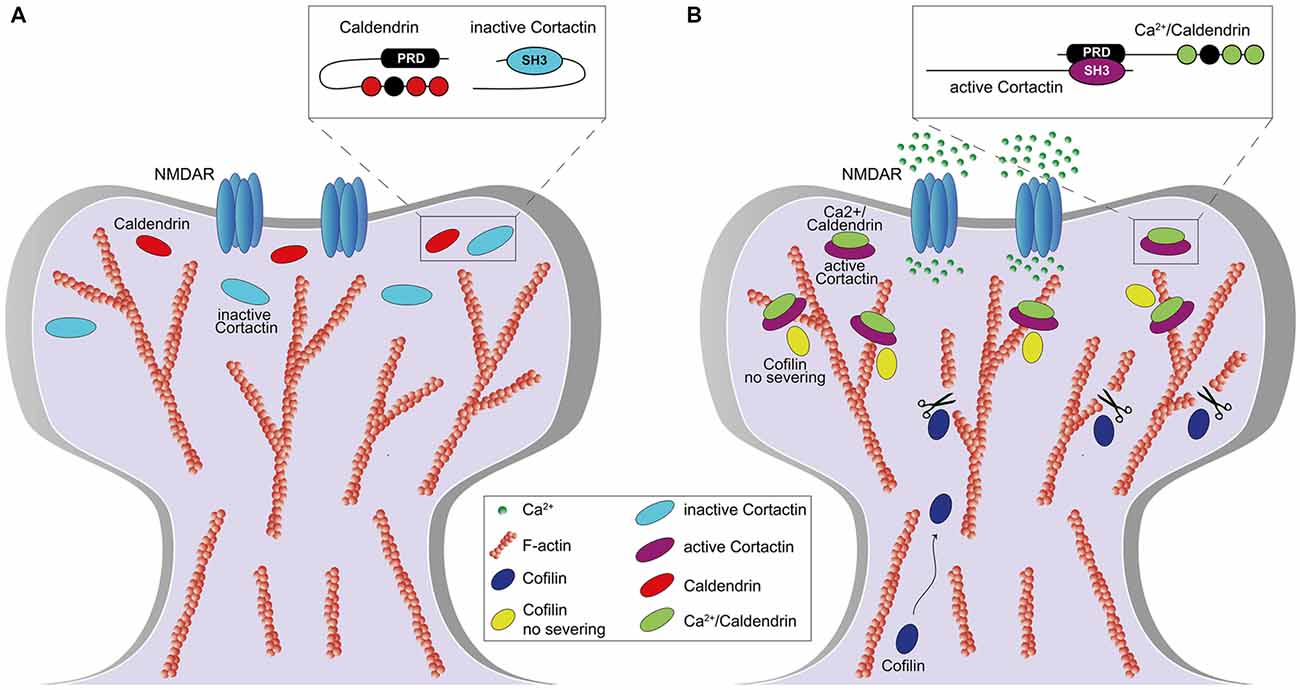
Figure 3. Role of caldendrin and cortactin in the stabilization of actin filaments in the initial phase of synaptic potentiation. (A) Under basal condition, caldendrin and cortactin are kept in an inactive state which prevents the exposure of caldendrin N-terminal proline-rich domain (PRD) and cortactin SH3-domain (A, inset). (B) Synaptic stimulation, via N-methyl-D-aspartate receptor (NMDAR) activation, leads to an influx of Ca2+ which is bound by caldendrin. Ca2+ binding results in a conformational change of caldendrin with subsequent exposure of the PRD which associates with the SH3-domain of cortactin, keeping cortactin in an active and open conformation (B, inset). Caldendrin-cortactin complex is then anchored to F-actin and prevents cofilin-mediated filament severing. At the same time, a rate of caldendrin-cortactin-free F-actin filaments are severed by cofilin, whose levels increase upon NMDARs activation, allowing generation of new filaments and reorganization of the spine.
Other Possible Cellular Functions of Calneurons in Neuronal Ca2+-Signaling
Compared to the multitude of CaM protein interactions, the interactome of nCaBPs and here in particular of calneurons is relatively small. In several studies an interaction with G-protein coupled receptors (GPCRs) was reported (Navarro et al., 2014; Angelats et al., 2018; Franco et al., 2018). It is well established in striatal neurons that [Ca2+]i impact modulation of A2AR-D2R heteromers that is mediated adenylyl-cyclase and MAP-kinases. Differential modulation is based on the interaction of the heteromer with NCS-1 and calneuron-1 at low and high [Ca2+]i, respectively (Navarro et al., 2016). The association with both Ca2+-binding proteins appears to differentially regulate subsequent allosteric interactions within the A2AR-D2R heteromer. Thus, binding of both proteins configures a unique cellular mechanism to integrate extracellular (adenosine and dopamine) and [Ca2+]i signals in order to elicit a specific downstream signaling event (Navarro et al., 2016). In the past years further interactions with either GPCR heteromers or heteromers of GPCR with glutamate receptors were reported (Angelats et al., 2018; Franco et al., 2018) and the emerging picture suggests that: (i) [Ca2+]i determines the binding affinity of different sensors; and that (ii) in case of cooperative binding differential intracellular responses can be elicited depending upon binding. What is clearly missing is an understanding of the cellular function of these interactions. This also holds true for work that shows an interaction between calneuron-1 and N-type calcium channels (Shih et al., 2009), an interaction that could play a role in the modulation of action potential firing or neurotransmitter release. Finally, calneuron-1 is highly expressed in aldosterone producing adenoma cells where it regulates the storage of Ca2+ into the ER (Kobuke et al., 2017). Overexpression of calneuron-1 leads to an increased Ca2+ level in the ER and aldosterone overproduction (Kobuke et al., 2017). A mechanistic explanation for this phenotype, however, is lacking.
On a Putative Role of Calneuron-1 and Caldendrin in Mental Disorders
The onset of schizophrenia usually takes place in young adulthood between the age of 20–30 (Thompson et al., 2004). It is widely believed that subtle developmental alterations in brain structure and connectivity in the interaction with environmental factors cause schizophrenia in early adulthood. The underlying processes have been also related to modifications of the epigenome. Three relatively recent studies from different groups propose that the human CALN1 is a candidate schizophrenia gene (Li et al., 2015; Xia et al., 2015; Roussos et al., 2016) but it is at present unclear how calneuron-1 function can be related to psychotic behavior. However, it has been shown that alteration in methylation levels on the CALN1 and the AUTS2 gene occur in schizophrenia patients (Wockner et al., 2014). A recently published study (Engmann et al., 2017) shows that the exposure to drugs of abuse, like cocaine, alters epigenetic patterns of certain brain regions due to chromatin modifications. Engmann et al. (2017) found that the methylation of the CALN1 promoter increases the expression of AUTS2, which is another known risk gene for schizophrenia (Zhang et al., 2014), and vice versa. They show that methylation of the AUTS2 or the CALN1 promoter enhances calneuron-1 expression although both genes are not in close proximity to each other. The distance of around 1542 Kb between both genes is bypassed by an epigenetic modification called chromatin looping, which enables regulatory elements such as enhancers, which are located relatively far from the transcription start site, to interact with the associated promoter regions using CCCTC-binding-factors (Sexton et al., 2009; Engmann et al., 2017). Interestingly, a meta-analysis of the genetic association studies came to the conclusion that calneuron-1 mRNA levels might be up-regulated in schizophrenia in the dorsolateral prefrontal cortex (Ohi et al., 2016). A potential mechanism might have been proposed by Engmann et al. (2017), whose results are indicating that an overexpression of CALN1 might lead to an overexpression of AUTS2, a well-known risk gene for schizophrenia, via chromatin looping. Alternatively, higher calneuron-1 protein levels might have independently an effect on cellular signaling that is related to psychotic behavior.
Interestingly, caldendrin protein levels are also regulated in human schizophrenia (Bernstein et al., 2007) and in mouse models of drug-induced psychosis (Smalla et al., 2009). Fewer caldendrin-immunopositive neurons are found in the left dorsolateral prefrontal cortex in schizophrenia patients, and it is tempting to speculate that synaptic and/or dendritic Ca2+-signaling is altered in schizophrenia due to a redistribution of the protein. Thus, it appears that the remaining pyramidal neurons expressing caldendrin exhibit higher protein levels. In summary, although it is at present unclear how exactly both proteins contribute to the pathophysiology of schizophrenia, the basic characterization of their unique cellular role of calneuron-1 might pave the way to understand how they are involved in schizophrenia.
Author Contributions
JM, CF, MK wrote the review article. CF designed the figures.
Funding
The work in the lab of MK is supported by grants from the Deutsche Forschungsgemeinschaft (DFG; Kr1879/5-1/6-1/SFB 779 TPB8), BMBF “Energi” FKZ: 01GQ1421B, The EU Joint Programme—Neurodegenerative Disease Research (JPND) project STAD and the Leibniz Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Angelats, E., Requesens, M., Aguinaga, D., Kreutz, M. R., Franco, R., and Navarro, G. (2018). Neuronal calcium and cAMP cross-talk mediated by cannabinoid CB1 receptor and EF-hand calcium sensor interactions. Front. Cell Dev. Biol. 6:67. doi: 10.3389/fcell.2018.00067
Bera, S., Raghuram, V., Mikhaylova, M., and Kreutz, M. R. (2016). A plasmid-based expression system to study protein-protein interactions at the Golgi in vivo. Anal. Biochem. 502, 50–52. doi: 10.1016/j.ab.2016.02.016
Bernstein, H., Sahin, J., Smalla, K., Gundelfinger, E., Bogerts, B., and Kreutz, M. (2007). A reduced number of cortical neurons show increased Caldendrin protein levels in chronic schizophrenia. Schizophr. Res. 96, 246–256. doi: 10.1016/j.schres.2007.05.038
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529. doi: 10.1038/nrm1155
Borgese, N., Brambillasca, S., and Colombo, S. (2007). How tails guide tail-anchored proteins to their destinations. Curr. Opin. Cell Biol. 19, 368–375. doi: 10.1016/j.ceb.2007.04.019
Borgese, N., Colombo, S., and Pedrazzini, E. (2003). The tale of tail-anchored proteins. J. Cell Biol. 161, 1013–1019. doi: 10.1083/jcb.200303069
Bosch, M., Castro, J., Saneyoshi, T., Matsuno, H., Sur, M., and Hayashi, Y. (2014). Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 82, 444–459. doi: 10.1016/j.neuron.2014.03.021
Burgoyne, R. D. (2007). Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 8, 182–193. doi: 10.1038/nrn2093
Cornelisse, L. N., van Elburg, R. A., Meredith, R. M., Yuste, R., and Mansvelder, H. D. (2007). High speed two-photon imaging of calcium dynamics in dendritic spines: consequences for spine calcium kinetics and buffer capacity. PLoS One 2:e1073. doi: 10.1371/journal.pone.0001073
Dick, I. E., Tadross, M. R., Liang, H., Tay, L. H., Yang, W., and Yue, D. T. (2008). A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature 451, 830–834. doi: 10.1038/nature06529
Dieterich, D. C., Karpova, A., Mikhaylova, M., Zdobnova, I., König, I., Landwehr, M., et al. (2008). Caldendrin-jacob: a protein liaison that couples NMDA receptor signalling to the nucleus. PLoS Biol. 6:e34. doi: 10.1371/journal.pbio.0060034
Engmann, O., Labonté, B., Mitchell, A., Bashtrykov, P., Calipari, E. S., Rosenbluh, C., et al. (2017). Cocaine-induced chromatin modifications associate with increased expression and three-dimensional looping of Auts2. Biol. Psychiatry 82, 794–805. doi: 10.1016/j.biopsych.2017.04.013
Faas, G. C., Raghavachari, S., Lisman, J. E., and Mody, I. (2011). Calmodulin as a direct detector of Ca2+ signals. Nat. Neurosci. 14, 301–304. doi: 10.1038/nn.2746
Fefeu, S., Biekofsky, R. R., Mccormick, J. E., Martin, S. R., Bayley, P. M., and Feeney, J. (2000). Calcium-induced refolding of the calmodulin v136g mutant studied by NMR spectroscopy: evidence for interaction between the two globular domains. Biochemistry 39, 15920–15931. doi: 10.1021/bi001772a
Few, A. P., Nanou, E., Scheuer, T., and Catterall, W. A. (2011). Molecular determinants of CaV2.1 channel regulation by calcium-binding protein-1. J. Biol. Chem. 286, 41917–41923. doi: 10.1074/jbc.m111.292417
Findeisen, F., and Minor, D. (2010). Crystal structure of the calcium sensor calcium-binding protein 1 (CaBP1). Structure 18, 1617–1631. doi: 10.2210/pdb3ox5/pdb
Findeisen, F., Rumpf, C. H., and Minor, D. L. Jr. (2013). Apo states of calmodulin and CaBP1 control CaV1 voltage-gated calcium channel function through direct competition for the IQ domain. J. Mol. Biol. 425, 3217–3234. doi: 10.1016/j.jmb.2013.06.024
Franco, R., Aguinaga, D., Reyes, I., Canela, E. I., Lillo, J., Tarutani, A., et al. (2018). N-methyl-D-aspartate receptor link to the MAP kinase pathway in cortical and hippocampal neurons and microglia is dependent on calcium sensors and is blocked by α-Synuclein, Tau, and Phospho-Tau in non-transgenic and transgenic APPSw,Ind mice. Front. Mol. Neurosci. 11:273. doi: 10.3389/fnmol.2018.00273
Fries, R., Reddy, P. P., Mikhaylova, M., Haverkamp, S., Wei, T., Müller, M., et al. (2010). Dynamic cellular translocation of caldendrin is facilitated by the Ca2+-myristoyl switch of recoverin. J. Neurochem. 113, 1150–1162. doi: 10.1111/j.1471-4159.2010.06676.x
Haeseleer, F., Sokal, I., Verlinde, C. L. M. J., Erdjument-Bromage, H., Tempst, P., Pronin, A. N., et al. (2000). Five members of a novel Ca2+-binding protein (CABP) subfamily with similarity to calmodulin. J. Biol. Chem. 275, 1247–1260. doi: 10.1074/jbc.275.2.1247
Haynes, L. P., Tepikin, A. V., and Burgoyne, R. D. (2004). Calcium-binding protein 1 is an inhibitor of agonist-evoked, inositol 1,4,5-trisphosphate-mediated calcium signaling. J. Biol. Chem. 279, 547–555. doi: 10.1074/jbc.m309617200
Heidarsson, P. O., Otazo, M. R., Bellucci, L., Mossa, A., Imparato, A., Paci, E., et al. (2013). Single-molecule folding mechanism of an EF-hand neuronal calcium sensor. Structure 21, 1812–1821. doi: 10.1016/j.str.2013.07.022
Hradsky, J., Bernstein, H.-G., Marunde, M., Mikhaylova, M., and Kreutz, M. R. (2015). Alternative splicing, expression and cellular localization of calneuron-1 in the rat and human brain. J. Histochem. Cytochem. 63, 793–804. doi: 10.1369/0022155415595841
Hradsky, J., Raghuram, V., Reddy, P. P., Navarro, G., Hupe, M., Casado, V., et al. (2011). Post-translational membrane insertion of tail-anchored transmembrane EF-hand Ca2+ sensor calneurons requires the TRC40/Asna1 protein chaperone. J. Biol. Chem. 286, 36762–36776. doi: 10.1074/jbc.M111.280339
Kasri, N. N., Bultynck, G., Sienaert, I., Callewaert, G., Erneux, C., Missiaen, L., et al. (2002). The role of calmodulin for inositol 1,4,5-trisphosphate receptor function. Biochim. Biophys. Acta 1600, 19–31. doi: 10.1016/S1570-9639(02)00440-5
Kasri, N. N., Holmes, A. M., Bultynck, G., Parys, J. B., Bootman, M. D., Rietdorf, K., et al. (2004). Regulation of InsP3 receptor activity by neuronal Ca2+-binding proteins. EMBO J. 23, 312–321. doi: 10.1038/sj.emboj.7600037
Keller, D. X., Franks, K. M., Bartol, T. M., and Sejnowski, T. J. (2008). Calmodulin activation by calcium transients in the postsynaptic density of dendritic spines. PLoS One 3:e2045. doi: 10.1371/journal.pone.0002045
Kim, K., Scholl, E., Liu, X., Shepherd, A., Haeseleer, F., and Lee, A. (2014). Localization and expression of CaBP1/caldendrin in the mouse brain. Neuroscience 268, 33–47. doi: 10.1016/j.neuroscience.2014.02.052
Kiran, U., Regur, P., Kreutz, M. R., Sharma, Y., and Chakraborty, A. (2017). Intermotif communication induces hierarchical Ca2+ filling of caldendrin. Biochemistry 56, 2467–2476. doi: 10.1021/acs.biochem.7b00132
Kobuke, K., Oki, K., Gomez-Sanchez, C. E., Gomez-Sanchez, E. P., Ohno, H., Itcho, K., et al. (2017). Calneuron 1 increased Ca2+ in the endoplasmic reticulum and aldosterone production in aldosterone-producing adenoma. Hypertension 71, 125–133. doi: 10.1161/hypertensionaha.117.10205
Landwehr, M., Redecker, P., Dieterich, D. C., Richter, K., Böckers, T. M., Gundelfinger, E. D., et al. (2003). Association of Caldendrin splice isoforms with secretory vesicles in neurohypophyseal axons and the pituitary. FEBS Lett. 547, 189–192. doi: 10.1016/s0014-5793(03)00713-0
Laube, G., Seidenbecher, C., Richter, K., Dieterich, D., Hoffmann, B., Landwehr, M., et al. (2002). The neuron-specific Ca2+-binding protein caldendrin: gene structure, splice isoforms, and expression in the rat central nervous system. Mol. Cell. Neurosci. 19, 459–475. doi: 10.1006/mcne.2001.1078
Leal, K., Mochida, S., Scheuer, T., and Catterall, W. A. (2012). Fine-tuning synaptic plasticity by modulation of CaV2.1 channels with Ca2+ sensor proteins. Proc. Natl. Acad. Sci. U S A 109, 17069–17074. doi: 10.1073/pnas.1215172109
Lee, A., Westenbroek, R. E., Haeseleer, F., Palczewski, K., Scheuer, T., and Catterall, W. A. (2002). Differential modulation of Cav2.1 channels by calmodulin and Ca2+-binding protein 1. Nat. Neurosci. 5, 210–217. doi: 10.1038/nn805
Li, C., Chan, J., Haeseleer, F., Mikoshiba, K., Palczewski, K., Ikura, M., et al. (2009). Structural insights into Ca2+-dependent regulation of inositol 1,4,5-trisphosphate receptors by CaBP1. J. Biol. Chem. 284, 2472–2481. doi: 10.1074/jbc.m806513200
Li, C., Enomoto, M., Rossi, A. M., Seo, M.-D., Rahman, T., Stathopulos, P. B., et al. (2013). CaBP1, a neuronal Ca2+ sensor protein, inhibits inositol trisphosphate receptors by clamping intersubunit interactions. Proc. Natl. Acad. Sci. U S A 110, 8507–8512. doi: 10.1073/pnas.1220847110
Li, Z., Xiang, Y., Chen, J., Li, Q., Shen, J., Liu, Y., et al. (2015). Loci with genome-wide associations with schizophrenia in the Han Chinese population. Br. J. Psychiatry 207, 490–494. doi: 10.1192/bjp.bp.114.150490
McCue, H. V., Burgoyne, R. D., and Haynes, L. P. (2011). Determination of the membrane topology of the small EF-hand Ca2+-sensing proteins CaBP7 and CaBP8. PLoS One 6:e17853. doi: 10.1371/journal.pone.0017853
McCue, H. V., Haynes, L. P., and Burgoyne, R. D. (2010). The diversity of calcium sensor proteins in the regulation of neuronal function. Cold Spring Harb. Perspect. Biol. 2:a004085. doi: 10.1101/cshperspect.a004085
Mikhaylova, M., Bär, J., van Bommel, B., Schätzle, P., YuanXiang, P., Raman, R., et al. (2018). Caldendrin directly couples postsynaptic calcium signals to actin remodeling in dendritic spines. Neuron 97, 1110.e14–1125.e14. doi: 10.1016/j.neuron.2018.01.046
Mikhaylova, M., Bera, S., Kobler, O., Frischknecht, R., and Kreutz, M. R. (2016). A dendritic golgi satellite between ERGIC and retromer. Cell Rep. 14, 189–199. doi: 10.1016/j.celrep.2015.12.024
Mikhaylova, M., Hradsky, J., and Kreutz, M. R. (2011). Between promiscuity and specificity: novel roles of EF-hand calcium sensors in neuronal Ca2+ signalling. J. Neurochem. 118, 695–713. doi: 10.1111/j.1471-4159.2011.07372.x
Mikhaylova, M., Reddy, P. P., Munsch, T., Landgraf, P., Suman, S. K., Smalla, K.-H., et al. (2009). Calneurons provide a calcium threshold for trans-Golgi network to plasma membrane trafficking. Proc. Natl. Acad. Sci. U S A 106, 9093–9098. doi: 10.1073/pnas.0903001106
Mikhaylova, M., Sharma, Y., Reissner, C., Nagel, F., Aravind, P., Rajini, B., et al. (2006). Neuronal Ca2+ signaling via caldendrin and calneurons. Biochim. Biophys. Acta 1763, 1229–1237. doi: 10.1016/j.bbamcr.2006.08.047
Nakajima, Y. (2011). Ca2+-dependent binding of calcium-binding protein 1 to presynaptic group III metabotropic glutamate receptors and blockage by phosphorylation of the receptors. Biochem. Biophys. Res. Commun. 412, 602–605. doi: 10.1016/j.bbrc.2011.08.006
Nanou, E., Lee, A., and Catterall, W. A. (2018). Control of excitation/inhibition balance in a hippocampal circuit by calcium sensor protein regulation of presynaptic calcium channels. J. Neurosci. 38, 4430–4440. doi: 10.1523/JNEUROSCI.0022-18.2018
Navarro, G., Aguinaga, D., Angelats, E., Medrano, M., Moreno, E., Mallol, J., et al. (2016). A significant role of the truncated ghrelin receptor GHS-R1b in ghrelin-induced signaling in neurons. J. Biol. Chem. 291, 13048–13062. doi: 10.1074/jbc.M116.715144
Navarro, G., Aguinaga, D., Moreno, E., Hradsky, J., Reddy, P. P., Cortés, A., et al. (2014). Intracellular calcium levels determine differential modulation of allosteric interactions within G protein-coupled receptor heteromers. Chem. Biol. 21, 1546–1556. doi: 10.1016/j.chembiol.2014.10.004
Ohi, K., Shimada, T., Nitta, Y., Kihara, H., Okubo, H., Uehara, T., et al. (2016). Schizophrenia risk variants in ITIH4 and CALN1 regulate gene expression in the dorsolateral prefrontal cortex. Psychiatr. Genet. 26, 142–143. doi: 10.1097/ypg.0000000000000127
Oz, S., Benmocha, A., Sasson, Y., Sachyani, D., Almagor, L., Lee, A., et al. (2013). Competitive and non-competitive regulation of calcium-dependent inactivation in CaV1.2 L-type Ca2+ channels by calmodulin and Ca2+ -binding protein 1. J. Biol. Chem. 288, 12680–12691. doi: 10.1074/jbc.m113.460949
Oz, S., Tsemakhovich, V., Christel, C. J., Lee, A., and Dascal, N. (2011). CaBP1 regulates voltage-dependent inactivation and activation of CaV1.2 (L-type) calcium channels. J. Biol. Chem. 286, 13945–13953. doi: 10.1074/jbc.M110.198424
Raghuram, V., Sharma, Y., and Kreutz, M. R. (2012). Ca2+ sensor proteins in dendritic spines: a race for Ca2+. Front. Mol. Neurosci. 5:61. doi: 10.3389/fnmol.2012.00061
Rajamanoharan, D., McCue, H. V., Burgoyne, R. D., and Haynes, L. P. (2015). Modulation of phosphatidylinositol 4-phosphate levels by CaBP7 controls cytokinesis in mammalian cells. Mol. Biol. Cell 26, 1428–1439. doi: 10.1091/mbc.e14-07-1243
Ranaghan, M. J., Kumar, R. P., Chakrabarti, K. S., Buosi, V., Kern, D., and Oprian, D. D. (2013). A highly conserved cysteine of neuronal calcium-sensing proteins controls cooperative binding of Ca2+ to recoverin. J. Biol. Chem. 288, 36160–36167. doi: 10.1074/jbc.M113.524355
Reddy, P. P., Raghuram, V., Hradsky, J., Spilker, C., Chakraborty, A., Sharma, Y., et al. (2014). Molecular dynamics of the neuronal EF-hand Ca2+-sensor caldendrin. PLoS One 9:e103186. doi: 10.1371/journal.pone.0103186
Roussos, P., Giakoumaki, S. G., Zouraraki, C., Fullard, J. F., Karagiorga, V.-E., Tsapakis, E.-M., et al. (2016). The relationship of common risk variants and polygenic risk for schizophrenia to sensorimotor gating. Biol. Psychiatry 79, 988–996. doi: 10.1016/j.biopsych.2015.06.019
Sala, C., and Segal, M. (2014). Dendritic spines: the locus of structural and functional plasticity. Physiol. Rev. 94, 141–188. doi: 10.1152/physrev.00012.2013
Seeger, C., Gorny, X., Reddy, P. P., Seidenbecher, C., and Danielson, U. H. (2012). Kinetic and mechanistic differences in the interactions between caldendrin and calmodulin with AKAP79 suggest different roles in synaptic function. J. Mol. Recognit. 25, 495–503. doi: 10.1002/jmr.2215
Seidenbecher, C. I., Landwehr, M., Smalla, K.-H., Kreutz, M., Dieterich, D. C., Zuschratter, W., et al. (2004). Caldendrin but not calmodulin binds to light chain 3 of MAP1A/B: an association with the microtubule cytoskeleton highlighting exclusive binding partners for neuronal Ca2+-sensor proteins. J. Mol. Biol. 336, 957–970. doi: 10.1016/j.jmb.2003.12.054
Seidenbecher, C. I., Langnaese, K., Sanmartí-Vila, L., Boeckers, T. M., Smalla, K.-H., Sabel, B. A., et al. (1998). Caldendrin, a novel neuronal calcium-binding protein confined to the somato-dendritic compartment. J. Biol. Chem. 273, 21324–21331. doi: 10.1074/jbc.273.33.21324
Sexton, T., Bantignies, F., and Cavalli, G. (2009). Genomic interactions: chromatin loops and gene meeting points in transcriptional regulation. Semin. Cell Dev. Biol. 20, 849–855. doi: 10.1016/j.semcdb.2009.06.004
Shih, P.-Y., Lin, C.-L., Cheng, P.-W., Liao, J.-H., and Pan, C.-Y. (2009). Calneuron I inhibits Ca2+ channel activity in bovine chromaffin cells. Biochem. Biophys. Res. Commun. 388, 549–553. doi: 10.1016/j.bbrc.2009.08.046
Smalla, K.-H., Sahin, J., Putzke, J., Tischmeyer, W., Gundelfinger, E. D., and Kreutz, M. R. (2009). Altered postsynaptic-density-levels of caldendrin in the para-chloroamphetamine-induced serotonin syndrome but not in the rat ketamine model of psychosis. Neurochem. Res. 34, 1405–1409. doi: 10.1007/s11064-009-9925-8
Tang, N., Lin, T., Yang, J., Foskett, J. K., and Ostap, E. M. (2007). CIB1 and CaBP1 bind to the myo1c regulatory domain. J. Muscle Res. Cell Motil. 28, 285–291. doi: 10.1007/s10974-007-9124-7
Thompson, J. L., Pogue-Geile, M. F., and Grace, A. A. (2004). Developmental pathology, dopamine, and stress: a model for the age of onset of schizophrenia symptoms. Schizophr. Bull. 30, 875–900. doi: 10.1093/oxfordjournals.schbul.a007139
Tippens, A. L., and Lee, A. (2007). Caldendrin, a neuron-specific modulator of Cav/1.2 (L-type) Ca2+ channels. J. Biol. Chem. 282, 8464–8473. doi: 10.1074/jbc.m611384200
Wingard, J. N., Chan, J., Bosanac, I., Haeseleer, F., Palczewski, K., Ikura, M., et al. (2005). Structural analysis of Mg2+ and Ca2+ binding to CaBP1, a neuron-specific regulator of calcium channels. J. Biol. Chem. 280, 37461–37470. doi: 10.1074/jbc.m508541200
Wockner, L. F., Noble, E. P., Lawford, B. R., Young, R. M., Morris, C. P., Whitehall, V. L. J., et al. (2014). Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl. Psychiatry 4:e339. doi: 10.1038/tp.2013.111
Wu, Y.-Q., Lin, X., Liu, C.-M., Jamrich, M., and Shaffer, L. G. (2001). Identification of a human brain-specific gene, calneuron 1, a new member of the calmodulin superfamily. Mol. Genet. Metab. 72, 343–350. doi: 10.1006/mgme.2001.3160
Xia, S., Zhou, X., Wang, T., Zhang, Q., Li, Q., Liu, Y., et al. (2015). Experimental validation of candidate schizophrenia gene CALN1 as a target for microRNA-137. Neurosci. Lett. 602, 110–114. doi: 10.1016/j.neulet.2015.07.001
Yang, T., Britt, J. K., Cintrón-Pérez, C. J., Vázquez-Rosa, E., Tobin, K. V., Stalker, G., et al. (2018). Ca2+-binding protein 1 regulates hippocampal-dependent memory and synaptic plasticity. Neuroscience 380, 90–102. doi: 10.1016/j.neuroscience.2018.04.004
Zhang, B., Xu, Y.-H., Wei, S.-G., Zhang, H.-B., Fu, D.-K., Feng, Z.-F., et al. (2014). Association study identifying a new susceptibility gene (AUTS2) for schizophrenia. Int. J. Mol. Sci. 15, 19406–19416. doi: 10.3390/ijms151119406
Zhou, H., Kim, S.-A., Kirk, E. A., Tippens, A. L., Sun, H., Haeseleer, F., et al. (2004). Ca2+-binding protein-1 facilitates and forms a postsynaptic complex with Cav1.2 (L-Type) Ca2+ channels. J. Neurosci. 24, 4698–4708. doi: 10.1523/JNEUROSCI.5523-03.2004
Keywords: golgi, trafficking, dendritic spines, F-actin, structural plasticity, CaV1.2, InsP3R
Citation: Mundhenk J, Fusi C and Kreutz MR (2019) Caldendrin and Calneurons—EF-Hand CaM-Like Calcium Sensors With Unique Features and Specialized Neuronal Functions. Front. Mol. Neurosci. 12:16. doi: 10.3389/fnmol.2019.00016
Received: 16 October 2018; Accepted: 17 January 2019;
Published: 06 February 2019.
Edited by:
Teresa Duda, Salus University, United StatesReviewed by:
Ivan Ivanovich Senin, Lomonosov Moscow State University, RussiaMaria E. Rubio, University of Pittsburgh, United States
Copyright © 2019 Mundhenk, Fusi and Kreutz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael R. Kreutz, kreutz@ifn-magdeburg.de