 Daniel Sobrido-Cameán
Daniel Sobrido-Cameán Antón Barreiro-Iglesias
Antón Barreiro-Iglesias- Department of Functional Biology, CIBUS, Faculty of Biology, Universidade de Santiago de Compostela, Santiago de Compostela, Spain
Spinal cord injury (SCI) causes the death of neurons and glial cells due to the initial mechanical forces (i.e., primary injury) and through a cascade of secondary molecular events (e.g., inflammation or excitotoxicity) that exacerbate cell death. The loss of neurons and glial cells that are not replaced after the injury is one of the main causes of disability after SCI. Evidence accumulated in last decades has shown that the activation of apoptotic mechanisms is one of the factors causing the death of intrinsic spinal cord (SC) cells following SCI. Although this is not as clear for brain descending neurons, some studies have also shown that apoptosis can be activated in the brain following SCI. There are two main apoptotic pathways, the extrinsic and the intrinsic pathways. Activation of caspase-8 is an important step in the initiation of the extrinsic pathway. Studies in rodents have shown that caspase-8 is activated in SC glial cells and neurons and that the Fas receptor plays a key role in its activation following a traumatic SCI. Recent work in the lamprey model of SCI has also shown the retrograde activation of caspase-8 in brain descending neurons following SCI. Here, we review our current knowledge on the role of caspase-8 and the Fas pathway in cell death following SCI. We also provide a perspective for future work on this process, like the importance of studying the possible contribution of Fas/caspase-8 signaling in the degeneration of brain neurons after SCI in mammals.
Introduction
Spinal cord injury (SCI) can cause permanent disability due to the dysfunction of motor, autonomic and sensory systems. There are also high economical costs associated with the care of SCI patients. In the USA, the lifetime cost of a SCI patient is between 1.1 and 4.6 million US dollars (National Spinal Cord Injury Statistical Center, 2016). So, it is of crucial importance to develop new and effective treatments for SCI patients. Nowadays, only a few treatments have been translated to the clinic: a treatment with methylprednisolone (which is still controversial), hypertensive therapy and early decompressive surgery (for a recent review see Ulndreaj et al., 2017). These treatments aim to stop further degeneration after SCI, but they only lead to limited improvements. One of the main causes of permanent deficits after SCI is due to the loss of cells (oligodendrocytes and neurons) that are not effectively replaced after the injury. Regeneration strategies are difficult to implement due to the complexity of the central nervous system; therefore, the development of neuroprotective therapies is one of the most promising strategies for clinical translation.
SCI has been divided in two stages, the primary and secondary injuries. The primary injury is caused by the mechanical forces of the traumatic event. Following the primary injury, a molecular cascade of secondary events is initiated, which expands the damage even to tissue that was not directly affected by the primary injury. Secondary injury events star within seconds of the occurrence of the primary injury and delay and progress over time. Inflammatory cells enter the injury site due to the disruption of the blood-spinal cord (SC) barrier and trigger the release of cytokines and reactive oxygen species (reviewed by Ahuja et al., 2017). Excitatory amino acids like glutamate are also massively released (Fernández-López et al., 2014, 2016) leading to elevated intracellular calcium levels. These processes cause the loss of cells by necrotic and apoptotic mechanisms. The final outcome of a SCI will depend on the extent of secondary damage; therefore, understanding the molecular pathways that lead to its progression will benefit the development of neuroprotective therapies for SCI patients.
Apoptosis is a process that occurs during development or aging and as a homeostatic mechanism to maintain cell populations in different tissues, but it is also activated after tissue damage. Cell death during secondary injury after SCI is caused in part by the activation of apoptotic mechanisms (Crowe et al., 1997; Shuman et al., 1997; Emery et al., 1998). There are two main apoptotic pathways: the extrinsic or death receptor pathway and the intrinsic or mitochondrial pathway. The extrinsic pathway involves the activation of death receptors, which leads to the activation of initiator caspases like caspase-8 or caspase-10. Several studies have shown the activation of caspase-8 in intrinsic SC cells following SCI in rodents (Casha et al., 2001, 2005; Keane et al., 2001; Takagi et al., 2003; Cantarella et al., 2010; Chen et al., 2011). More recently, work in lampreys has also shown that caspase-8 is retrogradely activated in identifiable descending brain neurons after SCI (Barreiro-Iglesias and Shifman, 2012, 2015; Barreiro-Iglesias et al., 2017). Here, we review our current knowledge on the role of caspase-8 in cell death after SCI. Since the activation of Fas receptors (also known as CD95 or APO-1) plays an important role in this process, we also focused our review on the role of this signaling pathway in caspase-8 activation following SCI. Finally, we propose new lines of work to advance our knowledge on the role of caspase-8 and Fas in cell death after SCI.
Activation of the Fas/Caspase-8 Apoptotic Pathway in the Spinal Cord
Procaspase-8 is an initiator caspase that can process itself after ligation with the Fas-tumor necrosis factor family of death receptors (Kischkel et al., 1995). After biding of the Fas-ligand [FasL (or CD95L)], the Fas receptor (a 45 kDa membrane receptor) forms a death-inducing signaling complex (DISC) with the adaptor protein FADD (a member of the death domain superfamily) and procaspase-8. Then, activated caspase-8 can initiate downstream cleavage of caspase-3, among other targets, by direct or mitochondrial-dependent mechanisms (see Figure 1A). Some studies have also shown that caspase-8 activation after SCI can be mediated through other members of the TNF receptor superfamily (Cantarella et al., 2010; Chen et al., 2011), but we have focused our review on the role of FasL/Fas in caspase-8 activation after SCI.
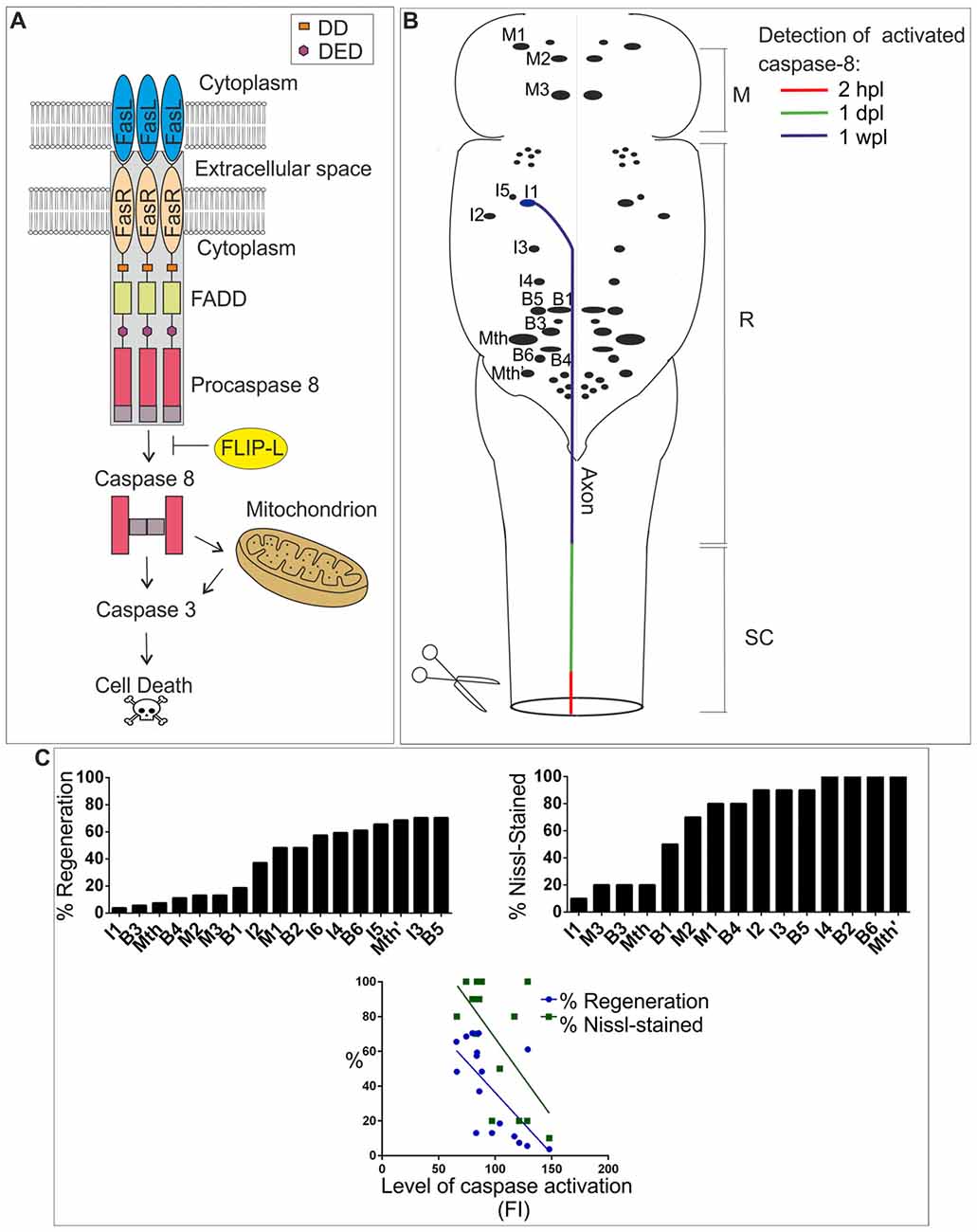
Figure 1. (A) FasL/Fas/caspase-8 signaling pathway. Binding of FasL (expressed in neurons, glial cells or immune system cells) to Fas (expressed in neurons or glial cells) induces oligomerization of the receptor, which causes the activation of the internal domain of Fas triggering FADD binding. The adaptor protein FADD binds to Fas via homophilic DD interactions. FADD recruits procaspase-8, which binds to FADD through the DED, causing the formation of death-inducing signaling complex (DISC; gray box). The formation of DISC is followed by cleavage of procaspase-8 into large and small subunits. Two large and two small subunits associate with each other to form an active caspase-8 heterodimer. The resulting mature caspase-8 is released to the cytosol and initiates downstream apoptosis directly by activating caspase-3 or indirectly through the mitochondrial pathway. FLIP-L can inhibit the activation of procaspase-8. (B) Schematic drawing of a dorsal view of the sea lamprey brainstem showing the location of identifiable descending neurons (for most neurons only the soma is represented). The I1 neuron is used as an example to show the progressive detection of activated caspase-8 after a complete spinal cord injury (SCI; from Barreiro-Iglesias and Shifman, 2015). Activated caspase-8 is detected first in the injured axon at the site of injury (within 2 h post-lesion, hpl), then in the axon at rostral SC levels (within 1 day post-lesion, dpl) and finally in the soma of descending neurons (1 week post-lesion, wpl). This timing of caspase-8 activation has been color-coded in the I1 neuron/axon. Rostral is to the top and the SCI site to the bottom. Abbreviations: M, Mesencephalon; R, Rhombencephalon; SC, Spinal cord. (C) The top graphs show the regenerative and survival abilities of identifiable descending neurons of lampreys. The regenerative ability is expressed as the percentage of times a given neuron regenerates its axon 5 mm below the site of injury 10 weeks after a complete SCI (from Jacobs et al., 1997). The survival ability is expressed as the percentage of times that a given neuron shows Nissl staining 1 year after a complete SCI (from Shifman et al., 2008). The bottom graph shows a significant correlation between the level of activated caspases (fluorescence intensity, FI) 2 wpl (from Barreiro-Iglesias et al., 2017) and the regenerative (from Jacobs et al., 1997) and survival abilities (from Shifman et al., 2008) of identifiable descending neurons. P-values of Pearson correlation are 0.0044 (Barreiro-Iglesias et al., 2017) and 0.0122, respectively.
One of the first reports showing that the Fas/caspase-8 pathway is activated after SC damage came from a study using a model of ischemic SCI (Matsushita et al., 2000). In this study, the authors developed a model of SC ischemia in mice by clamping the left subclavian artery. After ischemia, the number of Fas-positive neurons and the intensity of Fas-immunoreactivity increased. Also, ischemia induced the formation of a complex between Fas and procaspase-8 in the SC suggesting that ischemia induces the formation of DISC. Ischemia also induced and increase in procaspase-8 expression and caspase-8 cleavage/activation. Specifically, activated caspase-8 was detected in neurons (Matsushita et al., 2000). These authors did not establish how the ischemic damage leads to the formation of DISC and caspase-8 activation in neurons. But, interestingly, after cerebral ischemia there is an upregulation of Fas and the FasL (Martin-Villalba et al., 1999), suggesting that FasL release after SC ischemia could induce the formation of DISC and caspase-8 activation in neurons. This work has important implications for traumatic SCI, because the disruption of blood-vessels after SCI can also cause secondary ischemic damage.
The first reports demonstrating caspase-8 activation in intrinsic SC cells following a traumatic SCI came in 2001 from two studies by Casha et al. (2001) and Keane et al. (2001). The study by Keane et al. (2001) showed that a contusion injury at T9-T10 in rats leads to the appearance of caspase-8 immunoreactivity 6 h after the injury in neurons of the gray matter and in cells of the white matter (possibly oligodendrocytes). Immunoblots showed that this immunoreactivity correspond to the expression of the cleaved subunit of caspase-8 (Keane et al., 2001). In the same year, Casha et al. (2001) reported the first results showing the possible involvement of Fas receptors in cell death following a cervical SCI (the most common level of human SCI). These authors showed that, in rats, a clip compression C7-T1 SCI caused cell death in the SC. Apoptotic cells were mainly oligodendrocytes located along degenerating axons. Double immunohistochemistry with Fas and TUNEL revealed the presence of Fas-positive dying glia after the injury (Casha et al., 2001). Expression of FasL was observed in astrocytes and microglia. Interestingly, the appearance of Fas expression after the injury in dying glia correlated with increased levels of activated caspase-8 as revealed by western blots. Moreover, levels of FLIP-L (caspase-8 inhibitor; Figure 1) decreased after SCI at time points in which caspase-8 activation was observed (Casha et al., 2001). Similar results were later reported in mice after a T9-T10 contusion injury (Takagi et al., 2003). These authors showed that caspase-8 enzyme activity increased in the SC of mice after SCI (Takagi et al., 2003).
These earlier results suggested that activation of Fas after SCI could lead to activation of caspase-8 and cell death. However, the first true experimental demonstration showing that the activation of Fas leads to apoptosis following SCI came in 2004 with the studies by Demjen et al. (2004) and Yoshino et al. (2004). Demjen et al. (2004) showed that an acute treatment with neutralizing antibodies against FasL (CD95L) reduced neuronal apoptosis (TUNEL), promoted regeneration of corticospinal tract fibers and improved functional recovery in mice with a dorsal transection of the SC (two-thirds of the cord were transected) at T8-T9. Yoshino et al. (2004) also showed that locomotor recovery, after a contusion SCI, was improved in Fas-deficient mutant mice and that this correlated with reduced tissue damage and apoptosis. In Fas-deficient mice, fewer TUNEL positive cells undergoing apoptosis were observed (mainly neurons, although also oligodendrocytes and astrocytes). This correlated with the presence of Fas-expressing neurons after the injury in control mice and the presence of FasL-positive cells both in Fas-deficient and control mice (Yoshino et al., 2004). These functional studies confirmed that biding of the FasL to Fas receptors causes cell death after SCI and that inhibition of this signaling pathway could be a valuable therapeutic target for SCI patients.
Similar results were then obtained by Casha et al. (2005) using a model of T5-T6 clip compression SCI in mice. These authors detected post-traumatic apoptosis (activated caspase-8 and TUNEL) in neurons and oligodendrocytes. Apoptosis was reduced in FasLpr/Lpr mutant mice. However, in contrast to the results of Yoshino et al. (2004), the reduction in apoptosis was mainly observed in oligodendrocytes. Fas deficiency led to improved locomotor recovery after SCI, which was associated to increased axonal sparing and a significant improvement in white matter myelin (Casha et al., 2005). Whether Fas leads to caspase-8 activation and apoptotic death mainly in neurons or oligodendrocytes might depend on the type of injury (contusion/transection vs. clip compression, which causes a more severe ischemia).
Subsequent work has confirmed that neutralization of Fas signaling is beneficial for recovery after SCI (Ackery et al., 2006; Robins-Steele et al., 2012). Authors of these studies developed a treatment with soluble Fas receptors to improve recovery after SCI in rats. An immediate treatment with a soluble Fas receptor after a clip compression injury at C7-T1 in rats reduced the number of TUNEL positive cells 5 days post-injury and the expression of activated caspase-3 7 days post-injury (Ackery et al., 2006). This correlated with enhanced survival of neurons and oligodendrocytes, increased axonal integrity and improved behavioral recovery (Ackery et al., 2006). Results from this work were then confirmed by Robins-Steele et al. (2012). These authors showed that a delayed treatment (which is more clinically relevant) with a soluble Fas receptor 8–24 h after a clip compression injury at C7-T1 in rats enhances oligodendrocyte and neuronal survival, reduces cavity size and improves behavioral recovery (Robins-Steele et al., 2012).
A recent study has also shown that the transgenic overexpression of p45 (another member of the death domain superfamily) increases neuronal survival, decreases retraction of corticospinal tract fibers and improves functional recovery after a transection SCI at T9 in mice (Sung et al., 2013). p45 is able to form a complex with FADD attenuating FasL-induced caspase-8 activation and cell death caused by SCI (Sung et al., 2013).
The studies in rodent models have important implications for human SCI, because the Fas pathway is also activated in primates, including humans, after SCI (Jia et al., 2011; Yu and Fehlings, 2011). In rhesus monkeys, a T11 SC hemisection induced an increase in Fas and FasL immunoreactivity in the ventral horn at time points in which the number of apoptotic TUNEL positive cells also increased (Jia et al., 2011). A large number of Fas and FasL immunoreactive neurons and glial cells also accumulated at the injury epicenter in SCs from acutely injured human patients, while these were rarely observed in control or chronically injured SCs (Yu and Fehlings, 2011). This correlated with the appearance of TUNEL and active caspase positive cells in the SC of acutely injured patients. Moreover, double immunolabeling revealed the presence of Fas or FasL and caspase-3 positive cells and of Fas or FasL expressing macrophages/neutrophils (Yu and Fehlings, 2011). These studies highlight the importance of understanding apoptotic processes to develop effective therapies for patients with SCI. As shown in this section, neutralization of FasL/Fas signaling (antibodies or soluble receptors) leads to improvements in behavioral recovery after SCI in rodent models (Demjen et al., 2004; Ackery et al., 2006; Robins-Steele et al., 2012; Table 1). In addition, several potential treatments that have been effective in animal models reduced FasL/Fas signaling and/or caspase-8 activation (Table 1), indicating that this can be a key target when developing neuroprotective therapies for SCI patients. This includes therapies that have been translated to the clinic or that are in clinical trials for SCI patients (Table 1).
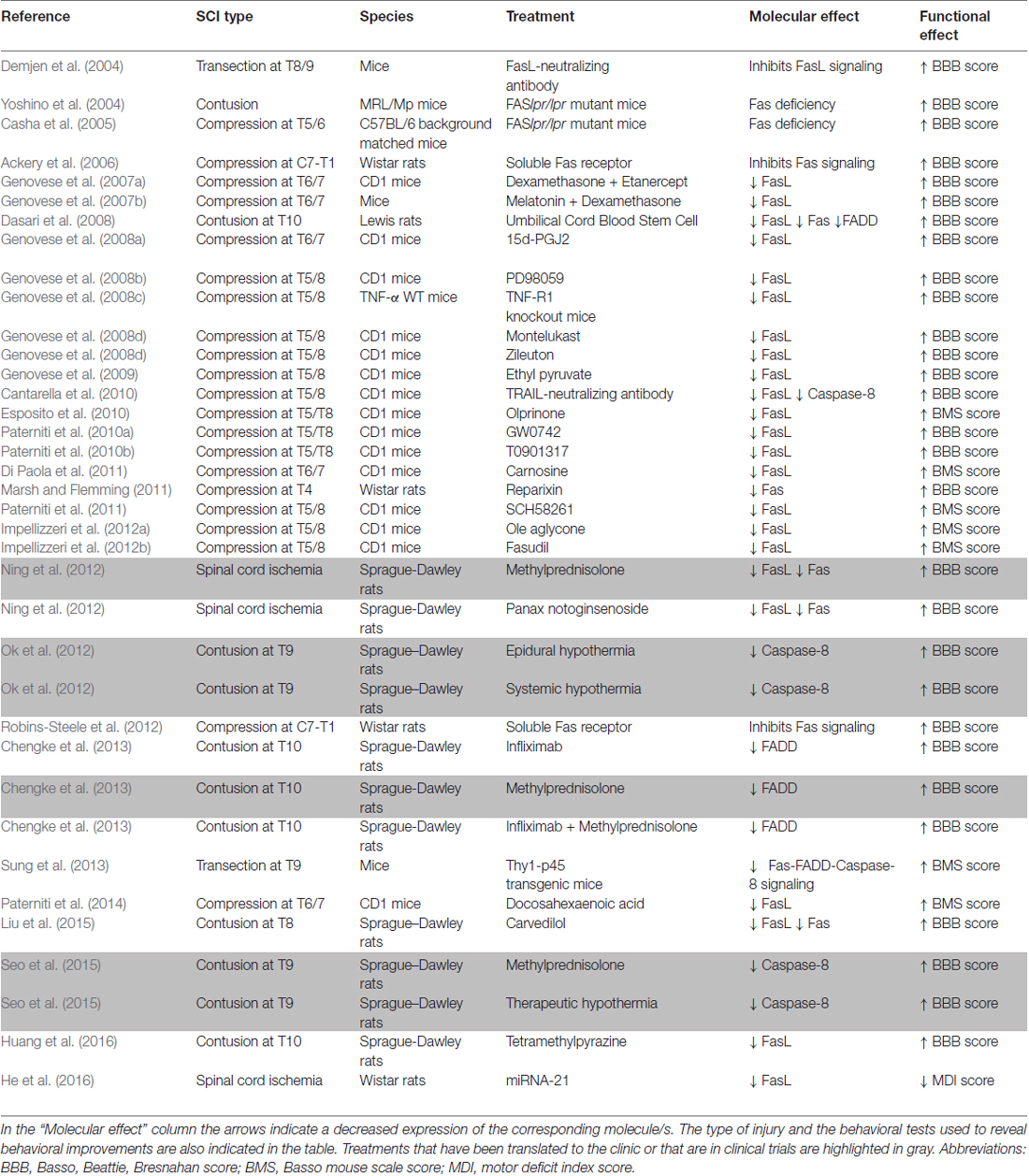
Table 1. Table showing treatments (genetic or pharmacological) used by different authors as potential therapies for SCI and that have been shown to reduce FasL/Fas signaling and/or caspase-8 activation in animal models.
Activation of Caspase-8 in the Brain
As shown in the previous section, most studies focused on the role of caspase-8 and Fas in apoptosis of intrinsic SC cells. But, some of these studies also showed that the manipulation of FasL/Fas signaling leads to a significant increase in the regeneration/preservation of descending axons (Demjen et al., 2004; Casha et al., 2005; Ackery et al., 2006; Sung et al., 2013). These results suggest that inhibition of Fas signaling could be beneficial to preserve brain descending neurons and innervation after SCI. However, even with this evidence, no study in mammalian models has yet looked at the expression of Fas receptors or activated caspase-8 in descending neurons of the brain after SCI. We should take into account that there is still controversy on the topic of cell death in the brain following SCI. Several studies have shown the death of brain neurons after SCI in mammals, including humans (Holmes and May, 1909; Feringa and Vahlsing, 1985; Fry et al., 2003; Hains et al., 2003; Wu et al., 2003; Lee et al., 2004; Klapka et al., 2005). However, two recent studies in rats did not find any evidence of the death of corticospinal neurons after SCI (Nielson et al., 2010, 2011). The study by Nielson et al. (2011) suggested that corticospinal neurons suffer atrophy after SCI but do not die. The death/atrophy of descending neurons appears to involve and apoptotic mechanism as revealed by the appearance of TUNEL staining (although TUNEL can also label necrotic cells in some instances) and activated caspase-3 immunoreactivity in descending neurons of the brain (Hains et al., 2003; Wu et al., 2003; Lee et al., 2004).
In contrast to mammals, lampreys show an amazing capacity for functional recovery following SCI. The regeneration of descending neurons is a key event in the recovery of swimming after SCI in lampreys (see Shifman et al., 2007; Rodicio and Barreiro-Iglesias, 2012). Regenerated descending axons of lampreys are able to establish new synapses with their target neurons below the site of injury and the recovery of function depends, among other events, on the re-establishment of these connections (see Shifman et al., 2007). However, even in lampreys not all descending neurons are able to regenerate their axon after a complete SCI. The lamprey brainstem contains several individually identifiable descending neurons (Figure 1B) that vary greatly in their regenerative abilities after SCI (Figure 1C; Davis and McClellan, 1994; Jacobs et al., 1997; Barreiro-Iglesias et al., 2014). Some of these neurons are classified as “good regenerators” (i.e., they regenerate their axon more than 55% of the times) and others are considered “bad regenerators” (i.e., they regenerate their axon less than 30% of the times; Figure 1C; Jacobs et al., 1997). In recent years, mounting evidence has confirmed that descending neurons of lampreys known to be “bad regenerators” suffer a process of delayed death and are also “poor survivors” after a complete SCI (Figure 1C; Shifman et al., 2008; Barreiro-Iglesias and Shifman, 2012, 2015; Busch and Morgan, 2012; Hu et al., 2013, 2017; Zhang et al., 2014; Barreiro-Iglesias, 2015; Fogerson et al., 2016; Barreiro-Iglesias et al., 2017). The occurrence of cell death in a subset of identifiable descending neurons after SCI in lampreys was confirmed based on the disappearance of Nissl staining (Figure 1C), the loss of neurofilament expression, the absence of labeling when using retrograde tracers (Shifman et al., 2008), and the early staining of these neurons with Fluoro-Jade C (Busch and Morgan, 2012; Barreiro-Iglesias et al., 2017), which is a marker for degenerating neurons. In addition, the appearance of TUNEL staining (Shifman et al., 2008; Hu et al., 2013) and activated caspases (Figure 1C; Barreiro-Iglesias and Shifman, 2012, 2015; Hu et al., 2013; Barreiro-Iglesias et al., 2017) in the soma of axotomized descending neurons suggests that their death after SCI is apoptotic. The detection of activated caspase-8 in the first 2 weeks after the injury (Figure 1B; Barreiro-Iglesias and Shifman, 2012; Barreiro-Iglesias et al., 2017) and the lack of cytochrome-c release from mitochondria (Barreiro-Iglesias et al., 2017) indicates that the extrinsic apoptotic pathway is activated in descending neurons of lampreys after SCI. Caspase-8 activation in the soma of descending neurons of lampreys is preceded by the activation of caspases in the axotomized axons at the lesion site within the first hours after the injury (Figure 1A; Barreiro-Iglesias and Shifman, 2015; Barreiro-Iglesias et al., 2017). Caspase activation is progressively detected in descending axons at higher spinal levels and finally in the soma of descending neurons after SCI (Figure 1A; Barreiro-Iglesias and Shifman, 2015; Barreiro-Iglesias et al., 2017), which indicates that the degenerative process is initiated in the damaged axon in the SC. Taxol, which improves recovery from SCI in mammalian models (Hellal et al., 2011), prevented the appearance of activated caspase-8 in the soma of descending neurons (Barreiro-Iglesias et al., 2017) of lampreys. This indicates that death signals (or caspase-8 itself) are retrogradely transported by microtubules to the soma of descending neurons after SCI in lampreys.
The work in lampreys suggests that activated caspase-8 could also play a role in the death of descending neurons of mammals after SCI. Interestingly, in olfactory neurons of mice, appearance of activated caspase-8 in the cell body after olfactory bulbectomy depends on microtubule-based retrograde transport of activated caspase-8 by its association with dynactin p150Glued (Carson et al., 2005), which might reveal conserved mechanisms with descending neurons of lampreys. In addition, the preservation of axonal projections in the SC after experimental inhibition of Fas signaling indicates that Fas receptors could play a role in the activation of caspase-8 in descending neurons after SCI.
Conclusion
Several reports have confirmed that the activation of caspase-8 and the extrinsic apoptotic pathway is one of the mechanisms causing cell death after SCI and that the FasL/Fas signaling pathway plays a key role in this process. Surprisingly, no study has yet attempted to directly inhibit caspase-8 after SCI. This experimental approach could be of interest, especially because caspase-8 activation is not only caused by Fas receptor activation, which could lead to further improvements in recovery from SCI. Also, another aim for future work, and based on the lamprey results, should be to investigate the possible activation of caspase-8 in descending neurons of mammals after SCI and the implication of Fas signaling and microtubule retrograde transport in this process. Finally, translation of all this knowledge to pre-clinical studies or even clinical trials is of obvious and crucial importance.
Author Contributions
AB-I wrote the manuscript with help from DS-C. DS-C prepared the figure and the table. All the work was supervised by AB-I.
Funding
This work was supported by a grant from the Spanish Ministry of Economy and Competitiveness and the European Regional Development Fund 2007–2013 (BFU2014-56300-P). AB-I was supported by a grant from the Xunta de Galicia (2016-PG008) and a grant from the crowdfunding platform Precipita (FECYT; Spanish Ministry of Economy and Competitiveness; grant number 2017-CP081).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the following individual donors of the crowdfunding campaign in Precipita: Emilio Río, Guillermo Vivar, Pablo Pérez, Jorge Férnandez, Ignacio Valiño, Pago de los Centenarios, Eva Candal, María del Pilar Balsa, Jorge Faraldo, Isabel Rodríguez-Moldes, José Manuel López, Juan José Pita, María E. Cameán, Jesús Torres, José Pumares, Verónica Rodríguez, Sara López, Tania Villares Balsa, Rocío Lizcano, José García, Ana M. Cereijo, María Pardo, Nerea Santamaría, Carolina Hernández, Jesús López and María Maneiro.
References
Ackery, A., Robins, S., and Fehlings, M. G. (2006). Inhibition of Fas-mediated apoptosis through administration of soluble Fas receptor improves functional outcome and reduces posttraumatic axonal degeneration after acute spinal cord injury. J. Neurotrauma 23, 604–616. doi: 10.1089/neu.2006.23.604
Ahuja, C. S., Wilson, J. R., Nori, S., Kotter, M. R., Druschel, C., Curt, A., et al. (2017). Traumatic spinal cord injury. Nat. Rev. Dis. Primers 3:17018. doi: 10.1038/nrdp.2017.18
Barreiro-Iglesias, A. (2015). “Bad regenerators” die after spinal cord injury: insights from lampreys. Neural Regen. Res. 10, 25–27. doi: 10.4103/1673-5374.150642
Barreiro-Iglesias, A., and Shifman, M. I. (2012). Use of fluorochrome-labeled inhibitors of caspases to detect neuronal apoptosis in the whole-mounted lamprey brain after spinal cord injury. Enzyme Res. 2012:835731. doi: 10.1155/2012/835731
Barreiro-Iglesias, A., and Shifman, M. I. (2015). Detection of activated caspase-8 in injured spinal axons by using fluorochrome-labeled inhibitors of caspases (FLICA FLICA). Methods Mol. Biol. 1254, 329–339. doi: 10.1007/978-1-4939-2152-2_23
Barreiro-Iglesias, A., Sobrido-Cameán, D., and Shifman, M. I. (2017). Retrograde activation of the extrinsic apoptotic pathway in spinal-projecting neurons after a complete spinal cord injury in lampreys. Biomed Res. Int. 2017:5953674. doi: 10.1155/2017/5953674
Barreiro-Iglesias, A., Zhang, G., Selzer, M. E., and Shifman, M. I. (2014). Complete spinal cord injury and brain dissection protocol for subsequent wholemount in situ hybridization in larval sea lamprey. J. Vis. Exp. 14:e51494. doi: 10.3791/51494
Busch, D. J., and Morgan, J. R. (2012). Synuclein accumulation is associated with cell-specific neuronal death after spinal cord injury. J. Comp. Neurol. 520, 1751–1771. doi: 10.1002/cne.23011
Cantarella, G., Di Benedetto, G., Scollo, M., Paterniti, I., Cuzzocrea, S., Bosco, P., et al. (2010). Neutralization of tumor necrosis factor-related apoptosis-inducing ligand reduces spinal cord injury damage in mice. Neuropsychopharmacology 35, 1302–1314. doi: 10.1038/npp.2009.234
Carson, C., Saleh, M., Fung, F. W., Nicholson, D. W., and Roskams, A. J. (2005). Axonal dynactin p150Glued transports caspase-8 to drive retrograde olfactory receptor neuron apoptosis. J. Neurosci. 25, 6092–6104. doi: 10.1523/JNEUROSCI.0707-05.2005
Casha, S., Yu, W. R., and Fehlings, M. G. (2001). Oligodendroglial apoptosis occurs along degenerating axons and is associated with FAS and p75 expression following spinal cord injury in the rat. Neuroscience 103, 203–218. doi: 10.1016/s0306-4522(00)00538-8
Casha, S., Yu, W. R., and Fehlings, M. G. (2005). FAS deficiency reduces apoptosis, spares axons and improves function after spinal cord injury. Exp. Neurol. 196, 390–400. doi: 10.1016/j.expneurol.2005.08.020
Chen, K. B., Uchida, K., Nakajima, H., Yayama, T., Hirai, T., Watanabe, S., et al. (2011). Tumor necrosis factor-α antagonist reduces apoptosis of neurons and oligodendroglia in rat spinal cord injury. Spine 36, 1350–1358. doi: 10.1097/BRS.0b013e3181f014ec
Chengke, L., Weiwei, L., Xiyang, W., Ping, W., Xiaoyang, P., Zhengquan, X., et al. (2013). Effect of infliximab combined with methylprednisolone on expressions of NF-κB, TRADD, and FADD in rat acute spinal cord injury. Spine 38, 861–869. doi: 10.1097/BRS.0b013e318294892c
Crowe, M. J., Bresnahan, J. C., Shuman, S. L., Masters, J. N., and Crowe, M. S. (1997). Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat. Med. 3, 73–76. doi: 10.1038/nm0197-73
Dasari, V. R., Spomar, D. G., Li, L., Gujrati, M., Rao, J. S., and Dinh, D. H. (2008). Umbilical cord blood stem cell mediated downregulation of fas improves functional recovery of rats after spinal cord injury. Neurochem. Res. 33, 134–149. doi: 10.1007/s11064-007-9426-6
Davis, G. R. Jr., and McClellan, A. D. (1994). Extent and time course of restoration of descending brainstem projections in spinal cord-transected lamprey. J. Comp. Neurol. 344, 65–82. doi: 10.1002/cne.903440106
Demjen, D., Klussmann, S., Kleber, S., Zuliani, C., Stieltjes, B., Metzger, C., et al. (2004). Neutralization of CD95 ligand promotes regeneration and functional recovery after spinal cord injury. Nat. Med. 10, 389–395. doi: 10.1038/nm1007
Di Paola, R., Impellizzeri, D., Salinaro, A. T., Mazzon, E., Bellia, F., Cavallaro, M., et al. (2011). Administration of carnosine in the treatment of acute spinal cord injury. Biochem. Pharmacol. 82, 1478–1489. doi: 10.1016/j.bcp.2011.07.074
Emery, E., Aldana, P., Bunge, M. B., Puckett, W., Srinivasan, A., Keane, R. W., et al. (1998). Apoptosis after traumatic human spinal cord injury. J. Neurosurg. 89, 911–920. doi: 10.3171/jns.1998.89.6.0911
Esposito, E., Mazzon, E., Paterniti, I., Impellizzeri, D., Bramanti, P., and Cuzzocrea, S. (2010). Olprinone attenuates the acute inflammatory response and apoptosis after spinal cord trauma in mice. PLoS One 5:e12170. doi: 10.1371/journal.pone.0012170
Feringa, E. R., and Vahlsing, H. L. (1985). Labeled corticospinal neurons one year after spinal cord transection. Neurosci. Lett. 58, 283–286. doi: 10.1016/0304-3940(85)90067-9
Fernández-López, B., Barreiro-Iglesias, A., and Rodicio, M. C. (2016). Anatomical recovery of the spinal glutamatergic system following a complete spinal cord injury in lampreys. Sci. Rep. 6:37786. doi: 10.1038/srep37786
Fernández-López, B., Valle-Maroto, S. M., Barreiro-Iglesias, A., and Rodicio, M. C. (2014). Neuronal release and successful astrocyte uptake of aminoacidergic neurotransmitters after spinal cord injury in lampreys. Glia 62, 1254–1269. doi: 10.1002/glia.22678
Fogerson, S. M., van Brummen, A. J., Busch, D. J., Allen, S. R., Roychaudhuri, R., Banks, S. M., et al. (2016). Reducing synuclein accumulation improves neuronal survival after spinal cord injury. Exp. Neurol. 278, 105–115. doi: 10.1016/j.expneurol.2016.02.004
Fry, E. J., Stolp, H. B., Lane, M. A., Dziegielewska, K. M., and Saunders, N. R. (2003). Regeneration of supraspinal axons after complete transection of the thoracic spinal cord in neonatal opossums (Monodelphis domestica). J. Comp. Neurol. 466, 422–444. doi: 10.1002/cne.10904
Genovese, T., Esposito, E., Mazzon, E., Di Paola, R., Meli, R., Caminiti, R., et al. (2009). Beneficial effects of ethyl pyruvate in a mouse model of spinal cord injury. Shock 32, 217–227. doi: 10.1097/SHK.0b013e31818d4073
Genovese, T., Esposito, E., Mazzon, E., Di Paola, R., Muia, C., Meli, R., et al. (2008a). Effect of cyclopentanone prostaglandin 15-deoxy-Δ12, 14PGJ2 on early functional recovery from experimental spinal cord injury. Shock 30, 142–152. doi: 10.1097/SHK.0b013e31815dd381
Genovese, T., Esposito, E., Mazzon, E., Muià, C., Di Paola, R., Meli, R., et al. (2008b). Evidence for the role of mitogen-activated protein kinase signaling pathways in the development of spinal cord injury. J. Pharmacol. Exp. Ther. 325, 100–114. doi: 10.1124/jpet.107.131060
Genovese, T., Mazzon, E., Crisafulli, C., Di Paola, R., Muià, C., Esposito, E., et al. (2008c). TNF-α blockage in a mouse model of SCI: evidence for improved outcome. Shock 29, 32–41. doi: 10.1097/shk.0b013e318059053a
Genovese, T., Rossi, A., Mazzon, E., Di Paola, R., Muià, C., Caminiti, R., et al. (2008d). Effects of zileuton and montelukast in mouse experimental spinal cord injury. Br. J. Pharmacol. 153, 568–582. doi: 10.1038/sj.bjp.0707577
Genovese, T., Mazzon, E., Crisafulli, C., Esposito, E., Di Paola, R., Muià, C., et al. (2007a). Combination of dexamethasone and etanercept reduces secondary damage in experimental spinal cord trauma. Neuroscience 150, 168–181. doi: 10.1016/j.neuroscience.2007.06.059
Genovese, T., Mazzon, E., Crisafulli, C., Esposito, E., Di Paola, R., Muià, C., et al. (2007b). Effects of combination of melatonin and dexamethasone on secondary injury in an experimental mice model of spinal cord trauma. J. Pineal Res. 43, 140–153. doi: 10.1111/j.1600-079x.2007.00454.x
Hains, B. C., Black, J. A., and Waxman, S. G. (2003). Primary cortical motor neurons undergo apoptosis after axotomizing spinal cord injury. J. Comp. Neurol. 462, 328–341. doi: 10.1002/cne.10733
He, F., Ren, Y., Shi, E., Liu, K., Yan, L., and Jiang, X. (2016). Overexpression of microRNA-21 protects spinal cords against transient ischemia. J. Thorac. Cardiovasc. Surg. 152, 1602–1608. doi: 10.1016/j.jtcvs.2016.07.065
Hellal, F., Hurtado, A., Ruschel, J., Flynn, K. C., Laskowski, C. J., Umlauf, M., et al. (2011). Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 331, 928–931. doi: 10.1126/science.1201148
Holmes, G., and May, W. P. (1909). On the exact origin of the pyramidal tracts in man and other mammals. Proc. R. Soc. Med. 2, 92–100. doi: 10.1093/brain/32.1.1
Hu, J., Zhang, G., Rodemer, W., Jin, L. Q., Shifman, M., and Selzer, M. E. (2017). The role of RhoA in retrograde neuronal death and axon regeneration after spinal cord injury. Neurobiol. Dis. 98, 25–35. doi: 10.1016/j.nbd.2016.11.006
Hu, J., Zhang, G., and Selzer, M. E. (2013). Activated caspase detection in living tissue combined with subsequent retrograde labeling, immunohistochemistry or in situ hybridization in whole-mounted lamprey brains. J. Neurosci. Methods 220, 92–98. doi: 10.1016/j.jneumeth.2013.08.016
Huang, J. H., Cao, Y., Zeng, L., Wang, G., Cao, M., Lu, H. B., et al. (2016). Tetramethylpyrazine enhances functional recovery after contusion spinal cord injury by modulation of MicroRNA-21, FasL, PDCD4 and PTEN expression. Brain Res. 1648, 35–45. doi: 10.1016/j.brainres.2016.07.023
Impellizzeri, D., Esposito, E., Mazzon, E., Paterniti, I., Di Paola, R., Bramanti, P., et al. (2012a). The effects of a polyphenol present in olive oil, oleuropein aglycone, in an experimental model of spinal cord injury in mice. Biochem. Pharmacol. 83, 1413–1426. doi: 10.1016/j.bcp.2012.02.001
Impellizzeri, D., Mazzon, E., Paterniti, I., Esposito, E., and Cuzzocrea, S. (2012b). Effect of fasudil, a selective inhibitor of Rho kinase activity, in the secondary injury associated with the experimental model of spinal cord trauma. J. Pharmacol. Exp. Ther. 343, 21–33. doi: 10.1124/jpet.111.191239
Jacobs, A. J., Swain, G. P., Snedeker, J. A., Pijak, D. S., Gladstone, L. J., and Selzer, M. E. (1997). Recovery of neurofilament expression selectively in regenerating reticulospinal neurons. J. Neurosci. 17, 5206–5220.
Jia, L., Yu, Z., Hui, L., Yu-Guang, G., Xin-Fu, Z., Chao, Y., et al. (2011). Fas and FasL expression in the spinal cord following cord hemisection in the monkey. Neurochem. Res. 36, 419–425. doi: 10.1007/s11064-010-0357-2
Keane, R. W., Kraydieh, S., Lotocki, G., Bethea, J. R., Krajewski, S., Reed, J. C., et al. (2001). Apoptotic and anti-apoptotic mechanisms following spinal cord injury. J. Neuropathol. Exp. Neurol. 60, 422–429. doi: 10.1093/jnen/60.5.422
Kischkel, F. C., Hellbardt, S., Behrmann, I., Germer, M., Pawlita, M., Krammer, P. H., et al. (1995). Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 14, 5579–5588.
Klapka, N., Hermanns, S., Straten, G., Masanneck, C., Duis, S., Hamers, F. P., et al. (2005). Suppression of fibrous scarring in spinal cord injuryof rat promotes long-distance regeneration of corticospinal tract axons, rescue of primary motoneurons in somatosensory cortex and significant functional recovery. Eur. J. Neurosci. 22, 3047–3058. doi: 10.1111/j.1460-9568.2005.04495.x
Lee, B. H., Lee, K. H., Kim, U. J., Sohn, J. H., Choi, S. S., Yi, I. G., et al. (2004). Injury in the spinal cord may produce cell death in the brain. Brain Res. 1020, 37–44. doi: 10.1016/j.brainres.2004.05.113
Liu, D., Huang, Y., Li, B., Jia, C., Liang, F., and Fu, Q. (2015). Carvedilol promotes neurological function, reduces bone loss and attenuates cell damage after acute spinal cord injury in rats. Clin. Exp. Pharmacol. Physiol. 42, 202–212. doi: 10.1111/1440-1681.12345
Marsh, D. R., and Flemming, J. M. P. (2011). Inhibition of CXCR1 and CXCR2 chemokine receptors attenuates acute inflammation, preserves gray matter and diminishes autonomic dysreflexia after spinal cord injury. Spinal Cord 49, 337–344. doi: 10.1038/sc.2010.127
Martin-Villalba, A., Herr, I., Jeremias, I., Hahne, M., Brandt, R., Vogel, J., et al. (1999). CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J. Neurosci. 19, 3809–3817.
Matsushita, K., Wu, Y., Qiu, J., Lang-Lazdunski, L., Hirt, L., Waeber, C., et al. (2000). Fas receptor and neuronal cell death after spinal cord ischemia. J. Neurosci. 20, 6879–6887.
National Spinal Cord Injury Statistical Center. (2016). Facts and Figures at a Glance. Birmingham, AL: University of Alabama at Birmingham.
Nielson, J. L., Sears-Kraxberger, I., Strong, M. K., Wong, J. K., Willenberg, R., and Steward, O. (2010). Unexpected survival of neurons of origin of the pyramidal tract after spinal cord injury. J. Neurosci. 30, 11516–11528. doi: 10.1523/JNEUROSCI.1433-10.2010
Nielson, J. L., Strong, M. K., and Steward, O. (2011). A reassessment of whether cortical motor neurons die following spinal cord injury. J. Comp. Neurol. 519, 2852–2869. doi: 10.1002/cne.22661
Ning, N., Dang, X., Bai, C., Zhang, C., and Wang, K. (2012). Panax notoginsenoside produces neuroprotective effects in rat model of acute spinal cord ischemia-reperfusion injury. J. Ethnopharmacol. 139, 504–512. doi: 10.1016/j.jep.2011.11.040
Ok, J. H., Kim, Y. H., and Ha, K. Y. (2012). Neuroprotective effects of hypothermia after spinal cord injury in rats: comparative study between epidural hypothermia and systemic hypothermia. Spine 37, 1551–1559. doi: 10.1097/BRS.0b013e31826ff7f1
Paterniti, I., Esposito, E., Mazzon, E., Galuppo, M., Di Paola, R., Bramanti, P., et al. (2010a). Evidence for the role of peroxisome proliferator-activated receptor-β/δ in the development of spinal cord injury. J. Pharmacol. Exp. Ther. 333, 465–477. doi: 10.1124/jpet.110.165605
Paterniti, I., Genovese, T., Mazzon, E., Crisafulli, C., Di Paola, R., Galuppo, M., et al. (2010b). Liver X receptor agonist treatment regulates inflammatory response after spinal cord trauma. J. Neurochem. 112, 611–624. doi: 10.1111/j.1471-4159.2009.06471.x
Paterniti, I., Impellizzeri, D., Di Paola, R., Esposito, E., Gladman, S., Yip, P., et al. (2014). Docosahexaenoic acid attenuates the early inflammatory response following spinal cord injury in mice: in-vivo and in-vitro studies. J. Neuroinflammation 11:6. doi: 10.1186/1742-2094-11-6
Paterniti, I., Melani, A., Cipriani, S., Corti, F., Mello, T., Mazzon, E., et al. (2011). Selective adenosine A2A receptor agonists and antagonists protect against spinal cord injury through peripheral and central effects. J. Neuroinflammation 8:31. doi: 10.1186/1742-2094-8-31
Robins-Steele, S., Nguyen, D. H., and Fehlings, M. G. (2012). The delayed post-injury administration of soluble fas receptor attenuates post-traumatic neural degeneration and enhances functional recovery after traumatic cervical spinal cord injury. J. Neurotrauma 29, 1586–1599. doi: 10.1089/neu.2011.2005
Rodicio, M. C., and Barreiro-Iglesias, A. (2012). Lampreys as an animal model in regeneration studies after spinal cord injury. Rev. Neurol. 55, 157–166.
Seo, J. Y., Kim, Y. H., Kim, J. W., Kim, S. I., and Ha, K. Y. (2015). Effects of therapeutic hypothermia on apoptosis and autophagy after spinal cord injury in rats. Spine 40, 883–890. doi: 10.1097/BRS.0000000000000845
Shifman, M. I., Jin, L. Q., and Selzer, M. E. (2007). “Regeneration in the lamprey spinal cord,” in Model Organisms in Spinal Cord Regeneration (Vol. 1), eds C. G. Becker and T. Becker (Weinheim: Wiley-VCH), 229–262.
Shifman, M. I., Zhang, G., and Selzer, M. E. (2008). Delayed death of identified reticulospinal neurons after spinal cord injury in lampreys. J. Comp. Neurol. 510, 269–282. doi: 10.1002/cne.21789
Shuman, S. L., Bresnahan, J. C., and Beattie, M. S. (1997). Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J. Neurosci. Res. 50, 798–808. doi: 10.1002/(sici)1097-4547(19971201)50:5<798::aid-jnr16>3.3.co;2-#
Sung, T. C., Chen, Z., Thuret, S., Vilar, M., Gage, F. H., Riek, R., et al. (2013). P45 forms a complex with FADD and promotes neuronal cell survival following spinal cord injury. PLoS One 8:e69286. doi: 10.1371/journal.pone.0069286
Takagi, T., Takayasu, M., Mizuno, M., Yoshimoto, M., and Yoshida, J. (2003). Caspase activation in neuronal and glial apoptosis following spinal cord injury in mice. Neurol. Med. Chir. 43, 20–30. doi: 10.2176/nmc.43.20
Ulndreaj, A., Badner, A., and Fehlings, M. G. (2017). Promising neuroprotective strategies for traumatic spinal cord injury with a focus on the differential effects among anatomical levels of injury. Research 6:1907. doi: 10.12688/f1000research.11633.1
Wu, K. L., Chan, S. H., Chao, Y. M., and Chan, J. Y. (2003). Expression of pro-inflammatory cytokine and caspase genes promotes neuronal apoptosis in pontine reticular formation after spinal cord transection. Neurobiol. Dis. 14, 19–31. doi: 10.1016/s0969-9961(03)00078-0
Yoshino, O., Matsuno, H., Nakamura, H., Yudoh, K., Abe, Y., Sawai, T., et al. (2004). The role of Fas-mediated apoptosis after traumatic spinal cord injury. Spine 29, 1394–1404. doi: 10.1097/01.brs.0000129894.34550.48
Yu, W. R., and Fehlings, M. G. (2011). Fas/FasL-mediated apoptosis and inflammation are key features of acute human spinal cord injury: implications for translational, clinical application. Acta Neuropathol. 122, 747–761. doi: 10.1007/s00401-011-0882-3
Keywords: first apoptosis signal receptor, apoptosis antigen 1, cluster of differentiation 95, tumor necrosis factor receptor superfamily member 6, caspase-8, Fas ligand, neuron, oligodendrocyte
Citation: Sobrido-Cameán D and Barreiro-Iglesias A (2018) Role of Caspase-8 and Fas in Cell Death After Spinal Cord Injury. Front. Mol. Neurosci. 11:101. doi: 10.3389/fnmol.2018.00101
Received: 01 February 2018; Accepted: 15 March 2018;
Published: 03 April 2018.
Edited by:
Andrew Paul Tosolini, University College London, United KingdomReviewed by:
Lee J. Martin, Johns Hopkins University, United StatesMichael G. Fehlings, Toronto Western Hospital, Canada
Copyright © 2018 Sobrido-Cameán and Barreiro-Iglesias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antón Barreiro-Iglesias, YW50b24uYmFycmVpcm9AdXNjLmVz