 Xiao-Cen Fan
Xiao-Cen Fan Su Fu
Su Fu Feng-Yu Liu
Feng-Yu Liu Shuang Cui
Shuang Cui Ming Yi
Ming Yi You Wan
You Wan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci., 22 March 2018
Sec. Pain Mechanisms and Modulators
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00085
This article is part of the Research TopicMolecular Mechanisms of NociceptionView all 14 articles
Previous experience of chronic pain causes enhanced responses to upcoming noxious events in both humans and animals, but the underlying mechanisms remain unclear. In the present study, we found that rats with complete Freund’s adjuvant (CFA)-induced chronic inflammatory pain experience exhibited aggravated pain responses to later formalin test. Enhanced neuronal activation upon formalin assaults and increased phosphorylated cAMP-response element binding protein (CREB) were observed in the prelimbic cortex (PL) of rats with chronic inflammatory pain experience, and inhibiting PL neuronal activities reversed the aggravated pain. Inflammatory pain experience induced persistent p38 mitogen-activated protein kinase (MAPK; p38) but not extracellular regulated protein kinase (ERK) or c-Jun N-terminal kinase (JNK) hyperphosphorylation in the PL. Inhibiting the p38 phosphorylation in PL reversed the aggravated nociceptive responses to formalin test and down-regulated enhanced phosphorylated CREB in the PL. Chemogenetics identified PL–periaqueductal gray (PAG) but not PL–nucleus accumbens (NAc) as a key pathway in inducing the aggravated formalin pain. Our results demonstrate that persistent hyperphosphorylation of p38 in the PL underlies aggravated nociceptive responses in rats with chronic inflammatory pain experience.
Chronic pain is one of the most prevalent clinical situations. The experience of chronic pain affects physiological states of the individual, even after the pain has perceptually recovered. A subject with chronic pain experience frequently shows enhanced responses to following noxious events, reflected in lower pain thresholds and increased pain ratings (Bachiocco et al., 1993; Lidow, 2002; Ren et al., 2004; Hermann et al., 2006; Wegner et al., 2015). In animal studies, enhanced formalin-evoked pain behaviors are also observed in adult rats with chronic inflammatory pain experience (Li et al., 2012). However, mechanisms underlying deteriorated pain responses following previous pain experience are not well illustrated.
Medial prefrontal cortex (mPFC) shows long-term morphological and functional changes in chronic pain (Apkarian et al., 2004; Metz et al., 2009; Seminowicz et al., 2009; Lim et al., 2016), which parallel long-term behavioral patterns such as anxiety, stress and depression (Negrón-Oyarzo et al., 2014; Fitzgerald et al., 2015; Moench and Wellman, 2015; Seo et al., 2017). Even when the chronic pain has been well treated, the neuroanatomical abnormality could only be partly reversible (Seminowicz et al., 2011). Prelimbic cortex (PL) is part of the rodent mPFC and plays a critical role in perceptual and emotional aspects of chronic pain (Baliki et al., 2012; Wang et al., 2015; Wu et al., 2016). Excitability of layer 4/5 pyramidal neurons in the PL increases in complete Freund’s adjuvant (CFA)-induced inflammatory mice (Wu et al., 2016), and bilateral lesions of the PL attenuate CFA-induced heat hyperalgesia (Wang et al., 2015). PL may regulate pain through its projections to a number of other brain areas (Kucyi et al., 2013; Khan et al., 2014; Yu et al., 2014; Lee et al., 2015; Vachon-Presseau et al., 2016), including amygdala, nucleus accumbens (NAc), hippocampus and in particular periaqueductal gray (PAG), a critical component of the descending pain modulatory system (Umana et al., 2017).
Several molecular signaling pathways underlie the altered neuronal activities in the PL in chronic pain. The mitogen-activated protein kinase (MAPK), including extracellular regulated protein kinase (ERK), p38 MAPK (p38) and c-Jun N-terminal kinase (JNK), is a family of serine/threonine protein kinases that transduces extracellular stimuli into intracellular post-translational and transcriptional responses (Seger and Krebs, 1995; Widmann et al., 1999; Johnson and Lapadat, 2002; Obata and Noguchi, 2004). Besides its role in chronic stress, depression and fear conditioning (Sherrin et al., 2011; Ferland et al., 2014; Pochwat et al., 2017), the MAPK also contributes to pain hypersensitivity by regulating neuronal plasticity and inflammatory responses in dorsal root ganglion (DRG), spinal cord and cerebral cortex (Impey et al., 1999; Ji and Woolf, 2001; Kumar et al., 2003; Gao and Ji, 2008; Ji et al., 2009). Chronic pain is accompanied by hyperphosphorylation of MAPKs in both peripheral and central nervous system (Zhuang et al., 2005; Crown et al., 2006, 2008; Cao et al., 2014), whereas inhibition of MAPKs impairs neuronal excitability and relieves pain (Hu and Gereau, 2003; Wynne, 2006; Toyoda et al., 2007; Zhang et al., 2007; Pucilowska et al., 2012).
Based on the findings above, we hypothesize that persistent functional alterations in the PL facilitate nociceptive responses to subsequent noxious exposure, even after the perceptual recovery of chronic pain. To test this hypothesis, we examined molecular changes of the PL in rats with experience of CFA-induced chronic inflammatory pain, and observed sustained PL hyper-reactivity which mediated the vulnerability to formalin pain test in these rats.
Adult male Sprague-Dawley rats (230–250 g at the beginning of experiments) were provided by the Department of Laboratory Animal Sciences, Peking University Health Science Center (Beijing, China). All animals were housed in standard cages with a 12-h alternating light/dark cycle and food and water available ad libitum. All experimental procedures were approved by the Animal Care and Use Committee of our University, according to the guidelines of the International Association for the Study of Pain. By the end of the experiment, euthanasia was performed with 1% pentobarbital sodium (1 ml/100 g, i.p.).
Following our previous protocol (Yue et al., 2017; Zhang C. et al., 2017), the rat was anesthetized with isoflurane. The plantar surface of left hindpaw was cleaned by 75% ethanol before a total of 100 μL CFA was injected intraplantarly. For control, an equal volume of normal saline was injected.
The rat was handled for 10 min, and adapted in a plexiglas box for 30 min per day for three consecutive days before the first measurement. Thermal or mechanical pain thresholds were measured as previously described (Chaplan et al., 1994; Zhang et al., 2016) while the rat was calm and awake. Paw withdrawal latencies (PWLs) to thermal stimuli were measured by a focused radiant heat (40 W of power) applied to either hindpaw (Hargreaves Method, IITC 390). PWLs were recorded three times and averaged as the thermal pain threshold. A cut-off value of 30 s was set to avoid any possible tissue injuries.
Fifty percent paw withdrawal thresholds (50% PWTs) to mechanical stimuli were measured by von Frey hairs (0.41–15.1 g; North Coast, Gilroy, CA, USA). The von Frey hair was applied to the central plantar surface of either hindpaw. The 50% PWTs were calculated by the “up and down” method as described by Chaplan et al. and in our lab (Chaplan et al., 1994; Zhang et al., 2016). Thermal hyperalgesia and mechanical allodynia were measured 1 day before and 1, 3, 7, 14, 21 and 28 days after CFA injection.
The rat was handled for 10 min and adapted in the hot plate for 10 min per day for three consecutive days before the test. Rats were placed individually onto the center of the hot plate (49°C) and the latency of the first sign of hind paw licking or jumping to avoid heating pain was recorded as an index of the nociceptive threshold (Luo et al., 2004; Yu et al., 2008). A cut-off value of 30 s was set to avoid any possible tissue injuries. The hot plate was cleaned by 75% ethanol between tests.
The rat was placed in a 100 × 100 × 50 cm box exposed to 50 lux illumination, with its activities videotaped for 10 min (Zhang M. et al., 2017; Zhang Y. et al., 2017). Time spent (C.Time) and distance traveled (C.Dis) in the central area (60 × 60 cm), and total distance traveled (T.Dis) in the field were measured using the SMART software (v2.5.21, Panlab, Harvard Apparatus). The box was cleaned by 75% ethanol between tests.
The elevated plus-maze test was carried out on the next day of the open field test (Li et al., 2017; Zhang M. et al., 2017). The maze was placed 50 cm above the floor in a 30 lux illuminated room and consisted of two open arms and two closed arms (48 × 8 cm, and 40 cm wall height for the closed arms). The rat was placed onto the center area, heading toward the same open arm, and videotaped in the following 10 min. Time spent (O.Time) and numbers of entries (O.Entries) into open arms and total arm entries (T.Entries) were analyzed. The maze was cleaned by 75% ethanol between tests.
Formalin test was performed 30 days after the CFA injection. Rats were handled for 10 min and adapted in a plexiglas chamber for 20 min per day for 3 days before test. The rat received an injection of 100 μL of 5% standard formalin solution into the plantar surface of right hindpaw (the opposite hindpaw of CFA/saline injection), with its behavior videotaped in the following 60 min. Time spent on licking and lifting the formalin injected paw were counted, and the formalin pain score was calculated as previously described: (time lifting + 2 × time licking)/total time (Yi et al., 2011; Zhang et al., 2014). The chamber was cleaned by 75% ethanol between tests.
Brains were removed 45 min after formalin injection for Western blotting and immunostaining, or 90 min after formalin injection for c-Fos immunostaining.
To test anxiety-like behaviors of rats after the formalin injection, open field and elevated plus-maze tests were performed 1 and 2 days after formalin injection, respectively.
Capsaicin test was performed 30 days after the CFA injection. The plantar surface of right hindpaw was cleaned by 75% ethanol before a total of 5 μg (0.1 μg/μl) capsaicin was injected intraplantarly (Chen et al., 2006). Evoked nociceptive responses were measured by focused radiant heat (as described above) 15, 30, 60, 90 and 120 min after capsaicin injection (Soliman et al., 2005).
The rat was anesthetized deeply with 1% sodium pentobarbital (0.5 ml/100 g, i.p.) and positioned in a stereotaxic frame (RWD, Shenzhen, China). A guide cannula (O.D. 0.48 mm/I.D. 0.34 mm, C.C 1.2 mm, RWD, Shenzhen, China) was implanted 1.5 mm above PL [anterior-posterior (AP) + 2.9 mm; medial lateral (ML) ± 0.6 mm from Bregma; dorsal-ventral (DV) −2.5 mm from brain surface] (Zhang et al., 2016). Four skull screws were used for securing the guide cannula to the skull surface with dental acrylic. The matching cap (0.5 mm below the guide cannula, RWD, Shenzhen, China) was inserted into the guide cannula. All animals were given at least 1 week for recovery from surgery before further experiments. The injection needle (1.5 mm below the guide cannula, RWD, Shenzhen, China) was used for microinjection with a polyethylene catheter connecting a micro-syringe. GABAAR agonist muscimol (1 μg/μl, 0.5 μl/side, Tocris Bioscience), p38 inhibitor SB203580 (1 μg/μl, 0.5 μl/side, Sigma-Aldrich) or vehicle (artificial cerebrospinal fluid, aCSF, 0.5 μl/side) was injected into PL of either side over 2 min. The injection needle was held on for at least 2 min to allow drug diffusion. The behavioral tests were performed 30 min after drug/vehicle injection. Rats with incorrect site of the guide cannula were excluded from analysis.
AAV5-CaMKIIα-hM4D(Gi)-mCherry and AAV5-CaMKIIα-mCherry viruses were packaged and purchased from the University of North Carolina Vector Core Facilities (Zhang Y. et al., 2017). AAV virus solution was microinjected into PL (AP +2.7/3.2 mm; ML ± 0.6 mm from Bregma; DV −2.5 mm from brain surface) with 0.5 μl/hole, 2 holes/side, at a speed of 0.1 μl/min after being anesthetized with 1% sodium pentobarbital (0.5 ml/100 g, i.p.). The needle was kept on the site for 3 min to allow for virus diffusion and gradually withdrawn over 1 min to prevent possible leakage from the needle track. The behavioral tests were performed 6 weeks after virus injection.
For local clozapine-N-Oxide (CNO) delivery, a guide cannula (O.D. 0.48 mm/I.D. 0.34 mm, C.C 1.6 mm, RWD, Shenzhen, China) was implanted 2 mm above ventrolateral periaqueductal gray matter (vlPAG; AP −7.8 mm; ML ± 0.8 mm from Bregma; DV −6.0 mm from brain surface), and guide cannula (O.D. 0.48 mm/I.D. 0.34 mm, C.C 3, 0 mm, RWD, Shenzhen, China) was implanted 2 mm above NAc core (AP +1.5 mm; ML ± 1.5 mm from Bregma; DV −7.7 mm from brain surface). CNO (1 mmol/L, 0.5 μl/side, Tocris Bioscience) was injected into vlPAG or NAc core slowly. Behavioral tests were performed 30 min after the CNO injection.
The rat brain was removed, embedded in optimum cutting temperature compound (catalog #0201 08926, Leica), and frozen in liquid nitrogen immediately. PL tissues were taken out by using No.9 puncture needles in a cryostat microtome according to the stereological location (Zheng et al., 2017). Protein was extracted by RIPA (C1053, Applygen Technologies Inc.) and mixed with a 6× loading buffer (DE0105, Beijing BioDee BioTech Corporation Ltd.). Thirty microgram protein in each hole was separated in 10% SDS-PAGE gel and transferred onto polyvinylidene fluoride membrane (ISEQ00010, Merck Millipore). The membrane was blocked with 5% bull serum albumin at room temperature for 1 h, then incubated with rabbit anti-p38 antibody (1:1000, 8690, Cell Signaling Technology), or anti-p-p38 antibody (1:1000, 4511, Cell Signaling Technology), or anti-cAMP-response element binding protein (CREB) antibody (1:1000, 9197, Cell Signaling Technology), or anti-p-CREB antibody (1:1000, 9198, Cell Signaling Technology), or anti-ERK antibody (1:1000, 4695, Cell Signaling Technology), or anti-p-ERK antibody (1:1000, 4370, Cell Signaling Technology), or anti-JNK antibody (1:1000, 9252, Cell Signaling Technology), anti-p-JNK antibody (1:1000, 9251, Cell Signaling Technology), anti-β-tubulin antibody (1:2000, 2128, Cell Signaling Technology) or anti-GAPDH (1:5000, 2118, Cell Signaling Technology) at 4°C for 20 h, washed in Tris-buffered saline and Tween-20 (TBST), and then incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (1:2000, 111–035–003, Jackson ImmunoResearch) at room temperature for 1 h. Protein bands were detected using Western blotting luminol reagent (sc-2048, Santa Cruz). Data were analyzed with the ImageJ software.
The rat was anesthetized with 1% pentobarbital sodium and intracardially perfused with 4% paraformaldehyde (PFA, in 0.1 M phosphate buffer, pH 7.4). The brain was post-fixed with 4% PFA for 12 h, and cryoprotected in 20% and 30% sucrose solutions in turn. 35 μm sections were sliced coronally using a cryostat microtome (Model 1950, Leica Instrument Co., Ltd.) throughout the entire PL (Zheng et al., 2017). Free-floating sections were washed in the phosphate buffered saline (PBS), blocked with a buffer containing 3% bull serum albumin and 0.3% triton X-100 for 1 h, and incubated with the following primary antibodies in 4°C for 24 h: rabbit anti-c-Fos antibody (1:300, sc-52, Santa Cruz), or rabbit anti-p-p38 antibody (1:500, 4511, Cell Signaling Technology), or goat anti-glutamate transporter (EAAC1) antibody (1:1000, AB1520, Merck Millipore), or mouse anti-GFAP antibody (1:1000, 3670, Cell Signaling Technology), or goat anti-Iba1 antibody (1:800, ab5076, Abcam) or mouse anti-GAD67 antibody (1:1000, ab26116, Abcam). Sections were then washed in PBS and incubated with secondary antibodies at room temperature for 90 min: Alexa Fluor 594-conjugated donkey anti-rabbit IgG (1:300, A21207, Invitrogen), or Alexa Fluor 488-conjugated donkey anti-goat IgG (1:300, A11055, Invitrogen) or Alexa Fluor 488-conjugated goat anti-mouse IgG (1:300, ab150113, Abcam). For p-p38 and glutamate transporter staining, sections were incubated in EDTA antigen retrieval solution at 98°C for 30 min to expose epitopes in DNA before blocked with serum. For co-staining of p-p38 and Iba1, antigen retrieval was not applied to avoid poor coloration of Iba1. Images were taken by a laser scanning confocal microscope (model FV1000, Olympus Co., Ltd.).
Data were presented as means ± SEM. Unpaired or paired two-tailed t tests and one-way analysis of variance (ANOVA) with Bonferroni post hoc tests were used for the comparison of two groups. Comparisons of two groups with different time points were performed using two-way ANOVA or ANOVA with repeated measures and Bonferroni post hoc test. The differences were calculated with software GraphPad Prism 5.0 and statistical significance was defined as p < 0.05.
To examine whether chronic pain experience would affect upcoming nociceptive assaults, we first established chronic inflammatory pain with intraplantar CFA injection. These animals showed significant thermal hyperalgesia (group effect: F(1,90) = 130.1, p < 0.001; time effect: F(6,90) = 29.27, p < 0.001; interaction: F(6,90) = 21.77, p < 0.001; Figure 1A) and mechanical allodynia (group effect: F(1,96) = 273.6, p < 0.001; time effect: F(6,96) = 65.37, p < 0.001; interaction: F(6,96) = 70.02, p < 0.001; Figure 1A), which recovered to the baseline level 28 days after CFA injection. PWLs and PWTs of the contralateral paw remained unchanged throughout this period (data not shown). Inflammatory rats exhibited significant paw swelling after CFA injection (on day 1: 3613.0 ± 100.6 vs. 1587.0 ± 60.9 mm3; on day 28: 2601.0 ± 49.8 vs. 2042.0 ± 56.9 mm3; CFA vs. Saline group).
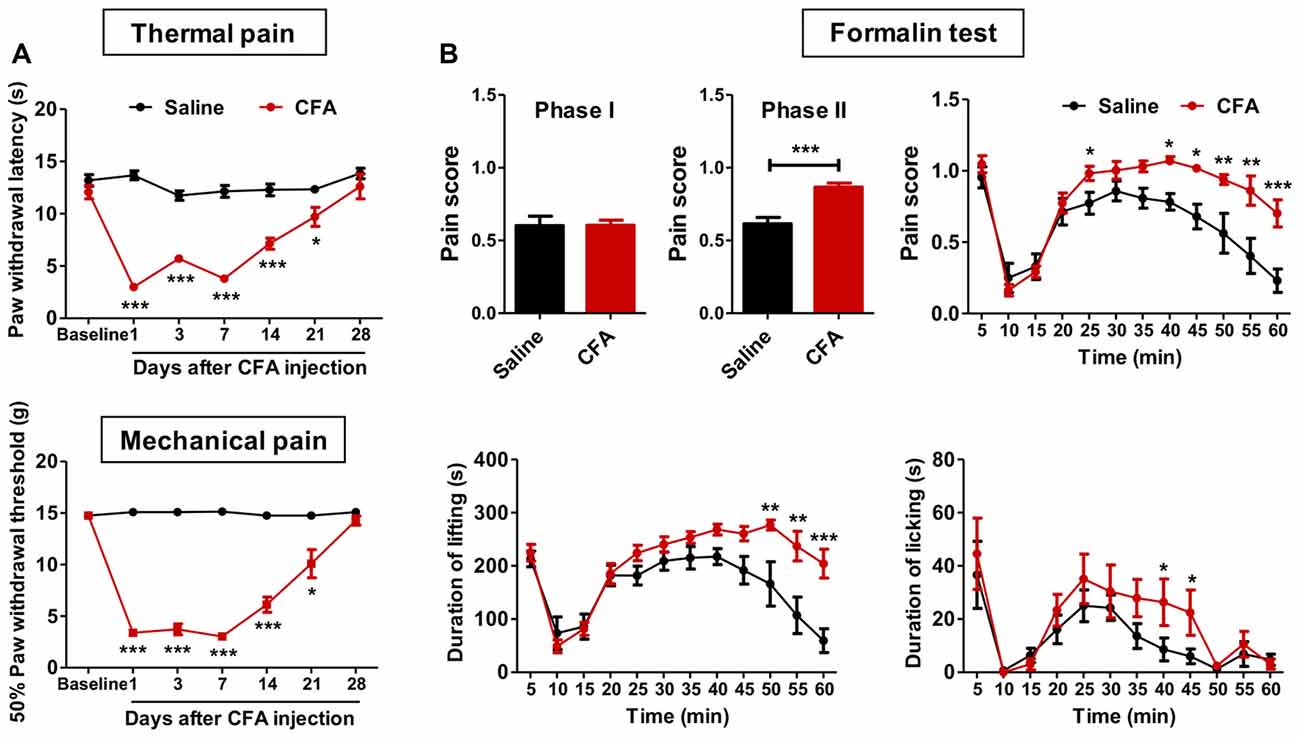
Figure 1. Aggravated formalin-induced pain in rats with chronic inflammatory pain experience. (A) Thermal hyperalgesia (top) and mechanical allodynia (bottom) in complete freund’s adjuvant (CFA)-induced chronic inflammatory pain recovered to the baseline level 28 days after CFA injection. n = 8 in each group. *p < 0.05, ***p < 0.001, CFA vs. Saline, analyses of variance (ANOVA) with repeated measures and Bonferroni post hoc test. (B) Increased pain scores in the phase II of the formalin test (top) in rats with chronic inflammatory pain experience. The elevated pain score involved increased durations of both paw lifting behavior (bottom, left) and paw licking behavior (bottom, right). n = 8 in each group. *p < 0.05, **p < 0.01, ***p < 0.001, CFA vs. Saline, t test, ANOVA with repeated measures and Bonferroni post hoc test.
Anxiety is a common co-morbidity of chronic pain (Zheng et al., 2017). To examine anxiety-like behaviors, we performed open field and elevated plus-maze tests on days 29 and 31 after CFA or saline injection, respectively. We did not observe significant differences between CFA and saline groups at this time point (open field: C.Time, t(16) = 0.38, p > 0.05; C.Dis, t(16) = 0.14, p > 0.05; T.Dis, t(16) = 1.92, p > 0.05; elevated plus-maze: O.Time, t(16) = 0.59, p > 0.05; O.Entries, t(16) = 0.14, p > 0.05; T.Entries, t(16) = 0.63, p > 0.05; Supplementary Figures S1A,B). These results indicate full recovery of thermal hyperalgesia, mechanical allodynia and anxiety-like behaviors in CFA-induced chronic inflammatory pain 28 days after CFA injection.
We next examined these animals’ responses to formalin and capsaicin tests. Rats with CFA pain experience showed significantly higher pain scores in the formalin test (phase I: t(15) = 0.04, p > 0.05; phase II: t(15) = 5.00, p < 0.001; Figure 1B), as revealed by increased duration of lifting time and licking time (lifting: group effect: F(1,165) = 12.03, p < 0.01; time effect: F(11,165) = 20.67, p < 0.001; interaction: F(11,165) = 4.10, p < 0.001; licking: group effect: F(1,165) = 1.79, p > 0.05; time effect: F(11,165) = 9.80, p < 0.001; interaction: F(11,165) = 0.77, p > 0.05; Figure 1B). Similarly, aggravated thermal hyperalgesia in the capsaicin test was also observed in rats with chronic pain experience (group effect: F(5,70) = 27.56, p < 0.01; baseline: t(14) = 0.03, p > 0.05; 15 min: t(14) = 2.56, p < 0.05; 30 min: t(14) = 2.65, p < 0.05; 60 min: t(14) = 3.40, p < 0.01; 90 min: t(14) = 5.10, p < 0.001; 120 min: t(14) = 3.53, p < 0.01; Supplementary Figure S2). However, no differences of anxiety-like behaviors were observed after formalin injection between CFA and saline treated rats (elevated plus-maze: O.Time, t(13) = 0.72, p > 0.05; O.Entries, t(13) = 0.23, p > 0.05; T.Entries, t(13) = 0.10, p > 0.05; open field: T.Dis, t(13) = 0.69, p > 0.05; Supplementary Figure S3).
Together, these results indicate aggravated nociceptive responses to following noxious assaults in rats with prior chronic pain experience. Given the similar findings from formalin and capsaicin tests, only the formalin test was performed in further experiments.
To examine whether PL participated in the observed aggravated nociceptive responses, we first performed c-Fos protein mapping after formalin injection. After saline injection, PL showed little c-Fos protein expression in rats with or without chronic pain experience. By contrast, formalin injection induced significant c-Fos expression in PL, especially in rats with chronic pain experience (F(3,85) = 159.5, p < 0.001; Figures 2A,B). The majority (93.70 ± 1.53%) of c-Fos positive neurons in PL co-labeled with EAAC1, a marker of excitatory glutamatergic neurons (Figure 2C).
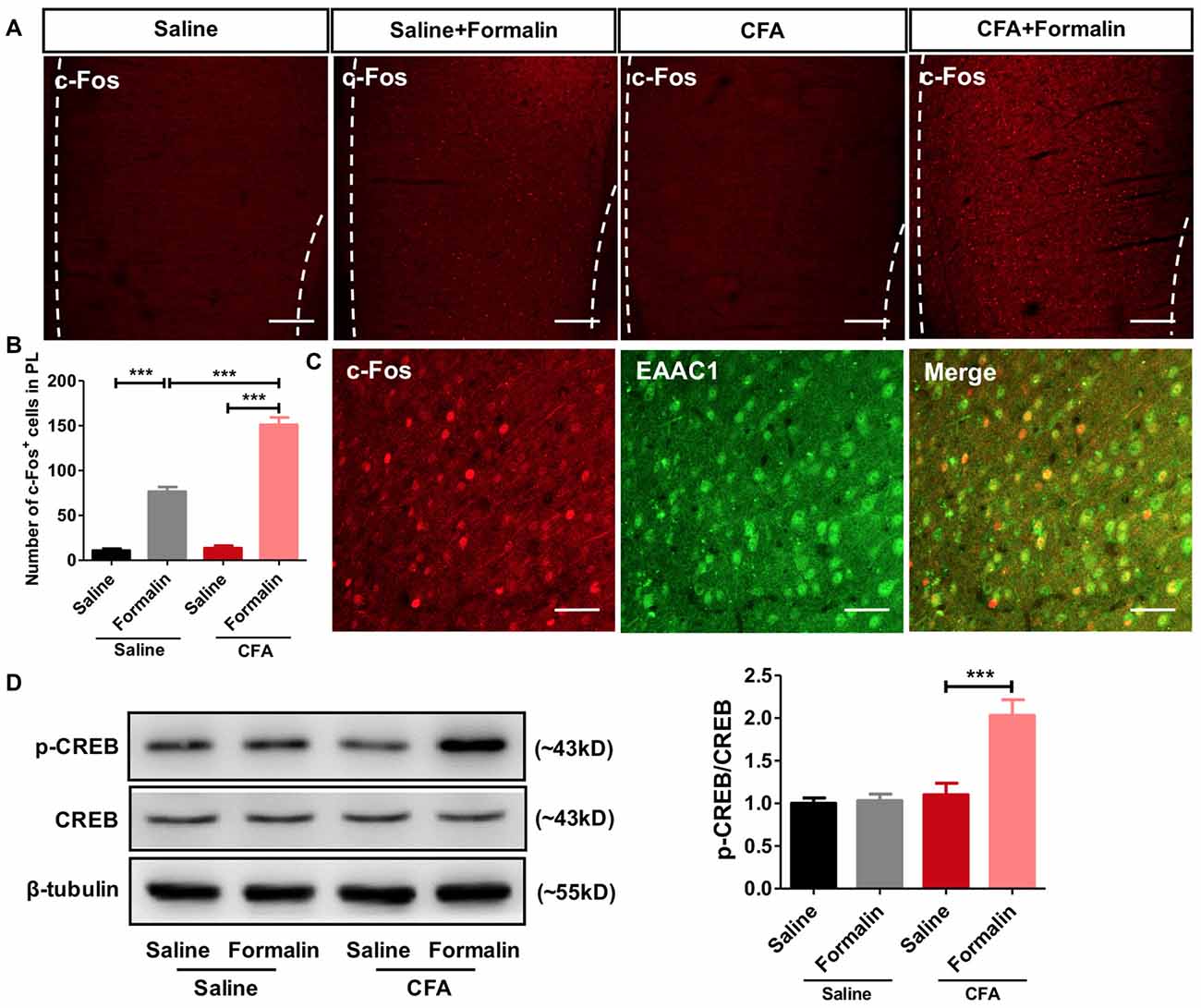
Figure 2. Enhanced activation of prelimbic cortex (PL) neurons to formalin pain in rats with chronic pain experience. (A) Representative immunofluorescence images showing c-Fos+ neurons in the PL of rats in CFA and saline groups, with or without formalin injection. Scale bars: 200 μm. (B) Quantitative analysis of images in (A). Formalin injection induced significant c-Fos expression in the PL, especially in rats with chronic pain experience. n = 6 in each group. ***p < 0.001, CFA+Formalin vs. Saline+Formalin, CFA vs. CFA+Formalin, Saline vs. Saline+Formalin, one-way ANOVA. (C) The majority (93.70 ± 1.53%) of c-Fos+ neurons (red) in PL in CFA+formalin group co-labeled with EAAC1+ neurons (green). Scale bars: 50 μm. (D) Increased PL p-CREB with formalin injection after chronic pain recovery. Chronic pain experience or formalin injection alone did not affect p-CREB expression in PL in rats. Representative Western blots of p-CREB, CREB and β-tubulin were shown above the corresponding histogram. n = 6 in each group. ***p < 0.001, CFA+Formalin vs. CFA+Saline, one-way ANOVA.
Meanwhile, we observed significantly up-regulated phosphorylation of CREB, a marker of neuronal activation, in PL of rats receiving formalin injection with chronic pain experience. However, formalin alone was not sufficient to elevate the content of p-CREB in the PL (F(3,26) = 15.45, p < 0.001; Figure 2D). These findings indicate stronger activation of PL by formalin pain in rats with chronic pain experience than in those without.
To examine whether PL contributed to aggravated nociceptive responses, we injected muscimol, a GABAA receptor agonist, into PL before the formalin test. Muscimol significantly relieved formalin-induced pain behaviors in rats with chronic pain experience, but not in those without (phase I: F(3,33) = 1.35, p > 0.05; phase II: F(3,33) = 11.62, p < 0.001; right: group effect: F(3,330) = 11.32, p < 0.001; time effect: F(11,330) = 55.07, p < 0.001; interaction: F(33,330) = 2.47, p < 0.001; Figure 3). These results indicate that PL contributes to aggravated nociceptive formalin pain responses in rats with chronic inflammatory pain experience.
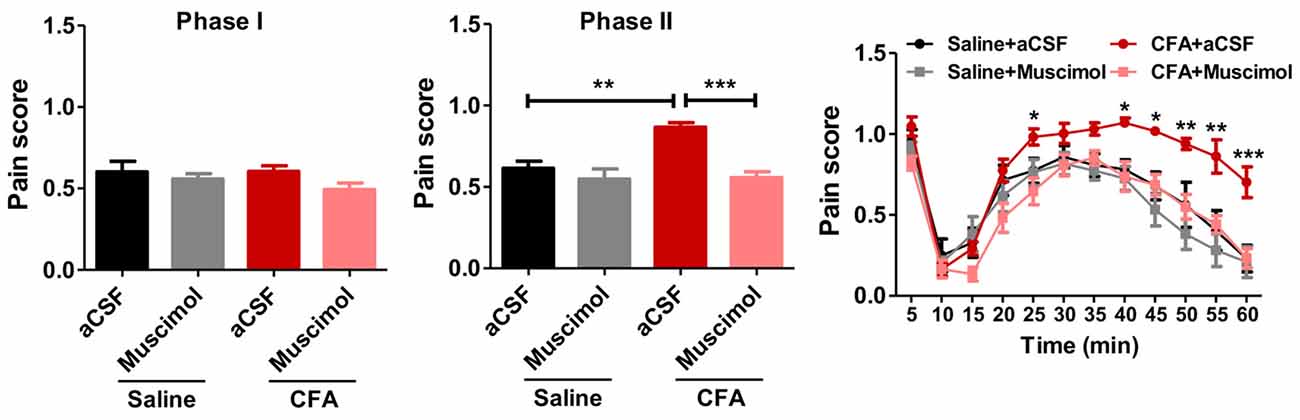
Figure 3. Inhibiting PL reverses aggravated formalin pain in rats with chronic pain experience. Inhibiting PL activities by muscimol relieved the aggravated phase II formalin pain in CFA (the middle column), but not saline (the right column) group. No significant differences in phase I of formalin pain were observed among groups (the left column). n = 8 in each group. In the middle column: ***p < 0.001, CFA+aCSF vs. CFA+Muscimol. **p < 0.01, CFA+aCSF vs. Saline+aCSF, one-way ANOVA. In the right column: *p < 0.05, **p < 0.01, ***p < 0.001, CFA+aCSF vs. CFA+Muscimol, ANOVA with repeated measures and Bonferroni post hoc test.
To explore molecular mechanisms of PL’s contribution to aggravated pain, we examined protein contents of MAPKs (including ERK, p38 and JNK) and their phosphorylation in bilateral PL. MAPKs are key players in nociceptive sensitization, which are activated by various second-messenger signal transduction cascades (Edelmayer et al., 2014). Western blotting showed significantly up-regulated phosphorylation of p38 in PL of rats with chronic pain experience after formalin injection (t(12) = 2.80, p < 0.05; Figure 4A). Indeed, the up-regulated phosphorylation of p38 was apparent in the PL of rats with chronic pain even without formalin injection (F(3,34) = 7.57, p < 0.001; Figure 4B). The up-regulated p-p38 was apparent 7 days after CFA injection (F(6,34) = 6.25, p < 0.001; Supplementary Figure S4), and lasted minimally 8 weeks (F(2,17) = 5.51, p < 0.05; Figure 4C). By contrast, no significant differences in the content of ERK, p-ERK, JNK or p-JNK were detected (p-ERK/ERK: F(3,33) = 1.24, p > 0.05; p-JNK/JNK: F(3,37) = 1.23, p > 0.05; Supplementary Figures S5A,B). Immunofluorescence showed that the majority (87.86 ± 0.84%) of p-p38 positive neurons in PL co-labeled with EAAC1 (Figure 4D), and only a small proportion (2.56 ± 0.51%) with GAD67, a marker of interneurons (Figure 4E). Few astrocytes or microglial cells (GFAP+ and Iba1+ cells, 1.23 ± 0.16% and 8.35 ± 0.66%, respectively) showed co-expression with p-p38 (Figures 4F,G). These findings indicate that previous chronic pain experience induces sustained hyperphosphorylation of p38 after pain recovery.
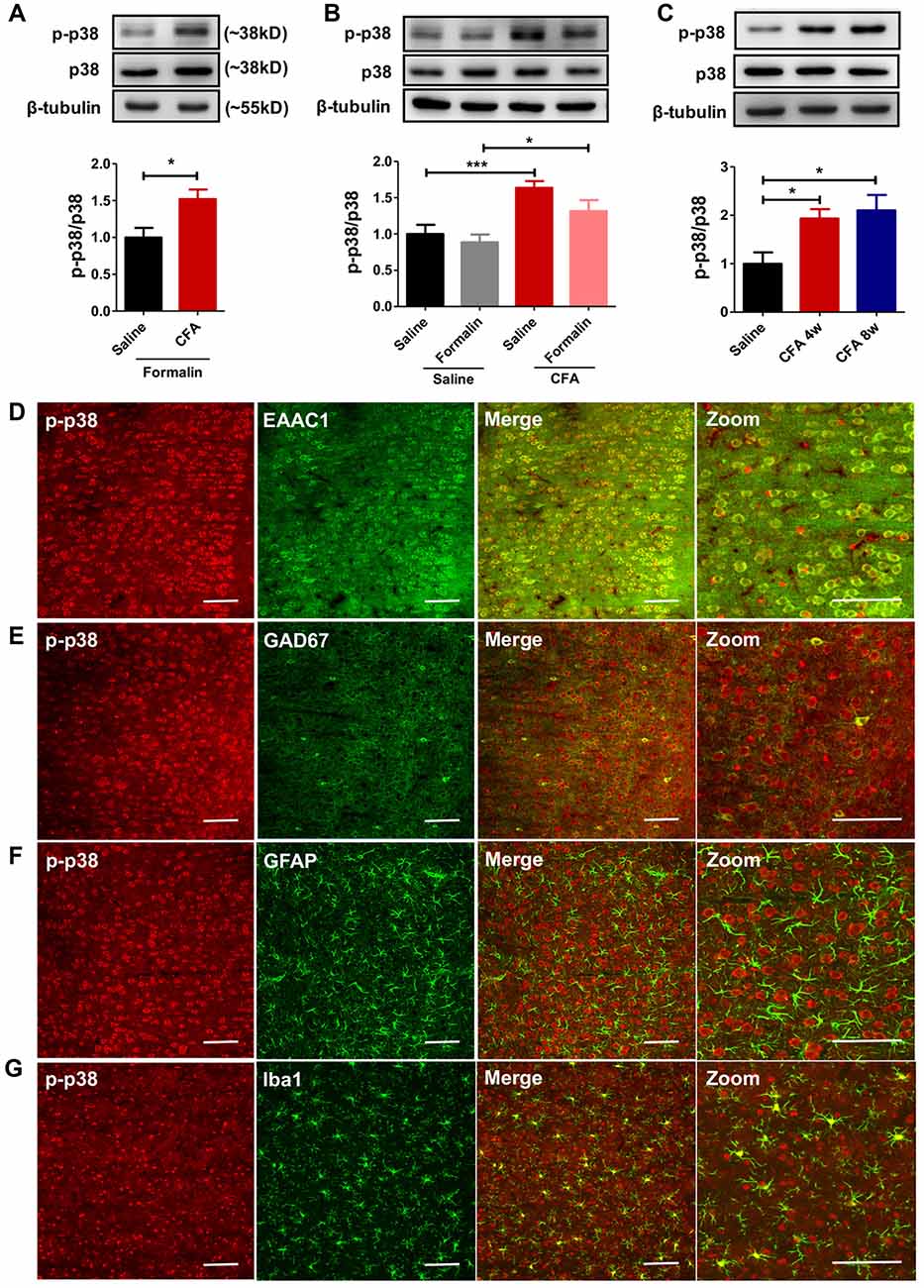
Figure 4. Persistent hyperphosphorylation of p38 accompanies aggravated formalin pain in rats with chronic pain experience. (A) Increased PL p-p38 after formalin injection in rats with chronic pain experience. Representative Western blots of p-p38, p38 and β-tubulin were shown above the corresponding histogram. n = 7 in each group. *p < 0.05, CFA+Formalin vs. Saline+Formalin, t test. (B) p-p38 in the PL increased in rats with chronic pain experience, with or without formalin injection. Formalin injection alone did not obviously influence the expression of p-p38 in PL. Representative Western blots of p-p38, p38 and β-tubulin were shown above the corresponding histogram. n = 8 in each group. ***p < 0.001, CFA vs. Saline, *p < 0.05, CFA+Formalin vs. Saline+Formalin, one-way ANOVA. (C) Hyperphosphorylation of p38 in PL maintained for at least 8 weeks after CFA injection. Representative Western blots of p-p38, p38 and β-tubulin were shown above the corresponding histogram. n = 6 in each group. *p < 0.05 CFA 1 m/CFA 2 m vs. Saline, one-way ANOVA. (D) Representative immunofluorescence images showing that the majority (87.86 ± 0.84%) of p-p38+ neurons (red) co-labeled with EAAC1+ neurons (green, a marker of glutamatergic neurons). Scale bars: 100 μm. (E) Representative immunofluorescence images showing that few (2.56 ± 0.51%) p-p38+ neurons (red) co-labeled with GAD67+ neurons (green, a marker of gabaergic neurons). Scale bars: 100 μm. (F,G) Representative immunofluorescence images showing that a small proportion of p-p38+ neurons (red) co-labeled with GFAP+ (1.23 ± 0.16%, green, a marker of astrocytes) and Iba1+ cells (8.35 ± 0.66%, green, a marker of microglia). Scale bars: 100 μm.
To examine whether the hyperphosphorylated p38 contributed to sensitized pain behaviors in rats with chronic pain experience, we micro-injected SB203580, a specific inhibitor of p38 phosphorylation, into bilateral PL 30 min before the formalin test (Figure 5A). SB203580 inhibited the hyperphosphorylation of p38 after chronic pain recovery (left: t(10) = 5.27, p < 0.001; right: t(10) = 4.17, p < 0.01; Figures 5B,C) and down-regulated formalin-induced p-CREB (t(6) = 2.72, p < 0.05; Figure 5D).
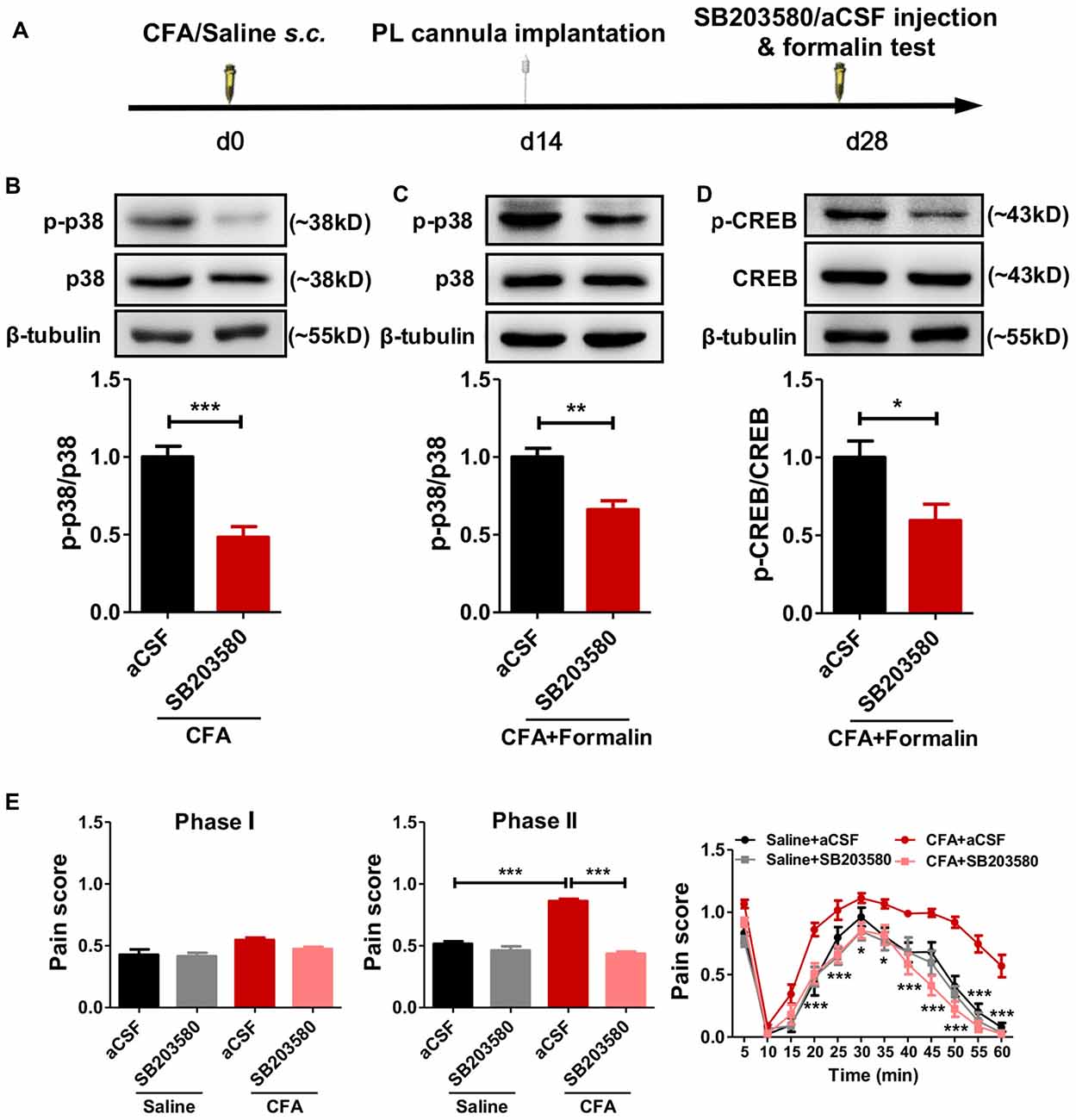
Figure 5. Inhibiting the phosphorylation of p38 reverses aggravated formalin pain in rats with chronic pain experience. (A) A diagram showing the experiment time line. SB203580/aCSF micro-injection and formalin test were performed 28 d after CFA/saline injection. The cannula implantation surgery was performed 2 weeks before behavior tests. (B,C) SB203580 micro-injection into PL inhibited the hyperphosphorylation of p38 in rats with chronic pain experience, without (B) or with formalin injection (C). Representative Western blots of p-p38, p38 and β-tubulin were shown above the corresponding histogram. n = 6 in each group. ***p < 0.001, CFA+aCSF vs. CFA+SB203580. **p < 0.01, CFA+aCSF+Formalin vs. CFA+SB203580+Formalin, t test. (D) SB203580 micro-injection into PL inhibited the hyperphosphorylation of cAMP-response element binding protein (CREB) after formalin injection in rats with chronic pain experience. Representative Western blots of p-CREB, CREB and β-tubulin were shown above the corresponding histogram. n = 6 in each group. *p < 0.05, CFA+aCSF+Formalin vs. CFA+SB203580+Formalin, t test. (E) Inhibiting the phosphorylation of p38 in PL by SB203580 reversed the aggravated phases II formalin pain in the CFA group, but not the saline group (the middle column). Detailed pain scores shown in every 5 min (the right column). n = 6 in each group. In the middle column: ***p < 0.001, Saline + aCSF/CFA+SB203580 vs. CFA+aCSF, one-way ANOVA. In the right column: *p < 0.05, ***p < 0.001, CFA+aCSF vs. CFA+SB203580, ANOVA with repeated measures and Bonferroni post hoc test.
Behaviorally, SB203580 reversed the aggravated formalin pain in rats with chronic pain experience, but not in those without (phase I: F(3,25) = 4.19, p < 0.05; phase II: F(3,28) = 83.37, p < 0.001; right: group effect: F(3,275) = 96.95, p < 0.001; time effect: F(11,275) = 95.58, p < 0.001; interaction: F(33,275) = 2.64, p < 0.001; Figure 5E).
These results suggest that persistent hyperphosphorylation of p38 in the PL contributes to the hypersensitized formalin pain in rats with chronic pain experience.
PL projects to several brain areas that modulate pain. We further investigated the neural pathways that aggravated formalin pain in rats with chronic pain experience (Figure 6A). We chemogenetically inhibited the PL–PAG pathway (Figure 6B), which is essential for descending pain modulation, and observed an relieved effect on the aggravated formalin pain in rats with chronic pain experience (phase I: F(3,31) = 5.07, p < 0.01; phase II: F(3,31) = 19.05, p < 0.001; right: group effect: F(3,308) = 19.04, p < 0.001; time effect: F(11,308) = 74.70, p < 0.001; interaction: F(33,308) = 2.23, p < 0.001; Figure 6C). By contrast, inhibiting the PL–PAG pathway did not affect physiological pain in rats with chronic pain experience (mCherry: t(21) = 1.11, p > 0.05; hM4Di: t(5) = 1.75, p > 0.05; Supplementary Figures S6A,B), nor the development of CFA induced chronic inflammatory pain (p > 0.05; Supplementary Figures S7A,B).
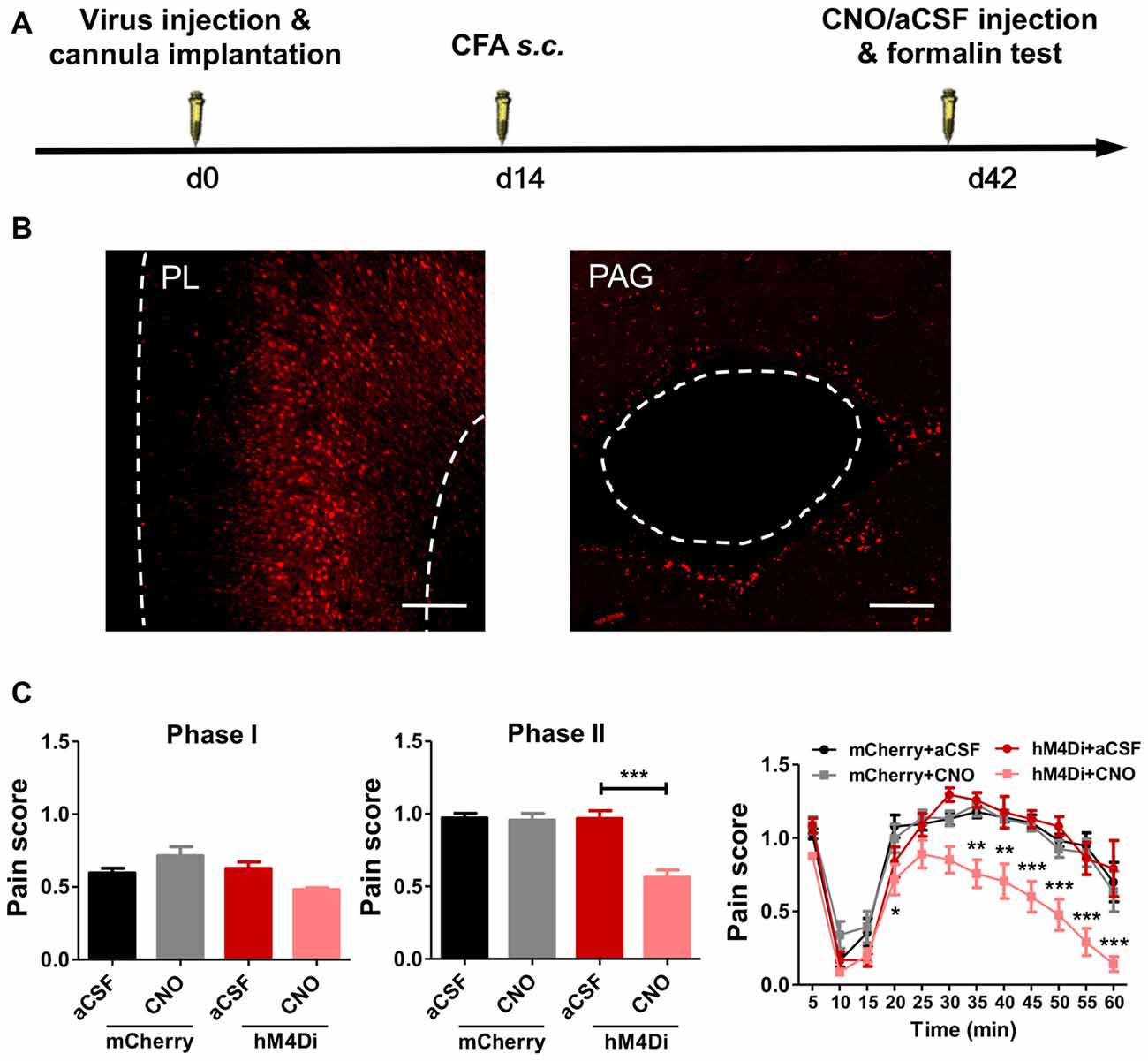
Figure 6. PL–periaqueductal gray (PAG) pathway mediates aggravated formalin pain in rats with chronic pain experience. (A) A diagram showing the experiment time line. The clozapine-N-Oxide (CNO)/aCSF micro-injection and formalin test were performed 42 days after virus injection and cannula implantation surgery. CFA injection was performed 28 days before formalin test. (B) AAV5-CaMKIIα-hM4D(Gi)-mCherry virus expression in PL (red, left), expression of hM4Di in PL projection in PAG (red, right). Scale bars: 200 μm. (C) Compared to the control, inhibiting PL–PAG pathway relieved the aggravated phases II formalin pain in the CFA group (the middle column). Detailed pain scores were shown in every 5 min (the right column). n = 8 in each group. In the middle column: ***p < 0.001, hM4Di+aCSF vs. hM4Di+CNO, one-way ANOVA. In the right column: *p < 0.05, **p < 0.01, ***p < 0.001, hM4Di+aCSF vs. hM4Di+CNO, ANOVA with repeated measures and Bonferroni post hoc test.
In contrast, chemogenetic inhibition of the PL–NAc core pathway did not relieve the aggravated formalin pain in rats with chronic pain experience (phase I: F(3,27) = 0.81, p > 0.05; phase II: F(3,28) = 0.52, p > 0.05; right: group effect: F(3,275) = 0.16, p > 0.05; time effect: F(11,275) = 76.15, p < 0.001; interaction: F(33,275) = 1.27, p > 0.05; Supplementary Figures S8A,B).
These findings indicate that PL–mediated aggravated formalin pain in rats with chronic pain experience is achieved through the PL–PAG pathway, but not the PL–NAc core pathway.
Prior pain experience alters future responses to painful stimuli in both human and animals (Bachiocco et al., 1993; Lidow, 2002; Ren et al., 2004; Hermann et al., 2006; Li et al., 2012; Wegner et al., 2015). In the present study, we identified PL as a key region in regulating the aggravated formalin-induced pain in rats after the recovery from CFA-induced chronic inflammatory pain. Rats with chronic pain experience showed elevated pain scores in the second, but not the first, phase of formalin pain. The second phase of formalin pain reflects the development of inflammation and central sensitization, and indicates the involvement of the central nervous system in the aggravated pain (Shibata et al., 1989; Vaccarino and Melzack, 1992).
Structural and functional changes in several brain regions persist long after pain recovery. For example, reduced gray matter densities in chronic back pain patients are observed in the middle cingulate gyrus, thalamus and prefrontal cortex (Ivo et al., 2013), and an increase of gray matter density is found in PAG, thalamus and cerebellum months after whiplash injury and after the headache subsided in most patients with posttraumatic headache (Obermann et al., 2009). Chronic back pain also alters the human brain chemistry: reduction of N-acetyl aspartate and glucose is demonstrated in the dorsolateral prefrontal cortex, but not cingulate, sensorimotor and other brain regions (Grachev et al., 2000). In animals, long-term neuropathic pain decreases frontal cortex volumes several months after nerve injury (Seminowicz et al., 2009), and increases basal dendrites of neurons and spine densities in the PL (Metz et al., 2009). These structural and functional changes are accompanied by altered synaptic plasticity and neuronal excitability (Metz et al., 2009; Baliki et al., 2014). Taken together, PL is a brain region particularly vulnerable to chronic pain, regardless of pain recovery or not (Grachev et al., 2000; Apkarian et al., 2004). Some evidences show that pain-induced functional and structural abnormalities in the PFC are at least partially reversible by effective pain treatment (Rodriguez-Raecke et al., 2009; Seminowicz et al., 2011). However, we demonstrated persistent hypersensitivity of PL neurons in rats with chronic pain experience to following pain assaults.
However, we do not consider PL to be the only brain region that contributes to the aggravated nociceptive responses in rats with experience of chronic inflammatory pain. Other areas such as anterior cingulate cortex (ACC) and infralimbic cortex (IL), insular cortex and thalamus are densely connected with PL and undergo plastic changes in chronic pain (Obermann et al., 2009; Ivo et al., 2013; Lin, 2014; Zhuo, 2016; Yue et al., 2017).
p38 is one of the major MAPK members crucial for generating pain hypersensitivity through transcription-dependent and -independent means (Ji and Woolf, 2001; Crown et al., 2006; Wynne, 2006; Toyoda et al., 2007; Ji et al., 2009). In both peripheral and central nervous system, such as spinal cord and ACC, p38 is activated following pain stimulations, and the activation lasts for more than 3 weeks after chronic constriction injury or spinal nerve ligation (Toyoda et al., 2007; Crown et al., 2008; Cao et al., 2014). Inhibiting p38 in spinal cord or ACC alleviates both subacute and chronic pain (Kumar et al., 2003; Crown et al., 2008; Cao et al., 2014). In the present study, p-p38+ neurons are mostly glutamatergic neurons in PL. Under persistent noxious stimulation, hyperphosphorylated p38 can be invoked to activate the downstream signal pathway, including p-CREB and c-Fos, and the activation of nociceptive neurons further sensitizes pain. Inhibiting the phosphorylation of p38 relieves the aggravated formalin pain in rats with chronic pain experience and down-regulates the phosphorylation of its downstream effector CREB and the neuronal activity. The persistent hyperphosphorylation of p38 in PL in rats with chronic pain experience does not directly contribute to pain responses unless a strong stimulus such as formalin injection is performed. These findings indicate that some endogenous inhibitors overlay sensitized p-p38 to keep the balanced “recovery” state.
One candidate of such inhibitors would be layer V pyramidal neurons in the IL, which directly innervate inhibitory interneurons in the PL (Saffari et al., 2016). In addition to pain (Baliki et al., 2012; Wang et al., 2015; Wu et al., 2016), PL also regulates other long-term cognitive behaviors including fear conditioning, working memory, drug addiction and chronic stress (Gisquet-Verrier and Delatour, 2006; Lasseter et al., 2010; Negrón-Oyarzo et al., 2014; Fitzgerald et al., 2015; Moench and Wellman, 2015; Seo et al., 2017). PL neuronal activities maintain freezing behaviors in fear conditioning, whereas extinction induces a novel inhibitory learning in the IL, which antagonize PL pyramidal neuronal activities through local interneurons (Ji and Neugebauer, 2012). These facts lead to an intriguing proposal that the recovery from chronic pain represents pain extinction, instead of simple oblivion of the pain memory (Apkarian et al., 2009; Yi and Zhang, 2011).
PL has direct projections to several nuclei, including ACC, NAc, amygdala and PAG. Previous studies have revealed roles of PL–NAc and PL–PAG pathways in regulating chronic pain (Kucyi et al., 2013; Yu et al., 2014; Lee et al., 2015). Activation of the PL to NAc core circuit inhibits persistent neuropathic pain and relieves the affective symptoms associated with chronic pain (Lee et al., 2015). Functional and structural connectivity between PAG and mPFC relates to individual differences in attention to pain (Kucyi et al., 2013). PAG is a critical component of the descending pain modulatory system, and usually exerts an inhibitory effect on nociceptive transmission (Umana et al., 2017). Evidence shows that glutamatergic projections from mPFC act to inhibit PAG function (Franklin et al., 2017). Pharmacogenetic inhibition of the projections from PL to PAG therefore disinhibits PAG, and in turn relieves the aggravated formalin-induced pain. The finding that paw lifting behaviors in response to formalin injection, indicating peripheral reflexes, increase in rat with chronic pain experience also supports the involvement of the descending pathway.
Different from the role of PAG in perceptual part of pain, NAc is more involved in pain affection, such as depression-like behaviors in the chronic neuropathic pain state (Goffer et al., 2013; LeBlanc et al., 2015; Navratilova et al., 2015; Kaneko et al., 2017). The present study shows that PL–PAG, but not PL–NAc, pathway regulates the aggravated formalin pain in rats with chronic pain experience, which reflects mostly aggravated pain sensation.
Patients with chronic pain have less PFC deactivation than controls during cognition or visual attention tasks (Baliki et al., 2008; Seminowicz et al., 2011), in keeping with our results that the PL is more strongly activated during the following noxious assault in rats with chronic pain experience. Previous work has identified the PL as a target for pain treatment. Repetitive transcranial magnetic stimulation, deep brain stimulation, cognitive-behavioral therapy or other therapies can lead abnormal function of prefrontal cortex to stable or normal state (Fierro et al., 2010; Seminowicz et al., 2013; Boccard et al., 2015), thus relieves the aggravated nociceptive responses in patients with chronic pain experience.
In conclusion, our present study demonstrates that persistent hyperphosphorylation of p38 in the PL underlies aggravated nociceptive responses in rats with chronic inflammatory pain experience, and indicates inhibiting the activity of PL as a promising option for treating hyperalgesia in patients with chronic pain experience.
X-CF, MY and YW designed experiments and wrote the manuscript; X-CF, SF, F-YL and SC performed the experiments and analyzed the data; YW and MY supervised the experiments.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by grants from the Ministry of Science and Technology of China (2014CB548200 and 2015CB554503), the National Natural Science Foundation of China (91732107, 81571067 and 81521063), the Beijing Natural Science Foundation (5182013) and the “111” Project of Ministry of Education of the People’s Republic of China.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2018.00085/full#supplementary-material
FIGURE S1 | Similar levels of anxiety-like behaviors in chronic pain rats 30 days after CFA injection. (A) Rats showed similar levels of anxiety-like behaviors in the open field test 30 days after CFA injection, indicated by similar time spent (C.Time, left) and distance traveled (C.Dis, middle) in the central area of the open field between groups. Total distance traveled (T.Dis, right) in the field were not affected either. n = 8 in each group. CFA vs. Saline, t test. (B) Rats showed similar levels of anxiety-like behaviors in the elevated plus-maze test 30 days after CFA injection, indicated by similar time spent (O.Time, left) and entries (O.Entries, middle) into the open arms between groups. Total arm entries (T.Entries, right) were not affected either. n = 8 in each group. CFA vs. Saline, t test.
FIGURE S2 | Aggravated capsaicin-induced pain in rats with chronic inflammatory pain experience. Rats with chronic inflammatory pain experience showed increased thermal hyperalgesia 15, 30, 60, 90 and 120 min after capsaicin injection. n = 8 in each group. p < 0.01, Saline+Capsaicin vs. CFA+Capsaicin, one-way ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001, Saline+Capsaicin vs. CFA+Capsaicin, t test.
FIGURE S3 | Similar levels of anxiety-like behaviors after formalin injection between CFA chronic pain group and saline control group. (A) Rats with chronic pain experience showed similar levels of anxiety-like behaviors in the elevated plus-maze test after formalin injection, indicated by similar time spent (O.Time, left) and entries (O.Entries, middle) into the open arms between groups. Total arm entries (T.Entries, right) were similar either. n = 8 in each group. CFA+Formalin vs. Saline+Formalin, t test. (B) Similar levels of locomotion after formalin injection between CFA and saline groups, indicated by similar total distance traveled (T.Dis) in the field. n = 8 in each group. CFA+Formalin vs. Saline+Formalin, t test.
FIGURE S4 | Increased levels of p-p38 in the PL after CFA injection. p-p38 in the PL increased in chronic pain rats 7 days after CFA injection, and maintained for at least 28 days. Representative Western blots of p-p38, p38 and β-tubulin were shown above the corresponding histogram. n = 5 in each group. ***p < 0.001, **p < 0.01, vs. Saline, one-way ANOVA.
FIGURE S5 | Chronic pain experience and formalin injection do not affect the phosphorylation of ERK1/2 and JNK in PL. (A) Similar levels of p-ERK1/2 in PL in rats with chronic pain experience. Formalin injection did not affect the phosphorylation of ERK1/2 in PL in rats with or without chronic pain experience. Representative Western blots of p-ERK1/2, ERK1/2 and β-tubulin were shown above the corresponding histogram. n = 8 in each group, one-way ANOVA. (B) Lack of significant changes of p-JNK in PL in rats with chronic pain experience. Formalin injection did not affect the phosphorylation of JNK in PL in rats with or without chronic pain experience. Representative Western blots of p-JNK, JNK and GAPDH were shown above the corresponding histogram. n = 8 in each group, one-way ANOVA.
FIGURE S6 | Inhibiting PL–PAG pathway does not affect physiological pain after chronic inflammatory pain recovery. (A) A diagram showing the time line of the experiment. The CNO/aCSF micro-injection and hot plate test were performed 28 days after CFA injection. Virus injection and cannula immplantation surgery were performed 14 days before CFA injection. (B) Compared to the control, inhibiting PL–PAG pathway did not affect physiological pain after chronic inflammatory pain recovery. n = 6 in each group. Before vs. After CNO injection, paired t test.
FIGURE S7 | Inhibiting PL–PAG pathway does not affect thermal hyperalgesia in the development of CFA-induced chronic inflammatory pain. (A) A diagram showing the experiment time line. CNO/aCSF micro-injection and thermal hyperalgesia test were performed 1 day before and 1, 3, 7, 14, 21 and 28 days after CFA injection. Virus injection and cannula implantation surgery were performed 6 weeks before CFA injection. (B) Compared to the control, inhibiting PL–PAG pathway did not influence thermal hyperalgesia in the development of CFA-induced chronic inflammatory pain. n = 7 in each group. Before vs. After CNO injection, paired t test.
FIGURE S8 | PL–NAc core pathway does not affect aggravated formalin pain in rats with chronic pain experience. (A) Confirmation of AAV5-CaMKIIα-hM4D(Gi)-mCherry virus expression in PL projection in NAc core (red). Scale bars: 100 μm. (B) Inhibiting PL–NAc core pathway did not relieve the aggravated formalin pain in either phase I (the left column) or phase II (the middle column) in CFA group. n = 8 in each group, one-way ANOVA. Detailed pain scores were shown in every 5 min (the right column). n = 8 in each group, ANOVA with repeated measures and Bonferroni post hoc test.
Apkarian, A. V., Baliki, M. N., and Geha, P. Y. (2009). Towards a theory of chronic pain. Prog. Neurobiol. 87, 81–97. doi: 10.1016/j.pneurobio.2008.09.018
Apkarian, A. V., Sosa, Y., Sonty, S., Levy, R. M., Harden, R. N., Parrish, T. B., et al. (2004). Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J. Neurosci. 24, 10410–10415. doi: 10.1523/JNEUROSCI.2541-04.2004
Bachiocco, V., Scesi, M., Morselli, A. M., and Carli, G. (1993). Individual pain history and familial pain tolerance models: relationships to post-surgical pain. Clin. J. Pain 9, 266–271. doi: 10.1097/00002508-199312000-00008
Baliki, M. N., Geha, P. Y., Apkarian, A. V., and Chialvo, D. R. (2008). Beyond feeling: chronic pain hurts the brain, disrupting the default-mode network dynamics. J. Neurosci. 28, 1398–1403. doi: 10.1523/JNEUROSCI.4123-07.2008
Baliki, M. N., Mansour, A. R., Baria, A. T., and Apkarian, A. V. (2014). Functional reorganization of the default mode network across chronic pain conditions. PLoS One 9:e106133. doi: 10.1371/journal.pone.0106133
Baliki, M. N., Petre, B., Torbey, S., Herrmann, K. M., Huang, L., Schnitzer, T. J., et al. (2012). Corticostriatal functional connectivity predicts transition to chronic back pain. Nat. Neurosci. 15, 1117–1119. doi: 10.1038/nn.3153
Boccard, S. G., Pereira, E. A., and Aziz, T. Z. (2015). Deep brain stimulation for chronic pain. J. Clin. Neurosci. 22, 1537–1543. doi: 10.1016/j.jocn.2015.04.005
Cao, H., Zang, K. K., Han, M., Zhao, Z. Q., Wu, G. C., and Zhang, Y. Q. (2014). Inhibition of p38 mitogen-activated protein kinase activation in the rostral anterior cingulate cortex attenuates pain-related negative emotion in rats. Brain Res. Bull. 107, 79–88. doi: 10.1016/j.brainresbull.2014.06.005
Chaplan, S. R., Bach, F. W., Pogrel, J. W., Chung, J. M., and Yaksh, T. L. (1994). Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63. doi: 10.1016/0165-0270(94)90144-9
Chen, S. C., Liu, B. C., Chen, C. W., and Wu, F. S. (2006). Intradermal pregnenolone sulfate attenuates capsaicin-induced nociception in rats. Biochem. Biophys. Res. Commun. 349, 626–633. doi: 10.1016/j.bbrc.2006.08.076
Crown, E. D., Gwak, Y. S., Ye, Z., Johnson, K. M., and Hulsebosch, C. E. (2008). Activation of p38 MAP kinase is involved in central neuropathic pain following spinal cord injury. Exp. Neurol. 213, 257–267. doi: 10.1016/j.expneurol.2008.05.025
Crown, E. D., Ye, Z., Johnson, K. M., Xu, G. Y., McAdoo, D. J., and Hulsebosch, C. E. (2006). Increases in the activated forms of ERK 1/2, p38 MAPK and CREB are correlated with the expression of at-level mechanical allodynia following spinal cord injury. Exp. Neurol. 199, 397–407. doi: 10.1016/j.expneurol.2006.01.003
Edelmayer, R. M., Brederson, J. D., Jarvis, M. F., and Bitner, R. S. (2014). Biochemical and pharmacological assessment of MAP-kinase signaling along pain pathways in experimental rodent models: a potential tool for the discovery of novel antinociceptive therapeutics. Biochem. Pharmacol. 87, 390–398. doi: 10.1016/j.bcp.2013.11.019
Ferland, C. L., Harris, E. P., Lam, M., and Schrader, L. A. (2014). Facilitation of the HPA axis to a novel acute stress following chronic stress exposure modulates histone acetylation and the ERK/MAPK pathway in the dentate gyrus of male rats. Endocrinology 155, 2942–2952. doi: 10.1210/en.2013-1918
Fierro, B., De Tommaso, M., Giglia, F., Giglia, G., Palermo, A., and Brighina, F. (2010). Repetitive transcranial magnetic stimulation (rTMS) of the dorsolateral prefrontal cortex (DLPFC) during capsaicin-induced pain: modulatory effects on motor cortex excitability. Exp. Brain Res. 203, 31–38. doi: 10.1007/s00221-010-2206-6
Fitzgerald, P. J., Giustino, T. F., Seemann, J. R., and Maren, S. (2015). Noradrenergic blockade stabilizes prefrontal activity and enables fear extinction under stress. Proc. Natl. Acad. Sci. U S A 112, E3729–E3737. doi: 10.1073/pnas.1500682112
Franklin, T. B., Silva, B. A., Perova, Z., Marrone, L., Masferrer, M. E., Zhan, Y., et al. (2017). Prefrontal cortical control of a brainstem social behavior circuit. Nat. Neurosci. 20, 260–270. doi: 10.1038/nn.4470
Gao, Y. J., and Ji, R. R. (2008). Activation of JNK pathway in persistent pain. Neurosci. Lett. 437, 180–183. doi: 10.1016/j.neulet.2008.03.017
Gisquet-Verrier, P., and Delatour, B. (2006). The role of the rat prelimbic/infralimbic cortex in working memory: not involved in the short-term maintenance but in monitoring and processing functions. Neuroscience 141, 585–596. doi: 10.1016/j.neuroscience.2006.04.009
Goffer, Y., Xu, D., Eberle, S. E., D’Amour, J., Lee, M., Tukey, D., et al. (2013). Calcium-permeable AMPA receptors in the nucleus accumbens regulate depression-like behaviors in the chronic neuropathic pain state. J. Neurosci. 33, 19034–19044. doi: 10.1523/JNEUROSCI.2454-13.2013
Grachev, I. D., Fredrickson, B. E., and Apkarian, A. V. (2000). Abnormal brain chemistry in chronic back pain: an in vivo proton magnetic resonance spectroscopy study. Pain 89, 7–18. doi: 10.1016/s0304-3959(00)00340-7
Hermann, C., Hohmeister, J., Demirakça, S., Zohsel, K., and Flor, H. (2006). Long-term alteration of pain sensitivity in school-aged children with early pain experiences. Pain 125, 278–285. doi: 10.1016/j.pain.2006.08.026
Hu, H. J., and Gereau, R. W. (2003). ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. II. Modulation of neuronal excitability. J. Neurophysiol. 90, 1680–1688. doi: 10.1152/jn.00341.2003
Impey, S., Obrietan, K., and Storm, D. R. (1999). Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron 23, 11–14. doi: 10.1016/S0896-6273(00)80747-3
Ivo, R., Nicklas, A., Dargel, J., Sobottke, R., Delank, K. S., Eysel, P., et al. (2013). Brain structural and psychometric alterations in chronic low back pain. Eur. Spine J. 22, 1958–1964. doi: 10.1007/s00586-013-2692-x
Ji, R. R., Gereau, R. W., Malcangio, M., and Strichartz, G. R. (2009). MAP kinase and pain. Brain Res. Rev. 60, 135–148. doi: 10.1016/j.brainresrev.2008.12.011
Ji, G., and Neugebauer, V. (2012). Modulation of medial prefrontal cortical activity using in vivo recordings and optogenetics. Mol. Brain 5:36. doi: 10.1186/1756-6606-5-36
Ji, R. R., and Woolf, C. J. (2001). Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol. Dis. 8, 1–10. doi: 10.1006/nbdi.2000.0360
Johnson, G. L., and Lapadat, R. (2002). Mitogen-activated protein kinase pathways mediated by ERK, JNK and p38 protein kinases. Science 298, 1911–1912. doi: 10.1126/science.1072682
Kaneko, H., Zhang, S., Sekiguchi, M., Nikaido, T., Makita, K., Kurata, J., et al. (2017). Dysfunction of nucleus accumbens is associated with psychiatric problems in patients with chronic low back pain: a functional magnetic resonance imaging study. Spine 42, 844–853. doi: 10.1097/BRS.0000000000001930
Khan, S. A., Keaser, M. L., Meiller, T. F., and Seminowicz, D. A. (2014). Altered structure and function in the hippocampus and medial prefrontal cortex in patients with burning mouth syndrome. Pain 155, 1472–1480. doi: 10.1016/j.pain.2014.04.022
Kucyi, A., Salomons, T. V., and Davis, K. D. (2013). Mind wandering away from pain dynamically engages antinociceptive and default mode brain networks. Proc. Natl. Acad. Sci. U S A 110, 18692–18697. doi: 10.1073/pnas.1312902110
Kumar, S., Boehm, J., and Lee, J. C. (2003). p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov. 2, 717–726. doi: 10.1038/nrd1177
Lasseter, H. C., Xie, X., Ramirez, D. R., and Fuchs, R. A. (2010). Prefrontal cortical regulation of drug seeking in animal models of drug relapse. Curr. Top. Behav. Neurosci. 3, 101–117. doi: 10.1007/7854_2009_19
LeBlanc, D. M., McGinn, M. A., Itoga, C. A., and Edwards, S. (2015). The affective dimension of pain as a risk factor for drug and alcohol addiction. Alcohol 49, 803–809. doi: 10.1016/j.alcohol.2015.04.005
Lee, M., Manders, T. R., Eberle, S. E., Su, C., D’Amour, J., Yang, R., et al. (2015). Activation of corticostriatal circuitry relieves chronic neuropathic pain. J. Neurosci. 35, 5247–5259. doi: 10.1523/JNEUROSCI.3494-14.2015
Li, M. J., Liu, L. Y., Chen, L., Cai, J., Wan, Y., and Xing, G. G. (2017). Chronic stress exacerbates neuropathic pain via the integration of stress-affect-related information with nociceptive information in the central nucleus of the amygdala. Pain 158, 717–739. doi: 10.1097/j.pain.0000000000000827
Li, S. G., Wang, J. Y., and Luo, F. (2012). Adult-age inflammatory pain experience enhances long-term pain vigilance in rats. PLoS One 7:e36767. doi: 10.1371/journal.pone.0036767
Lidow, M. S. (2002). Long-term effects of neonatal pain on nociceptive systems. Pain 99, 377–383. doi: 10.1016/s0304-3959(02)00258-0
Lim, M., Kim, J. S., Kim, D. J., and Chung, C. K. (2016). Increased low- and high-frequency oscillatory activity in the prefrontal cortex of fibromyalgia patients. Front. Hum. Neurosci. 10:111. doi: 10.3389/fnhum.2016.00111
Lin, C. S. (2014). Brain signature of chronic orofacial pain: a systematic review and meta-analysis on neuroimaging research of trigeminal neuropathic pain and temporomandibular joint disorders. PLoS One 9:e94300. doi: 10.1371/journal.pone.0094300
Luo, H., Cheng, J., Han, J. S., and Wan, Y. (2004). Change of vanilloid receptor 1 expression in dorsal root ganglion and spinal dorsal horn during inflammatory nociception induced by complete Freund’s adjuvant in rats. Neuroreport 15, 655–658. doi: 10.1097/00001756-200403220-00016
Metz, A. E., Yau, H. J., Centeno, M. V., Apkarian, A. V., and Martina, M. (2009). Morphological and functional reorganization of rat medial prefrontal cortex in neuropathic pain. Proc. Natl. Acad. Sci. U S A 106, 2423–2428. doi: 10.1073/pnas.0809897106
Moench, K. M., and Wellman, C. L. (2015). Stress-induced alterations in prefrontal dendritic spines: implications for post-traumatic stress disorder. Neurosci. Lett. 601, 41–45. doi: 10.1016/j.neulet.2014.12.035
Navratilova, E., Atcherley, C. W., and Porreca, F. (2015). Brain circuits encoding reward from pain relief. Trends Neurosci. 38, 741–750. doi: 10.1016/j.tins.2015.09.003
Negrón-Oyarzo, I., Pérez, M. A., Terreros, G., Muñoz, P., and Dagnino-Subiabre, A. (2014). Effects of chronic stress in adolescence on learned fear, anxiety and synaptic transmission in the rat prelimbic cortex. Behav. Brain Res. 259, 342–353. doi: 10.1016/j.bbr.2013.11.001
Obata, K., and Noguchi, K. (2004). MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci. 74, 2643–2653. doi: 10.1016/j.lfs.2004.01.007
Obermann, M., Nebel, K., Schumann, C., Holle, D., Gizewski, E. R., Maschke, M., et al. (2009). Gray matter changes related to chronic posttraumatic headache. Neurology 73, 978–983. doi: 10.1212/WNL.0b013e3181b8791a
Pochwat, B., Rafalo-Ulinska, A., Domin, H., Misztak, P., Nowak, G., and Szewczyk, B. (2017). Involvement of extracellular signal-regulated kinase (ERK) in the short and long-lasting antidepressant-like activity of NMDA receptor antagonists (zinc and Ro 25–6981) in the forced swim test in rats. Neuropharmacology 125, 333–342. doi: 10.1016/j.neuropharm.2017.08.006
Pucilowska, J., Puzerey, P. A., Karlo, J. C., Galán, R. F., and Landreth, G. E. (2012). Disrupted ERK signaling during cortical development leads to abnormal progenitor proliferation, neuronal and network excitability and behavior, modeling human neuro-cardio-facial-cutaneous and related syndromes. J. Neurosci. 32, 8663–8677. doi: 10.1523/JNEUROSCI.1107-12.2012
Ren, K., Anseloni, V., Zou, S. P., Wade, E. B., Novikova, S. I., Ennis, M., et al. (2004). Characterization of basal and re-inflammation-associated long-term alteration in pain responsivity following short-lasting neonatal local inflammatory insult. Pain 110, 588–596. doi: 10.1016/j.pain.2004.04.006
Rodriguez-Raecke, R., Niemeier, A., Ihle, K., Ruether, W., and May, A. (2009). Brain gray matter decrease in chronic pain is the consequence and not the cause of pain. J. Neurosci. 29, 13746–13750. doi: 10.1523/JNEUROSCI.3687-09.2009
Saffari, R., Teng, Z., Zhang, M., Kravchenko, M., Hohoff, C., Ambrée, O., et al. (2016). NPY+-, but not PV+-GABAergic neurons mediated long-range inhibition from infra- to prelimbic cortex. Transl. Psychiatry 6:e736. doi: 10.1038/tp.2016.7
Seger, R., and Krebs, E. G. (1995). The MAPK signaling cascade. FASEB J. 9, 726–735. doi: 10.1096/fasebj.9.9.7601337
Seminowicz, D. A., Laferriere, A. L., Millecamps, M., Yu, J. S., Coderre, T. J., and Bushnell, M. C. (2009). MRI structural brain changes associated with sensory and emotional function in a rat model of long-term neuropathic pain. Neuroimage 47, 1007–1014. doi: 10.1016/j.neuroimage.2009.05.068
Seminowicz, D. A., Shpaner, M., Keaser, M. L., Krauthamer, G. M., Mantegna, J., Dumas, J. A., et al. (2013). Cognitive-behavioral therapy increases prefrontal cortex gray matter in patients with chronic pain. J. Pain 14, 1573–1584. doi: 10.1016/j.jpain.2013.07.020
Seminowicz, D. A., Wideman, T. H., Naso, L., Hatami-Khoroushahi, Z., Fallatah, S., Ware, M. A., et al. (2011). Effective treatment of chronic low back pain in humans reverses abnormal brain anatomy and function. J. Neurosci. 31, 7540–7550. doi: 10.1523/JNEUROSCI.5280-10.2011
Seo, J. S., Wei, J., Qin, L., Kim, Y., Yan, Z., and Greengard, P. (2017). Cellular and molecular basis for stress-induced depression. Mol. Psychiatry 22, 1440–1447. doi: 10.1038/mp.2016.118
Sherrin, T., Blank, T., and Todorovic, C. (2011). c-Jun N-terminal kinases in memory and synaptic plasticity. Rev. Neurosci. 22, 403–410. doi: 10.1515/RNS.2011.032
Shibata, M., Ohkubo, T., Takahashi, H., and Inoki, R. (1989). Modified formalin test: characteristic biphasic pain response. Pain 38, 347–352. doi: 10.1016/0304-3959(89)90222-4
Soliman, A. C., Yu, J. S., and Coderre, T. J. (2005). mGlu and NMDA receptor contributions to capsaicin-induced thermal and mechanical hypersensitivity. Neuropharmacology 48, 325–332. doi: 10.1016/j.neuropharm.2004.10.014
Toyoda, H., Zhao, M. G., Xu, H., Wu, L. J., Ren, M., and Zhuo, M. (2007). Requirement of extracellular signal-regulated kinase/mitogen-activated protein kinase for long-term potentiation in adult mouse anterior cingulate cortex. Mol. Pain 3:36. doi: 10.1186/1744-8069-3-36
Umana, I. C., Daniele, C. A., Miller, B. A., Abburi, C., Gallagher, K., Brown, M. A., et al. (2017). Nicotinic modulation of descending pain control circuitry. Pain 158, 1938–1950. doi: 10.1097/j.pain.0000000000000993
Vaccarino, A. L., and Melzack, R. (1992). Temporal processes of formalin pain: differential role of the cingulum bundle, fornix pathway and medial bulboreticular formation. Pain 49, 257–271. doi: 10.1016/0304-3959(92)90150-a
Vachon-Presseau, E., Tetreault, P., Petre, B., Huang, L., Berger, S. E., Torbey, S., et al. (2016). Corticolimbic anatomical characteristics predetermine risk for chronic pain. Brain 139, 1958–1970. doi: 10.1093/brain/aww100
Wang, G. Q., Cen, C., Li, C., Cao, S., Wang, N., Zhou, Z., et al. (2015). Deactivation of excitatory neurons in the prelimbic cortex via Cdk5 promotes pain sensation and anxiety. Nat. Commun. 6:7660. doi: 10.1038/ncomms8660
Wegner, A., Elsenbruch, S., Rebernik, L., Roderigo, T., Engelbrecht, E., Jager, M., et al. (2015). Inflammation-induced pain sensitization in men and women: does sex matter in experimental endotoxemia? Pain 156, 1954–1964. doi: 10.1097/j.pain.0000000000000256
Widmann, C., Gibson, S., Jarpe, M. B., and Johnson, G. L. (1999). Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol. Rev. 79, 143–180. doi: 10.1152/physrev.1999.79.1.143
Wu, X. B., Liang, B., and Gao, Y. J. (2016). The increase of intrinsic excitability of layer V pyramidal cells in the prelimbic medial prefrontal cortex of adult mice after peripheral inflammation. Neurosci. Lett. 611, 40–45. doi: 10.1016/j.neulet.2015.11.030
Wynne, P. (2006). p38 mitogen-activated protein kinase: a novel modulator of hyperpolarization-activated cyclic nucleotide-gated channels and neuronal excitability. J. Neurosci. 26, 11253–11254. doi: 10.1523/jneurosci.3852-06.2006
Yi, M., and Zhang, H. (2011). Nociceptive memory in the brain: cortical mechanisms of chronic pain. J. Neurosci. 31, 13343–13345. doi: 10.1523/JNEUROSCI.3279-11.2011
Yi, M., Zhang, H., Lao, L., Xing, G. G., and Wan, Y. (2011). Anterior cingulate cortex is crucial for contra- but not ipsi-lateral electro-acupuncture in the formalin-induced inflammatory pain model of rats. Mol. Pain 7:61. doi: 10.1186/1744-8069-7-61
Yu, R., Gollub, R. L., Spaeth, R., Napadow, V., Wasan, A., and Kong, J. (2014). Disrupted functional connectivity of the periaqueductal gray in chronic low back pain. Neuroimage Clin. 6, 100–108. doi: 10.1016/j.nicl.2014.08.019
Yu, L., Yang, F., Luo, H., Liu, F. Y., Han, J. S., Xing, G. G., et al. (2008). The role of TRPV1 in different subtypes of dorsal root ganglion neurons in rat chronic inflammatory nociception induced by complete Freund’s adjuvant. Mol. Pain 4:61. doi: 10.1186/1744-8069-4-61
Yue, L., Ma, L. Y., Cui, S., Liu, F. Y., Yi, M., and Wan, Y. (2017). Brain-derived neurotrophic factor in the infralimbic cortex alleviates inflammatory pain. Neurosci. Lett. 655, 7–13. doi: 10.1016/j.neulet.2017.06.028
Zhang, Y., Cai, G., Ni, X., and Sun, J. (2007). The role of ERK activation in the neuronal excitability in the chronically compressed dorsal root ganglia. Neurosci. Lett. 419, 153–157. doi: 10.1016/j.neulet.2007.04.040
Zhang, C., Chen, R. X., Zhang, Y., Wang, J., Liu, F. Y., Cai, J., et al. (2017). Reduced GABAergic transmission in the ventrobasal thalamus contributes to thermal hyperalgesia in chronic inflammatory pain. Sci. Rep. 7:41439. doi: 10.1038/srep41439
Zhang, Y., Jiang, Y. Y., Shao, S., Zhang, C., Liu, F. Y., Wan, Y., et al. (2017). Inhibiting medial septal cholinergic neurons with DREADD alleviated anxiety-like behaviors in mice. Neurosci. Lett. 638, 139–144. doi: 10.1016/j.neulet.2016.12.010
Zhang, Y., Liu, F. Y., Liao, F. F., Wan, Y., and Yi, M. (2014). Exacerbation of tonic but not phasic pain by entorhinal cortex lesions. Neurosci. Lett. 581, 137–142. doi: 10.1016/j.neulet.2014.05.015
Zhang, M., Liu, J., Zhou, M. M., Wu, H., Hou, Y., Li, Y. F., et al. (2016). Elevated neurosteroids in the lateral thalamus relieve neuropathic pain in rats with spared nerve injury. Neurosci. Bull. 32, 311–322. doi: 10.1007/s12264-016-0044-7
Zhang, M., Liu, J., Zhou, M. M., Wu, H., Hou, Y., Li, Y. F., et al. (2017). Anxiolytic effects of hippocampal neurosteroids in normal and neuropathic rats with spared nerve injury. J. Neurochem. 141, 137–150. doi: 10.1111/jnc.13965
Zheng, J., Jiang, Y. Y., Xu, L. C., Ma, L. Y., Liu, F. Y., Cui, S., et al. (2017). Adult hippocampal neurogenesis along the dorsoventral axis contributes differentially to environmental enrichment combined with voluntary exercise in alleviating chronic inflammatory pain in mice. J. Neurosci. 37, 4145–4157. doi: 10.1523/JNEUROSCI.3333-16.2017
Zhuang, Z. Y., Gerner, P., Woolf, C. J., and Ji, R. R. (2005). ERK is sequentially activated in neurons, microglia and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain 114, 149–159. doi: 10.1016/j.pain.2004.12.022
Keywords: pain, chronic pain, prelimbic cortex, prefrontal cortex, p38
Citation: Fan X-C, Fu S, Liu F-Y, Cui S, Yi M and Wan Y (2018) Hypersensitivity of Prelimbic Cortex Neurons Contributes to Aggravated Nociceptive Responses in Rats With Experience of Chronic Inflammatory Pain. Front. Mol. Neurosci. 11:85. doi: 10.3389/fnmol.2018.00085
Received: 26 November 2017; Accepted: 05 March 2018;
Published: 22 March 2018.
Edited by:
Ildikó Rácz, Universitätsklinikum Bonn, GermanyReviewed by:
Esperanza Bas Infante, University of Miami, United StatesCopyright © 2018 Fan, Fu, Liu, Cui, Yi and Wan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Yi, bWluZ3lpQGJqbXUuZWR1LmNu
You Wan, eXdhbkBoc2MucGt1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.