 Paolo Remondelli
Paolo Remondelli Maurizio Renna
Maurizio Renna- 1Dipartimento di Medicina, Chirurgia e Odontoiatria “Scuola Medica Salernitana”, Università degli Studi di Salerno, Salerno, Italy
- 2Cambridge Institute for Medical Research, Department of Medical Genetics, Wellcome Trust, Addenbrooke’s Hospital, University of Cambridge, Cambridge, United Kingdom
In eukaryotic cells, the endoplasmic reticulum (ER) is the cell compartment involved in secretory protein translocation and quality control of secretory protein folding. Different conditions can alter ER function, resulting in the accumulation of unfolded or misfolded proteins within the ER lumen. Such a condition, known as ER stress, elicits an integrated adaptive response known as the unfolded protein response (UPR) that aims to restore proteostasis within the secretory pathway. Conversely, in prolonged cell stress or insufficient adaptive response, UPR signaling causes cell death. ER dysfunctions are involved and contribute to neuronal degeneration in several human diseases, including Alzheimer, Parkinson and Huntington disease and amyotrophic lateral sclerosis. The correlations between ER stress and its signal transduction pathway known as the UPR with neuropathological changes are well established. In addition, much evidence suggests that genetic or pharmacological modulation of UPR could represent an effective strategy for minimizing the progressive neuronal loss in neurodegenerative diseases. Here, we review recent results describing the main cellular mechanisms linking ER stress and UPR to neurodegeneration. Furthermore, we provide an up-to-date panoramic view of the currently pursued strategies for ameliorating the toxic effects of protein unfolding in disease by targeting the ER UPR pathway.
Introduction
The abnormal aggregation of misfolded proteins, which accumulate within the endoplasmic reticulum (ER) or the cytosol, characterizes several pathological conditions (Balch et al., 2008; Hartl et al., 2011).
Protein misfolding can result from genetic mutations affecting normal protein folding or derive from malfunction of the cytosolic or ER resident protein folding machinery. Many diseases result from misfolded proteins that accumulate within the ER, where they generate a stressful condition referred to as ER stress. Cells counteract ER stress by activating a group of signaling pathways termed the unfolded protein response (UPR) that coordinate a potent transcriptional program whose main purpose is restoring ER and cell function and ensuring cell survival. UPR improves folding control by increasing the expression of ER enzymes, chaperones and intracellular transporters with the aim of preventing protein aggregation and in parallel modulates the removal of misfolded proteins that would otherwise compromise cellular functions (Sitia and Braakman, 2003). Concomitantly, UPR temporarily down-regulates the protein translation rate to limit the amount of potential unfolded proteins entering the ER (Zhao and Ackerman, 2006). Moreover, UPR stimulates ER membrane synthesis and regulates the turnover of incorrectly folded proteins by fine-tuning the ER-associated protein degradation (ERAD) pathway (Meusser et al., 2005). Conversely, overtly severe or prolonged ER stress activates the UPR-dependent apoptotic cell death program (Zhang and Kaufman, 2006; Mori, 2009; Walter and Ron, 2011).
Although physically separate, there is an intimate interconnection between the protein folding machinery of the ER and cytosol. As such, increased misfolded proteins in the cytosol can compromise proteasome activity, inhibit ERAD and elicit an ER stress response, and compensatory activation of the autophagic pathway, both aimed at antagonizing the protein overload. Indeed, both proteasome activity and the autophagic pathway progressively decline in several neurodegenerative diseases resulting in proteotoxicity, chronic ER stress and ultimately lead to activation of the programmed cell death pathway (Schroder and Kaufman, 2005; Hetz et al., 2011).
Protein unfolding can be considered the initial event endorsing neurodegenerative diseases (Selkoe, 2003; Haass and Selkoe, 2007; Uversky, 2007; Makioka et al., 2010; Lindquist and Kelly, 2011; Martin et al., 2011; Roussel et al., 2013; Hetz and Mollereau, 2014; Valastyan and Lindquist, 2014) that are generally indicated as protein misfolding disorders (PMDs). Misfolded proteins are disease-specific. Among them, tau and β-amyloid (Aβ) cause Alzheimer disease (AD); α-synuclein (α-syn) mutations are a common hallmark of Parkinson disease (PD); the RNA binding proteins fused in sarcoma/translocated in sarcoma (FUS), ZNF and TAR DNA-binding protein 43 (TDP43) are associated with frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS); expanded polyglutamine (poly-Q) traits containing proteins cause Huntington disease (HD) and spinal cerebellar ataxias; prion proteins (PrP) are associated with Creutzfeldt-Jakob disease (CJD) (Prusiner, 1998; Knowles et al., 2014; Vidal et al., 2014).
Neuronal cells are particularly sensitive to protein misfolding in contrast to non-neuronal cells, in which cell duplications help to counteract misfolding during ER stress by repeatedly diluting unfolded peptides. By contrast, non-dividing post-mitotic neurons are completely dependent on UPR for survival. Consequently, if the misfolding is not removed and normal cell functions are not restored, UPR leads to apoptosis, leading to selective neuronal death.
In view of these observations, identifying molecules that can target UPR signaling components represents an important approach for discovering new therapies for neurodegenerative diseases. Genetic approaches such as gene therapy and RNA interference that can selectively modulate UPR in affected tissues are also under active investigation.
In this article, we review the role of UPR in the pathogenesis of neurological diseases and discuss recent progress in modulating ER stress, UPR and therefore ER homeostasis via pharmacological and gene therapy-based approaches.
The UPR Pathways
The UPR relies on the activation of three individual modules, each represented by an ER transmembrane protein, namely PRKR-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6) (Schroder and Kaufman, 2005). In normal conditions, PERK, IRE1, and ATF6 are kept inactive by physical interaction with the chaperone BIP/GRP78 (binding immunoglobulin protein/78-kDa glucose-regulated protein). In response to ER stress, BIP/GRP78 preferentially binds the unfolded polypeptides when they become abundant within the ER, thereby permitting PERK, IRE1, and ATF6 activation (schematically summarized in Figure 1).
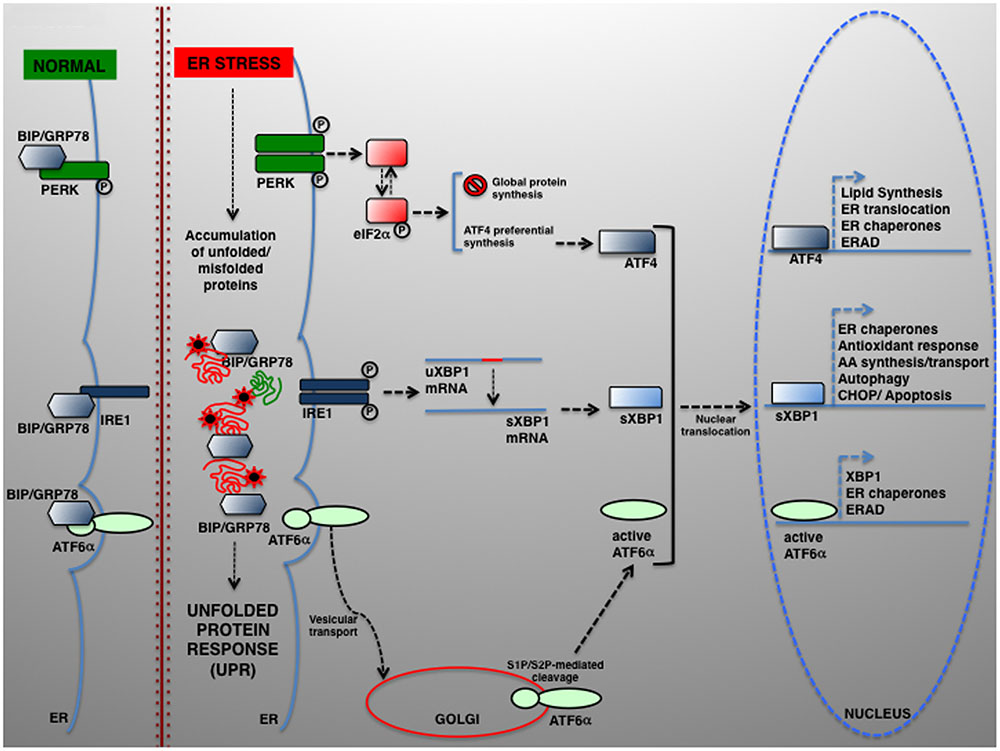
FIGURE 1. The UPR signaling pathways. In eukaryotic cells, a large portion of the total proteome is synthesized within the ER. Newly synthesized proteins can then be transported to their final destination along the secretory pathway. Several physiological and pathological conditions can hamper normal protein folding within the ER, which in turn determines the accumulation of unfolded or misfolded proteins and UPR activation. UPR is controlled by three transducers: IRE1, ATF6α, and PERK. In normal conditions, the luminal IRE1, ATF6α, and PERK regions are associated to and kept in the inactive state by the ER-resident chaperone BIP/GRP78. Upon ER stress, BIP/GRP78 dissociates and enables UPR activation. ATF6 is transported from the ER to the Golgi, where it undergoes proteolytic cleavage by S1P/S2P proteases. The cytosolic ATF6 fragment (active ATF6α) is ultimately translocated into the nucleus where it acts as a transcription factor of genes required for ERAD and modulates XBP1 transcription). ER stress also activates PERK, which phosphorylates eIF2α to attenuate protein translation. In parallel, preferential ATF4 translation drives the expression of ER chaperones and other genes controlling autophagy, redox control and nutrient metabolism. Under severe ER stress, the PERK–ATF4 pathway controls the proapoptotic genes, including CHOP, which leads to programmed cell death. Once activated, IRE1 operates the alternative splicing of XBP1. The active XBP1(s) up-regulates, among others, genes encoding for ER chaperones, ERAD components and proteins involved in the lipid biosynthesis pathways.
Upon BIP/GRP78 dissociation, PERK phosphorylates eukaryotic translation initiation factor 2α (eIF2α), which exerts an inhibitory effect on protein translation initiation (Harding et al., 1999). The reduction of protein synthesis affects the level of unfolded proteins, thus helping the ER folding machinery to act more efficiently. However, some proteins are preferentially translated and maintaining the synthesis of, among the others, ATF4, which activates key genes regulating protein folding, amino acid metabolism, redox control, and ERGIC-53, which regulates the ER-to-Golgi cargo protein trafficking (Blais et al., 2004; Spatuzza et al., 2004; Renna et al., 2006; Amodio et al., 2009).
BIP/GRP78 disassociation enables IRE1α activation, which retains both kinase and endoribonuclease activity, and the activation of its paralog IRE1β, for which PERK also induces translational repression by specifically controlling 28S rRNA turnover (Iwawaki et al., 2001). Instead, active IRE1α drives the mRNA splicing of a 26-nucleotide from X-box binding protein 1 (XBP1) (Calfon et al., 2002), which restores the translation of XBP1 that enhances the expression of UPR-dependent genes, including that for ER folding (calnexin, calreticulin), ERAD factors (HERPUD1 [homocysteine inducible ER protein with ubiquitin-like domain 1], EDEM1 [ER degradation-enhancing alpha-mannosidase-like protein 1]), autophagy components (ATG5, ATG12, BECN1 [beclin 1], UVRAG [UV radiation resistance-associated]) and redox metabolism (SOD1 [superoxide dismutase 1], catalase, TRX1 [thioredoxin]) (Acosta-Alvear et al., 2007; Liu et al., 2009).
The third UPR component is ATF6 p90. Upon release from BIP/GRP78, ATF6 is directed to the cis-Golgi, where it is proteolytically cleaved. The active ATF6 fragment (ATF6 p50 or nATF6) activates the transcription of not only BIP/GRP78, GRP94/GP96, and XBP1, but also the cell death activator CHOP, and other proteins such as the calcium pump SERCA (sarcoplasmic/ER calcium ATPase pump) and p58IPK/DNAJC3 (Haze et al., 1999; van Huizen et al., 2003; Renna et al., 2007; Ron and Walter, 2007).
However, in prolonged or severe ER stress UPR may lead to apoptosis (Zhang and Kaufman, 2006; Walter and Ron, 2011). A primary determinant of apoptosis is PERK–eIF2α–ATF4 signaling (Rutkowski et al., 2006), which culminates in the induction of CHOP/GADD153 (Zinszner et al., 1998), which eventually lead to apoptotic cell death (Ohoka et al., 2005; Puthalakath et al., 2007).
Under acute stress, IRE1α is also involved in apoptosis and programmed cell death. Here, IRE1α activation induces JNK (c-Jun N-terminal kinase) activation through the formation of a tripartite complex from IRE1, ASK1, and TNF receptor-associated factor 2 (TRAF2) (Nishitoh et al., 2002).
Thus, UPR plays either a protective or a lethal role in response to proteotoxicity. Intriguingly, UPR pathways may regulate the expression of several genes related to neurodegenerative disease. For example, XBP1 activates the expression of AD-related genes such as those encoding for subunits of the γ-secretase complex or cyclin-dependent kinase 5 (CDK5) (Acosta-Alvear et al., 2007) and can influence amyloid precursor protein (APP) processing (Yang et al., 1998; Domingues et al., 2007). Interestingly, a more recent report proposed a model describing a vicious circle in which the activation of JNK3 by ER stress in AD increased amyloid-β production, amplifying in turn the ER stress response (Yoon et al., 2012). Furthermore, the UPR PERK–eIF2α axis can improve APP processing through β-secretase 1 (O’Connor et al., 2008), whereas ATF4 can regulate presenilin expression and γ-secretase activity (Mitsuda et al., 2007; Ohta et al., 2011).
In PD, ATF4 regulates Parkin expression (Bouman et al., 2011); Parkin itself controls the transcription of another PD-associated gene (PARK7, [parkin RBR E3 ubiquitin protein ligase]) through XBP1 (Duplan et al., 2013).
To summarize, intracellular protein aggregation is a salient feature underlying various neurological diseases. The balance between protein generation and degradation is crucial for protein homeostasis. Intriguingly, while most aggregated proteins result in the common endpoint of neuronal impairment, each aggregated protein has a distinct mechanism of action. Therefore, distinct molecular mechanisms may converge and contribute, albeit to a different extent, to ER proteostasis imbalance and ultimately cause neurodegeneration (Figure 2).
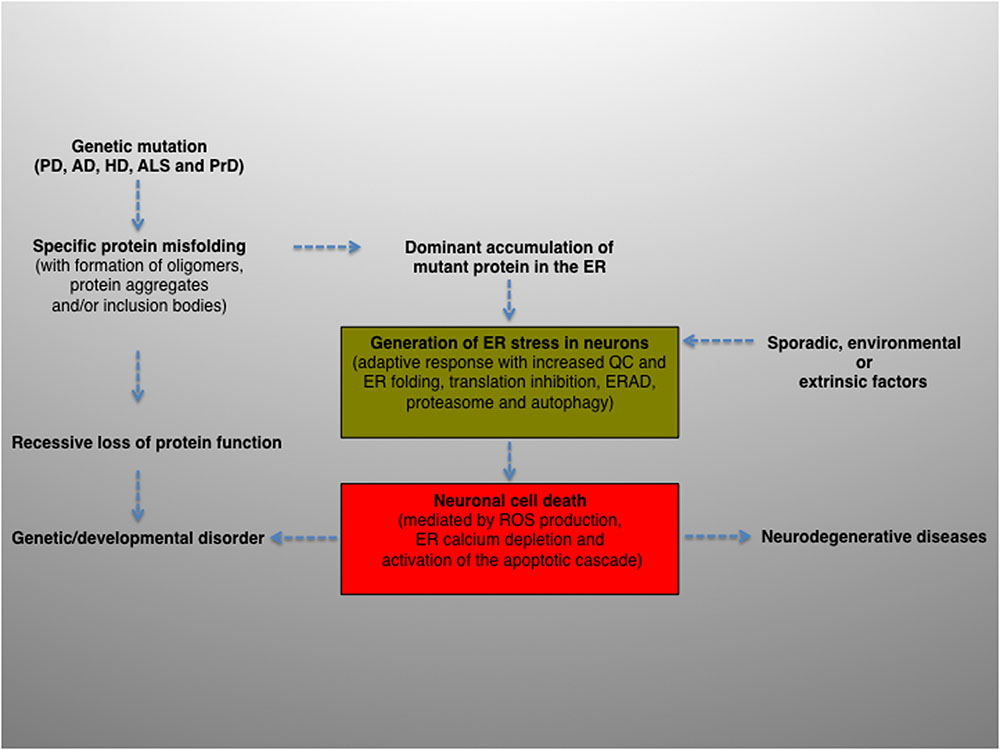
FIGURE 2. Schematic diagram depicting the potential role and contribution of ER stress in the pathogenesis of neurodegenerative diseases. ER activity is a major contributing factor to the overall biological fitness and homeostasis of eukaryotic cells. Consequently, ER stress and UPR are intimately interconnected with the pathogenesis of several human diseases and clinically relevant conditions. The activation of this pathway can derive from genetic or environmental factors, or both. For example, ER stress can result from a genetic mutation that causes a direct defect (i.e., folding, activity, post-translational modification, transport) of certain secretory proteins. More frequently, the progressive accumulation of misfolded and aggregation-prone mutant proteins with the formation of oligomers, aggregates and inclusion bodies represents a common feature of many age-related conditions, including neurodegenerative diseases. Neuronal protein aggregation can lead to recessive loss of function, which could contribute to the etiology of developmental and degenerative disorders. In this context, the accumulation of unfolded or misfolded proteins can affect the compartment at different levels, generating ER stress. The consequent activation of UPR will encompass an initial phase, characterized by an adaptive response aimed at alleviating the ER protein overload and preserving neuronal viability (green box), followed by a second, non-reversible response whereby the ER stress–mediated cell death program will ultimately be activated (red box), leading to neuronal loss. Finally, sporadic, extrinsic or environmental insults (e.g., physical injuries and neurotoxins) can perturb the ER, induce neuronal UPR and ultimately contribute to the pathogenesis of neurodegenerative diseases.
Crosstalk between UPR and Autophagy in Neurodegenerative Diseases
Increasing evidence suggests that compromised autophagy contributes to the pathogenesis of various neurodegenerative diseases. As many neurodegenerative disease-associated aggregate-prone proteins are autophagy substrates, the levels of these proteins increase in both the soluble and aggregated state when autophagy is compromised (Rubinsztein et al., 2012; Menzies et al., 2015). Furthermore, mitochondrial damage activates an autophagy pathway named mitophagy and it is linked to neurodegenerative diseases such as PD and ALS (Valente et al., 2004; Chung et al., 2016).
In PD, heterozygous mutation of GBA1 (glucocerebrosidase) is the most common known genetic risk factor. GBA1 encodes GBA, a lysosomal enzyme that cleaves the β-glucosyl linkage of glucosylceramide (GlcCer). Loss of GBA activity with resultant increased GlcCer levels led to increased α-syn levels in cultured neurons. Increased α-syn in turn inhibited lysosomal maturation and GBA activity, resulting in additional GlcCer accumulation and further α-syn accumulation (Mazzulli et al., 2011). Interestingly, in addition to inhibiting the autophagy-lysosomal pathway, GBA mutations activate UPR and lead to ER stress in fibroblasts and iPSC-derived DA neurons from patients with PD (Fernandes et al., 2016; Sanchez-Martinez et al., 2016). In contrast, in ALS pathogenesis intracellular aggregates correlate with autophagosome accumulation and decreased proteasome activity (Cheroni et al., 2009; Chen et al., 2012).
A significant number of studies indicate that UPR is intimately linked to autophagy (Kouroku et al., 2007; Adolph et al., 2013). As the continuously adjusted homeostatic balance between protein folding/synthesis and degradation is of particular importance in neuronal cells, autophagy has become a relevant target for disease intervention in most neurodegenerative diseases (Wong and Cuervo, 2010; Green and Levine, 2014). Many studies have proven that UPR manipulation has clear consequences in substrate degradation by autophagy. For example, IRE1 regulates mutant huntingtin (HTT) clearance and most studies have shown that IRE1 can control autophagy levels by recruiting the adaptor protein TRAF2 and activating MAPK8 (mitogen-activated protein kinase 8)/JNK (Fouillet et al., 2012). Another report indicates that poly-Q trait expression triggers PERK activation and the phosphorylation of eIF2α, activating autophagy possibly due to the up-regulation of ATG12, an essential autophagy regulator (Kouroku et al., 2007). Moreover, ATF4 induces ATG12, ATG5, and BECN1 expression, providing a direct link between UPR signaling and the control of autophagy (B’Chir et al., 2013).
Instead, XBP1 deficiency enhanced neuronal survival and the motor performance of the HD mice, with a mechanism of protection that does not appear strictly related to the ER stress response, but rather depending on up-regulated autophagy. Indeed, such regulation is independent of XBP1, but is due to the targeting of FOXO1 (forkhead box O1), a key transcription factor modulating autophagy in neurons that is regulated negatively by XBP1 in vivo (Zhou et al., 2011; Vidal et al., 2012; Zhao et al., 2013). Hence, these findings are indicative of critical crosstalk between the UPR IRE1–XBP1 arm and FOXO1–autophagy in HD neurons, providing a novel link between two major stress pathways and suggesting possible therapeutic benefits of targeting this pathway in a disease context.
Huntington disease is a late-onset neurodegenerative disease, characterized by specific mutations in the polyglutamine trait (poliQ expansion) contained in the first exon of the huntingtin protein. These mutations drastically change the hydrophobic properties of the protein, leading to the formation of large and insoluble inclusions within neurons (Arrasate and Finkbeiner, 2012). Even though toxic mHTT aggregates are primarily eliminated through autophagy, autophagy dysfunction is often observed in HD pathogenesis. In this context, the ectodermal-neural cortex 1 (ENC1) has been recently identified as a novel binding partner of p62/SQSTM1, which negatively regulates autophagy (Lee H. et al., 2016). Interestingly, under ER stress the expression of ENC1 and the ENC1/p62 interaction is enhanced by the IRE1/TRAF2/JNK pathway. Furthermore, ENC1 co-localizes with mHTT aggregates, thus inhibiting cargo recognition of p62 and leading to the accumulation and neurotoxicity of mtHTT aggregates. Accordingly, knockdown of ENC1 expression enhances co-localization of p62 with mtHTT aggregates, ameliorates the ER stress-induced impairment of autophagy and relieves neuronal cell death (Lee H. et al., 2016).
Growing evidence correlates defects in the autophagy system with ALS pathogenesis. ALS is predominantly sporadic, although a growing number of genes have been identified in the familial forms. ALS-associated mutations in TDP43, SOD1, FUS, and C9ORF72 (C9 open reading frame 72) (Watabe and Nakaki, 2004; Andersen and Al-Chalabi, 2011; Fecto and Siddique, 2011; Farg et al., 2014) result in protein misfolding, aggregate accumulation and is associated with ER stress (Dafinca et al., 2016). Furthermore, these intracellular aggregates correlate with autophagosome accumulation and decreased proteasome activity in the neurons and brain tissues from patients with ALS (Cheroni et al., 2009; Chen et al., 2012). Alterations in C9ORF72 are the most common cause of ALS and FTD and are linked by common pathological features (DeJesus-Hernandez et al., 2011). Wild-type C9ORF72 is involved in regulating endocytic transport and colocalises with the autophagic proteins in human motor neurons (Farg et al., 2014), and interacts with the autophagy receptors p62/SQSTM1 (sequestosome 1) and OPTN (optineurin) and affects autophagosome formation (Sellier et al., 2016). In a recent report the kinase HIPK2 has been identified as the essential link that promotes ER-stress-induced cell death via the IRE1α-ASK1-JNK pathway. In SOD1 mutant mice, loss of HIPK2 delays disease onset, reduces cell death in spinal motor neurons and improves survival. Conversely, blocking HIPK2 kinase activity protects motor neurons from TDP-43 cytotoxicity, revealing a previously unrecognized role of HIPK2 activation in ER-stress-mediated neurodegeneration and its potential role as a therapeutic target for ALS (Lee S. et al., 2016).
Overall, the current experimental evidence sheds light on how two fundamental homeostatic processes, i.e., the UPR pathway and autophagy, contribute to cellular stress management and provide evidence in favor of therapeutic strategies for manipulating UPR levels, which may have broad beneficial consequences for alleviating neurodegeneration.
Alteration of ER Proteostasis and Protein Trafficking as Possible Causes of Neurodegeneration
Global alteration in ER proteostasis can have an inhibitory effect on ER–Golgi trafficking (Renna et al., 2006; Amodio et al., 2009, 2013). As neuronal cells rely on the fitness of the secretory compartments for their function, impaired protein trafficking could also cause neurodegenerative disorders. Indeed, several findings suggest that ER retention of disease-related proteins can be decisive for neuronal cell death. This can occur in AD models (Katayama et al., 1999; Nakagawa et al., 2000) and in PD or familial CJD (Ugolino et al., 2011; Xu and Zhu, 2012).
In animal models and in the brain tissues from patients with PD (Colla et al., 2012a,b), mutant α-syn accumulates in the ER, where it is retained by BIP/GRP78 (Colla et al., 2012a) and can impair protein trafficking by affecting RAB1 (Ras-related protein Rab-1A) and SNAREs function at the ER–Golgi boundary (Cooper et al., 2006; Gitler et al., 2008; Thayanidhi et al., 2010). Similarly, mutant FUS and TDP43 interact with PDI (protein disulfide isomerase) both in experimental models and in tissues from patients with ALS (Farg et al., 2012; Walker et al., 2013).
In this context, envisaging that ER chaperones may act by reducing protein aggregation and therefore sustaining neuronal survival is conceivable (Atkin et al., 2006; Kikuchi et al., 2006; Walker et al., 2010).
Intriguingly, disease-related proteins that also accumulate in the cytosol, e.g., mutant SOD1 and HTT, can impair the ERAD machinery, induce ER stress and alter protein trafficking (Turner et al., 2005; Urushitani et al., 2006; Duennwald and Lindquist, 2008; Nishitoh et al., 2008; Yang et al., 2010; Hetz, 2012). A similar effect was reported for phosphorylated tau protein in AD models (Abisambra et al., 2013). Furthermore, cytosolic HTT accumulation can perturb different levels of the secretory pathway (del Toro et al., 2006; Vidal and Hetz, 2012). In addition, non-physiological protein–protein interactions can determine at different levels a number of distinct perturbations along the secretory pathway, generating chronic ER stress (Table 1). One example is the ALS-related mutant form of vesicle-associated protein B (VAPB) that accumulates and is retained within the ER, and the cargo receptor protein YIF1A (Yip1-interacting factor homolog A), which is normally localized along the secretory pathway in the ERGIC (Kuijpers et al., 2013). Moreover, an ALS-linked mutant VAPB variant affects the normal ER localization of quality control components (Moustaqim-Barrette et al., 2014). Finally, the expression of disease proteins such as VAPB and HTT can affect the three-dimensional organization of the ER tubular cisternae, ultimately contributing to the dysfunction of this compartment (Fasana et al., 2010). Furthermore, recent studies have identified mutations and a functional association between sigma non-opioid intracellular receptor 1 (SIGMAR1) and both FTD and ALS (Luty et al., 2010; Al-Saif et al., 2011). SIGMAR1 knockdown impairs ER–Golgi vesicular trafficking, ultimately leading to reduced autophagosome–lysosome fusion and reduced autophagy substrate degradation (Vollrath et al., 2014). Furthermore, it is worth mentioning how interplay between anterograde transport along the secretory pathway and the autophagosome/lysosome-dependent clearance of disease-causing proteins has also been reported in AD and HD experimental disease models (Lee et al., 2010; Renna et al., 2011).
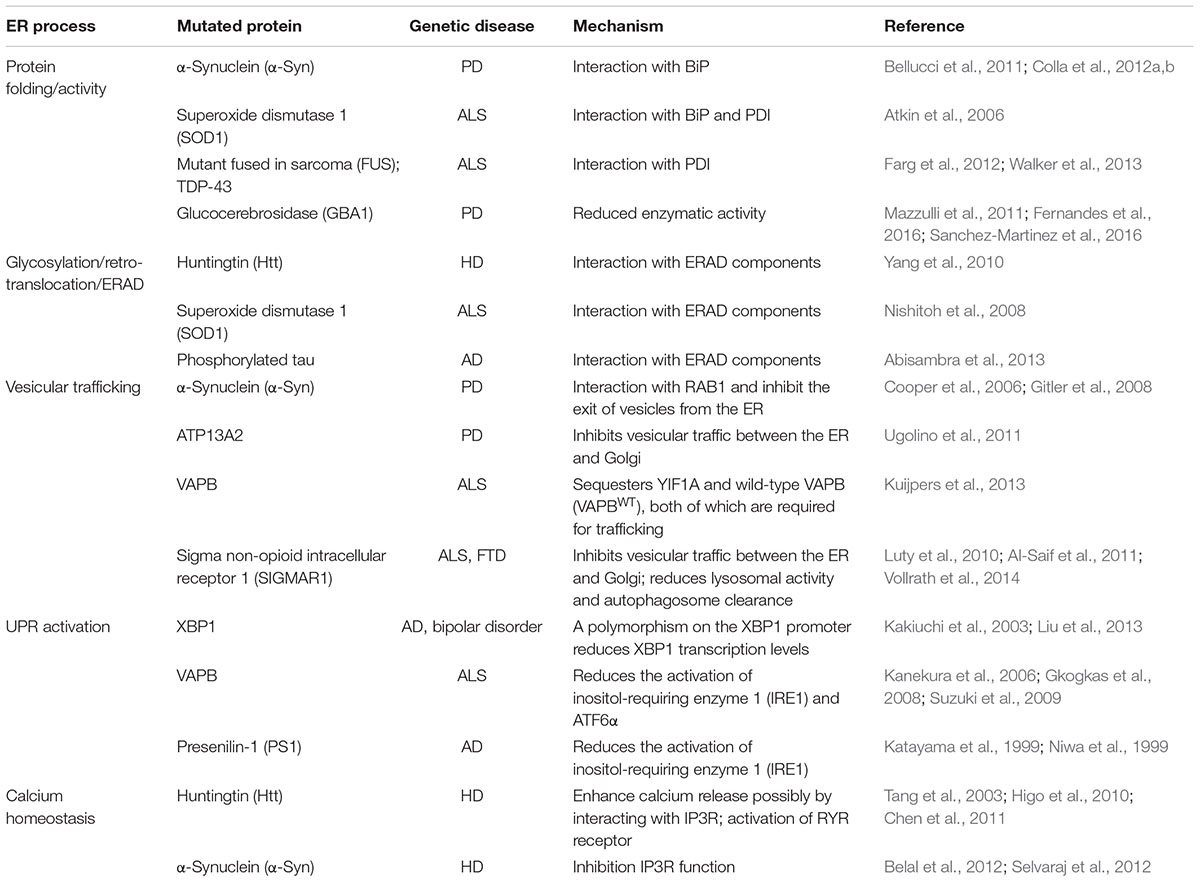
TABLE 1. Disturbance of ER homeostasis as a conduit to neurodegenerative diseases.
Hence, and despite the fact that the mechanisms have not been fully delineated, it is clear how the fine regulation of membrane trafficking events are intimately connected with general proteostasis of the ER. Consequently, these alterations can affect the ER and ultimately contribute to neurodegeneration.
To summarize, both experimental and clinical evidence support the causative role of three UPR branches in the pathogenesis of neurodegenerative diseases (Table 1). As UPR activation and impaired synaptic plasticity occur across a spectrum of neurodegenerative disorders, it presents an attractive therapeutic target in these increasingly prevalent diseases. However, based on the nature of the disease, the sequence of events mediated by the aggregated proteins can differ greatly.
For example, in AD, Aβ is the primary source of ER stress, whereas in PD accumulated α-syn first induces cellular responses such as reactive oxygen species (ROS) generation and oxidative stress, which in turn mediate secondary ER stress, leading to disease pathogenesis. Similarly, these studies suggest how the three individual UPR modules can exert different effects, ranging from an adaptive or protective response to apoptosis, depending on the disease. A better understanding of the specific response in mediating aberrant protein aggregation is thus critical for dissecting the mechanism(s) underlying disease pathogenesis and for future development of therapeutic targets. Nevertheless, as outlined in the following sections, the three arms of the UPR pathway represent potential targets for treating neurodegenerative diseases.
Impaired UPR Machinery as a Cause of Neurodegeneration
So far, few reports have provided evidence of a direct correlation between defective UPR effectors and neurodegeneration. Initial studies have suggested that presenilin-1 possibly inhibits IRE1 function (Katayama et al., 1999; Niwa et al., 1999), while XBP1 promoter polymorphism has been indicated as an AD risk factor (Liu et al., 2013) and for the development of bipolar disorders and schizophrenia in a genetic association study (Kakiuchi et al., 2003). Consistent with this, a more recent study has further reinforced the evidence that the IRE1/XBP1 signaling axis can exacerbate AD pathogenesis (Duran-Aniotz et al., 2017).
Interestingly, the existence of a possible role played by ER stress and UPR modulators in the pathogenesis of clinically relevant conditions such as bipolar disorder and schizophrenia is further highlighted by two other genetic association studies, where mutations or SNPs have been identified in the genes encoding the ER-resident chaperones calreticulin and GRP94/GP96 (Aghajani et al., 2006; Kakiuchi et al., 2007). Also, PDI variants (Kwok et al., 2013), UBQLN2 and p62/SQSTM1 mutations participate in ALS development (Deng et al., 2011; Teyssou et al., 2013). Consistent with this, the ER-resident chaperone BIP/GRP78 has been recently shown to be able to reduce protease-resistant PrP (PrPSc) accumulation and propagation in vitro and in vivo, suggesting that modulating its levels/activity may offer a novel opportunity for designing therapeutic approaches for these diseases (Park et al., 2017).
Furthermore, disease-associated proteins can interact with integral UPR components thus interfering with the signaling system. For example, the ALS-linked VAPB variant interacts with and inhibits ATF6 transcriptional activity (Gkogkas et al., 2008) and IRE1- and XBP1-mediated signaling activity, and ultimately influences cell susceptibility to ER stress (Kanekura et al., 2006; Suzuki et al., 2009). Conversely, mutant HTT expression selectively inhibits ATF6 but not the other UPR transducers (Fernandez-Fernandez et al., 2011). Similarly, manipulation of the PD-associated LRRK2 homolog in Caenorhabditis elegans led to altered BIP/GRP78 expression and high susceptibility to ER stress, while a mutant LRRK2 induced ER stress-dependent apoptotic cell death (Samann et al., 2009; Yuan et al., 2011).
Collectively, speculating how strategies aimed at alleviating ER stress and restoring ER proteostasis may have much potential benefit on the outcomes of neurodegenerative diseases is conceivable. However, they also imply how the possible relations between UPR impairment and the pathogenesis of some neurodegenerative diseases are complex and not necessarily univocal. On one hand, the precise mechanisms still require delineation in many cases. In particular, identifying the optimal target(s) to reduce ER stress in each neurodegenerative disease is of fundamental importance. On the other hand, it is worth mentioning that different experimental models can also account for the apparently contradictory (or unexpected) effects reported, implying that a thorough analysis of the pathological state is necessary for predicting and minimizing any possible side effect deriving from experimental manipulation of the UPR.
Impairment of ER Ca2+ Homeostasis and Er/Mitochondria Interplay in Neurodegenerative Diseases
Along with the mitochondria, the ER represents the major intracellular Ca2+ store. Many ER resident proteins require Ca2+ to preserve their function (Berridge et al., 2003; Krantic et al., 2005; Clapham, 2007). ER Ca2+ depletion leads to either down-regulation or overexpression of ER-resident factors (Amodio et al., 2011, 2016). Indeed, excessive Ca2+ efflux from ER stores has been reported in several pathologies of the central nervous system, including AD and PD (Mekahli et al., 2011; Rosenberg and Spitzer, 2011).
Despite the existence of different indirect means of inducing Ca2+ release from the ER via signaling events, direct ER Ca2+ channel perturbation has been associated with PMDs. For example, the non-physiological association between HTT and IP3R results in altered ER Ca2+ homeostasis (Tang et al., 2003; Higo et al., 2010). Similarly, RyR inhibition conferred significant neuroprotection in animal models of HD (Chen et al., 2011). In PD, α-syn over-expression impaired ER Ca2+ homeostasis and led to IP3R and RyR degradation (Belal et al., 2012). This observation agrees with the altered Ca2+ channel expression that has been described in both PD animal models and in tissues from patients with PD (Belal et al., 2012; Selvaraj et al., 2012). Finally, by sensitizing cells to ER stress, the over-expression of misfolded PrP drastically decreases ER Ca2+ availability (Torres et al., 2010).
Another important mechanism for regulating Ca2+ homeostasis is guaranteed by crosstalk between SERCA and store-operated Ca2+ entry (SOCE). In this context, the stromal interaction molecule proteins (STIMs) are fundamental SOCE regulators and are essential contributors to Ca2+ levels and homeostasis (Soboloff et al., 2012). Hence, and considering the high neuronal sensitivity to ER Ca2+ store perturbations, SOCE has been proposed as a potential target for gaining neuroprotection by reversing the ER stress-induced damage that often precedes neurodegeneration. Interestingly, a recent study has shown how STIM1 over-expression can reconstitute SOCE and confer protection against ER and oxidative stress induced by SERCA pharmacological blockade (Zhang and Thomas, 2016). These findings thus suggest that SOCE could indeed represent an additional mediator of neuroprotection, in particular under conditions where Ca2+ imbalance elicits ER stress (Zhang and Thomas, 2016).
Over previous years, ER and mitochondria contact sites have attracted much attention in the context of neurodegenerative disorders (McInnes, 2013). Neurons are critically dependent on mitochondria-associated membranes as a means of exchanging metabolites and signaling molecules between these organelles. Therefore, the processes affected by the ER and mitochondria contact sites are widely implicated in neurodegeneration, and an increasing number of disease-associated proteins have been reported to physically associate with the ER-mitochondria interface and cause structural and/or functional perturbations of this compartment (Krols et al., 2016; Erpapazoglou et al., 2017).
In AD Aβ affects both the ER and mitochondria and disrupts cellular Ca2+ homeostasis, and mitochondrial dysfunction is one of the main pathological events in AD. The mitochondria accumulate Aβ derived from the ER or Golgi compartment or from the ER–mitochondria contact sites, also known as mitochondria-associated ER-membranes (MAM) and synaptic mitochondria show very high vulnerability suggesting that the translocation of misfolded proteins to the mitochondrial membrane might play an important role in either triggering or perpetuating neurodegeneration (Schreiner et al., 2015).
In this context, MAM play a fundamental role in ER-mitochondria crosstalk, and mitochondria along with the UPR transducers and ER-localized Ca2+ receptors could represent a potential target for AD treatment (Volgyi et al., 2015; Penke et al., 2016).
Targeting UPR as a Potential Therapeutic Tool in Neurodegenerative Disorders
The UPR pathway can represent a valuable therapeutic target for controlling the ER stress response associated with a growing group of clinically relevant conditions, including neurodegenerative diseases. In recent years, many chemical compounds and small molecules capable of alleviating or inducing ER stress through different mechanisms have been identified (Wang and Kaufman, 2014) (summarized in Table 2).
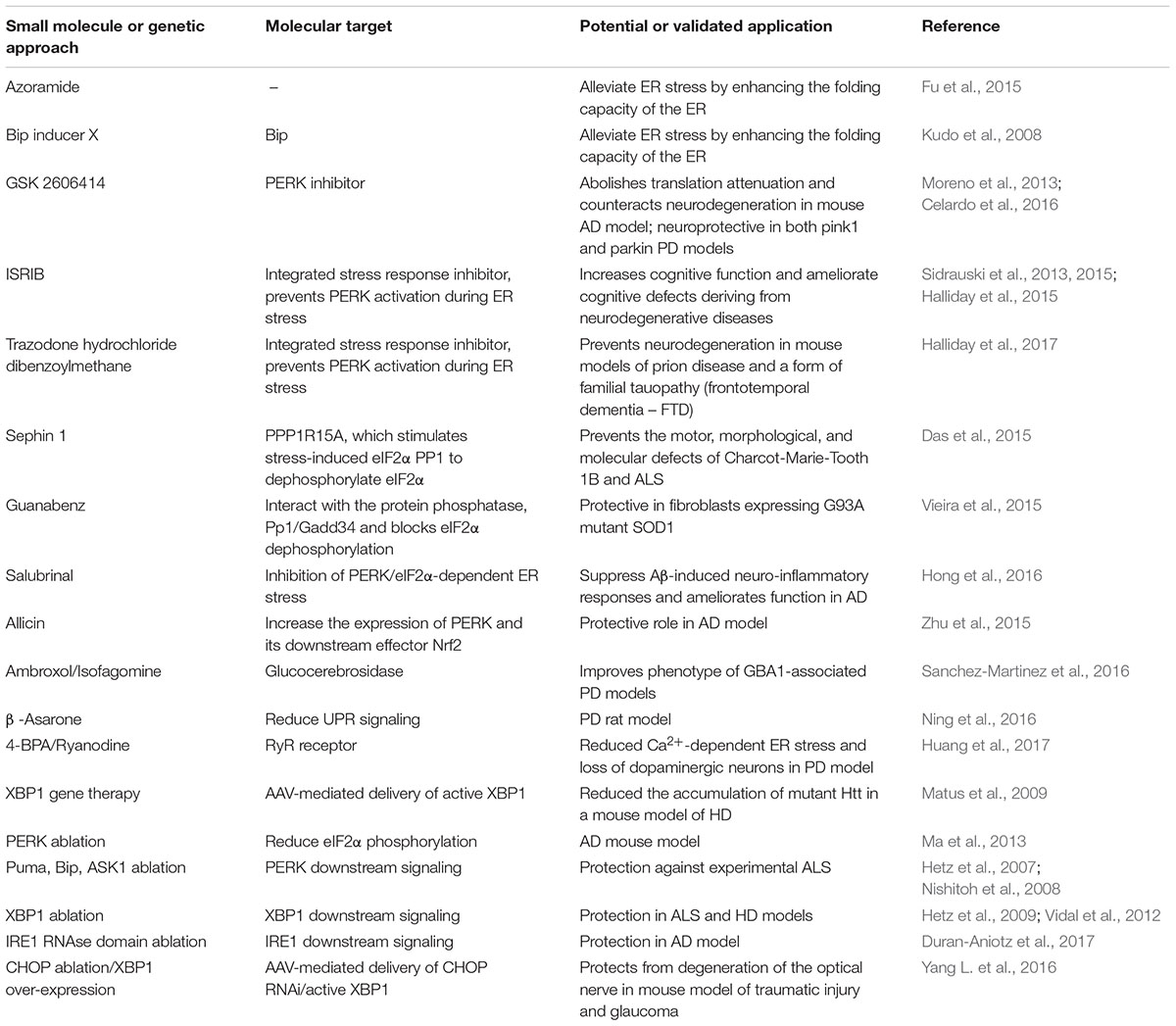
TABLE 2. ER stress and UPR pathways as therapeutic targets.
Chemical chaperones such as the antidiabetic compound azoramide (Fu et al., 2015) and BIP/GRP78 inducer X (BiX) (Kudo et al., 2008) can reduce ER stress by improving its folding capability. ER stress might also be relieved in vivo by gene therapy-based approaches. This is the case, for example, of active Xbp1, which substantially reduced the accumulation of mutant HTT in a HD mouse model (Matus et al., 2009).
Evidence from several experiments has revealed that the PERK/eIF2α phosphorylation axis can be considered the main pathway in the occurrence of neurodegenerative diseases, in which there is either lost or increased PERK function. Loss of PERK function due to PERK gene mutation occurs in a rare neurodegenerative disorder known as Wolcott–Rallison syndrome; immunohistochemical analyses have shown that the PERK deficiency generates peculiar pathological changes whose features resemble those observed in neurodegenerative conditions (Bruch et al., 2015).
Instead, the PERK signaling pathway is hyperactive in the majority of PMDs. Notably, a recent study recovered memory impairment via brain-specific ablation of PERK expression in an in vivo model of AD (Ma et al., 2013).
General protein synthesis attenuation has similar effects on neuronal functions, also via other kinases that can induce eIF2α phosphorylation. For example, GCN2 (general control non-derepressible 2 serine/threonine kinase 2), PKR (protein kinase RNA-activated) and HRI (heme-regulated eIF2α kinase) can also phosphorylate eIF2α at serine51, activate the selective translation of the BACE1 and ATF4 mRNAs and generate memory and synaptic plasticity repression. Moreover, a recent investigation suggested that phosphorylation of eIF2α in AD is modulated by PKR, rather than PERK (Lourenco et al., 2013). In an AD mouse model, GCN2 deficiency caused BACE1 and ATF4 over-expression and consequently greater formation of senile plaques. Moreover, PERK activation was considerably increased in response to GCN2 deletion, suggesting that eIF2α phosphorylation might induce senile plaque accumulation in AD brains by attenuating protein synthesis (Devi and Ohno, 2013).
Thus, PERK-specific inhibitors such as GSK2606414 abolished translation attenuation and counteract neurodegeneration in a mouse model of PrD-related neurodegeneration (Moreno et al., 2013). The high selectivity of GSK2606414 and its ability to cross the blood–brain barrier render such a molecule a promising therapeutic tool for neurodegenerative disorders. Therefore, they are becoming an attractive model to investigate the self-propagating properties of protein misfolding in neurodegenerative diseases, a common feature of AD, PD and ALS (Goedert et al., 2010; Soto, 2012).
Similarly, ISRIB (integrated stress response inhibitor) can prevent PERK activation under persistent ER stress (Sidrauski et al., 2013). ISRIB is a potent drug-like small molecule that retains the ability to inhibit eIF2α phosphorylation, and such an effect correlates well with increased cognitive function in rodents. Moreover, ISRIB inhibition of the PERK pathway significantly reduces ATF4 expression and consequently that of its target gene CHOP; a similar inhibitory effect of ISRIB has been shown on two other eIF2α kinases: GCN2 and HRI (Sidrauski et al., 2013). This observation is of particular importance as ATF4 is a repressor of memory and long-term synaptic plasticity. Moreover, recent studies have revealed that ISRIB activates eIF2β by stabilizing eIF2β-activated dimers, suggesting that by modulating eIF2β function, ISRIB is a promising instrument for ameliorating the cognitive defects that develop from neurodegenerative diseases (Sidrauski et al., 2015). ISRIB has also been proven effective in a mouse model of prion-related disease with reduced toxicity as compared to the PERK inhibitor GSK2606414 (Halliday et al., 2015). Consistent with this, a more recent study where a library of 1040 FDA-approved drugs was screened for novel disease modulators revealed how trazodone hydrochloride, a licensed antidepressant, and dibenzoylmethane, a compound currently under clinical trial as an anti-cancer drug, respectively, prevented emergence of the signs of brain cell damage in prion disease and restored memory in FTD mouse models (Halliday et al., 2017).
PPP1R15A is another eIF2α phosphorylation regulator that stimulates stress-induced eIF2α PP1 (serine/threonine protein phosphatase) to dephosphorylate eIF2α. Thus the inhibition of PPP1R15A, which prolongs eIF2α phosphorylation, could confer beneficial effects for treating PMD. Recently Sephin1, a small molecule derivative of the hypotensive drug guanabenz (GBZ) was synthesized. GBZ, a well-known α2 adrenergic receptor agonist, also interacts with PP1/GADD34, selectively blocking eIF2α dephosphorylation. Indeed, GBZ treatment protected fibroblasts bearing the SOD1 G93A mutation subjected to the ER stress (Vieira et al., 2015). As GBZ retains high affinity for α adrenergic receptors, its inhibition of UPR signaling cannot be considered exclusive. Further pharmacological studies have paved the way for the development of additional lead compounds. GBZ can bind and inhibit the PPP1R15A/PP1c complex, which is regulated in response to stress eIF2α phosphatase activity. This has led to the identification of Sephin1, which inhibits the PP1 regulatory subunit in vivo by specifically binding the PPP1R15A subunit of the complex. Moreover, Sephin1 prevented the molecular and neurological defects in Charcot-Marie-Tooth 1B and ALS models (Das et al., 2015).
Recent works in AD mouse models have suggested that a specific glutamate receptor (mGluR5) isoform may operate as a molecular target for extracellular Aβ–regulated long-term depression (LTD), synaptic plasticity and Aβ synaptic toxicity. In particular, the mGluR-LTD inhibition observed in animal models of AD could be antagonized by suppressing the PERK pathway. These data reinforce the concept that reversing the inhibition of protein synthesis mediated by the PERK/eIF2α axis may alleviate some cognitive deficits observed in, at least some, neurodegenerative conditions (Yang W. et al., 2016).
The PERK/eIF2α-dependent UPR branch is also involved in the Aβ-induced neuroinflammatory responses in astrocytes. In the same context, different compounds suppressed Aβ-induced neuroinflammation. For example, in Aβ-induced astrocytes, salubrinal inhibition of PERK/eIF2α-dependent ER stress and exposure to the neurosteroid progesterone (PG) significantly suppressed neuroinflammation by down-regulating PERK/eIF2α signaling (Hong et al., 2016). Natural compounds can also have protective activity in AD models. For example, allicin, a natural garlic extract, had a protective effect in a rat model of AD, as demonstrated by its ability to moderately increase the expression levels of both PERK and its downstream substrate NFR2 (Zhu et al., 2015).
By contrast, activation of the IRE1 UPR branch appears to benefit AD pathogenesis, as shown in an AD model of Drosophila melanogaster, in which Xbp1 over-expression prevented Aβ toxicity (Casas-Tinto et al., 2011), as in a similar study in C. elegans, indicating that XBP1 is involved in defense against Aβ toxicity, presumably by increased autophagy (Safra et al., 2013). However, a recent study has shown a positive correlation between IRE1/XBP1 signaling axis activity and AD pathogenesis (Duran-Aniotz et al., 2017). Contrary to the initial expectations that IRE1 signaling may protect against AD, genetic ablation of the RNAse domain of IRE1 in the nervous system significantly reduced APP expression and amyloid deposition, Aβ oligomer content and astrocyte activation. At molecular level, inhibiting IRE1 downstream signaling reduces the APP steady-state levels via its retention in the ER, followed by proteasome-mediated degradation.
Collectively, these findings reveal a so-far unanticipated role of IRE1 in AD pathogenesis, providing a novel therapeutic intervention target (Duran-Aniotz et al., 2017). Furthermore, inhibition of the IRE1/XBP1-mediated branch of ER stress by the natural compound β-asarone improved the phenotype in a rat model of PD (Ning et al., 2016).
In PD, PERK/eIF2α pathway activation represents the main cause of dopaminergic neuron loss in patients although there is little evidence of UPR activation from postmortem studies of patients with PD (Scheper and Hoozemans, 2015). Despite this, a recent work using a Parkinson-like disease mouse model has reported how sustained gene transfer–mediated ATF4 up-regulation in the dopaminergic neurons of the substantia nigra resulted in severe and caspase 3/7-dependent nigrostriatal degeneration, confirming that the PERK UPR branch plays an essential role in PD pathogenesis by activating dopaminergic neuronal loss (Gully et al., 2016). On the other hand, it is worth mentioning how genetic ATF6 deletion potentiates the susceptibility to PD-inducing neurotoxins in PD experimental mouse models (Egawa et al., 2011; Hashida et al., 2012), while Bip/Grp78 over-expression exerted neuroprotective effects (Gorbatyuk et al., 2012).
In PINK1- and Parkin-associated PD models, mitofusins cause enhanced ER stress signaling, acting as a molecular bridge interconnecting mitochondria damaged by mutant PINK1 and Parkin proteins to the ER membranes. PERK signaling suppression has a neuroprotective effect by reducing mitofusin contact with the ER. Furthermore, either pharmacological (the GSK2606414 PERK inhibitor) or genetic inhibition of the PERK pathway was beneficial in these experimental models of PD (Celardo et al., 2016).
Furthermore, a potential role for the RYR receptor in the dopaminergic neurons loss in the context of PD pathogenesis has been proposed in a recent study (Huang et al., 2017). In particular, the detrimental increase in cytoplasmic Ca2+ levels induced by 6-hydroxydopamine (6-OHDA) (a toxin capable to generate, in vitro and in vivo, some features of PD-associated neurodegeneration) can be antagonized by pretreatment with the ER stress inhibitor 4-PBA or ryanodine, which acts as a blocker on the eponymous receptor (RYR). Notably, the pretreatment with 4-PBA and ryanodine proved to be effective also in DA neurons derived from midbrain sections of 6-OHDA-treated rat, therefore suggesting suggest a potential therapeutic strategy for PD by inhibiting the RyRs Ca2+ channels on the ER (Huang et al., 2017).
Concerning more specifically the familial form of PD associated with GBA gene mutations, a promising study has revealed the efficacy of two small molecules with chaperone activity, namely ambroxol and isofagomine (Sanchez-Martinez et al., 2016). In particular, both chaperones increased GBA levels and activity in fly models and in fibroblasts from patients with PD. Treatment with both chaperones reduced ER stress and prevented motor function loss, proof of principle that small molecule chaperones can reverse mutant GBA-mediated ER stress in vivo and might prove effective for treating PD (Sanchez-Martinez et al., 2016).
Remarkably, in ALS, genetic UPR manipulation and pharmacological approaches have revealed complex involvement of the pathways, illuminating distinct outputs of specific UPR signaling branches in the same disease (Matus et al., 2011). Importantly, in ALS mouse models, chronic ER stress represents one of the earliest pathological events in motor neurons before the appearance of ALS symptoms (Saxena et al., 2009).
According to the experimental evidence collected so far, targeting PERK signaling exerts a dual role in ALS. Indeed, eIF2α phosphorylation has a protective effect (Saxena et al., 2009; Wang et al., 2011), whereas ATF4 over-expression and the consequential up-regulation of the proapoptotic factors CHOP and BIM has definite detrimental effects (Matus et al., 2013). Targeting XBP1 protects cells against ALS by up-regulating autophagy, which controls the elimination of mutant SOD1 aggregates (Hetz et al., 2009). Similar results were reported in an XBP1 knockout mouse model of HD, in which up-regulated FOXO1, an important transcription factor controlling the early-stage activation of several autophagy genes, was observed (Vidal et al., 2012).
Another example of PERK/eIF2α involvement is the prion-related disorders (PrDs), in which prion infection increases eIF2α phosphorylation, which in turn reduces the expression of relevant synaptic proteins (Moreno et al., 2012). More interestingly, in animal models, a PERK inhibitor counteracted the pathophysiological role of eIF2α phosphorylation, protecting animals against PrD-caused neurodegeneration (Moreno et al., 2013). By contrast, either XBP1 or caspase-12 depletion did not affect disease progression and pathophysiology in models of PrD infection (Steele et al., 2007; Hetz et al., 2008). Of further note, a recent work has shown how the AAV-mediated combinatorial modulation of CHOP and active XBP1s has proved beneficial in rescuing the neurodegeneration of the optical nerve associated to both traumatic injury and glaucoma, thus expanding the potential application of UPR modulation to other classes of neurodegenerative conditions (Yang L. et al., 2016).
Altogether, these findings reinforce the idea of the complexity of UPR and how this pathway can affect certain diseases, which are largely influenced by flexible and versatile overlap between the selective UPR signaling branches, their downstream modules, and a number of genetic and/or environmental co-contributing factors. Moreover, they illustrate and strongly inform the need for thorough and accurate evaluation of the contribution of each individual UPR signaling component in PMDs, which will aid the definition of optimal targets for therapeutic intervention.
Concluding Remarks and Future Directions
The identification of pharmacological therapies for neurodegenerative diseases remains an ambitious challenge in biomedical science and will become even more pressing in the future in view of the increased life expectancy of the aging population. The connections between ER stress, UPR and the pathogenesis of neurodegenerative disorders have been extensively studied in various disease models. These functional studies reinforce the concept that UPR can selectively modulate specific cellular events and this can in turn affect PMD progression through distinct mechanisms. Despite gaining numerous insights into the adaptive mechanisms for counterbalancing protein misfolding both at cellular and physiological level, many interesting aspects still have not been elucidated.
In this context, the role of UPR in the flexible control of ER proteostasis could be harnessed to find strategies aimed at attenuating or delaying the onset of neurodegenerative diseases. Indeed, prolonged albeit mild ER stress can induce an adaptive response, termed ER hormesis, which entails an UPR-dependent increase in ER proteostasis (Matus et al., 2012; Mollereau, 2013). However, additional studies have shown that this cellular response also involves autophagy, which decreases protein aggregates and cell death (Cullinan et al., 2003; Fouillet et al., 2012). For example, low dosages of tunicamycin attenuated neuronal degeneration by inducing the IRE1–XBP1 UPR axis (Fouillet et al., 2012). Similarly, neuronal ablation of Xbp1 delayed in an autophagy-dependent manner, disease progression associated with SOD1 or mutant HTT (Matus et al., 2009; Vidal et al., 2012). Thus, it is conceivable that enhancing the adaptive mechanisms by slight induction of specific UPR pathways may offer sufficient protection against neurotoxicity.
Collectively, these studies strongly reinforce the concept that strategies aimed at attenuating ER stress levels may impact neurodegenerative diseases positively. As such, better understanding of the pathological state for each specific clinical condition is critical if the aim is to target UPR for treating neurodegeneration. Therefore, further studies will be needed to: (a) identify the optimal targets for attenuating the ER stress response to each specific clinical condition; (b) develop novel drugs; (c) predict and define possible side effects derived from UPR perturbations.
In this regard, both novel molecules and repurposed compounds have in the past few years been proven effective in different models of neurodegenerative diseases (Hetz et al., 2013). Although these findings are all of great interest per se, the identification of additional mechanisms capable of driving and modulating UPR signaling in health and disease will shed further light on the regulation of such a fundamental biological response and will hopefully lead to the identification of novel therapeutic targets.
Author Contributions
MR and PR conceived the work, prepared the figures, wrote and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
MR would like to thank Prof. David C. Rubinsztein (Cambridge Institute for Medical Research, University of Cambridge) for the scientific and financial support (Wellcome Trust, Grant No.: 095317/Z/11/Z). MR and PR would like to thank Prof. Stefano Bonatti (Department of Molecular Medicine and Medical Biotechnology, University of Naples “Federico II”) for critical reading of the manuscript and for the helpful discussion. The revised version of the manuscript has been subjected to professional editing and proofreading service, provided by Cambridge Proofreading LLC (http://proofreading.org).
References
Abisambra, J. F., Jinwal, U. K., Blair, L. J., O’Leary, J. C. I. I. I., Li, Q., Brady, S., et al. (2013). Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 33, 9498–9507. doi: 10.1523/JNEUROSCI.5397-12.2013
Acosta-Alvear, D., Zhou, Y., Blais, A., Tsikitis, M., Lents, N. H., Arias, C., et al. (2007). XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 27, 53–66. doi: 10.1016/j.molcel.2007.06.011
Adolph, T. E., Tomczak, M. F., Niederreiter, L., Ko, H. J., Bock, J., Martinez-Naves, E., et al. (2013). Paneth cells as a site of origin for intestinal inflammation. Nature 503, 272–276. doi: 10.1038/nature12599
Aghajani, A., Rahimi, A., Fadai, F., Ebrahimi, A., Najmabadi, H., and Ohadi, M. (2006). A point mutation at the calreticulin gene core promoter conserved sequence in a case of schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141B, 294–295. doi: 10.1002/ajmg.b.30300
Al-Saif, A., Al-Mohanna, F., and Bohlega, S. (2011). A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 70, 913–919. doi: 10.1002/ana.22534
Amodio, G., Moltedo, O., Monteleone, F., D’Ambrosio, C., Scaloni, A., Remondelli, P., et al. (2011). Proteomic signatures in thapsigargin-treated hepatoma cells. Chem. Res. Toxicol. 24, 1215–1222. doi: 10.1021/tx200109y
Amodio, G., Renna, M., Paladino, S., Venturi, C., Tacchetti, C., Moltedo, O., et al. (2009). Endoplasmic Reticulum stress reduces the export from the ER and alters the architecture of post-ER compartments. Int. J. Biochem. Cell Biol. 41, 2511–2521. doi: 10.1016/j.biocel.2009.08.006
Amodio, G., Sasso, E., D’Ambrosio, C., Scaloni, A., Moltedo, O., Franceschelli, S., et al. (2016). Remondelli P. Identification of a microRNA (miR-663a) induced by ER stress and its target gene PLOD3 by a combined microRNome and proteome approach. Cell Biol. Toxicol. 32, 285–303. doi: 10.1007/s10565-016-9335-z
Amodio, G., Venditti, R., De Matteis, M. A., Moltedo, O., Pignataro, P., and Remondelli, P. (2013). Endoplasmic reticulum stress reduces COPII vesicle formation and modifies Sec23a cycling at ERESs. FEBS Lett. 587, 3261–3266. doi: 10.1016/j.febslet.2013.08.021
Andersen, P. M., and Al-Chalabi, A. (2011). Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 7, 603–615. doi: 10.1038/nrneurol.2011.150
Arrasate, M., and Finkbeiner, S. (2012). Protein aggregates in Huntington’s disease. Exp. Neurol. 238, 1–11. doi: 10.1016/j.expneurol.2011.12.013
Atkin, J. D., Farg, M. A., Turner, B. J., Tomas, D., Lysaght, J. A., Nunan, J., et al. (2006). Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem. 281, 30152–30165. doi: 10.1074/jbc.M603393200
Balch, W. E., Morimoto, R. I., Dillin, A., and Kelly, J. W. (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. doi: 10.1126/science.1141448
B’Chir, W., Maurin, A. C., Carraro, V., Averous, J., Jousse, C., Muranishi, Y., et al. (2013). The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41, 7683–7699. doi: 10.1093/nar/gkt563
Belal, C., Ameli, N. J., El Kommos, A., Bezalel, S., Al’Khafaji, A. M., Mughal, M. R., et al. (2012). The homocysteine-inducible endoplasmic reticulum (ER) stress protein Herp counteracts mutant α-synuclein-induced ER stress via the homeostatic regulation of ER-resident calcium release channel proteins. Hum. Mol. Genet. 21, 963–977. doi: 10.1093/hmg/ddr502
Bellucci, A., Navarria, L., Zaltieri, M., Falarti, E., Bodei, S., Sigala, S., et al. (2011). Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson’s disease. J. Neurochem. 116, 588–605. doi: 10.1111/j.1471-4159.2010.07143.x
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodeling. Nat. Rev. Mol. Cell Biol. 4, 517–529. doi: 10.1038/nrm1155
Blais, J. D., Filipenko, V., Bi, M., Harding, H. P., Ron, D., Koumenis, C., et al. (2004). Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol. Cell. Biol. 24, 7469–7482. doi: 10.1128/MCB.24.17.7469-7482.2004
Bouman, L., Schlierf, A., Lutz, A. K., Shan, J., Deinlein, A., Kast, J., et al. (2011). Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 18, 769–782. doi: 10.1038/cdd.2010.142
Bruch, J., Kurz, C., Vasiljevic, A., Nicolino, M., Arzberger, T., and Höglinger, G. U. (2015). Early Neurodegeneration in the Brain of a Child Without Functional PKR-like Endoplasmic Reticulum Kinase. J. Neuropathol. Exp. Neurol. 74, 850–857. doi: 10.1097/NEN.0000000000000224
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., et al. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96. doi: 10.1038/415092a
Casas-Tinto, S., Zhang, Y., Sanchez-Garcia, J., Gomez-Velazquez, M., Rincon-Limas, D. E., and Fernandez-Funez, P. (2011). The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet. 20, 2144–2160. doi: 10.1093/hmg/ddr100
Celardo, I., Costa, A. C., Lehmann, S., Jones, C., Wood, N., Mencacci, N. E., et al. (2016). Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 7, e2271. doi: 10.1038/cddis.2016.173
Chen, S., Zhang, X., Song, L., and Le, W. (2012). Autophagy dysregulation in amyotrophic lateral sclerosis. Brain Pathol. 22, 110–116. doi: 10.1111/j.1750-3639.2011.00546.x
Chen, X., Wu, J., Lvovskaya, S., Herndon, E., Supnet, C., and Bezprozvanny, I. (2011). Dantrolene is neuroprotective in Huntington’s disease transgenic mouse model. Mol. Neurodegener. 6, 81. doi: 10.1186/1750-1326-6-81
Cheroni, C., Marino, M., Tortarolo, M., Veglianese, P., De Biasi, S., Fontana, E., et al. (2009). Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 18, 82–96. doi: 10.1093/hmg/ddn319
Chung, S. Y., Kishinevsky, S., Mazzulli, J. R., Graziotto, J., Mrejeru, A., Mosharov, E. V., et al. (2016). Parkin and PINK1 Patient iPSC-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and alpha-synuclein accumulation. Stem Cell Rep. 7, 664–677. doi: 10.1016/j.stemcr.2016.08.012
Colla, E., Coune, P., Liu, Y., Pletnikova, O., Troncoso, J. C., Iwatsubo, T., et al. (2012a). Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 32, 3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012
Colla, E., Jensen, P. H., Pletnikova, O., Troncoso, J. C., Glabe, C., and Lee, M. K. (2012b). Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J. Neurosci. 32, 3301–3305. doi: 10.1523/JNEUROSCI.5368-11.2012
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., et al. (2006). α-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313, 324–328. doi: 10.1126/science.1129462
Cullinan, S. B., Zhang, D., Hannink, M., Arvisais, E., Kaufman, R. J., and Diehl, J. A. (2003). Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 23, 7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003
Dafinca, R., Scaber, J., Ababneh, N., Lalic, T., Weir, G., Christian, H., et al. (2016). C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis, and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis, and frontotemporal dementia. Stem Cells 34, 2063–2078. doi: 10.1002/stem.2388
Das, I., Krzyzosiak, A., Schneider, K., Wrabetz, L., D’Antonio, M., Barry, N., et al. (2015). Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 348, 239–242. doi: 10.1126/science.aaa4484
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
del Toro, D., Canals, J. M., Ginés, S., Kojima, M., Egea, G., and Alberch, J. (2006). Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J. Neurosci. 26, 12748–12757. doi: 10.1523/JNEUROSCI.3873-06.2006
Deng, H. X., Chen, W., Hong, S. T., Boycott, K. M., Gorrie, G. H., Siddique, N., et al. (2011). Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211–215. doi: 10.1038/nature10353
Devi, L., and Ohno, M. (2013). Deletion of the eIF2α Kinase GCN2 fails to rescue the memory decline associated with Alzheimer’s disease. PLoS ONE 8:e77335. doi: 10.1371/journal.pone.0077335
Domingues, S. C., Henriques, A. G., Wu, W., Da Cruz e Silva, E. F., and Da Cruz e Silva, O. A. (2007). Altered subcellular distribution of the Alzheimer’s amyloid precursor protein under stress conditions. Ann. N. Y. Acad. Sci 1096, 184–195. doi: 10.1196/annals.1397.085
Duennwald, M. L., and Lindquist, S. (2008). Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 22, 3308–3319. doi: 10.1101/gad.1673408
Duplan, E., Giaime, E., Viotti, J., Sévalle, J., Corti, O., Brice, A., et al. (2013). ER-stress-associated functional link between Parkin and DJ-1 via a transcriptional cascade involving the tumor suppressor p53 and the spliced X-box binding protein XBP-1. J. Cell Sci. 126, 2124–2133. doi: 10.1242/jcs.127340
Duran-Aniotz, C., Cornejo, V. H., Espinoza, S., Ardiles,ÁO., Medinas, D. B., Salazar, C., et al. (2017). IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. doi: 10.1007/s00401-017-1694-x [Epub ahead of print].
Egawa, N., Yamamoto, K., Inoue, H., Hikawa, R., Nishi, K., Mori, K., et al. (2011). The endoplasmic reticulum stress sensor, ATF6α, protects against neurotoxin-induced dopaminergic neuronal death. J. Biol. Chem. 286, 7947–7957. doi: 10.1074/jbc.M110.156430
Erpapazoglou, Z., Mouton-Liger, F., and Corti, O. (2017). From dysfunctional endoplasmic reticulum-mitochondria coupling to neurodegeneration. Neurochem. Int. doi: 10.1016/j.neuint.2017.03.021 [Epub ahead of print].
Farg, M. A., Soo, K. Y., Walker, A. K., Pham, H., Orian, J., Horne, M. K., et al. (2012). Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol. Aging 33, 2855–2868. doi: 10.1016/j.neurobiolaging.2012.02.009
Farg, M. A., Sundaramoorthy, V., Sultana, J. M., Yang, S., Atkinson, R. A., Levina, V., et al. (2014). C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 23, 3579–3595. doi: 10.1093/hmg/ddu068
Fasana, E., Fossati, M., Ruggiano, A., Brambillasca, S., Hoogenraad, C. C., Navone, F., et al. (2010). A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 24, 1419–1430. doi: 10.1096/fj.09-147850
Fecto, F., and Siddique, T. (2011). Making connections: pathology and genetics link amyotrophic lateral sclerosis with frontotemporal lobe dementia. J. Mol. Neurosci. 45, 663–675. doi: 10.1007/s12031-011-9637-9
Fernandes, H. J., Hartfield, E. M., Christian, H. C., Emmanoulidou, E., Zheng, Y., Booth, H., et al. (2016). ER stress and autophagic perturbations lead to elevated extracellular alpha-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 6, 342–356. doi: 10.1016/j.stemcr.2016.01.013
Fernandez-Fernandez, M. R., Ferrer, I., and Lucas, J. J. (2011). Impaired ATF6α processing, decreased Rheb and neuronal cell cycle re-entry in Huntington’s disease. Neurobiol. Dis. 41, 23–32. doi: 10.1016/j.nbd.2010.08.014
Fouillet, A., Levet, C., Virgone, A., Robin, M., Dourlen, P., Rieusset, J., et al. (2012). ER stress inhibits neuronal death by promoting autophagy. Autophagy 8, 915–926. doi: 10.4161/auto.19716
Fu, S., Yalcin, A., Lee, G. Y., Li, P., Fan, J., Arruda, A. P., et al. (2015). Phenotypic assays identify azoramide as a small-molecule modulator of the unfolded protein response with antidiabetic activity. Sci. Transl. Med. 7, 292ra98. doi: 10.1126/scitranslmed.aaa9134
Gitler, A. D., Bevis, B. J., Shorter, J., Strathearn, K. E., Hamamichi, S., Su, L. J., et al. (2008). The Parkinson’s disease protein α-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 145–150. doi: 10.1073/pnas.0710685105
Gkogkas, C., Middleton, S., Kremer, A. M., Wardrope, C., Hannah, M., Gillingwater, T. H., et al. (2008). VAPB interacts with and modulates the activity of ATF6. Hum. Mol. Genet. 17, 1517–1526. doi: 10.1093/hmg/ddn040
Goedert, M., Clavaguera, F., and Tolnay, M. (2010). The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 33, 317–325. doi: 10.1016/j.tins.2010.04.003
Gorbatyuk, M. S., Shabashvili, A., Chen, W., Meyers, C., Sullivan, L. F., Salganik, M., et al. (2012). Glucose regulated protein 78 diminishes α-synuclein neurotoxicity in a rat model of Parkinson disease. Mol. Ther. 20, 1327–1337. doi: 10.1038/mt.2012.28
Green, D. R., and Levine, B. (2014). To be or not to be? How selective autophagy and cell death govern cell fate. Cell 157, 65–75. doi: 10.1016/j.cell.2014.02.049
Gully, J. C., Sergeyev, V. G., Bhootada, Y., Mendez-Gomez, H., Meyers, C. A., Zolotukhin, S., et al. (2016). Up-regulation of activating transcription factor 4 induces severe loss of dopamine nigral neurons in a rat model of Parkinson’s disease. Neurosci. Lett. 627, 36–41. doi: 10.1016/j.neulet.2016.05.039
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Halliday, M., Radford, H., Sekine, Y., Moreno, J., Verity, N., le Quesne, J., et al. (2015). Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 6, e1672. doi: 10.1038/cddis.2015.49
Halliday, M., Radford, H., Zents, K. A. M., Molloy, C., Moreno, J. A., Verity, N. C., et al. (2017). Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 140, 1768–1783. doi: 10.1093/brain/awx074
Harding, H. P., Zhang, Y., and Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274. doi: 10.1038/16729
Hartl, F. U., Bracher, A., and Hayer-Hartl, M. (2011). Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. doi: 10.1038/nature10317
Hashida, K., Kitao, Y., Sudo, H., Awa, Y., Maeda, S., Mori, K., et al. (2012). ATF6α promotes astroglial activation and neuronal survival in a chronic mouse model of Parkinson’s disease. PLoS ONE 7:e47950. doi: 10.1371/journal.pone.0047950
Haze, K., Yoshida, H., Yanagi, H., Yura, T., and Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799. doi: 10.1091/mbc.10.11.3787
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Hetz, C., Chevet, E., and Harding, H. P. (2013). Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 12, 703–719. doi: 10.1038/nrd3976
Hetz, C., Lee, A. H., Gonzalez-Romero, D., Thielen, P., Castilla, J., Soto, C., et al. (2008). Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 757–762. doi: 10.1073/pnas.0711094105
Hetz, C., Martinon, F., Rodriguez, D., and Glimcher, L. H. (2011). The unfolded protein response: integrating stress signals through the stress sensor IRE1α. Physiol. Rev. 91, 1219–1243. doi: 10.1152/physrev.00001.2011
Hetz, C., and Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. doi: 10.1038/nrn3689
Hetz, C., Thielen, P., Fisher, J., Pasinelli, P., Brown, R. H., Korsmeyer, S., et al. (2007). The proapoptotic BCL-2 family member BIM mediates motoneuron loss in a model of amyotrophic lateral sclerosis. Cell Death Differ. 14, 1386–1389. doi: 10.1038/sj.cdd.4402166
Hetz, C., Thielen, P., Matus, S., Nassif, M., Court, F., Kiffin, R., et al. (2009). XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 23, 2294–2306. doi: 10.1101/gad.1830709
Higo, T., Hamada, K., Hisatsune, C., Nukina, N., Hashikawa, T., Hattori, M., et al. (2010). Mechanism of ER stress-induced brain damage by IP3 receptor. Neuron 68, 865–878. doi: 10.1016/j.neuron.2010.11.010
Hong, Y., Wang, X., Sun, S., Xue, G., Li, J., and Hou, Y. (2016). Progesterone exerts neuroprotective effects against Aβ-induced neuroinflammation by attenuating ER stress in astrocytes. Int. Immunopharmacol. 33, 83–89. doi: 10.1016/j.intimp.2016.02.002
Huang, L., Xue, Y., Feng, D., Yang, R., Nie, T., Zhu, G., et al. (2017). Blockade of RyRs in the ER Attenuates 6-OHDA-induced calcium overload, cellular hypo-excitability and apoptosis in dopaminergic neurons. Front. Cell Neurosci. 11:52. doi: 10.3389/fncel.2017.00052
Iwawaki, T., Hosoda, A., Okuda, T., Kamigori, Y., Nomura-Furuwatari, C., Kimata, Y., et al. (2001). Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat. Cell Biol. 3, 158–164. doi: 10.1038/35055065
Kakiuchi, C., Ishiwata, M., Nanko, S., Kunugi, H., Minabe, Y., Nakamura, K., et al. (2007). Association analysis of HSP90B1 with bipolar disorder. J. Hum. Genet. 52, 794–803. doi: 10.1007/s10038-007-0188-4
Kakiuchi, C., Iwamoto, K., Ishiwata, M., Bundo, M., Kasahara, T., Kusumi, I., et al. (2003). Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nat. Genet. 35, 171–175. doi: 10.1038/ng1235
Kanekura, K., Nishimoto, I., Aiso, S., and Matsuoka, M. (2006). Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8). J. Biol. Chem. 281, 30223–30233. doi: 10.1074/jbc.M605049200
Katayama, T., Imaizumi, K., Sato, N., Miyoshi, K., Kudo, T., Hitomi, J., et al. (1999). Presinilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat. Cell Biol. 1, 479–485. doi: 10.1038/70265
Kikuchi, H., Almer, G., Yamashita, S., Guégan, C., Nagai, M., Xu, Z., et al. (2006). Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci. U.S.A. 103, 6025–6030. doi: 10.1073/pnas.0509227103
Knowles, T. P., Vendruscolo, M., and Dobson, C. M. (2014). The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396. doi: 10.1038/nrm3810
Kouroku, Y., Fujita, E., Tanida, I., Ueno, T., Isoai, A., Kumagai, H., et al. (2007). ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 14, 230–239. doi: 10.1038/sj.cdd.4401984
Krantic, S., Mechawar, N., Reix, S., and Quirion, R. (2005). Molecular basis of programmed cell death involved in neurodegeneration. Trends Neurosci. 28, 670–676. doi: 10.1016/j.tins.2005.09.011
Krols, M., van Isterdael, G., Asselbergh, B., Kremer, A., Lippens, S., Timmerman, V., et al. (2016). Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol. 131, 505–523. doi: 10.1007/s00401-015-1528-7
Kudo, T., Kanemoto, S., Hara, H., Morimoto, N., Morihara, T., Kimura, R., et al. (2008). A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 15, 364–375. doi: 10.1038/sj.cdd.4402276
Kuijpers, M., Yu, K. L., Teuling, E., Akhmanova, A., Jaarsma, D., Hoogenraad, C. C., et al. (2013). The ALS8 protein VAPB interacts with the ER–Golgi recycling protein YIF1A and regulates membrane delivery into dendrites. EMBO J. 32, 2056–2072. doi: 10.1038/emboj.2013.131
Kwok, C. T., Morris, A. G., Frampton, J., Smith, B., Shaw, C. E., and de Belleroche, J. (2013). Association studies indicate that protein disulfide isomerase is a risk factor in amyotrophic lateral sclerosis. Free Radic. Biol. Med. 58, 81–86. doi: 10.1016/j.freeradbiomed.2013.01.001
Lee, H., Ahn, H. H., Lee, W., Oh, Y., Choi, H., Shim, S. M., et al. (2016). ENC1 modulates the aggregation and neurotoxicity of mutant huntingtin through p62 Under ER stress. Mol. Neurobiol. 53, 6620–6634. doi: 10.1007/s12035-015-9557-8
Lee, J. H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., and Peterhoff, C. M. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158. doi: 10.1016/j.cell.2010.05.008
Lee, S., Shang, Y., Redmond, S. A., Urisman, A., Tang, A. A., and Li, K. H. (2016). Activation of HIPK2 promotes ER stress-mediated neurodegeneration in amyotrophic lateral sclerosis. Neuron 91, 41–55. doi: 10.1016/j.neuron.2016.05.021
Lindquist, S. L., and Kelly, J. W. (2011). Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases: progress and prognosis. Cold Spring Harb. Perspect. Biol. 3:a004507. doi: 10.1101/cshperspect.a004507
Liu, S. Y., Wang, W., Cai, Z. Y., Yao, L. F., Chen, Z. W., Wang, C. Y., et al. (2013). Polymorphism -116C/G of human X-box-binding protein 1 promoter is associated with risk of Alzheimer’s disease. CNS Neurosci. Ther. 19, 229–234. doi: 10.1111/cns.12064
Liu, Y., Adachi, M., Zhao, S., Hareyama, M., Koong, A. C., Luo, D., et al. (2009). Preventing oxidative stress: a new role for XBP1. Cell Death. Differ. 16, 847–857. doi: 10.1038/cdd.2009.14
Lourenco, M. V., Clarke, J. R., Frozza, R. L., Bomfim, T. R., Forny-Germano, L., Batista, A. F., et al. (2013). TNF -α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β-amyloid oligomers in mice and monkeys. Cell Metab. 18, 831–843. doi: 10.1016/j.cmet.2013.11.002
Luty, A. A., Kwok, J. B., Dobson-Stone, C., Loy, C. T., Coupland, K. G., Karlstrom, H., et al. (2010). Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron disease. Ann. Neurol. 68, 639–649. doi: 10.1002/ana.22274
Ma, T., Trinh, M. A., Wexler, A. J., Bourbon, C., Gatti, E., Pierre, P., et al. (2013). Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 16, 1299–1305. doi: 10.1038/nn.3486
Makioka, K., Yamazaki, T., Fujita, Y., Takatama, M., Nakazato, Y., and Okamoto, K. (2010). Involvement of endoplasmic reticulum stress defined by activated unfolded protein response in multiple system atrophy. J. Neurol. Sci. 297, 60–65. doi: 10.1016/j.jns.2010.06.019
Martin, I., Dawson, V. L., and Dawson, T. M. (2011). Recent advances in the genetics of Parkinson’s disease. Annu. Rev. Genomics Hum. Genet. 12, 301–325. doi: 10.1146/annurev-genom-082410-101440
Matus, S., Castillo, K., and Hetz, C. (2012). Hormesis: protecting neurons against cellular stress in Parkinson disease. Autophagy 8, 997–1001. doi: 10.4161/auto.20748
Matus, S., Glimcher, L. H., and Hetz, C. (2011). Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Curr. Opin. Cell Biol. 23, 239–252. doi: 10.1016/j.ceb.2011.01.003
Matus, S., Lopez, E., Valenzuela, V., and Hetz, C. (2013). Functional role of the transcription factor ATF4 in the pathogenesis of amyotrophic lateral sclerosis. PLoS ONE 8:e66672. doi: 10.1371/journal.pone.0066672
Matus, S., Nassif, M., Glimcher, L. H., and Hetz, C. (2009). XBP-1 deficiency in the nervous system reveals a homeostatic switch to activate autophagy. Autophagy 5, 1226–1228. doi: 10.4161/auto.5.8.10247
Mazzulli, J. R., Xu, Y. H., Sun, Y., Knight, A. L., McLean, P. J., Caldwell, G. A., et al. (2011). Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. doi: 10.1016/j.cell.2011.06.001
McInnes, J. (2013). Insights on altered mitochondrial function and dynamics in the pathogenesis of neurodegeneration. Transl. Neurodegener. 2:12. doi: 10.1186/2047-9158-2-12
Mekahli, D., Bultynck, G., Parys, J. B., De Smedt, H., and Missiaen, L. (2011). Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 3:a004317. doi: 10.1101/cshperspect.a004317
Menzies, F. M., Fleming, A., and Rubinsztein, D. C. (2015). Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 16, 345–357. doi: 10.1038/nrn3961
Meusser, B., Hirsch, C., Jarosch, E., and Sommer, T. (2005). ERAD: the long road to destruction. Nat. Cell Biol. 7, 766–772. doi: 10.1038/ncb0805-766
Mitsuda, T., Hayakawa, Y., Itoh, M., Ohta, K., and Nakagawa, T. (2007). ATF4 regulates γ-secretase activity during amino acid imbalance. Biochem. Biophys. Res. Commun. 352, 722–727. doi: 10.1016/j.bbrc.2006.11.075
Mollereau, B. (2013). Establishing links between ER-hormesis and cancer. Mol. Cell Biol. 33, 2372–2374. doi: 10.1128/MCB.00315-13
Moreno, J. A., Halliday, M., Molloy, C., Radford, H., Verity, N., Axten, J. M., et al. (2013). Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 5, 206ra138. doi: 10.1126/scitranslmed.3006767
Moreno, J. A., Radford, H., Peretti, D., Steinert, J. R., Verity, N., Martin, M. G., et al. (2012). Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 485, 507–511. doi: 10.1038/nature11058
Mori, K. (2009). Signalling pathways in the unfolded protein response: development from yeast to mammals. J. Biochem. 146, 743–750. doi: 10.1093/jb/mvp166
Moustaqim-Barrette, A., Lin, Y. Q., Pradhan, S., Neely, G. G., Bellen, H. J., and Tsuda, H. (2014). The amyotrophic lateral sclerosis 8 protein, VAP, is required for ER protein quality control. Hum. Mol. Genet. 23, 1975–1989. doi: 10.1093/hmg/ddt594
Nakagawa, T., Zhu, H., Morishima, N., Li, E., Xu, J., Yankner, B. A., et al. (2000). Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 403, 98–103. doi: 10.1038/47513
Ning, B., Deng, M., Zhang, Q., Wang, N., and Fang, Y. (2016). β-Asarone Inhibits IRE1/XBP1 Endoplasmic Reticulum Stress Pathway in 6-OHDA-Induced Parkinsonian Rats. Neurochem. Res. 41, 2097–2101. doi: 10.1007/s11064-016-1922-0
Nishitoh, H., Kadowaki, H., Nagai, A., Maruyama, T., Yokota, T., Fukutomi, H., et al. (2008). ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 22, 1451–1464. doi: 10.1101/gad.1640108
Nishitoh, H., Matsuzawa, A., Tobiume, K., Saegusa, K., Takeda, K., Inoue, K., et al. (2002). ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 16, 1345–1355. doi: 10.1101/gad.992302
Niwa, M., Sidrauski, C., Kaufman, R. J., and Walter, P. (1999). A role for presenilin-1 in nuclear accumulation of Ire1 fragments and induction of the mammalian unfolded protein response. Cell 99, 691–702. doi: 10.1016/S0092-8674(00)81667-0
O’Connor, T., Sadleir, K. R., Maus, E., Velliquette, R. A., Zhao, J., Cole, S. L., et al. (2008). Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron 60, 988–1009. doi: 10.1016/j.neuron.2008.10.047
Ohoka, N., Yoshii, S., Hattori, T., Onozaki, K., and Hayashi, H. (2005). TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 24, 1243–1255. doi: 10.1038/sj.emboj.7600596
Ohta, K., Mizuno, A., Li, S., Itoh, M., Ueda, M., Ohta, E., et al. (2011). Endoplasmic reticulum stress enhances γ-secretase activity. Biochem. Biophys. Res. Commun. 416, 362–366. doi: 10.1016/j.bbrc.2011.11.042
Park, K. W., Eun Kim, G. M., Morales, R., Moreno-Gonzalez, I., Concha-Marambio, L., Lee, A. S., et al. (2017). The endoplasmic reticulum chaperone GRP78/BiP modulates prion propagation in vitro and in vivo. Sci. Rep. 7:44723. doi: 10.1038/srep44723
Penke, B., Bogár, F., and Fülöp, L. (2016). Protein folding and misfolding, endoplasmic reticulum stress in neurodegenerative diseases: in trace of novel drug targets. Curr. Protein Pept. Sci. 17, 169–182. doi: 10.2174/1389203716666151102104653
Prusiner, S. B. (1998). Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383. doi: 10.1073/pnas.95.23.13363
Puthalakath, H., O’Reilly, L. A., Gunn, P., Lee, L., Kelly, P. N., Huntington, N. D., et al. (2007). ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129, 1337–1349. doi: 10.1016/j.cell.2007.04.027
Renna, M., Caporaso, M. G., Bonatti, S., Kaufman, R. J., and Remondelli, P. (2007). Regulation of ERGIC-53 gene transcription in response to Endoplasmic Reticulum stress. J. Biol. Chem. 282, 22499–22512. doi: 10.1074/jbc.M703778200
Renna, M., Faraonio, R., Bonatti, S., De Stefano, D., Carnuccio, R., Tajana, G., et al. (2006). Nitric oxide-induced endoplasmic reticulum stress activates the expression of cargo receptor proteins and alters the glycoprotein transport to the Golgi complex. Int. J. Biochem. Cell Biol. 38, 2040–2048. doi: 10.1016/j.biocel.2006.05.016
Renna, M., Schaffner, C., Winslow, A. R., Menzies, F. M., Peden, A. A., Floto, R. A., et al. (2011). Autophagic substrate clearance requires activity of the Syntaxin-5 SNARE complex. J. Cell Sci. 124, 469–482. doi: 10.1242/jcs.076489
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. doi: 10.1038/nrm2199
Rosenberg, S. S., and Spitzer, N. C. (2011). Calcium signaling in neuronal development. Cold Spring Harb. Perspect. Biol. 3:a004259. doi: 10.1101/cshperspect.a004259
Roussel, B. D., Kruppa, A. J., Miranda, E., Crowther, D. C., Lomas, D. A., Marciniak, S. J., et al. (2013). Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 12, 105–118. doi: 10.1016/S1474-4422(12)70238-7
Rubinsztein, D. C., Codogno, P., and Levine, B. (2012). Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 11, 709–730. doi: 10.1038/nrd3802
Rutkowski, D. T., Arnold, S. M., Miller, C. N., Wu, J., Li, J., Gunnison, K. M., et al. (2006). Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 4:e374. doi: 10.1371/journal.pbio.0040374
Safra, M., Ben-Hamo, S., Kenyon, C., and Henis-Korenblit, S. (2013). The ire-1 ER stress-response pathway is required for normal secretory-protein metabolism in C. elegans. J. Cell Sci. 126, 4136–4146. doi: 10.1242/jcs.123000
Samann, J., Hegermann, J., von Gromoff, E., Eimer, S., Baumeister, R., Schmidt, E., et al. (2009). Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J. Biol. Chem. 284, 16482–16491. doi: 10.1074/jbc.M808255200
Sanchez-Martinez, A., Beavan, M., Gegg, M. E., Chau, K. Y., Whitworth, A. J., and Schapira, A. H. (2016). Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 6:31380. doi: 10.1038/srep31380
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi: 10.1038/nn.2297
Scheper, W., and Hoozemans, J. J. (2015). The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. 130, 315–331. doi: 10.1007/s00401-015-1462-8
Schreiner, B., Hedskog, L., Wiehager, B., and Ankarcrona, M. (2015). Amyloid-β(peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J. Alzheimers Dis. 43, 369–374. doi: 10.3233/JAD-132543
Schroder, M., and Kaufman, R. J. (2005). The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789. doi: 10.1146/annurev.biochem.73.011303.074134
Sellier, C., Campanari, M. L., Julie Corbier, C., Gaucherot, A., Kolb-Cheynel, I., Oulad-Abdelghani, M., et al. (2016). Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 35, 1276–1297. doi: 10.15252/embj.201593350
Selvaraj, S., Sun, Y., Watt, J. A., Wang, S., Lei, S., Birnbaumer, L., et al. (2012). Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Invest. 122, 1354–1367. doi: 10.1172/JCI61332
Sidrauski, C., Acosta-Alvear, D., Khoutorsky, A., Vedantham, P., Hearn, B. R., Li, H., et al. (2013). Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife 2:e00498. doi: 10.7554/eLife.00498
Sidrauski, C., McGeachy, A. M., Ingolia, N. T., and Walter, P. (2015). The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. eLife 4, e05033. doi: 10.7554/eLife.05033.131
Sitia, R., and Braakman, I. (2003). Quality control in the endoplasmic reticulum protein factory. Nature 426, 891–894. doi: 10.1038/nature02262
Soboloff, J., Rothberg, B. S., Madesh, M., and Gill, D. L. (2012). STIM proteins: dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 13, 549–565. doi: 10.1038/nrm3414
Soto, C. (2012). Transmissible proteins: expanding the prion heresy. Cell 149, 968–977. doi: 10.1016/j.cell.2012.05.007
Spatuzza, C., Renna, M., Faraonio, R., Cardinali, G., Martire, G., Bonatti, S., et al. (2004). Heat Shock induces preferential translation of ERGIC-53 and affects its recycling pathway. J. Biol. Chem. 279, 42535–42544. doi: 10.1074/jbc.M401860200
Steele, A. D., Hetz, C., Yi, C. H., Jackson, W. S., Borkowski, A. W., Yuan, J., et al. (2007). Prion pathogenesis is independent of caspase-12. Prion 1, 243–247. doi: 10.4161/pri.1.4.5551
Suzuki, H., Kanekura, K., Levine, T. P., Kohno, K., Olkkonen, V. M., Aiso, S., et al. (2009). ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J. Neurochem. 108, 973–985. doi: 10.1111/j.0022-3042.2008.05857.x
Tang, T. S., Tu, H., Chan, E. Y., Maximov, A., Wang, Z., Wellington, C. L., et al. (2003). Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39, 227–239. doi: 10.1016/S0896-6273(03)00366-0
Teyssou, E., Takeda, T., Lebon, V., Boillée, S., Doukouré, B., Bataillon, G., et al. (2013). Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol. 125, 511–522. doi: 10.1007/s00401-013-1090-0
Thayanidhi, N., Helm, J. R., Nycz, D. C., Bentley, M., Liang, Y., and Hay, J. C. (2010). α-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell 21, 1850–1863. doi: 10.1091/mbc.E09-09-0801
Torres, M., Castillo, K., Armisén, R., Stutzin, A., Soto, C., and Hetz, C. (2010). Prion protein misfolding affects calcium homeostasis and sensitizes cells to endoplasmic reticulum stress. PLoS ONE 5:e15658. doi: 10.1371/journal.pone.0015658
Turner, B. J., Atkin, J. D., Farg, M. A., Zang, D. W., Rembach, A., Lopes, E. C., et al. (2005). Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J. Neurosci. 25, 108–117. doi: 10.1523/JNEUROSCI.4253-04.2005
Ugolino, J., Fang, S., Kubisch, C., and Monteiro, M. J. (2011). Mutant Atp13a2 proteins involved in parkinsonism are degraded by ER-associated degradation and sensitize cells to ER-stress induced cell death. Hum. Mol. Genet. 20, 3565–3577. doi: 10.1093/hmg/ddr274
Urushitani, M., Sik, A., Sakurai, T., Nukina, N., Takahashi, R., and Julien, J. P. (2006). Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat. Neurosci. 9, 108–118. doi: 10.1038/nn1603
Uversky, V. N. (2007). Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J. Neurochem. 103, 17–37. doi: 10.1111/j.1471-4159.2007.04764.x
Valastyan, J. S., and Lindquist, S. (2014). Mechanisms of protein-folding diseases at a glance. Dis. Model Mech. 7, 9–14. doi: 10.1242/dmm.013474
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
van Huizen, R., Martindale, J. L., Gorospe, M., and Holbrook, N. J. (2003). P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. J. Biol. Chem. 278, 15558–15564. doi: 10.1074/jbc.M212074200
Vidal, R. L., Figueroa, A., Court, F. A., Thielen, P., Molina, C., Wirth, C., et al. (2012). Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 21, 2245–2262. doi: 10.1093/hmg/dds040
Vidal, R. L., and Hetz, C. (2012). Crosstalk between the UPR and autophagy pathway contributes to handling cellular stress in neurodegenerative disease. Autophagy 8, 970–972. doi: 10.4161/auto.20139
Vidal, R. L., Matus, S., Bargsted, L., and Hetz, C. (2014). Targeting autophagy in neurodegenerative diseases. Trends Pharmacol. Sci. 35, 583–591. doi: 10.1016/j.tips.2014.09.002
Vieira, F. G., Ping, Q., Moreno, A. J., Kidd, J. D., Thompson, K., Jiang, B., et al. (2015). Guanabenz treatment accelerates disease in a mutant SOD1 mouse model of ALS. PLoS ONE 10:e0135570. doi: 10.1371/journal.pone.0135570
Volgyi, K., Juhász, G., Kovacs, Z., and Penke, B. (2015). Dysfunction of Endoplasmic Reticulum (ER) and Mitochondria (MT) in Alzheimer’s Disease: the role of the ER-MT cross-talk. Curr. Alzheimer Res. 12, 655–672. doi: 10.2174/1567205012666150710095035
Vollrath, J. T., Sechi, A., Dreser, A., Katona, I., Wiemuth, D., Vervoorts, J., et al. (2014). Loss of function of the ALS protein SigR1 leads to ER pathology associated with defective autophagy and lipid raft disturbances. Cell Death Dis. 5, e1290. doi: 10.1038/cddis.2014.243
Walker, A. K., Farg, M. A., Bye, C. R., McLean, C. A., Horne, M. K., and Atkin, J. D. (2010). Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain 133, 105–116. doi: 10.1093/brain/awp267
Walker, A. K., Soo, K. Y., Sundaramoorthy, V., Parakh, S., Ma, Y., and Farg, M. A. (2013). ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS ONE 8:e81170. doi: 10.1371/journal.pone.0081170
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Wang, L., Popko, B., and Roos, R. P. (2011). The unfolded protein response in familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 20, 1008–1015. doi: 10.1093/hmg/ddq546
Wang, M., and Kaufman, R. J. (2014). The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 14, 581–597. doi: 10.1038/nrc3800
Watabe, M., and Nakaki, T. (2004). Rotenone induces apoptosis via activation of bad in human dopaminergic SH-SY5Y cells. J. Pharmacol. Exp. Ther. 311, 948–953. doi: 10.1124/jpet.104.071381
Wong, E., and Cuervo, A. M. (2010). Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 13, 805–811. doi: 10.1038/nn.2575
Xu, K., and Zhu, X. P. (2012). Endoplasmic reticulum stress and prion diseases. Rev. Neurosci. 23, 79–84. doi: 10.1515/rns.2011.062
Yang, H., Liu, C., Zhong, Y., Luo, S., Monteiro, M. J., and Fang, S. (2010). Huntingtin interacts with the cue domain of gp78 and inhibits gp78 binding to ubiquitin and p97/VCP. PLoS ONE 5:e8905. doi: 10.1371/journal.pone.0008905
Yang, L., Li, S., Miao, L., Huang, H., Liang, F., Teng, X., et al. (2016). Rescue of glaucomatous neurodegeneration by differentially modulating neuronal endoplasmic reticulum stress molecules. J. Neurosci. 36, 5891–5903. doi: 10.1523/JNEUROSCI.3709-15.2016
Yang, W., Zhou, X., Zimmermann, H. R., Cavener, D. R., Klann, E., and Ma, T. (2016). Repression of the eIF2α kinase PERK alleviates mGluR-LTD impairments in a mouse model of Alzheimer’s disease. Neurobiol. Aging 41, 19–24. doi: 10.1016/j.neurobiolaging.2016.02.005
Yang, Y., Turner, R. S., and Gaut, J. R. (1998). The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Aβ40 and Aβ42 secretion. J. Biol. Chem. 273, 25552–25555. doi: 10.1074/jbc.273.40.25552
Yoon, S. O., Park, D. J., Ryu, J. C., Ozer, H. G., Tep, C., Shin, Y. J., et al. (2012). JNK3 perpetuates metabolic stress induced by Aβ peptides. Neuron 75, 824–837. doi: 10.1016/j.neuron.2012.06.024
Yuan, Y., Cao, P., Smith, M. A., Kramp, K., Huang, Y., Hisamoto, N., et al. (2011). Dysregulated LRRK2 signaling in response to endoplasmic reticulum stress leads to dopaminergic neuron degeneration in C. elegans. PLoS ONE 6:e22354. doi: 10.1371/journal.pone.0022354
Zhang, C., and Thomas, T. W. (2016). Stromal Interaction Molecule 1 rescues store-operated calcium entry and protects NG115-401L cells against cell death induced by endoplasmic reticulum and mitochondrial oxidative stress. Neurochem. Int. 97, 137–145. doi: 10.1016/j.neuint.2016.04.002
Zhang, K., and Kaufman, R. J. (2006). Protein folding in the endoplasmic reticulum and the unfolded protein response. Handb. Exp. Pharmacol. 172, 69–91. doi: 10.1007/3-540-29717-0_3
Zhao, L., and Ackerman, S. L. (2006). Endoplasmic reticulum stress in health and disease. Curr. Opin. Cell Biol. 18, 444–452. doi: 10.1016/j.ceb.2006.06.005
Zhao, Y., Li, X., Cai, M. Y., Ma, K., Yang, J., Zhou, J., et al. (2013). XBP-1u suppresses autophagy by promoting the degradation of FoxO1 in cancer cells. Cell Res. 23, 491–507. doi: 10.1038/cr.2013.2
Zhou, Y., Lee, J., Reno, C. M., Sun, C., Park, S. W., Chung, J., et al. (2011). Regulation of glucose omeostasis through a XBP-1-FoxO1 interaction. Nat. Med. 17, 356–365. doi: 10.1038/nm.2293
Zhu, Y. F., Li, X. H., Yuan, Z. P., Li, C. Y., Tian, R. B., Jia, W., et al. (2015). Allicin improves endoplasmic reticulum stress-related cognitive deficits via PERK/Nrf2 antioxidative signaling pathway. Eur. J. Pharmacol. 762, 239–246. doi: 10.1016/j.ejphar.2015.06.002
Keywords: unfolded protein response, ER stress, protein misfolding disorders, neurodegenerative diseases, therapeutic targets
Citation: Remondelli P and Renna M (2017) The Endoplasmic Reticulum Unfolded Protein Response in Neurodegenerative Disorders and Its Potential Therapeutic Significance. Front. Mol. Neurosci. 10:187. doi: 10.3389/fnmol.2017.00187
Received: 22 January 2017; Accepted: 29 May 2017;
Published: 16 June 2017.
Edited by:
Angelo Poletti, Università degli Studi di Milano, ItalyReviewed by:
Bilal Malik, University College London, United KingdomCecilia Bucci, University of Salento, Italy
Copyright © 2017 Remondelli and Renna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Remondelli, cHJlbW9uZGVsbGlAdW5pc2EuaXQ= Maurizio Renna, bXI0NjZAY2FtLmFjLnVr