 Haijun Zhang
Haijun Zhang Longbin Zhang
Longbin Zhang Tao Sun1,3*
Tao Sun1,3*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 19 February 2018
Sec. Molecular Signalling and Pathways
Volume 11 - 2018 | https://doi.org/10.3389/fnmol.2018.00044
This article is part of the Research Topic Gene Expression in Neurons: Seeing is Believing View all 6 articles
Proper proliferation and differentiation of neural progenitors (NPs) in the developing cerebral cortex are critical for normal brain formation and function. Emerging evidence has shown the importance of microRNAs (miRNAs) in regulating cortical development and the etiology of neurological disorders. Here we show that miR-26 is co-expressed with its host gene Ctdsp in the mouse embryonic cortex. We demonstrate that similar to its host gene Ctdsp2, miR-26 positively regulates proliferation of NPs through controlling the cell-cycle progression, by using miR-26 overexpression and sponge approaches. On the contrary, miR-26 target gene Emx2 limits expansion of cortical NPs, and promotes transcription of miR-26 host gene Ctdsp. Our study suggests that miR-26, its target Emx2 and its host gene Ctdsp cohesively regulate proliferation of NPs during the mouse cortical development.
The precisely regulated proliferation, survival and differentiation of neural stem cells (NSCs) and neural progenitors (NPs) determine normal development of the mammalian cerebral cortex (Dehay and Kennedy, 2007; Rakic, 2007, 2009; Aguirre et al., 2010; Delaunay et al., 2017; Subramanian et al., 2017). During embryonic cortical development, NSCs first appear as the highly proliferative neuroepithelial cells lining the ventricular wall. They subsequently generate radial glial cells (RGCs) or outer radial glia (oRG), which undergo an asymmetric division (Bultje et al., 2009; Wang et al., 2011; Ostrem et al., 2014). RGCs are tightly regulated to maintain their own population while to produce intermediate progenitors (IPs) that reside in the subventricular zone (SVZ) and mature neurons that migrate into the cortical plate (CP; Guillemot, 2005; Kriegstein et al., 2006; Mizutani et al., 2007; Molyneaux et al., 2007; Molnár, 2011; Franco and Müller, 2013). Abnormal proliferation or/and differentiation, caused by prenatal or postnatal gene mutations and altered gene expression levels, is one of the critical factors for brain malformation (Chenn and Walsh, 2002; Piao et al., 2004; Sun and Hevner, 2014). Molecular mechanisms that regulate expression patterns and levels of critical genes that govern cortical development remain an exciting research topic.
The emerging evidence indicates essential roles of microRNAs (miRNAs) in diverse biological events, such as cell proliferation, differentiation, migration, apoptosis and tumorigenesis (Kremer-Tal et al., 2004; Bian and Sun, 2011; Nadim et al., 2017). miRNAs are ~22 nucleotide (nt) endogenous noncoding small RNAs acting in complex gene regulatory networks (Lee et al., 1993; Wightman et al., 1993; Cora et al., 2017). Mature miRNAs recognize a complementary sequence frequently in the 3′-untranslated region (3′UTR) of its target messenger RNA (mRNA) to affect mRNA stability and/or silence protein translation (Carthew and Sontheimer, 2009; Kim et al., 2009). Studies have shown that misregulation of miRNAs generated by cortical Dicer deletion causes significant cell death, loss of progenitors and abnormal differentiation (De Pietri Tonelli et al., 2008; Kawase-Koga et al., 2009, 2010; Hong et al., 2013). Knockout of specific miRNA has shown a significant impact of miRNAs in embryonic and adult neurogenesis (Shen et al., 2006; Aguirre et al., 2010; Krol et al., 2010; Mellios et al., 2011; Bian et al., 2013; Abdullah et al., 2016). Furthermore, dysfunction of miRNAs is associated with the pathogenesis of neurodevelopmental disorders, neurodegeneration diseases and affective mental disorders (Hugon and Paquet, 2008; Packer et al., 2008; Bian and Sun, 2011).
miRNA miR-26 has previously been described as a functional miRNA that is involved in various biological events such as cell proliferation, development of normal tissues and tumorigenesis (Gao and Liu, 2011). Interestingly, studies have shown a contradictive role of miR-26 as either a tumor suppressor or activator in different types of cancer via regulating cell proliferation and migration (Lu et al., 2011; Zhang et al., 2012; Tan et al., 2014; Du et al., 2015). Further studies have illustrated a regulatory role of miR-26 in G1/S-phase transition by concomitantly expressing with their host genes C-terminal domain RNA polymerase II small phosphatase (Ctdsp gene families; Zhu et al., 2012; Wang et al., 2016). Despite these reports in tumors, the role of miR-26 in cortical development has not been well explored.
This study demonstrates that miR-26 and its host gene Ctdsp are co-expressed in NPs in the mouse developing cortex, and they play a positive role in NP expansion. We show that Emx2 is a target gene of miR-26, and displays an opposing function in NP development, compared to miR-26. Moreover, Emx2 functions as a transcription activator to initiate expression of Ctdsp. Our results elucidate a regulatory loop of miR-26, their target gene Emx2 and host gene Ctdsp, which works cohesively to ensure proper development of NPs in the developing cortex.
The full-length sequence of Ctdsp2 with flanking regions was cloned from its cDNA and inserted into the backbone plasmids pCAGIG to construct the overexpression vectors of Ctdsp2. The Emx2 overexpressing constructor was achieved in the same strategy. For Ctdsp2 silencing, the Ctdsp2 specific short hairpin RNA (shCtdsp2) targeting 5′-GCCTGTTGAGGCAGCAGAAGC-3′ was cloned into the pSilencer vector. The Ctdsp2 knockdown efficiency by this vector was verified by real time reverse transcription PCR. The overexpression and knockdown plasmids of Emx2 was constructed as introduced above.
The mouse genomic sequence including miR-26a precursor was amplified by PCR, and cloned into pGEM-T (promega), following subcloned into the pCAGIG vector for in utero electroporation and into pcDNA3.1 (Invitrogen) for transfection, respectively. The following primers were used to amplify miR-26a: F-5′-GGACAAGAACCAGGAAGG-3′, and R-5′-GCTGCCTCCGCGTTCGC-3′. For miR-26a mutation construct, the wild-type miR-26a seed sequence 5′-UCAAGU-3′ was mutated to 5′-UGTTCU-3′ following the instruction of the QuikChange II Site-Directed Mutagenesis Kit (Agilent).
To knockdown the expression of miR-26a, miRNA sponge strategy was used according to previous description (Zhang et al., 2013; Pollock et al., 2014). Briefly, synthesis was operated to construct specific miR-26-related sponges, using forward and reverse sponge oligos (mmu-mir-26a-SP-F: 5′-AC TAGTGTTATCAGCCTATCCTGCTTACTTGAAGTTATCAG CCTATCCTGCTTACTTGAAGTTATCAGCCTATCCTGCTT ACTTGAATCTAGA-3′; mmu-mir-26a-SP-mut-F: 5′-ACTAG TGTTATCAGCCTATCCTGCTTAGTTCTAGTTATCAGCCT ATCCTGCTTAGTTCTAGTTATCAGCCTATCCTGCTTAGT TCTATCTAGA-3′) containing three bulged miR-26a, miR-26a with three mutations in the binding seed, or scrambled binding sites. Each miR-26 sponge contained multiple binding sequences complementary to mature miR-26. All sponges were flanked by the SpeI and XbaI cutting sites, and subcloned into 3′UTR of Pol II-driven green fluorescence protein (GFP) reporter gene, following by inserting into the pCBR conditional expression vector.
In situ hybridization for genes expression was performed on frozen sections using specific probes. Probes used in miRNA hybridization contain modified nucleotides that form a locked structure to stabilize LNA/RNA duplex, thus has been widely used to detect miRNA expression (Zhang and Yin, 2005; Elmen et al., 2008). After fixation with 4% paraformaldehyde (PFA), acetylation with acetylation buffer (1.3% Triethanolamine, 0.25% Acetic anhydride, 20 mM HCl), treatment with proteinase K (5 μg/ml, IBI Scientific) and pre-hybridization (1× SSC, 50% Formamide, 0.1 mg/ml Salmon Sperm DNA Solution, 1× Denhart, 5 mM EDTA, pH 7.5), brain sections were hybridized with DIG-labeled LNA probes at Tm-22°C overnight. After washing with pre-cooled wash buffer (1× SSC, 50% Formamide, 0.1% Tween-20) and 1× MABT, sections were blocked with blocking buffer (1× MABT, 2% Blocking solution, 20% heat-inactived sheep serum) and incubated with anti-DIG antibody (1:1500, Roche) at 4°C overnight. Brain sections were washed with 1× MABT and Staining buffer (0.1 M NaCl, 50 mM MgCl2, 0.1 M Tris-HCl, pH9.5), stained with BM purple (Roche) at room temperature until ideal intensity was reached. The miR-26 LNA probe was purchased from Exiqon with specific sequence (5′-UUCAAGUAAUCCAGGAUAGGCU-3′), the Ctdsp2 and Emx2 detective probe were reversed from the amplification of each mRNA using specific primer pairs (Ctdsp2: F-5′-TGCCTCCTGCTTCTCGTTAT-3′, R-5′-GGA CCTCGTGTGTGGAAACT-3′; Emx2: F-5′-TAGAGCACGCT TTTGAGAAGAACCA-3′, R-5′-TGAAACCATACTTTTACC TG-3′), respectively. Each probe was 3′- and 5′-end labeled with DIG–ddUTP.
Total RNA was isolated from the cerebral cortex of E12.5, E15.5 and P0 wild-type CD1 mice using the RNeasy Mini kit (Qiagen) according to the guideline’s instructions. All samples were treated with DNase to remove genomic DNA and reversely transcribed into cDNAs using a Random Hexamer primer (Roche). Three cDNA samples (10× dilution) of each cortex were used as templates to quantify the transcript of each Ctdsp2 or miR-26 precursors via quantitative real-time PCR (qRT-PCR) with paired primers. qRT-PCR was carried out using primers specific for Ctdsp2: 5′-GCATCTTACATCTTCCAC-3′ and 5′-TAGACATCATCAGTTCCA-3′; and universal primer: 5′-GCGAGCACAGAATAAATACGACTC-3′, together with specific primers for miR-26a-1: 5′-CCTATTCTTGGTTACTTG CACG-3′; miR-26a-2: 5′-CCTGTTCTTGATTACTTG TTTC-3′; miR-26b: 5′-TTCAAGTAAT TCAGGATAGGT-3′; and U6: 5′-CGCTTCGGCAGCACATATAC-3′ and 5′-GTGTCATCC TTGCGCAGGG-3′. The U6 was used as an internal standard. Relative transcript level of each gene was calculated as the ratio of its transcript in each RNA samples over that in the E12.5 RNA samples using the 2−ΔΔCt method (Livak and Schmittgen, 2001).
Total RNA was extracted and separated on 15% urea-PAGE gel, followed by transferred to positively charged nylon membrane (PerkinElmer, Waltham, MA, USA) using Trans-Blot SD Semi-Dry transfer Cell (Bio-Rad, Hercules, CA, USA). Oligonucleotide probes used for hybridization with miRNA were labeled with at their 5′ end. U6 was used as the loading control. The sequence of the probe was: miR-26a: 5′-ATTC AAGTTTTGAAACAGGTGTA-3′.
In utero electroporation was performed in E13.5 embryos according to the published protocol (Saito, 2006). Briefly, plasmid DNA was prepared using the EndoFree Plasmid Maxi Kit (Qiagen) according to manufacturer’s instructions, and diluted to 2 μg/μl. DNA solution was injected into the lateral ventricle of the cerebral cortex, and electroporated with five 50-ms pulses at 35V using an ECM830 electrosquareporator (BTX). Embryos were allowed to develop to E14.5. Animals with the brains electroporated, as detected by the GFP fluorescence under a fluorescent dissection scope (Leica, MZ16F), were selected for further analyses.
All experimental procedures involving animals were in accordance with the Guide for the Care and Use of Laboratory Animals (NIH publications Nos. 80-23, revised 1996) and were performed according to the institutional ethical guidelines for animal experiments.
Mouse Neuro2a cells were transfected using Lipofectamine 2000 (Invitrogen) using the manufacturer’s protocol. Plasmids were quantified by UV spectrophotometry and used for transfection in a 2:1 ratio (miRNA: target luciferase constructs). pGL4.13 firefly luciferase was used for 3′-UTRs of targets. pGL4.73 Renilla luciferase (Promega) was used as a transfection control. For transfections, Neuro2a were diluted in DMEM and plated into 24-well plates in triplicate at 1.5 × 104 cells/ 100 μL. Adherent cells were co-transfected with 100 ng/mL luciferase reporter containing the Emx2–3′-UTR and 50 nM pcDNA3.1 only (control), miR-26a mimics or miR-26amut. Each co-transfection was injected into pcDNA-iCre, miR-26a-SP, or miR-26a-SPmut expressing cells, respectively. After 48 h, luciferase was measured using the Dual-Luciferase Reporter Assay kit (Promega) using the manufacturer’s protocol and read on a Victor3 1420 multilabel counter (Perkin Elmer). All conditions were run in triplicate, and all experiments were repeated at least three times with similar results. Raw results for each condition were normalized for transfection efficiency as the ratio of Firefly luciferase to Renilla luciferase, and finally for each luciferase tested the empty vector control experiment was set to 1 for display.
For embryonic immunohistochemistry, the DNA constructs were injected into E13.5 embryos using in utero electroporation. To access proliferation of NP cells (NPCs) in developing cortex, one dose of bromodeoxyuridine (BrdU; 50 mg/g body weight) was administrated by intraperitoneal (I.P.) injection to DNA constructs-injected mice at 1 h or 1 day before sacrifice. Mouse embryonic brains were collected and fixed in 4% PFA in phosphate-buffered saline (PBS) over night, or incubated in 25%–30% sucrose in PBS, embedded in OCT and stored at −80°C until use. Brains were sectioned (14–16 μm) at a coronal plane using a cryostat. For antigen recovery, sections were incubated in heated (95–100°C) antigen recovery solution (1 mM EDTA, 5 mM Tris, pH 8.0) for 15–20 min, followed by 20–30 min of cooling treatment at 4°C. After being blocked in 10% normal goat serum (NGS) in PBS with 0.1% Tween-20 (PBT) for 1 h, sections were incubated with primary antibodies at 4°C overnight, then visualized after 1.5-h co-cultured with goat anti-rabbit IgG–Alexa- Fluor-488 and/or goat anti-mouse IgG–Alexa-Fluor-546 (1:300, Molecular Probes) at room temperature. Images were captured using a Leica digital camera under a fluorescent microscope (Leica DMI6000B) or a Zeiss confocal microscope.
Primary antibodies against the following antigens were used: BrdU (1:50, Developmental Studies Hybridoma Bank at University of Iowa (DSHB)), Ki67 (1:500, Abcam), Pax6 (1:500, Covance), Pax6 (1:15 DSHB), Tbr2 (1:500, Abcam), GFP (1:1000, Abcam, chicken), and GFP (1:1000, Rockland, rabbit).
Cell counting in the mouse cortical tissue was performed in a fixed area of 300 μm × 300 μm, a representative column of the cortical wall from coronal sections. All sections analyzed were selected from a similar medial point on the anterior-posterior axis. For each condition, at least three brains, and at least three images for each individual brain were counted.
Data were shown as mean ± SEM. One-way analysis of variance (ANOVA) with post hoc contrasts were used for statistical analysis. The results were considered significant at probability level less than 0.05.
To identify miRNAs that may function in neurogenesis, we performed expression profiles of miRNAs in the mouse cerebral cortex at embryonic day 12.5 (E12.5) and postnatal day 0 (P0) using microarray. The preliminary screen identified a miRNA, miR-26, which shows high expression at E12.5. Analyses of its seed sequence and genomic context indicated that the miR-26 family harbors two homologs, miR-26a and miR-26b, which are transcribed from three genomic loci, miR-26a-1, miR-26a-2 and miR-26b. Their seed sequences were highly conserved between species (Supplementary Figure S1A). Moreover, these loci reside in the introns of genes coding for Ctdspl in chromosome 9 (chr9), Ctdsp2 in chr10 and Ctdsp1 in chr1, respectively (Figure 1A).
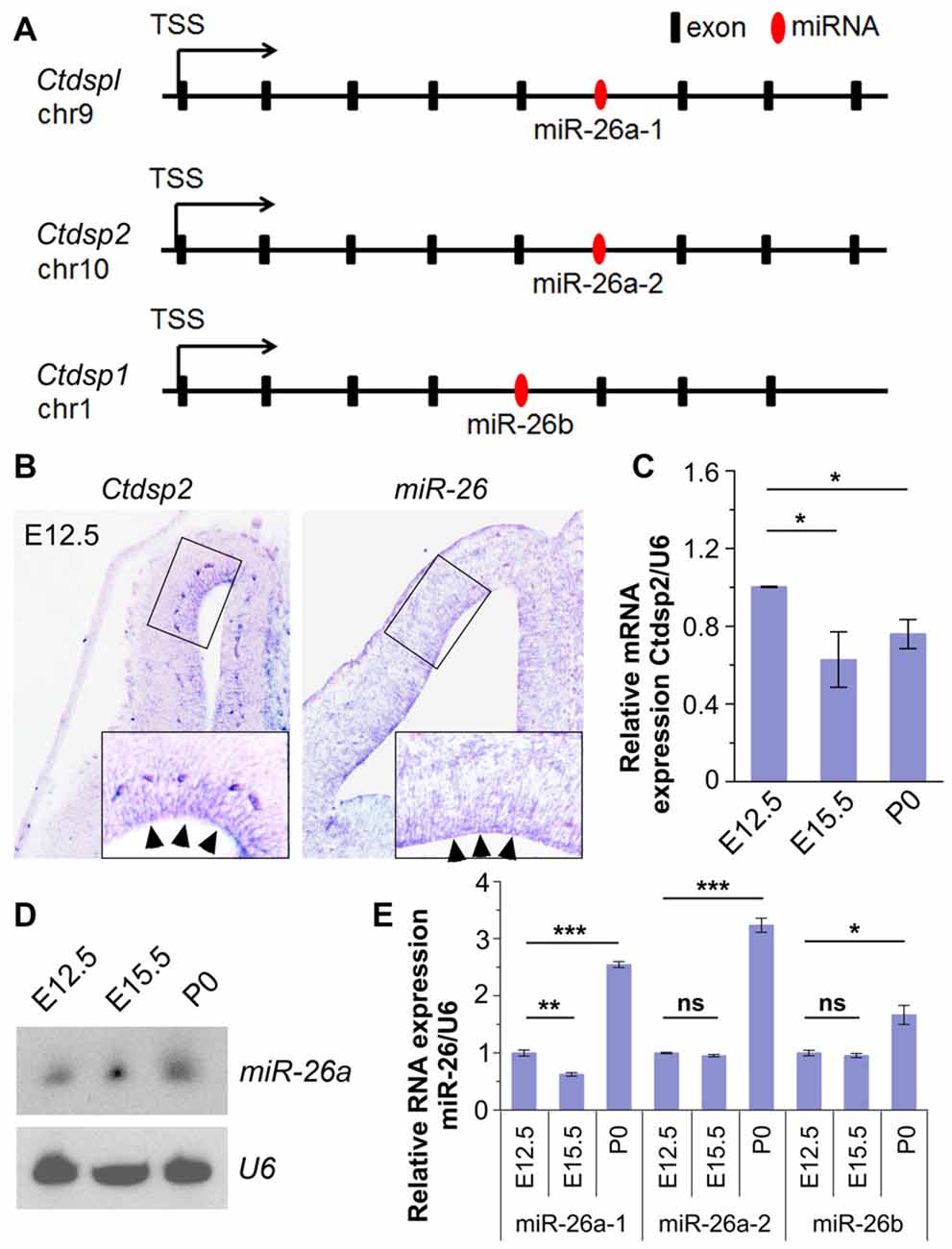
Figure 1. Expression patterns and levels of miR-26 and its host gene Ctdsp2 in the mouse developing cortex. (A) Schemes of exons and introns of Ctdspl, Ctdsp2 and Ctdsp1, and their intronic microRNA (miRNA) miR-26a-1, miR-26a-2 and miR-26b, respectively, in the mouse genome. (B) Ctdsp2 and miR-26a were both expressed in the ventricular zone (VZ; arrows). (C) Relative expression of Ctdsp2 gene vs. U6 in E12.5, E15.5 and P0 cortices, detected by quantitative real-time PCR (qRT-PCR). Ctdsp2 expression in the E12.5 cortex was set to 1. (D) miR-26a expression in different developmental stages, as detected by Northern blots. The ubiquitously expressed small non-coding RNA U6 was used as a loading control. (E) Relative expression levels of miR-26a-1, miR-26a-2 and miR-26b in E12.5, E15.5 and P0 cortices, detected by qRT-PCR. U6 was used as an internal control. miR-26 expression in the E12.5 cortex was set to 1. Values represent mean ± SEM. n > 3. *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant. Analysis of variance (ANOVA) with post hoc test was used.
We first examined whether miR-26a and its host gene Ctdsp2 are expressed concomitantly in the developing mouse cortices using in situ hybridization. The expression of Ctdsp2 and miR-26a was mainly aggregated in the VZ in E12.5 cortex, indicating co-expression of miR-26 with its host gene Ctdsp2 (Figure 1B). Sense probe for Ctdsp2 and scramble probe for miR-26 did not show detectable signals (Supplementary Figure S1B). qRT-PCR validated that the expression of Ctdsp2 is high in the cortex at E12.5, slightly decreased from E12.5 to E15.5, and is merely altered from E15.5 to P0 (Figure 1C).
We next examined miR-26 expression. Northern blotting assay of miR-26a demonstrated that the expression of miR-26a is slightly up-regulated from E12.5 to E15.5, and is maintained at P0 (Figure 1D). The qRT-PCR assays of three miR-26 precursors further confirmed miR-26 expression throughout cortical development (Figure 1E). The expression of precursor miR-26a-1 was down-regulated from E12.5 to E15.5, and significantly increased at P0. Moreover, precursors miR-26a-2 and miR-26b displayed similar expression levels at E12.5 and E15.5, and were upregulated at P0 (Figure 1E). These data indicate that miR-26 and their host genes display opposite expression patterns during cortical development.
Due to the conserved seed sequence of miR-26 family, the following study was focused predominantly on the mouse miR-26a-2 host gene Ctdsp2, and miR-26a that contains the same seed sequence of both miR-26a-1 and miR-26a-2.
Due to Ctdsp2 expression in the VZ of E12.5 mouse cortex, we examined the role of Ctdsp2 in regulation of NP development. We first overexpressed Ctdsp2 in NPs in the VZ using in utero electroporation at E13.5, and collected brain tissues at E14.5. A BrdU pulse was given 1 h before tissue collection to label dividing cells in the S-phase in a cell cycle. Ctdsp2 overexpression, compared to the pCAGIG control construct, caused significant increase of the percentage of BrdU+/GFP+ cells vs. GFP+ cells, indicating that Ctdsp2 plays a role in promoting proliferation of cortical NPs (Figure 2A). We further investigated whether the population of RGCs and IPs, which can be labeled by Pax6 or Tbr2, respectively, is affected by Ctdsp2 (Englund et al., 2005). Ctdsp2 overexpression induced the expansion of Pax6+ RGCs, but did not change that of Tbr2+ IPs (Figures 2B,C).
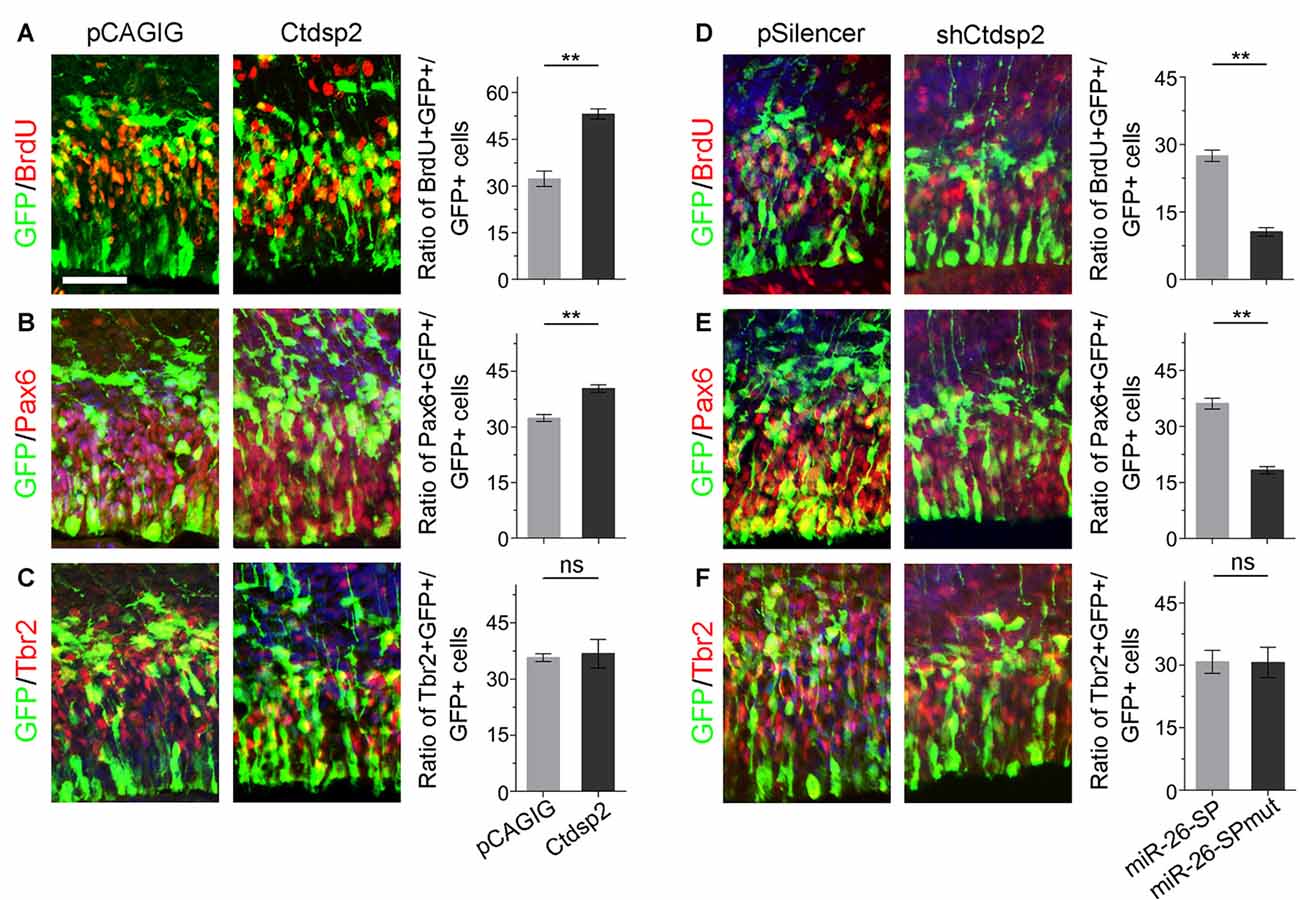
Figure 2. Ctdsp2 increases neural progenitor (NP) proliferation. (A–C) Overexpression of Ctdsp2, but not the control construct pCAGIG, increased the proportion of cells expressing both proliferative marker bromodeoxyuridine+ (BrdU+)/green fluorescence protein+ (GFP+) and radial glial cell (RGC) marker Pax6+/GFP+, but not intermediate progenitor (IP) marker Tbr2+/GFP+, in GFP-positive cells. (D–F) shRNA-mediated knockdown (shCtdsp2) of Ctdsp2 decreased the proportion of both BrdU+/GFP+ cells and Pax6+/GFP+ cells, but not Tbr2+/GFP+ cells in GFP-positive cells, compared to the control construct pSilencer. Values represent mean ± SEM. n = 9 sections from at least three brains. **P < 0.01; ns, not significant. ANOVA with post hoc test was used. Scale bar = 50 μm.
We next knocked down Ctdsp2 expression using short hairpin RNA (shRNA), shCtdsp2 (Supplementary Figure S2). Knockdown of Ctdsp2 expression resulted in a significant reduction of the percentage of BrdU+/GFP+ cells vs. GFP+ cells, compared to the pSilencer control (Figure 2D). Moreover, shCtdsp2 significantly blocked the expansion of RGCs, and did not affect IPs (Figures 2E,F). These results indicate that Ctdsp2 promotes expansion of RGCs.
We next evaluated the role of miR-26a in NP development via overexpressing a construct containing the mouse miR-26a precursor using in utero electroporation. miR-26a overexpression significantly increased the percentage of BrdU+/GFP+ cells vs. GFP+ cells, suggesting that miR-26 facilitates NP proliferation (Figures 3A,C). Moreover, overexpression of miR-26a promoted the population of RGCs, and had no effect on regulating the population of IPs (Figures 3A,D,F,G,I).
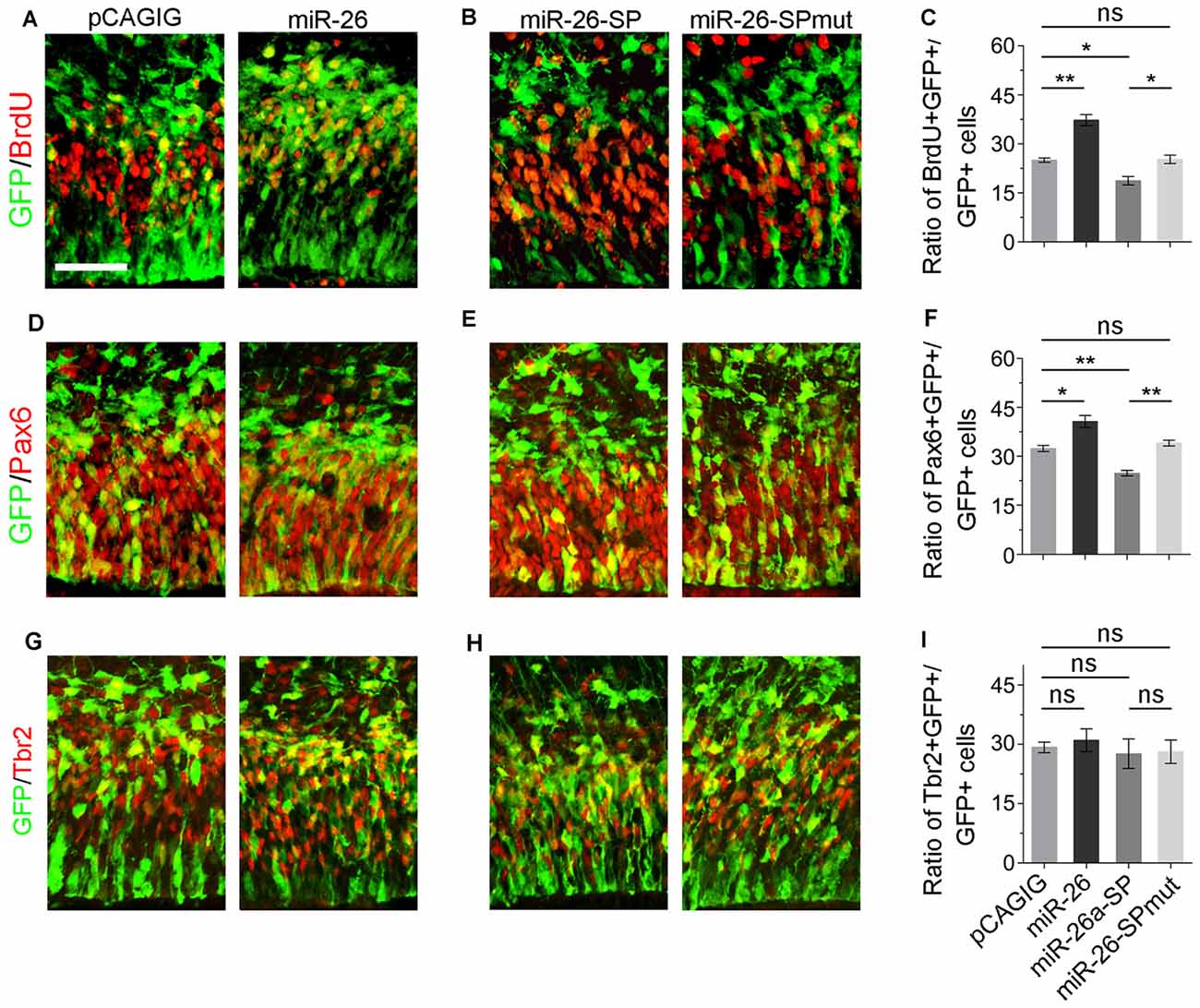
Figure 3. miR-26 promotes NP proliferation. (A,D,G) Overexpression of miR-26a, but not the control construct pCAGIG, proportion of cells expressing both proliferative marker BrdU+/GFP+ and RGC marker Pax6+/GFP+, but not IP marker Tbr2+/GFP+, in GFP-positive cells. (B,E,H) miRNA sponge-mediated knockdown (miR-26-SP), but not the mutated sponge (miR-26-SPmut), decreased proportion of cells expressing both proliferative marker BrdU+/GFP+ and RGC marker Pax6+/GFP+ in GFP-positive cells. (C,F,I) Ratio of BrdU+/GFP+, Pax6+/GFP+ or Tbr2+/GFP+ cells vs. GFP+ cells in the electroporated cortex. Values represent mean ± SEM. n = 9 sections from at least three brains. *P < 0.05; **P < 0.01; ns, not significant. ANOVA with post hoc test was used. Scale bar = 50 μm.
To knock down miR-26a expression, we designed and applied miR-26 sponge (miR-26a-SP; Supplementary Figure S3; Otaegi et al., 2011). Opposite to miR-26a overexpression, miR-26a-SP led to a pronounced reduction of the percentage of BrdU+/GFP+ cells vs. GFP+ cells, while mutation of miR-26 sponge (miR-26-SPmut) showed no effect (Figures 3B,C). Moreover, knockdown of miR-26a decreased the population of RGCs, and did not change the population of IPs (Figures 3E,F,H,I). Our results suggest that proper expression levels of miR-26 are essential for mediating the proliferation of NPs and maintaining the size of the RGC population in the embryonic cortex.
To further evaluate the mechanism of miR-26 function during neurogenesis, we analyzed its role in cell-cycle progression of NPs using in utero electroporation. Two parameters were quantified to indicate the status of cell-cycle. The labeling index was used to estimate the proportion of cells entering the cell cycle, by calculating BrdU incorporation in all cycling cells (Ki67+/GFP+). The cell-cycle-exit index was used to evaluate the proportion of cells exiting the cell cycle, by measuring the number of exiting cells within 24 h of BrdU incorporation (BrdU+/Ki67−/GFP+) vs. the total number of cycling cells (Ki67+/GFP+).
To analyze the labeling index, the embryonic cortex was electroporated at E13.5, and collected at E14.5. BrdU was injected 1 h before brain tissue collection (Figure 4A). miR-26a overexpression caused increased proportion of BrdU+/Ki67+ cells vs. GFP+ cells, compared to the control (Figures 4A,B). On the contrary, miR-26a knockdown using its sponge displayed a reduction of BrdU+/Ki67+ cells vs. GFP+ cells (Figures 4A,B). The cell-cycle labeling index analysis suggests that miR-26a promotes NPs reenter the cell cycle.
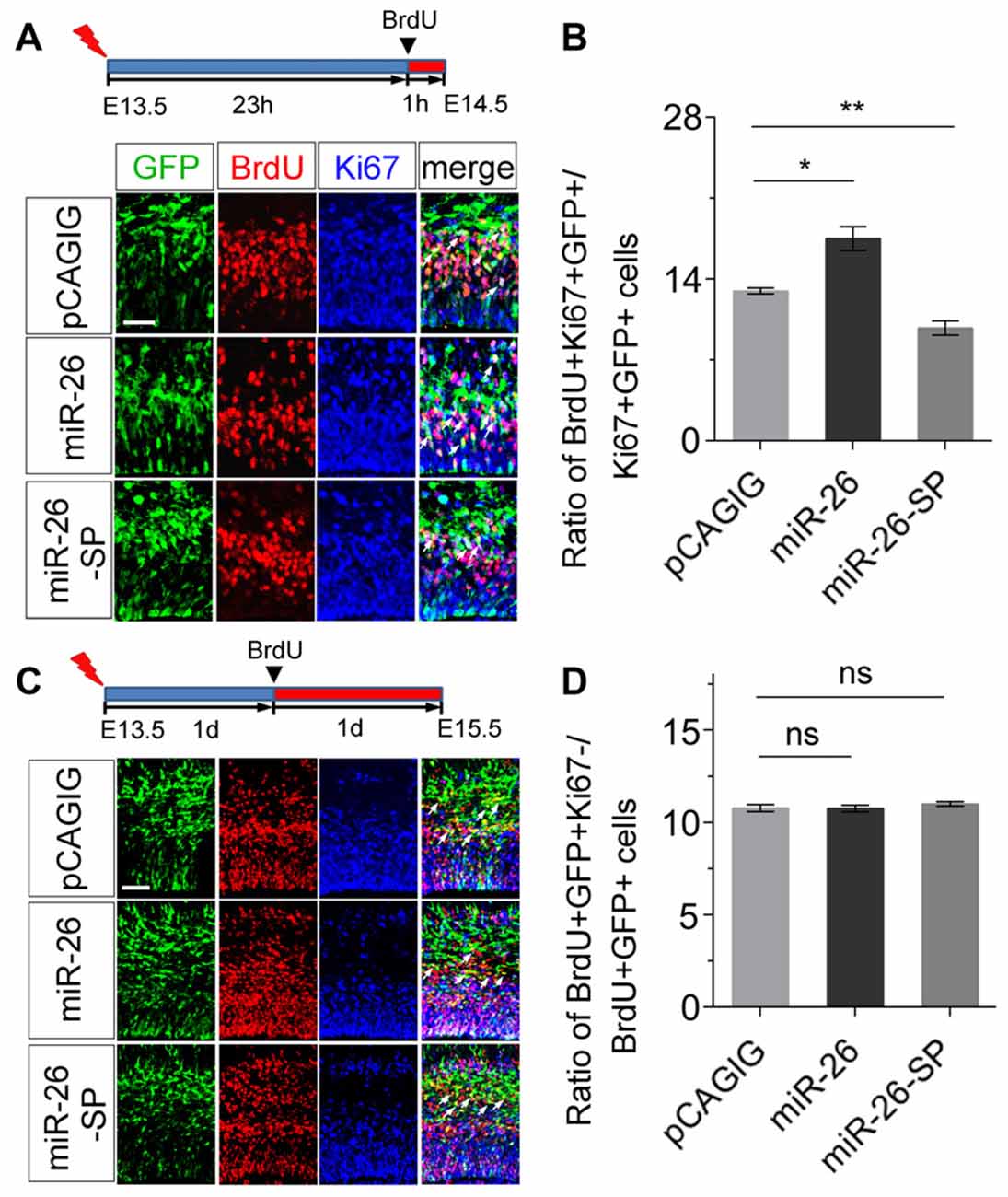
Figure 4. miR-26a regulates cell cycle progression in NPs. (A) Illustration of in utero electroporation and BrdU incorporation. Electroporation was performed at E13.5, followed by BrdU incorporation 23 hours (23 h) later, and tissues were analyzed at E14.5. BrdU+, Ki67+ and GFP+ cells were labeled and counted. (B) miR-26a overexpression increased the proportion of cells entering the S phase of the cell cycle, while miR-26a-SP decreased the proportion, by measuring the BrdU labeling index. (C) Electroporation was performed at E13.5, followed by BrdU incorporation 1 day (1d) later, and tissues were analyzed at E15.5. BrdU+, Ki67+ and GFP+ cells were labeled and counted. (D) Neither miR-26a nor miR-26-SP altered the proportion of cells exiting the cell cycle. Values represent mean ± SEM. n = 9 sections from at least three brains. *P < 0.05; **P < 0.01; ns, not significant. ANOVA with post hoc test was used. Scale bar = 50 μm.
To analyze the cell-cycle exit, the embryonic cortex was electroporated at E13.5, and collected at E15.5. BrdU was injected 1 day before brain tissue collection (Figure 4C). Neither miR-26a overexpression nor knockdown resulted in significant changes on the ratio of BrdU+/Ki67− cells vs. GFP+ cells, suggesting that miR-26a is not involved in the regulation of cell-cycle exit during the G1/S transition in NPs (Figures 4C,D).
miRNAs generally function via silencing coding genes. Our bioinformatic analysis predicted Emx2 (empty spiracles homeobox 2), which contains a binding site for miR-26a at the 3′UTR, as a potential targeted gene for miR-26 (Figure 5A). Previous studies suggest that miRNAs often have overlapping expression with their target genes in specific tissues (Hobert, 2007; Karres et al., 2007). We thus compared expression patterns of miR-26 and Emx2 in E12.5 and E15.5 mouse cortices using in situ hybridization. miR-26 expression was detected in the VZ in E12.5 cortices, and its expression in the VZ/SVZ was decreased in E15.5 cortices. Compared to miR-26a, Emx2 expression was observed in the VZ and SVZ in both E12.5 and E15.5 cortices with a higher and broader expression compared to that of miR-26, indicating overlapping but also distinct expression of miR-26 and Emx2 (Figure 5B).
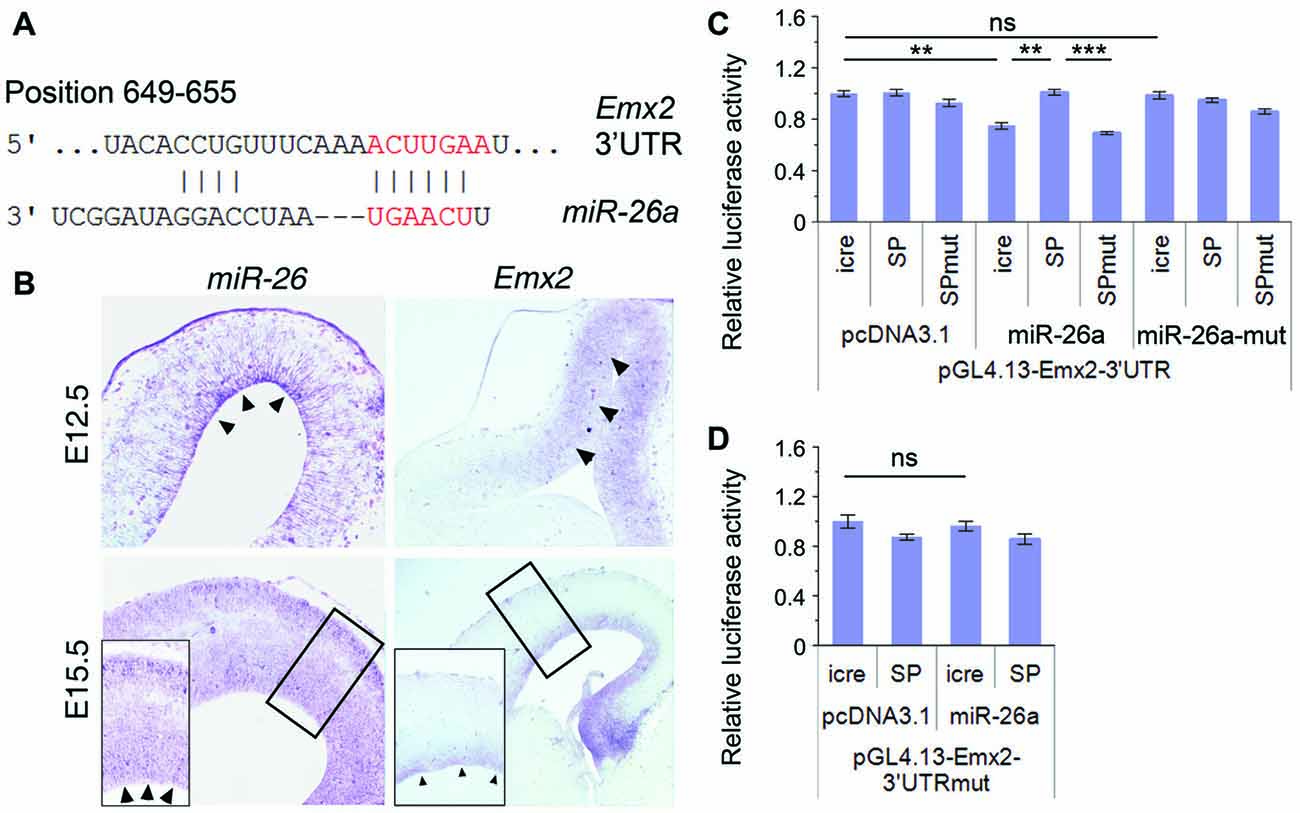
Figure 5. Emx2 is a target of miR-26a. (A) Emx2 3′ untranslated region (3′UTR) contains a binding site for miR-26. The seed sequence is shown in red. (B) miR-26 and Emx2 were co-expressed in the VZ and subventricular zone (SVZ; arrowheads) in E12.5 and E15.5 cortices, detected by in situ hybridization. Inset images show high power views of highlighted regions in E15.5 cortices. (C) miR-26a suppressed luciferase activities in the construct containing the Emx2–3′UTR, while miR-26-SP but not miR-26a-SPmut rescued the suppression. miR-26a-mut had no suppressing activity. (D) The mutation of miR-26 binding sites in the Emx2–3′UTR displayed no silencing activity by miR-26. n > 3. **P < 0.01; ***P < 0.001; ns, not significant. ANOVA with post hoc test was used.
To verify miR-26 targeting effect on Emx2, we performed luciferase assays by testing the Emx2 3′UTR and its mutation where miR-26 binding sites have been mutated. The luciferase activity in constructs containing the Emx2 3′UTR was notably repressed by miR-26a but not by miR-26 mutation (miR-26a-mut). Moreover, miR-26 sponge (miR-26-SP) could rescue miR-26 silencing effect on the Emx2 3′UTR, its mutation (miR-26-SPmut) showed no effect on the Emx2 3′UTR (Figure 5C). To further test the specificity of miR-26 on silencing the Emx2 3′UTR, we generated a mutant form in which the miR-26 binding sites in the Emx2 3′UTR are mutated, named Emx2 3′UTRmut. When it was co-expressed with miR-26, the luciferase activity did not show significant reduction, indicating that miR-26a failed to silence Emx2 3′UTRmut (Figure 5D). These results suggest that Emx2 is a specific putative target for miR-26a.
Due to the overlapping expression of miR-26a and its target Emx2 in the VZ and SVZ in the cortex, we examined whether Emx2 plays a similar or opposite role in NP development by overexpressing and silencing Emx2 at E13.5 (Supplementary Figure S2). Brain tissues were collected 24 h after in utero electroporation. Overexpression of Emx2, compared to the control, significantly decreased the percentage of BrdU+/GFP+ cells vs. GFP+ cells (Figures 6A,B). The population of Pax6+ RGCs was also decreased (Figures 6C,D). Conversely, knockdown of Emx2 using shEmx2 greatly increased the percentage of BrdU+/GFP+ NPs and RGCs (Figures 6A–D). Altered Emx2 expression did not cause detectable changes of the IP population (Figures 6E,F). Our results suggest that Emx2 plays a role in suppressing expansion of cortical NPs.
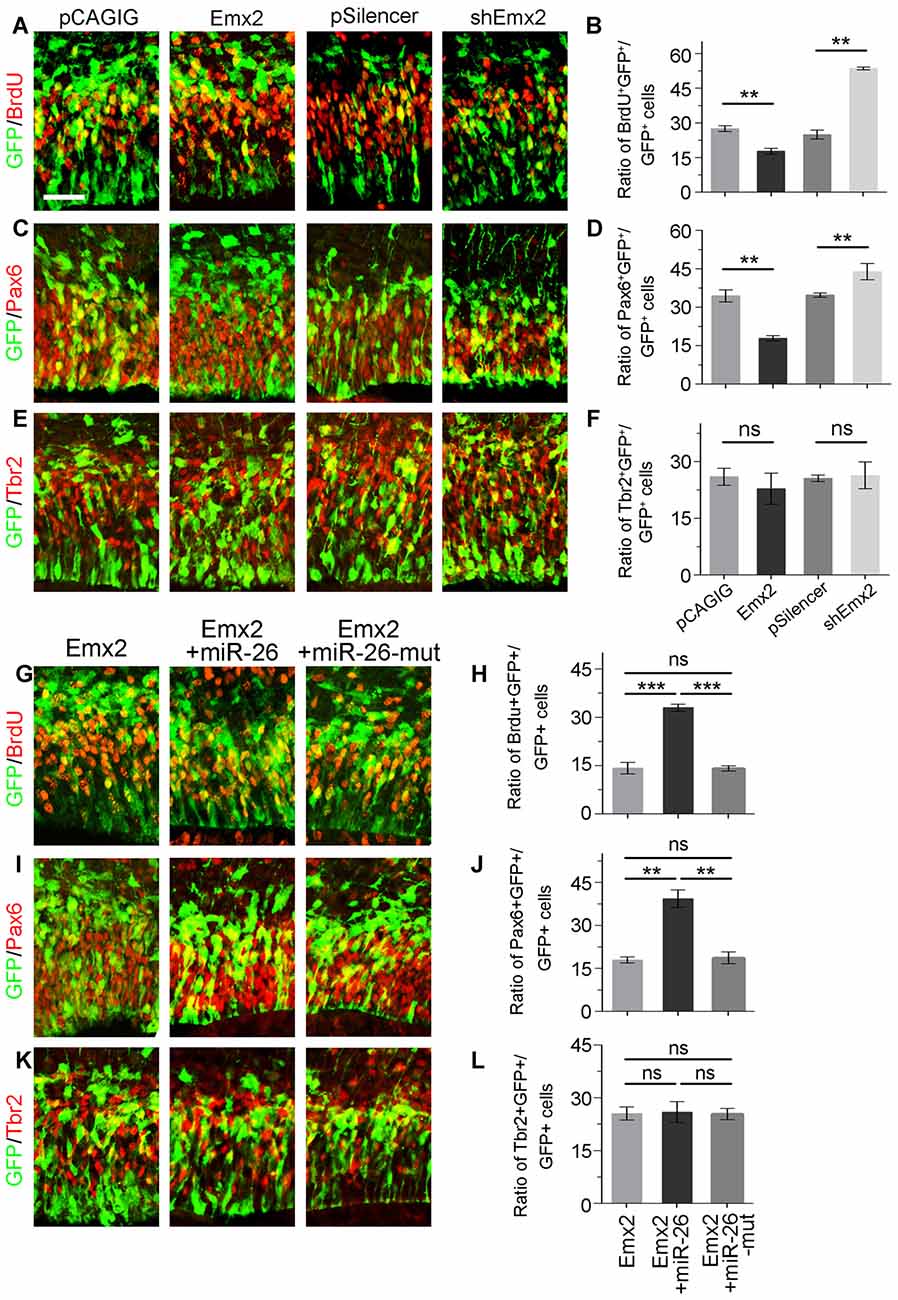
Figure 6. Emx2 is functionally inhibited by miR-26 in regulating NP proliferation. (A–F) Overexpression of Emx2, but not the control construct pCAGIG, decreased the proportion of cells expressing both proliferative marker BrdU+/GFP+ and RGC marker Pax6+/GFP+, but not IP marker Tbr2+/GFP+, in GFP-positive cells. short hairpin RNA (shRNA)-mediated knockdown (shEmx2) of Emx2 increased the proportion of both BrdU+/GFP+ cells and Pax6+/GFP+ cells, but not Tbr2+/GFP+ cells in GFP-positive cells, compared to the control construct pSilencer. (G–J) Emx2 expression suppressed the proportion of cells expressing both proliferative marker BrdU+/GFP+ and RGC marker Pax6+/GFP+ in GFP-positive cells. Co-expressing Emx2 with miR-26, but not miR-26-mut, dramatically reversed the suppression. (K,L) Emx2 expression did not alter the proportion of cells expressing IP marker Tbr2+/GFP+, in GFP-positive cells. Values represent mean ± SEM. n = 9 sections from at least three brains. **P < 0.01; ***P < 0.001; ns, not significant. ANOVA with post hoc test was used. Scale bar = 50 μm.
To further investigate the targeting interaction between miR-26a and Emx2 in NP development, miR-26a was co-electroporated with Emx2 full length cDNA that contains its 3′UTR in the E13.5 mouse cortex. Compared to Emx2 alone, co-electroporation of Emx2 with miR-26 caused a pronouncing elevation of the population of BrdU+/GFP+ cells vs. GFP+ cells and the population of Pax6+/GFP+ cells vs. GFP+ cells (Figures 6G,H). However, co-electroporation of Emx2 with miR-26 mutation in the seed sequence (miR-26-mut) did not change the proportion of BrdU+/GFP+ or Pax6+/GFP+ cells (Figures 6I,J). These results suggest that miR-26 specifically blocks the function of Emx2 in suppressing NP proliferation. Additionally, co-expression of Emx2 and miR-26 or miR-26-mut did not alter the proportion of Tbr2+/GFP+ cell populations, suggesting that Emx2 and miR-26 mostly regulate the RGC but not IP population (Figures 6K,L). Our results suggest a cohesive regulatory role of miR-26a and Emx2 in cortical NP expansion.
Several studies have shown that target genes of some miRNAs have a feedback regulation on miRNA expression (Bian and Sun, 2011). To investigate whether Emx2 might regulate expression of miR-26 host genes Ctdsp, we searched the promoter regions of three Ctdsp genes. Previous work has identified ATTA or TAAT as a binding motif for the transcription factor Emx2 (Mariani et al., 2012). Interestingly, 1 kb upstream regions of the transcription start site (TSS) of all three Ctdsp genes contained 1–3 Emx2 binding sites, suggesting that Emx2 may regulate Ctdsp expression, subsequently miR-26 expression, by binding to the Ctdsp promoter regions (Figure 7A). To further text Emx2 binding activities, 1 kb and 2 kb promoters of Ctdsp2 were cloned into a luciferase reporter vector, and were co-expressed with Emx2. The luciferase activities were significantly increased when Emx2 was co-expressed, compared to the control (Figure 7B). These results suggest that Emx2 may function as a transcription activator on Ctdsp gene expression and initiates miR-26 expression.
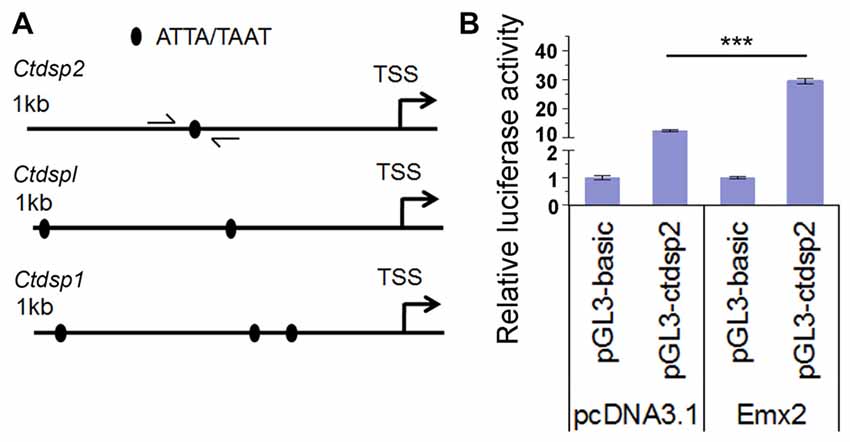
Figure 7. Emx2 promotes Ctdsp2 transcription. (A) The 1 kb upstream regions of the transcription start site (TSS) of Ctdsp genes contained the Emx2 binding sites. (B) Co-expressing Emx2 with 1 kb promoter of Ctdsp2 increased luciferase activities. n > 3, ***P < 0.001. ANOVA with post hoc test was used.
Dysregulation of proliferation, survival and differentiation of NPs causes malformation of cortical architecture, in turn the brain dysfunction, for example epilepsy and mental retardation (Molnár, 2011; Sun and Hevner, 2014). Revealing the regulatory mechanism of cortical development will contribute to a deep understanding of brain disorders and possible clinical therapies. The emerging evidence highlights critical roles of miRNAs in brain development and disorders (Livak and Schmittgen, 2001; Mellios and Sur, 2012; Mellios et al., 2017). In this study, we demonstrate that miR-26a, its target Emx2 and its host gene Ctdsp2 cohesively mediate NP proliferation and RGC expansion using a loop-regulatory mechanism during the mouse cortical development.
Proper development of the cortex relies on the proliferation and expansion of NPs, including RGCs and IPs, which are tightly regulated by precise expression patterns of both coding and noncoding RNAs (Rakic, 2009; Sun and Shi, 2015). Ctdsp2, also known as SCP2, is a phosphatase involved in regulating both signaling pathways and gene transcription (Yeo et al., 2003; Knockaert et al., 2006; Sapkota et al., 2006). Studies have shown that Ctdsp2 induces cell-cycle arrest and decreases the number of cells in S-phase in both human and mouse embryonic cells (Kashuba et al., 2004; Zhu et al., 2012; Kloet et al., 2015). Interestingly, our studies have shown an opposite role of Ctdsp2 in regulating the cell cycle to facilitate proliferation of NPs and expansion of RGCs in the mouse embryonic cortex. One possible reason for the conversed function of Ctdsp2 is likely due to an overlapping co-expression of its intronic miRNA miR-26. Studies have shown that most host genes are functionally associated with their intronic miRNAs (Woltering and Durston, 2008; Lund, 2010; Small et al., 2010). Expression of Ctdsp2 may concomitantly induce upregulation of miR-26 transcript level in the mouse cortex, and in turn promote NP proliferation.
Furthermore, miR-26 was previously reported to promote neuronal differentiation by suppressing Ctdsp in zebrafish (Dill et al., 2012; Han et al., 2012). Our studies have shown that miR-26-dependant regulation of the cell-cycle progression determines reentering but not exiting the cell-cycle in the NP population, which suggests a positive regulatory role in NP proliferation and expansion. These data indicate distinct roles of miR-26 between two species. Moreover, miR-26 can be either an activator or a suppressor of cell proliferation in different cell types in even the same species (Gao and Liu, 2011). Studies on miR-26 have also shown its inhibitory role in proliferation, migration and differentiation via targeting PFKFB3, EZH2 and other downstream genes (Lu et al., 2011; Du et al., 2015; Chen et al., 2016), and a positive role by targeting GSK-3beta, PTEN CHD1 and downstream genes (Huse et al., 2009; Kim et al., 2010; Zhang et al., 2012; Tan et al., 2014). Distinct functions of miR-26 are likely achieved through regulating different target genes in different cell types.
In the mouse cerebral cortex, we have identified miR-26a target gene Emx2, which functions in an opposite manner to miR-26a in regulating NP proliferation and RGC expansion. Studies have shown that Emx2 functionally interacts with specific transcript factors, like Sox2 and Pax6 in the developing cortex (Muzio et al., 2002a,b; Mariani et al., 2012). Notably, Emx2 cooperates with Pax6 in regulating neocortex development (Bishop et al., 2000; Bayatti et al., 2008). Emx2 regulates transcription levels of downstream genes associated with neuronal proliferation (Gangemi et al., 2006). Moreover, a report in Paralichthys olivaceus also indicated Emx2 as a target gene of miR-26a and miR-26b (Yin et al., 2015). In this study, we have demonstrated that Emx2 is a potential activator to facilitate the expression of miR-26 host gene Ctdsp, and subsequently the expression of its intronic miRNA miR-26.
Altogether, the concomitant expressions of miR-26 and their host genes Ctdsp play a positive role in NP proliferation and RGC expansion in the developing cortex. On the other hand, miR-26 target gene Emx2 negatively controls NP development and activates expression of Ctdsp, and further initiates miR-26 transcription, although the directly or indirectly regulatory function between Emx2 and Ctdsp requires further exploration in biological meanings. It is likely that miR-26 and its target Emx2 form a regulatory-loop via miR-26 host gene Ctdsp as an intermediator, in maintaining proper populations of NPs and RGCs during the cortical development. Our findings provide a new insight into the regulatory mechanism of miRNA miR-26-mediated NP development and cortical formation.
HZ and LZ contributed equally to this work. TS supervised the process of this work.
This work was supported by an R01-MH083680 grant from the National Institutes of Health (NIH)/NIMH (TS) and the National Natural Science Foundation of China (81471152, 31771141 and 81701132).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2018.00044/full#supplementary-material
Abdullah, A. I., Zhang, H., Nie, Y., Tang, W., and Sun, T. (2016). CDK7 and miR-210 Co-regulate cell-cycle progression of neural progenitors in the developing neocortex. Stem Cell Reports 7, 69–79. doi: 10.1016/j.stemcr.2016.06.005
Aguirre, A., Rubio, M. E., and Gallo, V. (2010). Notch and EGFR pathway interaction regulates neural stem cell number and self-renewal. Nature 467, 323–327. doi: 10.1038/nature09347
Bayatti, N., Sarma, S., Shaw, C., Eyre, J. A., Vouyiouklis, D. A., Lindsay, S., et al. (2008). Progressive loss of PAX6, TBR2, NEUROD and TBR1 mRNA gradients correlates with translocation of EMX2 to the cortical plate during human cortical development. Eur. J. Neurosci. 28, 1449–1456. doi: 10.1111/j.1460-9568.2008.06475.x
Bian, S., Hong, J., Li, Q., Schebelle, L., Pollock, A., Knauss, J. L., et al. (2013). MicroRNA cluster miR-17–92 regulates neural stem cell expansion and transition to intermediate progenitors in the developing mouse neocortex. Cell Rep. 3, 1398–1406. doi: 10.1016/j.celrep.2013.03.037
Bian, S., and Sun, T. (2011). Functions of noncoding RNAs in neural development and neurological diseases. Mol. Neurobiol. 44, 359–373. doi: 10.1007/s12035-011-8211-3
Bishop, K. M., Goudreau, G., and O’Leary, D. D. (2000). Regulation of area identity in the mammalian neocortex by Emx2 and Pax6. Science 288, 344–349. doi: 10.1126/science.288.5464.344
Bultje, R. S., Castaneda-Castellanos, D. R., Jan, L. Y., Jan, Y. N., Kriegstein, A. R., and Shi, S. H. (2009). Mammalian Par3 regulates progenitor cell asymmetric division via notch signaling in the developing neocortex. Neuron 63, 189–202. doi: 10.1016/j.neuron.2009.07.004
Carthew, R. W., and Sontheimer, E. J. (2009). Origins and mechanisms of miRNAs and siRNAs. Cell 136, 642–655. doi: 10.1016/j.cell.2009.01.035
Chen, C. Y., Chang, J. T., Ho, Y. F., and Shyu, A. B. (2016). MiR-26 down-regulates TNF-α/NF-kappaB signalling and IL-6 expression by silencing HMGA1 and MALT1. Nucleic Acids Res. 44, 3772–3787. doi: 10.1093/nar/gkw205
Chenn, A., and Walsh, C. A. (2002). Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297, 365–369. doi: 10.1126/science.1074192
Cora, D., Re, A., Caselle, M., and Bussolino, F. (2017). MicroRNA-mediated regulatory circuits: outlook and perspectives. Phys. Biol. 14:045001. doi: 10.1088/1478-3975/aa6f21
De Pietri Tonelli, D., Pulvers, J. N., Haffner, C., Murchison, E. P., Hannon, G. J., and Huttner, W. B. (2008). miRNAs are essential for survival and differentiation of newborn neurons but not for expansion of neural progenitors during early neurogenesis in the mouse embryonic neocortex. Development 135, 3911–3921. doi: 10.1242/dev.025080
Dehay, C., and Kennedy, H. (2007). Cell-cycle control and cortical development. Nat. Rev. Neurosci. 8, 438–450. doi: 10.1038/nrn2097
Delaunay, D., Kawaguchi, A., Dehay, C., and Matsuzaki, F. (2017). Division modes and physical asymmetry in cerebral cortex progenitors. Curr. Opin. Neurob. 42, 75–83. doi: 10.1016/j.conb.2016.11.009
Dill, H., Linder, B., Fehr, A., and Fischer, U. (2012). Intronic miR-26b controls neuronal differentiation by repressing its host transcript, ctdsp2. Genes Dev. 26, 25–30. doi: 10.1101/gad.177774.111
Du, J. Y., Wang, L. F., Wang, Q., and Yu, L. D. (2015). miR-26b inhibits proliferation, migration, invasion and apoptosis induction via the downregulation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 driven glycolysis in osteosarcoma cells. Oncol. Rep. 33, 1890–1898. doi: 10.3892/or.2015.3797
Elmen, J., Lindow, M., Schutz, S., Lawrence, M., Petri, A., Obad, S., et al. (2008). LNA-mediated microRNA silencing in non-human primates. Nature 452, 896–899. doi: 10.1038/nature06783
Englund, C., Fink, A., Lau, C., Pham, D., Daza, R. A., Bulfone, A., et al. (2005). Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J. Neurosci. 25, 247–251. doi: 10.1523/JNEUROSCI.2899-04.2005
Franco, S. J., and Müller, U. (2013). Shaping our minds: stem and progenitor cell diversity in the mammalian neocortex. Neuron 77, 19–34. doi: 10.1016/j.neuron.2012.12.022
Gangemi, R. M., Daga, A., Muzio, L., Marubbi, D., Cocozza, S., Perera, M., et al. (2006). Effects of Emx2 inactivation on the gene expression profile of neural precursors. Eur. J. Neurosci. 23, 325–334. doi: 10.1111/j.1460-9568.2005.04559.x
Gao, J., and Liu, Q. G. (2011). The role of miR-26 in tumors and normal tissues (Review). Oncol. Lett. 2, 1019–1023. doi: 10.3892/ol.2011.413
Guillemot, F. (2005). Cellular and molecular control of neurogenesis in the mammalian telencephalon. Curr. Opin. Cell Biol. 17, 639–647. doi: 10.1016/j.ceb.2005.09.006
Han, J., Denli, A. M., and Gage, F. H. (2012). The enemy within: intronic miR-26b represses its host gene, ctdsp2, to regulate neurogenesis. Genes Dev. 26, 6–10. doi: 10.1101/gad.184416.111
Hong, J., Zhang, H., Kawase-Koga, Y., and Sun, T. (2013). MicroRNA function is required for neurite outgrowth of mature neurons in the mouse postnatal cerebral cortex. Front. Cell. Neurosci. 7:151. doi: 10.3389/fncel.2013.00151
Hugon, J., and Paquet, C. (2008). Targeting miRNAs in Alzheimer’s disease. Expert Rev. Neurother. 8, 1615–1616. doi: 10.1586/14737175.8.11.1615
Huse, J. T., Brennan, C., Hambardzumyan, D., Wee, B., Pena, J., Rouhanifard, S. H., et al. (2009). The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 23, 1327–1337. doi: 10.1101/gad.1777409
Karres, J. S., Hilgers, V., Carrera, I., Treisman, J., and Cohen, S. M. (2007). The conserved microRNA miR-8 tunes atrophin levels to prevent neurodegeneration in Drosophila. Cell 131, 136–145. doi: 10.1016/j.cell.2007.09.020
Kashuba, V. I., Li, J., Wang, F., Senchenko, V. N., Protopopov, A., Malyukova, A., et al. (2004). RBSP3 (HYA22) is a tumor suppressor gene implicated in major epithelial malignancies. Proc. Natl. Acad. Sci. U S A 101, 4906–4911. doi: 10.1073/pnas.0401238101
Kawase-Koga, Y., Low, R., Otaegi, G., Pollock, A., Deng, H., Eisenhaber, F., et al. (2010). RNAase-III enzyme Dicer maintains signaling pathways for differentiation and survival in mouse cortical neural stem cells. J. Cell Sci. 123, 586–594. doi: 10.1242/jcs.059659
Kawase-Koga, Y., Otaegi, G., and Sun, T. (2009). Different timings of Dicer deletion affect neurogenesis and gliogenesis in the developing mouse central nervous system. Dev. Dyn. 238, 2800–2812. doi: 10.1002/dvdy.22109
Kim, V. N., Han, J., and Siomi, M. C. (2009). Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 10, 126–139. doi: 10.1038/nrm2632
Kim, H., Huang, W., Jiang, X., Pennicooke, B., Park, P. J., and Johnson, M. D. (2010). Integrative genome analysis reveals an oncomir/oncogene cluster regulating glioblastoma survivorship. Proc. Natl. Acad. Sci. U S A 107, 2183–2188. doi: 10.1073/pnas.0909896107
Kloet, D. E., Polderman, P. E., Eijkelenboom, A., Smits, L. M., van Triest, M. H., van den Berg, M. C., et al. (2015). FOXO target gene CTDSP2 regulates cell cycle progression through Ras and p21Cip1/Waf1. Biochem. J. 469, 289–298. doi: 10.1042/BJ20140831
Knockaert, M., Sapkota, G., Alarcón, C., Massagué, J., and Brivanlou, A. H. (2006). Unique players in the BMP pathway: small C-terminal domain phosphatases dephosphorylate Smad1 to attenuate BMP signaling. Proc. Natl. Acad. Sci. U S A 103, 11940–11945. doi: 10.1073/pnas.0605133103
Kremer-Tal, S., Reeves, H. L., Narla, G., Thung, S. N., Schwartz, M., Difeo, A., et al. (2004). Frequent inactivation of the tumor suppressor Kruppel-like factor 6 (KLF6) in hepatocellular carcinoma. Hepatology 40, 1047–1052. doi: 10.1002/hep.20460
Kriegstein, A., Noctor, S., and Martínez-Cerdeño, V. (2006). Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat. Rev. Neurosci. 7, 883–890. doi: 10.1038/nrn2008
Krol, J., Loedige, I., and Filipowicz, W. (2010). The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 11, 597–610. doi: 10.1038/nrg2843
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854. doi: 10.1016/0092-8674(93)90529-y
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, J., He, M. L., Wang, L., Chen, Y., Liu, X., Dong, Q., et al. (2011). MiR-26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Res. 71, 225–233. doi: 10.1158/0008-5472.CAN-10-1850
Lund, A. H. (2010). miR-10 in development and cancer. Cell Death Differ. 17, 209–214. doi: 10.1038/cdd.2009.58
Mariani, J., Favaro, R., Lancini, C., Vaccari, G., Ferri, A. L., Bertolini, J., et al. (2012). Emx2 is a dose-dependent negative regulator of Sox2 telencephalic enhancers. Nucleic Acids Res. 40, 6461–6476. doi: 10.1093/nar/gks295
Mellios, N., Feldman, D. A., Sheridan, S. D., Ip, J. P. K., Kwok, S., Amoah, S. K., et al. (2017). MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol. Psychiatry doi: 10.1038/mp.2017.86 [Epub ahead of print].
Mellios, N., Sugihara, H., Castro, J., Banerjee, A., Le, C., Kumar, A., et al. (2011). miR-132, an experience-dependent microRNA, is essential for visual cortex plasticity. Nat. Neurosci. 14, 1240–1242. doi: 10.1038/nn.2909
Mellios, N., and Sur, M. (2012). The emerging role of micrornas in schizophrenia and autism spectrum disorders. Front. Psychiatry 3:39. doi: 10.3389/fpsyt.2012.00039
Mizutani, K., Yoon, K., Dang, L., Tokunaga, A., and Gaiano, N. (2007). Differential Notch signalling distinguishes neural stem cells from intermediate progenitors. Nature 449, 351–355. doi: 10.1038/nature06090
Molnár, Z. (2011). Evolution of cerebral cortical development. Brain Behav. Evol. 78, 94–107. doi: 10.1159/000327325
Molyneaux, B. J., Arlotta, P., Menezes, J. R., and Macklis, J. D. (2007). Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 8, 427–437. doi: 10.1038/nrn2151
Muzio, L., Di Benedetto, B., Stoykova, A., Boncinelli, E., Gruss, P., and Mallamaci, A. (2002a). Conversion of cerebral cortex into basal ganglia in Emx2−/− Pax6Sey/Sey double-mutant mice. Nat. Neurosci. 5, 737–745. doi: 10.1038/nn892
Muzio, L., Di Benedetto, B., Stoykova, A., Boncinelli, E., Gruss, P., and Mallamaci, A. (2002b). Emx2 and Pax6 control regionalization of the pre-neuronogenic cortical primordium. Cereb. Cortex 12, 129–139. doi: 10.1093/cercor/12.2.129
Nadim, W. D., Simion, V., Benedetti, H., Pichon, C., Baril, P., and Morisset-Lopez, S. (2017). MicroRNAs in neurocognitive dysfunctions: new molecular targets for pharmacological treatments? Curr. Neuropharmacol. 15, 260–275. doi: 10.2174/1570159x14666160709001441
Ostrem, B. E., Lui, J. H., Gertz, C. C., and Kriegstein, A. R. (2014). Control of outer radial glial stem cell mitosis in the human brain. Cell Rep. 8, 656–664. doi: 10.1016/j.celrep.2014.06.058
Otaegi, G., Pollock, A., and Sun, T. (2011). An optimized sponge for microRNA miR-9 affects spinal motor neuron development in vivo. Front. Neurosci. 5:146. doi: 10.3389/fnins.2011.00146
Packer, A. N., Xing, Y., Harper, S. Q., Jones, L., and Davidson, B. L. (2008). The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. 28, 14341–14346. doi: 10.1523/JNEUROSCI.2390-08.2008
Piao, X., Hill, R. S., Bodell, A., Chang, B. S., Basel-Vanagaite, L., Straussberg, R., et al. (2004). G protein-coupled receptor-dependent development of human frontal cortex. Science 303, 2033–2036. doi: 10.1126/science.1092780
Pollock, A., Bian, S., Zhang, C., Chen, Z., and Sun, T. (2014). Growth of the developing cerebral cortex is controlled by microRNA-7 through the p53 pathway. Cell Rep. 7, 1184–1196. doi: 10.1016/j.celrep.2014.04.003
Rakic, P. (2007). The radial edifice of cortical architecture: from neuronal silhouettes to genetic engineering. Brain Res. Rev. 55, 204–219. doi: 10.1016/j.brainresrev.2007.02.010
Rakic, P. (2009). Evolution of the neocortex: a perspective from developmental biology. Nat. Rev. Neurosci. 10, 724–735. doi: 10.1038/nrn2719
Saito, T. (2006). In vivo electroporation in the embryonic mouse central nervous system. Nat. Protoc. 1, 1552–1558. doi: 10.1038/nprot.2006.276
Sapkota, G., Knockaert, M., Alarcon, C., Montalvo, E., Brivanlou, A. H., and Massague, J. (2006). Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-β pathways. J. Biol. Chem. 281, 40412–40419. doi: 10.1074/jbc.M610172200
Shen, Q., Wang, Y., Dimos, J. T., Fasano, C. A., Phoenix, T. N., Lemischka, I. R., et al. (2006). The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat. Neurosci. 9, 743–751. doi: 10.1038/nn1694
Small, E. M., Sutherland, L. B., Rajagopalan, K. N., Wang, S., and Olson, E. N. (2010). MicroRNA-218 regulates vascular patterning by modulation of Slit-Robo signaling. Circ. Res. 107, 1336–1344. doi: 10.1161/CIRCRESAHA.110.227926
Subramanian, L., Bershteyn, M., Paredes, M. F., and Kriegstein, A. R. (2017). Dynamic behaviour of human neuroepithelial cells in the developing forebrain. Nat. Commun. 8:14167. doi: 10.1038/ncomms14167
Sun, T., and Hevner, R. F. (2014). Growth and folding of the mammalian cerebral cortex: from molecules to malformations. Nat. Rev. Neurosci. 15, 217–232. doi: 10.1038/nrn3707
Sun, E., and Shi, Y. (2015). MicroRNAs: small molecules with big roles in neurodevelopment and diseases. Exp. Neurol. 268, 46–53. doi: 10.1016/j.expneurol.2014.08.005
Tan, S., Ding, K., Li, R., Zhang, W., Li, G., Kong, X., et al. (2014). Identification of miR-26 as a key mediator of estrogen stimulated cell proliferation by targeting CHD1, GREB1 and KPNA2. Breast Cancer Res. 16:R40. doi: 10.1186/bcr3644
Wang, H., Luo, J., Zhang, T., Tian, H., Ma, Y., Xu, H., et al. (2016). MicroRNA-26a/b and their host genes synergistically regulate triacylglycerol synthesis by targeting the INSIG1 gene. RNA Biol. 13, 500–510. doi: 10.1080/15476286.2016.1164365
Wang, X., Tsai, J. W., LaMonica, B., and Kriegstein, A. R. (2011). A new subtype of progenitor cell in the mouse embryonic neocortex. Nat. Neurosci. 14, 555–561. doi: 10.1038/nn.2807
Wightman, B., Ha, I., and Ruvkun, G. (1993). Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75, 855–862. doi: 10.1016/0092-8674(93)90530-4
Woltering, J. M., and Durston, A. J. (2008). MiR-10 represses HoxB1a and HoxB3a in zebrafish. PLoS One 3:e1396. doi: 10.1371/journal.pone.0001396
Yeo, M., Lin, P. S., Dahmus, M. E., and Gill, G. N. (2003). A novel RNA polymerase II C-terminal domain phosphatase that preferentially dephosphorylates serine 5. J. Biol. Chem. 278, 26078–26085. doi: 10.1074/jbc.M301791200
Yin, C., Zhang, J., Shi, Z., Sun, W., Zhang, H., and Fu, Y. (2015). Identification and expression of the target gene emx2 of miR-26a and miR-26b in Paralichthys olivaceus. Gene 570, 205–212. doi: 10.1016/j.gene.2015.06.030
Zhang, J., Han, C., and Wu, T. (2012). MicroRNA-26a promotes cholangiocarcinoma growth by activating β-catenin. Gastroenterology 143, 246.e8–256.e8. doi: 10.1053/j.gastro.2012.03.045
Zhang, H., Shykind, B., and Sun, T. (2013). Approaches to manipulating microRNAs in neurogenesis. Front. Neurosci. 6:196. doi: 10.3389/fnins.2012.00196
Zhang, H., and Yin, Z. S. (2005). Wnt signaling pathway and neural stem cells. Sheng Li Ke Xue Jin Zhan 36, 249–252. doi: 10.3321/j.issn:0559-7765.2005.03.014
Keywords: miR-26, Ctdsp, Emx2, neural progenitor, cell-cycle progression
Citation: Zhang H, Zhang L and Sun T (2018) Cohesive Regulation of Neural Progenitor Development by microRNA miR-26, Its Host Gene Ctdsp and Target Gene Emx2 in the Mouse Embryonic Cerebral Cortex. Front. Mol. Neurosci. 11:44. doi: 10.3389/fnmol.2018.00044
Received: 07 October 2017; Accepted: 02 February 2018;
Published: 19 February 2018.
Edited by:
Zhe Liu, Janelia Research Campus, United StatesReviewed by:
Carlos Vicario-Abejón, Consejo Superior de Investigaciones Científicas (CSIC), SpainCopyright © 2018 Zhang, Zhang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Sun, dGFvc3VuQGhxdS5lZHUuY24=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.