 Fiona Kerr
Fiona Kerr Ivana Bjedov
Ivana Bjedov Oyinkan Sofola-Adesakin
Oyinkan Sofola-Adesakin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci., 28 August 2018
Sec. Brain Disease Mechanisms
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00297
This article is part of the Research TopicLithium and NeurodegenerationView all 5 articles
Lithium has long been used for the treatment of psychiatric disorders, due to its robust beneficial effect as a mood stabilizing drug. Lithium’s effectiveness for improving neurological function is therefore well-described, stimulating the investigation of its potential use in several neurodegenerative conditions including Alzheimer’s (AD), Parkinson’s (PD) and Huntington’s (HD) diseases. A narrow therapeutic window for these effects, however, has led to concerted efforts to understand the molecular mechanisms of lithium action in the brain, in order to develop more selective treatments that harness its neuroprotective potential whilst limiting contraindications. Animal models have proven pivotal in these studies, with lithium displaying advantageous effects on behavior across species, including worms (C. elegans), zebrafish (Danio rerio), fruit flies (Drosophila melanogaster) and rodents. Due to their susceptibility to genetic manipulation, functional genomic analyses in these model organisms have provided evidence for the main molecular determinants of lithium action, including inhibition of inositol monophosphatase (IMPA) and glycogen synthase kinase-3 (GSK-3). Accumulating pre-clinical evidence has indeed provided a basis for research into the therapeutic use of lithium for the treatment of dementia, an area of medical priority due to its increasing global impact and lack of disease-modifying drugs. Although lithium has been extensively described to prevent AD-associated amyloid and tau pathologies, this review article will focus on generic mechanisms by which lithium preserves neuronal function and improves memory in animal models of dementia. Of these, evidence from worms, flies and mice points to GSK-3 as the most robust mediator of lithium’s neuro-protective effect, but it’s interaction with downstream pathways, including Wnt/β-catenin, CREB/brain-derived neurotrophic factor (BDNF), nuclear factor (erythroid-derived 2)-like 2 (Nrf2) and toll-like receptor 4 (TLR4)/nuclear factor-κB (NFκB), have identified multiple targets for development of drugs which harness lithium’s neurogenic, cytoprotective, synaptic maintenance, anti-oxidant, anti-inflammatory and protein homeostasis properties, in addition to more potent and selective GSK-3 inhibitors. Lithium, therefore, has advantages as a multi-functional therapy to combat the complex molecular pathology of dementia. Animal studies will be vital, however, for comparative analyses to determine which of these defense mechanisms are most required to slow-down cognitive decline in dementia, and whether combination therapies can synergize systems to exploit lithium’s neuro-protective power while avoiding deleterious toxicity.
Lithium has well-described clinical benefits as a mood-stabilizer, and accumulating pre-clinical evidence has provided a basis for research into its therapeutic use in the treatment of a range of neurodegenerative conditions, including Alzheimer’s (AD), Parkinson’s (PD) and Huntington’s (HD) diseases (De Ferrari et al., 2003; Noble et al., 2005; Sarkar et al., 2008; Chiu et al., 2011; Lieu et al., 2014). A narrow therapeutic window for these effects, however, has led to concerted efforts to understand the molecular mechanisms of lithium action in the central nervous system (CNS), in order to develop more selective treatments that harness its neuroprotective potential whilst limiting its toxic side-effects.
Animal models have proven pivotal in these studies, with lithium displaying advantageous effects on behavior across species, including worms (Caenorhabditis elegans; Farina et al., 2017), zebrafish (Danio Rerio; Nery et al., 2014), fruit flies (Drosophila melanogaster; Mudher et al., 2004; McBride et al., 2005; Sofola et al., 2010; Castillo-Quan et al., 2016) and rodents (O’Brien et al., 2004; King and Jope, 2013; Lu et al., 2015; Gelfo et al., 2017). Due to their susceptibility to genetic manipulation, these model organisms have provided evidence for the direct molecular determinants to which lithium’s neuro-protective effects are attributed, including inhibition of inositol monophosphatase (IMPA), glycogen synthase kinase-3 (GSK-3), and a plethora of down-stream targets that further exert neuroprotective potential. Lithium has an important role in cytoprotection particularly by preventing oxidative and neuro-inflammatory damage, maintaining protein homeostasis and enhancing neurogenesis and synaptic maintenance. These neuro-protective properties have beneficial effects across a range of animal models of neurodegeneration, and understanding the mechanisms underpinning these interactions has revealed new targets for the development of drugs to slow-down neuronal damage in these conditions.
Focussing on animal models of dementia, here we review the evidence for the molecular basis of the generic mechanisms by which lithium preserves neuronal function and improves memory. Animal studies will be vital, however, for comparative analyses to determine which of these defense mechanisms are most required to slow-down cognitive decline as disease progresses. We discuss the advantages of combination therapies to synergize the targets of lithium’s protective effects, in comparison with using lithium as a single multi-functional therapy to combat the complex molecular pathology of dementia.
Although the precise molecular determinants of lithium action remain unclear, evidence from model organisms have suggested that IMPA and GSK-3 are direct regulators of its beneficial effects on neuronal function (see Figure 1A). As originally posited by Berridge et al. (1989), the inositol depletion hypothesis of lithium action suggests that uncompetitive inhibition of IMPA and inositol polyphosphate 1-phosphatase (IPP) leads to a deficit in polyphosphoinositide (PIP) signaling, and subsequently inhibition of neuronal excitation, which may explain its beneficial effects as a mood stabilizer. Genetic mutation of IPP in Drosophila melanogaster (Acharya et al., 1998) or the IMPA homolog, ttx-7, in C. elegans (Tanizawa et al., 2006) has indeed shown that these enzymes play an important role in regulation of synaptic function in vivo, although this appears to be mediated by enhancing rather than suppressing synaptic transmission. Contrasting studies in mice, however, suggest that inositol depletion may not alter PIP levels in the CNS (Berry et al., 2004). Furthermore, IMPA1 deletion only partially mimics lithium’s effects on gene expression in the hippocampus (Damri et al., 2015) and IMPA2 deletion fails to phenocopy lithium’s protective effects against depression and anxiety-like behavior (Cryns et al., 2007). Hence the role of IMPA in mediating lithium’s function in the mammalian CNS is less clear.
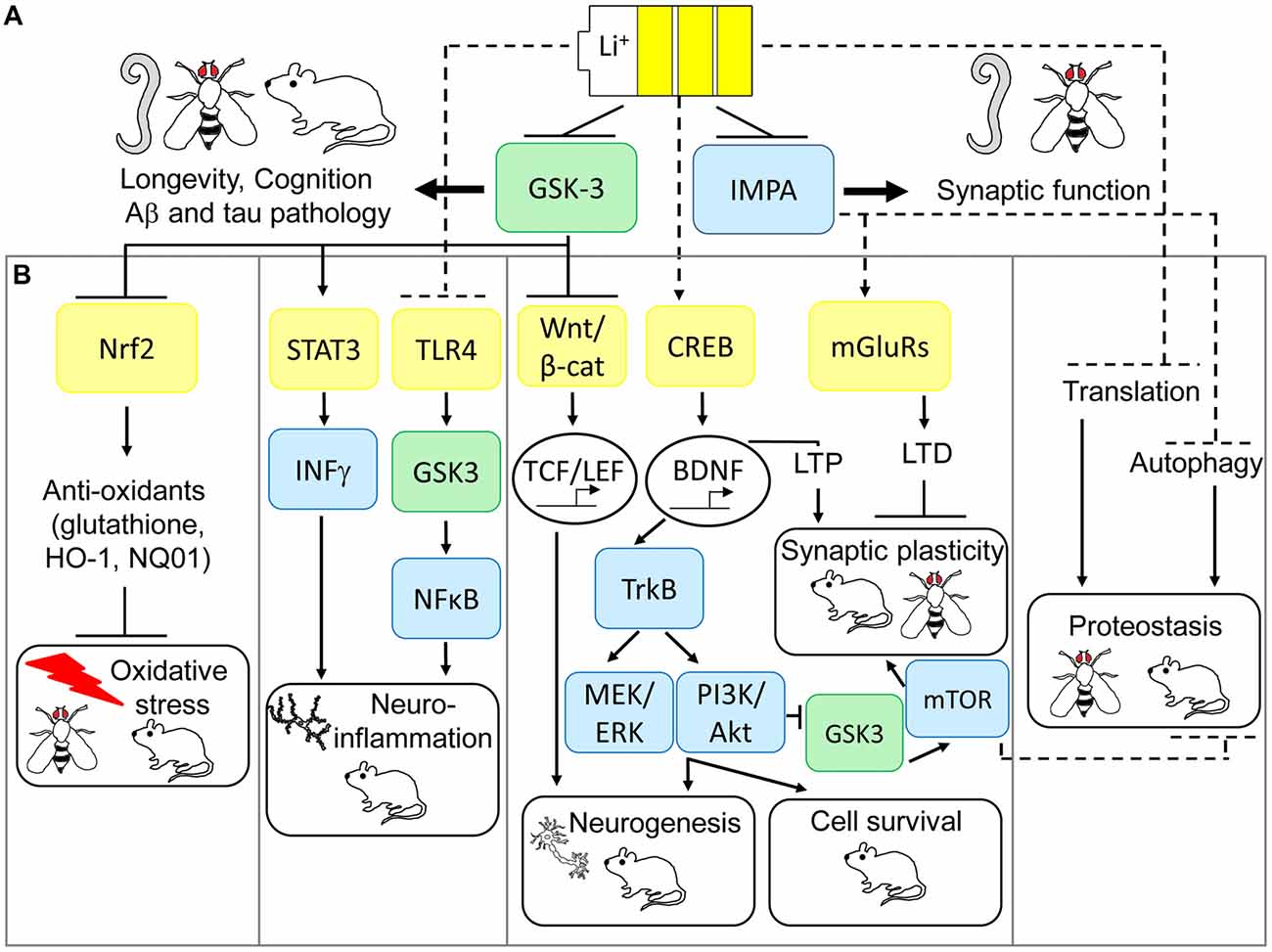
Figure 1. Multi-modal mechanisms of lithium-mediated neuronal protection in model organisms. (A) Glycogen synthase kinase-3 (GSK-3) and inositol monophosphatase (IMPA) are direct targets of lithium action in the central nervous system (CNS). Model organisms have revealed IMPA as a mediator of improved synaptic function in response to lithium, but IMPA1/2 mutations fail to consistently pheno-copy lithium’s protective effects on behavior in mice. GSK-3 appears to be a conserved mediator of lithium action, required for increased longevity, improved cognition and prevention of Alzheimer’s disease (AD) pathology across worm, fly and mice models of neurodegeneration. (B) Animal models have also uncovered several pathways which may preserve downstream neuro-protection processes in response to lithium via GSK-3-dependent and independent mechanisms. GSK-3 inhibition is an upstream regulator of lithium’s activation of nuclear factor (erythroid-derived 2)-like 2 (Nrf2) in preventing oxidative damage, inhibition of signal transducer and activator of transcription 3 (STAT3) to prevent neuro-inflammation, increased Wnt-dependent gene transcription to guide adult neurogenesis, and potentially prevention of protein synthesis by inhibiting translation. More recent evidence also suggests that lithium can exert neuro-protection through non-GSK-3-dependent anti-inflammatory effects on toll-like receptor 4 (TLR4), increased neurogenesis, cell survival and long term potentiation (LTP) via CREB-dependent transcription of brain-derived neurotrophic factor (BDNF) and prevention of long term depression (LTD) by inhibition of over-active metabotropic glutamate receptor (mGLuR)-dependent synaptic transmission. Finally, inhibition of IMPA mediates lithium-dependent activation of autophagy, by reducing inositol levels, thus maintaining protein turnover. Genetic and pharmacological modulation of these anti-oxidant, anti-inflammatory, neurogenesis, cell survival, synaptic plasticity and proteostasis signaling pathways prevents neurodegeneration and improves cognition in Drosophila and mouse models of AD, fronto-temporal dementia (FTD) and Fragile X syndrome.
GSK-3 is also non-competitively inhibited by lithium (Klein and Melton, 1996) and appears to be a more consistent regulator of lithium’s neuro-protective effects across species (Gurvich and Klein, 2002; Aghdam and Barger, 2007). GSK-3 is epistatically required for longevity in response to lithium in both C. elegans (McColl et al., 2008) and Drosophila (Castillo-Quan et al., 2016), and it’s mutation mimics lithium’s ability to alter exploratory behavior in mice (O’Brien et al., 2004, 2011). GSK-3 also plays a well-described role in the generation of AD-associated amyloid β (Aβ) and tau pathologies (Kremer et al., 2011), by promoting abnormal tau phosphorylation (Lucas et al., 2001) and increasing amyloid production (Phiel et al., 2003) and toxicity (DaRocha-Souto et al., 2012). Furthermore, GSK-3 inhibition protects against neuronal damage and cognitive decline in Drosophila (Mudher et al., 2004; Sofola et al., 2010) and rodent models of AD and fronto-temporal dementia (FTD; Serenó et al., 2009). As a pleiotropic enzyme, however, GSK-3 inhibition may exert its neuroprotective effects through diverse mechanisms, including maintenance of axonal transport and synaptic function, promoting adult neurogenesis, preventing apoptosis and reducing neuro-inflammation (Llorens-Martín et al., 2014). Building upon these observations, and as extensively reviewed (Eldar-Finkelman and Martinez, 2011), drug development efforts have aimed to discover new specific GSK-3 inhibitors with improved toxicity profiles for treatment of neurodegenerative disorders in comparison with lithium. These include ATP-competitive (indirubin, paullones, thiazoles, SB-216763 and SB-415286) and non-ATP-competitive (thiadiazolidindiones (TDZD-8, NP-12/tideglusib), and L803-mts) inhibitors, which have shown promising pre-clinical efficacy in improving cognition and protecting neurons in rodent models of AD and FTD. Of these, non-ATP-competitive GSK-3 inhibitors appear to have improved specificity and safety, which has led to Phase II clinical trials for the use of tideglusib in mild-moderate AD, as part of the ARGO study (Lovestone et al., 2015), and in progressive supranuclear palsy (PSP), as part of the TAUROS study (Tolosa et al., 2014). Although tideglusib proved safe for use in both AD and PSP patients, and some reduction in brain atrophy was observed in the TAUROS study, no significant clinical improvement in primary measures, ADAS-cog15 score or PSP rating scale, was reported in either of these trials. Based on the observation that mild AD patients in the ARGO study showed some cognitive improvement on lower doses of lithium (500 mg; Lovestone et al., 2015), suggesting a non-linear dose-response, future studies to examine the effects of tideglusib in patients at earlier stages and to optimize the most effective dose may be warranted.
Recent transcriptomic analyses across species, however, have revealed that lithium has wide-ranging cellular effects. For example by altering DNA replication, metabolism and endoplasmic reticulum (ER) genes in worms (McColl et al., 2008), translation and cellular detoxification genes in flies (Castillo-Quan et al., 2016), and neurogenesis, synaptic function, anti-apoptosis and anti-inflammatory genes in rat and mouse brain (Roux and Dosseto, 2017). This supports a multi-faceted mechanism of lithium-mediated neuroprotection, of relevance to many neurological conditions, through promotion of its cytoprotective, anti-oxidant, anti-inflammatory, protein homeostasis, neurogenic and synaptic maintenance properties. Alterations in these pathways may be an indirect consequence of GSK-3’s pleiotropic effects, but understanding the molecular basis of their modulation has revealed new selective targets for neuronal protection in dementia as we discuss below.
Oxidative damage is a common feature of dementia brain (Sultana and Butterfield, 2010; Iadecola, 2013), with reactive oxygen species (ROS) and peroxidized lipids and proteins accumulating early in the disease process (Sultana and Butterfield, 2010). This may be a result of aging related mitochondrial damage, hypoxia-induced ischemia or amyloid accumulation, and leads to wide-spread cellular damage through prevention of cytoprotective signaling, induction of apoptosis and neuro-inflammation (Guo et al., 2017).
Lithium prevents neuronal sensitivity to oxidative damage across species, including hyperoxia in Aβ-expressing flies (Kerr et al., 2017), cerebral ischemia and 3-nitropropionic acid (3-NP)-induced neurotoxicity in rats (Khan et al., 2015; Chen et al., 2016), and kainate and neuropeptide S-induced behavioral and neurological damage in mice (Rojo et al., 2008; Castro et al., 2009). Prevention of oxidative damage by lithium in these paradigms commonly correlates with reduced lipid peroxidation, as measured by thiobarbituric acid reactive substances (TBARS; Shao et al., 2005; Castro et al., 2009) or 4-hydroxynonenal (4-HNE) levels (Tan et al., 2012), protein carbonylation (Shao et al., 2005) and ROS production (Rojo et al., 2008). Conversely lithium treatment increases expression of anti-oxidant enzymes including catalase (Khan et al., 2015), heme-oxygenase-1 (HO-1; Khan et al., 2015; Chen et al., 2016) and NAD(P)H: quinone oxidoreductase 1 (NQ01; Chen et al., 2016) as well as restoring levels of glutathione and glutathione-s-transferases (GstD1, GstD2), an important mediator of neuronal protection against oxidative damage (Baxter and Hardingham, 2016), in Drosophila (Kasuya et al., 2009; Kerr et al., 2017) and rat (Khan et al., 2015) brain. Notably, the HO-1 inhibitor SnPP reversed the beneficial effects of lithium against 3-NP-induced oxidative stress and motor defects in rats (Khan et al., 2015), providing epistatic evidence that prevention of oxidative damage plays a key role in lithium-mediated neuronal protection in vivo.
GSK-3 inhibition parallels many of these anti-oxidant properties of lithium, with GSK-3 antisense oligonucleotides, short-interfering RNAs (siRNA) and specific GSK-3 inhibitors (SB216763, TDZD-8; Rojo et al., 2008; Chen et al., 2016) preventing oxidative damage in rat models of cerebral ischemia and the SAMP8 mouse model of aging-related AD (Farr et al., 2014; Chen et al., 2016). The transcription factor nuclear factor (erythroid-derived 2)-like 2 (Nrf2) has been proposed as a key molecular mediator of the antioxidant effects of lithium down-stream of GSK-3 as reviewed by Kanninen et al. (2011; see Figure 1B). Nrf2 responds to cellular stress, for example under oxidative or neuro-inflammatory conditions, which are common features of neurodegenerative diseases, by increasing expression of an array of antioxidant response element (ARE)-containing genes to counteract damage (Bruns et al., 2015). Lithium increases Nrf2 activity in Drosophila (Castillo-Quan et al., 2016; Kerr et al., 2017), and pheno-copies specific GSK3β inhibition (siRNA and SB216763, TDZD-8) in increasing Nrf2 nuclear translocation and activating transcription of Nrf2 target genes, HO-1 and NQ01, in rat and mouse brain (Rojo et al., 2008; Chen et al., 2016). Moreover, Nrf2 is genetically required for lifespan extension by lithium in flies (Castillo-Quan et al., 2016) and increased Nrf2 nuclear translocation correlates with reduced oxidative damage and improved cognition in SAMP8 mice following treatment with antisense GSK-3 (Farr et al., 2014). These further suggest a causal role for Nrf2 in mediating the neuroprotective effects of lithium and GSK-3 inhibitors in dementia models.
As a general regulator of neuronal protection, Nrf2 has become an attractive therapeutic target for the treatment of several neurodegenerative diseases including AD (Kanninen et al., 2009) and vascular dementia (Alfieri et al., 2011). Genetic activation of Nrf2 protects against neuronal and cognitive decline in Drosophila and mouse models of AD and PD (Kanninen et al., 2009; Barone et al., 2011; Kerr et al., 2017), and pharmacological Nrf2 activators, including triterpenoid compounds (CDDO-EA and CDDO-TFEA) and dimethyl fumarate (DMF), prevent oxidative damage and afford neuronal protection in mouse models of AD (Dumont et al., 2009), FTD (Cuadrado et al., 2018), amyotrophic lateral sclerosis (ALS; Neymotin et al., 2011), PD (Chen et al., 2009; Lastres-Becker et al., 2016) and cerebral ischemia (Fowler et al., 2017). Most classical Nrf2 activators are electrophilic agents which modify cysteine residues on Keap1, a negative regulator of Nrf2, disrupting their physical interaction and enabling Nrf2 to activate transcription. The non-selective nature of this mechanism, however, is thought to explain their toxicity, due to off-target effects, as observed in clinical trials (Abed et al., 2015). Recent drug discovery efforts have therefore focussed on developing direct disruptors of the Keap1-Nrf2 protein-protein interaction (PPI), with the aim of improving safety profiles (Abed et al., 2015; Wells, 2015). We and others have indeed reported promising results using small molecule and peptide Keap1-Nrf2 PPI disruptors in protecting against amyloid-induced synapto-toxicity in mouse primary neurons (Kerr et al., 2017) and neuronal protection in vivo using rat models of global cerebral ischemeia (Tu et al., 2015). Of note, our own studies using Drosophila and neuroblastoma cells suggest that lithium and GSK-3 inhibitors (TDZD8) are weak activators of Nrf2, relative to Keap1-Nrf2 disruption, in response to amyloid toxicity (Kerr et al., 2017). Combined GSK-3 inhibitor and Nrf2 activator therapies may be an advantage, as additive protective effects against Aβ toxicity are observed with Keap1 mutation and lithium treatment in the fly (Kerr et al., 2017) and DMF has recently been shown to prevent tau phosphorylation through inhibition of GSK-3 and neuronal damage by subsequent activation of Nrf2 through Keap1 dependent and independent mechanisms (Cuadrado et al., 2018). Hence further work is required to establish the relative importance of these pathways in maintaining Nrf2 activity in AD and other neurodegenerative diseases.
Alterations in microglia activation and inflammatory factors are an early feature in the brains of dementia patients, including AD and vascular dementia (Calsolaro and Edison, 2016). Moreover, neuronal inflammation directly correlates with decline in cognitive function across species, for example following bacterial infection in flies (Cao et al., 2013; Wu et al., 2017) and viral infection (Hosseini et al., 2018) in mice, suggesting a causal link between inflammatory dysregulation and memory impairment. Based on the increasing risk associated with immunity genes (including TREM2 and CD33) and early neuropathological detection of inflammation in late-onset AD (LOAD), emerging theories also suggest that chronic, aging-associated, inflammation may be an initiating factor leading to the disease-defining neuronal loss, amyloid and tau pathologies of Alzheimer’s (as reviewed in Nazem et al., 2015). Amyloid aggregates may then induce further microglial dysfunction exacerbating a vicious cycle of neuro-inflammation and progression of disease. Several rodent models of AD have been developed based on this inflammation hypothesis, including peripheral immune challenge with bacterial lipopolysaccharide (LPS; Pintado et al., 2012) or viral polyriboinosinic-polyribocytidilic acid (PolyI:C) proteins (Krstic et al., 2012), intracerebroventricular (i.c.v) injection of streptozotocin (STZ; Chen et al., 2013) and transgenic over-expression of p25 (Sundaram et al., 2012), resulting in neuro-inflammation, amyloid plaques, NFTs and cognitive dysfunction. Moreover, mice and rat models of traumatic brain injury (Yu et al., 2012) and cerebral ischemia (Kawabori and Yenari, 2015) result in neuro-inflammation, including increase in toll-like receptors (TLRs), microglial activation, and expression of pro-inflammatory enzymes (cyclo-oxygenase-2, COX-2) and cytokines (IL-1β, TNFα), in correlation with secondary cognitive defects comparable to TBI (Vincent et al., 2014) and vascular-related dementias (Kawabori and Yenari, 2015).
Alterations in such neuro-inflammatory factors have indeed been shown to correlate with cognitive improvement by lithium using these mammalian models. Studies using a rat LPS model of neuro-inflammation identified lithium as an anti-inflammatory agent through its prevention of neuro-inflammatory prostaglandin production, for example PGE2 and TXB2 (Basselin et al., 2007), and elevation of the anti-inflammatory docosanoid 17S-hydroxy-DHA (17-OH-DHA; Basselin et al., 2010). Consistent with these observations, lithium treatment also prevented traumatic brain injury and cerebral ischemia-induced microglial activation, in mouse and rat brain, and this associated with prevention of neuronal loss (Li et al., 2011; Yu et al., 2012) and anxiety-like behavior using an open-field test (Yu et al., 2012). Correlating with prevention of cognitive defects, lithium carbonate improved spatial reference and working memory in a rat intra-hippocampal injection model of AD and reversed reductions in anti-inflammatory (IL-4) and increases in pro-inflammatory (IL-1β, TNFα) cytokines in the frontal cortex and hippocampus (Budni et al., 2017).
The molecular mediators of lithium’s anti-inflammatory effects in dementia models are unclear, but correlative evidence suggest that GSK-3, toll-like receptor 4 (TLR4), signal transducer and activator of transcription (STAT) and nuclear factor-κB (NFκB) pathways may play a causal role (Jope et al., 2017; see Figure 1B). Pro-inflammatory agents such as LPS and 6-hydroxydopamine (6-OHDA) increase microglial GSK-3 activity in vitro (Yuskaitis and Jope, 2009; Green and Nolan, 2012). Conversely, both lithium and specific GSK-3 inhibitors prevented neuro-inflammatory IL-1β, IL-6, TNFα and nitric oxide (NO) production in rat microglia (Yuskaitis and Jope, 2009; Green and Nolan, 2012) and astrocytes (Wang et al., 2013), as well as in mouse hippocampal slice cultures in correlation with neuronal protection (Yuskaitis and Jope, 2009). Alterations in expression of TLR4 and its target transcription factor NFκB, a key regulatory pathway in microglial activation and cytokine production, correlated with lithium’s rescue of surgery-induced memory impairment in aged rats in association with reductions in hippocampal TNF-α and IL-1β (Lu et al., 2015). Additionally, using TLR4 knockout mice, Cheng et al. (2016) demonstrated that TLR4 was required for anxiety-induced GSK-3 activation in the hippocampus and subsequently that GSK-3 inhibition, using TDZD-8, alleviated increased cytokine and chemokine production and NFκB activation in this model. Alternatively, GSK-3 phosphorylates and activates STAT3 by promoting its association with the pro-inflammatory interferon-γ (IFN-γ) receptor in mouse primary astrocytes, by mechanisms which appear independent of TLR4; and lithium and TDZD-8 block these effects (Beurel and Jope, 2008). Together these data suggest that GSK-3 may mediate lithium’s anti-inflammatory and neuro-protective effects by independently modulating TLR4/NFκB and STAT3 signaling. However, further research is required to delineate these pathways by measuring the effects of altering TLR4, NFκB and STAT3 on lithium and GSK-3 inhibitor-mediated neuronal protection in animal models.
Although lithium’s prevention of neuro-inflammation may simply represent one of the multi-faceted mechanisms by which GSK-3 inhibition protects neurons, the above evidence also suggests that TLR4 is a direct target for lithium, upstream of GSK-3 (see Figure 1B). Genetic modification of TLR4 protected against anxiety-like behavior in mice (Cheng et al., 2016), as well as Aβ and oxidative stress-induced toxicity in primary mouse neurons (Tang et al., 2008). Conversely Aβ and 4-HNE increased TLR4 protein in mouse neurons, and TLR4 levels were altered in AD patient brain (Tang et al., 2008), further suggesting that this may represent an attractive therapeutic target for AD. Consistently, the naturally-occurring compound gx-50 prevented Aβ-induced microglial activation and neuro-inflammation (IL-1β, TNFα, NO, COX2) in rat microglia cultures and APP transgenic mouse brain, to levels comparable with TLR4 gene silencing, and this correlated with a rescue of Aβ-induced enhancement of TLR4 protein in vitro and in vivo (Shi et al., 2016). Moreover, small molecule antagonists of TLR4 have recently been developed (De Paola et al., 2016) and protect against neuro-toxicity using in vitro models of ALS (De Paola et al., 2016) and LPS-induced neuro-inflammation (Perrin-Cocon et al., 2017), suggesting that an investigation of the potential of these drugs in dementia models is warranted. Finally, targeted modulation of NFκB and STAT3 pathways may offer selective protection against neuro-inflammation in dementia, for example using natural and rationally-designed inhibitors of the NFκB pathway (as reviewed, Srinivasan and Lahiri, 2015) and small molecule STAT3 inhibitors which are effective in cancer models (Fletcher et al., 2011). This requires empirical comparison with the efficacy of lithium and GSK-3 inhibitors against neuronal protection using in vitro and in vivo models of AD and other dementias.
Protein homeostasis is maintained by balancing protein synthesis and degradation. Qualitative adaptation of the proteome, as well as the rapid removal of damaged and unfolded proteins, is essential for normal cell functioning and this is particularly important for non-diving cells, such as neurons, where damaged components cannot be excluded through cell division (Douglas and Dillin, 2010; Balchin et al., 2016). Interestingly, lithium impacts on several aspects of proteostasis, for example by reducing protein synthesis and increasing protein degradation through modulation of proteasomal activity and induction of autophagy. This may prove beneficial for many neurodegenerative diseases, including AD and other forms of dementia, either by enhancing clearance of abnormal protein aggregates, which are a common feature of these conditions, or by maintaining protein homeostasis and therefore preserving neuronal cell function.
Initial evidence for the effect of lithium on reduction of protein synthesis came from studies on the development of sea urchin embryos (Berg, 1968). Lithium treatment using clinically-relevant lithium doses (0.6–1.0 mM) was able to down-regulate the expression of the epsilon subunit of the initiation factor-2B (eIF2B) in rat brain suggesting down-regulation of protein synthesis (Bosetti et al., 2002). Interestingly, in vitro experiments using the SH-SY5Y human neuroblastoma cell line showed that lithium had a positive effect on translation through reduced inhibitory phosphorylation of elongation factor eEF2, while GSK-3 inhibitors had opposing effects on phospho-eEF2, thereby reducing translation (Karyo et al., 2010). Similarly, GSK-3 was a positive regulator of translation in breast cancer cells, via phosphorylation of 4E-BP1 and a concomitant increase in eIF4E-dependent protein synthesis (Shin et al., 2014). Using a different in vitro system, CHO.T cells, it was shown that GSK-3 negatively affects translation by mediating inhibitory phosphorylation of eIF2B (Welsh et al., 1998). These conflicting results may depend on the system studied and differences between in vitro and in vivo approaches, but lithium consistently reduces protein synthesis across species in models of neurological diseases. Low lithium concentrations decreased levels of cerebral protein synthesis to control levels in the Fmr1 KO mouse model of Fragile X syndrome, but had no effect on translation in wild-type mice (Liu et al., 2012). Lithium reduced protein synthesis, accompanied by increased longevity, in Drosophila models of AD (Sofola et al., 2010; Sofola-Adesakin et al., 2014), and experiments in yeast (Schizosaccharomyces pombe) confirmed the inhibitory effect of lithium on translation and its anti-aging effect as measured by chronological lifespan (Sofola-Adesakin et al., 2014). Although the precise molecular mechanisms remain unclear, several in vivo approaches therefore demonstrate reduced protein synthesis in response to lithium (Figure 1B). The benefits of this may include improved neuronal function by re-investing energy in cell maintenance processes, or delaying formation of protein aggregates by improving protein quality due to the increased availability of the protein folding and degradation machinery.
Lithium treatment is also increasingly linked to degradation pathways such as autophagy. Autophagy is a process whereby a portion of the cytoplasm is packaged into autophagosomes and subsequently, upon fusion with the lysosome, the content is degraded (Mizushima and Komatsu, 2011; Nikoletopoulou et al., 2015; Suzuki et al., 2017). Autophagy can degrade selective or random cargo and it is the only process that can degrade entire organelles (Mizushima and Komatsu, 2011). The main autophagy inducers are starvation and lack of growth factors which, through inhibition of mammalian target of rapamycin (mTOR) signaling, then trigger ULK1 complex (ULK1-ATG13-FIP200-ATG101) formation at Atg9-containing membranes and activation of the class 3 phosphatidylinositol-3-OH kinase (PI(3)K) complex (Jung et al., 2010). Significantly, autophagy can degrade protein aggregates and its reduction and defects have been consistently considered as contributing factors towards neurodegeneration (Tanaka and Matsuda, 2014; Frake et al., 2015). Moreover, knock-out mice that lack ATG5 or ATG7 autophagy genes in neurons, although viable, suffer from motor and behavioral defects, correlating with ubiquitinated protein aggregation and apoptosis (Hara et al., 2006; Komatsu et al., 2006). This suggests that insufficient autophagic clearance may directly cause neurodegeneration. Compounds that enhance autophagy, such as lithium, are therefore a promising therapeutic strategy for a variety of neurodegenerative conditions, such as AD, FTD, PD, HD and ALS, by removing toxic protein aggregates and/or improving mitochondrial health via mitophagy (Frake et al., 2015; Lionaki et al., 2015; Galluzzi et al., 2017; Menzies et al., 2017). For example, lithium treatment ameliorated motor dysfunction in mice overexpressing FTD-mutated human tau (P301L) via increased autophagy (Shimada et al., 2012). Low dose lithium also delayed disease in ALS patients (Fornai et al., 2008a), and neuroprotection by lithium in the SOD1G93A mouse model of ALS has been attributed to increased autophagy (Fornai et al., 2008a,b). Although some studies report no prevention of disease progression in this model (Pizzasegola et al., 2009). Numerous studies therefore report that lithium treatment is beneficial for a variety of neurodegenerative disorders owing to its capacity to enhance autophagy (Motoi et al., 2014). It is important to note, however, that there are exceptions where improvements in memory and decreased amyloid plaque formation following lithium treatment of mice expressing APPswe/PS1A246E associated with decreased autophagy (Zhang et al., 2011). These discrepancies may be explained by methodological differences in measuring autophagy or by variations in the lithium concentration used (Klionsky et al., 2016).
Interestingly, lithium has been shown to induce authophagy through mTOR-independent mechanisms (Figure 1B), more specifically via inhibition of IMPA leading to free inositol depletion and reduced myo-inositol-1,4,5-triphosphate (IP3) levels (Sarkar et al., 2005; Sarkar and Rubinsztein, 2006). This effect is replicated by the IMPA inhibitor L-690,330 (Atack et al., 1993) which enhanced the degradation of A53T α-synuclein and HDQ74 mutant huntingtin protein-expressing PC12 cells (Sarkar et al., 2005). GSK3β inhibition using SB216763 did not have any effect on autophagy induction or mutant HDQ74 protein clearance (Sarkar et al., 2005), providing further support that the effects of lithium on autophagy are GSK-3 independent. GSK3β inhibition can, in fact, have opposing effects and reduce autophagy through activation of the mTOR pathway. As these two lithium targets, IMPA and GSK3β, have independent but opposing effects on autophagy, combination of lithium with the mTOR inhibitor rapamycin, which has beneficial neuroprotective effects in Drosophila and mice models of AD and FTD (Khurana et al., 2006; Spilman et al., 2010; Ozcelik et al., 2013; Lin et al., 2017), maximized autophagy induction and clearance of mutant aggregate prone proteins more effectively than singular treatments using in vitro and Drosophila model of HD (Sarkar et al., 2008).
Lithium plays many crucial roles required for proper nervous system function, such as promoting neurogenesis, synaptic plasticity and cell survival (Chen et al., 2000; Schloesser et al., 2008; Kim and Thayer, 2009; see Figure 1B). Neurogenesis is vital for hippocampal plasticity, and persists into adulthood in the mammalian brain. It underlies learning and memory processes, and thus alterations in neurogenesis are implicated in neurodegenerative diseases such as AD (Taupin, 2008). Several animal studies have investigated neurogenesis in AD with rather conflicting results. For example, two studies that utilized APPSwe, Ind transgenic mice, expressing both the Swedish and Indiana amyloid precursor protein (APP) mutations but under different promoters, reported opposing phenotypic outcomes. One study reported an enhancement in neurogenesis in the hippocampus of transgenic mice relative to controls (Jin et al., 2004), while the other showed a reduction (Fiorentini et al., 2010). In the latter study, however, treatment of young mice with lithium, at 3 months of age, significantly increased neurogenesis in correlation with activation of Wnt signaling, as measured by increased nuclear β-catenin staining in newborn neurons (Fiorentini et al., 2010). Canonical Wnt signaling inhibits GSK3, thus enabling translocation of β-catenin to the nucleus and enhancing its association with the TCF/LEF family of transcription factors. Subsequent transcription of Wnt/β-catenin target genes leads to regulation of diverse processes that are critical for development of the mammalian CNS such as synapse plasticity and neurogenesis (Valvezan and Klein, 2012). Similarly, lithium pheno-copies the effects of Wnt signaling on GSK-3 inhibition and modulates neurogenesis and synaptic plasticity via activation of downstream β-catenin/TCF-LEF target genes (Fiorentini et al., 2010; Valvezan and Klein, 2012; Morris and Berk, 2016). Moreover, lithium is able to suppress astrogliogenesis (generation of new astroglia/astrocytes), through non-GSK-3-mediated mechanisms. Hence lithium may increase the neuronal fate of neuronal stem cells in two ways, by increasing neurogenesis and reducing astrogliogenesis (Zhu et al., 2011).
Cell survival by lithium is mediated by upregulation of basal adenylate cyclase activity, and thereby cyclic AMP (cAMP) and protein kinase A (PKA), which leads to CREB-dependent transcription of brain-derived neurotrophic factor (BDNF) and anti-apoptotic B cell lymphoma protein-2 (Bcl-2) genes (Chen and Chuang, 1999; Quiroz et al., 2010). Bcl-2 has been attributed to lithium’s anti-apoptotic effects, potentially by decreasing the pro-apoptotic Bax and p53 genes (Sugawara et al., 2010; Can et al., 2014). Bax promotes mitochondrial cytochrome c release subsequently promoting cytosolic activation of caspases and thus the degradation of specific protein substrates (Chen and Chuang, 1999). Bcl-2 counteracts these effects by preventing cytochrome c translocation and thus inhibiting Bax-induced caspase activation (Kluck et al., 1997). Alterations in the expression of Bcl-2 family members in AD brain have indicated a role for apoptosis in neuronal loss in this condition, but the role of caspase activation in mediating effects on AD pathology are unclear. Over-expression of Bcl-2 or neutralizing antibodies for Bax, however, prevented Aβ-induced death in vitro using primary neurons (Kudo et al., 2012). Lithium-mediated GSK-3 inhibition suppresses apoptosis (Chin et al., 2005), but it should be noted that the specific role of bcl-2 in mediating this effect is unclear as recent data has challenged alterations in its expression in response to lithium in light of optimal normalizing probes (Odeya et al., 2018).
Neurotrophins are also important mediators of cell survival, neurogenesis and synaptic maintenance. BDNF binds to tyrosine receptor kinase B (TrkB), both of which are widely expressed in the developing and adult mammalian brain, and activates phospholipase Cγ (PLCγ), phosphatidylinositol 3-kinase (PI3K), and extracellular signal-regulated kinase (ERK) pathways which are involved in neuronal differentiation, preventing apoptosis, promoting cell survival and maintaining synaptic plasticity (Tajes et al., 2009; Cunha et al., 2010). BDNF/TrkB-induced PI3K signaling leads to phosphorylation and inhibition of GSK-3, enabling the activation of downstream effectors, including mTOR, that play a key role in synaptic remodeling (Cunha et al., 2010; Morris and Berk, 2016). Moreover, BDNF plays an important role in the induction and maintenance of long term potentiation (LTP), synaptic transmission by acting at pre- and postsynaptic sites, and presynaptic release of the excitatory neurotransmitter glutamate (Jovanovic et al., 2000; Wurzelmann et al., 2017). Patients with bipolar disorder have significantly decreased BDNF in blood serum levels compared to healthy controls (Fernandes et al., 2015), and conversely lithium treatment is associated with increased serum BDNF protein in these patients (Gideons et al., 2017). Chronic lithium treatment has been shown to increase BDNF mRNA and protein expression across brain regions in rodent and mice models (Fukumoto et al., 2001; Hashimoto et al., 2002; Gideons et al., 2017), as well as in cortical neurons following acute administration. Indeed pre-treatment with either lithium or BDNF protected rat cerebral cortical neurons from glutamate excitotoxicity (Hashimoto et al., 2002), suggesting that BDNF is a key regulator of lithium-mediated neuroprotection. However, the therapeutic potential of BDNF is restricted, due to its relatively short half-life of less than 10 min, and inability to cross the blood-brain barrier (BBB; Wurzelmann et al., 2017). Small molecules that mimic BDNF’s function without its pharmacokinetic barriers would, therefore, be highly favorable. One such compound is 7,8-dihydroxyflavone (7,8-DHF), a selective TrkB agonist, which initiates activation of BDNF/TrkB signaling pathways, with a much longer half-life and greater permeability across the BBB than BDNF (Jang et al., 2010; Wurzelmann et al., 2017). Importantly, 7,8-DHF has been shown to have beneficial effects in cellular and animal models of AD, such as protecting primary neurons from Aβ-induced toxicity and promoting synaptogenesis (Zhang et al., 2014). Furthermore, chronic administration of 7,8-DHF prevented Aβ deposition, restored synaptic plasticity, and prevented memory deficits in AD transgenic mice (Zhang et al., 2014), suggesting that BDNF/TrkB signaling may have therapeutic potential in treatment of AD.
Glutamate is a major excitatory neurotransmitter in the CNS that plays important roles in synaptic plasticity and memory by initiating diverse signaling pathways including mTOR activation, phospholipase C, inositol triphosphate, ERK signaling and calcium release (Kumar et al., 2015; Ribeiro et al., 2017). Glutamate acts on ionotropic receptors such as N-methyl-D-asparate (NMDA), and metabotropic receptors to initiate an array of signaling responses (Kumar et al., 2015; Ribeiro et al., 2017). Metabotropic glutamate receptors (mGLuRs) are distributed throughout the CNS, and carry out multiple functions in maintaining synaptic transmission. Dysregulation of mGluRs have been implicated in several neurodegenerative diseases such as PD and AD, as well as diseases that affect intellectual capabilities including Fragile X Syndrome (Niswender and Conn, 2010; Ribeiro et al., 2017). Lithium attenuates the effects of hyperactive glutamate-mediated calcium signaling and has therefore been used as a therapy for conditions in which mGLuRs are dysfunctional (Sourial-Bassillious et al., 2009). For example, Fragile X mental retardation 1 (Fmr1) knockout mice have enhanced long term depression (LTD) in the hippocampus, due to increased activity of mGluR group I type 5 (Huber et al., 2002). Treating Fmr1 knockout mice with lithium improved hyperactivity and rescued cognitive impairment by restoring mGluR-LTD (Choi et al., 2011; Liu et al., 2011). Similarly in Drosophila fmr1 (dfmr1) mutant flies, lithium significantly improved mushroom body defects and restored learning and memory capabilities, as measured by altered courtship behavior, through a reduction in mGluR activity (McBride et al., 2005). Lithium affects signaling pathways that overlap with those regulated by mGluR. For example group 1 mGluRs mediate cerebellar mGluR-LTD by increasing inositol triphosphate (IP3) generation and releasing intracellular Ca2+ stores, and lithium is able to reverse these effects by inhibiting inositol levels (Berridge, 1993, 2009; Schloesser et al., 2008; Lüscher and Huber, 2010). Moreover, as previously mentioned, in lower model organisms loss of function mutations in the genes involved in inositol turnover, IMPA (ttx in C. elegans) and IPP (ipp in Drosophila), display defects in localization of synaptic components and synaptic transmission, respectively, which are phenocopied by lithium treatment (Acharya et al., 1998; Tanizawa et al., 2006). This strengthens the role of lithium in maintaining synaptic function by GSK-3 and IMPA-dependent mechanisms (see Figure 1B).
Although memantine, an NMDA antagonist, is currently used in symptomatic treatment of AD, long-term inhibition of this glutamate receptor may also lead to memory impairment due to its direct role in excitatory glutamatergic synaptic transmission. mGluR5 antagonists are therefore an attractive alternative, as mGluR5 plays a modulatory role in synaptic maintenance (Kumar et al., 2015). In particular, MPEP (2-Methyl-6-(phenylethynyl)pyridine) and MTEP(2-Methyl-4-thiazolyl)ethynyl)pyridine) are non-competitive mGluR antagonists that protect against neuronal toxicity in vitro, using primary neurons from AD transgenic mice, by preventing Aβ oligomer-induced dendritic spine loss (Um et al., 2013; Overk et al., 2014). Aβ oligomers interact with Prion protein (Prpc), and enhance the interaction between Prpc and mGluR for signal transmission (Um et al., 2013; Haas et al., 2017). MPEP and MTEP inhibit both glutamate and Aβ/Prpc signaling, while BMS-94923, a silent allosteric modulator does not alter glutamate signaling but rather inhibits the mGluR5-Prpc interaction (Haas et al., 2017). Furthermore, BMS-94923 is able to prevent Aβ oligomer-induced inhibition of synaptic plasticity, and rescues memory deficits in a transgenic mouse model of AD, and as such this drug has great therapeutic potential for AD treatment, without the adverse side effects from modulating glutamate (Haas et al., 2017).
Lithium therefore has multi-factorial neuro-protective effects, and drugs targeting several of these mechanisms can slow cognitive decline in animal models of dementia (see Table 1). Combining such therapies may synergize these protective properties whilst excluding the toxic peripheral side effects, due to renal COX2 activation, observed with long-term lithium treatment. Although specific GSK-3 inhibitors, such as tideglusib, have reached clinical trial and appear to be safe for human use, they have so-far not shown significant therapeutic benefit in mild-moderate AD and further studies are required to optimize dosing and to measure effects at earlier disease stages (Lovestone et al., 2015). GSK-3 inhibition however does not recapitulate all of the protective features of lithium (see Figure 1), and this raises the possibility that their combined use with drugs targeting non-GSK-3-dependent mechanisms of lithium action may exert more effective neuro-protection under accumulating damage in dementia. Few studies have investigated these interactions, but additive protective effects have been observed when combining lithium with rapamycin or Keap1 inhibition in Drosophila models of HD and AD respectively (Sarkar et al., 2008; Kerr et al., 2017). Although lithium enhances autophagy by inhibiting IMPA, as described above, GSK-3 inhibition limits this protective property of lithium by activating mTOR. Combining lithium with the mTOR inhibitor rapamycin therefore enhances autophagy leading to improved efficacy against huntingtin-induced proteo-toxicity in comparison to treatment with either compound alone (Sarkar et al., 2008). Our studies also indicate that lithium, and TDZD-8, protect against Aβ42 neuro-toxicity predominantly through Nrf2-independent mechanisms and that dual Keap1-Nrf2 disruption and lithium treatment enhance neuro-protection through non-overlapping mechanisms (Kerr et al., 2017). Further work is required to uncouple these mechanisms using specific GSK-3 inhibitors and to optimize conditions for supplementing GSK-3 dependent neuroprotection with rapamycin and Nrf2 activators, as well as testing their combination with anti-inflammatory (TLR4 antagonists), neurogenic (TrKB agonists) and synaptic maintenance (mGluR antagonists) compounds, which have individual beneficial effects in neuroprotection (Table 1).
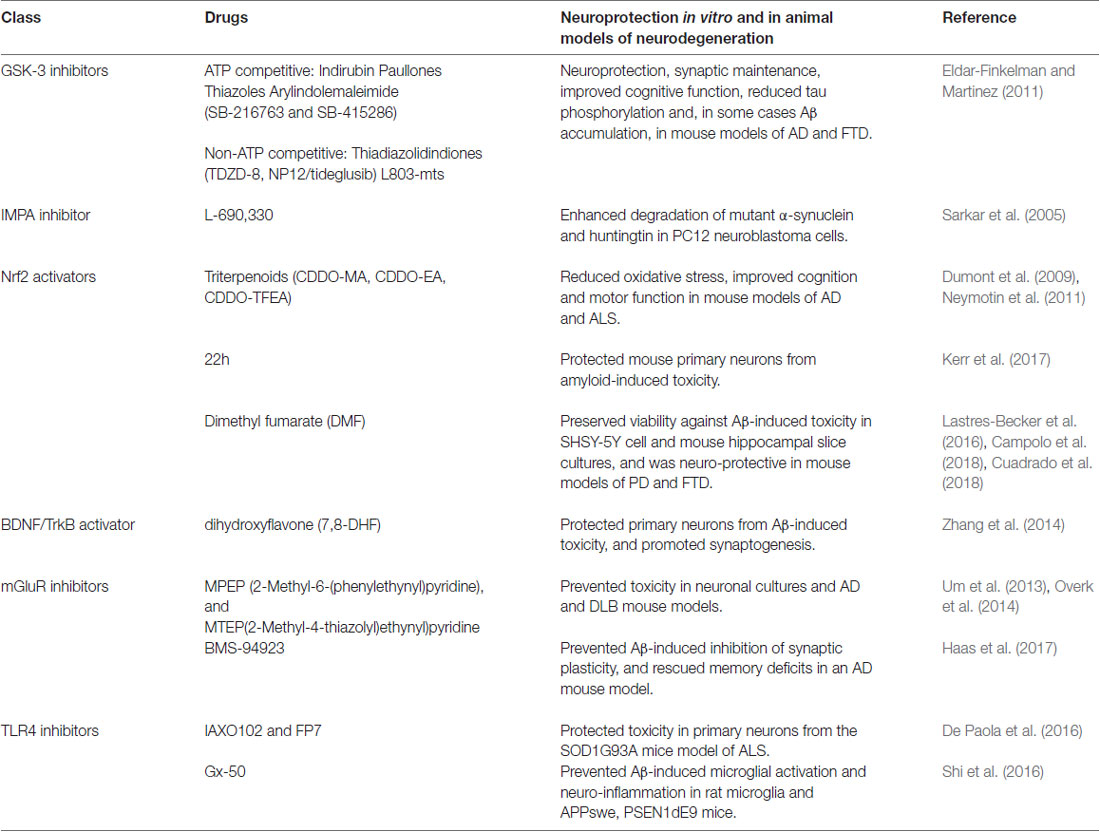
Table 1. Drugs targeting downstream mediators of lithium-mediated neuroprotection.
Alternatively, improved formulation and administration of lithium may better encapsulate the benefits of its neuroprotective properties in a single dementia therapy. Chronic (>15 months) microdose lithium administration has been suggested to stabilize memory impairment in AD patients (Nunes et al., 2013) and prevent neuropathology and cognitive decline in mice and rat models of AD with no observed toxicity (Nunes et al., 2015; Wilson et al., 2017). More recently LISPRO, a new ionic co-crystalized formulation of lithium salicylate and L-proline, has been reported to have improved brain penetrance and a safer pharmacokinetic profile over classical lithium chloride and carbonate salts (Habib et al., 2017). At therapeutically relevant doses, chronic LISPRO treatment prevented abnormal Aβ accumulation and tau phosphorylation, as well as neuronal and synaptic loss, in the Tg2576 mouse model of AD (Habib et al., 2017). Interestingly, this correlated with several of the established neuro-protective properties of lithium salts, including inhibition of GSK-3, enhanced autophagy, reduced neuro-inflammation and enhanced neurogenesis. Unlike lithium carbonate and lithium salicylate, however, acute or chronic LISPRO treatment did not enhance renal COX2 levels in vitro or in vivo, suggesting an improved safety profile by preventing the chronic renal toxicity observed using traditional lithium formulations.
The promise of lithium as a multi-functional therapy for the treatment of dementia therefore remains. Pre-clinical studies, across model organisms from flies to rodents, will prove vital however for systematic comparison of the effectiveness of combination treatments over new and microdose lithium formulations in slowing cognitive decline. Such analyses will guide the best approach for capturing the beneficial effects of lithium in the design of new disease-modifying therapies for clinical use in AD, as well as other neurodegenerative conditions for which lithium has shown favorable neuro-protective effects.
FK, IB and OS-A contributed equally to the conception, writing and editing of this review. FK and OS-A prepared the figures, tables and legends.
FK is a University-funded Research Fellow at Glasgow Caledonian University, and is supported by Tenovus Scotland (S18-11). IB is supported by a European Research Council Starting Grant (311331), Cancer Research UK, Royal Society Research Grant and The Bill Lyons foundation. OS-A is a University-funded lecturer at the University of Sussex.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Frontiers in Molecular Neuroscience for the opportunity to participate in this Research Topic.
Abed, D. A., Goldstein, M., Albanyan, H., Jin, H., and Hu, L. (2015). Discovery of direct inhibitors of Keap1-Nrf2 protein-protein interaction as potential therapeutic and preventive agents. Acta Pharm. Sin. B 5, 285–299. doi: 10.1016/j.apsb.2015.05.008
Acharya, J. K., Labarca, P., Delgado, R., Jalink, K., and Zuker, C. S. (1998). Synaptic defects and compensatory regulation of inositol metabolism in inositol polyphosphate 1-phosphatase mutants. Neuron 20, 1219–1229. doi: 10.1016/s0896-6273(00)80502-4
Aghdam, S. Y., and Barger, S. W. (2007). Glycogen synthase kinase-3 in neurodegeneration and neuroprotection: lessons from lithium. Curr. Alzheimer Res. 4, 21–31. doi: 10.2174/156720507779939832
Alfieri, A., Srivastava, S., Siow, R. C. M., Modo, M., Fraser, P. A., and Mann, G. E. (2011). Targeting the Nrf2-Keap1 antioxidant defence pathway for neurovascular protection in stroke. J. Physiol. 589, 4125–4136. doi: 10.1113/jphysiol.2011.210294
Atack, J. R., Cook, S. M., Watt, A. P., Fletcher, S. R., and Ragan, C. I. (1993). In vitro and In vivo inhibition of inositol monophosphatase by the bisphosphonate L-690,330. J. Neurochem. 60, 652–658. doi: 10.1111/j.1471-4159.1993.tb03197.x
Balchin, D., Hayer-Hartl, M., and Hartl, F. U. (2016). In vivo aspects of protein folding and quality control. Science 353:aac4354. doi: 10.1126/science.aac4354
Barone, M. C., Sykiotis, G. P., and Bohmann, D. (2011). Genetic activation of Nrf2 signaling is sufficient to ameliorate neurodegenerative phenotypes in a Drosophila model of Parkinson’s disease. Dis. Model. Mech. 4, 701–707. doi: 10.1242/dmm.007575
Basselin, M., Kim, H.-W., Chen, M., Ma, K., Rapoport, S. I., Murphy, R. C., et al. (2010). Lithium modifies brain arachidonic and docosahexaenoic metabolism in rat lipopolysaccharide model of neuroinflammation. J. Lipid Res. 51, 1049–1056. doi: 10.1194/jlr.m002469
Basselin, M., Villacreses, N. E., Lee, H.-J., Bell, J. M., and Rapoport, S. I. (2007). Chronic lithium administration attenuates up-regulated brain arachidonic acid metabolism in a rat model of neuroinflammation. J. Neurochem. 102, 761–772. doi: 10.1111/j.1471-4159.2007.04593.x
Baxter, P. S., and Hardingham, G. E. (2016). Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 100, 147–152. doi: 10.1016/j.freeradbiomed.2016.06.027
Berg, W. E. (1968). Effect of lithium on the rate of protein synthesis in the sea urchin embryo. Exp. Cell Res. 50, 133–139. doi: 10.1016/0014-4827(68)90401-1
Berridge, M. J. (1993). Inositol trisphosphate and calcium signalling. Nature 361, 315–325. doi: 10.1038/361315a0
Berridge, M. J. (2009). Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 1793, 933–940. doi: 10.1016/j.bbamcr.2008.10.005
Berridge, M. J., Downes, C. P., and Hanley, M. R. (1989). Neural and developmental actions of lithium: a unifying hypothesis. Cell 59, 411–419. doi: 10.1016/0092-8674(89)90026-3
Berry, G. T., Buccafusca, R., Greer, J. J., and Eccleston, E. (2004). Phosphoinositide deficiency due to inositol depletion is not a mechanism of lithium action in brain. Mol. Genet. Metab. 82, 87–92. doi: 10.1016/j.ymgme.2004.02.002
Beurel, E., and Jope, R. S. (2008). Differential regulation of STAT family members by glycogen synthase kinase-3. J. Biol. Chem. 283, 21934–21944. doi: 10.1074/jbc.m802481200
Bosetti, F., Seemann, R., and Rapoport, S. I. (2002). Chronic lithium chloride administration to rats decreases brain protein level of epsilon (ε) subunit of eukaryotic initiation factor-2B. Neurosci. Lett. 327, 71–73. doi: 10.1016/s0304-3940(02)00354-3
Bruns, D. R., Drake, J. C., Biela, L. M., Peelor, F. F., Miller, B. F., and Hamilton, K. L. (2015). Nrf2 signaling and the slowed aging phenotype: evidence from long-lived models. Oxid. Med. Cell. Longev. 2015:732596. doi: 10.1155/2015/732596
Budni, J., Feijó, D. P., Batista-Silva, H., Garcez, M. L., Mina, F., Belletini-Santos, T., et al. (2017). Lithium and memantine improve spatial memory impairment and neuroinflammation induced by β-amyloid 1–42 oligomers in rats. Neurobiol. Learn. Mem. 141, 84–92. doi: 10.1016/j.nlm.2017.03.017
Calsolaro, V., and Edison, P. (2016). Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 12, 719–732. doi: 10.1016/j.jalz.2016.02.010
Campolo, M., Casili, G., Lanza, M., Filippone, A., Paterniti, I., Cuzzocrea, S., et al. (2018). Multiple mechanisms of dimethyl fumarate in amyloid β-induced neurotoxicity in human neuronal cells. J. Cell. Mol. Med. 22, 1081–1094. doi: 10.1111/jcmm.13358
Can, A., Schulze, T. G., and Gould, T. D. (2014). Molecular actions and clinical pharmacogenetics of lithium therapy. Pharmacol. Biochem. Behav. 123, 3–16. doi: 10.1016/j.pbb.2014.02.004
Cao, Y., Chtarbanova, S., Petersen, A. J., and Ganetzky, B. (2013). Dnr1 mutations cause neurodegeneration in Drosophila by activating the innate immune response in the brain. Proc. Natl. Acad. Sci. U S A 110, E1752–E1760. doi: 10.1073/pnas.1306220110
Castillo-Quan, J. I., Li, L., Kinghorn, K. J., Ivanov, D. K., Tain, L. S., Slack, C., et al. (2016). Lithium promotes longevity through GSK3/NRF2-dependent hormesis. Cell Rep. 15, 638–650. doi: 10.1016/j.celrep.2016.03.041
Castro, A. A., Casagrande, T. S., Moretti, M., Constantino, L., Petronilho, F., Guerra, G. C. B., et al. (2009). Lithium attenuates behavioral and biochemical effects of neuropeptide S in mice. Peptides 30, 1914–1920. doi: 10.1016/j.peptides.2009.07.004
Chen, R. W., and Chuang, D. M. (1999). Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. A prominent role in neuroprotection against excitotoxicity. J. Biol. Chem. 274, 6039–6042. doi: 10.1074/jbc.274.10.6039
Chen, Y., Liang, Z., Blanchard, J., Dai, C.-L., Sun, S., Lee, M. H., et al. (2013). A non-transgenic mouse model (icv-STZ mouse) of Alzheimer’s disease: similarities to and differences from the transgenic model (3xTg-AD mouse). Mol. Neurobiol. 47, 711–725. doi: 10.1007/s12035-012-8375-5
Chen, X., Liu, Y., Zhu, J., Lei, S., Dong, Y., Li, L., et al. (2016). GSK-3β downregulates Nrf2 in cultured cortical neurons and in a rat model of cerebral ischemia-reperfusion. Sci. Rep. 6:20196. doi: 10.1038/srep20196
Chen, G., Rajkowska, G., Du, F., Seraji-Bozorgzad, N., and Manji, H. K. (2000). Enhancement of hippocampal neurogenesis by lithium. J. Neurochem. 75, 1729–1734. doi: 10.1046/j.1471-4159.2000.0751729.x
Chen, P.-C., Vargas, M. R., Pani, A. K., Smeyne, R. J., Johnson, D. A., Kan, Y. W., et al. (2009). Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: critical role for the astrocyte. Proc. Natl. Acad. Sci. U S A 106, 2933–2938. doi: 10.1073/pnas.0813361106
Cheng, Y., Pardo, M., Armini, R. S., Martinez, A., Mouhsine, H., Zagury, J.-F., et al. (2016). Stress-induced neuroinflammation is mediated by GSK3-dependent TLR4 signaling that promotes susceptibility to depression-like behavior. Brain Behav. Immun. 53, 207–222. doi: 10.1016/j.bbi.2015.12.012
Chin, P. C., Majdzadeh, N., and D’Mello, S. R. (2005). Inhibition of GSK3β is a common event in neuroprotection by different survival factors. Mol. Brain Res. 137, 193–201. doi: 10.1016/j.molbrainres.2005.03.004
Chiu, C.-T., Liu, G., Leeds, P., and Chuang, D.-M. (2011). Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington’s disease. Neuropsychopharmacology 36, 2406–2421. doi: 10.1038/npp.2011.128
Choi, C. H., Schoenfeld, B. P., Bell, A. J., Hinchey, P., Kollaros, M., Gertner, M. J., et al. (2011). Pharmacological reversal of synaptic plasticity deficits in the mouse model of Fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 1380, 106–119. doi: 10.1016/j.brainres.2010.11.032
Cryns, K., Shamir, A., Shapiro, J., Daneels, G., Goris, I., Van Craenendonck, H., et al. (2007). Lack of lithium-like behavioral and molecular effects in IMPA2 knockout mice. Neuropsychopharmacology 32, 881–891. doi: 10.1038/sj.npp.1301154
Cuadrado, A., Kügler, S., and Lastres-Becker, I. (2018). Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol. 14, 522–534. doi: 10.1016/j.redox.2017.10.010
Cunha, C., Brambilla, R., and Thomas, K. L. (2010). A simple role for BDNF in learning and memory? Front. Mol. Neurosci. 3:1. doi: 10.3389/neuro.02.001.2010
Damri, O., Sade, Y., Toker, L., Bersudsky, Y., Belmaker, R. H., Agam, G., et al. (2015). Molecular effects of lithium are partially mimicked by inositol-monophosphatase (IMPA)1 knockout mice in a brain region-dependent manner. Eur. Neuropsychopharmacol. 25, 425–434. doi: 10.1016/j.euroneuro.2014.06.012
DaRocha-Souto, B., Coma, M., Pérez-Nievas, B. G., Scotton, T. C., Siao, M., Sánchez-Ferrer, P., et al. (2012). Activation of glycogen synthase kinase-3 beta mediates β-amyloid induced neuritic damage in Alzheimer’s disease. Neurobiol. Dis. 45, 425–437. doi: 10.1016/j.nbd.2011.09.002
De Ferrari, G. V., Chacón, M. A., Barría, M. I., Garrido, J. L., Godoy, J. A., Olivares, G., et al. (2003). Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by β-amyloid fibrils. Mol. Psychiatry 8, 195–208. doi: 10.1038/sj.mp.4001208
De Paola, M., Sestito, S. E., Mariani, A., Memo, C., Fanelli, R., Freschi, M., et al. (2016). Synthetic and natural small molecule TLR4 antagonists inhibit motoneuron death in cultures from ALS mouse model. Pharmacol. Res. 103, 180–187. doi: 10.1016/j.phrs.2015.11.020
Douglas, P. M., and Dillin, A. (2010). Protein homeostasis and aging in neurodegeneration. J. Cell Biol. 190, 719–729. doi: 10.1083/jcb.201005144
Dumont, M., Wille, E., Calingasan, N. Y., Tampellini, D., Williams, C., Gouras, G. K., et al. (2009). Triterpenoid CDDO-methylamide improves memory and decreases amyloid plaques in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 109, 502–512. doi: 10.1111/j.1471-4159.2009.05970.x
Eldar-Finkelman, H., and Martinez, A. (2011). GSK-3 inhibitors: preclinical and clinical focus on CNS. Front. Mol. Neurosci. 4:32. doi: 10.3389/fnmol.2011.00032
Farina, F., Lambert, E., Commeau, L., Lejeune, F. X., Roudier, N., Fonte, C., et al. (2017). The stress response factor daf-16/FOXO is required for multiple compound families to prolong the function of neurons with Huntington’s disease. Sci. Rep. 7:4014. doi: 10.1038/s41598-017-04256-w
Farr, S. A., Ripley, J. L., Sultana, R., Zhang, Z., Niehoff, M. L., Platt, T. L., et al. (2014). Antisense oligonucleotide against GSK-3β in brain of SAMP8 mice improves learning and memory and decreases oxidative stress: involvement of transcription factor Nrf2 and implications for Alzheimer disease. Free Radic. Biol. Med. 67, 387–395. doi: 10.1016/j.freeradbiomed.2013.11.014
Fernandes, B. S., Molendijk, M. L., Köhler, C. A., Soares, J. C., Leite, C. M. G. S., Machado-Vieira, R., et al. (2015). Peripheral brain-derived neurotrophic factor (BDNF) as a biomarker in bipolar disorder: a meta-analysis of 52 studies. BMC Med. 13:289. doi: 10.1186/s12916-015-0529-7
Fiorentini, A., Rosi, M. C., Grossi, C., Luccarini, I., and Casamenti, F. (2010). Lithium improves hippocampal neurogenesis, neuropathology and cognitive functions in APP mice. PLoS One 5:e14382. doi: 10.1371/journal.pone.0014382
Fletcher, S., Page, B. D. G., Zhang, X., Yue, P., Li, Z. H., Sharmeen, S., et al. (2011). Antagonism of the Stat3-Stat3 protein dimer with salicylic acid based small molecules. ChemMedChem 6, 1459–1470. doi: 10.1002/cmdc.201100194
Fornai, F., Longone, P., Cafaro, L., Kastsiuchenka, O., Ferrucci, M., Manca, M. L., et al. (2008a). Lithium delays progression of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U S A 105, 2052–2057. doi: 10.1073/pnas.0708022105
Fornai, F., Longone, P., Ferrucci, M., Lenzi, P., Isidoro, C., Ruggieri, S., et al. (2008b). Autophagy and amyotrophic lateral sclerosis: the multiple roles of lithium. Autophagy 4, 527–530. doi: 10.4161/auto.5923
Fowler, J. H., McQueen, J., Holland, P. R., Manso, Y., Marangoni, M., Scott, F., et al. (2017). Dimethyl fumarate improves white matter function following severe hypoperfusion: involvement of microglia/macrophages and inflammatory mediators. J. Cereb. Blood Flow Metab. 38, 1354–1370. doi: 10.1177/0271678x17713105
Frake, R. A., Menzies, F. M., David, C., Invest, J. C., Frake, R. A., Ricketts, T., et al. (2015). Autophagy and neurodegeneration. J. Clin. Invest. 125, 65–74. doi: 10.1172/JCI73944
Fukumoto, T., Morinobu, S., Okamoto, Y., Kagaya, A., and Yamawaki, S. (2001). Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology 158, 100–106. doi: 10.1007/s002130100871
Galluzzi, L., Bravo-San Pedro, J. M., Levine, B., Green, D. R., and Kroemer, G. (2017). Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 16, 487–511. doi: 10.1038/nrd.2017.22
Gelfo, F., Cutuli, D., Nobili, A., De Bartolo, P., D’Amelio, M., Petrosini, L., et al. (2017). Chronic lithium treatment in a rat model of basal forebrain cholinergic depletion: effects on memory impairment and neurodegeneration. J. Alzheimer’s Dis. 56, 1505–1518. doi: 10.3233/jad-160892
Gideons, E. S., Lin, P. Y., Mahgoub, M., Kavalali, E. T., and Monteggia, L. M. (2017). Chronic lithium treatment elicits its antimanic effects via BDNF-TrkB dependent synaptic downscaling. Elife 6:e25480. doi: 10.7554/elife.25480
Green, H. F., and Nolan, Y. M. (2012). GSK-3 mediates the release of IL-1β, TNF-α and IL-10 from cortical glia. Neurochem. Int. 61, 666–671. doi: 10.1016/j.neuint.2012.07.003
Guo, J., Cheng, J., North, B. J., and Wei, W. (2017). Functional analyses of major cancer-related signaling pathways in Alzheimer’s disease etiology. Biochim. Biophys. Acta 1868, 341–358. doi: 10.1016/j.bbcan.2017.07.001
Gurvich, N., and Klein, P. S. (2002). Lithium and valproic acid: parallels and contrasts in diverse signaling contexts. Pharmacol. Ther. 96, 45–66. doi: 10.1016/s0163-7258(02)00299-1
Haas, L. T., Salazar, S. V., Smith, L. M., Zhao, H. R., Cox, T. O., Herber, C. S., et al. (2017). Silent allosteric modulation of mglur5 maintains glutamate signaling while rescuing Alzheimer’s mouse phenotypes. Cell Rep. 20, 76–88. doi: 10.1016/j.celrep.2017.06.023
Habib, A., Sawmiller, D., Li, S., Xiang, Y., Rongo, D., Tian, J., et al. (2017). LISPRO mitigates β-amyloid and associated pathologies in Alzheimer’s mice. Cell Death Dis. 8:e2880. doi: 10.1038/cddis.2017.279
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi: 10.1038/nature04724
Hashimoto, R., Takei, N., Shimazu, K., Christ, L., Lu, B., and Chuang, D. M. (2002). Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: an essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology 43, 1173–1179. doi: 10.1016/s0028-3908(02)00217-4
Hosseini, S., Wilk, E., Michaelsen-Preusse, K., Gerhauser, I., Baumgärtner, W., Geffers, R., et al. (2018). Long-term neuroinflammation induced by influenza a virus infection and the impact on hippocampal neuron morphology and function. J. Neurosci. 38, 3060–3080. doi: 10.1523/jneurosci.1740-17.2018
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U S A 99, 7746–7750. doi: 10.1073/pnas.122205699
Iadecola, C. (2013). The pathobiology of vascular dementia. Neuron 80, 844–866. doi: 10.1016/j.neuron.2013.10.008
Jang, S.-W., Liu, X., Yepes, M., Shepherd, K. R., Miller, G. W., Liu, Y., et al. (2010). A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. U S A 107, 2687–2692. doi: 10.1073/pnas.0913572107
Jin, K., Galvan, V., Xie, L., Mao, X. O., Gorostiza, O. F., Bredesen, D. E., et al. (2004). Enhanced neurogenesis in Alzheimer’s disease transgenic (PDGF-APPSw,Ind) mice. Proc. Natl. Acad. Sci. U S A 101, 13363–13367. doi: 10.1073/pnas.0403678101
Jope, R. S., Cheng, Y., Lowell, J. A., Worthen, R. J., Sitbon, Y. H., and Beurel, E. (2017). Stressed and inflamed, can GSK3 be blamed? Trends Biochem. Sci. 42, 180–192. doi: 10.1016/j.tibs.2016.10.009
Jovanovic, J. N., Czernik, A. J., Fienberg, A. A., Greengard, P., and Sihra, T. S. (2000). Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat. Neurosci. 3, 323–329. doi: 10.1038/73888
Jung, C. H., Ro, S.-H., Cao, J., Otto, N. M., and Kim, D.-H. (2010). mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295. doi: 10.1016/j.febslet.2010.01.017
Kanninen, K., Heikkinen, R., Malm, T., Rolova, T., Kuhmonen, S., Leinonen, H., et al. (2009). Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 106, 16505–16510. doi: 10.1073/pnas.0908397106
Kanninen, K., White, A. R., Koistinaho, J., and Malm, T. (2011). Targeting glycogen synthase kinase-3β for therapeutic benefit against oxidative stress in Alzheimer’s disease: involvement of the Nrf2-ARE pathway. Int. J. Alzheimers Dis. 2011:985085. doi: 10.4061/2011/985085
Karyo, R., Eskira, Y., Pinhasov, A., Belmaker, R. H., Agam, G., and Eldar-Finkelman Hagit, H. (2010). Identification of eukaryotic elongation factor-2 as a novel cellular target of lithium and glycogen synthase kinase-3. Mol. Cell. Neurosci. 45, 449–455. doi: 10.1016/j.mcn.2010.08.004
Kasuya, J., Kaas, G., and Kitamoto, T. (2009). Effects of lithium chloride on the gene expression profiles in Drosophila heads. Neurosci. Res. 64, 413–420. doi: 10.1016/j.neures.2009.04.015
Kawabori, M., and Yenari, M. A. (2015). Inflammatory responses in brain ischemia. Curr. Med. Chem. 22, 1258–1277. doi: 10.2174/0929867322666150209154036
Kerr, F., Sofola-Adesakin, O., Ivanov, D. K., Gatliff, J., Gomez Perez-Nievas, B., Bertrand, H. C., et al. (2017). Direct Keap1-Nrf2 disruption as a potential therapeutic target for Alzheimer’s disease. PLoS Genet. 13:e1006593. doi: 10.1371/journal.pgen.1006593
Khan, A., Jamwal, S., Bijjem, K. R. V., Prakash, A., and Kumar, P. (2015). Neuroprotective effect of hemeoxygenase-1/glycogen synthase kinase-3β modulators in 3-nitropropionic acid-induced neurotoxicity in rats. Neuroscience 287, 66–77. doi: 10.1016/j.neuroscience.2014.12.018
Khurana, V., Lu, Y., Steinhilb, M. L., Oldham, S., Shulman, J. M., and Feany, M. B. (2006). TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr. Biol. 16, 230–241. doi: 10.1016/j.cub.2005.12.042
Kim, H. J., and Thayer, S. A. (2009). Lithium increases synapse formation between hippocampal neurons by depleting phosphoinositides. Mol. Pharmacol. 75, 1021–1030. doi: 10.1124/mol.108.052357
King, M. K., and Jope, R. S. (2013). Lithium treatment alleviates impaired cognition in a mouse model of fragile X syndrome. Genes Brain Behav. 12, 723–731. doi: 10.1111/gbb.12071
Klein, P. S., and Melton, D. A. (1996). A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U S A 93, 8455–8459. doi: 10.1073/pnas.93.16.8455
Klionsky, D., Agholme, L., Agnello, M., Agostinis, P., Aguirre-ghiso, J. A., Ahn, H. J., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544. doi: 10.4161/auto.19496
Kluck, R. M., Bossy-Wetzel, E., Green, D. R., and Newmeyer, D. D. (1997). The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275, 1132–1136. doi: 10.1126/science.275.5303.1132
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J. I., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Kremer, A., Louis, J. V., Jaworski, T., and Van Leuven, F. (2011). GSK3 and Alzheimer’s disease: facts and fiction‥.. Front. Mol. Neurosci. 4:17. doi: 10.3389/fnmol.2011.00017
Krstic, D., Madhusudan, A., Doehner, J., Vogel, P., Notter, T., Imhof, C., et al. (2012). Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J. Neuroinflammation 9:151. doi: 10.1186/1742-2094-9-151
Kudo, W., Lee, H.-P., Smith, M. A., Zhu, X., Matsuyama, S., and Lee, H.-G. (2012). Inhibition of Bax protects neuronal cells from oligomeric Aβ neurotoxicity. Cell Death Dis. 3:e309. doi: 10.1038/cddis.2012.43
Kumar, A., Dhull, D. K., and Mishra, P. S. (2015). Therapeutic potential of mGluR5 targeting in Alzheimer’s disease. Front. Neurosci. 9:215. doi: 10.3389/fnins.2015.00215
Lastres-Becker, I., García-Yagüe, A. J., Scannevin, R. H., Casarejos, M. J., Kügler, S., Rábano, A., et al. (2016). Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson’s disease. Antioxid. Redox Signal. 25, 61–77. doi: 10.1089/ars.2015.6549
Li, H., Li, Q., Du, X., Sun, Y., Wang, X., Kroemer, G., et al. (2011). Lithium-mediated long-term neuroprotection in neonatal rat hypoxia-ischemia is associated with antiinflammatory effects and enhanced proliferation and survival of neural stem/progenitor cells. J. Cereb. Blood Flow Metab. 31, 2106–2115. doi: 10.1038/jcbfm.2011.75
Lieu, C. A., Dewey, C. M., Chinta, S. J., Rane, A., Rajagopalan, S., Batir, S., et al. (2014). Lithium prevents parkinsonian behavioral and striatal phenotypes in an aged parkin mutant transgenic mouse model. Brain Res. 1591, 111–117. doi: 10.1016/j.brainres.2014.10.032
Lin, A.-L., Jahrling, J. B., Zhang, W., DeRosa, N., Bakshi, V., Romero, P., et al. (2017). Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J. Cereb. Blood Flow Metab. 37, 217–226. doi: 10.1177/0271678x15621575
Lionaki, E., Markaki, M., Palikaras, K., and Tavernarakis, N. (2015). Mitochondria, autophagy and age-associated neurodegenerative diseases: new insights into a complex interplay. Biochim. Biophys. Acta 1847, 1412–1423. doi: 10.1016/j.bbabio.2015.04.010
Liu, Z.-H., Chuang, D.-M., and Smith, C. B. (2011). Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int. J. Neuropsychopharmacol. 14, 618–630. doi: 10.1017/s1461145710000520
Liu, Z. H., Huang, T., and Smith, C. B. (2012). Lithium reverses increased rates of cerebral protein synthesis in a mouse model of fragile X syndrome. Neurobiol. Dis. 45, 1145–1152. doi: 10.1016/j.nbd.2011.12.037
Llorens-Martín, M., Jurado, J., Hernández, F., and Avila, J. (2014). GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 7:46. doi: 10.3389/fnmol.2014.00046
Lovestone, S., Boada, M., Dubois, B., Hüll, M., Rinne, J. O., Huppertz, H.-J., et al. (2015). A Phase II trial of Tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 45, 75–88. doi: 10.3233/JAD-141959
Lu, S.-M., Gui, B., Dong, H.-Q., Zhang, X., Zhang, S.-S., Hu, L.-Q., et al. (2015). Prophylactic lithium alleviates splenectomy-induced cognitive dysfunction possibly by inhibiting hippocampal TLR4 activation in aged rats. Brain Res. Bull. 114, 31–41. doi: 10.1016/j.brainresbull.2015.02.008
Lucas, J. J., Hernández, F., Gómez-Ramos, P., Morán, M. A., Hen, R., and Avila, J. (2001). Decreased nuclear β-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3β conditional transgenic mice. EMBO J. 20, 27–39. doi: 10.1093/emboj/20.1.27
Lüscher, C., and Huber, K. M. (2010). Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459. doi: 10.1016/j.neuron.2010.01.016
McBride, S. M. J., Choi, C. H., Wang, Y., Liebelt, D., Braunstein, E., Ferreiro, D., et al. (2005). Pharmacological rescue of synaptic plasticity, courtship behavior and mushroom body defects in a Drosophila model of Fragile X syndrome. Neuron 45, 753–764. doi: 10.1016/j.neuron.2005.01.038
McColl, G., Killilea, D. W., Hubbard, A. E., Vantipalli, M. C., Melov, S., and Lithgow, G. J. (2008). Pharmacogenetic analysis of lithium-induced delayed aging in Caenorhabditis elegans. J. Biol. Chem. 283, 350–357. doi: 10.1074/jbc.m705028200
Menzies, F. M., Fleming, A., Caricasole, A., Bento, C. F., Andrews, S. P., Ashkenazi, A., et al. (2017). Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034. doi: 10.1016/j.neuron.2017.01.022
Mizushima, N., and Komatsu, M. (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. doi: 10.1016/j.cell.2011.10.026
Morris, G., and Berk, M. (2016). The putative use of lithium in Alzheimer’s disease. Curr. Alzheimer Res. 13, 853–861. doi: 10.2174/1567205013666160219113112
Motoi, Y., Shimada, K., Ishiguro, K., and Hattori, N. (2014). Lithium and autophagy. ACS Chem. Neurosci. 5, 434–442. doi: 10.1021/cn500056
Mudher, A., Shepherd, D., Newman, T. A., Mildren, P., Jukes, J. P., Squire, A., et al. (2004). GSK-3β inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol. Psychiatry 9, 522–530. doi: 10.1038/sj.mp.4001483
Nazem, A., Sankowski, R., Bacher, M., and Al-Abed, Y. (2015). Rodent models of neuroinflammation for Alzheimer’s disease. J. Neuroinflammation 12:74. doi: 10.1186/s12974-015-0291-y
Nery, L. R., Eltz, N. S., Hackman, C., Fonseca, R., Altenhofen, S., Guerra, H. N., et al. (2014). Brain intraventricular injection of amyloid-β in zebrafish embryo impairs cognition and increases tau phosphorylation, effects reversed by lithium. PLoS One 9:e105862. doi: 10.1371/journal.pone.0105862
Neymotin, A., Calingasan, N. Y., Wille, E., Naseri, N., Petri, S., Damiano, M., et al. (2011). Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 51, 88–96. doi: 10.1016/j.freeradbiomed.2011.03.027
Nikoletopoulou, V., Papandreou, M. E., and Tavernarakis, N. (2015). Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 22, 398–407. doi: 10.1038/cdd.2014.204
Niswender, C. M., and Conn, P. J. (2010). Metabotropic glutamate receptors: physiology, pharmacology and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322. doi: 10.1146/annurev.pharmtox.011008
Noble, W., Planel, E., Zehr, C., Olm, V., Meyerson, J., Suleman, F., et al. (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. U S A 102, 6990–6995. doi: 10.1073/pnas.0500466102
Nunes, M. A., Schöwe, N. M., Monteiro-Silva, K. C., Baraldi-Tornisielo, T., Souza, S. I. G., Balthazar, J., et al. (2015). Correction: chronic microdose lithium treatment prevented memory loss and neurohistopathological changes in a transgenic mouse model of Alzheimer’s disease. PLoS One 10:e0142267. doi: 10.1371/journal.pone.0145695
Nunes, M. A., Viel, T. A., and Buck, H. S. (2013). Microdose lithium treatment stabilized cognitive impairment in patients with Alzheimer’s disease. Curr. Alzheimer Res. 10, 104–107. doi: 10.2174/156720513804871354
O’Brien, W. T., Harper, A. D., Jové, F., Woodgett, J. R., Maretto, S., Piccolo, S., et al. (2004). Glycogen synthase kinase-3 haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 24, 6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004
O’Brien, W. T., Huang, J., Buccafusca, R., Garskof, J., Valvezan, A. J., Berry, G. T., et al. (2011). Glycogen synthase kinase-3 is essential for β-arrestin-2 complex formation and lithium-sensitive behaviors in mice. J. Clin. Invest. 121, 3756–3762. doi: 10.1172/JCI45194
Odeya, D., Galila, A., and Lilah, T. (2018). The observed alteration in BCL2 expression following lithium treatment is influenced by the choice of normalization method. Sci. Rep. 8:6399. doi: 10.1038/s41598-018-24546-1
Overk, C. R., Cartier, A., Shaked, G., Rockenstein, E., Ubhi, K., Spencer, B., et al. (2014). Hippocampal neuronal cells that accumulate α-synuclein fragments are more vulnerable to Aβ oligomer toxicity via mGluR5 - implications for dementia with lewy bodies. Mol. Neurodegener. 9:18. doi: 10.1186/1750-1326-9-18
Ozcelik, S., Fraser, G., Castets, P., Schaeffer, V., Skachokova, Z., Breu, K., et al. (2013). Rapamycin attenuates the progression of tau pathology in P301S Tau transgenic mice. PLoS One 8:e62459. doi: 10.1371/journal.pone.0062459
Perrin-Cocon, L., Aublin-Gex, A., Sestito, S. E., Shirey, K. A., Patel, M. C., André, P., et al. (2017). TLR4 antagonist FP7 inhibits LPS-induced cytokine production and glycolytic reprogramming in dendritic cells and protects mice from lethal influenza infection. Sci. Rep. 7:40791. doi: 10.1038/srep40791
Phiel, C. J., Wilson, C. A., Lee, V. M.-Y., and Klein, P. S. (2003). GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nature 423, 435–439. doi: 10.1038/nature01640
Pintado, C., Gavilán, M. P., Gavilán, E., García-Cuervo, L., Gutiérrez, A., Vitorica, J., et al. (2012). Lipopolysaccharide-induced neuroinflammation leads to the accumulation of ubiquitinated proteins and increases susceptibility to neurodegeneration induced by proteasome inhibition in rat hippocampus. J. Neuroinflammation 9:87. doi: 10.1186/1742-2094-9-87
Pizzasegola, C., Caron, I., Daleno, C., Ronchi, A., Minoia, C., Carrì, M. T., et al. (2009). Treatment with lithium carbonate does not improve disease progression in two different strains of SOD1 mutant mice. Amyotroph. Lateral Scler. 10, 221–228. doi: 10.1080/17482960902803440
Quiroz, J. A., Machado-Vieira, R., Zarate, C. A., and Manji, H. K. (2010). Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology 62, 50–60. doi: 10.1159/000314310
Ribeiro, F. M., Vieira, L. B., Pires, R. G. W., Olmo, R. P., and Ferguson, S. S. G. (2017). Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol. Res. 115, 179–191. doi: 10.1016/j.phrs.2016.11.013
Rojo, A. I., Rada, P., Egea, J., Rosa, A. O., López, M. G., and Cuadrado, A. (2008). Functional interference between glycogen synthase kinase-3 β and the transcription factor Nrf2 in protection against kainate-induced hippocampal celldeath. Mol. Cell. Neurosci. 39, 125–132. doi: 10.1016/j.mcn.2008.06.007
Roux, M., and Dosseto, A. (2017). From direct to indirect lithium targets: a comprehensive review of omics data. Metallomics 9, 1326–1351. doi: 10.1039/c7mt00203c
Sarkar, S., Floto, R. A., Berger, Z., Imarisio, S., Cordenier, A., Pasco, M., et al. (2005). Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 170, 1101–1111. doi: 10.1083/jcb.200504035
Sarkar, S., Krishna, G., Imarisio, S., Saiki, S., O’Kane, C. J., and Rubinsztein, D. C. (2008). A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum. Mol. Genet. 17, 170–178. doi: 10.1093/hmg/ddm294
Sarkar, S., and Rubinsztein, D. C. (2006). Inositol and IP3 levels regulate autophagy: biology and therapeutic speculations. Autophagy 2, 132–134. doi: 10.4161/auto.2387
Schloesser, R. J., Huang, J., Klein, P. S., and Manji, H. K. (2008). Cellular plasticity cascades in the pathophysiology and treatment of bipolar disorder. Neuropsychopharmacology 33, 110–133. doi: 10.1038/sj.npp.1301575
Serenó, L., Coma, M., Rodríguez, M., Sánchez-Ferrer, P., Sánchez, M. B., Gich, I., et al. (2009). A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 35, 359–367. doi: 10.1016/j.nbd.2009.05.025
Shao, L., Young, L. T., and Wang, J.-F. (2005). Chronic treatment with mood stabilizers lithium and valproate prevents excitotoxicity by inhibiting oxidative stress in rat cerebral cortical cells. Biol. Psychiatry 58, 879–884. doi: 10.1016/j.biopsych.2005.04.052
Shi, S., Liang, D., Chen, Y., Xie, Y., Wang, Y., Wang, L., et al. (2016). Gx-50 reduces β-amyloid-induced TNF-α, IL-1β, NO and PGE 2 expression and inhibits NF-κB signaling in a mouse model of Alzheimer’s disease. Eur. J. Immunol. 46, 665–676. doi: 10.1002/eji.201545855
Shimada, K., Motoi, Y., Ishiguro, K., Kambe, T., Matsumoto, S. E., Itaya, M., et al. (2012). Long-term oral lithium treatment attenuates motor disturbance in tauopathy model mice: implications of autophagy promotion. Neurobiol. Dis. 46, 101–108. doi: 10.1016/j.nbd.2011.12.050
Shin, S., Wolgamott, L., Tcherkezian, J., Vallabhapurapu, S., Yu, Y., Roux, P. P., et al. (2014). Glycogen synthase kinase-3β positively regulates protein synthesis and cell proliferation through the regulation of translation initiation factor 4E-binding protein 1. Oncogene 33, 1690–1699. doi: 10.1038/onc.2013.113
Sofola, O., Kerr, F., Rogers, I., Killick, R., Augustin, H., Gandy, C., et al. (2010). Inhibition of GSK-3 ameliorates Aβ pathology in an adult-onset Drosophila model of Alzheimer’s disease. PLoS Genet. 6:e1001087. doi: 10.1371/journal.pgen.1001087
Sofola-Adesakin, O., Castillo-Quan, J. I., Rallis, C., Tain, L. S., Bjedov, I., Rogers, I., et al. (2014). Lithium suppresses Aβ pathology by inhibiting translation in an adult Drosophila model of Alzheimer’s disease. Front. Aging Neurosci. 6:190. doi: 10.3389/fnagi.2014.00190
Sourial-Bassillious, N., Rydelius, P. A., Aperia, A., and Aizman, O. (2009). Glutamate-mediated calcium signaling: a potential target for lithium action. Neuroscience 161, 1126–1134. doi: 10.1016/j.neuroscience.2009.04.013
Spilman, P., Podlutskaya, N., Hart, M. J., Debnath, J., Gorostiza, O., Bredesen, D., et al. (2010). Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-β levels in a mouse model of Alzheimer’s disease. PLoS One 5:e9979. doi: 10.1371/journal.pone.0009979
Srinivasan, M., and Lahiri, D. K. (2015). Significance of NF-κB as a pivotal therapeutic target in the neurodegenerative pathologies of Alzheimer’s disease and multiple sclerosis. Expert Opin. Ther. Targets 19, 471–487. doi: 10.1517/14728222.2014.989834
Sugawara, H., Iwamoto, K., Bundo, M., Ishiwata, M., Ueda, J., Kakiuchi, C., et al. (2010). Effect of mood stabilizers on gene expression in lymphoblastoid cells. J. Neural Transm. 117, 155–164. doi: 10.1007/s00702-009-0340-8
Sultana, R., and Butterfield, D. A. (2010). Role of oxidative stress in the progression of Alzheimer’s disease. J. Alzheimers Dis. 19, 341–353. doi: 10.3233/JAD-2010-1222
Sundaram, J. R., Chan, E. S., Poore, C. P., Pareek, T. K., Cheong, W. F., Shui, G., et al. (2012). Cdk5/p25-induced cytosolic PLA2-mediated lysophosphatidylcholine production regulates neuroinflammation and triggers neurodegeneration. J. Neurosci. 32, 1020–1034. doi: 10.1523/JNEUROSCI.5177-11.2012
Suzuki, H., Osawa, T., Fujioka, Y., and Noda, N. N. (2017). Structural biology of the core autophagy machinery. Curr. Opin. Struct. Biol. 43, 10–17. doi: 10.1016/j.sbi.2016.09.010
Tajes, M., Yeste-Velasco, M., Zhu, X., Chou, S. P., Smith, M. A., Pallàs, M., et al. (2009). Activation of Akt by lithium: pro-survival pathways in aging. Mech. Ageing Dev. 130, 253–261. doi: 10.1016/j.mad.2008.12.006
Tan, H., Young, L. T., Shao, L., Che, Y., Honer, W. G., and Wang, J.-F. (2012). Mood stabilizer lithium inhibits amphetamine-increased 4-hydroxynonenal-protein adducts in rat frontal cortex. Int. J. Neuropsychopharmacol. 15, 1275–1285. doi: 10.1017/s1461145711001416
Tanaka, K., and Matsuda, N. (2014). Proteostasis and neurodegeneration: the roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta Mol. Cell Res. 1843, 197–204. doi: 10.1016/j.bbamcr.2013.03.012
Tang, S.-C., Lathia, J. D., Selvaraj, P. K., Jo, D.-G., Mughal, M. R., Cheng, A., et al. (2008). Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid β-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp. Neurol. 213, 114–121. doi: 10.1016/j.expneurol.2008.05.014
Tanizawa, Y., Kuhara, A., Inada, H., Kodama, E., Mizuno, T., and Mori, I. (2006). Inositol monophosphatase regulates localization of synaptic components and behavior in the mature nervous system of C. elegans. Genes Dev. 20, 3296–3310. doi: 10.1101/gad.1497806
Taupin, P. (2008). Adult neurogenesis pharmacology in neurological diseases and disorders. Expert Rev. Neurother. 8, 311–320. doi: 10.1586/14737175.8.2.311
Tolosa, E., Litvan, I., Höglinger, G. U., Burn, D., Lees, A., Andrés, M. V., et al. (2014). A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov. Disord. 29, 470–478. doi: 10.1002/mds.25824
Tu, J., Zhang, X., Zhu, Y., Dai, Y., Li, N., Yang, F., et al. (2015). Cell-permeable peptide targeting the Nrf2-Keap1 interaction: a potential novel therapy for global cerebral ischemia. J. Neurosci. 35, 14727–14739. doi: 10.1523/JNEUROSCI.1304-15.2015
Um, J. W., Kaufman, A. C., Kostylev, M., Heiss, J. K., Stagi, M., Takahashi, H., et al. (2013). Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer Aβ oligomer bound to cellular prion protein. Neuron 79, 887–902. doi: 10.1016/j.neuron.2013.06.036
Valvezan, A. J., and Klein, P. S. (2012). GSK-3 and Wnt signaling in neurogenesis and bipolar disorder. Front. Mol. Neurosci. 5:1. doi: 10.3389/fnmol.2012.00001
Vincent, A. S., Roebuck-Spencer, T. M., and Cernich, A. (2014). Cognitive changes and dementia risk after traumatic brain injury: implications for aging military personnel. Alzheimers Dement. 10, S174–S187. doi: 10.1016/j.jalz.2014.04.006
Wang, H.-M., Zhang, T., Li, Q., Huang, J.-K., Chen, R.-F., and Sun, X.-J. (2013). Inhibition of glycogen synthase kinase-3β by lithium chloride suppresses 6-hydroxydopamine-induced inflammatory response in primary cultured astrocytes. Neurochem. Int. 63, 345–353. doi: 10.1016/j.neuint.2013.07.003
Wells, G. (2015). Peptide and small molecule inhibitors of the Keap1-Nrf2 protein-protein interaction. Biochem. Soc. Trans. 43, 674–679. doi: 10.1042/bst20150051
Welsh, G. I., Miller, C. M., Loughlin, A. J., Price, N. T., and Proud, C. G. (1998). Regulation of eukaryotic initiation factor eIF2B: glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett. 421, 125–130. doi: 10.1016/s0014-5793(97)01548-2
Wilson, E. N., Do Carmo, S., Iulita, M. F., Hall, H., Ducatenzeiler, A., Marks, A. R., et al. (2017). BACE1 inhibition by microdose lithium formulation NP03 rescues memory loss and early stage amyloid neuropathology. Transl. Psychiatry 7:e1190. doi: 10.1038/tp.2017.169
Wu, S.-C., Cao, Z.-S., Chang, K.-M., and Juang, J.-L. (2017). Intestinal microbial dysbiosis aggravates the progression of Alzheimer’s disease in Drosophila. Nat. Commun. 8:24. doi: 10.1038/s41467-017-00040-6
Wurzelmann, M., Romeika, J., and Sun, D. (2017). Therapeutic potential of brain-derived neurotrophic factor (BDNF) and a small molecular mimics of BDNF for traumatic brain injury. Neural Regen. Res. 12, 7–12. doi: 10.4103/1673-5374.198964
Yu, F., Wang, Z., Tchantchou, F., Chiu, C.-T., Zhang, Y., and Chuang, D.-M. (2012). Lithium ameliorates neurodegeneration, suppresses neuroinflammation and improves behavioral performance in a mouse model of traumatic brain injury. J. Neurotrauma 29, 362–374. doi: 10.1089/neu.2011.1942
Yuskaitis, C. J., and Jope, R. S. (2009). Glycogen synthase kinase-3 regulates microglial migration, inflammation and inflammation-induced neurotoxicity. Cell. Signal. 21, 264–273. doi: 10.1016/j.cellsig.2008.10.014
Zhang, X., Heng, X., Li, T., Li, L., Yang, D., Zhang, X., et al. (2011). Long-term treatment with lithium alleviates memory deficits and reduces amyloid-beta production in an aged Alzheimer’s disease transgenic mouse model. J. Alzheimers Dis. 24, 739–749. doi: 10.3233/JAD-2011-101875
Zhang, Z., Liu, X., Schroeder, J. P., Chan, C.-B., Song, M., Yu, S. P., et al. (2014). 7,8-Dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 39, 638–650. doi: 10.1038/npp.2013.243
Keywords: lithium, dementia, GSK-3, oxidative damage, neuro-inflammation, proteostasis, neurogenesis, synaptic maintenance
Citation: Kerr F, Bjedov I and Sofola-Adesakin O (2018) Molecular Mechanisms of Lithium Action: Switching the Light on Multiple Targets for Dementia Using Animal Models. Front. Mol. Neurosci. 11:297. doi: 10.3389/fnmol.2018.00297
Received: 07 June 2018; Accepted: 03 August 2018;
Published: 28 August 2018.
Edited by:
Calum Sutherland, University of Dundee, United KingdomReviewed by:
Britta Johanna Eickholt, Charité Universitätsmedizin Berlin, GermanyCopyright © 2018 Kerr, Bjedov and Sofola-Adesakin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fiona Kerr, ZmlvbmEua2VycjJAZ2N1LmFjLnVr
Ivana Bjedov, aS5iamVkb3ZAdWNsLmFjLnVr
Oyinkan Sofola-Adesakin, by5hZGVzYWtpbkBzdXNzZXguYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.