 Angélique Boerboom
Angélique Boerboom Valérie Dion
Valérie Dion Alain Chariot
Alain Chariot Rachelle Franzen
Rachelle Franzen- 1GIGA-Neurosciences, University of Liège, Liège, Belgium
- 2GIGA-Molecular Biology of Diseases, University of Liège, Liège, Belgium
- 3Walloon Excellence in Lifesciences and Biotechnology (WELBIO), Wavre, Belgium
Schwann cell incredible plasticity is a hallmark of the utmost importance following nerve damage or in demyelinating neuropathies. After injury, Schwann cells undergo dedifferentiation before redifferentiating to promote nerve regeneration and complete functional recovery. This review updates and discusses the molecular mechanisms involved in the negative regulation of myelination as well as in the reprogramming of Schwann cells taking place early following nerve lesion to support repair. Significant advance has been made on signaling pathways and molecular components that regulate SC regenerative properties. These include for instance transcriptional regulators such as c-Jun or Notch, the MAPK and the Nrg1/ErbB2/3 pathways. This comprehensive overview ends with some therapeutical applications targeting factors that control Schwann cell plasticity and highlights the need to carefully modulate and balance this capacity to drive nerve repair.
Introduction
The peripheral nervous system (PNS) shows a surprising capacity of regeneration compared to the central nervous system (CNS). This ability of peripheral nerves to recover quickly following damage is to a large extent due to the remarkable plasticity of Schwann cells (SCs), the glial cells of the PNS (Jessen et al., 2015). SCs are derived from neural crest cells that differentiate into SC precursors and then into immature SCs between E12 and E15 in mice. Around birth, axonal sorting and myelination start in the peripheral nerves. Some SCs establish a 1:1 relationship with large-diameter axons, wrap them multiple times to form a thick and compact myelin sheath. Myelin-forming SCs allow the fast conduction of action potentials by insulating the axons. The non-myelinating SCs typically associate with several small-diameter axons to form Remak bundles. Nerve homeostasis, trophic support and myelin maintenance are other important functions of SCs in adulthood (Jessen and Mirsky, 2005).
Besides their major roles in normal nerve physiology, SCs play a key function for repair in many pathological conditions thanks to their striking plasticity (Zochodne, 2008). For example, after a peripheral nerve injury, they are capable of switching into a SC immature-like phenotype that drives nerve repair. Over the last decades, major progress has been made in unraveling molecular mechanisms and signaling pathways that drive SC dedifferentiation and regulate their plasticity. In this review, we will update and discuss the recent studies that identified molecular components involved in SC plasticity and their possible therapeutic implications.
Remarkable Plasticity and Regenerative Properties of Schwann Cells After Peripheral Nerve Injury
The unique plasticity of SCs has been extensively studied in transgenic animals after cut or crush injuries. Indeed, following a nerve lesion, quiescent, highly specialized myelinating and non-myelinating SCs reprogram into proliferative progenitor-like repair SCs that drive the entire regeneration process (Chen et al., 2007; Jessen and Mirsky, 2016). The reprogramming of SCs in physiopathological conditions is most of the time defined as dedifferentiation. However, transdifferentiation seems more appropriate, as repair SCs, besides re-expressing immature SC markers, exhibit completely different features (Arthur-Farraj et al., 2012). Indeed, the injury-induced conversion of mature SCs in regeneration-promoting cells is an active phenomenon. It involves a down-regulation of pro-myelinating genes including early growth response 2 (Egr-2, more frequently named Krox-20), POU domain class 3 transcription factor 1 (Pou3f1 or Oct-6), myelin protein zero (MPZ) or myelin basic protein (MBP) as well as an up-regulation of markers of immature, de-differentiated SCs such as c-Jun, low affinity neurotrophin receptor (p75NTR) or glial fibrillary acidic protein (GFAP) but also specific repair-supportive genes (Jessen and Mirsky, 2008). Following damage, the nerve undergoes a series of complex multicellular and molecular events in which SCs play a role of orchestrator (Figure 1).
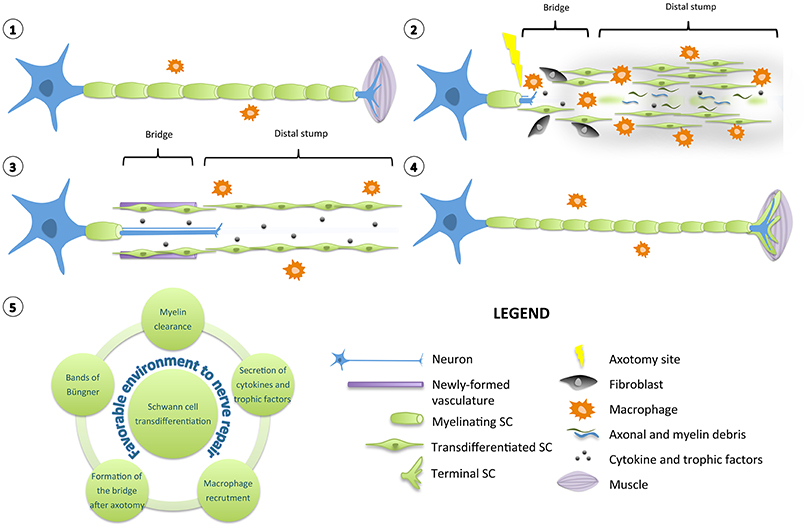
Figure 1. Diagram of Schwann cell response to nerve injury. (1) Schematic representation of a single neuron with myelinating SCs and resident macrophages. For simplification, the basal lamina around SCs is not shown. (2) After injury, the nerve distal to the injury site degenerates and undergoes a series of complex multicellular and molecular events in which SCs play a role of orchestrator. SCs transdifferentiate into repair-promoting cells, creating a permissive and favorable environment for nerve regeneration. SCs downregulate pro-myelinating genes and clear their myelin sheaths. They proliferate, secrete several pro-inflammatory cytokines and trophic factors that support glial and neuronal survival and regrowth. Axonal and myelin debris are also phagocyted by resident and blood-derived macrophages recruited by SCs. SCs interact with fibroblasts to build a bridge between the two stumps of the nerve over the lesion site. (3) Newly formed vasculature guides the SCs and the growing axons through the lesion site. In the distal stump, SCs align in tracts named bands of Büngner to provide a trophic and physical support for axons to regrow. (4) After axonal regeneration, transdifferentiated SCs readily exit the cell cycle, differentiate again and remyelinate the axons to support the complete functional recovery. However, the newly-formed myelin sheaths are most of the time shorter and thinner than expected based on axonal diameter. Specialized terminal SCs direct reinnervation by helping the axons to find their paths toward their initial targets. (5) Diagram displaying the various roles played by a transdifferentiated SC to create a favorable environment for nerve repair.
Quickly after nerve injury, damaged axons in the distal stump degenerate in an active process called Wallerian degeneration (Waller, 1850). Yet unidentified signals from damaged nerves induce the reprogramming of SCs. These downregulate pro-myelinating genes and start clearing their myelin sheaths through a mechanism of autophagy called myelinophagy (Gomez-Sanchez et al., 2015). Axonal and myelin debris are also phagocyted by resident and blood-derived macrophages recruited by SCs (Hirata and Kawabuchi, 2002; Lee et al., 2006; Barrette et al., 2008). An inflammatory reaction occurs: many blood cells invade the lesion site and secrete numerous cytokines and chemokines (Martini et al., 2008; Gaudet et al., 2011; Rotshenker, 2011). Following nerve axotomy, particularly, the basal lamina of SCs and the connective tissue are interrupted (Zochodne, 2008). A tissue bridge is formed between the two stumps of the nerve over the lesion site. Fibroblasts play a major role in building this bridge by interacting with SCs (Parrinello et al., 2010). Newly formed vasculature is also crucial to guide the SCs and the growing axons through the lesion site (Cattin et al., 2015). Many chemical and physical interactions happen between the actors present in the injured nerve, creating a permissive and favorable environment for regeneration (Cattin and Lloyd, 2016).
Irrespective of whether the injury is a crush or a cut, repair SCs in the distal stump proliferate, secrete several trophic factors that support glial and neuronal survival and regrowth including artemin, brain-derived neurotrophic factor (BDNF) or glial cell line-derived neurotrophic factor (GDNF) (Fernandez-Valle et al., 1995; Kim et al., 2000; Boyd and Gordon, 2003; Fontana et al., 2012). They also align in tracts named bands of Büngner and provide a trophic and physical support for axons to regrow and reinnervate correctly their targets (Weinberg and Spencer, 1978; Stoll and Müller, 1999). At the neuromuscular junction, specialized terminal SCs direct reinnervation by helping the axons to find their paths toward their appropriate sites (Son and Thompson, 1995). After axonal regeneration, repair SCs readily exit the cell cycle and differentiate again into myelinating and non-myelinating SCs to support the complete functional recovery. Nevertheless, most of the time, the newly-formed myelin sheaths are shorter and thinner than expected based on axonal diameter (Schröder, 1972).
Molecular Mechanisms that Modulate Schwann Cell Plasticity
The adaptive reprogramming of SCs after a nerve injury is a prerequisite for regeneration. Therefore, understanding the molecular components that regulate the phenotypic change of adult specialized SCs into repair-supportive SCs can lead to the development of new therapeutics that can boost repair in various peripheral neuropathies. In this section, we will review the most relevant molecular components or pathways that have been identified for controlling SC plasticity, the negative regulation of myelination and their response to nerve injury (Tables 1, 2; Figure 2).
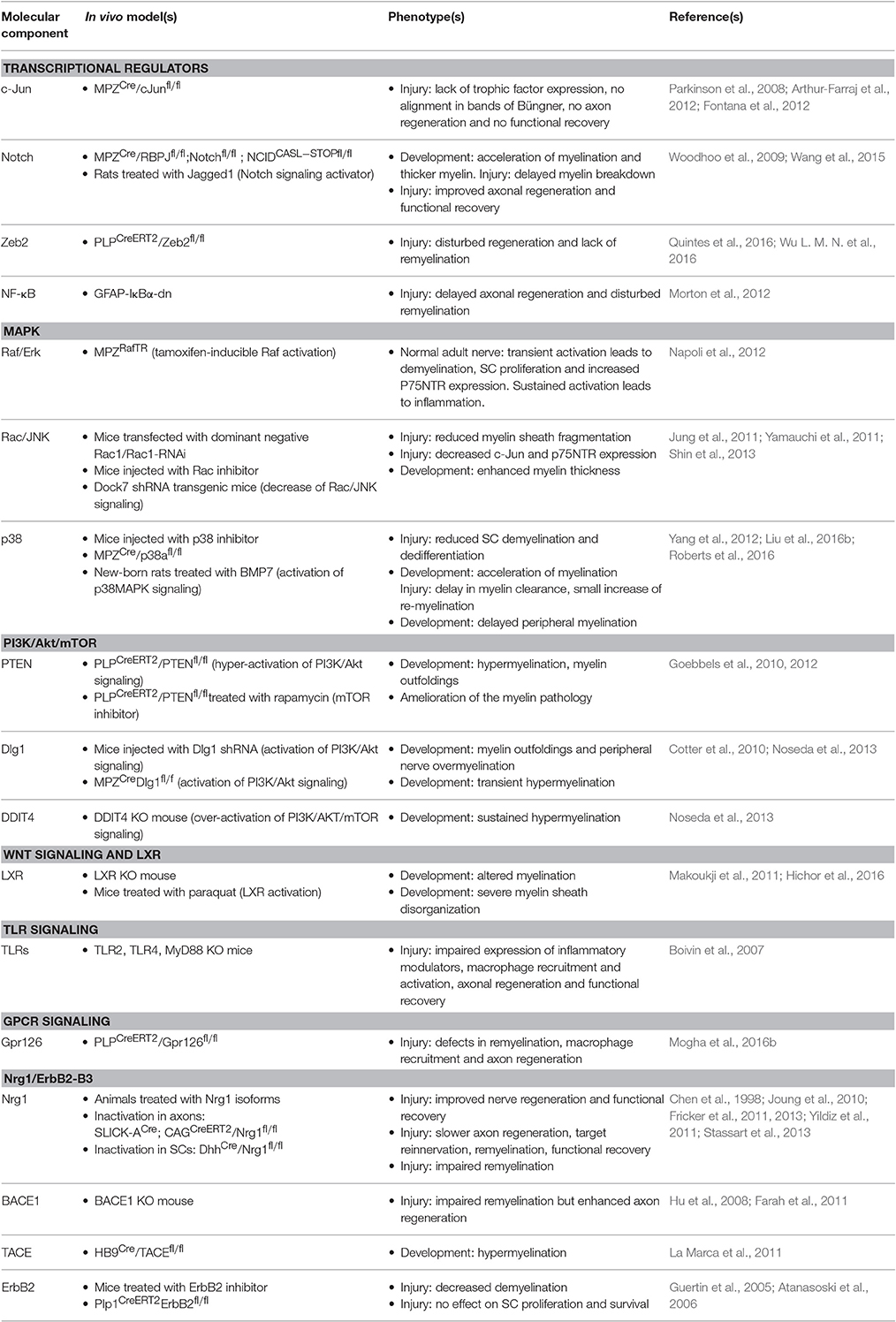
Table 1. Most relevant molecular components involved in Schwann cell plasticity, negative regulation of myelination and nerve repair.
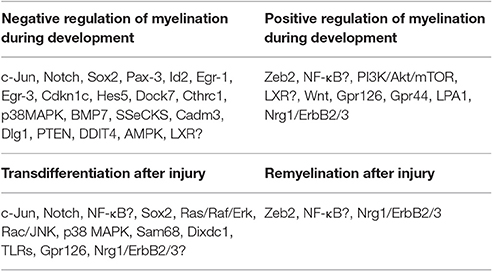
Table 2. Summary of the regulators and their roles in SCs.
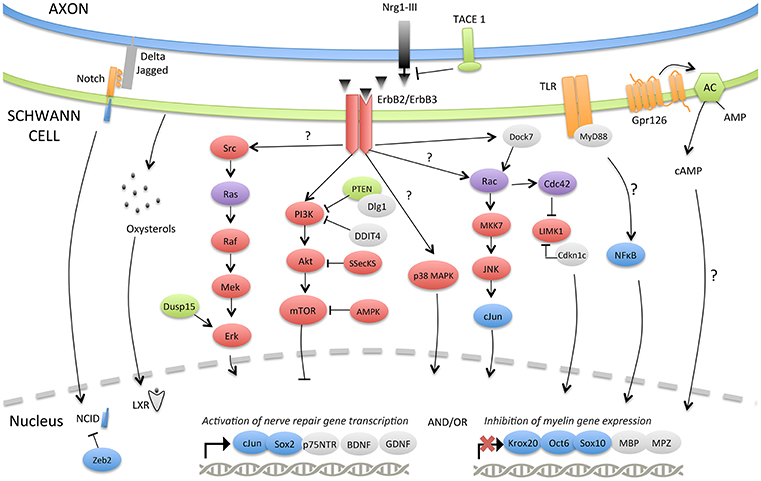
Figure 2. Molecular signaling pathways involved in SC plasticity, negative regulation of myelination and nerve repair. The injury-induced reprogramming of SCs in regeneration-promoting cells involves a down-regulation of pro-myelinating genes including Krox-20, Oct-6, Sox10, MPZ, or MBP as well as an up-regulation of markers characteristic of immature, de-differentiated SCs such as c-Jun and p75NTR but also specific repair-supportive proteins like BDNF or GDNF. The signaling pathways or molecular components activating (c-Jun, Notch, MAPKs, TLRs, Gpr126, NF-κB) or inhibiting (PI3K/Akt/mTOR) this reprogramming are represented. Some negative regulators of myelination during development (TACE1, LXRs, Dusp15, PTEN, Dlg1, DDIT4, Dock7, Zeb2) and possibly playing a role after injury are also shown.
Transcriptional Regulators
The Transcription Factor c-Jun
The transcription factor c-Jun does not appear to be essential during SC development but multiple arguments demonstrate its central function in SC reprogramming. Firstly, the time course of c-Jun expression supports its role in SC plasticity. Indeed, c-Jun is down-regulated post-natally during SC differentiation and myelination and is highly up-regulated under pathological conditions such as peripheral nerve injuries, demyelinating diseases or other peripheral neuropathies (Stewart, 1995; Parkinson et al., 2008; Hutton et al., 2011; Hantke et al., 2014; Klein et al., 2014). Secondly, c-Jun is a negative regulator of myelination and a cross-antagonist of Krox-20, one of the main pro-myelinating transcription factors. Deletion of c-Jun in SCs in vitro strikingly facilitates Krox-20-induced expression of MBP or MPZ. Conversely, forced expression of c-Jun inhibits the expression of myelin proteins. Moreover, the expression of c-Jun and Krox-20 is mutually exclusive: while immature SCs express relatively high levels of c-Jun and low levels of Krox-20, myelinating cells switch to high levels of Krox-20 and low levels of c-Jun (Parkinson et al., 2004, 2008). Thirdly, strong evidence in vivo exhibits the involvement of c-Jun in major aspects of the SC response following nerve injury. Myelination during development occurs normally in the c-Jun conditional knockout mice (Parkinson et al., 2008). However, the absence of c-Jun following a nerve lesion results in a failure of generating the repair SCs, leading to impaired axonal regeneration, target reinnervation and functional recovery. The myelin sheath degradation and clearance are also strikingly slowed in the absence of c-Jun in vitro and in vivo. Moreover, mutant SCs fail to form the Büngner regenerative tracks and to express the adhesion molecules and trophic factors critical for nerve repair (Parkinson et al., 2008; Arthur-Farraj et al., 2012; Fontana et al., 2012).
The role of c-Jun in SCs after nerve injury is to activate a repair program to support regeneration. The high levels of c-Jun found in different peripheral neuropathies that do not involve nerve injuries like Charcot-Marie Tooth (CMT) raises the question on the precise function of c-Jun in these diseases. Does c-Jun counteract the pathology, attenuate its symptoms and promote repair or does its up-regulation cause the disease? In two models of demyelinating diseases, CMT1A and CMT1X, c-Jun is increased in SCs that retain myelin differentiation (Hantke et al., 2014; Klein et al., 2014). The myelin sheaths are also relatively normal in a mouse mutant showing a 5–8 fold c-Jun overexpression in SCs (levels that are lower than those seen after a nerve cut though) (Jessen and Mirsky, 2016). Moreover, c-Jun and repair-associated genes are up-regulated in a mouse model in which the serine/threonine-protein kinase (LKB1, also called STK11) is inactivated in SCs and in which axons are damaged without overt demyelination (Beirowski et al., 2014). This suggests that moderated c-Jun up-regulation is compatible with myelination, does not necessarily provoke demyelination and that it is sufficient to activate the transcription of injury-related genes (Jessen and Mirsky, 2016). In this context, it is likely that the role of c-Jun in those diseases is to promote neuroprotection and repair. However, if c-Jun is activated in an uncontrolled and exacerbated manner, c-Jun could also be detrimental. For example, the increase of c-Jun expression in some human schwannomas may directly or indirectly be associated with the abnormal proliferation and the absence of myelin markers (Shivane et al., 2013).
Notch Signaling
Neurogenic locus notch homolog protein (Notch) is a transmembrane receptor protein that, upon ligand binding, is cleaved and generates an intracellular domain (NICD), which acts as a transcriptional regulator. Notch controls SC proliferation and promotes the generation of immature SCs from Schwann cell precursors (SCPs) in vivo but also acts as a negative regulator of myelination. It is down-regulated as myelination proceeds and its inactivation or over-activation leads to premature or delayed myelin formation, respectively. Moreover, the inhibition of Notch signaling in adult mice decelerates myelin breakdown following a nerve lesion (Woodhoo et al., 2009). The addition of recombinant jagged1, an activator of Notch, in rat nerves after injury improves nerve regeneration and functional recovery (Wang et al., 2015). This indicates that stimulation of Notch signaling in SCs could represent an interesting therapeutic strategy to efficiently promote nerve repair. However, aberrant activation of Notch signaling in SCs may also participate to the development of malignant peripheral nerve sheath tumors (Li et al., 2004).
The Transcriptional Repressor Zeb2
More recently, the function of a transcriptional repressor, Zeb2, has been investigated in SC myelination and nerve repair by two different teams (Quintes et al., 2016; Wu L. M. N. et al., 2016). They both showed that mice lacking Zeb2 in SCs develop a severe peripheral neuropathy, caused by a failure of SC maturation. Zeb2 transcriptional repression of negative regulators of myelination such as Notch and SRY-related HMG-box gene 2 (Sox2) is necessary for SC lineage progression. Although Zeb2 seems dispensable for the establishment of the repair SCs following nerve injury, its absence disrupts nerve regeneration and remyelination (Quintes et al., 2016). Mechanistically, Zeb2 controls SC differentiation and remyelination by recruiting histone deacetylases 1 and 2 (HDAC1/2) and NuRD complexes and by inhibiting a Notch–Hey2 signaling axis (Wu L. M. N. et al., 2016).
NF-κB
The nuclear factor κB (NF-κB) transcription factor regulates a lot of physiological processes and mediates the physiopathological inflammatory response in many diseases. Several studies suggest that NF-κB activation is essential for the differentiation and myelination of SCs in vitro (Nickols et al., 2003; Yoon et al., 2008; Limpert and Carter, 2010). Furthermore, it has been shown that the deacetylation of NF-κB mediated by HDAC 1/2 is necessary to control myelination in the PNS (Chen et al., 2011). Paradoxically, by using transgenic mice in which NF-κB activation is inhibited in SCs, Morton and colleagues demonstrated that NF-κB is dispensable for developmental myelination in vivo (Morton et al., 2013). However, they showed that the inhibition of NF-κB signaling in SCs following sciatic nerve crush transiently delays axonal regeneration and remyelination (Morton et al., 2012). This highlights again the differences between immature SCs during development and transdifferentiated repair-SCs. Importantly, NF-κB activity is robustly enhanced after peripheral nerve damage and in inflammatory neuropathies (Andorfer et al., 2001; Laura et al., 2006; Smith et al., 2009; Fu et al., 2010). The different NF-κB-induced transcriptional targets responsible for SC beneficial effects after nerve injury still have to be determined. Of note, the placental growth factor (PlGF) which is up-regulated by NF-κB in SCs following injury, is critical for SC proliferation, myelin sheath degradation, macrophage invasion and axonal regeneration (Chaballe et al., 2011). Targeting NF-κB or its downstream targets in SCs may offer new therapeutic strategies for PNS regeneration. Nevertheless, it is essential to keep in mind that aberrant activation of NF-κB may also lead to schwannoma (Ammoun et al., 2014; Dilwali et al., 2015).
Sox-2, Pax-3, Id2, Egr-1, Egr-3
Other factors that regulate gene transcription have been studied in SC dedifferentiation. These are most of the time negative regulators of myelination that functionally counteract positive regulators of myelination including Krox-20 or Oct-6 and are likely to have major roles after nerve damage (Jessen and Mirsky, 2008). For instance, the transcription regulators Sox-2, paired box protein 3 (Pax-3), DNA-binding protein inhibitor 2 (Id2) and early growth response proteins 1 and 3 (Egr-1 and Egr-3) could represent good candidates for regulating SC dedifferentiation in damaged nerves. As for c-Jun or Notch, they are inactive or expressed at low levels in myelinating cells and re-expressed at high levels under conditions that lead to SC dedifferentiation such as nerve injury (Kioussi et al., 1995; Nikam et al., 1995; Stewart et al., 1997; Le et al., 2005; Parkinson et al., 2008; Doddrell et al., 2012). In vitro, Sox2, Pax3, and Id2 decrease or increase myelin gene expression when overexpressed or depleted, respectively (Kioussi et al., 1995; Mager et al., 2008; Parkinson et al., 2008; Doddrell et al., 2012). Egr1 and Egr3 are both necessary to up-regulate p75NTR in SC primary cultures, known to be re-expressed in SCs following a nerve injury (Nikam et al., 1995; Topilko et al., 1997; Gao et al., 2007). It remains to be unraveled by in vivo evidence whether those different factors are required for SC response after nerve injury.
Besides its likely role in SC plasticity, it has been shown that Sox-2 is necessary in SCs for the formation of a nerve bridge after a complete transection. This process is mediated by an ephrin-B/EphB2 signaling between fibroblasts and SCs that initiates cell sorting, followed by directional collective cell migration of SCs out of the nerve stumps to guide axons across the injury wound (Parrinello et al., 2010).
MAPK Signaling
A growing number of studies provide evidence on the functions of mitogen activated protein kinase (MAPK) family proteins in the regulation of SC plasticity. Extracellular signal regulated kinase (Erk), c-jun N-terminal kinase (JNK) and p38 MAP kinase are rapidly and highly activated in the SCs of the distal stump following a nerve lesion and play specific, overlapping or complementary roles in the SC response after injury (Sheu et al., 2000; Harrisingh et al., 2004; Zrouri et al., 2004; Agthong et al., 2006; Parkinson et al., 2008; Lee et al., 2014; Ronchi et al., 2016).
Ras/Raf/Erk
Harrisingh and coworkers were the first ones to demonstrate the role of the Ras/Raf/Erk signaling in SC dedifferentiation (Harrisingh et al., 2004). They showed that the ectopic activation of Raf, an upstream activator of Erk, suppresses the differentiation of primary SCs induced by cyclic adenosine monophosphate (cAMP). Raf activation in cultured differentiated SCs also drives their dedifferentiation. Importantly, they demonstrated that sustained activation of the Ras/Raf/Erk pathway in myelinated dorsal root ganglion (DRG) neuron-SC co-cultures induces demyelination even in the presence of normal axon signaling. Consistent with this idea, another paper reported that the dual specificity phosphatase 15 (Dusp15) is necessary for full activation of Erk and that its activation represses the expression of several myelin genes including MBP (Rodríguez-Molina et al., 2017). While high and sustained activation of Ras/Raf/Erk signaling seems essential for SC plasticity, Ras over-activation might also be deleterious and lead to the development of SC-derived tumors in neurofibromatosis type 1 patients (Harrisingh et al., 2004). A strong activation of Erk has actually been reported in human primary schwannoma cells (Ammoun et al., 2008).
More recently, the strategy of Raf high activation has been used in vivo to determine whether this signaling was central in controlling SC plasticity (Napoli et al., 2012). Napoli et al. demonstrated that a tamoxifen-inducible Raf transgene in SCs is sufficient to induce the down-regulation of myelin proteins and the expression of dedifferentiation markers such as p75NTR, even in the absence of axonal damage. The duration of Raf activation, controlled by the period of tamoxifen treatment, determines the time frame of SC demyelination and proliferation. A prolonged tamoxifen treatment and activation of Raf in SCs also induces an inflammatory response, which includes the breakdown of the blood-nerve barrier and the infiltration of macrophages, which promote nerve regeneration.
Although the repression of Erk signaling with specific inhibitors blocks SC mitosis, cytokine expression and demyelination, it only prevents partially the induction of p75NTR or GDNF and the shutdown of myelin gene expression (Napoli et al., 2012; Shin et al., 2013). Thus, even if the sustained and pronounced activation of the Ras/Raf/Erk pathway is fundamental in regulating SC plasticity, this supports the idea that several independent mechanisms control multiple aspects of SC plasticity. Moreover, different studies showed that Erk activation is actually pro-myelinating and that its inhibition blocks SC differentiation and myelination in vivo (Grossmann et al., 2009; He et al., 2010; Newbern et al., 2011). Therefore, Erk signaling is necessary both for the differentiation of SCPs and for the dedifferentiation of adult SCs following a nerve injury. The reconciling explanation could be that distinct levels of Erk activity would define the state of SC differentiation. Low or basal activity would be required for SC differentiation while high Erk levels would drive dedifferentiation and proliferation (Napoli et al., 2012; Newbern and Snider, 2012).
Finally, the role of c-Jun as a downstream target of Erk signaling is an attractive idea but it is actually quite controversial and further in vivo studies are necessary to better understand the signaling mechanisms downstream of Erk in SC plasticity. Indeed, Syed et al. showed that Erk inhibition abolishes both the demyelination and the expression of c-Jun induced by Neuregulin 1 (Nrg1) treatment in SC-DRG neuron co-cultures (Syed et al., 2010). Additionally, the Lloyd group indicated that c-Jun is strongly up-regulated after Raf over-activation in SCs in vivo (Napoli et al., 2012). However, others reported that the induction of c-Jun and SC dedifferentiation triggered by cAMP removal in vitro depends on JNK rather than on Erk activity (Monje et al., 2010; Shin et al., 2013). Actually, other data revealed that Rac/JNK and Raf/Erk might be responsible for different complementary functions in SC plasticity. For example, microarray experiments showed that the expression of regeneration-associated genes, including p75NTR or GDNF is dependent on JNK but not on Erk signaling. In addition, it has been demonstrated that most Erk-dependent genes in dedifferentiated SCs do not require Rac activity (Shin et al., 2013).
Rac/JNK
JNK is another MAPK that plays important functions in SCs. It has been demonstrated that c-Jun activation by JNK is essential for SC migration and proliferation in vitro (Parkinson et al., 2001, 2004; Yamauchi et al., 2003). Parkinson and colleagues showed that overexpressing the MAPK kinase 7 (MKK7), a protein upstream of JNK, in SC primary cultures, inhibits myelin gene expression by elevating c-Jun, suggesting a role for JNK signaling in SC plasticity (Parkinson et al., 2008). This was confirmed by Monje et al. who revealed that a reduction of cAMP in vitro is sufficient to induce SC dedifferentiation through a JNK-dependent mechanism (Monje et al., 2010). More recently, it has also been demonstrated that the active Rac1 GTPase (Rac), an upstream regulator of MKK7 and JNK, inhibits SC differentiation. In SC cultures, Rac up-regulates c-Jun and down-regulates Krox-20 through the MKK7/JNK pathway, but not through the Raf/Erk pathway. Furthermore, MKK7 activation and the induction of c-Jun observed in sciatic nerves after axotomy were blocked by Rac inhibition (Shin et al., 2013). In addition, Rac activation has been observed in the distal stump of injured nerves, and specific Rac inhibition with a dominant negative Rac1 and Rac1-RNAi decelerates myelin sheath fragmentation. Rac would actually promote myelin sheath fragmentation by controlling actin polymerization in the Schmidt-Lantermann incisures of SCs (Jung et al., 2011).
The cyclin-dependent kinase inhibitor 1 (Cdkn1c), also called p57Kip2, is a protein first known to block G1/S transition during cell cycle that also regulates actin filament dynamics through the translocation of LIM domain kinase 1 (LIMK1), a downstream effector of Rac/Cdc42/Rho (Yokoo et al., 2003; Heinen et al., 2013). Knocking down Cdkn1c in cultured SCs leads to cell cycle exit, morphological changes and up-regulation of myelin genes. In SC-DRG neuron co-cultures, Cdkn1c depletion accelerates myelination. A global gene expression analysis also revealed a shift of the transcriptional expression program toward the pattern of differentiating SCs, strongly suggesting a role for Cdkn1c as an important negative regulator of myelination (Heinen et al., 2008). The same team also showed that Cdkn1c is a direct target of the enhancer of zeste homolog 2 (EZH2) and identify Hes5 as a new transcriptional repressor of myelin genes (Heinen et al., 2012). Indeed, EZH2 binds Cdkn1c promoter and its suppression results in a decrease of histone H3K27 trimethylation, an induction of Cdkn1c expression and a down-regulation of myelin genes in cultured primary SCs.
Yamauchi and co-workers reported that the atypical guanine-nucleotide exchange factor dedicator of cytokinesis protein 7 (Dock7), activates Rac/Cdc42/JNK signaling in SCs to positively regulate migration and negatively regulate differentiation and myelination (Yamauchi et al., 2008, 2011). Indeed, knocking down Dock7 in primary SCs results in an increase of cAMP-mediated differentiation and in a shorter duration of activation of Rac/Cdc42 and JNK while silencing Dock7 in mice increases myelin thickness.
Finally, collagen triple helix repeat containing 1 (Cthrc1) is another protein that has been linked to Rac activation in SCs. Apra and colleagues investigated Cthrc1 function in SCs both in vitro and in vivo (Apra et al., 2012). They showed that its silencing inhibits SC proliferation and promotes their migration and myelination in vitro. By using a transgenic mouse line, they demonstrated that the overexpression of Cthrc1 in SCs leads to a delayed myelination with SCs maintaining a proliferative state. More recently, Zhou et al identified miR-9 as an important negative regulator of SC migration targeting Cthrc1 after nerve injury (Zhou et al., 2014). The up-regulation of miR-9 decreases SC migration in culture and following nerve injury. miR-9 has also been shown to directly target Cthrc1, which in turn inactivates downstream Rac. Finally, miR-9 is down-regulated at a speed comparable to the rate of SC migration during nerve regeneration, suggesting that miR-9 down-regulation could be linked to the phenotypic modulation of SCs after nerve injury.
All these data confirm that Rac/JNK signaling is essential for SC plasticity, proliferation and migration following nerve injury. However, stimulating this pathway for nerve repair therapy must be cautioned as its upregulation has also been observed in primary human schwannoma cells (Kaempchen et al., 2003).
p38 MAPK
Yang and co-workers provided the most relevant data that suggest a role for p38 MAPK in SC plasticity and their response to injury (Yang et al., 2012). They reported that inhibition of p38 MAPK activity in mice blocks SC demyelination and dedifferentiation following nerve injury, despite ongoing axon degeneration. In myelinating co-cultures, they also showed that p38 MAPK mediates myelin breakdown induced by Nrg1. Finally, while the ectopic activation of p38 MAPK is sufficient to provoke myelin breakdown and c-Jun induction, its inhibition in SC-DRG neuron co-cultures promotes myelination. Roberts and collaborators recently investigated the in vivo function of the major p38 isoform in mouse conditional knockouts. In agreement with previous findings, the lack of p38alpha in SCS accelerates myelination during development, delays myelin clearance and slightly increases remyelination following injury. However, axonal regeneration and functional repair were not affected in the absence of p38alpha (Roberts et al., 2016).
A recent study on bone morphogenetic protein 7 (BMP7) in SCs revealed a role for this protein as a negative regulator of myelination through the activation of p38 MAPK signaling (Liu et al., 2016b). Liu and collaborators showed that BMP7 expression is inversely correlated with myelin gene expression during normal peripheral myelination. In addition, BMP7 treatment decreases cAMP-induced myelin gene expression and up-regulates c-Jun in primary SC cultures. By using p38 MAPK inhibitor, gene silencing and rescue experiments, they reported that p38 MAPK is responsible for BMP7-mediated suppression of myelin gene expression. Finally, when injected in newborn rats, BMP7 retards peripheral myelination.
PI3K/Akt/mTOR Signaling
Phosphatidylinositol 3-kinase (PI3K) signaling has been implicated in the regulation of proliferation, survival and differentiation of different cell types (Rameh and Cantley, 1999). The pathway is strongly activated in primary cultured SCs following treatment with soluble Nrg1 or contact with neurite membranes. Moreover, PI3K signaling is required for SC proliferation and survival in vitro (Maurel and Salzer, 2000; Li et al., 2001). Its inhibition affects the initiation of myelination but is not necessary for myelin maintenance in SC-neuron co-cultures (Maurel and Salzer, 2000). In addition, the selective activation of Akt in SCs by gene transfer results in an increased myelination in SC-DRG neuron co-cultures and allogenic nerve graft experiments. This last paper also demonstrated opposing functions for PI3K/Akt and Erk pathways in controlling SC myelination (Ogata et al., 2004). The imbalanced activation of the PI3K/Akt and Erk pathways has also been revealed in transgenic rodent models of CMT1A in which SCs develop persistent differentiation defects during early postnatal development (Fledrich et al., 2014). The transgenic expression of constitutively active Akt in SCs, however does not seem to disrupt PNS myelination in vivo (Flores et al., 2008).
Studies investigating the role of specific proteins indirectly indicated a role for PI3K/Akt signaling in the regulation of SC proliferation and differentiation after nerve injury. For example, Src-associated in mitosis of 68 kD (Sam68) has been suggested to promote SC proliferation by enhancing the PI3K/Akt pathway and to act on regeneration following sciatic nerve crush (Wu et al., 2016b). DIX domain containing-1 (Dixdc1), which is also linked to PI3K/Akt activation, promotes SC proliferation after sciatic nerve crush. Dixdc1 is up-regulated following nerve damage and its depletion leads to reduced PI3K/Akt activation and SC proliferation (Wu et al., 2016a). Consistently, 17β-estradiol has been shown to promote SC proliferation and early remyelination in the bridge after nerve transection through Akt activation (Chen et al., 2016a). Src-suppressed protein kinase C substrate (SSeCKS) has been indicated as a suppressor of SC differentiation and myelination by inhibiting Akt activation. SSeCKS is effectively down-regulated during SC differentiation and its depletion promotes Akt phosphorylation and myelin gene expression in cAMP-treated primary cultures or myelination when co-cultured with DRG neurons (Ji et al., 2010). SSeCKS is also essential for SC adhesion, spreading and migration (Yan et al., 2009). In addition, Chen and collaborators showed that cell adhesion molecule 3 (Cadm3) might act as a negative modulator of PNS myelination. Cadm3 interferes with PI3K/Akt activation and inhibits Schwann cell myelination in vitro (Chen M. S. et al., 2016).
The mammalian disks large homolog 1 (Dlg1) inhibits PI3K/Akt signaling via its interaction with the phosphatase and tensin homolog deleted on chromosome 10 (PTEN). This mechanism is necessary to transiently inhibit the axonal stimulation of myelination and to limit myelin sheath thickness. Myelin outfoldings and overmyelination followed later by pathologic demyelination are actually observed after Dlg1 silencing via RNA interference in mouse sciatic nerves (Cotter et al., 2010). Dlg1/PTEN functions and consequently Akt/mTOR signaling are also disturbed in different mouse models of demyelinating neuropathies and peripheral nerve sheath tumors (Bolino et al., 2004; Cotter et al., 2010; Goebbels et al., 2010, 2012; Keng et al., 2012). More specifically, Goebbels et al showed that mice lacking PTEN develop a tomaculous neuropathy characterized by focal hypermyelination and myelin outfoldings. Moreover, treating these mice with rapamycin, a mTOR inhibitor, significantly ameliorates the pathology (Goebbels et al., 2010, 2012). Another study on PTEN signaling showed that the hypermyelination observed in mice lacking Dlg1 is actually transitory. This study also identified DNA damage-inducible transcript 4 protein (DDIT4) as a sustained negative regulator of myelination. High DDIT4 expression in SCs precedes the peak of Dlg1 and Akt activity in peripheral nerves. Loss of DDIT4 expression both in vitro and in vivo results in sustained hypermyelination and enhanced PI3K/Akt/mTOR activation (Noseda et al., 2013). Therefore, it is likely that PI3K/Akt signaling has to be precisely regulated and balanced to induce SC differentiation or dedifferentiation.
mTOR is a signaling pathway that integrates a lot of signals (growth factors, nutrients, energy and oxygen) necessary for growth and proliferation (Norrmén and Suter, 2013). Sherman and collaborators demonstrated that the inactivation of mTOR in SCs provokes an arrest of myelination after axonal sorting and reduces axon growth (Sherman et al., 2012). More recently, it has been reported that the conditional ablation of raptor, an essential component of the mTORC1 complex, in SCs leads to hypomyelination and abnormal lipid biosynthesis (Norrmén et al., 2014). The pathways that converge to mTOR are numerous and include Nrg1/ErbB, PI3K/Akt, MAPKS or AMP-activated kinase (AMPK) signaling (Norrmén and Suter, 2013). AMPK is a heterotrimeric serine/threonine protein kinase acting as an inhibitor of mTOR and described as a crucial energy sensor important to maintain energy homeostasis (Hardie et al., 2012). Recently, Liu and colleagues revealed that AMPK is a critical negative regulator of SC myelination in the PNS (Liu et al., 2016a). They first showed that AMPK is gradually decreased as myelination proceeds. The high activation of AMPK decreases myelin gene expression and stimulates c-Jun expression in SCs whereas its inhibition or depletion induces myelin gene expression. The injection of an inhibitor of AMPK in newborn rats downregulates c-Jun expression, enhances lipid and protein synthesis and increases myelin gene expression and myelin sheath thickness.
Wnt Signaling and LXR
Wnt/β-catenin pathway has been demonstrated to be crucial for PNS development. Cell cultures and genetic experiments revealed that Wnt signaling is required for SC lineage progression, proliferation and myelination (Hari et al., 2002; Lee et al., 2004; Gess et al., 2008; Grigoryan et al., 2013). Recently, it has been proposed that interplay between Wnt pathway and liver X receptors (LXR) induces the fine-tuning of myelin gene expression (Makoukji et al., 2011). Oxysterols acting as ligands for LXR, are natural compounds that originate from the enzymatic oxidation of cholesterol. They are involved in cholesterol homeostasis and in the progression of different neurodegenerative disorders (Lütjohann et al., 2000; Russell, 2000; Leoni et al., 2002). Makoukji and co-workers provided evidence for the implication of LXR and oxysterols in the negative regulation of myelin gene expression (Makoukji et al., 2011). They showed that oxysterols are expressed in a SC line and in sciatic nerves and that they repress the gene expression of MPZ and PMP22 by a mechanism involving LXR α and β and the inhibition of the Wnt/β-catenin-dependent pathway. Unexpectedly, although myelin gene transcripts are up-regulated in mice lacking LXR α and β, presumably due to increased β-catenin activity, myelin protein expression is decreased and myelin sheath thickness is reduced. They conclude that this hypomyelination in the knockout animals may be the consequence of an altered cholesterol homeostasis and an inefficient myelin protein trafficking from the endoplasmic reticulum.
Recently, paraquat, a redox-active herbicide, has been shown to elicit an oxidative stress in the normal sciatic nerves of adult mice and a dramatic disorganization of myelin sheaths, causing severe locomotor and sensory deficits through a stimulation of the LXR pathway leading to reduced Wnt signaling (Hichor et al., 2016). Indeed, the authors provided evidence that paraquat alters myelin gene expression by activating LXR signaling and by preventing β-catenin recruitment on the myelin gene promoter. They also indicated that treating paraquat-exposed mice with lithium, a Wnt/β-catenin modulator acting as a GSK3 inhibitor, prevents the deleterious defects of paraquat on sciatic nerves. The effect of lithium chloride has also been studied after nerve injury. It accelerates axon and myelin debris clearance and remyelination by inhibiting GSK3 and increasing β-catenin levels (Chen et al., 2016b).
Toll-like Receptor Signaling
The Toll-like receptors (TLRs) are a class of proteins that play a key role in initiating the immune and inflammatory response. After nerve injury, damage-associated molecular patterns and ligands released from or expressed by activated or necrotic damaged cells result in the activation of TLRs in different cell types including SCs (Thakur et al., 2017). TLR3, TLR4, and TLR7 are constitutively expressed by SCs and TLR1 is upregulated following nerve injury, strongly suggesting a role for TLR signaling in SC-driven nerve repair (Goethals et al., 2010). In vitro, the addition of necrotic neurons to SCs activates their inflammatory response through TLR signaling. It increases their gene expression of inflammatory mediators such as tumor necrosis factor-alpha (TNF-α) or monocyte chemoattractant protein-1 (MCP-1) but this effect is reduced in SCs from mice lacking TLR3 (Lee et al., 2006). Boivin and collaborators provided in vivo evidence that TLR signaling is involved in WD and nerve regeneration following a nerve injury, possibly through NF-κB activation (Boivin et al., 2007). The early expression of inflammatory modulators such as MCP-1, macrophage recruitment and activation, axonal regeneration and functional recovery are impaired in the mice deficient in TLR signaling. The results are indeed similar in mice lacking TLR2, TLR4 or their adaptor, myeloid differentiation primary response gene 88 (MyD88) known to activate NF-κB. Interestingly, a single injection of TLR2 and TLR4 ligands at the site of the lesion speeds up myelin clearance and functional recovery. Besides its likely role in SC response to nerve injury, TLR signaling has also been linked to the development of neuropathic pain, making TLRs promising therapeutic targets (Thakur et al., 2017).
GPCR Signaling
To date, three different G protein-coupled receptors (GPCRs) have been demonstrated to be implicated in SC differentiation. These are Gpr126, Gpr44, and lysophosphatidic acid receptor 1 (LPA1) (Mogha et al., 2016a). Monk and colleagues were the first ones to show that a GPCR, Gpr126, is crucial for the initiation of SC myelination (Monk et al., 2009). The analysis of mice lacking LPA1 then revealed that this receptor is required for SC migration, axonal sorting and proper myelination (Anliker et al., 2013). Finally, Trimarco et al. provided in vitro and in vivo evidence for the involvement of Gpr44 and its ligand the prostaglandin D2 in PNS myelination (Trimarco et al., 2014). Very recently, Mogha and collaborators investigated the potentiel functions of Gpr126 in peripheral nerve injury and repair as its expression is maintained in adult SCs (Mogha et al., 2016b). By using an inducible conditional Gpr126 knockout model, they observed defects in remyelination, macrophage recruitment and axon regeneration following a nerve crush. Since approximately 30% of all approved drugs target GPCRs, Gpr126 may represent an attractive potential target to stimulate repair in myelinating disease or after nerve damage.
Nrg1/ErbB2/3 Signaling
Currently, the signals from damaged nerves that induce the reprogramming of SCs are not identified. Some argued that these can be linked to the Nrg1/ErbB pathway, one of the best described interdependent relationship between axons and SCs. Neuregulins form a large family of epidermal growth factor-like proteins that signal through the ErbB tyrosine kinase receptors (Falls, 2003). The Nrg1/ErbB signaling is first of all known for its critical functions in SC development (for review, Newbern and Birchmeier, 2010; Grigoryan and Birchmeier, 2015; Willem, 2016). Indeed, numerous genetic models have been generated to unravel fundamental functions of the Nrg1/ErbB signaling axis. Differential expression of Nrg1 isoforms, their proteolytic processing or ErbB receptor localization and trafficking are important parameters that control the Nrg1/ErbB pathway and its underlying functions. Depending on the developmental stage, the pathway significantly regulates SC survival, proliferation, migration, differentiation and myelination (Syroid et al., 1996; Morris et al., 1999; Leimeroth et al., 2002; Taveggia et al., 2005; Chen et al., 2006; Freidin et al., 2009; Newbern and Birchmeier, 2010). This functional diversity is presumably accomplished through the interaction of Nrg1/ErbB signaling with other pathways. For instance, Notch signaling, which is necessary for SCP proliferation and survival, increases the expression of ErbB2 in SCPs and their sensitivity to Nrg1 (Woodhoo et al., 2009). The tyrosine phosphatase Shp2 is another essential component of the Nrg-1/ErbB pathway that promotes SC proliferation and migration (Grossmann et al., 2009). Later in SC development, critical interactions between Nrg1/ErbB and cAMP or NF-κB signaling are necessary for SC differentiation and myelination (Limpert and Carter, 2010; Arthur-Farraj et al., 2011). Although the importance of Nrg1/ErbB signaling in SC development is well established, its role in adult SC plasticity is more controversial. In adulthood, the pathway seems dispensable for the maintenance of the myelin sheaths, as the absence of Nrg1 in peripheral axons or of ErbB2 in SCs has no effect on axon or myelin sheath integrity (Atanasoski et al., 2006; Fricker et al., 2011, 2013). However, increasing evidence indicates that Nrg1 is necessary for the SC response to nerve injury.
The fact that the Nrg1/ErbB system is selectively and highly regulated during peripheral nerve degeneration and regeneration represents a strong argument in favor of a role for this signaling pathway in SC plasticity (Carroll et al., 1997; Kwon et al., 1997; Ronchi et al., 2016). Recently, a study using a rat surgical model of delayed nerve repair showed that Nrg1 and some SC markers are highly down-regulated after chronic degeneration. Since long-term denervated Schwann cells are known to be partly responsible for delayed nerve repair, it suggests that Nrg1 plays an important function in SC activity after denervation (Ronchi et al., 2017). Guertin and colleagues showed that ErbB2 is activated in myelinating SCs after sciatic nerve axotomy and that its transient activation is sufficient to initiate SC demyelination in compartmentalized cell culture chambers. In addition, the treatment of rats with an ErbB2 inhibitor results in a reduction of demyelination after nerve transection (Guertin et al., 2005). Consistently, Syed et al demonstrated that a robust activation of ErbB2/3 via high concentration of soluble Nrg1 induces SC dedifferentiation and demyelination as well as increased c-Jun expression in primary cultures (Syed et al., 2010). Furthermore, in vitro experiments recapitulating nerve injuries provide evidence that Nrg1 treatment shows beneficial effects on SC migration and proliferation (Mahanthappa et al., 1996; Li et al., 1998). Finally, Lee and collaborators showed that Nrg1 on motor axons controls the terminal SC-mediated synapse elimination at the neuromuscular junctions (NMJs) during development. They also revealed that NMJs of adult transgenic mice overexpressing Nrg1 in motor axons exhibited continued remodeling, identifying Nrg1 as a molecular determinant for SC-driven neuromuscular synaptic plasticity (Lee et al., 2016).
Nrg1/ErbB signaling has also been linked to different pathways mentioned earlier for regulating SC plasticity. For example, Nrg1 has been proposed as the signal driving high Erk activation after injury (Napoli et al., 2012). High concentrations of Nrg1 indeed highly activates Erk signaling and induces SC dedifferentiation and demyelination (Syed et al., 2010; Newbern and Snider, 2012). In agreement with this, Tapinos and collaborators also showed that direct binding of Mycobacterium leprae onto ErbB2 receptors on SCs activates Erk signaling and promotes demyelination (Tapinos et al., 2006). Another possibility is that Nrg1 could trigger Rac/JNK activation. Indeed, the inhibition of ErbB2 signaling has been shown to prevent MKK7 activation, c-jun expression, and Rac-dependent gene transcription in cultures of sciatic nerve explants (Shin et al., 2013). ErbB2 also directly binds and activates the negative regulator of myelination, Dock7, upstream of Rac/JNK while the depletion of Dock7 attenuates Nrg1 effects in primary SC cultures (Yamauchi et al., 2008, 2011). Although these data suggest a role for Nrg1/ErbB signaling in SC dedifferentiation, in vivo studies revealed contradictory results. Atanasoski et al. showed that lack of ErbB2 in adult SCs does not affect their proliferation and survival after nerve injury, despite reduced levels of phosphorylated MAPK (Atanasoski et al., 2006). In addition, the absence of the axonal Nrg1 following a nerve crush does not disturb SC proliferation associated with nerve degeneration or the clearance of myelin debris by macrophages. Therefore, adult SCs would not require major Nrg1/ErbB signaling for their proliferation and survival after nerve damage, in contrast to what is observed in vitro and during development.
While the implication of Nrg1 signaling in the dedifferentiation of SCs is unclear, its involvement in myelination but also in remyelination after nerve injury is now well established. The transmembrane axonal isoforms Nrg1 type III signals in a juxtacrine manner through ErbB2/3 receptors on SCs and determines their myelin thickness, both during development and following a nerve lesion (Michailov et al., 2004; Taveggia et al., 2005; Fricker et al., 2011, 2013). The axonal Nrg1 is rate limiting but not essential for remyelination since compensations are observed at later stages (Fricker et al., 2013). All the same, myelin sheaths are often thinner after injury, suggesting an insufficent simulation by axonal signals (Schröder, 1972). Another mechanism is actually induced in SCs to supplement these poor neuronal cues. Nerve damage triggers the expression of soluble Nrg1 type I by SCs themselves. This particular autocrine and paracrine signaling in SCs is only required after nerve injury and promotes SC survival, redifferentiation and remyelination (Fricker and Bennett, 2011; Stassart et al., 2013).
Besides the importance of Nrg1/ErbB signaling in SC remyelination, it also plays a key role in repair processes after nerve injury (Fricker and Bennett, 2011). Fricker and colleagues showed that the absence of Nrg1 in adult axons after sciatic nerve crush results in defects of remyelination but also in slower axon regeneration, impaired muscle reinnervation and delayed functional recovery (Fricker et al., 2011, 2013). In a mouse model lacking β-Site amyloid precursor protein cleaving enzyme 1 (BACE1), a protein known to cleave and activate Nrg1 type III, SC remyelination is impaired and nerve repair is surprisingly accelerated (Hu et al., 2008). Indeed, knockout mice show an accelerated clearance of axonal and myelin debris from degenerated fibers, faster axonal regeneration and earlier reinnervation of neuromuscular junctions, compared to littermate controls (Farah et al., 2011). The enhanced myelin clearance might clearly be due to the initial hypomyelination observed in adult BACE1 KO sciatic nerves. However, how BACE1 deletion enhances peripheral regeneration still needs to be clarified. Another enzyme capable of cleaving the axonal Nrg1, counteracting BACE1 and interfering with SC differentiation is the tumor necrosis factor-α–converting enzyme (TACE, also named ADAM17). TACE depletion in SC-DRG neuron co-cultures increases myelination. The absence of TACE in motoneurons in vivo results in hypermyelination. The function of TACE is neuron autonomous since its knockdown in SCs does not affect myelination. Mechanistically, TACE cleaves Nrg1-III, which is no longer capable of activating PI3K signaling and inducing myelination (La Marca et al., 2011). Therefore, TACE can be considered as a modulator of NRG1 type III activity and as a negative regulator of SC myelination. The role of Nrg1 in peripheral regeneration has also been demonstrated in different studies examining the effects of Nrg1 treatment following nerve injury. The application of exogenous Nrg1 or strategies that elevates Nrg1 levels after nerve damage improve myelin clearance, axon regeneration, remyelination and functional recovery (Chen et al., 1998; Joung et al., 2010; Fricker and Bennett, 2011; Yildiz et al., 2011). Therefore, treatment with recombinant forms of Nrg1 may represent interesting therapeutic avenues to improve nerve repair following injury. It is however necessary to better understand the mechanisms by which Nrg1 treatment improves nerve regeneration.
Finally, it is essential to remember that the inappropriate overactivation of Nrg1/ErbB signaling pathway may be harmful in some circumstances and lead to demyelinating neuropathies or peripheral tumors. For instance, constitutive activation of the Nrg1/ErbB pathway promotes the proliferation of peripheral neuroepithelioma and neoplastic SC line (Frohnert et al., 2003; Fallon et al., 2004). Similarly, transgenic mice over-expressing Nrg1 in myelinating SCs develop hypertrophic neuropathies and malignant peripheral nerve sheath tumors (Huijbregts et al., 2003). In addition, leprosy, which is an important cause of demyelinating neuropathy in the world, is initiated by an excessive activation of ErbB2 by M. leprae (Tapinos et al., 2006).
Pathophysiological Implications and Therapeutic Strategies
Peripheral neuropathies are responsible for significant morbidity and decreased quality of life due to weakness, sensory loss and neuropathic pain. The causes are multiple: peripheral nerve traumatic injuries, inherited genetic diseases such as CMT, metabolic disorders including diabetes, infectious, toxic and inflammatory disorders (Zhou and Notterpek, 2016). Peripheral nerves have an impressive ability to regenerate compared to the CNS. However, the clinical recovery of patients suffering from peripheral neuropathies is often incomplete due to slow regeneration rate, target mis-reinnervation and lack of a long-term regeneration-supportive environment. After injury, peripheral axons regenerate at a very slow rate of approximately 1 mm per day, depending on the lesion site. Moreover, the regenerative capacity of the PNS decreases over time. Indeed, SCs become unsupportive to regeneration after long periods and they also exhibit diminished plasticity with age (Fu and Gordon, 1995; Zochodne, 2012; Painter et al., 2014). Of note, Joshi and coworkers recently demonstrated that SCs show an altered expression of c-Jun and Cdkn1c and that their pro-regenerative functions are diminished in a model of chronic inflammatory demyelinating polyneuropathy (CIDP) (Joshi et al., 2016). A better understanding of the signaling pathways that drive SC reprogramming and plasticity is of great interest and may offer new opportunities to enhance nerve repair. SCs are the main effectors of regeneration in many peripheral diseases and recent studies clearly positioned SCs at the hub for organizing the environment to clear myelin debris, promote axonal regrowth, remyelination and allow complete functional repair following injury (Zochodne, 2012; Kim et al., 2013).
The different signaling pathways and molecular components described in the present review represent interesting targets to boost the endogenous regeneration potential of SCs. For instance, the treatment with different Nrg1 isoforms or strategies to stimulate ErbB signaling have actually been shown to improve myelin clearance, axon regeneration, remyelination and functional recovery after nerve injury in different studies (Chen et al., 1998; Joung et al., 2010; Fricker and Bennett, 2011; Yildiz et al., 2011). In a CMT1A rat model, soluble Nrg1 therapy during early postnatal life prevents abnormal developmental demyelination and subsequent axon loss observed in adulthood (Fledrich et al., 2014). The overexpression of Nrg1 in mouse models of amyotrophic lateral sclerosis (ALS) and partial muscle denervation improves functional recovery by enhancing collateral reinnervation sprouting through both Akt and Erk pathways (Mancuso et al., 2016). Notch activation through the addition of a recombinant jagged1 after injury also promotes nerve regeneration and functional recovery (Wang et al., 2015). The inhibition of mTOR signaling with rapamycin in mice that lack PTEN and develop a tomaculous neuropathy significantly ameliorates the pathology (Goebbels et al., 2012). Finally, stimulating TLR signaling at the site of the lesion accelerates myelin clearance and functional recovery (Boivin et al., 2007).
Additional molecules, not directly linked to pathways analyzed in this review, have been shown to regulate SC differentiation/dedifferentiation and improve nerve repair. For example, Fingolimod, an agonist of the sphingosine-1-phosphate receptor (S1PR), previously known to affect SC migration and myelination in vitro and currently in clinical trial for the treatment of CIDP, has been demonstrated to modulate SC plasticity (Köhne et al., 2012; Heinen et al., 2015). Fingolimod activates different dedifferentiation markers such as c-Jun, increases the expression of growth factors, slows myelination and improves axon regeneration. Therefore, Fingolimod supports the generation of a repair promoting cellular phenotype and could be used for the treatment of peripheral nerve damages and diseases (Heinen et al., 2015). Its mechanism of action, however, still remains to be elucidated. Another recent example is the addition of TGF-β1 and forskolin directly on chronically injured nerves that can reactivate the growth-supportive environment through the reprogramming of chronically denervated SCs, the re-induction of their proliferation, and the re-expression of regeneration-associated proteins (Sulaiman and Nguyen, 2016). Immunoglobulins (IVIG) are another therapeutic strategy widely used to treat immune-mediated neuropathies such as CIDP. Tzekova and colleagues demonstrated that IVIG positively influence SC differentiation and maturation and increase their potential to induce axonal outgrowth (Tzekova et al., 2015).
Other approaches that are based on SC regenerative properties and improve nerve repair can also be addressed. First, exercise and electrical stimulation have been demonstrated to promote SC-driven peripheral nerve regeneration in both animal models and in human patients (Gordon and English, 2016). Second, transplantations of SCs from healthy nerves or engineered from different tissues thanks to the emergence of the induced pluripotent stem cell technology are another interesting method to support nerve healing (Zhou and Notterpek, 2016). Actually, numerous SC-based therapies also exist to promote repair in the lesioned CNS (Matsas et al., 2008). Third, there has been major progress in developing biomaterials that improve regeneration by providing trophic factors or by supporting the regenerative functions of transplanted cells in situ (Marquardt and Sakiyama-Elbert, 2013). For example, Liu and colleagues recently showed that Salidroside and tissue engineering using SCs and poly lactic-co-glycolic acid promote peripheral nerve regeneration and functional recovery after sciatic nerve transection in rats. Salidroside increases the proliferation and regenerative function of SCs, probably through the modulation of neurotrophic factors (Liu et al., 2017). Fourth, gene therapy that allows to deliver genetic material directly into somatic cells is another option to treat neuropathies that involve gene mutations or to improve nerve regeneration after injuries (Zhou and Notterpek, 2016). In hereditary diseases including CMTs, loss or gain of function mutations could be restored through the re-expression of the absent or non-functional protein or through the down-regulation of the mutant mRNA via RNA interference strategy, respectively. For example, Kagiava and coworkers recently demonstrated that the intrathecal injection of a lentiviral vector with a myelin-specific promoter restored the expression of a neuropathy-associated gene and rescued a model of demyelinating peripheral neuropathy (Kagiava et al., 2016). Also, other studies based on gene therapy in order to improve nerve regeneration used lentiviral vectors that potentiate the therapeutic functions of SCs transplanted in artificial nerves, in autografts or present in damaged nerves (Hoyng et al., 2015). Even if SCs are known to mainly support nerve repair through an enhanced expression of neurotrophic factors that stimulate axon regeneration and myelination, this review highlights the numerous additional roles and regenerative functions of SCs in nerve injuries and in demyelinating diseases. Therefore, targeting the molecular mechanisms that regulate SC reprogramming by gene therapy can be highly promising to stimulate nerve healing. In the future, the challenges will be to optimize gene therapy in order to specifically target SCs and to create safe and regulatable vectors to potentiate their regenerative functions in the appropriate timings. Lipid-based nanoparticles such as liposomes or extracellular vesicles are other valuable tools for the delivery of small molecules or genes that can modulate signaling pathways and promote nerve repair (Takeda and Xu, 2015). In order to selectively introduce material into SCs, Lee and colleagues developed phospholipid-based liposomes that can enter SCs when injected in tail vein and showed that the nanoparticles escape the endogenous degradative mechanisms in vitro (Lee et al., 2013). In the following years, it is expected that the improvements in cell and tissue engineering and in approaches that target SC capacity to promote regeneration will lead to the development of innovative and efficient PNS repair strategies.
The different therapeutic options that are built on or that stimulate SC plasticity represent new avenues for nerve regeneration but should be considered with prudence for different reasons. First, a prolonged stimulation of SC dedifferentiation could disrupt the remyelination of the regenerated axons, necessary for rapid nerve conduction and complete functional recovery. Also, the aberrant activation of some pathways including Notch, NF-κB, Ras/Raf/Erk, Rac/JNK, or Nrg1/ErbB can lead to abnormal cell proliferation and the development of peripheral nerve tumors. The better understanding of the molecular mechanisms regulating SC plasticity is thus necessary to set up therapies that modulate both temporally and quantitatively SC capacity to drive nerve repair without affecting remyelination and increasing the chances of triggering tumor development.
Conclusions
The remarkable capacity of regeneration of the PNS compared to the CNS is mainly due to the fantastic plasticity of SCs. The molecular components involved in the negative regulation of myelination and in the reprogramming of SCs in response to nerve injury provide important mechanistic insights and new therapeutic approaches into peripheral neuropathies as well as in peripheral nerve regeneration. However, this review demonstrates the complexity of the molecular signaling pathways and reveals that little is known about the interactions between them. Moreover, the temporal and quantitative activation of the different signals identified is of great importance but is not completely elucidated. Future studies are thus required to better understand the cross-talks, networks and activation patterns and timings between the different molecular mechanisms controlling SC plasticity.
Author Contributions
Manuscript writing: AB; Manuscript revision: VD, AC, and RF.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Grants from the FSR-FNRS, the Fonds Léon Fredericq, the Special Funds of the University of Liège, the Fondation Charcot and the Association Belge contre les Maladies Neuro-Musculaires.
Abbreviations
ALS, Amyotrophic lateral sclerosis; BACE1, Beta-Site amyloid precursor protein cleaving enzyme 1; BDNF, Brain-derived neurotrophic factor; BMP7, Bone morphogenetic protein 7; Cadm3, Cell adhesion molecule 3; cAMP, Cyclic adenosine monophosphate; Cdkn1c, Cyclin-dependent kinase inhibitor 1C; CIDP, Chronic inflammatory demyelinating polyneuropathy; CMT, Charcot-Marie-Tooth; Cthrc1, Collagen triple helix repeat containing 1; EZH2, Enhancer of zeste homolog 2; Dixdc1, DIX domain containing-1; DDIT4, DNA damage-inducible transcript 4 protein; Dlg1, Disks large homolog 1; Dock7, Dedicator of cytokinesis protein 7; DRG, Dorsal root ganglion; Dusp15, Dual specificity phosphatase 15; Egr-1, Early growth response protein 1; Egr-2/Krox-20Early growth response 2; Egr-3, Early growth response protein 3; Erk, Extracellular signal regulated kinase; GDNF, Glial cell-derived neurotrophic factor; GFAP, Glial fibrillary acidic protein; GPCR, G protein-coupled receptor; Id2, DNA-binding protein inhibitor 2; IVIG, Immunoglobulins; JNK, c-jun N-terminal kinase; LKB1/STK11, Liver kinase B1/ Serine/threonine-protein kinase 11; LPA1, Lysophosphatidic acid receptor 1; LXR, Liver X receptor; MAPK, Mitogen activated protein kinase; MBP, Myelin basic protein; MPZ, Myelin protein zero; MyD88, Myeloid differentiation primary response gene 88; NF-κB, Nuclear factor-kappa B; NMJ, Neuromuscular junction; Notch, Neurogenic locus notch homolog protein; Nrg1, Neuregulin 1; NuRD, Nucleosome remodeling and deacetylase complex; p75NTR, Low affinity neurotrophin receptor; Pax-3, Paired box protein 3; PI 3-kinase, Phosphatidylinositol 3-kinase; PMP22, Peripheral myelin protein 22; Pou3f1/Oct6, POU domain class 3 transcription factor / Octamer-binding factor 6; PTEN, Phosphatase and tensin homolog deleted on chromosome 10; S1PR, Sphingosine-1-phosphate receptor; Sam68, Src-associated in mitosis of 68 kD; Sox2, SRY-related HMG-box gene 2; SSeCKS, Src-suppressed protein kinace C substrate; STK11, Serine/threonine protein kinase; TACE, Tumor necrosis factor-α–converting enzyme; TGFβ, Transforming growth factor beta; Zeb2, Zinc-finger E-box-binding homeobox 2.
References
Agthong, S., Kaewsema, A., Tanomsridejchai, N., and Chentanez, V. (2006). Activation of MAPK ERK in peripheral nerve after injury. BMC Neurosci. 7:45. doi: 10.1186/1471-2202-7-45
Ammoun, S., Flaiz, C., Ristic, N., Schuldt, J., and Hanemann, C. O. (2008). Dissecting and targeting the growth factor-dependent and growth factor-independent extracellular signal-regulated kinase pathway in human schwannoma. Cancer Res. 68, 5236–5245. doi: 10.1158/0008-5472.CAN-07-5849
Ammoun, S., Provenzano, L., Zhou, L., Barczyk, M., Evans, K., Hilton, D. A., et al. (2014). Axl/Gas6/NFκB signalling in schwannoma pathological proliferation, adhesion and survival. Oncogene 33, 336–346. doi: 10.1038/onc.2012.587
Andorfer, B., Kieseier, B. C., Mathey, E., Armati, P., Pollard, J., Oka, N., et al. (2001). Expression and distribution of transcription factor NF-κB and inhibitor IκB in the inflamed peripheral nervous system. J. Neuroimmunol. 116, 226–232. doi: 10.1016/S0165-5728(01)00306-X
Anliker, B., Choi, J. W., Lin, M.-E., Gardell, S. E., Rivera, R. R., Kennedy, G., et al. (2013). Lysophosphatidic acid (LPA) and its receptor, LPA1, influence embryonic schwann cell migration, myelination, and cell-to-axon segregation. Glia 61, 2009–2022. doi: 10.1002/glia.22572
Apra, C., Richard, L., Coulpier, F., Blugeon, C., Gilardi-Hebenstreit, P., Vallat, J., et al. (2012). Cthrc1 is a negative regulator of myelination in Schwann cells. Glia 60, 393–403. doi: 10.1002/glia.22273
Arthur-Farraj, P. J., Latouche, M., Wilton, D. K., Quintes, S., Chabrol, E., Banerjee, A., et al. (2012). c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 75, 633–647. doi: 10.1016/j.neuron.2012.06.021
Arthur-Farraj, P., Wanek, K., Hantke, J., Davis, C. M., Jayakar, A., Parkinson, D. B., et al. (2011). Mouse schwann cells need both NRG1 and cyclic AMP to myelinate. Glia 59, 720–733. doi: 10.1002/glia.21144
Atanasoski, S., Scherer, S. S., Sirkowski, E., Leone, D., Garratt, A. N., Birchmeier, C., et al. (2006). ErbB2 signaling in Schwann cells is mostly dispensable for maintenance of myelinated peripheral nerves and proliferation of adult Schwann cells after injury. J. Neurosci. 26, 2124–2131. doi: 10.1523/JNEUROSCI.4594-05.2006
Barrette, B., Hébert, M.-A., Filali, M., Lafortune, K., Vallières, N., Gowing, G., et al. (2008). Requirement of myeloid cells for axon regeneration. J. Neurosci. 28, 9363–9376. doi: 10.1523/JNEUROSCI.1447-08.2008
Beirowski, B., Babetto, E., Golden, J. P., Chen, Y.-J., Yang, K., Gross, R. W., et al. (2014). Metabolic regulator LKB1 plays a crucial role in Schwann cell-mediated axon maintenance. Nat. Neurosci. 17, 1351–1361. doi: 10.1038/nn.3809
Boivin, A., Pineau, I., Barrette, B., Filali, M., Vallières, N., Rivest, S., et al. (2007). Toll-like receptor signaling is critical for wallerian degeneration and functional recovery after peripheral nerve injury. J. Neurosci. 27, 12565–12576. doi: 10.1523/JNEUROSCI.3027-07.2007
Bolino, A., Bolis, A., Previtali, S. C., Dina, G., Bussini, S., Dati, G., et al. (2004). Disruption of Mtmr2 produces CMT4B1-like neuropathy with myelin outfolding and impaired spermatogenesis. J. Cell Biol. 167, 711–721. doi: 10.1083/jcb.200407010
Boyd, J. G., and Gordon, T. (2003). Glial cell line-derived neurotrophic factor and brain-derived neurotrophic factor sustain the axonal regeneration of chronically axotomized motoneurons in vivo. Exp. Neurol. 183, 610–619. doi: 10.1016/S0014-4886(03)00183-3
Carroll, S. L., Miller, M. L., Frohnert, P. W., Kim, S. S., and Corbett, J. A. (1997). Expression of neuregulins and their putative receptors, ErbB2 and ErbB3, is induced during Wallerian degeneration. J. Neurosci. 17, 1642–1659.
Cattin, A.-L., Burden, J. J., Van Emmenis, L., Mackenzie, F. E., Hoving, J. J. A., Calavia, N. G., et al. (2015). Macrophage-induced blood vessels guide Schwann cell-mediated regeneration of peripheral nerves. Cell 162, 1127–1139. doi: 10.1016/j.cell.2015.07.021
Cattin, A.-L., and Lloyd, A. C. (2016). The multicellular complexity of peripheral nerve regeneration. Curr. Opin. Neurobiol. 39, 38–46. doi: 10.1016/j.conb.2016.04.005
Chaballe, L., Close, P., Sempels, M., Delstanche, S., Fanielle, J., Moons, L., et al. (2011). Involvement of placental growth factor in Wallerian degeneration. Glia 59, 379–396. doi: 10.1002/glia.21108
Chen, L. E., Liu, K., Seaber, A. V., Katragadda, S., Kirk, C., and Urbaniak, J. R. (1998). Recombinant human glial growth factor 2 (rhGGF2) improves functional recovery of crushed peripheral nerve (a double-blind study). Neurochem. Int. 33, 341–351. doi: 10.1016/S0197-0186(98)00037-0
Chen, M.-S., Kim, H., Jagot-Lacoussiere, L., and Maurel, P. (2016). Cadm3 (Necl-1) interferes with the activation of the PI3 kinase/Akt signaling cascade and inhibits Schwann cell myelination in vitro. Glia 64, 2247–2262. doi: 10.1002/glia.23072
Chen, S., Velardez, M. O., Warot, X., Yu, Z.-X., Miller, S. J., Cros, D., et al. (2006). Neuregulin 1-erbB signaling is necessary for normal myelination and sensory function. J. Neurosci. 26, 3079–3086. doi: 10.1523/JNEUROSCI.3785-05.2006
Chen, Y., Guo, W., Xu, L., Li, W., Cheng, M., Hu, Y., et al. (2016a). 17β-Estradiol promotes Schwann cell proliferation and differentiation, accelerating early remyelination in a mouse peripheral nerve injury model. BioMed Res. Int. 2016:7891202. doi: 10.1155/2016/7891202
Chen, Y., Wang, H., Yoon, S. O., Xu, X., Hottiger, M. O., Svaren, J., et al. (2011). HDAC-mediated deacetylation of NF-[κ] B is critical for Schwann cell myelination. Nat. Neurosci. 14, 437–441. doi: 10.1038/nn.2780
Chen, Y., Weng, J., Han, D., Chen, B., Ma, M., Yu, Y., et al. (2016b). GSK3β inhibition accelerates axon debris clearance and new axon remyelination. Am. J. Transl. Res. 8, 5410–5420.
Chen, Z.-L., Yu, W.-M., and Strickland, S. (2007). Peripheral Regeneration. Annu. Rev. Neurosci. 30, 209–233. doi: 10.1146/annurev.neuro.30.051606.094337
Cotter, L., Ozcelik, M., Jacob, C., Pereira, J. A., Locher, V., Baumann, R., et al. (2010). Dlg1-PTEN interaction regulates myelin thickness to prevent damaging peripheral nerve overmyelination. Science 328, 1415–1418. doi: 10.1126/science.1187735
Dilwali, S., Briët, M. C., Kao, S.-Y., Fujita, T., Landegger, L. D., Platt, M. P., et al. (2015). Preclinical validation of anti-nuclear factor-κ B therapy to inhibit human vestibular schwannoma growth. Mol. Oncol. 9, 1359–1370. doi: 10.1016/j.molonc.2015.03.009
Doddrell, R. D. S., Dun, X., Moate, R. M., Jessen, K. R., Mirsky, R., and Parkinson, D. B. (2012). Regulation of Schwann cell differentiation and proliferation by the Pax-3 transcription factor. Glia 60, 1269–1278. doi: 10.1002/glia.22346
Fallon, K. B., Havlioglu, N., Hamilton, L. H., Cheng, T. P. H., and Carroll, S. L. (2004). Constitutive activation of the neuregulin-1/erbB signaling pathway promotes the proliferation of a human peripheral neuroepithelioma cell line. J. Neurooncol. 66, 273–284. doi: 10.1023/B:NEON.0000014521.28294.84
Falls, D. L. (2003). Neuregulins: functions, forms, and signaling strategies. Exp. Cell Res. 284, 14–30. doi: 10.1016/S0014-4827(02)00102-7
Farah, M. H., Pan, B. H., Hoffman, P. N., Ferraris, D., Tsukamoto, T., Nguyen, T., et al. (2011). Reduced BACE1 activity enhances clearance of myelin debris and regeneration of axons in the injured peripheral nervous system. J. Neurosci. 31, 5744–5754. doi: 10.1523/JNEUROSCI.6810-10.2011
Fernandez-Valle, C., Bunge, R. P., and Bunge, M. B. (1995). Schwann cells degrade myelin and proliferate in the absence of macrophages: evidence from in vitro studies of Wallerian degeneration. J. Neurocytol. 24, 667–679. doi: 10.1007/BF01179817
Fledrich, R., Stassart, R. M., Klink, A., Rasch, L. M., Prukop, T., Haag, L., et al. (2014). Soluble neuregulin-1 modulates disease pathogenesis in rodent models of Charcot-Marie-Tooth disease 1A. Nat. Med. 20, 1055–1061. doi: 10.1038/nm.3664
Flores, A. I., Narayanan, S. P., Morse, E. N., Shick, H. E., Yin, X., Kidd, G., et al. (2008). Constitutively active Akt induces enhanced myelination in the CNS. J. Neurosci. 28, 7174–7183. doi: 10.1523/JNEUROSCI.0150-08.2008
Fontana, X., Hristova, M., Da Costa, C., Patodia, S., Thei, L., Makwana, M., et al. (2012). c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J. Cell Biol. 198, 127–141. doi: 10.1083/jcb.201205025
Freidin, M., Asche, S., Bargiello, T. A., Bennett, M. V. L., and Abrams, C. K. (2009). Connexin 32 increases the proliferative response of Schwann cells to neuregulin-1 (Nrg1). Proc. Natl. Acad. Sci. U.S.A. 106, 3567–3572. doi: 10.1073/pnas.0813413106
Fricker, F. R., Antunes-Martins, A., Galino, J., Paramsothy, R., La Russa, F., Perkins, J., et al. (2013). Axonal neuregulin 1 is a rate limiting but not essential factor for nerve remyelination. Brain 136, 2279–2297. doi: 10.1093/brain/awt148
Fricker, F. R., and Bennett, D. L. (2011). The role of neuregulin-1 in the response to nerve injury. Future Neurol. 6, 809–822. doi: 10.2217/fnl.11.45
Fricker, F. R., Lago, N., Balarajah, S., Tsantoulas, C., Tanna, S., Zhu, N., et al. (2011). Axonally derived neuregulin-1 is required for remyelination and regeneration after nerve injury in adulthood. J. Neurosci. 31, 3225–3233. doi: 10.1523/JNEUROSCI.2568-10.2011
Frohnert, P. W., Stonecypher, M. S., and Carroll, S. L. (2003). Constitutive activation of the neuregulin-1/ErbB receptor signaling pathway is essential for the proliferation of a neoplastic Schwann cell line. Glia 43, 104–118. doi: 10.1002/glia.10232
Fu, E. S., Zhang, Y. P., Sagen, J., Candiotti, K. A., Morton, P. D., Liebl, D. J., et al. (2010). Transgenic inhibition of glial NF-κB reduces pain behavior and inflammation after peripheral nerve injury. Pain 148, 509–518. doi: 10.1016/j.pain.2010.01.001
Fu, S. Y., and Gordon, T. (1995). Contributing factors to poor functional recovery after delayed nerve repair: prolonged axotomy. J. Neurosci. 15, 3876–3885.
Gao, X., Daugherty, R. L., and Tourtellotte, W. G. (2007). Regulation of low affinity neurotrophin receptor (p75NTR) by early growth response (Egr) transcriptional regulators. Mol. Cell. Neurosci. 36, 501–514. doi: 10.1016/j.mcn.2007.08.013
Gaudet, A. D., Popovich, P. G., and Ramer, M. S. (2011). Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J. Neuroinflammation 8:110. doi: 10.1186/1742-2094-8-110
Gess, B., Halfter, H., Kleffner, I., Monje, P., Athauda, G., Wood, P. M., et al. (2008). Inhibition of N-cadherin and beta-catenin function reduces axon-induced Schwann cell proliferation. J. Neurosci. Res. 86, 797–812. doi: 10.1002/jnr.21528
Goebbels, S., Oltrogge, J. H., Kemper, R., Heilmann, I., Bormuth, I., Wolfer, S., et al. (2010). Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. J. Neurosci. 30, 8953–8964. doi: 10.1523/JNEUROSCI.0219-10.2010
Goebbels, S., Oltrogge, J. H., Wolfer, S., Wieser, G. L., Nientiedt, T., Pieper, A., et al. (2012). Genetic disruption of Pten in a novel mouse model of tomaculous neuropathy. EMBO Mol. Med. 4, 486–499. doi: 10.1002/emmm.201200227
Goethals, S., Ydens, E., Timmerman, V., and Janssens, S. (2010). Toll-like receptor expression in the peripheral nerve. Glia 58, 1701–1709. doi: 10.1002/glia.21041
Gomez-Sanchez, J. A., Carty, L., Iruarrizaga-Lejarreta, M., Palomo-Irigoyen, M., Varela-Rey, M., Griffith, M., et al. (2015). Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J. Cell Biol. 210, 153–168. doi: 10.1083/jcb.201503019
Gordon, T., and English, A. W. (2016). Strategies to promote peripheral nerve regeneration: electrical stimulation and/or exercise. Eur. J. Neurosci. 43, 336–350. doi: 10.1111/ejn.13005
Grigoryan, T., and Birchmeier, W. (2015). Molecular signaling mechanisms of axon-glia communication in the peripheral nervous system. Bioessays 37, 502–513. doi: 10.1002/bies.201400172
Grigoryan, T., Stein, S., Qi, J., Wende, H., Garratt, A. N., Nave, K.-A., et al. (2013). Wnt/Rspondin/beta-catenin signals control axonal sorting and lineage progression in Schwann cell development. Proc. Natl. Acad. Sci. U.S.A. 110, 18174–18179. doi: 10.1073/pnas.1310490110
Grossmann, K. S., Wende, H., Paul, F. E., Cheret, C., Garratt, A. N., Zurborg, S., et al. (2009). The tyrosine phosphatase Shp2 (PTPN11) directs Neuregulin-1/ErbB signaling throughout Schwann cell development. Proc. Natl. Acad. Sci. U.S.A. 106, 16704–16709. doi: 10.1073/pnas.0904336106
Guertin, A. D., Zhang, D. P., Mak, K. S., Alberta, J. A., and Kim, H. A. (2005). Microanatomy of axon/glial signaling during Wallerian degeneration. J. Neurosci. 25, 3478–3487. doi: 10.1523/JNEUROSCI.3766-04.2005
Hantke, J., Carty, L., Wagstaff, L. J., Turmaine, M., Wilton, D. K., Quintes, S., et al. (2014). c-Jun activation in Schwann cells protects against loss of sensory axons in inherited neuropathy. Brain 137, 2922–2937. doi: 10.1093/brain/awu257
Hardie, D. G., Ross, F. A., and Hawley, S. A. (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262. doi: 10.1038/nrm3311
Hari, L., Brault, V., Kleber, M., Lee, H.-Y., Ille, F., Leimeroth, R., et al. (2002). Lineage-specific requirements of beta-catenin in neural crest development. J. Cell Biol. 159, 867–880. doi: 10.1083/jcb.200209039
Harrisingh, M. C., Perez-Nadales, E., Parkinson, D. B., Malcolm, D. S., Mudge, A. W., and Lloyd, A. C. (2004). The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. EMBO J. 23, 3061–3071. doi: 10.1038/sj.emboj.7600309
He, Y., Kim, J. Y., Dupree, J., Tewari, A., Melendez-Vasquez, C., Svaren, J., et al. (2010). Yy1 as a molecular link between neuregulin and transcriptional modulation of peripheral myelination. Nat. Neurosci. 13, 1472–1480. doi: 10.1038/nn.2686
Heinen, A., Beyer, F., Tzekova, N., Hartung, H.-P., and Küry, P. (2015). Fingolimod induces the transition to a nerve regeneration promoting Schwann cell phenotype. Exp. Neurol. 271, 25–35. doi: 10.1016/j.expneurol.2015.05.002
Heinen, A., Kremer, D., Göttle, P., Kruse, F., Hasse, B., Lehmann, H., et al. (2008). The cyclin-dependent kinase inhibitor p57kip2 is a negative regulator of Schwann cell differentiation and in vitro myelination. Proc. Natl. Acad. Sci. U.S.A. 105, 8748–8753. doi: 10.1073/pnas.0802659105
Heinen, A., Lehmann, H. C., and Küry, P. (2013). Negative regulators of Schwann cell differentiation—novel targets for peripheral nerve therapies? J. Clin. Immunol. 33, 18–26. doi: 10.1007/s10875-012-9786-9
Heinen, A., Tzekova, N., Graffmann, N., Torres, K. J., Uhrberg, M., Hartung, H.-P., et al. (2012). Histone methyltransferase enhancer of zeste homolog 2 regulates Schwann cell differentiation. Glia 60, 1696–1708. doi: 10.1002/glia.22388
Hichor, M., Sampathkumar, N. K., Montanaro, J., Borderie, D., Petit, P. X., Gorgievski, V., et al. (2016). Paraquat induces peripheral myelin disruption and locomotor defects: crosstalk with LXR and Wnt pathways. Antioxid. Redox Signal. doi: 10.1089/ars.2016.6711. [Epub ahead of print].
Hirata, K., and Kawabuchi, M. (2002). Myelin phagocytosis by macrophages and nonmacrophages during Wallerian degeneration. Microsc. Res. Tech. 57, 541–547. doi: 10.1002/jemt.10108
Hoyng, S. A., de Winter, F., Tannemaat, M. R., Blits, B., Malessy, M. J. A., and Verhaagen, J. (2015). Gene therapy and peripheral nerve repair: a perspective. Front. Mol. Neurosci. 8:32. doi: 10.3389/fnmol.2015.00032
Hu, X., He, W., Diaconu, C., Tang, X., Kidd, G. J., Macklin, W. B., et al. (2008). Genetic deletion of BACE1 in mice affects remyelination of sciatic nerves. FASEB J. 22, 2970–2980. doi: 10.1096/fj.08-106666
Huijbregts, R. P. H., Roth, K. A., Schmidt, R. E., and Carroll, S. L. (2003). Hypertrophic neuropathies and malignant peripheral nerve sheath tumors in transgenic mice overexpressing glial growth factor beta3 in myelinating Schwann cells. J. Neurosci. 23, 7269–7280.
Hutton, E. J., Carty, L., Laurá, M., Houlden, H., Lunn, M., Brandner, S., et al. (2011). c-Jun expression in human neuropathies: a pilot study. J. Peripher. Nerv. Syst. 16, 295–303. doi: 10.1111/j.1529-8027.2011.00360.x
Jessen, K. R., and Mirsky, R. (2005). The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 6, 671–682. doi: 10.1038/nrn1746
Jessen, K. R., and Mirsky, R. (2008). Negative regulation of myelination: relevance for development, injury, and demyelinating disease. Glia 56, 1552–1565. doi: 10.1002/glia.20761
Jessen, K. R., and Mirsky, R. (2016). The repair Schwann cell and its function in regenerating nerves. J. Physiol. 594, 3521–3531. doi: 10.1113/jp270874
Jessen, K. R., Mirsky, R., and Lloyd, A. C. (2015). Schwann cells: development and role in nerve repair. Cold Spring Harb. Perspect. Biol. 7:a020487. doi: 10.1101/cshperspect.a020487
Ji, Y., Tao, T., Cheng, C., Yang, H., Wang, Y., Yang, J., et al. (2010). SSeCKS is a suppressor in Schwann cell differentiation and myelination. Neurochem. Res. 35, 219–226. doi: 10.1007/s11064-009-0045-2
Joshi, A. R., Holtmann, L., Bobylev, I., Schneider, C., Ritter, C., Weis, J., et al. (2016). Loss of Schwann cell plasticity in chronic inflammatory demyelinating polyneuropathy (CIDP). J. Neuroinflammation 13, 255. doi: 10.1186/s12974-016-0711-7
Joung, I., Yoo, M., Woo, J. H., Chang, C. Y., Heo, H., and Kwon, Y. K. (2010). Secretion of EGF-like domain of heregulinbeta promotes axonal growth and functional recovery of injured sciatic nerve. Mol. Cells 30, 477–484. doi: 10.1007/s10059-010-0137-5
Jung, J., Cai, W., Lee, H. K., Pellegatta, M., Shin, Y. K., Jang, S. Y., et al. (2011). Actin polymerization is essential for myelin sheath fragmentation during Wallerian degeneration. J. Neurosci. 31, 2009–2015. doi: 10.1523/JNEUROSCI.4537-10.2011
Kaempchen, K., Mielke, K., Utermark, T., Langmesser, S., and Hanemann, C. O. (2003). Upregulation of the Rac1/JNK signaling pathway in primary human schwannoma cells. Hum. Mol. Genet. 12, 1211–1221. doi: 10.1093/hmg/ddg146
Kagiava, A., Sargiannidou, I., Theophilidis, G., Karaiskos, C., Richter, J., Bashiardes, S., et al. (2016). Intrathecal gene therapy rescues a model of demyelinating peripheral neuropathy. Proc. Natl. Acad. Sci. U.S.A. 113, E2421–E2429. doi: 10.1073/pnas.1522202113
Keng, V. W., Rahrmann, E. P., Watson, A. L., Tschida, B. R., Moertel, C. L., Jessen, W. J., et al. (2012). PTEN and NF1 inactivation in Schwann cells produces a severe phenotype in the peripheral nervous system that promotes the development and malignant progression of peripheral nerve sheath tumors. Cancer Res. 72, 3405–3413. doi: 10.1158/0008-5472.CAN-11-4092
Kim, H. A., Mindos, T., and Parkinson, D. B. (2013). Plastic fantastic: Schwann cells and repair of the peripheral nervous system. Stem Cells Transl. Med. 2, 553–557. doi: 10.5966/sctm.2013-0011
Kim, H. A., Pomeroy, S. L., Whoriskey, W., Pawlitzky, I., Benowitz, L. I., Sicinski, P., et al. (2000). A developmentally regulated switch directs regenerative growth of Schwann cells through cyclin D1. Neuron 26, 405–416. doi: 10.1016/S0896-6273(00)81173-3
Kioussi, C., Gross, M. K., and Gruss, P. (1995). Pax3: a paired domain gene as a regulator in PNS myelination. Neuron 15, 553–562. doi: 10.1016/0896-6273(95)90144-2
Klein, D., Groh, J., Wettmarshausen, J., and Martini, R. (2014). Nonuniform molecular features of myelinating Schwann cells in models for CMT1: distinct disease patterns are associated with NCAM and c-Jun upregulation. Glia 62, 736–750. doi: 10.1002/glia.22638
Köhne, A., Stettner, M., Jangouk, P., Dehmel, T., Hartung, H.-P., Lehmann, H. C., et al. (2012). Fingolimod impedes Schwann cell-mediated myelination: implications for the treatment of immune neuropathies? Arch. Neurol. 69, 1280–1289. doi: 10.1001/archneurol.2012.394
Kwon, Y. K., Bhattacharyya, A., Alberta, J. A., Giannobile, W. V., Cheon, K., Stiles, C. D., et al. (1997). Activation of ErbB2 during wallerian degeneration of sciatic nerve. J. Neurosci. 17, 8293–8299.
La Marca, R., Cerri, F., Horiuchi, K., Bachi, A., Feltri, M. L., Wrabetz, L., et al. (2011). TACE (ADAM17) inhibits Schwann cell myelination. Nat. Neurosci. 14, 857–865. doi: 10.1038/nn.2849
Laura, M., Mazzeo, A., Aguennouz, M., Santoro, M., Catania, M. A., Migliorato, A., et al. (2006). Immunolocalization and activation of nuclear factor-κB in the sciatic nerves of rats with experimental autoimmune neuritis. J. Neuroimmunol. 174, 32–38. doi: 10.1016/j.jneuroim.2006.01.004
Le, N., Nagarajan, R., Wang, J. Y. T., Araki, T., Schmidt, R. E., and Milbrandt, J. (2005). Analysis of congenital hypomyelinating Egr2Lo/Lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc. Natl. Acad. Sci. U.S.A. 102, 2596–2601. doi: 10.1073/pnas.0407836102
Lee, H., Jo, E.-K., Choi, S.-Y., Oh, S. B., Park, K., Soo Kim, J., et al. (2006). Necrotic neuronal cells induce inflammatory Schwann cell activation via TLR2 and TLR3: implication in Wallerian degeneration. Biochem. Biophys. Res. Commun. 350, 742–747. doi: 10.1016/j.bbrc.2006.09.108
Lee, H. J., Shin, Y. K., and Park, H. T. (2014). Mitogen activated protein kinase family proteins and c-jun signaling in injury-induced Schwann cell plasticity. Exp. Neurobiol. 23, 130–137. doi: 10.5607/en.2014.23.2.130
Lee, H.-Y., Kleber, M., Hari, L., Brault, V., Suter, U., Taketo, M. M., et al. (2004). Instructive role of Wnt/beta-catenin in sensory fate specification in neural crest stem cells. Science 303, 1020–1023. doi: 10.1126/science.1091611
Lee, S., Ashizawa, A. T., Kim, K. S., Falk, D. J., and Notterpek, L. (2013). Liposomes to target peripheral neurons and Schwann cells. PLoS ONE 8:e78724. doi: 10.1371/journal.pone.0078724
Lee, Y. I., Li, Y., Mikesh, M., Smith, I., Nave, K.-A., Schwab, M. H., et al. (2016). Neuregulin1 displayed on motor axons regulates terminal Schwann cell-mediated synapse elimination at developing neuromuscular junctions. Proc. Natl. Acad. Sci. U.S.A. 113, E479–E487. doi: 10.1073/pnas.1519156113
Leimeroth, R., Lobsiger, C., Lussi, A., Taylor, V., Suter, U., and Sommer, L. (2002). Membrane-bound neuregulin1 type III actively promotes Schwann cell differentiation of multipotent Progenitor cells. Dev. Biol. 246, 245–258. doi: 10.1006/dbio.2002.0670
Leoni, V., Masterman, T., Diczfalusy, U., De Luca, G., Hillert, J., and Björkhem, I. (2002). Changes in human plasma levels of the brain specific oxysterol 24S-hydroxycholesterol during progression of multiple sclerosis. Neurosci. Lett. 331, 163–166. doi: 10.1016/S0304-3940(02)00887-X
Li, H., Wigley, C., and Hall, S. M. (1998). Chronically denervated rat Schwann cells respond to GGF in vitro. Glia 24, 290–303.
Li, Y., Rao, P. K., Wen, R., Song, Y., Muir, D., Wallace, P., et al. (2004). Notch and Schwann cell transformation. Oncogene 23, 1146–1152. doi: 10.1038/sj.onc.1207068
Li, Y., Tennekoon, G. I., Birnbaum, M., Marchionni, M. A., and Rutkowski, J. L. (2001). Neuregulin signaling through a PI3K/Akt/Bad pathway in Schwann cell survival. Mol. Cell. Neurosci. 17, 761–767. doi: 10.1006/mcne.2000.0967
Limpert, A. S., and Carter, B. D. (2010). Axonal neuregulin 1 type III activates NF-κB in Schwann cells during myelin formation. J. Biol. Chem. 285, 16614–16622. doi: 10.1074/jbc.M109.098780
Liu, H., Lv, P., Zhu, Y., Wu, H., Zhang, K., Xu, F., et al. (2017). Salidroside promotes peripheral nerve regeneration based on tissue engineering strategy using Schwann cells and PLGA: in vitro and in vivo. Sci. Rep. 7:39869. doi: 10.1038/srep39869
Liu, X., Peng, S., Zhao, Y., Zhao, T., Wang, M., Luo, L., et al. (2016a). AMPK negatively regulates peripheral myelination via activation of c-Jun. Mol. Neurobiol. doi: 10.1007/s12035-016-9913-3. [Epub ahead of print].
Liu, X., Zhao, Y., Peng, S., Zhang, S., Wang, M., Chen, Y., et al. (2016b). BMP7 retards peripheral myelination by activating p38 MAPK in Schwann cells. Sci. Rep. 6:31049. doi: 10.1038/srep31049
Lütjohann, D., Papassotiropoulos, A., Björkhem, I., Locatelli, S., Bagli, M., Oehring, R. D., et al. (2000). Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J. Lipid Res. 41, 195–198.
Mager, G. M., Ward, R. M., Srinivasan, R., Jang, S.-W., Wrabetz, L., and Svaren, J. (2008). Active gene repression by the Egr2· NAB complex during peripheral nerve myelination. J. Biol. Chem. 283, 18187–18197. doi: 10.1074/jbc.M803330200
Mahanthappa, N. K., Anton, E. S., and Matthew, W. D. (1996). Glial growth factor 2, a soluble neuregulin, directly increases Schwann cell motility and indirectly promotes neurite outgrowth. J. Neurosci. 16, 4673–4683.
Makoukji, J., Meffre, D., Grenier, J., Liere, P., Lobaccaro, J.-M. A., Schumacher, M., et al. (2011). Interplay between LXR and Wnt/β-catenin signaling in the negative regulation of peripheral myelin genes by oxysterols. J. Neurosci. 31, 9620–9629. doi: 10.1523/JNEUROSCI.0761-11.2011
Mancuso, R., Martínez-Muriana, A., Leiva, T., Gregorio, D., Ariza, L., Morell, M., et al. (2016). Neuregulin-1 promotes functional improvement by enhancing collateral sprouting in SOD1(G93A) ALS mice and after partial muscle denervation. Neurobiol. Dis. 95, 168–178. doi: 10.1016/j.nbd.2016.07.023
Marquardt, L. M., and Sakiyama-Elbert, S. E. (2013). Engineering peripheral nerve repair. Curr. Opin. Biotechnol. 24, 887–892. doi: 10.1016/j.copbio.2013.05.006
Martini, R., Fischer, S., López-Vales, R., and David, S. (2008). Interactions between Schwann cells and macrophages in injury and inherited demyelinating disease. Glia 56, 1566–1577. doi: 10.1002/glia.20766
Matsas, R., Lavdas, A. A., Papastefanaki, F., and Thomaidou, D. (2008). Schwann cell transplantation for CNS repair. Curr. Med. Chem. 15, 151–160. doi: 10.2174/092986708783330593
Maurel, P., and Salzer, J. L. (2000). Axonal regulation of Schwann cell proliferation and survival and the initial events of myelination requires PI 3-kinase activity. J. Neurosci. 20, 4635–4645.
Michailov, G. V., Sereda, M. W., Brinkmann, B. G., Fischer, T. M., Haug, B., Birchmeier, C., et al. (2004). Axonal neuregulin-1 regulates myelin sheath thickness. Science 304, 700–703. doi: 10.1126/science.1095862
Mogha, A., D'Rozario, M., and Monk, K. R. (2016a). G protein-coupled receptors in myelinating glia. Trends Pharmacol. Sci. 37, 977–987. doi: 10.1016/j.tips.2016.09.002
Mogha, A., Harty, B., Carlin, D., Joseph, J., Sanchez, N., Suter, U., et al. (2016b). Gpr126/Adgrg6 has Schwann cell autonomous and nonautonomous functions in peripheral nerve injury and repair. J. Neurosci. 36, 12351–12367. doi: 10.1523/JNEUROSCI.3854-15.2016
Monje, P. V., Soto, J., Bacallao, K., and Wood, P. M. (2010). Schwann cell dedifferentiation is independent of mitogenic signaling and uncoupled to proliferation: role of cAMP and JNK in the maintenance of the differentiated state. J. Biol. Chem. 285, 31024–31036. doi: 10.1074/jbc.M110.116970
Monk, K. R., Naylor, S. G., Glenn, T. D., Mercurio, S., Perlin, J. R., Dominguez, C., et al. (2009). A G protein–coupled receptor is essential for Schwann cells to initiate myelination. Science 325, 1402–1405. doi: 10.1126/science.1173474
Morris, J. K., Lin, W., Hauser, C., Marchuk, Y., Getman, D., and Lee, K. F. (1999). Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron 23, 273–283. doi: 10.1016/S0896-6273(00)80779-5
Morton, P. D., Dellarole, A., Theus, M. H., Walters, W. M., Berge, S. S., and Bethea, J. R. (2013). Activation of NF-κB in Schwann cells is dispensable for myelination in vivo. J. Neurosci. 33, 9932–9936. doi: 10.1523/JNEUROSCI.2483-12.2013
Morton, P. D., Johnstone, J. T., Ramos, A. Y., Liebl, D. J., Bunge, M. B., and Bethea, J. R. (2012). Nuclear factor-κB activation in Schwann cells regulates regeneration and remyelination. Glia 60, 639–650. doi: 10.1002/glia.22297
Napoli, I., Noon, L. A., Ribeiro, S., Kerai, A. P., Parrinello, S., Rosenberg, L. H., et al. (2012). A central role for the ERK-signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration in vivo. Neuron 73, 729–742. doi: 10.1016/j.neuron.2011.11.031
Newbern, J., and Birchmeier, C. (2010). Nrg1/ErbB signaling networks in Schwann cell development and myelination. Semin. Cell Dev. Biol. 21, 922–928. doi: 10.1016/j.semcdb.2010.08.008
Newbern, J. M., Li, X., Shoemaker, S. E., Zhou, J., Zhong, J., Wu, Y., et al. (2011). Specific functions for ERK/MAPK signaling during PNS development. Neuron 69, 91–105. doi: 10.1016/j.neuron.2010.12.003
Newbern, J. M., and Snider, W. D. (2012). Bers-ERK Schwann cells coordinate nerve regeneration. Neuron 73, 623–626. doi: 10.1016/j.neuron.2012.02.002
Nickols, J. C., Valentine, W., Kanwal, S., and Carter, B. D. (2003). Activation of the transcription factor NF-κB in Schwann cells is required for peripheral myelin formation. Nat. Neurosci. 6, 161–167. doi: 10.1038/nn995
Nikam, S. S., Tennekoon, G. I., Christy, B. A., Yoshino, J. E., and Rutkowski, J. L. (1995). The zinc finger transcription factor Zif268/Egr-1 is essential for Schwann cell expression of the p75 NGF receptor. Mol. Cell. Neurosci. 6, 337–348. doi: 10.1006/mcne.1995.1026
Norrmén, C., Figlia, G., Lebrun-Julien, F., Pereira, J. A., Trötzmüller, M., Köfeler, H. C., et al. (2014). mTORC1 controls PNS myelination along the mTORC1-RXRγ-SREBP-lipid biosynthesis axis in Schwann cells. Cell Rep. 9, 646–660. doi: 10.1016/j.celrep.2014.09.001
Norrmén, C., and Suter, U. (2013). Akt/mTOR signalling in myelination. Biochem. Soc. Trans. 41, 944LP–950LP. doi: 10.1042/BST20130046
Noseda, R., Belin, S., Piguet, F., Vaccari, I., Scarlino, S., Brambilla, P., et al. (2013). DDIT4/REDD1/RTP801 is a novel negative regulator of Schwann cell myelination. J. Neurosci. 33, 15295–15305. doi: 10.1523/JNEUROSCI.2408-13.2013
Ogata, T., Iijima, S., Hoshikawa, S., Miura, T., Yamamoto, S., Oda, H., et al. (2004). Opposing extracellular signal-regulated kinase and Akt pathways control Schwann cell myelination. J. Neurosci. 24, 6724–6732. doi: 10.1523/JNEUROSCI.5520-03.2004
Painter, M. W., Brosius Lutz, A., Cheng, Y.-C., Latremoliere, A., Duong, K., Miller, C. M., et al. (2014). Diminished schwann cell repair responses underlie age-associated impaired axonal regeneration. Neuron 83, 331–343. doi: 10.1016/j.neuron.2014.06.016
Parkinson, D. B., Bhaskaran, A., Arthur-Farraj, P., Noon, L. A., Woodhoo, A., Lloyd, A. C., et al. (2008). c-Jun is a negative regulator of myelination. J. Cell Biol. 181, 625–637. doi: 10.1083/jcb.200803013
Parkinson, D. B., Bhaskaran, A., Droggiti, A., Dickinson, S., D'Antonio, M., Mirsky, R., et al. (2004). Krox-20 inhibits Jun-NH2-terminal kinase/c-Jun to control Schwann cell proliferation and death. J. Cell Biol. 164, 385–394. doi: 10.1083/jcb.200307132
Parkinson, D. B., Dong, Z., Bunting, H., Whitfield, J., Meier, C., Marie, H., et al. (2001). Transforming growth factor beta (TGFbeta) mediates Schwann cell death in vitro and in vivo: examination of c-Jun activation, interactions with survival signals, and the relationship of TGFbeta-mediated death to Schwann cell differentiation. J. Neurosci. 21, 8572–8585.
Parrinello, S., Napoli, I., Ribeiro, S., Wingfield Digby, P., Fedorova, M., Parkinson, D. B., et al. (2010). EphB signaling directs peripheral nerve regeneration through Sox2-dependent Schwann cell sorting. Cell 143, 145–155. doi: 10.1016/j.cell.2010.08.039
Quintes, S., Brinkmann, B. G., Ebert, M., Frob, F., Kungl, T., Arlt, F. A., et al. (2016). Zeb2 is essential for Schwann cell differentiation, myelination and nerve repair. Nat. Neurosci. 19, 1050–1059. doi: 10.1038/nn.4321
Rameh, L. E., and Cantley, L. C. (1999). The role of phosphoinositide 3-kinase lipid products in cell function. J. Biol. Chem. 274, 8347–8350. doi: 10.1074/jbc.274.13.8347
Roberts, S. L., Dun, X.-p., Dee, G., Gray, B., Mindos, T., and Parkinson, D. B. (2016). The role of p38alpha in Schwann cells in regulating peripheral nerve myelination and repair. J. Neurochem. doi: 10.1111/jnc.13929. [Epub ahead of print].
Rodríguez-Molina, J. F., Lopez-Anido, C., Ma, K. H., Zhang, C., Olson, T., Muth, K. N., et al. (2017). Dual specificity phosphatase 15 regulates Erk activation in Schwann cells. J. Neurochem. 140, 368–382. doi: 10.1111/jnc.13911
Ronchi, G., Cillino, M., Gambarotta, G., Fornasari, B. E., Raimondo, S., Pugliese, P., et al. (2017). Irreversible changes occurring in long-term denervated Schwann cells affect delayed nerve repair. J. Neurosurg. doi: 10.3171/2016.9.JNS16140. [Epub ahead of print].
Ronchi, G., Haastert-Talini, K., Fornasari, B. E., Perroteau, I., Geuna, S., and Gambarotta, G. (2016). The Neuregulin1/ErbB system is selectively regulated during peripheral nerve degeneration and regeneration. Eur. J. Neurosci. 43, 351–364. doi: 10.1111/ejn.12974
Rotshenker, S. (2011). Wallerian degeneration: the innate-immune response to traumatic nerve injury. J. Neuroinflammation 8:109. doi: 10.1186/1742-2094-8-109
Russell, D. W. (2000). Oxysterol biosynthetic enzymes. Biochim. Biophys. Acta 1529, 126–135. doi: 10.1016/S1388-1981(00)00142-6
Schröder, J. M. (1972). Altered ratio between axon diameter and myelin sheath thickness in regenerated nerve fibers. Brain Res. 45, 49–65. doi: 10.1016/0006-8993(72)90215-6
Sherman, D. L., Krols, M., Wu, L.-M. N., Grove, M., Nave, K.-A., Gangloff, Y.-G., et al. (2012). Arrest of myelination and reduced axon growth when Schwann cells lack mTOR. J. Neurosci. 32, 1817–1825. doi: 10.1523/JNEUROSCI.4814-11.2012
Sheu, J. Y., Kulhanek, D. J., and Eckenstein, F. P. (2000). Differential patterns of ERK and STAT3 phosphorylation after sciatic nerve transection in the rat. Exp. Neurol. 166, 392–402. doi: 10.1006/exnr.2000.7508
Shin, Y. K., Jang, S. Y., Park, J. Y., Park, S. Y., Lee, H. J., Suh, D. J., et al. (2013). The Neuregulin-Rac-MKK7 pathway regulates antagonistic c-jun/Krox20 expression in Schwann cell dedifferentiation. Glia 61, 892–904. doi: 10.1002/glia.22482
Shivane, A., Parkinson, D. B., Ammoun, S., and Hanemann, C. O. (2013). Expression of c-Jun and Sox-2 in human Schwannomas and traumatic neuromas. Histopathology 62, 651–656. doi: 10.1111/his.12062
Smith, D., Tweed, C., Fernyhough, P., and Glazner, G. W. (2009). Nuclear factor-κB activation in axons and Schwann cells in experimental sciatic nerve injury and its role in modulating axon regeneration: studies with etanercept. J. Neuropathol. Exp. Neurol. 68, 691–700. doi: 10.1097/NEN.0b013e3181a7c14e
Son, Y.-J., and Thompson, W. J. (1995). Schwann cell processes guide regeneration of peripheral axons. Neuron 14, 125–132. doi: 10.1016/0896-6273(95)90246-5
Stassart, R. M., Fledrich, R., Velanac, V., Brinkmann, B. G., Schwab, M. H., Meijer, D., et al. (2013). A role for Schwann cell-derived neuregulin-1 in remyelination. Nat. Neurosci. 16, 48–54. doi: 10.1038/nn.3281
Stewart, H. J. S. (1995). Expression of c-Jun, Jun B, Jun D and cAMP response element binding protein by Schwann cells and their precursors in vivo and in vitro. Eur. J. Neurosci. 7, 1366–1375. doi: 10.1111/j.1460-9568.1995.tb01128.x
Stewart, H. J. S., Zoidl, G., Rossner, M., Brennan, A., Zoidl, C., Nave, K. A., et al. (1997). Helix-loop-helix proteins in Schwann cells: a study of regulation and subcellular localization of Ids, REB, and E12/47 during embryonic and postnatal development. J. Neurosci. Res. 50, 684–701. doi: 10.1002/(SICI)1097-4547(19971201)50:5<684::AID-JNR6>3.0.CO;2-D
Stoll, G., and Müller, H. W. (1999). Nerve injury, axonal degeneration and neural regeneration: basic insights. Brain Pathol. 9, 313–325. doi: 10.1111/j.1750-3639.1999.tb00229.x
Sulaiman, W., and Nguyen, D. H. (2016). Transforming growth factor beta 1, a cytokine with regenerative functions. Neural Regen. Res. 11, 1549–1552. doi: 10.4103/1673-5374.193223
Syed, N., Reddy, K., Yang, D. P., Taveggia, C., Salzer, J. L., Maurel, P., et al. (2010). Soluble neuregulin-1 has bifunctional, concentration-dependent effects on Schwann cell myelination. J. Neurosci. 30, 6122–6131. doi: 10.1523/JNEUROSCI.1681-09.2010
Syroid, D. E., Maycox, P. R., Burrola, P. G., Liu, N., Wen, D., Lee, K. F., et al. (1996). Cell death in the Schwann cell lineage and its regulation by neuregulin. Proc. Natl. Acad. Sci. U.S.A. 93, 9229–9234. doi: 10.1073/pnas.93.17.9229
Takeda, Y. S., and Xu, Q. (2015). Synthetic and nature-derived lipid nanoparticles for neural regeneration. Neural Regen. Res. 10, 689–690. doi: 10.4103/1673-5374.156946
Tapinos, N., Ohnishi, M., and Rambukkana, A. (2006). ErbB2 receptor tyrosine kinase signaling mediates early demyelination induced by leprosy bacilli. Nat. Med. 12, 961–966. doi: 10.1038/nm1433
Taveggia, C., Zanazzi, G., Petrylak, A., Yano, H., Rosenbluth, J., Einheber, S., et al. (2005). Neuregulin-1 type III determines the ensheathment fate of axons. Neuron 47, 681–694. doi: 10.1016/j.neuron.2005.08.017
Thakur, K. K., Saini, J., Mahajan, K., Singh, D., Jayswal, D. P., Mishra, S., et al. (2017). Therapeutic implications of toll-like receptors in peripheral neuropathic pain. Pharmacol. Res. 115, 224–232. doi: 10.1016/j.phrs.2016.11.019
Topilko, P., Levi, G., Merlo, G., Mantero, S., Desmarquet, C., Mancardi, G., et al. (1997). Differential regulation of the zinc finger genes Krox-20 and Krox-24 (Egr-1) suggests antagonistic roles in Schwann cells. J. Neurosci. Res. 50, 702–712. doi: 10.1002/(SICI)1097-4547(19971201)50:5<702::AID-JNR7>3.0.CO;2-L
Trimarco, A., Forese, M. G., Alfieri, V., Lucente, A., Brambilla, P., Dina, G., et al. (2014). Prostaglandin D2 synthase/GPR44: a signaling axis in PNS myelination. Nat. Neurosci. 17, 1682–1692. doi: 10.1038/nn.3857
Tzekova, N., Heinen, A., Bunk, S., Hermann, C., Hartung, H.-P., Reipert, B., et al. (2015). Immunoglobulins stimulate cultured Schwann cell maturation and promote their potential to induce axonal outgrowth. J. Neuroinflammation 12, 107. doi: 10.1186/s12974-015-0331-7
Waller, A. (1850). Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Philos. Trans. R. Soc. Lond. 140, 423–429. doi: 10.1098/rstl.1850.0021
Wang, J., Ren, K.-Y., Wang, Y.-H., Kou, Y.-H., Zhang, P.-X., Peng, J.-P., et al. (2015). Effect of active Notch signaling system on the early repair of rat sciatic nerve injury. Artif. Cells Nanomed. Biotechnol. 43, 383–389. doi: 10.3109/21691401.2014.896372
Weinberg, H. J., and Spencer, P. S. (1978). The fate of Schwann cells isolated from axonal contact. J. Neurocytol. 7, 555–569. doi: 10.1007/BF01260889
Willem, M. (2016). Proteolytic processing of Neuregulin-1. Brain Res. Bull. 126, 178–182. doi: 10.1016/j.brainresbull.2016.07.003
Woodhoo, A., Alonso, M. B. D., Droggiti, A., Turmaine, M., D'Antonio, M., Parkinson, D. B., et al. (2009). Notch controls embryonic Schwann cell differentiation, postnatal myelination and adult plasticity. Nat. Neurosci. 12, 839–847. doi: 10.1038/nn.2323
Wu, L. M. N., Wang, J., Conidi, A., Zhao, C., Wang, H., Ford, Z., et al. (2016). Zeb2 recruits HDAC-NuRD to inhibit Notch and controls Schwann cell differentiation and remyelination. Nat. Neurosci. 19, 1060–1072. doi: 10.1038/nn.4322
Wu, W., Liu, Q., Liu, Y., Yu, Z., and Wang, Y. (2016a). Dixdc1 targets CyclinD1 and p21 via PI3K pathway activation to promote Schwann cell proliferation after sciatic nerve crush. Biochem. Biophys. Res. Commun. 478, 956–963. doi: 10.1016/j.bbrc.2016.08.058
Wu, W., Liu, Y., and Wang, Y. (2016b). Sam68 promotes Schwann cell proliferation by enhancing the PI3K/Akt pathway and acts on regeneration after sciatic nerve crush. Biochem. Biophys. Res. Commun. 473, 1045–1051. doi: 10.1016/j.bbrc.2016.04.013
Yamauchi, J., Chan, J. R., and Shooter, E. M. (2003). Neurotrophin 3 activation of TrkC induces Schwann cell migration through the c-Jun N-terminal kinase pathway. Proc. Natl. Acad. Sci. U.S.A. 100, 14421–14426. doi: 10.1073/pnas.2336152100
Yamauchi, J., Miyamoto, Y., Chan, J. R., and Tanoue, A. (2008). ErbB2 directly activates the exchange factor Dock7 to promote Schwann cell migration. J. Cell Biol. 181, 351–365. doi: 10.1083/jcb.200709033
Yamauchi, J., Miyamoto, Y., Hamasaki, H., Sanbe, A., Kusakawa, S., Nakamura, A., et al. (2011). The atypical Guanine-nucleotide exchange factor, dock7, negatively regulates Schwann cell differentiation and myelination. J. Neurosci. 31, 12579–12592. doi: 10.1523/JNEUROSCI.2738-11.2011
Yan, M., Cheng, C., Jiang, J., Liu, Y., Gao, Y., Guo, Z., et al. (2009). Essential role of SRC suppressed C kinase substrates in Schwann cells adhesion, spreading and migration. Neurochem. Res. 34, 1002–1010. doi: 10.1007/s11064-008-9869-4
Yang, D. P., Kim, J., Syed, N., Tung, Y., Bhaskaran, A., Mindos, T., et al. (2012). p38 MAPK activation promotes denervated Schwann cell phenotype and functions as a negative regulator of Schwann cell differentiation and myelination. J. Neurosci. 32, 7158–7168. doi: 10.1523/JNEUROSCI.5812-11.2012
Yildiz, M., Karlidag, T., Yalcin, S., Ozogul, C., Keles, E., Alpay, H. C., et al. (2011). Efficacy of glial growth factor and nerve growth factor on the recovery of traumatic facial paralysis. Eur. Arch. Otorhinolaryngol. 268, 1127–1133. doi: 10.1007/s00405-011-1492-3
Yokoo, T., Toyoshima, H., Miura, M., Wang, Y., Iida, K. T., Suzuki, H., et al. (2003). p57Kip2 regulates actin dynamics by binding and translocating LIM-kinase 1 to the nucleus. J. Biol. Chem. 278, 52919–52923. doi: 10.1074/jbc.M309334200
Yoon, C., Korade, Z., and Carter, B. D. (2008). Protein kinase A-induced phosphorylation of the p65 subunit of nuclear factor-κB promotes Schwann cell differentiation into a myelinating phenotype. J. Neurosci. 28, 3738–3746. doi: 10.1523/JNEUROSCI.4439-07.2008
Zhou, S., Gao, R., Hu, W., Qian, T., Wang, N., Ding, G., et al. (2014). MiR-9 inhibits Schwann cell migration by targeting Cthrc1 following sciatic nerve injury. J. Cell Sci. 127(Pt 5), 967–976. doi: 10.1242/jcs.131672
Zhou, Y., and Notterpek, L. (2016). Promoting peripheral myelin repair. Exp. Neurol. 283, 573–580. doi: 10.1016/j.expneurol.2016.04.007
Zochodne, D. W. (2008). Neurobiology of Peripheral Nerve Regeneration. Cambridge: Cambridge University Press.
Zochodne, D. W. (2012). The challenges and beauty of peripheral nerve regrowth. J. Peripher. Nerv. Syst. 17, 1–18. doi: 10.1111/j.1529-8027.2012.00378.x
Keywords: Schwann cell, plasticity, molecular mechanisms, nerve injury, peripheral neuropathy
Citation: Boerboom A, Dion V, Chariot A and Franzen R (2017) Molecular Mechanisms Involved in Schwann Cell Plasticity. Front. Mol. Neurosci. 10:38. doi: 10.3389/fnmol.2017.00038
Received: 22 December 2016; Accepted: 31 January 2017;
Published: 17 February 2017.
Edited by:
Margaret Su-chun Ho, Tongji University, ChinaReviewed by:
Xavier Navarro, Autonomous University of Barcelona, SpainMelissa R. Andrews, University of St Andrews, UK
Georg Zoidl, York University, Canada
Helmar Lehmann, University Hospital Cologne, Germany
Copyright © 2017 Boerboom, Dion, Chariot and Franzen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rachelle Franzen, rfranzen@ulg.ac.be