 Li Zhang
Li Zhang Handong Wang
Handong Wang- Department of Neurosurgery, Jinling Hospital, School of Medicine, Nanjing University, Nanjing, China
Traumatic brain injury (TBI) is one of the most devastating forms of brain injury. Many pathological mechanisms such as oxidative stress, apoptosis and inflammation all contribute to the secondary brain damage and poor outcomes of TBI. Current therapies are often ineffective and poorly tolerated, which drive the explore of new therapeutic targets for TBI. Autophagy is a highly conserved intracellular mechanism during evolution. It plays an important role in elimination abnormal intracellular proteins or organelles to maintain cell stability. Besides, autophagy has been researched in various models including TBI. Previous studies have deciphered that regulation of autophagy by different molecules and pathways could exhibit anti-oxidative stress, anti-apoptosis and anti-inflammation effects in TBI. Hence, autophagy is a promising target for further therapeutic development in TBI. The present review provides an overview of current knowledge about the mechanism of autophagy, the frequently used methods to monitor autophagy, the functions of autophagy in TBI as well as its potential molecular mechanisms based on the pharmacological regulation of autophagy.
Introduction
Traumatic brain injury (TBI) is one of the leading causes of disability and death in modern society, resulting in high medical costs (Brooks et al., 2013). It is defined as any head injury with traumatic etiology, such as penetrating or blunt trauma and non-accidental injury. The pathological process of TBI includes both primary and secondary brain injury. Although the primary brain damage is the major factor determining the patients’ outcomes, the secondary brain damage induced by multiple pathological processes, such as inflammation, cell death, apoptosis, oxidative stress and impaired calcium and iron homeostasis, provides the possibility for clinical intervention (Zhang and Wang, 2018). Despite the efforts on searching effective methods to attenuate the secondary brain injury, patients suffering with TBI always end up with poor prognosis (Sun et al., 2015). Therefore, new and effective strategies of treatment are urgently needed to reduce the heavy disease and economic burden.
Autophagy is a self-catabolic process by which cells conserve and recycle their organelles in a stressed or nutrient-deprived state (Levine and Kroemer, 2008). This process is essential to maintain the metabolism essential for cell survival under stress situations. However, dysfunction of autophagy is involved in multiple diseases, including infectious diseases, cancers and TBI (Lipinski et al., 2015; Byun et al., 2017). Therefore, clarifying the molecular mechanisms of this degradation process may contribute to develop novel treatment protocols for therapeutic purposes. In the present study, we summarize the process of autophagy and its role in TBI as well as the associated molecular mechanisms and its regulated agents.
The Process of Autophagy
Autophagy is a process that degrades cytoplasmic proteins and organelles. Generally, there are three types of autophagy that have been proposed up to now, including chaperon-mediated autophagy (CMA), microautophagy and macroautophagy. They distinguish from each other by how autophagic substrates are delivered to lysosome (Figure 1; Kaur and Debnath, 2015). CMA is only observed in mammalian cells. In CMA, autophagic substrates are directly translocated across the lysosomal membrane dependent on the Lys-Phe-Glu-Arg-Gln (KFERQ) motif, cytosolic chaperones of the heat-shock protein family and lysosomal-associated membrane protein 2 (LAMP2; Cuervo and Wong, 2014). Conversely, the delivery of autophagic substrates in microautophagy involves the direct invagination of lysosomal membrane (Mijaljica et al., 2011).
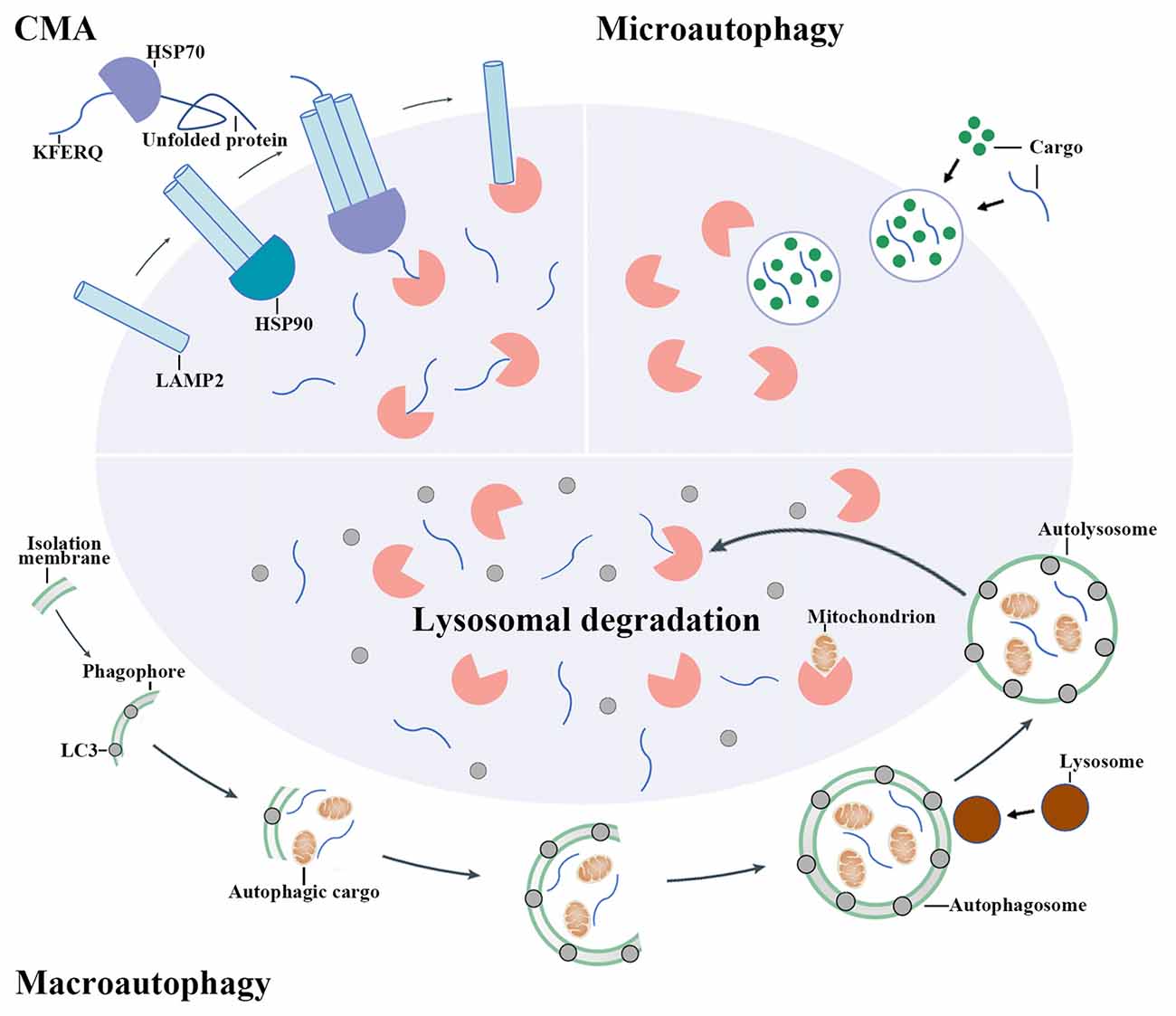
Figure 1. The mechanisms of autophagy pathway. There are three main types of autophagy including chaperon-mediated autophagy (CMA), microautophagy and macroautophagy. CMA involves the recognition of autophagic cargoes bearing a Lys-Phe-Glu-Arg-Gln (KFERQ) motif by heat shock proteins (HSPs), which is followed by the lysosomal-associated membrane protein 2 (LAMP2)-dependent translocation of chaperoned autophagic cargoes across the lysosomal membrane. By contrast, cargo delivery during microautophagy occurs upon the direct invagination of the lysosomal membrane. During macroautophagy, an isolation membrane encloses a portion of cytoplasm, forming a characteristic double-membraned organelle named autophagosome. Autophagosome then fuses with lysosome to form autolysosome and the cytoplasmic components are subsequently degraded by lysosomal enzymes.
Macroautophagy (hereafter simply referred to “autophagy”) is the most widely studied and best known type among these three pathways. When autophagy is activated, the cytoplasmic proteins or organelles are enclosed by an isolation membrane to form autophagosome. Autophagosome then fuses with lysosome to form autolysosome and the cytoplasmic proteins or organelles are subsequently degraded by lysosomal enzymes (Mizushima and Komatsu, 2011).
Methods for Monitoring Autophagy
Numerous reports have demonstrated that autophagy was activated in the brain after TBI. Accordingly, to exactly explore the role of autophagy in TBI, accurate methods to detect autophagy should be used. In this section, we summarize the frequently-used methods for detecting autophagy in TBI.
Transmission Electron Microscope (TEM)
Transmission electron microscope (TEM) has long been considered as the “gold standard” for identification of autophagic vesicles. It is one of the most precise methods to examine autophagy, which provides the “seeing is believing” data (Murakawa et al., 2015). Under TEM, autophagosome can be typically identified as a double membrane containing cytoplasmic materials. The cytoplasmic materials in autophagosome include various organelles, such as endoplasmic reticulum (ER) and mitochondrion (Ylä-Anttila et al., 2009). When autophagosome fuses with lysosomal vesicle to form autolysosome, the outer membrane of autophagosome fuses with the lysosome membrane. The cytoplasmic organelles, still surrounded by the inner membrane, are delivered to the lysosome lumen. This inner membrane of autophagosome is then degraded to allow the degradation of the materials. Therefore, at the stage of autolysosome, TEM usually observes a monolayer structure containing numerous cytoplasmic materials (Eskelinen, 2008).
There are also limitations of TEM. For example, instead of whole cell, ultrathin slices of cell, usually of 70–80 nm thickness, could be observed. Therefore, the sample size is very small and it is difficult to get a concept of the size and total volume of different compartments inside the cell (Ylä-Anttila et al., 2009). Besides, TEM requires expensive equipment and professional technology, and is also time spending.
Western Blot Assays of Autophagosome Marker Proteins
Beclin-1 is the mammalian ortholog of the yeast Apg6/Vps30 gene. It can promote the formation of autophagosome when overexpressed in mammalian cells (Liang et al., 1999). In addition, there is a lot of autophagy-related (ATG) proteins that mediate the activation of autophagy (Arroyo et al., 2014). Among them, the microtubule-associated protein light chain 3 (LC3) is widely used as a key marker for detection of autophagosome (Mizushima et al., 2010). LC3 is primarily synthesized in an unprocessed form, proLC3. When autophagy in activated, proLC3 is cleaved by ATG4 to form LC3-I, LC3-I then binds to phosphatidylethanolamine (PE) to become lipidated LC3 (LC3-II). Subsequently, LC3-II conjugates to both inner and outer membrane of autophagosome and contributes to the formation of autophagy. This process requires an ubiquitination-like reaction mediated by ATG3 and ATG7 (Kabeya et al., 2000). Therefore, the protein levels of Beclin-1, LC3, ATG3 and ATG7 detected by western blot are usually used to detect the formation of autophagy.
Fluorescence Microscopy
LC3-I distributes in the cell uniformly when autophagy levels are low. However, upon the induction of autophagy, LC3-I turns to LC3-II and binds to the membrane of autophagosome, which can be visualized and quantified by fluorescence microscopy through counting LC3 puncta (Dolman et al., 2013). Monitoring LC3 puncta can depend on either the signal of green fluorescent protein (GFP) tagged to LC3 or immunofluorescence using an anti-LC3 antibody (Yoshii and Mizushima, 2017). The main limitation of fluorescence microscopy is that although LC3 puncta reflects an image of ATG structures in a cell, it does not illustrate that the autophagosome would reach the final stage of degradation.
Autophagic Flux
Autophagy is initiated by the formation of autophagosome. Subsequently, autophagosome fuses with lysosome to form autolysosome and promotes cytoplasmic organoids degradation. This dynamic degradation process is named autophagy flux. There are generally three methods to detect autophagic flux, including LC3 turnover, autophagic substrate p62 degradation and tandem fluorescent-tagged LC3 (tfLC3) assay. At the stage of autophagic flux, the LC3-II is degraded by autolysosome. Thus, the lysosomal degradation of LC3-II reflects the progression of autophagic flux and assaying the expression of LC3-II in the presence of lysosomal inhibitors provides a reasonable way to monitor autophagic flux (Jiang and Mizushima, 2015). Moreover, p62, the adapter protein, promotes the ubiquitination of cytoplasmic organoids to autophagosome and degraded by autolysosome. Thus, the down-regulation of p62 suggests an occurrence of autophagic flux (Klionsky et al., 2016). In addition, a new autophagic flux observation method has been proposed by using a tandem monomeric red fluorescent protein (mRFP)-GFP-tfLC3. The GFP fluorescence signal is quenched in lysosome with an acidic compartment, whereas mRFP fluorescence signal remains its intensity in lysosome. Therefore, colocalization of GFP and mRFP fluorescence signal (yellow puncta) demonstrates that the tandem protein exists in an organelle which has not fused with lysosome, for example the autophagosome. Conversely, a single mRFP fluorescence signal without GFP (red puncta) demonstrates the translocation of tfLC3 to lysosome, that is, the formation of autolysosome. This novel system using tfLC3 allows a direct assessment of both autophagy induction and autophagy flux in the absence of any toxic inhibitors (Kimura et al., 2007).
The Dual Role of Autophagy in TBI
Autophagy was firstly reported to be activated after TBI by Diskin et al. (2005). They found that in a mouse modified weight-drop TBI model, Beclin-1 was significant up-regulated at 4 h and 24 h in the cortical site of injured brain post-TBI. Double staining of Beclin-1 and TUNEL indicated that most of the injured cells exhibited double staining. Subsequently, a great number of studies have demonstrated that autophagy was induced after TBI. For example, in a mouse weight-drop TBI model, the expression of Beclin-1 was increased at 1 h post-TBI and peaked at 6 h while the expression of LC3-II was up-regulated shortly after TBI and peaked at 48 h in the injured cortex and hippocampus. Instead, the expression of p62 was decreased at 24 and 48 h in the injured cortex and hippocampus after TBI (Luo et al., 2011). Moreover, in a rat moderate fluid percussion TBI model, both autophagosome and autolysosome were detected in the injured cortical neurons from 4 h to 15 days after TBI under TEM and the expression of LC3-II was also up-regulated from 4 h to 15 days post-TBI in the injured cortex (Liu et al., 2008). More importantly, the induction of autophagy after TBI was not only found in animal models but also confirmed in clinical trials. Clark et al. (2008) reported that both LC3-II and Beclin-1 were increased in the injured temporal lobe cortex in patients suffering TBI. Although autophagy enhancement after TBI has been found in both animal and human models, the role of autophagy in TBI was still controversial. Different studies may draw different or even opposite conclusions.
The Protective Role of Autophagy in TBI
The protective role of autophagy in TBI was firstly proposed by Erlich et al. (2007) using rapamycin. Rapamycin activate autophagy by inhibition of the phosphatidylinositide 3-kinases (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway (Heras-Sandoval et al., 2014). They found that rapamycin could increase the protein levels of Beclin-1 and induce an augmented autophagic response after TBI. Besides, administration of rapamycin improved neurobehavioral function, increased neuronal survival, reduced inflammation and gliosis in injured brain. Therefore, they concluded that rapamycin was neuroprotective following TBI by activation of autophagy. In another study, Zhang et al. (2008) demonstrated that within 1 day after TBI, there were few caspase-3 (+)/LC3 (+) overlapped cells. Whereas after d, the number of caspase-3 (+)/LC3 (+) overlapped cells significantly increased, indicating that autophagy was activated in apoptotic cells after TBI. Furthermore, the protective role of autophagy in TBI was also proposed by Sarkar et al. (2014). Results of their study indicated that impaired autophagy flux was involved in cell death and apoptosis following TBI. Additionally, many neuroprotective drugs have been suggested to attenuate TBI-induced secondary brain injury via activation of autophagy (Xu et al., 2014; Ding et al., 2015; Lin et al., 2016; Gao et al., 2017; Zhang et al., 2017).
The Detrimental Role of Autophagy in TBI
Support for the detrimental role of autophagy in TBI was initially according to the study conducted by Lai et al. (2008) Oxidative stress could induce autophagy after TBI (Scherz-Shouval et al., 2007), so they used an antioxidant, γ-glutamylcysteinyl ethyl ester (GCEE). Treatment of GCEE alleviated TBI-induced brain tissue loss and neuron death, increased antioxidant reserves, improved Morris-water maze performance and suppressed autophagy formation. Consequently, they speculated that autophagy played a detrimental role in TBI. Moreover, bafilomycin A1 (BafA1) and 3-methyladenine (3-MA), two autophagy inhibitors, were also used. Inhibition of autophagy by BafA1 or 3-MA attenuated behavioral outcome, reduced cell injury, lesion volume and apoptosis after TBI, supporting that autophagy was detrimental for TBI (Luo et al., 2011). Besides, it has been shown that treatment of ketamine could prevent TBI-induced inflammation and exert beneficial effects on memory and behavior by down-regulating Beclin-1 and LC3, suggesting that suppression of autophagy might be a potential therapy to attenuate functional deficits for TBI (Wang C. Q. et al., 2017). Consistent with these conclusions, there were also numerous studies confirming the detrimental role of autophagy in TBI (Feng et al., 2017a; Jiang et al., 2017; Liu et al., 2017; Shen et al., 2017; Tang et al., 2017).
The Regulation Molecules of Autophagy in TBI
Although the accurate role of autophagy in TBI was confused, one undisputable fact was that autophagy was activated after TBI and regulation of autophagy could provide neuroprotection by improvement of cognitive function, decrease of brain edema, protection of blood-brain barrier (BBB) function and suppression of apoptosis, inflammation and oxidative stress (Table 1). The detailed mechanisms mediating the activation of autophagy after TBI is unclear, some regulatory molecules have been suggested which may explain its activation in TBI. It has been shown that TBI could inhibit the phosphatidylinositide 3-kinases/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) pathway, activate forkhead box O 3a (FoxO3a), dynamin-related protein 1 (Drp1), nuclear factor erythroid 2-related factor 2/antioxidant response element (Nrf2/ARE) pathway and toll-like receptor 4 (TLR4)/nuclear factor kappa-light-chain-enhancer of activated B cells (TLR4/NF-κB) pathway. In addition, these molecules were in the upstream of autophagy and regulation of these molecules by TBI could promote the formation of autophagosome (Figure 2).
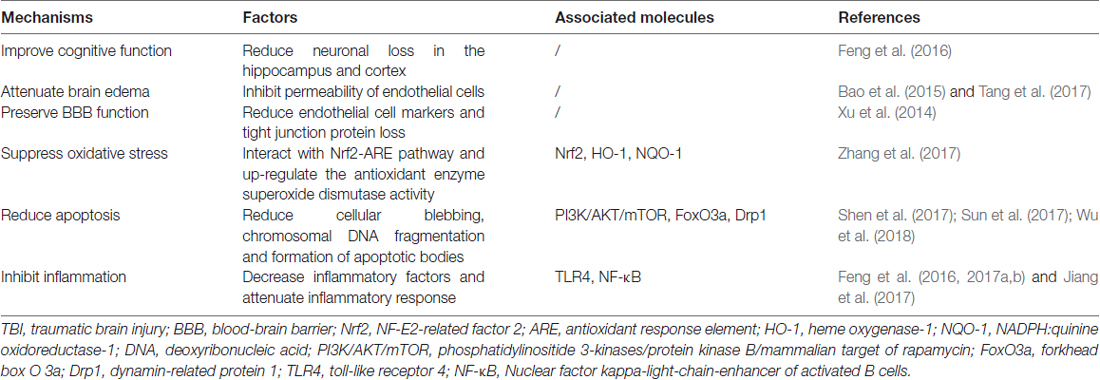
Table 1. Mechanisms of regulation of autophagy in TBI.
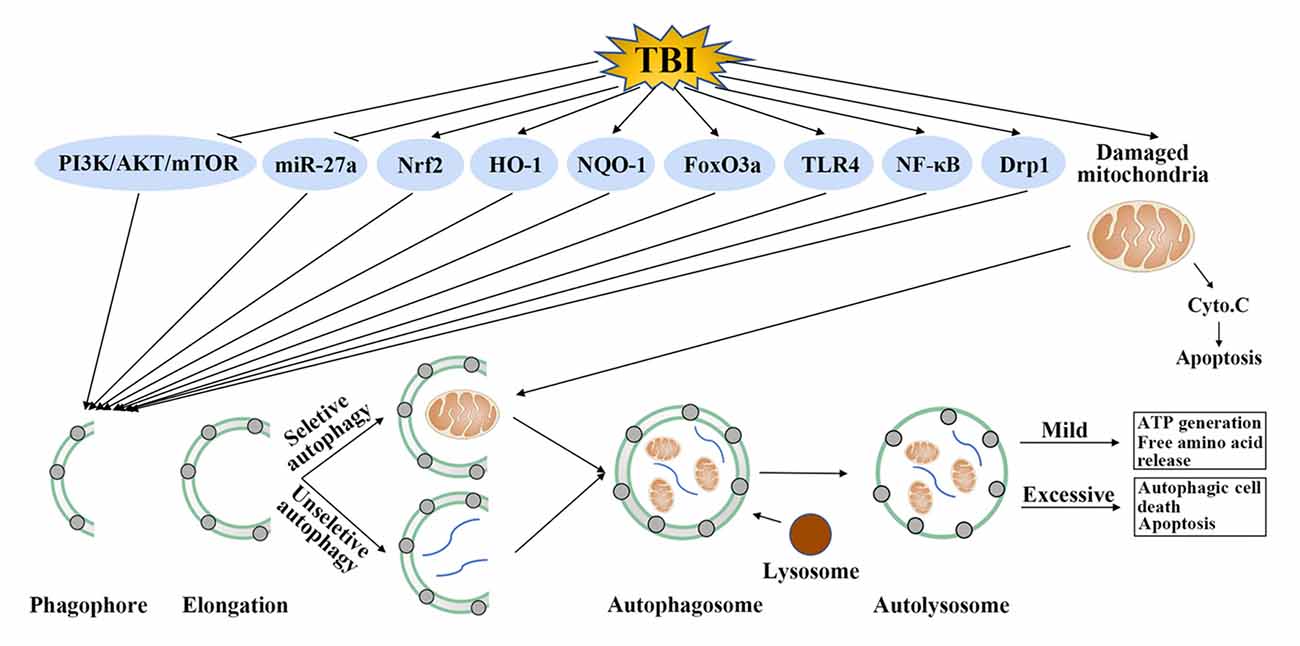
Figure 2. Possible autophagy signaling pathways in traumatic brain injury (TBI). TBI could inhibit phosphatidylinositide 3-kinases (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway and microRNA-27a (miR-27a), activate nuclear factor erythroid 2-related factor 2 (Nrf2), heme oxygenase-1 (HO-1), nicotinamide adenine dinucleotide phosphate, quinine oxidoreductase-1 (NQO-1), forkhead box O 3a (FoxO3a), toll-like receptor 4 (TLR4), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and dynamin-related protein 1 (Drp1). Regulation of these molecules by TBI further promotes the formation of autophagosome. This step requires unselective or selective targets, such as damaged mitochondria, for degradation. Mild autophagy leads to adenosine triphosphate (ATP) generation and free amino acid release, which are beneficial for TBI. Conversely, excessive autophagy results in autophagic cell death or apoptosis.
PI3K/AKT/mTOR
PI3K/AKT/mTOR signaling pathway is a crucial intracellular pathway in regulation of metabolism, inflammation, cell growth and survival (Huang T. et al., 2018). In addition, this pathway is imbalanced at the occurrence of brain injury (Li et al., 2016). Researches have offered compelling evidence to demonstrate that activation of the PI3K/AKT/mTOR pathway could reduce apoptosis by regulating autophagy in brain injury models (Huang et al., 2017; Lv et al., 2017), including TBI. It has been shown that TBI significantly increased the neurological injury, brain water content, neuron apoptosis and expression of Beclin-1 and LC3-II, while decreased the expression of Phosphorylated (p)-PI3K, p-AKT and p-mTOR in rats. Treatment with dexmedetomidine, a neuroprotective agent, attenuated brain injury, reduced the expression of Beclin-1 and LC3-II, and elevated the expression of the p-PI3K, p-AKT and p-mTOR. Whereas treatment with LY294002, a PI3K/AKT/mTOR pathway inhibitor, observed the opposite trends, indicating that activation of the PI3K/AKT/mTOR pathway provided neuroprotection in TBI via inhibition of autophagy (Shen et al., 2017).
The mechanisms of how PI3K/AKT/mTOR pathway regulates autophagy have been fully explained. mTOR is combined by two mTOR complexes, mTORC1 and mTORC2. mTORC1 (composed of mTOR, Raptor, GLβ and other proteins) is a key negative modulator of autophagy (Cuyàs et al., 2014) and PI3K/AKT pathway is a major regulator of mTORC1 (Manning and Cantley, 2007). AKT activates mTOR by phosphorylation of TSC2 at serine residue 939 (Nellist et al., 2002). Phosphorylated TSC2 subsequently results in the activation of Rheb and promotes mTOR activity (Long et al., 2005; Miyazaki et al., 2010). Therefore, inhibition of AKT suppresses mTOR activity, leads to dephosphorylation of autophagy and Beclin-1 regulator 1 (AMBRA1), activation of unc-51 like autophagy activating kinase 1/2 (ULK1/2) complex, phosphorylation of focal adhesion kinase family interacting protein 200 (FIP200) and finally initiation of autophagy (Wojcik, 2013).
Nrf2
Nrf2 is a basic leucine zipper redox-sensitive transcription factor that regulates the redox state of cell in harmful stresses (Villeneuve et al., 2010). Under normal conditions, Nrf2 is retained in the cytoplasm by Kelch-like ECH-associated protein 1 (Keap1; Kobayashi et al., 2004). However, under harmful conditions such as oxidative stress, Nrf2 dissociates from Keap1, translocates to the nucleus and activates numerous antioxidant enzymes such as malondialdehyde (MDA), glutathione peroxidase (GPx), heme oxygenase-1 (HO-1) and nicotinamide adenine dinucleotide phosphate, NQO-1 by binding to ARE (de Vries et al., 2008). To date, Nrf2 has been confirmed to provide neuroprotection in various central nervous system (CNS) diseases (Wang et al., 2007; Chen et al., 2011), including TBI (Yan et al., 2008).
There were also studies showing that Nrf2 could regulate autophagy (Li L. et al., 2015; Pajares et al., 2016). The regulation of autophagy by Nrf2 in TBI has been suggested by Zhang et al. (2017) They proposed that the Nrf2-autophagy pathway was activated after TBI to provide neuroprotection both in vivo and in vitro. However, Nrf2 failed to activate autophagy in Nrf2−/– mice after TBI. Moreover, they found that fucoxanthin, a marine carotenoid extraction from seaweeds, could alleviate TBI-induced brain injury by activation of the Nrf2-autophagy pathway.
But how Nrf2 regulated autophagy has not been fully understood. There were several explanations and these explanations were consistently associated with p62. p62 possess dual-binding sites for ubiquitin chains and LC3. It bound to ubiquitin chains via an ubiquitin-associated (UBA) domain and LC3 through an LC3-interacting region (LIR), leading to the initiation of autophagy (Noda et al., 2008). Moreover, another study indicated that Keap1 uncoupled from Nrf2 could bind to p62, interact with LC3 and transport the ubiquitin conjugate to autophagosome for degradation (Fan et al., 2010). The detailed mechanism of how Nrf2 regulated autophagy was unclear, further studies were needed to clarify it.
FoxO3a
FoxO3a belongs to the fork frame transcription factor family. It can regulate muscle atrophy, glucose metabolism and apoptosis in cells (Chaanine et al., 2016). Moreover, FoxO3a is expressed in the brain such as the cerebral cortex, hippocampus and cerebellum (Hoekman et al., 2006). Recent studies have revealed that FoxO3a participated in the damage of brain. It has been suggested that FoxO3a was involved in cerebral ischemia and promoted stroke, hence inhibition of FoxO3a could provide neuroprotection against ischemic injury (Yoo et al., 2012; Li D. et al., 2015). Furthermore, FoxO3a was implied in neuronal apoptosis in a rat subarachnoid hemorrhage (SAH) model (An et al., 2015). In addition, FoxO3a has also been confirmed to regulate autophagy. Activated FoxO3a could induce autophagy by directly increasing the transcription of Atgs, such as Beclin-1, LC3, Atg5 and Atg7 (Liu et al., 2015). Besides, FoxO3a could also avtivate autophagy indirectly. Phosphorylation of FoxO3a by AMP-activated protein kinase (AMPK) may repress transcription of S phase kinase-associated protein 2 (SKP2), which subsequently initiated autophagy formation (Sanchez et al., 2012).
FoxO3a also facilitated autophagy to decrease secondary injury after TBI. Knockdown of FoxO3a by small interfering ribonucleic acid (siRNA) significantly inhibited TBI-induced autophagy, thus reversing neuronal damage in the hippocampus and improving neurobehavioral dysfunctions. Whereas activation of autophagy showed the opposite effects (Sun et al., 2018).
FoxOs are a family of proteins that have been found to regulate various cellular functions (Zhou et al., 2012). Besides FoxO3a, other isoforms of FoxO, such as FoxO1, FoxO4 and FoxO6 also express in mammalian cells (Wang et al., 2014). Although they share overlapping structure and function, each member appears to have different tissue-dependent expression patterns and exert a specific biological role. In addition to FoxO3a, other isoforms of FoxO were also showed to regulate autophagy. For example, FoxO1 has been reported to mediate putative kinase 1 (PINK1) transcription and promote autophagy in response to mitochondrial oxidative stress in murine cardiomyocytes (Li W. et al., 2017). However, no reports so far have studied the effects of other FoxO isoforms except FoxO3a on autophagy in TBI models. Therefore, this is an interesting aspect worth exploring.
TLR4
TLR4 is the first reported mammalian TLR, which has been considered to play an crucial role in initiating the inflammatory reactions and ultimately resulting in neurological defcits in CNS (Wang C. et al., 2012; Fang et al., 2013). For example, the expression TLR4 significantly increased in brain after ICH, and knockout of TLR4 signifcantly ameliorated ICH-induced neurological impairments, cerebral edema and infammatory cytokines expression (Lin et al., 2012). Furthermore, the expression of TLR4 was up-regulated in transient cerebral ischemia, and TLR4-defcient decreased infarct volumes, improved neurobehavioral function and suppressed inflammation after ischemic brain injury (Hyakkoku et al., 2010). The mechanism of how TLR4 regulates inflammation attributes to its initiation of two parallel signaling pathways, the myeloid differentiation primary-response protein 88 (Myd88)/NF-κB pathway and the toll receptor associated activator of interferon (TRIF) pathway. These two signaling pathways subsequently activate transcription factors that regulate proinflammatory cytokine genes (Buchanan et al., 2010).
TLR4 has also been suggested to regulate autophagy in brain injury models such as TBI. Jiang et al. (2017) found that knockdown of TLR4 ameliorated neuroinfammatory response after TBI by inhibition of autophagy. In addition, many drugs such as resveratrol, resatorvid and apocynin have been reported to provide neuroprotection in TBI by inhibiting the TLR4-mediated autophagy pathway (Feng et al., 2016, 2017a,b). But how TLR4 enhanced autophagy remained unclear. An NF-κB binding site has been found in the promoter region of Beclin-1 genes. Therefore, activation of NF-κB by TLR4 may up-regulate Beclin-1 expression and promote autophagy (Copetti et al., 2009).
TLRs are a group of pattern recognition receptors present in cytoplasm and cell membrane, and can specifically recognize pathogen-associated molecular patterns. Interestingly, in a white matter injury (WMI) model, TLR3 was colocalized with the ER and autophagosome in ventral lateral posterior neurons, indicating that autophagy could be regulated by TLR3 (Vontell et al., 2015). Since TLR3 also participated in the process of inflammation (Liu et al., 2018), so whether inhibiton of TLR3 could suppress TBI-induced inflammation by modulation of autophagy was unclear, which required further researches.
Drp1
Drp1 is a dynamin-like GTPase shuttling between the mitochondrial and cytoplasm surface and it mediates mitochondrial fission by calcium-dependent dephosphorylation (Smirnova et al., 2001). Drp1 is highly expressed in brain neurons and has been investigated in Alzheimer’s disease, Parkinson’s diseases (PDs), stroke, epilepsy and TBI (Knott and Bossy-Wetzel, 2008; Qiu et al., 2013; Zuo et al., 2014; Kim et al., 2016). Recent researches showed that inhibition of Drp1 could decrease brain injury and apoptosis after TBI by maintaining mitochondrial functions (Wu et al., 2016).
Drp1 is also a crucial upstream protein of autophagy. When Drp1 is stimulated by reactive oxygen species (ROS) or damaged mitochondrial deoxyribonucleic acid (DNA), mitochondrion becomes depolarized and damage. The damaged mitochondrion is then recognized by autolysosome for degradation, this process is named mitophagy (Song et al., 2015). It has been suggested that Drp1 not only mediated BCL2/adenovirus E1B 19 kilodalton interacting protein-3 (BNIP3)-induced mitophagy in adult cardiomyocytes (Tanaka et al., 2010) but also participated in Parkin-induced mitophagy in mouse embryonic fibroblast (MEF) cells (Lee et al., 2011). Consistent with these results, one study demonstrated that suppression of Drp1 alleviated TBI-induced BBB disruption and apoptosis by inhibiting mitophagy.
Other Upstream Molecules of Autophagy Worth Studying in TBI
TBI is a complex disease involving many pathological processes. There are several molecular targets responsible for the secondary damage of TBI. Although the effects of molecules such as Nrf2 on autophagy have been widely described in TBI, the effects of other molecules such as long noncoding RNA (LncRNA) and BNIP3 on autophagy in TBI have not been fully explained so far.
LncRNA
LncRNA is an RNA molecule that is longer than 200 nucleotides and is not translated to a protein (Spizzo et al., 2012). LncRNAs were primarily regarded as transcriptional by-products. However, there was considerable evidence indicating that lncRNAs were invloved in many pathological processes (Batista and Chang, 2013). Indeed, the role of lncRNAs in TBI was definite. The expression of LncRNAs was significant changed in the injury brain after TBI (Zhong et al., 2016; Wang C. F. et al., 2017). Knockdown or overexpression of lncRNAs could suppress TBI-induced inflammation and apoptosis, resulting in better outcome in mice (Yu et al., 2017; Zhong et al., 2017).
Additionly, lncRNAs was emerging as new factors involved in autophagy in brain injury models. It has been revealed that down-regulation of metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) attenuated neuronal cell death by suppressing autophagy in cerebral ischemic stroke (Guo et al., 2017). Moreover, MALAT1 played a protective role against oxygen-glucose deprivation/reoxygenation-induced injury in brain microvascular endothelial cell (BMEC) by enhancing autophagy (Li Z. et al., 2017). Furthermore, lncRNA nuclear paraspeckle assembly transcript 1 (NEAT1) stabilized phosphatase and tensin homolog (PTEN)-induced PINK1 protein by promoting 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced autophagy in PD (Yan et al., 2018). Thus, it can be speculated that lncRNAs could also regulate autophagy in TBI. Further studies are needed to clarify it.
BNIP3
BNIP3 belongs to the unique family of death-inducing mitochondrial proteins (Chen et al., 1997). Under hypoxic conditions, BNIP3 is activated by transcriptional factor hypoxia inducible factor 1 (HIF-1) and promotes cell survival (Swiderek et al., 2013). BNIP3 has also been studied in TBI. It has been shown that inhibiton of BNIP3 by 2-methoxyestradiol (2ME2) could provide neuroprotection after TBI by inhibiton of secondary brain damage such as apoptosis and oxidative stress (Schaible et al., 2014).
The role of BNIP3 in autophagy is well established (Xin et al., 2011; Lu et al., 2016). BNIP3 can suppress mTOR by binding to and inhibiting Rheb, and subsequently induce autophagy (Zhang and Ney, 2011). Furthermore, BNIP3 can trigger the dissociation of Beclin-1 and B-cell lymphoma 2 (Bcl-2) by competing with Beclin-1 for binding to Bcl-2 to activate autophagy (Glick et al., 2010). So, it is necessary to examine whether BNIP3 could regulate autophagy in TBI in future studies.
Related Therapeutics Agents Targeting Autophagy in TBI
Numerous proof-of-principle studies have indicated that regulation of autophagy by pharmacological activators or inhibitors could attenuate TBI-induced brain injury in preclinical studies and represent a promising therapeutic approach for TBI. Therefore, we elucidate the autophagy activators and inhibitors used in TBI in this section (Table 2).
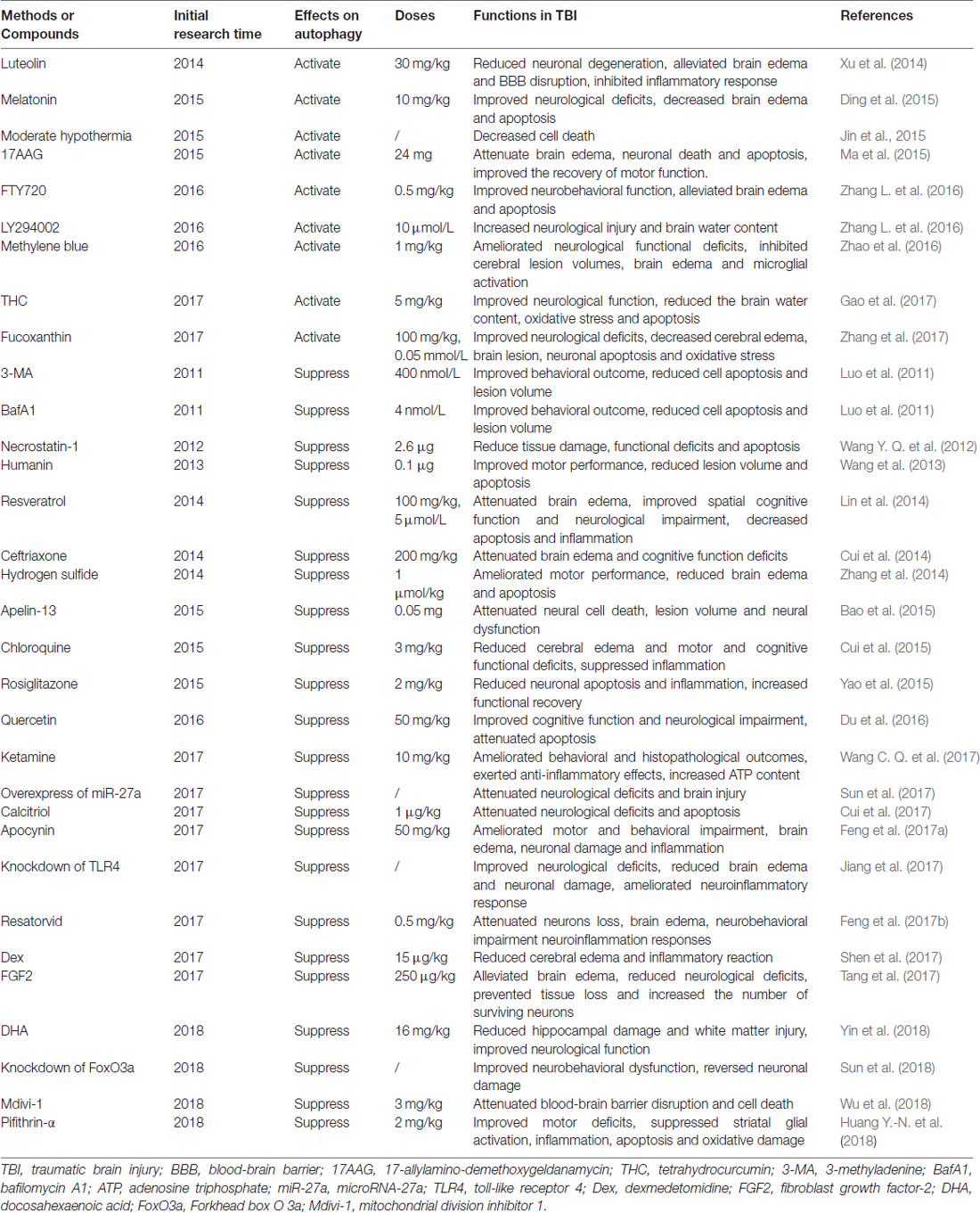
Table 2. Summary of therapeutics development targeting autophagy in TBI.
Autophay Inducers
Luteolin
Luteolin was shown to activate autophagy in TBI (Xu et al., 2014). Luteolin is a member of the flavonoid family and is abundant in vegetables and fruits such as broccoli, parsley and celery (Mencherini et al., 2007). Luteolin has a variety of pharmacological properties such as antioxidant, anti-inflammation and cancer preventive effects (Neuhouser, 2004). Besides, luteolin was reported to regulate autophagy in multiple models including TBI (Zhang B. C. et al., 2016; Cao et al., 2017). It has been shown that autophagy was protective after TBI and treatment of luteolin further enhanced autophagy by activating NF-κB, leading to decreased brain injury, inflammation and apoptosis following TBI (Xu et al., 2014).
Melatonin
Melatonin (N-acetyl 5-methoxytryptamine) was reported to activate autophagy in TBI (Ding et al., 2015). It is an autocrine hormone mainly produced by the pineal gland and regulates the sleepe cycle (Brzezinski, 1997). Melatonin can cross the BBB easily to provide neuroprotection (Cheung et al., 2006). Melatonin has been recognized as a powerful antioxidant that protect antioxidative enzymes from oxidative damage (Galano et al., 2013). In addition, studies have shown that melatonin activated autophagy in many models inclduing TBI (Chen et al., 2014; Kucharewicz et al., 2018; Pan et al., 2018). Activation of autophagy by melatonin was found to provide neuroprotection in TBI by suppression of inflammation and oxidative stress, suggesting that autophagy was beneficial for TBI (Ding et al., 2015).
Fucoxanthin
In 2017, fucoxanthin was proposed to exhibit neuroprotective effects after TBI by activation of autophagy (Zhang et al., 2017). Fucoxanthin is the most abundant marine carotenoid extraction in seaweeds and is considered as a powerful antioxidant (Sugawara et al., 2002). It has been proposed to exhibit a variety of pharmacological properties such as inhibiting tumor growth, repressing inflammation reaction and reducing oxidative stress by activation of autophagy (Hou et al., 2013; Moskalev et al., 2018). Furthermore, activation of autophagy was shown to provide neuroprotection after TBI and administration of fucoxanthin post-TBI could attenuated TBI-induced neurological defcits, cerebral edema, brain lesion, neuronal apoptosis and oxidative stress by further promoting autophagy (Zhang et al., 2017).
Tetrahydrocurcumin (THC)
Tetrahydrocurcumin (THC) is extracted from the roots of the Curcuma longa Linn. It owns antioxidant and anti-inflammatory activity in vitro and in vivo (Wu et al., 2014). Besides, THC could protect cerebral ischemia and neurodegenerative diseases against oxidative stress by modulation of autophagy (Mishra et al., 2011; Tyagi et al., 2012). Furthermore, the effects of THC on autophagy after TBI has also been investigated in 2017. Gao et al. (2017) found that THC improved neurological function, ameliorated cerebral edema, reduced oxidative stress and decreased the number of apoptotic neurons by activation of autophagy in a rat model of TBI, confirming the protective role of autophagy in autophagy.
Autopahgy Inhibitors
Necrostatin-1 (NEC-1)
As a special receptor-interacting protein-1 (RIP-1) inhibitor to depress necroptotic cell death, Necrostatin-1 (NEC-1) has been a hot topic of therapeutic agent in different models (Degterev et al., 2008). NEC-1 has been shown to improve functional outcomes and reduce the disrupture of brain tissue in TBI models (You et al., 2008). Moreover, previous studies have indicated that necroptosis was closely associated with autophagy and apoptosis, and thereby, suppression of necroptosis by NEC-1 may interfere with the process of autophagy and apoptosis. Rosenbaum et al. (2010) found that NEC-1 could decrease the expression of LC3-II after retinal ischemic. Furthermore, NEC-1 was found to inhibit autophagy in TBI in 2012. Wang Y. Q. et al. (2012) proposed that activation of autophagy could increase apoptosis after TBI and treatment of NEC-1 suppressed TBI-induced autophagy, leading to decreased apoptosis. These results indicated that autophagy played a detrimental role in TBI.
Apelin-13
Apelin-13 is the endogenous ligand of the APJ receptor. It is extracted from bovine stomachs (Tatemoto et al., 1998). Previous studies have shown that apelin-13 could attenuate postischemic cerebral edema and brain injury by suppressing apoptosis (Khaksari et al., 2012). Besides, apelin-13 could suppress glucose deprivation-induced cardiomyocyte autophagy (Jiao et al., 2013). The effects of apelin-13 on autophagy in TBI has also been confirmed in 2014. Bao et al. (2015) suggested that autophagy was activated and lead to secondary brain damage such as apoptosis after TBI. Adminstration of apelin-13 could reverse TBI-induced secondary brain damage by inhibiting autophagy.
Ketamine
Ketamine is usually used for starting and maintaining anesthesia (Green et al., 2011). Other functions of ketamine include sedation and acesodyne in intensive care (Zgaia et al., 2015). In addition to these effects, ketamine has been shown to provide neuroprotection for TBI patients by decreasing glutamate excitotoxicity and inflammatory factors (Chang et al., 2009; Bhutta et al., 2012). Moreover, in 2017, one study showed that autophagy promoted apoptosis and inflammation after TBI while treatment of ketamine could decrease autophagy by activation of the mTOR signaling pathway, thus ameliorating apoptosis and inflammation in TBI (Wang C. Q. et al., 2017).
Docosahexaenoic Acid (DHA)
Docosahexaenoic acid (DHA) is an omega-3 fatty acid that is a primary structural component of human brain. It can be extracted from fish oil and milk or synthesized by alpha-linolenic acid (Guesnet and Alessandri, 2011). DHA has been shown to provide neuroprotection by improving neurological deficits, decreasing infarct volume and reducing proapoptotic proteins (Belayev et al., 2009; Mayurasakorn et al., 2011). Furthermore, Yin et al. (2018) found that TBI significantly elevated the ATG preteins such as sequestosome 1 (SQSTM1/p62), lysosomal-associated membrane proteins 1 (Lamp1), Lamp2 and cathepsin D (Ctsd) in the rat hippocampusm, which led to decreased cognitive functions as well as both gray matter and white matter damages in rats. However, DHA treatment suppressed TBI-induced autophagy and reversed the hippocampal lysosomal biogenesis and function, suggesting that autophagy was detrimental for TBI and suppression of autophagy exhibited neuroprotective effects after TBI.
Other Autophagy Regulators
Recently, there were some other autophagy activators or inhibitors that have been proposed in TBI models such as pifithrin-α (PFT-α; Huang Y.-N. et al., 2018), apocynin (Feng et al., 2017a), trehalose (Portbury et al., 2017), dexmedetomidine (Shen et al., 2017), mitochondrial division inhibitor 1 (Mdivi-1; Wu et al., 2018) and so on (Wang et al., 2013; Cui et al., 2014, 2015, 2017; Lin et al., 2014; Zhang et al., 2014; Jin et al., 2015; Ma et al., 2015; Yao et al., 2015; Du et al., 2016; Zhang L. et al., 2016; Zhao et al., 2016; Sun et al., 2017). All these agents exerted neuroprotective effects in models of TBI, possibly by activation or inhibition of autophagy.
Possible Reasons for the Dual Role of Autophagy in TBI
The mixed results of these studies may be due to the activation degree of autophagy in TBI. Mild autophagy could lead to adenosine triphosphate (ATP) generation, which is beneficial for cell survival. Conversely, excessive autophagy may promote autophagic cell death or apoptosis (Hakumäki et al., 1999). Depending on different environment and stimulus of brain trauma, the activation degree of autophagy may also be different. Therefore, activation of mild autophagy or suppression of excessive autophagy could be both benefit for TBI.
In addition, the available studies exploring the role of autophagy in TBI relied on non-selective drugs that affected autophagy. Therefore, we must consider the effects of these drugs on other signaling pathways instead of the simple influence of autophagy. For example, 3-MA is a non-specific PI3K inhibitor, which may also regulate other pathways such as inflammation and apoptosis. As mentioned above, 3-MA was shown to decrease neuron cell death and improve neurological function after TBI. However, it was difficult to determine whether the protective role of 3-MA was due to its effects on inhibiting autophagy or inflammation or other pathways. Therefore, these studies drew diametrically opposing viewpoints as to the role of autophagy in TBI. The development of specific agents that regulated autophagy may help to clearly elucidate the role of autophagy in TBI. Meanwhile, with the help of molecular biology technology, the precise knockdown or knockout of ATG genes can be realized. Knockdown of the gene for encoding Beclin-1 protein has been achieved in a cerebral ischemia model (Xing et al., 2012). These are all directions of future researches.
Concluding Remarks
Autophagy plays an important role in TBI, it participates in a variety of cellular and molecular processes of TBI. In this review article, we describe the mechanism of autophagy, the functions of autophagy in TBI as well as some upstream moleculars and pharmacological regulators of autophagy involved in TBI. These observations make autophagy an attractive therapeutic target for developing new therapeutic strategies to achieve better outcomes for patients suffering from TBI.
Author Contributions
LZ wrote this review article. HW edited and revised it.
Funding
This work was supported by Grants from the National Natural Science Foundation of China (Grant Nos. 81672503 and 81702484).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
An, J. Y., Zhou, L. L., Sun, P., Pang, H. G., Li, D. D., Li, Y., et al. (2015). Role of the AMPK signaling pathway in early brain injury after subarachnoid hemorrhage in rats. Acta Neurochir. 157, 781–792. doi: 10.1007/s00701-015-2370-3
Arroyo, D. S., Gaviglio, E. A., Peralta Ramos, J. M., Bussi, C., Rodriguez-Galan, M. C., and Iribarren, P. (2014). Autophagy in inflammation, infection, neurodegeneration and cancer. Int. Immunopharmacol. 18, 55–65. doi: 10.1016/j.intimp.2013.11.001
Bao, H. J., Zhang, L., Han, W. C., and Dai, D. K. (2015). Apelin-13 attenuates traumatic brain injury-induced damage by suppressing autophagy. Neurochem. Res. 40, 89–97. doi: 10.1007/s11064-014-1469-x
Batista, P. J., and Chang, H. Y. (2013). Long noncoding RNAs: cellular address codes in development and disease. Cell 152, 1298–1307. doi: 10.1016/j.cell.2013.02.012
Belayev, L., Khoutorova, L., Atkins, K. D., and Bazan, N. G. (2009). Robust docosahexaenoic acid-mediated neuroprotection in a rat model of transient, focal cerebral ischemia. Stroke 40, 3121–3126. doi: 10.1161/strokeaha.109.555979
Bhutta, A. T., Schmitz, M. L., Swearingen, C., James, L. P., Wardbegnoche, W. L., Lindquist, D. M., et al. (2012). Ketamine as a neuroprotective and anti-inflammatory agent in children undergoing surgery on cardiopulmonary bypass: a pilot randomized, double-blind, placebo-controlled trial. Pediatr. Crit. Care Med. 13, 328–337. doi: 10.1097/PCC.0b013e31822f18f9
Brooks, J. C., Strauss, D. J., Shavelle, R. M., Paculdo, D. R., Hammond, F. M., and Harrison-Felix, C. L. (2013). Long-term disability and survival in traumatic brain injury: results from the National institute on disability and rehabilitation research model systems. Arch. Phys. Med. Rehabil. 94, 2203–2209. doi: 10.1016/j.apmr.2013.07.005
Brzezinski, A. (1997). Melatonin in humans. N. Engl. J. Med. 336, 186–195. doi: 10.1056/NEJM199701163360306
Buchanan, M. M., Hutchinson, M., Watkins, L. R., and Yin, H. (2010). Toll-like receptor 4 in CNS pathologies. J. Neurochem. 114, 13–27. doi: 10.1111/j.1471-4159.2010.06736.x
Byun, S., Lee, E., and Lee, K. W. (2017). Therapeutic implications of autophagy inducers in immunological disorders, infection, and cancer. Int. J. Mol. Sci. 18:E1959. doi: 10.3390/ijms18091959
Cao, Z., Zhang, H., Cai, X., Fang, W., Chai, D., Wen, Y., et al. (2017). Luteolin promotes cell apoptosis by inducing autophagy in hepatocellular carcinoma. Cell. Physiol. Biochem. 43, 1803–1812. doi: 10.1159/000484066
Chaanine, A. H., Kohlbrenner, E., Gamb, S. I., Guenzel, A. J., Klaus, K., Fayyaz, A. U., et al. (2016). FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Heart Circ. Physiol. 311, H1540–H1559. doi: 10.1152/ajpheart.00549.2016
Chang, Y., Lee, J. J., Hsieh, C. Y., Hsiao, G., Chou, D. S., and Sheu, J. R. (2009). Inhibitory effects of ketamine on lipopolysaccharide-induced microglial activation. Mediators Inflamm. 2009:705379. doi: 10.1155/2009/705379
Chen, G., Fang, Q., Zhang, J., Zhou, D., and Wang, Z. (2011). Role of the Nrf2-ARE pathway in early brain injury after experimental subarachnoid hemorrhage. J. Neurosci. Res. 89, 515–523. doi: 10.1002/jnr.22577
Chen, G., Ray, R., Dubik, D., Shi, L., Cizeau, J., Bleackley, R. C., et al. (1997). The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J. Exp. Med. 186, 1975–1983. doi: 10.1084/jem.186.12.1975
Chen, J., Wang, L., Wu, C., Hu, Q., Gu, C., Yan, F., et al. (2014). Melatonin-enhanced autophagy protects against neural apoptosis via a mitochondrial pathway in early brain injury following a subarachnoid hemorrhage. J. Pineal Res. 56, 12–19. doi: 10.1111/jpi.12086
Cheung, R. T., Tipoe, G. L., Tam, S., Ma, E. S., Zou, L. Y., and Chan, P. S. (2006). Preclinical evaluation of pharmacokinetics and safety of melatonin in propylene glycol for intravenous administration. J. Pineal Res. 41, 337–343. doi: 10.1111/j.1600-079x.2006.00372.x
Clark, R. S., Bayir, H., Chu, C. T., Alber, S. M., Kochanek, P. M., and Watkins, S. C. (2008). Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy 4, 88–90. doi: 10.4161/auto.5173
Copetti, T., Bertoli, C., Dalla, E., Demarchi, F., and Schneider, C. (2009). p65/RelA modulates BECN1 transcription and autophagy. Mol. Cell. Biol. 29, 2594–2608. doi: 10.1128/mcb.01396-08
Cuervo, A. M., and Wong, E. (2014). Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 24, 92–104. doi: 10.1038/cr.2013.153
Cui, C., Cui, Y., Gao, J., Sun, L., Wang, Y., Wang, K., et al. (2014). Neuroprotective effect of ceftriaxone in a rat model of traumatic brain injury. Neurol. Sci. 35, 695–700. doi: 10.1007/s10072-013-1585-4
Cui, C., Cui, J., Jin, F., Cui, Y., Li, R., Jiang, X., et al. (2017). Induction of the Vitamin D receptor attenuates autophagy dysfunction-mediated cell death following traumatic brain injury. Cell. Physiol. Biochem. 42, 1888–1896. doi: 10.1159/000479571
Cui, C. M., Gao, J. L., Cui, Y., Sun, L. Q., Wang, Y. C., Wang, K. J., et al. (2015). Chloroquine exerts neuroprotection following traumatic brain injury via suppression of inflammation and neuronal autophagic death. Mol. Med. Rep. 12, 2323–2328. doi: 10.3892/mmr.2015.3611
Cuyàs, E., Corominas-Faja, B., Joven, J., and Menendez, J. A. (2014). Cell cycle regulation by the nutrient-sensing mammalian target of rapamycin (mTOR) pathway. Methods Mol. Biol. 1170, 113–144. doi: 10.1007/978-1-4939-0888-2_7
de Vries, H. E., Witte, M., Hondius, D., Rozemuller, A. J., Drukarch, B., Hoozemans, J., et al. (2008). Nrf2-induced antioxidant protection: a promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic. Biol. Med. 45, 1375–1383. doi: 10.1016/j.freeradbiomed.2008.09.001
Degterev, A., Hitomi, J., Germscheid, M., Ch’en, I. L., Korkina, O., Teng, X., et al. (2008). Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321. doi: 10.1038/nchembio.83
Ding, K., Xu, J., Wang, H., Zhang, L., Wu, Y., and Li, T. (2015). Melatonin protects the brain from apoptosis by enhancement of autophagy after traumatic brain injury in mice. Neurochem. Int. 91, 46–54. doi: 10.1016/j.neuint.2015.10.008
Diskin, T., Tal-Or, P., Erlich, S., Mizrachy, L., Alexandrovich, A., Shohami, E., et al. (2005). Closed head injury induces upregulation of Beclin 1 at the cortical site of injury. J. Neurotrauma 22, 750–762. doi: 10.1089/neu.2005.22.750
Dolman, N. J., Chambers, K. M., Mandavilli, B., Batchelor, R. H., and Janes, M. S. (2013). Tools and techniques to measure mitophagy using fluorescence microscopy. Autophagy 9, 1653–1662. doi: 10.4161/auto.24001
Du, G., Zhao, Z., Chen, Y., Li, Z., Tian, Y., Liu, Z., et al. (2016). Quercetin attenuates neuronal autophagy and apoptosis in rat traumatic brain injury model via activation of PI3K/Akt signaling pathway. Neurol. Res. 38, 1012–1019. doi: 10.1080/01616412.2016.1240393
Erlich, S., Alexandrovich, A., Shohami, E., and Pinkas-Kramarski, R. (2007). Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol. Dis. 26, 86–93. doi: 10.1016/j.nbd.2006.12.003
Eskelinen, E. L. (2008). Fine structure of the autophagosome. Methods Mol. Biol. 445, 11–28. doi: 10.1007/978-1-59745-157-4_2
Fan, W., Tang, Z., Chen, D., Moughon, D., Ding, X., Chen, S., et al. (2010). Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy 6, 614–621. doi: 10.4161/auto.6.5.12189
Fang, H., Wang, P. F., Zhou, Y., Wang, Y. C., and Yang, Q. W. (2013). Toll-like receptor 4 signaling in intracerebral hemorrhage-induced inflammation and injury. J Neuroinflammation 10:27. doi: 10.1186/1742-2094-10-27
Feng, Y., Cui, Y., Gao, J. L., Li, M. H., Li, R., Jiang, X. H., et al. (2016). Resveratrol attenuates neuronal autophagy and inflammatory injury by inhibiting the TLR4/NF-κB signaling pathway in experimental traumatic brain injury. Int. J. Mol. Med. 37, 921–930. doi: 10.3892/ijmm.2016.2495
Feng, Y., Cui, C., Liu, X., Wu, Q., Hu, F., Zhang, H., et al. (2017a). Protective role of apocynin via suppression of neuronal autophagy and TLR4/NF-κB signaling pathway in a rat model of traumatic brain injury. Neurochem. Res. 42, 3296–3309. doi: 10.1007/s11064-017-2372-z
Feng, Y., Gao, J., Cui, Y., Li, M., Li, R., Cui, C., et al. (2017b). Neuroprotective effects of resatorvid against traumatic brain injury in rat: involvement of neuronal autophagy and TLR4 signaling pathway. Cell. Mol. Neurobiol. 37, 155–168. doi: 10.1007/s10571-016-0356-1
Galano, A., Tan, D. X., and Reiter, R. J. (2013). On the free radical scavenging activities of melatonin’s metabolites, AFMK and AMK. J. Pineal Res. 54, 245–257. doi: 10.1111/jpi.12010
Gao, Y., Zhuang, Z., Gao, S., Li, X., Zhang, Z., Ye, Z., et al. (2017). Tetrahydrocurcumin reduces oxidative stress-induced apoptosis via the mitochondrial apoptotic pathway by modulating autophagy in rats after traumatic brain injury. Am. J. Transl. Res. 9, 887–899.
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12. doi: 10.1002/path.2697
Green, S. M., Roback, M. G., Kennedy, R. M., and Krauss, B. (2011). Clinical practice guideline for emergency department ketamine dissociative sedation: 2011 update. Ann. Emerg. Med. 57, 449–461. doi: 10.1016/j.annemergmed.2010.11.030
Guesnet, P., and Alessandri, J. M. (2011). Docosahexaenoic acid (DHA) and the developing central nervous system (CNS) - Implications for dietary recommendations. Biochimie 93, 7–12. doi: 10.1016/j.biochi.2010.05.005
Guo, D., Ma, J., Yan, L., Li, T., Li, Z., Han, X., et al. (2017). Down-regulation of Lncrna MALAT1 attenuates neuronal cell death through suppressing Beclin1-dependent autophagy by regulating Mir-30a in cerebral ischemic stroke. Cell Physiol. Biochem. 43, 182–194. doi: 10.1159/000480337
Hakumäki, J. M., Poptani, H., Sandmair, A. M., Ylä-Herttuala, S., and Kauppinen, R. A. (1999). 1H MRS detects polyunsaturated fatty acid accumulation during gene therapy of glioma: implications for the in vivo detection of apoptosis. Nat. Med. 5, 1323–1327. doi: 10.1038/15279
Heras-Sandoval, D., Párez-Rojas, J. M., Hernández-Damián, J., and Pedraza-Chaverri, J. (2014). The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 26, 2694–2701. doi: 10.1016/j.cellsig.2014.08.019
Hoekman, M. F., Jacobs, F. M., Smidt, M. P., and Burbach, J. P. (2006). Spatial and temporal expression of FoxO transcription factors in the developing and adult murine brain. Gene Expr. Patterns 6, 134–140. doi: 10.1016/j.modgep.2005.07.003
Hou, L. L., Gao, C., Chen, L., Hu, G. Q., and Xie, S. Q. (2013). Essential role of autophagy in fucoxanthin-induced cytotoxicity to human epithelial cervical cancer HeLa cells. Acta Pharmacol. Sin. 34, 1403–1410. doi: 10.1038/aps.2013.90
Huang, L., Chen, C., Zhang, X., Li, X., Chen, Z., Yang, C., et al. (2017). Neuroprotective effect of curcumin against cerebral ischemia-reperfusion via mediating autophagy and inflammation. J. Mol. Neurosci. 64, 129–139. doi: 10.1007/s12031-017-1006-x
Huang, T., Wang, M., Huang, B., Chang, A., Liu, F., Zhang, Y., et al. (2018). Long noncoding RNAs in the mTOR signaling network: biomarkers and therapeutic targets. Apoptosis doi: 10.1007/s10495-018-1453-z [Epub ahead of print].
Huang, Y.-N., Yang, L. Y., Greig, N. H., Wang, Y. C., Lai, C. C., and Wang, J. Y. (2018). Neuroprotective effects of pifithrin-α against traumatic brain injury in the striatum through suppression of neuroinflammation, oxidative stress, autophagy, and apoptosis. Sci. Rep. 8:2368. doi: 10.1038/s41598-018-19654-x
Hyakkoku, K., Hamanaka, J., Tsuruma, K., Shimazawa, M., Tanaka, H., Uematsu, S., et al. (2010). Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience 171, 258–267. doi: 10.1016/j.neuroscience.2010.08.054
Jiang, P., and Mizushima, N. (2015). LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods 75, 13–18. doi: 10.1016/j.ymeth.2014.11.021
Jiang, H., Wang, Y., Liang, X., Xing, X., Xu, X., and Zhou, C. (2017). Toll-like receptor 4 knockdown attenuates brain damage and neuroinflammation after traumatic brain injury via inhibiting neuronal autophagy and astrocyte activation. Cell. Mol. Neurobiol. doi: 10.1007/s10571-017-0570-5 [Epub ahead of print].
Jiao, H., Zhang, Z., Ma, Q., Fu, W., and Liu, Z. (2013). Mechanism underlying the inhibitory effect of Apelin-13 on glucose deprivation-induced autophagy in rat cardiomyocytes. Exp. Ther. Med. 5, 797–802. doi: 10.3892/etm.2013.902
Jin, Y., Lin, Y., Feng, J. F., Jia, F., Gao, G., and Jiang, J. Y. (2015). Attenuation of cell death in injured cortex after post-traumatic brain injury moderate hypothermia: possible involvement of autophagy pathway. World Neurosurg 84, 420–430. doi: 10.1016/j.wneu.2015.03.039
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., et al. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728. doi: 10.1093/emboj/19.21.5720
Kaur, J., and Debnath, J. (2015). Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 16, 461–472. doi: 10.1038/nrm4024
Khaksari, M., Aboutaleb, N., Nasirinezhad, F., Vakili, A., and Madjd, Z. (2012). Apelin-13 protects the brain against ischemic reperfusion injury and cerebral edema in a transient model of focal cerebral ischemia. J. Mol. Neurosci. 48, 201–208. doi: 10.1007/s12031-012-9808-3
Kim, T., Mehta, S. L., Kaimal, B., Lyons, K., Dempsey, R. J., and Vemuganti, R. (2016). Poststroke induction of α-synuclein mediates ischemic brain damage. J. Neurosci. 36, 7055–7065. doi: 10.1523/JNEUROSCI.1241-16.2016
Kimura, S., Noda, T., and Yoshimori, T. (2007). Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460. doi: 10.4161/auto.4451
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. doi: 10.1080/15548627.2015.1100356
Knott, A. B., and Bossy-Wetzel, E. (2008). Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann. N Y Acad. Sci. 1147, 283–292. doi: 10.1196/annals.1427.030
Kobayashi, A., Kang, M. I., Okawa, H., Ohtsuji, M., Zenke, Y., Chiba, T., et al. (2004). Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24, 7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004
Kucharewicz, K., Dudkowska, M., Zawadzka, A., Ogrodnik, M., Szczepankiewicz, A. A., Czarnocki, Z., et al. (2018). Simultaneous induction and blockade of autophagy by a single agent. Cell Death Dis. 9:353. doi: 10.1038/s41419-018-0383-6
Lai, Y., Hickey, R. W., Chen, Y., Bayir, H., Sullivan, M. L., Chu, C. T., et al. (2008). Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant γ-glutamylcysteinyl ethyl ester. J. Cereb. Blood Flow Metab. 28, 540–550. doi: 10.1038/sj.jcbfm.9600551
Lee, Y., Lee, H. Y., Hanna, R. A., and Gustafsson, A. B. (2011). Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 301, H1924–H1931. doi: 10.1152/ajpheart.00368.2011
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. doi: 10.1016/j.cell.2007.12.018
Li, W., Du, M., Wang, Q., Ma, X., Wu, L., Guo, F., et al. (2017). FoxO1 promotes mitophagy in the podocytes of diabetic male mice via the PINK1/parkin pathway. Endocrinology 158, 2155–2167. doi: 10.1210/en.2016-1970
Li, Z., Li, J., and Tang, N. (2017). Long noncoding RNA Malat1 is a potent autophagy inducer protecting brain microvascular endothelial cells against oxygen-glucose deprivation/reoxygenation-induced injury by sponging miR-26b and upregulating ULK2 expression. Neuroscience 354, 1–10. doi: 10.1016/j.neuroscience.2017.04.017
Li, D., Li, X., Wu, J., Li, J., Zhang, L., Xiong, T., et al. (2015). Involvement of the JNK/FOXO3a/Bim pathway in neuronal apoptosis after hypoxic-ischemic brain damage in neonatal rats. PLoS One 10:e0132998. doi: 10.1371/journal.pone.0132998
Li, L., Tan, J., Miao, Y., Lei, P., and Zhang, Q. (2015). ROS and autophagy: interactions and molecular regulatory mechanisms. Cell. Mol. Neurobiol. 35, 615–621. doi: 10.1007/s10571-015-0166-x
Li, Y., Yang, W., Quinones-Hinojosa, A., Wang, B., Xu, S., Zhu, W., et al. (2016). Interference with protease-activated receptor 1 alleviates neuronal cell death induced by lipopolysaccharide-stimulated microglial cells through the PI3K/Akt pathway. Sci. Rep. 6:38247. doi: 10.1038/srep38247
Liang, X. H., Jackson, S., Seaman, M., Brown, K., Kempkes, B., Hibshoosh, H., et al. (1999). Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676. doi: 10.1038/45257
Lin, C., Chao, H., Li, Z., Xu, X., Liu, Y., Hou, L., et al. (2016). Melatonin attenuates traumatic brain injury-induced inflammation: a possible role for mitophagy. J. Pineal Res. 61, 177–186. doi: 10.1111/jpi.12337
Lin, C. J., Chen, T. H., Yang, L. Y., and Shih, C. M. (2014). Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis. 5:e1147. doi: 10.1038/cddis.2014.123
Lin, S., Yin, Q., Zhong, Q., Lv, F. L., Zhou, Y., Li, J. Q., et al. (2012). Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J. Neuroinflammation 9:46. doi: 10.1186/1742-2094-9-46
Lipinski, M. M., Wu, J., Faden, A. I., and Sarkar, C. (2015). Function and mechanisms of autophagy in brain and spinal cord trauma. Antioxid. Redox Signal. 23, 565–577. doi: 10.1089/ars.2015.6306
Liu, C. L., Chen, S., Dietrich, D., and Hu, B. R. (2008). Changes in autophagy after traumatic brain injury. J. Cereb. Blood Flow Metab. 28, 674–683. doi: 10.1038/sj.jcbfm.9600587
Liu, D., Chen, Q., Zhu, H., Gong, L., Huang, Y., Li, S., et al. (2018). Association of respiratory syncytial virus toll-like receptor 3-mediated immune response with COPD exacerbation frequency. Inflammation 41, 654–666. doi: 10.1007/s10753-017-0720-4
Liu, Y., Bao, Z., Xu, X., Chao, H., Lin, C., Li, Z., et al. (2017). Extracellular signal-regulated kinase/nuclear factor-erythroid2-like2/heme oxygenase-1 pathway-mediated mitophagy alleviates traumatic brain injury-induced intestinal mucosa damage and epithelial barrier dysfunction. J. Neurotrauma 34, 2119–2131. doi: 10.1089/neu.2016.4764
Liu, Z., Wang, Y., Zhao, S., Zhang, J., Wu, Y., and Zeng, S. (2015). Imidazole inhibits autophagy flux by blocking autophagic degradation and triggers apoptosis via increasing FoxO3a-Bim expression. Int. J. Oncol. 46, 721–731. doi: 10.3892/ijo.2014.2771
Long, X., Lin, Y., Ortiz-Vega, S., Yonezawa, K., and Avruch, J. (2005). Rheb binds and regulates the mTOR kinase. Curr. Biol. 15, 702–713. doi: 10.1016/j.cub.2005.02.053
Lu, J., Jiang, Z., Chen, Y., Zhou, C., and Chen, C. (2016). Knockout of programmed cell death 5 (PDCD5) gene attenuates neuron injury after middle cerebral artery occlusion in mice. Brain Res. 1650, 152–161. doi: 10.1016/j.brainres.2016.09.005
Luo, C. L., Li, B. X., Li, Q. Q., Chen, X. P., Sun, Y. X., Bao, H. J., et al. (2011). Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience 184, 54–63. doi: 10.1016/j.neuroscience.2011.03.021
Lv, B., Hua, T., Li, F., Han, J., Fang, J., Xu, L., et al. (2017). Hypoxia-inducible factor 1 α protects mesenchymal stem cells against oxygen-glucose deprivation-induced injury via autophagy induction and PI3K/AKT/mTOR signaling pathway. Am. J. Transl. Res. 9, 2492–2499.
Ma, L., Li, Z., Liu, Z., Li, M., Sui, D., Liu, Y., et al. (2015). 17AAG improves histological and functional outcomes in a rat CCI model through autophagy activation and apoptosis attenuation. Neurosci. Lett. 599, 1–6. doi: 10.1016/j.neulet.2015.05.004
Manning, B. D., and Cantley, L. C. (2007). AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274. doi: 10.1016/j.cell.2007.06.009
Mayurasakorn, K., Williams, J. J., Ten, V. S., and Deckelbaum, R. J. (2011). Docosahexaenoic acid: brain accretion and roles in neuroprotection after brain hypoxia and ischemia. Curr. Opin. Clin. Nutr. Metab. Care 14, 158–167. doi: 10.1097/MCO.0b013e328342cba5
Mencherini, T., Picerno, P., Scesa, C., and Aquino, R. (2007). Triterpene, antioxidant, and antimicrobial compounds from Melissa officinalis. J. Nat. Prod. 70, 1889–1894. doi: 10.1021/np070351s
Mijaljica, D., Prescott, M., and Devenish, R. J. (2011). Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 7, 673–682. doi: 10.4161/auto.7.7.14733
Mishra, S., Mishra, M., Seth, P., and Sharma, S. K. (2011). Tetrahydrocurcumin confers protection against amyloid β-induced toxicity. Neuroreport 22, 23–27. doi: 10.1097/WNR.0b013e328341e141
Miyazaki, M., McCarthy, J. J., and Esser, K. A. (2010). Insulin like growth factor-1-induced phosphorylation and altered distribution of tuberous sclerosis complex (TSC)1/TSC2 in C2C12 myotubes. FEBS J. 277, 2180–2191. doi: 10.1111/j.1742-4658.2010.07635.x
Mizushima, N., and Komatsu, M. (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. doi: 10.1016/j.cell.2011.10.026
Mizushima, N., Yoshimori, T., and Levine, B. (2010). Methods in mammalian autophagy research. Cell 140, 313–326. doi: 10.1016/j.cell.2010.01.028
Moskalev, A., Shaposhnikov, M., Zemskaya, N., Belyi, A., Dobrovolskaya, E., Patova, A., et al. (2018). Transcriptome analysis reveals mechanisms of geroprotective effects of fucoxanthin in Drosophila. BMC Genomics 19:77. doi: 10.1186/s12864-018-4471-x
Murakawa, T., Yamaguchi, O., Hashimoto, A., Hikoso, S., Takeda, T., Oka, T., et al. (2015). Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 6:7527. doi: 10.1038/ncomms8527
Nellist, M., Goedbloed, M. A., de Winter, C., Verhaaf, B., Jankie, A., Reuser, A. J., et al. (2002). Identification and characterization of the interaction between tuberin and 14–3-3ζ. J. Biol. Chem. 277, 39417–39424. doi: 10.1074/jbc.M204802200
Neuhouser, M. L. (2004). Dietary flavonoids and cancer risk: evidence from human population studies. Nutr. Cancer 50, 1–7. doi: 10.1207/s15327914nc5001_1
Noda, N. N., Kumeta, H., Nakatogawa, H., Satoo, K., Adachi, W., Ishii, J., et al. (2008). Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells 13, 1211–1218. doi: 10.1111/j.1365-2443.2008.01238.x
Pajares, M., Jiménez-Moreno, N., Garcüa-Yagüe, Á. J., Escoll, M., de Ceballos, M. L., Van Leuven, F., et al. (2016). Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 12, 1902–1916. doi: 10.1080/15548627.2016.1208889
Pan, P., Zhang, H., Su, L., Wang, X., and Liu, D. (2018). Melatonin balance the autophagy and apoptosis by regulating UCP2 in the LPS-induced cardiomyopathy. Molecules 23:E675. doi: 10.3390/molecules23030675
Portbury, S. D., Hare, D. J., Finkelstein, D. I., and Adlard, P. A. (2017). Trehalose improves traumatic brain injury-induced cognitive impairment. PLoS One 12:e0183683. doi: 10.1371/journal.pone.0183683
Qiu, X., Cao, L., Yang, X., Zhao, X., Liu, X., Han, Y., et al. (2013). Role of mitochondrial fission in neuronal injury in pilocarpine-induced epileptic rats. Neuroscience 245, 157–165. doi: 10.1016/j.neuroscience.2013.04.019
Rosenbaum, D. M., Degterev, A., David, J., Rosenbaum, P. S., Roth, S., Grotta, J. C., et al. (2010). Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J. Neurosci. Res. 88, 1569–1576. doi: 10.1002/jnr.22314
Sanchez, A. M., Csibi, A., Raibon, A., Cornille, K., Gay, S., Bernardi, H., et al. (2012). AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J. Cell. Biochem. 113, 695–710. doi: 10.1002/jcb.23399
Sarkar, C., Zhao, Z., Aungst, S., Sabirzhanov, B., Faden, A. I., and Lipinski, M. M. (2014). Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy 10, 2208–2222. doi: 10.4161/15548627.2014.981787
Schaible, E. V., Windschugl, J., Bobkiewicz, W., Kaburov, Y., Dangel, L., Kramer, T., et al. (2014). 2-Methoxyestradiol confers neuroprotection and inhibits a maladaptive HIF-1α response after traumatic brain injury in mice. J. Neurochem. 129, 940–954. doi: 10.1111/jnc.12708
Scherz-Shouval, R., Shvets, E., Fass, E., Shorer, H., Gil, L., and Elazar, Z. (2007). Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26, 1749–1760. doi: 10.1038/sj.emboj.7601623
Shen, M., Wang, S., Wen, X., Han, X. R., Wang, Y. J., Zhou, X. M., et al. (2017). Dexmedetomidine exerts neuroprotective effect via the activation of the PI3K/Akt/mTOR signaling pathway in rats with traumatic brain injury. Biomed. Pharmacother. 95, 885–893. doi: 10.1016/j.biopha.2017.08.125
Smirnova, E., Griparic, L., Shurland, D. L., and van der Bliek, A. M. (2001). Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12, 2245–2256. doi: 10.1091/mbc.12.8.2245
Song, Y., Ding, W., Xiao, Y., and Lu, K. J. (2015). The progress of mitophagy and related pathogenic mechanisms of the neurodegenerative diseases and tumor. Neurosci. J. 2015:543758. doi: 10.1155/2015/543758
Spizzo, R., Almeida, M. I., Colombatti, A., and Calin, G. A. (2012). Long non-coding RNAs and cancer: a new frontier of translational research? Oncogene 31, 4577–4587. doi: 10.1038/onc.2011.621
Sugawara, T., Baskaran, V., Tsuzuki, W., and Nagao, A. (2002). Brown algae fucoxanthin is hydrolyzed to fucoxanthinol during absorption by Caco-2 human intestinal cells and mice. J. Nutr. 132, 946–951. doi: 10.1093/jn/132.5.946
Sun, M., Zhao, Y., Gu, Y., and Zhang, Y. (2015). Protective effects of taurine against closed head injury in rats. J. Neurotrauma 32, 66–74. doi: 10.1089/neu.2012.2432
Sun, L., Zhao, M., Liu, M., Su, P., Zhang, J., Li, Y., et al. (2018). Suppression of FoxO3a attenuates neurobehavioral deficits after traumatic brain injury through inhibiting neuronal autophagy. Behav. Brain Res. 337, 271–279. doi: 10.1016/j.bbr.2017.08.042
Sun, L., Zhao, M., Wang, Y., Liu, A., Lv, M., Li, Y., et al. (2017). Neuroprotective effects of miR-27a against traumatic brain injury via suppressing FoxO3a-mediated neuronal autophagy. Biochem. Biophys. Res. Commun. 482, 1141–1147. doi: 10.1016/j.bbrc.2016.12.001
Swiderek, E., Kalas, W., Wysokinska, E., Pawlak, A., Rak, J., and Strzadala, L. (2013). The interplay between epigenetic silencing, oncogenic KRas and HIF-1 regulatory pathways in control of BNIP3 expression in human colorectal cancer cells. Biochem. Biophys. Res. Commun. 441, 707–712. doi: 10.1016/j.bbrc.2013.10.098
Tanaka, A., Cleland, M. M., Xu, S., Narendra, D. P., Suen, D. F., Karbowski, M., et al. (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380. doi: 10.1083/jcb.201007013
Tang, C., Shan, Y., Hu, Y., Fang, Z., Tong, Y., Chen, M., et al. (2017). FGF2 attenuates neural cell death via suppressing autophagy after rat mild traumatic brain injury. Stem Cells Int. 2017:2923182. doi: 10.1155/2017/2923182
Tatemoto, K., Hosoya, M., Habata, Y., Fujii, R., Kakegawa, T., Zou, M. X., et al. (1998). Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun. 251, 471–476. doi: 10.1006/bbrc.1998.9489
Tyagi, N., Qipshidze, N., Munjal, C., Vacek, J. C., Metreveli, N., Givvimani, S., et al. (2012). Tetrahydrocurcumin ameliorates homocysteinylated cytochrome-c mediated autophagy in hyperhomocysteinemia mice after cerebral ischemia. J. Mol. Neurosci. 47, 128–138. doi: 10.1007/s12031-011-9695-z
Villeneuve, N. F., Lau, A., and Zhang, D. D. (2010). Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: an insight into cullin-ring ubiquitin ligases. Antioxid. Redox Signal. 13, 1699–1712. doi: 10.1089/ars.2010.3211
Vontell, R., Supramaniam, V., Wyatt-Ashmead, J., Gressens, P., Rutherford, M., Hagberg, H., et al. (2015). Cellular mechanisms of toll-like receptor-3 activation in the thalamus are associated with white matter injury in the developing brain. J. Neuropathol. Exp. Neurol. 74, 273–285. doi: 10.1097/NEN.0000000000000172
Wang, J., Fields, J., Zhao, C., Langer, J., Thimmulappa, R. K., Kensler, T. W., et al. (2007). Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic. Biol. Med. 43, 408–414. doi: 10.1016/j.freeradbiomed.2007.04.020
Wang, C., Nie, H., Li, K., Zhang, Y. X., Yang, F., Li, C. B., et al. (2012). Curcumin inhibits HMGB1 releasing and attenuates concanavalin A-induced hepatitis in mice. Eur. J. Pharmacol. 697, 152–157. doi: 10.1016/j.ejphar.2012.09.050
Wang, Y. Q., Wang, L., Zhang, M. Y., Wang, T., Bao, H. J., Liu, W. L., et al. (2012). Necrostatin-1 suppresses autophagy and apoptosis in mice traumatic brain injury model. Neurochem. Res. 37, 1849–1858. doi: 10.1007/s11064-012-0791-4
Wang, C. Q., Ye, Y., Chen, F., Han, W. C., Sun, J. M., Lu, X., et al. (2017). Posttraumatic administration of a sub-anesthetic dose of ketamine exerts neuroprotection via attenuating inflammation and autophagy. Neuroscience 343, 30–38. doi: 10.1016/j.neuroscience.2016.11.029
Wang, C. F., Zhao, C. C., Weng, W. J., Lei, J., Lin, Y., Mao, Q., et al. (2017). Alteration in long non-coding RNA expression after traumatic brain injury in rats. J. Neurotrauma 34, 2100–2108. doi: 10.1089/neu.2016.4642
Wang, T., Zhang, L., Zhang, M., Bao, H., Liu, W., Wang, Y., et al. (2013). [Gly14]-Humanin reduces histopathology and improves functional outcome after traumatic brain injury in mice. Neuroscience 231, 70–81. doi: 10.1016/j.neuroscience.2012.11.019
Wang, Y., Zhou, Y., and Graves, D. T. (2014). FOXO transcription factors: their clinical significance and regulation. Biomed Res. Int. 2014:925350. doi: 10.1155/2014/925350
Wojcik, S. (2013). Crosstalk between autophagy and proteasome protein degradation systems: possible implications for cancer therapy. Folia Histochem. Cytobiol. 51, 249–264. doi: 10.5603/FHC.2013.0036
Wu, Q., Gao, C., Wang, H., Zhang, X., Li, Q., Gu, Z., et al. (2018). Mdivi-1 alleviates blood-brain barrier disruption and cell death in experimental traumatic brain injury by mitigating autophagy dysfunction and mitophagy activation. Int. J. Biochem. Cell Biol. 94, 44–55. doi: 10.1016/j.biocel.2017.11.007
Wu, J. C., Tsai, M. L., Lai, C. S., Wang, Y. J., Ho, C. T., and Pan, M. H. (2014). Chemopreventative effects of tetrahydrocurcumin on human diseases. Food Funct. 5, 12–17. doi: 10.1039/c3fo60370a
Wu, Q., Xia, S. X., Li, Q. Q., Gao, Y., Shen, X., Ma, L., et al. (2016). Mitochondrial division inhibitor 1 (Mdivi-1) offers neuroprotection through diminishing cell death and improving functional outcome in a mouse model of traumatic brain injury. Brain Res. 1630, 134–143. doi: 10.1016/j.brainres.2015.11.016
Xin, X. Y., Pan, J., Wang, X. Q., Ma, J. F., Ding, J. Q., Yang, G. Y., et al. (2011). 2-methoxyestradiol attenuates autophagy activation after global ischemia. Can. J. Neurol. Sci. 38, 631–638. doi: 10.1017/s031716710001218x
Xing, S., Zhang, Y., Li, J., Zhang, J., Li, Y., Dang, C., et al. (2012). Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy 8, 63–76. doi: 10.4161/auto.8.1.18217
Xu, J., Wang, H., Lu, X., Ding, K., Zhang, L., He, J., et al. (2014). Posttraumatic administration of luteolin protects mice from traumatic brain injury: implication of autophagy and inflammation. Brain Res. 1582, 237–246. doi: 10.1016/j.brainres.2014.07.042
Yan, W., Chen, Z.-Y., Chen, J.-Q., and Chen, H.-M. (2018). LncRNA NEAT1 promotes autophagy in MPTP-induced Parkinson’s disease through stabilizing PINK1 protein. Biochem. Biophys. Res. Commun. 496, 1019–1024. doi: 10.1016/j.bbrc.2017.12.149
Yan, W., Wang, H. D., Hu, Z. G., Wang, Q. F., and Yin, H. X. (2008). Activation of Nrf2-ARE pathway in brain after traumatic brain injury. Neurosci. Lett. 431, 150–154. doi: 10.1016/j.neulet.2007.11.060
Yao, J., Zheng, K., and Zhang, X. (2015). Rosiglitazone exerts neuroprotective effects via the suppression of neuronal autophagy and apoptosis in the cortex following traumatic brain injury. Mol. Med. Rep. 12, 6591–6597. doi: 10.3892/mmr.2015.4292
Yin, Y., Li, E., Sun, G., Yan, H. Q., Foley, L. M., Andrzejczuk, L. A., et al. (2018). Effects of DHA on hippocampal autophagy and lysosome function after traumatic brain injury. Mol. Neurobiol. 55, 2454–2470. doi: 10.1007/s12035-017-0504-8
Ylä-Anttila, P., Vihinen, H., Jokitalo, E., and Eskelinen, E. L. (2009). Monitoring autophagy by electron microscopy in Mammalian cells. Methods Enzymol. 452, 143–164. doi: 10.1016/s0076-6879(08)03610-0
Yoo, K. Y., Kwon, S. H., Lee, C. H., Yan, B., Park, J. H., Ahn, J. H., et al. (2012). FoxO3a changes in pyramidal neurons and expresses in non-pyramidal neurons and astrocytes in the gerbil hippocampal CA1 region after transient cerebral ischemia. Neurochem. Res. 37, 588–595. doi: 10.1007/s11064-011-0648-2
Yoshii, S. R., and Mizushima, N. (2017). Monitoring and measuring autophagy. Int. J. Mol. Sci. 18:E1865. doi: 10.3390/ijms18091865
You, Z., Savitz, S. I., Yang, J., Degterev, A., Yuan, J., Cuny, G. D., et al. (2008). Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 28, 1564–1573. doi: 10.1038/jcbfm.2008.44
Yu, Y., Cao, F., Ran, Q., and Wang, F. (2017). Long non-coding RNA Gm4419 promotes trauma-induced astrocyte apoptosis by targeting tumor necrosis factor α. Biochem. Biophys. Res. Commun. 491, 478–485. doi: 10.1016/j.bbrc.2017.07.021
Zgaia, A. O., Irimie, A., Sandesc, D., Vlad, C., Lisencu, C., Rogobete, A., et al. (2015). The role of ketamine in the treatment of chronic cancer pain. Clujul Med. 88, 457–461. doi: 10.15386/cjmed-500
Zhang, L., Ding, K., Wang, H., Wu, Y., and Xu, J. (2016). Traumatic brain injury-induced neuronal apoptosis is reduced through modulation of PI3K and autophagy pathways in mouse by FTY720. Cell. Mol. Neurobiol. 36, 131–142. doi: 10.1007/s10571-015-0227-1
Zhang, Y. B., Li, S. X., Chen, X. P., Yang, L., Zhang, Y. G., Liu, R., et al. (2008). Autophagy is activated and might protect neurons from degeneration after traumatic brain injury. Neurosci. Bull. 24, 143–149. doi: 10.1007/s12264-008-1108-0
Zhang, J., and Ney, P. A. (2011). Mechanisms and biology of B-cell leukemia/lymphoma 2/adenovirus E1B interacting protein 3 and Nip-like protein X. Antioxid. Redox Signal. 14, 1959–1969. doi: 10.1089/ars.2010.3772
Zhang, M., Shan, H., Chang, P., Wang, T., Dong, W., Chen, X., et al. (2014). Hydrogen sulfide offers neuroprotection on traumatic brain injury in parallel with reduced apoptosis and autophagy in mice. PLoS One 9:e87241. doi: 10.1371/journal.pone.0087241
Zhang, L., and Wang, H. (2018). Targeting the NF-E2-related factor 2 pathway: a novel strategy for traumatic brain injury. Mol. Neurobiol. 55, 1773–1785. doi: 10.1007/s12035-017-0456-z
Zhang, L., Wang, H., Fan, Y., Gao, Y., Li, X., Hu, Z., et al. (2017). Fucoxanthin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE and Nrf2-autophagy pathways. Sci. Rep. 7:46763. doi: 10.1038/srep46763
Zhang, B. C., Zhang, C. W., Wang, C., Pan, D. F., Xu, T. D., and Li, D. Y. (2016). Luteolin attenuates foam cell formation and apoptosis in Ox-LDL-stimulated macrophages by enhancing autophagy. Cell. Physiol. Biochem. 39, 2065–2076. doi: 10.1159/000447902
Zhao, M., Liang, F., Xu, H., Yan, W., and Zhang, J. (2016). Methylene blue exerts a neuroprotective effect against traumatic brain injury by promoting autophagy and inhibiting microglial activation. Mol. Med. Rep. 13, 13–20. doi: 10.3892/mmr.2015.4551
Zhong, J., Jiang, L., Cheng, C., Huang, Z., Zhang, H., Liu, H., et al. (2016). Altered expression of long non-coding RNA and mRNA in mouse cortex after traumatic brain injury. Brain Res. 1646, 589–600. doi: 10.1016/j.brainres.2016.07.002
Zhong, J., Jiang, L., Huang, Z., Zhang, H., Cheng, C., Liu, H., et al. (2017). The long non-coding RNA Neat1 is an important mediator of the therapeutic effect of bexarotene on traumatic brain injury in mice. Brain Behav. Immun. 65, 183–194. doi: 10.1016/j.bbi.2017.05.001
Zhou, J., Liao, W., Yang, J., Ma, K., Li, X., Wang, Y., et al. (2012). FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy 8, 1712–1723. doi: 10.4161/auto.21830
Keywords: traumatic brain injury, autophagy, methods, molecular mechanisms, pharmacological modulation
Citation: Zhang L and Wang H (2018) Autophagy in Traumatic Brain Injury: A New Target for Therapeutic Intervention. Front. Mol. Neurosci. 11:190. doi: 10.3389/fnmol.2018.00190
Received: 08 April 2018; Accepted: 15 May 2018;
Published: 05 June 2018.
Edited by:
Atanas G. Atanasov, Institute of Genetics and Animal Breeding (PAS), PolandReviewed by:
Denis Gris, Université de Sherbrooke, CanadaCathy W. Levenson, Florida State University, United States
Copyright © 2018 Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Handong Wang, bmpoZHdhbmdAaG90bWFpbC5jb20=