 Carlo Rinaldi
Carlo Rinaldi Imre Mäger
Imre Mäger Matthew J. Wood
Matthew J. Wood
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Mol. Neurosci. , 27 December 2016
Sec. Molecular Signalling and Pathways
Volume 9 - 2016 | https://doi.org/10.3389/fnmol.2016.00164
This article is part of the Research Topic The Role of the Protein Quality Control in Neurodegenerative Diseases View all 34 articles
The accumulation in neurons of aberrant protein species, the pathological hallmark of many neurodegenerative diseases, results from a global impairment of key cellular processes governing protein synthesis/degradation and repair mechanisms, also known as the proteostasis network (PN). The growing number of connections between dysfunction of this intricate network of pathways and diseases of the motor unit, where both motor neurons and muscle are primarily affected, has provided momentum to investigate the muscle- and motor neuron-specific response to physiological and pathological stressors and to explore the therapeutic opportunities that manipulation of this process may offer. Furthermore, these diseases offer an unparalleled opportunity to deepen our understanding of the molecular mechanisms behind the intertissue communication and transfer of signals of proteostasis. The most compelling aspect of these investigations is their immediate potential for therapeutic impact: targeting muscle to stem degeneration of the motor unit would represent a dramatic paradigm therapeutic shift for treating these devastating diseases. Here we will review the current state of the art of the research on the alterations of the PN in diseases of the motor unit and its potential to result in effective treatments for these devastating neuromuscular disorders.
The maintenance of proteome integrity is paramount for cellular and organismal health. Mammalian cells typically express about 20,000 different proteins, which, as they are newly synthesized, must adopt defined three-dimensional configurations to become functional (Balchin et al., 2016). These folded structures are unstable, and proteins, particularly those partially or entirely lacking ordered structure, can misfold under a number of stress conditions or unique metabolic challenges, such as those occurring during cancer or aging, and accumulate in toxic aggregates, the pathological hallmark of many human conditions, including Alzheimer’s and Parkinson’s disease (Balchin et al., 2016). The state of proteome functional balance is commonly referred as proteostasis and the cellular pathways involved in maintaining the integrity of the proteome are collectively referred to as the proteostasis network (PN; Balch et al., 2008). This network coordinates protein synthesis, folding, localization, and turnover and, given its fundamental biological role, requires a tight control within individual cells, tissues, and organs. The main players are chaperones and two proteolytic systems, the ubiquitin-proteasome and the lysosome-autophagy systems, overall comprising almost 1400 components in mammalian cells, which determine whether misfolded proteins will refold into their original stable conformation or whether they will instead be eliminated (Powers et al., 2009) (Figure 1). Chaperones are involved in all aspects of proteostasis: they promote the folding of newly synthesized proteins, their translocation across membranes, and the refolding of denatured substrates. Chaperones also prevent aggregation of metastable proteins by targeting misfolded proteins for degradation (Balchin et al., 2016) (Figure 1). In eukaryotic cells, two distinct chaperone networks exist: the chaperones linked to protein synthesis (CLIPS; Albanèse et al., 2010), which are linked to the translation machinery and assist folding of newly translated proteins, and the heat shock proteins (HSPs; Treweek et al., 2015), which can be induced by the transcription factor heat shock protein 1 (HSF1) and protect the proteome from stress. Fatally misfolded proteins are polyubiquitinated by an enzymatic E1/E2/E3 ubiquitination cascade and targeted for degradation by the proteasome, the major eukaryotic proteolytic pathway (Goldberg, 2003) (Figure 1). Misfolded proteins escaping proteolysis by the ubiquitine-proteasome system (UPS) can reach the lysosome through a membrane transporter in chaperone-mediated autophagy (CMA; Cuervo et al., 2004) or organize into oligomers and/or aggregates, which can be eliminated also by lysosomes through macroautophagy (MA; Jackson and Hewitt, 2016) (Figure 1). Numerous studies have highlighted the importance of lysosomal degradation of misfolded proteins in neurodegeneration: knock-down of autophagy genes, such as Atg5 and Atg7, promotes toxicity in mouse models (Hung et al., 2009; Pyo et al., 2013). Conversely, genetic or pharmacologic activation of autophagy has a protective role in a number of neurodegenerative diseases (Nah et al., 2015).
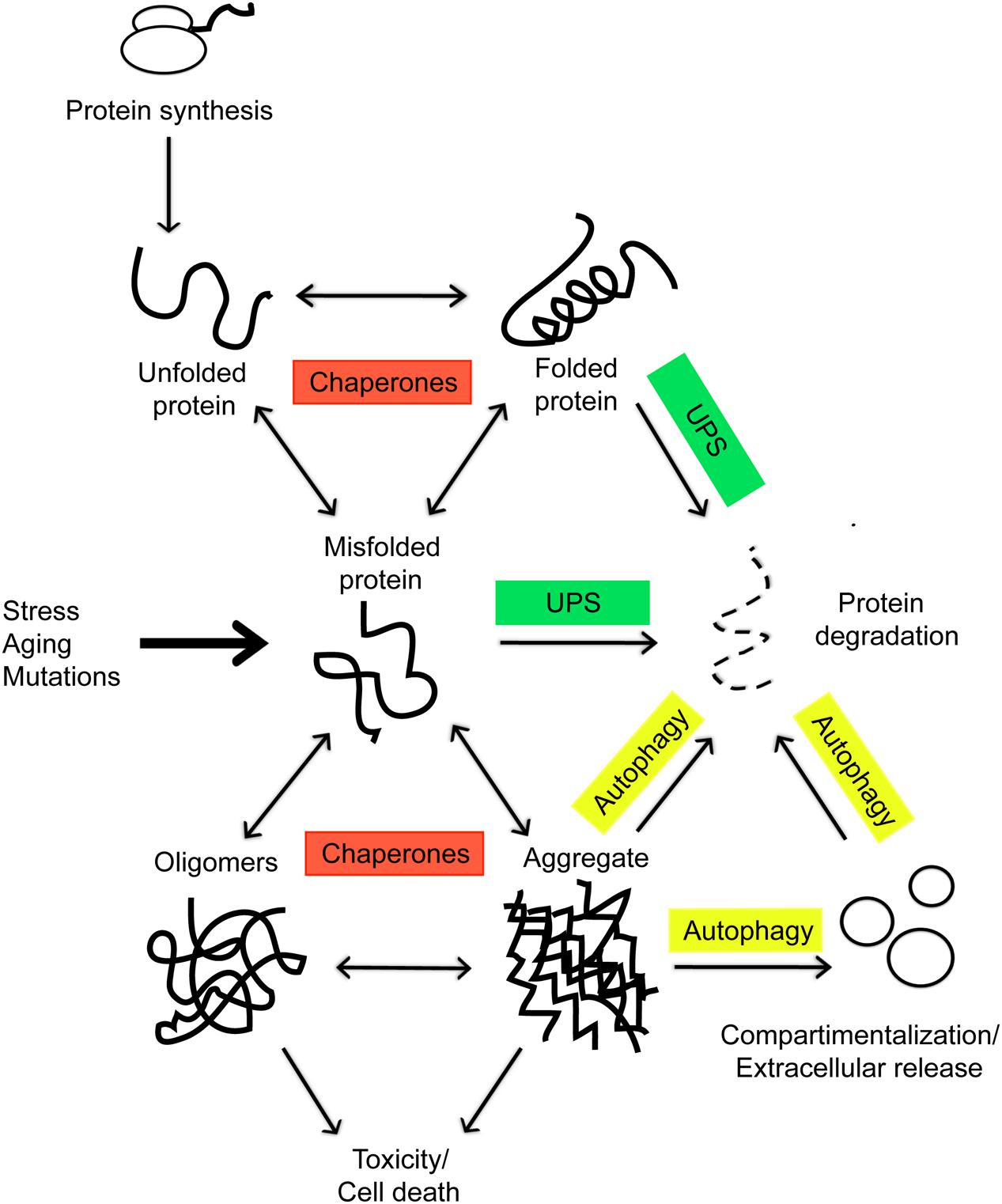
FIGURE 1. In eukaryotic cells, as nascent polypeptide chains emerge from ribosomes, they are met by the chaperones and assisted in the folding, which is necessary for a functional protein. Environmental stress, aging, and mutations can tilt the balance toward formation of misfolded proteins which tend to form toxic aggregates. These incorrectly folded species are detected by a quality-control mechanism and targeted for degradation by the cellular proteolytic systems or secreted via exosomes.
Another mechanism of clearance which is lately gaining increasing interest connected with autophagy is via exosomes, 30–140 nm membrane-bound particles defined by their origin from the endosomal pathway, which are emerging as pivotal players in cellular signaling in both normal physiology and disease states (Baixauli et al., 2014). Recent studies have shown that they can transfer insoluble aggregates to neighboring cells, resulting in a decreased toxic load on the proteome: whether the spreading of protein aggregates via exosomes could provide the molecular basis for the prion-like mechanism of disease propagation in neurodegeneration is certainly very intriguing (Yuyama et al., 2012; Schneider and Simons, 2013; Iguchi et al., 2016) and has the potential to be exploited for therapeutic and biomarker discovery (Boukouris and Mathivanan, 2015; Yuyama et al., 2015).
The PN is highly redundant and all the components are strongly interconnected: impairment of the UPS induces compensatory autophagy, whereas knockdown of autophagy components leads to the accumulation of proteasomal substrates (Korolchuk et al., 2010; Park and Cuervo, 2013).
Additionally, both proteasomal and lysosomal clearance pathways are tightly coupled to the chaperone systems via BAG-domain proteins and specific ubiquitin ligases, such as the cochaperone CHIP (Kabbage and Dickman, 2008). Interestingly, as organisms age, quality control and the cellular response to unfolded protein stress become compromised. Indeed, aging is considered the principal risk factor for the onset of a number of neurodegenerative disorders associated with aggregate deposition (Balchin et al., 2016). Researchers are challenged to develop an integrated view of the PN in health and disease in order to further understand the basic mechanisms of cell function and how loss of proteostasis leads to neurodegeneration.
A motor unit consists of one somatic efferent motor neuron and all of the muscle fibers that it innervates, a concept first introduced by Edward Liddell [1895–1981] and Charles Sherrington [1857–1952] (Duchateau and Enoka, 2011). Because an action potential generated by a motor neuron normally brings to threshold all the muscle fibers it has contact with, a single motor unit neuron constitutes the smallest power unit that can be activated to generate movement.
Many diseases of the motor unit are caused by destabilizing mutations that cause misfolding of the host protein, or alterations in components of the protein homeostasis and quality control machineries, overall suggesting a limited capacity to manage proteome alterations (Table 1). Motor neurons are uniquely sensitive to alterations in the PN due to their extreme polarization and post-mitotic nature: they require tight temporal and spatial regulation of protein translation and turnover and cannot rely on the dilution of toxic waste occurring during cell division (Tanaka and Matsuda, 2014). On the other side, systems of protein quality control and in particular fine-tuning the balance between protein synthesis and protein degradation are crucial for the maintenance of muscle mass. Excessive or defective activity of the autophagy-lysosome or ubiquitin-proteasome in acquired or inherited muscle disorders lead to detrimental effects on muscle homeostasis and function (Sandri, 2013). Like in other neurodegenerative diseases, the pathological hallmark of many diseases of the motor unit is the presence of ubiquitin-positive inclusions consisting of misfolded protein aggregates in the affected cells (Hasegawa and Arai, 2007). Several studies have shown that these aggregated structures or, more likely, their oligomeric precursors, directly exert neurotoxic effects by inducing proteotoxic stress and disturbing homeostasis (Ross and Poirier, 2005). Also large aggregates are believed to contribute to the toxicity by sequestering functional proteins and interfering with the UPS and the autophagic degradative pathway (Olzscha et al., 2011). Similarly to alterations in proteostasis in motor neurons, in sporadic and familial inclusion-body myositis (s-IBM), a highly prevalent adult-onset degenerative muscle disease, several proteins, including amyloid-β42 and its oligomers, and phosphorylated tau abnormally accumulate in aggregates, suggesting the presence of similar responses across tissues to proteotoxic insults (Mastaglia and Needham, 2015).
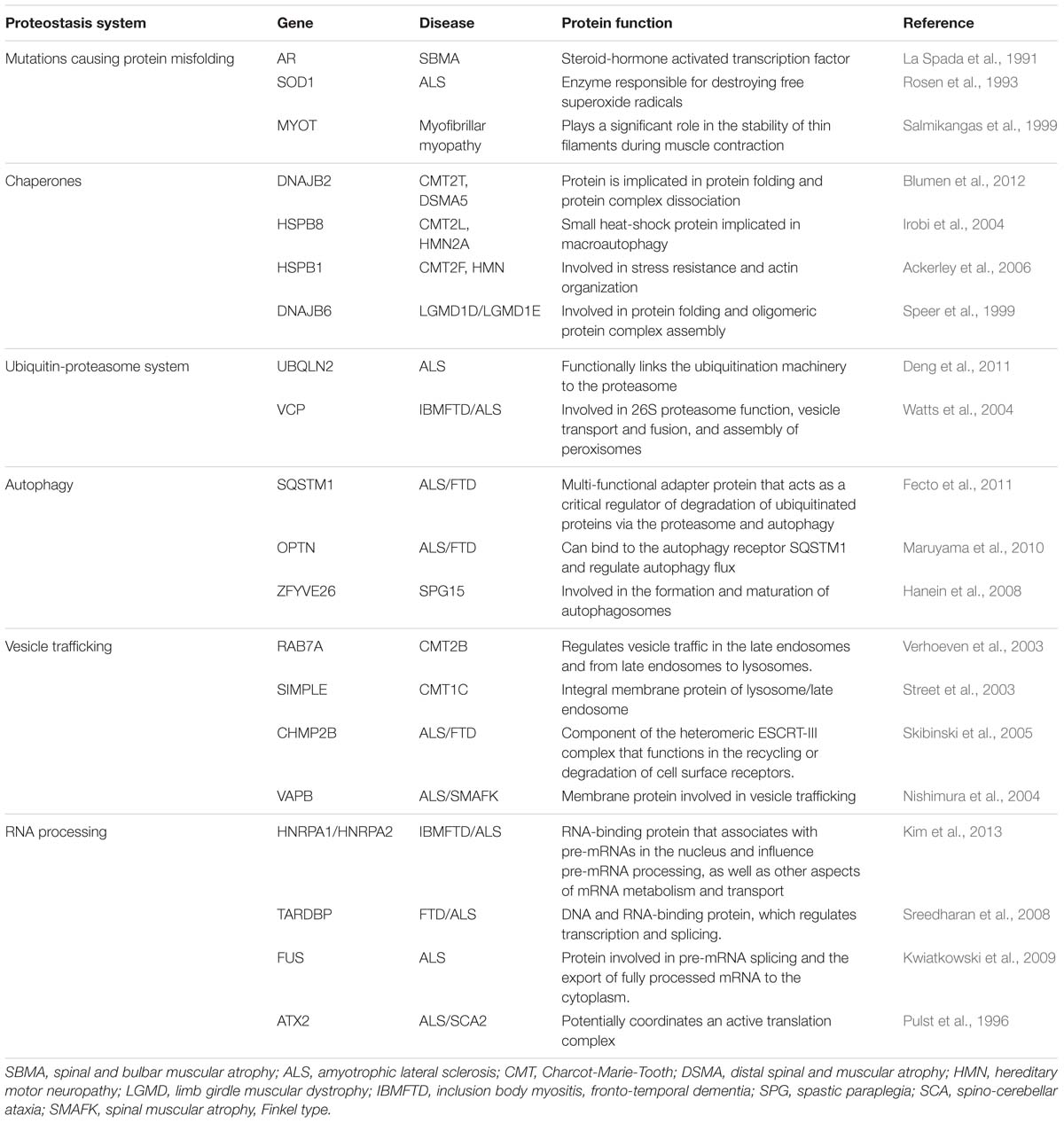
TABLE 1. Diseases of the motor unit associated with mutations in components of the proteostasis network.
Over the past decade, several observations in a group of neurodegenerative diseases have also highlighted important links between protein aggregation and RNA biology (Belzil et al., 2013). A conspicuous number of RNA binding proteins (RBPs) have been shown to be associated with amyotrophic lateral sclerosis (ALS) or other neuromuscular diseases: for example, mutations in RBPs such as hnRNPA1 and A2B1, TDP-43, and FUS are associated with ALS (Table 1) (Kabashi et al., 2008; Kim et al., 2013; Qiu et al., 2014). The current prevalent working model suggests that mutations in RBPs enhance dramatically their intrinsic tendency to form stable amyloid structures (Kim et al., 2013), which results in accumulation of ribonucleoproten (RNP) aggregates, disruption of RNA homeostasis or ‘ribostasis,’ and eventually cell death (Ramaswami et al., 2013). Because amyloid initiation is a stochastic event dependent on protein levels, any alteration that increases the local concentration of RBPs in stress granules, such as defects in clearance mechanisms due to VCP mutations, increases its frequency. This may explains why redistribution of the RBP TDP-43 from the nucleus to cytoplasmic inclusions is a hallmark pathological feature of most sporadic and familial forms of ALS and frontotemporal lobar degeneration (FTLD) (Winton et al., 2008). Recent evidence have also highlighted that components of the protein quality control system, such as Hsp70 proteins, survey RNP compositions by influencing mRNA biology (Gilks et al., 2004), suggesting that ribostasis is strongly coupled to the proteostatic state of the cell (Walters and Parker, 2015).
The lack of available treatments in the clinic combined with the increasing burden of neurodegenerative disorders in the aging population urgently require the development of effective therapeutic approaches to arrest, stabilize, or reverse the progression of these diseases. As a result of the intrinsic property of the PN to achieve and maintain proteome balance, a single PN modulator has the potential to restore proteostasis in multiple diseases of protein conformations, while both decreasing the levels of the toxic protein and alleviating its pathogenic effects (Balchin et al., 2016). This approach, alone or in synergistic combination with standard single-target therapies (i.e., antisense oligonucleotides), may be more suitable for the complex mechanisms of disease pathogenesis, therefore increasing the chances of success in clinical trials. The use of high throughput screening method to discover molecules able to restore the proteostatic imbalance is rapidly gaining momentum; therapeutic strategies that target the PN are now emerging as promising avenues for the treatment of diseases of protein conformation, based on recent evidence showing that manipulation of the PN in in vitro and in vivo models are able to suppress the toxicity associated with accumulation of misfolded proteins (Rivas et al., 2015). The ubiquitin proteasome system (UPS) and autophagy have a key role in proteostasis by clearance of proteins and removal of damaged and dysfunctional cellular components. These pathways are essential for normal physiology and development, and are also involved in numerous diseases including cancer and neurodegeneration. Not unexpectedly, these two proteostatic machineries are highly desirable therapeutic targets to improve diseases caused by a mechanism of proteotoxicity (Vidal René et al., 2014; Chondrogianni et al., 2015). Recently, a small-molecule inhibitor of USP14, a deubiquitinating enzyme that inhibits proteasomal degradation by removing the polyubiquitin chain from the client proteins, was shown to enhance degradation of aggregation-prone proteins, suggesting that manipulation of the ubiquitin-proteasome system could be an attractive target to promote the clearance of toxic protein (Lee et al., 2010). The curcumin analog ASC-JM17 ameliorates the disease phenotype in animal models of spinal and bulbar muscular atrophy (SBMA) by targeting the mutant protein, androgen receptor, for degradation via the UPS and simultaneously increasing oxidative stress resistance via activation of the Nrf1 and Nrf2 pathway (Bott et al., 2016). Targeting autophagy may also have therapeutic benefits, as compounds that increase protein turnover by induction of the autophagic activity mitigate neurodegeneration and enhance protein clearance in neuronal models of TDP-43 aggregation (Wang et al., 2012).
Another approach is represented by small molecules that modulate the activity of chaperone proteins, such as Hsp90 and Hsp70, which have essential roles in preventing aberrant protein–protein interactions and promoting correct protein folding (West et al., 2012). The appearance of aggregated proteins, as occurs in diseases of misfolded protein, indicates that the quality control pathways of the PN become compromised, leading to cellular dysfunction and death. Pharmacological agents that increase HSP levels by inducing the heat shock response, such as 17-AAG or arimoclomol have been shown to counteract the toxicity caused by accumulation of toxic proteins in models of SBMA, ALS, and IBM (Kieran et al., 2004; Waza et al., 2005; Ahmed et al., 2016). Interestingly, arimoclomol does not induce the heat shock response in unstressed cells, thereby avoiding the potential side effects associated with widespread heat shock response activation (Kieran et al., 2004; Ahmed et al., 2016). Despite the beneficial effects in preclinical models, several factors need to be considered in order to translate this approach into valuable therapeutic options: the high degree of integration makes manipulation of the PN a challenging task, where the risk of compromising the stability of other proteins will need to be addressed (Balch et al., 2008; Balchin et al., 2016). Also, for conformational diseases involving the motor unit, the proteostasis modulators will need to achieve targeted drug delivery in motor neurons and muscle. Researchers are challenged to develop a better understanding of the complex interplay between molecules, cells, and tissues in order to answer these questions and before moving these strategies into the clinic.
While the central components of protein quality control function autonomously within each cell, it has been recently demonstrated that the PN is also regulated by cell-non-autonomous signaling through communication between subcellular compartments and across different cells and tissues (van Oosten-Hawle et al., 2013). In the evolution from unicellularity to multicellularity, organisms have developed mechanisms of intercellular cooperation in order to protect tissues with intrinsic reduced buffering capacity, such as neurons, and promote a rapid, predictive, and adaptive response to proteotoxic insults (van Oosten-Hawle and Morimoto, 2014).
Most of the discoveries in non-cell-autonomous proteostasis have been made in invertebrates, although evidence of a similar process is starting to emerge also in mammals (Zeineddine and Yerbury, 2015). In Caenorhabditis elegans, it has been shown that the heat shock response of somatic cells is regulated by thermosensory neurons that detect temperature changes to control HSF-1 activity throughout the organism (Prahlad et al., 2008). These signaling responses are bidirectional and non-neuronal tissues such as gonad, intestine, or muscle tissue communicate conditions of altered proteostasis with each other and back to the nervous system independently of neural control via autocrine/paracrine and endocrine mechanisms. For example, expression of metastable myosin restricted to muscle cells of Caenorhabditis elegans induces a compensatory stress response not only in muscle but also in neuronal and intestinal cells, which relies upon transcriptional feedback regulated by the FoxA transcription factor PHA-4 (van Oosten-Hawle et al., 2013). The motor unit functionally largely relies on anterograde and retrograde signaling at the neuromuscular junction (NMJ): the nature of these signals, particularly from muscle to motor neurons, and the specific molecular mechanisms regulating NMJ structure, function and maintenance are only beginning to be elucidated (Yumoto et al., 2012), mainly due to the technical challenges of studying this complex interaction at the subcellular level. Diseases of the motor unit, such as SBMA and spinal muscular atrophy (SMA), where a mechanism of selective neuronal vulnerability has been challenged by recent evidence showing that therapies exclusively targeting muscle ameliorate the motor neuron pathology and rescue the disease phenotype in animal models (Boyer et al., 2013), represent an ideal working model to unravel the mechanisms of such interaction. Furthermore, the most compelling aspect of these findings is their potential for therapeutic impact: the possibility of manipulating proteostasis in muscle for stemming degeneration of motor neurons, which are less accessible for therapeutic intervention, would represent a dramatic paradigm shift for the treatment of these yet incurable devastating diseases.
Understanding how the signals of proteostasis are transmitted from one cell to another, how they are integrated in a network and which are the functional implications of such intercellular and intertissue communication in aging and diseases is crucial. In order to boost the organismal proteostasis capacity, it has been recently shown that under conditions of stress, cells can transfer cellular material ranging from stress signals to whole organelles, such as lysosomes, mitochondria, endosomes and lipid droplets through tunneling nanotubes, thin membranous, freely hovering channels with a diameter of 50–200 nm (Astanina et al., 2015; Burtey et al., 2015; Wang et al., 2016). Another mechanism of distant intercellular communication that is currently gaining considerable interest is via extracellular vesicles (Tkach and Théry, 2016). Recent studies suggest that exosomes mediate the non-cell-autonomous control of a number of physiological responses, including organismal proteostasis, by releasing their cargo (RNAs and proteins) to recipient cells and regulating the expression of target genes (Lee et al., 2012). In addition, (i) proteotoxic insults induce the exosome-mediated secretion of HSPs (Takeuchi et al., 2015; Kowal et al., 2016); (ii) adding chaperone-containing exosomes to cultured cells expressing aggregation-prone proteins restores the protein-folding environment in recipient cells (Takeuchi et al., 2015); and (iii) suppression of polyglutamine-mediated neurodegeneration by remote tissue-specific expression of Hsp40 in Drosophila depends on Ykt6, an R-SNARE protein necessary for exosome secretion (Takeuchi et al., 2015). Here we show that exosome or microvesicle proteomes derived from various cell types including myoblasts, platelets, endothelial cells and mesenchymal stem cells contain on average of 11.5% proteins related to protein folding, the ubiquitin proteasome, or autophagy (Figure 2), suggesting possible PN-related functions of extracellular vesicles as a whole or in their certain subtype. Upon understanding of their biology, it may be possible to use exosomes as vehicle for delivery of therapeutic factors to target tissues (El Andaloussi et al., 2013).
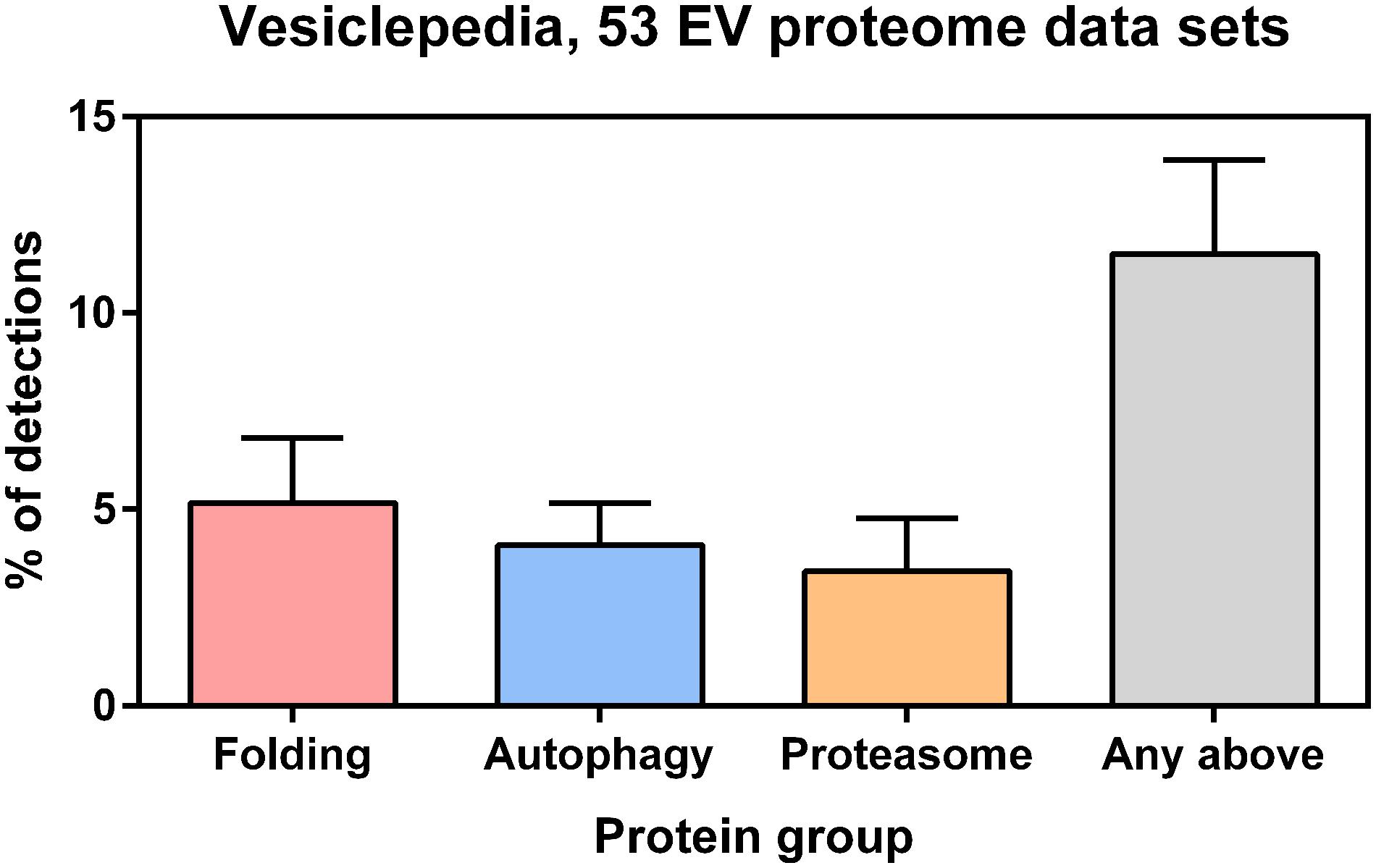
FIGURE 2. Protein annotations of specific Gene Ontology (GO) terms were downloaded from the QuickGO database (http://www.ebi.ac.uk/QuickGO/) and cross-referenced with selected EV proteomes from the Vesiclepedia database (http://microvesicles.org, PMID: 23271954). Only those experiments from Vesiclepedia that described exosome or microvesicle proteomes with >500 detected proteins from Homo sapiens or Mus musculus samples were included in the analysis. These selection criteria were matched by 53 studies in the Vesiclepedia database where EVs were derived from various cell types (e.g., cell lines, biofluids, primary tumor cells, lymphocytes, myoblasts, platelets, endothelial cells and mesenchymal stem cells). According to the Vesiclepedia database, on average 5.2% of EV proteins are related to protein folding (GO:0006457), 4.1% are related to autophagy (GO:0006914), and 3.4% are related to ubiquitin-dependent proteasomes (GO:0043161). Furthermore, on average 11.5% of EV proteins are related to proteins to any of those processes. Error bars represent standard deviation.
Given the close ties with proteostasis and diseases of the motor unit, insights into the mechanisms of protein quality control will ultimately impact research striving to decode the pathogenesis of these diseases. In recent years a better understanding of the intracellular systems that govern proteostasis has formed the rationale for numerous successful attempts to target cellular proteostasis to counteract the toxicity related to several neurodegenerative conditions in experimental models. Possible therapeutic approaches include drugs that inhibit toxic aggregate formation, activate protective mechanisms, or promote clearance of toxic aggregates, which might have both prophylactic and therapeutic benefits. Many unresolved issues still remain: understanding why cells occasionally fail to compartmentalize misfolded toxic species, allowing them to interfere with normal protein homeostasis, and how the information of the status of proteostasis is communicated between tissues will be instrumental in elucidating the etiology of these diseases. The possibility that exosomes may function in close relation with autophagy pathway to preserve protein and RNA homeostasis and to mediate the spreading of signals to surrounding cells in order to coordinate organismal systemic responses is intriguing and is a new exciting subject of investigation. Dissecting the complex interplay of proteostasis between different tissues using the motor unit as a working model offers the unparalleled opportunity to gain insight into the general mechanisms of protein quality control and has the potential to provide targets for treatment for these yet incurable conditions.
CR: Conception or design of the work, drafting the article, critical revision of the article. IM: Data collection, data analysis, and interpretation, drafting the article. MW: Critical revision of the article, final approval of the version to be published.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ackerley, S., Paul, A. J., Kalli, A., French, S., Davies, K. E., and Talbot, K. (2006). A mutation in the small heat-shock protein HSPB1 leading to distal hereditary motor neuronopathy disrupts neurofilament assembly and the axonal transport of specific cellular cargoes. Hum. Mol. Genet. 15, 347–354. doi: 10.1093/hmg/ddi452
Ahmed, M., Machado, P. M., Miller, A., Spicer, C., Herbelin, L., He J., et al. (2016). Targeting protein homeostasis in sporadic inclusion body myositis. Sci. Transl. Med. 8:331ra41. doi: 10.1126/scitranslmed.aad4583
Albanèse, V., Reissmann, S., and Frydman, J. (2010). A ribosome-anchored chaperone network that facilitates eukaryotic ribosome biogenesis. J. Cell Biol. 189, 69–81. doi: 10.1083/jcb.201001054
Astanina, K., Koch, M., Jüngst, C., Zumbusch, A., and Kiemer, A. K. (2015). Lipid droplets as a novel cargo of tunnelling nanotubes in endothelial cells. Sci. Rep. 5:11453. doi: 10.1038/srep11453
Baixauli, F., Lopez-Otin, C., and Mittelbrunn, M. (2014). Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 5:403. doi: 10.3389/fimmu.2014.00403
Balch, W. E., Morimoto, R. I., Dillin, A., and Kelly, J. W. (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. doi: 10.1126/science.1141448
Balchin, D., Hayer-Hartl, M., and Hartl, F. U. (2016). In vivo aspects of protein folding and quality control. Science 353:aac4354. doi: 10.1126/science.aac4354
Belzil, V. V., Gendron, T. F., and Petrucelli, L. (2013). RNA-mediated toxicity in neurodegenerative disease. Mol. Cell. Neurosci. 56, 406–419. doi: 10.1016/j.mcn.2012.12.006
Blumen, S. C., Astord, S., Robin, V., Vignaud, L., Toumi, N., Cieslik, A., et al. (2012). A rare recessive distal hereditary motor neuropathy with HSJ1 chaperone mutation. Ann. Neurol. 71, 509–519. doi: 10.1002/ana.22684
Bott, L. C., Badders, N. M., Chen, K., Harmison, G. G., Bautista, E., Shih, C. Y., et al. (2016). A small-molecule Nrf1 and Nrf2 activator mitigates polyglutamine toxicity in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 25, 1979–1989. doi: 10.1093/hmg/ddw073
Boukouris, S., and Mathivanan, S. (2015). Exosomes in Bodily Fluids Are a Highly Stable Resource of Disease Biomarkers. Proteomics Clin. Appl. 9, 358–367. doi: 10.1002/prca.201400114
Boyer, J. G., Ferrier, A., and Kothary, R. (2013). More than a bystander: the contributions of intrinsic skeletal muscle defects in motor neuron diseases. Front. Physiol. 4:356. doi: 10.3389/fphys.2013.00356
Burtey, A., Wagner, M., Hodneland, E., Skaftnesmo, K. O., Schoelermann, J., Mondragon, J. R., et al. (2015). Intercellular transfer of transferrin receptor by a contact-, Rab8-dependent mechanism involving tunneling nanotubes. FASEB J. 29, 4695–4712. doi: 10.1096/fj.14-268615
Chondrogianni, N., Voutetakis, K., Kapetanou, M., Delitsikou, V., Papaevgeniou, N., Sakellari, M., et al. (2015). Proteasome activation: an innovative promising approach for delaying aging and retarding age-related diseases. Ageing Res. Rev. 23(Pt A), 37–55. doi: 10.1016/j.arr.2014.12.003
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T., and Sulzer, D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. doi: 10.1126/science.1101738
Deng, H., Wenjie Chen, W., Hong, S. T., Boycott, K. M., Gorrie, G. H., Siddique, N., et al. (2011). Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211–215. doi: 10.1038/nature10353
Duchateau, J., and Enoka, R. M. (2011). Human motor unit recordings: origins and insight into the integrated motor system. Brain Res. 1409, 42–61. doi: 10.1016/j.brainres.2011.06.011
El Andaloussi, S., Mäger, I., Breakefield, X. O., and Wood, M. J. (2013). Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 12, 347–357. doi: 10.1038/nrd3978
Fecto, F., Yan, J., Vemula, S. P., Liu, E., Yang, Y., Chen, W., et al. (2011). “SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68, 1440–1446. doi: 10.1001/archneurol.2011.250
Gilks, N., Kedersha, N., Ayodele, M., Shen, L., Stoecklin, G., Dember, L. M., et al. (2004). “Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 15, 5383–5398. doi: 10.1091/mbc.E04-08-0715
Goldberg, A. L. (2003). Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899. doi: 10.1038/nature02263
Hanein, S., Martin, E., Boukhris, A., Byrne, P., Goizet, C., Hamri, A., et al. (2008). Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including kjellin syndrome. Am. J. Hum. Genet. 82, 992–1002. doi: 10.1016/j.ajhg.2008.03.004
Hasegawa, M., and Arai, T. (2007). [Component of ubiquitin-positive inclusions in ALS]. Brain Nerve 59, 1171–1177.
Hung, S. Y., Huang, W., Liou, H., and Fu, W. (2009). Autophagy protects neuron from abeta-induced cytotoxicity. Autophagy 5, 502–510. doi: 10.4161/auto.5.4.8096
Iguchi, Y., Eid, L., Parent, M., Soucy, G., Bareil, C., Riku, Y., et al. (2016). Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 139, 3187–3201. doi: 10.1093/brain/aww237
Irobi, J., Van Impe, K., Seeman, P., Jordanova, A., Dierick, I., Verpoorten, N., et al. (2004). Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat. Genet. 36, 597–601. doi: 10.1038/ng1328
Jackson, M. P., and Hewitt, E. W. (2016). Cellular proteostasis: degradation of misfolded proteins by lysosomes. Essays Biochem. 60, 173–180. doi: 10.1042/EBC20160005
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., McConkey, B. J., Vande Velde, C., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi: 10.1038/ng.132
Kabbage, M., and Dickman, M. B. (2008). The BAG proteins: a ubiquitous family of chaperone regulators. Cell Mol. Life. Sci. 65, 1390–1402. doi: 10.1007/s00018-008-7535-2
Kieran, D., Kalmar, B., Dick, J. R. T., Riddoch-Contreras, J., Burnstock, G., and Greensmith, L. (2004). Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in als mice. Nat. Med. 10, 402–405. doi: 10.1038/nm1021
Kim, H. J., Nam, C. K., Yong-Dong, W., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. doi: 10.1038/nature11922
Korolchuk, V. I., Menzies, F. M., and Rubinsztein, D. C. (2010). Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 584, 1393–1398. doi: 10.1016/j.febslet.2009.12.047
Kowal, J., Arras, G., Colombo, M., Jouve, M., Morath, J. P., Primdal-Bengtson, B., et al. (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. U.S.A. 113, E968–E977. doi: 10.1073/pnas.1521230113
Kwiatkowski, T. J., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
La Spada, A. R., Wilson, E. M., Lubahn, D. B., Harding, A. E., and Fischbeck, K. H. (1991). Androgen receptor gene mutations in x-linked spinal and bulbar muscular atrophy. Nature 352, 77–79. doi: 10.1038/352077a0
Lee, B. H., Jae Lee, M., Park, S., Oh, D. C., Elsasser, S., Chen, P. C., et al. (2010). Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467, 179–184. doi: 10.1038/nature09299
Lee, Y., El Andaloussi, S., and Wood, M. J. (2012). Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 21, R125–R134. doi: 10.1093/hmg/dds317
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.1038/nature08971
Mastaglia, F. L., and Needham, M. (2015). Inclusion body myositis: a review of clinical and genetic aspects, diagnostic criteria and therapeutic approaches. J. Clin. Neurosci. 22, 6–13. doi: 10.1016/j.jocn.2014.09.012
Nah, J., Yuan, J., and Jung, Y. K. (2015). Autophagy in neurodegenerative diseases: from mechanism to therapeutic approach. Mol. Cells 38, 381–389. doi: 10.14348/molcells.2015.0034
Nishimura, A. L., Mitne-Neto, M., Silva, H. C., Richieri-Costa, A., Middleton, S., Cascio, D., et al. (2004). “A Mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 822–831. doi: 10.1086/425287
Olzscha, H., Schermann, S. M., Woerner, A. C., Pinkert, S., Hecht, M. H., Tartaglia, G. G., et al. (2011). Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144, 67–78. doi: 10.1016/j.cell.2010.11.050
Park, C., and Cuervo, A. M. (2013). Selective autophagy: talking with the UPS. Cell Biochem. Biophys. 67, 3–13. doi: 10.1007/s12013-013-9623-7
Powers, E. T., Morimoto, R. I., Dillin, A., Kelly, J. W., and Balch, W. E. (2009). “Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 78, 959–991. doi: 10.1146/annurev.biochem.052308.114844
Prahlad, V., Cornelius, T., and Morimoto, R. I. (2008). Regulation of the cellular heat shock response in caenorhabditis elegans by thermosensory neurons. Science 320, 811–814. doi: 10.1126/science.1156093
Pulst, S. M., Nechiporuk, A., Nechiporuk, T., Gispert, S., Chen, X. N., Lopes-Cendes, I., et al. (1996). Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia Type 2. Nat. Genet. 14, 269–276. doi: 10.1038/ng1196-269
Pyo, J. O., Yoo, S. M., Ahn, H. H., Nah, J., Hong, S. H., Kam, T. I., et al. (2013). Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 4:2300. doi: 10.1038/ncomms3300
Qiu, H., Lee, S., Shang, Y., Wang, W., Au, K. F., Kamiya, S., et al. (2014). ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Invest. 124, 981–999. doi: 10.1172/JCI72723.DNA
Ramaswami, M., Taylor, J. P., and Parker, R. (2013). Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 154, 727–736. doi: 10.1016/j.cell.2013.07.038
Rivas, A., Vidal, R. L., and Hetz, C. (2015). “Targeting the unfolded protein response for disease intervention. Expert Opin. Ther. Targets 19, 1203–1218. doi: 10.1517/14728222.2015.1053869
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Ross, C. A., and Poirier, M. A. (2005). Opinion: What is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol. 6, 891–898. doi: 10.1038/nrm1742
Salmikangas, P., Mykkänen, O. M., Grönholm, M., Heiska, L., Kere, J., and Carpén, O. (1999). Myotilin, a novel sarcomeric protein with two ig-like domains, is encoded by a candidate gene for limb-girdle muscular dystrophy. Hum. Mol. Genet. 8, 1329–1336. doi: 10.1093/hmg/8.7.1329
Sandri, M. (2013). Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 45, 2121–2129. doi: 10.1016/j.biocel.2013.04.023
Schneider, A., and Simons, M. (2013). Exosomes: vesicular carriers for intercellular communication in neurodegenerative disorders. Cell Tissue Res. 352, 33–47. doi: 10.1007/s00441-012-1428-2
Skibinski, G., Parkinson, N. J., Brown, J. M., Chakrabarti, L., Lloyd, S. L., Hummerich, H., et al. (2005). Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37, 806–808. doi: 10.1038/ng1609
Speer, M. C., Vance, J. M., Grubber, J. M., Graham, F. L., Stajich, J. M., Viles, K. D., et al. (1999). Identification of a new autosomal dominant limb-girdle muscular dystrophy locus on chromosome 7. Am. J. Hum. Genet. 64, 556–562. doi: 10.1086/302252
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Street, V. A., Bennett, C. L., Goldy, J. D., Shirk, A. J., Kleopa, K. A., Tempel, B. L., et al. (2003). Mutation of a putative protein degradation gene LITAF/SIMPLE in charcot-marie-tooth disease 1C. Neurology 60, 22–26. doi: 10.1212/WNL.60.1.22
Takeuchi, T., Suzuki, M., Fujikake, N., Popiel, H. A., Kikuchi, H., Futaki, S., et al. (2015). Intercellular chaperone transmission via exosomes contributes to maintenance of protein homeostasis at the organismal level. Proc. Natl. Acad. Sci. U.S.A. 112, E2497–E2506. doi: 10.1073/pnas.1412651112
Tanaka, K., and Matsuda, N. (2014). “Proteostasis and neurodegeneration: the roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta 1843, 197–204. doi: 10.1016/j.bbamcr.2013.03.012
Tkach, M., and Théry, C. (2016). Communication by extracellular vesicles: where we are and where we need to go. Cell 164, 1226–1232. doi: 10.1016/j.cell.2016.01.043
Treweek, T. M., Meehan, S., Ecroyd, H., and Carver, J. A. (2015). Small heat-shock proteins: important players in regulating cellular proteostasis. Cell Mol. Life Sci. 72, 429–451. doi: 10.1007/s00018-014-1754-5
van Oosten-Hawle, P., and Morimoto, R. I. (2014). Organismal proteostasis: role of cell-nonautonomous regulation and transcellular chaperone signaling. Genes Dev. 28, 1533–1543. doi: 10.1101/gad.241125.114
van Oosten-Hawle, P., Porter, R. S., and Morimoto, R. I. (2013). Regulation of organismal proteostasis by transcellular chaperone signaling. Cell 153, 1366–1378. doi: 10.1016/j.cell.2013.05.015
Verhoeven, K., De Jonghe, P., Coen, K., Verpoorten, N., Auer-Grumbach, M., Kwon, J. M., et al. (2003). “Mutations in the small GTP-ase late endosomal protein RAB7 cause charcot-marie-tooth type 2B neuropathy. Am. J. Hum. Genet. 72, 722–727. doi: 10.1086/367847
Vidal René, L., Matus, S., Bargsted, L., and Hetz, C. (2014). Targeting autophagy in neurodegenerative diseases. Trends Pharmacol. Sci. 35, 583–591. doi: 10.1016/j.tips.2014.09.002
Walters, R. W., and Parker, R. (2015). Coupling of ribostasis and proteostasis: Hsp70 proteins in mRNA metabolism. Trends Biochem. Sci. 40, 552–559. doi: 10.1016/j.tibs.2015.08.004
Wang, I. F., Guo, B. S., Liu, Y. C., Wu, C. C., Yang, C. H., Tsai, K. J., et al. (2012). Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc. Natl. Acad. Sci. U.S.A. 109, 15024–15029. doi: 10.1073/pnas.1206362109
Wang, X., Yu, X., Xie, C., Tan, Z., Tian, Q., Zhu, D., et al. (2016). Rescue of brain function using tunneling nanotubes between neural stem cells and brain microvascular endothelial cells. Mol. Neurobiol. 53, 2480–2488. doi: 10.1007/s12035-015-9225-z
Watts, G. D. J., Wymer, J., Kovach, M., Mehta, S. G., Mumm, S., Darvish, D., et al. (2004). Inclusion body myopathy associated with paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381. doi: 10.1038/ng1332
Waza, M., Adachi, H., Katsuno, M., Minamiyama, M., Sang, C., Tanaka, F., et al. (2005). 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat. Med. 11, 1088–1095. doi: 10.1038/nm1298
West, J. D., Wang, Y., and Morano, K. A. (2012). Small molecule activators of the heat shock response: chemical properties, molecular targets, and therapeutic promise. Chem. Res. Toxicol. 25, 2036–2053. doi: 10.1021/tx300264x
Winton, M. J., Igaz, L. M., Wong, M. M., Kwong, L. K., Trojanowski, J. Q., and Lee, V. M. Y. (2008). Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 283, 13302–13309. doi: 10.1074/jbc.M800342200
Yumoto, N., Kim, N., and Burden, S. J. (2012). Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature 489, 438–442. doi: 10.1038/nature11348
Yuyama, K., Sun, H., Mitsutake, S., and Igarashi, Y. (2012). Sphingolipid-modulated exosome secretion promotes clearance of amyloid-β by microglia. J. Biol. Chem. 287, 10977–10989. doi: 10.1074/jbc.M111.324616
Yuyama, K., Sun, H., Usuki, S., Hanamatsu, H., Mioka, T., Kimura, N., et al. (2015). A potential function for neuronal exosomes: sequestering intracerebral amyloid-β peptide. FEBS Lett. 589, 84–88. doi: 10.1016/j.febslet.2014.11.027
Keywords: proteostasis, protein quality control, motor unit, intertissue communication, exosomes
Citation: Rinaldi C, Mäger I and Wood MJ (2016) Proteostasis and Diseases of the Motor Unit. Front. Mol. Neurosci. 9:164. doi: 10.3389/fnmol.2016.00164
Received: 06 November 2016; Accepted: 19 December 2016;
Published: 27 December 2016.
Edited by:
Oliver Stork, Otto von Guericke University Magdeburg, GermanyReviewed by:
Andrew P. Lieberman, University of Michigan, USACopyright © 2016 Rinaldi, Mäger and Wood. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlo Rinaldi, Y2FybG8ucmluYWxkaUBkcGFnLm94LmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.