 Małgorzata Drozd1,2
Małgorzata Drozd1,2 Barbara Bardoni
Barbara Bardoni Maria Capovilla
Maria Capovilla
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Neurosci. , 16 April 2018
Sec. Neuroplasticity and Development
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00124
This article is part of the Research Topic The Fragile X Syndrome – Where Do We Go? View all 19 articles
Intellectual disability (ID) and autism are hallmarks of Fragile X Syndrome (FXS), a hereditary neurodevelopmental disorder. The gene responsible for FXS is Fragile X Mental Retardation gene 1 (FMR1) encoding the Fragile X Mental Retardation Protein (FMRP), an RNA-binding protein involved in RNA metabolism and modulating the expression level of many targets. Most cases of FXS are caused by silencing of FMR1 due to CGG expansions in the 5′-UTR of the gene. Humans also carry the FXR1 and FXR2 paralogs of FMR1 while flies have only one FMR1 gene, here called dFMR1, sharing the same level of sequence homology with all three human genes, but functionally most similar to FMR1. This enables a much easier approach for FMR1 genetic studies. Drosophila has been widely used to investigate FMR1 functions at genetic, cellular, and molecular levels since dFMR1 mutants have many phenotypes in common with the wide spectrum of FMR1 functions that underlay the disease. In this review, we present very recent Drosophila studies investigating FMRP functions at genetic, cellular, molecular, and electrophysiological levels in addition to research on pharmacological treatments in the fly model. These studies have the potential to aid the discovery of pharmacological therapies for FXS.
The Fragile X Syndrome (FXS), previously known as Martin-Bell syndrome or marker X syndrome or FRAXA, is the first X-linked intellectual disability (ID) syndrome described involving a DNA alteration and the most frequent heritable monogenic form of ID (reviewed in Penagarikano et al., 2007; Santoro et al., 2012; Hayward et al., 2017). Human FXS patients present severe ID often accompanied by an increase in Autism Spectrum Disorder (ASD) traits and other phenotypes like delayed development, hyperactivity, attention deficit, hypersensitivity to sensorial stimuli, anxiety, aggression, sleep, cardiac disorders, and epileptic seizures (reviewed in Hagerman, 2002; Garber et al., 2008; Utari et al., 2010; Santoro et al., 2012; Hagerman et al., 2014; Kidd et al., 2014; Maurin et al., 2014; Schaefer et al., 2015; Dahlhaus, 2018). These abnormalities can be explained by defects in neuronal development and maturation. Some patients also present characteristic morphological facial traits, macrocephaly, flat feet, and male macroorchidism. The first morphological phenotype observed in FXS patients was the presence of abnormalities in the spines (Comery et al., 1997; Irwin et al., 2001). More recently, ElectroEncephaloGraphy (EEG) and Magnetic Resonance Imaging (MRI) have evidenced volume and Event Related Potential (ERP) defects in FXS patients (Devitt et al., 2015).
FXS was initially associated with an X-chromosome fragile site (an isochromatid gap in metaphase chromosomes) in position Xq27.3 (Harrison et al., 1983). In 1991, this site was mapped to a CGG trinucleotide expansion in the 5′ non-coding region of a gene named Fragile X Mental Retardation 1 (FMR1), the first gene associated with an X-linked ID (Verkerk et al., 1991). FMR1 is 38 kb long and transcribed in a 4.4 kb full length mRNA that encodes a 632 aa protein called Fragile X Mental Retardation Protein (FMRP). Through alternative splicing, at least 12 different isoforms of 67–80 kD are produced. The CGG repeats are polymorphic in the population ranging from 5 to 54 repeats in normal individuals to more than 200 (full mutation) in severely affected patients (reviewed in Hayward et al., 2017). The repeat expansion results in hypermethylation of the CGG repeat, of a 5′ CpG island, and of flanking promoter sequences causing the reduction or absence of FMR1 expression through an epigenetic mechanism involving FMR1 mRNA (Colak et al., 2014). Several deletions and point mutations leading to the production of non-functional proteins have also been described (Okray et al., 2015 and references therein). Individuals with 55–200 CGG repetitions (premutation) do not present FXS symptoms, but may develop two other disorders: Fragile-X Primary Ovarian Insufficiency (FXPOI) (reviewed in Sherman et al., 2014) or Fragile X Associated Tremor/Ataxia Syndrome (FXTAS) (reviewed in Hall et al., 2016; Dahlhaus, 2018). FXTAS has been modeled in Drosophila by overexpressing 90 rCGG repeats alone fused to GFP, which causes a neuron-specific degeneration and the formation of inclusions (Jin et al., 2003; Qurashi et al., 2012).
In mammals, FMRP is nearly ubiquitous, present mainly in neurons (particularly in the cortex, hippocampus, and Purkinje cells) and in testes and absent from muscles and the heart (Devys et al., 1993). FMRP has two paralogs: Fragile X Related 1 (FXR1) and Fragile X Related 2 (FXR2). While FXR2 has a distribution comparable to that of FMRP, some isoforms of FXR1 display a specific expression in brain while other isoforms are only present in muscle and heart (Khandjian et al., 1998; Bechara et al., 2007). These three proteins are members of the same family, namely the Related Fragile X Protein family, and are RNA-binding proteins mainly localized in the cytoplasm, although they carry a Nuclear Localization Signal (NLS) and a Nuclear Exportation Signal (NES) (Bardoni et al., 2000). Indeed, some isoforms of FMRP have also been localized in the nucleus (Eberhart et al., 1996; Bardoni et al., 1997). Collectively, these results have suggested that the three FXR proteins are able to shuttle between nucleus and cytoplasm to export their target mRNAs.
Three RNA-binding sequence motifs are the hallmarks of FMRP that may explain its function, i.e., two K homology (KH) domains and one Arginine-Glycine-Glycine (RGG) box. The main function of FMRP is to regulate translation and indeed it has been found associated with polyribosomes in different cell lines and, importantly, in the brain (Khandjian et al., 2004). Although FMRP mainly acts as a repressor, an activator function has been observed (reviewed in Maurin et al., 2014). Many methods have been used to identify FMRP targets (reviewed in Maurin et al., 2014; Davis and Broadie, 2017; Hayward et al., 2017): binding to biotinylated RNAs (Ashley et al., 1993), Cross-Link Immuno Precipitation (CLIP) (Darnell et al., 2011; Ascano et al., 2012; Anderson et al., 2016), Systematic Evolution of Ligands by Exponential Enrichment (SELEX) (Chen et al., 2003), yeast two-hybrid system (Ma et al., 2016), yeast three-hybrid system (Dolzhanskaya et al., 2003), and Antibody-Positioned RNA Amplification (APRA) (Miyashiro et al., 2003). Many of the identified targets have been involved in autism, other neuronal pathologies or gonadal development and many of them encode synaptic proteins (reviewed in Maurin et al., 2014). Finally, FMRP has been linked to the microRNA (miRNA) and Piwi-interacting RNA (piRNA) pathways in Mammals, Drosophila, and zebrafish (reviewed in Kelley et al., 2012; Specchia et al., 2017).
The first model of FXS was the mammalian mouse model (The Dutch-Belgian Fragile X Consortium, 1994; Mientjes et al., 2006), which recapitulates some major patients' phenotypes (Dahlhaus, 2018 and references therein). However, ever since then, also research on Drosophila melanogaster has brought important knowledge on the basic mechanisms underlying FMRP function. The Drosophila homolog of FMR1 was first identified in 2000 (Wan et al., 2000) and named dfmr1. Over the years, it has been called by many different names that are listed in the Drosophila database FlyBase (http://flybase.org/reports/FBrf0174476.html). It is now named Fmr1 with a capital F, meaning that it has been identified through the human homolog FMR1. Here, we will call it dFMR1 to distinguish it from the mouse gene (Fmr1). FlyBase names the protein Fmr1, but here we will call it dFMRP with the “d” indicating Drosophila. The dFMR1 gene exhibits high sequence homology with all three human genes (FMRP, FXR1, and FXR2; Zhang et al., 2001; Coffee et al., 2010), but is most functionally related to FMR1 (Coffee et al., 2010; see below). dFMR1 is 8.7 kb long and transcribed in many different mRNAs of 2–4 kb encoding many different proteins of different sizes (http://flybase.org/reports/FBgn0028734.html). All functional domains are highly conserved with the two KH domains being 75% identical and 85% similar between dFMR1 and hFMR1 (Wan et al., 2000).
The gene expression of dFMR1 in embryos was explored soon after its cloning and observed in the Central Nervous System (CNS), in the somatic musculature, in pole cells, in the gut and in the gonads (Wan et al., 2000; Zhang et al., 2001; Schenck et al., 2002). In Figure 1, we show the expression of dFMR1 at stage 14 by in situ hybridization with a full-length probe using the Tyramide Signal Amplification (TSA) (Tevy et al., 2014). High levels of expression are found in the brain (Figure 1A, arrowhead), in the CNS (Figure 1A, arrow) and in muscle precursors (Figure 1B), confirming the previously described pattern of dFMR1 expression at this stage through a sensitive method.
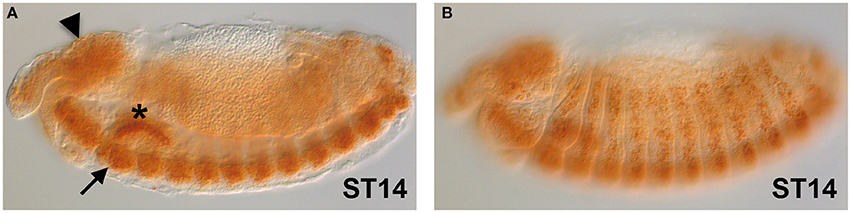
Figure 1. dFMR1 expression in stage 14 Drosophila embryos. (A) Lateral view of a stage 14 embryo (middle focus) showing expression in the brain (arrowhead) and in the CNS (arrow). The salivary gland (asterisk) is non-specific background. (B) Lateral view of the same stage 14 embryo (surface focus) showing expression in several muscle precursors. The dFMR1 anti-sense probe was synthesized from the full length EST-clone LD09557 (Drosophila Genomics Resource Center, Bloomington, IN, USA) linearized with EcoRI and transcribed with the T7 RNA polymerase using the Riboprobe Combination System kit (Promega, Madison, WI) and the DIG RNA Labeling Mix (Roche, Indianapolis, IN). In situ hybridization was performed as in Tevy et al. (2014) except that SA-HRP and TSA were diluted at 1:250. Images were acquired at the SPIBOC imaging platform of the Institut Sophia Agrobiotech (Sophia Antipolis, France) on an Axioplan II microscope using the ZEN software (Carl Zeiss, Germany).
In embryos, dFMRP has been localized in the brain, ventral nerve cord, and mesoderm (Zhang et al., 2001; Schenck et al., 2002; Dolzhanskaya et al., 2003; Ma et al., 2016), muscle attachment sites and dendritic tips of chordotonal organs, dendrites of the trachea-innervating neurons (Schenck et al., 2002), developing egg chambers (Zarnescu et al., 2005; Pepper et al., 2009), and in punctate cytoplasmic structures in cleavage-stage embryos in association with cytoplasmic RNP bodies (Monzo et al., 2006; Papoulas et al., 2010). In larvae, dFMRP has been detected in the CNS, the PNS, the eye disk, the testis, at low levels in muscles, in the Mushroom Bodies (MBs, the Drosophila learning and memory center; Schenck et al., 2002) and in dendritic arborization (DA) neurons (Lee et al., 2003). dFMRP is found exclusively in the cytoplasm of the soma of neurons (Zhang et al., 2001). Finally, in adults dFMRP has been found in the central brain and eyes (Zhang et al., 2001), in pupal and adult brain neurons (Morales et al., 2002), in specific cells of the adult brain (Morales et al., 2002), in antennal lobe projection neurons (Sudhakaran et al., 2014), and in the cell bodies of specific neurons of the MBs (Pan et al., 2004). Although the main tissue in which dFMRP is present is the CNS, these studies suggest that this protein also has non-neuronal functions, most of which still have to be dissected. Indeed, dFMR1 has been involved by FlyBase (http://flybase.org/reports/FBgn0028734.html) in 58 biological processes, which are summarized in Table 1.

Table 1. Main phenotypes of loss of function dFMR1 mutants.
It is interesting to note that the phenotypes of dFMR1 mutants largely recall the pathological symptoms of FXS patients. For instance, some FXS patients have delayed motor development, which can be compared to uncoordinated behavior of flies measured by flight or climbing assays. Moreover, olfactory learning and courtship conditioning of Drosophila can serve to test learning and memory behaviors that are often impaired in FXS patients. Also the changes in neuron structure observed in FXS patients are mimicked in the fly model (reviewed in van Alphen and van Swinderen, 2013). The first dFMR1 mutants were generated soon after its cloning (Zhang et al., 2001) and resulted viable and fertile as in humans. Nevertheless, mutant viability is highly sensitive to the genetic background, such that dFMR1 mutants can become fully lethal in some backgrounds (see Morales et al., 2002) in a generation-dependent manner, a phenomenon that requires further study. These original alleles were loss-of-function excisions of hypomorphic EP insertions (producing over-expression of the gene in which they are inserted) presenting phenotypes upon over-expression in the eye (Zhang et al., 2001). Other null mutants were obtained by excision of other EP elements (Dockendorff et al., 2002; Inoue et al., 2002) or by EMS mutagenesis (Lee et al., 2003). Altogether, dFMR1 mutants show defects in many different biological functions listed in Table 1. Over-expression studies in flies have also provided evidence for understating the mis-functions of mutant human FMRP as in the case of the assessment of a neomorphic function for a frameshift FMRP mutant (Okray et al., 2015). In summary, dFMR1 mainly plays a crucial role in synaptic plasticity and this affects many neuronal processes that are important for fly behavior.
RNA-mediated interference (RNAi) enables the knockdown of a gene of interest at the post-transcriptional stage (Fire et al., 1998). Combining the RNAi with the UAS/GAL4 system (Brand and Perrimon, 1993) in flies makes it possible to down-regulate gene expression in certain tissues and/or at a desired stage of development. Using this combined mechanism, the promoter-GAL4 fusion drives the expression of the RNAi hairpin fragment under the control of UAS sequences (Piccin et al., 2001). Tissue- or stage-specific expression is achieved by the use of specific GAL4 drivers. For instance, elav-GAL4 is usually used to trigger pan-neuronal expression, mef2-GAL4 for pan-muscular expression and Act5c-GAL4 for ubiquitous expression. Since 2007, the Vienna Drosophila Resource Center (VDRC; http://stockcenter.vdrc.at/control/main; Dietzl et al., 2007) has established many RNAi lines of D. melanogaster. These comprise around 12,671 (91%) of Drosophila protein-coding genes, making it the largest collection of RNAi lines for all model systems. Currently, there are three different types of UAS-RNAi stocks available: GD and KK with long hairpins and shRNA with short hairpin micro RNAs. The existence of such a broad array of RNAi lines provides many experimental benefits for biological studies, including those focusing on FMRP. For example, tissue-specific RNAi studies uncovered the role of dFMRP in early developmental stages of fly larvae. It was shown that dFMRP regulates glial-dependent proper timing of neuroblast reactivation during brain development (Callan et al., 2012). In another study, RNAi knockdown of Torsin revealed its involvement in locomotion. Torsin probably works together with dFMR1 to regulate synaptic plasticity and dFMR1 expression is altered in Torsin mutant flies (Nguyen et al., 2016). By creating the cardiac-specific dFMR1 RNAi knockdown, dFmr1 was also shown to be involved in regulating heart rate during development using the climbing assay. In this simple assay, adults are placed in a vial and tapped down. The time by which they reach the height of 5 cm is then measured (Novak et al., 2015). Some FMR1 patients present cardiac defects (Kidd et al., 2014) and changes in FMRP levels have been associated with structural and functional defects in zebrafish and mice (Mientjes et al., 2004; Van't Padje et al., 2009). On the contrary, in Drosophila no structural defects have been found, suggesting that dFMR1 and FMR1 regulate distinct targets. In a different study, hemocyte-specific dFMR1 knockdown by RNAi causes a defect in immune cell phagocytosis of bacteria and increases Drosophila sensitivity to bacterial infections, suggesting that dFMR1 is involved in the regulation of phagocytosis (O'Connor et al., 2017; reviewed in Logan, 2017). This study can provide further insights into the engagement of the immune system in FXS pathogenesis, especially considering the fact that some FXS patients exhibit defects in this system (O'Connor et al., 2017 and references therein). Aberrations in calcium homeostasis are connected with changes in neuron structure that probably cause the learning and memory deficits seen in FXS patients (Tessier and Broadie, 2011 and references therein). This was supported by RNAi knockdown of dFMR1 during a critical period of development, which proved the importance of the dFMRP role in regulating calcium signaling in the learning and memory circuitry (Doll and Broadie, 2016). These studies highlight the value of the Drosophila system in detailed phenotypic analyses of FMRP function.
NeuroMuscular Junctions (NMJs) of Drosophila are simple synapses that resemble those present in the Vertebrate CNS. Thus, they are a good model for the study of synapses (reviewed in Menon et al., 2013). The neuromuscular system of Drosophila contains 32 motor neurons in each abdominal hemisegment. NMJ synapses show developmental and functional plasticity. They are large and individually specified, enabling their visualization. NMJs are composed of branches and three types of boutons (I, II, and III) that are oval structures hosting synapses differing in size, shape, physiology, and in the amount of sub-synaptic reticulum surrounding them. Type I boutons are glutamatergic and have been the focus of FXS studies. Immunohistological staining can be used to visualize these structures and observe different NMJ phenotypes. dFMR1 is expressed pre-synaptically in motor neurons (Zhang et al., 2001), but post-synaptically in muscles (Zhang et al., 2001; Schenck et al., 2002). In dFMR1 mutants, NMJs display increased synapse arborization and branching, increased synaptic bouton numbers, and elevated neurotransmission, whereas larvae over-expressing dFMR1 show the opposite phenotypes (Zhang et al., 2001). These phenotypes recall the dendritic spine over-growth observed in mammalian mutants and in FXS patients (Irwin et al., 2001).
In addition to defects in NMJ synaptic architecture in neurons, dFMR1 mutants also exhibit fecundity and testes dysfunctions, which can be used to evaluate non-neuronal requirements (Zhang et al., 2004). Coffee et al. examined the evolutionary conservation of FMR1 and its paralogs in the Drosophila FXS model at the neuronal and non-neuronal levels (Coffee et al., 2010). In this study, out of the three human genes, only FMR1 turned out to be able to restore the normal number of boutons in dFMR1 null mutants. On the other hand, all three homologs rescued the sterility and testicular phenotypes. These results indicate that in neurons FMR1 has a unique evolutionarily conserved role. In contrast, in non-neuronal tissues FMR1, hFXR1, and hFXR2 are able to substitute each other (Coffee et al., 2010).
The larval NMJ has proved a powerful system to study genetic interactions occurring in the actin remodeling pathway that is altered in mammalian FMRP-null neurons (Castets et al., 2005; Pyronneau et al., 2017). FMRP has been shown to interact with Cytoplasmic FMRP Interacting Protein 1 (CYFIP1) in vitro (Schenck et al., 2001, 2003) and also in both mammals (Schenck et al., 2001) and Drosophila (Schenck et al., 2003; Abekhoukh et al., 2017). CYFIP1 is part of the WAVE regulatory complex (WRC) along with five other proteins involved in actin polymerization (reviewed in Cory and Ridley, 2002; Takenawa and Suetsugu, 2007). Human CYFIP1 has been linked to neurodevelopmental disorders such as ID, autism, schizophrenia, epilepsy, and Burnside-Butler (15q11.2 BP1-BP2 micro-deletion) syndrome (Madrigal et al., 2012; Waltes et al., 2014; Huang, 2015; Wang et al., 2015). Abekhoukh et al. (2017) utilized the fine genetic tools of Drosophila to investigate the genetic interactions between dFMRP and dCYFIP1. Through loss- and gain-of-functions studies, the authors showed that dFMR1 and dCYFIP1 have opposing functions on larval NMJ length: dFMRP represses while dCYFIP1 promotes synaptic growth at the NMJ in gene dosage studies using the presynaptic elav-Gal4 driver. A rescue of the reciprocal NMJ length phenotypes is observed in double homozygous mutant animals. It should be noted that double homozygous mice are lethal, thus preventing a similar epistatic genetic analysis in the mouse model. Here, the advantage of Drosophila for synaptic plasticity and actin studies is that specific parameters can easily be monitored. Since dFMR1 and CYFIP1 are candidates for ID and autism, these studies on the fly pave the way to deeper and more refined studies in mice and in humans.
The first Drosophila target of dFMRP was the gene futsch, identified by RNA immunoprecipitation (Zhang et al., 2001). This gene encodes a microtubule-associated protein with homology to mammalian MAP1B. futsch and dFMR1 have opposite phenotypes (undergrowth and overgrowth of synaptic boutons, respectively) and dFMRP binds futsch mRNA, negatively regulating its translation (Zhang et al., 2001). Binding of dFMRP was also found for the mRNA of the actin monomer binding protein profilin (encoded by chickadee; Reeve et al., 2005) and of the small GTPase Rac1 (Lee et al., 2003), whose loss- and gain-of-functions also have opposite phenotypes to those of dFMRP that are rescued by over-expression of both genes. dFMRP also binds the mRNAs of BMPR2 (Kashima et al., 2016), DSCAM (Cvetkovska et al., 2013), and the Ca2+/calmodulin-dependent protein kinase II (CaMKII) mRNA together with Ataxin-2 (Sudhakaran et al., 2014), confirming the involvement of dFMR1 in the Ca2+ pathway. Nevertheless, there is still a need for high throughput studies to identify novel dFMRP targets.
MBs consist of bilateral clusters of ~2,500 neurons in the fly brain. In MBs, there are three types of intrinsic neurons: αβ, α′β′, and γ (Crittenden et al., 1998). α′β′ are a prerequisite for gaining olfactory memory, whereas αβ are required to retrieve memory (Krashes et al., 2007). MBs play a major role in olfactory learning and memory in Drosophila. Odor and courtship-based tests are frequently used to assess memory dysfunctions in this system (reviewed in Weisz et al., 2015). MBs are also involved in visual context generalization, information processing, locomotion, sleep, courtship conditioning, and choice behavior. The MB axons in dFMR1 null mutants show architectural defects such as an increase in volume and branching, as well as abnormalities in synapse formation (Pan et al., 2004; Chang et al., 2008). dFMR1 null mutants show morphological MB defects in the lobes, the most frequent of which is the failure of β-lobe arrest at the brain midline (Michel et al., 2004 and see Table 1), whereas over-expression of dFMR1 causes the opposite phenotype (Reeve et al., 2005). These MB malformations can be restored through pharmacological treatment, but they are not indispensable for restoring memory (McBride et al., 2005). It has been shown that dFMR1 expression requirements differ between lobe types in MBs. Simultaneous expression of dFMR1 in α, β, and γ lobes is essential for learning skills. The lack of dFMR1 expression in α and β lobes is sufficient to impair associative olfactory learning and memory, whereas the knock-down of dFMR1 only in γ lobes does not exhibit detrimental effects on learning (Kanellopoulos et al., 2012). The MBs have also been the focus of calcium-signaling studies using a transgenic GCaMP calcium sensor (Doll and Broadie, 2016 and references therein). The MB neurons have been shown to be involved in activity-dependent processes during critical period development thanks to their ability to respond when illuminated by a blue light (see below; reviewed in Doll and Broadie, 2014). All the dFMR1 phenotypes in the MBs correlate with learning and memory dysfunctions and make the FXS Drosophila model very appealing because of its easy in vivo analysis and the wide range of tools that have been developed (Ugur et al., 2016; Chow and Reiter, 2017).
Electrophysiological techniques have been used in Drosophila to study the effect of dFMR1 loss-of-function and over-expression on synaptic transmission. Two-Electrode Voltage Clamp (TEVC) studies at NMJs in Drosophila showed that FMRP has clear pre-synaptic functions (Zhang et al., 2001), although photoreceptor synaptic transmission is normal in the mutants (Morales et al., 2002). Through TEVC recordings, Gatto and Broadie (2008) showed that presynaptic dFMR1 expression in dFMR1 mutants rescues the defects in NMJ structure, but not in neurotransmission, suggesting that dFMRP also has post-synaptic functions. On the other hand, in mice, FMRP was first shown to act post-synaptically (Huber et al., 2002) and only recently has also been found to play a pre-synaptic role in signal transmission (Koga et al., 2015; Myrick et al., 2015; Zhang et al., 2015) as in the fly. The possibility to target gene expression to specific cells through the UAS/GAL4 system (Brand and Perrimon, 1993), the wide availability of genetic mutants and the precise spatial and temporal resolutions make electrophysiology a very informative technique for FXS modeling in flies, and a useful complement to the advanced imaging studies described below.
Optogenetics is an in vivo technique that uses light to measure neuronal activity in living tissues and has also been exploited in the FXS Drosophila model. For example, dFMRP has been shown to play a cell-specific role in the regulation of activity-dependent calcium transients that is restricted to the early critical period (Tessier and Broadie, 2011; Doll and Broadie, 2016). Using optogenetic stimulation, it was recently established that dFMR1 mutants show increased circuit excitability (probably due to reduced GABAergic lateral inhibition; Franco et al., 2017) and that dFMRP is required for the activity-dependent regulation of synaptic connectivity (Doll et al., 2017). These data may explain the deficits in olfactory behaviors and the hyper-excitation found in FXS patients. Drosophila has been used to study even sleep patterns in dFMR1 mutants by electrophysiology and optogenetics. dFMR1 mutants show deeper night-like sleep during the day (van Alphen et al., 2013), which is likely because of the FMRP function in synaptic remodeling. It is clear that, through Drosophila, the cellular and physiological processes involved in FXS pathology can be studied at a deeper level than in any other model system. In addition, flies have been used for pharmacological studies on FXS through their complex behaviors like for example olfactory learning and memory, courtship, circadian rhythm, crawling and sleep (see Table 1 and below).
Drosophila is not the only animal model used to study FXS. Here, we report some examples of other animal models that can help in FXS studies. Mammalian mouse and rat models have been predominantly used for FXS studies. Two different mouse (The Dutch-Belgian Fragile X Consortium, 1994; Mientjes et al., 2006) and rat (Hamilton et al., 2014; Tian et al., 2017) KO models have been generated. It should be noted that, in mice, the full CGG expansion does not cause methylation and Fmr1 silencing (Brouwer et al., 2007) as it does in humans. Thus, all FXS studies in mice have been carried out using the KO animal (The Dutch-Belgian Fragile X Consortium, 1994; Mientjes et al., 2006). In general, the Fmr1-KO mouse is considered to be a good model for this disorder since it recapitulates most FXS phenotypes (deficits in learning and memory, hyperactivity, altered volumes of some brain regions, altered morphology of dendritic spines, and increased size of testis) and because it allows genetic experimentation. Also KO mutant rats present behavioral abnormalities related to ASD-like altered patterns of social interaction (Tian et al., 2017) and social play behavior (Hamilton et al., 2014), defects in visual attention (Berzhanskaya et al., 2016), and speech and auditory dysfunctions (Engineer et al., 2014). Rats have provided an excellent model for neuroscience and pharmacology as they have bigger brains than mice, are easier to train, can learn sophisticated behaviors and have an elaborated social repertoire; however, they are more expensive and much less genetically amenable than mice and flies. These recent studies on Fmr1 KO mice and rats show that both of these models are useful to study the complex phenotypes of FXS patients. Nevertheless, although Vertebrate model organisms provide more precise insights into the human disease pathogenesis, they are much more difficult than flies to maintain, more time-consuming and incur considerable expenses. In addition, performing experiments on Vertebrate models is much more restrictive in the context of animal laws and often triggers ethical issues.
Other animal models have been generated to understand the physiopathology of FXS, such as the zebrafish, a small fresh water fish endogenous to South-East Asia, the frog Xenopus laevis and the marine mollusk Aplysia californica. Although zebrafish fmr1 mutants generated by morpholino knock-down showed gross morphological defects in neurons (Tucker et al., 2006), true genetic null alleles later obtained by random mutagenesis resulted viable, fertile, and of normal morphology (den Broeder et al., 2009) likely due to off-targeting effects of the morpholino technology. Importantly, mutant zebrafish exhibit hyperactivity, learning deficits, impaired anxiety, and increased social behaviors like shoaling (Ng et al., 2013; Kim et al., 2014; Wu et al., 2017) maybe because of hyperactivity as in mice (Sorensen et al., 2015). The frog fmr1 mutant was also obtained by morpholino KO and showed behavioral (Truszkowski et al., 2016) and FMRP level-sensitive neuronal defects (Faulkner et al., 2015). Finally, in Aplysia, basic neurobiological studies have evidenced the pre- and post-synaptic control of plasticity regulating long-term memory and a functional interaction with the Na+-activated K+ channel (KNa) Slack (reviewed in Abrams, 2012). Overall, these simpler systems have also facilitated further insights into the mechanisms of FXS pathology.
In comparison to vertebrate models, the more complex human behaviors do not always correspond to those of flies and many neurological diseases can only be modeled in certain aspects. This is mainly due to the fact that Drosophila and humans differ in anatomy and despite of having many orthologs in common their pathways exhibit many differences. In addition, although drug administration is much simpler in Drosophila, the potential toxicity is much tougher to predict in humans because of significant metabolic differences and complexities (Pandey and Nichols, 2011).
Since the Drosophila dFMR1 phenotypes recapitulate the patients' symptoms, this model has been used to develop pharmacological treatments for the disease. One reason is that drugs can easily be administered in the standard fly food in which larvae feed and grow. This food is composed of cornmeal, agar and yeast and requires boiling. Recently, the Formula 4–24 (Carolina Biological Supply Company) medium has been exploited because it does not require heating and can simply be dissolved in room temperature water so that even heat-sensitive drugs can be tested (Kashima et al., 2017). Simple feeding has been used in the articles reported below, but many other methods to feed embryos, larvae or adults have been developed (reviewed in Pandey and Nichols, 2011). Another reason is that Drosophila does not carry vessels, but all of its organs bathe in hemolymph, which circulates thanks to a tubular heart in an open circulatory system. The fly blood/brain barrier is only made of a thin layer of glial cells presenting septate and gap junctions contrary to the Vertebrate one that is composed of glial cells and of endothelial cells forming tight junctions. Thus, the fly blood/brain barrier is simpler and allows for a better pharmacokinetic penetration (Pinsonneault et al., 2011; Limmer et al., 2014). Here, we discuss some examples of drugs successfully tested on flies to cure dFMR1 phenotypes that affect the different pathways listed below.
Glutamate is the main excitatory neurotransmitter in the CNS. Metabotropic glutamate receptors (mGluRs) at synapses control Long Term Depression (LTD), which mediates synaptic plasticity, thus weakening the synaptic response to stimuli. mGluR-dependent LTD was initially found to be enhanced in FMR1 mutant mice (Huber et al., 2002) and later in Drosophila (McBride et al., 2005). After the establishment of the “mGluR theory” of FXS by Bear et al. (2004), it was in Drosophila that inhibition of mGluR signaling was first shown to alleviate the behavioral fly phenotypes mimicking human FXS symptoms (McBride et al., 2005). In addition, a rescue of the defects in the fibers crossing the β-lobe of the MBs was observed in the treated dFMR1 mutants. Interestingly, adult therapy is sufficient to restore normal courtship behavior and short-term memory, but not β-lobe crossing, suggesting that other morphological defects are responsible for the memory defects (McBride et al., 2005). Since then, much research has been carried out to pharmacologically correct this defect both in mice (reviewed in Hagerman et al., 2014) and in flies (Choi et al., 2010; Kanellopoulos et al., 2012). dFMR1 mutants have less vigorous courtship behavior, learn normally, but forget. Treatments of larvae and adults either with the non-competitive mGluR antagonist 2-methyl-6-(phenylethynyl)pyridine (MPEP), three competitive mGluR antagonists or LiCl rescue naive courtship behavior, immediate recall memory and short-term memory of dFMR1 mutants (Choi et al., 2010). Subsequently, Choi et al. (2010) compared naive courtship, locomotion, olfactory capabilities, learning, and memory between 20 day-old flies and 5 day-old flies. They found that the inhibitors of the Glutamate receptor pathway and Lithium rescue these phenotypes of dFMR1 mutants to different degrees. The best rescue is obtained when the treatment is applied at both larval and adult stages. Learning is defective only in old flies, without the involvement of cell death. Treatment with four different mGluR inhibitors or LiCl exclusively during development rescues the learning, but not the courtship defects. Despite these successful treatments in pre-clinical trials, all clinical trials carried out so far have failed, although others are currently underway (https://clinicaltrials.gov). One reason could be the timing of the treatment or the need to combine different drugs since many other molecular mechanisms have been evoked as causative aspects of this disease. Importantly, in addition to FXS, many other neurodevelopmental disorders are correlated with defects in mGluR signaling.
As opposed to glutamate, GABA is the main inhibitory neurotransmitter in the brain. It functions through Gamma-AminoButyric Acid (GABA)A and GABAB receptors (R) that are fast-acting inotropic and metabotropic receptors, respectively. A decrease in GABAA R expression and defects in GABAergic signaling were found in the FMR1 Knock Out (KO) mouse (Idrissi et al., 2005; D'Hulst et al., 2006; Gantois et al., 2006; Sabanov et al., 2017). GABAB R has been linked to mGluR signaling and its reduction associated with autism (reviewed in Hagerman et al., 2014). Many clinical treatments targeted to the GABAergic pathway have been attempted or are underway (reviewed in Braat and Kooy, 2015). Altered GABAergic circuitry, like depressed glutamic acid decarboxylase (GAD) levels, was also found in the Drosophila FXS model, particularly in neurons expressing dFMR1 (D'Hulst et al., 2006; Gatto et al., 2014). In this system, through a fine mapping method of Mosaic Analysis with a Repressible Cell Marker (MARCM), Gatto et al. (2014) showed that dFMR1 is not required for GABAergic neuron survival, but it regulates the architecture of GABergic neurons innervating the MBs. In addition, dFMR1 GABAergic neurons display elevated calcium signaling (Gatto et al., 2014). To further underline the critical role of the GABA pathway in defining the FXR phenotype in the fly, it is worth mentioning that the down regulation of the GABA receptors in projection neurons of the antennal lobe observed in dFMR1 mutants is sufficient to produce olfactory behavioral defects (Franco et al., 2017).
Thanks to the toxicity of glutamate for the dFMR1 mutant, Chang et al. (2008) were able to carry out an unbiased screen for small molecules that can rescue the lethality of glutamate-treated dFMR1 mutants, using the mGluR5 non-competitive antagonist MPEP as a positive control. The active compounds belong to biochemical pathways not targeting mGluR signaling, namely the GABAergic, the muscarinic, the serotonin, and hormone-related pathways. Notably, GABA and MPEP treatments were also able to rescue the β-lobe crossing in the MBs and the courtship behavior phenotypes. Indeed, a decrease in GABAA R expression and defects in GABAergic signaling were found in the FMR1 KO mouse (Sabanov et al., 2017 and references therein) and in the Drosophila FXS model, particularly in neurons expressing dFMR1 (Gatto et al., 2014). This is the first chemical screen for FXS in flies and was made possible by the easily scorable phenotype of lethality, which revealed novel pathways implicated in FXS (Chang et al., 2008). GABAB R has been linked to mGluR signaling and its reduction associated with autism (reviewed in Hagerman et al., 2014). Many clinical treatments targeting the GABAergic pathway have been attempted or are underway (reviewed in Braat and Kooy, 2015).
Another pathway that has been shown to be involved in FXS alterations is the Bone Morphogenetic Protein (BMP) signal transduction pathway (Kashima et al., 2016), controlling a number of developmental processes including nervous system development (Liu and Niswander, 2005). This pathway has been linked to anxiety and object exploration in mice (McBrayer et al., 2015). It was previously established that the LIM domain kinase 1 (LIMK1) co-localizes with the BMP type II receptor (BMPR2) in the neuronal terminals (Lee-Hoeflich et al., 2004). These two proteins bind to each other leading to activation of the LIMK1 catalytic activity. Once activated by BMPR2, LIMK1 promotes neural growth and dendritogenesis through phosphorylation and inhibition of cofilin (Meng et al., 2002; Lee-Hoeflich et al., 2004). The C-terminal domain (CTD) of BMPR2 plays a role as a repressor of BMPR2 translation and as an activator of LIMK1 function. Kashima et al. (2016) found that FMRP binds to the CTD of the BMPR2 and inhibits the translation of full-length BMPR2 in humans and mice. These data have been genetically confirmed in the Drosophila model. In the NMJs of dFMR1 mutants, synaptic boutons are over-grown, whereas they are under-grown in mutants for the BMPR2 homolog wishful thinking (wit) (see also Eaton and Davis, 2005). Double wit, dFMR1 mutants show a number of boutons as in single wit mutants, indicating that wit is negatively regulated by dFMR1. These results obtained in the fly encouraged the authors to carry out pharmacological studies in mice that revealed the Fmr1 rescue potential of a LIMK1 inhibitor (Kashima et al., 2016). These cellular phenotypes correlate with the crawling behavior of Drosophila larvae, which has been used as a simple genetic and pharmacological screening tool for FXS treatment (Kashima et al., 2017). dFMR1 mutant larvae crawl faster than wild-type larvae: heterozygotes for three different dFMR1 alleles show increased larval locomotor activity correlated with an increased bouton number at the NMJ of muscle 6/7 in A3. This phenotype is rescued by a loss-of-function allele of wit in heterozygosis suggesting that it is due to an up-regulation of the BMPR2 homolog. The oral treatment with a pharmacological inhibitor of LIMK1 (downstream target of BMPR2, see above) restores the number of boutons in dFMR1 mutants. Using a newly developed larval crawling assay and a sophisticated algorithm (LarvaTrack) for drug screening, the locomotion defects (distance and velocity) of dFMR1 larvae were used as a readout of the larval bouton number phenotype (Kashima et al., 2017). In this case, LIMK1 inhibitors and puromycin rescue the dFMR1 locomotion phenotypes in correlation with the bouton number. Hyperactivity and anxiety of Fmr1-KO mice are also ameliorated by treatment with a LIMK inhibitor, showing that this specific and simple assay developed in flies has considerable potential in the assessment of new drug therapies. Indeed, all of these results can be applied to the human condition because an increase in BMPR2 is observed in the prefrontal cortexes of FXS patients (Kashima et al., 2016), paving the way to drug trials in humans.
Neuronal Calcium Sensor 1 (NCS-1) and its Drosophila homolog Frequenin (Frq) 2 are Ca2+-binding proteins that play a role in the control of the synapse number: loss-of-function mutations increase the synapse number and over-expression decreases it. Human NCS-1 is involved in schizophrenia (Koh et al., 2003; Torres et al., 2009) and autism (Piton et al., 2008). Frq2 physically interacts with the guanine nucleotide-exchange factor Ric8a and the structure of their interaction has been resolved (Romero-Pozuelo et al., 2014). The Ric8a protein is localized at larval motor neuron terminals and is particularly abundant in boutons. Ric8a knockdown causes a reduction in the number of synapses, thus producing a phenotype opposite to that of Freq2 with Frq2 acting as a negative regulator of Ric8a in the control of the synapse number (Romero-Pozuelo et al., 2014). On the other hand, as in humans and mice (Irwin et al., 2001; Hutsler and Zhang, 2010), loss of dFMR1 causes an increase in the synapse number and neuron volume (see Table 1). Mansilla et al. (2017) identified the aminophenothiazine derivative FD44 as a potent inhibitor of the Drosophila NCS-1/Ric8a interaction and showed that treatment of dFMR1 mutant adults and larvae with this drug reduces their synapse number and volume at the glutamatergic NMJ of larval muscle fibers 6/7 of A3. In addition, FD44 restores normal associative learning. This indicates that, in the dFMR1 mutant, the interaction between NCS-1 and Ric8a is unbalanced, probably because dFMRP controls Frq2 transcription (Tessier and Broadie, 2011). Consistently, over-expression of both NCS-1 and Ric8a restores a normal number of synapses (Romero-Pozuelo et al., 2014). FD44 is an interesting drug because it is small and able to cross the blood/brain barrier and because the structure of the interaction with its target proteins has been clearly illustrated (Mansilla et al., 2017).
The Down Syndrome Cell Adhesion Molecule (DSCAM) is a conserved protein the levels of which have been found to be elevated in many brain disorders including FXS. This has been confirmed in the mouse and Drosophila models where it has been shown that FMRP binds the Dscam mRNA and down-regulates its translation (Darnell et al., 2001; Cvetkovska et al., 2013; Kim et al., 2013). Dscam encodes a transmembrane protein that is a member of the immunoglobulin superfamily of cell adhesion molecules involved in self-avoidance, synaptic target selection, and axon guidance. In pre-synaptic terminals, Dscam has been shown to interact with Abelson tyrosine kinase (Abl), which mediates the exaggerated presynaptic terminal growth followed by Dscam over-expression probably because it is activated by DSCAM (Sterne et al., 2015). As multiple FDA-approved Abl inhibitors were available, Sterne et al. (2015) tested whether ABL inhibition could restore normal presynaptic terminal growth in larvae over-expressing Dscam or mutant for dFMR1. The nilotinib inhibitor was found to have this property, which suggests that ABL is as a promising therapeutic target for FXS.
Insulin and insulin-like growth factors (IGF) are evolutionarily highly conserved proteins that play an important role in growth and metabolism, but also in neurogenesis through their role in neuronal stem-cell homeostasis (reviewed in Lee et al., 2016; Ali et al., 2018). In Drosophila, expression of dFMR1 specifically in the insulin-producing cells (IPSs) or in the whole nervous system is able to rescue the defective free-running locomotor rhythmicity of dFMR1 mutants, as well as short-term and long-term memory defects in olfactory learning tests (Monyak et al., 2016). Protein levels of the major insulin-like peptide DILP2 are elevated in the IPCs of dFMR1 mutants, suggesting a post-transcriptional control, and markers of the insulin pathway are up-regulated. Through fine genetic manipulations aimed at reducing insulin signaling (IS), the authors were also able to obtain a rescue of the circadian rhythmicity of dFMR1 flies. These data are consistent with the fact that FMR1 is expressed in the IPSs of the mammalian pancreas and that insulin and IGFs have been involved in many neurological events, including synaptogenesis and progenitor cell growth (reviewed in Fernandez and Torres-Alemán, 2012). Metformin is a drug used to cure type 2 diabetes that acts as a sensitizer of IS signaling by increasing Phosphatase and TENsin homolog protein (PTEN) expression, AMP-activated protein Kinase (AMPK) activation and decreasing Target Of Rapamycin (TOR) signaling. Treatment of dFMR1 mutants with metformin restores normal short-term courtship memory and normal olfactory long-term memory (Monyak et al., 2016). Using GAL80, the temperature-sensitive repressor of GAL4, Monyak et al. (2016) also tested the precise stage at which reduction of IS is necessary to rescue the different behavioral phenotypes of dFMR1 mutants. While lowering IS during adulthood is sufficient to rescue learning and memory in the olfactory-based paradigm, reduction during the pupal stages is indispensable to rescue circadian behavior. This may mean that dFMRP is required at different developmental steps to temporally modulate IS. The weight of KO Fmr1 mice is significantly increased compared to wild-type (Dölen et al., 2007) and some FXS patients are obese (Nowicki et al., 2007) and show elevated IS through the mTor pathway in blood and brain (Hoeffer et al., 2012), implying that mis-regulation of the insulin pathway is likely to be one cause of the disease and a promising target for therapy (reviewed in Castagnola et al., 2017).
Individuals affected by FXS show a broad spectrum of clinical presentations, with a great variability of signs, symptoms and severity levels (Hagerman, 2002). Indeed, due to the high number of FMR1 targets, FXS can be considered as a multifactorial disorder. The possibility to study null dFMR1 flies and mice brought significant advantages for understanding the functional roles of FMRP. These systems made it possible to decrypt several pathways responsible for phenotypes recalling human symptoms, such as cognitive and learning deficits. In comparison to the other animal models, Drosophila is less expensive and easier to maintain, lays many eggs that enable to perform genetic screens, has a shorter lifespan and generation time, a smaller entirely sequenced genome (Adams et al., 2000) and sophisticated genetic tools to study human diseases like the easiness to make transgenics or to carry out in vivo functional studies (reviewed in Wangler et al., 2015; Ugur et al., 2016; Chow and Reiter, 2017). The fly genome has many orthologs displaying similar roles to human disease genes (Fortini et al., 2000; Doronkin and Reiter, 2008) that can be potential targets for functional and therapeutic studies. dFMR1 mutants exhibit defects that resemble those observed in human FXS patients, and flies and humans share many pathways that are altered in the disease and likely responsible for a specific set of phenotypes. Studies on FXS in Drosophila have set a paradigm to validate drug targets and gain a deeper insight into their molecular mechanisms for future research on FXS and other neurodevelopmental diseases. This is also because the dissection of the correlation between pathways and genotypes can be more easily realized in the fly than in any other model system.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are very grateful to the Drosophila Genomics Resource Center for the dFMR1 cDNA and to FlyBase for always useful informations. We thank Angela Algeri and Sara Castagnola for their comments on the manuscript, Arnaud Fernandez and Thomas Maurin for valuable discussions, Viviana Trezza for sharing her expertise on the rat model, and Olivier Pierre from SPIBOC for imaging. This study was supported by INSERM, CNRS, CNRS LIA NEOGENEX, ANR-11-LABX-0028-01, ANR-12-BSV4-0020, ANR-12-SVSE8-0022, ANR-15-CE16-0015, and FRM DEQ20140329490 to BB; Monaco Against Autism Foundation to BB and MC. MD is recipient of a Signalife- LabEx Program international Ph.D. fellowship. This review is dedicated to Kathy Matthews, co-founder of the Bloomington Drosophila Stock Center and of FlyBase.
Abekhoukh, S., Sahin, H. B., Grossi, M., Zongaro, S., Maurin, T., Madrigal, I., et al. (2017). New insights into the regulatory function of CYFIP1 in the context of WAVE- and FMRP-containing complexes. Dis. Model. Mech. 10, 463–474. doi: 10.1242/dmm.025809
Abrams, T. W. (2012). Studies on Aplysia neurons suggest treatments for chronic human disorders. Curr. Biol. 22, R705–R711. doi: 10.1016/j.cub.2012.08.011
Adams, M. D., Celniker, S. E., Holt, R. A., Evans, C. A., Gocayne, J. D., Amanatides, P. G., et al. (2000). The genome sequence of Drosophila melanogaster. Science 287, 2185–2195. doi: 10.1126/science.287.5461.2185
Adewoye, A. B., Kyriacou, C. P., and Tauber, E. (2015). Identification and functional analysis of early gene expression induced by circadian light-resetting in Drosophila. BMC Genomics 16:570. doi: 10.1186/s12864-015-1787-7
Ali, D., Abuelreich, S., Alkeraishan, N., Shwish, N. B., Hamam, R., Kassem, M., et al. (2018). Multiple intracellular signaling pathways orchestrate adipocytic differentiation of human bone marrow stromal stem cells. Biosci. Rep. 38:BSR20171252. doi: 10.1042/BSR20171252
Anderson, B. R., Chopra, P., Suhl, J. A., Warren, S. T., and Bassell, G. J. (2016). Identification of consensus binding sites clarifies FMRP binding determinants. Nucleic Acids Res. 44, 6649–6659. doi: 10.1093/nar/gkw593
Andlauer, T. F. M., Scholz-Kornehl, S., Tian, R., Kirchner, M., Babikir, H. A., Depner, H., et al. (2014). Drep-2 is a novel synaptic protein important for learning and memory. Elife 3:e03895. doi: 10.7554/eLife.03895
Ascano, M., Mukherjee, N., Bandaru, P., Miller, J. B., Nusbaum, J. D., Corcoran, D. L., et al. (2012). FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492, 382–386. doi: 10.1038/nature11737
Ashley, C. T., Wilkinson, K. D., Reines, D., and Warren, S. T. (1993). FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262, 563–566. doi: 10.1126/science.7692601
Banerjee, P., Nayar, S., Hebbar, S., Fox, C. F., Jacobs, M. C., Park, J. H., et al. (2007). Substitution of critical isoleucines in the KH domains of Drosophila fragile X protein results in partial loss-of-function phenotypes. Genetics 175, 1241–1250. doi: 10.1534/genetics.106.068908
Banerjee, P., Schoenfeld, B. P., Bell, A. J., Choi, C. H., Bradley, M. P., Hinchey, P., et al. (2010). Short- and long-term memory are modulated by multiple isoforms of the fragile X mental retardation protein. J. Neurosci. 30, 6782–6792. doi: 10.1523/JNEUROSCI.6369-09.2010
Bardoni, B., Mandel, J. L., and Fisch, G. S. (2000). FMR1 gene and fragile X syndrome. Am. J. Med. Genet. 97, 153–163. doi: 10.1002/1096-8628(200022)97:2<153::AID-AJMG7>3.0.CO;2-M
Bardoni, B., Sittler, A., Shen, Y., and Mandel, J. L. (1997). Analysis of domains affecting intracellular localization of the FMRP protein. Neurobiol. Dis. 4, 329–336. doi: 10.1006/nbdi.1997.0142
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. doi: 10.1016/j.tins.2004.04.009
Bechara, E., Davidovic, L., Melko, M., Bensaid, M., Tremblay, S., Grosgeorge, J., et al. (2007). Fragile X related protein 1 isoforms differentially modulate the affinity of fragile X mental retardation protein for G-quartet RNA structure. Nucleic Acids Res. 35, 299–306. doi: 10.1093/nar/gkl1021
Beerman, R. W., and Jongens, T. A. (2011). A non-canonical start codon in the Drosophila fragile X gene yields two functional isoforms. Neuroscience 181, 48–66. doi: 10.1016/j.neuroscience.2011.02.029
Berzhanskaya, J., Phillips, M. A., Shen, J., and Colonnese, M. T. (2016). Sensory hypo-excitability in a rat model of fetal development in fragile X syndrome. Sci. Rep. 6:30769. doi: 10.1038/srep30769
Bhogal, B., Jepson, J. E., Savva, Y. A., Pepper, A. S.-R., Reenan, R. A., and Jongens, T. A. (2011). Modulation of dADAR-dependent RNA editing by the Drosophila fragile X mental retardation protein. Nat. Neurosci. 14, 1517–1524. doi: 10.1038/nn.2950
Bolduc, F. V., Bell, K., Cox, H., Broadie, K. S., and Tully, T. (2008). Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat. Neurosci. 11, 1143–1145. doi: 10.1038/nn.2175
Bolduc, F. V., Valente, D., Nguyen, A. T., Mitra, P. P., and Tully, T. (2010). An assay for social interaction in Drosophila fragile X mutants. Fly 4, 216–225. doi: 10.4161/fly.4.3.12280
Bozzetti, M. P., Specchia, V., Cattenoz, P. B., Laneve, P., Geusa, A., Sahin, H. B., et al. (2015). The Drosophila fragile X mental retardation protein participates in the piRNA pathway. J. Cell Sci. 128, 2070–2084. doi: 10.1242/jcs.161810
Braat, S., and Kooy, R. F. (2015). Insights into GABAAergic system deficits in fragile X syndrome lead to clinical trials. Neuropharmacology 88, 48–54. doi: 10.1016/j.neuropharm.2014.06.028
Brand, A. H., and Perrimon, N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415.
Brouwer, J. R., Mientjes, E. J., Bakker, C. E., Nieuwenhuizen, I. M., Severijnen, L. A., Van der Linde, H. C., et al. (2007). Elevated Fmr1 mRNA levels and reduced protein expression in a mouse model with an unmethylated Fragile X full mutation. Exp. Cell Res. 313, 244–253. doi: 10.1016/j.yexcr.2006.10.002
Bushey, D., Tononi, G., and Cirelli, C. (2009). The Drosophila fragile X mental retardation gene regulates sleep need. J. Neurosci. 29, 1948–1961. doi: 10.1523/JNEUROSCI.4830-08.2009
Callan, M. A., Cabernard, C., Heck, J., Luois, S., Doe, C. Q., and Zarnescu, D. C. (2010). Fragile X protein controls neural stem cell proliferation in the Drosophila brain. Hum. Mol. Genet. 19, 3068–3079. doi: 10.1093/hmg/ddq213
Callan, M. A., Clements, N., Ahrendt, N., and Zarnescu, D. C. (2012). Fragile X Protein is required for inhibition of insulin signaling and regulates glial-dependent neuroblast reactivation in the developing brain. Brain Res. 1462, 151–161. doi: 10.1016/j.brainres.2012.03.042
Castagnola, S., Bardoni, B., and Maurin, T. (2017). The search for an effective therapy to treat fragile X syndrome: dream or reality? Front. Synaptic Neurosci. 9:15. doi: 10.3389/fnsyn.2017.00015
Castets, M., Schaeffer, C., Bechara, E., Schenck, A., Khandjian, E. W., Luche, S., et al. (2005). FMRP interferes with the Rac1 pathway and controls actin cytoskeleton dynamics in murine fibroblasts. Hum. Mol. Genet. 14, 835–844. doi: 10.1093/hmg/ddi077
Cavolo, S. L., Bulgari, D., Deitcher, D. L., and Levitan, E. S. (2016). Activity induces Fmr1-sensitive synaptic capture of anterograde circulating neuropeptide vesicles. J. Neurosci. 36, 11781–11787. doi: 10.1523/JNEUROSCI.2212-16.2016
Chang, S., Bray, S. M., Li, Z., Zarnescu, D. C., He, C., Jin, P., et al. (2008). Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat. Chem. Biol. 4, 256–263. doi: 10.1038/nchembio.78
Chen, L., Yun, S. W., Seto, J., Liu, W., and Toth, M. (2003). The fragile X mental retardation protein binds and regulates a novel class of mRNAs containing U rich target sequences. Neuroscience 120, 1005–1017. doi: 10.1016/S0306-4522(03)00406-8
Choi, C. H., McBride, S. M. J., Schoenfeld, B. P., Liebelt, D. A., Ferreiro, D., Ferrick, N. J., et al. (2010). Age-dependent cognitive impairment in a Drosophila fragile X model and its pharmacological rescue. Biogerontology 11, 347–362. doi: 10.1007/s10522-009-9259-6
Choi, C. H., Schoenfeld, B. P., Weisz, E. D., Bell, A. J., Chambers, D. B., Hinchey, J., et al. (2015). PDE-4 inhibition rescues aberrant synaptic plasticity in Drosophila and mouse models of fragile X syndrome. J. Neurosci. 35, 396–408. doi: 10.1523/JNEUROSCI.1356-12.2015
Chow, C. Y., and Reiter, L. T. (2017). Etiology of human genetic disease on the fly. Trends Genetics 33, 391–398. doi: 10.1016/j.tig.2017.03.007
Coffee, R. L., Tessier, C. R., Woodruff, E. A., and Broadie, K. (2010). Fragile X mental retardation protein has a unique, evolutionarily conserved neuronal function not shared with FXR1P or FXR2P. Dis. Model. Mech. 3, 471–485. doi: 10.1242/dmm.004598
Colak, D., Zaninovic, N., Cohen, M. S., Rosenwaks, Z., Yang, W.-Y., Gerhardt, J., et al. (2014). Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343, 1002–1005. doi: 10.1126/science.1245831
Comery, T. A., Harris, J. B., Willems, P. J., Oostra, B. A., Irwin, S. A., Weiler, I. J., et al. (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc. Natl. Acad. Sci U.S.A. 94, 5401–5404. doi: 10.1073/pnas.94.10.5401
Cory, G. O. C., and Ridley, A. J. (2002). Cell motility: braking WAVEs. Nature 418, 732–733. doi: 10.1038/418732a
Costa, A., Wang, Y., Dockendorff, T. C., Erdjument-Bromage, H., Tempst, P., Schedl, P., et al. (2005). The Drosophila fragile X protein functions as a negative regulator in the orb autoregulatory pathway. Dev. Cell 8, 331–342. doi: 10.1016/j.devcel.2005.01.011
Coyne, A. N., Yamada, S. B., Siddegowda, B. B., Estes, P. S., Zaepfel, B. L., Johannesmeyer, J. S., et al. (2015). Fragile X protein mitigates TDP-43 toxicity by remodeling RNA granules and restoring translation. Hum. Mol. Genet. 24, 6886–6898. doi: 10.1093/hmg/ddv389
Crittenden, J. R., Skoulakis, E. M., Han, K. A., Kalderon, D., and Davis, R. L. (1998). Tripartite mushroom body architecture revealed by antigenic markers. Learn. Mem. 5, 38–51.
Cvetkovska, V., Hibbert, A. D., Emran, F., and Chen, B. E. (2013). Overexpression of Down syndrome cell adhesion molecule impairs precise synaptic targeting. Nat. Neurosci. 16, 677–682. doi: 10.1038/nn.3396
Cziko, A. M. J., McCann, C. T., Howlett, I. C., Barbee, S. A., Duncan, R. P., Luedemann, R., et al. (2009). Genetic modifiers of dFMR1 encode RNA granule components in Drosophila. Genetics 182, 1051–1060. doi: 10.1534/genetics.109.103234
Dahlhaus, R. (2018). Of men and mice: modeling the fragile X syndrome. of men and mice: modeling the fragile X syndrome. Front. Mol. Neurosci. 11:41. doi: 10.3389/fnmol.2018.00041
Darnell, J. C., Jensen, K. B., Jin, P., Brown, V., Warren, S. T., and Darnell, R. B. (2001). Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107, 489–499. doi: 10.1016/S0092-8674(01)00566-9
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y. S., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
Davis, J. K., and Broadie, K. (2017). Multifarious functions of the fragile X mental retardation protein. Trends Genet. 33, 703–714. doi: 10.1016/j.tig.2017.07.008
den Broeder, M. J., van der Linde, H., Brouwer, J. R., Oostra, B. A., Willemsen, R., and Ketting, R. F. (2009). Generation and characterization of FMR1 knockout zebrafish. PLoS ONE 4:e7910. doi: 10.1371/journal.pone.0007910
Deshpande, G., Calhoun, G., and Schedl, P. (2006). The drosophila fragile X protein dFMR1 is required during early embryogenesis for pole cell formation and rapid nuclear division cycles. Genetics 174, 1287–1298. doi: 10.1534/genetics.106.062414
Devitt, N. M., Gallagher, L., and Reilly, R. B. (2015). Autism Spectrum Disorder (ASD) and Fragile X syndrome (FXS): two overlapping disorders reviewed through electroencephalography-what can be interpreted from the available information? Brain Sci. 5, 92–117. doi: 10.3390/brainsci5020092
Devys, D., Lutz, Y., Rouyer, N., Bellocq, J. P., and Mandel, J. L. (1993). The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet. 4, 335–340. doi: 10.1038/ng0893-335
D'Hulst, C., De Geest, N., Reeve, S. P., Van Dam, D., De Deyn, P. P., Hassan, B. A., et al. (2006). Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 1121, 238–245. doi: 10.1016/j.brainres.2006.08.115
Dietzl, G., Chen, D., Schnorrer, F., Su, K.-C., Barinova, Y., Fellner, M., et al. (2007). A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151–156. doi: 10.1038/nature05954
Dockendorff, T. C., Su, H. S., McBride, S. M. J., Yang, Z., Choi, C. H., Siwicki, K. K., et al. (2002). Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 34, 973–984. doi: 10.1016/S0896-6273(02)00724-9
Dölen, G., Osterweil, E., Rao, B. S. S., Smith, G. B., Auerbach, B. D., Chattarji, S., et al. (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962. doi: 10.1016/j.neuron.2007.12.001
Doll, C. A., and Broadie, K. (2014). Impaired activity-dependent neural circuit assembly and refinement in autism spectrum disorder genetic models. Front. Cell. Neurosci. 8:30. doi: 10.3389/fncel.2014.00030
Doll, C. A., and Broadie, K. (2015). Activity-dependent FMRP requirements in development of the neural circuitry of learning and memory. Development 142, 1346–1356. doi: 10.1242/dev.117127
Doll, C. A., and Broadie, K. (2016). Neuron class-specific requirements for fragile X mental retardation protein in critical period development of calcium signaling in learning and memory circuitry. Neurobiol. Dis. 89, 76–87. doi: 10.1016/j.nbd.2016.02.006
Doll, C. A., Vita, D. J., and Broadie, K. (2017). Fragile X mental retardation protein requirements in activity-dependent critical period neural circuit refinement. Curr. Biol. 27, 2318–2330.e3. doi: 10.1016/j.cub.2017.06.046
Dolzhanskaya, N., Sung, Y. J., Conti, J., Currie, J. R., and Denman, R. B. (2003). The fragile X mental retardation protein interacts with U-rich RNAs in a yeast three-hybrid system. Biochem. Biophys. Res. Commun. 305, 434–441. doi: 10.1016/S0006-291X(03)00766-6
Doronkin, S., and Reiter, L. T. (2008). Drosophila orthologues to human disease genes: an update on progress. Prog. Nucleic Acid Res. Mol. Biol. 82, 1–32. doi: 10.1016/S0079-6603(08)00001-9
Eaton, B. A., and Davis, G. W. (2005). LIM Kinase1 controls synaptic stability downstream of the type II BMP receptor. Neuron 47, 695–708. doi: 10.1016/j.neuron.2005.08.010
Eberhart, D. E., Malter, H. E., Feng, Y., and Warren, S. T. (1996). The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum. Mol. Genet. 5, 1083–1091. doi: 10.1093/hmg/5.8.1083
Engineer, C. T., Centanni, T. M., Im, K. W., Rahebi, K. C., Buell, E. P., and Kilgard, M. P. (2014). Degraded speech sound processing in a rat model of fragile X syndrome. Brain Res. 1564, 72–84. doi: 10.1016/j.brainres.2014.03.049
Epstein, A. M., Bauer, C. R., Ho, A., Bosco, G., and Zarnescu, D. C. (2009). Drosophila Fragile X protein controls cellular proliferation by regulating cbl levels in the ovary. Dev. Biol. 330, 83–92. doi: 10.1016/j.ydbio.2009.03.011
Faulkner, R. L., Wishard, T. J., Thompson, C. K., Liu, H.-H., and Cline, H. T. (2015). FMRP regulates neurogenesis in vivo in Xenopus laevis tadpoles. eNeuro 2:e0055. doi: 10.1523/ENEURO.0055-14.2014
Fernandez, A. M., and Torres-Alemán, I. (2012). The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 13, 225–239. doi: 10.1038/nrn3209
Fire, A., Xu, S., Montgomery, M. K., Kostas, S. A., Driver, S. E., and Mello, C. C. (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811. doi: 10.1038/35888
Fortini, M., Skupski, M., Boguski, M., and Hariharan, I. (2000). A survey of human disease gene counterparts in the Drosophila genome. J. Cell Biol. 150, F23–F30. doi: 10.1083/jcb.150.2.F23
Franco, L. M., Okray, Z., Linneweber, G. A., Hassan, B. A., and Yaksi, E. (2017). Reduced lateral inhibition impairs olfactory computations and behaviors in a Drosophila model of fragile X syndrome. Curr. Biol. 27, 1111–1123. doi: 10.1016/j.cub.2017.02.065
Friedman, S. H., Dani, N., Rushton, E., and Broadie, K. (2013). Fragile X mental retardation protein regulates trans-synaptic signaling in Drosophila. Dis. Model. Mech. 6, 1400–1413. doi: 10.1242/dmm.012229
Gantois, I., Vandesompele, J., Speleman, F., Reyniers, E., D'Hooge, R., Severijnen, L.-A., et al. (2006). Expression profiling suggests underexpression of the GABA(A) receptor subunit delta in the fragile X knockout mouse model. Neurobiol. Dis. 21, 346–357. doi: 10.1016/j.nbd.2005.07.017
Garber, K. B., Visootsak, J., and Warren, S. T. (2008). Fragile X syndrome. Eur. J. Hum. Genet. 16, 666–672. doi: 10.1038/ejhg.2008.61
Gatto, C. L., and Broadie, K. (2008). Temporal requirements of the fragile X mental retardation protein in the regulation of synaptic structure. Development 135, 2637–2648. doi: 10.1242/dev.022244
Gatto, C. L., and Broadie, K. (2009). Temporal requirements of the fragile X mental retardation protein in modulating circadian clock circuit synaptic architecture. Front. Neural Circuits 3:8. doi: 10.3389/neuro.04.008.2009
Gatto, C. L., and Broadie, K. (2011). Fragile X mental retardation protein is required for programmed cell death and clearance of developmentally-transient peptidergic neurons. Dev. Biol. 356, 291–307. doi: 10.1016/j.ydbio.2011.05.001
Gatto, C. L., Pereira, D., and Broadie, K. (2014). GABAergic circuit dysfunction in the Drosophila fragile X syndrome model. Neurobiol. Dis. 65, 142–159. doi: 10.1016/j.nbd.2014.01.008
Gross, C., Chang, C.-W., Kelly, S. M., Bhattacharya, A., McBride, S. M. J., Danielson, S. W., et al. (2015). Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell Rep. 11, 727–736. doi: 10.1016/j.celrep.2015.03.060
Günther, M. N., Nettesheim, G., and Shubeita, G. T. (2016). Quantifying and predicting Drosophila larvae crawling phenotypes. Sci. Rep. 6:27972. doi: 10.1038/srep27972
Hagerman, R. J. (2002). “The physical and behavioral phenotype,” in Fragile X Syndrome, Diagnosis, Treatment, and Research, eds R. J. Hagerman and P. J. Hagerman (Baltimore, MD: John Hopkins University Press), 3–109.
Hagerman, R. J., Portes, D. V., Gasparini, F., Jacquemont, S., and Gomez-Mancilla, B. (2014). Translating molecular advances in fragile X syndrome into therapy: a review. J. Clin. Psychiatry 75, e294–e307. doi: 10.4088/JCP.13r08714
Hall, D. A., Robertson, E., Shelton, A. L., Losh, M. C., Milà, M., Moreno, E. G., et al. (2016). Update on the clinical, radiographic, and neurobehavioral manifestations in FXTAS and FMR1 Premutation Carriers. Cerebellum 15, 578–586. doi: 10.1007/s12311-016-0799-4
Hamilton, S. M., Green, J. R., Veeraragavan, S., Yuva, L., McCoy, A., Wu, Y., et al. (2014). Fmr1 and Nlgn3 knockout rats: novel tools for investigating autism spectrum disorders. Behav. Neurosci. 128, 103–109. doi: 10.1037/a0035988
Harrison, C. J., Jack, E. M., Allen, T. D., and Harris, R. (1983). The fragile X: a scanning electron microscope study. J. Med. Genet. 20, 280–285. doi: 10.1136/jmg.20.4.280
Hayward, B. E., Kumari, D., and Usdin, K. (2017). Recent advances in assays for the fragile X-related disorders. Hum. Genet. 136, 1313–1327. doi: 10.1007/s00439-017-1840-5
Hoeffer, C. A., Sanchez, E., Hagerman, R. J., Mu, Y., Nguyen, D. V., Wong, H., et al. (2012). Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 11, 332–341. doi: 10.1111/j.1601-183X.2012.00768.x
Huang, Y. (2015). Up-regulated cytoplasmic FMRP-interacting protein 1 in intractable temporal lobe epilepsy patients and a rat model. Int. J. Neurosci. 126, 542–551 doi: 10.3109/00207454.2015.1038711
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 7746–7750. doi: 10.1073/pnas.122205699
Hutsler, J. J., and Zhang, H. (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94. doi: 10.1016/j.brainres.2009.09.120
Idrissi, E. L. A., Ding, X.-H., Scalia, J., Trenkner, E., Brown, W. T., and Dobkin, C. (2005). Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci. Lett. 377, 141–146. doi: 10.1016/j.neulet.2004.11.087
Inoue, S., Shimoda, M., Nishinokubi, I., Siomi, M. C., Okamura, M., Nakamura, A., et al. (2002). A role for the Drosophila fragile X-related gene in circadian output. Curr. Biol. 12, 1331–1335. doi: 10.1016/S0960-9822(02)01036-9
Irwin, S. A., Patel, B., Idupulapati, M., Harris, J. B., Crisostomo, R. A., Larsen, B. P., et al. (2001). Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am. J. Med. Genet. 98, 161–167. doi: 10.1002/1096-8628(20010115)98:2<161::AID-AJMG1025>3.0.CO;2-B
Jiang, F., Lu, F., Li, P., Liu, W., Zhao, L., Wang, Q., et al. (2016). Drosophila homolog of FMRP maintains genome integrity by interacting with piwi. J. Genet. Genomics 43, 11–24. doi: 10.1016/j.jgg.2015.11.001
Jin, P., Zarnescu, D. C., Zhang, F., Pearson, C. E., Lucchesi, J. C., Moses, K., et al. (2003). Repeats in Drosophila. Neuron 39, 739–747. doi: 10.1016/S0896-6273(03)00533-6
Kacsoh, B. Z., Bozler, J., Hodge, S., Ramaswami, M., and Bosco, G. (2015a). A novel paradigm for nonassociative long-term memory in Drosophila: predator-induced changes in oviposition behavior. Genetics 199, 1143–1157. doi: 10.1534/genetics.114.172221
Kacsoh, B. Z., Bozler, J., Ramaswami, M., and Bosco, G. (2015b). Social communication of predator-induced changes in Drosophila behavior and germ line physiology. Elife 13:4. doi: 10.7554/eLife.07423
Kanellopoulos, A. K., Semelidou, O., Kotini, A. G., Anezaki, M., and Skoulakis, E. M. C. (2012). Learning and memory deficits consequent to reduction of the fragile X mental retardation protein result from metabotropic glutamate receptor-mediated inhibition of cAMP signaling in Drosophila. J. Neurosci. 32, 13111–13124. doi: 10.1523/JNEUROSCI.1347-12.2012
Kashima, R., Redmond, P. L., Ghatpande, P., Roy, S., Kornberg, T. B., Hanke, T., et al. (2017). Hyperactive locomotion in a Drosophila model is a functional readout for the synaptic abnormalities underlying fragile X syndrome. Sci. Signal. 10:eaai8133. doi: 10.1126/scisignal.aai8133
Kashima, R., Roy, S., Ascano, M., Martinez-Cerdeno, V., Ariza-Torres, J., Kim, S., et al. (2016). Augmented noncanonical BMP type II receptor signaling mediates the synaptic abnormality of fragile X syndrome. Sci. Signal. 9:ra58. doi: 10.1126/scisignal.aaf6060
Kelley, K., Chang, S.-J. E., and Lin, S.-L. (2012). Mechanism of repeat-associated microRNAs in fragile X syndrome. Neural Plast. 2012:104796. doi: 10.1155/2012/104796
Kennedy, T., and Broadie, K. (2017). Fragile X mental retardation protein restricts small dye iontophoresis entry into central neurons. J. Neurosci. 37, 9844–9858. doi: 10.1523/JNEUROSCI.0723-17.2017
Khandjian, E. W., Bardoni, B., Corbin, F., Sittler, A., Giroux, S., Heitz, D., et al. (1998). Novel isoforms of the fragile X related protein FXR1P are expressed during myogenesis. Hum. Mol. Genet. 7, 2121–2128.
Khandjian, E. W., Huot, M.-E., Tremblay, S., Davidovic, L., Mazroui, R., and Bardoni, B. (2004). Biochemical evidence for the association of fragile X mental retardation protein with brain polyribosomal ribonucleoparticles. Proc. Natl. Acad. Sci. U.S.A. 101, 13357–13362. doi: 10.1073/pnas.0405398101
Kidd, S. A., Lachiewicz, A., Barbouth, D., Blitz, R. K., Delahunty, C., McBrien, D., et al. (2014). Fragile X syndrome: a review of associated medical problems. Pediatrics 134, 995–1005. doi: 10.1542/peds.2013-4301
Kim, J. H., Wang, X., Coolon, R., and Ye, B. (2013). Dscam expression levels determine presynaptic arbor sizes in Drosophila sensory neurons. Neuron 78, 827–838. doi: 10.1016/j.neuron.2013.05.020
Kim, L., He, L., Maaswinkel, H., Zhu, L., Sirotkin, H., and Weng, W. (2014). Anxiety, hyperactivity and stereotypy in a zebrafish model of fragile X syndrome and autism spectrum disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 55, 40–49. doi: 10.1016/j.pnpbp.2014.03.007
Koga, K., Liu, M.-G., Qiu, S., Song, Q., O'Den, G., Chen, T., et al. (2015). Impaired presynaptic long-term potentiation in the anterior cingulate cortex of Fmr1 knock-out mice. J. Neurosci. 35, 2033–2043. doi: 10.1523/JNEUROSCI.2644-14.2015
Koh, P. O., Undie, A. S., Kabbani, N., Levenson, R., Goldman-Rakic, P. S., and Lidow, M. S. (2003). Up-regulation of neuronal calcium sensor-1 (NCS-1) in the prefrontal cortex of schizophrenic and bipolar patients. Proc. Natl. Acad. Sci. U.S.A. 100, 313–317. doi: 10.1073/pnas.232693499
Krashes, M. J., Keene, A. C., Leung, B., Armstrong, J. D., and Waddell, S. (2007). Sequential use of mushroom body neuron subsets during Drosophila odor memory processing. Neuron 53, 103–115. doi: 10.1016/j.neuron.2006.11.021
Lee, A., Li, W., Xu, K., Bogert, B. A., Su, K., and Gao, F.-B. (2003). Control of dendritic development by the Drosophila fragile X-related gene involves the small GTPase Rac1. Development 130, 5543–5552. doi: 10.1242/dev.00792
Lee, S. H., Zabolotny, J. M., Huang, H., Lee, H., and Kim, Y. B. (2016). Insulin in the nervous system and the mind: functions in metabolism, memory, and mood. Mol. Metab. 5, 589–601. doi: 10.1016/j.molmet.2016.06.011
Lee-Hoeflich, S. T., Causing, C. G., Podkowa, M., Zhao, X., Wrana, J. L., and Attisano, L. (2004). Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J. 23, 4792–4801. doi: 10.1038/sj.emboj.7600418
Limmer, S., Weiler, A., Volkenhoff, A., Babatz, F., and Klämbt, C. (2014). The Drosophila blood-brain barrier: development and function of a glial endothelium. Front. Neurosci. 8:41. doi: 10.3389/fnins.2014.00365
Liu, A., and Niswander, L. A. (2005). Bone morphogenetic protein signalling and vertebrate nervous system development. Nat. Rev. Neurosci. 6, 945–954. doi: 10.1038/nrn1805
Logan, M. A. (2017). Fragile phagocytes: FMRP positively regulates engulfment activity. J. Cell Biol. 216, 531–533. doi: 10.1083/jcb.201702034
Ma, Y., Tian, S., Wang, Z., Wang, C., Chen, X., Li, W., et al. (2016). CMP-N-acetylneuraminic acid synthetase interacts with fragile X related protein 1. Mol. Med. Rep. 14, 1501–1508. doi: 10.3892/mmr.2016.5438
Madrigal, I., Rodríguez-Revenga, L., Xunclà, M., and Milà, M. (2012). 15q11.2 microdeletion and FMR1 premutation in a family with intellectual disabilities and autism. Gene 508, 92–95. doi: 10.1016/j.gene.2012.07.023
Mansilla, A., Chaves-Sanjuan, A., Campillo, N. E., Semelidou, O., Martínez-González, L., Infantes, L., et al. (2017). Interference of the complex between NCS-1 and Ric8a with phenothiazines regulates synaptic function and is an approach for fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 114, E999–E1008. doi: 10.1073/pnas.1611089114
Martinez, V. G., Javadi, C. S., Ngo, E., Ngo, L., Lagow, R. D., and Zhang, B. (2007). Age-related changes in climbing behavior and neural circuit physiology in Drosophila. Dev. Neurobiol. 67, 778–791. doi: 10.1002/dneu.20388
Maurin, T., Zongaro, S., and Bardoni, B. (2014). Fragile X syndrome: from molecular pathology to therapy. Neurosci. Biobehav. Rev. 46(Pt 2), 242–255. doi: 10.1016/j.neubiorev.2014.01.006
McBrayer, Z. L., Dimova, J., Pisansky, M. T., Sun, M., Beppu, H., Gewirtz, J. C., et al. (2015). Forebrain-specific loss of BMPRII in mice reduces anxiety and increases object exploration. PLoS ONE 10:e0139860. doi: 10.1371/journal.pone.0139860
McBride, S. M. J., Choi, C. H., Wang, Y., Liebelt, D., Braunstein, E., Ferreiro, D., et al. (2005). Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron 45, 753–764. doi: 10.1016/j.neuron.2005.01.038
Megosh, H. B., Cox, D. N., Campbell, C., and Lin, H. (2006). The role of PIWI and the miRNA machinery in Drosophila germline determination. Curr. Biol. 16, 1884–1894. doi: 10.1016/j.cub.2006.08.051
Meng, Y., Zhang, Y., Tregoubov, V., Janus, C., Cruz, L., Jackson, M., et al. (2002). Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 35, 121–133. doi: 10.1016/S0896-6273(02)00758-4
Menon, K. P., Carrillo, R. A., and Zinn, K. (2013). Development and plasticity of the Drosophila larval neuromuscular junction. Wiley Interdiscip. Rev. Dev. Biol. 2, 647–670. doi: 10.1002/wdev.108
Michel, C. I., Kraft, R., and Restifo, L. L. (2004). Defective neuronal development in the mushroom bodies of Drosophila fragile X mental retardation 1 mutants. J. Neurosci. 24, 5798–5809. doi: 10.1523/JNEUROSCI.1102-04.2004
Mientjes, E. J., Nieuwenhuizen, I., Kirkpatrick, L., Zu, T., Hoogeveen-Westerveld, M., Severijnen, L., et al. (2006). The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol. Dis. 21, 549–555. doi: 10.1016/j.nbd.2005.08.019
Mientjes, E. J., Willemsen, R., Kirkpatrick, L. L., Nieuwenhuizen, I. M., Hoogeveen-Westerveld, M., Verweij, M., et al. (2004). Fxr1 knockout mice show a striated muscle phenotype: implications for Fxr1p function in vivo. Hum. Mol. Genet. 13, 1291–1302. doi: 10.1093/hmg/ddh150
Miyashiro, K. Y., Beckel-Mitchener, A., Purk, T. P., Becker, K. G., Barret, T., Liu, L., et al. (2003). RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 37, 417–431. doi: 10.1016/S0896-6273(03)00034-5
Monyak, R. E., Emerson, D., Schoenfeld, B. P., Zheng, X., Chambers, D. B., Rosenfelt, C., et al. (2016). Insulin signaling misregulation underlies circadian and cognitive deficits in a Drosophila fragile X model. Mol. Psychiatry 22, 1140–1148. doi: 10.1038/mp.2016.51
Monzo, K., Papoulas, O., Cantin, G. T., Wang, Y., Yates, J. R., and Sisson, J. C. (2006). Fragile X mental retardation protein controls trailer hitch expression and cleavage furrow formation in Drosophila embryos. Proc. Natl. Acad. Sci. U.S.A. 103, 18160–18165. doi: 10.1073/pnas.0606508103
Morales, J., Hiesinger, P. R., Schroeder, A. J., Kume, K., Verstreken, P., Jackson, F. R., et al. (2002). Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron 34, 961–972. doi: 10.1016/S0896-6273(02)00731-6
Myrick, L. K., Deng, P.-Y., Hashimoto, H., Oh, Y. M., Cho, Y., Poidevin, M. J., et al. (2015). Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc. Natl. Acad. Sci. U.S.A. 112, 949–956. doi: 10.1073/pnas.1423094112
Ng, M.-C., Yang, Y.-L., and Lu, K.-T. (2013). Behavioral and synaptic circuit features in a zebrafish model of fragile X syndrome. PLoS ONE 8:e51456. doi: 10.1371/journal.pone.0051456
Nguyen, P., Seo, J. B., Ahn, H.-M., and Koh, Y. H. (2016). Drosophila torsin protein regulates motor control and stress sensitivity and forms a complex with Fragile-X mental retardation protein. Neural Plast. 2016:6762086. doi: 10.1155/2016/6762086
Novak, S. M., Joardar, A., Gregorio, C. C., and Zarnescu, D. C. (2015). Regulation of heart rate in drosophila via fragile X mental retardation protein. PLoS ONE 10:e0142836. doi: 10.1371/journal.pone.0142836
Nowicki, S. T., Tassone, F., Ono, M. Y., Ferranti, J., Croquette, M. F., Goodlin-Jones, B., et al. (2007). The Prader-Willi phenotype of fragile X syndrome. J. Dev. Behav. Pediatr. 28, 133–138. doi: 10.1097/01.DBP.0000267563.18952.c9
O'Connor, R. M., Stone, E. F., Wayne, C. R., Marcinkevicius, E. V., Ulgherait, M., Delventhal, R., et al. (2017). A Drosophila model of Fragile X syndrome exhibits defects in phagocytosis by innate immune cells. J. Cell Biol. 216, 595–605. doi: 10.1083/jcb.201607093
Okray, Z., de Esch, C. E. F., Van Esch, H., Devriendt, K., Claeys, A., Yan, J., et al. (2015). A novel fragile X syndrome mutation reveals a conserved role for the carboxy-terminus in FMRP localization and function. EMBO Mol. Med. 7, 423–437. doi: 10.15252/emmm.201404576
Pan, L., Zhang, Y. Q., Woodruff, E., and Broadie, K. (2004). The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr. Biol. 14, 1863–1870. doi: 10.1016/j.cub.2004.09.085
Pandey, U. B., and Nichols, C. D. (2011). Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol. Rev. 63, 441–436. doi: 10.1124/pr.110.003293
Papoulas, O., Monzo, K. F., Cantin, G. T., Ruse, C., Yates, J. R., Ryu, Y. H., et al. (2010). dFMRP and Caprin, translational regulators of synaptic plasticity, control the cell cycle at the Drosophila mid-blastula transition. Development 137, 4201–4209. doi: 10.1242/dev.055046
Penagarikano, O., Mulle, J. G., and Warren, S. T. (2007). The pathophysiology of fragile x syndrome. Annu. Rev. Genomics Hum. Genet. 8, 109–129. doi: 10.1146/annurev.genom.8.080706.092249
Pepper, A. S.-R., Beerman, R. W., Bhogal, B., and Jongens, T. A. (2009). Argonaute2 suppresses Drosophila fragile X expression preventing neurogenesis and oogenesis defects. PLoS ONE 4:e7618. doi: 10.1371/journal.pone.0007618
Piccin, A., Salameh, A., Benna, C., Sandrelli, F., Mazzotta, G., Zordan, M., et al. (2001). Efficient and heritable functional knock-out of an adult phenotype in Drosophila using a GAL4-driven hairpin RNA incorporating a heterologous spacer. Nucleic Acids Res. 29, e55–e55. doi: 10.1093/nar/29.12.e55
Pinsonneault, R. L., Mayer, N., Mayer, F., Tegegn, N., and Bainton, R. J. (2011). Novel models for studying the blood-brain and blood-eye barriers in Drosophila. Methods Mol. Biol. 686, 357–369. doi: 10.1007/978-1-60761-938-3_17
Piton, A., Michaud, J. L., Peng, H., Aradhya, S., Gauthier, J., Mottron, L., et al. (2008). Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum. Mol. Genet. 17, 3965–3974. doi: 10.1093/hmg/ddn300
Pyronneau, A., He, Q., Hwang, J.-Y., Porch, M., Contractor, A., and Zukin, R. S. (2017). Aberrant Rac1-cofilin signaling mediates defects in dendritic spines, synaptic function, and sensory perception in fragile X syndrome. Sci. Signal. 10:eaan0852. doi: 10.1126/scisignal.aan0852
Qurashi, A., Liu, H., Ray, L., Nelson, D. L., Duan, R., and Jin, P. (2012). Chemical screen reveals small molecules suppressing fragile X premutation rCGG repeat-mediated neurodegeneration in Drosophila. Hum. Mol. Genet. 21, 2068–2075. doi: 10.1093/hmg/dds024
Reeve, S. P., Bassetto, L., Genova, G. K., Kleyner, Y., Leyssen, M., Jackson, F. R., et al. (2005). The Drosophila fragile X mental retardation protein controls actin dynamics by directly regulating profilin in the brain. Curr. Biol. 15, 1156–1163. doi: 10.1016/j.cub.2005.05.050
Reeve, S. P., Lin, X., Sahin, B. H., Jiang, F., Yao, A., Liu, Z., et al. (2008). Mutational analysis establishes a critical role for the N terminus of fragile X mental retardation protein FMRP. J. Neurosci. 28, 3221–3226. doi: 10.1523/JNEUROSCI.5528-07.2008
Romero-Pozuelo, J., Dason, J. S., Mansilla, A., Baños-Mateos, S., Sardina, J. L., Chaves-Sanjuan, A., et al. (2014). The guanine-exchange factor Ric8a binds to the Ca2+ sensor NCS-1 to regulate synapse number and neurotransmitter release. J. Cell Sci. 127, 4246–4259. doi: 10.1242/jcs.152603
Sabanov, V., Braat, S., D'Andrea, L., Willemsen, R., Zeidler, S., Rooms, L., et al. (2017). Impaired GABAergic inhibition in the hippocampus of Fmr1 knockout mice. Neuropharmacology 116, 71–81. doi: 10.1016/j.neuropharm.2016.12.010
Santoro, M. R., Bray, S. M., and Warren, S. T. (2012). Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu. Rev. Pathol. 7, 219–245. doi: 10.1146/annurev-pathol-011811-132457
Schaefer, T. L., Davenport, M. H., and Erickson, C. A. (2015). Emerging pharmacologic treatment options for fragile X syndrome. Appl. Clin. Genet. 8, 75–93. doi: 10.2147/TACG.S35673
Schenck, A., Bardoni, B., Langmann, C., Harden, N., Mandel, J., and Giangrande, A. (2003). CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron 38, 887–898. doi: 10.1016/S0896-6273(03)00354-4
Schenck, A., Bardoni, B., Moro, A., Bagni, C., and Mandel, J. L. (2001). A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc. Natl. Acad. Sci. U.S.A. 98, 8844–8849. doi: 10.1073/pnas.151231598
Schenck, A., Van de Bor, V., Bardoni, B., and Giangrande, A. (2002). Novel features of dFMR1, the Drosophila orthologue of the fragile X mental retardation protein. Neurobiol. Dis. 11, 53–63. doi: 10.1006/nbdi.2002.0510
Sherman, S. L., Curnow, E. C., Easley, C. A., Jin, P., Hukema, R. K., Tejada, M. I., et al. (2014). Use of model systems to understand the etiology of fragile X-associated primary ovarian insufficiency (FXPOI). J. Neurodev. Disord. 6:26. doi: 10.1186/1866-1955-6-26
Siller, S. S., and Broadie, K. (2011). Neural circuit architecture defects in a Drosophila model of Fragile X syndrome are alleviated by minocycline treatment and genetic removal of matrix metalloproteinase. Dis. Model. Mech. 4, 673–685. doi: 10.1242/dmm.008045
Sorensen, E. M., Bertelsen, F., Weikop, P., Skovborg, M. M., Banke, T., Drasbek, K. R., et al. (2015). Hyperactivity and lack of social discrimination in the adolescent Fmr1 knockout mouse. Behav. Pharmacol. 26, 733–740. doi: 10.1097/FBP.0000000000000152
Specchia, V., D'Attis, S., Puricella, A., and Bozzetti, M. P. (2017). dFmr1 plays roles in small RNA pathways of Drosophila melanogaster. Int. J. Mol. Sci. 18:E1066. doi: 10.3390/ijms18051066
Sterne, G. R., Kim, J. H., and Ye, B. (2015). Dysregulated Dscam levels act through Abelson tyrosine kinase to enlarge presynaptic arbors. Elife 4:e05196. doi: 10.7554/eLife.05196
Sudhakaran, I. P., Hillebrand, J., Dervan, A., Das, S., Holohan, E. E., Hülsmeier, J., et al. (2014). FMRP and Ataxin-2 function together in long-term olfactory habituation and neuronal translational control. Proc. Natl. Acad. Sci. U.S.A. 111, E99–E108. doi: 10.1073/pnas.1309543111
Takenawa, T., and Suetsugu, S. (2007). The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 8, 37–48. doi: 10.1038/nrm2069
Tauber, J. M., Vanlandingham, P. A., and Zhang, B. (2011). Elevated levels of the vesicular monoamine transporter and a novel repetitive behavior in the Drosophila model of fragile X syndrome. PLoS ONE 6:e27100. doi: 10.1371/journal.pone.0027100
Tessier, C. R., and Broadie, K. (2008). Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development 135, 1547–1557. doi: 10.1242/dev.015867
Tessier, C. R., and Broadie, K. (2011). The fragile X mental retardation protein developmentally regulates the strength and fidelity of calcium signaling in Drosophila mushroom body neurons. Neurobiol. Dis. 41, 147–159. doi: 10.1016/j.nbd.2010.09.002
Tevy, M. F., Seyres, D., Traina, C., Perrin, L., and Capovilla, M. (2014). Ndae1 expression and regulation in Drosophila embryos. PLoS ONE 9:e92956. doi: 10.1371/journal.pone.0092956
The Dutch-Belgian Fragile X Consortium (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 78, 23–33.
Tian, Y., Yang, C., Shang, S., Cai, Y., Deng, X., Zhang, J., et al. (2017). Loss of FMRP impaired hippocampal long-term plasticity and spatial learning in rats. Front. Mol. Neurosci. 10:269. doi: 10.3389/fnmol.2017.00269
Torres, K. C. L., Souza, B. R., Miranda, D. M., Sampaio, A. M., Nicolato, R., Neves, F. S., et al. (2009). Expression of neuronal calcium sensor-1 (NCS-1) is decreased in leukocytes of schizophrenia and bipolar disorder patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 33, 229–234. doi: 10.1016/j.pnpbp.2008.11.011
Truszkowski, T. L. S., James, E. J., Hasan, M., Wishard, T. J., Liu, Z., Pratt, K. G., et al. (2016). Fragile X mental retardation protein knockdown in the developing Xenopus tadpole optic tectum results in enhanced feedforward inhibition and behavioral deficits. Neural Dev. 11:14. doi: 10.1186/s13064-016-0069-7
Tucker, B., Richards, R. I., and Lardelli, M. (2006). Contribution of mGluR and Fmr1 functional pathways to neurite morphogenesis, craniofacial development and fragile X syndrome. Hum. Mol. Genet. 15, 3446–3458. doi: 10.1093/hmg/ddl422
Ugur, B., Chen, K., and Bellen, H. J. (2016). Drosophila tools and assays for the study of human diseases. Dis. Model. Mech. 9, 235–244. doi: 10.1242/dmm.023762
Utari, A., Adams, E., Berry-Kravis, E., Chavez, A., Scaggs, F., Ngotran, L., et al. (2010). Aging in fragile X syndrome. J. Neurodev. Disord. 2, 70–76. doi: 10.1007/s11689-010-9047-2
van Alphen, B., and van Swinderen, B. (2013). Drosophila strategies to study psychiatric disorders. Brain Res. Bull. 92, 1–11. doi: 10.1016/j.brainresbull.2011.09.007
van Alphen, B., Yap, M. H. W., Kirszenblat, L., Kottler, B., and van Swinderen, B. (2013). A dynamic deep sleep stage in Drosophila. J. Neurosci. 33, 6917–6927. doi: 10.1523/JNEUROSCI.0061-13.2013
Van't Padje, S., Chaudhry, B., Severijnen, L. A., van der Linde, H. C., Mientjes, E. J., Oostra, B. A., et al. (2009). Reduction in fragile X related 1 protein causes cardiomyopathy and muscular dystrophy in zebrafish. J. Exp. Biol. 212, 2564–2570. doi: 10.1242/jeb.032532.
Verkerk, A. J., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P., Pizzuti, A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. doi: 10.1016/0092-8674(91)90397-H
Waltes, R., Duketis, E., Knapp, M., Anney, R. J. L., Huguet, G., Schlitt, S., et al. (2014). Common variants in genes of the postsynaptic FMRP signalling pathway are risk factors for autism spectrum disorders. Hum. Genet. 133, 781–792. doi: 10.1007/s00439-013-1416-y
Wan, L., Dockendorff, T. C., Jongens, T. A., and Dreyfuss, G. (2000). Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol. Cell. Biol. 20, 8536–8547. doi: 10.1128/MCB.20.22.8536-8547.2000
Wang, J., Tao, Y., Song, F., Sun, Y., Ott, J., and Saffen, D. (2015). Common regulatory variants of CYFIP1 contribute to susceptibility for Autism Spectrum Disorder (ASD) and Classical Autism. Ann. Hum. Genet. 79, 329–340. doi: 10.1111/ahg.12121
Wangler, M. F., Yamamoto, S., and Bellen, H. J. (2015). Fruit flies in biomedical research. Genetics 199, 639–653. doi: 10.1534/genetics.114.171785
Weisz, E. D., Monyak, R. E., and Jongens, T. A. (2015). Deciphering discord: how Drosophila research has enhanced our understanding of the importance of FMRP in different spatial and temporal contexts. Exp. Neurol. 274, 14–24. doi: 10.1016/j.expneurol.2015.05.015
Wu, Y.-J., Hsu, M.-T., Ng, M.-C., Amstislavskaya, T. G., Tikhonova, M. A., Yang, Y.-L., et al. (2017). Fragile X mental retardation-1 knockout zebrafish shows precocious development in social behavior. Zebrafish 14, 438–443. doi: 10.1089/zeb.2017.1446
Xu, K., Bogert, B. A., Li, W., Su, K., Lee, A., and Gao, F.-B. (2004). The fragile X-related gene affects the crawling behavior of Drosophila larvae by regulating the mRNA level of the DEG/ENaC protein pickpocket1. Curr. Biol. 14, 1025–1034. doi: 10.1016/j.cub.2004.05.055
Xu, S., Poidevin, M., Han, E., Bi, J., and Jin, P. (2012). Circadian rhythm-dependent alterations of gene expression in Drosophila brain lacking fragile X mental retardation protein. PLoS ONE 7:e37937. doi: 10.1371/journal.pone.0037937
Xu, X.-L., Li, Y., Wang, F., and Gao, F.-B. (2008). The steady-state level of the nervous-system-specific microRNA-124a is regulated by dFMR1 in Drosophila. J. Neurosci. 28, 11883–11889. doi: 10.1523/JNEUROSCI.4114-08.2008
Yang, Y., Xu, S., Xia, L., Wang, J., Wen, S., Jin, P., et al. (2009). The bantam microRNA is associated with drosophila fragile X mental retardation protein and regulates the fate of germline stem cells. PLoS Genet. 5:e1000444. doi: 10.1371/journal.pgen.1000444
Zarnescu, D. C., Jin, P., Betschinger, J., Nakamoto, M., Wang, Y., Dockendorff, T. C., et al. (2005). Fragile X protein functions with lgl and the par complex in flies and mice. Dev. Cell 8, 43–52. doi: 10.1016/j.devcel.2004.10.020
Zhang, P., Abdelmohsen, K., Liu, Y., Tominaga-Yamanaka, K., Yoon, J.-H., Ioannis, G., et al. (2015). Novel RNA- and FMRP-binding protein TRF2-S regulates axonal mRNA transport and presynaptic plasticity. Nat. Commun. 6:8888. doi: 10.1038/ncomms9888
Zhang, W., Cheng, Y., Li, Y., Chen, Z., Jin, P., and Chen, D. (2014). A feed-forward mechanism involving Drosophila fragile X mental retardation protein triggers a replication stress-induced DNA damage response. Hum. Mol. Genet. 23, 5188–5196. doi: 10.1093/hmg/ddu241
Zhang, Y. Q., Bailey, A. M., Matthies, H. J., Renden, R. B., Smith, M. A., Speese, S. D., et al. (2001). Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107, 591–603. doi: 10.1016/S0092-8674(01)00589-X
Keywords: Fragile X Syndrome, FMR1, Fragile X Mental Retardation Protein, Drosophila, dFRM1, neuromuscular junction, mushroom bodies, behavior
Citation: Drozd M, Bardoni B and Capovilla M (2018) Modeling Fragile X Syndrome in Drosophila. Front. Mol. Neurosci. 11:124. doi: 10.3389/fnmol.2018.00124
Received: 29 November 2017; Accepted: 29 March 2018;
Published: 16 April 2018.
Edited by:
Regina Dahlhaus, Friedrich-Alexander-Universität Erlangen-Nürnberg, GermanyReviewed by:
Alicia Mansilla, Instituto Ramón y Cajal de Investigación Sanitaria, SpainCopyright © 2018 Drozd, Bardoni and Capovilla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Bardoni, YmFyZG9uaUBpcG1jLmNucnMuZnI=
Maria Capovilla, bWFyaWEuY2Fwb3ZpbGxhQGlwbWMuY25ycy5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.