 Ning Cheng
Ning Cheng Jong M. Rho
Jong M. Rho Susan A. Masino
Susan A. Masino
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 21 February 2017
Sec. Neuroplasticity and Development
Volume 10 - 2017 | https://doi.org/10.3389/fnmol.2017.00034
This article is part of the Research Topic Metabolic control of brain homeostasis View all 17 articles
Autism spectrum disorder (ASD) is characterized by deficits in sociability and communication, and increased repetitive and/or restrictive behaviors. While the etio-pathogenesis of ASD is unknown, clinical manifestations are diverse and many possible genetic and environmental factors have been implicated. As such, it has been a great challenge to identify key neurobiological mechanisms and to develop effective treatments. Current therapies focus on co-morbid conditions (such as epileptic seizures and sleep disturbances) and there is no cure for the core symptoms. Recent studies have increasingly implicated mitochondrial dysfunction in ASD. The fact that mitochondria are an integral part of diverse cellular functions and are susceptible to many insults could explain how a wide range of factors can contribute to a consistent behavioral phenotype in ASD. Meanwhile, the high-fat, low-carbohydrate ketogenic diet (KD), used for nearly a century to treat medically intractable epilepsy, has been shown to enhance mitochondrial function through a multiplicity of mechanisms and affect additional molecular targets that may address symptoms and comorbidities of ASD. Here, we review the evidence for the use of metabolism-based therapies such as the KD in the treatment of ASD as well as emerging co-morbid models of epilepsy and autism. Future research directions aimed at validating such therapeutic approaches and identifying additional and novel mechanistic targets are also discussed.
Autism spectrum disorder (ASD) is characterized by persistent deficits in sociability and communication, as well as restricted and repetitive patterns of behavior and interests (DiCicco-Bloom et al., 2006; Llaneza et al., 2010; Lai et al., 2014). The term “spectrum” refers to the wide range of symptoms and levels of impairment that can occur in individuals with ASD. Beyond these core behavioral symptoms, ASD is increasingly shown to affect the gastrointestinal, immune, hepatic, and endocrine systems (Goines and Van de Water, 2010; Patterson, 2011; Hsiao, 2013; Frye et al., 2015; Mayer et al., 2015). Common co-morbidities include neurologic, psychiatric and physical conditions: neurologic comorbidities include epilepsy, sleep impairment, sensory abnormalities, and delays and/or deficits in motor function; psychiatric conditions such as depression, anxiety, irritability and attention deficit hyperactivity disorder; and physical health issues such as chronic gastrointestinal disturbance. The co-occurrence rate of one or more non-ASD developmental diagnoses is as high as 83% (Levy et al., 2010).
ASD occurs in all racial, ethnic, and socioeconomic groups and is highly prevalent. It affects tens of millions individuals worldwide and costs millions of US dollars on average to support an affected individual during his/her lifespan (Buescher et al., 2014). In the U.S., the incidence of ASD is 1 in 68 children (1 in 42 boys and 1 in 189 girls) based on data released by the Centers for Disease Control and Prevention (CDC) in 2014. The prevalence appears to be on the rise (a 10-fold increase in 40 years), and is explained only in part by improved diagnosis and awareness (Hansen et al., 2015). Developmental delay in ASD can be detected as early as 6 months of age, a critical time for the development of higher-order social, emotional, and communications functions (Courchesne et al., 2007); the importance of early intervention is recognized (Orinstein et al., 2014). However, on average, children are not diagnosed until after 4 years of age (CDC, 2014), even though patients can now be reliably diagnosed at 2 years of age (Lord et al., 2006; Kleinman et al., 2008).
Given that ASD has broad and heterogeneous manifestations, and has been associated with a plethora of possible etiological factors (both genetic and environmental), ASD remains a clinical and broad-spectrum diagnosis. In most cases, ASD is diagnosed without any defined etiology. A dearth of knowledge about underlying causes has limited the ability to develop and mobilize effective treatments, and currently only co-morbid manifestations of the disorder can be alleviated. The hope is that reducing co-morbidities such as epileptic seizures, psychiatric disturbances, hyperactivity, sleep problems and digestive issues (DiCicco-Bloom et al., 2006; Llaneza et al., 2010; Lai et al., 2014) will improve overall function and reduce the severity of ASD symptoms (Kohane et al., 2012; Frye and Rossignol, 2016).
Genetic susceptibility factors and environmental influences (and likely often both) contribute to ASD (Chaste and Leboyer, 2012; Sandin et al., 2014; Tordjman et al., 2014; Kim and Leventhal, 2015). Genome screening and sequencing has identified rare chromosomal abnormalities and copy number variations, as well as hundreds of rare gene mutations associated with autism (Devlin and Scherer, 2012; Huguet et al., 2013; Jeste and Geschwind, 2014; Baker and Jeste, 2015). A small number of these genetic changes appear highly penetrant and sufficient to cause autism. However, most genetic factors only increase the risk to varying degrees, and likely combine with additional influences such as advanced parental age at time of conception, adverse metabolic conditions and/or maternal illness during pregnancy, birth complications, and exposure to toxins and/or drugs during early brain development (Stromland et al., 1994; Durkin et al., 2008; Gardener et al., 2011; Krakowiak et al., 2012; Christensen et al., 2013). Not surprisingly, the molecular pathways implicated in ASD are also highly complex and diverse, and include synaptic dysfunction and plasticity of various neurotransmitter systems, transcriptional regulation and chromatin remodeling, protein translation and modification, neuroimmunological modulation, and mitochondrial function (Veenstra-Vanderweele et al., 2004; Bourgeron, 2015; De Rubeis and Buxbaum, 2015; Kopp et al., 2015; Loke et al., 2015; Mahfouz et al., 2015; Nelson and Valakh, 2015; Subramanian et al., 2015; de la Torre-Ubieta et al., 2016; Wen et al., 2016).
Given such extreme etiological diversity, it is reasonable to hypothesize that perturbation of a common nexus can precipitate the behavioral hallmarks of ASD (Geschwind, 2008; Berg and Geschwind, 2012). Identifying such a common factor would provide novel insights into the development of ASD. Further, targeting this pathway could lead to selective therapeutic approaches that might enhance efficacy and address core symptoms. One possibility is mitochondrial function, which is integral to many cellular pathways. In addition to its well-known role as the “powerhouse of the cell,” producing the bulk of the cellular energy, mitochondria are also critically involved in cellular metabolism, intracellular calcium signaling, generation of reactive oxygen species (ROS), and apoptosis (Suen et al., 2008; Murphy, 2009; Palmieri et al., 2010; Antico Arciuch et al., 2012; Rizzuto et al., 2012), as well as in the regulation of innate and adaptive immunity (Weinberg et al., 2015). For example, mitochondria carry out both cleavage and synthesis of glycine (Kikuchi et al., 2008), which is the ligand of glycine receptors. These receptors are chloride channels that mediate inhibitory neurotransmission in the adult nervous system. However, they are highly expressed in the embryonic brain and mediate excitatory neurotransmission, and are believed to promote cortical interneuron migration and generation of excitatory projection neurons (Pilorge et al., 2016). Interestingly, recent genetic and functional studies have identified a role of abnormal glycinergic signaling in ASD (Pilorge et al., 2016). Furthermore, mitochondria are known to be affected by many of the same endogenous and exogenous risk factors of ASD, such as toxins, drugs, immune activation, and metabolic disturbances (Frye and Rossignol, 2011). Thus, elucidating the role of mitochondrial dysfunction in ASD may help unify our understanding of this complex disorder.
Mitochondria play a particularly vital role in the central nervous system. The brain has very high energy demands, consuming approximately 20% of calories while accounting for only 2% of total body weight (Raichle and Gusnard, 2002), and demanding a great amount of adenosine triphosphate (ATP) to maintain ionic gradients essential for neurotransmission and plasticity (Harris et al., 2012). In addition, mitochondria are involved in the proliferation, differentiation and maturation of neural stem cells, formation of dendritic processes, developmental and synaptic plasticity, and cell survival and death (Li et al., 2004; Kann and Kovacs, 2007; Mattson et al., 2008; Kimura and Murakami, 2014; Xavier et al., 2016). Thus, it is not surprising that multiple lines of evidence in both human and animal models support a role for mitochondrial dysfunction in the etiology of ASD (Haas, 2010; Dhillon et al., 2011; Frye and Rossignol, 2011; Rossignol and Frye, 2012; Legido et al., 2013).
The prevalence of mitochondrial disease in the ASD population is estimated to be about 5.0%, 500 times higher than that found in the general population (≈0.01%). The prevalence of abnormal metabolic biomarkers is even higher, suggesting that as many as 30% of children with ASD may experience metabolic abnormalities: almost one-third of autistic children have documented elevations in plasma lactate and/or the lactate-to-pyruvate ratio, and the levels of many other mitochondrial biomarkers (pyruvate, carnitine, and ubiquinone) are significantly different between ASD and controls (Rossignol and Frye, 2012). In addition, several genes known to regulate mitochondrial function are clearly autism-risk genes. These include SLC25A12 (Ramoz et al., 2004; Segurado et al., 2005; Silverman et al., 2008; Turunen et al., 2008; Kim et al., 2011), which encodes the predominant form of mitochondrial aspartate/glutamate carrier (however, also see Correia et al., 2006; Palmieri et al., 2010). These carriers participate in a wide range of mitochondrial functions, including control of respiration, calcium signaling and antioxidant defense, as well as glutamate-mediated excitotoxicity (Amoedo et al., 2016). Furthermore, TMLHE, (trimethyllysine hydroxylase epsilon), which encodes the first enzyme in carnitine biosynthesis, has also been associated with ASD (Celestino-Soper et al., 2012; Nava et al., 2012). It has been well established that carnitines are involved in mitochondrial transport of long-chain fatty acids and play an important role in maintaining normal mitochondrial function (Bremer, 1983). In addition, the gene encoding an inner mitochondrial membrane protease-like protein (IMMP2L) may help regulate susceptibility to ASD (Maestrini et al., 2010). It is important to note that metabolic and mitochondrial dysfunction may not exist in all patients with ASD, and biomarkers to identify this impairment would be advantageous in developing personalized treatment.
In parallel with clinical findings, many animal models of ASD also display mitochondrial dysfunction, including those based on susceptibility genes such as MECP2, UBE3A, TSC, and FOXG1. Mitochondrial dysfunction has also been observed in animal models of ASD induced by environmental risk factors such as maternal immune activation and exposure to propionic acid or valproic acid (VPA). Current evidence linking mitochondrial perturbations to ASD and the corresponding references are summarized in Tables 1, 2.
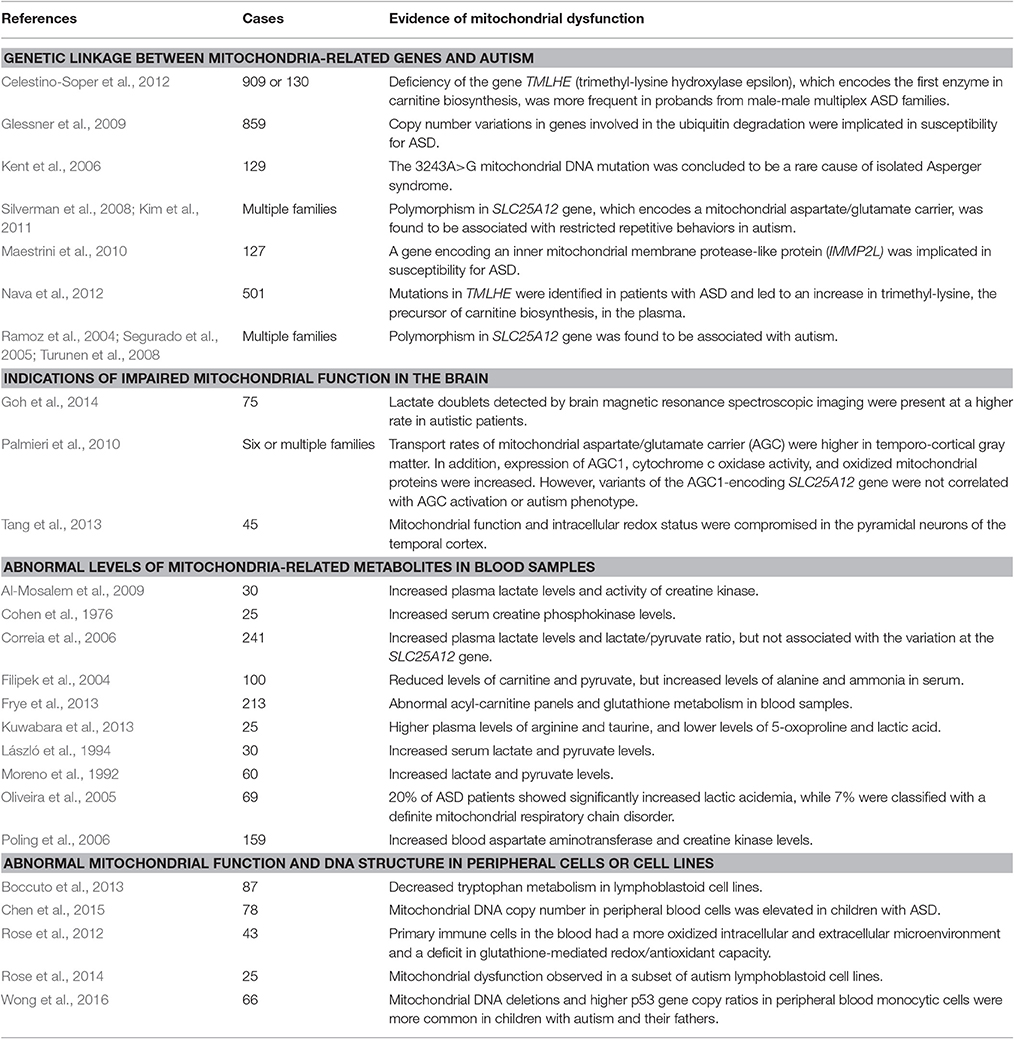
Table 1. Studies showing linkage between ASD and mitochondrial dysfunction in ASD patients (only those reporting more than 25 subjects are included in this table).
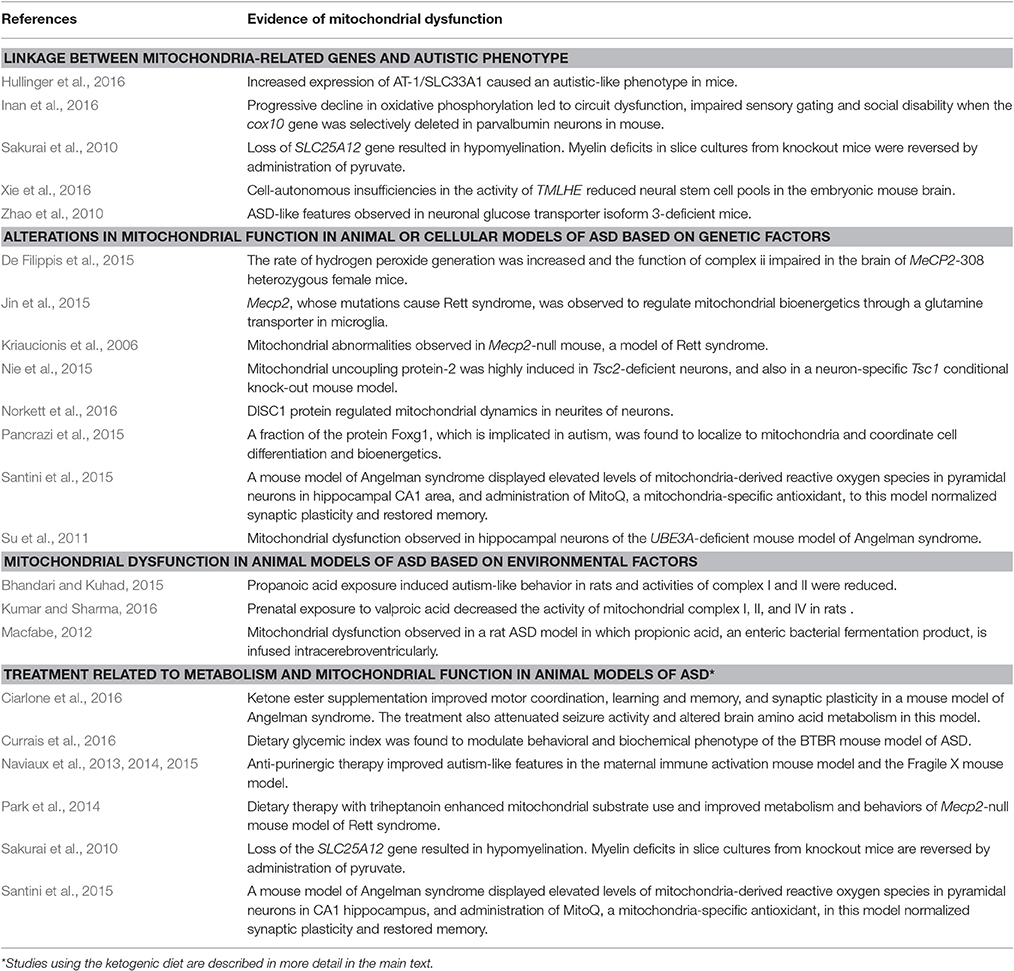
Table 2. Studies showing association between ASD and mitochondrial dysfunction in animal models of ASD.
Common co-morbidities of ASD also suggest metabolic and mitochondrial dysfunction. One of the most significant co-morbidities is epilepsy, with a prevalence of 5–38% in children with ASD—much higher than the 1–2% prevalence in the general population (Frye, 2015). Seizures also occur in 35–60% of individuals with biochemically-confirmed mitochondrial disease (Rahman, 2012), suggesting there may be a common etio-pathology. Similarly, gastrointestinal dysfunction, a frequent comorbidity of ASD (Chaidez et al., 2014), is also common in mitochondrial disease (Frye et al., 2015).
Taken together, we believe that mitochondria act as a central nexus responding to and regulating many domains of cellular biology that have been implicated in ASD. Given the prevalence of metabolic/mitochondrial dysfunction in ASD, options for metabolic therapy should be explored. Below we review some of the emerging clinical and research evidence that metabolic therapy and improved mitochondrial function can ameliorate ASD symptoms and comorbidities.
A metabolic therapy in use for decades is the ketogenic diet (KD), a high-fat, low-carbohydrate diet—a remarkably effective non-pharmacological treatment for medically intractable epilepsy (Neal et al., 2008). Based on historical observations that either fasting or starvation rendered anti-seizure effects, the KD was designed to reproduce the biochemical changes seen in these physiological states (Masino and Rho, 2012). Recently, various dietary and metabolic therapies have been attempted in a wider variety of neurological diseases including ASD, Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, sleep disorders, multiple sclerosis, brain trauma, stroke, pain, Huntington's disease and brain cancer (Ruskin et al., 2009; Stafstrom and Rho, 2012; Napoli et al., 2014). Although generally limited in scope, clinical studies thus far showed promising results in conditions such as Alzheimer's disease and ASD and are discussed in more detail below. In addition, research using animal models has pointed to a common mechanism of regulating energy metabolism to afford neuroprotective effects (Stafstrom and Rho, 2012).
Overall, recent clinical and laboratory evidence suggests that the KD may have positive effects in ASD. The complex pathophysiology of ASD and the diversity of mechanisms mobilized by dietary therapy combine to make identifying the key molecular mechanisms challenging, but a number of candidates are emerging. Two hallmark biochemical features after the KD treatment are increased ketone body production by the liver through fatty acid oxidation and reduced blood glucose levels (Stafstrom and Rho, 2012). More specific metabolic effects, such as increases in specific polyunsaturated fatty acids, might regulate neuronal membrane excitability (Voskuyl and Vreugdenhil, 2001), reduce inflammation (Cullingford, 2008; Jeong et al., 2011), or decrease the production of ROS by mitochondria (Kim do and Rho, 2008). Additionally, ketone bodies themselves have been shown to possess neuroprotective properties through improved bioenergetics - raising ATP levels and reducing ROS production through enhancement of NADH oxidation and inhibition of mitochondrial permeability transition (Kim do et al., 2007, 2015); related to this, a KD has also been shown to stimulate mitochondrial biogenesis (Bough et al., 2006; Ahola-Erkkila et al., 2010). In parallel, reduced glycolysis can suppress seizures, improve mitochondrial function, decrease oxidative stress, reduce activity of pro-apoptotic factors, and inhibit inflammatory mediators such as interleukins and tumor necrosis factor alpha (Garriga-Canut et al., 2006; Maalouf et al., 2009). The KD has also been proposed to increase adenosine (a product of extracellular ATP dephosphorylation); ATP and adenosine are purines with pleiotropic neuromodulatory and neuroprotective roles proposed to underlie in part the diet's clinical efficacy (Masino and Geiger, 2008; Masino et al., 2009, 2010). Separately, increased adenosine has been proposed to reduce symptoms and comorbidities of ASD (Masino et al., 2013). In addition, the KD has been reported to regulate energy-sensing pathways such as those involving the insulin-like growth factor and the mammalian target of rapamycin (McDaniel et al., 2011; Gano et al., 2014). Epigenetic regulation is a new but potentially important mechanism as well (Boison, 2016).
Given the effects of the KD and its substrates (e.g., ketone bodies, fatty acids) on cognitive and behavioral functioning, it is reasonable to speculate that this diet would induce changes in synaptic morphology and function. Studies have shown that the KD can modulate excitability through actions on potassium ion channels (Tigerholm et al., 2012; Lutas and Yellen, 2013) and glutamatergic synaptic transmission (Xu et al., 2006; Juge et al., 2010; Lutas and Yellen, 2013; Chang et al., 2016), as well as possible regulation of GABA production (Yudkoff et al., 2007). In addition, the KD or its metabolic mediators can induce changes in synaptic vesicular cycling (Hrynevich et al., 2016), hippocampal mossy fiber sprouting (Muller-Schwarze et al., 1999), and both age- and region-dependent changes in synaptic morphology (Balietti et al., 2008, 2009).
Collectively, evidence thus far indicates that the KD affords broad neuroprotective effects, and hence, it is reasonable to hypothesize that this diet could prove to be beneficial for individuals with ASD.
To date, there have been limited clinical trials involving treatment of ASD patients with metabolic therapy using variants of a KD. The first report was a pilot prospective study in autistic children aged between 4 and 10 years carried out by Evangeliou and colleagues; they applied an intermittent modified medium-chain triglyceride (MCT) diet (Evangeliou et al., 2003). Most of the 18 patients who adhered to the diet improved based on the Childhood Autism Rating Scale (CARS) and several additional clinical parameters. Significant (i.e., >12 units of decrease in CARS) and average (>8–12 units of decrease in CARS) improvement was recorded in two and eight patients, respectively, while minor (2–8 units of decrease in CARS) improvement was reported in the remaining eight patients. More recently, Spilioti and colleagues reported the effects of KD treatment in a group of Greek children with ASD aged between 3.5 and 6 years (Spilioti et al., 2013). Of the 6 patients who implemented the diet successfully, significant and average improvement was recorded in one and two patients, respectively, and minor improvement was reported in the remaining three patients.
The diet is also effective in reducing common comorbidities of ASD such as seizures, not surprisingly, but also improved cognition and behavior. A pilot retrospective study analyzed outcomes in children prescribed the KD to treat epileptic seizures; among these children, some also had autistic symptoms and abnormal behaviors. Children assigned in the KD group were currently on the diet and had been for at least 6 months; children assigned in the non-KD group stopped the diet at least 2 months prior. Fewer abnormal behaviors and significant behavioral improvement were found in the KD group, and behavioral improvement was not correlated with seizure control (Masino et al., 2011). More recently a randomized control trial showed improved cognition, mood and behavior—particularly reduced anxiety—in children prescribed the KD for refractory epilepsy. These behavioral benefits were also unrelated to seizure control (IJff et al., 2016). In a remarkable case study, Herbert and Buckley reported on a 12-year-old child with comorbid autism and epilepsy treated with a gluten- and casein-free KD (fats composed mostly of MCTs) (Herbert and Buckley, 2013). In addition to a significant reduction in seizures, the diet resolved morbid obesity and improved cognitive and behavioral function. Over the course of several years following initial diagnosis, the child's CARS score decreased from 49 to 17, representing a change from severe autism to a non-autistic state, and her intelligence quotient increased by 70 points.
In summary, clinical evidence to date remains limited, but results from the aforementioned studies show promise that metabolic therapy with several different versions of a KD can improve symptoms of ASD and can also improve cognition and behavior—the latter benefits that can facilitate optimal outcomes in ASD. In patients with diagnosed ASD, greater than 50% of autism patients who received this metabolic therapy showed moderate-to-significant clinical improvement, while the remainder displayed minor improvement. At present, more larger-scale clinical studies are required. Meanwhile, as mentioned earlier, metabolic and mitochondrial dysfunction may represent only a subgroup of the ASD population. Thus, it would be important to determine the relation between the effects of the KD and metabolic/genetic profile of ASD patients.
Due to the complexity of ASD, investigators have developed and employed numerous animal models. Some have clear metabolic underpinnings, underscoring the link between metabolic dysfunction and symptoms of autism. Metabolic therapy with a KD and/or a restricted diet has already been examined in several models. In agreement with the aforementioned clinical studies, reports in animal models have been positive. The ASD models tested with metabolic therapy discussed here include genetic disorders that mirror clinical conditions, induced ASD that models environmental conditions found to increase ASD risk in humans, and behavioral ASD models with unknown etiologies that recapitulate all or some of the core symptoms, and may or may not have comorbid seizures.
As one genetic example, succinic semialdehyde dehydrogenase (SSADH) deficiency is a rare autosomal recessive condition that results in mild-to-moderate mental retardation, disproportionate language dysfunction, seizures, hypotonia, hyporeflexia, hallucinations, and autistic behaviors (Pearl et al., 2003). In an animal model of SSADH deficiency, the SSADH knockout mouse, Nylen and colleagues found that KD treatment normalized electroencephalogram (EEG) activity and restored miniature inhibitory post-synaptic currents recorded in CA1 pyramidal cells using hippocampal slices. In contrast, there were no significant differences between the groups in terms of miniature excitatory post-synaptic currents. Behaviorally, KD-treated mutant animals experienced significantly fewer seizures compared to mutant animals fed the control diet (Nylen et al., 2008).
Metabolic therapy with dietary restriction (either a standard diet or KD) was tested in another clinically relevant genetic model of Rett syndrome. Rett syndrome is a neurodevelopmental disorder characterized by normal early maturation, followed by a slowing of development, impairment of motor functions, seizure susceptibility, and intellectual disability. In most cases, Rett syndrome is caused by mutations in the methyl-CpG-binding protein 2 (MECP2) gene (Amir et al., 1999). Children with Rett syndrome often exhibit autistic-like behaviors in the early stages of the disease (Percy, 2011). Mantis and colleagues found that Mecp2 mutant mice performed significantly worse in assays of motor function and anxiety compared to wild-type control animals, and restriction of either standard diet or the KD improved motor behavior and reduced anxiety in these mutant animals (Mantis et al., 2009). There is also limited clinical evidence for anti-seizure efficacy and improved behavior after KD treatment in Rett syndrome (Liebhaber et al., 2003).
Most cases of ASD have unknown genetic underpinnings (Gaugler et al., 2014), and models of unidentified etiology have been characterized with behavioral tests assessing autistic symptomatology. The BTBR T+tf/J (BTBR) inbred mouse strain is one of the most clinically relevant animal models of autism; it was identified in an extensive effort to characterize ASD-like behaviors in ten inbred mouse strains (Moy et al., 2007) and displays all the core behavioral features that define the disorder (Moy et al., 2007; McFarlane et al., 2008; Meyza et al., 2013; Ruskin et al., 2013; Smith et al., 2014; Ellegood and Crawley, 2015). BTBR mice display deficits in social interaction and communication assays and exhibit repetitive and stereotyped behaviors. During the relatively short time since its discovery as an ASD model, the BTBR strain has been increasingly used to study the etiology and to uncover potential interventions for ASD (Moy et al., 2007; McFarlane et al., 2008; Llaneza et al., 2010; Ruskin et al., 2013; Cheng et al., 2016; Mychasiuk and Rho, 2016; Newell et al., 2016). Ruskin and colleagues reported that the BTBR mice showed decreased sociability in the three-chamber test, decreased self-directed repetitive behavior, and improved social communication in a food preference assay after being fed a KD (Ruskin et al., 2013). In addition, the authors showed that the behavioral improvements were probably not related to any anti-seizure effect of the diet, because no spontaneous seizures or abnormal EEG features were observed in the BTBR animals.
Interestingly, a recent study showed that gut microbiota composition of cecal and fecal samples was significantly altered in BTBR mice compared to B6 animals (Newell et al., 2016). In addition, a KD decreased total host bacterial abundance in both sample types, and in the BTBR animals counteracted a low Firmicutes to Bacteroidetes ratio, which is commonly observed in patients with ASD (Finegold et al., 2010; De Angelis et al., 2013). Related to these findings, it has been shown that dietary glycemic index, which is a measure of how much the carbohydrate in a food item affects blood glucose level, modulates behavioral and biochemical phenotype in the BTBR mice (Currais et al., 2016). These data support the idea that in the context of genetic predisposition to ASD, diet could potentially alter the expression of the disorders.
More recently, Ruskin et al. also showed KD-induced behavioral improvements in the EL mouse, a model of comorbid ASD-associated behaviors and progressive spontaneous epilepsy. Mice (of both sexes) were fed a KD for 3 weeks after weaning and prior to the age of onset of the seizure phenotype. Sociability improved and repetititve behaviors decreased; intriguingly, these effects were more pronounced in females. Also, some behavioral benefits were observed in females even when a more liberal dietary formulation was applied (Ruskin et al., 2016).
As mentioned above, environmental factors also contribute to the risk of developing ASD. In this regard, exposure to exogenous chemicals is best exemplified by VPA use during pregnancy. VPA is a pharmacological anticonvulsant used in humans primarily for the treatment of epilepsy and migraine, and epidemiological studies have shown that use of VPA during pregnancy is associated with an increased risk of ASD in the offspring (Bromley et al., 2013; Christensen et al., 2013). The VPA exposure model is one of the most frequently studied models of autism (Chomiak et al., 2013; Roullet et al., 2013) since it exhibits many similar structural and behavioral features seen in ASD individuals. Ahn and colleagues found that KD treatment recovered part of the play behavior of juvenile rats exposed to VPA prenatally (Ahn et al., 2014). Interestingly, the authors also found that prenatal exposure to VPA altered mitochondrial respiration, and the KD was able to partially restore this. A recent study in VPA-treated mice also found improved social behavior (Castro et al., 2016).
Taken together, there is increasing evidence for the beneficial effects of the KD in different animal models of ASD. However, clear evidence for converging mechanistic links remains hypothetical, and few fundamental mechanistic studies have been conducted to date in either animal models or human ASD tissues. Thus, there is a need for further studies utilizing diverse animal models and incorporating comprehensive behavioral assays to elucidate common molecular pathways in ASD and to validate the positive effects of the KD observed thus far in animal models. Equally important, studies aimed at identifying the mechanisms relevant to such models of ASD are required to optimize treatment, discover novel therapeutic targets, and ultimately provide key insights to the neurobiology of ASD. Borrowing from the rich literature on the KD in epilepsy, shifts in energy metabolism, the direct actions of the ketone bodies on the mitochondria, neuromodulatory functions of ATP and adenosine, regulation of excitation/inhibition balance, and epigenetic effects of the diet are among the promising candidate mechanisms.
At present, there is strong evidence that mitochondrial and metabolic dysfunction may underlie the complex pathophysiology of ASD. Precise mechanisms remain elusive and many questions remain unanswered: Is mitochondrial dysfunction a cause or a consequence of ASD? In which specific organs and cell types is it most relevant? Can addressing mitochondrial and metabolic disturbances directly help at least a subgroup of patients with ASD for more targeted treatments that will ameliorate the diverse symptom complex? Thus far, the KD is a proven metabolic therapy for medically intractable epilepsy, but there are only limited data for its use in ASD and a rudimentary understanding of how the KD may exert positive behavioral effects. The optimum formulation of the KD needs to be established and may be different for ASD compared to epilepsy. Proper efforts to address these fundamental questions and to identify molecular mechanisms and biomarkers will require the collective and collaborative efforts of many, including basic, translational and clinical researchers, as well as investigators with diverse expertise in multi-organ dysfunction, metabolism, and genetic and environmental risk factors. The ultimate reward could be a major breakthrough in understanding its causes and developing much-needed broadly effective therapies for ASD—and in particular, treatments that address core symptoms.
All authors have drafted and revised the manuscript together and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work is supported by Alberta Children's Hospital Research Foundation grant to JR, and NIH grant NS066392 to SM.
Ahn, Y., Narous, M., Tobias, R., Rho, J. M., and Mychasiuk, R. (2014). The ketogenic diet modifies social and metabolic alterations identified in the prenatal valproic acid model of autism spectrum disorder. Dev. Neurosci. 36, 371–380. doi: 10.1159/000362645
Ahola-Erkkila, S., Carroll, C. J., Peltola-Mjosund, K., Tulkki, V., Mattila, I., Seppanen-Laakso, T., et al. (2010). Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum. Mol. Genet. 19, 1974–1984. doi: 10.1093/hmg/ddq076
Al-Mosalem, O. A., El-Ansary, A., Attas, O., and Al-Ayadhi, L. (2009). Metabolic biomarkers related to energy metabolism in Saudi autistic children. Clin. Biochem. 42, 949–957. doi: 10.1016/j.clinbiochem.2009.04.006
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Amoedo, N. D., Punzi, G., Obre, E., Lacombe, D., De Grassi, A., Pierri, C. L., et al. (2016). AGC1/2, the mitochondrial aspartate-glutamate carriers. Biochim. Biophys. Acta 1863, 2394–2412. doi: 10.1016/j.bbamcr.2016.04.011
Antico Arciuch, V. G., Elguero, M. E., Poderoso, J. J., and Carreras, M. C. (2012). Mitochondrial regulation of cell cycle and proliferation. Antioxid. Redox Signal. 16, 1150–1180. doi: 10.1089/ars.2011.4085
Baker, E., and Jeste, S. S. (2015). Diagnosis and management of autism spectrum disorder in the era of genomics: rare disorders can pave the way for targeted treatments. Pediatr. Clin. North Am. 62, 607–618. doi: 10.1016/j.pcl.2015.03.003
Balietti, M., Fattoretti, P., Giorgetti, B., Casoli, T., Di Stefano, G., Platano, D., et al. (2009). Effect of two medium chain triglycerides-supplemented diets on synaptic morphology in the cerebellar cortex of late-adult rats. Microsc. Res. Tech. 72, 933–938. doi: 10.1002/jemt.20737
Balietti, M., Giorgetti, B., Fattoretti, P., Grossi, Y., Di Stefano, G., Casoli, T., et al. (2008). Ketogenic diets cause opposing changes in synaptic morphology in CA1 hippocampus and dentate gyrus of late-adult rats. Rejuvenation Res. 11, 631–640. doi: 10.1089/rej.2007.0650
Berg, J. M., and Geschwind, D. H. (2012). Autism genetics: searching for specificity and convergence. Genome Biol. 13:247. doi: 10.1186/gb-2012-13-7-247
Bhandari, R., and Kuhad, A. (2015). Neuropsychopharmacotherapeutic efficacy of curcumin in experimental paradigm of autism spectrum disorders. Life Sci. 141, 156–169. doi: 10.1016/j.lfs.2015.09.012
Boccuto, L., Chen, C.-F., Pittman, A. R., Skinner, C. D., McCartney, H. J., Jones, K., et al. (2013). Decreased tryptophan metabolism in patients with autism spectrum disorders. Mol. Autism 4:16. doi: 10.1186/2040-2392-4-16
Boison, D. (2016). The biochemistry and epigenetics of epilepsy: focus on adenosine and glycine. Front. Mol. Neurosci. 9:26. doi: 10.3389/fnmol.2016.00026
Bough, K. J., Wetherington, J., Hassel, B., Pare, J. F., Gawryluk, J. W., Greene, J. G., et al. (2006). Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 60, 223–235. doi: 10.1002/ana.20899
Bourgeron, T. (2015). From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci. 16, 551–563. doi: 10.1038/nrn3992
Bromley, R. L., Mawer, G. E., Briggs, M., Cheyne, C., Clayton-Smith, J., Garcia-Finana, M., et al. (2013). The prevalence of neurodevelopmental disorders in children prenatally exposed to antiepileptic drugs. J. Neurol. Neurosurg. Psychiatr. 84, 637–643. doi: 10.1136/jnnp-2012-304270
Buescher, A. V., Cidav, Z., Knapp, M., and Mandell, D. S. (2014). Costs of autism spectrum disorders in the United Kingdom and the United States. JAMA Pediatr. 168, 721–728. doi: 10.1001/jamapediatrics.2014.210
Castro, K., Baronio, D., Perry, I. S., dos Santos Riesgo, R., and Gottfried, C. (2016). The effect of ketogenic diet in an animal model of autism induced by prenatal exposure to valproic acid. Nutr. Neurosci. doi: 10.1080/1028415X.2015.1133029. [Epub ahead of print].
CDC (2014). Prevalence of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2010. Morb. Mort. Wkly Rep. Surveil. Summ. 63, 1–21.
Celestino-Soper, P. B. S., Violante, S., Crawford, E. L., Luo, R., Lionel, A. C., Delaby, E., et al. (2012). A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc. Natl. Acad. Sci. U.S.A. 109, 7974–7981. doi: 10.1073/pnas.1120210109
Chaidez, V., Hansen, R. L., and Hertz-Picciotto, I. (2014). Gastrointestinal problems in children with autism, developmental delays or typical development. J. Autism Dev. Disord. 44, 1117–1127. doi: 10.1007/s10803-013-1973-x
Chang, P., Augustin, K., Boddum, K., Williams, S., Sun, M., Terschak, J. A., et al. (2016). Seizure control by decanoic acid through direct AMPA receptor inhibition. Brain 139(Pt 2), 431–443. doi: 10.1093/brain/awv325
Chaste, P., and Leboyer, M. (2012). Autism risk factors: genes, environment, and gene-environment interactions. Dialogues Clin. Neurosci. 14, 281–292.
Chen, S., Li, Z., He, Y., Zhang, F., Li, H., Liao, Y., et al. (2015). Elevated mitochondrial DNA copy number in peripheral blood cells is associated with childhood autism. BMC Psychiatry 15:50. doi: 10.1186/s12888-015-0432-y
Cheng, N., Khanbabaei, M., Murari, K., and Rho, J. M. (2016). Disruption of visual circuit formation and refinement in a mouse model of autism. Autism Res. doi: 10.1002/aur.1687. [Epub ahead of print].
Chomiak, T., Turner, N., and Hu, B. (2013). What we have learned about autism spectrum disorder from valproic acid. Patholog. Res. Int. 2013:712758. doi: 10.1155/2013/712758
Christensen, J., Gronborg, T. K., Sorensen, M. J., Schendel, D., Parner, E. T., Pedersen, L. H., et al. (2013). Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 309, 1696–1703. doi: 10.1001/jama.2013.2270
Ciarlone, S. L., Grieco, J. C., D'Agostino, D. P., and Weeber, E. J. (2016). Ketone ester supplementation attenuates seizure activity, and improves behavior and hippocampal synaptic plasticity in an Angelman syndrome mouse model. Neurobiol. Dis. 96, 38–46. doi: 10.1016/j.nbd.2016.08.002
Cohen, D. J., Johnson, W., Caparulo, B. K., and Young, J. G. (1976). Creatine phosphokinase levels in children with severe developmental disturbances. Arch. Gen. Psychiatry 33, 683–686. doi: 10.1001/archpsyc.1976.01770060025004
Correia, C., Coutinho, A. M., Diogo, L., Grazina, M., Marques, C., Miguel, T., et al. (2006). Brief report: high frequency of biochemical markers for mitochondrial dysfunction in autism: no association with the mitochondrial aspartate/glutamate carrier SLC25A12 gene. J. Autism Dev. Disord. 36, 1137–1140. doi: 10.1007/s10803-006-0138-6
Courchesne, E., Pierce, K., Schumann, C. M., Redcay, E., Buckwalter, J. A., Kennedy, D. P., et al. (2007). Mapping early brain development in autism. Neuron 56, 399–413. doi: 10.1016/j.neuron.2007.10.016
Cullingford, T. (2008). Peroxisome proliferator-activated receptor alpha and the ketogenic diet. Epilepsia 49(Suppl. 8), 70–72. doi: 10.1111/j.1528-1167.2008.01840.x
Currais, A., Farrokhi, C., Dargusch, R., Goujon-Svrzic, M., and Maher, P. (2016). Dietary glycemic index modulates the behavioral and biochemical abnormalities associated with autism spectrum disorder. Mol. Psychiatry 21, 426–436. doi: 10.1038/mp.2015.64
De Angelis, M., Piccolo, M., Vannini, L., Siragusa, S., De Giacomo, A., Serrazzanetti, D. I., et al. (2013). Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS ONE 8:e76993. doi: 10.1371/journal.pone.0076993
De Filippis, B., Valenti, D., de Bari, L., De Rasmo, D., Musto, M., Fabbri, A., et al. (2015). Mitochondrial free radical overproduction due to respiratory chain impairment in the brain of a mouse model of Rett syndrome: protective effect of CNF1. Free Radic. Biol. Med. 83, 167–177. doi: 10.1016/j.freeradbiomed.2015.02.014
de la Torre-Ubieta, L., Won, H., Stein, J. L., and Geschwind, D. H. (2016). Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 22, 345–361. doi: 10.1038/nm.4071
De Rubeis, S., and Buxbaum, J. D. (2015). Genetics and genomics of autism spectrum disorder: embracing complexity. Hum. Mol. Genet. 24, R24–R31. doi: 10.1093/hmg/ddv273
Devlin, B., and Scherer, S. W. (2012). Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 22, 229–237. doi: 10.1016/j.gde.2012.03.002
Dhillon, S., Hellings, J. A., and Butler, M. G. (2011). Genetics and mitochondrial abnormalities in autism spectrum disorders: a review. Curr. Genomics 12, 322–332. doi: 10.2174/138920211796429745
DiCicco-Bloom, E., Lord, C., Zwaigenbaum, L., Courchesne, E., Dager, S. R., Schmitz, C., et al. (2006). The developmental neurobiology of autism spectrum disorder. J. Neurosci. 26, 6897–6906. doi: 10.1523/JNEUROSCI.1712-06.2006
Durkin, M. S., Maenner, M. J., Newschaffer, C. J., Lee, L. C., Cunniff, C. M., Daniels, J. L., et al. (2008). Advanced parental age and the risk of autism spectrum disorder. Am. J. Epidemiol. 168, 1268–1276. doi: 10.1093/aje/kwn250
Ellegood, J., and Crawley, J. N. (2015). Behavioral and neuroanatomical phenotypes in mouse models of autism. Neurotherapeutics 12, 521–533. doi: 10.1007/s13311-015-0360-z
Evangeliou, A., Vlachonikolis, I., Mihailidou, H., Spilioti, M., Skarpalezou, A., Makaronas, N., et al. (2003). Application of a ketogenic diet in children with autistic behavior: pilot study. J. Child Neurol. 18, 113–118. doi: 10.1177/08830738030180020501
Filipek, P. A., Juranek, J., Nguyen, M. T., Cummings, C., and Gargus, J. J. (2004). Relative carnitine deficiency in autism. J. Autism Dev. Disord. 34, 615–623. doi: 10.1007/s10803-004-5283-1
Finegold, S. M., Dowd, S. E., Gontcharova, V., Liu, C., Henley, K. E., Wolcott, R. D., et al. (2010). Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 16, 444–453. doi: 10.1016/j.anaerobe.2010.06.008
Frye, R. E. (2015). Metabolic and mitochondrial disorders associated with epilepsy in children with autism spectrum disorder. Epilepsy Behav. 47, 147–157. doi: 10.1016/j.yebeh.2014.08.134
Frye, R. E., Melnyk, S., and Macfabe, D. F. (2013). Unique acyl-carnitine profiles are potential biomarkers for acquired mitochondrial disease in autism spectrum disorder. Transl. Psychiatry 3:e220. doi: 10.1038/tp.2012.143
Frye, R. E., Rose, S., Slattery, J., and MacFabe, D. F. (2015). Gastrointestinal dysfunction in autism spectrum disorder: the role of the mitochondria and the enteric microbiome. Microb. Ecol. Health Dis. 26:27458. doi: 10.3402/mehd.v26.27458
Frye, R. E., and Rossignol, D. A. (2011). Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatric Res. 69(5 Pt 2), 41R–47R. doi: 10.1203/PDR.0b013e318212f16b
Frye, R. E., and Rossignol, D. A. (2016). Identification and treatment of pathophysiological comorbidities of autism spectrum disorder to achieve optimal outcomes. Clin. Med. Insights Pediatrics 10, 43–56. doi: 10.4137/CMPed.S38337
Gano, L. B., Patel, M., and Rho, J. M. (2014). Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 55, 2211–2228. doi: 10.1194/jlr.R048975
Gardener, H., Spiegelman, D., and Buka, S. L. (2011). Perinatal and neonatal risk factors for autism: a comprehensive meta-analysis. Pediatrics 128, 344–355. doi: 10.1542/peds.2010-1036
Garriga-Canut, M., Schoenike, B., Qazi, R., Bergendahl, K., Daley, T. J., Pfender, R. M., et al. (2006). 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat. Neurosci. 9, 1382–1387. doi: 10.1038/nn1791
Gaugler, T., Klei, L., Sanders, S. J., Bodea, C. A., Goldberg, A. P., Lee, A. B., et al. (2014). Most genetic risk for autism resides with common variation. Nat. Genet. 46, 881–885. doi: 10.1038/ng.3039
Geschwind, D. H. (2008). Autism: many genes, common pathways? Cell 135, 391–395. doi: 10.1016/j.cell.2008.10.016
Glessner, J. T., Wang, K., Cai, G., Korvatska, O., Kim, C. E., Wood, S., et al. (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. doi: 10.1038/nature07953
Goh, S., Dong, Z., Zhang, Y., DiMauro, S., and Peterson, B. S. (2014). Mitochondrial dysfunction as a neurobiological subtype of autism spectrum disorder: evidence from brain imaging. JAMA Psychiatry 71, 665–671. doi: 10.1001/jamapsychiatry.2014.179
Goines, P., and Van de Water, J. (2010). The immune system's role in the biology of autism. Curr. Opin. Neurol. 23, 111–117. doi: 10.1097/WCO.0b013e3283373514
Haas, R. H. (2010). Autism and mitochondrial disease. Dev. Disabil. Res. Rev. 16, 144–153. doi: 10.1002/ddrr.112
Hansen, S. N., Schendel, D. E., and Parner, E. T. (2015). Explaining the increase in the prevalence of autism spectrum disorders: the proportion attributable to changes in reporting practices. JAMA Pediatr. 169, 56–62. doi: 10.1001/jamapediatrics.2014.1893
Harris, J. J., Jolivet, R., and Attwell, D. (2012). Synaptic energy use and supply. Neuron 75, 762–777. doi: 10.1016/j.neuron.2012.08.019
Herbert, M. R., and Buckley, J. A. (2013). Autism and dietary therapy: case report and review of the literature. J. Child Neurol. 28, 975–982. doi: 10.1177/0883073813488668
Hrynevich, S. V., Waseem, T. V., Hebert, A., Pellerin, L., and Fedorovich, S. V. (2016). β-Hydroxybutyrate supports synaptic vesicle cycling but reduces endocytosis and exocytosis in rat brain synaptosomes. Neurochem. Int. 93, 73–81. doi: 10.1016/j.neuint.2015.12.014
Hsiao, E. Y. (2013). Immune dysregulation in autism spectrum disorder. Int. Rev. Neurobiol. 113, 269–302. doi: 10.1016/B978-0-12-418700-9.00009-5
Huguet, G., Ey, E., and Bourgeron, T. (2013). The genetic landscapes of autism spectrum disorders. Annu. Rev. Genomics Hum. Genet. 14, 191–213. doi: 10.1146/annurev-genom-091212-153431
Hullinger, R., Li, M., Wang, J., Peng, Y., Dowell, J. A., Bomba-Warczak, E., et al. (2016). Increased expression of AT-1/SLC33A1 causes an autistic-like phenotype in mice by affecting dendritic branching and spine formation. J. Exp. Med. 213, 1267–1284. doi: 10.1084/jem.20151776
IJff, D. M., Postulart, D., Lambrechts, D. A., Majoie, M. H., de Kinderen, R. J., Hendriksen, J. G., et al. (2016). Cognitive and behavioral impact of the ketogenic diet in children and adolescents with refractory epilepsy: a randomized controlled trial. Epilepsy Behav. 60, 153–157. doi: 10.1016/j.yebeh.2016.04.033
Inan, M., Zhao, M., Manuszak, M., Karakaya, C., Rajadhyaksha, A. M., Pickel, V. M., et al. (2016). Energy deficit in parvalbumin neurons leads to circuit dysfunction, impaired sensory gating and social disability. Neurobiol. Dis. 93, 35–46. doi: 10.1016/j.nbd.2016.04.004
Jeong, E. A., Jeon, B. T., Shin, H. J., Kim, N., Lee, D. H., Kim, H. J., et al. (2011). Ketogenic diet-induced peroxisome proliferator-activated receptor-γ activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp. Neurol. 232, 195–202. doi: 10.1016/j.expneurol.2011.09.001
Jeste, S. S., and Geschwind, D. H. (2014). Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat. Rev. Neurol. 10, 74–81. doi: 10.1038/nrneurol.2013.278
Jin, L.-W., Horiuchi, M., Wulff, H., Liu, X.-B., Cortopassi, G. A., Erickson, J. D., et al. (2015). Dysregulation of glutamine transporter SNAT1 in Rett syndrome microglia: a mechanism for mitochondrial dysfunction and neurotoxicity. J. Neurosci. 35, 2516–2529. doi: 10.1523/JNEUROSCI.2778-14.2015
Juge, N., Gray, J. A., Omote, H., Miyaji, T., Inoue, T., Hara, C., et al. (2010). Metabolic control of vesicular glutamate transport and release. Neuron 68, 99–112. doi: 10.1016/j.neuron.2010.09.002
Kann, O., and Kovacs, R. (2007). Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 292, C641–C657. doi: 10.1152/ajpcell.00222.2006
Kent, L., Lambert, C., Pyle, A., Elliott, H., Wheelwright, S., Baron-Cohen, S., et al. (2006). The mitochondrial DNA A3243A>G mutation must be an infrequent cause of Asperger syndrome. J. Pediatr. 149, 280–281. doi: 10.1016/j.jpeds.2005.08.060
Kikuchi, G., Motokawa, Y., Yoshida, T., and Hiraga, K. (2008). Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proc. Jpn Acad. B Phys. Biol. Sci. 84, 246–263. doi: 10.2183/pjab.84.246
Kim, S. J., Silva, R. M., Flores, C. G., Jacob, S., Guter, S., Valcante, G., et al. (2011). A quantitative association study of SLC25A12 and restricted repetitive behavior traits in autism spectrum disorders. Mol. Autism 2:8. doi: 10.1186/2040-2392-2-8
Kim, Y. S., and Leventhal, B. L. (2015). Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biol. Psychiatry 77, 66–74. doi: 10.1016/j.biopsych.2014.11.001
Kim do, Y., Davis, L. M., Sullivan, P. G., Maalouf, M., Simeone, T. A., van Brederode, J., et al. (2007). Ketone bodies are protective against oxidative stress in neocortical neurons. J. Neurochem. 101, 1316–1326. doi: 10.1111/j.1471-4159.2007.04483.x
Kim do, Y., and Rho, J. M. (2008). The ketogenic diet and epilepsy. Curr. Opin. Clin. Nutr. Metab. Care 11, 113–120. doi: 10.1097/MCO.0b013e3282f44c06
Kim do, Y., Simeone, K. A., Simeone, T. A., Pandya, J. D., Wilke, J. C., Ahn, Y., et al. (2015). Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann. Neurol. 78, 77–87. doi: 10.1002/ana.24424
Kimura, T., and Murakami, F. (2014). Evidence that dendritic mitochondria negatively regulate dendritic branching in pyramidal neurons in the neocortex. J. Neurosci. 34, 6938–6951. doi: 10.1523/JNEUROSCI.5095-13.2014
Kleinman, J. M., Ventola, P. E., Pandey, J., Verbalis, A. D., Barton, M., Hodgson, S., et al. (2008). Diagnostic stability in very young children with autism spectrum disorders. J. Autism Dev. Disord. 38, 606–615. doi: 10.1007/s10803-007-0427-8
Kohane, I. S., McMurry, A., Weber, G., MacFadden, D., Rappaport, L., Kunkel, L., et al. (2012). The co-morbidity burden of children and young adults with autism spectrum disorders. PLoS ONE 7:e33224. doi: 10.1371/journal.pone.0033224
Kopp, N., Climer, S., and Dougherty, J. D. (2015). Moving from capstones toward cornerstones: successes and challenges in applying systems biology to identify mechanisms of autism spectrum disorders. Front. Genet. 6:301. doi: 10.3389/fgene.2015.00301
Krakowiak, P., Walker, C. K., Bremer, A. A., Baker, A. S., Ozonoff, S., Hansen, R. L., et al. (2012). Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics 129, e1121–e1128. doi: 10.1542/peds.2011-2583
Kriaucionis, S., Paterson, A., Curtis, J., Guy, J., Macleod, N., and Bird, A. (2006). Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol. Cell. Biol. 26, 5033–5042. doi: 10.1128/MCB.01665-05
Kumar, H., and Sharma, B. (2016). Memantine ameliorates autistic behavior, biochemistry & blood brain barrier impairments in rats. Brain Res. Bull. 124, 27–39. doi: 10.1016/j.brainresbull.2016.03.013
Kuwabara, H., Yamasue, H., Koike, S., Inoue, H., Kawakubo, Y., Kuroda, M., et al. (2013). Altered metabolites in the plasma of autism spectrum disorder: a capillary electrophoresis time-of-flight mass spectroscopy study. PLoS ONE 8:e73814. doi: 10.1371/journal.pone.0073814
Lai, M. C., Lombardo, M. V., and Baron-Cohen, S. (2014). Autism. Lancet 383, 896–910. doi: 10.1016/S0140-6736(13)61539-1
László, A., Horváth, E., Eck, E., and Fekete, M. (1994). Serum serotonin, lactate and pyruvate levels in infantile autistic children. Clin. Chim. Acta 229, 205–207. doi: 10.1016/0009-8981(94)90243-7
Legido, A., Jethva, R., and Goldenthal, M. J. (2013). Mitochondrial dysfunction in autism. Semin. Pediatr. Neurol. 20, 163–175. doi: 10.1016/j.spen.2013.10.008
Levy, S. E., Giarelli, E., Lee, L. C., Schieve, L. A., Kirby, R. S., Cunniff, C., et al. (2010). Autism spectrum disorder and co-occurring developmental, psychiatric, and medical conditions among children in multiple populations of the United States. J. Dev. Behav. Pediatr. 31, 267–275. doi: 10.1097/DBP.0b013e3181d5d03b
Li, Z., Okamoto, K., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. doi: 10.1016/j.cell.2004.11.003
Liebhaber, G. M., Riemann, E., and Baumeister, F. A. (2003). Ketogenic diet in Rett syndrome. J. Child Neurol. 18, 74–75. doi: 10.1177/08830738030180011801
Llaneza, D. C., DeLuke, S. V., Batista, M., Crawley, J. N., Christodulu, K. V., and Frye, C. A. (2010). Communication, interventions, and scientific advances in autism: a commentary. Physiol. Behav. 100, 268–276. doi: 10.1016/j.physbeh.2010.01.003
Loke, Y. J., Hannan, A. J., and Craig, J. M. (2015). The role of epigenetic change in autism spectrum disorders. Front. Neurol. 6:107. doi: 10.3389/fneur.2015.00107
Lord, C., Risi, S., DiLavore, P. S., Shulman, C., Thurm, A., and Pickles, A. (2006). Autism from 2 to 9 years of age. Arch. Gen. Psychiatry 63, 694–701. doi: 10.1001/archpsyc.63.6.694
Lutas, A., and Yellen, G. (2013). The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. 36, 32–40. doi: 10.1016/j.tins.2012.11.005
Maalouf, M., Rho, J. M., and Mattson, M. P. (2009). The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res. Rev. 59, 293–315. doi: 10.1016/j.brainresrev.2008.09.002
Macfabe, D. F. (2012). Short-chain fatty acid fermentation products of the gut microbiome: implications in autism spectrum disorders. Microb. Ecol. Health Dis. 23:19260. doi: 10.3402/mehd.v23i0.19260
Maestrini, E., Pagnamenta, A. T., Lamb, J. A., Bacchelli, E., Sykes, N. H., Sousa, I., et al. (2010). High-density SNP association study and copy number variation analysis of the AUTS1 and AUTS5 loci implicate the IMMP2L-DOCK4 gene region in autism susceptibility. Mol. Psychiatry 15, 954–968. doi: 10.1038/mp.2009.34
Mahfouz, A., Ziats, M. N., Rennert, O. M., Lelieveldt, B. P., and Reinders, M. J. (2015). Shared pathways among autism candidate genes determined by co-expression network analysis of the developing human brain transcriptome. J. Mol. Neurosci. 57, 580–594. doi: 10.1007/s12031-015-0641-3
Mantis, J. G., Fritz, C. L., Marsh, J., Heinrichs, S. C., and Seyfried, T. N. (2009). Improvement in motor and exploratory behavior in Rett syndrome mice with restricted ketogenic and standard diets. Epilepsy Behav. 15, 133–141. doi: 10.1016/j.yebeh.2009.02.038
Masino, S. A., and Geiger, J. D. (2008). Are purines mediators of the anticonvulsant/neuroprotective effects of ketogenic diets? Trends Neurosci. 31, 273–278. doi: 10.1016/j.tins.2008.02.009
Masino, S. A., Kawamura, M. Jr., Cote, J. L., Williams, R. B., and Ruskin, D. N. (2013). Adenosine and autism: a spectrum of opportunities. Neuropharmacology 68, 116–121. doi: 10.1016/j.neuropharm.2012.08.013
Masino, S. A., Kawamura, M. Jr., Ruskin, D. N., Gawryluk, J., Chen, X., and Geiger, J. D. (2010). Purines and the anti-epileptic actions of ketogenic diets. Open Neurosci. J. 4, 58–63. doi: 10.2174/1874082001004010058
Masino, S. A., Kawamura, M., Wasser, C. D., Pomeroy, L. T., and Ruskin, D. N. (2009). Adenosine, ketogenic diet and epilepsy: the emerging therapeutic relationship between metabolism and brain activity. Curr. Neuropharmacol. 7, 257–268. doi: 10.2174/157015909789152164
Masino, S. A., and Rho, J. M. (2012). “Mechanisms of ketogenic diet action,” in Jasper's Basic Mechanisms of the Epilepsies, 4th Edn., eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen, and A. V. Delgado-Escueta (Bethesda, MD: Oxford University Press).
Masino, S. A., Svedova, J., Kawamura, M., DiMario, F. D., and Eigsti, I. M. (2011). “Adenosine and autism -recent research and a new perspective,” in Autism—a Neurodevelopmental Journey from Genes to Behaviour, ed V. Eapen (InTech). Available online at: http://www.intechopen.com/books/autism-a-neurodevelopmental-journey-from-genes-to-behaviour/adenosine-and-autism-recent-research-and-a-new-perspective
Mattson, M. P., Gleichmann, M., and Cheng, A. (2008). Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766. doi: 10.1016/j.neuron.2008.10.010
Mayer, E. A., Tillisch, K., and Gupta, A. (2015). Gut/brain axis and the microbiota. J. Clin. Invest. 125, 926–938. doi: 10.1172/JCI76304
McDaniel, S. S., Rensing, N. R., Thio, L. L., Yamada, K. A., and Wong, M. (2011). The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia 52, e7–e11. doi: 10.1111/j.1528-1167.2011.02981.x
McFarlane, H. G., Kusek, G. K., Yang, M., Phoenix, J. L., Bolivar, V. J., and Crawley, J. N. (2008). Autism-like behavioral phenotypes in BTBR T+tf/J mice. Genes, brain, and behavior 7, 152–163. doi: 10.1111/j.1601-183X.2007.00330.x
Meyza, K. Z., Defensor, E. B., Jensen, A. L., Corley, M. J., Pearson, B. L., Pobbe, R. L., et al. (2013). The BTBR T+tf /J mouse model for autism spectrum disorders-in search of biomarkers. Behav. Brain Res. 251, 25–34. doi: 10.1016/j.bbr.2012.07.021
Moreno, H., Borjas, L., Arrieta, A., Saez, L., Prassad, A., Estevez, J., et al. (1992). [Clinical heterogeneity of the autistic syndrome: a study of 60 families]. Invest. Clin. 33, 13–31.
Moy, S. S., Nadler, J. J., Young, N. B., Perez, A., Holloway, L. P., Barbaro, R. P., et al. (2007). Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav. Brain Res. 176, 4–20. doi: 10.1016/j.bbr.2006.07.030
Muller-Schwarze, A. B., Tandon, P., Liu, Z., Yang, Y., Holmes, G. L., and Stafstrom, C. E. (1999). Ketogenic diet reduces spontaneous seizures and mossy fiber sprouting in the kainic acid model. Neuroreport 10, 1517–1522. doi: 10.1097/00001756-199905140-00023
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Mychasiuk, R., and Rho, J. M. (2016). Genetic modifications associated with ketogenic diet treatment in the BTBRT+Tf/J mouse model of autism spectrum disorder. Autism Res. doi: 10.1002/aur.1682. [Epub ahead of print].
Napoli, E., Duenas, N., and Giulivi, C. (2014). Potential therapeutic use of the ketogenic diet in autism spectrum disorders. Front. Pediatrics 2:69. doi: 10.3389/fped.2014.00069
Nava, C., Lamari, F., Héron, D., Mignot, C., Rastetter, A., Keren, B., et al. (2012). Analysis of the chromosome X exome in patients with autism spectrum disorders identified novel candidate genes, including TMLHE. Transl. Psychiatry 2, e179. doi: 10.1038/tp.2012.102
Naviaux, R. K., Zolkipli, Z., Wang, L., Nakayama, T., Naviaux, J. C., Le, T. P., et al. (2013). Antipurinergic therapy corrects the autism-like features in the poly(IC) mouse model. PLoS ONE 8:e57380. doi: 10.1371/journal.pone.0057380
Naviaux, J. C., Schuchbauer, M. A., Li, K., Wang, L., Risbrough, V. B., Powell, S. B., et al. (2014). Reversal of autism-like behaviors and metabolism in adult mice with single-dose antipurinergic therapy. Transl. Psychiatry 4, e400. doi: 10.1038/tp.2014.33
Naviaux, J. C., Wang, L., Li, K., Bright, A. T., Alaynick, W. A., Williams, K. R., et al. (2015). Antipurinergic therapy corrects the autism-like features in the Fragile X (Fmr1 knockout) mouse model. Mol. Autism 6:1. doi: 10.1186/2040-2392-6-1
Neal, E. G., Chaffe, H., Schwartz, R. H., Lawson, M. S., Edwards, N., Fitzsimmons, G., et al. (2008). The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 7, 500–506. doi: 10.1016/S1474-4422(08)70092-9
Nelson, S. B., and Valakh, V. (2015). Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698. doi: 10.1016/j.neuron.2015.07.033
Newell, C., Bomhof, M. R., Reimer, R. A., Hittel, D. S., Rho, J. M., and Shearer, J. (2016). Ketogenic diet modifies the gut microbiota in a murine model of autism spectrum disorder. Mol. Autism 7:37. doi: 10.1186/s13229-016-0099-3
Nie, D., Chen, Z., Ebrahimi-Fakhari, D., Di Nardo, A., Julich, K., Robson, V. K., et al. (2015). The stress-induced Atf3-gelsolin cascade underlies dendritic spine deficits in neuronal models of tuberous sclerosis complex. J. Neurosci. 35, 10762–10772. doi: 10.1523/JNEUROSCI.4796-14.2015
Norkett, R., Modi, S., Birsa, N., Atkin, T. A., Ivankovic, D., Pathania, M., et al. (2016). DISC1-dependent regulation of mitochondrial dynamics controls the morphogenesis of complex neuronal dendrites. J. Biol. Chem. 291, 613–629. doi: 10.1074/jbc.M115.699447
Nylen, K., Velazquez, J. L., Likhodii, S. S., Cortez, M. A., Shen, L., Leshchenko, Y., et al. (2008). A ketogenic diet rescues the murine succinic semialdehyde dehydrogenase deficient phenotype. Exp. Neurol. 210, 449–457. doi: 10.1016/j.expneurol.2007.11.015
Oliveira, G., Diogo, L., Grazina, M., Garcia, P., Ataíde, A., Marques, C., et al. (2005). Mitochondrial dysfunction in autism spectrum disorders: a population-based study. Dev. Med. Child Neurol. 47, 185–189. doi: 10.1017/S0012162205000332
Orinstein, A. J., Helt, M., Troyb, E., Tyson, K. E., Barton, M. L., Eigsti, I. M., et al. (2014). Intervention for optimal outcome in children and adolescents with a history of autism. J. Dev. Behav. Pediatrics 35, 247–256. doi: 10.1097/DBP.0000000000000037
Palmieri, L., Papaleo, V., Porcelli, V., Scarcia, P., Gaita, L., Sacco, R., et al. (2010). Altered calcium homeostasis in autism-spectrum disorders: evidence from biochemical and genetic studies of the mitochondrial aspartate/glutamate carrier AGC1. Mol. Psychiatry 15, 38–52. doi: 10.1038/mp.2008.63
Pancrazi, L., Di Benedetto, G., Colombaioni, L., Della Sala, G., Testa, G., Olimpico, F., et al. (2015). Foxg1 localizes to mitochondria and coordinates cell differentiation and bioenergetics. Proc. Natl. Acad. Sci. U.S.A. 112, 13910–13915. doi: 10.1073/pnas.1515190112
Park, M. J., Aja, S., Li, Q., Degano, A. L., Penati, J., Zhuo, J., et al. (2014). Anaplerotic triheptanoin diet enhances mitochondrial substrate use to remodel the metabolome and improve lifespan, motor function, and sociability in MeCP2-null mice. PLoS ONE 9:e109527. doi: 10.1371/journal.pone.0109527
Patterson, P. H. (2011). Maternal infection and immune involvement in autism. Trends Mol. Med. 17, 389–394. doi: 10.1016/j.molmed.2011.03.001
Pearl, P. L., Gibson, K. M., Acosta, M. T., Vezina, L. G., Theodore, W. H., Rogawski, M. A., et al. (2003). Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology 60, 1413–1417. doi: 10.1212/01.WNL.0000059549.70717.80
Percy, A. K. (2011). Rett syndrome: exploring the autism link. Arch. Neurol. 68, 985–989. doi: 10.1001/archneurol.2011.149
Pilorge, M., Fassier, C., Le Corronc, H., Potey, A., Bai, J., De Gois, S., et al. (2016). Genetic and functional analyses demonstrate a role for abnormal glycinergic signaling in autism. Mol. Psychiatry 21, 936–945. doi: 10.1038/mp.2015.139
Poling, J. S., Frye, R. E., Shoffner, J., and Zimmerman, A. W. (2006). Developmental regression and mitochondrial dysfunction in a child with autism. J. Child Neurol. 21, 170–172. doi: 10.1177/08830738060210021401
Rahman, S. (2012). Mitochondrial disease and epilepsy. Dev. Med. Child Neurol. 54, 397–406. doi: 10.1111/j.1469-8749.2011.04214.x
Raichle, M. E., and Gusnard, D. A. (2002). Appraising the brain's energy budget. Proc. Natl. Acad. Sci. U.S.A. 99, 10237–10239. doi: 10.1073/pnas.172399499
Ramoz, N., Reichert, J. G., Smith, C. J., Silverman, J. M., Bespalova, I. N., Davis, K. L., et al. (2004). Linkage and association of the mitochondrial aspartate/glutamate carrier SLC25A12 gene with autism. Am. J. Psychiatry 161, 662–669. doi: 10.1176/appi.ajp.161.4.662
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578. doi: 10.1038/nrm3412
Rose, S., Melnyk, S., Trusty, T. A., Pavliv, O., Seidel, L., Li, J., et al. (2012). Intracellular and extracellular redox status and free radical generation in primary immune cells from children with autism. Autism Res. Treat. 2012:986519. doi: 10.1155/2012/986519
Rose, S., Frye, R. E., Slattery, J., Wynne, R., Tippett, M., Pavliv, O., et al. (2014). Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. PLoS ONE 9:e85436. doi: 10.1371/journal.pone.0085436
Rossignol, D. A., and Frye, R. E. (2012). Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol. Psychiatry 17, 290–314. doi: 10.1038/mp.2010.136
Roullet, F. I., Lai, J. K., and Foster, J. A. (2013). In utero exposure to valproic acid and autism–a current review of clinical and animal studies. Neurotoxicol. Teratol. 36, 47–56. doi: 10.1016/j.ntt.2013.01.004
Ruskin, D. N., Fortin, J. A., Bisnauth, S. N., and Masino, S. A. (2016). Ketogenic diets improve behaviors associated with autism spectrum disorder in a sex-specific manner in the EL mouse. Physiol. Behav. 168, 138–145. doi: 10.1016/j.physbeh.2016.10.023
Ruskin, D. N., Kawamura, M., and Masino, S. A. (2009). Reduced pain and inflammation in juvenile and adult rats fed a ketogenic diet. PLoS ONE 4:e8349. doi: 10.1371/journal.pone.0008349
Ruskin, D. N., Svedova, J., Cote, J. L., Sandau, U., Rho, J. M., Kawamura, M. Jr., et al. (2013). Ketogenic diet improves core symptoms of autism in BTBR mice. PLoS ONE 8:e65021. doi: 10.1371/journal.pone.0065021
Sakurai, T., Ramoz, N., Barreto, M., Gazdoiu, M., Takahashi, N., Gertner, M., et al. (2010). Slc25a12 disruption alters myelination and neurofilaments: a model for a hypomyelination syndrome and childhood neurodevelopmental disorders. Biol. Psychiatry 67, 887–894. doi: 10.1016/j.biopsych.2009.08.042
Sandin, S., Lichtenstein, P., Kuja-Halkola, R., Larsson, H., Hultman, C. M., and Reichenberg, A. (2014). The familial risk of autism. JAMA 311, 1770–1777. doi: 10.1001/jama.2014.4144
Santini, E., Turner, K. L., Ramaraj, A. B., Murphy, M. P., Klann, E., and Kaphzan, H. (2015). Mitochondrial superoxide contributes to hippocampal synaptic dysfunction and memory deficits in Angelman syndrome model mice. J. Neurosci. 35, 16213–16220. doi: 10.1523/JNEUROSCI.2246-15.2015
Segurado, R., Conroy, J., Meally, E., Fitzgerald, M., Gill, M., and Gallagher, L. (2005). Confirmation of association between autism and the mitochondrial aspartate/glutamate carrier SLC25A12 gene on chromosome 2q31. Am. J. Psychiatry 162, 2182–2184. doi: 10.1176/appi.ajp.162.11.2182
Silverman, J. M., Buxbaum, J. D., Ramoz, N., Schmeidler, J., Reichenberg, A., Hollander, E., et al. (2008). Autism-related routines and rituals associated with a mitochondrial aspartate/glutamate carrier SLC25A12 polymorphism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147, 408–410. doi: 10.1002/ajmg.b.30614
Smith, J. D., Rho, J. M., Masino, S. A., and Mychasiuk, R. (2014). Inchworming: a novel motor stereotypy in the BTBR T+ Itpr3tf/J mouse model of autism. J. Visual. Exp. 50791. doi: 10.3791/50791
Spilioti, M., Evangeliou, A. E., Tramma, D., Theodoridou, Z., Metaxas, S., Michailidi, E., et al. (2013). Evidence for treatable inborn errors of metabolism in a cohort of 187 Greek patients with autism spectrum disorder (ASD). Front. Hum. Neurosci. 7:858. doi: 10.3389/fnhum.2013.00858
Stafstrom, C. E., and Rho, J. M. (2012). The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front. Pharmacol. 3:59. doi: 10.3389/fphar.2012.00059
Stromland, K., Nordin, V., Miller, M., Akerstrom, B., and Gillberg, C. (1994). Autism in thalidomide embryopathy: a population study. Dev. Med. Child Neurol. 36, 351–356. doi: 10.1111/j.1469-8749.1994.tb11856.x
Su, H., Fan, W., Coskun, P. E., Vesa, J., Gold, J.-A., Jiang, Y.-H., et al. (2011). Mitochondrial dysfunction in CA1 hippocampal neurons of the UBE3A deficient mouse model for Angelman syndrome. Neurosci. Lett. 487, 129–133. doi: 10.1016/j.neulet.2009.06.079
Subramanian, M., Timmerman, C. K., Schwartz, J. L., Pham, D. L., and Meffert, M. K. (2015). Characterizing autism spectrum disorders by key biochemical pathways. Front. Neurosci. 9:313. doi: 10.3389/fnins.2015.00313
Suen, D. F., Norris, K. L., and Youle, R. J. (2008). Mitochondrial dynamics and apoptosis. Genes Dev. 22, 1577–1590. doi: 10.1101/gad.1658508
Tang, G., Gutierrez Rios, P., Kuo, S.-H., Akman, H. O., Rosoklija, G., Tanji, K., et al. (2013). Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol. Dis. 54, 349–361. doi: 10.1016/j.nbd.2013.01.006
Tigerholm, J., Borjesson, S. I., Lundberg, L., Elinder, F., and Fransen, E. (2012). Dampening of hyperexcitability in CA1 pyramidal neurons by polyunsaturated fatty acids acting on voltage-gated ion channels. PLoS ONE 7:e44388. doi: 10.1371/journal.pone.0044388
Tordjman, S., Somogyi, E., Coulon, N., Kermarrec, S., Cohen, D., Bronsard, G., et al. (2014). Gene x Environment interactions in autism spectrum disorders: role of epigenetic mechanisms. Front. Psychiatry 5:53. doi: 10.3389/fpsyt.2014.00053
Turunen, J. A., Rehnstrom, K., Kilpinen, H., Kuokkanen, M., Kempas, E., and Ylisaukko-Oja, T. (2008). Mitochondrial aspartate/glutamate carrier SLC25A12 gene is associated with autism. Autism Res. 1, 189–192. doi: 10.1002/aur.25
Veenstra-Vanderweele, J., Christian, S. L., and Cook, E. H. Jr. (2004). Autism as a paradigmatic complex genetic disorder. Annu. Rev. Genomics Hum. Genet. 5, 379–405. doi: 10.1146/annurev.genom.5.061903.180050
Voskuyl, R. A., and Vreugdenhil, M. (2001). “Effects of essential fatty acids on voltage-regulated ionic channels and seizure thresholds in animals,” in Fatty Acids: Physiological and Behavioral Functions, eds D. Mostofsky, S. Yehuda Jr., and N. Salem (Totowa, NJ: Humana Press), 63–78.
Weinberg, S. E., Sena, L. A., and Chandel, N. S. (2015). Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417. doi: 10.1016/j.immuni.2015.02.002
Wen, Y., Alshikho, M. J., and Herbert, M. R. (2016). Pathway network analyses for autism reveal multisystem involvement, major overlaps with other diseases and convergence upon MAPK and calcium signaling. PLoS ONE 11:e0153329. doi: 10.1371/journal.pone.0153329
Wong, S., Napoli, E., Krakowiak, P., Tassone, F., Hertz-Picciotto, I., and Giulivi, C. (2016). Role of p53, mitochondrial DNA deletions, and paternal age in autism: a case-control study. Pediatrics 137:e20151888. doi: 10.1542/peds.2015-1888
Xavier, J. M., Rodrigues, C. M. P., and Solá, S. (2016). Mitochondria: major regulators of neural development. Neuroscientist 22, 346–358. doi: 10.1177/1073858415585472
Xie, Z., Jones, A., Deeney, J. T., Hur, S. K., and Bankaitis, V. A. (2016). Inborn Errors of Long-Chain fatty acid β-oxidation link neural stem cell self-renewal to autism. Cell Rep. 14, 991–999. doi: 10.1016/j.celrep.2016.01.004
Xu, X. P., Sun, R. P., and Jin, R. F. (2006). Effect of ketogenic diet on hippocampus mossy fiber sprouting and GluR5 expression in kainic acid induced rat model. Chinese Med. J. 119, 1925–1929.
Yudkoff, M., Daikhin, Y., Melo, T. M., Nissim, I., Sonnewald, U., and Nissim, I. (2007). The ketogenic diet and brain metabolism of amino acids: relationship to the anticonvulsant effect. Annu. Rev. Nutr. 27, 415–430. doi: 10.1146/annurev.nutr.27.061406.093722
Keywords: autism spectrum disorder, ketogenic diet, metabolism, mitochondria, therapeutics, epilepsy, co-morbidity, mechanism
Citation: Cheng N, Rho JM and Masino SA (2017) Metabolic Dysfunction Underlying Autism Spectrum Disorder and Potential Treatment Approaches. Front. Mol. Neurosci. 10:34. doi: 10.3389/fnmol.2017.00034
Received: 20 September 2016; Accepted: 30 January 2017;
Published: 21 February 2017.
Edited by:
Kimberly Raab-Graham, Wake Forest School of Medicine, USAReviewed by:
Michael J. Schmeisser, Otto-von-Guericke University Magdeburg, GermanyCopyright © 2017 Cheng, Rho and Masino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ning Cheng, bmNoZW5nQHVjYWxnYXJ5LmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.