 Ahmed Eltokhi
Ahmed Eltokhi Gudrun Rappold
Gudrun Rappold Rolf Sprengel
Rolf Sprengel- 1Max Planck Research Group “Molecular Neurobiology”, Max Planck Institute for Medical Research, Heidelberg, Germany
- 2Department of Human Molecular Genetics, Institute of Human Genetics, Heidelberg University, Heidelberg, Germany
- 3Institute for Anatomy and Cell Biology, Heidelberg University, Heidelberg, Germany
The SHANK scaffolding proteins are important organizers for signaling proteins in the postsynapse of excitatory neurons. The functional significance of SHANK proteins becomes apparent by the wide spectrum of neurodevelopmental and neuropsychiatric disorders associated with SHANK variants in human patients. A similar diversity of neuropsychiatric-like phenotypes is described for numerous Shank2 and Shank3 knockout (KO) mouse lines. In this review, we will focus on and discuss the experimental results obtained from different, but genetically related and therefore comparable, Shank2 mouse models. First, we will describe the distinct SHANK2 variant-mediated neurodevelopmental and neuropsychiatric disorders in human patients. Then we will discuss the current knowledge of the expressed SHANK2 isoforms in the mouse, and we will describe the genetic strategies used for generating three conventional and seven conditional Shank2 mouse lines. The distinct impairments i.e., autistic-like and mania-like behavior and the alterations on the molecular, electrophysiological and behavioral levels will be compared between the different Shank2 mouse models. We will present our view as to why in these mouse models a spectrum of phenotypes can arise from similar Shank2 gene manipulations and how Shank2 mutant mice can be used and should be analyzed on the behavioral level in future research.
Introduction
Gene variants of the multi-domain postsynaptic scaffolding proteins included in the SHANK family (also known as ProSAP) are significantly associated with autism spectrum disorders (ASD). After the first publication of a SHANK3 variant in human patients with ASD (Durand et al., 2007), numerous studies confirmed the close link between SHANK variants and ASD and intellectual disability (ID; see https://gene.sfari.org). In humans, three different genes, SHANK 1, 2 and 3 produce several SHANK isoforms via internal promoters and alternative splice products (Lim et al., 1999; for review see Sala et al., 2015). SHANK proteins are master scaffolding proteins in the postsynapse of excitatory neurons. There, they are critically involved in the morphogenesis of spines (Sala et al., 2001). Within the spine, SHANK proteins form a net-like matrix structure (Baron et al., 2006; Hayashi et al., 2009) that serves as a scaffold for the organization of other postsynaptic proteins including N-methyl-D-aspartate receptors (NMDAR), L-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR) and the metabotropic glutamate receptors (mGluRs; Sheng and Kim, 2011; Jacob and Weinberg, 2015; for review see Verpelli et al., 2012). The longest isoforms of SHANK1–3 contain several protein-protein interaction sites: the N-terminal ankyrin repeat-containing (ANK) domain, the Src homology 3 (SH3) domain, the PSD-95/Discs large/zona occludens (PDZ) domain, the proline-rich region (PRR) and the sterile alpha motif (SAM) in the C-terminal domain. The N-terminal ANK domain interacts with the cytoskeletal protein alpha-fodrin (Böckers et al., 2001) and the SH3 domain binds to glutamate receptor-interacting protein (GRIP), which is important for AMPAR trafficking (Lu and Ziff, 2005). The PDZ site in SHANKs can bind to the adaptor protein guanylate kinase-associated protein (GKAP; Naisbitt et al., 1999). GKAP itself can bind to the postsynaptic density molecule 95 (PSD95; Naisbitt et al., 1997) that is in contact with the glutamate gated ion channels, NMDAR (Kornau et al., 1995) and AMPAR (Kim et al., 2001; Uemura et al., 2004; for review see Boeckers et al., 2002). Most clearly the interaction of SHANK and Homer proteins via the PRR was noted (Naisbitt et al., 1999; Tu et al., 1999). This interaction allows the coupling of mGluRs to Ca2+ release from endoplasmic reticulum (ER) in the postsynaptic density. Cortactin, a molecule involved in actin polymerization, was also shown to bind to PRR (Ammer and Weed, 2008). The C-terminal SAM domain serves as a Zn2+ dependent dimerization domain for SHANKs (Baron et al., 2006; Gundelfinger et al., 2006).
This complex multidomain structure of the SHANK proteins and the diversity of SHANK1–3 isoforms might be one reason for the broad spectrum of SHANK variant-associated neurodevelopmental and neuropsychiatric disorders. Distinct SHANK mutations can yield different protein products, with a specific combination of protein domains, different from that of the WT protein. This, in turn, can affect the potential protein-protein interactions, thus altering the role of the protein in neurons. In ASD patients, chromosomal deletions, translocations, gene duplications, gene fragment deletions and coding mutations in all three SHANK genes were identified (see https://gene.sfari.org) but the simple haploinsufficiency caused by the mutation is unlikely to explain the broad spectrum of phenotypes. In order to dissect the causal link between SHANK variants and the underlying molecular mechanisms of neurodevelopmental and neuropsychiatric disorders, numerous mouse models for “shankopathies” were generated (Table 1, Figure 1). For SHANK3, 14 mouse lines, generated with different gene-targeted strategies, are published (Figure 1). Interestingly, the behavioral analysis of the Shank3 knockout (KO) mouse lines revealed differences in social and repetitive behavior as well as in cognitive functions, similar to the distinct neuropsychiatric phenotypes of different patients. These behavioral inconsistencies were explained partially by the residual SHANK3 isoforms that could be expressed from the targeted Shank3 gene in mice (Bozdagi et al., 2010; Peca et al., 2011; Yang et al., 2012) as discussed in detail in a recent review (Monteiro and Feng, 2017). However, in all 10 different Shank2 KO mouse lines, the gene KO strategies were very similar leading to protein products truncated within the PDZ domain or the PRR, both present in all SHANK2 isoforms of the WT mice (Figure 1). Nevertheless, the phenotype of the three conventional and seven conditional Shank2 KO mouse lines diverged between the different lines. This phenotypic variability is reminiscent of the fact that in human patients, SHANK2 variants are not only significantly associated with ASD or ID, but also with schizophrenia (SCZ), a correlation that cannot be stated as strong for SHANK3 variants (Peykov et al., 2015; Homann et al., 2016; de Sena Cortabitarte et al., 2017).
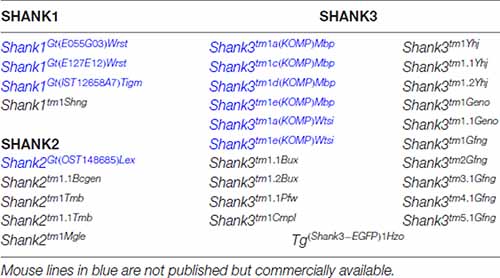
Table 1. Genetically modified mouse lines encoding gene-targeted mutations of endogenous Shank genes and one transgenic mouse line expressing a SHANK3-GFP that are made available to public in the mouse genome informatics database (http://www.informatics.jax.org).
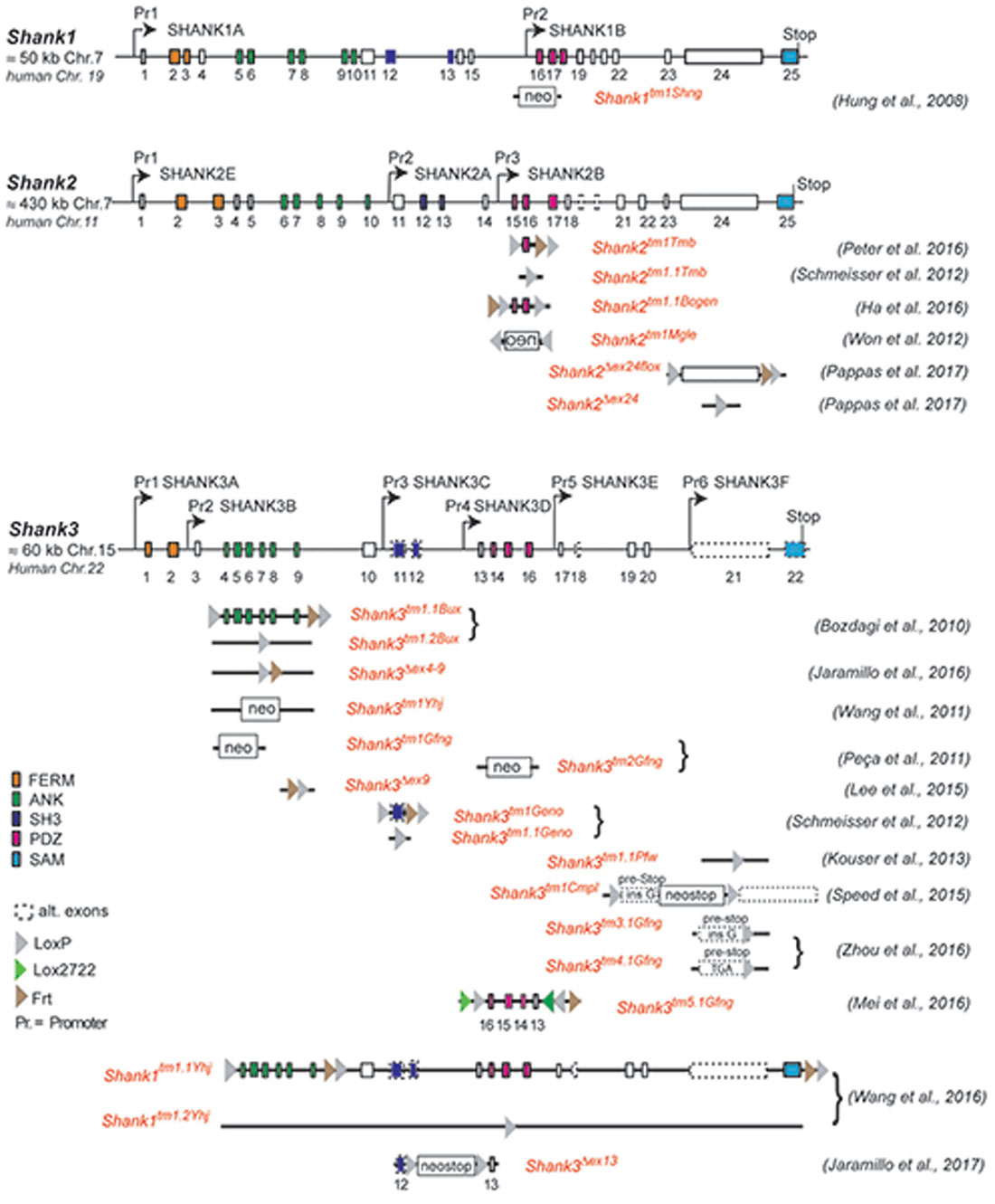
Figure 1. Gene structure and gene segment deletions in gene-targeted Shank mouse models. Structures of the mouse Shank1, Shank2 and Shank3 genes are depicted. Exons are given in rectangles; alternative spliced exons are in dashed lines. Positions of promoters (Pr) for the expression of the different isoforms of the Shank1–3 gene loci are indicated. Positions of the neomycin (neo) selection marker, loxP, lox2722 and Frt sites in the targeted alleles are indicated. Targeted gene segments flanked by two loxP or two lox2722 elements can be removed or inverted by tissue-specific expression of Cre to generate a conditional Shank knockout (KO) or Shank knockin mouse models. neo = selection marker, sense orientation; oen = neo selection marker, reverse orientation. References for the first publication of the mouse lines are given. FERM (F for 4.1 protein, E for ezrin, R for radixin and M for moesin), ANK (ankyrin repeat domain), SH3 (Src homology 3), PDZ (PSD-95/Discs large/zona occludens), SAM (sterile alpha motife). (Hung et al., 2008; Bozdagi et al., 2010; Peca et al., 2011; Wang et al., 2011, 2016; Schmeisser et al., 2012; Won et al., 2012; Kouser et al., 2013; Lee J. et al., 2015; Speed et al., 2015; Ha et al., 2016; Jaramillo et al., 2016, 2017; Mei et al., 2016; Peter et al., 2016; Zhou et al., 2016; Pappas et al., 2017).
Shank2 Gene Variants in Neurodevelopmental and Neuropsychiatric Disorders
After the first identification of SHANK2 gene mutations in patients with ASD and ID (Berkel et al., 2010), several other publications described further variations in the SHANK2 gene locus in patients with neuropsychiatric disorders. They identified SHANK2 variations including truncations, missense mutations, gene deletions and mutations in the SHANK2 promoter regions; all of these findings solidified the causal link of SHANK2 variants to ASD and ID (Pinto et al., 2010; Wischmeijer et al., 2011; Leblond et al., 2012, 2014; Prasad et al., 2012; Chilian et al., 2013; Schluth-Bolard et al., 2013; Bowling et al., 2017; Yuen et al., 2017). Interestingly, an association between SHANK2 gene mutations and SCZ was described in Peykov et al. (2015). By sequencing the SHANK2 gene in 481 SCZ patients and 659 unaffected individuals, Peykov et al. (2015) identified several non-synonymous variants exclusively in SCZ patients (Supplementary Table S1). This association was confirmed by a study reporting seven siblings in a family with SCZ spectrum disorders carrying a missense variant in the SHANK2 gene (Homann et al., 2016).
How Divergent Phenotypes Can Arise From Different SHANK2 Variants
Mutations in Different Protein Interaction Domains
As outlined above, the comprehensive analysis of patients harboring SHANK2 alterations with neurodevelopmental and neuropsychiatric disorders revealed a wide range of phenotypic expression, with various symptoms of ASD, ID, and SCZ. Moreover, the severity of SHANK2-associated disorders and cognitive deficiency is highly variable among patients. Imprecise clinical diagnoses and the clinical data from different clinics might explain this inconsistency. However, the SHANK2 variants as such also need to be considered as one reason for such variability. Different mutations in the SHANK2 gene can lead to increased or decreased expression of the SHANK2 protein, dysfunctional SHANK2 protein domains or truncated protein products lacking protein interaction domains. This can, in turn, alter protein-protein interactions and the organization of the postsynaptic protein network. Therefore, different SHANK2 mutations can lead to different alterations in molecular and cellular processes in the neurons, resulting in a wide range of behavioral phenotypes.
This perspective finds support in the rescue experiment of SHANK2 knockdown by overexpression of human SHANK2 variants in rat primary cultures (Berkel et al., 2012). The SHANK2A(R462X) variant accumulated in the soma and the dendrites, whereas overexpressed SHANK2A-WT selectively located in dendritic spines (Berkel et al., 2012). The SHANK2A(T1127M) and SHANK2A(L1008_P1009dup) variants were confined into synapses with lower SHANK2A immunosignal compared to SHANK2A-WT (Berkel et al., 2012), indicating that various mutations can lead to a decrease in the total final amount of SHANK2A present at the synapse, which could be an important factor in the pathophysiology of the neuropsychiatric disorder. Furthermore, the dendritic spine volume was only significantly increased by the overexpression of SHANK2A-WT, whereas all three SHANK2A variants (R462X, T1127M, L1008_P1009dup) did not have the potential to increase the spine volume. Similarly, the SHANK2 knockdown, which provoked a reduced spine volume, an increased number of immature spines and a dendritic complexity near the cell body, was rescued to different degrees when SHANK2A variants were co-expressed with the SHANK2 knockdown shRNA in rat primary neurons thus suggesting a dosage effect in the case of SHANK2. The SHANK2A-WT and SHANK2A(L1008_P1009dup) were able to rescue the dendritic spine and the dendritic arbor development but SHANK2A(R462X) and SHANK2A(T1127M) could not rescue the reduced spine volume. Consistently, SHANK2A(R462X) failed to rescue the enhanced growth of dendrites to a normal level, whereas the SHANK2A(T1127M) and SHANK2A(L1008_P1009dup) were able to rescue the phenotype similar to the SHANK2A-WT. From these cell culture studies, it can be concluded that the SHANK2 gene is involved in at least two separate functions: the control of dendrite morphology and the regulation of spine size.
Genetic and Epigenetic Modulations
In addition to the nature of SHANK2 variants, other genetic, epigenetic and environmental factors have a strong influence on the expression of the SHANK2 variant-mediated neuropsychiatric disorders. For example, the same SHANK2 variants were found in patients with different clinical features (Table 2). SHANK2(K535R) and SHANK2(P587S) variants were each identified in one ASD patient and in one patient with ID (Berkel et al., 2010). Similarly, SHANK2(A577V), previously misnamed as A578V, was associated with different characteristics of SCZ spectrum disorders in seven brothers (Homann et al., 2016). SHANK2(S610Y) was found in two patients: one with ID and the other with catatonic SCZ (Berkel et al., 2010; Peykov et al., 2015). The SHANK2(A1731S) variant was present in four patients with different subtypes of SCZ spectrum disorders (Peykov et al., 2015). In addition, some synonymous variants appear in patients with ASD, ID and SCZ, but not in healthy controls (Berkel et al., 2010; Rauch et al., 2012; Peykov et al., 2015; Supplementary Table S1). The influence of other genes on the phenotypic expression of the SHANK2 variants became most obvious in patients with an inherited SHANK2 variant-associated ASD or SCZ, which had not been diagnosed in their parents. The aforementioned SHANK2(A577V) variant found in seven schizophrenic male brothers, for example, was inherited from a healthy mother (Homann et al., 2016). This was also the case for five different SHANK2 variants: T438M, P1144L, V1608I, L1646M and A1731S (Peykov et al., 2015) and for several other variants identified in ASD patients (Supplementary Table S1) suggesting that sexually dimorphic pathways have an effect on the penetrance of these variants. Together, all these findings strongly support the proposed multiple hit model of neurodevelopmental and neuropsychiatric disorders (Leblond et al., 2012). Additionally, epigenetic factors most likely affect the penetrance of SHANK2 variants. A DNA hypermethylation value of 5 CpG positions within the SHANK2 gene was found in a male patient with ID and developmental delay (Kolarova et al., 2015), suggesting that SHANK2 expression is sensitive to the DNA methylation pattern. Other epigenetic mechanisms such as histone acetylation are expected to regulate the expression of the SHANK2 gene in an isoform-specific manner. Furthermore, one long non-coding variant associated to SHANK2 has been found to be up-regulated in blood samples of ASD patients (Wang et al., 2015).
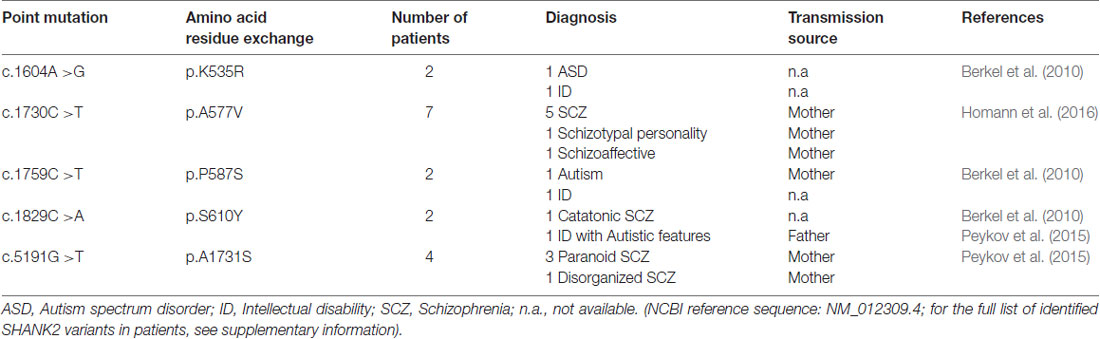
Table 2. Five single point mutations in the coding region (c.) of the SHANK2 gene leading to five amino acid residue exchanges (p.) that are associated with different neuropsychiatric disorders.
Shank2 Isoforms in Mice
For the design and the possible impact of Shank2 gene manipulation in mice, the detailed knowledge of the different SHANK2 isoforms and their expression pattern in the mouse brain is a prerequisite. As Shank1–3 can express many different SHANK isoforms using several promoters and alternative splicing (Figure 1), the different SHANK isoforms are proposed to have different functions during developmental stages (Jiang and Ehlers, 2013). According to the NCBI database, 21 putative isoforms are predicted to be expressed by the Shank2 gene locus on chromosome 7 in mice. Cloned rat cDNAs for the SHANK2 isoforms, SHANK2E, SHANK2A and SHANK2B, provide experimental evidence that these three isoforms are differentially expressed in the rat brain (Figure 2; Boeckers et al., 1999a,b; Han et al., 2006). Three different promoters are recruited for the expression of SHANK2E, SHANK2A and SHANK2B. The 5’ located promoter is used to generate the largest isoform, SHANK2E. The SHANK2E encoding transcript contains a 5’-untranslated exon, which is designated as exon 1. The promoters for the initiation of Shank2a and Shank2b transcripts are located in introns 10 and 14, respectively. For the translation of Shank2a and Shank2b transcripts, intron-located translational start codons open the translational reading frames (Figure 2).

Figure 2. SHANK2 isoforms in the rat evaluated by direct cDNA cloning. (Top) Schematic view of different conserved protein domains in SHANK2 isoforms. (Bottom) Amino acid residue sequence alignment of the SHANK2E (AY298755; Han et al., 2006), SHANK2A (AJ249562; Boeckers et al., 1999a) and SHANK2B (AF060116 or AJ131899; Du et al., 1998; Boeckers et al., 1999b) isoforms. (Bottom) Amino acid residue sequence alignment of the SHANK2E (AY298755; Han et al., 2006), SHANK2A (AJ249562; Boeckers et al., 1999a) and SHANK2B (AF060116 or AJ131899; Du et al., 1998; Boeckers et al., 1999b) isoforms. The SHANK2 protein domains are color-coded. The positions of the introns in the coding regions are indicated by vertical black lines. Exons are numbered starting with exon 2 as the first translated exon. N-terminal amino acid residues of isoform A and B are given in red. The sequences inside the brown rectangles are absent in some other Shank2 transcripts. The N-terminal sequence of isoform A is encoded by the three terminal end of intron 10. The N-terminal sequence of isoform B is encoded in intron 14. Isoform C which is found in humans cannot be detected in mouse or rat mRNAs.
The mouse Shank2 gene contains 25 exons. The alternative splicing of exon 19, 20 and 23 and the internal alternative splice donor site in exon 22 can lead to additional isoforms of SHANK2E, SHANK2A and SHANK2B. Using rat brain and human brain-derived cDNAs, the presence and absence of the alternatively spliced Shank2a exons were experimentally verified in rats and humans. The transcripts with exons 19, 20 and 23 seem to be underrepresented in peripheral tissues, whereas they seem to be dominantly expressed in the brain (Böckers et al., 2004; Leblond et al., 2012). It is important to mention that the short SHANK2C isoform, which is obtained by an alternative splicing event of exon 22 in humans (Leblond et al., 2012), cannot be detected in the transcriptom of mice and rats. The description of the presence or absence of exon 19 and 20 in the NCBI database-predicted 21 SHANK2 isoforms in mice is inconsistent and needs to be verified. This might explain why different numbers of exons in the Shank2 gene loci are given in different genome browsers (e.g., in Ensemble: ENSMUSG00000037541, the mouse Shank2 gene is composed of 23 exons).
As shown in Figure 2, SHANK2E is the full-length SHANK2 isoform containing an ANK domain, SH3 domain, PDZ domain, PRR and SAM domain, which is located on the SHANK2 C-terminal synaptic targeting elements (Böckers et al., 2004; Boeckers et al., 2005). In the brain, SHANK2E is expressed at different levels in different regions with the highest expression in the cerebellum (Leblond et al., 2012). The shortest SHANK2 isoform, SHANK2B (named as SHANK2C in Monteiro and Feng, 2017), contains just the PDZ, PRR and SAM domains (Figure 2). As determined by mRNA analysis in humans and rats, SHANK2 expression is strongest in the brain, but SHANK2 mRNA can also be PCR-amplified in most peripheral tissues with the exception of skeletal muscles and heart (Leblond et al., 2012). The SHANK2B RT-PCR products confirmed the differential SHANK2B expression in all human brain regions (Leblond et al., 2012), a finding that had already been well documented in rats by classical in situ RNA hybridization studies showing brain region-specific as well as developmentally controlled expression of SHANK1–3 (Böckers et al., 2004). According to this study, SHANK2 is co-expressed with SHANK1 during the early days of development, whereas SHANK3 reaches its maximum expression at postnatal day P16. By using the laminar organization of the cerebellum and the hippocampus, the authors showed convincingly that: (i) Shank1 and Shank2 mRNA co-exist in Purkinje cells (PCs) and their dendrites, whereas Shank3 mRNA was found only in the cerebellar granular cells; and (ii) Shank2 mRNA expression in the hippocampus is restricted to the cell layer of principal neurons, while the signals for the Shank1 and Shank3 mRNA can be detected in the molecular layers as well (Böckers et al., 2004).
In biochemical studies using rats, SHANK2 and SHANK3 seemed to be essential elements for the proper organization of the PSD. They localize to the PSD via their SAM domain and interactions with Zn2+ ions, which can be enhanced by the activity and synaptic release of Zn2+. In contrast, SHANK1 is recruited to the PSD by a pre-formed scaffold via its PDZ domain (Baron et al., 2006; Gundelfinger et al., 2006; Grabrucker et al., 2011). Thus, all SHANKs can respond to activity and synaptic events that might underlie learning and memory. However, SHANK1 is lacking the capability to localize to immature/inactive synapses, which seems to be SAM domain/Zn2+-dependent (Schmeisser and Verpelli, 2016). During neuronal development, the subcellular distribution of SHANK2 proteins changes and immunoelectron microscopy studies revealed that SHANK2 accumulation shifts in the developing, cortical nuerons. At postnatal day P5, SHANK is accumulated in the lamellipodium, then shifts into the cytoplasm of the cell bodies and in the growing neurites at P8, and finally to the PSD at P10, which supports the idea that it may play a role in multiple cell biological frameworks of neurons (Boeckers et al., 1999a). Interestingly, on the electrophysiological level, SHANK1 and SHANK2, but not SHANK3 virus-mediated knockdown reduced the number of spines and the AMPAR responses at CA3-to-CA1 synapses in acute hippocampal slice cultures (Shi et al., 2017). Due to this functional divergence of SHANK proteins, it is not surprising that in ASD patients, SHANK2 and SHANK3 mutations are over-represented (Leblond et al., 2014).
Modeling Shank2 Mutations in Mice
The multiple genetic, epigenetic and environmental factors affecting the phenotypic expression of ASD and SCZ disorders are a major issue when we try to understand the molecular, anatomical, physiological and behavioral phenotypes of SHANK2 variants. Analyzing genetically comparable inbred mice should at least minimize the environmental and epigenetic components when mice are raised and housed under comparable conditions. Observations of male and female single-housed or cohort-housed mice with identical or mixed genotypes, and either with conditional gene deletions or gene overexpression, offer multiple options to investigate different aspects of the neurodevelopmental and neuropsychiatric disorders in an animal model. Already in 2007, Crawley and Tordjman proposed several behavioral tasks that are suitable for the analysis of autistic behavior in mice (Crawley, 2007; Tordjman et al., 2007).
Distinct Phenotypes of the Different Shank2 Knockout Mice
The proposal of Crawley and Tordjman likely promoted the generation of several gene-targeted mutations in mice modeling human SHANK1, 2 and 3 mutations-associated ASD (Figure 1). In 2012, the first two Shank2 KO mouse models (Shank2−/−) were generated. Both mouse models resembled two PDZ domain-microdeletions found in two human patients (Berkel et al., 2010). Mice of both lines showed comparable cognitive and social impairments. In Shank2Δex16 mice (named Shank2Δex7), exon 16 was deleted (Schmeisser et al., 2012). In Shank2Δex15-16 mice (named Shank2Δex6-7), the exons 15-16 encoding gene segment was replaced by a loxP site flanked inverse-oriented neomycin resistance (neo) selection marker (Won et al., 2012). Although no residual SHANK2 expression was detected in homozygous Shank2Δex16 or Shank2Δex15-16 mice as determined by immunoblots and immunohistology, and although the genetic background of the two mouse lines was similar (C57Bl/6N and C57Bl/6J), significant differences on the molecular, electrophysiological, synaptic composition and anatomical levels of Shank2Δex16 or Shank2Δex15-16 were evident (Table 3, Supplementary Table S2). The impairment of synaptic plasticity at hippocampal CA1 synapses showed opposite results in the two mouse lines and the NMDAR response was increased in Shank2Δex16 but decreased in Shank2Δex15–16 mice. In addition, the spine number and spine density were reduced in CA1 pyramidal cells of Shank2Δex16, whereas Shank2Δex15-16 mice showed normal spine numbers and density (Table 3, Supplementary Table S2).
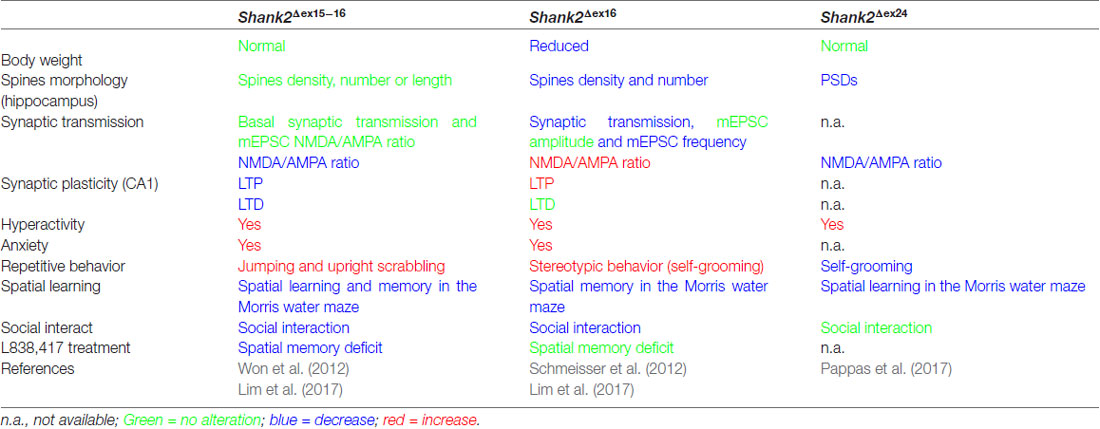
Table 3. Distinct endophenotypes in genetically very similar gene-manipulated, conventional Shank2 knockout mice.
In 2017, Lim et al. (2017) substantiated the distinct phenotypes of Shank2Δex16 and Shank2Δex15-16 mice by a direct comparison of the two mouse lines in a mixed C57Bl/6N × C57Bl/6J background. The differences in gene expression (Supplementary Table S2) and synaptic properties such as the AMPA/NMDA ratio and long-term potentiation (LTP; Neves et al., 2008) as well as differences in the inhibitory signaling were directly correlated to the Shank2 Δex16 and Δex15-16 gene-targeted mutations. The Shank2Δex15-16 mice exhibited a decreased GABAAR-mediated inhibition, most likely caused by the reduced expression of the GABAR gene, Gabra2. Interestingly, a similar discrepancy between the phenotypic expression of Shank2 Δex16 and Δex15-16 gene deletions was found when the two gene deletions were restricted to cerebellar PCs (Ha et al., 2016; Peter et al., 2016; Supplementary Table S2). These mouse lines were generated by combining the floxed Shank2 ex16 or the floxed Shank2 ex15-16 gene, with transgenic Cre-expressing mouse lines that used the PC specific Pcp2 promoter, also called L7 promoter, for PC specific inactivation of the Shank2 gene (Figure 1). Shank2Δex15-16–Pcp2-Cre mice showed impaired motor coordination and learning in the Erasmus ladder test, but normal motor performance in the rotarod test. However, they exhibited neither repetitive behavior nor social interaction deficits, but only a mild anxiety-like behavior. In contrast, Shank2Δex16–L7–Cre mice showed normal motor performance in the Erasmus ladder with no anxiety-like behavior, but deficits in social interaction as well as social novelty recognition. Thus, similar inactivation of the Shank2 gene in cerebellar PCs produced distinct phenotypes at the molecular and up to the behavioral level. Similarly in two recent mouse models restricting the Δex15-16 deletion in two specific cell types; one in excitatory neurons (Shank2Δex15-16–CaMK2a-Cre) and the other in GABAergic inhibitory neurons (Shank2Δex15-16–Viaat-Cre, Kim et al., 2018), Shank2Δex15-16–CaMK2a-Cr and Shank2Δex15-16–Viaat-Cre mice showed differences on the electrophysiological level by reduced mEPSC frequency in hippocampal CA1 neurons in Shank2Δex15-16–CaMK2a-Cre but not in Shank2Δex15-16–Viaat-Cre mice. Shank2Δex15-16–CaMK2a-Cre mice showed increased anxiety-like but normal repetitive behaviors in comparison to Shank2Δex15-16–Viaat-Cre mice, which showed no anxiety-like but increased repetitive behaviors (Supplementary Table S2). Thus, in Shank2 KO mice, a residual expression of truncated proteins or mRNAs, which differs between the Shank2 mouse lines (Figure 3), might disturb the structure or the flexibility of the SHANK scaffold at different levels and thus the flexibility of the postsynaptic protein organization. This impaired synaptic function might vary in different neurons or even be different in various synapses of the same neuron. These finding in cell type specific Shank2 KO mice underlines observations that very similar SHANK2 mutations can lead to a different phenotype, as noticed by the wide behavioral spectrum of ASD described for patients with mutations in the SHANK2 genes (Supplementary Table S1).
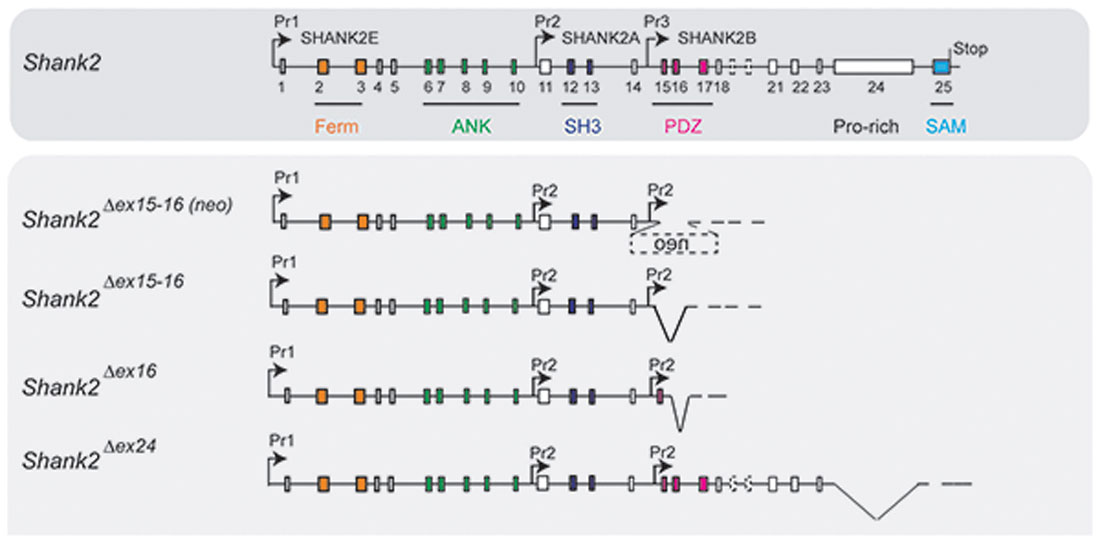
Figure 3. Putative pre-mRNA transcripts in homozygous Shank2 KO mice. Schematic view of the Shank2 gene and the putative pre-mRNA that can be expressed by the four different gene-targeted alleles of the Shank2 mouse models with very distinct phenotypes. Symbols are as in Figure 2.
In 2017, a third conventional Shank2 KO mouse was generated by the out-of-frame deletion of Shank2 exon 24, which deleted the PRR region (Pappas et al., 2017). These Shank2Δex24 KO mice exhibited a pronounced behavioral difference compared to the published ASD-like behavior of Shank2Δex16 and Shank2Δex15-16 (Table 3, Supplementary Table S2). Shank2Δex24 mice showed a bipolar-associated mania-like behavior. Shank2Δex24 mice were hyperactive in the home cage and open field test, showed a decrease in repetitive behaviors such as self-grooming, had no social preference in the social affiliation test nor did they show a SCZ-like behavior. Instead, Shank2Δex24 mice showed anhedonia-like behavior and disturbed circadian rhythms. Some of these unique behavioral impairments persisted even in conditional Shank2Δex24 mice (Supplementary Table S2; Pappas et al., 2017). When exon 24 of Shank2 was removed in cerebellar PCs, Shank2Δex24-Pcp2-Cre mice showed an impaired motor performance in the rotarod (Pappas et al., 2017). The Cre-mediated removal of exon 24 in the neocortex in Shank2Δex24-Emx1-Cre was correlated with mania-like behavior, hyperactivity, impaired spatial memory and a surprisingly significant social preference in female mice (Pappas et al., 2017). The developmental contribution of the Shank2Δex24 was unraveled by the conditional deletion of exon 24 in forebrain postnatally, using the developmentally-controlled promoter for the alpha subunit of Ca2+-calmodulin dependent protein kinase II (CaMKII) for Cre expression. Thus, when exon 24 was deleted efficiently in 3 weeks old Shank2Δex24-CaMKI2a-Cre mice, they didn’t show any hyperactivity, which was used as signature for the mania-like behavior of the global Shank2Δex24 mice (Pappas et al., 2017).
How Can the Distinct Phenotypes of the Different Shank2 Knockout Mice Be Explained?
The heterogeneity in the phenotypic expression of all three conventional and seven conditional Shank2-“deficient” mouse models indicated that there is no clearly defined SHANK2 deficiency-mediated phenotype. Similar to human patients, each Shank2 KO mouse line showed a distinct neuropsychiatric-like phenotype, which was dependent on the specific type of Shank2 mutation. In contrast to humans, where genetic and environmental factors cannot be excluded as cofactors for the penetrance of abnormal social behavior, the common social experience of experimental mouse cohorts can largely exclude environmental factors. Therefore, it is more likely that similar genetic modulations still express different truncated SHANK2 proteins at different levels (Figure 3), which might disturb the balanced co-expression of both SHANK1 and SHANK3 isoforms and exert more different dominant-negative effects on other protein interacting partners, that can be recognized by different behavioral outcomes. Experimental evidence for disturbed or unbalanced expression of SHANK proteins was provided by the virus-mediated overexpression in mice of the truncated SHANK2A(R462X) variant (Berkel et al., 2012), initially identified in one ASD patient (Berkel et al., 2010; Supplementary Table S2). Neurons in hippocampal cultures showed impaired spine development, characterized by their filopodia-like structure. In CA1 cells from acute hippocampal slices, reduced mEPSC amplitudes could be recorded which were correlated to impaired cognitive function, demonstrating that the truncated SHANK2 proteins can disturb the physiological function of the coordinated SHANK expression in the CNS.
Thus, as already described for similar Shank3 KO mouse models, the Shank2 KO lines are characterized by functionally distinct molecular, electrophysiological and neurophysiological phenotypes. In particular, the opposing effect of the disturbed excitatory and inhibitory responses of CA1 pyramidal cells in Shank2Δex15-16 and Shank2Δex16 was unexpected and still is not well understood. Already small differences in the genetic background could be responsible for this drastic difference. Moreover, it might be interesting to find out whether female and male mice or littermates with the very same Shank2 KO mutation show different neuropsychiatric-like behavior, as described in human patients for four and seven siblings, who inherited the SHANK2 mutation from their healthy mothers (Table 2). The careful comparative analysis of individual Shank2 KO littermates of the same KO mouse line might identify epigenetic and environmental ASD or SCZ-facilitating factors; although this is a major and long-lasting endeavor.
Insight Into ASD Pathogenesis and Treatment From Shank2 Knockout Mouse Models
The finding of distinct phenotypes of genetically very similar Shank2 KO mice finds further strong support by the pharmacological treatment strategies of the different Shank2 KO mice. In the Shank2Δex15-16 mice, the hippocampal NMDAR function in brain slices was decreased (Won et al., 2012). The treatment of Shank2Δex15-16 with D-cycloserine, a partial agonist at the glycine-binding site of NMDARs, increased the NMDAR responses and rescued the social impairments of Shank2Δex15-16 mice. Similarly, the administration of a positive allosteric modulator of mGluR5, 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl benzamide (CDPPB), which enhances the NMDAR function via mGluR5 activation, rescued the NMDAR signaling impairments, as measured in hippocampal CA1 cells. The CDPPB-mediated recovery of NMDAR function in Shank2Δex15-16 mice was correlated with normalized social interaction of CDPPB treated Shank2Δex15-16 mice. However, hyperactivity and anxiety-like behaviors were not recovered (Won et al., 2012).
A different approach to activate the NMDAR function in Shank2Δex15-16 mice was carried out by inducing postsynaptic Zn2+ elevation using a Zn2+ chelator, clioquinol (Lee E. J. et al., 2015). Zn2+ is mainly derived from presynaptic pools and activates NMDAR through postsynaptic activation of the tyrosine kinase Src (Manzerra et al., 2001). Treating Shank2Δex15-16 mice with clioquinol enhanced social interaction, but did not had any effect on social novelty recognition, anxiety-like behavior, hyperactivity and repetitive behavior. The functional NMDAR rescue was demonstrated by restoring NMDAR signaling at hippocampal (SC-CA1) synapses (Lee E. J. et al., 2015). Although the pharmacological treatment of Shank2Δex15-16 mice was carried out 30–120 min before the behavioral tests, and even though the recovery of the NMDAR function was shown at hippocampal synapses, other brain regions, e.g., the oxytocin neurons in the hypothalamus, might also be involved in producing social incompetence and could be good candidate targets for pharmacological therapy.
In addition to the stimulation of the glutamatergic system, the enhancement of the inhibitory synaptic activity can reverse genetically-based ASD-associated deficits. In Shank2Δex15-16 mice, Gabra2 expression on RNA level as well as GABAA receptor-mediated inhibitory neurotransmission in CA1 hippocampal slices were reduced in Shank2Δex15-16, but not in Shank2Δex16 mice. Treatment with an allosteric modulator of GABAA receptor, L838,417, reversed the spatial memory deficits of the Shank2Δex15-16 mice, but not those of Shank2Δex16 mice (Lim et al., 2017). However, L838,417 had no significant effects on anxiety-like behavior or social impairment in Shank2Δex15-16 mice. Whether or not long or short-term treatment in young and adult mice will have a persistent rescue effect remains to be investigated. The pharmacological rescues showed that the identification of the underlying electrophysiological mechanism for the type of ASD is mandatory for a successful treatment. Since a detailed electrophysiological differential diagnosis of ASD patients is not feasible, the Shank mutant mice are the best accessible tools to dissect different ASD endophenotypes of SHANK mutations at the molecular, anatomical, electrophysiological and behavioral levels.
Behavioral Test Battery for Shank2 Mouse Models
The widely varying phenotypes of Shank2 mouse models for ASD and other neuropsychiatric disorders can be grouped into several categories: molecular, anatomical, hormonal, immunological, neurophysiological and behavioral. The detailed description within all six categories is crucial for the putative therapeutic treatment which, if successful, will likely to alleviate only some of ASD, SCZ or cognitive phenotypes. Standardized molecular, anatomical and neuronal activity analyses are straightforward and comparable between different studies, at least when the analyzed mutant and control mice are derived from the same colony, are age-matched, of the same gender, and when the genetic background of the mice used in different studies is similar. A major issue is comparing behavioral phenotypes determined in different laboratories (Schellinck et al., 2010), which can be a reason for the difference in behavior between the Shank2 mouse models.
For the comparable description of ASD and other neuropsychiatric disorder phenotypes in Shank2 mouse models, it is obligatory to exclude gross physical abnormalities (Crawley, 2007). General health, body weight and neurological reflexes including eye blink, ear reflex and whisker twitch should be investigated. The three-stage SHIRPA procedure (Immelmann and Beer, 1989) can be performed to assess mice phenotypes for obvious physical, behavioral and morphological impairments before testing social behavior and cognition. In the best case, a complete ethogram of the Shank2 mutant mouse line, before and after genetic or pharmacological rescue, should be determined to cover the expected wide spectrum of phenotypes. If only a partial ethogram is provided, at least the full range of social and cognitive performance tests should be presented. As suggested (Crawley, 2007; Tordjman et al., 2007), a consistent “multi-trait” analysis of cognitive and social behavior is necessary for the dissection of the clinically heterogeneous ASD spectrum in the different mouse models.
First Simple Behavioral Tests
For Shank2 mouse models, the first behavioral observations can already be started when the mouse is still used to live in its home cage. The burrowing or marble burying tests are indicators for anxiety and obsessive-compulsive disorders (Broekkamp et al., 1986; Borsini et al., 2002; Deacon, 2006; Thomas et al., 2009; Angoa-Pérez et al., 2013). The nest building test (Deacon, 2012) is considered a potential endophenotype relevant to the negative symptoms of SCZ (Amann et al., 2010) and depression. The unbiased physical performance and activity, as well as circadian rhythm of mice, can be recorded in the home cage (LABORAS system from Metris) and the running wheel; motor coordination in rotarod and Erasmus ladder test (Dunham and Miya, 1957; Van Der Giessen et al., 2008; Vandeputte et al., 2010; Luong et al., 2011). For anxiety testing, which is considered one of the most important co-morbidities of neuropsychiatric disorders, the zero-maze, elevated zero-maze and elevated plus-maze (Handley and Mithani, 1984; Pellow et al., 1985; Lister, 1987; Shepherd et al., 1994; Heisler et al., 1998; Cook et al., 2001) and the dark-light compartment (Crawley and Goodwin, 1980) can be used.
The Four Most Important Diagnostic Symptoms Need to Be Investigated
The behavioral test battery of Shank2 mouse models should focus primarily on diagnostic symptoms of ASD: abnormal social interactions, deficits in communication, high levels of repetitive behaviors and cognitive dysfunction. However, to express cognitive and social competence in a behavioral test, anxiety and other stress factors have to be minimized. Pre-handling of the mice for at least 1 week is obligatory before testing the social behavior and memory (Fridgeirsdottir et al., 2014). In addition, increased stress by different housing conditions might affect the results (Kamakura et al., 2016). During the handling phase, repetitive behaviors (self-grooming, jumping and climbing) can be recorded in the LABORAS, and repetitive digging behavior in the marble burying assay. Social ability is usually assessed in the three-chambered box (Moy et al., 2004), the partition test apparatus (Kudryavtseva, 2003) and reciprocal social interaction in a novel arena (Silverman et al., 2010). When communicating information, olfactory stimulations as well as vocalizations in the ultrasonic ranges are used to enhance social bonding between mice, which can be evaluated by the olfactory habituation/dishabituation test (Wesson et al., 2008) and by measuring ultrasonic vocalizations (USV). As a measure of cognitive function, the spatial working reference memory can be tested in the T-maze and spatial reference memory in the Morris water maze, Y- or Radial-maze. The simple puzzle box is testing the goal-oriented performance of mice, which can give early hints of cognitive functions (Ben Abdallah et al., 2011). Social transmission of food preference has been used as a method for studying memory by several laboratories (Wrenn, 2004).
Additional Behavioral Assays Analogous to Other Symptoms of Neuropsychiatric Disorders
Patients with neuropsychiatric disorders suffer from a wide range of symptoms which can be measured in Shank2 mice to some extent. Depression in mice can be noticed in the forced swim test, the tail-suspension tests (Porsolt et al., 1977; Steru et al., 1985) and by anhedonia in the sucrose preference test (Papp et al., 1991; Tye et al., 2013). Deficits in sensory processing can be considered a symptom of SCZ, which can be evaluated by the Pre-Pulse Inhibition (PPI) of Acoustic Startle Response (ASR; Braff and Geyer, 1990; Swerdlow et al., 1994). Other symptoms of SCZ, such as delusions and auditory hallucinations, still cannot be fully assessed in mice (Chadman et al., 2009). If necessary, learning and memorizing fear can be examined as the last step in the test battery. However, the analysis of contextual and cued fear learning (Maren, 2001) or the passive avoidance test (Gray and McNaughton, 2000) are usually not performed with Shank2 mouse models (for the summary of tests see Table 4).

Table 4. Summary of behavioral tests that can be used in the test battery for Shank2 mouse models and other mice with potential neuropsychiatric-like phenotype.
Conclusion
The different alterations in synaptic responses, synaptic plasticity, social interaction and exploration behavior, as well as therapeutic responses in Shank2 KO mice emphasize that a balanced, physiologically-controlled expression of SHANKs is necessary for the appropriate organization of postsynaptic proteins and receptors at excitatory synapses. In addition, the comparison between Shank2 KO mouse lines strongly suggests that truncated SHANK2 isoforms or their mRNAs disrupt the homeostasis of SHANK proteins and their function as major scaffolding organizers in the postsynaptic matrix. Different Shank2 mutations might yield different protein products with potentially contrasting effects at the synapse (e.g., increasing or decreasing its strength), thus leading to a diversity of phenotypes. However, epigenetic factors as well as inconsistent analysis of the various Shank2 mouse lines cannot yet be completely ruled out.
Perspective
Understanding the detailed function of the multitude of SHANK isoforms is one of the biggest challenges in cognitive neuroscience. The disturbed balance of different SHANK proteins within individual postsynapses significantly affects the plasticity of synapses, possibly by changing the composition of the postsynaptic scaffold, which translates to a rearrangement of the glutamate receptors and measurable alterations in the glutamatergic signaling. For in vivo analysis of certain SHANK2 isoforms in mice, certain exons or SHANK internal promoters could be eliminated or certain SHANK isoforms could be overexpressed by viruses or transgenes. New genetic engineering techniques can generate relatively quickly novel Shank2 mouse lines. However, in future studies with novel constitutive and conditional Shank2 mouse lines, the expression of the residual truncated isoforms should be analyzed in more details, and the full battery of behavioral analysis covering ASD, SCZ, mania and Attention Deficit Hyperactivity Disorder (ADHD) has to be applied, since the existing Shank2 mouse models showed very distinct, unpredictable phenotype despite very similar genetic manipulations. In addition, males and females and real littermates need to be compared in detail to unravel epigenetic, environmental and gender effects on the expression of the phenotypes. More importantly, the current Shank2 mouse models provided first insights into molecular and physiological changes underlying ASD-related symptoms. The Shank2 mutant mice showed that ASD-related symptoms can be diminished by pharmacological treatment of the mutant mice, which can guide future therapeutic strategies for ASD patients. Now the application of spatio-temporal removal of SHANK2 using cell-type specific Cre or CreERT2 mice can identify those neuronal connections and circuits that have the strongest impact on the expression of the ASD-like phenotype. Moreover, conditional Shank2 KO mice can help to dissect the neurodevelopmental vs. the transient deficiencies mediated by the SHANK depletion. Once the most crucial neuronal circuits and cell types are identified, those neuronal populations can be targeted and analyzed in great detail in behaving mice by novel physiological technologies, e.g., multicellular recordings and optogenetics, which allow the recording, visualization and manipulation of neuronal activity ensembles during social interaction, repetitive behavior, vocalization and cognition, or even during different phases of sleep. Due to the very distinct phenotypes of different Shank2 mouse lines, it will be exciting to see how their altered neuronal network activity is correlated with the impaired social behavior.
Author Contributions
All authors significantly contributed to the manuscript. AE and RS wrote the first draft and provided the figures, tables and supplementary information. GR critically revised the manuscript.
Funding
This work was supported by a grant from the Ingeborg Ständer Foundation to RS. AE received a PhD fellowship from Heidelberg Bioscience International Graduate School (HBIGS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Trey Chiu and Flavia-Bianca Cristian for proofreading. We would also like to sincerely thank Shaimaa Madbouly for her constant support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2018.00240/full#supplementary-material
References
Amann, L. C., Gandal, M. J., Halene, T. B., Ehrlichman, R. S., White, S. L., McCarren, H. S., et al. (2010). Mouse behavioral endophenotypes for schizophrenia. Brain Res. Bull. 83, 147–161. doi: 10.1016/j.brainresbull.2010.04.008
Ammer, A. G., and Weed, S. A. (2008). Cortactin branches out: roles in regulating protrusive actin dynamics. Cell Motil. Cytoskeleton 65, 687–707. doi: 10.1002/cm.20296
Angoa-Pérez, M., Kane, M. J., Briggs, D. I., Francescutti, D. M., and Kuhn, D. M. (2013). Marble burying and nestlet shredding as tests of repetitive, compulsive-like behaviors in mice. J. Vis. Exp. 82:50978. doi: 10.3791/50978
Baron, M. K., Boeckers, T. M., Vaida, B., Faham, S., Gingery, M., Sawaya, M. R., et al. (2006). An architectural framework that may lie at the core of the postsynaptic density. Science 311, 531–535. doi: 10.1126/science.1118995
Ben Abdallah, N. M., Fuss, J., Trusel, M., Galsworthy, M. J., Bobsin, K., Colacicco, G., et al. (2011). The puzzle box as a simple and efficient behavioral test for exploring impairments of general cognition and executive functions in mouse models of schizophrenia. Exp. Neurol. 227, 42–52. doi: 10.1016/j.expneurol.2010.09.008
Berkel, S., Marshall, C. R., Weiss, B., Howe, J., Roeth, R., Moog, U., et al. (2010). Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 42, 489–491. doi: 10.1038/ng.589
Berkel, S., Tang, W., Trevino, M., Vogt, M., Obenhaus, H. A., Gass, P., et al. (2012). Inherited and de novo SHANK2 variants associated with autism spectrum disorder impair neuronal morphogenesis and physiology. Hum. Mol. Genet. 21, 344–357. doi: 10.1093/hmg/ddr470
Boeckers, T. M., Bockmann, J., Kreutz, M. R., and Gundelfinger, E. D. (2002). ProSAP/Shank proteins—a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J. Neurochem. 81, 903–910. doi: 10.1046/j.1471-4159.2002.00931.x
Boeckers, T. M., Kreutz, M. R., Winter, C., Zuschratter, W., Smalla, K. H., Sanmarti-Vila, L., et al. (1999a). Proline-rich synapse-associated protein-1/cortactin binding protein 1 (ProSAP1/CortBP1) is a PDZ-domain protein highly enriched in the postsynaptic density. J. Neurosci. 19, 6506–6518. doi: 10.1523/jneurosci.19-15-06506.1999
Boeckers, T. M., Winter, C., Smalla, K. H., Kreutz, M. R., Bockmann, J., Seidenbecher, C., et al. (1999b). Proline-rich synapse-associated proteins ProSAP1 and ProSAP2 interact with synaptic proteins of the SAPAP/GKAP family. Biochem. Biophys. Res. Commun. 264, 247–252. doi: 10.1006/bbrc.1999.1489
Boeckers, T. M., Liedtke, T., Spilker, C., Dresbach, T., Bockmann, J., Kreutz, M. R., et al. (2005). C-terminal synaptic targeting elements for postsynaptic density proteins ProSAP1/Shank2 and ProSAP2/Shank3. J. Neurochem. 92, 519–524. doi: 10.1111/j.1471-4159.2004.02910.x
Böckers, T. M., Mameza, M. G., Kreutz, M. R., Bockmann, J., Weise, C., Buck, F., et al. (2001). Synaptic scaffolding proteins in rat brain. Ankyrin repeats of the multidomain Shank protein family interact with the cytoskeletal protein α-fodrin. J. Biol. Chem. 276, 40104–40112. doi: 10.1074/jbc.m102454200
Böckers, T. M., Segger-Junius, M., Iglauer, P., Bockmann, J., Gundelfinger, E. D., Kreutz, M. R., et al. (2004). Differential expression and dendritic transcript localization of Shank family members: identification of a dendritic targeting element in the 3′ untranslated region of Shank1 mRNA. Mol. Cell. Neurosci. 26, 182–190. doi: 10.1016/j.mcn.2004.01.009
Borsini, F., Podhorna, J., and Marazziti, D. (2002). Do animal models of anxiety predict anxiolytic-like effects of antidepressants? Psychopharmacology 163, 121–141. doi: 10.1007/s00213-002-1155-6
Bowling, K. M., Thompson, M. L., Amaral, M. D., Finnila, C. R., Hiatt, S. M., Engel, K. L., et al. (2017). Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 9:43. doi: 10.1186/s13073-017-0433-1
Bozdagi, O., Sakurai, T., Papapetrou, D., Wang, X., Dickstein, D. L., Takahashi, N., et al. (2010). Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction and social communication. Mol. Autism 1:15. doi: 10.1186/2040-2392-1-15
Braff, D. L., and Geyer, M. A. (1990). Sensorimotor gating and schizophrenia. Human and animal model studies. Arch. Gen. Psychiatry 47, 181–188. doi: 10.1001/archpsyc.1990.01810140081011
Broekkamp, C. L., Rijk, H. W., Joly-Gelouin, D., and Lloyd, K. L. (1986). Major tranquillizers can be distinguished from minor tranquillizers on the basis of effects on marble burying and swim-induced grooming in mice. Eur. J. Pharmacol. 126, 223–229. doi: 10.1016/0014-2999(86)90051-8
Chadman, K. K., Yang, M., and Crawley, J. N. (2009). Criteria for validating mouse models of psychiatric diseases. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 1–11. doi: 10.1002/ajmg.b.30777
Chilian, B., Abdollahpour, H., Bierhals, T., Haltrich, I., Fekete, G., Nagel, I., et al. (2013). Dysfunction of SHANK2 and CHRNA7 in a patient with intellectual disability and language impairment supports genetic epistasis of the two loci. Clin. Genet. 84, 560–565. doi: 10.1111/cge.12105
Cook, M. N., Williams, R. W., and Flaherty, L. (2001). Anxiety-related behaviors in the elevated zero-maze are affected by genetic factors and retinal degeneration. Behav. Neurosci. 115, 468–476. doi: 10.1037//0735-7044.115.2.468
Crawley, J., and Goodwin, F. K. (1980). Preliminary report of a simple animal behavior model for the anxiolytic effects of benzodiazepines. Pharmacol. Biochem. Behav. 13, 167–170. doi: 10.1016/0091-3057(80)90067-2
Crawley, J. N. (2007). Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol. 17, 448–459. doi: 10.1111/j.1750-3639.2007.00096.x
Deacon, R. M. (2012). Assessing burrowing, nest construction, and hoarding in mice. J. Vis. Exp. 59:e2607. doi: 10.3791/2607
Deacon, R. M. (2006). Digging and marble burying in mice: simple methods for in vivo identification of biological impacts. Nat. Protoc. 1, 122–124. doi: 10.1038/nprot.2006.20
de Sena Cortabitarte, A., Degenhardt, F., Strohmaier, J., Lang, M., Weiss, B., Roeth, R., et al. (2017). Investigation of SHANK3 in schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 174, 390–398. doi: 10.1002/ajmg.b.32528
Du, Y., Weed, S. A., Xiong, W. C., Marshall, T. D., and Parsons, J. T. (1998). Identification of a novel cortactin SH3 domain-binding protein and its localization to growth cones of cultured neurons. Mol. Cell. Biol. 18, 5838–5851. doi: 10.1128/mcb.18.10.5838
Dunham, N. W., and Miya, T. S. (1957). A note on a simple apparatus for detecting neurological deficit in rats and mice. J. Am. Pharm. Assoc. Am. Pharm. Assoc. 46, 208–209. doi: 10.1002/jps.3030460322
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Fridgeirsdottir, G. A., Hillered, L., and Clausen, F. (2014). Escalated handling of young C57BL/6 mice results in altered Morris water maze performance. Ups. J. Med. Sci. 119, 1–9. doi: 10.3109/03009734.2013.847511
Grabrucker, A. M., Knight, M. J., Proepper, C., Bockmann, J., Joubert, M., Rowan, M., et al. (2011). Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J. 30, 569–581. doi: 10.1038/emboj.2010.336
Gray, J. A., and McNaughton, N. (2000). The Neuropsychology of Anxiety: An Enquiry into the Functions of the Septo-Hippocampal System. 2nd Edn. New York, NY: Oxford University Press.
Gundelfinger, E. D., Boeckers, T. M., Baron, M. K., and Bowie, J. U. (2006). A role for zinc in postsynaptic density asSAMbly and plasticity? Trends Biochem. Sci. 31, 366–373. doi: 10.1016/j.tibs.2006.05.007
Ha, S., Lee, D., Cho, Y. S., Chung, C., Yoo, Y. E., Kim, J., et al. (2016). Cerebellar Shank2 regulates excitatory synapse density, motor coordination and specific repetitive and anxiety-like behaviors. J. Neurosci. 36, 12129–12143. doi: 10.1523/jneurosci.1849-16.2016
Han, W., Kim, K. H., Jo, M. J., Lee, J. H., Yang, J., Doctor, R. B., et al. (2006). Shank2 associates with and regulates Na+/H+ exchanger 3. J. Biol. Chem. 281, 1461–1469. doi: 10.1074/jbc.M509786200
Handley, S. L., and Mithani, S. (1984). Effects of α-adrenoceptor agonists and antagonists in a maze-exploration model of ‘fear’-motivated behaviour. Naunyn Schmiedebergs Arch. Pharmacol. 327, 1–5. doi: 10.1007/bf00504983
Hayashi, M. K., Tang, C., Verpelli, C., Narayanan, R., Stearns, M. H., Xu, R. M., et al. (2009). The postsynaptic density proteins Homer and Shank form a polymeric network structure. Cell 137, 159–171. doi: 10.1016/j.cell.2009.01.050
Heisler, L. K., Chu, H. M., Brennan, T. J., Danao, J. A., Bajwa, P., Parsons, L. H., et al. (1998). Elevated anxiety and antidepressant-like responses in serotonin 5-HT1A receptor mutant mice. Proc. Natl. Acad. Sci. U S A 95, 15049–15054. doi: 10.1073/pnas.95.25.15049
Homann, O. R., Misura, K., Lamas, E., Sandrock, R. W., Nelson, P., McDonough, S. I., et al. (2016). Whole-genome sequencing in multiplex families with psychoses reveals mutations in the SHANK2 and SMARCA1 genes segregating with illness. Mol. Psychiatry 21, 1690–1695. doi: 10.1038/mp.2016.24
Hung, A. Y., Futai, K., Sala, C., Valtschanoff, J. G., Ryu, J., Woodworth, M. A., et al. (2008). Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J. Neurosci. 28, 1697–1708. doi: 10.1523/jneurosci.3032-07.2008
Immelmann, K., and Beer, C. (1989). A Dictionary of Ethology. Cambridge, MA: Harvard University Press.
Jacob, A. L., and Weinberg, R. J. (2015). The organization of AMPA receptor subunits at the postsynaptic membrane. Hippocampus 25, 798–812. doi: 10.1002/hipo.22404
Jaramillo, T. C., Speed, H. E., Xuan, Z., Reimers, J. M., Escamilla, C. O., Weaver, T. P., et al. (2017). Novel Shank3 mutant exhibits behaviors with face validity for autism and altered striatal and hippocampal function. Autism Res. 10, 42–65. doi: 10.1002/aur.1664
Jaramillo, T. C., Speed, H. E., Xuan, Z., Reimers, J. M., Liu, S., and Powell, C. M. (2016). Altered striatal synaptic function and abnormal behaviour in Shank3 Exon4–9 deletion mouse model of autism. Autism Res. 9, 350–375. doi: 10.1002/aur.1529
Jiang, Y. H., and Ehlers, M. D. (2013). Modeling autism by SHANK gene mutations in mice. Neuron 78, 8–27. doi: 10.1016/j.neuron.2013.03.016
Kamakura, R., Kovalainen, M., Leppaluoto, J., Herzig, K. H., and Makela, K. A. (2016). The effects of group and single housing and automated animal monitoring on urinary corticosterone levels in male C57BL/6 mice. Physiol. Rep. 4:e12703. doi: 10.14814/phy2.12703
Kim, C. H., Chung, H. J., Lee, H. K., and Huganir, R. L. (2001). Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc. Natl. Acad. Sci. U S A 98, 11725–11730. doi: 10.1073/pnas.211132798
Kim, R., Kim, J., Chung, C., Ha, S., Lee, S., Lee, E., et al. (2018). Cell-type-specific shank2 deletion in mice leads to differential synaptic and behavioral phenotypes. J. Neurosci. 38, 4076–4092. doi: 10.1523/jneurosci.2684-17.2018
Kolarova, J., Tangen, I., Bens, S., Gillessen-Kaesbach, G., Gutwein, J., Kautza, M., et al. (2015). Array-based DNA methylation analysis in individuals with developmental delay/intellectual disability and normal molecular karyotype. Eur. J. Med. Genet. 58, 419–425. doi: 10.1016/j.ejmg.2015.05.001
Kornau, H. C., Schenker, L. T., Kennedy, M. B., and Seeburg, P. H. (1995). Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 269, 1737–1740. doi: 10.1126/science.7569905
Kouser, M., Speed, H. E., Dewey, C. M., Reimers, J. M., Widman, A. J., Gupta, N., et al. (2013). Loss of predominant Shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. J. Neurosci. 33, 18448–18468. doi: 10.1523/jneurosci.3017-13.2013
Kudryavtseva, N. N. (2003). Use of the “partition” test in behavioral and pharmacological experiments. Neurosci. Behav. Physiol. 33, 461–471. doi: 10.1023/A:1023411217051
Leblond, C. S., Heinrich, J., Delorme, R., Proepper, C., Betancur, C., Huguet, G., et al. (2012). Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 8:e1002521. doi: 10.1371/journal.pgen.1002521
Leblond, C. S., Nava, C., Polge, A., Gauthier, J., Huguet, G., Lumbroso, S., et al. (2014). Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: a gradient of severity in cognitive impairments. PLoS Genet. 10:e1004580. doi: 10.1371/journal.pgen.1004580
Lee, J., Chung, C., Ha, S., Lee, D., Kim, D. Y., Kim, H., et al. (2015). Shank3-mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing, and spatial memory deficit. Front. Cell. Neurosci. 9:94. doi: 10.3389/fncel.2015.00094
Lee, E. J., Lee, H., Huang, T. N., Chung, C., Shin, W., Kim, K., et al. (2015). Trans-synaptic zinc mobilization improves social interaction in two mouse models of autism through NMDAR activation. Nat. Commun. 6:7168. doi: 10.1038/ncomms8168
Lim, S., Naisbitt, S., Yoon, J., Hwang, J. I., Suh, P. G., Sheng, M., et al. (1999). Characterization of the Shank family of synaptic proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J. Biol. Chem. 274, 29510–29518. doi: 10.1074/jbc.274.41.29510
Lim, C. S., Kim, H., Yu, N. K., Kang, S. J., Kim, T., Ko, H. G., et al. (2017). Enhancing inhibitory synaptic function reverses spatial memory deficits in Shank2 mutant mice. Neuropharmacology 112, 104–112. doi: 10.1016/j.neuropharm.2016.08.016
Lister, R. G. (1987). The use of a plus-maze to measure anxiety in the mouse. Psychopharmacology 92, 180–185. doi: 10.1007/bf00177912
Lu, W., and Ziff, E. B. (2005). PICK1 interacts with ABP/GRIP to regulate AMPA receptor trafficking. Neuron 47, 407–421. doi: 10.1016/j.neuron.2005.07.006
Luong, T. N., Carlisle, H. J., Southwell, A., and Patterson, P. H. (2011). Assessment of motor balance and coordination in mice using the balance beam. J. Vis. Exp. 49:2376. doi: 10.3791/2376
Manzerra, P., Behrens, M. M., Canzoniero, L. M., Wang, X. Q., Heidinger, V., Ichinose, T., et al. (2001). Zinc induces a Src family kinase-mediated up-regulation of NMDA receptor activity and excitotoxicity. Proc. Natl. Acad. Sci. U S A 98, 11055–11061. doi: 10.1073/pnas.191353598
Maren, S. (2001). Neurobiology of Pavlovian fear conditioning. Annu. Rev. Neurosci. 24, 897–931. doi: 10.1146/annurev.neuro.24.1.897
Mei, Y., Monteiro, P., Zhou, Y., Kim, J. A., Gao, X., Fu, Z., et al. (2016). Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484. doi: 10.1038/nature16971
Monteiro, P., and Feng, G. (2017). SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 18, 147–157. doi: 10.1038/nrn.2016.183
Moy, S. S., Nadler, J. J., Perez, A., Barbaro, R. P., Johns, J. M., Magnuson, T. R., et al. (2004). Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 3, 287–302. doi: 10.1111/j.1601-1848.2004.00076.x
Naisbitt, S., Kim, E., Tu, J. C., Xiao, B., Sala, C., Valtschanoff, J., et al. (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23, 569–582. doi: 10.1016/s0896-6273(00)80809-0
Naisbitt, S., Kim, E., Weinberg, R. J., Rao, A., Yang, F. C., Craig, A. M., et al. (1997). Characterization of guanylate kinase-associated protein, a postsynaptic density protein at excitatory synapses that interacts directly with postsynaptic density-95/synapse-associated protein 90. J. Neurosci. 17, 5687–5696. doi: 10.1523/JNEUROSCI.17-15-05687.1997
Neves, G., Cooke, S. F., and Bliss, T. V. (2008). Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat. Rev. Neurosci. 9, 65–75. doi: 10.1038/nrn2303
Papp, M., Willner, P., and Muscat, R. (1991). An animal model of anhedonia: attenuation of sucrose consumption and place preference conditioning by chronic unpredictable mild stress. Psychopharmacology 104, 255–259. doi: 10.1007/bf02244188
Pappas, A. L., Bey, A. L., Wang, X., Rossi, M., Kim, Y. H., Yan, H., et al. (2017). Deficiency of Shank2 causes mania-like behavior that responds to mood stabilizers. JCI Insight 2:e92052. doi: 10.1172/jci.insight.92052
Peca, J., Feliciano, C., Ting, J. T., Wang, W., Wells, M. F., Venkatraman, T. N., et al. (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. doi: 10.1038/nature09965
Pellow, S., Chopin, P., File, S. E., and Briley, M. (1985). Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J. Neurosci. Methods 14, 149–167. doi: 10.1016/0165-0270(85)90031-7
Peter, S., Ten Brinke, M. M., Stedehouder, J., Reinelt, C. M., Wu, B., Zhou, H., et al. (2016). Dysfunctional cerebellar Purkinje cells contribute to autism-like behaviour in Shank2-deficient mice. Nat. Commun. 7:12627. doi: 10.1038/ncomms12627
Peykov, S., Berkel, S., Schoen, M., Weiss, K., Degenhardt, F., Strohmaier, J., et al. (2015). Identification and functional characterization of rare SHANK2 variants in schizophrenia. Mol. Psychiatry 20, 1489–1498. doi: 10.1038/mp.2014.172
Pinto, D., Pagnamenta, A. T., Klei, L., Anney, R., Merico, D., Regan, R., et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372. doi: 10.1038/nature09146
Porsolt, R. D., Bertin, A., and Jalfre, M. (1977). Behavioral despair in mice: a primary screening test for antidepressants. Arch. Int. Pharmacodyn. Ther. 229, 327–336.
Prasad, A., Merico, D., Thiruvahindrapuram, B., Wei, J., Lionel, A. C., Sato, D., et al. (2012). A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 2, 1665–1685. doi: 10.1534/g3.112.004689
Rauch, A., Wieczorek, D., Graf, E., Wieland, T., Endele, S., Schwarzmayr, T., et al. (2012). Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380, 1674–1682. doi: 10.1016/S0140-6736(12)61480-9
Sala, C., Piëch, V., Wilson, N. R., Passafaro, M., Liu, G., and Sheng, M. (2001). Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31, 115–130. doi: 10.1016/s0896-6273(01)00339-7
Sala, C., Vicidomini, C., Bigi, I., Mossa, A., and Verpelli, C. (2015). Shank synaptic scaffold proteins: keys to understanding the pathogenesis of autism and other synaptic disorders. J. Neurochem. 135, 849–858. doi: 10.1111/jnc.13232
Schellinck, H. M., Cyr, D. P., and Brown, R. E. (2010). “Chapter 7—how many ways can mouse behavioral experiments go wrong? Confounding variables in mouse models of neurodegenerative diseases and how to control them,” in Advances in the Study of Behavior, eds H. J. Brockmann, T. J. Roper, M. Naguib, K. E. Wynne-Edwards, J. C. Mitani and L. W. Simmons (Burlington, MA: Academic Press), 255–366.
Schluth-Bolard, C., Labalme, A., Cordier, M. P., Till, M., Nadeau, G., Tevissen, H., et al. (2013). Breakpoint mapping by next generation sequencing reveals causative gene disruption in patients carrying apparently balanced chromosome rearrangements with intellectual deficiency and/or congenital malformations. J. Med. Genet. 50, 144–150. doi: 10.1136/jmedgenet-2012-101351
Schmeisser, M. J., Ey, E., Wegener, S., Bockmann, J., Stempel, A. V., Kuebler, A., et al. (2012). Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486, 256–260. doi: 10.1038/nature11015
Schmeisser, M. J., and Verpelli, C. (2016). “Chapter 10—SHANK mutations in intellectual disability and autism spectrum disorder,” in Neuronal and Synaptic Dysfunction in Autism Spectrum Disorder and Intellectual Disability, eds C. Sala and C. Verpelli (San Diego, CA: Academic Press), 151–160.
Sheng, M., and Kim, E. (2011). The postsynaptic organization of synapses. Cold Spring Harb. Perspect. Biol. 3:a005678. doi: 10.1101/cshperspect.a005678
Shepherd, J. K., Grewal, S. S., Fletcher, A., Bill, D. J., and Dourish, C. T. (1994). Behavioural and pharmacological characterisation of the elevated “zero-maze” as an animal model of anxiety. Psychopharmacology 116, 56–64. doi: 10.1007/bf02244871
Shi, R., Redman, P., Ghose, D., Liu, Y., Ren, X., Ding, L. J., et al. (2017). Shank proteins differentially regulate synaptic transmission. eNeuro 4:ENEURO.0163-15.2017. doi: 10.1523/ENEURO.0163-15.2017
Silverman, J. L., Yang, M., Lord, C., and Crawley, J. N. (2010). Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 11, 490–502. doi: 10.1038/nrn2851
Speed, H. E., Kouser, M., Xuan, Z., Reimers, J. M., Ochoa, C. F., Gupta, N., et al. (2015). Autism-associated insertion mutation (InsG) of Shank3 exon 21 causes impaired synaptic transmission and behavioral deficits. J. Neurosci. 35, 9648–9665. doi: 10.1523/JNEUROSCI.3125-14.2015
Steru, L., Chermat, R., Thierry, B., and Simon, P. (1985). The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology 85, 367–370. doi: 10.1007/bf00428203
Swerdlow, N. R., Braff, D. L., Taaid, N., and Geyer, M. A. (1994). Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch. Gen. Psychiatry 51, 139–154. doi: 10.1001/archpsyc.1994.03950020063007
Thomas, A., Burant, A., Bui, N., Graham, D., Yuva-Paylor, L. A., and Paylor, R. (2009). Marble burying reflects a repetitive and perseverative behavior more than novelty-induced anxiety. Psychopharmacology 204, 361–373. doi: 10.1007/s00213-009-1466-y
Tordjman, S., Drapier, D., Bonnot, O., Graignic, R., Fortes, S., Cohen, D., et al. (2007). Animal models relevant to schizophrenia and autism: validity and limitations. Behav. Genet. 37, 61–78. doi: 10.1007/s10519-006-9120-5
Tu, J. C., Xiao, B., Naisbitt, S., Yuan, J. P., Petralia, R. S., Brakeman, P., et al. (1999). Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23, 583–592. doi: 10.1016/s0896-6273(00)80810-7
Tye, K. M., Mirzabekov, J. J., Warden, M. R., Ferenczi, E. A., Tsai, H. C., Finkelstein, J., et al. (2013). Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature 493, 537–541. doi: 10.1038/nature11740
Uemura, T., Mori, H., and Mishina, M. (2004). Direct interaction of GluRdelta2 with Shank scaffold proteins in cerebellar Purkinje cells. Mol. Cell. Neurosci. 26, 330–341. doi: 10.1016/j.mcn.2004.02.007
Van Der Giessen, R. S., Koekkoek, S. K., van Dorp, S., De Gruijl, J. R., Cupido, A., Khosrovani, S., et al. (2008). Role of olivary electrical coupling in cerebellar motor learning. Neuron 58, 599–612. doi: 10.1016/j.neuron.2008.03.016
Vandeputte, C., Taymans, J. M., Casteels, C., Coun, F., Ni, Y., Van Laere, K., et al. (2010). Automated quantitative gait analysis in animal models of movement disorders. BMC Neurosci. 11:92. doi: 10.1186/1471-2202-11-92
Verpelli, C., Schmeisser, M. J., Sala, C., and Boeckers, T. M. (2012). Scaffold proteins at the postsynaptic density. Adv. Exp. Med. Biol. 970, 29–61. doi: 10.1007/978-3-7091-0932-8_2
Wang, X., Bey, A. L., Katz, B. M., Badea, A., Kim, N., David, L. K., et al. (2016). Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun. 7:11459. doi: 10.1038/ncomms11459
Wang, X., McCoy, P. A., Rodriguiz, R. M., Pan, Y., Je, H. S., Roberts, A. C., et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20, 3093–3108. doi: 10.1093/hmg/ddr212
Wang, Y., Zhao, X., Ju, W., Flory, M., Zhong, J., Jiang, S., et al. (2015). Genome-wide differential expression of synaptic long noncoding RNAs in autism spectrum disorder. Transl. Psychiatry 5:e660. doi: 10.1038/tp.2015.144
Wesson, D. W., Donahou, T. N., Johnson, M. O., and Wachowiak, M. (2008). Sniffing behavior of mice during performance in odor-guided tasks. Chem. Senses 33, 581–596. doi: 10.1093/chemse/bjn029
Wischmeijer, A., Magini, P., Giorda, R., Gnoli, M., Ciccone, R., Cecconi, L., et al. (2011). Olfactory receptor-related duplicons mediate a microdeletion at 11q13.2q13.4 associated with a syndromic phenotype. Mol. Syndromol. 1, 176–184. doi: 10.1159/000322054
Won, H., Lee, H. R., Gee, H. Y., Mah, W., Kim, J. I., Lee, J., et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265. doi: 10.1038/nature11208
Wrenn, C. C. (2004). Social transmission of food preference in mice. Curr. Protoc. Neurosci. 8:8.5G. doi: 10.1002/0471142301.ns0805gs28
Yang, M., Bozdagi, O., Scattoni, M. L., Wöhr, M., Roullet, F. I., Katz, A. M., et al. (2012). Reduced excitatory neurotransmission and mild autism-relevant phenotypes in adolescent Shank3 null mutant mice. J. Neurosci. 32, 6525–6541. doi: 10.1523/JNEUROSCI.6107-11.2012
Yuen, R. K. C., Merico, D., Bookman, M., L Howe, J., Thiruvahindrapuram, B., Patel, R. V., et al. (2017). Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 20, 602–611. doi: 10.1038/nn.4524
Keywords: SHANK2 domains, SHANK2 isoforms, SHANK2 gene variants, autism spectrum disorder, intellectual disability, schizophrenia, Shank2 knockout mice, behavioral tests
Citation: Eltokhi A, Rappold G and Sprengel R (2018) Distinct Phenotypes of Shank2 Mouse Models Reflect Neuropsychiatric Spectrum Disorders of Human Patients With SHANK2 Variants. Front. Mol. Neurosci. 11:240. doi: 10.3389/fnmol.2018.00240
Received: 31 March 2018; Accepted: 21 June 2018;
Published: 19 July 2018.
Edited by:
Eunjoon Kim, Institute for Basic Science (IBS), South KoreaReviewed by:
Andreas Martin Grabrucker, University of Limerick, IrelandChiara Verpelli, Istituto di Neuroscienze (IN), Italy
Copyright © 2018 Eltokhi, Rappold and Sprengel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rolf Sprengel, cm9sZi5zcHJlbmdlbEBtcGltZi1oZWlkZWxiZXJnLm1wZy5kZQ==