 Paul Heo1
Paul Heo1 Dae-Hyuk Kweon
Dae-Hyuk Kweon
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 31 March 2017
Sec. Molecular Signalling and Pathways
Volume 10 - 2017 | https://doi.org/10.3389/fnmol.2017.00093
This article is part of the Research Topic The Molecular Mechanism Behind Synaptic Transmission View all 22 articles
Neurotransmitters are released within a millisecond after Ca2+ arrives at an active zone. However, the vesicle fusion pathway underlying this synchronous release is yet to be understood. At the center of controversy is whether hemifusion, in which outer leaflets are merged while inner leaflets are still separated, is an on-pathway or off-pathway product of Ca2+-triggered exocytosis. Using the single vesicle fusion assay, we recently demonstrated that hemifusion is an on-pathway intermediate that immediately proceeds to full fusion upon Ca2+ triggering. It has been shown that the flavonoid myricetin arrests soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE)-mediated vesicle fusion at hemifusion, but that the hemifused vesicles spontaneously convert to full fusion when the myricetin clamp is removed by the enzyme laccase. In the present study, we visualized SNARE-mediated hemifusion between two SNARE-reconstituted giant unilamellar vesicles (GUVs) arrested by myricetin. The large size of the GUVs enabled us to directly image the hemifusion between them. When two merging GUVs were labeled with different fluorescent dyes, GUV pairs showed asymmetric fluorescence intensities depending on the position on the GUV pair consistent with what is expected for hemifusion. The flow of lipids from one vesicle to the other was revealed with fluorescence recovery after photobleaching (FRAP), indicating that the two membranes had hemifused. These results support the hypothesis that hemifusion may be the molecular status that primes Ca2+-triggered millisecond exocytosis. This study represents the first imaging of SNARE-driven hemifusion between GUVs.
Membrane fusion constitutes the final step in the secretion and cargo transfer pathways between cellular compartments (Südhof and Rizo, 2011; Scheller, 2013; Rothman, 2014), and it is also essential in many cellular processes, including autophagy (Wang et al., 2016). When two membranes are approaching each other for fusion, free energy is required to overcome electrostatic repulsive forces, steric hindrances and the hydration force between two membranes. The soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE) proteins comprise the molecular fusion machine. SNARE proteins provide the free energy required for fusion during the formation of a parallel four-helix bundle called the SNARE complex (Poirier et al., 1998; Sutton et al., 1998). In neurons, there are three SNARE proteins: syntaxin 1a (Stx1) and synaptosome-associated protein 25 (SNAP-25) on the plasma membrane and vesicle-associated membrane protein 2 or Syb2 (VAMP2) on the vesicle membrane. These three SNARE proteins constitute the minimal machinery for fusion between the synaptic vesicle and the presynaptic plasma membrane (Weber et al., 1998). Overwhelming evidence favors the zippering hypothesis, in which SNARE complex formation starts from N-termini and zippers progressively towards the membranes (Melia et al., 2002; Matos et al., 2003; Gao et al., 2012; Lou and Shin, 2016).
Many membrane fusion processes proceed via several sequential intermediates (Kozlov and Markin, 1983; Chernomordik and Kozlov, 2003; Chernomordik et al., 2006; Jahn and Scheller, 2006). When two membranes approach each other, they become locally connected by forming a hemifusion stalk. Proximal leaflets of bilayers are fused, but distal leaflets are separated at this stage. Hemifusion is also shown to be an on-pathway intermediate in SNARE-mediated membrane fusion (Lu et al., 2005; Xu et al., 2005). Subsequently, the stalk expands radially into a hemifusion diaphragm with the distal leaflets remaining separated, though it is possible that hemifusion expansion results in a dead-end product in Ca2+-triggered exocytosis (Diao et al., 2012; Hernandez et al., 2012). Finally, a fusion pore is opened within the hemifusion diaphragm, directly from the hemifusion stalk or from a point of membrane contact. Although hemifusion is considered to be an essential fusion intermediate, its experimental verification and characterization in biological membranes has been very difficult, yielding contradictory results (Zampighi et al., 2006; Wong et al., 2007; Fernández-Busnadiego et al., 2010; Zhao et al., 2016). After identification of the SNARE complex assembly and membrane fusion intermediate, Jahn and Scheller (2006) proposed that straining of lipids at the edge of an extended docking zone initiates fusion (Hernandez et al., 2012). Another cryo-electron microscopy study showed that Ca2+-triggered immediate fusion starts from a point-contact between membranes and proceeds to full fusion without discernible hemifusion intermediates (Diao et al., 2012). In both studies, stable hemifusion diaphragms were kinetically trapped and represented an off-pathway product. However, a study using super-resolution stimulated emission depletion microscopy observed membrane hemifusion directly in live chromaffin cells in real time (Zhao et al., 2016). An Ω-shaped hemifusion structure was observed in the live cells, and it was found that even the ‘kiss-and-run’ model can be explained by the competition between transitions of hemifusion/hemi-fission to full fusion or to full fission. Recently, we also showed that a stable hemifusion state can proceed to complete fusion and form a fusion pore almost synchronously with Ca2+ triggering (Heo et al., 2016). When a small molecule flavonoid (myricetin) that halts SNARE zippering in the middle (Yang et al., 2010) was removed from the SNARE complex intermediate with the enzyme laccase, hemifusion proceeded to full fusion. The speed of Ca2+-triggered fusion was comparable to that in neurons, and the pattern of release was reminiscent of the synchronous and asynchronous release of neuroexocytosis depending on the stage of Ca2+ arrival in the reconstituted systems (Heo et al., 2016). These results provide clear evidence that the hemifusion state is the bona fide intermediate enabling millisecond exocytosis.
Hemifusion mediated by SNARE proteins and myricetin was analyzed again using dynamic light scattering (DLS) spectroscopy (Yang et al., 2015). We verified hemifusion between vesicles in the presence of myricetin by simulating vesicle hydrodynamic radius changes during fusion and by cleaving SNARE proteins with proteinase K. In the present study, we aimed to visualize hemifusion between individual giant unilamellar vesicles (GUVs). GUVs are excellent objects for fluorescence microscopy visualization and analysis because GUV dimensions are larger than light microscopy’s intrinsic resolution limit. The mean diameter of GUVs is tens of μm, for which mean and dispersion values are strictly dependent on the method of GUV preparation. GUV size can be made comparable to the plasma membrane of a variety of cells. Membrane fusion processes, including the existence of the protein-free hemifusion diaphragm as a fusion intermediate, can be visualized using GUV and fluorescence microscopy (Lei and MacDonald, 2003; Heuvingh et al., 2004; Nikolaus et al., 2010). GUVs were also elegantly used to visualize the molecular interplay between membranes, accessory proteins and SNARE proteins (Bacia et al., 2004; Tareste et al., 2008; Hui et al., 2009; Malsam et al., 2012). Here, the hemifusion induced by SNARE proteins and myricetin was evaluated with confocal microscopy to directly visualize SNARE-mediated hemifusion.
Neuronal SNAREs from Rattus norvegicus: SNAP-25 (amino acids 1–206), Syb2 (amino acids 1–116) and Stx1 (amino acids 1–288) were expressed in Escherichia coli CodonPlus-RIL (DE3) and purified by a glutathione S-transferase (GST) tag system. In brief, cell pellets were resuspended in PBS (pH 7.4) supplemented with 2 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride, 2 mM ethylenediaminetetraacetic acid (EDTA) and 2 mM dithiothreitol (DTT). After sonication, the supernatant was mixed with GST-agarose beads at 4°C for 3 h. Excess PBS was used for washing, and each protein of interest was eluted in thrombin cleavage buffer (TCB, 50 mM Tris-HCl and 150 mM NaCl, pH 8.0). For transmembrane proteins, 0.2% Triton X-100 and 0.05% Tween 20 were added to PBS for the lysis and washing steps, and subsequently 1% n-octyl-beta-D-glucopyranoside (OG) was added to TCB instead of Triton X-100 at the elution step. All purified proteins were analyzed by SDS-PAGE and the Bradford assay.
We used a conventional SNARE reconstitution method to make proteoliposomes. 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dioleoyl-sn-glycero-3-phospho-Lserine (DOPS), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (NBD) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissaminerhodamine B sulfonyl, Rhod) were purchased from Avanti Polar Lipids Inc. A lipid mixture composed of PC:PS (95:5) was dried with nitrogen gas and further dried under vacuum overnight. For fluorescent GUVs, fluorescent lipids were incorporated into the mixture at the expense of PC. Large unilamellar vesicles (LUV) were formed by extruding the hydrated lipid mixtures through polycarbonate filters with a 100-nm pore size. SNARE proteins were mixed with liposomes at the indicated lipid/SNARE ratio while keeping the 0.8% OG concentration. The proteoliposomes were diluted with dialysis buffer (25 mM HEPES, 150 mM NaCl, pH 7.4) and dialyzed against 2 L dialysis buffer overnight at 4°C. Residual OG in samples was eliminated by SM2 Bio-Beads at 4°C for 30 mins. To compare reconstitution efficiency on GUVs depending on the lipid-to-protein (LP) ratio, SNARE proteins labeled with Alexa 647 dye through an amine reaction were used.
The GUVs with SNARE proteins were generated by the electroformation method based on previous reports with modifications (Bacia et al., 2004; Tareste et al., 2008; Hui et al., 2009; Malsam et al., 2012). The process began after preparation of SNARE-embedded LUVs. After obtaining LUV pellets by centrifugation at 100,000 g at 4°C for 2 h, each pellet was resuspended in 10 mL low salt buffer (5 mM HEPES, 5 mM NaCl, pH 7.4). A droplet of the solution was applied to an indium tin oxide (ITO)-coated glass slide. After drying the lipid droplet, two ITO slides were assembled to form a 5 × 5 × 2 mm chamber (Supplementary Figure S1). The dried films were rehydrated with 200 mM sucrose while applying a sinusoidal electric field at 0.01 V and 10 Hz for 10 mins. The electric field was increased gradually by 0.1 V per 5 min up to 1.2 V, while maintaining the 10 Hz frequency. Then, 1.2 V (at 10 Hz) was applied for 6 h. Finally, 3 V (at 10 Hz) was applied for 15 min to detach the GUVs from the ITO slide. The SNARE-containing GUV mixture was incubated in 200 mM glucose solution to remove aberrant lipid aggregates. The efficiency of SNARE-containing GUV formation was estimated using fluorescent dye-labeled SNARE proteins and fluorescent lipids. GUVs were used for assays within 1 day of their formation. Note that residual OG strongly inhibited the formation of a dried film on the ITO slides and subsequent GUV formation. GUVs with no membrane proteins were generated following the same protocol as above, except that 2 μl of the lipid mixture in chloroform was directly spread on the ITO slides.
For membrane fusion assays, 5 μl of each t- and v-GUV preparation containing the binary acceptor complex (Syx1 and SNAP-25) and Syb2, respectively, were mixed with 5 μl buffer (25 mM HEPES, 400 mM KCl, 1 mM DTT, pH 7.4) and incubated at 37°C for 40 min. The GUVs deposited on glass slides were observed through a confocal laser-scanning microscope (LSM 700, Zeiss, Germany) equipped with a C-Apochromat 63×/1.2 water immersion objective. After focusing on the focal plane on the bottle-neck of a hemifused GUV pair, the fluorescence intensity distribution was obtained from the points on the line which cross the center of bottle-neck and meets the vesicles at opposite positons. The fluorescence intensities were normalized by the maximum fluorescence intensity. The fluorescence recovery after photobleaching (FRAP) assay was performed following the manufacturer’s instructions. After bleaching the regions of interest (ROI) at 100% power, fluorescence recovery was observed at the same power as before bleaching (~10% of maximum). ROI was ~10 μm in diameter to select entire single GUV. Further image analysis was performed with ZEN 2010 LSM software and ImageJ software (National Institutes of Health). The fluorescence recovery curve was fitted to an exponential decay function f(t) = A(1-exp(−t/τ)), where, A is the fraction of mobile component, t is the time passed after photobleaching and τ is the time constant. The lateral diffusion coefficient of lipids was calculated with following equation D = r2/4τ, where, D is diffusion coefficient, r is the radius of the photobleached GUV, and τ is the characteristic diffusion time.
GUVs containing SNARE proteins were prepared through the electroformation method. To make sure that SNARE proteins were incorporated into GUVs, the SNARE proteins and/or GUVs were labeled with fluorescent dyes. First, Syb2 labeled with Alexa 647 was reconstituted into GUVs containing NBD. We observed many GUVs with the Alexa 647 fluorescent signal and the co-localized NBD signal (Figure 1A), indicating that Syb2-containing GUVs were well formed. Next, Stx1 labeled with Alexa 647 was reconstituted into the non-fluorescent GUVs to exclude the possibility that the fluorescence of circles derives from inadequate filtration of lipid fluorescence. We expected to observe the Alexa 647 signal on circles if the t-GUVs were formed as planned. Otherwise, no fluorescence or any signal from aggregates was expected. Though several amorphous dots representing protein aggregates were present, we found many circles representing GUVs containing Stx1 (Figure 1B).
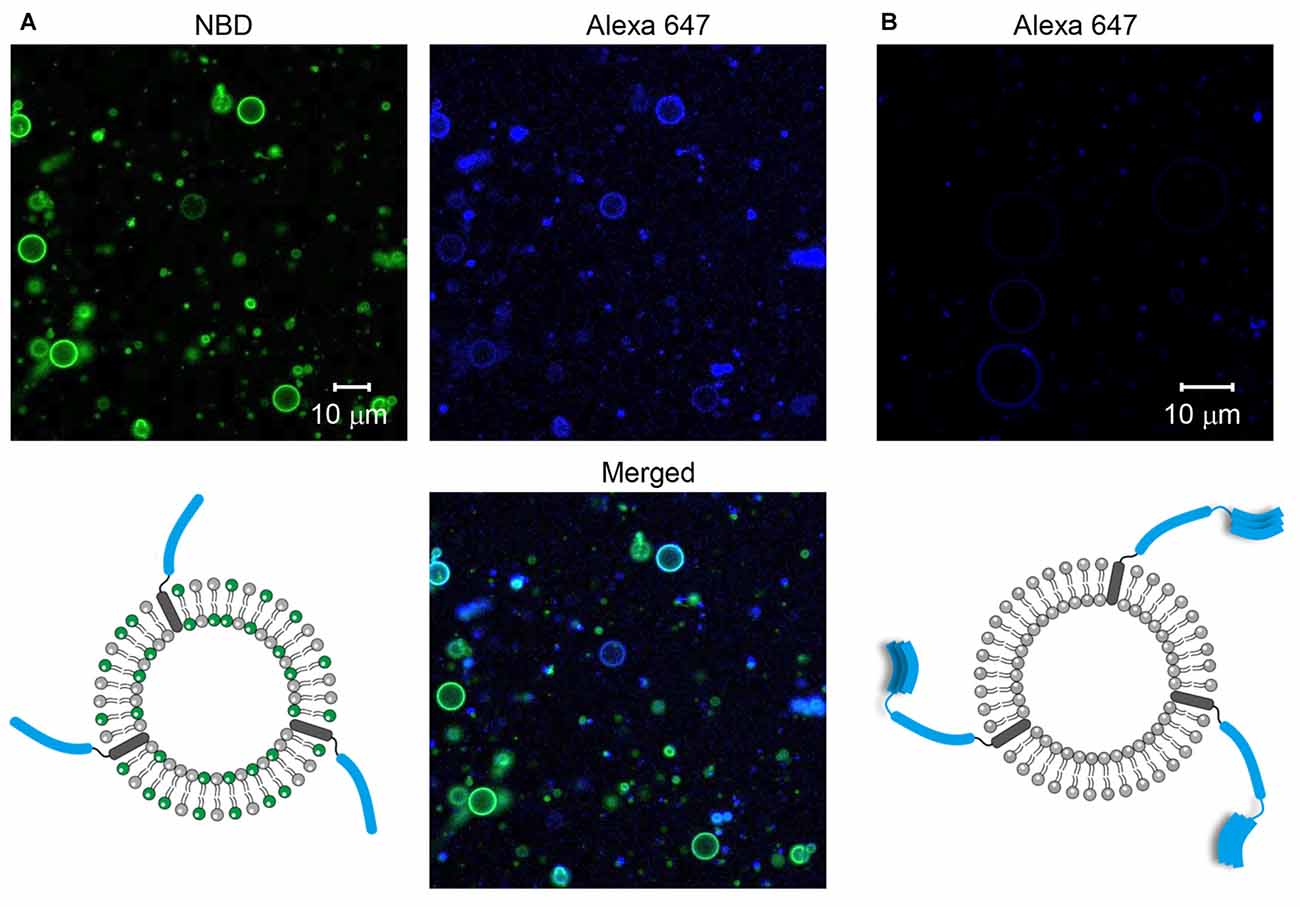
Figure 1. Formation of soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE)-containing giant unilamellar vesicles (GUVs). (A) Syb2 labeled with Alexa 647 was reconstituted into GUVs containing nitro-2-1,3-benzoxadiazol-4-yl (NBD). (B) Syntaxin 1a (Stx1) labeled with Alexa 647 was reconstituted into GUVs that were not labeled with fluorescent dyes.
We found that GUVs were not well formed when we tried to incorporate SNARE proteins above a certain limit. Because too few SNARE proteins on a GUV may not induce efficient GUV-GUV fusion, we tested several lipid-to-SNARE protein (LP) ratios to find an optimal condition for the efficient formation of SNARE-containing GUVs (Table 1). The efficiency of GUV formation was inversely proportional to the concentration of SNARE proteins. When the LP ratio was below 200 for Syb2 and below 500 for Stx1, we did not observe any GUV. In contrast, GUVs did not contain any SNARE proteins when the LP ratio was 4000, though many GUVs were observed. The 1000 LP ratio was optimal for both Stx1 and Syb2. For example, when Syb2 was incorporated into GUVs at an LP ratio of 1000, we observed tens of GUVs in a single focal plane, and approximately 73% of the GUVs contained fluorescently-labeled SNARE proteins (Table 1). Thus, we used an LP ratio of 1000 for further experiments.
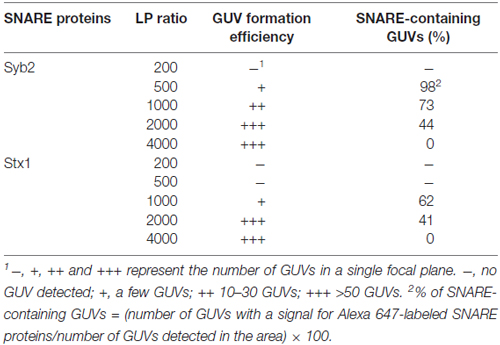
Table 1. The effect of lipid-to-protein (LP) ratio on the formation of giant unilamellar vesicles (GUVs) and reconstitution yields.
T-GUVs and v-GUVs containing the binary t-SNARE complex (Stx1 and SNAP-25) and Syb2, respectively, were separately prepared following the procedure described above at an LP ratio of 1000. T-GUVs and v-GUVs were labeled with rhodamine and NBD, respectively. After mixing equal amounts of t- and v-GUVs, the mixture was incubated at 37°C for 40 min. While major populations were unfused, we found that ~10% of the GUVs exhibited both NBD and rhodamine fluorescence, which is expected to happen when the two vesicles are fully fused (Figures 2A, Supplementary Figure S2). On the other hand, no vesicle exhibited both fluorescent signals when GUVs containing only fluorescent dyes (but no SNARE proteins) were mixed together (Figure 2B). This clearly indicated that GUVs did not spontaneously fuse in the absence of SNARE proteins. Thus, SNARE proteins reconstituted on GUVs mediated GUV-GUV fusion.
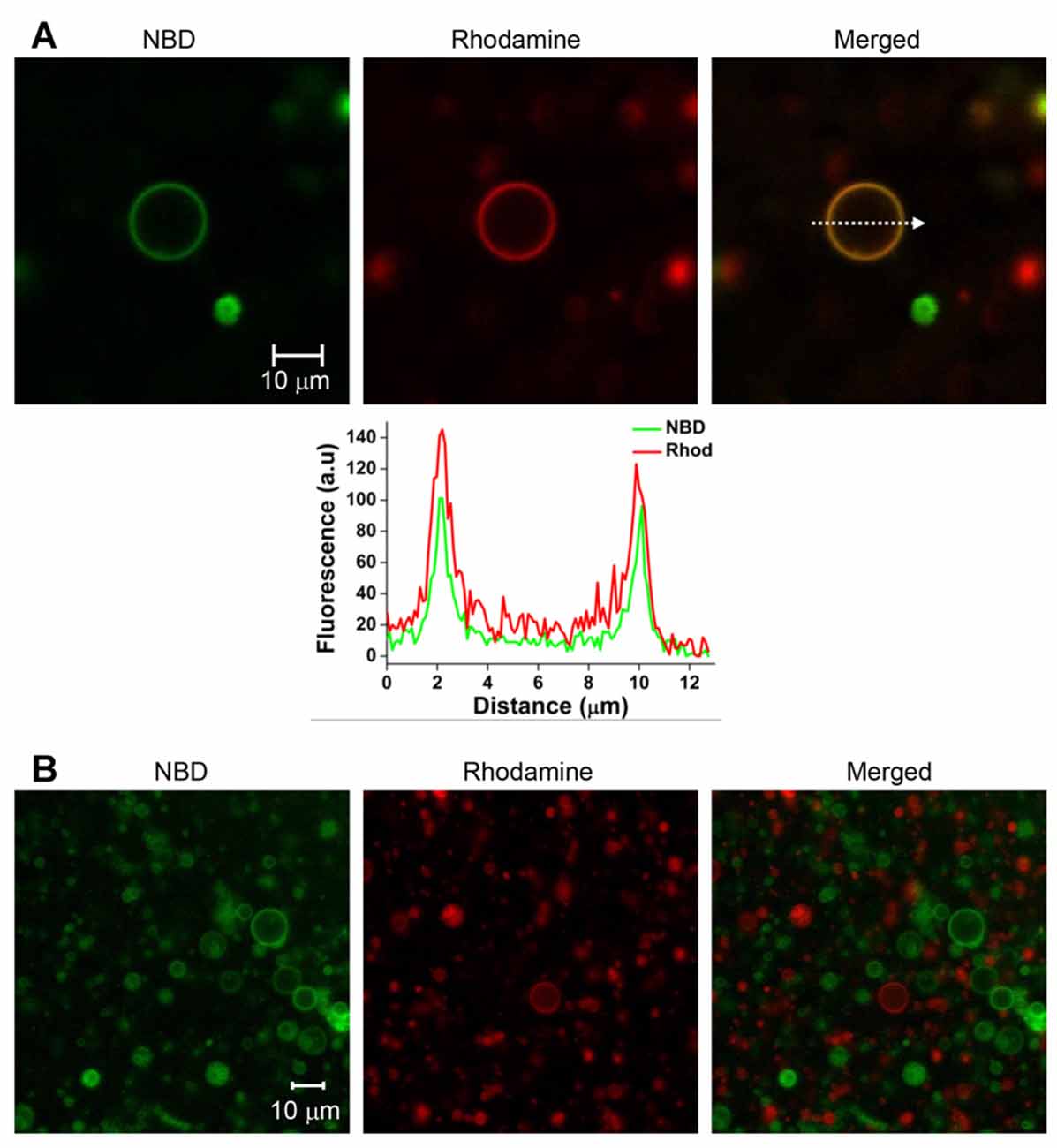
Figure 2. SNARE-mediated full fusion between v- and t-GUVs. (A) Representative images of fully fused GUVs. V-GUVs and t-GUVs were labeled with 3 mol % NBD and rhodamine, respectively. A fluorescence intensity profile (lower panel) was obtained, shown on the line indicated by an arrow in the merged GUV image. (B) Mixture of GUVs without SNARE proteins.
We tested whether the membrane fusion intermediate arrested by myricetin was hemifused or not. It was expected that hemifused vesicles would show strong fluorescence intensities for both NBD and rhodamine near the stalk. Other regions of t-GUVs were expected to show a high rhodamine intensity with a lower NBD fluorescence intensity than that observed in the v-GUVs, and vice versa (Figure 3A, upper panel). If lipids were not mixed but the membranes were merely closely apposed, NBD fluorescence would not be detected in t-GUVs, and vice versa for v-GUVs (Figure 3A, lower panel).
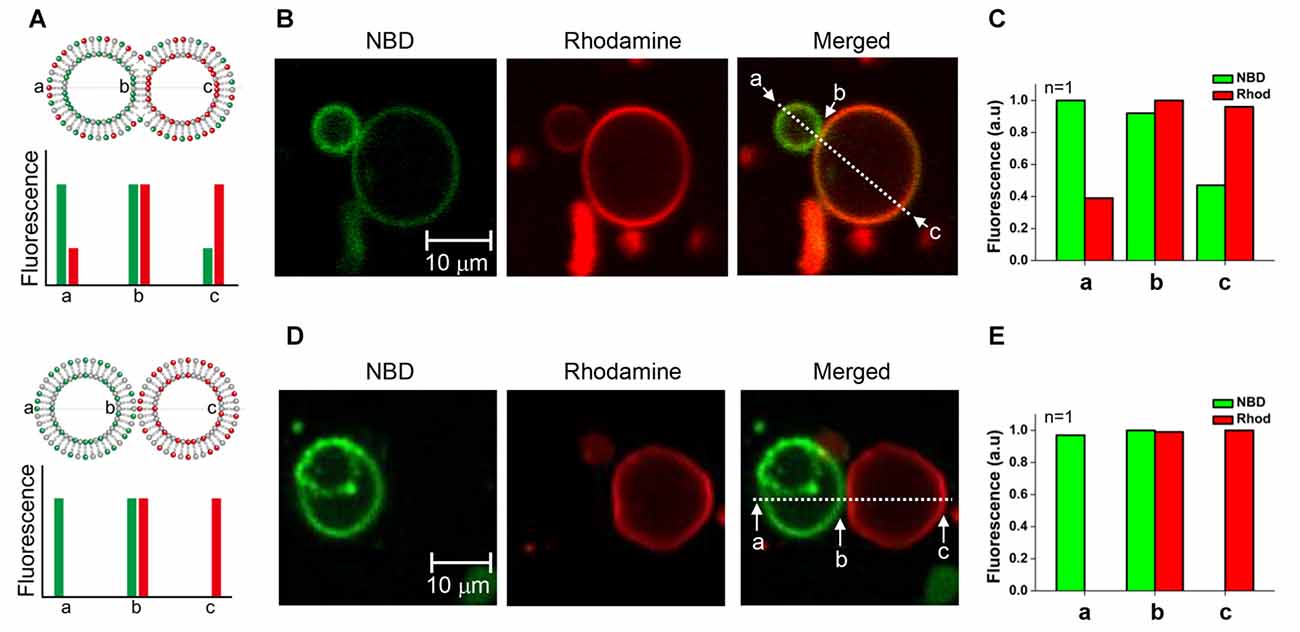
Figure 3. Comparison of the hemifused vesicle pair and the docked vesicle pair. (A) Schematic showing asymmetric fluorescence intensities depending on the location in the vesicle pair. (B) The fluorescently labeled v-GUVs (NBD, green) and t-GUVs (rhodamine, red) were mixed and incubated at 37°C for 40 min in the presence 1 μM myricetin. (C) The distribution of fluorescence intensity was measured at various regions of each GUV pair. (D) The fluorescently labeled v-GUVs and t-GUVs were mixed and incubated at 4°C for 60 min before taking the image. (E) The distribution of fluorescence intensity at various regions of the docked GUV pair.
The mixture of t- and v-GUVs was incubated at 37°C for 40 min in the presence of 1 μM myricetin. We observed hourglass-shaped vesicles forming in the presence of myricetin (Figures 3B, Supplementary Figure S3). One vesicle showed strong NBD fluorescence, while the other showed rhodamine fluorescence; this obviously represented the signal for the v- and t-GUVs, respectively. However, each vesicle also showed the fluorescence signal of the other vesicle, although the intensity was low. The intensity of each fluorescent signal was analyzed at three different positions: one near the stalk (designated b), one on the v-GUV (designated a) and one on the t-GUV (designated c). The asymmetry of the fluorescence intensities of NBD and rhodamine was dependent on the location of the GUV pair, and was consistent with what we expected for the hemifused GUVs (Figure 3C). On the other hand, GUV pairs were also observed when the mixture of t- and v-GUVs was incubated at 4°C (Figure 3D), which was a condition in which membrane fusion did not happen while docking of vesicles was allowed. Though the shape was similar to the hemifused vesicle pair, the fluorescent signals of NBD and rhodamine were not detected on opposite vesicles (Figure 3E). This result suggested that the hourglass-shaped GUV pairs enriched in the presence of myricetin were hemifused.
Hemifusion between the vesicles in the GUV pair arrested by myricetin was confirmed with a FRAP assay. If two GUV outer leaflets are connected continuously, lipid molecules of the outer leaflets will diffuse laterally, leading to recovery of fluorescence after photobleaching. In contrast, diffusion of lipids from one GUV to another is not allowed if the GUV pair is simply docked, but bilayer leaflets are not connected between the two GUVs.
After hemifused GUV pairs were prepared in the presence of myricetin, GUV-GUV pairs that looked like hourglasses were selected. Hemifusion between a pair of GUVs was identified based on the fluorescence asymmetry as described above (Figure 4A). After photobleaching, the entire NBD fluorescence in the GUV at the right-hand side was measured as a function of time. The NBD fluorescence gradually recovered over time, suggesting that the NBD of the GUV on the left-hand side moved to the GUV on the right-hand side (Figure 4B). This result clearly indicated the GUV pair was hemifused. The lateral diffusion coefficient of the NBD between GUVs was calculated from the kinetics of fluorescence recovery (Figure 4C). It was determined to be 0.18 ± 0.03 μm2/s from 3 independent GUV pairs. The average decay constant τ was 71 s when the radius of a photobleached GUV was 7.15 μm. When the rhodamine fluorescence in one vesicle of the GUV pair was photobleached instead, fluorescence was recovered within a few minutes, consistent with NBD photobleaching (Figure 4D). These results suggested that the GUV pairs treated with myricetin were hemifused to allow lipid diffusion through the continuous outer leaflets of the two GUVs.
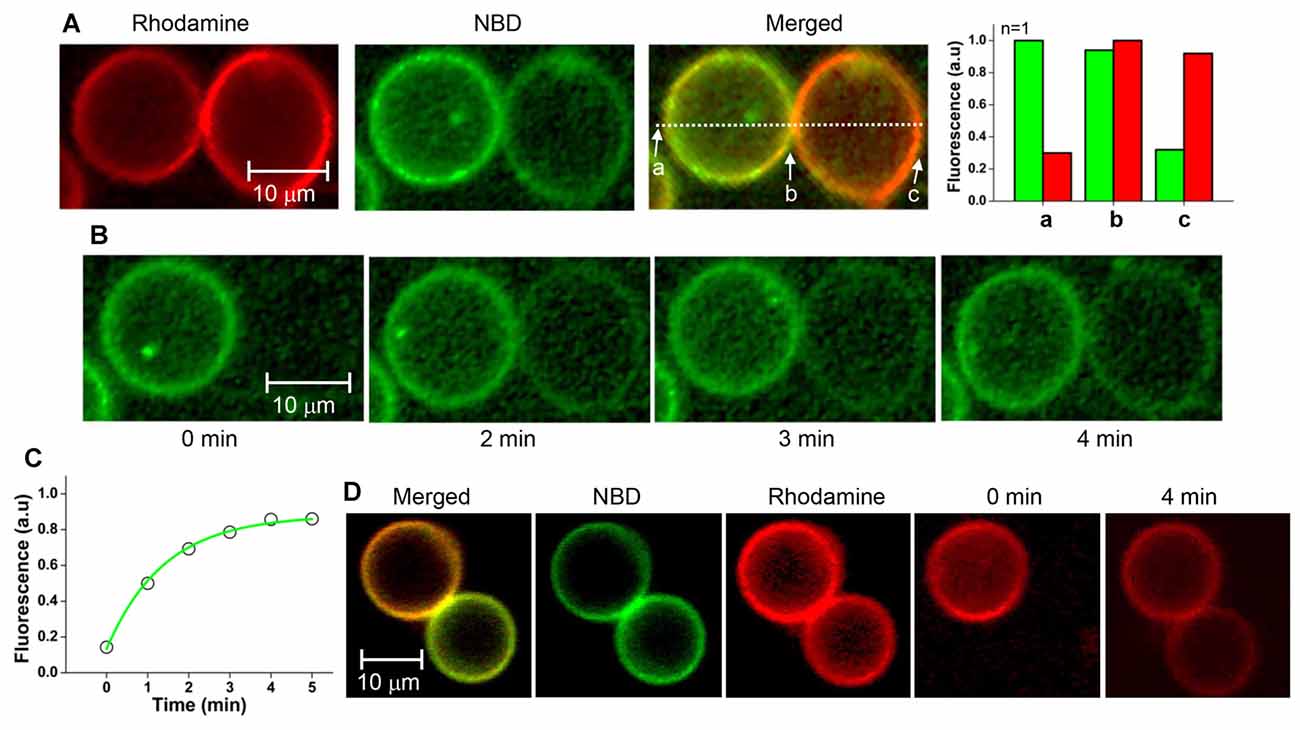
Figure 4. Fluorescence recovery after photobleaching (FRAP) assay of the GUV pair arrested by myricetin. (A) A GUV pair arrested by myricetin was selected, and the fluorescence intensity asymmetry dependent on the location was confirmed. (B) Representative images taken during FRAP experiments. The entire single v-GUV (the GUV on the right-hand side) was bleached, and its fluorescence recovery was monitored for 5 min. (C) Fluorescence recovery kinetics. (D) Representative FRAP images taken after t-GUV bleaching.
GUVs are sufficiently large to be viewed using optical or fluorescence microscopy, and as such they are excellent samples to directly visualize and analyze the individual membrane fusion process. However, membrane protein incorporation, size control, and molecule encapsulation inside the GUVs are still challenging, although the electroformation-based method is relatively reproducible for protein-free GUV formation (Yamashita et al., 2002; Limozin et al., 2003; Chiantia et al., 2011; Dezi et al., 2013). It is likely that these difficulties have limited direct visualization of SNARE-driven fusion between GUVs, even in studies that have made use of GUVs for the analysis of membrane fusion (Bacia et al., 2004; Tareste et al., 2008; Hui et al., 2009; Nikolaus et al., 2010; Malsam et al., 2012). In the present study, all t- and v-SNARE proteins were successfully reconstituted in GUVs by optimizing the LP ratio and the GUV formation procedure. Reconstituted GUVs containing SNARE proteins enabled us to analyze membrane fusion intermediates occurring during GUV-GUV fusion with a fluorescence microscope.
The copy number of Syb2 in a synaptic vesicle with a 42-nm diameter is ~70, which corresponds to an LP ratio of ~176 in reconstituted proteoliposomes (Takamori et al., 2006; Ji et al., 2010). This LP ratio also corresponds to 5 × 106 copies of Syb2 in a v-GUV with a diameter of 10 μm. However, we found that such a high protein density did not allow efficient GUV formation. Rather, higher LP ratios enabled more efficient GUV formation, though the probability that the GUVs contained SNARE proteins was lowered. GUV formation efficiency and SNARE incorporation appeared to be somewhat incompatible. We found that an LP ratio of 1000 and 2000 was optimal for both Syb2 and Stx1.
Our FRAP assay revealed a lipid diffusion coefficient of 0.18 μm2/s at 25°C. The diffusion constant of POPC (which was also used in our experiments) in multilamellar vesicles at 25°C is 7–10 μm2/s (Gaede and Gawrisch, 2003). This value is much smaller than the values for the cortical granule membrane and the plasma membrane (Wong et al., 2007). This small diffusion constant of lipids indicates the hemifused GUV pair does not share a wide area, and that only a small region is merged. The small shared area is most likely the bottleneck of the lateral lipid movement. It is not likely that the reconstituted SNARE proteins directly hindered the flow of lipid molecules because the protein density is too low to restrict lipid diffusion at such a low LP ratio as 1000. But, it is also possible that SNARE proteins that induced hemifusion do not dissipate from the stalk of hemifusion, restricting the lateral diffusion of lipids from one vesicle to the other (Chernomordik et al., 1998). Regardless of the exact reason for the small diffusion constant, it suggests that hemifusion arrested by myricetin does not expand to a wide area (i.e., the hemifusion diaphragm).
We previously suggested that N-terminal half zippering might drive hemifusion. SNARE complex formation was arrested at the half-zippered state by a flavonoid (myricetin), and it was found that the membrane fusion intermediate arrested by myricetin corresponded to hemifusion in proteoliposome fusion. Though it is yet unclear how only half-zippering of the SNARE complex induced hemifusion, hemifusion arrested by myricetin could be converted to full fusion when the myricetin was removed from the SNARE complex by the enzyme laccase. The hemifusion was metastable, and Ca2+ could trigger immediate full fusion and content mixing. FRET-based bulk lipid-mixing assays (Yang et al., 2010), FRET-based single vesicle-vesicle lipid-mixing assays (Heo et al., 2016), FRET-based single vesicle-vesicle content-mixing assays (Heo et al., 2016), and DLS-based hydrodynamic radius change assays (Yang et al., 2015) were employed to investigate all of the features mentioned above. In the present study, it was shown that GUV-GUV fusion was also arrested in the hemifusion state by myricetin. This result suggested that hemifusion is a bona fide intermediate leading to fusion pore opening, and serves as the primed state for Ca2+-triggered millisecond exocytosis (Heo et al., 2016).
PH and D-HK devised the experiment. PH and J-BP performed the experiments. PH, Y-KS and D-HK wrote the article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by a grant from the Korea Healthcare Technology R&D Project, Ministry of Health and Welfare, South Korea (Grant No: HN14C01010000), and by the National Institutes of Health (GM05290 and 5U54GM0 87519). The Advanced Biomass R&D Center (ABC) of Korea Grant (2011-0031359) funded by the Ministry of Science, ICT and Future Planning.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnmol.2017.00093/full#supplementary-material
FIGURE S1 | Assembly of ITO slides.
FIGURE S2 | Representative images of fully fused GUVs. V-GUVs and t-GUVs were labeled with 3 mol % NBD and rhodamine, respectively. Fluorescence intensity profile (lower panel) was obtained on the line indicated by an arrow in the merged GUV image.
FIGURE S3 | Hemifused vesicle pairs exhibiting fluorescence intensity asymmetry. (A) Images were taken in the same manner as in Figure 3, but in different experiments. The fluorescently labeled v-GUV (NBD, green) and t-GUV (rhodamine, red) were mixed and incubated at 37°C for 40 min in the presence 1 μM myricetin. The distribution of fluorescent intensity was measured at various regions of GUV pair. (B) Number of GUVs observed in this study. Major population (80%–90%) of GUVs in a focal plane is unfused free t- or v-GUVs even after optimization of GUV reconstitution scheme because of low probability of GUV-GUV fusion. Tens of glass slides from several independent experiments were used to obtain significant numbers of GUVs. We observed 161 and 117 GUVs in the absence and presence of 1 μM myricetin, respectively. Circles exhibiting both NBD and rhodamine fluorescence were counted as fully fused vesicles. Hourglass-shaped GUV pairs were counted by eye. Hemifusion was confirmed for 7 GUV pairs out of 79 hourglass-shaped GUV pairs by analyzing fluorescence intensity asymmetry. (C) Averaged fluorescence intensities which are asymmetrical depending on the locations on the GUV pair.
Bacia, K., Schuette, C. G., Kahya, N., Jahn, R., and Schwille, P. (2004). SNAREs prefer liquid-disordered over “raft” (liquid-ordered) domains when reconstituted into giant unilamellar vesicles. J. Biol. Chem. 279, 37951–37955. doi: 10.1074/jbc.M407020200
Chernomordik, L. V., Frolov, V. A., Leikina, E., Bronk, P., and Zimmerberg, J. (1998). The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion and lipidic fusion pore formation. J. Cell Biol. 140, 1369–1382. doi: 10.1083/jcb.140.6.1369
Chernomordik, L. V., and Kozlov, M. M. (2003). Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 72, 175–207. doi: 10.1146/annurev.biochem.72.121801.161504
Chernomordik, L. V., Zimmerberg, J., and Kozlov, M. M. (2006). Membranes of the world unite! J. Cell Biol. 175, 201–207. doi: 10.1083/jcb.200607083
Chiantia, S., Schwille, P., Klymchenko, A. S., and London, E. (2011). Asymmetric GUVs prepared by MβCD-mediated lipid exchange: an FCS study. Biophys. J. 100, L1–L3. doi: 10.1016/j.bpj.2010.11.051
Dezi, M., Di Cicco, A., Bassereau, P., and Lévy, D. (2013). Detergent-mediated incorporation of transmembrane proteins in giant unilamellar vesicles with controlled physiological contents. Proc. Natl. Acad. Sci. U S A 110, 7276–7281. doi: 10.1073/pnas.1303857110
Diao, J., Grob, P., Cipriano, D. J., Kyoung, M., Zhang, Y., Shah, S., et al. (2012). Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. Elife 1:e00109. doi: 10.7554/eLife.00109
Fernández-Busnadiego, R., Zuber, B., Maurer, U. E., Cyrklaff, M., Baumeister, W., and Lučić, V. (2010). Quantitative analysis of the native presynaptic cytomatrix by cryoelectron tomography. J. Cell Biol. 188, 145–156. doi: 10.1083/jcb.200908082
Gaede, H. C., and Gawrisch, K. (2003). Lateral diffusion rates of lipid, water, and a hydrophobic drug in a multilamellar liposome. Biophys. J. 85, 1734–1740. doi: 10.1016/s0006-3495(03)74603-7
Gao, Y., Zorman, S., Gundersen, G., Xi, Z., Ma, L., Sirinakis, G., et al. (2012). Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science 337, 1340–1343. doi: 10.1126/science.1224492
Heo, P., Yang, Y., Han, K. Y., Kong, B., Shin, J. H., Jung, Y., et al. (2016). A chemical controller of SNARE-driven membrane fusion that primes vesicles for Ca2+-triggered millisecond exocytosis. J. Am. Chem. Soc. 138, 4512–4521. doi: 10.1021/jacs.5b13449
Hernandez, J. M., Stein, A., Behrmann, E., Riedel, D., Cypionka, A., Farsi, Z., et al. (2012). Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science 336, 1581–1584. doi: 10.1126/science.1221976
Heuvingh, J., Pincet, F., and Cribier, S. (2004). Hemifusion and fusion of giant vesicles induced by reduction of inter-membrane distance. Eur. Phys. J. E Soft Matter 14, 269–276. doi: 10.1140/epje/i2003-10151-2
Hui, E. F., Johnson, C. P., Yao, J., Dunning, F. M., and Chapman, E. R. (2009). Synaptotagmin-mediated bending of the target membrane is a critical step in Ca2+-regulated fusion. Cell 138, 709–721. doi: 10.1016/j.cell.2009.05.049
Jahn, R., and Scheller, R. H. (2006). SNAREs—engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7, 631–643. doi: 10.1038/nrm2002
Ji, H., Coleman, J., Yang, R., Melia, T. J., Rothman, J. E., and Tareste, D. (2010). Protein determinants of SNARE-mediated lipid mixing. Biophys. J. 99, 553–560. doi: 10.1016/j.bpj.2010.04.060
Kozlov, M. M., and Markin, V. S. (1983). [Possible mechanism of membrane fusion]. Biofizika 28, 242–247.
Lei, G., and MacDonald, R. C. (2003). Lipid bilayer vesicle fusion: intermediates captured by high-speed microfluorescence spectroscopy. Biophys. J. 85, 1585–1599. doi: 10.1016/s0006-3495(03)74590-1
Limozin, L., Bärmann, M., and Sackmann, E. (2003). On the organization of self-assembled actin networks in giant vesicles. Eur. Phys. J. E Soft Matter 10, 319–330. doi: 10.1140/epje/i2002-10118-9
Lu, X. B., Zhang, F., McNew, J. A., and Shin, Y. K. (2005). Membrane fusion induced by neuronal SNAREs transits through hemifusion. J. Biol. Chem. 280, 30538–30541. doi: 10.1074/jbc.M506862200
Malsam, J., Parisotto, D., Bharat, T. A., Scheutzow, A., Krause, J. M., Briggs, J. A., et al. (2012). Complexin arrests a pool of docked vesicles for fast Ca2+-dependent release. EMBO J. 31, 3270–3281. doi: 10.1038/emboj.2012.164
Matos, M. F., Mukherjee, K., Chen, X., Rizo, J., and Südhof, T. C. (2003). Evidence for SNARE zippering during Ca2+-triggered exocytosis in PC12 cells. Neuropharmacology 45, 777–786. doi: 10.1016/s0028-3908(03)00318-6
Melia, T. J., Weber, T., McNew, J. A., Fisher, L. E., Johnston, R. J., Parlati, F., et al. (2002). Regulation of membrane fusion by the membrane-proximal coil of the t-SNARE during zippering of SNAREpins. J. Cell Biol. 158, 929–940. doi: 10.1083/jcb.200112081
Nikolaus, J., Stöckl, M., Langosch, D., Volkmer, R., and Herrmann, A. (2010). Direct visualization of large and protein-free hemifusion diaphragms. Biophys. J. 98, 1192–1199. doi: 10.1016/j.bpj.2009.11.042
Poirier, M. A., Xiao, W., Macosko, J. C., Chan, C., Shin, Y. K., and Bennett, M. K. (1998). The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat. Struct. Biol. 5, 765–769. doi: 10.1038/1799
Rothman, J. E. (2014). The principle of membrane fusion in the cell (Nobel lecture). Angew. Chem. Int. Ed Engl. 53, 12676–12694. doi: 10.1002/anie.201402380
Scheller, R. H. (2013). In search of the molecular mechanism of intracellular membrane fusion and neurotransmitter release. Nat. Med. 19, 1232–1235. doi: 10.1038/nm.3339
Südhof, T. C., and Rizo, J. (2011). Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol. 3:a005637. doi: 10.1101/cshperspect.a005637
Sutton, R. B., Fasshauer, D., Jahn, R., and Brunger, A. T. (1998). Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395, 347–353. doi: 10.1038/26412
Takamori, S., Holt, M., Stenius, K., Lemke, E. A., Grønborg, M., Riedel, D., et al. (2006). Molecular anatomy of a trafficking organelle. Cell 127, 831–846. doi: 10.1016/j.cell.2006.10.030
Tareste, D., Shen, J., Melia, T. J., and Rothman, J. E. (2008). SNAREpin/Munc18 promotes adhesion and fusion of large vesicles to giant membranes. Proc. Natl. Acad. Sci. U S A 105, 2380–2385. doi: 10.1073/pnas.0712125105
Wang, Y., Li, L., Hou, C., Lai, Y., Long, J., Liu, J., et al. (2016). SNARE-mediated membrane fusion in autophagy. Semin. Cell Dev. Biol. 60, 97–104. doi: 10.1016/j.semcdb.2016.07.009
Weber, T., Zemelman, B. V., McNew, J. A., Westermann, B., Gmachl, M., Parlati, F., et al. (1998). SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772. doi: 10.1016/s0092-8674(00)81404-x
Wong, J. L., Koppel, D. E., Cowan, A. E., and Wessel, G. M. (2007). Membrane hemifusion is a stable intermediate of exocytosis. Dev. Cell 12, 653–659. doi: 10.1016/j.devcel.2007.02.007
Xu, Y. B., Zhang, F., Su, Z. L., McNew, J. A., and Shin, Y. K. (2005). Hemifusion in SNARE-mediated membrane fusion. Nat. Struct. Mol. Biol. 12, 417–422. doi: 10.1038/nsmb921
Yamashita, Y., Oka, M., Tanaka, T., and Yamazaki, M. (2002). A new method for the preparation of giant liposomes in high salt concentrations and growth of protein microcrystals in them. Biochim. Biophys. Acta 1561, 129–134. doi: 10.1016/s0005-2736(02)00338-3
Yang, Y., Heo, P., Kong, B., Park, J. B., Jung, Y. H., Shin, J., et al. (2015). Dynamic light scattering analysis of SNARE-driven membrane fusion and the effects of SNARE-binding flavonoids. Biochem. Biophys. Res. Commun. 465, 864–870. doi: 10.1016/j.bbrc.2015.08.111
Yang, Y., Shin, J. Y., Oh, J. M., Jung, C. H., Hwang, Y., Kim, S., et al. (2010). Dissection of SNARE-driven membrane fusion and neuroexocytosis by wedging small hydrophobic molecules into the SNARE zipper. Proc. Natl. Acad. Sci. U S A 107, 22145–22150. doi: 10.1073/pnas.1006899108
Zampighi, G. A., Zampighi, L. M., Fain, N., Lanzavecchia, S., Simon, S. A., and Wright, E. M. (2006). Conical electron tomography of a chemical synapse: vesicles docked to the active zone are hemi-fused. Biophys. J. 91, 2910–2918. doi: 10.1529/biophysj.106.084814
Keywords: SNARE, membrane fusion, hemifusion, myricetin, calcium, neurotransmitter release
Citation: Heo P, Park J-B, Shin Y-K and Kweon D-H (2017) Visualization of SNARE-Mediated Hemifusion between Giant Unilamellar Vesicles Arrested by Myricetin. Front. Mol. Neurosci. 10:93. doi: 10.3389/fnmol.2017.00093
Received: 29 December 2016; Accepted: 20 March 2017;
Published: 31 March 2017.
Edited by:
Cong Ma, Huazhong University of Science and Technology, ChinaReviewed by:
Masahiro Kono, Medical University of South Carolina, USACopyright © 2017 Heo, Park, Shin and Kweon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dae-Hyuk Kweon, ZGhrd2VvbkBza2t1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.