 Ruey-Horng Shih
Ruey-Horng Shih Chen-Yu Wang
Chen-Yu Wang Chuen-Mao Yang
Chuen-Mao Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Neurosci. , 18 December 2015
Sec. Molecular Signalling and Pathways
Volume 8 - 2015 | https://doi.org/10.3389/fnmol.2015.00077
This article is part of the Research Topic NF-kappaB signalling pathway in Neurological diseases View all 7 articles
The NF-κB (nuclear factor κ-light-chain-enhancer of activated B cells) transcription factor family is a pleiotropic regulator of many cellular signaling pathways, providing a mechanism for the cells in response to a wide variety of stimuli linking to inflammation. The stimulated cells will be regulated by not only the canonical but also non-canonical NF-κB pathways. To initiate both of these pathways, IκB-degradation triggers NF-κB release and the nuclear translocated-heterodimer (or homodimer) can associate with the κB sites of promoter to regulate the gene transcriptions. NF-κB ubiquitously expresses in neurons and the constitutive NF-κB activation is associated with processing of neuronal information. NF-κB can regulate the transcription of genes such as chemokines, cytokines, proinflammatory enzymes, adhesion molecules, proinflammatory transcription factors, and other factors to modulate the neuronal survival. In neuronal insult, NF-κB constitutively active in neuron cell bodies can protect neurons against different injuries and regulate the neuronal inflammatory reactions. Besides neurons, NF-κB transcription factors are abundant in glial cells and cerebral blood vessels and the diverse functions of NF-κB also regulate the inflammatory reaction around the neuronal environment. NF-κB transcription factors are abundant in the brain and exhibit diverse functions. Several central nerve system (CNS) diseases are linked to NF-κB activated by inflammatory mediators. The RelA and c-Rel expression produce opposite effects on neuronal survival. Importantly, c-Rel expression in CNS plays a critical role in anti-apoptosis and reduces the age-related behaviors. Moreover, the different subunits of NF-κB dimer formation can modulate the neuroninflammation, neuronal protection, or neurotoxicity. The diverse functions of NF-κB depend on the subunits of the NF-κB dimer-formation which enable us to develop a therapeutic approach to neuroinflammation based on a new concept of inflammation as a strategic tool in neuronal cells. However, the detail role of NF-κB in neuroinflammation, remains to be clarified. In the present article, we provide an updated review of the current state of our knowledge about relationship between NF-κB and neuroinflammation.
NF-κB exerts effects on almost all cell types in the body, playing an important function in inflammation, immune responses, cell cycle, and cell survival (Sen and Baltimore, 1986; Li and Verma, 2002; Kaltschmidt et al., 2005; Mattson, 2005; Ledoux and Perkins, 2014). NF-κB has been recognized as a member of Rel family of transcription factors. In mammals, there are five different members to compose the NF-κB family: p65 (RelA), RelB, c-Rel, p50/p105 (NF-κB1), and p52/p100 (NF-κB2) which have the similar amino acid sequence, the RHD (Rel homology domain, over approximate 300 amino acids) of these proteins (Chen and Greene, 2004). The activated NF-κB subunits will assemble to form the homo-or hetero-dimerized transcription factor complexes displaying the DNA-binding ability and transactivation potentials. The most widely studied form of NF-κB is a heterodimer of the p50 and p65 subunits and is a potent activator of gene transcription (Schmitz and Baeuerle, 1991). NF-κB is activated by a wide variety of agents including viruses, bacterial toxins such as lipopolysaccharide (LPS), UV light, oxidative stresses such as free radicals and cigarette smoke, inflammatory stimuli, cytokines, carcinogens, tumor promoters, and various mitogens (Baeuerle and Henkel, 1994; Baldwin, 1996). NF-κB regulates the expression of almost 500 different genes, including enzymes [e.g., cyclooxygenase (COX)-2, 5-lipoxygenase (LOX), and inducible NO synthase (iNOS)], cytokines [such as interleukin (IL)-1, IL-6, IL-8, chemokines, and tumor necrosis factor (TNF)], adhesion molecules, cell cycle regulatory molecules, and angiogenic factors (Duh et al., 1989; Kaltschmidt et al., 1993; Ahn and Aggarwal, 2005; Gupta et al., 2010a,b). The activation of NF-κB, especially the constitutively activated NF-κB in chronic inflammatory patients, has been found the critical linkage with a wide variety of human diseases, including asthma, atherosclerosis, AIDS, Alzheimer’s disease (AD), Parkinson’s disease (PD), rheumatoid arthritis, cancer, diabetes, and osteoporosis which belong to autoimmune/inflammatory diseases (Vallabhapurapu and Karin, 2009; Gupta et al., 2010b). The opposite, several native or artificial agents such as Th2 cytokines (IL-4, IL-13, and IL-10), interferons, endocrine hormones (LH, HCG, MSH, and GH), phytochemicals, corticosteroids, and immunosuppressive drugs, are known to block the specific signaling transductions and suppress NF-κB activation (Ahn and Aggarwal, 2005). Therefore, regulation and dysregulation of NF-κB play a key role in diseases control.
Based on the previous studies, NF-κB is activated via two distinct kinase-dependent pathways, the classical/canonical NF-κB pathway and the alternative/non-canonical NF-κB pathway. The most extensively studied NF-κB activation pathway is the canonical pathway (Figure 1, modified from Noort et al., 2015), which can be mediated through activation of a variety of cell surface receptors, including IL-1 receptor, Toll-like receptors (TLRs), and TNF receptor, in response to pro-inflammatory mediators like IL-1, LPS, and TNF, as well as via triggering of the T-cell receptor or B-cell receptor. The inactive NF-κB resides in the cytoplasm and associates or links with the natural biological inhibitor IκB. The NF-κB function and nuclear translocation ability are sequestered in the cytoplasm and nuclear compartments, respectively (Verma et al., 1995; Baeuerle and Baltimore, 1996). The IκB family members include IκBα, IκBβ, p105/IκBγ (precursor of p50), p100 (precursor of p52), and IκBε (Li and Nabel, 1997; Whiteside et al., 1997). Each shares a series of ankyrin repeats which sequester NF-κB in the cytosol by masking its nuclear localization signal (NLS) and also prevents NF-κB from binding to DNA by masking its DNA binding domain. Treatment of cells with various stimuli activates IκB kinase complex, for example, leading to the phosphorylation of serines 32 and 36 of IκBα or serines 19 and 23 of IκBβ (DiDonato et al., 1997; Mercurio et al., 1997; Regnier et al., 1997; Zandi et al., 1997). These phosphorylation events target IκB for ubiquitin-dependent degradation through the 26S proteasome complex, resulting in the release and nuclear translocation of NF-κB (Finco and Baldwin, 1995; Thanos and Maniatis, 1995). Briefly, NF-κB is expressed ubiquitously in the cytoplasm of almost all cell types. The activated NF-κB will translocate from cytoplasm to nucleus and the NF-κB-dimer can bind to the κB site of promoter. In this classical pathway, inhibitor of κB kinase (IKK)β is required for NF-κB activation (Tak et al., 2001), whereas IKKα is redundant (Vallabhapurapu and Karin, 2009). However, the canonical NF-κB pathway is essential for both acute and chronic inflammatory responses. Moreover, this pathway is implicated in cell proliferation and survival, demonstrated by constitutively active NF-κB signaling in many tissues (Ben-Neriah and Karin, 2011).
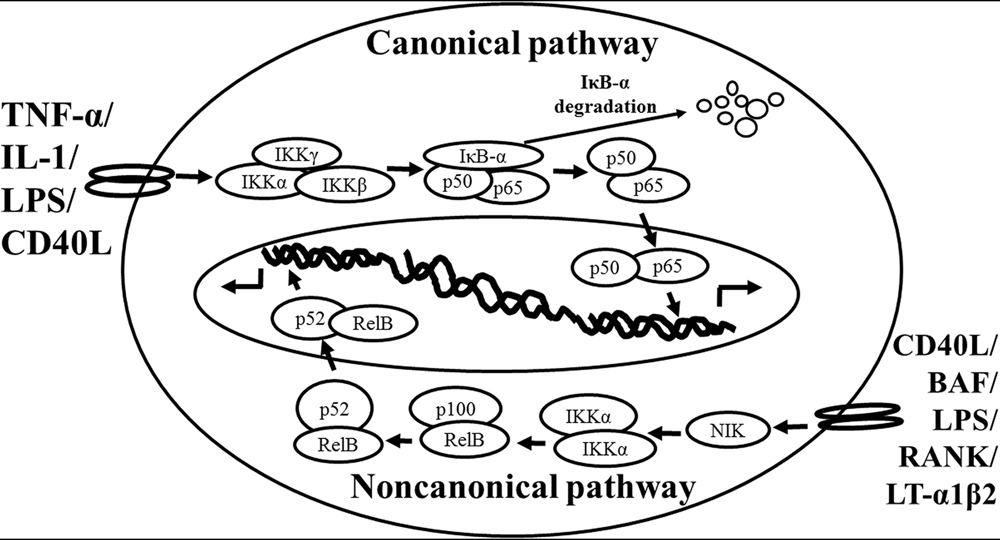
FIGURE 1. Schematic representation of the canonical and non-canonical nuclear factor (NF)-κB activation pathways. The canonical NF-κB pathway (upper) can be activated by a wide range of various stimuli, including tumor necrosis factor (TNF)-α, interlukin (IL)-1, lipopolysaccharide (LPS), and Toll-like receptors ligand (such as CD40L). Initiation of the canonical pathway via Toll-like receptor or cytokine receptor signaling depends on the inhibitor of κB kinase (IKK) complex, which is composed of the kinases IKKα and IKKβ, and the regulatory subunit IKKγ (NEMO). Activated IKK phosphorylates the inhibitory subunit IκBα leading to its degradation. The released NF-κB dimers (p50-p65) translocate to the nucleus and bind to κB site of chromosome to induce transcription of NF-κB targeted genes. The non-canonical pathway (lower) is activated by specific stimuli including B cell activating factor (BAF) belonging to the TNF family receptor, LPS, lymphotoxin (LT) α1β2, receptor activator of NF-κB (RANK), and CD40L. NF-κB inducing kinase (NIK) is stabilized. When stimulated, NIK is activated and recruits IKKα to the p100 complex to phosphorylate p100, leading to p100 ubiquitination. P52, the processing product of p100, generates the activated p52/RelB NF-κB complex, which is able to translocate to the nucleus and induce the downstream gene expressions.
The non-canonical NF-κB pathway (Figure 1), can be triggered by the activation of members of the TNF-receptor superfamily including B cell activating factor (BAF), belonging to the TNF family receptor, CD40, lymphotoxin β (LTβ) receptor, and receptor activator of NF-κB (RANK). Of note, these receptors trigger not only the non-canonical NF-κB pathway, but also the canonical pathway, simultaneously. The non-canonical NF-κB pathway is strictly dependent on IKKα homodimers and unlike the canonical pathway, the IKKβ or IKKγ is not involved in the IκB phosphorylation (Sun, 2012). To regulate the non-canonical pathway, expression of NF-κB inducing kinase (NIK) plays a role as the most important kinase. In the steady state, TNF receptor-associated factor (TRAF)3 mediates recruitment of NIK to TRAF2, which leads to NIK ubiquitination and continuous degradation. Consequently, endogenous levels of NIK are very low and the NF-κB complex is retained in the cytoplasm and kept inactive. Upon activation of the non-canonical NF-κB pathway, TRAF2 induces proteolysis of TRAF3. Degradation of TRAF3 prevents targeting of newly synthesized NIK, resulting in NIK release and accumulation. Subsequently, NIK induces p100 phosphorylation by IKKα homodimers and partial degradation to release p52. Next, mainly p52-RelB heterodimers translocate to the nucleus, leading to transcription of target genes. Whereas canonical NF-κB activation is rapid and independent of protein synthesis, non-canonical NF-κB activation requires NIK synthesis and accumulation. Consequently, the kinetics of this pathway are considerably slower (Vallabhapurapu and Karin, 2009; Sun, 2012).
There are cross-talks between these two pathways. IKKα has, for instance, been described to also have nuclear functions and serve as a regulator of canonical NF-κB-dependent gene expression through control of promoter-associated histone phosphorylation exposed to cytokines (Anest et al., 2003; Yamamoto et al., 2003). It has been demonstrated that the activated canonical pathway not only initiated the signal transduction of NF-κB but also suppressed basal non-canonical signaling in immune cells (Gray et al., 2014). Interestingly, under certain circumstances and other stimuli (including TNF) can also activate non-canonical NF-κB signaling in specific cell types (Zhang et al., 2014), and IKKα is critical for interferon-α production induced by TLR 7 and 9 (Hoshino et al., 2006).
The expression of NF-κB transcription factors is abundant in the brain. The basal levels of NF-κB expression have been identified in the brain where their amounts are higher than those of peripheral tissues. Several lines of evidence indicate that constitutively activated NF-κB is found in glutamatergic neurons of the central nervous system (CNS), such as cerebral cortex (layers 2, 4, and 5) and hippocampus (granule cells and pyramidal neurons of CA1 and CA3; Kaltschmidt et al., 1993, 1994, 1995). A number of studies also show constitutive NF-κB activity in various rodent brain regions such as amygdala, cerebral cortex, cerebellum, hippocampus, hypothalamus, and olfactory lobes (Schmidt-Ullrich et al., 1996). Among members of NF-κB, all of the complexes of c-Rel/p65, p50/p65 heterodimer, and p50 homodimers are detected in the developing rat brain (Bakalkin et al., 1993). While to analyze the distribution of NF-κB, the released p65 and p50 NF-κB subunits are abundantly expressed in neurons. Moreover, p50/p65 heterodimers are located in the cell nucleus and exhibit constitutive activity in the adult brain (Kaltschmidt et al., 2005; Meffert and Baltimore, 2005). In the developed rodent brain, the p50/p65 heterodimeric variant of NF-κB is converted to the major κB-binding complex (Schmidt-Ullrich et al., 1996; Meffert et al., 2003). It is important for the neuronal physiological characteristics, for example, constitutive NF-κB activity in glutamatergic neurons of the hippocampus and cerebral cortex can be suppressed by N-methyl-D-aspartate, and to a lesser extent AMPA, glutamate receptor antagonists, as well as L-type Ca2+ channel blockers (Lilienbaum and Israel, 2003; Meffert et al., 2003). These studies suggest that constitutive NF-κB activity is modulated by physiological basal synaptic transmission. However, inducible NF-κB is detected in synapses, glutamatergic stimulation activates retrograde transport of p65 protein from synapses to the cell nucleus (Kaltschmidt et al., 1993; Meberg et al., 1996; Meffert et al., 2003). Thus, NF-κB is involved in translation of short-lasting synaptic signals to persistent changes in gene expression (Wellmann et al., 2001; Meffert et al., 2003). The activated IKK ant it’s product, phosphorylated IκBα, were detected within the axon initial segment, the site where action potentials are generated (Schultz et al., 2006), suggesting that constitutive NF-κB activation is involved in the processing of neuronal information.
At the molecular level, inflammation is regulated by numerous molecules and factors, including adhesion molecules [intercellular adhesion molecule (ICAM-1), vascular cell adhesion molecule (VCAM)-1, endothelial-leukocyte adhesion molecule (ELAM)-1], chemokines (such as monocyte chemoattractant protein 1, IL-8), cytokines (IL-1, IL-2, IL-6, IL-12, TNF-α, TNF-β), signal transducer and activator of transcription (STAT)-3, proinflammatory enzymes [COX-2, 5-LOX, 12-LOX, matrix metalloproteinases (MMPs), prostate-specific antigen (PSA), C-reactive protein], vascular endothelial growth factor (VEGF), and proinflammatory transcription factors NF-κB (Aggarwal, 2004). Among these mediators, NF-κB is the central regulator of inflammation (Lukiw and Bazan, 1998; Aggarwal, 2004; Ahn and Aggarwal, 2005). For example: IL-1β treatment can induce COX-2 expression in canine tracheal smooth muscle cells (Yang et al., 2002) and ICAM-1 expression in human rheumatoid arthritis synovial fibroblasts (Yang et al., 2010), respectively. LPS, to mimic the bacterial infection, and endothelin-1 also can induce COX-2 and PGE2 expression in mouse brain microvascular endothelial (bEnd.3) cells (Shih and Yang, 2010; Lin et al., 2013). TNF-α can induce ICAM-1 expression in osteoblast-like MC3T3-E1 cells (Tsai et al., 2014). All of these target proteins syntheses are mediated through NF-κB-dependent signaling pathway.
NF-κB has been shown to activate more than 500 genes, which are implicated in inflammation related responses (Gupta et al., 2010a,b). The NF-κB family is suggested to be the most extensively studied target in inflammation issue for its critical role (Chen and Greene, 2004; Lin and Karin, 2007). In neuroinflammation, NF-κB can be transiently activated by various stimuli, like acute alcohol exposure, which induces neuroinflammatory responses in mice (Yakovleva et al., 2011). The role of NF-κB is critical in the regulation of neuroinflammation-associated disease pathogenesis (Niranjan, 2013).
In the CNS, NF-κB transcription factors are key players in a number of physiological processes such as neurogenesis (Koo et al., 2010), neuritogenesis (Rolls et al., 2007), and synaptic plasticity which related to learning and memory (Levenson et al., 2004; O’Riordan et al., 2006; Ahn et al., 2008). A number of studies also provide evidence that activation of NF-κB protects neurons against the different injuries such as excitotoxicity (Mattson, 2005), and oxidative stress (Sarnico et al., 2009b), as well as amyloid β peptide toxicity (Barger et al., 1995; Kaltschmidt et al., 1997) and exerts as a cellular defense program. Apoptotic cortical neurons have been observed to be rescued by overexpression of p65, while enhanced damage by IκB super-repressor or dominant negative NF-κB-inducing kinase (NIK; Bhakar et al., 2002). NF-κB is constitutively active, and involved in neuronal injury as well as neuroprotection in neuron cell bodies, however, NF-κB is present in a latent form at the synapse. Only when NF-κB is activated, it can be transported to the neuron cell nucleus (Yakovleva et al., 2011).
Besides neurons, the roles of NF-κB in astroglia/microglia have been studied in relation to brain injury (O’Neill and Kaltschmidt, 1997; Block et al., 2007; Kaltschmidt and Kaltschmidt, 2009). Briefly, NF-κB is present in a latent form in glia of naive animals (Schmidt-Ullrich et al., 1996; Bhakar et al., 2002). NF-κB may be activated under pathological conditions such as exposure to HIV-1 Tat or amyloid β peptide (Aβ) leading to the production of nitric oxide (Akama et al., 1998; El-Hage et al., 2008). It has been shown that glia responses to injury triggered by endogenous ligands for TLR and TLR signaling are mediated through the NF-κB (Akira and Takeda, 2004). Moreover, inhibition of astroglial NF-κB signaling leads to reduced chemokine expression and leukocyte entry into the injured CNS (Brambilla et al., 2005; Khorooshi et al., 2008). NF-κB has been shown to play the regulatory role of astrocytes on immune and inflammatory responses (Farina et al., 2007). Microgliosis is a common pathologically neurodegenerative disorder. Microglial activation of NF-κB plays a central role associated with the release reactive oxygen species and proinflammatory cytokines (such as IL-1β, interferon-γ, and TNF-α) that can cause secondary neurotoxicity (Kaltschmidt et al., 1993; Block et al., 2007). Briefly, in glia, NF-κB is inducible and regulates inflammatory processes that exacerbate inflammation-induced neurodegeneration (Yakovleva et al., 2011). NF-κB has been also demonstrated as a major signal transducer affecting cellular permeability, endocytosis, and intracellular trafficking at the level of the blood–brain barrier (Stone et al., 2011). Activation of NF-κB signaling by LPS has been shown to induce inflammatory target protein COX-2 and PGE2 production leading to cerebral vascular inflammation (Pan et al., 2010; Shih and Yang, 2010). All of above studies show that NF-κB transcription factors are abundant in the brain where they have diverse functions among neurons, glia, and cerebral blood vessels.
Constitutive activation of NF-κB is detected mostly in glutamatergic neurons. NF-κB in glia has a lower basal activity and is highly inducible, which plays a crucial role in brain inflammation (Kaltschmidt and Kaltschmidt, 2009). A role of glial NF-κB in pain research has attracted more attention. Pain signaling can arise from the activation of specific high-threshold PNS neurons (nociceptors) and could serve as a sensing mechanism to prevent further injury. In clinic, pain signaling can arise not only from damage to the nervous system (neuropathic pain), but also from chronic inflammation (inflammatory pain). Interestingly, an impairment of acute and inflammatory nociception has been revealed in p50-/- mice in a previous study (Niederberger et al., 2007). Moreover, inhibition of astroglial NF-κB can reduce inflammation and therefore improve functional recovery after spinal cord injury (Brambilla et al., 2005). All of these data suggests that NF-κB plays a crucial role on inflammatory pain in CNS.
In recent years, NF-κB dysregulation has been shown to link to neurodegenerative mechanisms that occur in brain during trauma or ischemia (Bethea et al., 1998; Schneider et al., 1999), as well as in the brain of patients suffered by PD (Hunot et al., 1997; Ghosh et al., 2007) or AD (Boissiere et al., 1997; Kaltschmidt et al., 1997; Lukiw and Bazan, 1998). These CNS diseases are associated with neuroinflammatory mediators. More evidence has shown that the neuronal response to external stimuli relies on a differential activation of NF-κB dimers. RelA or c-Rel expression produces opposite effects on neuron survival (Pizzi et al., 2002, 2005b; Sarnico et al., 2009b).
Among the members of NF-κB, the RelA subunit, composing the activated p50/RelA dimer, and its post-transcriptional modifications play a pivotal role in the onset of neurodegenerative processes triggered by ischemic insults (Inta et al., 2006; Sarnico et al., 2009a,b) as well as glutamate (Pizzi et al., 2002) or Aβ toxicity (Pizzi et al., 2005b; Inta et al., 2006; Lanzillotta et al., 2010). In ischemic stroke, activated RelA induces the expression of the 1B isoform of the divalent metal transporter-1(1B/DMT1) which can exert as an upstream response for iron accumulation and contributing to neuronal cell death after injury (Ingrassia et al., 2012). Notably, RelA is demonstrated as a most contributing subunit in degenerative changes associated with senescence in a mice model (Tilstra et al., 2012).
RelA has been demonstrated to contributing to neuronal cell death, while the overexpression of c-Rel factor can limit the cell death. The c-Rel factor is reduced in neurons exposed to oxygen–glucose deprivation (OGD), interestingly, the overexpression of c-Rel prevents neuronal loss in cortical neurons exposed to OGD. This protective effect involves in increasing the transcription of Bcl-xL gene (Pizzi et al., 2009; Sarnico et al., 2009a,b). Similarly, knocking down c-Rel expression exacerbated neuronal susceptibility to OGD-mediated damage (Pizzi et al., 2009). Further, knocking out c-Rel expression appeared insensitive to neuroprotective activity of leptin, a c-Rel inducer capable to limit cortical damage in wild-type mice and mice brain ischemia (Valerio et al., 2006, 2009). Therefore, the c-Rel subunit within activated NF-κB dimers also counteracts the ischemic injury acting as an innate mechanism of neuroprotection (Sarnico et al., 2009a,b). In addition, overexpression of c-Rel in cultured neurons promotes anti-apoptotic effects by inducing the transcription of manganese superoxide dismutase (MnSOD; Chen et al., 2000; Bernard et al., 2001; Pizzi et al., 2005a). On the viewpoint of disease events, the deficiency of c-Rel induces an age-related behavioral Parkinsonism in mice, with degeneration of nigral dopaminergic (DA) neurons and development of a PD-like neuropathology (Baiguera et al., 2012). Recent evidence has shown that activation of NF-κB drives the systemic and brain aging processes in mice (Adler et al., 2007; Zhang et al., 2013). In brain ischemic tissue of mice subjected to permanent middle cerebral artery occlusion (MCAO) and in primary cortical neurons exposed to OGD, NF-κB followed a pattern of increasing p50/RelA dimmer (Crack et al., 2006; Inta et al., 2006) and decreasing c-Rel-containing dimmers (Sarnico et al., 2009b). Inhibition of c-Rel-containing dimers and activation of p50/RelA are key events in the pathogenesis of brain injury. These data strongly suggested that NF-κB transcription factors have diverse functions that depend on the composition of the NF-κB complex (Lanzillotta et al., 2015).
The role of NF-κB is critical in the regulation of neuroinflammation-associated disease pathogenesis. NF-κB transcription factors are abundant and constitutive activation in brain where they have diverse functions among neurons, glia, and cerebral blood vessels. These functional diversions are dependent on the recruitment of components of the NF-κB dimer formation. Especially, c-Rel containing NF-κB dimers can induce the Bcl-xL and MnSOD expression and exert as anti-apoptotic effects while the pro-apoptotic effect elicited by NF-κB p50/RelA dimer. The imbalance of NF-κB dimer formation between RelA and c-Rel might result in the pathological process in certain neurons. The roles of NF-κB in neurological damage have been illustrated in Figure 2. The detail signal transduction pathways in different compositions of the NF-κB complex remain to be clarified. Obviously much more work is required to elucidate the role of NF-κB in the neuroinflammatory signaling pathways, which in turn will enable us to devise a therapeutic approach to neuroinflammation based on a new concept of inflammation as a strategic tool by which inflammatory neuronal cells can be made more susceptible to drugs than normal cells. By understanding the signal transduction pathways mediating the induction of NF-κB in neuronal cells, it may be possible to manipulate these diseases for therapeutic gain.
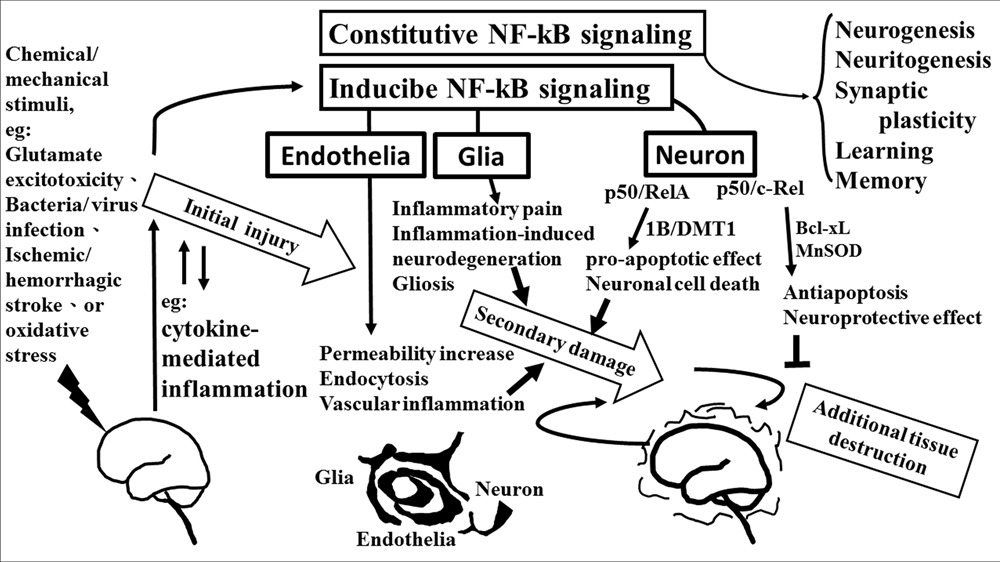
FIGURE 2. The role of NF-κB in neurological damage. Chemical/mechanical stimulation (such as glutamate excitotoxicity, bacteria/virus infection, ischemic/hemorrhagic stroke, or oxidative stress) to the brain/spinal cord tissue results in initial injury, including glutamate neuron-excitotoxicity, and cytokine-mediated inflammation which increase oxidative stress linking to neuroinflammatory response. NF-κB transcription factors are abundant in the brain where they have diverse functions between neuron, glia, and cerebral blood vessels. Constitutive NF-κB transduction factors are responsible for neurogenesis, neuritogenesis, synaptic plasticity, learning, and memory. Either glial or endothelial inducible NF-κB activation was implicated in neuroinflammation-associated pathogenesis related to secondary neuronal damage, while p50/RelA and p50/c-Rel subunit within activated NF-κB dimers play different roles on neuronal pathogenesis in neuron. The p50/RelA enhances damage by inducing the expression of the 1B isoform of the divalent metal transporter-1(1B/DMT1), p50/c-Rel protects against the damage by increasing the transcription of gene of Bcl-Xl or MnSOD. NF-κB transcription factors have diverse functions that depend on the composition of the NF-κB complex and cell types.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by MOST 104-2320-B-182-010 from Ministry of Science and Technology, Taiwan; EMRPD1E1641 from Ministry of Education, Taiwan; and CMRPD1C0103, CMRPD1B0383, CMRPD1C0563, CMRPD1B0332, and CMRPD1E0341 from Chang Gung Medical Research Foundation, Taiwan.
Adler, A. S., Sinha, S., Kawahara, T. L., Zhang, J. Y., Segal, E., and Chang, H. Y. (2007). Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 21, 3244–3257. doi: 10.1101/gad.1588507
Aggarwal, B. B. (2004). Nuclear factor-kappaB: the enemy within. Cancer Cell 6, 203–208. doi: 10.1016/j.ccr.2004.09.003
Ahn, H. J., Hernandez, C. M., Levenson, J. M., Lubin, F. D., Liou, H. C., and Sweatt, J. D. (2008). c-Rel, an NF-kappaB family transcription factor, is required for hippocampal long-term synaptic plasticity and memory formation. Learn. Mem. 15, 539–549. doi: 10.1101/lm.866408
Ahn, K. S., and Aggarwal, B. B. (2005). Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann. N. Y. Acad. Sci. 1056, 218–233. doi: 10.1196/annals.1352.026
Akama, K. T., Albanese, C., Pestell, R. G., and Van Eldik, L. J. (1998). Amyloid beta-peptide stimulates nitric oxide production in astrocytes through an NFkappaB-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 95, 5795–5800. doi: 10.1073/pnas.95.10.5795
Akira, S., and Takeda, K. (2004). Functions of toll-like receptors: lessons from KO mice. C. R. Biol. 327, 581–589. doi: 10.1016/j.crvi.2004.04.002
Anest, V., Hanson, J. L., Cogswell, P. C., Steinbrecher, K. A., Strahl, B. D., and Baldwin, A. S. (2003). A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature 423, 659–663. doi: 10.1038/nature01648
Baeuerle, P. A., and Baltimore, D. (1996). NF-kappa B: ten years after. Cell 87, 13–20. doi: 10.1016/S0092-8674(00)81318-5
Baeuerle, P. A., and Henkel, T. (1994). Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 12, 141–179. doi: 10.1146/annurev.iy.12.040194.001041
Baiguera, C., Alghisi, M., Pinna, A., Bellucci, A., De Luca, M. A., Frau, L., et al. (2012). Late-onset Parkinsonism in NFkappaB/c-Rel-deficient mice. Brain 135, 2750–2765. doi: 10.1093/brain/aws193
Bakalkin, G., Yakovleva, T., and Terenius, L. (1993). NF-kappa B-like factors in the murine brain. Developmentally-regulated and tissue-specific expression. Brain. Res. Mol. Brain Res. 20, 137–146. doi: 10.1016/0169-328X(93)90119-A
Baldwin, A. S. Jr. (1996). The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu. Rev. Immunol. 14, 649–683. doi: 10.1146/annurev.immunol.14.1.649
Barger, S. W., Horster, D., Furukawa, K., Goodman, Y., Krieglstein, J., and Mattson, M. P. (1995). Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc. Natl. Acad. Sci. U.S.A. 92, 9328–9332. doi: 10.1073/pnas.92.20.9328
Ben-Neriah, Y., and Karin, M. (2011). Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat. Immunol. 12, 715–723. doi: 10.1038/ni.2060
Bernard, D., Quatannens, B., Begue, A., Vandenbunder, B., and Abbadie, C. (2001). Antiproliferative and antiapoptotic effects of crel may occur within the same cells via the up-regulation of manganese superoxide dismutase. Cancer Res. 61, 2656–2664. doi: 10.1016/S0092-8674(00)81318-5
Bethea, J. R., Castro, M., Keane, R. W., Lee, T. T., Dietrich, W. D., and Yezierski, R. P. (1998). Traumatic spinal cord injury induces nuclear factor-kappaB activation. J. Neurosci. 18, 3251–3260. doi: 10.1016/S0092-8674(00)81318-5
Bhakar, A. L., Tannis, L. L., Zeindler, C., Russo, M. P., Jobin, C., Park, D. S., et al. (2002). Constitutive nuclear factor-kappa B activity is required for central neuron survival. J. Neurosci. 22, 8466–8475. doi: 10.1146/annurev.iy.12.040194.001041
Block, M. L., Zecca, L., and Hong, J. S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. doi: 10.1038/nrn2038
Boissiere, F., Hunot, S., Faucheux, B., Duyckaerts, C., Hauw, J. J., Agid, Y., et al. (1997). Nuclear translocation of NF-kappaB in cholinergic neurons of patients with Alzheimer’s disease. Neuroreport 8, 2849–2852. doi: 10.1097/00001756-199709080-00009
Brambilla, R., Bracchi-Ricard, V., Hu, W. H., Frydel, B., Bramwell, A., Karmally, S., et al. (2005). Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med. 202, 145–156. doi: 10.1084/jem.20041918
Chen, C., Edelstein, L. C., and Gelinas, C. (2000). The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol. Cell. Biol. 20, 2687–2695. doi: 10.1128/MCB.20.8.2687-2695.2000
Chen, L. F., and Greene, W. C. (2004). Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 5, 392–401. doi: 10.1038/nrm1368
Crack, P. J., Taylor, J. M., Ali, U., Mansell, A., and Hertzog, P. J. (2006). Potential contribution of NF-kappaB in neuronal cell death in the glutathione peroxidase-1 knockout mouse in response to ischemia-reperfusion injury. Stroke 37, 1533–1538. doi: 10.1161/01.STR.0000221708.17159.64
DiDonato, J. A., Hayakawa, M., Rothwarf, D. M., Zandi, E., and Karin, M. (1997). A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 388, 548–554. doi: 10.1038/41493
Duh, E. J., Maury, W. J., Folks, T. M., Fauci, A. S., and Rabson, A. B. (1989). Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc. Natl. Acad. Sci. U.S.A. 86, 5974–5978. doi: 10.1073/pnas.86.15.5974
El-Hage, N., Bruce-Keller, A. J., Yakovleva, T., Bazov, I., Bakalkin, G., Knapp, P. E., et al. (2008). Morphine exacerbates HIV-1 Tat-induced cytokine production in astrocytes through convergent effects on [Ca(2+)](i), NF-kappaB trafficking and transcription. PLoS ONE 3, e4093. doi: 10.1371/journal.pone.0004093
Farina, C., Aloisi, F., and Meinl, E. (2007). Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28, 138–145. doi: 10.1016/j.it.2007.01.005
Finco, T. S., and Baldwin, A. S. (1995). Mechanistic aspects of NF-kappa B regulation: the emerging role of phosphorylation and proteolysis. Immunity 3, 263–272. doi: 10.1016/1074-7613(95)90112-4
Ghosh, A., Roy, A., Liu, X., Kordower, J. H., Mufson, E. J., Hartley, D. M., et al. (2007). Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 104, 18754–18759. doi: 10.1073/pnas.0704908104
Gray, C. M., Remouchamps, C., Mccorkell, K. A., Solt, L. A., Dejardin, E., Orange, J. S., et al. (2014). Noncanonical NF-kappaB signaling is limited by classical NF-kappaB activity. Sci. Signal. 7, ra13. doi: 10.1126/scisignal.2004557
Gupta, S. C., Kim, J. H., Prasad, S., and Aggarwal, B. B. (2010a). Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev 29, 405–434. doi: 10.1007/s10555-010-9235-2
Gupta, S. C., Sundaram, C., Reuter, S., and Aggarwal, B. B. (2010b). Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 1799, 775–787. doi: 10.1016/j.bbagrm.2010.05.004
Hoshino, K., Sugiyama, T., Matsumoto, M., Tanaka, T., Saito, M., Hemmi, H., et al. (2006). IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature 440, 949–953. doi: 10.1038/nature04641
Hunot, S., Brugg, B., Ricard, D., Michel, P. P., Muriel, M. P., Ruberg, M., et al. (1997). Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 94, 7531–7536. doi: 10.1073/pnas.94.14.7531
Ingrassia, R., Lanzillotta, A., Sarnico, I., Benarese, M., Blasi, F., Borgese, L., et al. (2012). 1B/(-)IRE DMT1 expression during brain ischemia contributes to cell death mediated by NF-kappaB/RelA acetylation at Lys310. PLoS ONE 7, e38019. doi: 10.1371/journal.pone.0038019
Inta, I., Paxian, S., Maegele, I., Zhang, W., Pizzi, M., Spano, P., et al. (2006). Bim and Noxa are candidates to mediate the deleterious effect of the NF-kappa B subunit RelA in cerebral ischemia. J. Neurosci. 26, 12896–12903. doi: 10.1523/JNEUROSCI.3670-06.2006
Kaltschmidt, B., and Kaltschmidt, C. (2009). NF-kappaB in the nervous system. Cold Spring Harb. Perspect. Biol. 1, a001271. doi: 10.1101/cshperspect.a001271
Kaltschmidt, B., Uherek, M., Volk, B., Baeuerle, P. A., and Kaltschmidt, C. (1997). Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 94, 2642–2647. doi: 10.1073/pnas.94.6.2642
Kaltschmidt, B., Widera, D., and Kaltschmidt, C. (2005). Signaling via NF-kappaB in the nervous system. Biochim. Biophys. Acta 1745, 287–299. doi: 10.1016/j.bbamcr.2005.05.009
Kaltschmidt, C., Kaltschmidt, B., and Baeuerle, P. A. (1993). Brain synapses contain inducible forms of the transcription factor NF-kappa B. Mech. Dev. 43, 135–147. doi: 10.1016/0925-4773(93)90031-R
Kaltschmidt, C., Kaltschmidt, B., and Baeuerle, P. A. (1995). Stimulation of ionotropic glutamate receptors activates transcription factor NF-kappa B in primary neurons. Proc. Natl. Acad. Sci. U.S.A. 92, 9618–9622. doi: 10.1073/pnas.92.21.9618
Kaltschmidt, C., Kaltschmidt, B., Neumann, H., Wekerle, H., and Baeuerle, P. A. (1994). Constitutive NF-kappa B activity in neurons. Mol. Cell. Biol. 14, 3981–3992. doi: 10.1128/MCB.14.6.3981
Khorooshi, R., Babcock, A. A., and Owens, T. (2008). NF-kappaB-driven STAT2 and CCL2 expression in astrocytes in response to brain injury. J. Immunol. 181, 7284–7291. doi: 10.4049/jimmunol.181.10.7284
Koo, J. W., Russo, S. J., Ferguson, D., Nestler, E. J., and Duman, R. S. (2010). Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc. Natl. Acad. Sci. U.S.A. 107, 2669–2674. doi: 10.1073/pnas.0910658107
Lanzillotta, A., Porrini, V., Bellucci, A., Benarese, M., Branca, C., Parrella, E., et al. (2015). NF-kappaB in Innate Neuroprotection and Age-Related Neurodegenerative Diseases. Front Neurol 6, 98. doi: 10.3389/fneur.2015.00098
Lanzillotta, A., Sarnico, I., Ingrassia, R., Boroni, F., Branca, C., Benarese, M., et al. (2010). The acetylation of RelA in Lys310 dictates the NF-kappaB-dependent response in post-ischemic injury. Cell Death Dis 1, e96. doi: 10.1038/cddis.2010.76
Ledoux, A. C., and Perkins, N. D. (2014). NF-kappaB and the cell cycle. Biochem. Soc. Trans. 42, 76–81. doi: 10.1042/BST20130156
Levenson, J. M., Choi, S., Lee, S. Y., Cao, Y. A., Ahn, H. J., Worley, K. C., et al. (2004). A bioinformatics analysis of memory consolidation reveals involvement of the transcription factor c-rel. J. Neurosci. 24, 3933–3943. doi: 10.1523/JNEUROSCI.5646-03.2004
Li, Q., and Verma, I. M. (2002). NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2, 725–734. doi: 10.1038/nri910
Li, Z., and Nabel, G. J. (1997). A new member of the I kappaB protein family, I kappaB epsilon, inhibits RelA (p65)-mediated NF-kappaB transcription. Mol. Cell. Biol. 17, 6184–6190. doi: 10.1128/MCB.17.10.6184
Lilienbaum, A., and Israel, A. (2003). From calcium to NF-kappa B signaling pathways in neurons. Mol. Cell. Biol. 23, 2680–2698. doi: 10.1128/MCB.23.8.2680-2698.2003
Lin, C. C., Hsieh, H. L., Shih, R. H., Chi, P. L., Cheng, S. E., and Yang, C. M. (2013). Up-regulation of COX-2/PGE2 by endothelin-1 via MAPK-dependent NF-kappaB pathway in mouse brain microvascular endothelial cells. Cell Commun Signal 11, 8. doi: 10.1186/1478-811X-11-8
Lin, W. W., and Karin, M. (2007). A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Invest. 117, 1175–1183. doi: 10.1172/JCI31537
Lukiw, W. J., and Bazan, N. G. (1998). Strong nuclear factor-kappaB-DNA binding parallels cyclooxygenase-2 gene transcription in aging and in sporadic Alzheimer’s disease superior temporal lobe neocortex. J. Neurosci. Res. 53, 583–592. doi: 10.1002/(SICI)1097-4547(19980901)53:5<583::AID-JNR8>3.0.CO;2-5
Mattson, M. P. (2005). NF-kappaB in the survival and plasticity of neurons. Neurochem. Res. 30, 883–893. doi: 10.1007/s11064-005-6961-x
Meberg, P. J., Kinney, W. R., Valcourt, E. G., and Routtenberg, A. (1996). Gene expression of the transcription factor NF-kappa B in hippocampus: regulation by synaptic activity. Brain Res. Mol. Brain Res. 38, 179–190. doi: 10.1016/0169-328X(95)00229-L
Meffert, M. K., and Baltimore, D. (2005). Physiological functions for brain NF-kappaB. Trends Neurosci 28, 37–43. doi: 10.1016/j.tins.2004.11.002
Meffert, M. K., Chang, J. M., Wiltgen, B. J., Fanselow, M. S., and Baltimore, D. (2003). NF-kappa B functions in synaptic signaling and behavior. Nat. Neurosci. 6, 1072–1078. doi: 10.1038/nn1110
Mercurio, F., Zhu, H., Murray, B. W., Shevchenko, A., Bennett, B. L., Li, J., et al. (1997). IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278, 860–866. doi: 10.1126/science.278.5339.860
Niederberger, E., Schmidtko, A., Gao, W., Kuhlein, H., Ehnert, C., and Geisslinger, G. (2007). Impaired acute and inflammatory nociception in mice lacking the p50 subunit of NF-kappaB. Eur. J. Pharmacol 559, 55–60. doi: 10.1016/j.ejphar.2006.11.074
Niranjan, R. (2013). Molecular basis of etiological implications in Alzheimer’s disease: focus on neuroinflammation. Mol. Neurobiol. 48, 412–428. doi: 10.1007/s12035-013-8428-4
Noort, A. R., Tak, P. P., and Tas, S. W. (2015). Noncanonical NF-kappaB signaling in rheumatoid arthritis: Dr Jekyll and Mr Hyde? Arthritis Res. Ther. 17:15. doi: 10.1186/s13075-015-0527-3
O’Neill, L. A., and Kaltschmidt, C. (1997). NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends in neurosciences 20, 252–258. doi: 10.1016/S0166-2236(96)01035-1
O’Riordan, K. J., Huang, I. C., Pizzi, M., Spano, P., Boroni, F., Egli, R., et al. (2006). Regulation of nuclear factor kappaB in the hippocampus by group I metabotropic glutamate receptors. J. Neurosci. 26, 4870–4879. doi: 10.1523/JNEUROSCI.4527-05.2006
Pan, W., Yu, C., Hsuchou, H., and Kastin, A. J. (2010). The role of cerebral vascular NFkappaB in LPS-induced inflammation: differential regulation of efflux transporter and transporting cytokine receptors. Cell Physiol. Biochem 25, 623–630.
Pizzi, M., Boroni, F., Bianchetti, A., Moraitis, C., Sarnico, I., Benarese, M., et al. (2002). Expression of functional NR1/NR2B-type NMDA receptors in neuronally differentiated SK-N-SH human cell line. Eur. J. Neurosci. 16, 2342–2350. doi: 10.1046/j.1460-9568.2002.02403.x
Pizzi, M., Sarnico, I., Boroni, F., Benarese, M., Steimberg, N., Mazzoleni, G., et al. (2005a). NF-kappaB factor c-Rel mediates neuroprotection elicited by mGlu5 receptor agonists against amyloid beta-peptide toxicity. Cell Death. Differ 12, 761–772. doi: 10.1038/sj.cdd.4401598
Pizzi, M., Sarnico, I., Boroni, F., Benetti, A., Benarese, M., and Spano, P. F. (2005b). Inhibition of IkappaBalpha phosphorylation prevents glutamate-induced NF-kappaB activation and neuronal cell death. Acta Neurochir. Suppl. 93, 59–63. doi: 10.1007/3-211-27577-0_8
Pizzi, M., Sarnico, I., Lanzillotta, A., Battistin, L., and Spano, P. (2009). Post-ischemic brain damage: NF-kappaB dimer heterogeneity as a molecular determinant of neuron vulnerability. FEBS J. 276, 27–35. doi: 10.1111/j.1742-4658.2008.06767.x
Regnier, C. H., Song, H. Y., Gao, X., Goeddel, D. V., Cao, Z., and Rothe, M. (1997). Identification and characterization of an IkappaB kinase. Cell 90, 373–383. doi: 10.1016/S0092-8674(00)80344-X
Rolls, A., Shechter, R., London, A., Ziv, Y., Ronen, A., et al. (2007). Toll-like receptors modulate adult hippocampal neurogenesis. Nat. Cell Biol. 9, 1081–1088. doi: 10.1038/ncb1629
Sarnico, I., Lanzillotta, A., Benarese, M., Alghisi, M., Baiguera, C., Battistin, L., et al. (2009a). NF-kappaB dimers in the regulation of neuronal survival. Int. Rev. Neurobiol. 85, 351–362. doi: 10.1016/S0074-7742(09)85024-1
Sarnico, I., Lanzillotta, A., Boroni, F., Benarese, M., Alghisi, M., Schwaninger, M., et al. (2009b). NF-kappaB p50/RelA and c-Rel-containing dimers: opposite regulators of neuron vulnerability to ischaemia. J. Neurochem. 108, 475–485. doi: 10.1111/j.1471-4159.2008.05783.x
Schmidt-Ullrich, R., Memet, S., Lilienbaum, A., Feuillard, J., Raphael, M., and Israel, A. (1996). NF-kappaB activity in transgenic mice: developmental regulation and tissue specificity. Development 122, 2117–2128. doi: 10.1101/cshperspect.a001271
Schmitz, M. L., and Baeuerle, P. A. (1991). The p65 subunit is responsible for the strong transcription activating potential of NF-kappa B. EMBO J. 10, 3805–3817. doi: 10.1101/cshperspect.a001271
Schneider, A., Martin-Villalba, A., Weih, F., Vogel, J., Wirth, T., and Schwaninger, M. (1999). NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat. Med. 5, 554–559. doi: 10.1038/6458
Schultz, C., Konig, H. G., Del Turco, D., Politi, C., Eckert, G. P., Ghebremedhin, E., et al. (2006). Coincident enrichment of phosphorylated IkappaBalpha, activated IKK, and phosphorylated p65 in the axon initial segment of neurons. Mol. Cell. Neurosci. 33, 68–80. doi: 10.1016/j.mcn.2006.06.008
Sen, R., and Baltimore, D. (1986). Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell 47, 921–928. doi: 10.1016/0092-8674(86)90807-X
Shih, R. H., and Yang, C. M. (2010). Induction of heme oxygenase-1 attenuates lipopolysaccharide-induced cyclooxygenase-2 expression in mouse brain endothelial cells. J Neuroinflammation 7, 86. doi: 10.1186/1742-2094-7-86
Stone, K. P., Kastin, A. J., and Pan, W. (2011). NFkB is an unexpected major mediator of interleukin-15 signaling in cerebral endothelia. Cell Physiol. Biochem 28, 115–124. doi: 10.1159/000331720
Sun, S. C. (2012). The noncanonical NF-kappaB pathway. Immunol. Rev. 246, 125–140. doi: 10.1111/j.1600-065X.2011.01088.x
Tak, P. P., Gerlag, D. M., Aupperle, K. R., Van De Geest, D. A., Overbeek, M., Bennett, B. L., et al. (2001). Inhibitor of nuclear factor kappaB kinase beta is a key regulator of synovial inflammation. Arthritis Rheum. 44, 1897–1907. doi: 10.1002/1529-0131(200108)44:8<1897::AID-ART328>3.0.CO;2-4
Thanos, D., and Maniatis, T. (1995). NF-kappa B: a lesson in family values. Cell 80, 529–532. doi: 10.1016/0092-8674(95)90506-5
Tilstra, J. S., Robinson, A. R., Wang, J., Gregg, S. Q., Clauson, C. L., Reay, D. P., et al. (2012). NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Invest. 122, 2601–2612. doi: 10.1172/JCI45785
Tsai, C. L., Chen, W. C., Hsieh, H. L., Chi, P. L., Hsiao, L. D., and Yang, C. M. (2014). TNF-alpha induces matrix metalloproteinase-9-dependent soluble intercellular adhesion molecule-1 release via TRAF2-mediated MAPKs and NF-kappaB activation in osteoblast-like MC3T3-E1 cells. J. Biomed. Sci. 21, 12. doi: 10.1186/1423-0127-21-12
Valerio, A., Boroni, F., Benarese, M., Sarnico, I., Ghisi, V., Bresciani, L. G., et al. (2006). NF-kappaB pathway: a target for preventing beta-amyloid (Abeta)-induced neuronal damage and Abeta42 production. Eur. J. Neurosci. 23, 1711–1720. doi: 10.1111/j.1460-9568.2006.04722.x
Valerio, A., Dossena, M., Bertolotti, P., Boroni, F., Sarnico, I., Faraco, G., et al. (2009). Leptin is induced in the ischemic cerebral cortex and exerts neuroprotection through NF-kappaB/c-Rel-dependent transcription. Stroke 40, 610–617. doi: 10.1161/STROKEAHA.108.528588
Vallabhapurapu, S., and Karin, M. (2009). Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 27, 693–733. doi: 10.1146/annurev.immunol.021908.132641
Verma, I. M., Stevenson, J. K., Schwarz, E. M., Van Antwerp, D., and Miyamoto, S. (1995). Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev. 9, 2723–2735. doi: 10.1101/gad.9.22.2723
Wellmann, H., Kaltschmidt, B., and Kaltschmidt, C. (2001). Retrograde transport of transcription factor NF-kappa B in living neurons. J. Biol. Chem. 276, 11821–11829. doi: 10.1074/jbc.M009253200
Whiteside, S. T., Epinat, J. C., Rice, N. R., and Israel, A. (1997). I kappa B epsilon, a novel member of the I kappa B family, controls RelA and cRel NF-kappa B activity. EMBO J. 16, 1413–1426. doi: 10.1093/emboj/16.6.1413
Yakovleva, T., Bazov, I., Watanabe, H., Hauser, K. F., and Bakalkin, G. (2011). Transcriptional control of maladaptive and protective responses in alcoholics: a role of the NF-kappaB system. Brain Behav. Immun. 25(Suppl. 1), S29–S38. doi: 10.1016/j.bbi.2010.12.019
Yamamoto, Y., Verma, U. N., Prajapati, S., Kwak, Y. T., and Gaynor, R. B. (2003). Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature 423, 655–659. doi: 10.1038/nature01576
Yang, C. M., Chien, C. S., Hsiao, L. D., Luo, S. F., and Wang, C. C. (2002). Interleukin-1beta-induced cyclooxygenase-2 expression is mediated through activation of p42/44 and p38 MAPKS, and NF-kappaB pathways in canine tracheal smooth muscle cells. Cell. Signal. 14, 899–911. doi: 10.1016/S0898-6568(02)00037-2
Yang, C. M., Luo, S. F., Hsieh, H. L., Chi, P. L., Lin, C. C., Wu, C. C., et al. (2010). Interleukin-1beta induces ICAM-1 expression enhancing leukocyte adhesion in human rheumatoid arthritis synovial fibroblasts: involvement of ERK, JNK, AP-1, and NF-kappaB. J. Cell. Physiol. 224, 516–526. doi: 10.1002/jcp.22153
Zandi, E., Rothwarf, D. M., Delhase, M., Hayakawa, M., and Karin, M. (1997). The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 91, 243–252. doi: 10.1016/S0092-8674(00)80406-7
Zhang, G., Li, J., Purkayastha, S., Tang, Y., Zhang, H., Yin, Y., et al. (2013). Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature 497, 211–216. doi: 10.1038/nature12143
Keywords: NF-kappaB, neuroinflammation, neuroprotection, adhesion molecules, proinflammatory transcription factors
Citation: Shih R-H, Wang C-Y and Yang C-M (2015) NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 8:77. doi: 10.3389/fnmol.2015.00077
Received: 13 August 2015; Accepted: 30 November 2015;
Published: 18 December 2015.
Edited by:
Jun Yan, University of Queensland, AustraliaReviewed by:
Walter J. Lukiw, Louisiana State University Health Sciences Center, USACopyright © 2015 Shih, Wang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chuen-Mao Yang, Y2h1ZW5tYW9AbWFpbC5jZ3UuZWR1LnR3
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.