 Catalina Sakai
Catalina Sakai Sundas Ijaz
Sundas Ijaz Ellen J. Hoffman
Ellen J. Hoffman- 1Child Study Center, Program on Neurogenetics, Yale School of Medicine, Yale University, New Haven, CT, United States
- 2Department of Neuroscience, Yale School of Medicine, Yale University, New Haven, CT, United States
Zebrafish are increasingly being utilized as a model system to investigate the function of the growing list of risk genes associated with neurodevelopmental disorders. This is due in large part to the unique features of zebrafish that make them an optimal system for this purpose, including rapid, external development of transparent embryos, which enable the direct visualization of the developing nervous system during early stages, large progenies, which provide considerable tractability for performing high-throughput pharmacological screens to identify small molecule suppressors of simple behavioral phenotypes, and ease of genetic manipulation, which has been greatly facilitated by the advent of CRISPR/Cas9 gene editing technologies. This review article focuses on studies that have harnessed these advantages of the zebrafish system for the functional analysis of genes that are strongly associated with the following neurodevelopmental disorders: autism spectrum disorders (ASD), epilepsy, intellectual disability (ID) and schizophrenia. We focus primarily on studies describing early morphological and behavioral phenotypes during embryonic and larval stages resulting from loss of risk gene function. We highlight insights into basic mechanisms of risk gene function gained from these studies as well as limitations of studies to date. Finally, we discuss advances in in vivo neural circuit imaging in zebrafish, which promise to transform research using the zebrafish model by illuminating novel circuit-level mechanisms with relevance to neurodevelopmental disorders.
Introduction
In recent years, there has been growing interest in the use of zebrafish as a model system for the functional analysis of genes in neurodevelopmental disorders, which are a group of disorders characterized by alterations in behavior, cognition, communication, and/or motor function during development (American Psychiatric Association, 2013). This is due in large part to the unique features of this system, which offer distinct advantages over more traditional model systems (McCammon and Sive, 2015; Ijaz and Hoffman, 2016; Kozol et al., 2016). For example, zebrafish have transparent embryos that develop externally and rapidly, allowing for the direct visualization of neurodevelopmental processes and neural activity in an intact, functioning nervous system. In addition, zebrafish are highly tractable and produce large progenies, which facilitate the conduct of high-throughput pharmacological screens at a scale that would not be feasible in rodent models. Further, with advances in CRISPR/Cas9 gene-editing techniques in zebrafish (Hwang et al., 2013; Moreno-Mateos et al., 2015), it is now possible to generate zebrafish mutants carrying loss-of-function mutations in a gene of interest relatively rapidly and at a low cost. Given this ease of genetic manipulation, zebrafish are emerging as an optimal system for modeling the growing list of risk genes in neurodevelopmental disorders, keeping pace with the rapid rate of gene discovery in these disorders (Allen et al., 2013; Purcell et al., 2014; Sanders et al., 2015). Therefore, zebrafish have considerable potential for advancing our understanding of the roles of risk genes in the developing brain and elucidating basic biological mechanisms underlying neurodevelopmental disorders.
Despite the limitations of modeling human disorders in zebrafish, given their evolutionary divergence, several lines of evidence point to a remarkable degree of conservation, suggesting that studies in zebrafish are likely to have translational relevance to humans. First, at a structural level, zebrafish have the same major subdivisions of a vertebrate brain as mammals—forebrain, midbrain, hindbrain and spinal cord (Guo, 2009) While there are notable structural differences, such as the development of the telencephalon, which forms by a different process in zebrafish (eversion) than mammals (invagination), many brain regions in zebrafish and mammals, including the thalamus, optic tectum and cerebellum, display structural homology and are reviewed in detail in Kozol et al. (2016). In addition, early developmental genes share similar expression patterns in the brains of zebrafish and mammals, and the major neurotransmitter systems in the mammalian brain, including GABA, glutamate, dopamine, norepinephrine, serotonin, histamine and acetylcholine, are present in zebrafish (Guo, 2009). Second, there is evidence for conservation of pharmacological pathways (Burgess and Granato, 2007b; Renier et al., 2007; Rihel et al., 2010). For example, a large-scale screen of psychoactive compounds found that drugs targeting conserved neurotransmitter systems elicit similar effects on sleep in zebrafish and mammals (Rihel et al., 2010). Third, approximately 80% of risk genes associated with human disorders have an orthologous version in zebrafish, revealing considerable genetic conservation (Howe et al., 2013). Fourth, there is evidence that the neural circuits underlying basic behaviors, such as acoustic startle, prepulse inhibition, sleep and arousal, are conserved, suggesting that findings in zebrafish are likely to be relevant to our understanding of related circuits in mammals (Prober et al., 2006; Burgess and Granato, 2007a, b; Schoonheim et al., 2010; Lovett-Barron et al., 2017). These studies highlight the potential of zebrafish, with their optical transparency and amenability to whole-brain, in vivo imaging, for elucidating the roles of risk genes in neurodevelopmental processes and neural circuit function.
In this review article, we will focus on genetic models of the following neurodevelopmental disorders: autism spectrum disorders (ASD), intellectual disability (ID), epilepsy and schizophrenia, in zebrafish. Specifically, we will focus on early phenotypes (morphological and behavioral), rather than adult behaviors, which have been addressed in detail in other reviews (Meshalkina et al., 2018; Shams et al., 2018). We will highlight insights into basic mechanisms of risk gene function and potential drug candidates identified in zebrafish models, as well as the limitations of studies to date. Finally, we will discuss advances in functional imaging of zebrafish brain activity, which has the potential to illuminate new roles for risk genes in conserved neural circuits.
Advances in Gene Targeting Methods in Zebrafish
Zebrafish first emerged as an optimal model system for studying vertebrate development through their use in large-scale forward genetics screens, leading to the discovery of hundreds of genes involved in early developmental processes (Granato and Nusslein-Volhard, 1996). Until recently, one of the challenges of using zebrafish as a model for reverse genetics was the limited availability of methods for generating mutants in a gene of interest. While mouse “knockouts” are generated by isolating embryonic stem cells, related methods are more challenging in zebrafish. For this reason, generating zebrafish mutants relied for a long time on Targeted Induced Local Lesions in Genomes (TILLING), which involves screening thousands of zebrafish carrying random mutations induced by the chemical N-ethyl N-nitrosourea (ENU) to identify a damaging mutation in a target gene (Moens et al., 2008). Limitations of TILLING include the time-consuming process of screening zebrafish libraries and the relatively low likelihood of identifying the desired mutation, though more recent large-scale ENU and retroviral mutagenesis projects using next-generation sequencing have improved the efficiency of this approach (Kettleborough et al., 2013; Varshney et al., 2013; Pan et al., 2015). Because zebrafish have a duplicated genome with at least two orthologs of many human risk genes that display sub-functionalization (Kozol et al., 2016), the lack of methods for rapidly generating targeted mutations greatly restricted the use of zebrafish as a genetic model.
However, the introduction of targeted nuclease technologies, including zinc finger nucleases (ZFN) and transcription activator-like effector nucleases (TALEN), transformed the field, enabling the rapid induction of damaging, heritable mutations in a gene of interest in zebrafish (Doyon et al., 2008; Meng et al., 2008; Sander et al., 2011; Dahlem et al., 2012). ZFNs and TALENs are chimeric fusion proteins designed to bind to a target site within a gene and produce double-stranded breaks that are repaired inefficiently by non-homologous end joining, resulting in insertion-deletion mutations. Despite their advantages over TILLING, a number of limitations prevented their widespread use, including challenges in predicting the efficiency of gene disruption and identifying a target site within an early exon of a gene of interest, along with the high cost of commercially available ZFNs. While TALENs improved upon many of these features, offering increased flexibility and lower cost, both methods were soon supplanted by CRISPRs.
Clustered regularly interspaced short palindromic repeats (CRISPRs) hijack an adaptive immune mechanism used by bacteria for protection against viruses (Jinek et al., 2012). To generate zebrafish mutants, a single guide RNA (sgRNA) recognizing a target genomic sequence is introduced into zebrafish embryos along with mRNA encoding the enzyme, Cas9 (Hwang et al., 2013). Following sgRNA-directed cleavage of DNA by Cas9, inefficient non-homologous end joining leads to insertion-deletion mutations, as with ZFNs and TALENs, though CRISPRs offer superior flexibility and efficiency over these earlier gene-editing methods (Hwang et al., 2013). Further, their low cost and ease of use have made this technology accessible to most laboratories, facilitating the rapid generation of zebrafish mutants and leading to a paradigm-shift in the use of zebrafish as a reverse genetics tool. While a limited number of studies to date have used ZFNs or TALENs to generate zebrafish mutants of genes associated with neurodevelopmental disorders, it is likely that a growing number of studies will harness CRISPRs for this purpose in the near future.
In contrast, the majority of zebrafish studies of neurodevelopmental disorder-associated genes to date have used morpholinos, which are modified antisense oligonucleotides that cause a transient “knockdown” of target gene expression by blocking mRNA splicing or translation (Nasevicius and Ekker, 2000; Draper et al., 2001). Given their low cost, ease of use, and until recently, the limited availability of methods for rapidly generating genetic mutants, morpholinos have been a commonly used method for analyzing gene function in early development in zebrafish. However, morpholinos have several notable drawbacks, including their transient effects, which limit the ability to investigate gene function beyond early stages, as well as their tendency to induce off-target effects (Eisen and Smith, 2008). For example, some morpholinos activate p53 via an unknown mechanism, resulting in widespread apoptosis, which may lead to nonspecific phenotypes, such as changes in head size or brain structure (Robu et al., 2007; Eisen and Smith, 2008). In addition, a growing number of studies are finding a lack of concordance between the phenotypes of genetic mutants and morpholino-induced knockdowns of the same gene, which in most cases are due to the off-target effects of morpholinos (Kok et al., 2015; Lawson, 2016). Therefore, morpholino-induced phenotypes must be interpreted with caution.
Given these limitations, early guidelines were established for confirming the specificity of morpholino-induced phenotypes, including: (i) using two morpholinos targeting distinct sites; and (ii) demonstrating rescue of the phenotype by introducing mRNA lacking the morpholino target site (Eisen and Smith, 2008). With the careful use of these controls, morpholinos have been used successfully to investigate the early developmental roles of genes in zebrafish (Eisen and Smith, 2008). However, many experiments fail to follow these guidelines (Lawson, 2016). Further, studies have found discrepancies between morphant and mutant phenotypes even when these guidelines have been followed (Stainier et al., 2017).
To address these issues, a group of leaders in the zebrafish scientific community recently established new guidelines for the use of morpholinos, requiring that all morpholino-induced phenotypes be confirmed in genetic mutants where possible, which is now feasible due to CRISPRs (Stainier et al., 2017). These guidelines further state that morpholinos may continue to be used for rapid gene “knockdown” only in cases where the phenotype under investigation has been recapitulated in a mutant (Stainier et al., 2017). While there are cases where genetic compensation has been shown to occur in mutants (Rossi et al., 2015; El-Brolosy and Stainier, 2017), the current guidelines require demonstrating the absence of the morpholino-induced phenotype in the mutant background as evidence for compensation (Stainier et al., 2017). Moving forward, following these guidelines will be critical, particularly for the interpretation of phenotypes in zebrafish models of neurodevelopmental disorders, given that morpholinos alone can induce nonspecific neural effects (Shams et al., 2018). At the same time, as mutants become the “gold standard” for reverse genetics studies in zebrafish, it will be equally important to confirm that germline mutations result in loss-of-function (Shams et al., 2018).
Zebrafish Models of Neurodevelopmental Disorders
Here, we discuss findings from studies that used zebrafish to investigate the function of genes that are strongly associated with ASD, epilepsy, ID and schizophrenia, or to screen for the functionality of newly identified risk genes or variants (Tables 1–4). Importantly, the genetics of these disorders is complex, likely involving hundreds of risk genes, and is characterized by considerable pleiotropy, with the same genes or genomic regions conferring risk to a range of disorders (State and Šestan, 2012). For clarity, we have categorized genes in Tables 1–4 by the disorder to which they are most closely associated in the literature, noting overlapping associations where applicable, though studies of the biological functions of these genes are likely to be relevant across diagnostic boundaries. While zebrafish have also been used to study other neurodevelopmental disorders, including attention-deficit/hyperactivity disorder (ADHD; Lange et al., 2012a,b), Bardet-Biedl syndrome (BBS; Zaghloul et al., 2010; Heon et al., 2016; Lindstrand et al., 2016), and maple syrup urine disease (MSUD; Friedrich et al., 2012), we focus on ASD, epilepsy, ID and schizophrenia, which have been the subject of most zebrafish studies to date and highlight the advantages of this system for the functional analysis of risk genes. Moreover, while an increasing number of studies are investigating complex behaviors in adult zebrafish, such as social behaviors, it is important to observe that there are limitations of face validity, such that it is not possible to recapitulate fully the symptoms of neurodevelopmental disorders in zebrafish (or any animal model), and this is not a prerequisite for demonstrating the relevance of the model. Therefore, in this review article, we focus our discussion on embryonic and larval phenotypes, which highlight the unique strengths of the zebrafish system for illuminating conserved roles of risk genes in basic biological pathways and brain circuits underlying simple behaviors, which are likely to have translational relevance.
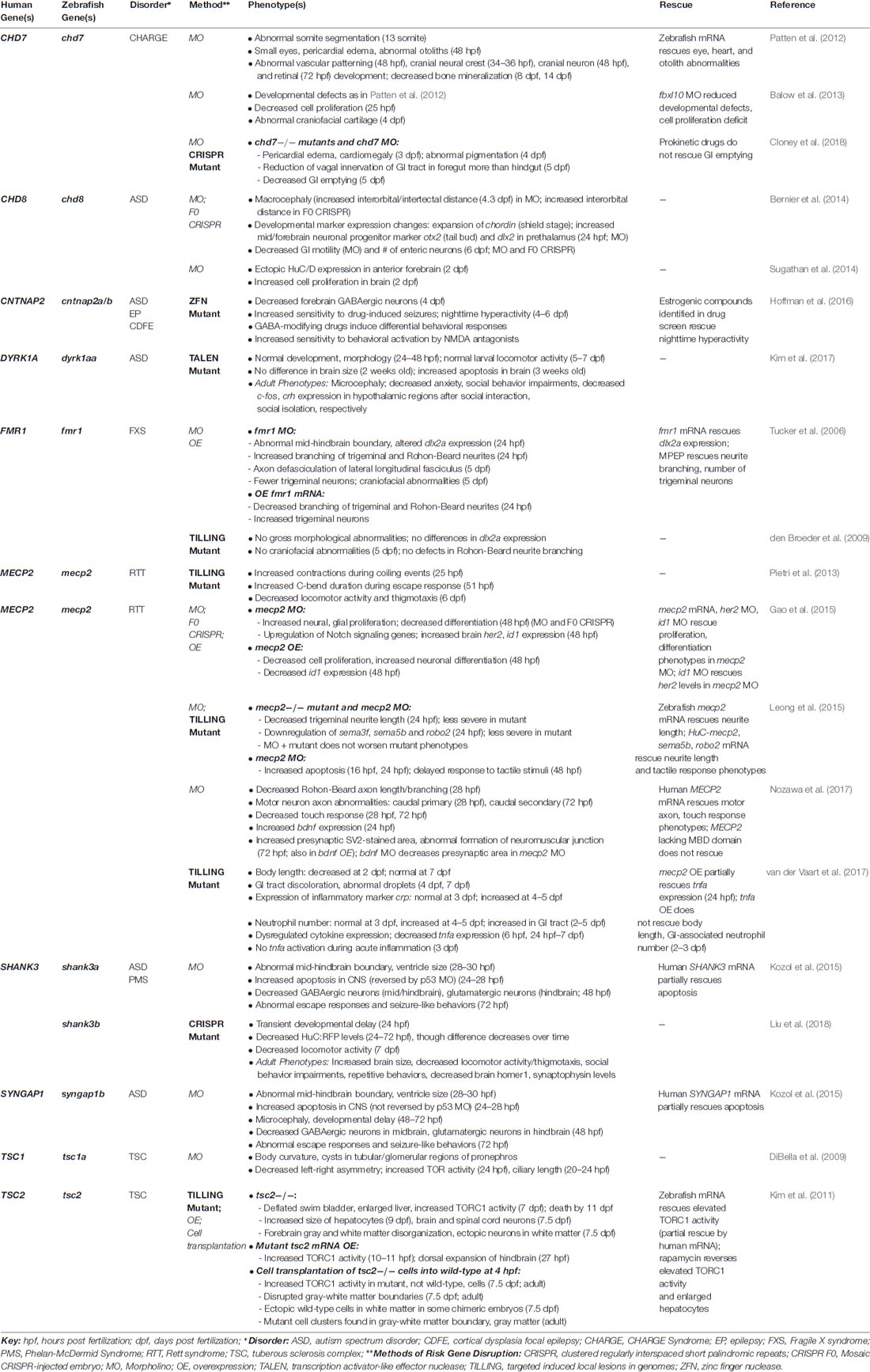
Table 1. Zebrafish models of autism spectrum disorders (ASD)-associated genes and genetic syndromes.
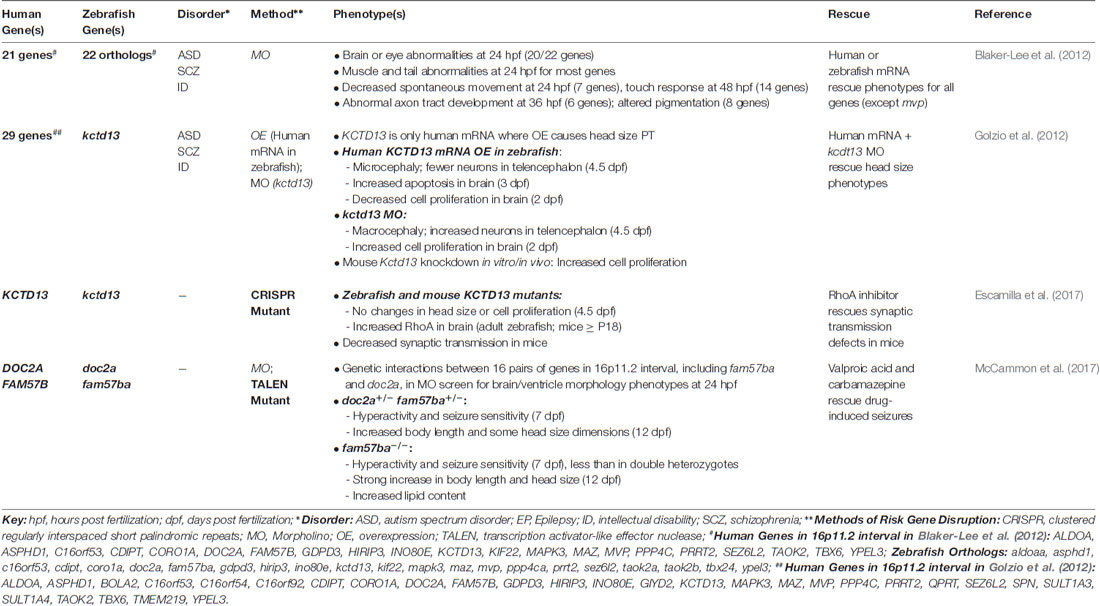
Table 2. Zebrafish models of genes in 16p11.2 interval.
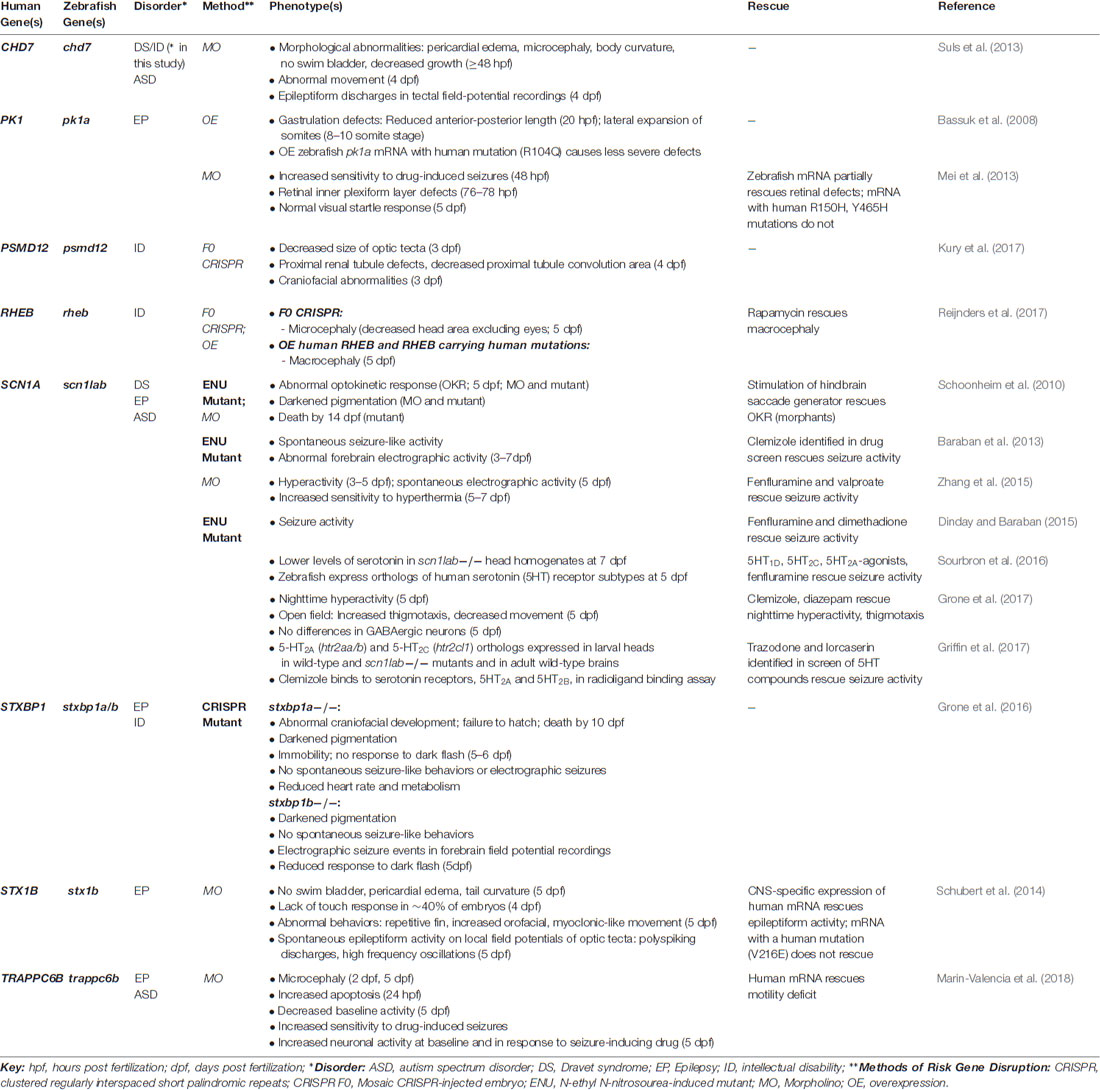
Table 3. Zebrafish models of epilepsy and intellectual disability-associated genes.
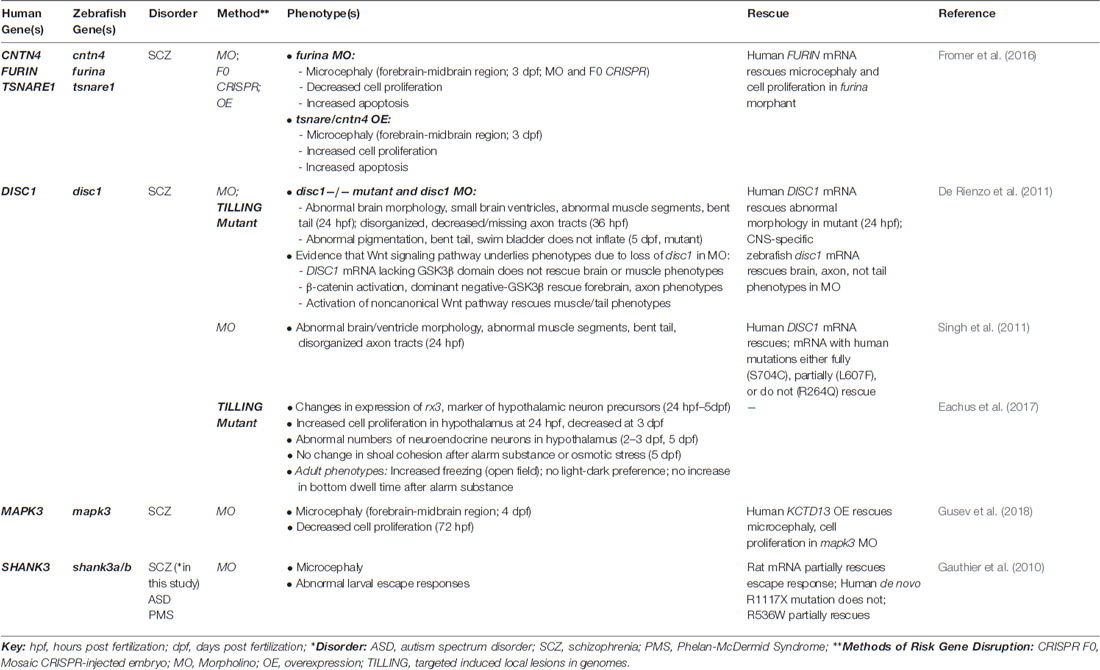
Table 4. Zebrafish models of schizophrenia-associated genes.
Autism Spectrum Disorder
ASDs are a devastating group of neurodevelopmental disorders characterized by marked impairments in social behavior and communication, and by the presence of restricted, repetitive behaviors (American Psychiatric Association, 2013). In recent years, large-scale, whole-exome sequencing studies have led to a rapidly expanding list of reliable, “high confidence” ASD risk genes, which are beginning to reveal common biological mechanisms (Willsey et al., 2013; De Rubeis et al., 2014; Iossifov et al., 2014; Sanders et al., 2015). Despite this progress, how the disruption of these genes leads to the alteration of specific cell types and neural pathways during early stages of brain development remains poorly understood. A growing number of studies have used zebrafish models to investigate the function of “high confidence” ASD risk genes and genes linked to ASD-associated syndromes, such as Fragile X syndrome, tuberous sclerosis complex, Rett syndrome, and CHARGE syndrome (Table 1), as well as genes found in the 16p11.2 chromosomal interval (Table 2), where copy number variants (CNVs) have been associated with ASD, ID and schizophrenia (Kumar et al., 2008; Marshall et al., 2008; Weiss et al., 2008; McCarthy et al., 2009).
Recent studies of zebrafish models of ASD risk genes are beginning to shed light on relevant neurobiological mechanisms. For example, because excitatory-inhibitory imbalance has been implicated as a potential mechanism underlying ASD (and epilepsy; Rubenstein and Merzenich, 2003), some studies have investigated the extent to which disruption of ASD risk genes alters inhibitory GABAergic and excitatory glutamatergic neurons during early brain development using transgenic lines that label these cell populations. For example, Kozol et al. (2015) found that knockdown of two of the zebrafish orthologs of the ASD-associated genes, SHANK3 and SYNGAP1, led to fewer GABAergic neurons in the midbrain (and hindbrain for shank3a) and glutamatergic neurons in the hindbrain. In addition, our group found that zebrafish mutants of both orthologs of the ASD- and epilepsy-linked gene, CNTNAP2, display deficits in forebrain GABAergic neurons, but lack regional deficits in glutamatergic neurons (Hoffman et al., 2016). Interestingly, GABAergic deficits were also found in mouse knockouts of Cntnap2 (Penagarikano et al., 2011), suggesting that this gene affects conserved pathways in fish and mice.
In addition, given that differences in head and brain size, particularly macrocephaly, and changes in neuron number or organization have been described in ASDs (Courchesne et al., 2011; Stoner et al., 2014), a number of morpholino-based studies have examined the effects of decreased ASD risk gene expression on related phenotypes. For example, reduced expression of most zebrafish orthologs of genes in the 16p11.2 chromosomal interval led to structural brain abnormalities at 24 h post fertilization (hpf), including smaller brain ventricles, altered midbrain-hindbrain boundary, or a straight midbrain (Blaker-Lee et al., 2012). In addition, reduced expression of shank3a and syngap1b, led to alterations in the midbrain-hindbrain boundary, ventricle size, and microcephaly (Kozol et al., 2015). Further, knockdown of the “high confidence” ASD risk gene, CHD8, was associated with macrocephaly and increased cell proliferation in the brain (Bernier et al., 2014; Sugathan et al., 2014). However, because morpholinos themselves can cause nonspecific neural phenotypes, confirmation of these phenotypes in germline mutants is a critical next step.
Several studies have also investigated behavioral phenotypes in zebrafish larvae as a means of elucidating how risk gene disruption leads to alterations in simple behaviors. For example, zebrafish mutants of MECP2, the gene responsible for Rett syndrome, display decreased locomotor activity, reduced thigmotaxis (wall preference), and longer touch-evoked escape responses, indicating that loss of mecp2 affects embryonic and larval behaviors (Pietri et al., 2013). In addition, shank3a and syngap1b morphant larvae display abnormal escape responses (Kozol et al., 2015), while shank3b mutant larvae (lacking the function of the other zebrafish ortholog of SHANK3) exhibit reduced locomotor activity (Liu et al., 2018). Mutant larvae of scn1lab, an ortholog of the ASD- and epilepsy-associated genes, SCN1A and SCN2A, exhibit spontaneous seizures (discussed in the “Epilepsy” section), as well as nighttime hyperactivity and increased thigmotaxis (Grone et al., 2017). cntnap2ab mutants also display nighttime hyperactivity and increased sensitivity to drug-induced seizures (Hoffman et al., 2016). Further, double heterozygous mutants of two genes in the 16p11.2 interval, doc2a and fam57ba, show greater hyperactivity and drug-induced seizure sensitivity than single homozygous mutants of each gene, suggesting a genetic interaction (McCammon et al., 2017).
An important advantage of studying larval behavioral phenotypes is their amenability to high-throughput quantitative assays and small molecule screens (Prober et al., 2006; Kokel et al., 2010; Rihel et al., 2010). We capitalized on this approach to investigate rest-wake activity in zebrafish cntnap2ab mutants, which display nighttime hyperactivity (Hoffman et al., 2016). By comparing the behavioral “fingerprint” of cntnap2ab mutants across a range of rest-wake behavioral parameters with a database of the responses of wild-type fish to over 550 psychoactive compounds (Rihel et al., 2010), we predicted compounds that might rescue the mutant behavioral phenotype and tested a select group of these compounds to identify suppressors (Hoffman et al., 2016). Intriguingly, we found that estrogenic compounds selectively suppress nighttime hyperactivity in cntnap2ab mutants, revealing a new neurochemical pathway not previously associated with this gene (Hoffman et al., 2016). In this way, pharmaco-behavioral profiling of zebrafish ASD risk gene mutants represent a promising first-pass screening approach to identify potential pharmacological candidates for further investigation.
Zebrafish mutants also provide an opportunity to investigate risk gene function over the course of development from embryonic stages through adulthood. This is particularly relevant for ASD, where many risk genes are highly expressed in the human brain during embryonic and fetal stages (State and Šestan, 2012). Interestingly, two recent studies found that zebrafish mutants of ASD risk genes display distinct phenotypes at different developmental stages. First, zebrafish mutants of one ortholog of DYRK1A, a “high confidence” ASD risk gene located in the Down syndrome critical region, developed normally with no gross morphological or locomotor abnormalities during embryonic and larval stages, yet displayed increased apoptosis in the brain at 3 weeks old, and microcephaly and behavioral abnormalities in adulthood (Kim et al., 2017). Second, mutants of shank3b, the second ortholog of SHANK3, exhibited transient developmental delay at 24 hpf and fewer CNS neurons at 24–72 hpf, yet this difference diminished over time (Liu et al., 2018). In contrast, adult shank3b mutants display increased brain size and behavioral deficits (Liu et al., 2018). These studies highlight the importance of assessing phenotypes along a developmental trajectory.
With regard to ASD-associated syndromes, several studies have investigated signaling pathways in zebrafish models of Rett syndrome. For example, one study found that mecp2 knockdown led to increased proliferation of neural precursors and decreased neuronal differentiation, which were reversed by simultaneously knocking down id1 or her2, implicating Id1-HER2 signaling as a downstream pathway (Gao et al., 2015). Another study tracked inflammatory phenotypes in mecp2 mutants over the course of development, finding decreased expression of the proinflammatory cytokine, tnfa, as early as 6 hpf, while differences in other cytokines emerge later (van der Vaart et al., 2017). This study further highlights the relevance of assessing phenotypes along a developmental trajectory. Reduction of mecp2 expression was also associated with abnormalities in both sensory and motor axon outgrowth (Leong et al., 2015; Nozawa et al., 2017). In addition, (Leong et al., 2015) found that the axon guidance molecules, sema5b and robo2, are downregulated in mecp2 morphants and mutants, and that co-expression of these genes in morphants rescues decreased trigeminal neurite length and delayed touch responses. Of note, the structural and gene expression phenotypes were more severe in morphants than mutants, but were not worsened by the introduction of the morpholino in mutant embryos, suggesting there may be compensation (Leong et al., 2015). This underscores the importance of directly comparing mutant and morphant phenotypes.
However, zebrafish models of Fragile X syndrome provide a cautionary tale regarding the off-target effects of morpholinos. That is, morpholino-induced knockdown of fmr1 was associated with multiple neurodevelopmental phenotypes, including alterations in the midbrain-hindbrain boundary, abnormal neurite branching, and craniofacial abnormalities (Tucker et al., 2006). However, none of these phenotypes was replicated in two lines of fmr1 mutants, which lacked Fmr protein expression by western blot (den Broeder et al., 2009). KCTD13 offers another example where morphant phenotypes did not replicate in a mutant. KCTD13 is found in the 16p11.2 chromosomal interval, where deletions are associated with macrocephaly, ASD and ID, and duplications with microcephaly, ASD and schizophrenia. Consistent with this association, Golzio et al. (2012) found that morpholino-induced kctd13 knockdown and overexpression of human KCTD13 led to reciprocal phenotypes of macrocephaly and microcephaly, respectively, as well as related changes in cell proliferation in the brains of zebrafish larvae, implicating this gene as a potential driver of head size phenotypes (Golzio et al., 2012). However, a recent study did not identify differences in head size or cell proliferation in zebrafish (or mouse) mutants of KCTD13 (Escamilla et al., 2017). These studies underscore the importance of validating morpholino-induced phenotypes in genetic mutants.
Zebrafish models of ASD-associated syndromes also provide evidence for the conservation of molecular pathways. For example, both mutants and morphants of CHD7, the gene that is associated with most cases of CHARGE syndrome, display pericardial edema and cardiac abnormalities, consistent with cardiac abnormalities found in affected individuals (Patten et al., 2012; Balow et al., 2013; Cloney et al., 2018). In addition, chd7 mutants (and morphants) display decreased GI emptying (Cloney et al., 2018), while chd8 morphants showed reduced GI motility (Bernier et al., 2014), which may be relevant to GI symptoms in individuals carrying mutations in these genes. Further, zebrafish models of tuberous sclerosis complex, including tsc1a morphants and tsc2 mutants, display increased TORC activity (DiBella et al., 2009; Kim et al., 2011). Consistent with findings in mammals, rapamycin reverses elevated TORC1 activity in tsc2 mutants (Kim et al., 2011). Interestingly, by transplanting cells from tsc2 to wild-type embryos at the blastula stage, Kim et al. (2011) showed that increased TORC1 activity is cell autonomous, but that mutant cells also induce non-cell autonomous effects, leading to the ectopic localization of wild-type cells in the white matter. Transplanted mutant cells were also found in abnormal clusters at the gray-white matter boundary in adult brains, suggestive of brain hamartomas found in individuals with this disorder (Kim et al., 2011). Together, these studies highlight the potential of zebrafish models of ASD risk genes to reveal conserved pathways with translational relevance to mammals.
Epilepsy
Epilepsy is a common neurological condition characterized by recurrent seizures (Myers and Mefford, 2015). There has been considerable progress in risk gene discovery in epilepsy from studies of Mendelian syndromes in large family pedigrees, as well as through the identification of de novo single nucleotide variants and CNVs in affected individuals (Hildebrand et al., 2013). Here, we highlight studies using zebrafish models of genetic epilepsy syndromes (Table 3). Zebrafish offer several advantages in this regard. First, zebrafish larvae display robust, seizure-like behaviors, including rapid burst-like and circling movements, following exposure to the GABA-A antagonist, pentylenetetrazol (PTZ; Baraban et al., 2005), providing a quantifiable readout of seizure susceptibility. That is, increased sensitivity to PTZ-induced seizures has been shown in zebrafish models of epilepsy and ASD (Mei et al., 2013; Hoffman et al., 2016; McCammon et al., 2017). Second, zebrafish mutants of epilepsy-associated genes have been shown to exhibit spontaneous seizures as larvae (Baraban et al., 2013; Grone et al., 2016). Third, both drug-induced and spontaneous locomotor seizures are associated with electrographic seizures (Baraban et al., 2005, 2013), and are readily quantifiable in high-throughput assays (Baraban et al., 2013; Hong et al., 2016; Fuller et al., 2018), making zebrafish an optimal model for drug discovery in epilepsy syndromes.
In particular, zebrafish mutants of scn1lab have been used as a model of Dravet syndrome, a severe, intractable form of epilepsy, which in most cases is caused by mutations in SCN1A (Baraban et al., 2013). Homozygous scn1lab mutants display spontaneous seizures beginning at 4 days post fertilization (dpf), as well as electrographic seizures in forebrain extracellular field recordings, which worsen from 3–7 dpf (Baraban et al., 2013). Interestingly, scn1lab mutants were first identified in a forward genetic screen of ENU-mutagenized fish due to their inability to sustain saccadic eye movements during the optokinetic response (OKR; Schoonheim et al., 2010). These mutants have an abnormal pigmentation pattern and die by 14 dpf (Schoonheim et al., 2010). Using these mutants, Baraban et al. (2013) performed a high-throughput screen of 320 compounds, and identified clemizole, a U.S. Food and Drug Administration-approved drug and antihistamine, as a suppressor of both seizure-like behaviors and electrographic seizures. A subsequent study found that clemizole has activity at 5-HT2A and 5-HT2B receptors in a radioligand binding assay, suggesting that a serotonergic mechanism may be responsible for its anti-epileptic activity (Griffin et al., 2017). Interestingly, fenfluramine, an inducer of serotonin (5-hydroxytrypamine, 5-HT) release, which was found to have some efficacy in improving seizures in individuals with Dravet syndrome (Ceulemans et al., 2012), also reduced seizure activity in zebrafish scn1lab mutants and morphants, suggesting conservation of pharmacological pathways (Dinday and Baraban, 2015; Zhang et al., 2015; Sourbron et al., 2016).
Based on the serotonergic mechanism of fenfluramine, (Sourbron et al., 2016) tested selective 5-HT receptor agonists in scn1lab mutants and found that 5-HT1D, 5-HT2C, and 5-HT2A agonists reverse electrographic seizure activity. Also, Griffin et al. (2017) screened a library of 5-HT-modulating compounds and found that lorcaserin and trazodone rescued seizure activity. Through a compassionate use program, lorcaserin was subsequently prescribed to five patients with Dravet syndrome with intractable seizures. While these patients experienced an initial decrease in seizure frequency, seizures returned to baseline after 3 months in most patients (Griffin et al., 2017). Clearly, larger, double-blind, placebo-controlled trials are needed to fully assess the efficacy of this medication. Another important consideration is how to accurately translate effective dosages between systems. Nonetheless, these studies highlight the strengths of zebrafish as a first-pass screening approach for identifying potential anti-epileptic drug candidates. Indeed, as more epilepsy-associated genes are identified in human studies, it is likely that zebrafish models will continue to be instrumental in this regard. For example, zebrafish mutants of stxbp1b, an ortholog of STXBP1, which is associated with epileptic encephalopathy syndromes, display electrographic seizures at baseline, suggesting this may be a useful model for these syndromes (Grone et al., 2016).
Screening Risk Genes Associated With Epilepsy and Intellectual Disability
Zebrafish have also been used a genetic tool for rapidly screening the functionality of novel genes and rare variants identified in human genetics studies of epilepsy, ID and other neurodevelopmental disorders (Table 3, Bassuk et al., 2008; Gauthier et al., 2010; Suls et al., 2013; Schubert et al., 2014; Kury et al., 2017; Reijnders et al., 2017; Marin-Valencia et al., 2018). These studies assess the extent to which wild-type mRNA or mRNA carrying rare variants identified in affected individuals reverses morpholino-induced or CRISPR F0 phenotypes, providing an in vivo readout of the effect of the mutation on gene function. For example, morpholino-induced knockdown of STX1B, which was identified by linkage analysis in large pedigrees as carrying damaging mutations in individuals with epilepsy, caused electrographic seizures (Schubert et al., 2014). These seizures were reduced by CNS-specific expression of human STX1B mRNA, but not mRNA carrying a patient mutation, demonstrating that this variant represents a loss-of-function. In addition, morpholino-induced knockdown of TRAPPC6B, which was identified as a risk gene by linkage analysis and homozygosity mapping in individuals with epilepsy, microcephaly, and ASD from consanguineous families, led to increased baseline neural activity and sensitivity to PTZ-induced seizures in zebrafish larvae (Marin-Valencia et al., 2018).
Another approach to assess the functionality of newly identified human genes or rare variants is overexpression in zebrafish embryos. For example, overexpression of wild-type pk1a, the zebrafish ortholog of PK1A, caused a more severe phenotype than overexpressing mRNA carrying a mutation identified in individuals with progressive myoclonic epilepsy, suggesting that the mutation alters the in vivo function of this gene (Bassuk et al., 2008). In addition, overexpression of mRNA encoding human RHEB and versions of the gene containing two missense mutations identified in individuals with ID and macrocephaly, caused macrocephaly in zebrafish larvae, while F0 CRISPR mosaics of this gene displayed microcephaly, suggesting these variants may represent a gain-of-function (Reijnders et al., 2017).
There are several points to consider in the use of zebrafish for screening variants identified in human genetics studies. First, while in vivo rescue or overexpression screens may be informative regarding the biological function of an identified variant, the identification of nonspecific neural phenotypes, particularly morpholino-based phenotypes, are not sufficient to establish causation of an identified gene or variant and should not be used a substitute for strong evidence from human genetic studies. Second, while the presence of a phenotype in CRISPR F0 mosaics provides additional support for specificity, it is important to demonstrate in a stable mutant line that the phenotype results from loss of gene function and not nonspecific effects in F0-injected embryos. Third, with advances in CRISPR technology (Auer et al., 2014; Kimura et al., 2014), it will be increasingly feasible to rapidly generate not only loss-of-function mutations in a gene of interest, but “knock-in” models of specific patient mutations, which will be particularly informative given the pleiotropy of genes associated with neurodevelopmental disorders.
Schizophrenia
Schizophrenia is a psychotic disorder characterized by hallucinations, delusions and disorganized thought processes or behavior, as well as diminished affect, energy and motivation, which severely impacts overall functioning (American Psychiatric Association, 2013). The genetics of schizophrenia are complex, with over 100 common variants identified by genome-wide association studies (GWAS; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014), and rare damaging variants and CNVs contributing to risk according to a polygenic model (Walsh et al., 2008; Purcell et al., 2014). This genetic architecture complicates the functional analysis of risk variants in schizophrenia (Fromer et al., 2016). Here, we discuss several studies that used zebrafish to analyze the function of schizophrenia-associated genes (Table 4).
While schizophrenia is highly polygenic, DISC1 is an example of a rare schizophrenia-associated gene, which was discovered in a large Scottish family where a balanced chromosomal translocation segregated with schizophrenia and other psychiatric disorders (schizoaffective disorder, bipolar disorder, major depressive disorder; Millar et al., 2000). Some studies have used zebrafish to investigate DISC1 function. For example, De Rienzo et al. (2011) found that disc1 mutants and morphants display abnormal brain morphology at early developmental stages, including small brain ventricles. Full-length human DISC1 mRNA rescued structural brain phenotypes in morphants, while DISC1 lacking the GSK3β binding domain did not, suggesting that that loss of Wnt signaling is responsible for these phenotypes. In a subsequent study, Singh et al. (2011) showed that two common variants in DISC1 identified in individuals with schizophrenia and bipolar disorder that lacked Wnt signaling activity in an in vitro assay were unable to rescue structural brain phenotypes in disc1 morphants, further implicating Wnt signaling as an important pathway downstream of DISC1. Another study found evidence for altered hypothalamic development as well as stress responses in zebrafish disc1 mutants (Eachus et al., 2017). Interestingly, this study found variable phenotypes in two disc1 mutant lines, such as differences in the time course of expression changes in markers of hypothalamic precursors, even though both lines carry mutations that induce early premature stop codons. This suggests that the specific location of a mutation may alter the expression of a phenotype in mutants. Also, while this study used one of the same mutants as the previous study, no morphological defects were identified in disc1 homozygous mutants, suggesting that background variation may alter the expression of phenotypes in genetic mutants (Eachus et al., 2017). Together, these studies highlight insights into DISC1 function gained from zebrafish models.
Additional studies have used zebrafish to rapidly assess the effect of changes in the expression of schizophrenia candidate genes implicated in human genetics studies. By comparing risk variants identified by GWAS with RNA sequencing data from post-mortem brain samples from individuals with schizophrenia, Fromer et al. (2016) identified genomic loci where risk variants might contribute to observed changes in gene expression. By altering the expression of three implicated genes in zebrafish in the same direction as the RNA sequencing result from human brain tissue, this study found that morpholino-induced knockdown of the downregulated gene, furina, led to microcephaly and decreased cell proliferation, which was rescued by introducing human FURIN mRNA, while overexpression of the upregulated genes, tsnare and cntn4, led to microcephaly and increased cell proliferation (Fromer et al., 2016). Another study combining GWAS and human gene expression data identified MAPK3, which is found in the 16p11.2 interval, as a schizophrenia susceptibility gene. Morpholino-induced knockdown of mapk3 caused microcephaly, which was reversed by overexpression of human KCTD13, another gene in the 16p11.2 interval (Gusev et al., 2018). As discussed earlier, while nonspecific neural phenotypes induced by morpholinos, overexpression, or CRISPR F0 mosaics, may be suggestive of a functional effect, replication of these findings in a stable mutant line is necessary for validation.
Future Directions: Functional Imaging of Neural Circuits
Most studies of zebrafish models of neurodevelopmental disorders to date have focused primarily on early morphological and simple behavioral phenotypes. However, recent advances in functional imaging are likely to transform these studies in the near future, allowing for the assessment of circuit-level phenotypes resulting from risk gene disruption. Progress in brain imaging is due in large part to the development of genetically-encoded calcium indicators (GECIs), such as GCaMP, which provide a rapid readout of activity at the level of a single neuron (Chen et al., 2013). GCaMP can be expressed transgenically in a subset of neurons or throughout the brain of larval zebrafish, which is an ideal system for monitoring neural activity. By harnessing advances in imaging technologies, including two-photon and light-sheet microscopy, a number of studies are beginning to dissect neural circuit mechanisms in the developing zebrafish brain (Ahrens et al., 2013; Portugues et al., 2014; Bianco and Engert, 2015; Dunn et al., 2016; Filosa et al., 2016; Naumann et al., 2016; Thompson et al., 2016). Together with the transparency and relative simplicity of the larval zebrafish brain, these technologies are likely to have considerable translational potential for revealing mechanisms by which the disruption of risk genes leads to alterations in signaling networks in the developing vertebrate brain, resulting in simple behavioral phenotypes.
For example, several studies have used two-photon microscopy to record brain activity in response to visually-evoked stimuli. Two-photon microscopy is a point-scanning method that provides excellent spatial resolution, but is more limited in its imaging speed (Keller and Ahrens, 2015). To image brain activity, zebrafish are immobilized in agarose, while visual stimuli are projected onto a screen to the side or below the fish while brain activity is recorded. Portugues et al. (2014) used this approach to investigate the circuitry underlying the OKR, a reflexive series of eye movements induced by a rotating drum of alternating light and dark stripes. By simultaneously recording brain activity, eye and tail movements during stimulus exposure, this study identified a stereotyped pattern of brain activity that occurs during the OKR, and found that activity in specific brain regions correlates with sensory or motor signals. In addition, Filosa et al. (2016) used two-photon imaging to interrogate the neural circuitry governing feeding behavior. Interestingly, this study showed that hunger not only makes zebrafish more likely to pursue visual stimuli that resemble their food, but increases the responsiveness of specific cells in the optic tectum to these food-like stimuli, providing a neural correlate for the observed behavior. Other studies have also used two-photon microscopy to examine behavioral circuits, such as those involved in prey capture, predator responses, responses to visual and olfactory stimuli, and the optomotor response, a reflexive behavior that occurs following a perceived change in whole-field motion (Dreosti et al., 2014; Bianco and Engert, 2015; Dunn et al., 2016; Naumann et al., 2016).
In addition, a growing number of studies have used light-sheet fluorescence microscopy for functional imaging of the zebrafish brain. Light-sheet microscopy, which uses a thin “sheet” of light to illuminate samples, offers superior speed over two-photon microscopy (Keller and Ahrens, 2015). For example, this method was used to successfully image over 80% of the neurons in the larval zebrafish brain in approximately 1.3 s (Ahrens et al., 2013). Given its speed, light-sheet imaging was used to perform continuous whole-brain activity recordings at baseline, revealing functional networks of correlated activity in the zebrafish brain (Ahrens et al., 2013). Light-sheet imaging has also been used to study brain activity following exposure to various stimuli. For example, Thompson et al. (2016) found that distinct clusters of neurons in the optic tectum respond to visual, auditory and water flow stimuli, and provide evidence for integration in the processing of these stimuli. One drawback of light-sheet imaging is the potential for retinal activation by the light “sheet” itself (Keller and Ahrens, 2015). Two-photon imaging offers an alternative in this regard, because it provides stimulation outside of the visible range of zebrafish. Another approach is to position multiple light sheets to avoid direct retinal stimulation (Vladimirov et al., 2014). Further, functional imaging in general generates considerably large datasets, which may be difficult to analyze, though computational algorithms have been developed to address this challenge (Keller and Ahrens, 2015).
At the same time, these functional imaging techniques are technically challenging, not high-throughput, and often require immobilizing the fish. To address these limitations, Randlett et al. (2015) developed a technique called mitogen-activated protein kinase (MAP)-mapping, in which fixed brain tissue is stained for phosphorylated extracellular-signaling-regulated kinase (pERK), a marker of active neurons, and then imaged using confocal microscopy. To identify regions of differential activity, images are mapped onto a zebrafish brain atlas (Z-Brain). This approach can be used to obtain a readout of whole-brain activity in freely moving zebrafish either at baseline or in response to a stimulus or drug (Randlett et al., 2015). Another method, developed by Lovett-Barron et al. (2017), called MultiMAP, combines two-photon imaging of zebrafish during exposure to visual stimuli with immunostaining for neuronal cell types. By integrating the functional imaging and immunostained datasets, this method allows for the identification of the specific cell types that were active during a behavioral task. Intriguingly, using this approach, (Lovett-Barron et al., 2017) identified the neuromodulatory cell types controlling alertness in zebrafish, and found that manipulation of related cell types in mice induces similar behavioral effects, providing remarkable evidence for conservation of behavioral circuits in fish and mammals (Lovett-Barron et al., 2017). Therefore, findings in zebrafish models of neurodevelopmental disorders are likely to have translational relevance for understanding related circuits in mammals. Together, these technologies offer considerable promise for illuminating circuit-level mechanisms in zebrafish models of neurodevelopmental disorders.
Conclusion
Zebrafish have critical advantages as a model system for investigating the function of genes associated with neurodevelopmental disorders. A growing number of studies are beginning to capitalize on their unique features to illuminate neurobiological and pharmacological pathways underlying ASD, epilepsy, ID and schizophrenia. These studies have utilized the transparency, tractability and throughout of the zebrafish model to identify the effects of loss of risk gene function on the development of specific neuron populations, molecular pathways, and simple behaviors, all of which can be leveraged to screen for novel small molecule suppressors. While many studies to date have used morpholino knockdown technology, which is prone to off-target effects and should not be used as a “standalone tool” (Lawson, 2016), it is important moving forward that the field commit to using genetic mutants to confirm morpolino-induced phenotypes, which is particularly essential for neural phenotypes. While the advent of CRISPR technology has made this goal increasingly feasible, confirming that CRISPR-generated mutants result in loss of function is equally critical. Because splice-site mutations may lead to alternative transcripts that reverse deleterious mutations (Anderson et al., 2017), targeting CRISPRs within a conserved exon, removing most of a target gene using multiple CRISPRs, and demonstrating loss of protein by western blot are recommended steps.
At the same time, one of the challenges in analyzing the function of the growing list of risk genes associated with neurodevelopmental disorders is determining which phenotypes are likely to be relevant to the pathophysiology of these disorders (State and Šestan, 2012). Assessing phenotypes over a developmental time course from embryonic to adult stages will likely provide key insights into when and where risk genes play important roles. In addition, investigating the effect of specific mutations identified in affected individuals by introducing or “knocking in” these mutations using CRISPR will also be instrumental in elucidating how particular variants affect neural development. Moreover, future studies capitalizing on the strengths of zebrafish as a first-pass, high-throughput screening approach have the potential to reveal novel pharmacological candidates for further investigation in these disorders. Given the evidence for conservation of pharmacological and circuit-level pathways in zebrafish and mammals (Rihel et al., 2010; Lovett-Barron et al., 2017), it is likely that these studies will have translational relevance, though testing compounds identified in zebrafish in rodent models will be an important next step prior to clinical trials in humans. Furthermore, advances in in vivo calcium imaging in zebrafish represent an exciting new avenue for investigating the circuit-level roles of risk genes with translational relevance. Taken together, zebrafish represent a promising model system for the discovery of novel biological pathways, pharmacological candidates, and circuit mechanisms with relevance to neurodevelopmental disorders.
Author Contributions
CS, SI and EH contributed to the conceptualization, literature review and writing of this article.
Funding
This work was supported by the National Institutes of Health grant R01MH116002, the Kavli Foundation, National Genetics Foundation, Simons Foundation, Spector Fund and the Swebilius Foundation (EH).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahrens, M. B., Orger, M. B., Robson, D. N., Li, J. M., and Keller, P. J. (2013). Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat. Methods 10, 413–420. doi: 10.1038/nmeth.2434
American Psychiatric Association. (2013). Diagnostic and Statistical Manual of Mental Disorders. 5th Edn. Arlington, VA: American Psychiatric Publishing.
Anderson, J. L., Mulligan, T. S., Shen, M. C., Wang, H., Scahill, C. M., Tan, F. J., et al. (2017). mRNA processing in mutant zebrafish lines generated by chemical and CRISPR-mediated mutagenesis produces unexpected transcripts that escape nonsense-mediated decay. PLoS Genet. 13:e1007105. doi: 10.1371/journal.pgen.1007105
Auer, T. O., Duroure, K., De Cian, A., Concordet, J. P., and Del Bene, F. (2014). Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res. 24, 142–153. doi: 10.1101/gr.161638.113
Balow, S. A., Pierce, L. X., Zentner, G. E., Conrad, P. A., Davis, S., Sabaawy, H. E., et al. (2013). Knockdown of fbxl10/kdm2bb rescues chd7 morphant phenotype in a zebrafish model of CHARGE syndrome. Dev. Biol. 382, 57–69. doi: 10.1016/j.ydbio.2013.07.026
Baraban, S. C., Dinday, M. T., and Hortopan, G. A. (2013). Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Commun. 4:2410. doi: 10.1038/ncomms3410
Baraban, S. C., Taylor, M. R., Castro, P. A., and Baier, H. (2005). Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience 131, 759–768. doi: 10.1016/j.neuroscience.2004.11.031
Bassuk, A. G., Wallace, R. H., Buhr, A., Buller, A. R., Afawi, Z., Shimojo, M., et al. (2008). A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome. Am. J. Hum. Genet. 83, 572–581. doi: 10.1016/j.ajhg.2008.10.003
Bernier, R., Golzio, C., Xiong, B., Stessman, H. A., Coe, B. P., Penn, O., et al. (2014). Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276. doi: 10.1016/j.cell.2014.06.017
Bianco, I. H., and Engert, F. (2015). Visuomotor transformations underlying hunting behavior in zebrafish. Curr. Biol. 25, 831–846. doi: 10.1016/j.cub.2015.01.042
Blaker-Lee, A., Gupta, S., McCammon, J. M., De Rienzo, G., and Sive, H. (2012). Zebrafish homologs of genes within 16p11.2, a genomic region associated with brain disorders, are active during brain development and include two deletion dosage sensor genes. Dis. Model Mech. 5, 834–851. doi: 10.1242/dmm.009944
Burgess, H. A., and Granato, M. (2007a). Modulation of locomotor activity in larval zebrafish during light adaptation. J. Exp. Biol. 210, 2526–2539. doi: 10.1242/jeb.003939
Burgess, H. A., and Granato, M. (2007b). Sensorimotor gating in larval zebrafish. J. Neurosci. 27, 4984–4994. doi: 10.1523/jneurosci.0615-07.2007
Ceulemans, B., Boel, M., Leyssens, K., Van Rossem, C., Neels, P., Jorens, P. G., et al. (2012). Successful use of fenfluramine as an add-on treatment for Dravet syndrome. Epilepsia 53, 1131–1139. doi: 10.1111/j.1528-1167.2012.03495.x
Chen, T. W., Wardill, T. J., Sun, Y., Pulver, S. R., Renninger, S. L., Baohan, A., et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. doi: 10.1038/nature12354
Cloney, K., Steele, S. L., Stoyek, M. R., Croll, R. P., Smith, F. M., Prykhozhij, S. V., et al. (2018). Etiology and functional validation of gastrointestinal motility dysfunction in a zebrafish model of CHARGE syndrome. FEBS J. 285, 2125–2140. doi: 10.1111/febs.14473
Courchesne, E., Campbell, K., and Solso, S. (2011). Brain growth across the life span in autism: age-specific changes in anatomical pathology. Brain Res. 1380, 138–145. doi: 10.1016/j.brainres.2010.09.101
Dahlem, T. J., Hoshijima, K., Jurynec, M. J., Gunther, D., Starker, C. G., Locke, A. S., et al. (2012). Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet. 8:e1002861. doi: 10.1371/journal.pgen.1002861
De Rienzo, G., Bishop, J. A., Mao, Y., Pan, L., Ma, T. P., Moens, C. B., et al. (2011). Disc1 regulates both β-catenin-mediated and noncanonical Wnt signaling during vertebrate embryogenesis. FASEB J. 25, 4184–4197. doi: 10.1096/fj.11-186239
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
den Broeder, M. J., van der Linde, H., Brouwer, J. R., Oostra, B. A., Willemsen, R., and Ketting, R. F. (2009). Generation and characterization of FMR1 knockout zebrafish. PLoS One 4:e7910. doi: 10.1371/journal.pone.0007910
DiBella, L. M., Park, A., and Sun, Z. (2009). Zebrafish Tsc1 reveals functional interactions between the cilium and the TOR pathway. Hum. Mol. Genet. 18, 595–606. doi: 10.1093/hmg/ddn384
Dinday, M. T., and Baraban, S. C. (2015). Large-scale phenotype-based antiepileptic drug screening in a zebrafish model of dravet syndrome (1,2,3). eNeuro 2:ENEURO.0068-15.2015. doi: 10.1523/eneuro.0068-15.2015
Doyon, Y., McCammon, J. M., Miller, J. C., Faraji, F., Ngo, C., Katibah, G. E., et al. (2008). Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat. Biotechnol. 26, 702–708. doi: 10.1038/nbt1409
Draper, B. W., Morcos, P. A., and Kimmel, C. B. (2001). Inhibition of zebrafish fgf8 pre-mRNA splicing with morpholino oligos: a quantifiable method for gene knockdown. Genesis 30, 154–156. doi: 10.1002/gene.1053
Dreosti, E., Vendrell Llopis, N., Carl, M., Yaksi, E., and Wilson, S. W. (2014). Left-right asymmetry is required for the habenulae to respond to both visual and olfactory stimuli. Curr. Biol. 24, 440–445. doi: 10.1016/j.cub.2014.01.016
Dunn, T. W., Gebhardt, C., Naumann, E. A., Riegler, C., Ahrens, M. B., Engert, F., et al. (2016). Neural circuits underlying visually evoked escapes in larval zebrafish. Neuron 89, 613–628. doi: 10.1016/j.neuron.2015.12.021
Eachus, H., Bright, C., Cunliffe, V. T., Placzek, M., Wood, J. D., and Watt, P. J. (2017). Disrupted-in-Schizophrenia-1 is essential for normal hypothalamic-pituitary-interrenal (HPI) axis function. Hum. Mol. Genet. 26, 1992–2005. doi: 10.1093/hmg/ddx076
Eisen, J. S., and Smith, J. C. (2008). Controlling morpholino experiments: don’t stop making antisense. Development 135, 1735–1743. doi: 10.1242/dev.001115
El-Brolosy, M. A., and Stainier, D. Y. R. (2017). Genetic compensation: a phenomenon in search of mechanisms. PLoS Genet. 13:e1006780. doi: 10.1371/journal.pgen.1006780
Epi4K Consortium, Epilepsy Phenome/Genome, Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., et al. (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi: 10.1038/nature12439
Escamilla, C. O., Filonova, I., Walker, A. K., Xuan, Z. X., Holehonnur, R., Espinosa, F., et al. (2017). Kctd13 deletion reduces synaptic transmission via increased RhoA. Nature 551, 227–231. doi: 10.1038/nature24470
Filosa, A., Barker, A. J., Dal Maschio, M., and Baier, H. (2016). Feeding state modulates behavioral choice and processing of prey stimuli in the zebrafish tectum. Neuron 90, 596–608. doi: 10.1016/j.neuron.2016.03.014
Friedrich, T., Lambert, A. M., Masino, M. A., and Downes, G. B. (2012). Mutation of zebrafish dihydrolipoamide branched-chain transacylase E2 results in motor dysfunction and models maple syrup urine disease. Dis. Model Mech. 5, 248–258. doi: 10.1242/dmm.008383
Fromer, M., Roussos, P., Sieberts, S. K., Johnson, J. S., Kavanagh, D. H., Perumal, T. M., et al. (2016). Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 19, 1442–1453. doi: 10.1038/nn.4399
Fuller, T. D., Westfall, T. A., Das, T., Dawson, D. V., and Slusarski, D. C. (2018). High-throughput behavioral assay to investigate seizure sensitivity in zebrafish implicates ZFHX3 in epilepsy. J. Neurogenet. 32, 92–105. doi: 10.1080/01677063.2018.1445247
Gao, H., Bu, Y., Wu, Q., Wang, X., Chang, N., Lei, L., et al. (2015). Mecp2 regulates neural cell differentiation by suppressing the Id1 to Her2 axis in zebrafish. J. Cell Sci. 128, 2340–2350. doi: 10.1242/jcs.167874
Gauthier, J., Champagne, N., Lafreniere, R. G., Xiong, L., Spiegelman, D., Brustein, E., et al. (2010). De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. U S A 107, 7863–7868. doi: 10.1073/pnas.0906232107
Golzio, C., Willer, J., Talkowski, M. E., Oh, E. C., Taniguchi, Y., Jacquemont, S., et al. (2012). KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 485, 363–367. doi: 10.1038/nature11091
Granato, M., and Nusslein-Volhard, C. (1996). Fishing for genes controlling development. Curr. Opin. Genet. Dev. 6, 461–468. doi: 10.1016/s0959-437x(96)80068-2
Griffin, A., Hamling, K. R., Knupp, K., Hong, S., Lee, L. P., and Baraban, S. C. (2017). Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain 140, 669–683. doi: 10.1093/brain/aww342
Grone, B. P., Marchese, M., Hamling, K. R., Kumar, M. G., Krasniak, C. S., Sicca, F., et al. (2016). Epilepsy, behavioral abnormalities and physiological comorbidities in syntaxin-binding protein 1 (STXBP1) mutant zebrafish. PLoS One 11:e0151148. doi: 10.1371/journal.pone.0151148
Grone, B. P., Qu, T., and Baraban, S. C. (2017). Behavioral comorbidities and drug treatments in a zebrafish scn1lab model of dravet syndrome. eNeuro 4:ENEURO.0066-17.2017. doi: 10.1523/eneuro.0066-17.2017
Guo, S. (2009). Using zebrafish to assess the impact of drugs on neural development and function. Expert Opin. Drug Discov. 4, 715–726. doi: 10.1517/17460440902988464
Gusev, A., Mancuso, N., Won, H., Kousi, M., Finucane, H. K., Reshef, Y., et al. (2018). Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat. Genet. 50, 538–548. doi: 10.1038/s41588-018-0092-1
Heon, E., Kim, G., Qin, S., Garrison, J. E., Tavares, E., Vincent, A., et al. (2016). Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Hum. Mol. Genet. 25, 2283–2294. doi: 10.1093/hmg/ddw096
Hildebrand, M. S., Dahl, H. H., Damiano, J. A., Smith, R. J., Scheffer, I. E., and Berkovic, S. F. (2013). Recent advances in the molecular genetics of epilepsy. J. Med. Genet. 50, 271–279. doi: 10.1136/jmedgenet-2012-101448
Hoffman, E. J., Turner, K. J., Fernandez, J. M., Cifuentes, D., Ghosh, M., Ijaz, S., et al. (2016). Estrogens suppress a behavioral phenotype in zebrafish mutants of the autism risk gene, CNTNAP2. Neuron 89, 725–733. doi: 10.1016/j.neuron.2015.12.039
Hong, S., Lee, P., Baraban, S. C., and Lee, L. P. (2016). A novel long-term, multi-channel and non-invasive electrophysiology platform for zebrafish. Sci. Rep. 6:28248. doi: 10.1038/srep28248
Howe, K., Clark, M. D., Torroja, C. F., Torrance, J., Berthelot, C., Muffato, M., et al. (2013). The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503. doi: 10.1038/nature12111
Hwang, W. Y., Fu, Y., Reyon, D., Maeder, M. L., Tsai, S. Q., Sander, J. D., et al. (2013). Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227–229. doi: 10.1038/nbt.2501
Ijaz, S., and Hoffman, E. J. (2016). Zebrafish: a translational model system for studying neuropsychiatric disorders. J. Am. Acad. Child Adolesc. Psychiatry 55, 746–748. doi: 10.1016/j.jaac.2016.06.008
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Keller, P. J., and Ahrens, M. B. (2015). Visualizing whole-brain activity and development at the single-cell level using light-sheet microscopy. Neuron 85, 462–483. doi: 10.1016/j.neuron.2014.12.039
Kettleborough, R. N., Busch-Nentwich, E. M., Harvey, S. A., Dooley, C. M., de Bruijn, E., van Eeden, F., et al. (2013). A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature 496, 494–497. doi: 10.1038/nature11992
Kim, O. H., Cho, H. J., Han, E., Hong, T. I., Ariyasiri, K., Choi, J. H., et al. (2017). Zebrafish knockout of Down syndrome gene, DYRK1A, shows social impairments relevant to autism. Mol. Autism 8:50. doi: 10.1186/s13229-017-0168-2
Kim, S. H., Speirs, C. K., Solnica-Krezel, L., and Ess, K. C. (2011). Zebrafish model of tuberous sclerosis complex reveals cell-autonomous and non-cell-autonomous functions of mutant tuberin. Dis. Model. Mech. 4, 255–267. doi: 10.1242/dmm.005587
Kimura, Y., Hisano, Y., Kawahara, A., and Higashijima, S. (2014). Efficient generation of knock-in transgenic zebrafish carrying reporter/driver genes by CRISPR/Cas9-mediated genome engineering. Sci. Rep. 4:6545. doi: 10.1038/srep06545
Kok, F. O., Shin, M., Ni, C. W., Gupta, A., Grosse, A. S., van Impel, A., et al. (2015). Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 32, 97–108. doi: 10.1016/j.devcel.2014.11.018
Kokel, D., Bryan, J., Laggner, C., White, R., Cheung, C. Y., Mateus, R., et al. (2010). Rapid behavior-based identification of neuroactive small molecules in the zebrafish. Nat. Chem. Biol. 6, 231–237. doi: 10.1038/nchembio.307
Kozol, R. A., Abrams, A. J., James, D. M., Buglo, E., Yan, Q., and Dallman, J. E. (2016). Function over form: modeling groups of inherited neurological conditions in zebrafish. Front. Mol. Neurosci. 9:55. doi: 10.3389/fnmol.2016.00055
Kozol, R. A., Cukier, H. N., Zou, B., Mayo, V., De Rubeis, S., Cai, G., et al. (2015). Two knockdown models of the autism genes SYNGAP1 and SHANK3 in zebrafish produce similar behavioral phenotypes associated with embryonic disruptions of brain morphogenesis. Hum. Mol. Genet. 24, 4006–4023. doi: 10.1093/hmg/ddv138
Kumar, R. A., KaraMohamed, S., Sudi, J., Conrad, D. F., Brune, C., Badner, J. A., et al. (2008). Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 17, 628–638. doi: 10.1093/hmg/ddm376
Kury, S., Besnard, T., Ebstein, F., Khan, T. N., Gambin, T., Douglas, J., et al. (2017). De novo disruption of the proteasome regulatory subunit PSMD12 causes a syndromic neurodevelopmental disorder. Am. J. Hum. Genet. 100, 352–363. doi: 10.1016/j.ajhg.2017.01.003
Lange, M., Norton, W., Coolen, M., Chaminade, M., Merker, S., Proft, F., et al. (2012a). The ADHD-linked gene Lphn3.1 controls locomotor activity and impulsivity in zebrafish. Mol. Psychiatry 17:855. doi: 10.1038/mp.2012.119
Lange, M., Norton, W., Coolen, M., Chaminade, M., Merker, S., Proft, F., et al. (2012b). The ADHD-susceptibility gene lphn3.1 modulates dopaminergic neuron formation and locomotor activity during zebrafish development. Mol. Psychiatry 17, 946–954. doi: 10.1038/mp.2012.29
Lawson, N. D. (2016). Reverse genetics in zebrafish: mutants, morphants, and moving forward. Trends Cell Biol. 26, 77–79. doi: 10.1016/j.tcb.2015.11.005
Leong, W. Y., Lim, Z. H., Korzh, V., Pietri, T., and Goh, E. L. (2015). Methyl-CpG binding protein 2 (Mecp2) regulates sensory function through Sema5b and Robo2. Front. Cell. Neurosci. 9:481. doi: 10.3389/fncel.2015.00481
Lindstrand, A., Frangakis, S., Carvalho, C. M., Richardson, E. B., McFadden, K. A., Willer, J. R., et al. (2016). Copy-number variation contributes to the mutational load of bardet-biedl syndrome. Am. J. Hum. Genet. 99, 318–336. doi: 10.1016/j.ajhg.2015.04.023
Liu, C. X., Li, C. Y., Hu, C. C., Wang, Y., Lin, J., Jiang, Y. H., et al. (2018). CRISPR/Cas9-induced shank3b mutant zebrafish display autism-like behaviors. Mol. Autism 9:23. doi: 10.1186/s13229-018-0204-x
Lovett-Barron, M., Andalman, A. S., Allen, W. E., Vesuna, S., Kauvar, I., Burns, V. M., et al. (2017). Ancestral circuits for the coordinated modulation of brain state. Cell 171, 1411.e17–1423.e17. doi: 10.1016/j.cell.2017.10.021
Marin-Valencia, I., Novarino, G., Johansen, A., Rosti, B., Issa, M. Y., Musaev, D., et al. (2018). A homozygous founder mutation in TRAPPC6B associates with a neurodevelopmental disorder characterised by microcephaly, epilepsy and autistic features. J. Med. Genet. 55, 48–54. doi: 10.1136/jmedgenet-2017-104627
Marshall, C. R., Noor, A., Vincent, J. B., Lionel, A. C., Feuk, L., Skaug, J., et al. (2008). Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 82, 477–488. doi: 10.1016/j.ajhg.2007.12.009
McCammon, J. M., Blaker-Lee, A., Chen, X., and Sive, H. (2017). The 16p11.2 homologs fam57ba and doc2a generate certain brain and body phenotypes. Hum. Mol. Genet. 26, 3699–3712. doi: 10.1093/hmg/ddx255
McCammon, J. M., and Sive, H. (2015). Challenges in understanding psychiatric disorders and developing therapeutics: a role for zebrafish. Dis. Model. Mech. 8, 647–656. doi: 10.1242/dmm.019620
McCarthy, S. E., Makarov, V., Kirov, G., Addington, A. M., McClellan, J., Yoon, S., et al. (2009). Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 41, 1223–1227. doi: 10.1038/ng.474
Mei, X., Wu, S., Bassuk, A. G., and Slusarski, D. C. (2013). Mechanisms of prickle1a function in zebrafish epilepsy and retinal neurogenesis. Dis. Model. Mech. 6, 679–688. doi: 10.1242/dmm.010793
Meng, X., Noyes, M. B., Zhu, L. J., Lawson, N. D., and Wolfe, S. A. (2008). Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat. Biotechnol. 26, 695–701. doi: 10.1038/nbt1398
Meshalkina, D. A., M, N. K., E, V. K., Collier, A. D., Echevarria, D. J., Abreu, M. S., et al. (2018). Zebrafish models of autism spectrum disorder. Exp. Neurol. 299, 207–216. doi: 10.1016/j.expneurol.2017.02.004
Millar, J. K., Wilson-Annan, J. C., Anderson, S., Christie, S., Taylor, M. S., Semple, C. A., et al. (2000). Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 9, 1415–1423. doi: 10.1093/hmg/9.9.1415
Moens, C. B., Donn, T. M., Wolf-Saxon, E. R., and Ma, T. P. (2008). Reverse genetics in zebrafish by TILLING. Brief. Funct. Genomics Proteomic. 7, 454–459. doi: 10.1093/bfgp/eln046
Moreno-Mateos, M. A., Vejnar, C. E., Beaudoin, J. D., Fernandez, J. P., Mis, E. K., Khokha, M. K., et al. (2015). CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 12, 982–988. doi: 10.1038/nmeth.3543
Myers, C. T., and Mefford, H. C. (2015). Advancing epilepsy genetics in the genomic era. Genome Med. 7:91. doi: 10.1186/s13073-015-0214-7
Nasevicius, A., and Ekker, S. C. (2000). Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 26, 216–220. doi: 10.1038/79951
Naumann, E. A., Fitzgerald, J. E., Dunn, T. W., Rihel, J., Sompolinsky, H., and Engert, F. (2016). From whole-brain data to functional circuit models: the zebrafish optomotor response. Cell 167, 947.e20–960.e20. doi: 10.1016/j.cell.2016.10.019
Nozawa, K., Lin, Y., Kubodera, R., Shimizu, Y., Tanaka, H., and Ohshima, T. (2017). Zebrafish Mecp2 is required for proper axonal elongation of motor neurons and synapse formation. Dev. Neurobiol. 77, 1101–1113. doi: 10.1002/dneu.22498
Pan, L., Shah, A. N., Phelps, I. G., Doherty, D., Johnson, E. A., and Moens, C. B. (2015). Rapid identification and recovery of ENU-induced mutations with next-generation sequencing and Paired-End Low-Error analysis. BMC Genomics 16:83. doi: 10.1186/s12864-015-1263-4
Patten, S. A., Jacobs-McDaniels, N. L., Zaouter, C., Drapeau, P., Albertson, R. C., and Moldovan, F. (2012). Role of Chd7 in zebrafish: a model for CHARGE syndrome. PLoS One 7:e31650. doi: 10.1371/journal.pone.0031650
Penagarikano, O., Abrahams, B. S., Herman, E. I., Winden, K. D., Gdalyahu, A., Dong, H., et al. (2011). Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246. doi: 10.1016/j.cell.2011.08.040
Pietri, T., Roman, A. C., Guyon, N., Romano, S. A., Washbourne, P., Moens, C. B., et al. (2013). The first mecp2-null zebrafish model shows altered motor behaviors. Front. Neural Circuits 7:118. doi: 10.3389/fncir.2013.00118
Portugues, R., Feierstein, C. E., Engert, F., and Orger, M. B. (2014). Whole-brain activity maps reveal stereotyped, distributed networks for visuomotor behavior. Neuron 81, 1328–1343. doi: 10.1016/j.neuron.2014.01.019
Prober, D. A., Rihel, J., Onah, A. A., Sung, R. J., and Schier, A. F. (2006). Hypocretin/orexin overexpression induces an insomnia-like phenotype in zebrafish. J. Neurosci. 26, 13400–13410. doi: 10.1523/JNEUROSCI.4332-06.2006
Purcell, S. M., Moran, J. L., Fromer, M., Ruderfer, D., Solovieff, N., Roussos, P., et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. doi: 10.1038/nature12975
Randlett, O., Wee, C. L., Naumann, E. A., Nnaemeka, O., Schoppik, D., Fitzgerald, J. E., et al. (2015). Whole-brain activity mapping onto a zebrafish brain atlas. Nat. Methods 12, 1039–1046. doi: 10.1038/nmeth.3581
Reijnders, M. R. F., Kousi, M., van Woerden, G. M., Klein, M., Bralten, J., Mancini, G. M. S., et al. (2017). Variation in a range of mTOR-related genes associates with intracranial volume and intellectual disability. Nat. Commun. 8:1052. doi: 10.1038/s41467-017-00933-6
Renier, C., Faraco, J. H., Bourgin, P., Motley, T., Bonaventure, P., Rosa, F., et al. (2007). Genomic and functional conservation of sedative-hypnotic targets in the zebrafish. Pharmacogenet Genomics 17, 237–253. doi: 10.1097/fpc.0b013e3280119d62
Rihel, J., Prober, D. A., Arvanites, A., Lam, K., Zimmerman, S., Jang, S., et al. (2010). Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science 327, 348–351. doi: 10.1126/science.1183090
Robu, M. E., Larson, J. D., Nasevicius, A., Beiraghi, S., Brenner, C., Farber, S. A., et al. (2007). p53 activation by knockdown technologies. PLoS Genet. 3:e78. doi: 10.1371/journal.pgen.0030078
Rossi, A., Kontarakis, Z., Gerri, C., Nolte, H., Hölper, S., Krüger, M., et al. (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230–233. doi: 10.1038/nature14580
Rubenstein, J. L., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267. doi: 10.1034/j.1601-183x.2003.00037.x
Sander, J. D., Cade, L., Khayter, C., Reyon, D., Peterson, R. T., Joung, J. K., et al. (2011). Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat. Biotechnol. 29, 697–698. doi: 10.1038/nbt.1934
Sanders, S. J., He, X., Willsey, A. J., Ercan-Sencicek, A. G., Samocha, K. E., Cicek, A. E., et al. (2015). Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233. doi: 10.1016/j.neuron.2015.09.016
Schizophrenia Working Group of the Psychiatric Genomics Consortium. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Schoonheim, P. J., Arrenberg, A. B., Del Bene, F., and Baier, H. (2010). Optogenetic localization and genetic perturbation of saccade-generating neurons in zebrafish. J. Neurosci. 30, 7111–7120. doi: 10.1523/JNEUROSCI.5193-09.2010
Schubert, J., Siekierska, A., Langlois, M., May, P., Huneau, C., Becker, F., et al. (2014). Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat. Genet. 46, 1327–1332. doi: 10.1038/ng.3130
Shams, S., Rihel, J., Ortiz, J. G., and Gerlai, R. (2018). The zebrafish as a promising tool for modeling human brain disorders: a review based upon an IBNS symposium. Neurosci. Biobehav. Rev. 85, 176–190. doi: 10.1016/j.neubiorev.2017.09.002
Singh, K. K., De Rienzo, G., Drane, L., Mao, Y., Flood, Z., Madison, J., et al. (2011). Common DISC1 polymorphisms disrupt Wnt/GSK3β signaling and brain development. Neuron 72, 545–558. doi: 10.1016/j.neuron.2011.09.030
Sourbron, J., Schneider, H., Kecskes, A., Liu, Y., Buening, E. M., Lagae, L., et al. (2016). Serotonergic modulation as effective treatment for dravet syndrome in a zebrafish mutant model. ACS Chem. Neurosci. 7, 588–598. doi: 10.1021/acschemneuro.5b00342
Stainier, D. Y. R., Raz, E., Lawson, N. D., Ekker, S. C., Burdine, R. D., Eisen, J. S., et al. (2017). Guidelines for morpholino use in zebrafish. PLoS Genet. 13:e1007000. doi: 10.1371/journal.pgen.1007000
State, M. W., and Šestan, N. (2012). Neuroscience. The emerging biology of autism spectrum disorders. Science 337, 1301–1303. doi: 10.1126/science.1224989
Stoner, R., Chow, M. L., Boyle, M. P., Sunkin, S. M., Mouton, P. R., Roy, S., et al. (2014). Patches of disorganization in the neocortex of children with autism. N. Engl. J. Med. 370, 1209–1219. doi: 10.1056/NEJMoa1307491
Sugathan, A., Biagioli, M., Golzio, C., Erdin, S., Blumenthal, I., Manavalan, P., et al. (2014). CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. U S A 111, E4468–E4477. doi: 10.1073/pnas.1405266111
Suls, A., Jaehn, J. A., Kecskes, A., Weber, Y., Weckhuysen, S., Craiu, D. C., et al. (2013). De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am. J. Hum. Genet. 93, 967–975. doi: 10.1016/j.ajhg.2013.09.017
Thompson, A. W., Vanwalleghem, G. C., Heap, L. A., and Scott, E. K. (2016). Functional profiles of visual-, auditory-, and water flow-responsive neurons in the zebrafish tectum. Curr. Biol. 26, 743–754. doi: 10.1016/j.cub.2016.01.041
Tucker, B., Richards, R. I., and Lardelli, M. (2006). Contribution of mGluR and Fmr1 functional pathways to neurite morphogenesis, craniofacial development and fragile X syndrome. Hum. Mol. Genet. 15, 3446–3458. doi: 10.1093/hmg/ddl422
van der Vaart, M., Svoboda, O., Weijts, B. G., Espín-Palazón, R., Sapp, V., Pietri, T., et al. (2017). Mecp2 regulates tnfa during zebrafish embryonic development and acute inflammation. Dis. Model. Mech. 10, 1439–1451. doi: 10.1242/dmm.026922
Varshney, G. K., Lu, J., Gildea, D. E., Huang, H., Pei, W., Yang, Z., et al. (2013). A large-scale zebrafish gene knockout resource for the genome-wide study of gene function. Genome Res. 23, 727–735. doi: 10.1101/gr.151464.112
Vladimirov, N., Mu, Y., Kawashima, T., Bennett, D. V., Yang, C. T., Looger, L. L., et al. (2014). Light-sheet functional imaging in fictively behaving zebrafish. Nature methods 11, 883–884. doi: 10.1038/nmeth.3040
Walsh, T., McClellan, J. M., McCarthy, S. E., Addington, A. M., Pierce, S. B., Cooper, G. M., et al. (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543. doi: 10.1126/science.1155174
Weiss, L. A., Shen, Y., Korn, J. M., Arking, D. E., Miller, D. T., Fossdal, R., et al. (2008). Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 358, 667–675. doi: 10.1056/NEJMoa075974
Willsey, A. J., Sanders, S. J., Li, M., Dong, S., Tebbenkamp, A. T., Muhle, R. A., et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. doi: 10.1016/j.cell.2013.10.020
Zaghloul, N. A., Liu, Y., Gerdes, J. M., Gascue, C., Oh, E. C., Leitch, C. C., et al. (2010). Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. U S A 107, 10602–10607. doi: 10.1073/pnas.1000219107
Zhang, Y., Kecskes, A., Copmans, D., Langlois, M., Crawford, A. D., Ceulemans, B., et al. (2015). Pharmacological characterization of an antisense knockdown zebrafish model of Dravet syndrome: inhibition of epileptic seizures by the serotonin agonist fenfluramine. PLoS One 10:e0125898. doi: 10.1371/journal.pone.0125898
Keywords: zebrafish, neurodevelopmental disorders, autism spectrum disorders, epilepsy, schizophrenia, model system, genetics, neural circuits
Citation: Sakai C, Ijaz S and Hoffman EJ (2018) Zebrafish Models of Neurodevelopmental Disorders: Past, Present, and Future. Front. Mol. Neurosci. 11:294. doi: 10.3389/fnmol.2018.00294
Received: 15 May 2018; Accepted: 03 August 2018;
Published: 29 August 2018.
Edited by:
Edna Grünblatt, Universität Zürich, SwitzerlandReviewed by:
Matthias Carl, Università di Trento, ItalyMerlin Lange, RIKEN Brain Science Institute (BSI), Japan
Ruxandra Bachmann-Gagescu, Universität Zürich, Switzerland
Copyright © 2018 Sakai, Ijaz and Hoffman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ellen J. Hoffman, ZWxsZW4uaG9mZm1hbkB5YWxlLmVkdQ==