 Xin-Yi Chen
Xin-Yi Chen Yi-Feng Du2
Yi-Feng Du2 Lei Chen
Lei Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 11 January 2019
Sec. Brain Disease Mechanisms
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00493
Alzheimer's disease (AD) is an age-related neurodegenerative disorder characterized by cognitive deficits and neuronal loss. Deposition of beta-amyloid peptide (Aβ) causes neurotoxicity through the formation of plaques in brains of Alzheimer's disease. Numerous studies have indicated that the neuropeptides including ghrelin, neurotensin, pituitary adenylate cyclase-activating polypeptide (PACAP), neuropeptide Y, substance P and orexin are closely related to the pathophysiology of Alzheimer's disease. The levels of neuropeptides and their receptors change in Alzheimer's disease. These neuropeptides exert neuroprotective roles mainly through preventing Aβ accumulation, increasing neuronal glucose transport, increasing the production of neurotrophins, inhibiting endoplasmic reticulum stress and autophagy, modulating potassium channel activity and hippocampal long-term potentiation. Therefore, the neuropeptides may function as potential drug targets in the prevention and cure of Alzheimer's disease.
Alzheimer's disease (AD) is an age-related neurodegenerative disorder which is clinically characterized by cognitive deficits, memory impairment, disorientation, and behavioral issues (Burns and Iliffe, 2009). Commonly the Alzheimer's disease begins in people over the age of 65 years. The neuropathological features in Alzheimer's disease are amyloid plaques, neurofibrillary tangles, and neuronal loss (Tiraboschi et al., 2004). Beta-amyloid peptide (Aβ) oligomers have been reported to be the primary pathogenic forms of Aβ, which change the structure of synapses and eventually disrupt neuronal communication (Lacor et al., 2007). However, the pathogenesis remains unknown.
Neuropeptides are molecules which function as endogenous active substances within central nervous system and peripheral nervous system. Neuropeptides play important roles in a wide range of brain functions, including food intake, metabolism, reproduction, social behaviors, reward, learning and memory, sleep and wakefulness. Recent studies revealed that many neuropeptides including ghrelin, neurotensin, pituitary adenylate cyclase-activating polypeptide (PACAP), neuropeptide Y, substance P, and orexin may be associated with the pathophysiology and potential therapy of Alzheimer's disease. In this article, we review the recent advances about the involvement of neuropeptides in Alzheimer's disease.
Ghrelin, a 28-amino acid brain-gut peptide, activates the growth hormone secretagogue receptors which are expressed widely in the brain (Guan et al., 1997). Recent studies revealed that the receptor expression is developmental, with a stronger staining in the early stages and a weaker expression in the later stages of development (Lattuada et al., 2013). Ghrelin exerts critical roles in the regulation of energy homeostasis, neuroendocrine and neurodegenerative processes, especially in higher brain functions, such as learning and memory consolidation (Spitznagel et al., 2010; Rak-Mardyla, 2013; Murray et al., 2014; Panagopoulos and Ralevski, 2014; Jiao et al., 2017). Ghrelin is also involved in mitochondrial respiration and neuroprotection, which can be developed as biomarkers or drug targets for prevention and treatment of neurological disorders, including Parkinson's disease, stroke, epilepsy and Alzheimer's disease (dos Santos et al., 2013a; Shi et al., 2014, 2017; Stoyanova, 2014).
Ghrelin has been demonstrated to have a close relationship with Alzheimer's disease. One of the single nucleotide polymorphisms of the ghrelin gene, rs4684677 (Leu90Gln), has been proved to be associated with the onset age of Alzheimer's disease (Shibata et al., 2011). Early study revealed that the level of serum ghrelin is negatively related to several cognitive domains in older adults (Spitznagel et al., 2010). The level of the functional form of ghrelin, acylated ghrelin, is associated with Alzheimer's disease risk factors and mild cognitive impairment (Gahete et al., 2010; Cao et al., 2018). However, a significant reduction of ghrelin mRNA was observed in the temporal gyrus of Alzheimer's disease patients, suggesting the possible involvement of ghrelin in the cognitive deficit of Alzheimer's disease (Gahete et al., 2010).
Administration of ghrelin or ghrelin agonist decreases the level of Aβ and attenuates Alzheimer's disease-related cognitive impairment (Gahete et al., 2010, 2011; Dhurandhar et al., 2013; Kunath et al., 2015). Recent in vitro study demonstrated that pre-administration of ghrelin or ghrelin analog protects SH-SY5Y cellular models of Alzheimer's disease against methylglyoxal-induced toxicity and apoptosis, suggesting the potential treatment for neurodegenerative disorders (Cecarini et al., 2016; Popelová et al., 2018). Ghrelin exerts neuroprotective effects through different mechanisms. Central application of acylated ghrelin prevents Aβ-induced impairments of memory and energy and glucose metabolisms, probably through increase of AMPK and GSK phosphorylation and decrease of tau phosphorylation (Kang et al., 2015). Further study revealed that acylated ghrelin also blunts Aβ-induced depression of long-term potentiation (LTP) in hippocampus and therefore prevents impairments of recognition and spatial orientation (Santos et al., 2017). Ghrelin ameliorates neurogenesis impairment in hippocampus and improves memory deficits by prevention of synaptic degeneration including cholinergic fiber loss (Moon et al., 2011, 2014). Recent studies revealed that ghrelin exerts protective effects against Aβ-induced toxicity via preventing superoxide production, calcium elevation and mitochondrial membrane depolarization (Martins et al., 2013; Gomes et al., 2014). In addition, ghrelin restores the proteasome functionality in Alzheimer's disease and thus contributes to the elimination of toxic aggregates (Cecarini et al., 2016). It is known that brain insulin resistance is closely related to the cognitive impairment and neurodegeneration, particularly within Alzheimer's disease. Recently, it is demonstrated that ghrelin increases neuronal glucose uptake and improves tau hyperphosphrylation by activating Akt and GSK-3 beta phosphorylation in cultured hippocampal neurons (Chen et al., 2010).
In summary, the levels of ghrelin change in Alzheimer's disease. The functional form of ghrelin, acylated ghrelin, increases in mild cognitive impairment. A single nucleotide polymorphism of ghrelin gene is associated with the onset age of Alzheimer's disease. Ghrelin protects against Aβ-induced neurotoxicity through multiple pathways. Ghrelin prevents calcium elevation, superoxide production and mitochondrial membrane depolarization. Moreover, ghrelin increases neuronal glucose uptake and improves Aβ-induced deterioration of memory through activating AMPK and GSK phosphorylation and decreasing tau phosphorylation. Furthermore, ghrelin prevents cholinergic synaptic degeneration. Therefore, ghrelin is considered as a potential drug in the treatment of Alzheimer's disease (Figure 1).
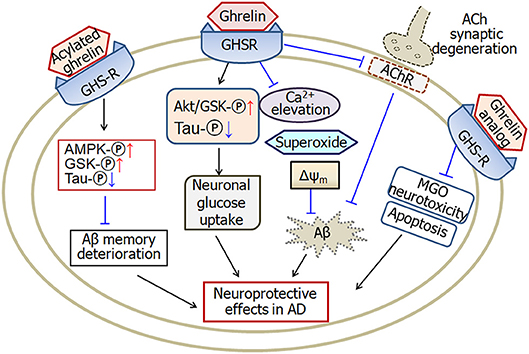
Figure 1. A schematic diagram describing the possible pathways of ghrelin-induced neuroprotective effects in Alzheimer's disease. Ghrelin protects against Aβ-induced neurotoxicity through prevention of calcium elevation, superoxide production and mitochondrial membrane depolarization. Ghrelin also increases neuronal glucose uptake by activating Akt/GSK phosphorylation and improving tau hyperphosphrylation. Moreover, ghrelin prevents cholinergic synaptic degeneration and therefore protects against Aβ-induced memory deficits. Acylated ghrelin improves Aβ-induced deterioration of memory through increase of AMPK and GSK phosphorylation and decrease of tau phosphorylation. Analog of ghrelin protects against MGO-induced neurotoxicity and apoptosis in cellular models of Alzheimer's disease. GHSR, growth hormone secretagogue receptors, also known as ghrelin receptors; Δψm, mitochondrial membrane potential; MGO, methylglyoxal; ACh, acetylcholine; AMPK, adenosine 5′-monophosphate (AMP)-activated protein kinase; GSK, glycogen synthase kinase; AChR, cholinergic receptors; -℗, phosphrylation. The internal and external circles represent the inner and outer leaflets of the cellular membrane. The dotted line in ACh synapse represents the degenerated synapse.
Neurotensin is a tridecapeptide which binds with two neurotensin receptors, type-1 and type-2, in the brain. Neurotensin plays multiple effects in central nervous system and is involved in the pathophysiology of several central nervous system disorders, including schizophrenia (Garver et al., 1991; Kinkead and Nemeroff, 2004), Parkinson's disease (Bissette et al., 1985; Fernandez et al., 1994), as well as Alzheimer's disease (Constantinidis et al., 1983; Struble et al., 1987; Gahete et al., 2010; Xiao et al., 2014). The levels of neurotensin and neurotensin receptors change in several brain regions of Alzheimer's disease patients. Neurotensin influences the formation of senile plaques and therefore is associated with the pathogenesis of Alzheimer's disease through different pathways (Figure 2).
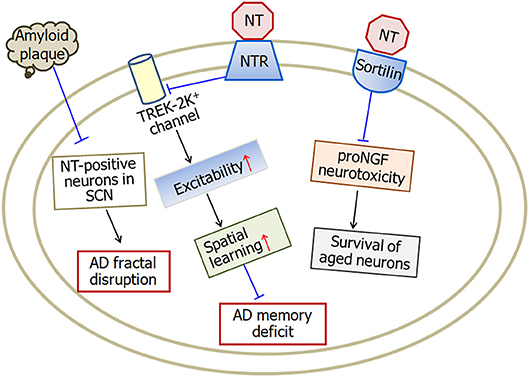
Figure 2. A model illustrating the neuroprotective effects of neurotensin in Alzheimer's disease. Neurotensin increases the excitability of neurons by inhibiting TREK-2K+ channel, which therefore improves memory status in Alzheimer's disease mice. Being a sortilin ligand, neurotensin rescues the survival of aged neurons through blocking sortilin-induced proNGF neurotoxicity. In addition, amyloid plaque density in the occipital cortex is negatively associated with neurotensin-positive neurons in the suprachiasmatic nucleus suggesting the involvement of neurotensin in fractal activity disruption in Alzheimer's disease. NT, neurotensin; NTR, neurotensin receptors; SCN, suprachiasmatic nucleus. The internal and external circles represent the inner and outer leaflets of the cellular membrane.
Previous studies revealed a decreased level of neurotensin in the septum (Ferrier et al., 1983), suprachiasmatic nucleus (Hu et al., 2013) and amygdala (Benzing et al., 1990), and a low level of neurotensin receptors in the entorhinal area (Jansen et al., 1990), dentate gyrus (Rowe et al., 2006) and temporal gyrus (Gahete et al., 2010) of Alzheimer's disease. In Alzheimer's disease patients, the amyloid plaque density in the occipital cortex is negatively associated with the neurotensin-positive neurons in the suprachiasmatic nucleus, i.e., the more plaques, the fewer neurotensin-positive neurons, which suggests the involvement of neurotensin in fractal activity disruption in Alzheimer's disease (Hu et al., 2013). The levels of amygdala neurotensin in males are significantly higher than that in females (Biggins et al., 1983), which suggests that the intrinsic function of the amygdala may be associated with hormonal regulation of other sex-dependent mechanism. For example, Skup et al. (2011) reported that the males and the females show different patterns in amygdala volume decline over time. Previous studies revealed that men exhibit greater volumes and neuronal densities in amygdala (Giedd et al., 1996; Goldstein et al., 2001; Witte et al., 2010). Furthermore, the density of neurotensin and acetylcholine containing fibers is reduced dramatically in the regions of amygdala with greatest senile plaques (Benzing et al., 1992, 1993). However, in the case of high plaque non-demented, no significant reduction of neurotensinergic fibers and other neurotransmitter fibers was observed in the amygdala (Benzing et al., 1993).
It is well-known that the entorhinal cortex is a crucial brain region involved in the earliest pathological change of Alzheimer's disease (Mann, 1989; Beach et al., 1997). Recently, Xiao et al. (2014) found that neurotensin persistently increases the spontaneous firing rate of neurons in the entorhinal cortex. This facilitation is mediated by neurotensin type 1 receptor and TREK-2K+ channels. Further behavioral studies revealed that activation of neurotensin type 1 receptors enhances spatial learning and therefore improves memory status in APP/PS1 Alzheimer's disease mice model. Recent electrophysiological studies further demonstrated that neurotensin increases glutamate release and spontaneous firing rate of dentate gyrus through both presynaptic and post-synaptic neurotensin receptors, respectively (Zhang et al., 2015, 2016).
It has recently been shown that the precursor form of nerve growth factor (proNGF) forming complex with p75 and sortilin contributes to neuronal death in basal forebrain neurons (Al-Shawi et al., 2007, 2008). Sortilin plays an important role in proNGF-mediated neurotoxicity. As a sortilin ligand, neurotensin blocks the sortilin-induced effects of proNGF and further rescues the survival of old neurons (Al-Shawi et al., 2008).
In summary, the levels of neurotensin and neurotensin receptors decrease in many brain areas of Alzheimer's disease. The amyloid plaque density in the occipital cortex is negatively associated with the neurotensin-positive neurons in the suprachiasmatic nucleus. Neurotensin increases the excitability of entorhinal cortex neurons and therefore improves spatial learning and memory. Furthermore, neurotensin rescues the survival of aged neurons through blocking sortilin-induced proNGF neurotoxicity.
PACAP belongs to the superfamily of secretin/glucagon/vasoactive intestinal polypeptide (Miyata et al., 1989). High level of PACAP is expressed in the hypothalamus, hippocampus, cerebellum and several brainstem nuclei (Hannibal, 2002). By activating three types of receptors (PAC1, VPAC1, and VPAC2), PACAP functions as a neurohormone, neurotransmitter, or neurotrophic factor in central nervous system (Lee and Seo, 2014). There is growing evidence (Reglodi et al., 2011) suggests that PACAP is closely associated with the pathology of Alzheimer's disease (Figure 3).
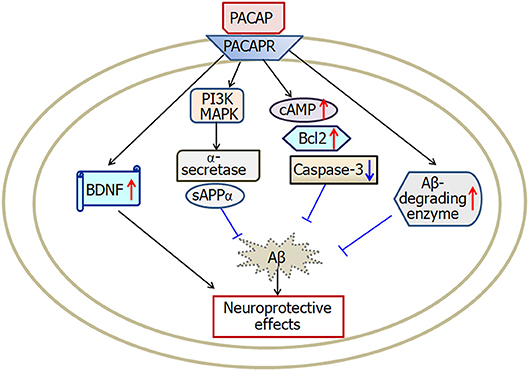
Figure 3. A scheme describing the possible mechanisms of PACAP-induced neuroprotective effects in Alzheimer's disease. PACAP protects against Aβ-induced neurotoxicity by activation of cAMP, BDNF, Bcl-2, Aβ-degrading enzyme and deactivation of caspase-3. Furthermore, PACAP increases α-secretase activation and then enhances secretion of sAPPα through both the MAPK and PI3K pathways. PACAP, pituitary adenylate cyclase-activating polypeptide; PACAPR, pituitary adenylate cyclase-activating polypeptide receptors; BDNF, brain-derived neurotrophic factor. The internal and external circles represent the inner and outer leaflets of the cellular membrane.
By using three different mouse models of Alzheimer's disease, early study showed downregulation of PACAP genes (Wu et al., 2006). Moreover, the PACAP levels are reduced in brain areas including entorhinal cortex, middle temporal gyrus, superior frontal gyrus, and primary visual cortex in Alzheimer's disease patients (Han et al., 2014a). Further study revealed that the lower PACAP levels are correlated with higher amyloid burden, tau protein, and the declined recognition memory with aging (Han et al., 2014b; An et al., 2017).
It is demonstrated that PACAP exerts neuroprotective effects through multiple mechanisms in Alzheimer's disease patients and mouse models (Vaudry et al., 2004; Rat et al., 2011). Onoue et al. (2002) first demonstrated that PACAP protects against Aβ-induced neuronal toxicity. Treatment with PACAP rescues 80% of Aβ-induced reduction of cell viability through increase of cAMP and deactivation of caspase-3 in PC12 cells. Intranasal application of PACAP increases the levels of brain-derived neurotrophic factor (BDNF) and antiapoptotic Bcl-2 protein. In addition, PACAP also increases the level of Aβ-degrading enzyme (Rat et al., 2011). Therefore, intranasal application of PACAP could be a useful therapeutic approach in treating Alzheimer's disease. It is known that, in the non-amyloidogenic pathway, the α-secretase cleaves the amyloid precursor protein and prevents amyloid plaque formation (Postina, 2012). Activation of PAC1 receptors increases APPs alpha secretion and enhances α-secretase cleavage of APP. Furthermore, both the MAPK and PI3K pathways are involved in PACAP-mediated α-secretase activation (Kojro et al., 2006).
Ginsenoside is a major component of the traditional herb ginseng which has been proved to exert neurotrophic and neuroprotective effects via preventing neuronal degeneration. Ginsenosides Rh2 increases the gene expression of PACAP in astrocytes of the brain and therefore ameliorates Aβ-induced growth inhibition of astrocytes (Shieh et al., 2008).
In summary, the level of PACAP decreases in brain areas of Alzheimer's disease patients and mouse models, which is correlated with higher amyloid burden, tau protein, and the declined recognition memory. PACAP protects against Aβ-induced neuronal toxicity through activation of cAMP, Bcl2, BDNF, Aβ-degrading enzyme, and deactivation of caspase-3. In addition, PACAP enhances α-secretase cleavage of APP via both MAPK and PI3K pathways.
Neuropeptide Y is a 36-amino acid neuropeptide. Neuropeptide Y receptors are classified into five subtypes: Y1, Y2, Y4, Y5, and Y6 (Hsieh et al., 2013; Mittapalli and Roberts, 2014; Pérez-Fernández et al., 2014). Neuropeptide Y plays functions associated with modulation of food ingestion, mood, learning and memory (dos Santos et al., 2013b) and also plays an important role in neuroprotection against neurodegenerative diseases (Figure 4).
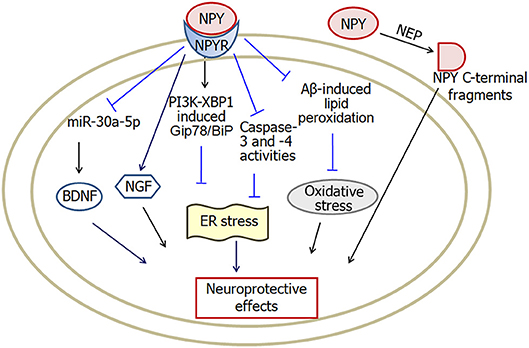
Figure 4. A model showing the possible pathways of neuropeptide Y-induced neuroprotective effects in Alzheimer's disease. Neuropeptide Y inhibits Aβ-induced lipid peroxidation and prevents intracellular oxidative stress. Activation of PI3K-XBP1 pathway may also be involved in neuropeptide Y-induced neuroprotection against endoplasmic reticulum stress. Moreover, both NGF and BDNF are involved in neuropeptide Y-induced neuroprotective effects. In addition, NEP cleaves neuropeptide Y into C-terminal fragments, which protect against the neurodegenerative pathology in Alzheimer's disease. NPY, neuropeptide Y; NPYR, neuropeptide Y receptors; NEP, neutral endopeptidase; ER, endoplasmic reticulum; BDNF, brain-derived neurotrophic factor; NGF, nerve growth factor. The internal and external circles represent the inner and outer leaflets of the cellular membrane.
The expression of neuropeptide Y changes under neurodegenerative diseases including Alzheimer's disease (Duarte-Neves et al., 2016). An anomalous high expression level of neuropeptide Y with aging was detected in the hippocampal circuits of mouse model of Alzheimer's disease (Diez et al., 2003; Krezymon et al., 2013). Recently, Mahar et al. (2017) reported that the number of neuropeptide Y immunoreactive hippocampal interneurons reduces in presymptomatic TgCRND8 Alzheimer's disease mouse model.
Neuropeptide Y has neuroprotective effects in Alzheimer's disease (Croce et al., 2011, 2012, 2013; Angelucci et al., 2014; Duarte-Neves et al., 2016; Spencer et al., 2016). Intracerebroventricular application of neuropeptide Y prevents Aβ1−40-induced depressive-like symptoms and spatial memory impairments through Y2 receptors. Neuropeptide Y produces the neuroprotection through inhibition of Aβ-induced lipid peroxidation, indicating the involvement of prevention of intracellular oxidative stress (dos Santos et al., 2013b). Moreover, neuropeptide Y induces protective effects against endoplasmic reticulum stress-mediated cell loss through activation of PI3K-XBP1-induced Gip78/BiP pathway as well as inhibition of caspase-3 and caspase-4 activities (Lee et al., 2018). Neuropeptide Y-induced neuroprotective effects are also associated with the production of neurotrophin family (Croce et al., 2011, 2012, 2013; Angelucci et al., 2014). Pretreatment with neuropeptide Y protects neurons against Aβ neurotoxicity which is accompanied by an increased intracellular level of NGF (Croce et al., 2012) and BDNF (Croce et al., 2013). Moreover, decrease of miR-30-5p (a membrane of miR-30a family regulating BDNF tuning expression) levels is involved in neuropeptide Y-induced modulation of BDNF.
The neutral endopeptidase (NEP) neprilysin is an Aβ degrading enzyme which is therefore involved in the pathogenesis of Alzheimer's disease. However, NEP has also been proved to cleave neuropeptide Y into C-terminal fragments. The NEP-produced C-terminal fragments of neuropeptide Y attenuate the neurodegenerative process in both transgenic Alzheimer's disease mice and Aβ treated human neurons (Rose et al., 2009). It is well-known that Aβ accumulation, senile plaque formation and neuronal dysfunction contribute to the cognitive declination in Alzheimer's disease. However, weight loss is an early sign of Alzheimer's disease. It is reported that high level of Aβ potentially devastates hypothalamic arcuate neuropeptide Y neurons and downregulates the leptin state in the early disease process, which may lead to the weight loss (dos Santos et al., 2013b; Ishii et al., 2014).
In summary, the level of neuropeptide Y increases significantly in hippocampus of mouse Alzheimer's disease models. Neuropeptide Y exerts neuroprotective effects through multiple pathways. Neuropeptide Y mitigates endoplasmic reticulum stress-induced neuronal cell death through activation of PI3K-XBP1-induced Gip78/BiP pathway and inhibition of caspase-3 and caspase-4 activities. Furthermore, neuropeptide Y suppresses oxidative stress via inhibition of Aβ-induced lipid peroxidation. In addition, neuropeptide Y plays neuroprotection via increasing the levels of BDNF and NGF.
The tachykinin family includes substance P, neurokinin A and neurokinin B. By activating neurokinin receptors, tachykinin plays important roles including pain, depression, nausea and emesis (Mantyh, 2002). In addition, tachykinin protects against the neurotoxic processes of Alzheimer's disease by multiple pathways (Severini et al., 2016).
Early studies revealed that the expression of substance P changes in different brain regions of Alzheimer's disease. The level of substance P decreases in the cortex, hippocampus and striatum in Alzheimer's disease patients and animal models (Bouras et al., 1990; Quigley and Kowall, 1991; Nag et al., 1999; Ahmed et al., 2004), but increases in the pallidum and substantia nigra (Bouras et al., 1990). However, Willis et al. (2007) showed that in old age of Alzheimer's disease transgenic mice, the substance P-immunoreactivity exists in astrocytes of hippocampus and thalamus. In late onset Alzheimer's disease patients, the level of substance P in cerebrospinal fluid increases significantly (Rösler et al., 2001). Furthermore, the increased level of substance P is positively associated with the level of Aβ1−42 in Alzheimer's disease patients (Johansson et al., 2015). In addition to the change of substance P, the activity of neuropeptidases to metabolize substance P also alters in Alzheimer's disease. Waters and David (1995) demonstrated that the activity of neuropeptidases is decreased in the temporal cortex of senile dementia of the Alzheimer's disease, which therefore increases the metabolic half-life of substance P.
Substance P exerts neuroprotective effects in a variety of in vitro and in vivo studies (Figure 5). Potassium channel dysfunction is a possible mechanism underlying the pathophysiology of Alzheimer's disease. Some kinds of voltage-gated potassium channels are involved in substance P-induced neuroprotective effects. Application of substance P prevents Aβ-induced impairment of cognitive processes through inhibition of Aβ-induced overexpression of potassium channel subunits (Campolongo et al., 2013), as well as Aβ-induced enhancement of A-type K+ currents (Pieri et al., 2010). Substance P also protects cerebellar granule cells against Aβ-induced apoptosis through inhibition of caspase-3-induced PARP-1 cleavage (Pieri et al., 2010). In addition, substance P plays non-amyloidogenic effect through decreasing Aβ1−42, increasing sAPPα and α-secretase activity (Marolda et al., 2012). Aβ25−35 reduces the expression of substance P in hippocampus before the neuronal loss of Alzheimer's disease. Memantine, a non-competitive NMDA receptor antagonist, attenuates Aβ25−35-induced decrease of substance P (Arif et al., 2009). Furthermore, in ibotenic acid-treated Alzheimer's disease model, memantine treatment recovers the decreased substance P expression (Ahmed et al., 2004). Recently, Fernandes et al. (2018) reported that a substance P receptor antagonist attenuates aluminum-induced spatial memory deficit probably through blockade of substance P-mediated neuroinflammation. It is known that the dysfunction of metal ions, such as copper is a feature of Alzheimer's disease. Neurokinin B protects against copper-induced calcium channel opening (Russino et al., 2013), as well as the synaptic homeostasis (Grosas et al., 2014).
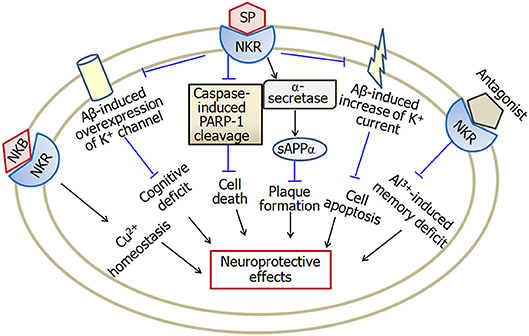
Figure 5. A model describing the multiple effects of substance P in Alzheimer's disease. Substance P inhibits Aβ-induced overexpression of K+ channel and Aβ-induced increase of K+ current, and therefore attenuates cognitive deficit and apoptosis in Alzheimer's disease. Furthermore, substance P exerts neuroprotective effects through inhibition of caspase-3-induced PARP-1 cleavage and enhancement of α-secretase activity. Neurokinin B plays a role in copper homeostasis. However, substance P receptor antagonist attenuates aluminum-induced spatial memory deficit probably through blockade of substance P-mediated neuroinflammation. SP, substance P; NKR, neurokinin receptors; PARP-1, poly ADP-ribose polymerase-1; sAPPα, soluble amyloid precursor protein α; NKB, neurokinin B; Cu, copper; Al, aluminum. The internal and external circles represent the inner and outer leaflets of the cellular membrane.
In summary, the levels of substance P decrease in brain regions including cortex and hippocampus, but increase in cerebrospinal fluid of late onset Alzheimer's disease patients. Substance P exerts neuroprotective effects through inhibition of Aβ-induced over- expression and activity of K+ channel. Furthermore, substance P inhibits caspase-3-induced PARP-1 cleavage and increases α-secretase activity, and is therefore involved in neuroprotection in Alzheimer's disease.
Orexin is a hypothalamic neuropeptide which plays an important role in maintaining wakefulness. It is well-known that sleep disturbances are common clinical symptom in neurodegenerative disorders. Recently people focus on the involvement of orexinergic system in the pathophysiology, especially sleep disturbances, of Alzheimer's disease (Ferini-Strambi, 2014; Malkki, 2014; Liguori, 2017; Liguori et al., 2017). Early morphological study revealed that the orexinergic neurons are reduced significantly in postmortem hypothalamus of Alzheimer's disease patients (Fronczek et al., 2012). However, it is demonstrated recently that the level of orexin-A in cerebrospinal fluid is higher in Alzheimer's disease patients which is positively associated with Alzheimer's disease biomarkers including Aβ and Tau (Liguori et al., 2016; Osorio et al., 2016; Gabelle et al., 2017). In both Alzheimer's disease patients and Aβ42 treated-SH-SY5Y cells, amyloid deposition and tau phosphorylation decrease the expression of orexin-1 and orexin-2 receptors (Davies et al., 2015). In vivo microdialysis revealed that infusion of orexin-A increases the amount of Aβ in brain interstitial fluid, while orexin receptor antagonist suppresses Aβ levels and reduces Aβ plaque deposition in the cortex (Kang et al., 2009). In Aβ-treated microglial cells, both orexin-A and orexin-B reduce the uptake of Aβ through downregulation of phagocytosis regulating molecules including PI3K, Akt and p38-MAPK. In addition, orexins suppress autophagosome-lysosome fusion and lead to impaired Aβ degradation (An et al., 2017). Furthermore, the orexin receptors 2 gene, rs2653349 polymorphism, is likely to be a risk factor of Alzheimer's disease (Gallone et al., 2014). In APP/PS1 transgenic Alzheimer's disease mice, orexin gene knock out markedly decreases the amount of Aβ pathology, while rescue of orexinergic neurons increases the amount of Aβ pathology in the brain (Roh et al., 2014). Recent study revealed that orexinergic system modulates both the hippocampal clock and clock-controlled-genes, Bace1 and Bace2. Both the two genes are correlated with the production of Aβ (Ma et al., 2016).
In addition to the inhibition of Aβ uptake and degradation in microglial cells (An et al., 2017), orexin may also exert neuroprotective effects. Orexinergic neuron degeneration impairs long-term social memory in mice (Yang et al., 2013). Intracerebroventricular microinjection of orexin-A improves memory in SAMP8 Alzheimer disease mice (Jaeger et al., 2002). Orexin receptors have been demonstrated to exert neuroprotective effects in Alzheimer's disease via heterodimerization with GPR103. ERK is a key molecule in the prevention of neurodegeneration. Treatment with orexins induces ERK1/2 phosphorylation suggesting the neuroprotection of the heterodimerization. Microarray showed that orexin-A augments NF-κB signaling and orexin-B up-regulates PI3K-Akt and Jak-STAT signaling, respectively (Davies et al., 2015).
The hippocampus is a crucial brain region that plays important roles in learning and memory. Intra-hippocampal administration of orexin-A attenuates pain-induced impairment of learning and memory (Raoof et al., 2015), while blocking orexin receptors in hippocampal CA1 regions impairs spatial memory retrieval (Akbari et al., 2006, 2008). It has been demonstrated that activation of cholinergic transmission increases the neuronal excitability of CA1 pyramidal neurons and therefore enhances hippocampus-dependent learning in Alzheimer disease animal model (Disterhoft and Oh, 2006). Moreover, decreasing the firing rate of CA1 pyramidal neurons by potassium channel activator impairs associative learning in rats (McKay et al., 2012). Therefore, our recent study that orexin-A increases the firing rate of hippocampal CA1 neurons may provide direct in vivo electrophysiological evidence for the possible involvement of orexin-A in Alzheimer disease (Chen et al., 2017).
In summary, Aβ deposition and tau phosphorylation decrease the expression of orexin and the receptors in hypothalamus, while the levels of orexins in cerebrospinal fluid increase in Alzheimer's disease patients. In microglial cells, orexins increase Aβ level through suppression of Aβ uptake and degradation. However, in cells forming OXRs and GPR103 heterodimers, orexins exert neuroprotective effects through upregulation of NF-κB, PI3K-Akt, Jak-STAT signaling, as well as ERK1/2 phosphorylation. Figure 6 illustrated the complicated effects of orexin in Alzheimer's disease.
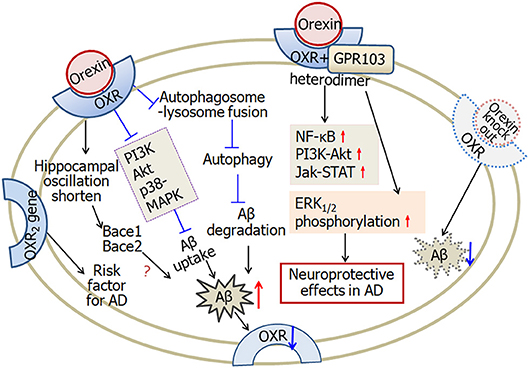
Figure 6. A scheme describing the complicated effects of orexin in Alzheimer's disease. Firstly, in microglial cells, orexin suppresses autophagosome-lysosome fusion process, leading to impaired Aβ degradation. Furthermore, orexin suppresses Aβ uptake through downregulating phagocytosis regulating molecules, such as PI3K, Akt, and p38-MAPK. Aβ-plaque formation and tau hyper-phosphorylation decrease the expression of orexin receptors in Alzheimer's disease. Secondly, orexin receptors and GPR103 form functional heterodimers. Orexin augments NF-κB, PI3K-Akt, Jak-STAT signaling and induces ERK1/2 phosphorylation, and therefore is involved in neuroprotective functions. Finally, one of the orexin receptor 2 gene is likely a risk factor for Alzheimer's disease. In APP/PS1 Alzheimer's disease mice, orexin gene knock out decreases the amount of Aβ. Orexin modulates the hippocampal oscillation and the expression of clock-controlled-genes, Bace1 and Bace2, which are associated with the production of Aβ. OXR, orexin receptors; AD, Alzheimer's disease. The internal and external circles represent the inner and outer leaflets of the cellular membrane.
In this review article, we provide a description of recent advances of neuropeptides including ghrelin, neurotensin, PACAP, neuropeptide Y, substance P and orexin in Alzheimer's disease. Based on current studies, the levels of these neuropeptides and their receptors change in Alzheimer's disease (Table 1). Neuropeptides exert significant neuroprotective effects against Aβ-induced neuronal toxicity (Table 2). Multiple intracellular mechanisms, including activation of α-secretases and Aβ-degrading enzyme, production of neurotrophins, increase of neuronal glucose uptake, upregulation of NF-κB, PI3K-Akt, MAPK, Jak-STAT, ERK1/2 phosphorylation and downregulation of caspase-3, inhibition of endoplasmic reticulum stress and autophagy, modulation of LTP and potassium channel activity, appear to be involved in neuropeptide-induced neuroprotection in Alzheimer's disease. As the levels of neuropeptides and the receptors may change before Aβ deposition and neuronal loss, as well as positively/negatively correlate with cognitive impairment, future studies would focus on detection of neuropeptides as biomarkers in early diagnosis and treatment evaluation of Alzheimer's disease. Furthermore, for the notable neuroprotective effects, further researches are needed to explore the potential use of neuropeptides, especially via convenient administration method like intranasal application, in the prevention and cure of Alzheimer's disease.
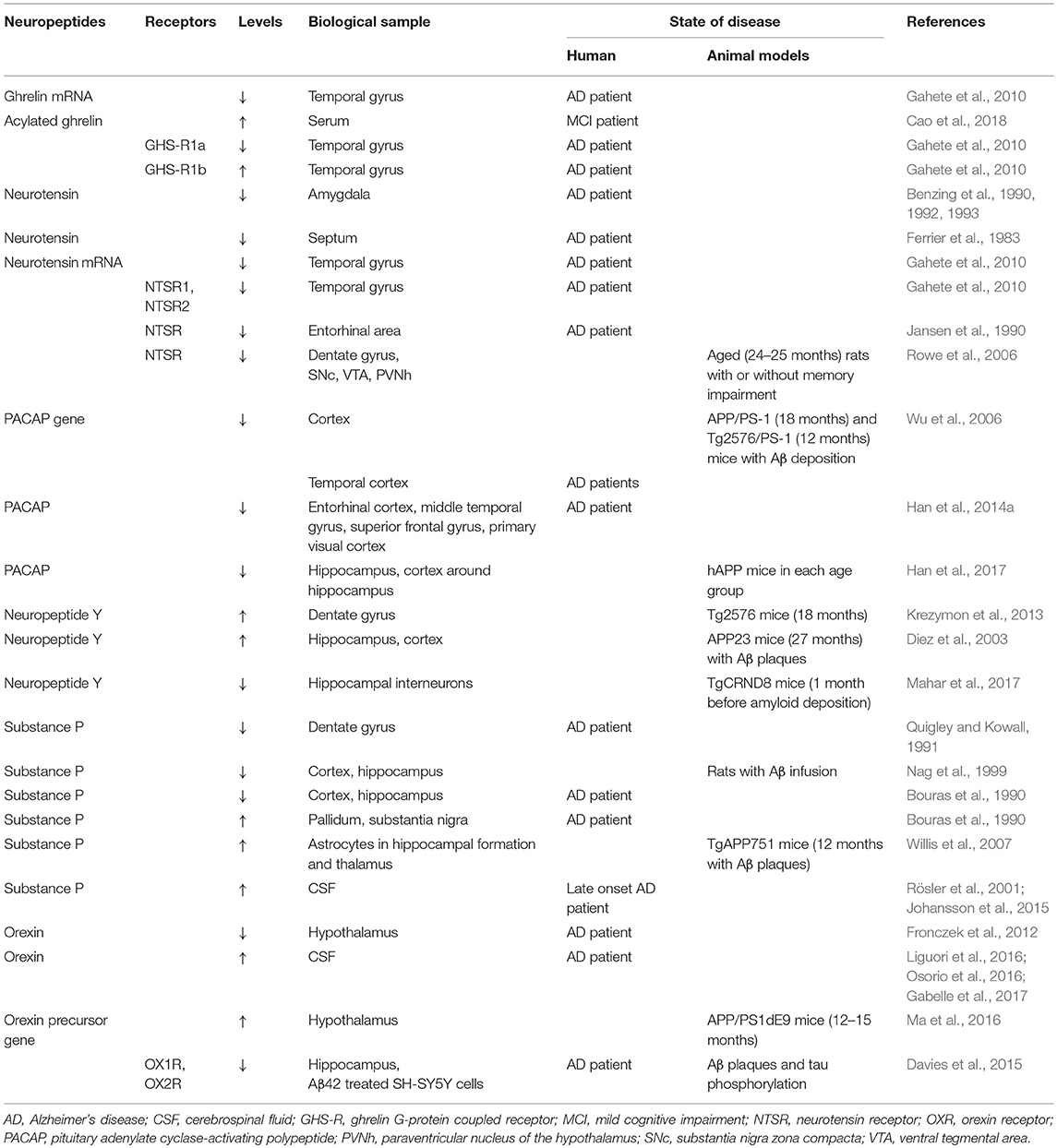
Table 1. Changes in the level of neuropeptides and receptors in Alzheimer's disease.
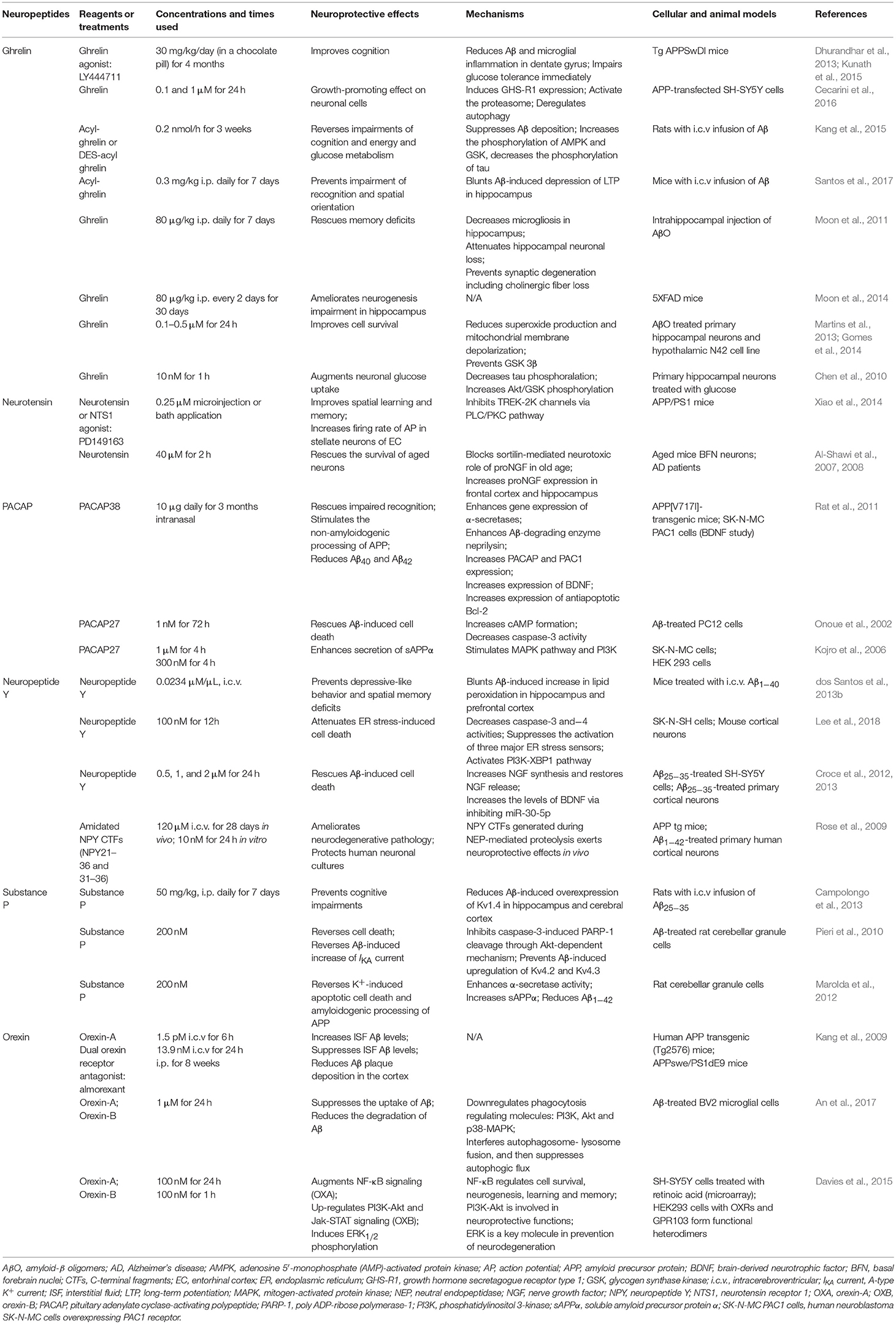
Table 2. Neuroprotective effects and possible mechanisms of neuropeptides in Alzheimer's disease.
X-YC wrote the manuscript. Y-FD and LC revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by grants from the National Natural Science Foundation of China (31671076, 81772448, 81200872), and Taishan Scholars Construction Project.
Ahmed, M. M., Hoshino, H., Chikuma, T., Yamada, M., and Kato, T. (2004). Effect of memantine on the levels of glial cells, neuropeptides, and peptide-degrading enzymes in rat brain regions of ibotenic acid-treated Alzheimer's disease model. Neuroscience 126, 639–649. doi: 10.1016/j.neuroscience.2004.04.024
Akbari, E., Motamedi, F., Naghdi, N., and Noorbakhshnia, M. (2008). The effect of antagonization of orexin 1 receptors in CA1 and dentate gyrus regions on memory processing in passive avoidance task. Behav. Brain Res. 187, 172–177. doi: 10.1016/j.bbr.2007.09.019
Akbari, E., Naghdi, N., and Motamedi, F. (2006). Functional inactivation of orexin 1 receptors in CA1 region impairs acquisition, consolidation and retrieval in Morris water maze task. Behav. Brain Res. 173, 47–52. doi: 10.1016/j.bbr.2006.05.028
Al-Shawi, R., Hafner, A., Chun, S., Raza, S., Crutcher, K., Thrasivoulou, C., et al. (2007). ProNGF, sortilin, and age-related neurodegeneration. Ann. N. Y. Acad. Sci. 1119, 208–215. doi: 10.1196/annals.1404.024
Al-Shawi, R., Hafner, A., Olsen, J., Chun, S., Raza, S., Thrasivoulou, C., et al. (2008). Neurotoxic and neurotrophic roles of proNGF and the receptor sortilin in the adult and ageing nervous system. Eur. J. Neurosci. 27, 2103–2114. doi: 10.1111/j.1460-9568.2008.06152.x
An, H., Cho, M. H., Kim, D. H., Chung, S., and Yoon, S. Y. (2017). Orexin impairs the phagocytosis and degradation of amyloid-β fibrils by microglial cells. J. Alzheimers Dis. 58, 253–261. doi: 10.3233/JAD-170108
Angelucci, F., Gelfo, F., Fiore, M., Croce, N., Mathé, A. A., Bernardini, S., et al. (2014). The effect of neuropeptide Y on cell survival and neurotrophin expression in in-vitro models of Alzheimer's disease. Can. J. Physiol. Pharmacol. 92, 621–630. doi: 10.1139/cjpp-2014-0099
Arif, M., Chikuma, T., Ahmed, M. M., Nakazato, M., Smith, M. A., and Kato, T. (2009). Effects of memantine on soluble Alphabeta(25-35)-induced changes in peptidergic and glial cells in Alzheimer's disease model rat brain regions. Neuroscience 164, 1199–1209. doi: 10.1016/j.neuroscience.2009.08.063
Beach, T. G., Honer, W. G., and Hughes, L. H. (1997). Cholinergic fibre loss associated with diffuse plaques in the non-demented elderly: the preclinical stage of Alzheimer's disease? Acta Neuropathol. 93, 146–153. doi: 10.1007/s004010050595
Benzing, W. C., Mufson, E. J., and Armstrong, D. M. (1993). Immunocytochemical distribution of peptidergic and cholinergic fibers in the human amygdala: their depletion in Alzheimer's disease and morphologic alteration in non-demented elderly with numerous senile plaques. Brain Res. 625, 125–138. doi: 10.1016/0006-8993(93)90145-d
Benzing, W. C., Mufson, E. J., Jennes, L., and Armstrong, D. M. (1990). Reduction of neurotensin immunoreactivity in the amygdala in Alzheimer's disease. Brain Res. 537, 298–302. doi: 10.1016/0006-8993(90)90372-I
Benzing, W. C., Mufson, E. J., Jennes, L., Stopa, E. G., and Armstrong, D. M. (1992). Distribution of neurotensin immunoreactivity within the human amygdaloid complex: a comparison with acetylcholinesterase- and Nissl-stained tissue sections. J. Comp. Neurol. 317, 283–297. doi: 10.1002/cne.903170306
Biggins, J. A., Perry, E. K., McDermott, J. R., Smith, A. I., Perry, R. H., and Edwardson, J. A. (1983). Post mortem levels of thyrotropin-releasing hormone and neurotensin in the amygdala in Alzheimer's disease, schizophrenia and depression. J. Neurol. Sci. 58, 117–122. doi: 10.1016/0022-510X(83)90114-4
Bissette, G., Nemeroff, C. B., Decker, M. W., Kizer, J. S., Agid, Y., and Javoy-Agid, F. (1985). Alterations in regional brain concentrations of neurotensin and bombesin in Parkinson's disease. Ann. Neurol. 17, 324–328. doi: 10.1002/ana.410170403
Bouras, C., Vallet, P. G., Hof, P. R., Charnay, Y., Golaz, J., and Constantinidis, J. (1990). Substance P immunoreactivity in Alzheimer disease: a study in cases presenting symmetric or asymmetric cortical atrophy. Alzheimer Dis. Assoc. Disord. 4, 24–34. doi: 10.1097/00002093-19904010000003
Campolongo, P., Ratano, P., Ciotti, M. T., Florenzano, F., Nori, S. L., Marolda, R., et al. (2013). Systemic administration of substance P recovers beta amyloid-induced cognitive deficits in rat: involvement of Kv potassium channels. PLoS ONE 8:e78036. doi: 10.1371/journal.pone.0078036
Cao, X., Zhu, M., He, Y., Chu, W., Du, Y., and Du, H. (2018). Increased serum acylated ghrelin levels in patients with mild cognitive impairment. J. Alzheimers Dis. 61, 545–552. doi: 10.3233/JAD-170721
Cecarini, V., Bonfili, L., Cuccioloni, M., Keller, J. N., Bruce-Keller, A. J., and Eleuteri, A. M. (2016). Effects of ghrelin on the proteolytic pathways of Alzheimer's disease neuronal cells. Mol. Neurobiol. 53, 3168–3178. doi: 10.1007/s12035-015-9227-x
Chen, X. Y., Chen, L., and Du, Y. F. (2017). Orexin-A increases the firing activity of hippocampal CA1 neurons through orexin-1 receptors. J. Neurosci. Res. 95, 1415–1426. doi: 10.1002/jnr.23975
Chen, Y., Cao, C. P., Li, C. R., Wang, W., Zhang, D., Han, L. L., et al. (2010). Ghrelin modulates insulin sensitivity and tau phosphorylation in high glucose-induced hippocampal neurons. Biol. Pharm. Bull. 33, 1165–1169. doi: 10.1248/bpb.33.1165
Constantinidis, J., Bouras, C., and Richard, J. (1983). Putative peptide neurotransmitters in human neuropathology: a review of topography and clinical implications. Clin. Neuropathol. 2, 47–54.
Croce, N., Ciotti, M. T., Gelfo, F., Cortelli, S., Federici, G., Caltagirone, C., et al. (2012). Neuropeptide Y protects rat cortical neurons against β-amyloid toxicity and re-establishes synthesis and release of nerve growth factor. ACS Chem. Neurosci. 3, 312–318. doi: 10.1021/cn200127e
Croce, N., Dinallo, V., Ricci, V., Federici, G., Caltagirone, C., Bernardini, S., et al. (2011). Neuroprotective effect of neuropeptide Y against β-amyloid 25-35 toxicity in SH-SY5Y neuroblastoma cells is associated with increased neurotrophin production. Neurodegener. Dis. 8, 300–309. doi: 10.1159/000323468
Croce, N., Gelfo, F., Ciotti, M. T., Federici, G., Caltagirone, C., Bernardini, S., et al. (2013). NPY modulates miR-30a-5p and BDNF in opposite direction in an in vitro model of Alzheimer disease: a possible role in neuroprotection? Mol. Cell Biochem. 376, 189–195. doi: 10.1007/s11010-013-1567-0
Davies, J., Chen, J., Pink, R., Carter, D., Saunders, N., Sotiriadis, G., et al. (2015). Orexin receptors exert a neuroprotective effect in Alzheimer's disease (AD) via heterodimerization with GPR103. Sci. Rep. 5:12584. doi: 10.1038/srep12584
Dhurandhar, E. J., Allison, D. B., van Groen, T., and Kadish, I. (2013). Hunger in the absence of caloric restriction improves cognition and attenuates Alzheimer's disease pathology in a mouse model. PLoS ONE 8:e60437. doi: 10.1371/journal.pone.0060437
Diez, M., Danner, S., Frey, P., Sommer, B., Staufenbiel, M., Wiederhold, K. H., et al. (2003). Neuropeptide alterations in the hippocampal formation and cortex of transgenic mice overexpressing beta-amyloid precursor protein (APP) with the Swedish double mutation (APP23). Neurobiol. Dis. 14, 579–594. doi: 10.1016/j.nbd.2003.08.003
Disterhoft, J. F., and Oh, M. M. (2006). Pharmacological and molecular enhancement of learning in aging and Alzheimer's disease. J. Physiol. Paris 99, 180–192. doi: 10.1016/j.jphysparis.2005.12.079
dos Santos, V. V., Rodrigues, A. L., De Lima, T. C., de Barioglio, S. R., Raisman-Vozari, R., and Prediger, R. D. (2013a). Ghrelin as a neuroprotective and palliative agent in Alzheimer's and Parkinson's disease. Curr. Pharm. Des. 19, 6773–6790. doi: 10.2174/13816128113199990411
dos Santos, V. V., Santos, D. B., Lach, G., Rodrigues, A. L., Farina, M., De Lima, T. C., et al. (2013b). Neuropeptide Y (NPY) prevents depressive-like behavior, spatial memory deficits and oxidative stress following amyloid-β [Aβ(1-40)] administration in mice. Behav. Brain Res. 244, 107–115. doi: 10.1016/j.bbr.2013.01.039
Duarte-Neves, J., Pereira de Almeida, L., and Cavadas, C. (2016). Neuropeptide Y (NPY) as a therapeutic target for neurodegenerative diseases. Neurobiol. Dis. 95, 210–224. doi: 10.1016/j.nbd.2016.07.022
Ferini-Strambi, L. (2014). Possible role of orexin in the pathogenesis of Alzheimer disease. JAMA Neurol. 71, 1478–1480. doi: 10.1001/jamaneurol.2014.2819
Fernandes, J., Mudgal, J., Rao, C. M., Arora, D., Basu Mallik, S., Pai, K. S. R., et al. (2018). N-acetyl-L-tryptophan, a substance-P receptor antagonist attenuates aluminum-induced spatial memory deficit in rats. Toxicol. Mech. Methods 28, 328–334. doi: 10.1080/15376516.2017.1411412
Fernandez, A., de Ceballos, M. L., Jenner, P., and Marsden, C. D. (1994). Neurotensin, substance P, delta and mu opioid receptors are decreased in basal ganglia of Parkinson's disease patients. Neuroscience 61, 73–79. doi: 10.1016/0306-4522(94)90061-2
Ferrier, I. N., Cross, A. J., Johnson, J. A., Roberts, G. W., Crow, T. J., Corsellis, J. A., et al. (1983). Neuropeptides in Alzheimer type dementia. J. Neurol. Sci. 62, 159–170. doi: 10.1016/0022-510X(83)90196-X
Fronczek, R., van Geest, S., Frölich, M., Overeem, S., Roelandse, F. W., Lammers, G. J., et al. (2012). Hypocretin (orexin) loss in Alzheimer's disease. Neurobiol. Aging 33, 1642–1650. doi: 10.1016/j.neurobiolaging.2011.03.014
Gabelle, A., Jaussent, I., Hirtz, C., Vialaret, J., Navucet, S., Grasselli, C., et al. (2017). Cerebrospinal fluid levels of orexin-A and histamine, and sleep profile within the Alzheimer process. Neurobiol. Aging 53, 59–66. doi: 10.1016/j.neurobiolaging.2017.01.011
Gahete, M. D., Córdoba-Chacón, J., Kineman, R. D., Luque, R. M., and Castaño, J. P. (2011). Role of ghrelin system in neuroprotection and cognitive functions: implications in Alzheimer's disease. Peptides 32, 2225–2228. doi: 10.1016/j.peptides.2011.09.019
Gahete, M. D., Rubio, A., Córdoba-Chacón, J., Gracia-Navarro, F., Kineman, R. D., Avila, J., et al. (2010). Expression of the ghrelin and neurotensin systems is altered in the temporal lobe of Alzheimer's disease patients. J. Alzheimers Dis. 22, 819–828. doi: 10.3233/JAD-2010-100873
Gallone, S., Boschi, S., Rubino, E., De Martino, P., Scarpini, E., Galimberti, D., et al. (2014). Is HCRTR2 a genetic risk factor for Alzheimer's disease? Dement. Geriatr. Cogn. Disord. 38, 245–253. doi: 10.1159/000359964
Garver, D. L., Bissette, G., Yao, J. K., and Nemeroff, C. B. (1991). Relation of CSF neurotensin concentrations to symptoms and drug response of psychotic patients. Am. J. Psychiatry 148, 484–488. doi: 10.1176/ajp.148.4.484
Giedd, J. N., Snell, J. W., Lange, N., Rajapakse, J. C., Casey, B. J., Kozuch, P. L., et al. (1996). Quantitative magnetic resonance imaging of human brain development: ages 4–18. Cereb. Cortex 6, 551–560. doi: 10.1093/cercor/6.4.551
Goldstein, J., Seidman, L., Horton, N., Makris, N., Kennedy, D., Caviness, V., et al. (2001). Normal sexual dimorphism of the adult human brain assessed by in vivo magnetic resonance imaging. Cereb. Cortex 11, 490–497. doi: 10.1093/cercor/11.6.490
Gomes, S., Martins, I., Fonseca, A. C., Oliveira, C. R., Resende, R., and Pereira, C. M. (2014). Protective effect of leptin and ghrelin against toxicity induced by amyloid-β oligomers in a hypothalamic cell line. J. Neuroendocrinol. 26, 176–185. doi: 10.1111/jne.12138
Grosas, A. B., Kalimuthu, P., Smith, A. C., Williams, P. A., Millar, T. J., Bernhardt, P. V., et al. (2014). The tachykinin peptide neurokinin B binds copper(I) and silver(I) and undergoes quasi-reversible electrochemistry: towards a new function for the peptide in the brain. Neurochem. Int. 70, 1–9. doi: 10.1016/j.neuint.2014.03.002
Guan, X. M., Yu, H., Palyha, O. C., McKee, K. K., Feighner, S. D., Sirinathsinghji, D. J., et al. (1997). Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res. Mol. Brain Res. 48, 23–29. doi: 10.1016/S0169-328X(97)00071-5
Han, P., Liang, W., Baxter, L. C., Yin, J., Tang, Z., Beach, T. G., et al. (2014a). Pituitary adenylate cyclase-activating polypeptide is reduced in Alzheimer disease. Neurology 82, 1724–1728. doi: 10.1212/wnl.0000000000000417
Han, P., Nielsen, M., Song, M., Yin, J., Permenter, M. R., Vogt, J. A., et al. (2017). The impact of aging on brain pituitary adenylate cyclase activating polypeptide, pathology and cognition in mice and rhesus macaques. Front. Aging Neurosci. 9:180. doi: 10.3389/fnagi.2017.00180
Han, P., Tang, Z., Yin, J., Maalouf, M., Beach, T. G., Reiman, E. M., et al. (2014b). Pituitary adenylate cyclase-activating polypeptide protects against β-amyloid toxicity. Neurobiol. Aging 35, 2064–2071. doi: 10.1016/j.neurobiolaging.2014.03.022
Hannibal, J. (2002). Pituitary adenylate cyclase-activating peptide in the rat central nervous system: an immunohistochemical and in situ hybridization study. J. Comp. Neurol. 453, 389–417. doi: 10.1002/cne.10418
Hsieh, Y. S., Chen, P. N., Yu, C. H., Liao, J. M., and Kuo, D. Y. (2013). The neuropeptide Y Y1 receptor knockdown modulates activator protein 1-involved feeding behavior in amphetamine-treated rats. Mol. Brain 6:46. doi: 10.1186/1756-6606-6-46
Hu, K., Harper, D. G., Shea, S. A., Stopa, E. G., and Scheer, F. A. (2013). Noninvasive fractal biomarker of clock neurotransmitter disturbance in humans with dementia. Sci. Rep. 3:2229. doi: 10.1038/srep02229
Ishii, M., Wang, G., Racchumi, G., Dyke, J. P., and Iadecola, C. (2014). Transgenic mice overexpressing amyloid precursor protein exhibit early metabolic deficits and a pathologically low leptin state associated with hypothalamic dysfunction in arcuate neuropeptide Y neurons. J. Neurosci. 34, 9096–9106. doi: 10.1523/jneurosci.0872-14.2014
Jaeger, L. B., Farr, S. A., Banks, W. A., and Morley, J. E. (2002). Effects of orexin-A on memory processing. Peptides 23, 1683–1688. doi: 10.1016/s0196-9781(02)00110-9
Jansen, K. L., Faull, R. L., Dragunow, M., and Synek, B. L. (1990). Alzheimer's disease: changes in hippocampal N-methyl-D-aspartate, quisqualate, neurotensin, adenosine, benzodiazepine, serotonin and opioid receptors–an autoradiographic study. Neuroscience 39, 613–627. doi: 10.1016/0306-4522(90)90246-Z
Jiao, Q., Du, X., Li, Y., Gong, B., Shi, L., Tang, T., et al. (2017). The neurological effects of ghrelin in brain diseases: Beyond metabolic functions. Neurosci. Biobehav. Rev. 73, 98–111. doi: 10.1016/j.neubiorev.2016.12.010
Johansson, P., Almqvist, E. G., Wallin, A., Johansson, J. O., Andreasson, U., Blennow, K., et al. (2015). Cerebrospinal fluid substance P concentrations are elevated in patients with Alzheimer's disease. Neurosci. Lett. 609, 58–62. doi: 10.1016/j.neulet.2015.10.006
Kang, J. E., Lim, M. M., Bateman, R. J., Lee, J. J., Smyth, L. P., Cirrito, J. R., et al. (2009). Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. doi: 10.1126/science.1180962
Kang, S., Moon, N. R., Kim, D. S., Kim, S. H., and Park, S. (2015). Central acylated ghrelin improves memory function and hippocampal AMPK activation and partly reverses the impairment of energy and glucose metabolism in rats infused with β-amyloid. Peptides 71, 84–93. doi: 10.1016/j.peptides.2015.07.005
Kinkead, B., and Nemeroff, C. B. (2004). Neurotensin, schizophrenia, and antipsychotic drug action. Int. Rev. Neurobiol. 59, 327–349. doi: 10.1016/S0074-7742(04)59013-X
Kojro, E., Postina, R., Buro, C., Meiringer, C., Gehrig-Burger, K., and Fahrenholz, F. (2006). The neuropeptide PACAP promotes the alpha-secretase pathway for processing the Alzheimer amyloid precursor protein. FASEB J. 20, 512–514. doi: 10.1096/fj.05-4812fje
Krezymon, A., Richetin, K., Halley, H., Roybon, L., Lassalle, J. M., Francès, B., et al. (2013). Modifications of hippocampal circuits and early disruption of adult neurogenesis in the tg2576 mouse model of Alzheimer's disease. PLoS ONE 8:e76497. doi: 10.1371/journal.pone.0076497
Kunath, N., van Groen, T., Allison, D. B., Kumar, A., Dozier-Sharpe, M., and Kadish, I. (2015). Ghrelin agonist does not foster insulin resistance but improves cognition in an Alzheimer's disease mouse model. Sci. Rep. 5:11452. doi: 10.1038/srep11452
Lacor, P. N., Buniel, M. C., Furlow, P. W., Clemente, A. S., Velasco, P. T., Wood, M., et al. (2007). Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J. Neurosci. 27, 796–807. doi: 10.1523/jneurosci.3501-06.2007
Lattuada, D., Crotta, K., Tonna, N., Casnici, C., Benfante, R., Fornasari, D., et al. (2013). The expression of GHS-R in primary neurons is dependent upon maturation stage and regional localization. PLoS ONE 8:e64183. doi: 10.1371/journal.pone.0064183
Lee, D. Y., Hong, S. H., Kim, B., Lee, D. S., Yu, K., and Lee, K. S. (2018). Neuropeptide Y mitigates ER stress-induced neuronal cell death by activating the PI3K-XBP1 pathway. Eur. J. Cell Biol. 97, 339–348. doi: 10.1016/j.ejcb.2018.04.003
Lee, E. H., and Seo, S. R. (2014). Neuroprotective roles of pituitary adenylate cyclase-activating polypeptide in neurodegenerative diseases. BMB Rep. 47, 369–375. doi: 10.5483/BMBRep.2014.47.7.086
Liguori, C. (2017). Orexin and Alzheimer's disease. Curr. Top. Behav. Neurosci. 33, 305–322. doi: 10.1007/7854_2016_50
Liguori, C., Chiaravalloti, A., Nuccetelli, M., Izzi, F., Sancesario, G., Cimini, A., et al. (2017). Hypothalamic dysfunction is related to sleep impairment and CSF biomarkers in Alzheimer's disease. J. Neurol. 264, 2215–2223. doi: 10.1007/s00415-017-8613-x
Liguori, C., Nuccetelli, M., Izzi, F., Sancesario, G., Romigi, A., Martorana, A., et al. (2016). Rapid eye movement sleep disruption and sleep fragmentation are associated with increased orexin-A cerebrospinal-fluid levels in mild cognitive impairment due to Alzheimer's disease. Neurobiol. Aging 40, 120–126. doi: 10.1016/j.neurobiolaging.2016.01.007
Ma, Z., Jiang, W., and Zhang, E. E. (2016). Orexin signaling regulates both the hippocampal clock and the circadian oscillation of Alzheimer's disease-risk genes. Sci. Rep. 6:36035. doi: 10.1038/srep36035
Mahar, I., Albuquerque, M. S., Mondragon-Rodriguez, S., Cavanagh, C., Davoli, M. A., Chabot, J. G., et al. (2017). Phenotypic alterations in hippocampal NPY- and PV-expressing interneurons in a presymptomatic transgenic mouse model of Alzheimer's disease. Front. Aging Neurosci. 8:327. doi: 10.3389/fnagi.2016.00327
Malkki, H. (2014). Alzheimer disease: increased orexin level correlates with sleep disruption and cognitive decline in Alzheimer disease. Nat. Rev. Neurol. 10:672. doi: 10.1038/nrneurol.2014.209
Mann, D. M. (1989). The pathogenesis and progression of the pathological changes of Alzheimer's disease. Ann. Med. 21, 133–136.
Mantyh, P. W. (2002). Neurobiology of substance P and the NK1 receptor. J. Clin. Psychiatry 63 (Suppl. 11), 6–10.
Marolda, R., Ciotti, M. T., Matrone, C., Possenti, R., Calissano, P., Cavallaro, S., et al. (2012). Substance P activates ADAM9 mRNA expression and induces α-secretase-mediated amyloid precursor protein cleavage. Neuropharmacology 62, 1954–1963. doi: 10.1016/j.neuropharm.2011.12.025
Martins, I., Gomes, S., Costa, R. O., Otvos, L., Oliveira, C. R., Resende, R., et al. (2013). Leptin and ghrelin prevent hippocampal dysfunction induced by Aβ oligomers. Neuroscience 241, 41–51. doi: 10.1016/j.neuroscience.2013.02.062
McKay, B. M., Oh, M. M., Galvez, R., Burgdorf, J., Kroes, R. A., and Weiss, C. (2012). Increasing SK2 channel activity impairs associative learning. J. Neurophysiol. 108, 863–870. doi: 10.1152/jn.00025.2012
Mittapalli, G. K., and Roberts, E. (2014). Ligands of the neuropeptide Y Y2 receptor. Bioorg. Med. Chem. Lett. 24, 430–441. doi: 10.1016/j.bmcl.2013.11.061
Miyata, A., Arimura, A., Dahl, R. R., Minamino, N., Uehara, A., Jiang, L., et al. (1989). Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem. Biophys. Res. Commun. 164, 567–574. doi: 10.1016/0006-291X(89)91757-9
Moon, M., Cha, M. Y., and Mook-Jung, I. (2014). Impaired hippocampal neurogenesis and its enhancement with ghrelin in 5XFAD mice. J. Alzheimers Dis. 41, 233–241. doi: 10.3233/jad-132417
Moon, M., Choi, J. G., Nam, D. W., Hong, H. S., Choi, Y. J., Oh, M. S., et al. (2011). Ghrelin ameliorates cognitive dysfunction and neurodegeneration in intrahippocampal amyloid-β1-42 oligomer-injected mice. J. Alzheimers Dis. 23, 147–159. doi: 10.3233/jad-2010-101263
Murray, S., Tulloch, A., Gold, M. S., and Avena, N. M. (2014). Hormonal and neural mechanisms of food reward, eating behaviour and obesity. Nat. Rev. Endocrinol. 10, 540–552. doi: 10.1038/nrendo.2014.91
Nag, S., Yee, B. K., and Tang, F. (1999). Reduction in somatostatin and substance P levels and choline acetyltransferase activity in the cortex and hippocampus of the rat after chronic intracerebroventricular infusion of beta-amyloid (1-40). Brain Res. Bull. 50, 251–262. doi: 10.1016/S0361-9230(99)00196-3
Onoue, S., Endo, K., Ohshima, K., Yajima, T., and Kashimoto, K. (2002). The neuropeptide PACAP attenuates beta-amyloid (1-42)-induced toxicity in PC12 cells. Peptides 23, 1471–1478. doi: 10.1016/S0196-9781(02)00085-2
Osorio, R. S., Ducca, E. L., Wohlleber, M. E., Tanzi, E. B., Gumb, T., Twumasi, A., et al. (2016). Orexin-A is associated with increases in cerebrospinal fluid phosphorylated-tau in cognitively normal elderly subjects. Sleep 39, 1253–1260. doi: 10.5665/sleep.5846
Panagopoulos, V. N., and Ralevski, E. (2014). The role of ghrelin in addiction: a review. Psychopharmacology (Berl). 231, 2725–2740. doi: 10.1007/s00213-014-3640-0
Pérez-Fernández, J., Megías, M., and Pombal, M. A. (2014). Cloning, phylogeny, and regional expression of a Y5 receptor mRNA in the brain of the sea lamprey (Petromyzon marinus). J. Comp. Neurol. 522, 1132–1154. doi: 10.1002/cne.23481
Pieri, M., Amadoro, G., Carunchio, I., Ciotti, M. T., Quaresima, S., Florenzano, F., et al. (2010). SP protects cerebellar granule cells against beta-amyloid-induced apoptosis by down-regulation and reduced activity of Kv4 potassium channels. Neuropharmacology 58, 268–276. doi: 10.1016/j.neuropharm.2009.06.029
Popelová, A., Kákonová, A., Hrubá, L., Kuneš, J., Maletínská, L., and Železná, B. (2018). Potential neuroprotective and anti-apoptotic properties of a long-lasting stable analog of ghrelin: an in vitro study using SH-SY5Y cells. Physiol. Res. 67, 339–346.
Postina, R. (2012). Activation of α-secretase cleavage. J. Neurochem. 120 Suppl. 1, 46–54. doi: 10.1111/j.1471-4159.2011.07459.x
Quigley, B. J. Jr., and Kowall, N. W. (1991). Substance P-like immunoreactive neurons are depleted in Alzheimer's disease cerebral cortex. Neuroscience 41, 41–60. doi: 10.1016/0306-4522(91)90199-X
Rak-Mardyla, A. (2013). Ghrelin role in hypothalamus-pituitary-ovarian axis. J. Physiol. Pharmacol. 64, 695–704.
Raoof, R., Esmaeili-Mahani, S., Abbasnejad, M., Raoof, M., Sheibani, V., Kooshki, R., et al. (2015). Changes in hippocampal orexin 1 receptor expression involved in tooth pain-induced learning and memory impairment in rats. Neuropeptides 50, 9–16. doi: 10.1016/j.npep.2015.03.002
Rat, D., Schmitt, U., Tippmann, F., Dewachter, I., Theunis, C., Wieczerzak, E., et al. (2011). Neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) slows down Alzheimer's disease-like pathology in amyloid precursor protein-transgenic mice. FASEB J. 25, 3208–3218. doi: 10.1096/fj.10-180133
Reglodi, D., Kiss, P., Lubics, A., and Tamas, A. (2011). Review on the protective effects of PACAP in models of neurodegenerative diseases in vitro and in vivo. Curr. Pharm. Des. 17, 962–972. doi: 10.2174/138161211795589355
Roh, J. H., Jiang, H., Finn, M. B., Stewart, F. R., Mahan, T. E., Cirrito, J. R., et al. (2014). Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer's disease. J. Exp. Med. 211, 2487–2496. doi: 10.1084/jem.20141788
Rose, J. B., Crews, L., Rockenstein, E., Adame, A., Mante, M., Hersh, L. B., et al. (2009). Neuropeptide Y fragments derived from neprilysin processing are neuroprotective in a transgenic model of Alzheimer's disease. J. Neurosci. 29, 1115–1125. doi: 10.1523/jneurosci.4220-08.2009
Rösler, N., Wichart, I., and Jellinger, K. A. (2001). Ex vivo lumbar and post mortem ventricular cerebrospinal fluid substance P-immunoreactivity in Alzheimer's disease patients. Neurosci. Lett. 299, 117–120. doi: 10.1016/S0304-3940(01)01514-2
Rowe, W. B., Kar, S., Meaney, M. J., and Quirion, R. (2006). Neurotensin receptor levels as a function of brain aging and cognitive performance in the Morris water maze task in the rat. Peptides 27, 2415–2423. doi: 10.1016/j.peptides.2006.03.036
Russino, D., McDonald, E., Hejazi, L., Hanson, G. R., and Jones, C. E. (2013). The tachykinin peptide neurokinin B binds copper forming an unusual [CuII(NKB)2] complex and inhibits copper uptake into 1321N1 astrocytoma cells. ACS Chem. Neurosci. 4, 1371–1381. doi: 10.1021/cn4000988
Santos, V. V., Stark, R., Rial, D., Silva, H. B., Bayliss, J. A., Lemus, M. B., et al. (2017). Acyl ghrelin improves cognition, synaptic plasticity deficits and neuroinflammation following amyloid β (Aβ1-40) administration in mice. J. Neuroendocrinol. 29,1–11. doi: 10.1111/jne.12476
Severini, C., Petrella, C., and Calissano, P. (2016). Substance P and Alzheimer's disease: emerging novel roles. Curr. Alzheimer Res. 13, 964–972. doi: 10.2174/1567205013666160401114039
Shi, L., Du, X., Jiang, H., and Xie, J. (2017). Ghrelin and neurodegenerative disorders-a review. Mol. Neurobiol. 54, 1144–1155. doi: 10.1007/s12035-016-9729-1
Shi, L. M., Jiang, H., and Xie, J. X. (2014). Neuroprotective effects of ghrelin in Parkinsons disease and Alzheimer's disease. Sheng Li Ke Xue Jin Zhan. 45, 140–144.
Shibata, N., Ohnuma, T., Kuerban, B., Komatsu, M., and Arai, H. (2011). Genetic association between ghrelin polymorphisms and Alzheimer's disease in a Japanese population. Dement. Geriatr. Cogn. Disord. 32, 178–181. doi: 10.1159/000333075
Shieh, P. C., Tsao, C. W., Li, J. S., Wu, H. T., Wen, Y. J., Kou, D. H., et al. (2008). Role of pituitary adenylate cyclase-activating polypeptide (PACAP) in the action of ginsenoside Rh2 against beta-amyloid-induced inhibition of rat brain astrocytes. Neurosci. Lett. 434, 1–5. doi: 10.1016/j.neulet.2007.12.032
Skup, M., Zhu, H., Wang, Y., Giovanello, K. S., Lin, J. A., Shen, D., et al. (2011). Sex differences in grey matter atrophy patterns among AD and aMCI patients: results from ADNI. Neuroimage 56, 890–906. doi: 10.1016/j.neuroimage.2011.02.060
Spencer, B., Potkar, R., Metcalf, J., Thrin, I., Adame, A., Rockenstein, E., et al. (2016). Systemic central nervous system (CNS)-targeted delivery of neuropeptide Y (NPY) reduces neurodegeneration and increases neural precursor cell proliferation in a mouse model of Alzheimer disease. J. Biol. Chem. 291, 1905–1920. doi: 10.1074/jbc.M115.678185
Spitznagel, M. B., Benitez, A., Updegraff, J., Potter, V., Alexander, T., Glickman, E., et al. (2010). Serum ghrelin is inversely associated with cognitive function in a sample of non-demented elderly. Psychiatry Clin. Neurosci. 64, 608–611. doi: 10.1111/j.1440-1819.2010.02145.x
Stoyanova, I. I. (2014). Ghrelin: a link between ageing, metabolism and neurodegenerative disorders. Neurobiol. Dis. 72(Pt A), 72–83. doi: 10.1016/j.nbd.2014.08.026
Struble, R. G., Powers, R. E., Casanova, M. F., Kitt, C. A., Brown, E. C., and Price, D. L. (1987). Neuropeptidergic systems in plaques of Alzheimer's disease. J. Neuropathol. Exp. Neurol. 46, 567–584. doi: 10.1097/00005072-198709000-00006
Tiraboschi, P., Hansen, L. A., Thal, L. J., and Corey-Bloom, J. (2004). The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology 62, 1984–1989. doi: 10.1212/01.wnl.0000129697.01779
Vaudry, D., Cottet-Rousselle, C., Basille, M., Falluel-Morel, A., Fournier, A., Vaudry, H., et al. (2004). Pituitary adenylate cyclase-activating polypeptide inhibits caspase-3 activity but does not protect cerebellar granule neurons against beta-amyloid (25-35)-induced apoptosis. Regul. Pept. 123, 43–49. doi: 10.1016/j.regpep.2004.05.025
Waters, S. M., and David, T. P. (1995). Alterations of substance P metabolism and neuropeptidases in Alzheimer's disease. J. Gerontol. A. Biol. Sci. Med. Sci. 50, B315–B319. doi: 10.1093/gerona/50a.5.b315
Willis, M., Hutter-Paier, B., Wietzorrek, G., Windisch, M., Humpel, C., Knaus, H. G., et al. (2007). Localization and expression of substance P in transgenic mice overexpressing human APP751 with the London (V717I) and Swedish (K670M/N671L) mutations. Brain Res. 1143, 199–207. doi: 10.1016/j.brainres.2007.01.080
Witte, A. V., Savli, M., Holik, A., Kasper, S., and Lanzenberger, R. (2010). Regional sex differences in grey matter volume are associated with sex hormones in the young adult human brain. Neuroimage 49, 1205–1212. doi: 10.1016/j.neuroimage.2009.09.046
Wu, Z. L., Ciallella, J. R., Flood, D. G., O'Kane, T. M., Bozyczko-Coyne, D., and Savage, M. J. (2006). Comparative analysis of cortical gene expression in mouse models of Alzheimer's disease. Neurobiol. Aging 27, 377–386. doi: 10.1016/j.neurobiolaging.2005.02.010
Xiao, Z., Cilz, N. I., Kurada, L., Hu, B., Yang, C., Wada, E., et al. (2014). Activation of neurotensin receptor 1 facilitates neuronal excitability and spatial learning and memory in the entorhinal cortex: beneficial actions in an Alzheimer's disease model. J. Neurosci. 34, 7027–7042. doi: 10.1523/jneurosci.0408-14.2014
Yang, L., Zou, B., Xiong, X., Pascual, C., Xie, J., Malik, A., et al. (2013). Hypocretin/orexin neurons contribute to hippocampusdependent social memory and synaptic plasticity in mice. J. Neurosci. 33, 5275–5284. doi: 10.1523/jneurosci.3200-12.2013
Zhang, H., Dong, H., Cilz, N. I., Kurada, L., Hu, B., Wada, E., et al. (2016). Neurotensinergic excitation of dentate gyrus granule cells via Gαq-coupled inhibition of TASK-3 channels. Cereb. Cortex 26, 977–990. doi: 10.1093/cercor/bhu267
Keywords: neuropeptide, Alzheimer's disease, ghrelin, neurotensin, pituitary adenylate cyclase-activating polypeptide, neuropeptide Y, substance P, orexin
Citation: Chen X-Y, Du Y-F and Chen L (2019) Neuropeptides Exert Neuroprotective Effects in Alzheimer's Disease. Front. Mol. Neurosci. 11:493. doi: 10.3389/fnmol.2018.00493
Received: 05 May 2018; Accepted: 21 December 2018;
Published: 11 January 2019.
Edited by:
Christian Gonzalez-Billault, Universidad de Chile, ChileReviewed by:
Luis Gerardo Aguayo, Universidad de Concepción, ChileCopyright © 2019 Chen, Du and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Chen, Y2hlbmxlaXFkQDE2My5jb20=
†Present Address: Xin-Yi Chen, Department of Medicine and Therapeutics, The Chinese University of Hong Kong, Hong Kong, Hong Kong
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.