 Jian Jing Siew1,2
Jian Jing Siew1,2 Yijuang Chern1,2*
Yijuang Chern1,2*- 1Taiwan International Graduate Program in Molecular Medicine, National Yang-Ming University and Academia Sinica, Taipei, Taiwan
- 2Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan
Microglia are the innate sentinels of the central nervous system (CNS) and are responsible for the homeostasis and immune defense of the CNS. Under the influence of the local environment and cell-cell interaction, microglia exhibit a multidimensional and context-dependent phenotypes that can be cytotoxic and neuroprotective. Recent studies suggest that microglia express multitudinous types of lectins, including galectins, Siglecs, mannose-binding lectins (MBLs) and other glycan binding proteins. Because most studies that examine lectins focus on the peripheral system, the functions of lectins have not been critically investigated in the CNS. In addition, the types of brain cells that contribute to the altered levels of lectins present in diseases are often unclear. In this review, we will discuss how galectins, Siglecs, selectins and MBLs contribute to the dynamic functions of microglia. The interacting ligands of these lectins are complex glycoconjugates, which consist of glycoproteins and glycolipids that are expressed on microglia or surrounding cells. The current understanding of the heterogeneity and functions of glycans in the brain is limited. Galectins are a group of pleotropic proteins that recognize both β-galactoside-containing glycans and non- β-galactoside-containing proteins. The function and regulation of galectins have been implicated in immunomodulation, neuroinflammation, apoptosis, phagocytosis and oxidative bursts. Most Siglecs are expressed at a low level on the plasma membrane and bind to sialic acid residues for immunosurveillance and cell-cell communication. Siglecs are classified based on their inhibitory and activatory downstream signaling properties. Inhibitory Siglecs negatively regulate microglia activation upon recognizing the intact sialic acid patterns and vice versa. MBLs are expressed upon infection in cytoplasm and can be secreted in order to recognize molecules containing terminal mannose as an innate immune defense machinery. Most importantly, multiple studies have reported dysregulation of lectins in neurological disorders. Here, we reviewed recent studies on microglial lectins and their functions in CNS health and disease, and suggest that these lectin families are novel, potent therapeutic targets for neurological diseases.
Introduction
The central nervous system (CNS) consists of neurons and glial cells such as microglia, astrocytes and oligodendrocytes. Microglia are the CNS counterpart of macrophages and differ from other types of glial cells in terms of their origin, morphology and functions (Kettenmann et al., 2011). They are widely distributed throughout the brain and spinal cord, and constitute 5%–10% of total brain cells depending on the region (Lawson et al., 1990).
Microglia play multiple roles in healthy and diseased brains. During development, microglia regulate the number of neuronal cells by triggering programmed cell death and actively phagocytose the apoptotic neurons (de la Rosa and de Pablo, 2000; Bessis et al., 2007). In addition, microglia are capable of promoting neurogenesis by secreting trophic factors (such as nerve growth factor (NGF) and insulin-like growth factor 1 (IGF-1; Frielingsdorf et al., 2007; Thored et al., 2009). In addition to regulating the number of neurons, microglia also strengthen and prune excessive synapses to ensure that neuronal circuits are connected correctly (Paolicelli et al., 2011). Hence, microglia have been implicated in synaptic plasticity.
As the immune sentinels of the CNS, microglia are responsible for detecting and swiftly responding to infection, injury, molecules released by damaged cells and misfolded proteins (Davalos et al., 2005; Nimmerjahn et al., 2005). In this context, microglia are believed to play exacerbating roles in the inflammatory response of neurological diseases, including multiple sclerosis (MS; Politis et al., 2012), ischemic stroke (Patel et al., 2013), Alzheimer’s disease (AD; Fuhrmann et al., 2010), Parkinson’s disease (PD; Subramaniam and Federoff, 2017), amyotrophic lateral sclerosis (ALS; Brites and Vaz, 2014), Huntington’s disease (HD; Hsiao et al., 2014) and virus infection in the brain (Cenker et al., 2017). Hence, elucidating the machinery that regulates the multidimensional phenotypes of microglia is critical for understanding neurological diseases and developing effective therapeutic interventions.
Microglia express various types of lectins that functionally regulate their dynamic phenotypes. These lectins include galectins, Siglecs, C-type lectins (such as mannose-binding lectins, MBLs) and other glycan binding proteins. Some of these lectins are expressed constitutively, while others are expressed upon stimulation. For instance, microglia generally do not express galectin-3, but its expression can be up-regulated by inflammatory mediators (Burguillos et al., 2015). Galectins have multiple cellular localizations and different functions. For instance, galectin-1, -3 and -9 have been reported to exist in the nucleus, cytoplasm, plasma membrane and extracellular matrix. The cell types in the CNS that express galectins and their cellular localization and functions are summarized in Table 1. Contrary to galectins, all CNS Siglecs (except Siglec-4) are localized on the plasma membrane of microglia. Details about the CNS Siglecs are summarized in Table 2. Similar to Siglecs, selectins are localized on the plasma membrane of endothelial cells. In contrast, MBL is found in the cytoplasm of all brain cells in disease conditions and can be secreted into the extracellular matrix. Details on these C-type lectins are summarized in Table 3. The cell surface contains a complex layer of glycosylated molecules, such as glycoproteins and glycolipids that are recognized by these lectins and store important information for cell-cell communication and response (Brandley and Schnaar, 1986; Groves, 2013; Letschert et al., 2014). Classification of lectins is based on the homology of amino acid sequences and specificity of glycan structures. Their functions have been reviewed in detail elsewhere (Varki et al., 2009; Fujimoto et al., 2014). In this review, we focus on the role of lectins and how they affect the functions of microglia in the CNS. Tables 1–3 summarizes the major lectins that have been reported in the CNS so far.
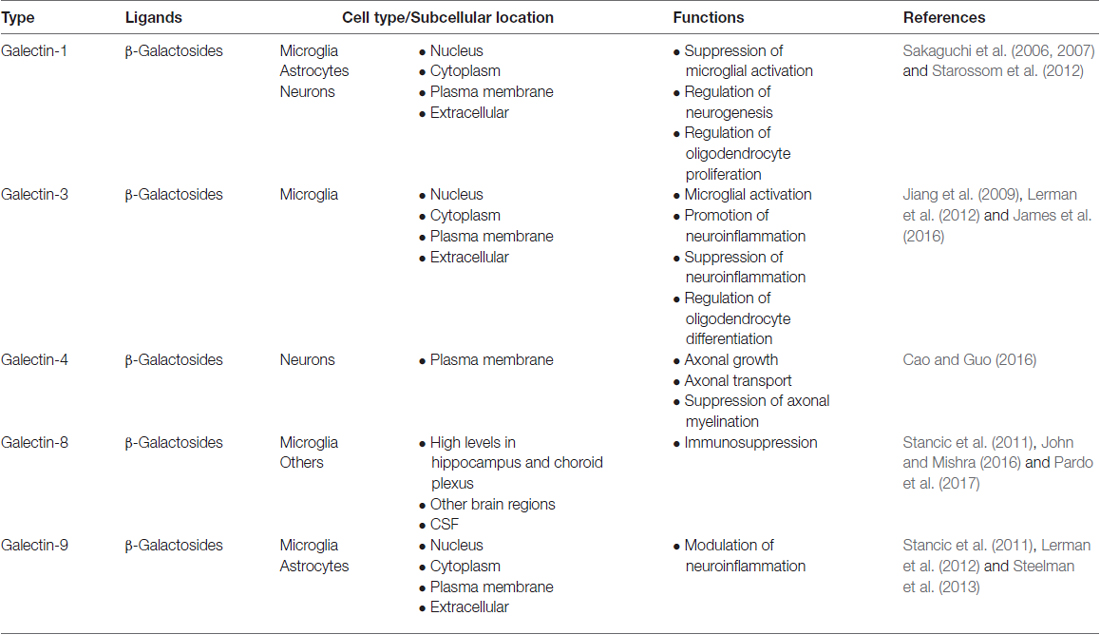
Table 1. Summary of galectins in central nervous system (CNS).
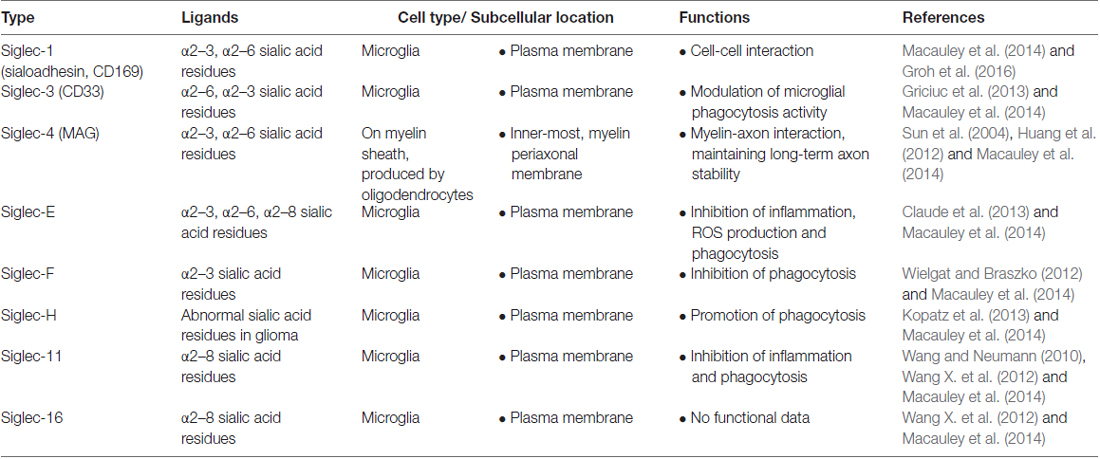
Table 2. Summary of Siglecs in CNS.
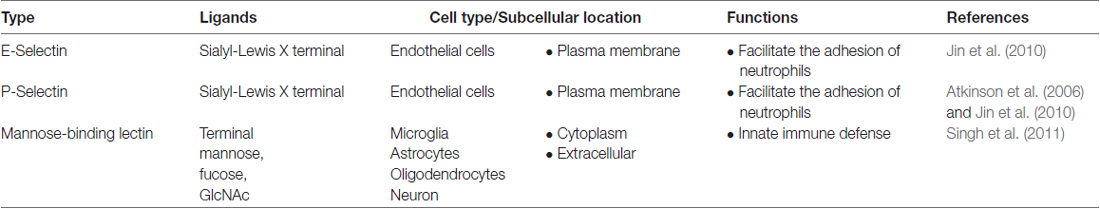
Table 3. Summary of C-type lectins in CNS.
Galectins
Galectins represent a family of lectins characterized by conserved carbohydrate-recognition domains (CRDs) and bind to β-galactosides with different specificities and affinities (Liu and Rabinovich, 2005). Through the conserved CRDs, 15 galectins have been identified; they can be categorized into three groups: (1) proto, or one CRD; (2) tandem repeats, or two distinct CRDs in tandem; and (3) chimera, consisting of CRD with unusual tandem repeat of glycine- and proline-rich short stretches (Kasai and Hirabayashi, 1996). The prototype galectins include galectins-1, -2, -5, -7, -10, -11, -13, -14 and -15, while the tandem repeats type consists of galectins -4, -6, -8, -9 and -12; and galectin-3 is the only galectin classified as chimera type. Among these galectins, only galectins -1, -2, -3, -4, -7, -8, -9, -10, -12 and -13 are expressed in humans (Chang et al., 2017). Furthermore, many of these galectins have differentially spliced forms that may be expressed in different tissues. For instance, galectins-8 and -9 have seven and three isoforms, respectively (Zhang et al., 2009; Troncoso et al., 2014). In particular, the galectin-9M and 9S isoforms positively promote the expression of E-selectin, while the galectin-9L isoform suppresses the level of E-selectin (Zhang et al., 2009). These isoforms are critical in regulating important cellular activity. The mechanisms involved in coordinating the expression of these isoforms and the functions they serve are still in need of extensive exploration. To date, no specific surface receptor for galectins has been reported. Nonetheless, galectins can bind to and interact with various glycoproteins and cell membrane glycoconjugates via their carbohydrate moieties and trigger a cascade of transmembrane signaling events (Liu and Rabinovich, 2005; Wan and Liu, 2016). In addition, galectins have non-carbohydrate binding partners and function intracellularly to regulate gene transcription, mRNA splicing, cell growth, apoptosis and immune responses (Liu et al., 2002; Wang J. L. et al., 2012). For instance, intracellular galectin-1 and galectin-3 interact with Gemin4 to facilitate the assembly of spliceosomes (Park et al., 2001). The functions of galectins in the peripheral system have been extensively studied. To date, only galectins-1, -3, -4, -8 and -9 have been found in the CNS. Table 4 summarizes the role of galectins in neurological disorders.
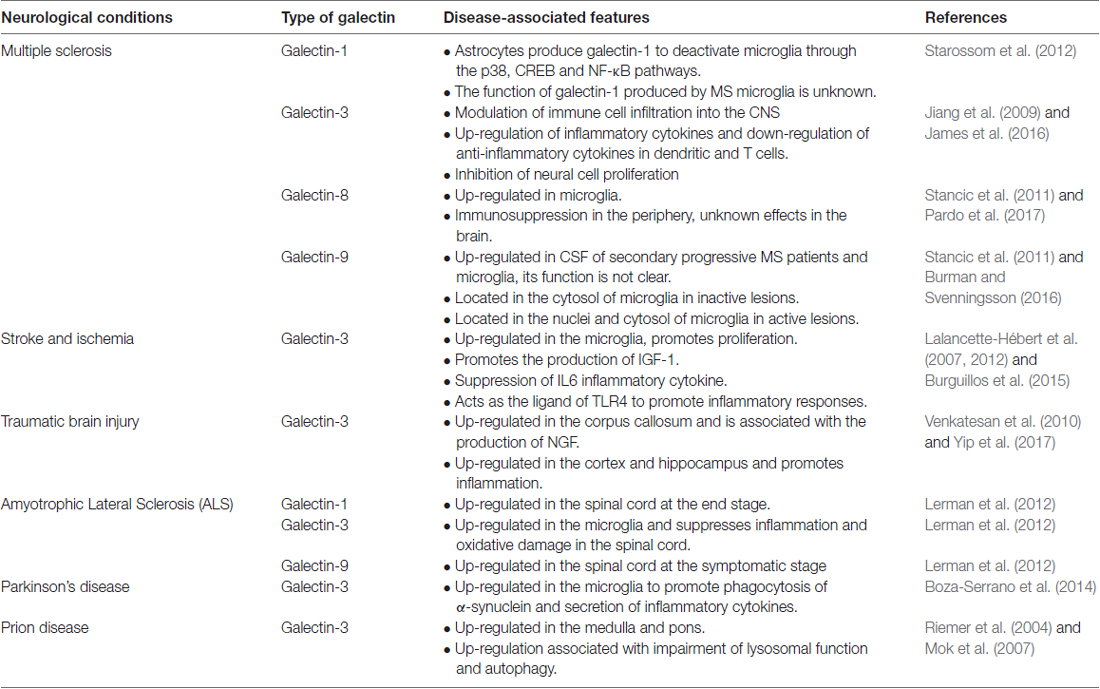
Table 4. Summary of the role of galectins in neurological disease.
Galectins and Myelination
During the maturation of neurons, axons are myelinated in a specific manner that leaves certain parts of the segments unmyelinated; these unmyelinated nodes are marked by contactin-1 (Çolakoğlu et al., 2014; Figure 1). Beginning in development, the expression of galectin-4 is different in each cortical layer and brain region. Galectin-4 is absent in the striatum and cortical layer VI, which are heavily myelinated, while its level remains high in cortical layers II to III, which contain neurons with less myelination at the mature stage. In particular, galectin-4 is found to cover these unmyelinated segments, colocalize with contactin-1 and inhibit oligodendrocytes from depositing myelin on these segments (Díez-Revuelta et al., 2017). Additionally, galectin-4 is secreted by neurons and inhibits the maturation of oligodendrocytes (Stancic et al., 2012). In contrast, astrocytes and microglia are involved in regulating and supporting myelination in different manners. In particular, astrocytes promote the proliferation of oligodendrocytes, while microglia induce the differentiation of oligodendrocytes, as demonstrated by culturing oligodendrocytes with conditioned medium from the two cell types (Pang et al., 2013). In addition, galectin-1, which is secreted mainly by astrocytes, shift microglia from an inflammatory to an anti-inflammatory status, which subsequently drives oligodendrocytes toward the myelination process (Starossom et al., 2012; Miron et al., 2013; Sirko et al., 2015). In parallel with these findings, galectin-3 controls the integrity of myelin in vivo, as demonstrated by the loosely wrapped myelin structures in galectin-3-depleted mice (Pasquini et al., 2011). Furthermore, in the cuprizone-induced demyelination mouse model, the level of galectin-3 is increased in microglia, which promotes the differentiation of oligodendrocytes for remyelination. The abovementioned machinery is impaired in galectin-3-depleted mice (Hoyos et al., 2014, 2016).
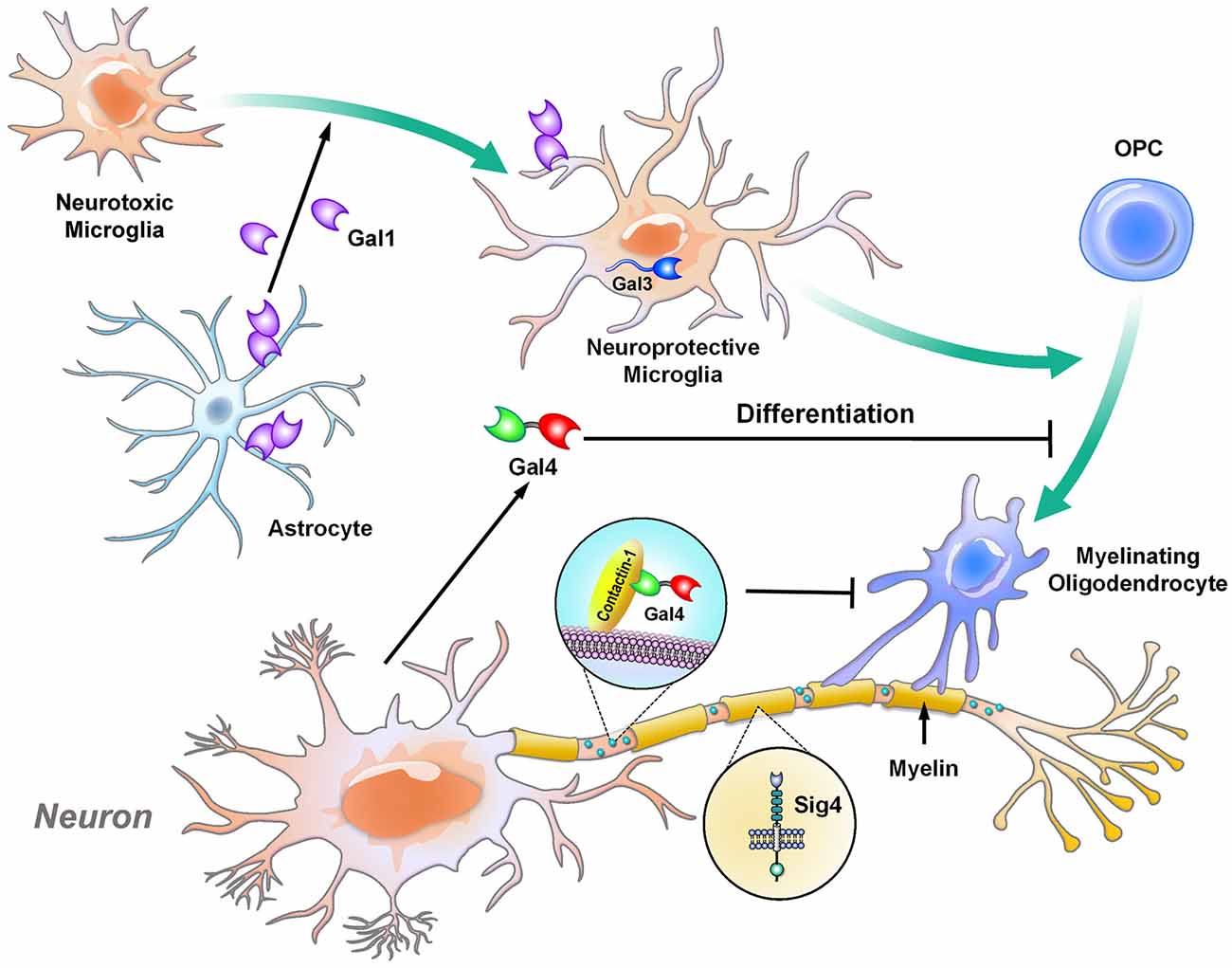
Figure 1. Galectins and Siglec-4 contribute to the glia-mediated regulation of neuronal myelination. Oligodendrocytes are the cells responsible for myelination in the central nervous system (CNS). Under normal conditions, galectin-4 is delivered to the unmyelinated axonal regions and co-localizes with contactin-1 (a marker for nodes of Ranvier). The presence of galectin-4 inhibits oligodendrocytes from myelinating the region (Çolakoğlu et al., 2014). In addition, soluble galectin-4 inhibits the maturation of oligodendrocytes (Stancic et al., 2012). Siglec-4 is produced by oligodendrocytes and is expressed in the myelin sheath (Sun et al., 2004; Huang et al., 2012). Astrocytes and microglia promote the proliferation and differentiation, respectively, of oligodendrocytes. In particular, astrocytes secrete galectin-1 to drive microglia toward neuroprotective phenotype, while these microglia trigger the differentiation of oligodendrocytes in a galectin-3-dependent manner (Hoyos et al., 2014, 2016). Gal, galectin; Sig, Siglec.
Galectins in Multiple Sclerosis (MS)
MS is a chronic inflammatory and autoimmune neurological disease marked by damage to the myelin that surrounds the axons in the CNS (Goldenberg, 2012). The pathogenesis of MS can be divided into two processes: (1) the overactivated immune system damages the myelin sheath and axon; and (2) remyelination is impaired (Dendrou et al., 2015). Both microglia and macrophages are chronically activated throughout the progression of MS and contribute to the inflammatory and demyelination processes (Fischer et al., 2012). Importantly, galectins-1, -3, -8 and -9 are reported to be up-regulated in human MS white matter samples. Immunohistochemical staining reveals that galectin-1 is up-regulated in microglia and macrophages, while its quantity is decreased in the astrocytes, at the lesioned area. The reduction of astrocytic galectin-1 may be due to the enhanced release of galectin-1 by astrocytes and may play an important role in MS progression. Conversely, the levels of galectins-8 and -9 are increased in microglia and macrophages located in active MS lesions. Interestingly, galectin-9 alters cellular localization in MS microglia upon activation. In inactive lesion, galectin-9 is located in the cytoplasmic region of microglia, while it is present in both the nuclei and the cytosol of microglia in active lesions (Stancic et al., 2011). The functional relevance of this nuclear localization of galectin-9 remains elusive and requires further investigation.
To date, the most widely used animal model for the study of MS pathogenesis has been the experimental autoimmune encephalomyelitis (EAE) model (Steinman and Zamvil, 2006). In the EAE model, the expression of galectin-1 is up-regulated, with the highest level at the peak of the disease. Further analysis shows that galectin-1 is highly expressed in astrocytes (GFAP+ cells) and moderately expressed in a subset of T cells (CD4+ cells) and microglia/macrophages (CD11b+ cells) during the acute phase. Interestingly, astrocytes, but not other cell types, continue to express galectin-1 during the chronic phase. One of the specific binding targets of the secreted galectin-1 consists of the core 2 O-glycans of CD45 on the cell surface of microglia. Treatment of the pro-inflammatory microglia with galectin-1 inhibits the activation of the p38, CREB and NF-κB pathways (Figure 2). In the absence of galectin-1, MS progresses much more rapidly. More microglia with inflammatory phenotype and much more severe demyelination can be observed in the galectin-1 depleted EAE model (Starossom et al., 2012). These studies suggest a beneficial role of galectin-1 in the EAE model.
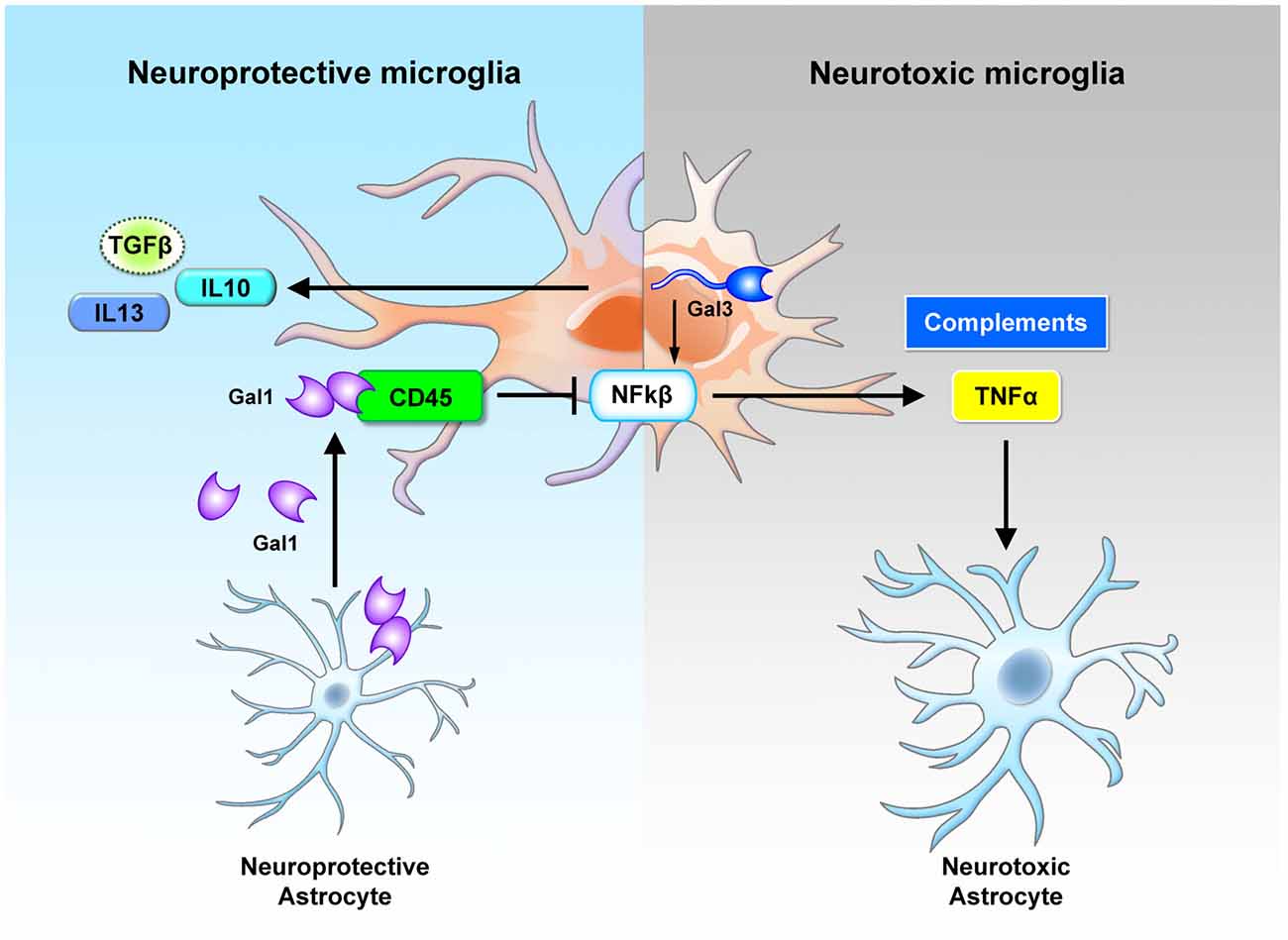
Figure 2. Cross-regulation between astrocytes and microglia by galectin-1 and galectin-3. Neuroprotective astrocytes secrete galectin-1 to inhibit the neurotoxic activation of microglia. The secreted galectin-1 binds with CD45 on the microglia surface, suppresses NF-κB activation and promotes the polarization of neuroprotective microglia (Starossom et al., 2012). Neurotoxic microglia express various types of lectins. In particular, the up-regulated galectin-3 promotes inflammation response through NF-κB activation (Boza-Serrano et al., 2014). These microglial cells secrete inflammatory cytokines (TNFα) and complements that trigger the activation of neurotoxic astrocytes (Liddelow et al., 2017). Gal, galectin; Sig, Siglec.
The level of galectin-3 is also up-regulated in the phagocytosing microglia in areas of demyelination in the EAE model (Reichert and Rotshenker, 1999; Stancic et al., 2011). Further studies suggested that the onset and severity of EAE were markedly reduced in galectin-3 KO mice (Jiang et al., 2009). In a demyelination experimental model of MS, generated by infection with Theiler’s murine encephalomyelitis virus (TMEV), the level of galectin-3 is also up-regulated in microglia residing in the subventricular zone (the neurogenetic niche that is markedly inflamed in MS and its murine models) and the cerebral cortex. In this model, genetic removal of galectin-3 greatly normalizes the immune response observed in the SVZ, reduces the migration of immune cells into the SVZ, and increases the number of progenitors in the corpus callosum (James et al., 2016). These lines of evidence suggest that galectin-3 may play an essential role in MS.
Galectin-8 can be detected in the cerebrospinal fluid (CSF) of MS patients. Genetic removal of galectin-8 hastens the onset of the disease and worsens the progression. Daily treatment with galectin-8 significantly delays the disease progression of EAE. In particular, galectin-8 inhibits the activity of Th17-positive cells to induce inflammation by influencing cells toward apoptosis (Pardo et al., 2017). Similarly, galectin-9 can also be detected in the CSF of secondary progressive MS patients (Burman and Svenningsson, 2016). Compared to the peripheral nervous system, the roles of galectin-8 and galectin-9 in the CNS (such as their expression profiles and their functions in different brain cells) have not been extensively explored and are worth further investigation.
Galectins in Stroke and Ischemia
Galectin-3 is up-regulated in microglia after stroke and induces the expression of IGF-1 (Lalancette-Hébert et al., 2007). Importantly, galectin-3 interacts directly with IGF-1 to promote post-ischemic microglial proliferation. These microglia are suggested to serve as the reservoir of neurotrophic factors, such as IGF-1. Genetic depletion of galectin-3 significantly reduces microglial activation and proliferation; these effects are associated with larger ischemic lesion size, decreased IGF-1 levels and neuronal death. Moreover, there is a twofold increase in IL6 in the ischemic brains of galectin-3-depleted mice (Lalancette-Hébert et al., 2012). These studies suggest that the presence of galectin-3 promotes alternative activation of microglia toward an anti-inflammatory status after stroke. Nonetheless, in a separate study, secreted galectin-3 was shown to act as a ligand for the toll-like receptor 4 (TLR4) and promote inflammatory response in a murine neuroinflammatory LPS model and in the brain of stroke patients. Compared to wild-type controls, galectin-3-knockout (KO) mice have elevated neuronal survival in the hippocampus after ischemia, suggesting that galectin-3 prolongs microglia at the pro-inflammatory status (Burguillos et al., 2015). Consistent with a detrimental role, galectin-3 is found to be up-regulated in the CA1 and CA2 hippocampal regions following transient ischemia. The expression of galectin-3 in these regions is associated with neuronal death and can be prevented by intra-ischemic hypothermia treatment (Satoh et al., 2011; Hisamatsu et al., 2016). Moreover, in the permanent middle cerebral artery occlusion (pMCAO) model, the lack of galectin-3 rescues significant neuronal loss. Treatment with sera collected from pMCAO-treated WT mice also induces increased death of enteric neurons compared with sera collected from pMCAO-treated galectin-3 KO mice. The death of these neurons can be prevented by inhibition of TAK1 and AMPK (transforming growth factor and beta-activated kinase 1 and AMP activated kinase, respectively; Cheng et al., 2016). Collectively, the inflammatory roles of galectin-3 are diverse and may depend on the following factors: the type of ischemic insult, the stage of disease/trauma and the subcellular localization.
Galectins in Traumatic Brain Injury (TBI)
Traumatic Brain Injury (TBI) is clinically categorized into three groups, namely, mild, moderate and severe. Its effects can be further divided into primary and secondary injuries. Primary injuries (such as direct damage to the brain parenchyma that may shear axons) are usually irreversible. On the other hand, secondary injuries are the detrimental effects resulting from the primary injury (such as inflammation and ischemia; Blennow et al., 2016). In response to TBI, in order to restore the normal brain environment, microglia rapidly migrate to the site of injury to remove damaged and dead cells. In a mouse closed-skull model of TBI, the level of galectin-3 increased in the corpus callosum for about 1 month following injury, with the peak at 24 h after injury. The galectin-3- positive microglia engulf damaged axons and produce NGF that facilitates the repair of damaged neurons (Venkatesan et al., 2010). In another study using the controlled cortical impact (CCI) model of head injury, galectin-3 is also up-regulated in the microglia but binds with TLR4 to promote inflammation in the cortex and hippocampus. Treatment with a galectin-3-neutralizing antibody reduces the expression of pro-inflammatory genes IL1β, IL6 and NOS2 while promoting the expression of Ym1, Arg1 and TGFβ. Consistent with a damaging role of galectin-3, galectin-3-deficient mice show less neuronal death and microglial activation in a CCI model (Yip et al., 2017). These data suggest that the role of microglial galectin-3 in TBI can be complex depending on the severity and type of trauma.
Galectins in Amyotrophic Lateral Sclerosis (ALS)
ALS is a rare, progressive and fatal neurodegenerative disease. Multiple genes have been identified as being associated with familial ALS, but those genes explain only approximately 10% of cases. Nearly 90% of ALS cases are sporadic (Abhinav et al., 2007). Up-regulation of plasma galectin-3 is detected in ALS patients with limb onset or disease duration longer than 12 months. The level of plasma galectin-3 is correlated with the duration of disease. Female patients exhibit higher increase of galectin-3 in the plasma than male patients, although the reason is still unknown (Yan et al., 2016). A study using the B6SJL SOD1(G93A) transgenic mouse model of familial ALS (Smittkamp et al., 2008) revealed that microglia undergo a transition stage from the protective phenotype in early-stage ALS to the detrimental phenotype in end-stage ALS. Furthermore, early-stage microglia promote the survival of motor neurons in a co-culture system, while end-stage microglia trigger neuronal death (Liao et al., 2012).
In particular, galectin-1 is increased in the spinal cord of SOD1 mice, while galectins-3 and -9 are increased in SOD1 mice and sporadic ALS patients. The alterations in these galectins during ALS progression are different. Specifically, the amount of galectin-3 can be detected in microglia of spinal cord at the presymptomatic stage, and its level continues to increase until the end stage. Conversely, the expression of galectin-9 is increased at the symptomatic stage, while the levels of galectin-1 is up-regulated only at the end stage (Lerman et al., 2012). The types of cells that contribute to the higher levels of galectin-1 and -9 in the spinal cord are currently unknown.
Nonetheless, it is of great interest to observe that treatment of SOD1 mice with recombinant galectin-1 significantly enhances the survival of motor neurons, delays the onset of disease, improves motor performance, and prolongs the lifespan of ALS mice. Because galectin-1 treatment has no specific effect on isolated primary neurons, the therapeutic effect of galectin-1 may be mediated by non-neuronal cells (Chang-Hong et al., 2005). In particular, microglia and macrophages are among the target cells of galectin-1. For example, treatment with galectin-1 induces macrophages to produce an axonal regeneration-promoting factor that is critical to the survival of degenerating motor neurons (Horie et al., 2004). Overall, although the underlying mechanism remain to be clarified, galectin-1 is a potential therapeutic target for ALS.
Another interesting drug target for ALS is galectin-3. Genetic depletion of galectin-3 in SOD1 mice accelerates disease progression (i.e., a shorter life span and faster motor function impairment) with a significant increase in microglial activation, along with higher inflammatory response and oxidative damage (Lerman et al., 2012). In the CNS of healthy rats, the basal expression of galectin-3 is relatively higher in the spinal cord than in the cortex. Flow cytometry analysis detects at least two major populations of microglia (namely, galectin-3- positive and negative) in the spinal cord of SOD1(G93A) rats after disease onset. Compared to microglia in wild-type rats, microglia in the spinal cord of SOD1(G93A) rats express significantly higher levels of galectin-3 from early to late stage of the disease. The levels of both neurotoxic and neuroprotective molecules (i.e., TNFα, IL6, arginase-1, IL10 and BDNF) in the late stage of the disease are lower, compared with in the early stage. Intriguingly, these alterations only appeared in the spinal cord but not in the cortex, suggesting that the function of galectin-3 and the features of microglia are different in the spinal cord and cortex (Nikodemova et al., 2014). Collectively, these findings suggest that microglial galectin-3 plays a protective role in ALS, which appears different from most of the neurodegenerative diseases where the role of galectin-3 has been investigated so far; further investigation is required.
Galectin-3 in Parkinson’s Disease (PD)
The pathology of PD is characterized by the degeneration of dopaminergic neurons in the substantia nigra and widespread Lewy bodies that contain α-synuclein (α-Syn) aggregates. Accumulated evidence suggest that neurons can release α-Syn to neighboring astrocytes and microglia (Kim et al., 2013). Microglia would take up α-Syn either by phagocytosis or endocytosis. Treatment of a BV2 microglial cell line with different forms of α-Syn (monomer and aggregates) promotes microglial activation as assessed by the levels of iNOS and pro-inflammatory cytokines (TNFα, IL2 and IL12). Importantly, knockdown of galectin-3 or treatment with a galectin-3 inhibitor abolishes the effect of α-Syn on the release of iNOS and pro-inflammatory cytokines. Inhibition of galectin-3 reduces the phagocytic capability of microglia to take up exogenous α-Syn, while treatment with recombinant galectin-3 promotes phagocytosis. Injection of oligomeric forms of α-Syn into the olfactory bulb also significantly enhances the expression of galectin-3 and the phagocytotic activity of microglia. These findings suggest that exposure of microglia to α-Syn causes up-regulation of galectin-3 and subsequently enhances phagocytosis (Boza-Serrano et al., 2014).
The glycan binding targets of galectin (i.e., β-galactosides) are located in the plasma membrane outer leaflet and the luminal side of the intracellular vesicles. Cytosolic galectins (e.g., galectin-3) therefore have no access to β-galactosides. In a neuronal model (SY5Y cells), treatments with α-Syn trigger the formation of galectin-3 puncta because the endocytosed α-Syn cannot be effectively degraded and triggers cellular stress (e.g., elevated ROS), leading to vesicle rupture (Flavin et al., 2017). Exposure of surface glycans from the ruptured vesicular membranes to the cytoplasm recruits galectin-3, which forms galectin-3 puncta (Freeman et al., 2013; Flavin et al., 2017). Such endosomal and lysosomal ruptures not only expose the internal compartment of the vesicles but also release into the cytoplasm substances (e.g., cathepsin B) that need to be confined inside the vesicle, potentially inducing the mitochondrial dysfunction and inflammation that are the common features of PD. It is likely that cell-to-cell transmission of cytosolic protein aggregates (e.g., α-Syn) may induce cellular stress that causes vesicle rupture and the formation of galectin-3 puncta as illustrated in Figure 3. Similar to galectin-8 (Thurston et al., 2012), galectin-3 may serve as a cellular danger receptor that senses detrimental conditions during neuronal degeneration. Further investigation is required to determine whether the formation of galectin-3 puncta at these ruptured vesicles affects cellular functions.
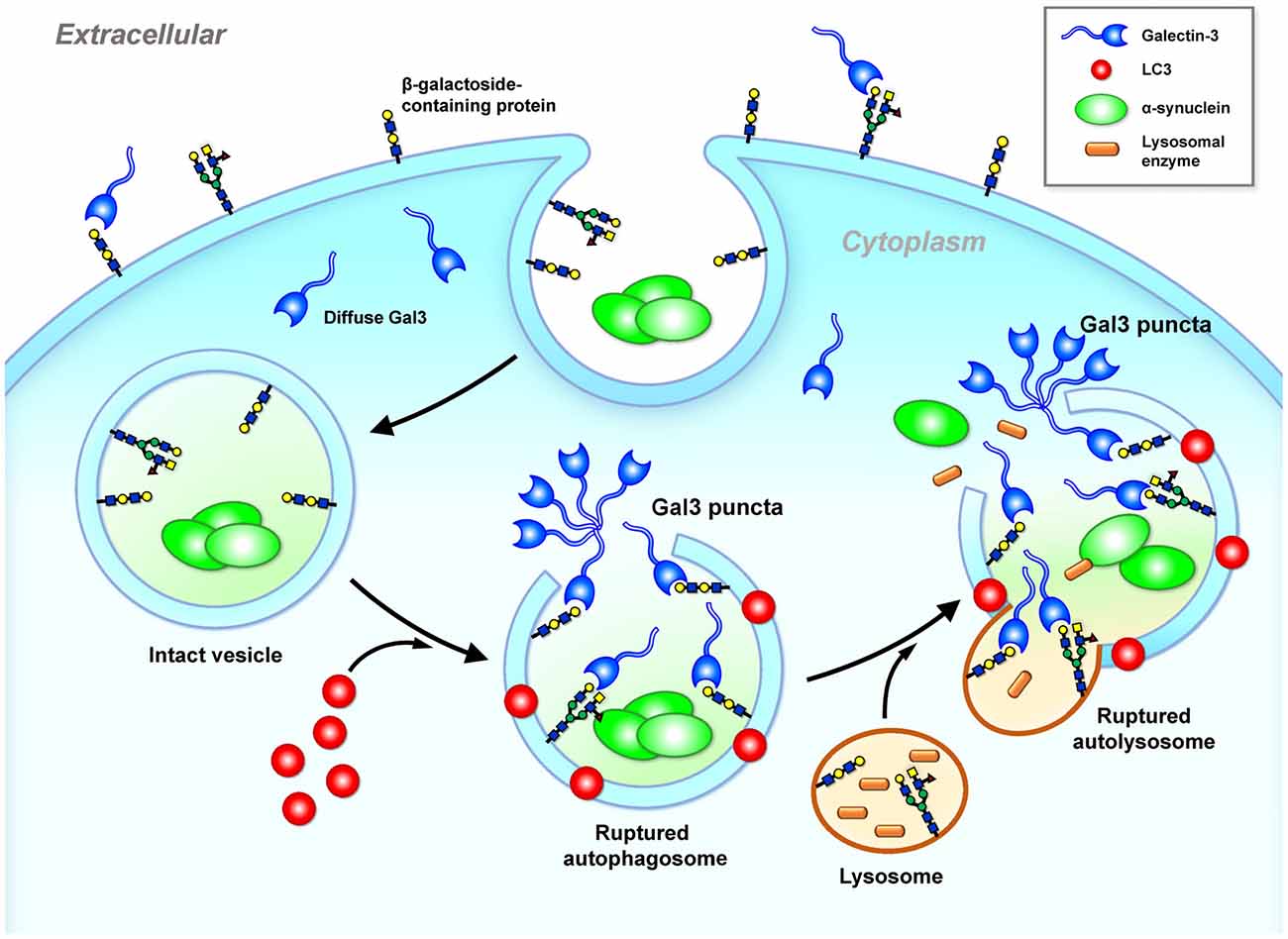
Figure 3. Galectin-3 is a sensor of ruptured intracellular vesicles. Galectin-3 recognizes and binds to β-galactosides. β-galactosides containing proteins are located in the plasma membrane outer leaflet and the luminal side of intact intracellular vesicles (e.g., lysosome, autophagosome and autolysosome). Once these vesicles leak or rupture under stress, galectin-3 gains access and binds to β-galactosides located inside of ruptured vesicles and forms puncta (Freeman et al., 2013; Flavin et al., 2017). Gal3, galectin-3; α-Syn, α-synuclein.
Galectin-3 in Prion Diseases
Prion diseases consist of a group of neurodegenerative disorders that are caused by misfolded proteins called prions. Prions are infectious and the disease can occur in a sporadic, genetic or acquired manner. Genetic prion disease is caused by the mutation of prion-related protein gene (PRPN; Geschwind, 2015). In the scrapie-infected mouse model of prion disease, gene array analysis of the brain reveals 114 altered mRNA levels, and many of the identified genes were related to immune and stress responses. Among the genes with altered expression, the mRNA of galectin-3 is significantly up-regulated in the medulla and pons (Riemer et al., 2004). In a separate study, the protein level of galectin-3 was also found to be increased in scrapie-infected brains. Because galectin-3 KO mice with scrapie infection survive longer than the scrapie-infected wild-type mice and because the levels of a lysosomal activation marker (LAMP-2) and autophagy-related proteins (i.e., Beclin-1 and Atg5) are significantly decreased in the scrapie-infected galectin-3 KO mice, it is possible that galectin-3 may play a detrimental role by damaging the integrity of lysosomes and suppressing autophagy in prion disease (Mok et al., 2007).
Siglecs
Siglecs (sialic acid-binding immunoglobulin-like lectins) represent the best-characterized subgroup of I-type lectins, which contain the immunoglobulin domain that recognizes structural diversity of carbohydrates (Crocker et al., 2007). In general, Siglecs are expressed on plasma membrane, containing an amino terminal V-set immunoglobulin domain that binds sialic acid and a number of C2-set immunoglobulin domains. As their full name suggests, Siglecs recognize sialic acid residues (best described as N-acetyl-neuraminic acids, Neu5Ac) that are presented on glycoproteins and glycolipids with distinct yet overlapping specificities. The presence of C-2 α-hydroxyl group at the terminal of sialic acid enables covalent linkage to other hydroxyl groups on the C-3 and C-6 positions to form the α2–3 and α2–6 linkages, respectively. Enzymes that are responsible for generating such linkages are named sialyltransferases. There are multiples types of sialyltransferases that produce a variety of linkages that can be recognized by Siglecs with different specificities.
Currently, up to 16 Siglecs have been identified and can be categorized based on their sequence similarity, conservation among murine and human and the regulatory properties (i.e., inhibitory or activatory; for review, see Pillai et al., 2012; Macauley et al., 2014). In brief, Siglec-1 (sialoadhesin), Siglec-2 (CD22), Siglec-4 (myelin-associated glycoprotein, MAG) and Siglec-15 are conserved across mammals. Conversely, a variety of Siglecs-3 (CD33)- related Siglecs have been found across mammals. The human CD33-related Siglecs include Siglecs-3, -5, -6, -7, -8, -9, -10, -11, -12, -14 and -16, while the murine CD33-related Siglecs consist of Siglecs-3, -E, -F, -G and -H (Macauley et al., 2014). Most Siglecs (excluding Siglec-1 and MAG) contain immunoreceptor tyrosine-based inhibitory motifs (ITIMs) at the cytoplasmic regions to recruit SH2 domain-containing protein tyrosine phosphatase SHP1 and SHP2; these Siglecs are classified as inhibitory Siglecs. On the other hand, Siglecs-14, -15 and -16 contain the immunoreceptor tyrosine-based activation motifs (ITAMs) on the adaptor of DAP12 to recruit spleen tyrosine kinase (SYK); these Siglecs are classified as activatory Siglecs.
Siglecs are expressed specifically on the cells from hematopoietic lineage, with the exceptions of MAG on oligodendrocytes and Siglec-6 on placental trophoblasts (Macauley et al., 2014). Knowledge of Siglecs is collected mainly from the peripheral system, while Siglecs that are reported to exist in the CNS include sialoadhesin, CD33, MAG, Siglec-E, Siglec-F, Siglec-H, Siglec-11 and Siglec-16. Siglecs are generally absent or scarce under normal physiological condition and are induced to express by various stimuli that activate microglia. These Siglecs, with exception of MAG that is located in oligodendrocytes, are expressed on microglia in the brain. Table 5 summarizes the role of Siglecs in neurological disorders.
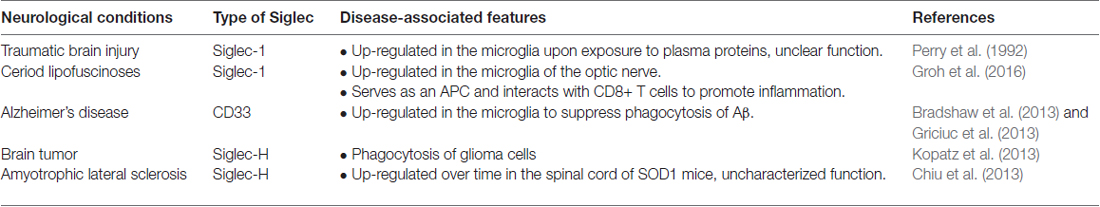
Table 5. Summary of the role of Siglecs in neurological disease.
Siglec-1 (Sialoadhesin, CD169) During the Breakdown of the BBB
Siglec-1 does not have the ITIM or ITAM at its cytoplasmic regions but contains a long immunoglobulin domain that allows the binding domain to reach a distal site from the membrane. Therefore, Siglec-1 is thought to be important for cell-cell adhesion and cell-pathogen recognition (Crocker et al., 1994). Siglec-1 is present in peripheral blood mononuclear phagocytes. Nevertheless, its expression in microglia is rather controversial and may be due to the exposure to plasma proteins upon the breakdown of the blood-brain barrier (BBB) during brain trauma (e.g., TBI model, Perry et al., 1992). Similarly, during retinal degeneration, activated microglia may express Siglec-1 after the breakdown of the blood-retinal barrier (Hughes et al., 2003). Whether these Siglec-1-expressing microglia are originated in the brain or blood-derived cells (i.e., macrophages) that migrate into the brain and their functions are not clear at this time.
In ceroid lipofuscinosis (CLN), a disease that is characterized by lysosomal storage dysfunction, neurodegeneration and early death, the expression of Siglec-1 is significantly up-regulated. These Siglec-1-expressing microglia serve as antigen-presenting cells (APCs) to interact with CD8+ T cells and promote the pathology of the disease. In Siglec-1-deficient CLN mice, microglia express significantly lower inflammatory cytokines (e.g., IL1β and TNFα) and higher level of anti-inflammatory cytokines (e.g., TGFβ). In addition, Siglec-1-deficient CLN mice exhibit reduced axonal degeneration and longer lifespan, supporting a harmful role of Siglec-1 (Groh et al., 2016). Hence, antagonizing the function of Siglec-1 appears as a potential therapeutic approach during the breakdown of the BBB from disease or injury.
Siglec-3 (CD33) in AD
AD is a chronic neurodegenerative disorder that affects up to 30% of the population over 65 years old (Masters et al., 2015). The pathology of the disease is characterized by intracellular neurofibrillary tangles and failure to clear extracellular amyloid-β (Aβ) peptides from the brain. Most patients with AD have the sporadic form, while a small portion of patients carry genes that cause ineffective clearing of Aβ. Recent genome-wide association studies have identified CD33 as a genetic risk factor of AD (Hollingworth et al., 2011; Naj et al., 2011). The risk allele rs3865444 is associated with higher expression of CD33 on the cell surface without affecting the number of microglial cells. Moreover, these CD33-immunoreactive microglia exhibit a positive correlation with Aβ aggregates and plaque burden in the brain of AD patients (Bradshaw et al., 2013; Griciuc et al., 2013). Further in vitro studies confirmed the presence of CD33 with the risk allele rs3865444, which significantly suppresses the phagocytosis capability of microglia. Interestingly, the levels of CD33 with the minor allele of rs3865444 single nucleotide polymorphism (SNP) are markedly reduced. Similarly, primary microglia derived from CD33 KO mice showed greater uptake of Aβ than the control cells, while the rates of Aβ degradation are unaffected. Moreover, the KO of CD33 in the APP/PS1 mouse model significantly reduces Aβ plaques in the cortex and hippocampus (Griciuc et al., 2013). Therefore, tuning the capability of microglia to take up and degrade Aβ through inhibition of CD33 activity may provide a novel therapeutic intervention for AD. The regulation of microglial phagocytosis by CD33 is summarized in Figure 4.
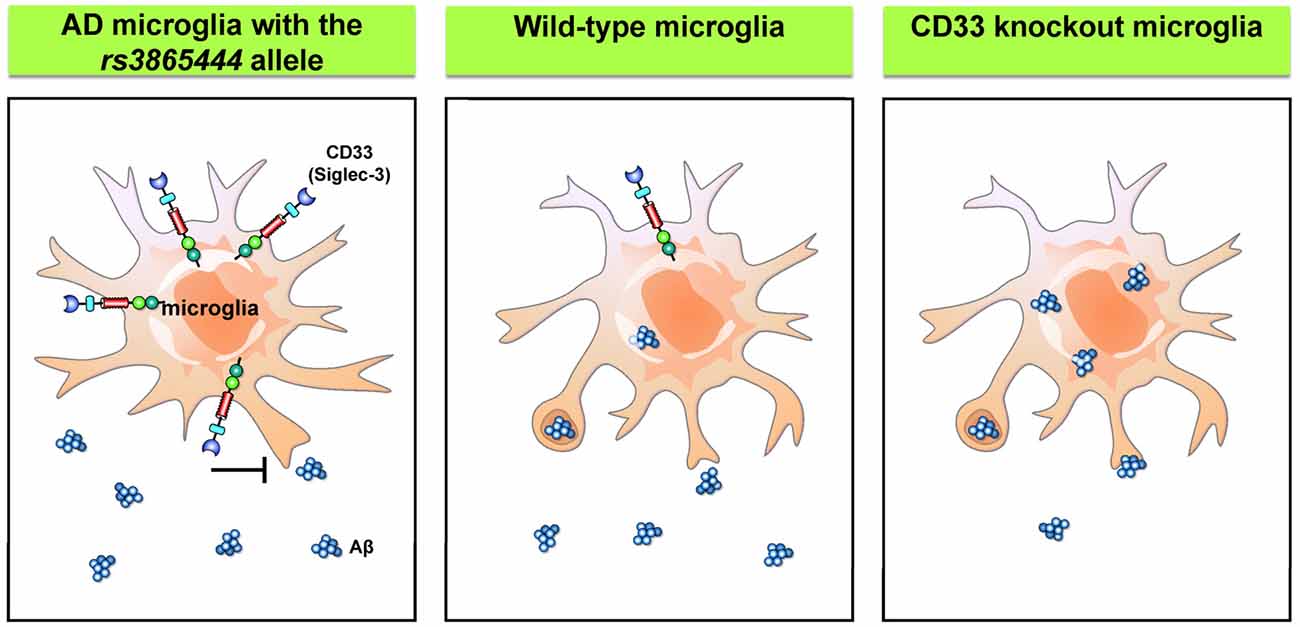
Figure 4. Siglecs regulate microglial phagocytosis. Wild-type microglia express a low level of CD33/Siglec 3, a member of the Siglec family. The risk allele rs3865444 was originally identified in patients with Alzheimer’s disease (AD) and is associated with higher levels of CD33 that suppress the microglial phagocytosis. Conversely, knockdown of CD33 significantly improves the phagocytosis capability of microglia (Hollingworth et al., 2011; Naj et al., 2011; Griciuc et al., 2013). Aβ, amyloid β.
Siglec-H in Brain Tumors
Siglec-H is expressed only in murine microglia and plasmacytoid dendritic cells (pDCs), while they are absent in human monocytes (Zhang et al., 2006; Kopatz et al., 2013). In contrast to most Siglecs, Siglec-H is an activatory Siglec with complex functions. Siglec-H suppresses the downstream signaling of TLR9 to produce interferon-α in pDCs (Takagi et al., 2011; Puttur et al., 2013). Although Siglec-H is known to be expressed in microglia, its ligands and functions are only now being discovered, as it does not bind to the typical sialic acid-containing glycoproteins or glycolipids of healthy cells (Varki and Angata, 2006; Varki, 2009). Treatment of a microglial cell line and primary microglia with interferon-γ significantly enhances inflammatory phenotypes (e.g., up-regulation of iNOS, CXCL10 and TNFα) and up-regulation of Siglec-H. These Siglec-H-enriched microglia exhibit high phagocytosis activity that can be suppressed by down-regulation of Siglec-H. Importantly, Siglec-H binds to and triggers the engulfment of glioma cell lines (namely, GL261 and SMA560) but not normal astrocytes, splenocytes, or fibroblasts. It is possible that microglial Siglec-H may function to monitor the surface sialylation profiles present on tumor cells in the brain (Kopatz et al., 2013). These studies identified the functions of Siglec-H in the brain. However, whether enhancing Siglec-H activity is a potent approach to target brain tumor required further investigation.
Siglec-H in ALS
Microglia share many markers with peripheral monocytes and macrophages. Transcriptome analysis reveals that Siglec-H is the third highest gene (out of 29 identified genes) that can distinguish microglia from monocytes and macrophages. The level of Siglec-H is also up-regulated in the spinal cord of an ALS mouse model (SOD1; Chiu et al., 2013). The function of Siglec-H in ALS remains unclear.
C-Type Lectins
C-type lectins are a group of proteins that bind to glycan in a calcium-dependent manner. The proteins contain a C-type lectin domain (CTLD), which is also present in many proteins that do not recognize or bind to any glycan. C-type lectins and protein with CTLD are found in all organism and can be categorized to at least 17 groups based on their domain architecture, such as selectins, collectins, etc. The classification of C-type lectins and their roles in the peripheral immunity have been reviewed extensively elsewhere (Sancho and Reis e Sousa, 2012; Dambuza and Brown, 2015; Yan et al., 2015). In this review, we focus on a few C-type lectins (Table 6) that have been implicated in neurological disorder.
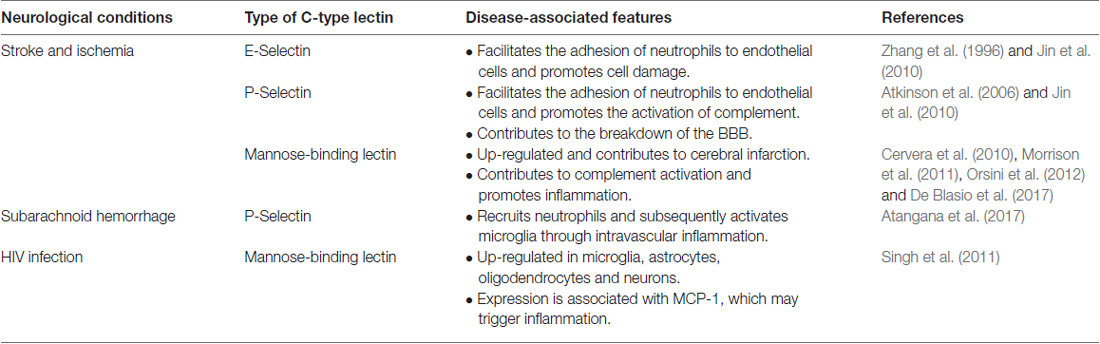
Table 6. Summary of the role of C-type lectins in neurological disease.
Selectins
Selectins are single-chain transmembrane glycoproteins of the cell adhesion molecule family. They are highly homologous proteins, which contain a CTLD, an epidermal growth factor-like domain, a few consensus repeats composed of sushi domain, a transmembrane domain and an intracellular cytoplasmic tail. The CTLD of selectins recognizes sialyl Lewis X antigens (sialylated and fucosylated glycoprotein and glycolipid structures; Ley, 2003; Angiari and Constantin, 2013). Three types of selectins (i.e., E-, L- and P-Selectins) have being identified so far. In the brain, selectins (particularly the E- and P-type) are located in endothelial cells and are responsible for controlling leukocyte infiltration and the BBB permeability during trauma or acute inflammation (Tang et al., 1996; Carvalho-Tavares et al., 2000; Bernardes-Silva et al., 2001). This is an important aspect because accumulation and adhesion of leukocytes to the endothelial cells are the critical steps that cause cell death at the ischemic regions (Kataoka et al., 2004; Langer and Chavakis, 2009).
In the animal model of focal cerebral ischemia and reperfusion injury, E-Selectins are induced and expressed on the endothelial cells (Wang et al., 1995; Zhang et al., 1996). Because E-Selectins bind sialyl Lewis X glycans enriched in neutrophils (Lasky, 1992) and the administration of sialyl Lewis X significantly decreases the recruitment of neutrophils to the ischemic region and decrease the infarct volume (Zhang et al., 1996), E-Selectins appear to be an important component of ischemia pathogenesis. Another intriguing observation is that the polysialylated E-Selectin Ligand-1 (ESL1, a ligand for E-Selectins) was found to be enriched in the Golgi compartments of microglia. Brain injuries translocate ESL1 to the cell surfaces and therefore trigger the binding of E-Selectins with microglia (Werneburg et al., 2016). Binding of E-Selectins to myeloid cells (i.e., neutrophils) has been demonstrated to regulate cell homing to sites of inflammation (Mondal et al., 2016), while the effect of binding of E-Selectins to microglia remains to be determined.
The expression of P-Selectin in endothelial cells is also up-regulated in a complement C3-dependent manner in an ischemia mouse model (Atkinson et al., 2006). Both P-Selectin deficiency and P-Selectin inhibition protect mice from the cerebral injury following ischemia (Zhou et al., 2000; Atkinson et al., 2006). Up-regulation of P-Selectin also contributes to the transient breakdown of BBB following ischemia (Jin et al., 2010). It is interesting to note that the level of P-Selectin is significantly up-regulated in the experimental subarachnoid hemorrhage (eSAH) model as well. The presence of P-Selectins on endothelial cells recruit neutrophils and microglia to the site of injury. It is likely that these microglia are activated by the inflammatory cytokines released by the neutrophils promoted by intravascular inflammation (Schneider et al., 2015; Atangana et al., 2017). In addition, induction of eSAH in mice lacking the P-Selectin glycoprotein ligand-1 (PSGL1; a P-Selectin ligand) results in significant reductions in the endothelial-neutrophil interactions, the number of activated microglia and the extent of neuronal death (Atangana et al., 2017). Collectively, these studies suggest that P-Selectin is likely to play a detrimental role in neurological traumas by facilitating the microglia-mediated neuroinflammation. Figure 5 summarizes the roles of E-selectins and P-selectins in brain ischemia.
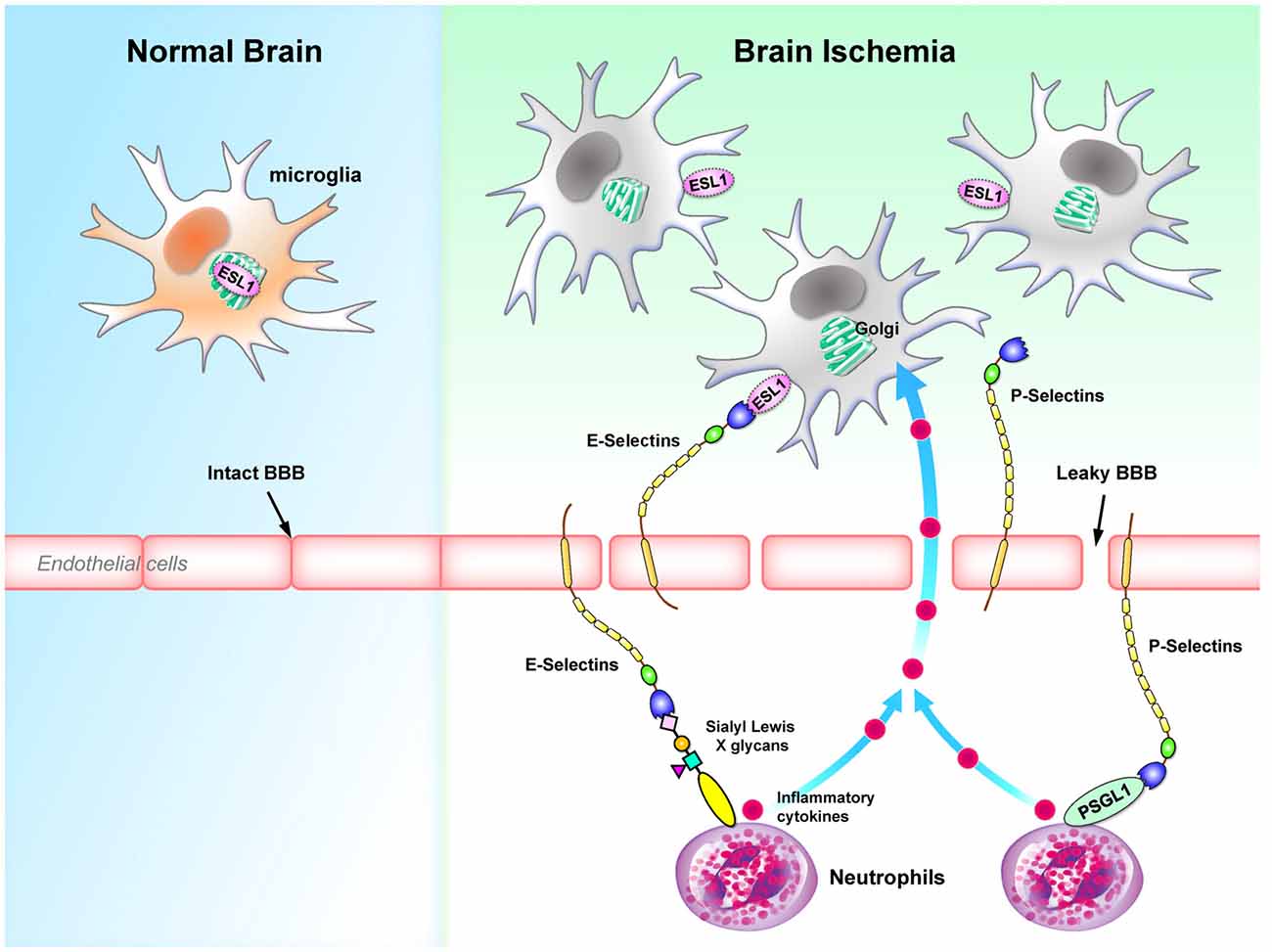
Figure 5. Selectins regulates microglial activation and homing to sites of inflammation during brain ischemia. E-selectins and P-selectins that bind to Sialyl Lewis X glycans are up-regulated in endothelial cells during brain ischemia. Binding of selectins serves as a homing mechanism to retain Sialyl Lewis X glycans-containing cells close to endothelial cells. In particular, ESL1 located in the Golgi compartments of microglia is translocated to the plasma membrane and binds to E-selectins on the surface of endothelial cells (Werneburg et al., 2016). In contrast, P-selectins cause a transient leakage of BBB (Jin et al., 2010). P-selectins bind to PSGL1 on neutrophils, which secrete inflammatory cytokines and further activate microglia in the CNS (Schneider et al., 2015; Atangana et al., 2017). BBB, blood-brain barrier; ESL1, E-selectin ligand-1; PSGL1, P-selectin glycoprotein ligand-1.
Mannose-Binding Lectins (MBLs)
MBLs, also called mannan-binding lectins, are from the collectin class of the C-type lectin family and are important components of the innate immune system. These proteins contain a cysteine-rich N-terminal region, a collagenous domain, a short α-helical coiled-coil domain and a CRD at the C-terminal. It is important to note that CRDs of MBL recognize glycans on the surface of pathogens as well as cryptic self-antigens that are modified in disease conditions or injuries (Ip et al., 2009). They exist in oligomeric forms (i.e., from dimer to hexamer) and recognizes mannose, fucose and N-acetyl-glucosamine (GlcNAc), but not sialic acid and galactose (Drickamer, 1992; Weis et al., 1992; Yokota et al., 1995). MBLs participate in a range of biological functions including the recognition of pathogen invasion, activation of complement system, phagocytosis and mediation of inflammatory signaling (Ip et al., 2009; Auriti et al., 2017).
Activation of complement system is one of the main causes for brain damage during ischemia and stroke. MBL has been implicated to participate in the activation of complement system via the lectin pathway (Morrison et al., 2011). MBL-KO mice are protected from transient and permanent ischemic injuries with the reduction of infarct volume and sensorimotor impairments. Blocking MBL using an anti-MBL-A antibody also produce similar results (Orsini et al., 2012; Longhi et al., 2014). Consistently, ischemic stroke patients, who have MBL-low genotypes and lower MBL in their serum, are associated with a favorable stroke outcome and decreased levels of proinflammatory cytokines (e.g., TNFα and IL6). These patients with lower MBL levels (<100 ng/ml) are likely to recover from movement disabilities in less than 3 months (Cervera et al., 2010; Osthoff et al., 2011). Collectively, effective inhibition of MBL may reduce the damage caused by ischemic stroke.
In patients infected by HIV, viruses can enter the CNS and continue to replicate within the brain. Infection of HIV in the brain therefore causes a chronic inflammatory response and a spectrum of neurological dysfunction (Saylor et al., 2016). By studying post-mortem brain tissues, it was shown that the expression of MBL is up-regulated in the HIV- infected brains. Interestingly, the expression of MBL is detected in all brain cells, including microglia, astrocytes, oligodendrocytes and neurons. In particular, axons in the HIV-infected brain show high levels of MBL, while the level of MBL is associated with the level of monocytes chemoattractant protein-1 (MCP-1 or CCL2; Singh et al., 2011). MCP-1 is the key chemokine that triggers the migration of microglia or macrophages (Deshmane et al., 2009; Hinojosa et al., 2011). Hence, the expression of MBL is suggested to be associated with the activation of complement and neuroinflammation, which subsequently cause neuronal damage in the HIV-infected brain.
Conclusion and Future Perspectives
Accumulated evidence suggest that dysregulation of lectins is a critical pathogenetic factor in neurological diseases. The altered expressions of lectins on microglia affect the phenotypes of microglia (Yip et al., 2017). Importantly, the aberrant expression of lectins on the surrounding cells can also trigger the activation of microglia (Atangana et al., 2017). These may independently or synergistically lead to the chronic pathogenic inflammatory response that causes neuronal dysfunctions or death. Likewise, these activated microglia may also secrete inflammatory cytokines and complement that will influence the activity of astrocytes to produce neurotoxic factors (Liddelow et al., 2017).
One major area that still requires clarification is the specific type of cells that express these lectins. Many lectins are not expressed in the healthy CNS, nevertheless are induced to express or up-regulated in disease condition. It is crucial to identify the specific types of cells that show aberrant expression of lectins and their functional effect on the cells themselves or the surrounding cells. For instance, galectin-3 is expressed virtually on activated microglia in various neurological disorders in vivo, while it is expressed in various cell lines derived from brain glial cells in vitro (Pasquini et al., 2011). These studies raise the questions of why cell lines express galectin-3 even under normal conditions. There is a possibility that galectin-3 is involved in the immortalization and proliferation of cell lines, as it does in tumor cells (i.e., pancreatic cancer cells, thyroid carcinoma cells; Song et al., 2012; Cardoso et al., 2016). At the same time, it is also of interest to investigate whether other glial cells (astrocytes and oligodendrocytes) in abnormal conditions also express galectin-3 in vivo. Meanwhile, Galectin-4 is expressed in non-myelinated region of axons and Siglec-4 is expressed specifically on oligodendrocytes to regulate myelination (Figure 1). Contrary to Galectins that can be expressed in various types of cells, all CD33-related Siglecs are expressed specifically on myeloid- origin cells, namely, microglia in the CNS. Selectins are mainly on endothelial cells and their expressions directly affects the integrity of BBB and the activity of nearby microglia. MBL has been reported to increase in stroke and ischemia, while the sources of MBL are elusive. It is unclear whether MBLs are produced by brain cells or migrate from the peripheral nervous system due to the leakage of the BBB. Interestingly, in HIV-infected brain, all brain cells show positive signals for MBL, indicating brain cells are capable of producing MBL upon stimulation. Further understandings on how lectins affect microglia and its interaction with other brain cells certainly would broaden our view on neurodegenerative diseases and raise new questions as well. Are these lectins druggable targets for neurological disease? Would pharmacological inhibition or activation of these lectins be effective in treating neurological diseases? Notably, these inhibitors/activators also need to have the capacity to cross the BBB to treat brain disorders.
The concept of lectin-based therapeutics deploys the specific targets of lectins or the delivery of drugs that interacts with lectins to the target site. For example, Galectin-3 has been implicated in numerous neurological disorders and diseases in the peripheral nervous system. Pharmacological suppression of galectin-3 appears to be a promising approach. Specifically, TD139 that binds to galectin-3 and inhibit its functions have passed the clinical phase Ib/IIa trials for treating idiopathic pulmonary fibrosis (http://ClinicalTrials.gov identifier, NCT02257177). Although this development is encouraging, the ability of TD139 to cross the BBB is poor and therefore limits its application to neurological diseases with abnormal up-regulation of Gal3. Further modification of TD139 to increase its permeability across the BBB may pave the way for the development of therapeutic intervention for brain diseases.
On the other hand, lectin-based therapeutics may enhance the exposure of drugs to the targeted cells/sites, and reduce the dosage and side effects. Moreover, internalization of drug also increases the uptake of therapeutic agents with low cellular permeability. The decoration of drug enveloped with surface sialic acid ligands can directly activate Siglecs downstream signaling or deploy their internalization properties. For example, nanoparticles coated with di(α2–8) N-acetylneuraminic acid effectively target Siglec-7 and Siglec-9 to exert anti-inflammatory effects of macrophages in a human ex vivo lung perfusion model (Spence et al., 2015). Furthermore, liposomal nanoparticles that are coated with high-affinity CD22 ligands result in substantial uptake of the nanoparticles by B cell lymphomas that express the endocytic receptor CD22 (Chen et al., 2012).
Collectively, emerging studies have shown that modulating the lectins activity via direct interference in the lectin itself or the receptor-ligand interaction in CNS is a promising area for further exploration. Currently, treatment of neurological disorders using a lectin-based approach has not been envisaged, likely owing to an incomplete understanding of the roles of lectins in the CNS and the limited ability of drugs to cross the BBB. A greater understanding of microglial lectins may facilitate the development of novel therapeutic interventions for patients with neurological disorders.
Author Contributions
JJS designed and wrote the manuscript. YC designed and edited the manuscript.
Funding
This work was supported by Academia Sinica and Ministry of Science and Technology, Taiwan (MOST 103-2321-B-001068, MOST 106-0210-01-15-02, MOST 107-0210-01-19-01).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the Medical Art Room of Institute of Biomedical Sciences at Academia Sinica for the assistance in preparing the illustration.
References
Abhinav, K., Stanton, B., Johnston, C., Hardstaff, J., Orrell, R. W., Howard, R., et al. (2007). Amyotrophic lateral sclerosis in South-East England: a population-based study. The South-East England register for amyotrophic lateral sclerosis (SEALS Registry). Neuroepidemiology 29, 44–48. doi: 10.1159/000108917
Angiari, S., and Constantin, G. (2013). Selectins and their ligands as potential immunotherapeutic targets in neurological diseases. Immunotherapy 5, 1207–1220. doi: 10.2217/imt.13.122
Atangana, E., Schneider, U. C., Blecharz, K., Magrini, S., Wagner, J., Nieminen-Kelha, M., et al. (2017). Intravascular inflammation triggers intracerebral activated microglia and contributes to secondary brain injury after experimental subarachnoid hemorrhage (eSAH). Transl. Stroke Res. 8, 144–156. doi: 10.1007/s12975-016-0485-3
Atkinson, C., Zhu, H., Qiao, F., Varela, J. C., Yu, J., Song, H., et al. (2006). Complement-dependent P-selectin expression and injury following ischemic stroke. J. Immunol. 177, 7266–7274. doi: 10.4049/jimmunol.177.10.7266
Auriti, C., Prencipe, G., Moriondo, M., Bersani, I., Bertaina, C., Mondì, V., et al. (2017). Mannose-binding lectin: biologic characteristics and role in the susceptibility to infections and ischemia-reperfusion related injury in critically Ill neonates. J. Immunol. Res. 2017:7045630. doi: 10.1155/2017/7045630
Bernardes-Silva, M., Anthony, D. C., Issekutz, A. C., and Perry, V. H. (2001). Recruitment of neutrophils across the blood-brain barrier: the role of E- and P-selectins. J. Cereb. Blood Flow Metab. 21, 1115–1124. doi: 10.1097/00004647-200109000-00009
Bessis, A., Béchade, C., Bernard, D., and Roumier, A. (2007). Microglial control of neuronal death and synaptic properties. Glia 55, 233–238. doi: 10.1002/glia.20459
Blennow, K., Brody, D. L., Kochanek, P. M., Levin, H., McKee, A., Ribbers, G. M., et al. (2016). Traumatic brain injuries. Nat. Rev. Dis. Primers 2:16084. doi: 10.1038/nrdp.2016.84
Boza-Serrano, A., Reyes, J. F., Rey, N. L., Leffler, H., Bousset, L., Nilsson, U., et al. (2014). The role of Galectin-3 in α-synuclein-induced microglial activation. Acta Neuropathol. Commun. 2:156. doi: 10.1186/s40478-014-0156-0
Bradshaw, E. M., Chibnik, L. B., Keenan, B. T., Ottoboni, L., Raj, T., Tang, A., et al. (2013). CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat. Neurosci. 16, 848–850. doi: 10.1038/nn.3435
Brandley, B. K., and Schnaar, R. L. (1986). Cell-surface carbohydrates in cell recognition and response. J. Leukoc. Biol. 40, 97–111. doi: 10.1002/jlb.40.1.97
Brites, D., and Vaz, A. R. (2014). Microglia centered pathogenesis in ALS: insights in cell interconnectivity. Front. Cell. Neurosci. 8:117. doi: 10.3389/fncel.2014.00117
Burguillos, M. A., Svensson, M., Schulte, T., Boza-Serrano, A., Garcia-Quintanilla, A., Kavanagh, E., et al. (2015). Microglia-secreted galectin-3 acts as a toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. 10, 1626–1638. doi: 10.1016/j.celrep.2015.02.012
Burman, J., and Svenningsson, A. (2016). Cerebrospinal fluid concentration of Galectin-9 is increased in secondary progressive multiple sclerosis. J. Neuroimmunol. 292, 40–44. doi: 10.1016/j.jneuroim.2016.01.008
Cao, Z. Q., and Guo, X. L. (2016). The role of galectin-4 in physiology and diseases. Protein Cell 7, 314–324. doi: 10.1007/s13238-016-0262-9
Cardoso, A. C., Andrade, L. N., Bustos, S. O., and Chammas, R. (2016). Galectin-3 determines tumor cell adaptive strategies in stressed tumor microenvironments. Front. Oncol. 6:127. doi: 10.3389/fonc.2016.00127
Carvalho-Tavares, J., Hickey, M. J., Hutchison, J., Michaud, J., Sutcliffe, I. T., and Kubes, P. (2000). A role for platelets and endothelial selectins in tumor necrosis factor-α-induced leukocyte recruitment in the brain microvasculature. Circ. Res. 87, 1141–1148. doi: 10.1161/01.res.87.12.1141
Cenker, J. J., Stultz, R. D., and McDonald, D. (2017). Brain microglial cells are highly susceptible to HIV-1 infection and spread. AIDS Res. Hum. Retroviruses 33, 1155–1165. doi: 10.1089/aid.2017.0004
Cervera, A., Planas, A. M., Justicia, C., Urra, X., Jensenius, J. C., Torres, F., et al. (2010). Genetically-defined deficiency of mannose-binding lectin is associated with protection after experimental stroke in mice and outcome in human stroke. PLoS One 5:e8433. doi: 10.1371/journal.pone.0008433
Chang, W. A., Tsai, M. J., Kuo, P. L., and Hung, J. Y. (2017). Role of galectins in lung cancer. Oncol. Lett. 14, 5077–5084. doi: 10.3892/ol.2017.6882
Chang-Hong, R., Wada, M., Koyama, S., Kimura, H., Arawaka, S., Kawanami, T., et al. (2005). Neuroprotective effect of oxidized galectin-1 in a transgenic mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 194, 203–211. doi: 10.1016/j.expneurol.2005.02.011
Chen, W. C., Sigal, D. S., Saven, A., and Paulson, J. C. (2012). Targeting B lymphoma with nanoparticles bearing glycan ligands of CD22. Leuk. Lymphoma 53, 208–210. doi: 10.3109/10428194.2011.604755
Cheng, X., Boza-Serrano, A., Turesson, M. F., Deierborg, T., Ekblad, E., and Voss, U. (2016). Galectin-3 causes enteric neuronal loss in mice after left sided permanent middle cerebral artery occlusion, a model of stroke. Sci. Rep. 6:32893. doi: 10.1038/srep32893
Chiu, I. M., Morimoto, E. T., Goodarzi, H., Liao, J. T., O’Keeffe, S., Phatnani, H. P., et al. (2013). A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 4, 385–401. doi: 10.1016/j.celrep.2013.06.018
Claude, J., Linnartz-Gerlach, B., Kudin, A. P., Kunz, W. S., and Neumann, H. (2013). Microglial CD33-related Siglec-E inhibits neurotoxicity by preventing the phagocytosis-associated oxidative burst. J. Neurosci. 33, 18270–18276. doi: 10.1523/jneurosci.2211-13.2013
Çolakoğlu, G., Bergstrom-Tyrberg, U., Berglund, E. O., and Ranscht, B. (2014). Contactin-1 regulates myelination and nodal/paranodal domain organization in the central nervous system. Proc. Natl. Acad. Sci. U S A 111, E394–E403. doi: 10.1073/pnas.1313769110
Crocker, P. R., Mucklow, S., Bouckson, V., McWilliam, A., Willis, A. C., Gordon, S., et al. (1994). Sialoadhesin, a macrophage sialic acid binding receptor for haemopoietic cells with 17 immunoglobulin-like domains. EMBO J. 13, 4490–4503.
Crocker, P. R., Paulson, J. C., and Varki, A. (2007). Siglecs and their roles in the immune system. Nat. Rev. Immunol. 7, 255–266. doi: 10.1038/nri2056
Dambuza, I. M., and Brown, G. D. (2015). C-type lectins in immunity: recent developments. Curr. Opin. Immunol. 32, 21–27. doi: 10.1016/j.coi.2014.12.002
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
De Blasio, D., Fumagalli, S., Longhi, L., Orsini, F., Palmioli, A., Stravalaci, M., et al. (2017). Pharmacological inhibition of mannose-binding lectin ameliorates neurobehavioral dysfunction following experimental traumatic brain injury. J. Cereb. Blood Flow Metab. 37, 938–950. doi: 10.1177/0271678x16647397
de la Rosa, E. J., and de Pablo, F. (2000). Cell death in early neural development: beyond the neurotrophic theory. Trends Neurosci. 23, 454–458. doi: 10.1016/s0166-2236(00)01628-3
Dendrou, C. A., Fugger, L., and Friese, M. A. (2015). Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 15, 545–558. doi: 10.1038/nri3871
Deshmane, S. L., Kremlev, S., Amini, S., and Sawaya, B. E. (2009). Monocyte chemoattractant protein-1 (MCP-1): an overview. J. Interferon Cytokine Res. 29, 313–326. doi: 10.1089/jir.2008.0027
Díez-Revuelta, N., Higuero, A. M., Velasco, S., Peñas-de-la-Iglesia, M., Gabius, H. J., and Abad-Rodríguez, J. (2017). Neurons define non-myelinated axon segments by the regulation of galectin-4-containing axon membrane domains. Sci. Rep. 7:12246. doi: 10.1101/115758
Drickamer, K. (1992). Engineering galactose-binding activity into a C-type mannose-binding protein. Nature 360, 183–186. doi: 10.1038/360183a0
Fischer, M. T., Sharma, R., Lim, J. L., Haider, L., Frischer, J. M., Drexhage, J., et al. (2012). NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 135, 886–899. doi: 10.1093/brain/aws012
Flavin, W. P., Bousset, L., Green, Z. C., Chu, Y., Skarpathiotis, S., Chaney, M. J., et al. (2017). Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 134, 629–653. doi: 10.1007/s00401-017-1722-x
Freeman, D., Cedillos, R., Choyke, S., Lukic, Z., McGuire, K., Marvin, S., et al. (2013). α-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One 8:e62143. doi: 10.1371/journal.pone.0062143
Frielingsdorf, H., Simpson, D. R., Thal, L. J., and Pizzo, D. P. (2007). Nerve growth factor promotes survival of new neurons in the adult hippocampus. Neurobiol. Dis. 26, 47–55. doi: 10.1016/j.nbd.2006.11.015
Fuhrmann, M., Bittner, T., Jung, C. K., Burgold, S., Page, R. M., Mitteregger, G., et al. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 13, 411–413. doi: 10.1038/nn.2511
Fujimoto, Z., Tateno, H., and Hirabayashi, J. (2014). Lectin structures: classification based on the 3-D structures. Methods Mol. Biol. 1200, 579–606. doi: 10.1007/978-1-4939-1292-6_46
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., Mullin, K., et al. (2013). Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid β. Neuron 78, 631–643. doi: 10.1016/j.neuron.2013.04.014
Groh, J., Ribechini, E., Stadler, D., Schilling, T., Lutz, M. B., and Martini, R. (2016). Sialoadhesin promotes neuroinflammation-related disease progression in two mouse models of CLN disease. Glia 64, 792–809. doi: 10.1002/glia.22962
Groves, J. T. (2013). Cell membranes: glycans’ imprints. Nat. Mater. 12, 96–97. doi: 10.1038/nmat3555
Hinojosa, A. E., Garcia-Bueno, B., Leza, J. C., and Madrigal, J. L. (2011). CCL2/MCP-1 modulation of microglial activation and proliferation. J. Neuroinflammation 8:77. doi: 10.1186/1742-2094-8-77
Hisamatsu, K., Niwa, M., Kobayashi, K., Miyazaki, T., Hirata, A., Hatano, Y., et al. (2016). Galectin-3 expression in hippocampal CA2 following transient forebrain ischemia and its inhibition by hypothermia or antiapoptotic agents. Neuroreport 27, 311–317. doi: 10.1097/wnr.0000000000000538
Hollingworth, P., Harold, D., Sims, R., Gerrish, A., Lambert, J. C., Carrasquillo, M. M., et al. (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 43, 429–435. doi: 10.1038/ng.803
Horie, H., Kadoya, T., Hikawa, N., Sango, K., Inoue, H., Takeshita, K., et al. (2004). Oxidized galectin-1 stimulates macrophages to promote axonal regeneration in peripheral nerves after axotomy. J. Neurosci. 24, 1873–1880. doi: 10.1523/jneurosci.4483-03.2004
Hoyos, H. C., Marder, M., Ulrich, R., Gudi, V., Stangel, M., Rabinovich, G. A., et al. (2016). The role of galectin-3: from oligodendroglial differentiation and myelination to demyelination and remyelination processes in a cuprizone-induced demyelination model. Adv. Exp. Med. Biol. 949, 311–332. doi: 10.1007/978-3-319-40764-7_15
Hoyos, H. C., Rinaldi, M., Mendez-Huergo, S. P., Marder, M., Rabinovich, G. A., Pasquini, J. M., et al. (2014). Galectin-3 controls the response of microglial cells to limit cuprizone-induced demyelination. Neurobiol. Dis. 62, 441–455. doi: 10.1016/j.nbd.2013.10.023
Hsiao, H. Y., Chiu, F. L., Chen, C. M., Wu, Y. R., Chen, H. M., Chen, Y. C., et al. (2014). Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum. Mol. Genet. 23, 4328–4344. doi: 10.1093/hmg/ddu151
Huang, J. Y., Wang, Y. X., Gu, W. L., Fu, S. L., Li, Y., Huang, L. D., et al. (2012). Expression and function of myelin-associated proteins and their common receptor NgR on oligodendrocyte progenitor cells. Brain Res. 1437, 1–15. doi: 10.1016/j.brainres.2011.12.008
Hughes, E. H., Schlichtenbrede, F. C., Murphy, C. C., Sarra, G. M., Luthert, P. J., Ali, R. R., et al. (2003). Generation of activated sialoadhesin-positive microglia during retinal degeneration. Invest. Ophthalmol. Vis. Sci. 44, 2229–2234. doi: 10.1167/iovs.02-0824
Ip, W. K., Takahashi, K., Ezekowitz, R. A., and Stuart, L. M. (2009). Mannose-binding lectin and innate immunity. Immunol. Rev. 230, 9–21. doi: 10.1111/j.1600-065X.2009.00789.x
James, R. E., Hillis, J., Adorján, I., Gration, B., Mundim, M. V., Iqbal, A. J., et al. (2016). Loss of galectin-3 decreases the number of immune cells in the subventricular zone and restores proliferation in a viral model of multiple sclerosis. Glia 64, 105–121. doi: 10.1002/glia.22906
Jiang, H. R., Al Rasebi, Z., Mensah-Brown, E., Shahin, A., Xu, D., Goodyear, C. S., et al. (2009). Galectin-3 deficiency reduces the severity of experimental autoimmune encephalomyelitis. J. Immunol. 182, 1167–1173. doi: 10.4049/jimmunol.182.2.1167
Jin, A. Y., Tuor, U. I., Rushforth, D., Kaur, J., Muller, R. N., Petterson, J. L., et al. (2010). Reduced blood brain barrier breakdown in P-selectin deficient mice following transient ischemic stroke: a future therapeutic target for treatment of stroke. BMC Neurosci. 11:12. doi: 10.1186/1471-2202-11-12
John, S., and Mishra, R. (2016). mRNA transcriptomics of galectins unveils heterogeneous organization in mouse and human brain. Front. Mol. Neurosci. 9:139. doi: 10.3389/fnmol.2016.00139
Kasai, K., and Hirabayashi, J. (1996). Galectins: a family of animal lectins that decipher glycocodes. J. Biochem. 119, 1–8. doi: 10.1093/oxfordjournals.jbchem.a021192
Kataoka, H., Kim, S. W., and Plesnila, N. (2004). Leukocyte-endothelium interactions during permanent focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 24, 668–676. doi: 10.1097/01.wcb.0000117812.35136.5b
Kettenmann, H., Hanisch, U. K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Kim, C., Ho, D. H., Suk, J. E., You, S., Michael, S., Kang, J., et al. (2013). Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 4:1562. doi: 10.1038/ncomms2534
Kopatz, J., Beutner, C., Welle, K., Bodea, L. G., Reinhardt, J., Claude, J., et al. (2013). Siglec-h on activated microglia for recognition and engulfment of glioma cells. Glia 61, 1122–1133. doi: 10.1002/glia.22501
Lalancette-Hébert, M., Gowing, G., Simard, A., Weng, Y. C., and Kriz, J. (2007). Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 27, 2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007
Lalancette-Hébert, M., Swarup, V., Beaulieu, J. M., Bohacek, I., Abdelhamid, E., Weng, Y. C., et al. (2012). Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J. Neurosci. 32, 10383–10395. doi: 10.1523/JNEUROSCI.1498-12.2012
Langer, H. F., and Chavakis, T. (2009). Leukocyte-endothelial interactions in inflammation. J. Cell. Mol. Med. 13, 1211–1220. doi: 10.1111/j.1582-4934.2009.00811.x
Lasky, L. A. (1992). Selectins: interpreters of cell-specific carbohydrate information during inflammation. Science 258, 964–969. doi: 10.1126/science.1439808
Lawson, L. J., Perry, V. H., Dri, P., and Gordon, S. (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170. doi: 10.1016/0306-4522(90)90229-w
Lerman, B. J., Hoffman, E. P., Sutherland, M. L., Bouri, K., Hsu, D. K., Liu, F. T., et al. (2012). Deletion of galectin-3 exacerbates microglial activation and accelerates disease progression and demise in a SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Brain Behav. 2, 563–575. doi: 10.1002/brb3.75
Letschert, S., Gohler, A., Franke, C., Bertleff-Zieschang, N., Memmel, E., Doose, S., et al. (2014). Super-resolution imaging of plasma membrane glycans. Angew. Chem. Int. Ed. Engl. 53, 10921–10924. doi: 10.1002/anie.201406045
Ley, K. (2003). The role of selectins in inflammation and disease. Trends Mol. Med. 9, 263–268. doi: 10.1016/s1471-4914(03)00071-6
Liao, B., Zhao, W., Beers, D. R., Henkel, J. S., and Appel, S. H. (2012). Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 237, 147–152. doi: 10.1016/j.expneurol.2012.06.011
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Liu, F. T., Patterson, R. J., and Wang, J. L. (2002). Intracellular functions of galectins. Biochim. Biophys. Acta 1572, 263–273. doi: 10.1016/s0304-4165(02)00313-6
Liu, F. T., and Rabinovich, G. A. (2005). Galectins as modulators of tumour progression. Nat. Rev. Cancer 5, 29–41. doi: 10.1038/nrc1527
Longhi, L., Orsini, F., De Blasio, D., Fumagalli, S., Ortolano, F., Locatelli, M., et al. (2014). Mannose-binding lectin is expressed after clinical and experimental traumatic brain injury and its deletion is protective. Crit. Care Med. 42, 1910–1918. doi: 10.1097/CCM.0000000000000399
Macauley, M. S., Crocker, P. R., and Paulson, J. C. (2014). Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 14, 653–666. doi: 10.1038/nri3737
Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., and Cummings, J. L. (2015). Alzheimer’s disease. Nat. Rev. Dis. Primers 1:15056. doi: 10.1038/nrdp.2015.56
Miron, V. E., Boyd, A., Zhao, J. W., Yuen, T. J., Ruckh, J. M., Shadrach, J. L., et al. (2013). M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 16, 1211–1218. doi: 10.1038/nn.3469
Mok, S. W., Riemer, C., Madela, K., Hsu, D. K., Liu, F. T., Gultner, S., et al. (2007). Role of galectin-3 in prion infections of the CNS. Biochem. Biophys. Res. Commun. 359, 672–678. doi: 10.1016/j.bbrc.2007.05.163
Mondal, N., Stolfa, G., Antonopoulos, A., Zhu, Y., Wang, S. S., Buffone, A. Jr., et al. (2016). Glycosphingolipids on human myeloid cells stabilize E-selectin-dependent rolling in the multistep leukocyte adhesion cascade. Arterioscler. Thromb. Vasc. Biol. 36, 718–727. doi: 10.1161/ATVBAHA.115.306748
Morrison, H., Frye, J., Davis-Gorman, G., Funk, J., McDonagh, P., Stahl, G., et al. (2011). The contribution of mannose binding lectin to reperfusion injury after ischemic stroke. Curr. Neurovasc. Res. 8, 52–63. doi: 10.2174/156720211794520260
Naj, A. C., Jun, G., Beecham, G. W., Wang, L. S., Vardarajan, B. N., Buros, J., et al. (2011). Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 43, 436–441. doi: 10.1038/ng.801
Nikodemova, M., Small, A. L., Smith, S. M., Mitchell, G. S., and Watters, J. J. (2014). Spinal but not cortical microglia acquire an atypical phenotype with high VEGF, galectin-3 and osteopontin, and blunted inflammatory responses in ALS rats. Neurobiol. Dis. 69, 43–53. doi: 10.1016/j.nbd.2013.11.009
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Orsini, F., Villa, P., Parrella, S., Zangari, R., Zanier, E. R., Gesuete, R., et al. (2012). Targeting mannose-binding lectin confers long-lasting protection with a surprisingly wide therapeutic window in cerebral ischemia. Circulation 126, 1484–1494. doi: 10.1161/CIRCULATIONAHA.112.103051
Osthoff, M., Katan, M., Fluri, F., Schuetz, P., Bingisser, R., Kappos, L., et al. (2011). Mannose-binding lectin deficiency is associated with smaller infarction size and favorable outcome in ischemic stroke patients. PLoS One 6:e21338. doi: 10.1371/journal.pone.0021338
Pang, Y., Fan, L. W., Tien, L. T., Dai, X., Zheng, B., Cai, Z., et al. (2013). Differential roles of astrocyte and microglia in supporting oligodendrocyte development and myelination in vitro. Brain Behav. 3, 503–514. doi: 10.1002/brb3.152
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Pardo, E., Cárcamo, C., Uribe-San Martin, R., Ciampi, E., Segovia-Miranda, F., Curkovic-Pena, C., et al. (2017). Galectin-8 as an immunosuppressor in experimental autoimmune encephalomyelitis and a target of human early prognostic antibodies in multiple sclerosis. PLoS One 12:e0177472. doi: 10.1371/journal.pone.0177472
Park, J. W., Voss, P. G., Grabski, S., Wang, J. L., and Patterson, R. J. (2001). Association of galectin-1 and galectin-3 with Gemin4 in complexes containing the SMN protein. Nucleic Acids Res. 29, 3595–3602. doi: 10.1093/nar/29.17.3595
Pasquini, L. A., Millet, V., Hoyos, H. C., Giannoni, J. P., Croci, D. O., Marder, M., et al. (2011). Galectin-3 drives oligodendrocyte differentiation to control myelin integrity and function. Cell Death Differ. 18, 1746–1756. doi: 10.1038/cdd.2011.40
Patel, A. R., Ritzel, R., McCullough, L. D., and Liu, F. (2013). Microglia and ischemic stroke: a double-edged sword. Int. J. Physiol. Pathophysiol. Pharmacol. 5, 73–90.
Perry, V. H., Crocker, P. R., and Gordon, S. (1992). The blood-brain barrier regulates the expression of a macrophage sialic acid-binding receptor on microglia. J. Cell Sci. 101, 201–207.
Pillai, S., Netravali, I. A., Cariappa, A., and Mattoo, H. (2012). Siglecs and immune regulation. Annu. Rev. Immunol. 30, 357–392. doi: 10.1146/annurev-immunol-020711-075018
Politis, M., Giannetti, P., Su, P., Turkheimer, F., Keihaninejad, S., Wu, K., et al. (2012). Increased PK11195 PET binding in the cortex of patients with MS correlates with disability. Neurology 79, 523–530. doi: 10.1212/WNL.0b013e3182635645
Puttur, F., Arnold-Schrauf, C., Lahl, K., Solmaz, G., Lindenberg, M., Mayer, C. T., et al. (2013). Absence of Siglec-H in MCMV infection elevates interferon α production but does not enhance viral clearance. PLoS Pathog. 9:e1003648. doi: 10.1371/journal.ppat.1003648
Reichert, F., and Rotshenker, S. (1999). Galectin-3/MAC-2 in experimental allergic encephalomyelitis. Exp. Neurol. 160, 508–514. doi: 10.1006/exnr.1999.7229
Riemer, C., Neidhold, S., Burwinkel, M., Schwarz, A., Schultz, J., Krätzschmar, J., et al. (2004). Gene expression profiling of scrapie-infected brain tissue. Biochem. Biophys. Res. Commun. 323, 556–564. doi: 10.1016/j.bbrc.2004.08.124
Sakaguchi, M., Imaizumi, Y., and Okano, H. (2007). Expression and function of galectin-1 in adult neural stem cells. Cell. Mol. Life Sci. 64, 1254–1258. doi: 10.1007/s00018-007-6476-5
Sakaguchi, M., Shingo, T., Shimazaki, T., Okano, H. J., Shiwa, M., Ishibashi, S., et al. (2006). A carbohydrate-binding protein, Galectin-1, promotes proliferation of adult neural stem cells. Proc. Natl. Acad. Sci. U S A 103, 7112–7117. doi: 10.1073/pnas.0508793103
Sancho, D., and Reis e Sousa, C. (2012). Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu. Rev. Immunol. 30, 491–529. doi: 10.1146/annurev-immunol-031210-101352
Satoh, K., Niwa, M., Goda, W., Binh, N. H., Nakashima, M., Takamatsu, M., et al. (2011). Galectin-3 expression in delayed neuronal death of hippocampal CA1 following transient forebrain ischemia and its inhibition by hypothermia. Brain Res. 1382, 266–274. doi: 10.1016/j.brainres.2011.01.049
Saylor, D., Dickens, A. M., Sacktor, N., Haughey, N., Slusher, B., Pletnikov, M., et al. (2016). HIV-associated neurocognitive disorder—pathogenesis and prospects for treatment. Nat. Rev. Neurol. 12:309. doi: 10.1038/nrneurol.2016.53
Schneider, U. C., Davids, A. M., Brandenburg, S., Müller, A., Elke, A., Magrini, S., et al. (2015). Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol. 130, 215–231. doi: 10.1007/s00401-015-1440-1
Singh, K. K., Nathamu, S., Adame, A., Alire, T. U., Dumaop, W., Gouaux, B., et al. (2011). Expression of mannose binding lectin in HIV-1-infected brain: implications for HIV-related neuronal damage and neuroAIDS. Neurobehav. HIV Med. 3, 41–52. doi: 10.2147/nbhiv.s19969
Sirko, S., Irmler, M., Gascon, S., Bek, S., Schneider, S., Dimou, L., et al. (2015). Astrocyte reactivity after brain injury-: the role of galectins 1 and 3. Glia 63, 2340–2361. doi: 10.1002/glia.22898
Smittkamp, S. E., Brown, J. W., and Stanford, J. A. (2008). Time-course and characterization of orolingual motor deficits in B6SJL-Tg(SOD1–G93A)1Gur/J mice. Neuroscience 151, 613–621. doi: 10.1016/j.neuroscience.2007.10.017
Song, S., Ji, B., Ramachandran, V., Wang, H., Hafley, M., Logsdon, C., et al. (2012). Overexpressed galectin-3 in pancreatic cancer induces cell proliferation and invasion by binding Ras and activating Ras signaling. PLoS One 7:e42699. doi: 10.1371/journal.pone.0042699
Spence, S., Greene, M. K., Fay, F., Hams, E., Saunders, S. P., Hamid, U., et al. (2015). Targeting Siglecs with a sialic acid-decorated nanoparticle abrogates inflammation. Sci. Transl. Med. 7:303ra140. doi: 10.1126/scitranslmed.aab3459
Stancic, M., Slijepcevic, D., Nomden, A., Vos, M. J., de Jonge, J. C., Sikkema, A. H., et al. (2012). Galectin-4, a novel neuronal regulator of myelination. Glia 60, 919–935. doi: 10.1002/glia.22324
Stancic, M., van Horssen, J., Thijssen, V. L., Gabius, H. J., van der Valk, P., Hoekstra, D., et al. (2011). Increased expression of distinct galectins in multiple sclerosis lesions. Neuropathol. Appl. Neurobiol. 37, 654–671. doi: 10.1111/j.1365-2990.2011.01184.x
Starossom, S. C., Mascanfroni, I. D., Imitola, J., Cao, L., Raddassi, K., Hernandez, S. F., et al. (2012). Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity 37, 249–263. doi: 10.1016/j.immuni.2012.05.023
Steelman, A. J., Smith, R. III., Welsh, C. J., and Li, J. (2013). Galectin-9 protein is up-regulated in astrocytes by tumor necrosis factor and promotes encephalitogenic T-cell apoptosis. J. Biol. Chem. 288, 23776–23787. doi: 10.1074/jbc.M113.451658
Steinman, L., and Zamvil, S. S. (2006). How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann. Neurol. 60, 12–21. doi: 10.1002/ana.20913
Subramaniam, S. R., and Federoff, H. J. (2017). Targeting microglial activation states as a therapeutic avenue in Parkinson’s disease. Front. Aging Neurosci. 9:176. doi: 10.3389/fnagi.2017.00176
Sun, J., Shaper, N. L., Itonori, S., Heffer-Lauc, M., Sheikh, K. A., and Schnaar, R. L. (2004). Myelin-associated glycoprotein (Siglec-4) expression is progressively and selectively decreased in the brains of mice lacking complex gangliosides. Glycobiology 14, 851–857. doi: 10.1093/glycob/cwh107
Takagi, H., Fukaya, T., Eizumi, K., Sato, Y., Sato, K., Shibazaki, A., et al. (2011). Plasmacytoid dendritic cells are crucial for the initiation of inflammation and T cell immunity in vivo. Immunity 35, 958–971. doi: 10.1016/j.immuni.2011.10.014
Tang, T., Frenette, P. S., Hynes, R. O., Wagner, D. D., and Mayadas, T. N. (1996). Cytokine-induced meningitis is dramatically attenuated in mice deficient in endothelial selectins. J. Clin. Invest. 97, 2485–2490. doi: 10.1172/jci118695
Thored, P., Heldmann, U., Gomes-Leal, W., Gisler, R., Darsalia, V., Taneera, J., et al. (2009). Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia 57, 835–849. doi: 10.1002/glia.20810
Thurston, T. L., Wandel, M. P., von Muhlinen, N., Foeglein, A., and Randow, F. (2012). Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418. doi: 10.1038/nature10744
Troncoso, M. F., Ferragut, F., Bacigalupo, M. L., Cárdenas Delgado, V. M., Nugnes, L. G., Gentilini, L., et al. (2014). Galectin-8: a matricellular lectin with key roles in angiogenesis. Glycobiology 24, 907–914. doi: 10.1093/glycob/cwu054
Varki, A. (2009). Natural ligands for CD33-related Siglecs? Glycobiology 19, 810–812. doi: 10.1093/glycob/cwp063
Varki, A., and Angata, T. (2006). Siglecs—the major subfamily of I-type lectins. Glycobiology 16, 1R–27R. doi: 10.1093/glycob/cwj008
Varki, A., Etzler, M. E., Cummings, R. D., and Esko, J. D. (2009). “Discovery and classification of glycan-binding proteins,” in Essentials of Glycobiology, 2nd Edn. eds A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, et al. (New York, NY: Cold Spring Harbor), 1–22.
Venkatesan, C., Chrzaszcz, M., Choi, N., and Wainwright, M. S. (2010). Chronic upregulation of activated microglia immunoreactive for galectin-3/Mac-2 and nerve growth factor following diffuse axonal injury. J. Neuroinflammation 7:32. doi: 10.1186/1742-2094-7-32
Wan, L., and Liu, F.-T. (2016). Galectin-3 and inflammation. Glycobiol. Insights 6, 1–9. doi: 10.4137/gbi.s13921
Wang, J. L., Haudek, K. C., Voss, P. G., Patterson, R. J., and Arnoys, E. J. (2012). “Intracellular galectins: platforms for assembly of macromolecular complexes,” in Galectins and Disease Implications for Targeted Therapeutics, eds A. A. Klyosov and P. G. Traber (Washington, DC: ACS Symposium Series), 81–93. doi: 10.1021/bk-2012-1115.ch004
Wang, X., Mitra, N., Cruz, P., Deng, L., NISC Comparative Sequencing Program, Varki, N., et al. (2012). Evolution of siglec-11 and siglec-16 genes in hominins. Mol. Biol. Evol. 29, 2073–2086. doi: 10.1093/molbev/mss077
Wang, Y., and Neumann, H. (2010). Alleviation of neurotoxicity by microglial human Siglec-11. J. Neurosci. 30, 3482–3488. doi: 10.1523/JNEUROSCI.3940-09.2010
Wang, X., Yue, T. L., Barone, F. C., and Feuerstein, G. Z. (1995). Demonstration of increased endothelial-leukocyte adhesion molecule-1 mRNA expression in rat ischemic cortex. Stroke 26, 1665–1668; discussion 1668–1669. doi: 10.1161/01.str.26.9.1665
Weis, W. I., Drickamer, K., and Hendrickson, W. A. (1992). Structure of a C-type mannose-binding protein complexed with an oligosaccharide. Nature 360, 127–134. doi: 10.1038/360127a0
Werneburg, S., Buettner, F. F., Erben, L., Mathews, M., Neumann, H., Muhlenhoff, M., et al. (2016). Polysialylation and lipopolysaccharide-induced shedding of E-selectin ligand-1 and neuropilin-2 by microglia and THP-1 macrophages. Glia 64, 1314–1330. doi: 10.1002/glia.23004
Wielgat, P., and Braszko, J. J. (2012). The participation of sialic acids in microglia-neuron interactions. Cell. Immunol. 273, 17–22. doi: 10.1016/j.cellimm.2011.12.002
Yan, H., Kamiya, T., Suabjakyong, P., and Tsuji, N. M. (2015). Targeting C-type lectin receptors for cancer immunity. Front. Immunol. 6:408. doi: 10.3389/fimmu.2015.00408
Yan, J., Xu, Y., Zhang, L., Zhao, H., Jin, L., Liu, W. G., et al. (2016). Increased expressions of plasma galectin-3 in patients with amyotrophic lateral sclerosis. Chin. Med. J. 129, 2797–2803. doi: 10.4103/0366-6999.194656
Yip, P. K., Carrillo-Jimenez, A., King, P., Vilalta, A., Nomura, K., Chau, C. C., et al. (2017). Galectin-3 released in response to traumatic brain injury acts as an alarmin orchestrating brain immune response and promoting neurodegeneration. Sci. Rep. 7:41689. doi: 10.1038/srep41689
Yokota, Y., Arai, T., and Kawasaki, T. (1995). Oligomeric structures required for complement activation of serum mannan-binding proteins. J. Biochem. 117, 414–419. doi: 10.1093/jb/117.2.414
Zhang, R. L., Chopp, M., Zhang, Z. G., Phillips, M. L., Rosenbloom, C. L., Cruz, R., et al. (1996). E-selectin in focal cerebral ischemia and reperfusion in the rat. J. Cereb. Blood Flow Metab. 16, 1126–1136. doi: 10.1097/00004647-199611000-00006
Zhang, J., Raper, A., Sugita, N., Hingorani, R., Salio, M., Palmowski, M. J., et al. (2006). Characterization of Siglec-H as a novel endocytic receptor expressed on murine plasmacytoid dendritic cell precursors. Blood 107, 3600–3608. doi: 10.1182/blood-2005-09-3842
Zhang, F., Zheng, M., Qu, Y., Li, J., Ji, J., Feng, B., et al. (2009). Different roles of galectin-9 isoforms in modulating E-selectin expression and adhesion function in LoVo colon carcinoma cells. Mol. Biol Rep. 36, 823–830. doi: 10.1007/s11033-008-9251-2
Keywords: microglia, galectin, Siglec, selectin, mannose-binding lectin, neurological disease, neurodegeneration
Citation: Siew JJ and Chern Y (2018) Microglial Lectins in Health and Neurological Diseases. Front. Mol. Neurosci. 11:158. doi: 10.3389/fnmol.2018.00158
Received: 27 January 2018; Accepted: 25 April 2018;
Published: 14 May 2018.
Edited by:
Margaret Su-chun Ho, ShanghaiTech University, ChinaReviewed by:
Marina Bentivoglio, University of Verona, ItalySavina Apolloni, Fondazione Santa Lucia (IRCCS), Italy
Copyright © 2018 Siew and Chern. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yijuang Chern, Ym15Y2hlcm5AaWJtcy5zaW5pY2EuZWR1LnR3