 Rongfang He1,2†
Rongfang He1,2† Juan Zhang
Juan Zhang Weizhong Wang
Weizhong Wang Miaoling Li
Miaoling Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Neurosci. , 16 July 2018
Sec. Molecular Signalling and Pathways
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00213
This article is part of the Research Topic The Molecular Mechanism Behind Synaptic Transmission View all 22 articles
Action potential (AP) induces presynaptic membrane depolarization and subsequent opening of Ca2+ channels, and then triggers neurotransmitter release at the active zone of presynaptic terminal. Presynaptic Ca2+ channels and SNARE proteins (SNAREs) interactions form a large signal transfer complex, which are core components for exocytosis. Ca2+ channels serve to regulate the activity of Ca2+ channels through direct binding and indirect activation of active zone proteins and SNAREs. The activation of Ca2+ channels promotes synaptic vesicle recruitment, docking, priming, fusion and neurotransmission release. Intracellular calcium increase is a key step for the initiation of vesicle fusion. Various voltage-gated calcium channel (VGCC) subtypes exert different physiological functions. Until now, it has not been clear how different subtypes of calcium channels integrally regulate the release of neurotransmitters within 200 μs of the AP arriving at the active zone of synaptic terminal. In this mini review, we provide a brief overview of the structure and physiological function of Ca2+ channel subtypes, interactions of Ca2+ channels and SNAREs in neurotransmitter release, and dynamic fine-tune Ca2+ channel activities by G proteins (Gβγ), multiple protein kinases and Ca2+ sensor (CaS) proteins.
Influx of Ca2+ through presynaptic calcium channels into presynaptic terminals at active zone is a crucial step in synaptic vesicle exocytosis and rapid neurotransmitter release (Catterall, 2011). Interactions of Ca2+ channel and soluble N-ethyl-maleimide-sensitive factor attachment protein receptor (SNAREs) complex contribute to reduce the distance between vesicles and the presynaptic membrane (Catterall and Few, 2008). The close distance provides a spatial structure that can ensure triggering of the fast neurotransmitter release within milliseconds of the action potential (AP) arriving at the synaptic terminal (Südhof, 2013; Mochida, 2017). Changes in the kinetic properties of Ca2+ channels (such as channels open, close, inactivate and so on) directly or indirectly induce modulation of the exocytosis of the synaptic vesicle, and subsequently modulate the release of neurotransmitters in a negative or positive way (Atlas, 2013). Multiple subtypes of Ca2+ channels are present in the nervous system with diverse physiological functions (Mochida, 2018). Furthermore, a single neuron also contains different types of Ca2+ channel isoforms. Thus, the channel isoforms play a key role in integral regulation of the synaptic vesicle exocytosis. Until now, it has not been clear how the Ca2+ channel isoforms coordinate well and accurately regulate fast neurotransmitter release at synaptic terminals. In this mini review, we focus on the molecular structures and regulatory mechanisms of multiple Ca2+ channel isoforms, and the interactions of Ca2+ channels and SNAREs involving vesicles fusion and neurotransmitter releases.
The diverse subtypes of Ca2+ channels display different biological structures and distribution in the nervous system. The diversity of channels corroborates its different physiological functions.
According to the unique electrophysiological and pharmacological properties, voltage-gated calcium channel (VGCC) have been classified into N-, P/Q-, R-, L- and T-type (Ertel et al., 2000). N-, P/Q-, R- and L-type is termed as high-voltage activated Ca2+ channel, while T-type is low-voltage activated Ca2+ channel (<−40 mV). High-voltage activated Ca2+ channels are composed of the pore-forming Cavα1 and four auxiliary subunits (Cavα2, Cavβ, Cavγ and Cavδ; Catterall, 2000), while T-type contains only the Cavα1 subunit (Cavα1G, Cavα1H and Cavα1I; Figures 1A,B). The neuronal Cavα1 subunit (190–250 kDa) is the largest and main subunit, which is composed of about 2000 amino-acid residues. The molecular weights of α2, β, γ and δ subunits are 143 kDa, 53–70 kDa, 30 kDa and 24–27 kDa, respectively. The Cavα1 contains four homologous domains (I–IV; Figure 1C), and each domain of Cavα1 is comprised of six transmembrane α helices (S1–S6). The transmembrane S5–S6 segments form a p loop, and the S1–S4 segments serve as the voltage sensor (Yu et al., 2005). Diversity of Cavα1 isoforms determine the channel subtypes. Ten different types of Ca2+ channels have been identified (Yu and Catterall, 2004). The Cavα1 subunit genes are classified as Cav1.1–1.4 (L-type), Cav2.1–2.3 (P/Q-, N-, and R-type) and Cav3.1–3.3 (T-type; Figure 1B), each of them belongs to CACNA1x gene families. N-type and P/Q-type Ca2+ channels are the main Ca2+ channels in nerve terminals and play an important role in fine-tuning of rapid neurotransmitter release at synaptic terminals (Ariel et al., 2012). The R-type (Cav2.3) Ca2+ channels are present in the peripheral nervous system (PNS) and central nervous system (CNS). Though R-type Ca2+ channels are not the main Ca2+ channels, they are also involved in presynaptic plasticity and neurotransmitter release (Breustedt et al., 2003; Dietrich et al., 2003; Naidoo et al., 2010). T-type Ca2+ channel present in peripheral, central synapses and neuroendocrine cells, play a key role on tuning of basal neurosecretion near resting potential with a mild stimulation (Lambert et al., 2014).
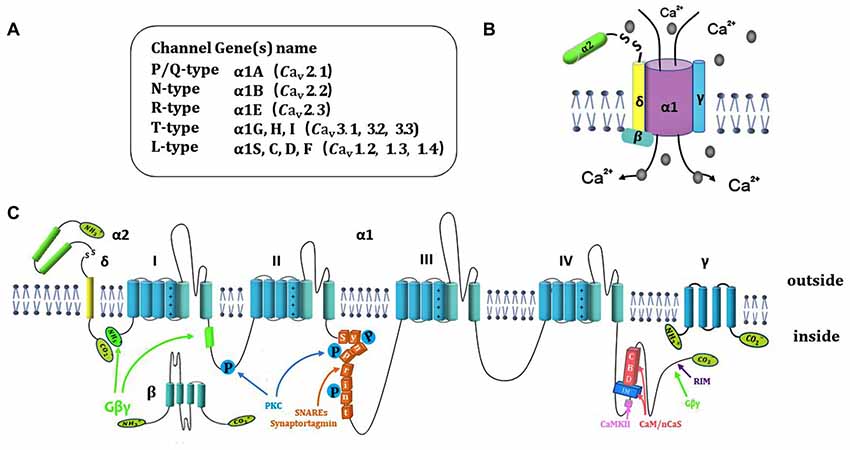
Figure 1. Ca2+ channel structure, organization and interaction with regulatory proteins. (A) Summary of 10 different subtypes of Ca2+ channel. (B) Representation of subunits composition of Ca2+ channels and auxiliary subunits. (C) The subunit consists of four homologous domains (I–IV), auxiliary subunits and interaction of Ca2+ channel subunits with regulatory proteins include SNARE proteins, Gβγ, kinase and CaS proteins.
The auxiliary subunits of Ca2+ channels include α2, β, γ and δ subunits (Figure 1A). The α2 and δ subunits are encoded by the same gene that bind together with disulfide linkage to form α2-δ subunit complex. α2-δ subunit exerts a role of increased calcium current and upregulation of gene expression. The interactions of α2-δ subunit with extracellular Cavα1 modulate the binding of divalent cations (Cantí et al., 2005). α2δ-1 isoform is encoded by the gene Cacna2d1. The interaction of α2δ-1 and NMDA receptors significantly increased in neuropathic pain (Dolphin, 2012; Patel et al., 2013). Chen et al. (2018) demonstrate that gabapentin reduces neuropathic pain by inhibiting of the interaction between the C terminus of α2δ-1 and NMDA receptors. The whole β subunit is located in cytoplasm. The functional role of β subunit is to ensure the α1 subunit binding to the plasma membrane and prevent it trafficking to the endoplasmic reticulum. β subunit regulates membrane protein expression and gating of Ca2+ channels (Arikkath and Campbell, 2003). The γ subunit comprises of four transmembrane α helices, that can slightly reduce Ca2+ current density and change kinetic properties by interacting with the Cavα1 subunit (Osten and Stern-Bach, 2006). The specific polypeptide toxins from snail and spider venoms, block multiple Ca2+ channel subtypes. ω-conotoxin GVIA (ω-Cgtx GVIA) blocks the N-type channels irreversibly in central nervous system (CNS) and Peripheral nervous system (PNS; Prashanth et al., 2014). ω-agatoxin IVA (ω-AgaIVA) blocks the Cav2.1 (P-type) and ω-Aga IVA blocks the Cav2.1 (Q-type) with a lower affinity in CNS (Arranz-Tagarro et al., 2014; Ricoy and Frerking, 2014). ω-conotox in MVIIC is a toxin from the venom of marine conus snail, which targets Cav2.1 with high affinity and targets Cav2.2 with low affinity (Catterall et al., 2005). α-conotox in Vc1.1 does not affect Cav2.1 but strongly inhibits Cav2.3 Ca2+ channels through GABAB receptor (Berecki et al., 2014). Cav2.3 Ca2+ channels were potently blocked by Zn2+ (IC50 = 0.78 ± 0.07 μmol/L; Traboulsie et al., 2007). The tetraline derivative of mibefradil and the peptide blocker of scorpion toxin kurtoxin have been evaluated as potential Cav3 Ca2+ channel inhibitors in CNS and PNS (Chuang et al., 1998).
The diversity of Cavα1 and auxiliary subunits confirms distinct molecular structures, synaptic properties and distributions that are involved in the regulation of various physiology functions in neurotransmitter release. P-/Q-type Ca2+ channels mediated GABA release in the most of GABA releasing inhibitory neurons (Lonchamp et al., 2009). Glutamate-release is often mediated by integrated interactions of P-/Q- and N-type Ca2+ channels in the vast majority of glutamatergic cortical and cerebellar synapses (Ladera et al., 2009). Furthermore, P-/Q-type Ca2+ channels decrease fusion pore stability and trigger vesicle fusion, N-type and L-type Ca2+ channels slow down fusion pore expansion (Ardiles et al., 2007). In the axon terminal, P-/Q- type Ca2+ channels are close to the release zone than other Ca2+ channels in various synapses. As a result, P-/Q- type Ca2+ channels (Cav2.1) may lead to higher local presynaptic Ca2+ concentrations and frequently co-localized with synaptotagmin-containing vesicle clusters, whereas the N-type channel (Cav2.2) and R-type channel (Cav2.3) are only partially involved in vesicle clusters (Wu et al., 1999). The significant role of N-type Ca2+ channels is involved in neurotransmitter release in cortical and hippocampal synapses. L- and T-type Ca2+ channels are involved in neurotransmission release in various retinal neurons. T-type Ca2+ channels play a crucial role in neurotransmitter release and its regulation in special reciprocal synapses. Functionally, P-/Q-type Ca2+ channels may be mainly related to fast, synchronous exocytosis, and N-type Ca2+ channels may contribute to exocytosis in neurons processing information, P-/Q-type Ca2+ channels have been shown to be more efficient in neurotransmitter release than N-type Ca2+ channels in most investigated synapses, as in entorhinal stellate neurons, different inhibitory interneurons, cerebrocortical synapses or cerebellar parallel fiber terminals (Ladera et al., 2009).
Ca2+ entry through presynaptic Ca2+ channels can trigger vesicle fusion by assembly of the SNARE proteins complex [t-SNARE proteins syntaxin-1 and SNAP-25, v-SNARE protein synaptobrevin (VAMP)] (Südhof, 2004; Bao et al., 2018; Figure 2A). SNARE function is widely reported to be associated with the processing of physiology and pathophysiology. It is reported that modifying SNARE function through regulating exocytosis can provoke metabolic diseases such as obesity (Valladolid-Acebes et al., 2015), which is improved by many therapies such as exercise training (Ramos-Miguel et al., 2015; Roh and So, 2017; Roh et al., 2017). The release of neurotransmitter requires localization of both calcium channels and synaptic vesicle proteins to the presynaptic active zone (Südhof, 2012). Rab3 interacting molecules (RIM) localizes in active zone (Figure 2B), which contain an N-terminal zinc finger domain, a central PDZ domain, C-terminal C2A and C2B domain and a conserved sequence between the two C-terminal domains (Wang and Südhof, 2003). RIM plays an essential role for synaptic vesicle docking and priming (Deng et al., 2011; Han et al., 2011). Munc13-1 is a large multidomain protein in active zone that plays a central role in synaptic vesicle priming (Brose et al., 1995; Augustin et al., 1999; Fukuda, 2003). The interaction of SNARE and SM (sec1/Munc18) proteins control the millisecond timescale presynaptic fusion after AP. Before priming, the Munc13 C2A-domain forms a constitutive homodimer (inactive state; Figure 2C). When Munc13 transforms from inactive state to an active state, Munc13-1 switches from a homodimer to a heterodimer (Munc13-1-RIM), may regulate synaptic vesicle priming (Lu et al., 2006). RIM-binding proteins (RIM-BPs) are also large multidomain proteins (~200 kDa) in active zone, that tightly bind to RIM. PDZ-domain of both RIM and RIM-BPs bind to Ca2+-channels for tethering Ca2+ channels to an active zone (Han et al., 2011; Kaeser et al., 2011). Deletion of RIM or RIM-BP (Liu et al., 2011; Kaeser et al., 2012) causes loss of Ca2+ channels from active zone and decreases Ca2+ entry. The central PDZ-domain of RIM can bind directly with N-type and P/Q-type, without binding with L-type Ca2+ channels (Kaeser et al., 2011). RIM that lacks the PDZ-domain exhibits loss binding abilities with Cav channels. Cav channels are recruited to active zone for synaptic vesicle fusion by a tripartite complex formation (RIM, RIM-BP and the C-terminal tail of Ca2+ channels) that needs assistance by Munc13-1. Munc13-1-RIM heterodimer formation is a key component for fusion. Furthermore, the C2B domain of RIM can modulate Ca2+ channel activation (Kaeser et al., 2012). Recently, it was reported that Munc13, independent with Munc18, promotes the syntaxin-1-synaptobrevin complex formation during the assembly of the triplet SNARE complex. Interaction with Munc18 and Munc13 contributes to syntaxin/SNAP-25 complex formation (Lai et al., 2017).
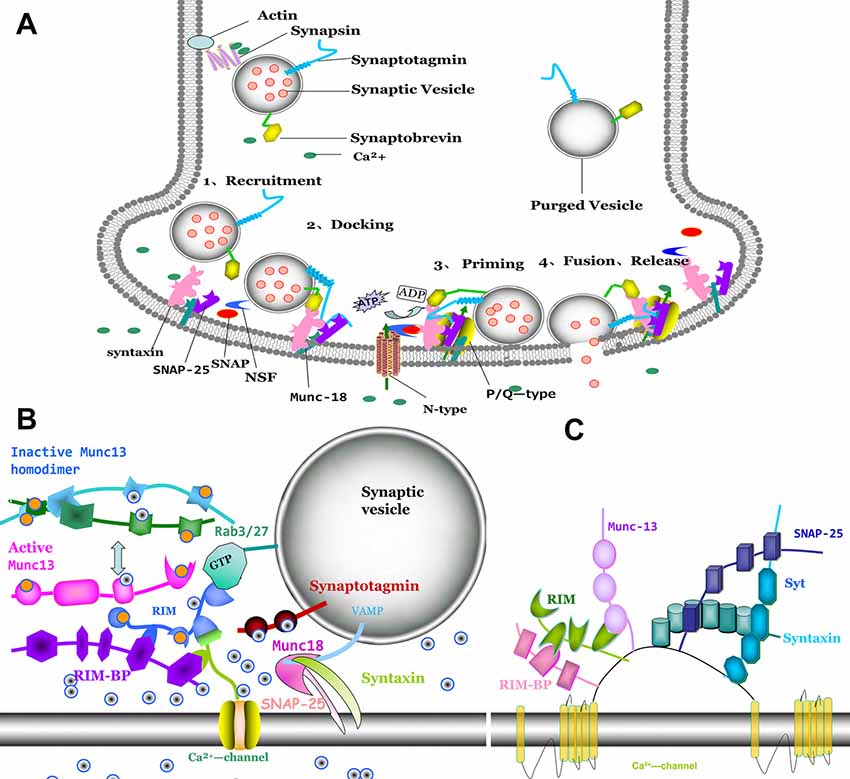
Figure 2. Molecular model of synaptic vesicle fusion machinery, interactions of active zone proteins, presynaptic Ca2+ channel and SNAREs. (A) The process of vesicle fusion: 1. synaptic vesicle recruiting to active zone; 2. synaptic vesicle docking at the presynaptic membrane and with SNAREs complex conformation. 3. priming of synaptic vesicle on presynaptic membrane; and 4. fusion pore to open and with neurotransmitter release. (B) Direct interaction of Cav2 α1 subunits with proteins Rab3 interacting molecules (RIM), RIM-binding protein (RIM-BP), Munc-13 and SNAREs (syntaxin, SNAP-25 and synaptobrevin). (C) RIM binding to RIM-BP induced Munc13 from inactive homodimer to active heterodimer, which promoted Sec1/Munc18-1 (SM) protein dissociated with syntaxin-1. Syntaxin-1 changes from closed formation to open formation. Syntaxin-1 and SNAP-25 interacted with synaptobrevin to form SNAREs. Ca2+ entry through Ca2+ channel induced interaction with synaptotagmin, which trigger vesicle fusion.
Before forming the SNARE complex (Figure 2A), syntaxin-1 is presented in a closed conformation by interaction with SM proteins which cannot promote SNARE complex formation. When the zinc-finger of RIM binds to the C2A domain of Munc13, Munc13 is activated by homodimer dissociation. Subsequently, the activation of Munc13 drags RIM closer to the presynaptic membrane. Ma et al. (2011) have demonstrated that Munc13 can accelerate the transfer from the closed syntaxin-1-Munc18-1 heterodimer to an open syntaxin-1 for promoting SNARE complex formation. SM proteins are fundamental for synaptic vesicle trafficking. However, another study reported that the SM proteins exert no effect on spontaneous fusion and Ca2+-triggered fusion with SNAREs, complexin-1 and syt-1 (Zhang et al., 2015). Stable SNAREs complex provide energy for membrane fusion (Weber et al., 1998).
Complexin is a small soluble protein that controls (activates or suppresses) the trigger-release and spontaneous release (Fernández-Chacón et al., 2001; Pang et al., 2006; Mohrmann et al., 2015; Yu et al., 2018). The central helix of complexin binds to the interface of the v- and t-SNAREs close to the membrane (Fernández-Chacón et al., 2001; Chen et al., 2002; Tang et al., 2006). Complexin displays an activated effect in fast synchronous release and an inhibited effect in spontaneous release (Maximov et al., 2009; Kaeser-Woo et al., 2012). The synchronous function of complexin-1 is promoted by interactions with the SNARE complex at the N-terminal, whereas the suppressive action of spontaneous fusion is involved in binding with the C-terminal domain of complexin-1, but not the N-terminal domain (Lai et al., 2014). Lai et al. (2016) have demonstrated the mechanism that the N-terminal domain of the complexin can independently modulate the interaction of presynaptic membrane and the SNAREs. Furthermore, Gong et al. (2016) have revealed that the C-terminal domain is pivotal for regulation of spontaneous release and suppression of Ca2+-independent fusion in a curvature-dependent phase. Misplacement of complexin to the plasma membrane increases the variableness and the mean decay time constant of synchronization with NMDA-type glutamate receptor initiated postsynaptic currents.
Synaptotagmin (syt) is a Ca2+ sensor that can evoke fast and synchronous neurotransmitter release (Xu et al., 2007). Syt-1 contains two homologous Ca2+ sensor modules: C2 domains (C2A and C2B) and transmembrane domain. Syt-1, Syt-2 and Syt-9 bind to Ca2+ to promote synchronous transmitter release, while Syt-7 evokes a asynchronous transmitter release (Bacaj et al., 2013; Brewer et al., 2015; Zhou et al., 2015; Pérez-Lara et al., 2016).
Ca2+ binding to syt abolishes the complexin clamp and triggers synaptic vesicle fusion. Recent study has revealed that complexin may regulate fusion in cooperation with Syt. Syt1-SNARE and complexin-SNARE cooperate to activate synchronous release and regulate synchronous release after the AP has arrived at the synaptic terminal (Jorquera et al., 2012; Dhara et al., 2014). Recent study has demonstrated that the tripartite SNARE complexin-syt-1 complex at a synaptic vesicle docking site exerts an open state for trigger fusion. Interaction of interfaces are fundamental for Ca2+-triggered neurotransmitter release. Disruption of tripartite interface cannot trigger neurotransmitter release, although the primary interface is intact. It implied that both the primary and tripartite interfaces are required for Ca2+-triggered synaptic vesicle fusion (Akyuz et al., 2013; Gipson et al., 2017). Before the Ca2+ trigger, syt interacts with SNARE proteins in the targeted membrane to prevent SNARE complex assembly (Chicka et al., 2008). Ca2+ entry through Ca2+ channels increases the affinity of syt-1 with syntaxin-1 for approximately two orders of magnitude (Chapman et al., 1995; Bhalla et al., 2006). Munc13, notably, enhances the transforms from the Munc18-1 syntaxin-1 complex to the SNARE complex (Ma et al., 2011) that can open the closed form of the SNARE protein (Lu et al., 2006; Kaeser et al., 2011). NSF (Sec18) and α-SNAP (Sec17) form a molecular chaperone for dynamic modulation of the disassembly of cis-SNAREs. Recently, Song et al. (2017) demonstrated that Sec17 residue K159 contributes to enhance the synaptic vesicle fusion. Furthermore, Sec18 can augment the interactions of Sec17 and trans-SNARE (Schwartz et al., 2017; Song et al., 2017). Lai et al. (2013) also have found that syt1 and Ca2+ are required for pore formation and expansion. Furthermore, SNAREs alone are sufficient in promoting membrane hemifusion.
Presynaptic VGCCs have been classified into three super families (Cav1, Cav2 and Cav3). Cav2 (P/Q-, N- and R-type) are the dominant channel subtypes for fast presynaptic transmitter. Cav2.2 (N-type) interacts with active zone proteins (RIM, RIM-BP) and SNAREs to regulate the vesicle fusion. RIM-C2A and RIM-C2B bind the pore-forming subunit of N-type Cav channels in a Ca2+-independent manner that weakly interacts with the Cav1.2 (L-type, α1c), but do not interact with the Cav1.3 (L-type, α1D). Furthermore, RIM (C2 domain) enhances the interaction with syt-1 when intracellular Ca2+ concentration is increased. Removal of RIM domain heavily reduces the channel current and number of docking vesicles resulting in decreasing Ca2+ channel coupling with vesicle. The central PDZ-domain of RIM interacts with the C-terminal of presynaptic N- and P/Q-type Ca2+-channels, with no act on L-type Ca2+ channels. Deletion of RIM inhibits most neurotransmitter release due to impairing the synaptic priming and decreasing the Ca2+ channels localization in presynaptic membrane (Kaeser et al., 2011; Han et al., 2015). It is well-known that vesicle priming and Ca2+ influx do not require RIM C2B domains. Recently, studies have found that C2 domains of RIM do not bind to Ca2+, but bind to PIP2. PIP2 binding to RIM C2B domains exerts a critical role for vesicle priming and Ca2+ channel tethering to PIP2 containing targeted membranes (de Jong et al., 2018).
Active zone scaffold protein Bassoon directly binds to RIM-BP to modulate synaptic vesicle docking via an indirect contact with Cav2.1. Genetic deletion of Bassoon or an acute interference with Bassoon RIM-BP interaction reduces synaptic amount of CaV2.1, which gently regulates P/Q-type Ca2+ current to trigger synaptic transmission (Davydova et al., 2014). Both genetic ablation of Bassoon or interference of the link between Bassoon and RIM-BP reduced the numbers of Cav2.1 in active zone, decelerated AP-triggered neurotransmitter release and impaired the synaptic transmission. Cav2.2 current was increased for compensation for Cav2.1-induced decreases (Acuna et al., 2015). RIMs-mediated vesicle priming is not produced by coupling with Munc13, whereas it is directly activated by Munc13. Zn2+ finger domain of RIM binds to Munc13 to promote vesicle priming, thereby dissociating Munc13 from heterodimer to homodimer and promotes priming in Munc13-deficient synapses. Hence, homodimer of Munc13 inhibits priming, and RIM activates priming by disrupting Munc13 homodimer (Deng et al., 2011). At rod photoreceptor ribbon synapses, RIM causes a dramatic loss of Ca2+ entry through Cav1.4 channels and reduces trigger release. RIM induces Ca2+ entry, which in turn promotes release by modulating Cav1.4 channel opening (Grabner et al., 2015). Alternative splicing (exons of 44 and 47) of Cav2.1α1 (P/Q-type) induces gene variants of the C-terminal region (CTD) of Cav2.1. The two exons interact with RIM (1α and 2α), impair the binding of CTD with RIM and implied suppressive effect of RIM on voltage-dependent inactivation (Hirano et al., 2017). Syntaxin, SNAP-25 and syt-1 possess specific “synprint” binding site interaction with CaV2.1 and CaV2.2 at the intracellular loop linking domains II and III (LII-III; Figure 1C). Diversity of VGCC types display distinct tissue specificity, subcellular localizations, kinetics performance and amount of Ca2+ influx. CaV2.1 is the most abundant expression in neurons (Cabañes, 2008; Catterall and Few, 2008; Davies et al., 2011; Jahn and Fasshauer, 2012; Davydova et al., 2014; Wang and Augustine, 2014; Chai et al., 2017; Silva et al., 2017). GSK-3β displays inhibitory effects in presynaptic vesicle exocytosis by phosphorylating CaV2.1 and disturbing SNARE complex formation. A mutation in the first intracellular loop of CaV2.1 prevents interaction with SNARE proteins and impair SNAREs complex formation. SNAREs proper interact with synprint site to help vesicles docking near the Ca2+ entry pathway, and modulate steady-state inactivation of Cav2.1 (Serra et al., 2018). R-type (Cav2.3) channels are localized at the presynaptic terminal and trigger neurotransmitter release by enhancing presynaptic Ca2+ levels. Wu et al. (1999) reported that R-type (Cav2.3) Ca2+ channels contributed to about 26% of the total Ca2+ current during a medial nucleus of the trapezoid body presynaptic AP, but display a lower efficacy than other types of Ca2+ channels. R-type Ca2+ channels are also involved in fast synaptic excitation (Naidoo et al., 2010). Recently, researchers revealed that R-type Ca2+ channels linked with NOS to induce NO release by controlling gastrointestinal smooth muscle relaxation in the guinea pig ileum via a purine transmitter (Rodriguez-Tapia et al., 2017).
Aplysia pleural sensory neurons are involved in the forms of presynaptic plasticity. The Aplysia CaV2α1 subunit EF-hand tyrosine Y1501 are targets for modulation by GPCRs through Src kinase. The heterosynaptic depression of the CaV2 channel current is inhibited when channel is combined with a Y-F mutation at the conserved Src phosphorylation. It implies that the inhibition of the Cav2 calcium current is partially, at least, responsible for the inhibition of neurotransmitter release with heterosynaptic depression (Dunn et al., 2018). Ca2+ channels are also involved in nerve injury. Lu et al. (2018) first demonstrated that lycopene depress glutamate release through inhibition of voltage-dependent Ca2+ entry (N-type and P/Q-type channels) and protein kinase C in rat cerebrocortical nerve terminals and not by intracellular Ca2+ release.
The CaV3 family CaV3.1(α1G), CaV3.2(α1H), and CaV3.3(α1I) mediate T-type Ca2+ currents. T-type channels have been revealed to regulate neurotransmitter release in central, peripheral synapses and neuroendocrine cells that modulate basal neurosecretion close to resting potential with mild stimulations. Although T-type channels have no directly binding peptide (no synprint binding site), Cav3.2 channels interact with syntaxin 1A and SNAP-25. The interactions form nanodomains that can be regulated transiently and low voltages controlling neural activity and neuroendocrine. Interaction of T-type channels, secretory vesicles, and SNAREs form a nanodomains complex. T-type Ca2+ channels can directly interact with SNAREs (syntaxin 1A-Cav3.2-SNAP25) to control exocytosis. It is clear that T-type channels contribute to synaptic transmission in neurons and neuroendocrine cells under conditions of rest and mild stimulation. T-type Ca2+ channels are also involved in the development of a neuropathic pain. T-type Ca2+ channel subunit CaVα2δ interaction with the extracellular matrix protein thrombospondin-4 (TSP4) contributes to initiate, but not for the maintenance of excitatory synaptogenesis. Treatment with gabapentin blocks the early pain state but does not reverse the delayed state. It implies that early intervention with gabapentin may prevent the development of injury-induced chronic pain, one of the reasons is that CaVα2δ1/TSP4 initiates abnormal synapse formation (Yu et al., 2018).
Interestingly, Diao et al. (2013) have found that native presynaptic protein α-Synuclein (α-Syn) has little effect on Ca2+-triggered synaptic fusion efficiency or kinetics in neurotransmitter releases. On the contrary, α-Syn plays a key role in clustering of v-vesicles. Parkinson’s disease induces α-Syn mutant at A30P. Pathogenic α-Syn reduces the clustering ability that resulted in affecting neurotransmission (Diao et al., 2013). Furthermore, N-terminal acetylation can significantly decrease α-Syn oligomerization that can preserve its native conformation against pathological aggregation (Bu et al., 2017).
The activity of presynaptic calcium channels is also modulated by βγ-subunits of G proteins (Gβγ), protein kinases (PKC, CaMKII) and Ca2+ sensor (CaS) proteins. Gβγ negatively regulates the neurotransmitter release by inhibition of CaV2 (P/Q- and N-type) Ca2+ channels in synaptic terminals. Gβγ directly binds to CaV2.2α1 at the N-terminal45–55 (Cantí et al., 1999), the intracellular loop domains between I and II (LI-II) at 377393 (Zamponi et al., 1997) and the C-terminus at 22572336 (Li et al., 2004). Only the N-terminal can suppress CaV2 channels activity. The site at the N terminus and intracellular loop (LI-II) produces a more potent effect (Stephens and Mochida, 2005; Figure 1C). Furthermore, it has also been demonstrated that the CaV2.2 alternative splicing isoform, e37a, exerts an increase in the expression of N-type Ca2+ channels and also increases the channel opening compared to Cav2.2 channels that contain e37b (Castiglioni et al., 2006). Injection of N-terminal or a I-II loop interaction domain peptide into sympathetic superior cervical ganglion (SCG) neurons attenuates noradrenaline-initiated G protein regulation, and reduces synaptic transmission, and decreases Ca2+ current density. Furthermore, mutation at N-terminal abolishes the inhibitory effects of the N-terminal peptide (Bucci et al., 2011). Gβγ binding to N-terminal and loop I–II of CaV2.2 contributes to regulate the function of CaV2.2. Interestingly, the SNARE protein syntaxin 1A co-localizes with Ca2+ channels and Gβγ. Co-expression of syntaxin 1A with N-type channels induces tonic inhibition mediated by Gβγ (Jarvis et al., 2000). Nevertheless, syntaxin 1B does not display such effect (Lü et al., 2001). It is suggested that the spatial localization of the G protein-synaprint-CaV2.2 complex is critical for neurotransmitter release (Yoon et al., 2008). The synaptic protein cysteine string protein promotes interaction between G proteins and the synprint site on CaV2.2 channel for enhancing neurotransmitter release (Figure 2C).
Protein kinases (such as PKC and CaMKII) are localized in presynaptic terminals that can phosphorylate both Ca2+ channels and SNAREs (Figure 2C). PKC and CaMKII-phosphorylation of Ca2+ channels at the synprint site induce forceful inhibition of its binding to syntaxin-1A and SNAP-25 (Yokoyama et al., 1997). Phosphorylation of Ca2+ channels at the synprint by PKC is located at serines 774 and 898 which resulted in modulating the interaction with syntaxin-1A and SNAP-25. However, PKC phosphorylation failed to dissociate CaV2.2/syntaxin 1A complexes. Auxiliary subunits of Cav2.2 also participate in regulation of the function of Cav2.2 channel and then modulate the transmitter release. The acetyl-β-methylcholine (MCh) or PKC isozymes (βII or ε) are unable to potentiate Cav2.2 current in the presence of CaVβ subunits. Cavβ subunits complete suppression of the interactions between PKC and Ser/Thr sites of Cav2.2α1 subunits (Thr-422, Ser-425, Ser-1757, Ser-2108 and ser-2132; Rajagopal et al., 2014). The mutation of PKC sites (Thr-422, Ser-1757 and Ser2132) can abolish MCh potentiation on Cav2.2α1 currents. The stimulatory sites at Thr-422, Ser-2108 or Ser-2132 and inhibitory sites at Ser-425 of Cav2.2α1 are identified by binding to PKCs βII and ε subunits. Whereas, the stimulatory sites at Thr-365, Ser-1995 and Ser-2011 and the inhibitory sites at Ser-369of Cav2.3α1 subunits are homologous with Cav2.2α1. The stimulatory effects of PKC at the site of Thr-365 or Ser-1995 were fully offset by inhibitory site at Ser-369. PKC cannot inhibit the effects via the coexistence with Thr-365 and Ser-1995 (Rajagopal et al., 2017).
The phosphorylation of Core-conserved residues inside the SNARE domain can suppress vesicle fusion. Studies revealed that secretory protein VAMP8 phosphorylation by PKC at multiple residues in the SNARE domain mediated vesicle fusion, where protein kinase activation decreases and phosphatase activation increases the capacity of VAMP8 (Malmersjö et al., 2016).
CaMKII potently inhibits the interactions between syntaxin-1A and SNAP25 by phosphorylation at Ser 784 and 896 (Yokoyama et al., 2005). Each site of phosphorylation modulates syntaxin-1 and SNAP-25 binding to the synprint site. PKC or CaMKII phosphorylates Cavα1 at the synprint sites that manipulates a biochemical switch for controlling the interaction of synprint and SNAREs. It implied that switch role provides a potential functional link between neurotransmitter release and protein phosphorylation for tethering and docking synaptic vesicle in an optimal position to respond to the Ca2+ signal from presynaptic Ca2+ channels (Catterall and Few, 2008).
In neurons, multiple Ca2+ sensor (CaS) proteins are involved in neuronal Ca2+ signaling transmitter. The distance between voltage-gate Ca2+ channels and CaS for exocytosis determines the timing and probability of neurotransmitter release (Nakamura et al., 2018). Calmodulin (CaM) is one of the members of a subfamilies of CaS proteins. Vesicle protein synaptotagmin is also a CaS protein for fast neurotransmission. Interactions of Ca2+/CaM binding to the CaM-binding domain (CBD) and IQ-like motif (IM) of CaV2.1 contribute to facilitate and inactivate Cav2.1 channels. Mutation of the motifs of CBD and IM prevents synaptic facilitation. Nanou et al. (2018) demonstrate a direct link between regulation of CaV2.1 channels and short-term synaptic plasticity in native hippocampal excitatory and inhibitory synapses. CaBP1 and VILIP-2 are neurospecific CaM-like CaS proteins that potently modulate CaV2.1 channels function. Ca2+-binding protein (CaBP1), Visinin-like protein 2 (VILIP-2) and neuronal calcium sensor-1 (NCS-1) are the key CaS proteins for synaptic transmission. CaBP1 is highly expressed in the brain and retina, and co-localized in the CBD of Cav2.1α1 (Lee et al., 2002). CaBP1 binds to CBD in a Ca2+ independent profile. Leal et al. (2012) demonstrated that CaBP1 performed a blockade effect on Ca2+-dependent facilitation of Cav2.1, and reduced facilitation of synaptic transmission in superior cervical ganglion neurons. Nanou et al. (2018) also demonstrated CaBP1/caldendrin as the CaS protein interacting with CaV2.1 channels to mediate rapid synaptic depression in the inhibitory hippocampal synapses. On the contrary, VILIP-2 blocked Ca2+-dependent inactivation of CaV2.1 current, and notably reduced synaptic depression and showed increasing facilitation. VILIP-2 is highly expressed in neocortex and hippocampus, and plays a complementary effect on CaBP1. These studies reveal that CaBP1 and VILIP-2 bind to the same site with opposite effects on Cav2.1. The integrated effect contributes to modulating short-term synaptic plasticity (Leal et al., 2012; Catterall et al., 2013). The N-terminal myristoylation site and EF-hand motifs of CaBP1 and VILIP-2 determine their differential regulated role on CaV2.1 channels. CaS proteins serve as bidirectional switch that fine-tune the relationships of CaV and synaptic transmission. Thereby, the balance between facilitation and depression is a key role on neurotransmitter release (Leal et al., 2012).
Neuronal calcium sensor-1 (NCS-1) has been also shown to enhance synaptic facilitation. NCS-1 directly interacts with IQ-like motif and CBD site at the C-terminal domain of CaV2.1. NCS-1 reduces Ca2+-dependent inactivation of Cav2.1 through interaction with the IQ-like motif and CBD. NCS-1 modulates Ca2+ current amplitude or kinetics activity. These studies indicate that NCS-1 directly binds to CaV2.1 to serve short-term synaptic facilitation and confirm that CaS proteins are crucial in fine-tuning short-term synaptic plasticity (Yan et al., 2014).
RH, JZ, YY and LZ contributed to the review of the literature, and editing of the manuscript. WW and ML wrote the draft manuscript. All authors read and approved the submission.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the undergraduate students Zenan Fan and Xiaoli Gao for drawing figures of this manuscript.
Acuna, C., Liu, X., Gonzalez, A., and Sudhof, T. C. (2015). RIM-BPs mediate tight coupling of action potentials to Ca2+-triggered neurotransmitter release. Neuron 87, 1234–1247. doi: 10.1016/j.neuron.2015.08.027
Akyuz, N., Altman, R. B., Blanchard, S. C., and Boudker, O. (2013). Transport dynamics in a glutamate transporter homologue. Nature 502, 114–118. doi: 10.1038/nature12265
Ardiles, A. O., González-Jamett, A. M., Maripillán, J., Naranjo, D., Caviedes, P., and Cardenas, A. M. (2007). Calcium channel subtypes differentially regulate fusion pore stability and expansion. J. Neurochem. 103, 1574–1581. doi: 10.1111/j.1471-4159.2007.04871.x
Ariel, P., Hoppa, M. B., and Ryan, T. A. (2012). Intrinsic variability in Pv, RRP size, Ca2+ channel repertoire, and presynaptic potentiation in individual synaptic boutons. Front. Synaptic Neurosci. 4:9. doi: 10.3389/fnsyn.2012.00009
Arikkath, J., and Campbell, K. P. (2003). Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr. Opin. Neurobiol. 13, 298–307. doi: 10.1016/s0959-4388(03)00066-7
Arranz-Tagarro, J. A., de los Rios, C., Garcia, A. G., and Padin, J. F. (2014). Recent patents on calcium channel blockers: emphasis on CNS diseases. Expert Opin. Ther. Pat. 24, 959–977. doi: 10.1517/13543776.2014.940892
Atlas, D. (2013). The voltage-gated calcium channel functions as the molecular switch of synaptic transmission. Annu. Rev. Biochem. 82, 607–635. doi: 10.1146/annurev-biochem-080411-121438
Augustin, I., Rosenmund, C., Sudhof, T. C., and Brose, N. (1999). Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 400, 457–461. doi: 10.1038/22768
Bacaj, T., Wu, D., Yang, X., Morishita, W., Zhou, P., Xu, W., et al. (2013). Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80, 947–959. doi: 10.1016/j.neuron.2013.10.026
Bao, H., Das, D., Courtney, N. A., Jiang, Y., Briguglio, J. S., Lou, X., et al. (2018). Dynamics and number of trans-SNARE complexes determine nascent fusion pore properties. Nature 554, 260–263. doi: 10.1038/nature25481
Berecki, G., McArthur, J. R., Cuny, H., Clark, R. J., and Adams, D. J. (2014). Differential Cav2.1 and Cav2.3 channel inhibition by baclofen and α-conotoxin Vc1.1 via GABAB receptor activation. J. Gen. Physiol. 143, 465–479. doi: 10.1085/jgp.201311104
Bhalla, A., Chicka, M. C., Tucker, W. C., and Chapman, E. R. (2006). Ca2+-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat. Struct. Mol. Biol. 13, 323–330. doi: 10.1038/nsmb1076
Breustedt, J., Vogt, K. E., Miller, R. J., Nicoll, R. A., and Schmitz, D. (2003). α1E-containing Ca2+ channels are involved in synaptic plasticity. Proc. Natl. Acad. Sci. U S A 100, 12450–12455. doi: 10.1073/pnas.2035117100
Brewer, K. D., Bacaj, T., Cavalli, A., Camilloni, C., Swarbrick, J. D., Liu, J., et al. (2015). Dynamic binding mode of a Synaptotagmin-1-SNARE complex in solution. Nat. Struct. Mol. Biol. 22, 555–564. doi: 10.1038/nsmb.3035
Brose, N., Hofmann, K., Hata, Y., and Sudhof, T. C. (1995). Mammalian homologues of Caenorhabditis elegans unc-13 gene define novel family of C2-domain proteins. J. Biol. Chem. 270, 25273–25280. doi: 10.1074/jbc.270.42.25273
Bu, B., Tong, X., Li, D., Hu, Y., He, W., Zhao, C., et al. (2017). N-terminal acetylation preserves α-synuclein from oligomerization by blocking intermolecular hydrogen bonds. ACS Chem. Neurosci. 8, 2145–2151. doi: 10.1021/acschemneuro.7b00250
Bucci, G., Mochida, S., and Stephens, G. J. (2011). Inhibition of synaptic transmission and G protein modulation by synthetic CaV2.2 Ca2+ channel peptides. J. Physiol. 589, 3085–3101. doi: 10.1113/jphysiol.2010.204735
Cabañes, F. J. (2008). Mycoses and zoonoses: cryptococcus spp. Rev. Iberoam. Micol. 25, S1–S3. doi: 10.1016/S1130-1406(08)70018-4
Cantí, C., Nieto-Rostro, M., Foucault, I., Heblich, F., Wratten, J., Richards, M. W., et al. (2005). The metal-ion-dependent adhesion site in the Von Willebrand factor-A domain of α2δ subunits is key to trafficking voltage-gated Ca2+ channels. Proc. Natl. Acad. Sci. U S A 102, 11230–11235. doi: 10.1073/pnas.0504183102
Cantí, C., Page, K. M., Stephens, G. J., and Dolphin, A. C. (1999). Identification of residues in the N terminus of α1B critical for inhibition of the voltage-dependent calcium channel by Gβ γ. J. Neurosci. 19, 6855–6864. doi: 10.1523/JNEUROSCI.19-16-06855.1999
Castiglioni, A. J., Raingo, J., and Lipscombe, D. (2006). Alternative splicing in the C-terminus of CaV2.2 controls expression and gating of N-type calcium channels. J. Physiol. 576, 119–134. doi: 10.1113/jphysiol.2006.115030
Catterall, W. A. (2000). Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16, 521–555. doi: 10.1146/annurev.cellbio.16.1.521
Catterall, W. A. (2011). Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 3:a003947. doi: 10.1101/cshperspect.a003947
Catterall, W. A., and Few, A. P. (2008). Calcium channel regulation and presynaptic plasticity. Neuron 59, 882–901. doi: 10.1016/j.neuron.2008.09.005
Catterall, W. A., Leal, K., and Nanou, E. (2013). Calcium channels and short-term synaptic plasticity. J. Biol. Chem. 288, 10742–10749. doi: 10.1074/jbc.R112.411645
Catterall, W. A., Perez-Reyes, E., Snutch, T. P., and Striessnig, J. (2005). International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 57, 411–425. doi: 10.1124/pr.57.4.5
Chai, Z., Wang, C., Huang, R., Wang, Y., Zhang, X., Wu, Q., et al. (2017). CaV2.2 gates calcium-independent but voltage-dependent secretion in mammalian sensory neurons. Neuron 96, 1317.e4–1326.e4. doi: 10.1016/j.neuron.2017.10.028
Chapman, E. R., Hanson, P. I., An, S., and Jahn, R. (1995). Ca2+ regulates the interaction between synaptotagmin and syntaxin 1. J. Biol. Chem. 270, 23667–23671. doi: 10.1074/jbc.270.40.23667
Chen, J., Li, L., Chen, S. R., Chen, H., Xie, J. D., Sirrieh, R. E., et al. (2018). The α2δ-1-NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Rep. 22, 2307–2321. doi: 10.1016/j.celrep.2018.02.021
Chen, X., Tomchick, D. R., Kovrigin, E., Araç, D., Machius, M., Südhof, T. C., et al. (2002). Three-dimensional structure of the complexin/SNARE complex. Neuron 33, 397–409. doi: 10.2210/pdb1kil/pdb
Chicka, M. C., Hui, E., Liu, H., and Chapman, E. R. (2008). Synaptotagmin arrests the SNARE complex before triggering fast, efficient membrane fusion in response to Ca2+. Nat. Struct. Mol. Biol. 15, 827–835. doi: 10.1038/nsmb.1463
Chuang, R. S., Jaffe, H., Cribbs, L., Perez-Reyes, E., and Swartz, K. J. (1998). Inhibition of T-type voltage-gated calcium channels by a new scorpion toxin. Nat. Neurosci. 1, 668–674. doi: 10.1038/3669
Davies, J. N., Jarvis, S. E., and Zamponi, G. W. (2011). Bipartite syntaxin 1A interactions mediate CaV2.2 calcium channel regulation. Biochem. Biophys. Res. Commun. 411, 562–568. doi: 10.1016/j.bbrc.2011.06.185
Davydova, D., Marini, C., King, C., Klueva, J., Bischof, F., Romorini, S., et al. (2014). Bassoon specifically controls presynaptic P/Q-type Ca2+ channels via RIM-binding protein. Neuron 82, 181–194. doi: 10.1016/j.neuron.2014.02.012
de Jong, A. P. H., Roggero, C. M., Ho, M. R., Wong, M. Y., Brautigam, C. A., Rizo, J., et al. (2018). RIM C2B domains target presynaptic active zone functions to PIP2-containing membranes. Neuron 98, 335.e7–349.e7. doi: 10.1016/j.neuron.2018.03.011
Deng, L., Kaeser, P. S., Xu, W., and Südhof, T. C. (2011). RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron 69, 317–331. doi: 10.1016/j.neuron.2011.01.005
Dhara, M., Yarzagaray, A., Schwarz, Y., Dutta, S., Grabner, C., Moghadam, P. K., et al. (2014). Complexin synchronizes primed vesicle exocytosis and regulates fusion pore dynamics. J. Cell Biol. 204, 1123–1140. doi: 10.1083/jcb.201311085
Diao, J., Burré, J., Vivona, S., Cipriano, D. J., Sharma, M., Kyoung, M., et al. (2013). Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. Elife 2:e00592. doi: 10.7554/eLife.00592
Dietrich, D., Kirschstein, T., Kukley, M., Pereverzev, A., von der Brelie, C., Schneider, T., et al. (2003). Functional specialization of presynaptic Cav2.3 Ca2+ channels. Neuron 39, 483–496. doi: 10.1016/s0896-6273(03)00430-6
Dolphin, A. C. (2012). Calcium channel auxiliary α2δ and β subunits: trafficking and one step beyond. Nat. Rev. Neurosci. 13, 542–555. doi: 10.1038/nrn3311
Dunn, T. W., Fan, X., Ase, A. R., Séguéla, P., and Sossin, W. S. (2018). The CaV2α1 EF-hand F helix tyrosine, a highly conserved locus for GPCR inhibition of CaV2 channels. Sci. Rep. 8:3263. doi: 10.1038/s41598-018-21586-5
Ertel, E. A., Campbell, K. P., Harpold, M. M., Hofmann, F., Mori, Y., Perez-Reyes, E., et al. (2000). Nomenclature of voltage-gated calcium channels. Neuron 25, 533–535. doi: 10.1016/S0896-6273(00)81057-0
Fernández-Chacón, R., Königstorfer, A., Gerber, S. H., García, J., Matos, M. F., Stevens, C. F., et al. (2001). Synaptotagmin I functions as a calcium regulator of release probability. Nature 410, 41–49. doi: 10.1038/35065004
Fukuda, M. (2003). Distinct Rab binding specificity of Rim1, Rim2, rabphilin, and Noc2. Identification of a critical determinant of Rab3A/Rab27A recognition by Rim2. J. Biol. Chem. 278, 15373–15380. doi: 10.1074/jbc.M212341200
Gipson, P., Fukuda, Y., Danev, R., Lai, Y., Chen, D. H., Baumeister, W., et al. (2017). Morphologies of synaptic protein membrane fusion interfaces. Proc. Natl. Acad. Sci. U S A 114, 9110–9115. doi: 10.1073/pnas.1708492114
Gong, J., Lai, Y., Li, X., Wang, M., Leitz, J., Hu, Y., et al. (2016). C-terminal domain of mammalian complexin-1 localizes to highly curved membranes. Proc. Natl. Acad. Sci. U S A 113, E7590–E7599. doi: 10.1073/pnas.1609917113
Grabner, C. P., Gandini, M. A., Rehak, R., Le, Y., Zamponi, G. W., and Schmitz, F. (2015). RIM1/2-mediated facilitation of Cav1.4 channel opening is required for Ca2+-stimulated release in mouse rod photoreceptors. J. Neurosci. 35, 13133–13147. doi: 10.1523/JNEUROSCI.0658-15.2015
Han, Y., Babai, N., Kaeser, P., Südhof, T. C., and Schneggenburger, R. (2015). RIM1 and RIM2 redundantly determine Ca2+ channel density and readily releasable pool size at a large hindbrain synapse. J. Neurophysiol. 113, 255–263. doi: 10.1152/jn.00488.2014
Han, Y., Kaeser, P. S., Südhof, T. C., and Schneggenburger, R. (2011). RIM determines Ca2+ channel density and vesicle docking at the presynaptic active zone. Neuron 69, 304–316. doi: 10.1016/j.neuron.2010.12.014
Hirano, M., Takada, Y., Wong, C. F., Yamaguchi, K., Kotani, H., Kurokawa, T., et al. (2017). C-terminal splice variants of P/Q-type Ca2+ channel CaV2.1 α1 subunits are differentially regulated by Rab3-interacting molecule proteins. J. Biol. Chem. 292, 9365–9381. doi: 10.1074/jbc.M117.778829
Jahn, R., and Fasshauer, D. (2012). Molecular machines governing exocytosis of synaptic vesicles. Nature 490, 201–207. doi: 10.1038/nature11320
Jarvis, S. E., Magga, J. M., Beedle, A. M., Braun, J. E., and Zamponi, G. W. (2000). G protein modulation of N-type calcium channels is facilitated by physical interactions between syntaxin 1A and Gβγ. J. Biol. Chem. 275, 6388–6394. doi: 10.1074/jbc.275.9.6388
Jorquera, R. A., Huntwork-Rodriguez, S., Akbergenova, Y., Cho, R. W., and Littleton, J. T. (2012). Complexin controls spontaneous and evoked neurotransmitter release by regulating the timing and properties of synaptotagmin activity. J. Neurosci. 32, 18234–18245. doi: 10.1523/JNEUROSCI.3212-12.2012
Kaeser, P. S., Deng, L., Fan, M., and Sudhof, T. C. (2012). RIM genes differentially contribute to organizing presynaptic release sites. Proc. Natl. Acad. Sci. U S A 109, 11830–11835. doi: 10.1073/pnas.1209318109
Kaeser, P. S., Deng, L., Wang, Y., Dulubova, I., Liu, X., Rizo, J., et al. (2011). RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell 144, 282–295. doi: 10.1016/j.cell.2010.12.029
Kaeser-Woo, Y. J., Yang, X., and Südhof, T. C. (2012). C-terminal complexin sequence is selectively required for clamping and priming but not for Ca2+ triggering of synaptic exocytosis. J. Neurosci. 32, 2877–2885. doi: 10.1523/JNEUROSCI.3360-11.2012
Ladera, C., Martin, R., Bartolomé-Martín, D., Torres, M., and Sánchez-Prieto, J. (2009). Partial compensation for N-type Ca2+ channel loss by P/Q-type Ca2+ channels underlines the differential release properties supported by these channels at cerebrocortical nerve terminals. Eur. J. Neurosci. 29, 1131–1140. doi: 10.1111/j.1460-9568.2009.06675.x
Lai, Y., Choi, U. B., Leitz, J., Rhee, H. J., Lee, C., Altas, B., et al. (2017). Molecular mechanisms of synaptic vesicle priming by Munc13 and Munc18. Neuron 95, 591.e10–607.e10. doi: 10.1016/j.neuron.2017.07.004
Lai, Y., Choi, U. B., Zhang, Y., Zhao, M., Pfuetzner, R. A., Wang, A. L., et al. (2016). N-terminal domain of complexin independently activates calcium-triggered fusion. Proc. Natl. Acad. Sci. U S A 113, E4698–E4707. doi: 10.1073/pnas.1604348113
Lai, Y., Diao, J., Cipriano, D. J., Zhang, Y., Pfuetzner, R. A., Padolina, M. S., et al. (2014). Complexin inhibits spontaneous release and synchronizes Ca2+-triggered synaptic vesicle fusion by distinct mechanisms. Elife 3:e03756. doi: 10.7554/eLife.03756
Lai, Y., Diao, J., Liu, Y., Ishitsuka, Y., Su, Z., Schulten, K., et al. (2013). Fusion pore formation and expansion induced by Ca2+ and synaptotagmin 1. Proc. Natl. Acad. Sci. U S A 110, 1333–1338. doi: 10.1073/pnas.1218818110
Lambert, R. C., Bessaih, T., Crunelli, V., and Leresche, N. (2014). The many faces of T-type calcium channels. Pflugers Arch. 466, 415–423. doi: 10.1007/s00424-013-1353-6
Leal, K., Mochida, S., Scheuer, T., and Catterall, W. A. (2012). Fine-tuning synaptic plasticity by modulation of CaV2.1 channels with Ca2+ sensor proteins. Proc. Natl. Acad. Sci. U S A 109, 17069–17074. doi: 10.1073/pnas.1215172109
Lee, A., Westenbroek, R. E., Haeseleer, F., Palczewski, K., Scheuer, T., and Catterall, W. A. (2002). Differential modulation of Cav2.1 channels by calmodulin and Ca2+-binding protein 1. Nat. Neurosci. 5, 210–217. doi: 10.1038/nn805
Li, B., Zhong, H., Scheuer, T., and Catterall, W. A. (2004). Functional role of a C-terminal Gβγ-binding domain of Cav2.2 channels. Mol. Pharmacol. 66, 761–769. doi: 10.1124/mol.66.3
Liu, K. S., Siebert, M., Mertel, S., Knoche, E., Wegener, S., Wichmann, C., et al. (2011). RIM-binding protein, a central part of the active zone, is essential for neurotransmitter release. Science 334, 1565–1569. doi: 10.1126/science.1212991
Lonchamp, E., Dupont, J. L., Doussau, F., Shin, H. S., Poulain, B., and Bossu, J. L. (2009). Deletion of Cav2.1(α1A) subunit of Ca2+-channels impairs synaptic GABA and glutamate release in the mouse cerebellar cortex in cultured slices. Eur. J. Neurosci. 30, 2293–2307. doi: 10.1111/j.1460-9568.2009.07023.x
Lü, Q., AtKisson, M. S., Jarvis, S. E., Feng, Z. P., Zamponi, G. W., and Dunlap, K. (2001). Syntaxin 1A supports voltage-dependent inhibition of α1B Ca2+ channels by Gβγ in chick sensory neurons. J. Neurosci. 21, 2949–2957. doi: 10.1523/JNEUROSCI.21-09-02949.2001
Lu, C. W., Hung, C. F., Jean, W. H., Lin, T. Y., Huang, S. K., and Wang, S. J. (2018). Lycopene depresses glutamate release through inhibition of voltage-dependent Ca2+ entry and protein kinase C in rat cerebrocortical nerve terminals. Can. J. Physiol. Pharmacol. 96, 479–484. doi: 10.1139/cjpp-2017-0520
Lu, J., Machius, M., Dulubova, I., Dai, H., Sudhof, T. C., Tomchick, D. R., et al. (2006). Structural basis for a Munc13-1 homodimer to Munc13-1/RIM heterodimer switch. PLoS Biol. 4:e192. doi: 10.2210/pdb2cjt/pdb
Ma, C., Li, W., Xu, Y., and Rizo, J. (2011). Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat. Struct. Mol. Biol. 18, 542–549. doi: 10.1038/nsmb.2047
Malmersjö, S., Di Palma, S., Diao, J., Lai, Y., Pfuetzner, R. A., Wang, A. L., et al. (2016). Phosphorylation of residues inside the SNARE complex suppresses secretory vesicle fusion. EMBO J. 35, 1810–1821. doi: 10.15252/embj.201694071
Maximov, A., Tang, J., Yang, X., Pang, Z. P., and Sudhof, T. C. (2009). Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science 323, 516–521. doi: 10.1126/science.1166505
Mochida, S. (2017). Millisecond Ca2+ dynamics activate multiple protein cascades for synaptic vesicle control. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 93, 802–820. doi: 10.2183/pjab.93.050
Mochida, S. (2018). Presynaptic calcium channels. Neurosci. Res. 127, 33–44. doi: 10.1016/j.neures.2017.09.012
Mohrmann, R., Dhara, M., and Bruns, D. (2015). Complexins: small but capable. Cell. Mol. Life Sci. 72, 4221–4235. doi: 10.1007/s00018-015-1998-8
Naidoo, V., Dai, X., and Galligan, J. J. (2010). R-type Ca2+ channels contribute to fast synaptic excitation and action potentials in subsets of myenteric neurons in the guinea pig intestine. Neurogastroenterol. Motil. 22, e353–e363. doi: 10.1111/j.1365-2982.2010.01596.x
Nakamura, Y., Reva, M., and DiGregorio, D. A. (2018). Variations in Ca2+ influx can alter chelator-based estimates of Ca2+ channel-synaptic vesicle coupling distance. J. Neurosci. 38, 3971–3987. doi: 10.1523/JNEUROSCI.2061-17.2018
Nanou, E., Lee, A., and Catterall, W. A. (2018). Control of excitation/inhibition balance in a hippocampal circuit by calcium sensor protein regulation of presynaptic calcium channels. J. Neurosci. 38, 4430–4440. doi: 10.1523/JNEUROSCI.0022-18.2018
Osten, P., and Stern-Bach, Y. (2006). Learning from stargazin: the mouse, the phenotype and the unexpected. Curr. Opin. Neurobiol. 16, 275–280. doi: 10.1016/j.conb.2006.04.002
Pang, Z. P., Shin, O. H., Meyer, A. C., Rosenmund, C., and Südhof, T. C. (2006). A gain-of-function mutation in synaptotagmin-1 reveals a critical role of Ca2+-dependent soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex binding in synaptic exocytosis. J. Neurosci. 26, 12556–12565. doi: 10.1523/JNEUROSCI.3804-06.2006
Patel, R., Bauer, C. S., Nieto-Rostro, M., Margas, W., Ferron, L., Chaggar, K., et al. (2013). α2δ-1 gene deletion affects somatosensory neuron function and delays mechanical hypersensitivity in response to peripheral nerve damage. J. Neurosci. 33, 16412–16426. doi: 10.1523/JNEUROSCI.1026-13.2013
Pérez-Lara, Á., Thapa, A., Nyenhuis, S. B., Nyenhuis, D. A., Halder, P., Tietzel, M., et al. (2016). PtdInsP2 and PtdSer cooperate to trap synaptotagmin-1 to the plasma membrane in the presence of calcium. Elife 5:e15886. doi: 10.7554/eLife.15886
Prashanth, J. R., Brust, A., Jin, A. H., Alewood, P. F., Dutertre, S., and Lewis, R. J. (2014). Cone snail venomics: from novel biology to novel therapeutics. Future Med. Chem. 6, 1659–1675. doi: 10.4155/fmc.14.99
Rajagopal, S., Burton, B. K., Fields, B. L., El, I. O., and Kamatchi, G. L. (2017). Stimulatory and inhibitory effects of PKC isozymes are mediated by serine/threonine PKC sites of the Cav2.3α1 subunits. Arch. Biochem. Biophys. 621, 24–30. doi: 10.1016/j.abb.2017.04.002
Rajagopal, S., Fields, B. L., Burton, B. K., On, C., Reeder, A. A., and Kamatchi, G. L. (2014). Inhibition of protein kinase C (PKC) response of voltage-gated calcium (Cav)2.2 channels expressed in Xenopus oocytes by Cavβ subunits. Neuroscience 280, 1–9. doi: 10.1016/j.neuroscience.2014.08.049
Ramos-Miguel, A., Honer, W. G., Boyda, H. N., Sawada, K., Beasley, C. L., Procyshyn, R. M., et al. (2015). Exercise prevents downregulation of hippocampal presynaptic proteins following olanzapine-elicited metabolic dysregulation in rats: Distinct roles of inhibitory and excitatory terminals. Neuroscience 301, 298–311. doi: 10.1016/j.neuroscience.2015.06.022
Ricoy, U. M., and Frerking, M. E. (2014). Distinct roles for Cav2.1–2.3 in activity-dependent synaptic dynamics. J. Neurophysiol. 111, 2404–2413. doi: 10.1152/jn.00335.2013
Rodriguez-Tapia, E. S., Naidoo, V., DeVries, M., Perez-Medina, A., and Galligan, J. J. (2017). R-Type Ca2+ channels couple to inhibitory neurotransmission to the longitudinal muscle in the guinea-pig ileum. Exp. Physiol. 102, 299–313. doi: 10.1113/EP086027
Roh, H.-T., and So, W.-Y. (2017). The effects of aerobic exercise training on oxidant-antioxidant balance, neurotrophic factor levels, and blood-brain barrier function in obese and non-obese men. J. Sport Health Sci. 6, 447–453. doi: 10.1016/j.jshs.2016.07.006
Roh, H.-T., Cho, S.-Y., and So, W.-Y. (2017). Obesity promotes oxidative stress and exacerbates blood-brain barrier disruption after high-intensity exercise. J. Sport Health Sci. 6, 225–230. doi: 10.1016/j.jshs.2016.06.005
Schwartz, M. L., Nickerson, D. P., Lobingier, B. T., Plemel, R. L., Duan, M., Angers, C. G., et al. (2017). Sec17 (α-SNAP) and an SM-tethering complex regulate the outcome of SNARE zippering in vitro and in vivo. Elife 6:e27396. doi: 10.7554/eLife.27396
Serra, S. A., Gene, G. G., Elorza-Vidal, X., and Fernández-Fernández, J. M. (2018). Cross talk between β subunits, intracellular Ca2+ signaling and SNAREs in the modulation of CaV2.1 channel steady-state inactivation. Physiol. Rep. 6:e13557. doi: 10.14814/phy2.13557
Silva, F. R., Miranda, A. S., Santos, R. P. M., Olmo, I. G., Zamponi, G. W., Dobransky, T., et al. (2017). N-type Ca2+ channels are affected by full-length mutant huntingtin expression in a mouse model of Huntington’s disease. Neurobiol. Aging 55, 1–10. doi: 10.1016/j.neurobiolaging.2017.03.015
Song, H., Orr, A., Duan, M., Merz, A. J., and Wickner, W. (2017). Sec17/Sec18 act twice, enhancing membrane fusion and then disassembling cis-SNARE complexes. Elife 6:e26646. doi: 10.7554/eLife.26646
Stephens, G. J., and Mochida, S. (2005). G protein βγ subunits mediate presynaptic inhibition of transmitter release from rat superior cervical ganglion neurones in culture. J. Physiol. 563, 765–776. doi: 10.1113/jphysiol.2004.080192
Südhof, T. C. (2004). The synaptic vesicle cycle. Annu. Rev. Neurosci. 27, 509–547. doi: 10.1146/annurev.neuro.26.041002.131412
Südhof, T. C. (2012). The presynaptic active zone. Neuron 75, 11–25. doi: 10.1016/j.neuron.2012.06.012
Südhof, T. C. (2013). Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690. doi: 10.1016/j.neuron.2013.10.022
Tang, J., Maximov, A., Shin, O. H., Dai, H., Rizo, J., and Südhof, T. C. (2006). A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126, 1175–1187. doi: 10.1016/j.cell.2006.08.030
Traboulsie, A., Chemin, J., Chevalier, M., Quignard, J. F., Nargeot, J., and Lory, P. (2007). Subunit-specific modulation of T-type calcium channels by zinc. J. Physiol. 578, 159–171. doi: 10.1113/jphysiol.2006.114496
Valladolid-Acebes, I., Daraio, T., Brismar, K., Harkany, T., Ögren, S. O., Hökfelt, T. G., et al. (2015). Replacing SNAP-25b with SNAP-25a expression results in metabolic disease. Proc. Natl. Acad. Sci. U S A 112, E4326–E4335. doi: 10.1073/pnas.1511951112
Wang, L. Y., and Augustine, G. J. (2014). Presynaptic nanodomains: a tale of two synapses. Front. Cell. Neurosci. 8:455. doi: 10.3389/fncel.2014.00455
Wang, Y., and Südhof, T. C. (2003). Genomic definition of RIM proteins: evolutionary amplification of a family of synaptic regulatory proteins. Genomics 81, 126–137. doi: 10.1016/s0888-7543(02)00024-1
Weber, T., Zemelman, B. V., McNew, J. A., Westermann, B., Gmachl, M., Parlati, F., et al. (1998). SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772. doi: 10.1016/s0092-8674(00)81404-x
Wu, L. G., Westenbroek, R. E., Borst, J. G., Catterall, W. A., and Sakmann, B. (1999). Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J. Neurosci. 19, 726–736. doi: 10.1523/JNEUROSCI.19-02-00726.1999
Xu, J., Mashimo, T., and Sudhof, T. C. (2007). Synaptotagmin-1, -2, and -9: Ca2+ sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 54, 567–581. doi: 10.1016/j.neuron.2007.05.004
Yan, J., Leal, K., Magupalli, V. G., Nanou, E., Martinez, G. Q., Scheuer, T., et al. (2014). Modulation of CaV2.1 channels by neuronal calcium sensor-1 induces short-term synaptic facilitation. Mol. Cell. Neurosci. 63, 124–131. doi: 10.1016/j.mcn.2014.11.001
Yokoyama, C. T., Myers, S. J., Fu, J., Mockus, S. M., Scheuer, T., and Catterall, W. A. (2005). Mechanism of SNARE protein binding and regulation of Cav2 channels by phosphorylation of the synaptic protein interaction site. Mol. Cell. Neurosci. 28, 1–17. doi: 10.1016/j.mcn.2004.08.019
Yokoyama, C. T., Sheng, Z. H., and Catterall, W. A. (1997). Phosphorylation of the synaptic protein interaction site on N-type calcium channels inhibits interactions with SNARE proteins. J. Neurosci. 17, 6929–6938. doi: 10.1523/JNEUROSCI.17-18-06929.1997
Yoon, E. J., Hamm, H. E., and Currie, K. P. (2008). G protein βγ subunits modulate the number and nature of exocytotic fusion events in adrenal chromaffin cells independent of calcium entry. J. Neurophysiol. 100, 2929–2939. doi: 10.1152/jn.90839.2008
Yu, F. H., and Catterall, W. A. (2004). The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci. STKE 2004:re15. doi: 10.1126/stke.2532004re15
Yu, Y., Chen, S., Mo, X., Gong, J., Li, C., and Yang, X. (2018). Accessory and central α-helices of complexin selectively activate Ca2+ triggering of synaptic exocytosis. Front. Mol. Neurosci. 11:61. doi: 10.3389/fnmol.2018.00061
Yu, Y. P., Gong, N., Kweon, T. D., Vo, B., and Luo, Z. D. (2018). Gabapentin prevents synaptogenesis between sensory and spinal cord neurons induced by thrombospondin-4 acting on pre-synaptic Cav α2 δ1 subunits and involving T-type Ca2+ channels. Br. J. Pharmacol. 175, 2348–2361. doi: 10.1111/bph.14149
Yu, F. H., Yarov-Yarovoy, V., Gutman, G. A., and Catterall, W. A. (2005). Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 57, 387–395. doi: 10.1124/pr.57.4.13
Zamponi, G. W., Bourinet, E., Nelson, D., Nargeot, J., and Snutch, T. P. (1997). Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature 385, 442–446. doi: 10.1038/385442a0
Zhang, Y., Diao, J., Colbert, K. N., Lai, Y., Pfuetzner, R. A., Padolina, M. S., et al. (2015). Munc18a does not alter fusion rates mediated by neuronal SNAREs, synaptotagmin, and complexin. J. Biol. Chem. 290, 10518–10534. doi: 10.1074/jbc.M114.630772
Keywords: Ca2+ channel subtypes, SNAREs, Ca2+ sensor, active zone, membrane fusion, neurotransmitter release
Citation: He R, Zhang J, Yu Y, Jizi L, Wang W and Li M (2018) New Insights Into Interactions of Presynaptic Calcium Channel Subtypes and SNARE Proteins in Neurotransmitter Release. Front. Mol. Neurosci. 11:213. doi: 10.3389/fnmol.2018.00213
Received: 12 April 2018; Accepted: 30 May 2018;
Published: 16 July 2018.
Edited by:
Jiajie Diao, University of Cincinnati, United StatesReviewed by:
Guo-Xing Zhang, Soochow University, ChinaCopyright © 2018 He, Zhang, Yu, Jizi, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weizhong Wang, d2FuZ3d6NjhAaG90bWFpbC5jb20=
Miaoling Li, bGltaWFvbGluZ2NjQHN3bXUuZWR1LmNu
† These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.