 Allison M. Barry
Allison M. Barry Julia R. Sondermann
Julia R. Sondermann Jan-Hendrik Sondermann
Jan-Hendrik Sondermann David Gomez-Varela
David Gomez-Varela Manuela Schmidt
Manuela Schmidt
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 14 August 2018
Sec. Pain Mechanisms and Modulators
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00259
To obtain a thorough understanding of chronic pain, large-scale molecular mapping of the pain axis at the protein level is necessary, but has not yet been achieved. We applied quantitative proteome profiling to build a comprehensive protein compendium of three regions of the pain neuraxis in mice: the sciatic nerve (SN), the dorsal root ganglia (DRG), and the spinal cord (SC). Furthermore, extensive bioinformatics analysis enabled us to reveal unique protein subsets which are specifically enriched in the peripheral nervous system (PNS) and SC. The immense value of these datasets for the scientific community is highlighted by validation experiments, where we monitored protein network dynamics during neuropathic pain. Here, we resolved profound region-specific differences and distinct changes of PNS-enriched proteins under pathological conditions. Overall, we provide a unique and validated systems biology proteome resource (summarized in our online database painproteome.em.mpg.de), which facilitates mechanistic insights into somatosensory biology and chronic pain—a prerequisite for the identification of novel therapeutic targets.
Chronic pain is a complex maladaptive disease. It has a high incidence and is a major cause of long term disability (Breivik, 2013). Despite prominent scientific advances, our knowledge of the mechanistic underpinnings of chronic pain syndromes remains unsatisfactory (Costigan et al., 2009; Price and Gold, 2017). Effective treatment faces major challenges, largely arising from the fact that therapeutic interventions target widely expressed molecules in multiple tissues, including the central nervous system (Borsook et al., 2014; Price and Gold, 2017). Their modulation is accompanied by severe side effects (Patapoutian et al., 2009; Price and Gold, 2017). Central side effects can be attenuated by selectively targeting the PNS (i.e., afferent fibers, DRG), e.g., by local injections (intranerval, intraganglionic, and epidural). This facilitates effective and safer treatment options (Puljak et al., 2009; Liem et al., 2013; Krames, 2014; Berta et al., 2017b). Interestingly, several studies have demonstrated a crucial role of primary afferent input for the maintenance of chronic pain, e.g., neuropathic pain (Sapunar et al., 2012; Liem et al., 2013; Haroutounian et al., 2014; Krames, 2014), and phantom limb pain (Vaso et al., 2014). While the existence of a peripheral “pain” generator is becoming increasingly accepted, associated changes in molecules and protein networks remain to be fully elucidated (Costigan, 2012; Borsook et al., 2014).
During the past decade, enormous progress has been made toward the identification of molecules implicated in chronic pain. These studies have yielded important insights into genetic variants (Diatchenko et al., 2007, 2013; van Hecke et al., 2015; Zorina-Lichtenwalter et al., 2016; Parisien et al., 2017; Peng et al., 2017) and transcriptome alterations (Grace et al., 2012; Antunes-Martins et al., 2013; Simonetti et al., 2013; Jeong et al., 2016) relevant for chronic pain. Yet, the interpretation of these results is often not straightforward. In particular, genome/transcriptome-based techniques suffer from a limited correspondence between genomic/transcriptomic and proteomic content (Schwanhausser et al., 2011; Sharma et al., 2015; Liu et al., 2016).
As proteins are the functional building blocks of cells, they strongly influence the status-quo of a cell, tissue or even organism. Several proteome studies have thus been conducted on chronic pain (Huang et al., 2008; Olausson et al., 2012, 2015; Sui et al., 2014; Vacca et al., 2014). We are still lacking, however, an integrated systems-level view of protein network organization and dynamics (Alberts, 1998; Rouwette et al., 2015) in the somatosensory system. According to emerging network medicine efforts, the identification of cellular networks and in particular, distinct hubs of disease modules within these networks (Chapman et al., 2008; Barabási et al., 2011; Antunes-Martins et al., 2013; Borsook and Kalso, 2013; Chen et al., 2015), may hold great potential for diagnosis, patient stratification, and design of selective therapeutic interventions for chronic pain conditions (Gereau et al., 2014; Baron et al., 2017; Price and Gold, 2017).
We have previously demonstrated the potential of an emerging mass spectrometry-based proteomic technique (so-called data-independent acquisition mass spectrometry, DIA-MS) to reproducibly profile proteome changes in DRG of two mouse models of chronic pain (Rouwette et al., 2016). Here we considerably extended our approach to build a comprehensive protein compendium (>5000 proteins in total) of three regions at the beginning of the pain neuraxis: the sciatic nerve (SN), the DRG, and the spinal cord (SC). We compared this data to tissue proteomes of the central nervous system which enabled us to define “region-enriched” proteomes. The immense value of these defined region-enriched proteomes becomes apparent by (i) the inclusion of many proteins known to be involved in pain in mice and humans, (ii) the identification of uncharacterized proteins in a tissue-dependent manner, and (iii) the results of our validation experiments resolving profound differences among analyzed tissues and distinct modulations of PNS-enriched proteins during neuropathic pain. Taken together, our findings provide the scientific community with a unique protein-centric systems biology framework for studying the PNS and SC in normal and pathological states. All presented data are summarized in a freely accessible online database at painproteome.em.mpg.de.
All experiments were performed by observing standard institutional biosecurity and institutional safety procedures. All workplaces and involved personnel were approved for biosafety level 1 (BSL-1) procedures.
Adult C57Bl/6J male mice (6–8 weeks old at the time of surgery) were bred in-house and kept in a temperature- and humidity-controlled environment under a 12 h light/dark cycle with food and water provided ad libitum. All animal experiments were approved and carried out in compliance with institutional guidelines and guidelines of the Landesamt für Verbraucherschutz und Lebensmittelsicherheit of Lower Saxony, Germany (3392-42502-04-13/1077).
The spared nerve injury (SNI) paradigm was induced according to standard protocols as described previously (Decosterd and Woolf, 2000; Minett et al., 2013; Rouwette et al., 2016). Mice were anesthetized with isoflurane (4% induction, 1.8% maintenance) and the SN and its branches, the common peroneal, tibial, and sural nerves, were exposed. Common peroneal and tibial nerves were ligated with a 6.0 silk suture (Braun) and transected distal to the ligation, removing 2–3 mm of each nerve. Care was taken to avoid damaging the sural nerve. Sham surgery was performed in a similar manner without ligating and transecting the nerves. The surgical procedure lasted approximately 15 min. Analgesia during surgery was achieved by Buprenorphine (0.07 mg/kg body weight; Guarnieri et al., 2012). Postsurgical analgesia was achieved by subcutaneous injection of Carprofen (5 mg/kg body weight) for up to 3 days, as necessary. Mechanical sensitivity was assessed 2 days before and 26 days after surgery with a dynamic plantar aesthesiometer (Ugo Basile), as described (Rouwette et al., 2016). Behavioral tests were performed in the light phase in a blinded manner. Mice with neuropathic pain, however, could be easily identified upon the first measurements due to the pronounced mechanical hypersensitivity. All 12 mice which underwent SNI surgery exhibited evident mechanical hypersensitivity (manifested as reduced withdrawal latencies) but were otherwise indistinguishable from Sham surgery mice. None of the 12 mice which underwent Sham surgery exhibited mechanical hypersensitivity. All mice were euthanized with CO2 for tissue collection 28 days after surgery.
The following tissues were isolated after Sham surgery or SNI: ipsilateral sciatic nerve, ipsilateral DRG (L3-L5), ipsilateral SC (L3-L5; a reliable and consistent separation of dorsal and ventral horn was technically not feasible), amygdala, anterior cingulate cortex, prefrontal cortex, and cerebellum.
Tissues were isolated immediately after CO2 euthanization, snap-frozen in liquid nitrogen, and stored at −80°C. Tissue from 4 mice were pooled to obtain 3 replicates per tissue and per condition. Protein isolation was performed as described previously (Bruderer et al., 2017b). In brief, the frozen tissue was homogenized in 4% SDS lysis buffer (4% SDS in 100 mM Tris, 10 mM DTT, 5% glycerol, complete protease inhibitor cocktail (Roche), pH 7.5). Following, the homogenate was incubated at 70°C for 10 min. To remove cell debris, the homogenate was centrifuged at 10,000 × g for 5 min at room temperature. The supernatant corresponds to the whole cell lysates. Subsequently, proteins were precipitated by the addition of 5 × volume pre-chilled acetone (Roth) and incubated for 2 h at −20°C. The protein precipitate was centrifuged at 14,000 × g for 30 min, the pellet washed with ice cold 80% ethanol (AppliedChem), and centrifuged again at 14,000 × g for 30 min. The proteins were air-dried, re-suspended in 2% SDS lysis buffer, and ultimately analyzed by DIA-MS.
To generate the spectral library, i.e., a compendium of all detectable peptides and their physicochemical information necessary to uniquely identify peptides in subsequent DIA-MS experiments, membrane and cytosolic fractions of the whole cell lysate were prepared for one replicate per condition of each tissue. To this end, the whole cell lysate was subjected to ultracentrifugation at 100,000 × g for 1 h at 18°C (TLA 100.3 rotor, Beckman Coulter). The supernatant (cytosolic fraction) and the dissolved pellet (membrane fraction; in 2% SDS lysis buffer) were subjected to protein precipitation as described above.
Sample preparation, pan-mouse spectral library generation, and DIA-MS were performed by Biognosys (Zurich, Switzerland) as previously reported (Bruderer et al., 2015, 2017b; Rouwette et al., 2016). Data were analyzed with Spectronaut Pulsar (Biognosys) with precursor and protein FDR set at 0.01. Further analyses were performed with R (https://www.r-project.org/). Upon publication, all DIA-MS data, and the pan-mouse library will be publicly available in the Peptide Atlas (www.peptideatlas.org) and in our database painproteome.em.mpg.de.
To examine reproducibility across samples, regression analyses were performed. Protein intensities were calculated per protein per replicate, as a mean intensity across peptides. Pearson's correlation coefficients were subsequently calculated for samples within each tissue, and kernel density estimates were used to visualize sample distribution. Initial analyses were performed with R, and subsequent plotting done with Python (Python Software Foundation, https://www.python.org/). Unsupervised hierarchical clustering was performed in Perseus v1.5.6.0 (Tyanova et al., 2016). The clustering was done with Euclidean distances and average linkage on mean protein intensities (per replicate), which had been log2 transformed and z-score normalized. For visualization purposes (i.e., to help find proteins showing similar expression), row clusters with an arbitrary chosen distance threshold of 3.4 were presented in different colors.
For each control (Sham) tissue, peptide intensities were combined across all three replicates for a given protein identifier. Mean intensity, standard deviation, and the number of peptides were calculated. A positive protein detection refers to at least one peptide in all three Sham replicates. We opted to include all proteins meeting these criteria (i.e., including proteins only identified by a single peptide across all three Sham replicates) to be able (i) to provide the most comprehensive analysis of tissue proteomes and (ii) to perform the most stringent comparisons across tissues possible (see below for the definition of tissue-enriched proteomes). To prevent redundancy in our protein list, as well as facilitate literature comparisons (described below), only protein identifiers corresponding to reviewed Uniprot entries (http://www.uniprot.org) were used for subsequent analysis. Additionally, detected keratin contamination was removed from the analysis. This analysis was repeated using a spectral library designed in silico from the mouse brain proteome data published by Sharma and colleagues (Sharma et al., 2015).
To calculate expression changes in the SNI model, log2 intensity ratios were first calculated between SNI and Sham tissues at a peptide level. For each peptide, three ratios were calculated: SNI Replicate 1/Sham Replicate 1, SNI Replicate 2/Sham Replicate 2, SNI Replicate 3/Sham Replicate 3. Hence, peptides were only considered if they are detected across all three Sham replicates and all three SNI replicates. Mean log2 ratios were subsequently calculated for each protein ID, and one-sample t-tests were performed. After filtering for reviewed Uniprot IDs, p-values were adjusted using the Benjamini-Hochberg (BH) procedure (R: p.adjust, method = “BH”). Regulated proteins were defined as having a BH-adjusted p-value q < 0.05. If the regulation of a protein could only be established based on a single peptide across all six samples, it was considered a “low confidence regulated hit” and is highlighted as such in Supplementary Table 8 (according to latest considerations regarding the quantitation based on single-peptide hits, please see tutorial at http://www.mcponline.org/site/misc/ParisReport_Final.xhtml). In keeping with the Sham data, only reviewed Uniprot entries were considered and keratin contaminations were removed.
In the interest of establishing a PNS-enriched proteome, DRG and SN data were compared to four brain tissues (ACC, AMY, PFC, and CER). A protein was considered to be PNS-enriched if either criteria was met: (A) protein was detected in DRG and/or SN but none of the brain tissues, or (B) 10-fold enrichment (one order of magnitude) in at least one PNS tissue compared to brain regions. To calculate the latter, protein abundance was compared between each PNS-tissue and the maximum abundance detected in a brain region. A 1-tailed Welch's t-test was used to determine significance (p < 0.05). Only proteins which were more than 10-fold enriched with a p < 0.05 were considered PNS-enriched for further analysis. To strengthen our confidence in the PNS-enriched proteome, we then compared this initial PNS-enriched list to data analyzed using the in silico library designed from the mouse brain proteome published by Sharma and colleagues (Sharma et al., 2015). Any protein which was detected in a brain tissue of this secondary analysis was removed from the PNS-enriched list, unless it met our 10-fold enrichment criteria. Again, proteins identified by a single peptide across replicates were included to enable the most stringent tissue comparison possible.
An SC-enriched proteome was produced by mirroring the aforementioned criteria: non-detected in any brain region or 10-fold enriched (p < 0.05), with subsequent comparison to data analyzed using the in silico library designed from the mouse brain proteome data published by Sharma and colleagues (Sharma et al., 2015).
To assess the quality of our datasets, as well as the translational applications, our data were compared to relevant previously published genomic, transcriptomic, and proteomic datasets. For each comparison, identifiers were uploaded to Uniprot and converted to reviewed mouse Uniprot identifiers. This conversion provided a framework for genomic and transcriptomic, as well as human data to be matched to our mouse-generated proteome. Of note, the conversion to reviewed Uniprot identifiers from previously published data resulted in a loss of some identifiers, occasionally reducing the total number of proteins used for the comparison. Even so, this standardization allowed for a comparison not otherwise possible. Percentage overlap was calculated between these lists and our datasets (e.g., all proteins, PNS-enriched, SC-enriched, proteins regulated in SNI/Sham, etc.). For comparison to the Human Protein Atlas (Uhlen et al., 2015; Thul et al., 2017), data were accessed in July 2017, corresponding to Protein Atlas version 16.1. Both “approved” and “supported” proteins at expression scores of “low,” “medium,” and “high” were included. Full details about the annotations can be found here: https://www.proteinatlas.org/about/assays+annotation. In essence, reliably detected proteins were examined across all tissues. Proteins with uncertain reliability scores and those “not detected” were excluded for a given tissue. All tissues were compiled for the analysis, as no data from PNS tissue was included in the Atlas. Identifiers were then uploaded to Uniprot and converted, as described above.
Previously, our group has reported changes in the DRG membrane proteome upon SNI and inflammatory pain induced by CFA injection (Rouwette et al., 2016). These data were reanalyzed with our updated generated pan-mouse library. Proteins regulated in either pain model were subsequently compared to determine commonly-regulated proteins.
Protein lists (per tissue: Sham and SNI/Sham, PNS-enriched, SC-enriched) were uploaded to IPA® (Qiagen) for further analysis. For SNI/Sham data, only significantly regulated proteins q < 0.05 (BH-adjusted p-value) were considered.
Protein lists (per tissue: Sham and SNI/Sham, PNS-enriched, SC-enriched) were uploaded to the web interfaces DAVID (david.ncifcrf.gov; Huang da et al., 2009) and STRING (string-db.org; Franceschini et al., 2013). We reported only significant enrichments (FDR < 0.05) in cellular component (CC) or biological process (BP).
The goal of our study was twofold. On one hand, we aimed to comprehensively define the adult mouse proteome of the SN, DRG, and SC. On the other hand, we envisioned using this information to enable the trustful monitoring of proteome alterations during a chronic pain condition. To this end, we chose the widely employed spared-nerve-injury (SNI)-model of neuropathic pain (Decosterd and Woolf, 2000) and performed a comparison to Sham-operated control mice (please see Methods for details on mice).
Our workflow is outlined in Figure 1, and ranges from the induction of neuropathic pain, via DIA-MS to an in-depth data and pathway analysis.
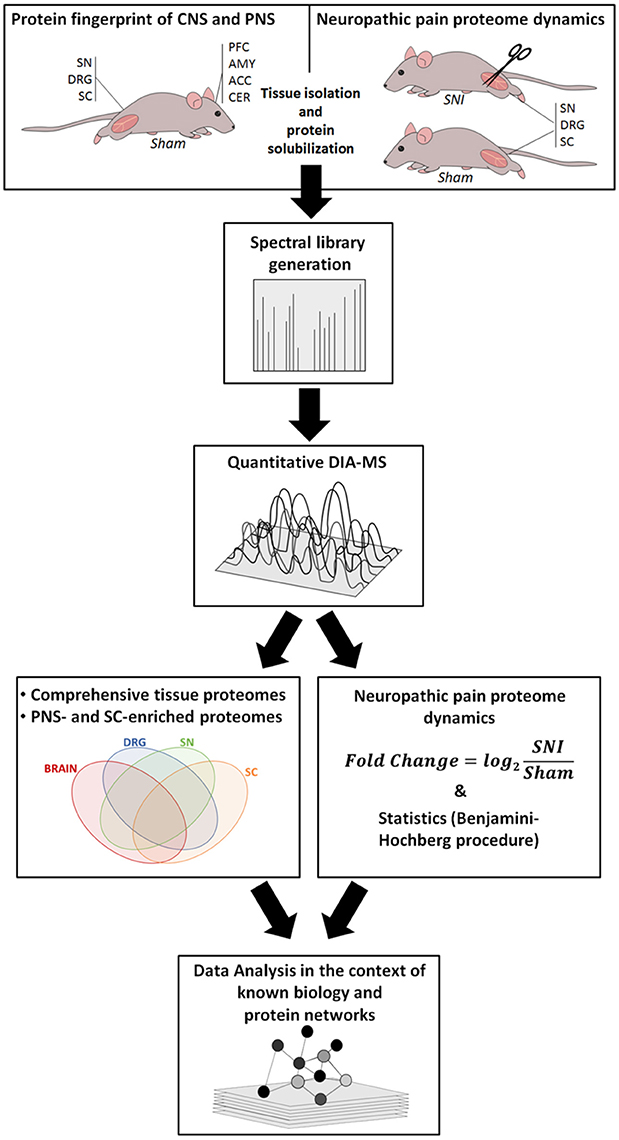
Figure 1. Overview of our integrated workflow: from Sham-operated mice and the SNI model via mouse behavior, protein solubilization, quantitative DIA-MS and data analysis to protein network dynamics. ACC, anterior cingulate cortex; AMY, amygdala; CER, cerebellum; DRG, dorsal root ganglia; PFC, prefrontal cortex; SC, spinal cord; SN, sciatic nerve.
Initially, we focused on Sham-operated mice as controls to define region-resolved proteomes (Figure 1 left). We prepared lysates of the SN, DRG, and SC. For the sake of comparative analysis between the PNS (SN and DRG) and CNS, we additionally prepared lysates of four major brain regions: the prefrontal cortex (PFC), the anterior cingulate cortex (ACC), the amygdala (AMY), and the cerebellum (CER). The first three CNS regions were chosen based on their implication in chronic pain states. The CER was chosen based on a recent study showing that the DRG transcriptome resembled the cerebellum more than any other brain region (Parisien et al., 2017).
Using mass spectrometry, we first generated a spectral library according to previously established procedures (Rouwette et al., 2016; Figure 1). Next, we assembled a pan-mouse peptide spectral library (pan-mouse library) by combining individual tissue libraries obtained in this study with our previously published libraries (Rouwette et al., 2016; Bruderer et al., 2017a,b). Taken together, this pan-mouse library contains peptides corresponding to 6205 proteins (complete list: Supplementary Table 1). These include 3008 proteins predicted to be localized to the membrane according to gene ontology (GO) analysis for cellular compartment, GO_CC (BH-corrected p-value: 5,2 × 10−173) using the web interface DAVID (Huang da et al., 2009). Among these, 254 are annotated to be involved in ion transport (GO analysis for biological process, GO_BP; BH-corrected p-value: 1,8 × 10−7) including ion channels and receptors with prominent functions in nociceptive processing, e.g., voltage-dependent calcium (Cav) and sodium (Nav) channel subunits, members of the transient receptor potential (TRP) family, and glutamate receptors (complete list: Supplementary Table 1). We have recently shown that mouse spectral libraries can be applied for protein profiling across species, i.e., also in humans (Bruderer et al., 2017b). Based on its comprehensive protein coverage, our pan-mouse library can be used for the standardized interrogation of diverse mouse and human datasets in pain research. It contains information of 31% of the genes represented in the Human Pain Genes Database (HPGDB, https://humanpaingenetics.org/hpgdb/; Meloto et al., 2017) as well as 45% of genes identified in pain-related transgenic knockout studies summarized in the Pain Genes Database (PGD, http://www.jbldesign.com/jmogil/enter.html; Lacroix-Fralish et al., 2007; complete list: Supplementary Table 1). Therefore, our pan-mouse library represents a valuable resource to profile these pain-related proteins and others in human and rodent tissues.
Next, we performed DIA-MS experiments according to published procedures (Figure 1 workflow; Bruderer et al., 2015, 2017a,b). This enabled us to quantify >5,000 proteins across DIA-MS runs. Applying very stringent criteria for reproducibility of protein identification across all replicates (please see Methods for details) resulted in highly reproducible profiling of 3141-4103 proteins depending on the tissue (Figures 2A–C; Supplementary Figure 2; complete list: Supplementary Table 2). While the vast majority of proteins were expressed across regions, differentially expressed proteins could be determined when comparing the brain proteome with SC and PNS proteomes (Figure 2C; complete list: Supplementary Table 2). Among the latter, we observed a significant overlap of SC and DRG proteomes (Figure 2C), which is anatomically supported given that spinal roots (central branches of DRG axons) innervate the SC dorsal horn. Hierarchical clustering across individual replicates further supports the similarity of the DRG and SC proteomes (Figure 2A). Together, this data prompted us to analyze the SC separately from other CNS (i.e., brain) areas.
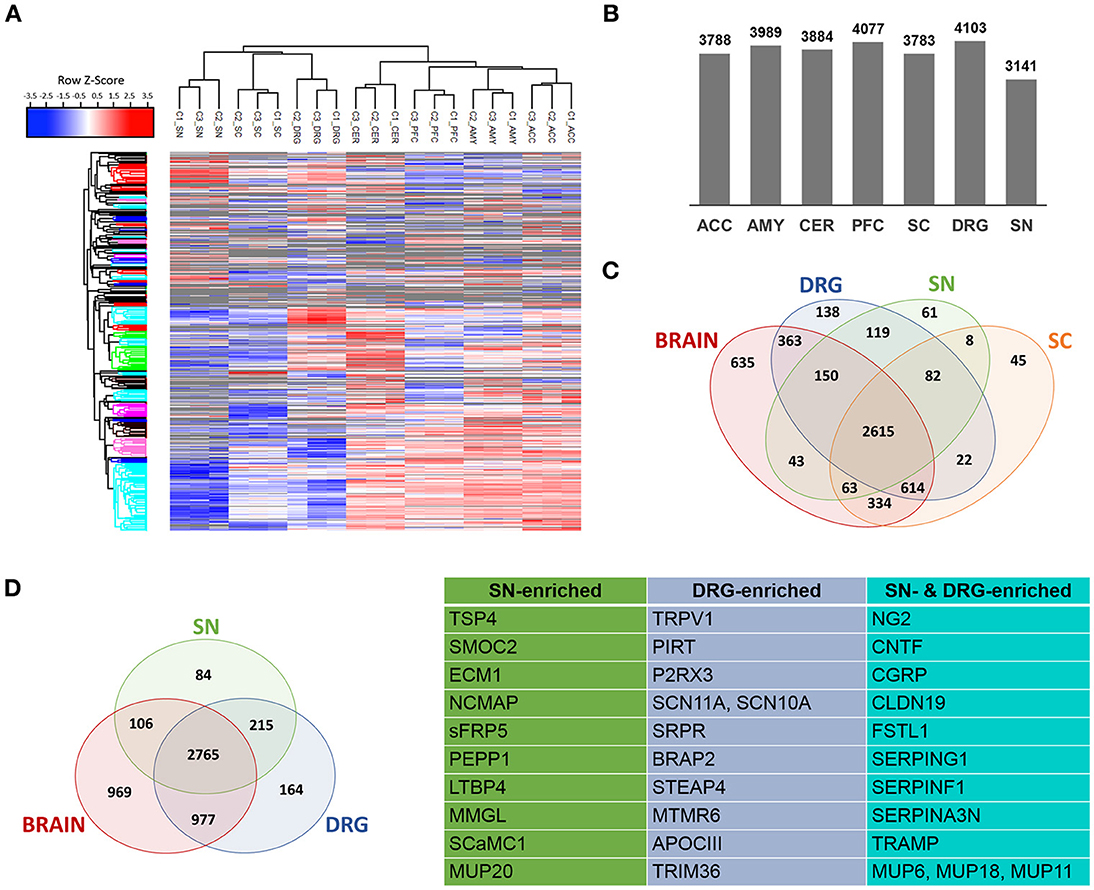
Figure 2. Determination of PNS- and SC-enriched proteomes. (A) Unsupervised clustering of z-scored normalized protein intensities from all Sham replicates (C1-C3; 4 mice each) across the 7 analyzed regions of the PNS and CNS. High experimental reproducibility becomes apparent. Furthermore, the clustering shows expected similarities between related tissues, e.g., PNS tissues and spinal cord (SC) are more closely clustered together than the 4 brain regions. The different colors of the y-axis dendrogram visualize exemplary clusters of proteins. (B) Number of proteins consistently detected across tissues. (C) Venn diagram illustrates differentially expressed and shared proteins across SN, DRG, SC, and brain regions. (D) A quantitative comparative analysis reveals that 299 and 379 proteins are enriched (> 10-fold) in SN and DRG proteomes (i.e., PNS-enriched) compared to the brain proteome. Further, tissue-specific expression of 84 (SN) and 164 (DRG) proteins can be observed. The table lists examples of PNS-enriched proteins according to their expression in SN, DRG, or both.
We then performed pathway analysis with IPA® to determine enriched canonical and disease pathways in each PNS, SC, and brain region analyzed. IPA® results recalled known global and region-specific features, demonstrating the quality and biological significance of identified proteomes (complete list: Supplementary Table 3). Among the top pathways highly enriched in the PNS compared to brain areas were acute phase response signaling, LXR/RXR (liver x receptors/retinoid x receptors) activation, the complement system, and the superpathway of cholesterol biosynthesis. Proteins belonging to these pathways are indicated in Supplementary Table 3. Conversely, glutamate, GABA, and dopamine receptor signaling, as well as synaptic long term potentiation/depression were found to be enriched in analyzed brain tissues as expected. Moreover, disease/functional pathways such as cognition, learning, seizures, plasticity of synapse, locomotion, and schizophrenia were highly represented in brain tissues compared to tissues of the PNS (complete list: Supplementary Table 3). In contrast, PNS and SC proteomes showed enrichment of proteins involved in demyelinating neuropathy, fatty acid metabolism, myopathy, and chronic skin disorder. Within the PNS we detected differences attributable to the diverse biology of SN and DRG, such as the overrepresentation of proteins associated with (de)myelination, psoriasis, and chronic inflammatory disorder, as well as extracellular matrix and adhesion in the SN vs. DRG.
To objectively define PNS- and SC-enriched proteomes, we performed a quantitative and subtractive cross-comparison with the identified brain proteome, applying stringent selection criteria (please see Methods for details). First, protein enrichment in the PNS or SC was defined by > 10-fold enrichment of protein abundance. As a second filter, we used data published by Sharma and colleagues (Sharma et al., 2015), which represents the most extensive mouse brain proteome to date. We then again excluded every protein from our PNS or SC-enriched protein compendium that was not >10-fold enriched over any brain tissue using the in silico library designed from data published by Sharma and colleagues.
This procedure yielded the definition of 463 PNS-enriched proteins, 164 of which were preferably expressed in DRG and 84 in SN (Figure 2D; complete list: Supplementary Table 4). Among these proteins are several well-known key PNS proteins, confirming the validity of our results. These include hallmark ion channels of sensory neurons, such as the voltage-gated sodium channels SCN11A and SCN10A, the TRP ion channel TRPV1 and its interaction partner PIRT, and the purinergic ion channel P2RX3. Moreover, the SN proteome featured prominent myelin proteins, e.g., proteolipidprotein 2 (PLP2), peripheral myelin protein 2 (PMP2) and NCMAP, as well as several other candidates with reported involvement in pain or PNS neurobiology: thrombospondin-4 (TSP4), ciliary neurotrophic factor (CNTF), Calcitonin gene-related peptide 1 (CGRP), follistatin 1 (FSTL1), and the Schwann cell tight junction protein claudin 19 (CLDN19; Miyamoto et al., 2005; Li et al., 2011a,b; Kim et al., 2012). The latter four are shared among DRG and SN.
More interestingly, we detected many proteins which are hardly characterized within the PNS. To illustrate, the tripartite motif-containing 36 protein (TRIM36) is an E3 ubiquitin-protein ligase which may bind microtubules and is involved in cell cycle regulation (Miyajima et al., 2009). Interestingly, its transcript has been found to be enriched in mouse and human DRG (Ray et al., 2018). We further identified C-type lectin domain family 10A (MMGL; Zizzari et al., 2015), which acts as an immune system modulator, particularly during inflammation. Members of the serpin family of serine protease inhibitors such as SERPINA3N, SERPING1, and SERPINF1 were also found to be enriched in the PNS. SERPINs are known to exhibit multiple functions from controlling the complement system to acute inflammatory immune responses (Gettins, 2002). SERPINF1 is notable as it lacks the protease inhibitory activity and is known to be antiangiogenic and neurotrophic (Gettins et al., 2002; Rychli et al., 2009). The role of SERPING1 and F1 in the PNS is unknown, but worthwhile investigating: Vicuna and colleagues have recently shown the prominent role of DRG-derived SERPINA3N in promoting early phases of neuropathic pain upon SNI (Vicuna et al., 2015).
Another prominent group, which is enriched in SN and has some overlap with the DRG, are proteins associated with the extracellular matrix (ECM). Among the enriched ECM proteins in our PNS-resolved proteomes, we identified: Chondroitin sulfate proteoglycan 4 (NG2; Fukushi et al., 2004), known to contribute to cell migration, neurite outgrowth and angiogenesis; latent transforming growth factor beta binding protein 4 (LTBP4; Lamar et al., 2016), involved in muscular dystrophy through the activity control of TGF-beta; dermatopontin (TRAMP; Krishnaswamy et al., 2014), a stabilizer of collagen fibrils facilitating wound healing; SPARC related modular calcium binding 2 (SMOC2; Vannahme et al., 2003), implicated in angiogenesis and matrix assembly facilitating wound healing; and extracellular matrix protein 1 (ECM1; Anderson et al., 2009) with functions in tumor biology, skin integrity, and inflammatory bowel disease.
We performed a similar analysis to define an SC-enriched proteome (in comparison to the brain proteome) yielding 74 proteins (complete list: Supplementary Table 5). The SC-enriched proteome contains the purinergic ion channel P2RX7 and other proteins known to be involved in nociception or pain, including Fibroblast Growth Factor Receptor 1 (FGFR-1) and S100 Calcium Binding Protein A8 (S100A8; Jamieson et al., 2014). Like in our PNS-enriched list, proteins with largely unexplored function in the SC can also be found. These include: Tax1-binding protein 3 (TIP-1; Kanamori et al., 2003) with effects on Wnt-signaling via catenin inhibition; microfibril-associated glycoprotein 4 (MFAP4; Koyanagi et al., 2016), an extracellular integrin ligand with glucocorticoid-dependent diurnal oscillations in the SC; and zinc finger protein (DPF3; Lange et al., 2008) implicated in developmental chromatin remodeling.
To determine the quality of our analysis, we assessed various aspects of resolved PNS and SC proteomes. Considering the importance of membrane proteins as drug targets (Dib-Hajj et al., 2009; Patapoutian et al., 2009; Lunn, 2010; Raouf et al., 2010; Antunes-Martins et al., 2013; Jamieson et al., 2014), we performed a GO-based enrichment analysis for cellular compartment using the web interface DAVID (Huang da et al., 2009). This revealed that membrane proteins are well-represented in these proteomes: membrane proteins constitute roughly 50% of proteins across analyzed proteomes, approximately 40% of which are predicted to be localized to the plasma membrane (PM) [SC: 50% (1897/3779) of which 40% (764/1897) at PM; DRG: 49.2% (2017/3817) of which 37.8% (764/2017) at PM; SN: 49.5% (1551/3136) of which 40.0% (621/1551) at PM].
Next, we compared our datasets with published reports on the molecular composition of the PNS and SC (complete list: Supplementary Table 6). With few published proteome studies on these tissues, we also included transcriptome data from DRG, SN, and SC in our comparison. However, comparisons between transcriptomes and proteomes often show major discrepancies due to the well-known limited predictability of mRNA alterations for protein levels (Schwanhausser et al., 2011; Sharma et al., 2015; Liu et al., 2016). These limitations need to be considered when interpreting data. Nonetheless, our results demonstrate a significant overlap with previous -omics studies on SC, SN, and DRG essentially validating our datasets (Figure 3A). Our SN proteome comprises >70% of the published rat SN membrane proteome (Lu et al., 2009) and the mouse myelin proteome (Patzig et al., 2011). Moreover, overlapping proteins represent 10% (31/299) and 35% (106/299) of the SN-enriched proteome as defined above, respectively. A comparison of our SC data with the published rat SC membrane proteome (Lu et al., 2009) and the published human SC proteome (Bruderer et al., 2017b) yielded similar results, i.e., strong overlaps of >70% (Figure 3A; complete list: Supplementary Table 6).
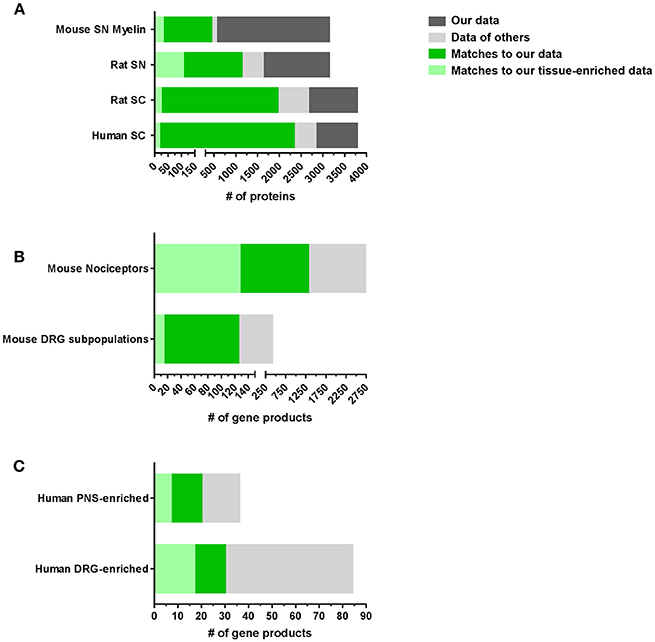
Figure 3. Comparison of PNS and SC proteomes with previously published reports in mice and humans. (A) Comparison of our SN or SC proteomes with published proteome studies: SN mouse myelin proteome (Patzig et al., 2011), SN rat membrane proteome (Lu et al., 2009), SC rat membrane proteome (Lu et al., 2009), and SC human proteome (Bruderer et al., 2017b). (B) Comparison of our DRG proteomes with published DRG transcriptome/RNA-seq studies: mouse DRG nociceptors (Thakur et al., 2014) and mouse DRG neuronal subpopulations (Usoskin et al., 2015). (C) Comparison of our PNS and DRG proteomes with studies on the human PNS-enriched transcriptome (Sapio et al., 2016) and the human DRG-enriched transcriptome (Ray et al., 2018).
Remarkably, we also observed pronounced similarities between the DRG proteome identified here and previous mRNA-based studies on purified nociceptors (Thakur et al., 2014) and sensory neuron subtypes of DRG (Usoskin et al., 2015; Figure 3B). For example, 31% (1306/4103) of all identified proteins in DRG and 33% (127/379) of DRG-enriched proteins match nociceptor mRNA (Thakur et al., 2014), of which we obtain a 47% (1306/2733) coverage on protein level (Figure 3B). Even more, our DRG proteome list contains information on 29% (125/417 annotated mRNAs) of the characteristic top 50 members across most DRG subpopulations (Usoskin et al., 2015; Figure 3B) including neurofilament-rich myelinated neurons (NF), peptidergic nociceptors (PEP), nonpeptidergic nociceptors (NP), and tyrosine hydroxylase-expressing neurons (TH; results for neuronal subpopulations: Supplementary Figure 3 and Supplementary Table 6). Given that we used lysates of whole DRG containing glia and immune cells alongside neurons, this analysis strongly suggests that a substantial part of our dataset represents the neuronal proteome of DRG. The lists of all comparisons and overlapping proteins can be found in Supplementary Table 6. These findings validate our results and support their significance for further hypothesis-driven analysis at the protein level across cell types. They also highlight their utility for discovering uncharacterized neuronal and non-neuronal proteins.
The motivation underlying high-throughput methodologies is to identify novel molecular and mechanistic insights into complex pathological processes. Therefore, we aimed at critically examining whether our proteome datasets could also provide a resource to explore the molecular set-up of painful pathologies across species to enable translational work. First, we investigated the coverage of genes and gene products reported to underlie painful pathologies in humans and rodents. Again, we leveraged previous data mining efforts and compared our data with the Human Pain Gene Database [HPGDB, (Meloto et al., 2017)] and with pain-related transgenic knockout studies summarized in the Pain Genes Database [PGD, (Lacroix-Fralish et al., 2007)] (complete list: Supplementary Table 7). Indeed, many pain-related gene products are represented in our proteome datasets, strongly supporting the utility of our data for investigating pathological processes associated with pain: specifically, our PNS proteome includes 17% of the HPGDB (23/129; 9 defined as PNS-enriched) and 30% of the PGD (133/435; 31 defined as PNS-enriched ones). Similar values were obtained for the SC proteome, with 16% of the HPGDB (21/129; 2 defined as SC-enriched) and 26% of the PGD (116/435; 2 defined as SC-enriched).
Considering that side effects on key bodily functions limit current therapeutic options of many painful disorders, defining a PNS-enriched set of proteins in a body-wide manner is of immense importance (Patapoutian et al., 2009; Borsook et al., 2014; Berta et al., 2017b; Ray et al., 2018). Excellent recent studies have systematically investigated this on the transcriptome level by comparing human and mouse datasets (Sapio et al., 2016; Ray et al., 2018). They reported a PNS-enriched and DRG-enriched transcriptome, respectively, which is conserved between mouse and humans. Intriguingly, we observed a significant overlap with our PNS-enriched datasets (complete list: Supplementary Table 7): Our full PNS proteome comprises 75% (27/36) of reported human PNS-enriched gene products (Sapio et al., 2016) including 19% (7/36) in our list of PNS-enriched proteins, such as PNS hallmark ion channels SCN11A, SCN10A, and TRPV1 (Figure 3C). Moreover, among the 84 conserved and DRG-enriched transcripts described by Ray and colleagues (Ray et al., 2018), we detected 35% (30/84) at the protein level in DRG. Of these 30 proteins, 17 match to our DRG-enriched dataset and 20 to our PNS-enriched dataset including aforementioned hallmark proteins of the PNS (Figure 3C; complete list: Supplementary Table 7).
Despite these matches, we recognize that our proteome data (i) lacks information on 65% (54/84 reported by Ray and colleagues; Ray et al., 2018) of conserved and DRG-enriched transcripts, and (ii) on the other hand, does not confirm the DRG-enrichment of others at the protein level. To illustrate the latter, we identified 13 proteins in our brain proteome (Supplementary Table 2), including stathmin 2 (STMN2), tubulin-beta 3 (TUBB3), collagen 28 (COL28A1), protein prune homolog 2 (PRUNE2), and known DRG marker proteins like advillin (AVIL) and peripherin (PRPH; complete list: Supplementary Table 7). Reasons for the missing proteins in our data are likely related to well-known limitations of proteomics approaches in detecting low abundant proteins in complex samples; notably for transmembrane proteins. On the other hand, many studies have demonstrated a prominent mismatch between transcriptome and proteome data due to posttranslational modifications, altered protein stability, and cellular buffering mechanisms (Schwanhausser et al., 2011; Sharma et al., 2015; Liu et al., 2016). Hence, mismatches in defining protein-enrichment may arise.
To address this, in part, we extended our definition of PNS-enriched proteins with respect to their body-wide expression pattern in humans at the protein level. To this end, we employed the exhaustive immunohistochemistry-based dataset available in the Human Protein Atlas (Uhlen et al., 2015; Thul et al., 2017), which contains protein expression data on 30 different tissues, but excludes the PNS and SC. We scrutinized our mouse PNS-enriched proteome data for those proteins that did not match the Human Protein Atlas. Notably, we uncovered 31% (142/463) as putative PNS-enriched candidate proteins in humans given the previously shown good agreement between sensory neurons in mice and humans (Sapio et al., 2016; Lopes et al., 2017; Ray et al., 2018). Validating our results, this list contains well known PNS proteins (TRPV1, PIRT, SCN11A, SCN10A, P2RX3, CLDN19, CNTF) as well as diverse myelin proteins of the SN, like noncompact myelin-associated protein (MP11), myelin P2 protein (MYP2), and myelin protein P0 (MYP0; complete list: Table 1). The list also encompasses 14 candidates reported by Ray and colleagues (Ray et al., 2018) to be DRG-enriched and conserved in humans (labeled with * in Table 1).
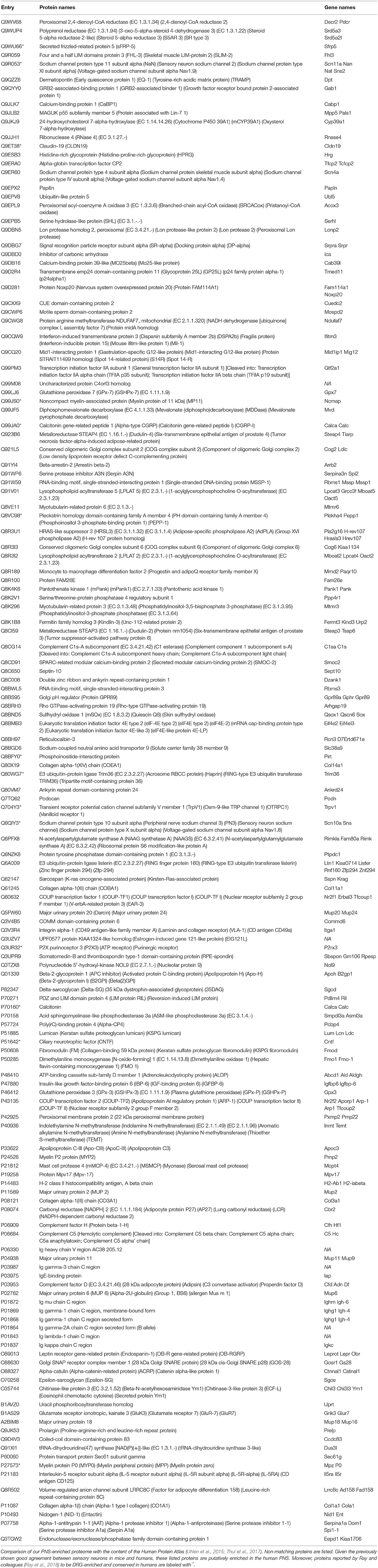
Table 1. List of putative PNS-enriched candidate proteins in humans.
More importantly, our analysis highlighted many likely human PNS-enriched proteins whose functional relevance in the PNS is not well understood. Diverse collagens (COBA1, COEA1, CO1A1, CO3A1), along with their receptors (integrin alpha-1) and their binding proteins (fibromodulin) appear to be enriched in the PNS. Together with dermatopontin and lumican they are involved in GO_BP pathways of extracellular matrix and collagen fibril organization, respectively (enrichment FDR: 0.00038 and 0.00258). We also observed a significant enrichment of proteins implicated in GO_BP pathway of lipid metabolic processes (enrichment FDR: 0.037). These include diverse enzymes (e.g., branched-chain acyl-CoA oxidase, lysophospholipid acyltransferases 2 and 5, acid sphingomyelinase-like phosphodiesterase 3a and members of the myotubularin-related protein family). They catalyze lipid mediators with known roles in signaling cascades relevant for chronic pain such as sphingolipids (Patti et al., 2012) and lysophosphatidylcholine (LPC; Inoue et al., 2008), encouraging functional studies on these enzymes. Interestingly, our analysis also suggests several major urinary proteins (MUPs) to be enriched in the PNS. MUPs are known to bind lipophilic pheromones and are excreted in urine to mediate social communication in rodents (Hurst et al., 2001). However, their additional (patho) physiological functions are unclear and may be related to GO-BP lipid storage (enrichment FDR: 0.00731) and glucose metabolism (enrichment FDR: 0.037; Hui et al., 2009; Zhou et al., 2009). Moreover, the relationship between MUPs and neuropathic pain still needs to be determined.
Taken together, our PNS-enriched proteome will serve as a valuable resource for translational studies: This is encouraged by the high similarity between the mouse and human PNS (Sapio et al., 2016; Lopes et al., 2017; Ray et al., 2018) as well as our recent demonstration that mouse spectral libraries can be applied for protein profiling of human tissues (Bruderer et al., 2017b). Future work will require validated antibodies to reliably determine the localization of here identified PNS-enriched proteins in mouse and human. In particular, proteins which have not been characterized in the PNS before may represent novel players in peripheral pathologies, including pain, and should be queried for their functional role.
Thus far, we have demonstrated that PNS and SC proteomes can be comprehensively defined, and can also be employed to interrogate - at the protein level - the molecular set-up of tissues and pathologies across species. Next, we applied our DIA-MS analysis workflow to an ongoing endeavor in the field of chronic pain: The definition of a protein signature of chronic pain (Costigan, 2012). Knowledge of such a protein signature can be exploited for mechanism-oriented research and, ultimately, may offer opportunities for novel therapeutic approaches (Gereau et al., 2014; Baron et al., 2017). To this end we employed the widely-used SNI model, which mimics clinically relevant features including hypersensitivity to mechanical stimuli (tactile allodynia; Decosterd and Woolf, 2000). All SNI mice included in this study exhibited pronounced hypersensitivity to mechanical stimulation at 4 weeks, in contrast to Sham operated mice (Supplementary Figure 1).
We quantitatively compared our PNS and SC datasets obtained by DIA-MS from control (Sham surgery) mice with those which underwent SNI. We again had highest demands toward reproducibility and applied very stringent analysis criteria: proteins were only included in our analysis if their peptides were consistently detected in all six replicates, i.e., in three Sham and in three SNI replicates for a given tissue (4 mice/replicate, totaling 12 mice/condition; please see Methods for details). The heatmap of protein abundance across tissues and correlation analyses can be found in Supplementary Figure 4. Overall, more than 93% of proteins detected in control tissues were consistently quantified in all six replicates (complete list: Supplementary Table 8). These include 400 PNS-enriched proteins and 41 SC-enriched proteins. At this point it is unclear why the detection of SC-enriched proteins across all six replicates yielded only 55% (41/74) of proteins identified in control replicates described above (Supplementary Table 8). This contrasts with the high reproducibility of DIA-MS in PNS tissues and might hint at a massive downregulation of SC-enriched proteins below the detection limit in SNI samples.
Upon statistical comparison of these data (please see Methods for details), we observed distinct and tissue-specific changes after SNI (Figures 4A–C; complete list: Supplementary Table 8). Most regulated proteins were in the SN (1026/2939, 34%) followed by DRG (715/3828, 18%) and SC (53/3584, 1.4%; Figures 4A–C). For the latter, more than half of the proteins were shared with the PNS as expected from aforementioned data (Figure 4D). Strikingly, 29% (114/400; 58 in DRG and 88 in SN; overlap in 32 proteins) of our PNS-enriched proteome showed significant alterations during neuropathic pain (Figure 4E). Among these, were 32 proteins that do not match the content of the Human Protein Atlas (Uhlen et al., 2015; Thul et al., 2017), thus, candidates considered to be putatively enriched in the human PNS (Table 2). These include several proteins involved in ECM integrity (e.g., some collagens, fibromodulin, and lumican).
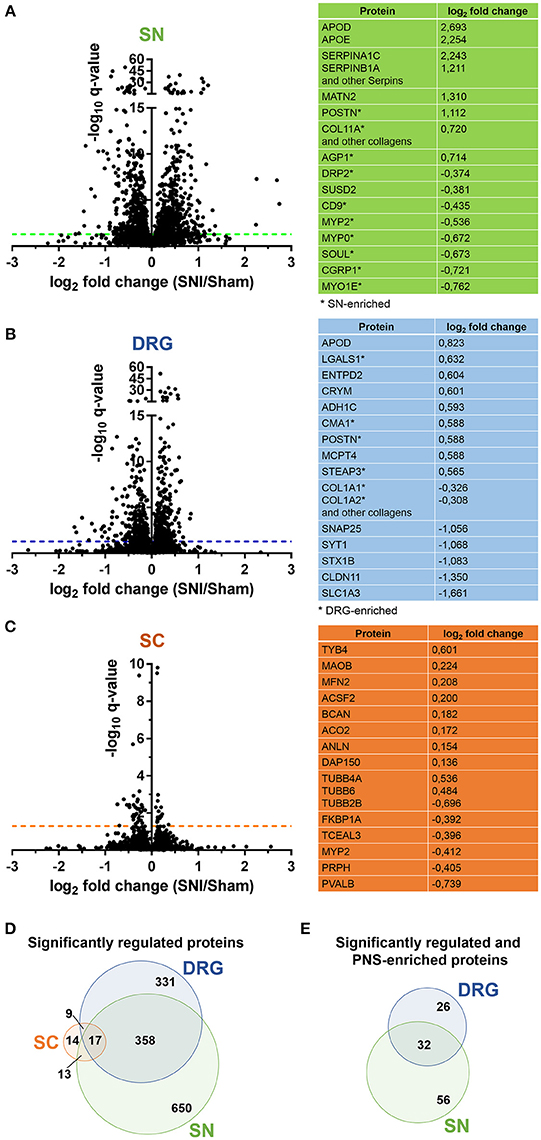
Figure 4. Proteome dynamics in the SNI model of neuropathic pain. (A–C) Volcano plots show the regulation of members of SN, DRG, and SC proteomes in Sham mice versus SNI mice. –log10 q-value is plotted against log2 fold change (SNI/Sham). The dotted horizontal line represents the cutoff for significance, q < 0.05. Tables list examples of regulated candidates in each region and respective log2 fold changes (complete list: Supplementary Table 8). (D) Venn diagram illustrates regulated (q < 0.05) proteins across SN, DRG, and SC proteomes. (E) Venn diagram illustrates regulated (q < 0.05) and differentially expressed proteins of the PNS-enriched proteome. In all panels the q-value represents the Benjamini-Hochberg-corrected p-value.
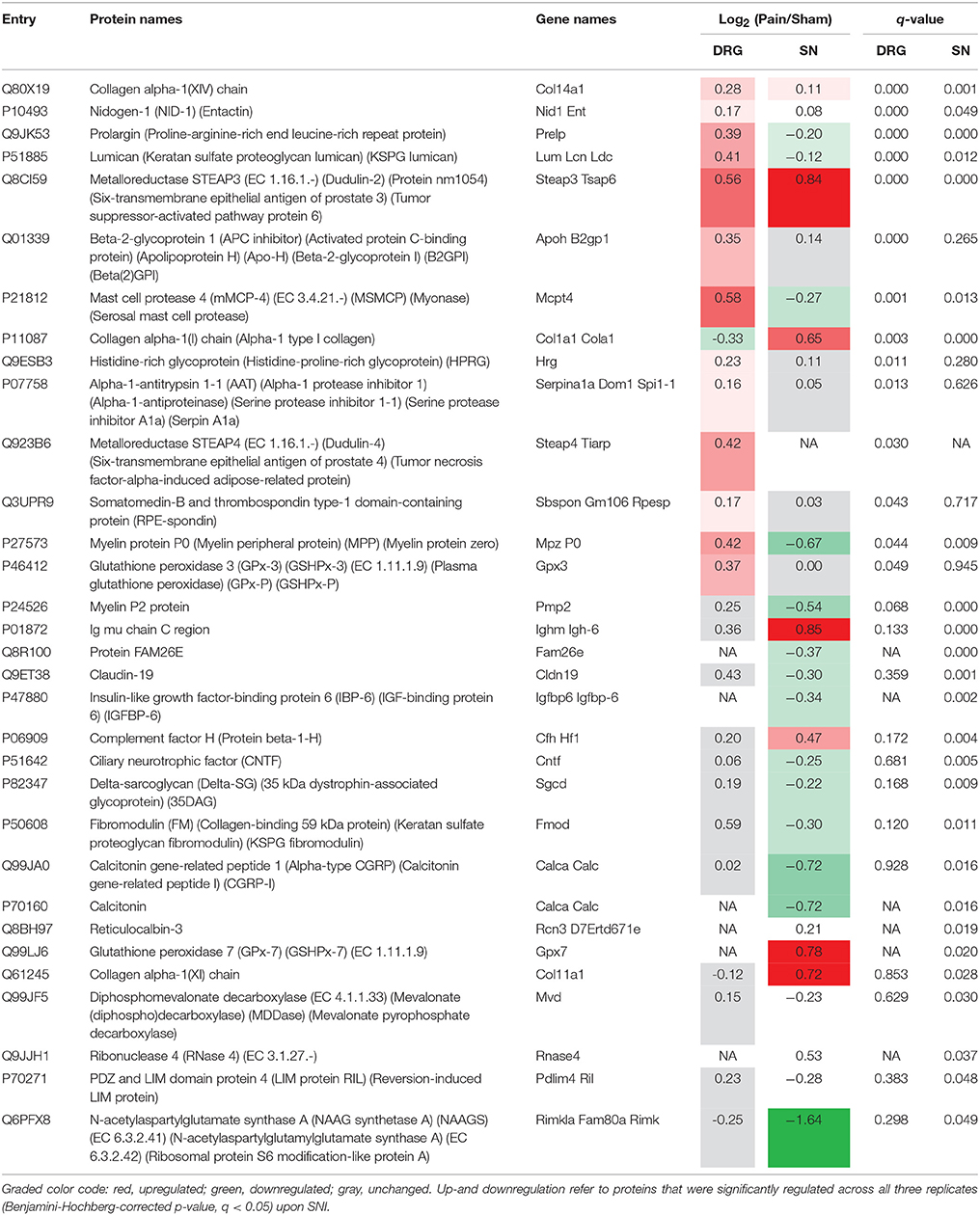
Table 2. List of putative PNS-enriched candidate proteins in humans that are regulated upon SNI.
Supporting our findings, a comparison with the catalog of protein-protein interactions involved in neuropathic pain (Jamieson et al., 2014), the PGD (Lacroix-Fralish et al., 2007), and the HPGDB (Meloto et al., 2017) revealed links of many SNI-regulated proteins to painful conditions in mouse and human. These include well-known pain-related gene products like nerve growth factor receptor (NGFR), annexin A2 (ANXA2), alpha2/delta subunit 1 of the voltage-dependent calcium channel (CACNA2d1), calcitonin gene-related peptide 1 (CGRP1 or CALCA), thrombospondin 4 (THBS4), and follistatin-like 1 (FSTL1; complete list: Supplementary Table 9).
More importantly, we found largely uncharacterized regulations. Several members of the aforementioned serpin family of serine protease inhibitors are upregulated in SN (e.g., SERPINA1C SERPINB6, SERPINC1, SERPINF1) or in DRG and SN (e.g., SERPINB1A, SERPING1, SERPINH1). GO-BP classification implicate these regulated SERPINs in acute inflammatory responses, in processes related to wound healing and, in general, related to the regulation of stress responses. Periostin (PSTN) is also prominently upregulated in SN and DRG in addition to being enriched in the PNS. There, it is likely secreted from dermal fibroblasts (Murota et al., 2017). It plays a role in cell adhesion and ECM integrity with a strong impact on inflammatory pathologies of the skin (e.g., atopic dermatitis) as well as the bowels (Koh et al., 2016; Murota et al., 2017).
Moreover, we have identified prominent protein downregulations after SNI. Dystrophin-related protein 2 (DRP-2) is among PNS-enriched proteins which are downregulated in SN. It is crucial for myelination in Schwann cells (Sherman et al., 2001) and, thus, has been linked to demyelinating neuropathies in both mouse and human (Sherman et al., 2001; Brennan et al., 2015). In the SC, a top-downregulated candidate is the low molecular weight protein thymosin beta4 (TYB4). Given its anti-inflammatory and angiogenic functions, TYB4 has consistently been shown to facilitate the repair and regeneration of diverse tissues and diabetic neuropathy mice (Philp and Kleinman, 2010; Wang et al., 2012). Hence, further exploration of TYB4 in the context of chronic pain may uncover an analgesic potential.
Our results suggest a prominent regulation of candidate proteins with roles in inflammatory and immune processes upon SNI. This supports the notion that the immune system and inflammation are critical players underlying SNI pathologies (Krames, 2014; Ji et al., 2016; Sommer, 2016; Price and Gold, 2017; Cobos et al., 2018). Thus, we exploited the benefits of DIA-MS to retrospectively extend the analysis of our previously published datasets on chronic pain, specifically on the regulation of the DRG membrane proteome during inflammatory (upon injection of CFA) and neuropathic pain (SNI-model; Rouwette et al., 2016). As explained above (Figure 1) DIA-MS data are analyzed by means of spectral libraries. Hence, the depth and quality of the spectral library largely determine the proteome coverage one can obtain by a given experiment. The pan-mouse library generated here contains approximately 2-fold more proteins than our previously published spectral library (Rouwette et al., 2016). We employed this superior pan-mouse library to retrospectively extend our insights on pain-induced changes within the DRG membrane proteome.
Compared to our previous study (Rouwette et al., 2016), we identified dozens of additional proteins which appear to be regulated in only one of the two chronic pain models (CFA: 235, previously: 64; SNI: 156, previously 77). To give some examples, microsomal glutathione S-transferase 3 (MGST3) is downregulated during inflammatory pain. This protein belongs to a large family of peroxidase proteins involved in the production of leukotrienes and prostaglandin E (Bresell et al., 2005; Hayes et al., 2005). Notably, Hirai et al. (Hirai et al., 2017) have identified MGST3 through RNA sequencing after sciatic nerve entrapment (SNE) injury in rats, although it's role has not yet been examined in detail. Another example protein, Cancer susceptibility 4 (CASC4), is upregulated during inflammatory pain. As its name suggests, CASC4 is a proto-oncogene associated with breast and ovarian cancers (Anczuków and Krainer, 2015; Anczuków et al., 2015). It has yet to be actively discussed in the context of pain, but its transcript was found to be downregulated after spinal nerve transection (http://www.painnetworks.org:/ SNT_L5vsNAIVE_exonArrays). During neuropathic pain, galactin-1 (LGALS1) is upregulated. LGALS1 is a widely expressed lectin in a family of proteins implicated in inflammation, endometriosis, and cancer (Vergetaki et al., 2014). In the context of pain, it has been shown to mediate the severity of endometriosis (Vergetaki et al., 2014), as well as potentiate neuropathic pain in the dorsal horn (Imbe et al., 2003). Similarly, low-density lipoprotein receptor-related protein (LRP1) is found to be upregulated upon SNI. LRP1 is a large, multi-ligand signaling receptor which has been discussed in numerous contexts, ranging from cancer to oligodendritic cholesterol homeostasis to ECM remodeling (Etique et al., 2013; Lin et al., 2017). It has previously been considered as an “injury detection receptor” in addition to having a role in axonal regeneration after spinal injury (Yoon et al., 2013). The detection of LRP1 modulation after SNI in this retrospective analysis further hints at an important role after nerve injury.
Furthermore, we identified five times more proteins which are regulated in both pain models (55 proteins, previously: 11; Figure 5; complete list: Supplementary Table 10). While extensive work is needed to assess the functional relevance of these 55 shared proteins, their regulation in two etiologically different chronic pain models suggests a role as universal players in chronic pain. Here, we discuss some examples in more detail: One notable example is the alpha 2 isoform of Na+/K+ ATPase (ATP1A2). ATP1A2 is highly expressed in the brain, and has been implicated in cortical spreading depression and migraines (Unekawa et al., 2017). In fact, ATP1A2 represents one of the known susceptibility loci of familial hemiplegic migraine in humans (Friedrich et al., 2016). We also found many hits uncharacterized in the context of chronic pain which may warrant further study. Among them, we identified 2′,3′-cyclic-nucleotide 3'-phosphodiesterase (CNP), a myelin protein upregulated in both pain models, which is highly expressed in oligodendrocytes and Schwann cells (de Monasterio-Schrader et al., 2012). CNP is thought to play a role in RNA metabolism (Gravel et al., 2009) and has also been linked to neuroinflammation in Alzheimer's disease, Down Syndrome, and schizophrenia (Barley et al., 2009). Recent work suggested that its role in axonal support is biased toward sensory axons in the PNS (http://hdl.handle.net/11858/00-1735-0000-0023-964D-8) with details remaining to be explored. Interestingly, the macrophage mannose receptor 1 (MMR or MRC1) appears to be differentially regulated across both pain models (CFA down- and SNI upregulated). MMR is a cell surface receptor involved in host immune responses (Schuette et al., 2016). It contributes to the suppressive state of macrophages (Schuette et al., 2016). As such, MMR is upregulated by interleukin 4 upon nerve injury, which correlates with the amelioration of behavioral hypersensitivity (Kiguchi et al., 2015). Its differential regulation in the CFA- and SNI-model likely reflects its immunomodulatory role in macrophages and deserves further investigations.
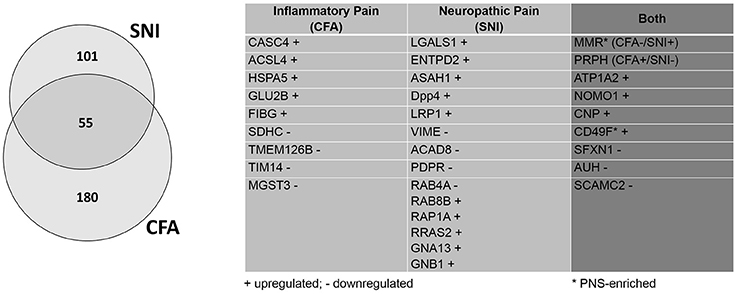
Figure 5. Retrospective analysis of DRG membrane proteome dynamics in two chronic pain models. Application of our pan-mouse library for retrospective analysis of our recently published datasets on mean abundance changes of DRG membrane proteins in the SNI-model of neuropathic pain and the CFA-model of inflammatory pain (Rouwette et al., 2016). Venn diagram illustrates the proportions of significantly regulated (across all three replicates; Benjamini-Hochberg-corrected p-value, q < 0.05) DRG membrane proteins. The table lists examples of regulated (+, upregulated; -, downregulated) candidates across both pain models (complete list: Supplementary Table 10).
In contrast to our current study on whole DRG, our previous work was performed on membrane-enriched DRG samples (Rouwette et al., 2016). As such, this retrospective analysis is not exhaustive. Nevertheless, it is highly valuable for deciphering the regulation of protein levels at membranes compared to whole-tissue data (the latter reflecting the overall cellular abundance of proteins). To highlight an example, several G-protein signaling components (e.g., Ras-related proteins: RAB8B RAB4A, RAP1A, RRAS2; guanine nucleotide-binding protein subunit alpha-13, GNA13) display changes upon SNI in the DRG membrane proteome (Supplementary Table 10), which could not be detected in whole tissue lysates. This suggests that subcellular proteome analysis can provide additional and insightful knowledge about the regulation of proteins during pathological conditions.
From a technical point of view, we significantly increased the depth of our previous data using the pan-mouse library generated here without requiring new tissue samples. This provides a proof-of-principle to how DIA-MS can open new avenues toward standardizing and continuously improving proteome data in the pain community.
Rarely do proteins exert their functions alone. Rather, they are embedded in protein complexes and larger protein networks which enable the regulation and plasticity of cellular pathways (Alberts, 1998; Rouwette et al., 2015). Knowledge of such protein networks may therefore contribute to a mechanistic understanding of pathophysiological processes underlying SNI. This is in line with emerging approaches in network medicine for modulating diseases (Chapman et al., 2008; Antunes-Martins et al., 2013; Borsook and Kalso, 2013). We thus inquired whether significantly regulated proteins upon SNI cluster in distinct networks and signaling pathways.
Using IPA® we first broadly determined the interactions of regulated proteins within known, experimentally verified networks across all three examined tissues (Krämer A et al., 2014). Membership of regulated proteins in these networks is scored based on the in-built Fisher's exact test by IPA® (Supplementary Tables 11–13, which list members of each network). Besides shared networks, we detected prominent regional specificity of protein regulation within these networks. For example, several collagens and integrins implicated in ECM integrity are co-clustered with others in a network relevant for dermatological and inflammatory diseases (Network #20, Supplementary Table 11). Network analysis highlights their regulation upon SNI in a region-specific manner (Figure 6): strong upregulation of COL1A1, COL1A2, COL4A1, and COL4A2 in SN contrasts their downregulation in DRG. They are unchanged in SC. These data lend further support to the implication of ECM signaling in PNS processes associated with SNI.
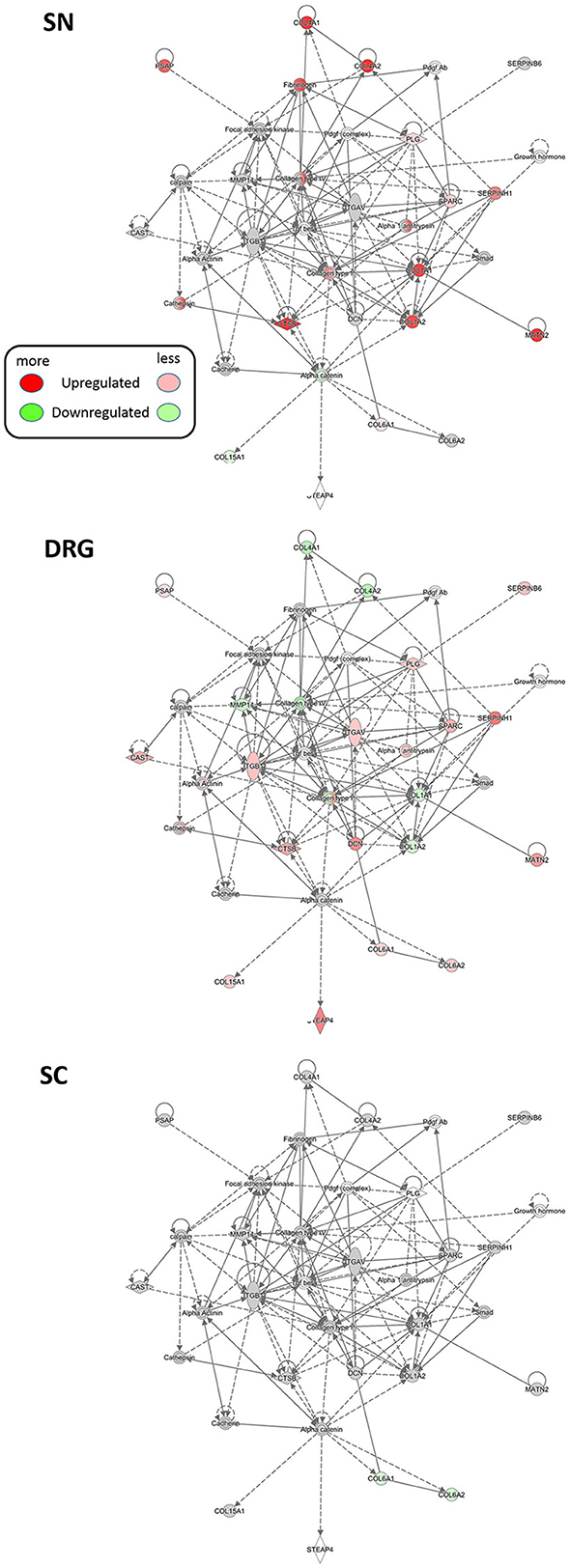
Figure 6. Region-resolved changes upon SNI in a protein network implicated in dermatological and inflammatory diseases (Network #20, Supplementary Table 11). Differential regulation of mean protein abundance across SN, DRG, and SC becomes apparent and is visualized by a graded color code. Node color code: red, upregulated; green, downregulated; gray, unchanged; white, not detected. Predicted relationship among nodes is indicated by dashed lines. All graphs are created by IPA®. Up-and downregulation refer to proteins that were significantly regulated across all three replicates (Benjamini-Hochberg-corrected p-value, q < 0.05) upon SNI.
We additionally critically assessed the possibility that our data can provide novel network-level insights into the pathology of proteins already described to be involved in pain. To this end, we turned to the regulation of several members of the sodium-potassium ATPase (NKA) family as an example. NKAs are significantly downregulated in our PNS dataset upon SNI. Two NKA members, ATP1A1A and ATP1B1, were reported to set sensory neuron excitability and to be controlled by follistatin-like protein 1 (FSTL1; Li et al., 2011b). Interestingly, a reduction of this FSTL1-NKA system in primary afferents correlated with neuropathic pain behaviors in rodents (Li et al., 2011a). In line with these findings, we not only observed an overall downregulation of the FSTL1-NKA system upon SNI, but could also visualize associated and partially modulated proteins within the same network in a region-dependent manner (Figure 7). These include the additional downregulation of several other NKA family members (A3, B3, and A2) in SN. Moreover, IPA® causal analysis (Krämer A et al., 2014) further suggests an interaction and inhibition of spectrin family members in the PNS compared to the SC.
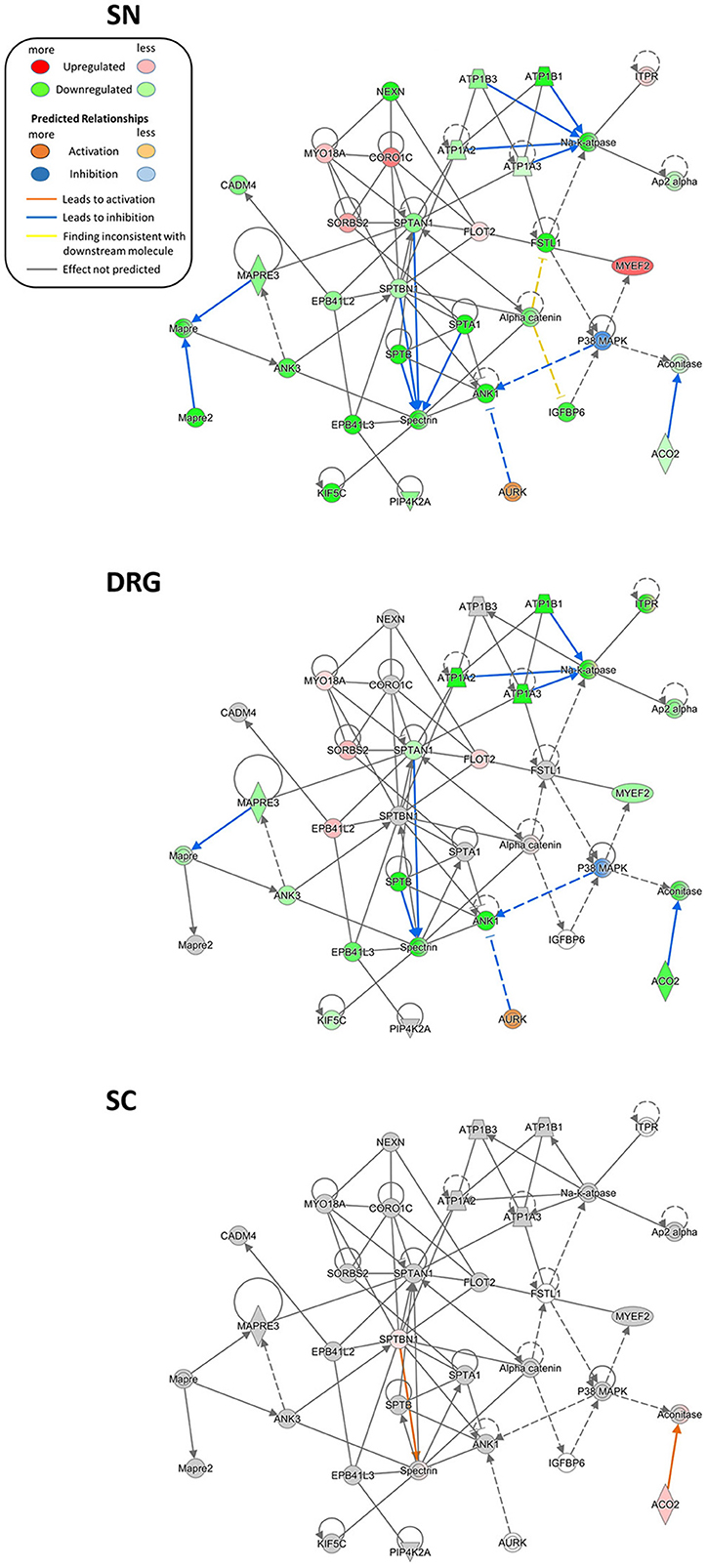
Figure 7. Region-resolved changes in the FSTL1-NKA-protein network upon SNI. Differential regulation of mean protein abundance across SN, DRG, and SC becomes apparent and is visualized by a graded color code. Node color code: red, upregulated; green, downregulated; gray, unchanged; white, not detected. Moreover, causal network analysis predicts differences in activation (line color orange) and inhibition (line color blue) of distinct connections between members of this network. Predicted relationship among nodes is indicated by dashed lines. All graphs are created by IPA®. Up-and downregulation refer to proteins that were significantly regulated across all three replicates (Benjamini-Hochberg-corrected p-value, q < 0.05) upon SNI.
We then went on to employ IPA® to interrogate regulated proteins associated with canonical cellular pathways. Overall, we detected changes in diverse pathways, many of which were differentially modulated in SN compared to DRG or SC (Figure 8A left). A notable example are interconnected pathways of mitochondrial dysfunction, oxidative stress response, oxidative phosphorylation and TCA cycle, known to underlie the diverse pathology of chronic pain (Baloh, 2008; Akude et al., 2011; Flatters, 2015). Figure 8A presents a heatmap comparing mean relative abundance changes of proteins involved in mitochondrial dysfunction across analyzed regions, with DRG being the most affected. These results are schematically visualized in Figure 8B.
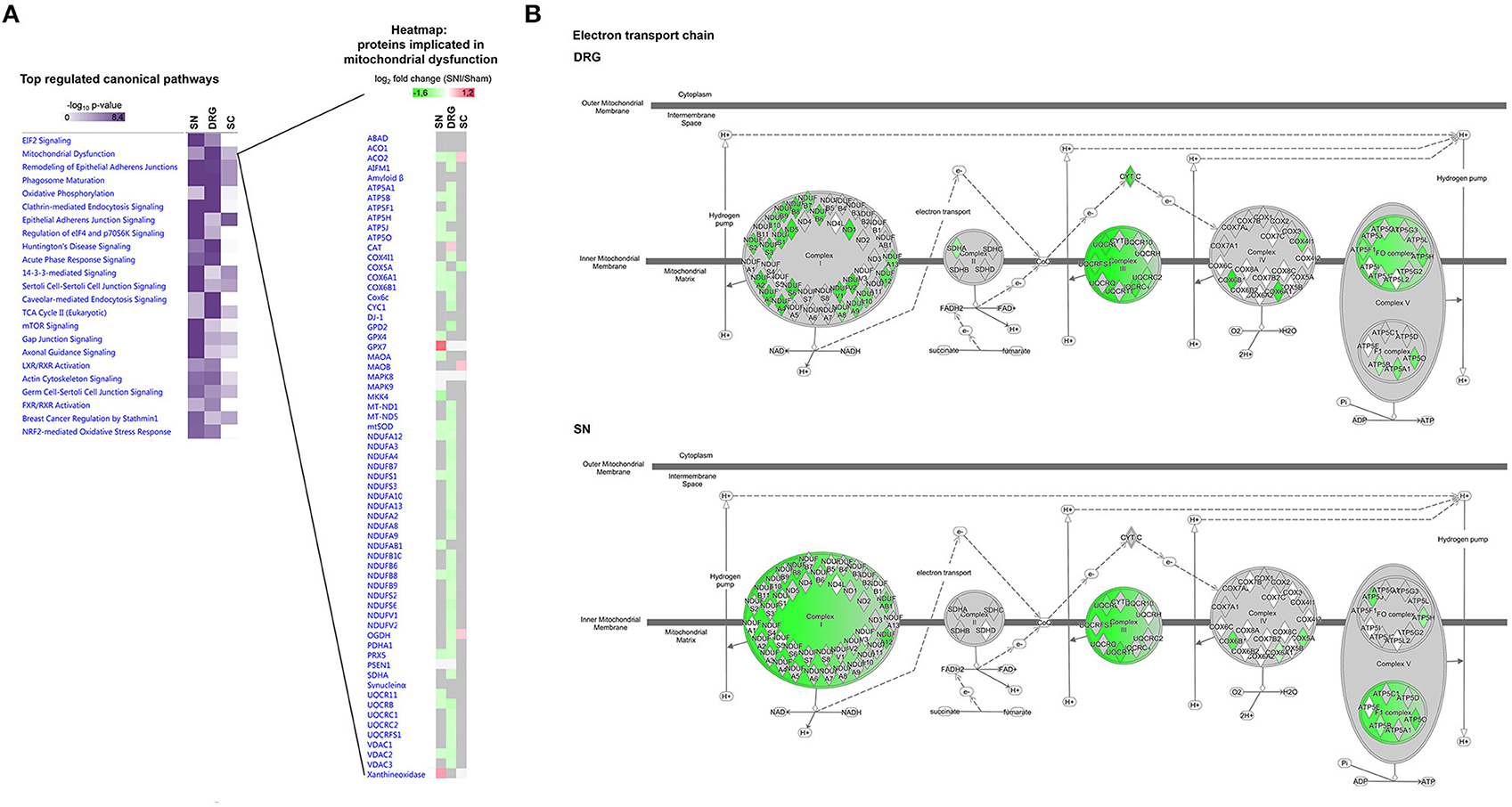
Figure 8. Region-resolved regulation of canonical pathways upon SNI. (A) Left, annotation matrix of pathway enrichment analysis for proteins regulated across all three replicates upon SNI in SN, DRG, and SC (scored based on the in-built Fisher's exact test by IPA® using the –log10 p-value). Right, heatmap depicts mean abundance changes (log2 fold change SNI/Sham; across all three replicates) of regulated proteins implicated in “mitochondrial dysfunction.” Color code: red, upregulated; green, downregulated; gray, unchanged; white, not detected. (B) Schematic representation and comparison of differentially regulated proteins in the electron transport chain (DRG versus SN). Mean protein abundance changes of significantly regulated proteins across all replicates are coded by a color gradient. Color code: red, upregulated; green, downregulated; gray, unchanged; white, not detected. All matrices and graphs are created by IPA®. Up-and downregulation refer to proteins that were significantly regulated across all three replicates (Benjamini-Hochberg-corrected p-value, q < 0.05) upon SNI.
Importantly, the prominent effect of SNI on mitochondrial proteins in DRG is in line with our retrospective analysis of the DRG membrane proteome. This was obtained from an independent cohort of mice in our previous study (Supplementary Table 10; Rouwette et al., 2016). Remarkably, thus, we found differences in the regulation of protein levels between data from DRG membranes compared to whole DRG. In DRG membrane samples we measured an upregulation of many proteins belonging to the mitochondrial respiratory chain (e.g., NDUFV1, NDUFS4, NDUFS5, NDUFA5, and NDUFS3; Supplementary Table 10) in contrast to their downregulation in whole DRG (Supplementary Table 8). These data may provide valuable insights into subcompartmental regulation, particularly regarding mitochondria. Ninety-nine percent of mitochondrial proteins are encoded by nuclear genes, thus their synthesis at cytosolic ribosomes, their cytosolic abundance, and their subsequent import into mitochondria are crucial for organelle function (Schmidt et al., 2010; Wiedemann and Pfanner, 2017). Protein import occurs in response to a variety of conditions, including oxidative stress, and is considered to contribute to mitochondrial dysfunction associated with diverse human diseases (MacKenzie and Payne, 2007). Currently, we can only speculate as to how here detected abundance changes may affect mitochondrial dysfunction. Nonetheless, our data build on previous work to suggest defects in mitochondrial import and membrane assembly play a role in neuropathic pain.
To obtain a thorough understanding of chronic pain, system wide mapping of the pain axis at the protein level is necessary. This has not yet been achieved. This is largely due to the fact that the technology required to reproducibly and quantitatively compare proteomes has emerged only recently. Here, we built upon these technical advances and offer a comprehensive protein compendium of three regions at the beginning of the pain neuraxis—the SN, DRG, and SC. Our results yield unprecedented insights into the proteome composition of these regions and their dynamics during neuropathic pain. We further provide a thus far unique dataset on proteins and protein networks that are highly enriched in the PNS and SC. Consequently, our study fills an eminent gap in the pain field and serves as a stepping stone for defining protein disease signatures and novel therapeutic targets of chronic pain.
Chronic pain is an extremely complex disease involving a myriad of molecular changes (Costigan et al., 2009; Patapoutian et al., 2009; Sommer, 2016; Price and Gold, 2017). This is likely why treatments interfering with single proteins exhibit only limited efficacy. Emerging medical research initiatives aim for a “mechanism-based” approach toward pain therapy (Borsook and Kalso, 2013; Gilron et al., 2013; Treede et al., 2013; Sommer, 2016; Vardeh et al., 2016; Baron et al., 2017). A prerequisite for this is a thorough, systems biology understanding of the complex and dynamic molecular interplay contributing to chronic pain (Chapman et al., 2008; Antunes-Martins et al., 2013; Borsook and Kalso, 2013).
Extensive genomic (Diatchenko et al., 2005, 2007, 2013; Sorge et al., 2012; Zorina-Lichtenwalter et al., 2016), epigenomic (Doehring et al., 2011; Denk and McMahon, 2012; Niederberger, 2014), and transcriptomic studies (Grace et al., 2012; Simonetti et al., 2013; Jeong et al., 2016) have provided highly valuable insights into the changes associated with painful pathologies in rodents and patients. Nonetheless, their predictability of phenotypes is limited (Mogil, 2012; Geyer et al., 2016; Liu et al., 2016; Sommer, 2016). This is especially the case for transient and/or dynamic biological processes, given the existence of various cellular buffering mechanisms (Liu et al., 2016). In contrast, the proteome constitutes a highly valuable source of molecular information for monitoring phenotypes - including disease onset and progression (Chen et al., 2015; Ebhardt et al., 2015; Sajic et al., 2015; Gazerani and Vinterhoj, 2016; Geyer et al., 2016, 2017). This advantage of standardized proteome analysis is extensively exploited in oncological research to define robust molecular tumor signatures (Hüttenhain et al., 2012; Cerciello et al., 2013; Takadate et al., 2013). Thus far, similar system-wide studies are lacking in the context of pain. In the pain field, most work published relies on gel-based approaches (typically 2D-PAGE) followed by data-dependent acquisition mass spectrometry (DDA-MS), often referred to as shotgun proteomics (Domon and Aebersold, 2010; Michalski et al., 2011). These methods have laid the foundation for proteomic studies of chronic pain (Huang et al., 2008; Olausson et al., 2012, 2015; Vacca et al., 2014). They are also known for their limitations regarding biased sampling, protein losses, and low reproducibility between studies (Domon and Aebersold, 2010; Michalski et al., 2011). Emerging data-independent acquisition mass spectrometry (DIA-MS; Venable et al., 2004; Panchaud et al., 2009; Carvalho et al., 2010) helps to overcome some of these limitations. In essence, DIA-MS enables the identification and quantification of proteins on a large scale and in a standardized and reproducible manner (Domon and Aebersold, 2010; Sajic et al., 2015). Therefore, it is ideally suited to interrogate pathology-related proteome changes (Hüttenhain et al., 2012; Cerciello et al., 2013).
Using DIA-MS, our previous study on the membrane proteome of DRG provided a glimpse of differential and pain-model specific changes in distinct cellular protein networks (Rouwette et al., 2016). Here, we extended our previous study and monitored global proteome changes in the SN, DRG, and SC. A wealth of previous work has shown that the PNS is (i) a significant driver of chronic pain, and (ii) offers higher therapeutic accessibility than brain areas implicated in pain (Puljak et al., 2009; Liem et al., 2013; Krames, 2014; Berta et al., 2017b). Consequently, recent therapeutic strategies aim to selectively target peripheral sensory neurons for pain relief (Sapunar et al., 2012; Liem et al., 2013; Haroutounian et al., 2014; Krames, 2014; Berta et al., 2017b). Despite its apparent clinical importance, the protein composition of the PNS has not been thoroughly investigated. Our study fills this gap and defines for the first time a set of PNS-enriched proteins in reference to two independent but highly similar and large-scale datasets of the mouse brain (our own results and results of others; Sharma et al., 2015). In addition, we cross-correlated these data with the Human Protein Atlas harboring protein information on more than 30 human tissues. This enabled us to define PNS-enriched protein signatures with high utility for translational research.
Strikingly, 32 of these putative PNS-enriched candidate proteins in humans displayed significant regulation in the SNI-model (Table 2). Among these, several are implicated in ECM integrity. The ECM is crucial for signal transduction and communication between extra- and intracellular spaces (Manninen and Varjosalo, 2017). Additionally, it is modulated in response to diverse stimuli. The ECM has been implicated in tissue homeostasis, as well as pathological processes triggered by nerve injury in the SC (Bartus et al., 2014; Tajerian and Clark, 2015). In chronic pain conditions, there is accumulating evidence for the role of ECM changes in hypersensitivity and long lasting maladaptation, particularly by affecting synaptic plasticity in the CNS (Tajerian and Clark, 2015). Yet, details about the role of its protein constituents for PNS (patho-) physiology remain to be investigated. Here, we reveal pronounced abundance changes of distinct proteins implicated in ECM integrity in our DRG and SN datasets. Collectively, these findings strongly suggest the involvement of ECM signaling in processes associated with SNI, and may warrant detailed studies on here identified regulated candidates.
Another notable feature of observed SNI-induced dynamics within the PNS is the high representation of proteins involved in immune signaling and inflammation (Figure 4 and Supplementary Table 8). This is in line with the evolving notion that DRG and SN harbor diverse immune cells (e.g., macrophages and T cells) with critical functions for normal physiology and during chronic pain (Krames, 2014; Ji et al., 2016; Sommer, 2016; Price and Gold, 2017; Cobos et al., 2018). In fact, a recent study demonstrated mechanistic differences in the contribution of immune cells to SNI-induced phenotypes: While they are largely dispensable for cold allodynia, immune cells appear to be critical for the development of tactile hypersensitivity likely via their interplay with sensory neurons (Cobos et al., 2018). Interestingly, our data reveal a significant overrepresentation of proteins associated with IPA® pathways of acute phase response signaling and chronic inflammatory disorders in PNS-enriched proteomes (Supplementary Table 3). Many of these are regulated upon SNI (e.g., diverse SERPINS; Figure 4 and Supplementary Table 8) and associated with other regulated proteins in distinct protein networks (Figure 6 and Supplementary Tables 11–13). Moreover, our retrospective analysis finds similarities in protein regulation during neuropathic and inflammatory pain (e.g., CNP, ATP1A2, NOMO1; Figure 5 and Supplementary Table 10). Hence, we uncover candidates for mechanistic follow-up studies on their role for immune and inflammatory signaling in the SNI-model.
Furthermore, we observe marked alterations in canonical pathways related to mitochondrial dysfunction (Figure 8). The importance of mitochondria in chronic pain is increasingly recognized (Baloh, 2008; Akude et al., 2011; Flatters, 2015). Our data extend existing knowledge by providing unprecedented and region-resolved details about alterations in specific mitochondrial proteins during chronic pain, particularly in the PNS. Interestingly, a recent transcriptome study showed that specifically injured neurons activate oxidative stress responses as early as 7 days after SNI (Berta et al., 2017a) suggesting that in our study a substantial part of observed changes may be derived from injured neurons.
In summary, we report here pronounced and differential dynamics upon SNI by looking at the proteome. On one hand, this knowledge will contribute to our understanding of diverse (patho-) physiological processes underlying the SNI-model (Decosterd and Woolf, 2000): not only chronic pain, but also tissue injury, activation of immune cells, and nerve de-/regeneration. On the other hand, our findings open opportunities for targeting regulated proteins and associated protein networks in treatments of chronic pain.
Of note, in order to reduce the number of mice and samples needed for this study in observation of the 3 Rs of animal research, we used mice 28 days after Sham-surgery as controls to define tissue-resolved proteomes. While we cannot exclude proteome changes induced by Sham-surgery lasting for 28 days thereafter, several of our findings support the idea that our region-resolved proteomes largely reflect their “normal” protein content: (i) the here identified brain proteome is highly similar to previously published data in mice (Sharma et al., 2015), and (ii) the here identified PNS, SC proteomes, and region-enriched proteomes are in high accordance with legacy data on the molecular-set up of these tissues (Figure 3).
A common concern of tissue-based proteomics (and transcriptomics alike) experiments is the mixture of different cell types contained in each tissue. Therefore, the identified proteome alterations may be contributed by any cell type of the studied tissue. Further, changes of a distinct protein in one cell population may be masked by opposing changes in others. For example, our results in DRG likely reflect the proteome of diverse subpopulations of sensory neurons (Usoskin et al., 2015), immune cells, glia cells, and endothelial cells, many of which with prominent roles in painful pathologies (Krames, 2014; Ji et al., 2016; Sommer, 2016; Price and Gold, 2017). At this point we cannot judge the relative contributions of cell types to our DRG datasets; however, a comparison with existing transcriptome data of purified nociceptors (Thakur et al., 2014) and DRG subpopulations (Usoskin et al., 2015) enabled us to roughly estimate the proportion of neuronally expressed proteins to ~50%. These included known hallmark proteins of sensory DRG neurons, such as distinct ion channels and their interaction partners.
Another problem of tissue-based -omics arises from the intermingling of injured (dissected) and uninjured neurons in DRG and SN upon SNI. Moreover, fibers which were not primarily injured by the SNI surgery have been shown to be affected as well, exhibiting ectopic firing and contributing to the pathology (Costigan et al., 2009; Berta et al., 2017a). Recent transcriptome studies have elegantly compared injured with uninjured fibers (Hu et al., 2016; Berta et al., 2017a). Interestingly, it appears that such changes extend far beyond transcriptional regulation and need to be analyzed on the protein level, as well (Hu et al., 2016; Berta et al., 2017a).
Hence, future efforts should focus on efficiently labeling neuronal subpopulations in vivo. This would enable their separation followed by differential proteome analysis. Ideally, one would like to analyze single cell populations. So far, the analysis of single cell types is not amenable to comprehensive proteome profiling due to restrictions in sample amount and known limitations of profiling low abundant proteins in complex samples. Mass spectrometry workflows are rapidly evolving to improve both sensitivity and reproducibility. These advances involve novel acquisition strategies and computational analysis tools to maximize the amount of protein information obtained (Schubert et al., 2015; Tsou et al., 2015; Geyer et al., 2016; Bruderer et al., 2017a,b), all of which will open new possibilities in comprehensive proteome profiling of chronic pain.
Overall, we reveal dynamic changes of distinct protein networks during chronic pain (Figure 6 and Supplementary Tables 11–13; Figures 7, 8). This is highly relevant for state-of-the-art strategies in finding novel drug targets. As mentioned, therapeutic strategies targeting single proteins lack efficiency and often—depending on their organismal role—specificity to chronic pain (Borsook et al., 2014). The system-wide identification of critical hubs within defined protein networks, however, holds the promising potential to generate combinatorial therapeutic interventions, ideally less likely to cause severe side effects (Chapman et al., 2008; Barabási et al., 2011; Antunes-Martins et al., 2013; Borsook and Kalso, 2013; Chen et al., 2015). In order to promote a systems view on pain, Perkins and colleagues implemented “PainNetworks” (Perkins et al., 2013), Jamieson and colleagues summarized the “pain interactome” (Jamieson et al., 2014) and recent studies systematically mapped the DRG/PNS-enriched transcriptome by contrasting it to body-wide gene expression (Sapio et al., 2016; Ray et al., 2018). We now complement these efforts and provide a freely-available and searchable database (painproteome.em.mpg.de) harboring all data described in this work, i.e., region-resolved proteomes as well as SNI-associated proteome changes. In addition to encouraging future experimental investigations, these data may be used to test the application of designated network theory algorithms to find disease modules in large datasets (Zhang et al., 2009; Barabási et al., 2011; Shi et al., 2015; Kitsak et al., 2016). Examples of network theory impact include insightful work on cardiovascular diseases, cancer, and diabetes summarized elsewhere (Oti et al., 2006; Barabási et al., 2011; Zhou et al., 2014; Kitsak et al., 2016).
In conclusion, we have shown that the analysis of proteins and protein networks can promote novel insights into disease features of chronic pain. This may facilitate the development of mechanism-based therapies, which are urgently required for chronic pain syndromes (Gereau et al., 2014; Baron et al., 2017). We are confident that the pain community will quickly integrate such a protein-centric and systems-inspired view. To expedite this process, we comprehensively characterized the proteome and its pathology-associated dynamics along the beginning of the pain axis. Our results provide the necessary foundation for future studies examining the protein signature of chronic pain syndromes in different regions and in time.
All relevant data are contained within the manuscript: All datasets (generated and analyzed ones) for this study are included in the manuscript and the Supplementary Files. Moreover, all data is freely available in the searchable online database painproteome.em.mpg.de. Raw DIA-MS data was deposited in the PeptideAtlas repository (http://www.peptideatlas.org/).
AMB performed the comparative and bioinformatics analysis of data and helped write the manuscript. JRS performed behavior experiments and biochemistry and helped write the manuscript. J-HS developed the database with the help of JRS. DG-V conceived the project and supervised the analysis of data. MS conceived, designed, and guided all parts of the project, and wrote the manuscript.
This work was supported by the Deutsche Forschungsgemeinschaft (Emmy Noether-Program: SCHM 2533/2-1 to MS; research grants: SCHM 2533/4-1 to MS, GO 2481/3-1 to DGV), the Max Planck Society (Ph.D. fellowship to JRS, student IMPRS fellowship to AMB), a Ph.D. studentship from the Wellcome Trust to AMB, and the Max von Frey-Prize 2015 awarded to MS by the German Pain Society (DGSS) and Astellas Pharma.
MS received travel support by the German Pain Society (DGSS), which was sponsored by Astellas Pharma GmbH (Germany). DG-V received research support from Biognosys AG (Zurich, Switzerland).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the team of the MPI animal facility for their excellent work. We are also grateful to the MPI IT-team for their help with the database.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2018.00259/full#supplementary-material
Supplementary Figure 1. Quantification of unilateral (ipsilateral, ipsi) mechanical allodynia induced by SNI. The graph depicts average withdrawal latencies to innocuous mechanical stimulation of ipsi- and contralateral (contra, untreated control) paws before (pre) and 4 weeks (4 wk) after Sham and SNI-surgery, respectively. Mechanical allodynia is only observed 4 weeks after surgery in SNI-treated animals as indicated by a reduction of the withdrawal thresholds of ipsi paws compared to contra paws (N = 12 animals per condition). One-way ANOVA followed by Holm-Sidak's multiple comparison test, ****p < 0.0001 of corresponding ipsi vs. contra paws of SNI-treated animals.
Supplementary Figure 2. Regression analysis across Sham sample replicates. Sham replicates (control, C1-C3) are plotted per tissue, with scatter plots and Pearson's correlation coefficient shown per interaction. Protein intensity distributions (per replicate) are shown on the diagonal as kernel density estimates.
Supplementary Figure 3. Representation of gene products (top 50) of indicated sensory neuron subpopulations published by Usoskin et al. (2015) in our DRG proteome of Sham mice.
Supplementary Figure 4. DIA-MS: comparison of Sham with SNI samples. (A) Unsupervised clustering of z-scored normalized protein intensities from all replicates of SN, DRG, and SC tissue across SNI (pain, P1-P3) and Sham (control, C1-C3) replicates. The difference of replicate C2_DRG to other DRG samples is unexpected and likely explained by variability in sample preparation. Different colors of the y-axis dendrogram visualize exemplary clusters of proteins. (B) Regression analysis across SNI (pain, P1-P3) and Sham (control, C1-C3) replicates. Replicates are plotted per tissue (SN, DRG, and SC) with Pearson's correlation coefficient shown for each interaction.
Supplementary Table 1. The pan-mouse library.
Supplementary Table 2. DIA-MS results of proteins identified in tissues of Sham mice.
Supplementary Table 3. Pathway analysis in tissues of Sham mice.
Supplementary Table 4. List of proteins enriched in the PNS of Sham mice compared to brain areas.
Supplementary Table 5. List of proteins enriched in the SC of Sham mice compared to brain areas.
Supplementary Table 6. Comparisons of published studies with our PNS proteome, PNS-enriched proteome, SC proteome, and SC-enriched proteome.
Supplementary Table 7. Comparisons of our data to PGD, HPGDB and conserved DRG-enriched transcripts.
Supplementary Table 8. Proteins significantly regulated upon SNI within the PNS proteome, PNS-enriched proteome, SC proteome, and SC-enriched proteome.
Supplementary Table 9. Comparisons of published studies with proteins significantly regulated upon SNI within the PNS proteome and the PNS-enriched proteome.
Supplementary Table 10. Retrospective analysis of data on the DRG membrane proteome previously published by our laboratory.
Supplementary Table 11. Top biological networks harboring DRG proteins significantly regulated upon SNI.
Supplementary Table 12. Top biological networks harboring SN proteins significantly regulated upon SNI.
Supplementary Table 13. Top biological networks harboring SC proteins significantly regulated upon SNI.
Akude, E., Zherebitskaya, E., Chowdhury, S. K., Smith, D. R., Dobrowsky, R. T., and Fernyhough, P. (2011). Diminished superoxide generation is associated with respiratory chain dysfunction and changes in the mitochondrial proteome of sensory neurons from diabetic rats. Diabetes 60, 288–297. doi: 10.2337/db10-0818
Alberts, B. (1998). The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell 92, 291–294.
Anczuków, O., Akerman, M., Clery, A., Wu, J., Shen, C., Shirole, N. H., et al. (2015). SRSF1-regulated alternative splicing in breast cancer. Mol. Cell 60, 105–117. doi: 10.1016/j.molcel.2015.09.005
Anczuków, O., and Krainer, A. R. (2015). The spliceosome, a potential Achilles heel of MYC-driven tumors. Genome Med. 7:107. doi: 10.1186/s13073-015-0234-3
Anderson, C. A., Massey, D. C., Barrett, J. C., Prescott, N. J., Tremelling, M., Fisher, S. A., et al. (2009). Investigation of Crohn's disease risk loci in ulcerative colitis further defines their molecular relationship. Gastroenterology 136, 523–529 e3. doi: 10.1053/j.gastro.2008.10.032
Antunes-Martins, A., Perkins, J. R., Lees, J., Hildebrandt, T., Orengo, C., and Bennett, D. L. (2013). Systems biology approaches to finding novel pain mediators. Wiley Interdiscip. Rev. Syst. Biol. Med. 5, 11–35. doi: 10.1002/wsbm.1192
Baloh, R. H. (2008). Mitochondrial dynamics and peripheral neuropathy. Neuroscientist 14, 12–18. doi: 10.1177/1073858407307354
Barabási, A. L., Gulbahce, N., and Loscalzo, J. (2011). Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 12, 56–68. doi: 10.1038/nrg2918
Barley, K., Dracheva, S., and Byne, W. (2009). Subcortical oligodendrocyte- and astrocyte-associated gene expression in subjects with schizophrenia, major depression and bipolar disorder. Schizophr. Res. 112, 54–64. doi: 10.1016/j.schres.2009.04.019
Baron, R., Maier, C., Attal, N., Binder, A., Bouhassira, D., Cruccu, G., et al. (2017). Peripheral neuropathic pain: a mechanism-related organizing principle based on sensory profiles. Pain 158, 261–272. doi: 10.1097/j.pain.0000000000000753
Bartus, K., James, N. D., Didangelos, A., Bosch, K. D., Verhaagen, J., Yanez-Munoz, R. J., et al. (2014). Large-scale chondroitin sulfate proteoglycan digestion with chondroitinase gene therapy leads to reduced pathology and modulates macrophage phenotype following spinal cord contusion injury. J. Neurosci. 34, 4822–4836. doi: 10.1523/JNEUROSCI.4369-13.2014
Berta, T., Perrin, F. E., Pertin, M., Tonello, R., Liu, Y. C., Chamessian, A., et al. (2017a). Gene expression profiling of cutaneous injured and non-injured nociceptors in SNI animal model of neuropathic pain. Sci. Rep. 7:9367. doi: 10.1038/s41598-017-08865-3
Berta, T., Qadri, Y., Tan, P. H., and Ji, R. R. (2017b). Targeting dorsal root ganglia and primary sensory neurons for the treatment of chronic pain. Expert Opin. Ther. Targets 21, 695–703. doi: 10.1080/14728222.2017.1328057
Borsook, D., and Kalso, E. (2013). Transforming pain medicine: adapting to science and society. Eur. J. Pain 17, 1109–1125. doi: 10.1002/j.1532-2149.2013.00297.x
Borsook, D., Hargreaves, R., Bountra, C., and Porreca, F. (2014). Lost but making progress–Where will new analgesic drugs come from? Sci. Transl. Med. 6:249sr243. doi: 10.1126/scitranslmed.3008320
Breivik, H. (2013). European pain management discussion forum. J. Pain Palliat. Care Pharmacother. 27, 190–191. doi: 10.3109/15360288.2013.788607
Brennan, K. M., Bai, Y., Pisciotta, C., Wang, S., Feely, S. M., Hoegger, M., et al. (2015). Absence of dystrophin related protein-2 disrupts cajal bands in a patient with charcot-marie-tooth disease. Neuromuscul. Disord. 25, 786–793. doi: 10.1016/j.nmd.2015.07.001
Bresell, A., Weinander, R., Lundqvist, G., Raza, H., Shimoji, M., Sun, T. H., et al. (2005). Bioinformatic and enzymatic characterization of the MAPEG superfamily. FEBS J. 272, 1688–1703. doi: 10.1111/j.1742-4658.2005.04596.x
Bruderer, R., Bernhardt, O. M., Gandhi, T., Miladinovic, S. M., Cheng, L. Y., Messner, S., et al. (2015). Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics 14, 1400–1410. doi: 10.1074/mcp.M114.044305
Bruderer, R., Bernhardt, O. M., Gandhi, T., Xuan, Y., Sondermann, J., Schmidt, M., et al. (2017a). Optimization of experimental parameters in data-independent mass spectrometry significantly increases depth and reproducibility of results. Mol. Cell. Proteomics 16, 2296–2309. doi: 10.1074/mcp.RA117.000314
Bruderer, R., Sondermann, J., Tsou, C. C., Barrantes-Freer, A., Stadelmann, C., Nesvizhskii, A. I., et al. (2017b). New targeted approaches for the quantification of data-independent acquisition mass spectrometry. Proteomics 17:1700021. doi: 10.1002/pmic.201700021
Carvalho, P. C., Han, X., Xu, T., Cociorva, D., Carvalho Mda, G., Barbosa, V. C., et al. (2010). XDIA: improving on the label-free data-independent analysis. Bioinformatics 26, 847–848. doi: 10.1093/bioinformatics/btq031
Cerciello, F., Choi, M., Nicastri, A., Bausch-Fluck, D., Ziegler, A., Vitek, O., et al. (2013). Identification of a seven glycopeptide signature for malignant pleural mesothelioma in human serum by selected reaction monitoring. Clin. Proteomics 10:16. doi: 10.1186/1559-0275-10-16
Chapman, C. R., Tuckett, R. P., and Song, C. W. (2008). Pain and stress in a systems perspective: reciprocal neural, endocrine, and immune interactions. J. Pain 9, 122–145. doi: 10.1016/j.jpain.2007.09.006
Chen, H., Zhu, Z., Zhu, Y., Wang, J., Mei, Y., and Cheng, Y. (2015). Pathway mapping and development of disease-specific biomarkers: protein-based network biomarkers. J. Cell. Mol. Med. 19, 297–314. doi: 10.1111/jcmm.12447
Cobos, E. J., Nickerson, C. A., Gao, F., Chandran, V., Bravo-Caparros, I., Gonzalez-Cano, R., et al. (2018). Mechanistic differences in neuropathic pain modalities revealed by correlating behavior with global expression profiling. Cell Rep. 22, 1301–1312. doi: 10.1016/j.celrep.2018.01.006
Costigan, M., Scholz, J., and Woolf, C. J. (2009). Neuropathic pain: a maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 32, 1–32. doi: 10.1146/annurev.neuro.051508.135531
de Monasterio-Schrader, P., Jahn, O., Tenzer, S., Wichert, S. P., Patzig, J., and Werner, H. B. (2012). Systematic approaches to central nervous system myelin. Cell. Mol. Life Sci. 69, 2879–2894. doi: 10.1007/s00018-012-0958-9
Decosterd, I., and Woolf, C. J. (2000). Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158. doi: 10.1016/S0304-3959(00)00276-1
Denk, F., and McMahon, S. B. (2012). Chronic pain: emerging evidence for the involvement of epigenetics. Neuron 73, 435–444. doi: 10.1016/j.neuron.2012.01.012
Diatchenko, L., Fillingim, R. B., Smith, S. B., and Maixner, W. (2013). The phenotypic and genetic signatures of common musculoskeletal pain conditions. Nat. Rev. Rheumatol. 9, 340–350. doi: 10.1038/nrrheum.2013.43
Diatchenko, L., Nackley, A. G., Tchivileva, I. E., Shabalina, S. A., and Maixner, W. (2007). Genetic architecture of human pain perception. Trends Genet. 23, 605–613. doi: 10.1016/j.tig.2007.09.004
Diatchenko, L., Slade, G. D., Nackley, A. G., Bhalang, K., Sigurdsson, A., Belfer, I., et al. (2005). Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum. Mol. Genet. 14, 135–143. doi: 10.1093/hmg/ddi013
Dib-Hajj, S. D., Black, J. A., and Waxman, S. G. (2009). Voltage-gated sodium channels: therapeutic targets for pain. Pain Med. 10, 1260–1269. doi: 10.1111/j.1526-4637.2009.00719.x
Doehring, A., Geisslinger, G., and Lotsch, J. (2011). Epigenetics in pain and analgesia: an imminent research field. Eur. J. Pain 15, 11–16. doi: 10.1016/j.ejpain.2010.06.004
Domon, B., and Aebersold, R. (2010). Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721. doi: 10.1038/nbt.1661
Ebhardt, H. A., Root, A., Sander, C., and Aebersold, R. (2015). Applications of targeted proteomics in systems biology and translational medicine. Proteomics 15, 3193–3208. doi: 10.1002/pmic.201500004
Etique, N., Verzeaux, L., Dedieu, S., and Emonard, H. (2013). LRP-1: a checkpoint for the extracellular matrix proteolysis. Biomed Res. Int. 2013:152163. doi: 10.1155/2013/152163
Flatters, S. J. (2015). The contribution of mitochondria to sensory processing and pain. Prog. Mol. Biol. Transl. Sci. 131, 119–146. doi: 10.1016/bs.pmbts.2014.12.004
Franceschini, A., Szklarczyk, D., Frankild, S., Kuhn, M., Simonovic, M., Roth, A., et al. (2013). STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815. doi: 10.1093/nar/gks1094
Friedrich, T., Tavraz, N. N., and Junghans, C. (2016). ATP1A2 mutations in migraine: seeing through the facets of an ion pump onto the neurobiology of disease. Front. Physiol. 7:239. doi: 10.3389/fphys.2016.00239
Fukushi, J., Makagiansar, I. T., and Stallcup, W. B. (2004). NG2 proteoglycan promotes endothelial cell motility and angiogenesis via engagement of galectin-3 and alpha3beta1 integrin. Mol. Biol. Cell 15, 3580–3590. doi: 10.1091/mbc.e04-03-0236
Gazerani, P., and Vinterhoj, H. S. H. (2016). ‘Omics’: an emerging field in pain research and management. Future Neurol. 11, 255–265. doi: 10.2217/fnl-2016-0018
Gereau, R. W., Sluka, K. A., Maixner, W., Savage, S. R., Price, T. J., Murinson, B. B., et al. (2014). A pain research agenda for the 21st century. J. Pain 15, 1203–1214. doi: 10.1016/j.jpain.2014.09.004
Gettins, P. G. (2002). Serpin structure, mechanism, and function. Chem. Rev. 102, 4751–4804. doi: 10.1021/cr010170
Gettins, P. G., Simonovic, M., and Volz, K. (2002). Pigment epithelium-derived factor (PEDF), a serpin with potent anti-angiogenic and neurite outgrowth-promoting properties. Biol. Chem. 383, 1677–1682. doi: 10.1515/BC.2002.188
Geyer, P. E., Holdt, L. M., Teupser, D., and Mann, M. (2017). Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 13:942. doi: 10.15252/msb.20156297
Geyer, P. E., Kulak, N. A., Pichler, G., Holdt, L. M., Teupser, D., and Mann, M. (2016). Plasma proteome profiling to assess human health and disease. Cell Syst. 2, 185–195. doi: 10.1016/j.cels.2016.02.015
Gilron, I., Jensen, T. S., and Dickenson, A. H. (2013). Combination pharmacotherapy for management of chronic pain: from bench to bedside. Lancet Neurol. 12, 1084–1095. doi: 10.1016/S1474-4422(13)70193-5
Grace, P. M., Hurley, D., Barratt, D. T., Tsykin, A., Watkins, L. R., Rolan, P. E., et al. (2012). Harnessing pain heterogeneity and RNA transcriptome to identify blood-based pain biomarkers: a novel correlational study design and bioinformatics approach in a graded chronic constriction injury model. J. Neurochem. 122, 976–994. doi: 10.1111/j.1471-4159.2012.07833.x
Gravel, M., Robert, F., Kottis, V., Gallouzi, I. E., Pelletier, J., and Braun, P. E. (2009). 2′,3′-Cyclic nucleotide 3′-phosphodiesterase: a novel RNA-binding protein that inhibits protein synthesis. J. Neurosci. Res. 87, 1069–1079. doi: 10.1002/jnr.21939
Guarnieri, M., Brayton, C., DeTolla, L., Forbes-McBean, N., Sarabia-Estrada, R., and Zadnik, P. (2012). Safety and efficacy of buprenorphine for analgesia in laboratory mice and rats. Lab. Anim. 41, 337–343. doi: 10.1038/laban.152
Haroutounian, S., Nikolajsen, L., Bendtsen, T. F., Finnerup, N. B., Kristensen, A. D., Hasselstrom, J. B., et al. (2014). Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain 155, 1272–1279. doi: 10.1016/j.pain.2014.03.022
Hayes, J. D., Flanagan, J. U., and Jowsey, I. R. (2005). Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 45, 51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857
Hirai, T., Mulpuri, Y., Cheng, Y., Xia, Z., Li, W., Ruangsri, S., et al. (2017). Aberrant plasticity of peripheral sensory axons in a painful neuropathy. Sci. Rep. 7:3407. doi: 10.1038/s41598-017-03390-9
Hu, G., Huang, K., Hu, Y., Du, G., Xue, Z., Zhu, X., et al. (2016). Single-cell RNA-seq reveals distinct injury responses in different types of DRG sensory neurons. Sci. Rep. 6:31851. doi: 10.1038/srep31851
Huang da, W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi: 10.1038/nprot.2008.211
Huang, H. L., Cendan, C. M., Roza, C., Okuse, K., Cramer, R., Timms, J. F., et al. (2008). Proteomic profiling of neuromas reveals alterations in protein composition and local protein synthesis in hyper-excitable nerves. Mol. Pain 4:33. doi: 10.1186/1744-8069-4-33
Hui, X., Zhu, W., Wang, Y., Lam, K. S., Zhang, J., Wu, D., et al. (2009). Major urinary protein-1 increases energy expenditure and improves glucose intolerance through enhancing mitochondrial function in skeletal muscle of diabetic mice. J. Biol. Chem. 284, 14050–14057. doi: 10.1074/jbc.M109.001107
Hurst, J. L., Payne, C. E., Nevison, C. M., Marie, A. D., Humphries, R. E., Robertson, D. H., et al. (2001). Individual recognition in mice mediated by major urinary proteins. Nature 414, 631–634. doi: 10.1038/414631a
Hüttenhain, R., Soste, M., Selevsek, N., Rost, H., Sethi, A., Carapito, C., et al. (2012). Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci. Transl. Med. 4:142ra194. doi: 10.1126/scitranslmed.3003989
Imbe, H., Okamoto, K., Kadoya, T., Horie, H., and Senba, E. (2003). Galectin-1 is involved in the potentiation of neuropathic pain in the dorsal horn. Brain Res. 993, 72–83. doi: 10.1016/j.brainres.2003.08.064
Inoue, M., Xie, W., Matsushita, Y., Chun, J., Aoki, J., and Ueda, H. (2008). Lysophosphatidylcholine induces neuropathic pain through an action of autotaxin to generate lysophosphatidic acid. Neuroscience 152, 296–298. doi: 10.1016/j.neuroscience.2007.12.041
Jamieson, D. G., Moss, A., Kennedy, M., Jones, S., Nenadic, G., Robertson, D. L., et al. (2014). The pain interactome: connecting pain-specific protein interactions. Pain 155, 2243–2252. doi: 10.1016/j.pain.2014.06.020
Jeong, H., Na, Y. J., Lee, K., Kim, Y. H., Lee, Y., Kang, M., et al. (2016). High-resolution transcriptome analysis reveals neuropathic pain gene-expression signatures in spinal microglia after nerve injury. Pain 157, 964–976. doi: 10.1097/j.pain.0000000000000470
Ji, R. R., Chamessian, A., and Zhang, Y. Q. (2016). Pain regulation by non-neuronal cells and inflammation. Science 354, 572–577. doi: 10.1126/science.aaf8924
Kanamori, M., Sandy, P., Marzinotto, S., Benetti, R., Kai, C., Hayashizaki, Y., et al. (2003). The PDZ protein tax-interacting protein-1 inhibits beta-catenin transcriptional activity and growth of colorectal cancer cells. J. Biol. Chem. 278, 38758–38764. doi: 10.1074/jbc.M306324200
Kiguchi, N., Kobayashi, Y., Saika, F., Sakaguchi, H., Maeda, T., and Kishioka, S. (2015). Peripheral interleukin-4 ameliorates inflammatory macrophage-dependent neuropathic pain. Pain 156, 684–693. doi: 10.1097/j.pain.0000000000000097
Kim, D. S., Li, K. W., Boroujerdi, A., Peter Yu, Y., Zhou, C. Y., Deng, P., et al. (2012). Thrombospondin-4 contributes to spinal sensitization and neuropathic pain states. J. Neurosci. 32, 8977–8987. doi: 10.1523/JNEUROSCI.6494-11.2012
Kitsak, M., Sharma, A., Menche, J., Guney, E., Ghiassian, S. D., Loscalzo, J., et al. (2016). Tissue specificity of human disease module. Sci. Rep. 6:35241. doi: 10.1038/srep35241
Koh, S. J., Choi, Y., Kim, B. G., Lee, K. L., Kim, D. W., Kim, J. H., et al. (2016). Matricellular protein periostin mediates intestinal inflammation through the activation of nuclear factor kappab signaling. PLoS ONE 11:e0149652. doi: 10.1371/journal.pone.0149652
Koyanagi, S., Kusunose, N., Taniguchi, M., Akamine, T., Kanado, Y., Ozono, Y., et al. (2016). Glucocorticoid regulation of ATP release from spinal astrocytes underlies diurnal exacerbation of neuropathic mechanical allodynia. Nat. Commun. 7:13102. doi: 10.1038/ncomms13102
Krämer, A, Green, J., Pollard, J. Jr., and Tugendreich, S. (2014). Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530. doi: 10.1093/bioinformatics/btt703
Krames, E. S. (2014). The role of the dorsal root ganglion in the development of neuropathic pain. Pain Med. 15, 1669–1685. doi: 10.1111/pme.12413
Krishnaswamy, V. R., Manikandan, M., Munirajan, A. K., Vijayaraghavan, D., and Korrapati, P. S. (2014). Expression and integrity of dermatopontin in chronic cutaneous wounds: a crucial factor in impaired wound healing. Cell Tissue Res. 358, 833–841. doi: 10.1007/s00441-014-2000-z
Lacroix-Fralish, M. L., Ledoux, J. B., and Mogil, J. S. (2007). The pain genes database: an interactive web browser of pain-related transgenic knockout studies. Pain 131:3.e1–4. doi: 10.1016/j.pain.2007.04.041
Lamar, K. M., Bogdanovich, S., Gardner, B. B., Gao, Q. Q., Miller, T., Earley, J. U., et al. (2016). Overexpression of latent TGFbeta binding protein 4 in muscle ameliorates muscular dystrophy through myostatin and TGFbeta. PLoS Genet. 12:e1006019. doi: 10.1371/journal.pgen.1006019
Lange, M., Kaynak, B., Forster, U. B., Tonjes, M., Fischer, J. J., Grimm, C., et al. (2008). Regulation of muscle development by DPF3, a novel histone acetylation and methylation reader of the BAF chromatin remodeling complex. Genes Dev. 22, 2370–2384. doi: 10.1101/gad.471408
Li, K. C., Wang, F., Zhong, Y. Q., Lu, Y. J., Wang, Q., Zhang, F. X., et al. (2011a). Reduction of follistatin-like 1 in primary afferent neurons contributes to neuropathic pain hypersensitivity. Cell Res. 21, 697–699. doi: 10.1038/cr.2011.43
Li, K. C., Zhang, F. X., Li, C. L., Wang, F., Yu, M. Y., Zhong, Y. Q., et al. (2011b). Follistatin-like 1 suppresses sensory afferent transmission by activating Na+,K+-ATPase. Neuron 69, 974–987. doi: 10.1016/j.neuron.2011.01.022
Liem, L., Russo, M., Huygen, F. J., Van Buyten, J. P., Smet, I., Verrills, P., et al. (2013). A multicenter, prospective trial to assess the safety and performance of the spinal modulation dorsal root ganglion neurostimulator system in the treatment of chronic pain. Neuromodulation 16, 471–482; discussion 482. doi: 10.1111/ner.12072
Lin, J. P., Mironova, Y. A., Shrager, P., and Giger, R. J. (2017). LRP1 regulates peroxisome biogenesis and cholesterol homeostasis in oligodendrocytes and is required for proper CNS myelin development and repair. Elife 6:30498. doi: 10.7554/eLife.30498
Liu, Y., Beyer, A., and Aebersold, R. (2016). On the dependency of cellular protein levels on mRNA abundance. Cell 165, 535–550. doi: 10.1016/j.cell.2016.03.014
Lopes, D. M., Denk, F., and McMahon, S. B. (2017). The molecular fingerprint of dorsal root and trigeminal ganglion neurons. Front. Mol. Neurosci. 10:304. doi: 10.3389/fnmol.2017.00304
Lu, A., Wisniewski, J. R., and Mann, M. (2009). Comparative proteomic profiling of membrane proteins in rat cerebellum, spinal cord, and sciatic nerve. J. Proteome Res. 8, 2418–2425. doi: 10.1021/pr8010364
Lunn, C. A. (2010). Membrane proteins as drug targets. Preface. Prog. Mol. Biol. Transl. Sci. 91, 1–247. doi: 10.1016/S1877-1173(10)91012-0
MacKenzie, J. A., and Payne, R. M. (2007). Mitochondrial protein import and human health and disease. Biochim. Biophys. Acta 1772, 509–523. doi: 10.1016/j.bbadis.2006.12.002
Manninen, A., and Varjosalo, M. (2017). A proteomics view on integrin-mediated adhesions. Proteomics 17:1600022. doi: 10.1002/pmic.201600022
Meloto, C. B., Benavides, R., Lichtenwalter, R. N., Wen, X., Tugarinov, N., Zorina-Lichtenwalter, K., et al. (2017). The Human Pain Genetics Database (HPGDB): a resource dedicated to human pain genetics research. Pain 159, 749–763. doi: 10.1097/j.pain.0000000000001135
Michalski, A., Cox, J., and Mann, M. (2011). More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J. Proteome Res. 10, 1785–1793. doi: 10.1021/pr101060v
Minett, M. S., Quick, K., and Wood, J. N. (2013). Behavioral measures of pain thresholds. Curr. Protoc. Mouse Biol. 1, 383–412. doi: 10.1002/9780470942390.mo110116
Miyajima, N., Maruyama, S., Nonomura, K., and Hatakeyama, S. (2009). TRIM36 interacts with the kinetochore protein CENP-H and delays cell cycle progression. Biochem. Biophys. Res. Commun. 381, 383–387. doi: 10.1016/j.bbrc.2009.02.059
Miyamoto, T., Morita, K., Takemoto, D., Takeuchi, K., Kitano, Y., Miyakawa, T., et al. (2005). Tight junctions in Schwann cells of peripheral myelinated axons: a lesson from claudin-19-deficient mice. J. Cell Biol. 169, 527–538. doi: 10.1083/jcb.200501154
Mogil, J. S. (2012). Pain genetics: past, present and future. Trends Genet. 28, 258–266. doi: 10.1016/j.tig.2012.02.004
Murota, H., Lingli, Y., and Katayama, I. (2017). Periostin in the pathogenesis of skin diseases. Cell. Mol. Life Sci. 74, 4321–4328. doi: 10.1007/s00018-017-2647-1
Niederberger, E. (2014). [Epigenetics and pain]. Anaesthesist 63, 63–69. doi: 10.1007/s00101-013-2274-7
Olausson, P., Gerdle, B., Ghafouri, N., Larsson, B., and Ghafouri, B. (2012). Identification of proteins from interstitium of trapezius muscle in women with chronic myalgia using microdialysis in combination with proteomics. PLoS ONE 7:e52560. doi: 10.1371/journal.pone.0052560
Olausson, P., Gerdle, B., Ghafouri, N., Sjostrom, D., Blixt, E., and Ghafouri, B. (2015). Protein alterations in women with chronic widespread pain–An explorative proteomic study of the trapezius muscle. Sci. Rep. 5:11894. doi: 10.1038/srep11894
Oti, M., Snel, B., Huynen, M. A., and Brunner, H. G. (2006). Predicting disease genes using protein-protein interactions. J. Med. Genet. 43, 691–698. doi: 10.1136/jmg.2006.041376
Panchaud, A., Scherl, A., Shaffer, S. A., von Haller, P. D., Kulasekara, H. D., Miller, S. I., et al. (2009). Precursor acquisition independent from ion count: how to dive deeper into the proteomics ocean. Anal. Chem. 81, 6481–6488. doi: 10.1021/ac900888s
Parisien, M., Khoury, S., Chabot-Dore, A. J., Sotocinal, S. G., Slade, G. D., Smith, S. B., et al. (2017). Effect of human genetic variability on gene expression in dorsal root ganglia and association with pain phenotypes. Cell Rep. 19, 1940–1952. doi: 10.1016/j.celrep.2017.05.018
Patapoutian, A., Tate, S., and Woolf, C. J. (2009). Transient receptor potential channels: targeting pain at the source. Nat. Rev. Drug Discov. 8, 55–68. doi: 10.1038/nrd2757
Patti, G. J., Yanes, O., Shriver, L. P., Courade, J. P., Tautenhahn, R., Manchester, M., et al. (2012). Metabolomics implicates altered sphingolipids in chronic pain of neuropathic origin. Nat. Chem. Biol. 8, 232–234. doi: 10.1038/nchembio.767
Patzig, J., Jahn, O., Tenzer, S., Wichert, S. P., de Monasterio-Schrader, P., Rosfa, S., et al. (2011). Quantitative and integrative proteome analysis of peripheral nerve myelin identifies novel myelin proteins and candidate neuropathy loci. J. Neurosci. 31, 16369–16386. doi: 10.1523/JNEUROSCI.4016-11.2011
Peng, C., Li, L., Zhang, M. D., Bengtsson Gonzales, C., Parisien, M., Belfer, I., et al. (2017). miR-183 cluster scales mechanical pain sensitivity by regulating basal and neuropathic pain genes. Science 356, 1168–1171. doi: 10.1126/science.aam7671
Perkins, J. R., Lees, J., Antunes-Martins, A., Diboun, I., McMahon, S. B., Bennett, D. L., et al. (2013). PainNetworks: a web-based resource for the visualisation of pain-related genes in the context of their network associations. Pain 154, e2581–e2512. doi: 10.1016/j.pain.2013.09.003
Philp, D., and Kleinman, H. K. (2010). Animal studies with thymosin beta, a multifunctional tissue repair and regeneration peptide. Ann. N. Y. Acad. Sci. 1194, 81–86. doi: 10.1111/j.1749-6632.2010.05479.x
Price, T. J., and Gold, M. S. (2017). From mechanism to cure: renewing the goal to eliminate the disease of pain. Pain Med. doi: 10.1093/pm/pnx108. [Epub ahead of print].
Puljak, L., Kojundzic, S. L., Hogan, Q. H., and Sapunar, D. (2009). Lidocaine injection into the rat dorsal root ganglion causes neuroinflammation. Anesth. Analg. 108, 1021–1026. doi: 10.1213/ane.0b013e318193873e
Raouf, R., Quick, K., and Wood, J. N. (2010). Pain as a channelopathy. J. Clin. Invest. 120, 3745–3752. doi: 10.1172/JCI43158
Ray, P., Torck, A., Quigley, L., Wangzhou, A., Neiman, M., Rao, C., et al. (2018). Comparative transcriptome profiling of the human and mouse dorsal root ganglia: an RNA-seq-based resource for pain and sensory neuroscience research. Pain 159, 1325–1345. doi: 10.1097/j.pain.0000000000001217
Rouwette, T., Avenali, L., Sondermann, J., Narayanan, P., Gomez-Varela, D., and Schmidt, M. (2015). Modulation of nociceptive ion channels and receptors via protein-protein interactions: implications for pain relief. Channels 9, 175–185. doi: 10.1080/19336950.2015.1051270
Rouwette, T., Sondermann, J., Avenali, L., Gomez-Varela, D., and Schmidt, M. (2016). Standardized profiling of the membrane-enriched proteome of mouse dorsal root ganglia (DRG) provides novel insights into chronic pain. Mol. Cell. Proteomics 15, 2152–2168. doi: 10.1074/mcp.M116.058966
Rychli, K., Huber, K., and Wojta, J. (2009). Pigment epithelium-derived factor (PEDF) as a therapeutic target in cardiovascular disease. Expert Opin. Ther. Targets 13, 1295–1302. doi: 10.1517/14728220903241641
Sajic, T., Liu, Y., and Aebersold, R. (2015). Using data-independent, high-resolution mass spectrometry in protein biomarker research: perspectives and clinical applications. Proteomics Clin. Appl. 9, 307–321. doi: 10.1002/prca.201400117
Sapio, M. R., Goswami, S. C., Gross, J. R., Mannes, A. J., and Iadarola, M. J. (2016). Transcriptomic analyses of genes and tissues in inherited sensory neuropathies. Exp. Neurol. 283(Pt A), 375–395. doi: 10.1016/j.expneurol.2016.06.023
Sapunar, D., Kostic, S., Banozic, A., and Puljak, L. (2012). Dorsal root ganglion - a potential new therapeutic target for neuropathic pain. J. Pain Res. 5, 31–38. doi: 10.2147/JPR.S26603
Schmidt, O., Pfanner, N., and Meisinger, C. (2010). Mitochondrial protein import: from proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 11, 655–667. doi: 10.1038/nrm2959
Schubert, O. T., Gillet, L. C., Collins, B. C., Navarro, P., Rosenberger, G., Wolski, W. E., et al. (2015). Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat. Protoc. 10, 426–441. doi: 10.1038/nprot.2015.015
Schuette, V., Embgenbroich, M., Ulas, T., Welz, M., Schulte-Schrepping, J., Draffehn, A. M., et al. (2016). Mannose receptor induces T-cell tolerance via inhibition of CD45 and up-regulation of CTLA-4. Proc. Natl. Acad. Sci. U.S.A. 113, 10649–10654. doi: 10.1073/pnas.1605885113
Schwanhausser, B., Busse, D., Li, N., Dittmar, G., Schuchhardt, J., Wolf, J., et al. (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342. doi: 10.1038/nature10098
Sharma, K., Schmitt, S., Bergner, C. G., Tyanova, S., Kannaiyan, N., Manrique-Hoyos, N., et al. (2015). Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 18, 1819–1831. doi: 10.1038/nn.4160
Sherman, D. L., Fabrizi, C., Gillespie, C. S., and Brophy, P. J. (2001). Specific disruption of a schwann cell dystrophin-related protein complex in a demyelinating neuropathy. Neuron 30, 677–687. doi: 10.1016/S0896-6273(01)00327-0
Shi, X., Barnes, R. O., Chen, L., Shajahan-Haq, A. N., Hilakivi-Clarke, L., Clarke, R., et al. (2015). BMRF-Net: a software tool for identification of protein interaction subnetworks by a bagging Markov random field-based method. Bioinformatics 31, 2412–2414. doi: 10.1093/bioinformatics/btv137
Simonetti, M., Hagenston, A. M., Vardeh, D., Freitag, H. E., Mauceri, D., Lu, J., et al. (2013). Nuclear calcium signaling in spinal neurons drives a genomic program required for persistent inflammatory pain. Neuron 77, 43–57. doi: 10.1016/j.neuron.2012.10.037
Sommer, C. (2016). Exploring pain pathophysiology in patients. Science 354, 588–592. doi: 10.1126/science.aaf8935
Sorge, R. E., Trang, T., Dorfman, R., Smith, S. B., Beggs, S., Ritchie, J., et al. (2012). Genetically determined P2X7 receptor pore formation regulates variability in chronic pain sensitivity. Nat. Med. 18, 595–599. doi: 10.1038/nm.2710
Sui, P., Watanabe, H., Ossipov, M. H., Bakalkin, G., Artemenko, K., and Bergquist, J. (2014). Proteomics of neuropathic pain: proteins and signaling pathways affected in a rat model. J. Proteome Res. 13, 3957–3965. doi: 10.1021/pr500241q
Tajerian, M., and Clark, J. D. (2015). The role of the extracellular matrix in chronic pain following injury. Pain 156, 366–370. doi: 10.1097/01.j.pain.0000460323.80020.9d
Takadate, T., Onogawa, T., Fukuda, T., Motoi, F., Suzuki, T., Fujii, K., et al. (2013). Novel prognostic protein markers of resectable pancreatic cancer identified by coupled shotgun and targeted proteomics using formalin-fixed paraffin-embedded tissues. Int. J. Cancer 132, 1368–1382. doi: 10.1002/ijc.27797
Thakur, M., Crow, M., Richards, N., Davey, G. I., Levine, E., Kelleher, J. H., et al. (2014). Defining the nociceptor transcriptome. Front. Mol. Neurosci. 7:87. doi: 10.3389/fnmol.2014.00087
Thul, P. J., Akesson, L., Wiking, M., Mahdessian, D., Geladaki, A., Ait Blal, H., et al. (2017). A subcellular map of the human proteome. Science 356:eaal3321. doi: 10.1126/science.aal3321
Treede, R. D., Wagner, T., Kern, K. U., Husstedt, I. W., Arendt, G., Birklein, F., et al. (2013). Mechanism- and experience-based strategies to optimize treatment response to the capsaicin 8% cutaneous patch in patients with localized neuropathic pain. Curr. Med. Res. Opin. 29, 527–538. doi: 10.1185/03007995.2013.781019
Tsou, C. C., Avtonomov, D., Larsen, B., Tucholska, M., Choi, H., Gingras, A. C., et al. (2015). DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 12, 258–264. doi: 10.1038/nmeth.3255
Tyanova, S., Temu, T., Sinitcyn, P., Carlson, A., Hein, M. Y., Geiger, T., et al. (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. doi: 10.1038/nmeth.3901
Uhlen, M., Fagerberg, L., Hallstrom, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347:1260419. doi: 10.1126/science.1260419
Unekawa, M., Ikeda, K., Tomita, Y., Kawakami, K., and Suzuki, N. (2017). Enhanced susceptibility to cortical spreading depression in two types of Na+,K+-ATPase α2 subunit-deficient mice as a model of familial hemiplegic migraine 2. Cephalalgia doi: 10.1177/0333102417738249. [Epub ahead of print].
Usoskin, D., Furlan, A., Islam, S., Abdo, H., Lonnerberg, P., Lou, D., et al. (2015). Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 18, 145–153. doi: 10.1038/nn.3881
Vacca, V., Marinelli, S., Pieroni, L., Urbani, A., Luvisetto, S., and Pavone, F. (2014). Higher pain perception and lack of recovery from neuropathic pain in females: a behavioural, immunohistochemical, and proteomic investigation on sex-related differences in mice. Pain 155, 388–402. doi: 10.1016/j.pain.2013.10.027
van Hecke, O., Kamerman, P. R., Attal, N., Baron, R., Bjornsdottir, G., Bennett, D. L., et al. (2015). Neuropathic pain phenotyping by international consensus (NeuroPPIC) for genetic studies: a NeuPSIG systematic review, Delphi survey, and expert panel recommendations. Pain 156, 2337–2353. doi: 10.1097/j.pain.0000000000000335
Vannahme, C., Gosling, S., Paulsson, M., Maurer, P., and Hartmann, U. (2003). Characterization of SMOC-2, a modular extracellular calcium-binding protein. Biochem. J. 373(Pt 3), 805–814. doi: 10.1042/BJ20030532
Vardeh, D., Mannion, R. J., and Woolf, C. J. (2016). Toward a mechanism-based approach to pain diagnosis. J. Pain 17, T50–T69. doi: 10.1016/j.jpain.2016.03.001
Vaso, A., Adahan, H. M., Gjika, A., Zahaj, S., Zhurda, T., Vyshka, G., et al. (2014). Peripheral nervous system origin of phantom limb pain. Pain 155, 1384–1391. doi: 10.1016/j.pain.2014.04.018
Venable, J. D., Dong, M. Q., Wohlschlegel, J., Dillin, A., and Yates, J. R. (2004). Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods 1, 39–45. doi: 10.1038/nmeth705
Vergetaki, A., Jeschke, U., Vrekoussis, T., Taliouri, E., Sabatini, L., Papakonstanti, E. A., et al. (2014). Galectin-1 overexpression in endometriosis and its regulation by neuropeptides (CRH, UCN) indicating its important role in reproduction and inflammation. PLoS ONE 9:e114229. doi: 10.1371/journal.pone.0114229
Vicuna, L., Strochlic, D. E., Latremoliere, A., Bali, K. K., Simonetti, M., Husainie, D., et al. (2015). The serine protease inhibitor SerpinA3N attenuates neuropathic pain by inhibiting T cell-derived leukocyte elastase. Nat. Med. 21, 518–523. doi: 10.1038/nm.3852
Wang, L., Chopp, M., Szalad, A., Liu, Z., Lu, M., Zhang, L., et al. (2012). Thymosin beta4 promotes the recovery of peripheral neuropathy in type II diabetic mice. Neurobiol. Dis. 48, 546–555. doi: 10.1016/j.nbd.2012.08.002
Wiedemann, N., and Pfanner, N. (2017). Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 86, 685–714. doi: 10.1146/annurev-biochem-060815-014352
Yoon, C., Van Niekerk, E. A., Henry, K., Ishikawa, T., Orita, S., Tuszynski, M. H., et al. (2013). Low-density lipoprotein receptor-related protein 1 (LRP1)-dependent cell signaling promotes axonal regeneration. J. Biol. Chem. 288, 26557–26568. doi: 10.1074/jbc.M113.478552
Zhang, B., Li, H., Riggins, R. B., Zhan, M., Xuan, J., Zhang, Z., et al. (2009). Differential dependency network analysis to identify condition-specific topological changes in biological networks. Bioinformatics 25, 526–532. doi: 10.1093/bioinformatics/btn660
Zhou, X., Menche, J., Barabasi, A. L., and Sharma, A. (2014). Human symptoms-disease network. Nat. Commun. 5:4212. doi: 10.1038/ncomms5212
Zhou, Y., Jiang, L., and Rui, L. (2009). Identification of MUP1 as a regulator for glucose and lipid metabolism in mice. J. Biol. Chem. 284, 11152–11159. doi: 10.1074/jbc.M900754200
Zizzari, I. G., Martufi, P., Battisti, F., Rahimi, H., Caponnetto, S., Bellati, F., et al. (2015). The Macrophage Galactose-Type C-Type Lectin (MGL) Modulates Regulatory T Cell Functions. PLoS ONE 10:e0132617. doi: 10.1371/journal.pone.0132617
Keywords: chronic pain, DIA-MS, proteomics, systems biology, neuropathic pain, dorsal root ganglia, sciatic nerve, spinal cord
Citation: Barry AM, Sondermann JR, Sondermann J-H, Gomez-Varela D and Schmidt M (2018) Region-Resolved Quantitative Proteome Profiling Reveals Molecular Dynamics Associated With Chronic Pain in the PNS and Spinal Cord. Front. Mol. Neurosci. 11:259. doi: 10.3389/fnmol.2018.00259
Received: 19 March 2018; Accepted: 10 July 2018;
Published: 14 August 2018.
Edited by:
Volker Eulenburg, Klinik und Poliklinik für Anästhesiologie und Intensivtherapie, Universitätsklinikum Leipzig, GermanyReviewed by:
Michaela Kress, Innsbruck Medical University, AustriaCopyright © 2018 Barry, Sondermann, Sondermann, Gomez-Varela and Schmidt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuela Schmidt, bXNjaG1pZHRAZW0ubXBnLmRl
David Gomez-Varela, Z29tZXpAZW0ubXBnLmRl
†Present Address: Allison M. Barry, Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, United Kingdom
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.