 Danilo B. Medinas
Danilo B. Medinas Jose V. González1,2,3
Jose V. González1,2,3 Claudio Hetz
Claudio Hetz
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Mol. Neurosci. , 05 July 2017
Sec. Brain Disease Mechanisms
Volume 10 - 2017 | https://doi.org/10.3389/fnmol.2017.00216
This article is part of the Research Topic The Role of the Protein Quality Control in Neurodegenerative Diseases View all 34 articles
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the progressive loss of motoneurons and paralysis. The mechanisms underlying neuronal degeneration in ALS are starting to be elucidated, highlighting disturbances in motoneuron proteostasis. Endoplasmic reticulum (ER) stress has emerged as an early pathogenic event underlying motoneuron vulnerability and denervation in ALS. Maintenance of ER proteostasis is controlled by a dynamic signaling network known as the unfolded protein response (UPR). Inositol-requiring enzyme 1 (IRE1) is an ER-located kinase and endoribonuclease that operates as a major ER stress transducer, mediating the establishment of adaptive and pro-apoptotic programs. Here we discuss current evidence supporting the role of ER stress in motoneuron demise in ALS and build the rational to target IRE1 to ameliorate neurodegeneration.
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that affects motoneurons of cerebral cortex, brainstem and spinal cord, leading to muscle weakness, paralysis and premature death within 3–5 years after diagnosis (Turner et al., 2013; Peters et al., 2015). Besides motoneuron loss, the intracellular accumulation of protein inclusions of different compositions is a hallmark of ALS (Turner et al., 2013; Peters et al., 2015; Ruegsegger and Saxena, 2016). The hereditary forms of the disease account for approximately 5%–10% of total cases and are termed familial ALS (fALS), caused by mutations in different genes such as SOD1, TARDBP, FUS and hexanucleotide repeat expansions in C9orf72 (Turner et al., 2013; Leblond et al., 2014; Peters et al., 2015). Interestingly, the corresponding mutant proteins and (repeat-associated non-ATG translated, RAN) dipeptides form protein oligomers and aggregates, leading to impaired proteostasis with resultant motoneuron dysfunction and death (Turner et al., 2013; Peters et al., 2015; Ruegsegger and Saxena, 2016). In sporadic cases of ALS (sALS), misfolding and aggregation of the same proteins in the absence of mutations suggest common pathogenic mechanisms in fALS and sALS (Neumann et al., 2006; Bosco et al., 2010; Farg et al., 2012).
The development of genetic models of ALS has enabled dissection of disease course at histological, cellular and molecular levels (Philips and Rothstein, 2015). Although multiple mechanisms are proposed to drive ALS (Taylor et al., 2016), several recent unbiased studies in mutant SOD1 transgenic mice and induced pluripotent stem cell (iPSC)-derived patient motoneurons have identified endoplasmic reticulum (ER) stress as an early and transversal pathogenic mechanism underlying selective vulnerability of motoneurons in ALS (Saxena et al., 2009; Kiskinis et al., 2014; Filézac de L’Etang et al., 2015; Sun et al., 2015). ER stress is a condition generated by abnormal levels of misfolded proteins in the ER lumen, engaging a signal transduction pathway termed the unfolded protein response (UPR). The UPR operates as a central controller of cell fate, mediating initial adaptive responses to restore proteostasis through various mechanisms including transcriptional and translational regulation, enhancement of protein quality control mechanisms, degradation of abnormal proteins, among other outputs (Hetz, 2012). The UPR is a binary pathway that shifts its signaling toward a terminal phase to eliminate irreversibly damaged cells through apoptosis (Walter and Ron, 2011). The adaptive UPR is marked by rapid inhibition of protein translation due to the phosphorylation of the eukaryotic initiation factor 2α (eIF2α), in addition to transcriptional induction of chaperones, foldases, protein quality control and degradation systems, lipid biosynthesis, among others. Under pathological conditions of chronic ER stress as observed in numerous neurodegenerative diseases (Hetz and Mollereau, 2014; Scheper and Hoozemans, 2015; Smith and Mallucci, 2016), the terminal UPR engages pro-inflammatory and apoptotic cascades leading to cell death (Urra et al., 2013; Oakes and Papa, 2015).
The UPR transduces information about protein folding status from ER lumen to cytosol and nucleus through the action of various type-I ER transmembrane proteins that respond to the accumulation of misfolded proteins. These sensors reprogram the transcriptional and translational profile of the cell by a concerted action of transcription factors, phosphorylation events and RNA processing (Hetz et al., 2015). The mammalian UPR relies on three stress transducers, named activating transcription factor 6 (ATF6), protein kinase R (PKR)-like ER kinase (PERK) and inositol-requiring enzyme 1 (IRE1), being IRE1 the most conserved sensor from yeast to human (Wang and Kaufman, 2016). IRE1 is a kinase and endoribonuclease that upon ER stress is activated by dimerization and auto-transphosphorylation to catalyze the unconventional splicing of X-box binding protein 1 (XBP1) mRNA (Figure 1), thus leading to production of a potent transcription factor termed XBP1s (Hetz et al., 2015). During the adaptive UPR, XBP1s induces expression of ER chaperones and co-factors, ER-associated protein degradation (ERAD) components and lipid biosynthesis to increase the protein folding and quality control capacity (Walter and Ron, 2011). When ER stress is chronic, IRE1 is overactivated through assembly into high-order oligomers and reduces its substrate specificity to catalyze degradation of mRNA and microRNAs (Figure 1), an activity termed Regulated IRE1-dependent Decay (RIDD; Maurel et al., 2014). The activation of RIDD depletes ER components and reflects the terminal UPR directing cell fate towards apoptosis by directly controlling the stability of microRNAs, apoptosis genes and pro-inflammatory factors (Hollien and Weissman, 2006; Han et al., 2009; Hollien et al., 2009; Lerner et al., 2012; Ghosh et al., 2014). Furthermore, IRE1 can interact with cytosolic components, including adaptor proteins, to fine-tune UPR outputs in a dynamic fashion (Figure 1), comprising a protein platform termed “UPRosome” (Hetz and Glimcher, 2009). For instance, IRE1 can be coupled to JNK and NF-κB pathways through adaptor proteins to induce apoptosis upon prolonged ER stress (Urano et al., 2000; Hu et al., 2006). Thus, IRE1 signaling governs adjustment of proteostasis through XBP1-dependent transcriptional control, turning into a pro-degenerative effector when proteostasis cannot be recovered, engaging a variety of downstream pro-inflammatory and apoptotic regulators (Figure 1).
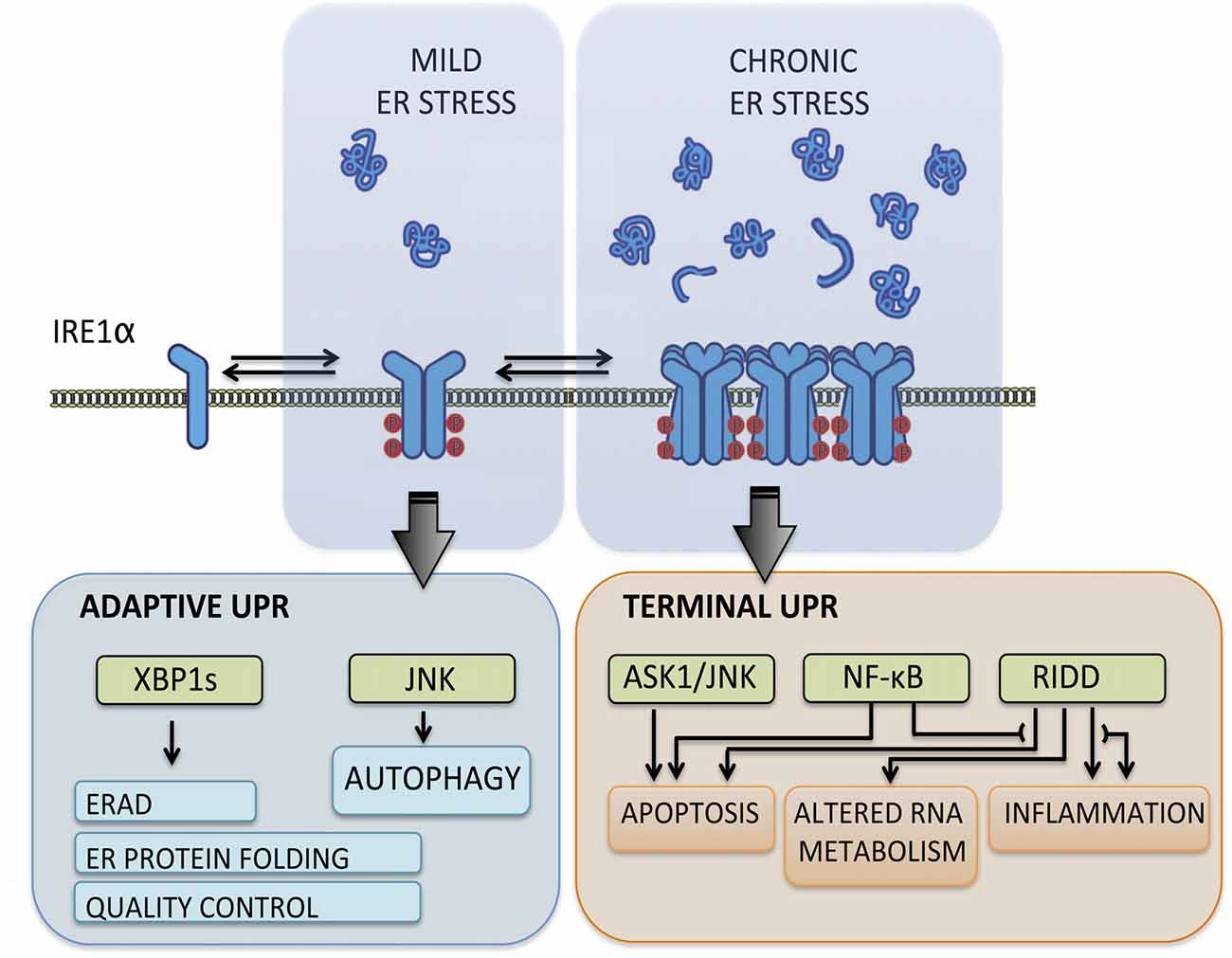
Figure 1. Inositol-requiringenzyme 1 (IRE1) signaling outputs. Under transient and mild endoplasmic reticulum (ER) stress, IRE1 undergoes dimerization and auto-transphosphorylation activating RNase activity and production of the potent transcription factor spliced X-box binding protein 1 (XBP1s), which induces adaptive programs to reduce protein misfolding, mediated by the upregulation of genes involved in protein folding, quality control, ER-associated protein degradation (ERAD) components and lipid biosynthesis. When ER stress is irremediable and chronic, IRE1 assembles into a scaffold platform for activation of ASK1-JNK and NF-κB pathways, which cause induction of apoptosis and modulates inflammation and autophagy levels. Furthermore, IRE1 overactivation decreases its RNase specificity and induces Regulated IRE1-dependent Decay (RIDD) activity, which degrades mRNA and microRNA and contributes to cell demise by depleting ER components and inducing pro-inflammatory and apoptotic factors.
The involvement of ER stress on ALS pathogenesis has been inferred from multiple studies in patient post-mortem tissue and iPSC-derived motoneurons, as well as animal models of disease (see examples in Atkin et al., 2006, 2008; Ilieva et al., 2007; Hetz et al., 2009; Ito et al., 2009; Saxena et al., 2009; Sasaki, 2010; Matus et al., 2013; Kiskinis et al., 2014). A landmark study developed a comparative gene expression profiling of vulnerable and resistant motoneurons, identifying ER stress as the earliest pathological event in mutant SOD1 mice occurring before any denervation is detected (Saxena et al., 2009). On a recent follow-up study, the Saxena’s group reported altered ER chaperone network underlying differential susceptibility of motoneurons in ALS (Filézac de L’Etang et al., 2015). Briefly, the BiP co-chaperone SIL1 was found enriched in resistant while progressively reduced in vulnerable motoneurons over disease course, and SIL1 overexpression using adeno-associated virus was proven to be neuroprotective (Filézac de L’Etang et al., 2015). Using ribosome profiling of motoneurons and glia in vivo, the Cleveland’s group indicated that ER stress is a major pathological signature of motoneurons, and may mediate cell autonomous neurodegeneration cascades in mutant SOD1 models (Sun et al., 2015). Additional findings support the concept that motoneurons are selectively vulnerable to perturbations to ER function. For instance, deletion of one calreticulin allele, an essential ER chaperone, led to exacerbated muscle weakness and denervation in the mutant SOD1 mouse model, accelerating the progression of the disease (Bernard-Marissal et al., 2012). Importantly, motoneuron dysfunction due to deficiency of calreticulin did not involve increased motoneuron loss, suggesting a role of ER chaperone network at early stages of ALS leading to muscle denervation (Bernard-Marissal et al., 2012). We recently provided genetic evidence supporting the concept that alteration in the ER folding network may be part of the etiology of the disease. Targeted sequencing of ALS cases identified point mutations in two protein disulfide isomerases (PDI) family members, ERp57 (also known as Grp58 or PDIA3) and PDIA1 (also termed PDI) (Gonzalez-Perez et al., 2015). Functional studies indicated that perturbation in the activity of these foldases alters neuromuscular junction structure and function, possibly involving abnormal synthesis of synaptic proteins (Woehlbier et al., 2016). These studies suggested that disrupted ER folding capacity underlays early ALS stages that result in muscle denervation.
The rise of iPSC technology enabled the study of patient motoneurons expressing mutant proteins at endogenous levels (Matus et al., 2014; de Boer and Eggan, 2015). An elegant study using iPSC-derived patient motoneurons harboring a SOD1 mutation discovered that basal physiological levels of ER stress is an intrinsic property of motoneurons linked to their electrical activity (Kiskinis et al., 2014). Furthermore, the exacerbated ER stress in patient motoneurons leads to hyperexcitability, and knock down of XBP1s affords significant neuroprotection (Kiskinis et al., 2014). This concept was also corroborated in motoneurons from patients carrying repeat expansions in c9orf72 and VCP mutations (Kiskinis et al., 2014; Dafinca et al., 2016; Hall et al., 2017), evidencing the transversal role of ER stress in disease etiology. Indeed, we described abnormaly higher levels of the UPR transcription factors ATF4 and XBP1s in post-mortem tissue of sALS cases. To determine the significance of the UPR to ALS pathogenesis in vivo, our group performed genetic manipulation of XBP1 or ATF4 in mutant SOD1 mice to assess the functional impact on disease course (Hetz et al., 2009; Matus et al., 2013). Genetic ablation of ATF4 increased the lifespan of mutant SOD1 mice possibly due to attenuated expression of the apoptotic factors CHOP and BIM (Matus et al., 2013). On the other hand, conditional deletion of XBP1s in the nervous system delayed disease onset and extended lifespan of mutant SOD1 mice due to a homeostatic link between the UPR and the autophagy pathway (Hetz et al., 2009). We found that upon ablation of XBP1 expression in mutant SOD1 mice, motoneurons up-regulated the autophagy pathway thus boosting degradation of toxic SOD1 aggregates and slowing disease progression (Hetz et al., 2009). This unexpected effect revealed that the proteostatic networks could be reprogramed through homeostatic responses with potential therapeutic benefits. Using pharmacological approach, we and others provided evidence suggesting that activation of autophagy with pharmacological agents reduce mutant SOD1 and TDP43 aggregates, increasing healthspan (Castillo et al., 2013; Zhang et al., 2014).
The IRE1-XBP1 axis is proposed to play a role in other diseases as well. For instance, we found that deletion of XBP1 in transgenic models of Huntington’s disease (HD) also leads to neuroprotection associated to autophagy induction, enhancing degradation of mutant huntingtin (Vidal et al., 2012). Recently, we have showed that phosphorylation of IRE1 directly correlates with Alzheimer’s disease (AD) progression using histological analysis of post-mortem human brain tissue (Duran-Aniotz et al., 2017). Remarkably, we discovered that activation of IRE1 exacerbates AD pathology by enhancing amyloid precursor protein (APP) expression (Duran-Aniotz et al., 2017). The specific deletion of the RNase domain of IRE1 strongly decreased the deposition of amyloid plaques and neuroinflammation on an AD mouse model. These effects resulted in improved neuronal plasticity and cognitive function (Duran-Aniotz et al., 2017). IRE1 activation also enhances the progression of glioma and its signaling inhibition strongly reduces the growth of tumors in vivo (Obacz et al., 2017). Similar findings were reported in other forms of cancer, motivating the screening of small molecules that inhibit IRE1 activity for disease treatment (Hetz et al., 2013; see below). In contrast, many other studies using gain of function to deliver XBP1s into the nervous system using gene therapy revealed important neuroprotective effects in various disease models due to artifical enforcement of adaptive programs governed by the UPR (reviewed in Valenzuela et al., 2016). A complex scenario is emerging where the IRE1-XBP1 pathway can play physiological roles in the nervous system related to neuronal function and plasticity (Martínez et al., 2016) and promote distinct outcomes under pathological conditions depending on the neurodegenerative insult.
The participation of IRE1 signaling in motoneurons demise in ALS has been reported in mutant SOD1 mice (Nishitoh et al., 2008; Lee et al., 2016). Ichijo’s group showed that mutant SOD1 contributes to ER stress by blocking ERAD through a specific interaction with Derlin-1, an ER membrane protein participating in the retro-translocation machinery (Nishitoh et al., 2008). Mutant SOD1 possibly alters Derlin-1 function, leading to impaired ERAD, resulting as a consequence in ER stress and activation of the IRE1-ASK1 apoptotic module. Remarkably, genetic deletion of ASK1 spared motoneurons and delayed disease progression in mutant SOD1 mice (Nishitoh et al., 2008). A recent study extended the role of IRE1 in ALS by identifying homeodomain interacting protein kinase 2 (HIPK2) as an essential component of the IRE1-ASK1 apoptotic cascade, leading to JNK activation under ER stress (Lee et al., 2016). Targeting HIPK2 in mutant SOD1 mice delayed disease onset and prolonged survival due to attenuated motoneuron loss (Lee et al., 2016). Relevantly, activation of HIPK2 positively correlated with TDP-43 proteinopathy in familial C9orf72 and sporadic ALS cases, implicating a broad role of ER stress and IRE1 signaling in ALS pathogenesis (Lee et al., 2016).
The current state of the field supports a pathogenic role of IRE1 pathway in ALS through activation of cell death programs, besides the possible effects of XBP1s deficiency in enhancing the activity of the autophagy pathway whereas reducing hyperexcitability in motoneurons (Nishitoh et al., 2008; Hetz et al., 2009; Kiskinis et al., 2014; Lee et al., 2016). Moreover, we reason that the sustained activation of IRE1 under chronic ER stress in motoneurons may also lead to exacerbated RIDD through induction of high-order oligomers, potentially causing excessive degradation of mRNA and down-regulation of essential components necessary to sustain motoneuron homeostasis. Indeed, the discovery of ALS genes involved in transcriptional regulation highlighted altered RNA metabolism as a relevant disease mechanism (Peters et al., 2015). In the context of ALS, IRE1 can be envisioned as a hub for integrating proteostasis and RNA metabolism at organelle level, underlying neurodegenerative cascades when these major cellular systems are significantly compromised. It is also conceivable that the deleterious activity of IRE1 may extend beyond motoneurons by enhancing astro- and microgliosis, thus promoting neuroinflammation (Boillée et al., 2006). In fact, the activity of IRE1 has been shown to enhance immune responses, promoting cytokine production in macrophages, in addition to modulate the activity of dendritic cells (Martinon and Glimcher, 2011).
The involvement of ER stress and IRE1 signaling in a myriad of diseases has fostered the development of different categories of compounds targeting IRE1 (Figure 2A; Hetz et al., 2013; Maly and Papa, 2014). Most IRE1 inhibitors target the activity of the RNase domain, including 4μ8c, MKC-3946 and STF-083110 (Hetz et al., 2013; Figure 2B). Such compounds indiscriminately shut off XBP1 splicing and RIDD activity, offering an interesting approach where complete inhibition of IRE1 pathway is desired. As suggested by many studies, the abrogation of IRE1 activity in CNS may probably not be the optimal intervention since basal levels of XBP1s appear to be needed for motoneuron homeostasis (Kiskinis et al., 2014), in addition to neuronal plasticity involved in memory and learning-associated processes (Martínez et al., 2016). The recent development of highly potent monoselective IRE1 inhibitors termed Kinase Inhibitors RNase Attenuators (KIRA) brings about the possibility to fine-tune IRE1 activity in vivo (Ghosh et al., 2014). KIRA compounds act by breaking down IRE1 oligomers thus preferentially blocking terminal IRE1 RIDD activity over XBP1 splicing depending on the dose used (Figures 2A,B). Importantly, KIRA6 has proven efficacious in preserving photoreceptor viability in ER stress-induced retinal degeneration and sustain pancreatic β-cell function in the aggressive Akita diabetic model (Ghosh et al., 2014). More recently, KIRAs were shown to provide protection in models of autoimmune diabetes (Morita et al., 2017). KIRAs did not trigger any evident side effects after systemic administration. It is proposed that KIRAs may specifically target distinct oligomerization states of IRE1, which may selectively affect pathological levels of RIDD triggered by hyperactive IRE1, allowing the signaling through the beneficial effects driven by XBP1s. The optimized use of KIRA compounds in ALS is expected to prevent or halt neurodegeneration by blunting terminal UPR in motoneurons, shifting deleterious RIDD activity to attenuated XBP1 splicing at levels contributing to homeostasis.
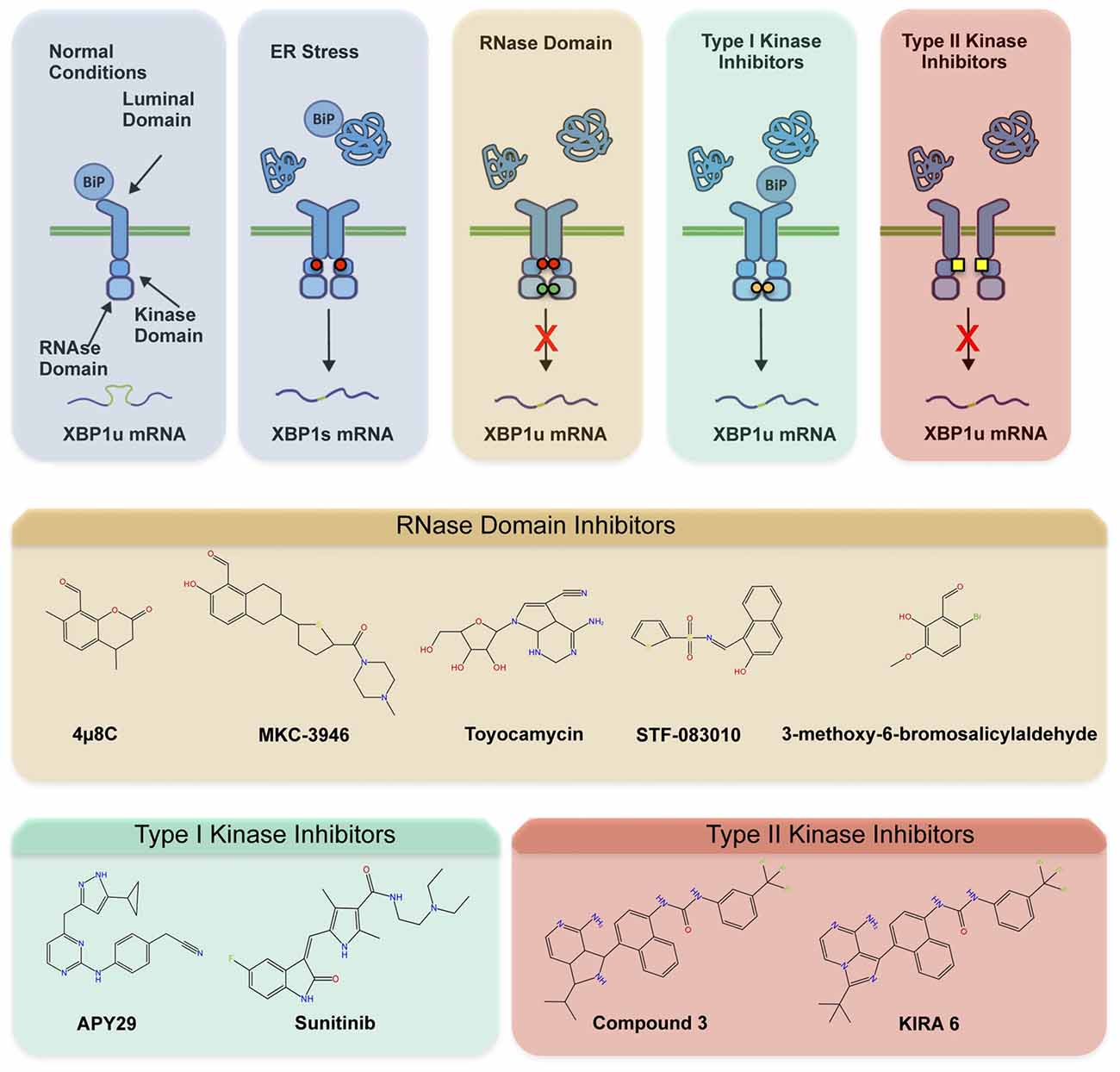
Figure 2. Pharmacological modulation of IRE1. Different classes of inhibitors target distinct IRE1 domains and differentially modulate RNase activity and oligomeric states. (A) In the first step of activation, IRE1 suffers dimerization and auto-transphosphorylation to activate its RNase activity and initiate the unconventional splicing of XBP1, in addition to low RIDD activity. Subsequently, IRE1 can form high-order oligomers to potentiate RIDD and catalyze degradation of a select pool of mRNAs and microRNAs. The compounds directly targeting the RNase domain (e.g., 4μ8c, MKC-3946, STF-083110) completely inhibit XBP1 splicing and RIDD without interfering with kinase activity or oligomeric states. The Type I kinase inhibitors (APY29, Sunitinib) prevent auto-transphosphorylation but promote RNase activity and oligomerization by generating conformational changes. The Type II inhibitors (Kinase Inhibitors RNase Attenuators, KIRAs) affect both kinase and RNase activities, possibly altering oligomerization through allosteric interactions. (B) Chemical structure of IRE1 inhibitors by mode of action is presented.
In addition to pathology, the UPR has essential roles in various organs, highlighting specialized secretory cells that require a developed ER for their proper function (Cornejo et al., 2013). This is why serious adverse side effects are predicted of the systemic and long-term administration of UPR-targeting drugs (Dufey et al., 2014). However, the recent generation of a conditional knockout mouse for IRE1 in the nervous system demonstrated that it is devoid of any gross spontaneous phenotype. Similarly, the full deletion of IRE1, except in the placenta, generates a viable animal with minor defects in secretory organs (Iwawaki et al., 2009, 2010). Taken together, these observations suggest that blocking the pathway in neurons with small molecules may be safe.
In summary, the compelling evidence in the literature linking ER stress to ALS pathogenesis and the continuous development of specific small molecules to modulate IRE1 activity may offer the opportunity to investigate novel therapeutic approaches to treat this devastating disease. Further research in mouse models of ALS is needed to address the relative contribution of IRE1 signaling in motoneurons and glia to disease progression, in addition to define its mRNA targets under pathological conditions in the nervous system.
DBM performed bibliographical research, and wrote and reviewed the article. JVG and PF performed bibliographical research, prepared figures and reviewed the article. CH performed bibliographical research and wrote and reviewed the article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was funded by Congressionally Directed Medical Research Programs, ALSRP Therapeutic Idea Award AL150111, Muscular Dystrophy Association 382453, FONDAP program 15150012 and Millennium Institute P09-015-F (CH) and Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT) no. 11150579 (DBM). We also thank the support from Michael J. Fox Foundation for Parkinson’s Research—Target Validation grant No. 9277, FONDEF ID16I10223, FONDEF D11E1007, FONDECYT no. 1140549, US Office of Naval Research-Global (ONR-G) N62909-16-1-2003, US Air Force Office of Scientific Research FA9550-16-1-0384, European Commission R&D, MSCA-RISE #734749 and CONICYT-Brazil 441921/2016-7 (CH).
Atkin, J. D., Farg, M. A., Turner, B. J., Tomas, D., Lysaght, J. A., Nunan, J., et al. (2006). Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem. 281, 30152–30165. doi: 10.1074/jbc.M603393200
Atkin, J. D., Farg, M. A., Walker, A. K., McLean, C., Tomas, D., and Horne, M. K. (2008). Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 30, 400–407. doi: 10.1016/j.nbd.2008.02.009
Bernard-Marissal, N., Moumen, A., Sunyach, C., Pellegrino, C., Dudley, K., Henderson, C. E., et al. (2012). Reduced calreticulin levels link endoplasmic reticulum stress and fas-triggered cell death in motoneurons vulnerable to ALS. J. Neurosci. 32, 4901–4912. doi: 10.1523/JNEUROSCI.5431-11.2012
Boillée, S., Vande Velde, C., and Cleveland, D. W. (2006). ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59. doi: 10.1016/j.neuron.2006.09.018
Bosco, D. A., Morfini, G., Karabacak, N. M., Song, Y., Gros-Louis, F., Pasinelli, P., et al. (2010). Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403. doi: 10.1038/nn.2660
Castillo, K., Nassif, M., Valenzuela, V., Rojas, F., Matus, S., Mercado, G., et al. (2013). Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 9, 1308–1320. doi: 10.4161/auto.25188
Cornejo, V. H., Pihán, P., Vidal, R. L., and Hetz, C. (2013). Role of the unfolded protein response in organ physiology: lessons from mouse models. IUBMB Life 65, 962–975. doi: 10.1002/iub.1224
Dafinca, R., Scaber, J., Ababneh, N., Lalic, T., Weir, G., Christian, H., et al. (2016). C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal demen. Stem Cells 34, 2063–2078. doi: 10.1002/stem.2388
de Boer, A. S., and Eggan, K. (2015). A perspective on stem cell modeling of amyotrophic lateral sclerosis. Cell Cycle 14, 3679–3688. doi: 10.1080/15384101.2015.1093712
Dufey, E., Sepúlveda, D., Rojas-Rivera, D., and Hetz, C. (2014). Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 1. An overview. Am. J. Physiol. Cell Physiol. 307, C582–C594. doi: 10.1152/ajpcell.00258.2014
Duran-Aniotz, C., Cornejo, V. H., Espinoza, S., Ardiles, Á. O., Medinas, D. B., Salazar, C., et al. (2017). IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. doi: 10.1007/s00401-017-1694-x [Epub ahead of print].
Farg, M. A., Soo, K. Y., Walker, A. K., Pham, H., Orian, J., Horne, M. K., et al. (2012). Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol. Aging 33, 2855–2868. doi: 10.1016/j.neurobiolaging.2012.02.009
Filézac de L’Etang, A., Maharjan, N., Cordeiro Braña, M., Ruegsegger, C., Rehmann, R., Goswami, A., et al. (2015). Marinesco-Sjögren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nat. Neurosci. 18, 227–238. doi: 10.1038/nn.3903
Ghosh, R., Wang, L., Wang, E. S., Perera, B. G. K., Igbaria, A., Morita, S., et al. (2014). Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 158, 534–548. doi: 10.1016/j.cell.2014.07.002
Gonzalez-Perez, P., Woehlbier, U., Chian, R.-J., Sapp, P., Rouleau, G. A., Leblond, C. S., et al. (2015). Identification of rare protein disulfide isomerase gene variants in amyotrophic lateral sclerosis patients. Gene 566, 158–165. doi: 10.1016/j.gene.2015.04.035
Hall, C. E., Yao, Z., Choi, M., Tyzack, G. E., Serio, A., Luisier, R., et al. (2017). Progressive motor neuron pathology and the role of astrocytes in a human stem cell model of VCP-related ALS. Cell Rep. 19, 1739–1749. doi: 10.1016/j.celrep.2017.05.024
Han, D., Lerner, A. G., Vande Walle, L., Upton, J. P., Xu, W., Hagen, A., et al. (2009). IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138, 562–575. doi: 10.1016/j.cell.2009.07.017
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Hetz, C., Chevet, E., and Harding, H. P. (2013). Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 12, 703–719. doi: 10.1038/nrd3976
Hetz, C., Chevet, E., and Oakes, S. A. (2015). Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 829–838. doi: 10.1038/ncb3184
Hetz, C., and Glimcher, L. H. (2009). Fine-tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol. Cell 35, 551–561. doi: 10.1016/j.molcel.2009.08.021
Hetz, C., and Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. doi: 10.1038/nrn3689
Hetz, C., Thielen, P., Matus, S., Nassif, M., Court, F., Kiffin, R., et al. (2009). XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 23, 2294–2306. doi: 10.1101/gad.1830709
Hollien, J., Lin, J. H., Li, H., Stevens, N., Walter, P., and Weissman, J. S. (2009). Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 186, 323–331. doi: 10.1083/jcb.200903014
Hollien, J., and Weissman, J. S. (2006). Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107. doi: 10.1126/science.1129631
Hu, P., Han, Z., Couvillon, A. D., Kaufman, R. J., and Exton, J. H. (2006). Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α-mediated NF-κB activation and down-regulation of TRAF2 expression. Mol. Cell Biol. 26, 3071–3084. doi: 10.1128/mcb.26.8.3071-3084.2006
Ilieva, E. V., Ayala, V., Jové, M., Dalfó, E., Cacabelos, D., Povedano, M., et al. (2007). Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 130, 3111–3123. doi: 10.1093/brain/awm190
Ito, Y., Yamada, M., Tanaka, H., Aida, K., Tsuruma, K., Shimazawa, M., et al. (2009). Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis. 36, 470–476. doi: 10.1016/j.nbd.2009.08.013
Iwawaki, T., Akai, R., and Kohno, K. (2010). IRE1α disruption causes histological abnormality of exocrine tissues, increase of blood glucose level and decrease of serum immunoglobulin level. PLoS One 5:e13052. doi: 10.1371/journal.pone.0013052
Iwawaki, T., Akai, R., Yamanaka, S., and Kohno, K. (2009). Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl. Acad. Sci. U S A 106, 16657–16662. doi: 10.1073/pnas.0903775106
Kiskinis, E., Sandoe, J., Williams, L. A., Boulting, G. L., Moccia, R., Wainger, B. J., et al. (2014). Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14, 781–795. doi: 10.1016/j.stem.2014.03.004
Leblond, C. S., Kaneb, H. M., Dion, P. A., and Rouleau, G. A. (2014). Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp. Neurol. 262, 91–101. doi: 10.1016/j.expneurol.2014.04.013
Lee, S., Shang, Y., Redmond, S. A., Urisman, A., Tang, A. A., Li, K. H., et al. (2016). Activation of HIPK2 promotes er stress-mediated neurodegeneration in amyotrophic lateral sclerosis. Neuron 91, 41–55. doi: 10.1016/j.neuron.2016.05.021
Lerner, A. G., Upton, J.-P., Praveen, P. V. K., Ghosh, R., Nakagawa, Y., Igbaria, A., et al. (2012). IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 16, 250–264. doi: 10.1016/j.cmet.2012.07.007
Maly, D. J., and Papa, F. R. (2014). Druggable sensors of the unfolded protein response. Nat. Chem. Biol. 10, 892–901. doi: 10.1038/nchembio.1664
Martinon, F., and Glimcher, L. H. (2011). Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Curr. Opin. Immunol. 23, 35–40. doi: 10.1016/j.coi.2010.10.016
Martínez, G., Vidal, R. L., Mardones, P., Serrano, F. G., Ardiles, A. O., Wirth, C., et al. (2016). Regulation of memory formation by the transcription factor XBP1. Cell Rep. 14, 1382–1394. doi: 10.1016/j.celrep.2016.01.028
Matus, S., Lopez, E., Valenzuela, V., Nassif, M., and Hetz, C. (2013). Functional contribution of the transcription factor ATF4 to the pathogenesis of amyotrophic lateral sclerosis. PLoS One 8:e66672. doi: 10.1371/journal.pone.0066672
Matus, S., Medinas, D. B., and Hetz, C. (2014). Common ground: stem cell approaches find shared pathways underlying ALS. Cell Stem Cell 14, 697–699. doi: 10.1016/j.stem.2014.05.001
Maurel, M., Chevet, E., Tavernier, J., and Gerlo, S. (2014). Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 39, 245–254. doi: 10.1016/j.tibs.2014.02.008
Morita, S., Villalta, S. A., Feldman, H. C., Register, A. C., Rosenthal, W., Hoffmann-Petersen, I. T., et al. (2017). Targeting ABL-IRE1α signaling spares ER-stressed pancreatic β cells to reverse autoimmune diabetes. Cell Metab. 25, 883–897.e8. doi: 10.1016/j.cmet.2017.03.018
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nishitoh, H., Kadowaki, H., Nagai, A., Maruyama, T., Yokota, T., Fukutomi, H., et al. (2008). ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 22, 1451–1464. doi: 10.1101/gad.1640108
Oakes, S. A., and Papa, F. R. (2015). The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 10, 173–194. doi: 10.1146/annurev-pathol-012513-104649
Obacz, J., Avril, T., Le Reste, P.-J., Urra, H., Quillien, V., Hetz, C., et al. (2017). Endoplasmic reticulum proteostasis in glioblastoma-From molecular mechanisms to therapeutic perspectives. Sci. Signal. 10:eaal2323. doi: 10.1126/scisignal.aal2323
Peters, O. M., Ghasemi, M., and Brown, R. H. (2015). Emerging mechanisms of molecular pathology in ALS. J. Clin. Invest. 125:2548. doi: 10.1172/JCI82693
Philips, T., and Rothstein, J. D. (2015). Rodent models of amyotrophic lateral sclerosis. Curr. Protoc. Pharmacol. 69, 5.67.1–5.67.21. doi: 10.1002/0471141755.ph0567s69
Ruegsegger, C., and Saxena, S. (2016). Proteostasis impairment in ALS. Brain Res. 1648, 571–579. doi: 10.1016/j.brainres.2016.03.032
Sasaki, S. (2010). Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 69, 346–355. doi: 10.1097/NEN.0b013e3181d44992
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi: 10.1038/nn.2297
Scheper, W., and Hoozemans, J. J. M. (2015). The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. 130, 315–331. doi: 10.1007/s00401-015-1462-8
Smith, H. L., and Mallucci, G. R. (2016). The unfolded protein response: mechanisms and therapy of neurodegeneration. Brain 139, 2113–2121. doi: 10.1093/brain/aww101
Sun, S., Sun, Y., Ling, S.-C., Ferraiuolo, L., McAlonis-Downes, M., Zou, Y., et al. (2015). Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc. Natl. Acad. Sci. U S A 112, E6993–E7002. doi: 10.1073/pnas.1520639112
Taylor, J. P., Brown, R. H., and Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Turner, M. R., Hardiman, O., Benatar, M., Brooks, B. R., Chio, A., de Carvalho, M., et al. (2013). Controversies and priorities in amyotrophic lateral sclerosis. Lancet. Neurol. 12, 310–322. doi: 10.1016/S1474-4422(13)70036-X
Urano, F., Wang, X., Bertolotti, A., Zhang, Y., Chung, P., Harding, H. P., et al. (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666. doi: 10.1126/science.287.5453.664
Urra, H., Dufey, E., Lisbona, F., Rojas-Rivera, D., and Hetz, C. (2013). When ER stress reaches a dead end. Biochim. Biophys. Acta 1833, 3507–3517. doi: 10.1016/j.bbamcr.2013.07.024
Valenzuela, V., Martínez, G., Duran-Aniotz, C., and Hetz, C. (2016). Gene therapy to target ER stress in brain diseases. Brain Res. 1648, 561–570. doi: 10.1016/j.brainres.2016.04.064
Vidal, R. L., Figueroa, A., Court, F. A., Thielen, P., Molina, C., Wirth, C., et al. (2012). Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 21, 2245–2262. doi: 10.1093/hmg/dds040
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Wang, M., and Kaufman, R. J. (2016). Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326–335. doi: 10.1038/nature17041
Woehlbier, U., Colombo, A., Saaranen, M. J., Pérez, V., Ojeda, J., Bustos, F. J., et al. (2016). ALS-linked protein disulfide isomerase variants cause motor dysfunction. EMBO J. 35, 845–865. doi: 10.15252/embj.201592224
Keywords: ALS, ER stress, UPR, IRE1α, protein aggregation
Citation: Medinas DB, González JV, Falcon P and Hetz C (2017) Fine-Tuning ER Stress Signal Transducers to Treat Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 10:216. doi: 10.3389/fnmol.2017.00216
Received: 15 May 2017; Accepted: 19 June 2017;
Published: 05 July 2017.
Edited by:
Angelo Poletti, Università degli Studi di Milano, ItalyReviewed by:
Maruf M. U. Ali, Imperial College London, United KingdomCopyright © 2017 Medinas, González, Falcon and Hetz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Danilo B. Medinas, ZG1lZGluYXNAbWVkLnVjaGlsZS5jbA==
Claudio Hetz, Y2hldHpAbWVkLnVjaGlsZS5jbA==; Y2hldHpAaHNwaC5oYXJ2YXJkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.