 Jada H. Vaden
Jada H. Vaden Jennifer A. Watson
Jennifer A. Watson Alan D. Howard
Alan D. Howard Ping-Chung Chen
Ping-Chung Chen Julie A. Wilson
Julie A. Wilson Scott M. Wilson
Scott M. Wilson
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mol. Neurosci. , 23 April 2015
Sec. Molecular Signalling and Pathways
Volume 8 - 2015 | https://doi.org/10.3389/fnmol.2015.00011
This article is part of the Research Topic Ubiquitin and the Brain: Roles of Proteolysis in the Normal and Abnormal Nervous System View all 19 articles
Ubiquitin-specific protease 14 (USP14) is a major deubiquitinating enzyme and a key determinant of neuromuscular junction (NMJ) structure and function. We have previously reported dramatic ubiquitin depletion in the nervous systems of the USP14-deficient ataxia (axJ) mice and demonstrated that transgenic ubiquitin overexpression partially rescues the axJ neuromuscular phenotype. However, later work has shown that ubiquitin overexpression does not correct the axJ deficits in hippocampal short term plasticity, and that transgenic expression of a catalytically inactive form of USP14 in the nervous system mimics the neuromuscular phenotype observed in the axJ mice, but causes a only a modest reduction of free ubiquitin. Instead, increased ubiquitin conjugates and aberrant activation of pJNK are observed in the nervous systems of the USP14 catalytic mutant mice. In this report, we demonstrate that restoring free ubiquitin levels in the USP14 catalytic mutant mice improved NMJ structure and reduced pJNK accumulation in motor neuron terminals, but had a negative impact on measures of NMJ function, such as motor performance and muscle development. Transgenic expression of ubiquitin had a dose-dependent effect on NMJ function in wild type mice: moderate levels of overexpression improved NMJ function while more robust ubiquitin overexpression reduced muscle development and motor coordination. Combined, these results suggest that maintenance of free ubiquitin levels by USP14 contributes to NMJ structure, but that USP14 regulates NMJ function through a separate pathway.
Ubiquitination can regulate a diverse array of cellular pathways depending on the number of ubiquitin monomers conjugated onto a protein and the internal lysine residue used to form the ubiquitin chain. These pathways regulate protein stability (Hershko and Ciechanover, 1998), activity (Schmukle and Walczak, 2012; Humphrey et al., 2013; Zhou et al., 2013), and localization (Tanno and Komada, 2013). The accumulation of ubiquitin-positive aggregates is a hallmark of neurodegenerative diseases (Perry et al., 1987; Lowe et al., 1988; Jara et al., 2013), suggesting that sequestration of ubiquitin, and the consequent reduction of ubiquitin availability, could contribute to neuronal dysfunction. This suggestion is supported by the hypothalamic dysfunction and neurodegeneration observed in mice lacking the ubiquitin gene Ubb (Ryu et al., 2008).
Although ubiquitin is encoded by four genes in the mammalian genome (Lund et al., 1985; Wiborg et al., 1985; Baker and Board, 1987, 1991; Finley et al., 1989; Redman and Rechsteiner, 1989), the equilibrium between ubiquitin conjugated onto proteins and free ubiquitin is largely controlled by the opposing actions of ubiquitin ligases and deubiquitinating enzymes (DUBs; Hallengren et al., 2013). We have previously shown that loss of the proteasome-associated DUB ubiquitin-specific protease 14 (USP14) in the ataxia (axJ) mice results in perinatal lethality, reduced muscle development, and structural and functional defects at the neuromuscular junction (NMJ). Loss of USP14 also severely alters ubiquitin homeostasis by causing a dramatic depletion of free ubiquitin in the brain and spinal cord (Anderson et al., 2005; Chen et al., 2009). Ubiquitin levels were most severely affected at the synapse, suggesting that the NMJ deficits in the axJ mice are due to ubiquitin depletion (Chen et al., 2009). In fact, neuronal-specific transgenic expression of ubiquitin corrects the reduced muscle development, altered NMJ structure, reduced synaptic transmission, and perinatal lethality observed in the axJ mice (Chen et al., 2011). Although these findings suggest a role for USP14’s in the maintenance of free ubiquitin pools in the nervous system, later work showed ubiquitin complementation does not rescue the deficits in hippocampal short-term plasticity observed in the axJ mice (Walters et al., 2014).
Furthermore, we recently reported that transgenic expression of a dominant-negative, catalytically inactive USP14 species in the murine nervous system recreates many of the essential phenotypes observed in the axJ mice, including deficits in NMJ structure, poor motor performance, and reduced muscle development (Vaden et al., 2015). In contrast to the axJ mice, however, the USP14 catalytic mutant mice have a normal lifespan and only a modest decrease in free ubiquitin. Instead, the spinal cords of the USP14 catalytic mutant mice show an increase in levels of ubiquitin conjugates linked through lysine 63 (K63), which are known to promote kinase activation (Yang et al., 2010; Humphrey et al., 2013). Consistent with this finding, the USP14 catalytic mutant mice have increased activation of the ubiquitin-dependent mixed lineage kinase 3 (MLK3) pathway, which signals through c-Jun N-terminal kinase (JNK; Vaden et al., 2015). As expected from the well-documented role of pJNK in rearranging synaptic architecture in invertebrates (Etter et al., 2005; Collins et al., 2006; Drerup and Nechiporuk, 2013), in vivo inhibition of JNK significantly improved NMJ structure and muscle development in the USP14 catalytic mutant mice (Vaden et al., 2015).
These findings led us to hypothesize that, in addition to maintaining ubiquitin homeostasis, USP14’s catalytic activity is required for the termination of ubiquitin signaling cascades and, consequently, that the increase in ubiquitin conjugates observed in the spinal cords of the USP14 catalytic mutant mice can alter neuronal function independent of the reduction of free ubiquitin. To directly test this hypothesis, we generated mice expressing both catalytically inactive USP14 (TgCA) and ubiquitin (TgUb) under the neuronal Thy1.2 promoter. The spinal cords of the resulting TgCA, TgUb mice had control levels of free ubiquitin and a nearly twofold increase in ubiquitin conjugates. The motor performance and muscle development of the double transgenic TgCA,TgUb mice were reduced compared to both TgCA mice and controls, underlining the need for USP14-dependent deubiquitination events in the nervous system. However, despite the detrimental effects of ubiquitin complementation on the overall phenotype of the TgCA mice, the structure of the NMJ was improved, and the amount of pJNK-positive pathology was decreased, in the TgCA,TgUb mice compared to TgCA mice. Additionally, we found that overexpression of ubiquitin had a bidirectional, dose-dependent effect on muscle mass, motor function, and measures of synaptic transmission even in wild type mice where USP14’s activity was intact. These data suggest that ubiquitin complementation in axJ mice indirectly corrects the functional deficits caused by loss of USP14 and demonstrate that increased protein ubiquitination alters motor function.
Wild type C57BL/6J, Thy1-YFP mice (16JRS, Jackson Laboratories, Bar Harbor, ME, USA), transgenic mice expressing USP14 (TgUsp14) catalytically inactive USP14 (TgCA; previously referred to as TgUsp14CA), ubiquitin (TgUb), or both catalytically inactive USP14 and ubiquitin (TgCA,TgUb) have been maintained in our breeding colony at the University of Alabama at Birmingham, which is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. We have previously described the generation of the TgUsp14 (Crimmins et al., 2006), and TgUb (Chen et al., 2011), and TgCA mice (Vaden et al., 2015), and all transgenes are expressed from the neuronal Thy1.2 promoter. Generation of TgUb mice resulted in two different founder lines with differing ubiquitin expression, TgUb-H (high expresser) and TgUb (low expresser). Double transgenic (TgCA,TgUb) mice were generated by breeding male TgCA mice to female TgUb mice. All mouse lines were maintained on a C57BL/6J background and transgenic mice were heterozygous for the transgene(s) of interest. Research was conducted without bias toward the sex of animals used for each study, and equal numbers of male and female mice were used. All research complied with the United States Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals, and adhered to principles stated in the Guide for the Care and Use of Laboratory Animals, United States National Research Council. In addition, all experiments were carried out with the approval of the University of Alabama at Birmingham’s Institutional Animal Care and Use Committee.
Mice were deeply anesthetized with isoflurane prior to rapid decapitation. Spinal cords were removed and homogenized in modified RIPA buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.5 mM EGTA, 1 mM EDTA, 0.5% SDS, 1% Triton X-100, and 1% sodium deoxycholate. Complete protease inhibitor (Roche, Indianapolis, IN, USA), phosphatase inhibitor cocktail III (Sigma Aldrich, St. Louis, MO, USA), and 50 μM PR-619 (inhibitor of DUBs, Life Sensors, Malvern, PA, USA) were added to the homogenization buffer per manufacturer instructions. Following homogenization, samples were sonicated and centrifuged at 17,000 × g for 10 min at 4°C. The supernatants were removed and stored immediately at -80°C. Protein concentrations were determined using the bicinchoninic acid (BCA) protein assay kit from Pierce (Rockford, IL, USA).
Proteins were resolved on 10% Tris-glycine gels and transferred onto nitrocellulose membranes. BSA (2%) in PBS containing 0.1% NP-40 was used to block the membranes and dilute the primary and secondary antibodies. HRP-conjugated secondary antibodies (Southern Biotechnology Associates, Birmingham, AL, USA) and SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL, USA) were used for detection. For detection of ubiquitin, proteins were resolved on 4–12% NuPage Tris-bis gels (Life Technologies, Grand Island, NY, USA) and transferred onto PVDF membranes. Membranes were treated with 0.1% glutaraldehyde in PBS for 20 min prior to blocking in PBS containing 2% BSA and 0.1% NP-40. The secondary antibody was diluted into PBS containing 1% non-fat dry milk and 0.1% NP-40. For quantitation, all blots were scanned using a Hewlett-Packard Scanjet 3970, and band density was quantified with UN-SCAN-IT gel digitizing software (Silk Scientific, Inc., Orem, UT, USA).
The following antibodies were used: USP14 (Anderson et al., 2005), β-tubulin (Developmental Studies Hybridoma Bank, Iowa City, IA, USA); Ubiquitin (UAB Hybridoma Facility, Birmingham, AL, USA); pJNK, and JNK (Cell Signaling Technology, Danvers, MA, USA).
Animals were handled 1 day prior to open field testing. Locomotor activity was measured in an open field chamber (43.2 cm × 43.2 cm × 30.5 cm) for 15 min by an automated video tracking system (Med Associated, St. Albans, VT, USA). The first 5 min were not analyzed to account for habituation to the chamber.
Motor coordination was tested by placing mice on a rotating rod (ENV-575, Med Associates), which accelerated from 3.5 to 35 rpm over a 5-min period. Latency to fall was recorded over three trials, each separated by 1 h, and the individual trials for each animal were averaged.
Total RNA was isolated from gastrocnemius muscles or spinal cords using RNA-STAT60 (Tel-Test, Friendswood, TX, USA) and reverse transcribed using the Superscript VILO cDNA synthesis kit (Life Technologies) per manufacturer instructions. Individual gene assays were purchased from Applied Biosystems for each of the RNAs analyzed: AChR-α (Mm00431629_m1), AChR-ε (Mm00437411_m1), and AChR-γ (Mm00437419_m1). ΔΔCt values were generated using Gapdh (Mm99999915_g1) as an internal standard. All values are reported as mean ± SEM of at least three different animals per genotype, run in triplicate.
Whole mount immunostaining of the tibialis anterior (TA) muscle was performed as described (Vaden et al., 2015). Briefly, the TA muscle was immersed in ice-cold PBS containing 2% PFA for 1 h following dissection and immediately teased into thin bundles. Muscle bundles were then transferred to PBS containing 1% PFA and 1% Triton (PBS-T) and incubated overnight at 4°C with constant rocking. To improve visualization of axons and ultra-terminal sprouting, all mice used for NMJ immunostaining carried the Thy1-YFP transgene in addition to the transgene(s) of interest. Endplates were labeled by a 1 h room temperature incubation with rhodamine-conjugated α-bungarotoxin (α-BTX) and prepared for antibody application by a 1 h incubation in a blocking buffer containing 2% bovine serum albumin and 4% normal goat serum in PBS-T. For pJNK immunostaining, muscle bundles were incubated with primary antibody (pJNK, ##81E11, Cell Signaling Technology) for 5 days at 4°C with constant rocking. All images were captured using a Zeiss LSM 510 Meta confocal microscope (Carl Zeiss, Oberkochen, Germany). Endplate size was determined by tracing the circumference of the α-BTX-positive post-synaptic AChR cluster and computing area using ImageJ software (NIH, Bethesda, MD, USA).
All analyses were carried out using GraphPad Prism version for 6.0 for Mac OS X (GraphPad Software, La Jolla, CA, USA). For scaled variables, significant differences among genotypes were determined with a one-way ANOVA after assumptions for normality were verified with a D’Agostino and Pearson omnibus normality test with alpha set to 0.05. Post hoc comparisons were made with independent samples t-tests with Bonferroni-corrected alpha. In samples in which one or more genotype did not meet the assumption of normality, significant differences were determined with a Kruskal–Wallis test and post hoc comparisons were made with Mann–Whitney tests with Bonferroni-corrected alpha.
To assess the interaction of USP14 with cellular ubiquitin pools, we performed immunoblot analysis on spinal cord extracts from wild type, TgCA, and axJ mice (Figure 1A). TgUsp14 mice, which overexpress wild type USP14 in the nervous system (Crimmins et al., 2006), were included as a control. As previously reported (Anderson et al., 2005; Chen et al., 2009), loss of USP14 in the axJ mice resulted in significant decreases in free ubiquitin levels, whereas neuronal expression of catalytically inactive USP14 in the TgCA mice led to only a modest reduction in free ubiquitin levels (Figures 1A,C). Instead, loss of USP14’s catalytic activity led to the retention of more proteins in a ubiquitinated state, as indicated by the increase in ubiquitin conjugates observed in the spinal cords of TgCA mice compared to controls (Figures 1A,B). We also observed both an increase in free ubiquitin and a reduction in ubiquitin conjugates in the spinal cords of the mice overexpressing wild type USP14 (TgUsp14), suggesting that USP14’s DUB activity makes a significant contribution to protein deubiquitination.
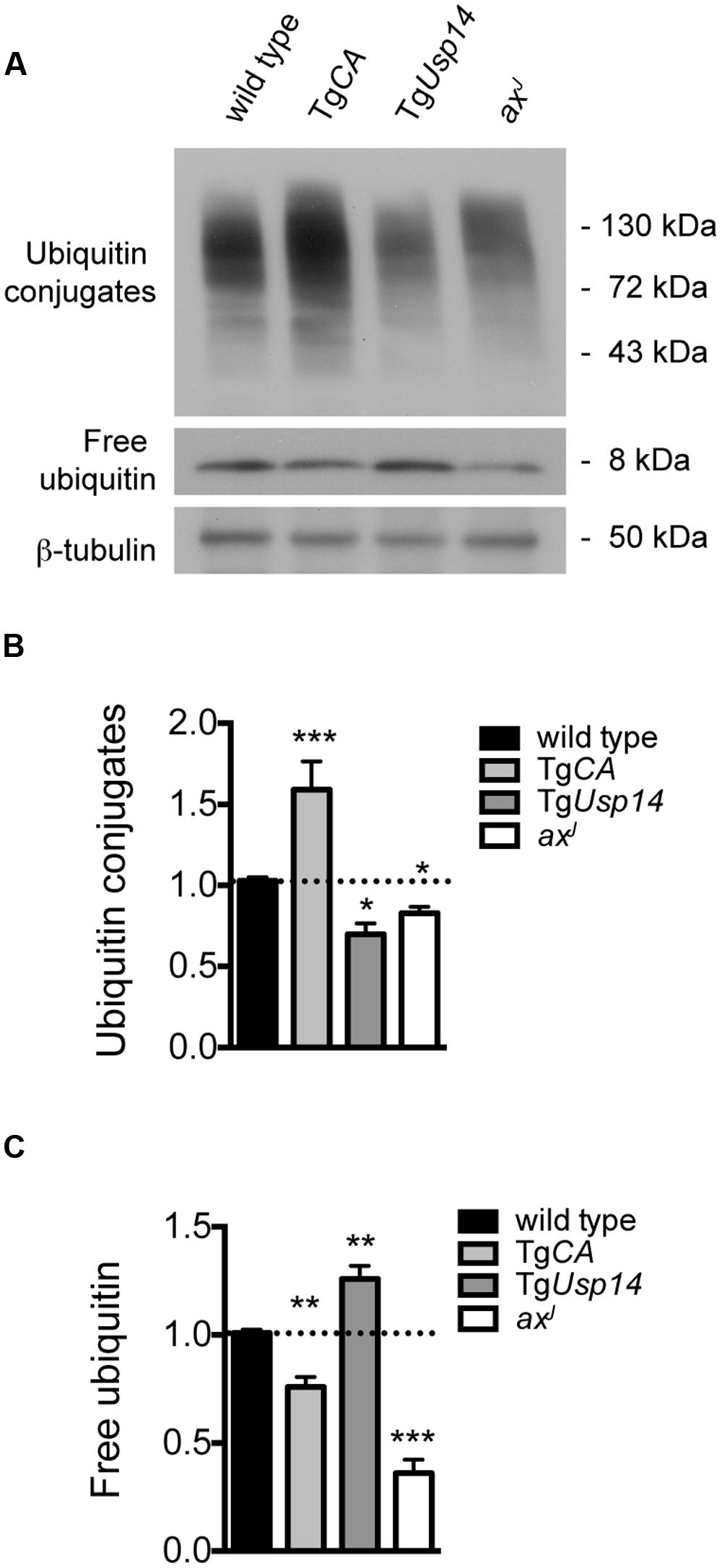
FIGURE 1. Effects of manipulating USP14 expression on neuronal ubiquitin pools. (A) Representative immunoblot of spinal cord lysates taken from 4-weeks-old wild type, TgCA, TgUsp14, and axJ mice, probed for ubiquitin. β-tubulin was used as a loading control. (B) Quantitation of the levels of ubiquitin conjugates, normalized to wild type levels; [F(3,30) = 20.35, p < 0.0001, one-way ANOVA]. (C) Quantitation of the levels of free ubiquitin, normalized to wild type levels; [F(3,30) = 59.15, p < 0.0001, one-way ANOVA]. Symbols represent unpaired t-tests compared against wild type and corrected for multiple comparisons with a Bonferroni adjustment, where ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. n = at least three animals per genotype, run in duplicate.
To test the hypothesis that increased ubiquitin conjugates, and not depletion of free ubiquitin, cause the motor neuron dysfunction in TgCA mice, we restored free ubiquitin levels in the TgCA mice by generating double transgenic mice expressing both TgUb and TgCA under the neuronal Thy1.2 promoter. The resulting TgCA,TgUb mice expressed wild type levels of free ubiquitin as well as an increase in ubiquitin conjugates above what was observed in the TgCA mice (Figures 2A–C). Consistent with our previous report (Vaden et al., 2015), there was no difference in body mass between 4-weeks-old TgCA mice and controls (Figure 2D), but there was a significant reduction in the mass of the gastrocnemius muscles in the TgCA mice compared to controls (Figure 2E). While transgenic expression of ubiquitin increased body and muscle mass in the axJ mice (Chen et al., 2011), both parameters were significantly reduced in the TgCA,TgUb mice compared to both control and TgCA mice (Figures 2D,E). Furthermore, whereas rotarod performance in axJTgUb mice is improved over axJ mice (Chen et al., 2011), the TgCA,TgUb mice did not perform better than TgCA mice in this assay (Figure 2F). In fact, when we measured motor function in the less demanding open field assay, the TgCA,TgUb mice showed reduced ambulatory distance (Figure 2G) and velocity (Figure 2H) compared to both wild type and TgCA mice. In contrast, ubiquitin overexpression alone, in TgUb mice, caused increased body and gastrocnemius mass (Figures 2D,E) and ambulatory distance in the open field assay (Figure 2G) compared to controls.
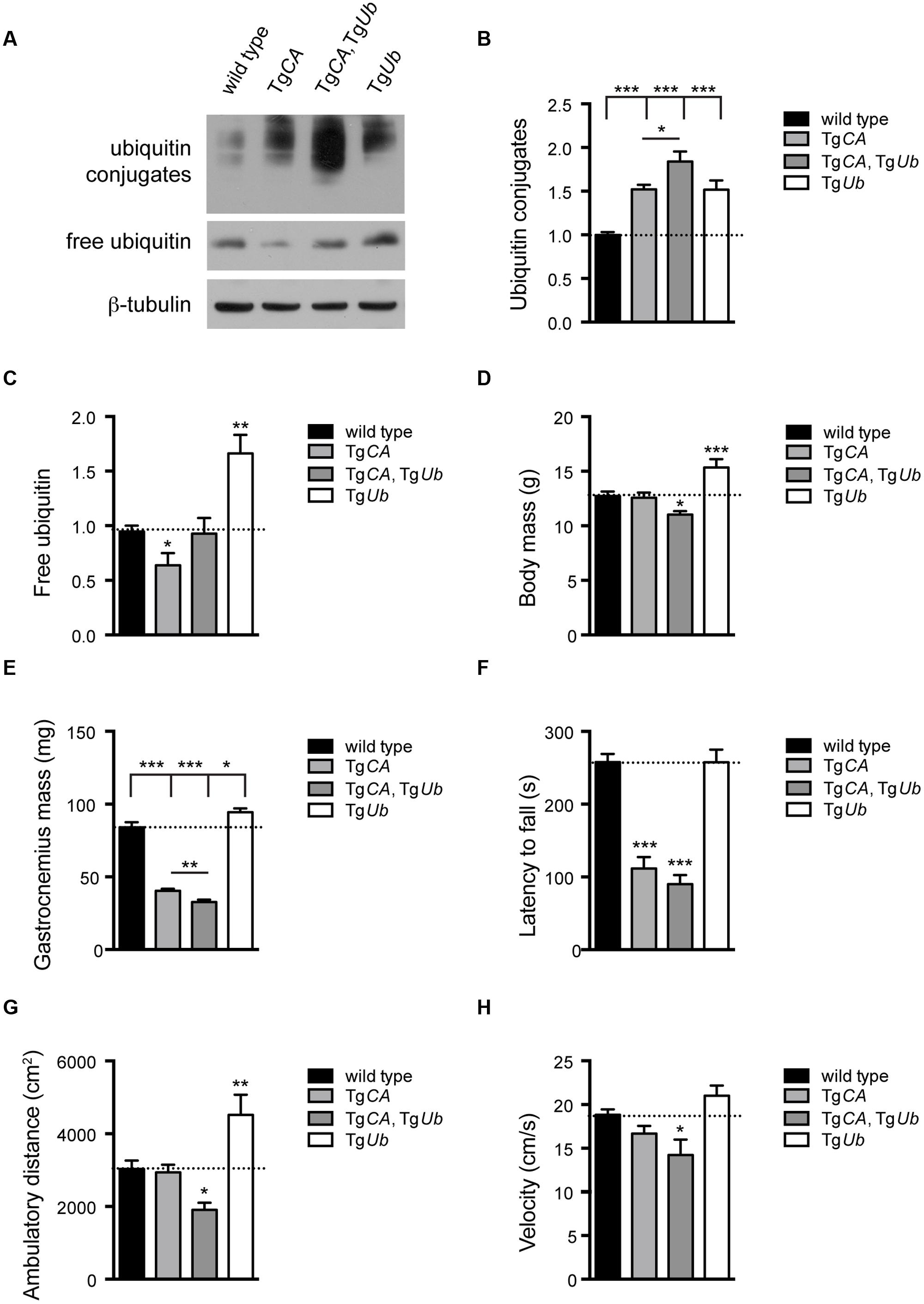
FIGURE 2. Ubiquitin complementation does not rescue body mass, muscle mass, or motor deficits in TgCA mice. (A) Representative immunoblots of ubiquitin and USP14 from spinal cords of 4-weeks-old wild type; TgCA, TgCA, TgUb, and TgUb mice. β-tubulin was used as a loading control. (B) Quantitation of the levels of ubiquitin conjugates, normalized to wild type levels; [F(3,28) = 17.36, p < 0.0001, one-way ANOVA]. (C) Quantitation of the levels of free ubiquitin, normalized to wild type levels; [F(3,12) = 11.70, p < 0.001, one-way ANOVA]. (D) Body mass [F(3,56) = 9.03, p < 0.0001, one-way ANOVA] and (E) gastrocnemius muscle mass [F(3,70) = 94.72, p < 0.0001, one-way ANOVA] of 4-weeks-old mice. n = at least 12 animals per genotype. (F) Latency to fall from beam during a rotarod assay [F(3,26) = 38.36; p < 0.0001, one-way ANOVA]. (G) Total ambulatory distance [F(3,29) = 9.26; p < 0.001, one-way ANOVA] and (H) velocity [F(3,26) = 6.56; p < 0.01, one-way ANOVA] during 10 min open field assay. For (F–H), n = at least five animals per genotype. All data are shown as mean ± SEM. Symbols represent unpaired t-tests compared against wild type and corrected for multiple comparisons with a Bonferroni adjustment, where ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. In (B,C), an additional unpaired t-test with Bonferroni adjustment was used to compare TgCA and TgCA,TgUb mice.
We have previously shown that deficits in motor function and NMJ synaptic transmission are inversely related to muscle AChR transcript levels, and, in particular, that expression of the fetal AChR-γ subunit is dramatically increased in adult animals with motor and synaptic deficits (Chen et al., 2009, 2011; Vaden et al., 2015). As predicted by their poor open field performance, AChR-α, -ε, and -γ transcripts were significantly increased in TgCA,TgUb mice over all other genotypes, including TgCA (Figures 3A–C). Finally, there was a significant decrease in AChR-γ transcripts in TgUb mice compared to controls (Figure 3C), and a trend toward decreased AChR-α and -ε transcripts (Figures 3A,B). Together with our previous report, these data demonstrate that increased ubiquitin expression in the presence of catalytically inactive USP14 (in the TgCA,TgUb mice) has drastically different effects than when USP14 is absent (axJTgUb mice).
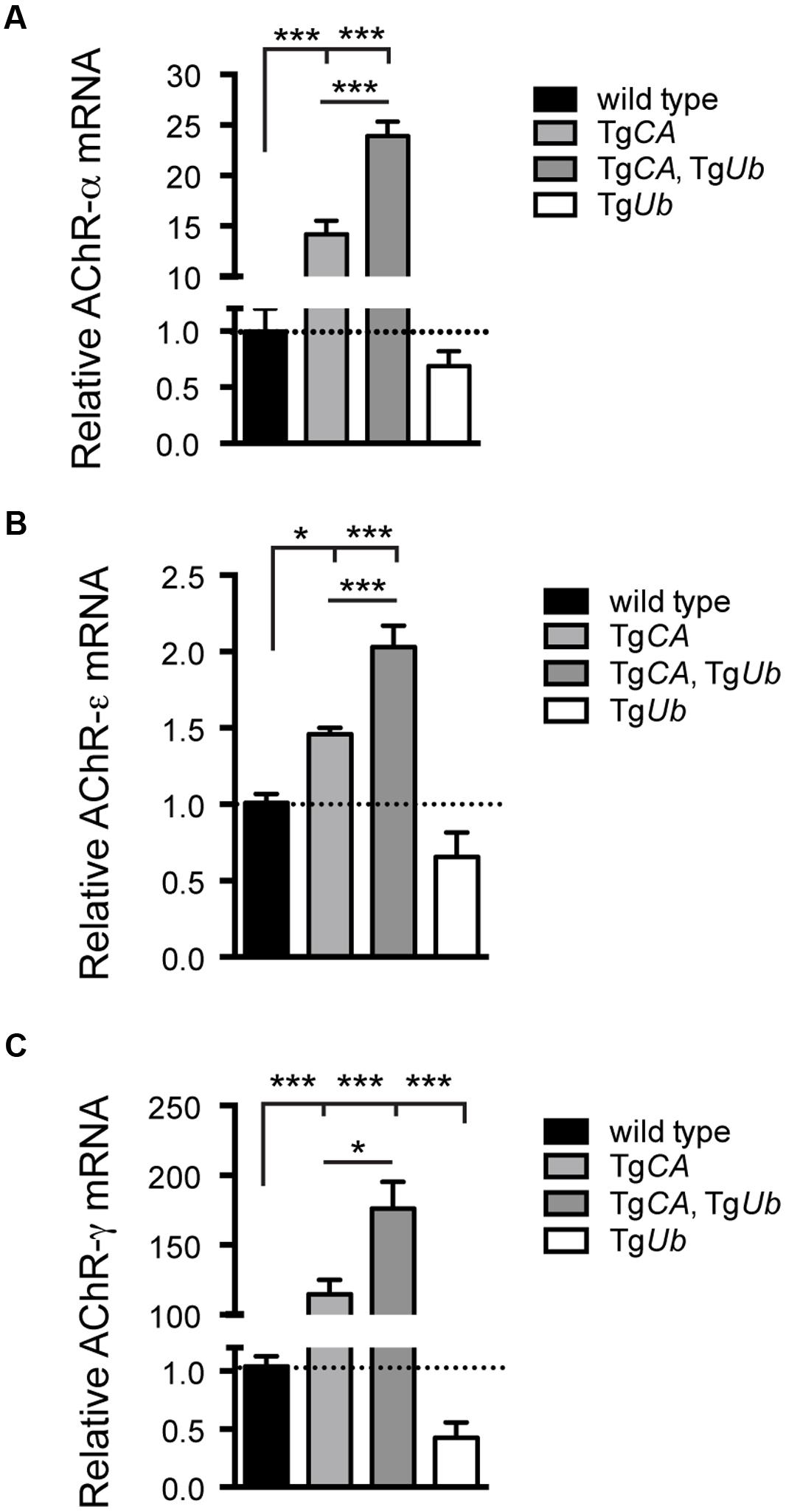
FIGURE 3. Consequences of ubiquitin overexpression on AChR transcript levels in TgCA mice. qPCR analysis of AChR (A) –α [F(3,22) = 133.1; p < 0.0001, one-way ANOVA], (B) –ε [F(3,28) = 30.03; p < 0.0001, one-way ANOVA], and (C) –γ [F(3,19) = 35.56; p < 0.0001, one-way ANOVA] subunit mRNA abundance in 4-weeks-old wild type; TgCA; TgCA,TgUb, and TgUb mice expressed as relative fold change from wild type. n = at least four animals per genotype, run in triplicate. Data are shown as mean ± SEM. Symbols represent unpaired t-tests corrected for multiple comparisons with a Bonferroni adjustment where ∗p < 0.05, ∗∗∗p < 0.001.
Loss of USP14’s catalytic activity leads to a greater increase in ubiquitin conjugates in TgCA,TgUb spinal cords than ubiquitin overexpression alone causes in the spinal cords of TgUb mice (Figure 2). To determine whether increased protein ubiquitination in the nervous system could alter neuromuscular function independently of reduced USP14 activity, we compared muscle mass, AChR expression, and rotarod performance in the TgUb mice used in previous experiments with another transgenic founder line (TgUb-H) that has higher levels of ubiquitin expression (Figures 4A–C). The gastrocnemius muscles of 4- to 6-weeks-old female TgUb mice were significantly larger than those of controls (Figure 4D). In contrast, the increased ubiquitin expression in the TgUb-H mice resulted in significantly decreased gastrocnemius mass compared to controls (Figure 4D). The same was true for male wild type, TgUb, and TgUb-H mice (data not shown). Further, we found that TgUb-H mice had a significant increase in the abundance of fetal AChR-γ transcripts (Figure 4E), a marker of synaptic deficits and motor dysfunction in adult axJ animals (Chen et al., 2009, 2011). Finally, whereas overexpression of ubiquitin in the TgUb mice had no effect on rotarod performance, increased levels of ubiquitin in the TgUb-H mice caused them to fall from the rotating beam more quickly than controls (Figure 4F), and display an abnormal gait while performing this task (data not shown). Together, these data show that ubiquitin has dose-dependent effects on motor function and muscle development.

FIGURE 4. Dose-dependent effects of ubiquitin on muscle development and motor function. (A) Representative immunoblots of free and conjugated ubiquitin in spinal cords from two lines of transgenic mice over-expressing ubiquitin under the neuronal Thy1.2 promoter. β-tubulin was used as a loading control. (B) Quantitation of the levels of ubiquitin conjugates, normalized to wild type levels; [F(2,39) = 10.10, p < 0.001, one-way ANOVA]. (C) Quantitation of the levels of free ubiquitin, normalized to wild type levels; [F(2,38) = 24.01, p < 0.0001, one-way ANOVA]. (D) Comparison of gastrocnemius muscle mass from 4- to 5-weeks-old female mice; [F(2,23) = 89.83, p < 0.0001, one-way ANOVA]. The same effect was observed in male mice; [F(2,19) = 17.35, p < 0.0001, one-way ANOVA]. (E) Comparison of AChR-γ transcript abundance, normalized to wild type [F(2,12) = 185.5, p < 0.0001]. For (B–E) symbols represent unpaired t-tests compared against wild type and corrected for multiple comparisons with a Bonferroni adjustment where ∗∗p < 0.01, ∗∗∗p < 0.001. (F) Latency to fall from beam during a rotarod assay (H = 12.22, 1 df, p < 0.01, Kruskal–Wallis test), symbol represents Mann–Whitney test compared against wild type and corrected for multiple comparisons with a Bonferroni adjustment where ∗∗∗p < 0.001. For (D–F), n = at least five animals per genotype. All data are shown as mean ± SEM.
We have previously reported that the NMJs of 4- to 6-weeks-old USP14-deficient axJ mice have poor motor endplate arborization, swollen presynaptic terminals, and ultra-terminal sprouting, and that these deficits are corrected in axJTgUb mice (Chen et al., 2011). Because TgCA mice recapitulate these axJ NMJ deficits (Vaden et al., 2015), we investigated whether NMJ structure was improved in TgCA,TgUb mice (Figure 5A). Whereas 56% of TgCA NMJs had presynaptic swellings, ubiquitin overexpression improved NMJ structure and only 11% of TgCA,TgUb terminals were swollen (Figure 5B). Similarly, we observed ultra-terminal or ultra-axonal sprouting in 50% of the terminals of the TgCA mice compared to only 17% in the TgCA,TgUb mice (Figure 5C). Terminal swelling and sprouting were seen only rarely in wild type and TgUb mice (Figures 5B,C). We found a significant effect of genotype on endplate area, with smaller, more plaque-like endplates in the TgCA and TgCA,TgUb mice than wild type and TgUb mice (Figure 5D), consistent with the increase in AChR mRNA abundance observed in these mice (Figure 3).
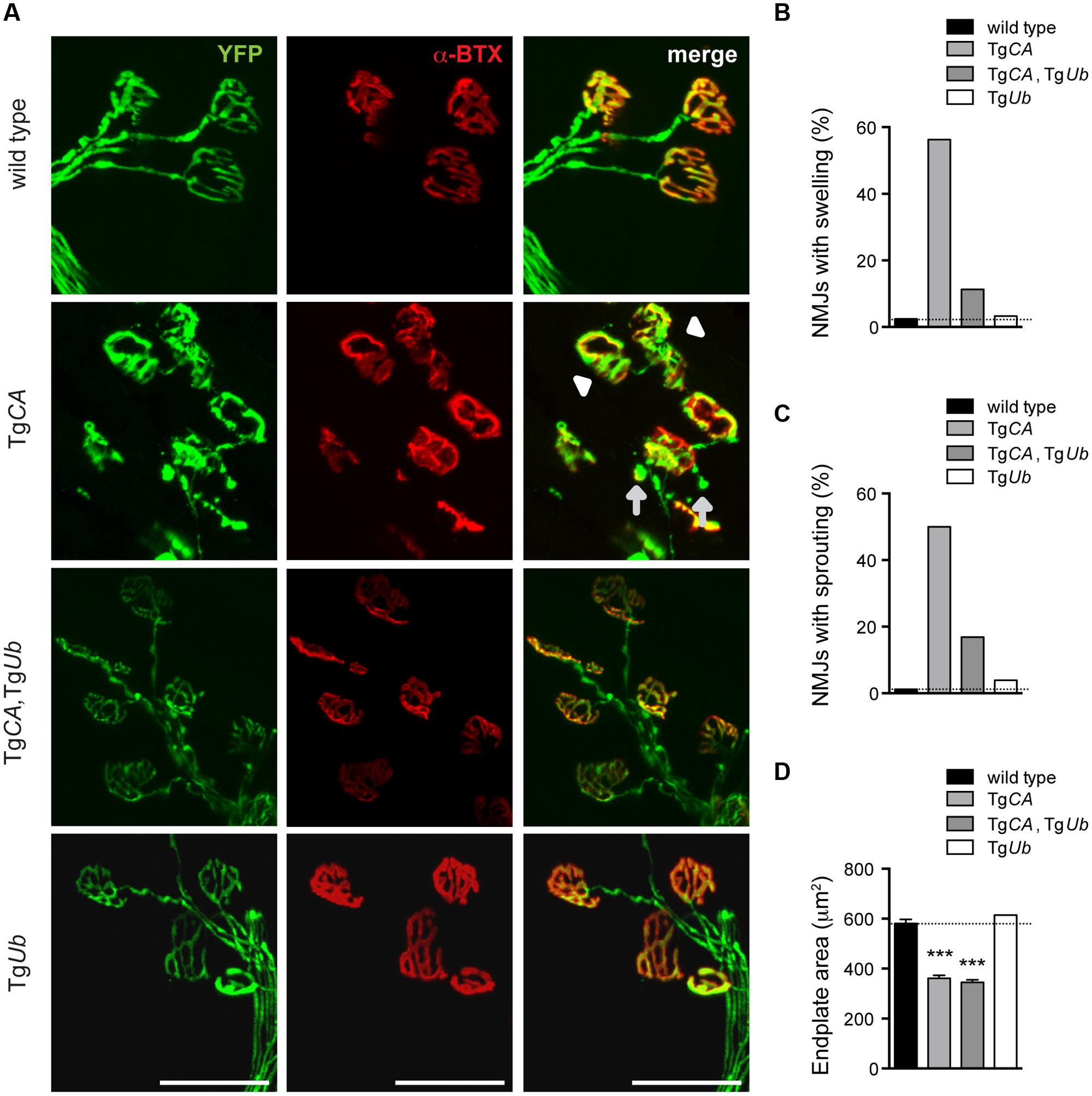
FIGURE 5. Ubiquitin complementation improves NMJ structure in TgCA mice. (A) Whole-mount immunostaining of tibialis anterior (TA) muscles from 4- to 5-weeks-old wild type; TgCA, TgCA, TgUb, and TgUb mice expressing YFP (green) under the neuronal Thy1.2 promoter. AChR clusters were visualized with rhodamine-conjugated α-bungarotoxin (α-BTX, red). Arrowhead indicates terminal swelling and arrows indicate ultra-terminal sprouting. Scale bar represents 50 μM. (B) Percent of NMJs with presynaptic swellings or (C) sproutings. (D) Quantitation of α-BTX-positive endplate area shown as mean ± SEM. [F(3,564) = 104.60; p < 0.0001, one-way ANOVA]. Symbols in (C) represent unpaired t-tests compared against wild type and corrected for multiple comparisons with a Bonferroni adjustment, where ∗∗∗p < 0.001. For (B–D), n = at least 130 NMJs from five animals.
We recently reported that pJNK is a prominent component of the terminal swellings and sproutings in TgCA mice, and that the extent of this pathology is reduced by treatment with the JNK inhibitor SP600125 (Vaden et al., 2015). Because ubiquitin complementation improved the structure of the TgCA endplates (Figure 5), we investigated whether this reduction in pathology was associated with a reduction of pJNK at the NMJ. As previously reported, we observed strong pJNK immunostaining in the presynaptic terminals of TgCA mice (Figure 6A): 55.8% of terminals contained pJNK-positive swellings (Figure 6B) and 43.5% of terminals contained pJNK-positive ultra-terminal sprouts (Figure 6C). In contrast, ubiquitin overexpression in the TgCA,TgUb mice reduced the pathology, and only 8.3% of terminals contained pJNK-positive swelling or sprouting (Figures 6B,C). pJNK-positive pathology was negligible in wild type mice and absent in TgUb mice. However, when we immunoblotted spinal cord lysates from wild type, TgCA, TgCA,TgUb, and TgUb mice with a pJNK-specific antibody, we found that the levels of pJNK in TgCA,TgUb mice were elevated over the levels observed in TgCA mice, and that spinal cord lysates of both genotypes had significantly more pJNK than spinal cord lysates from wild type mice (Figures 7A,B). There was no change in total JNK abundance in the TgCA or TgCA,TgUb mice compared to controls, indicating that USP14 and ubiquitin regulate JNK activation and not JNK stability. We also observed a significant decrease in the abundance of pJNK, but not total JNK, in TgUb spinal cord lysates compared to controls (Figures 7A,B). A schematic summarizing the levels of pJNK abundance observed in the spinal cords, distal motor neuron axons, and NMJs of wild type, TgCA, and TgCA,TgUb mice is shown in Figure 7C.
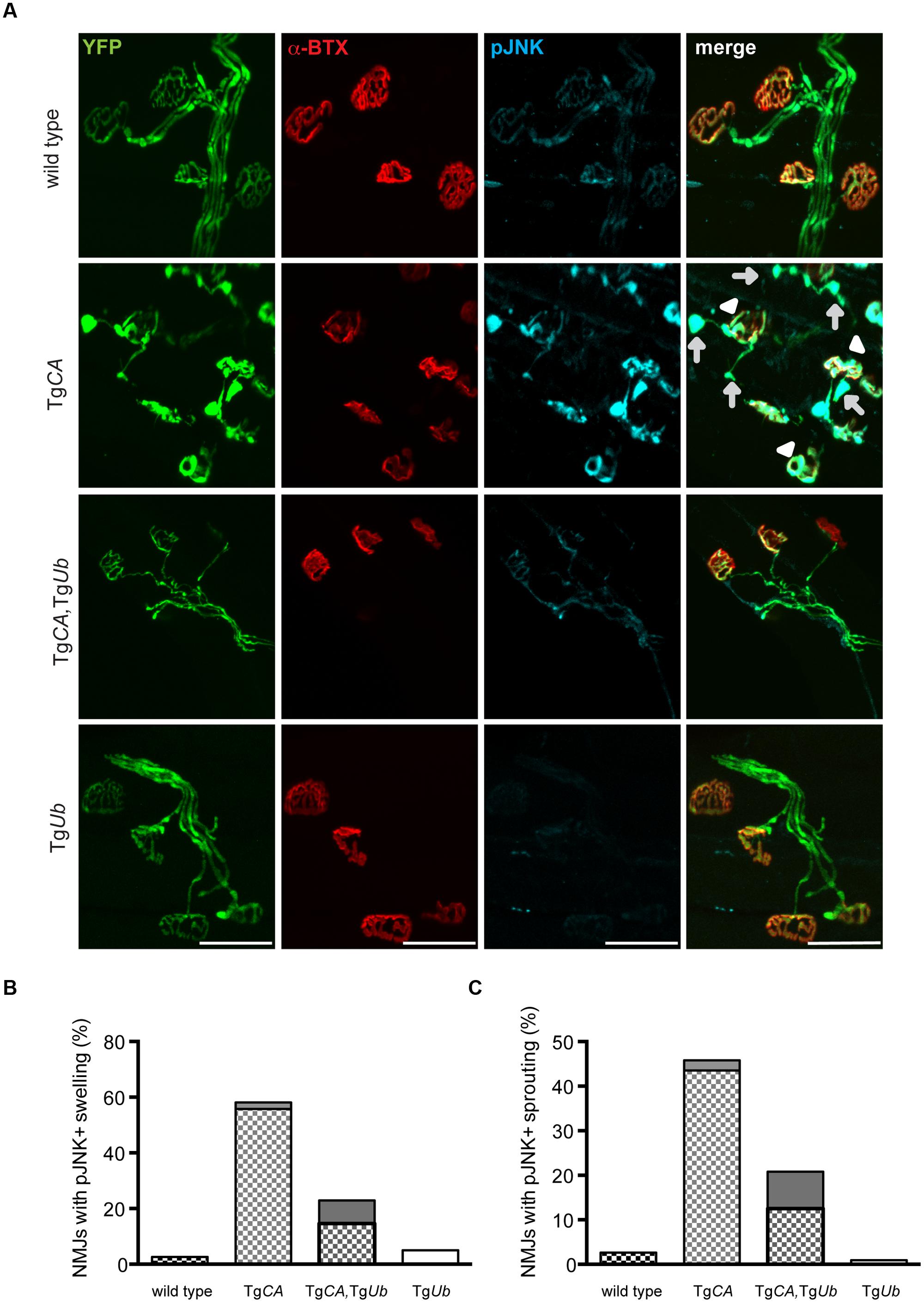
FIGURE 6. Ubiquitin complementation reduces pJNK levels at the NMJs of TgCA mice. (A) Whole-mount immunostaining of TA muscles from 4- to 5-weeks-old wild type; TgCA, TgCA, TgUb, and TgUb mice expressing YFP (green) under the neuronal Thy1.2 promoter using an antibody against pJNK (light blue). Endplates were visualized with rhodamine-conjugated α-bungarotoxin (α-BTX, red). Arrowheads indicate pJNK-positive terminal swelling and arrows indicate pJNK-positive ultra-terminal sprouting. Scale bar represents 50 μM. Quantitation of (B) presynaptic swellings or (C) ultra-terminal sproutings. The patterned portion of each bar represents pJNK-positive swellings and the height of the bar represents the total percent of swollen NMJs. n = at least 60 NMJs from three animals per genotype.
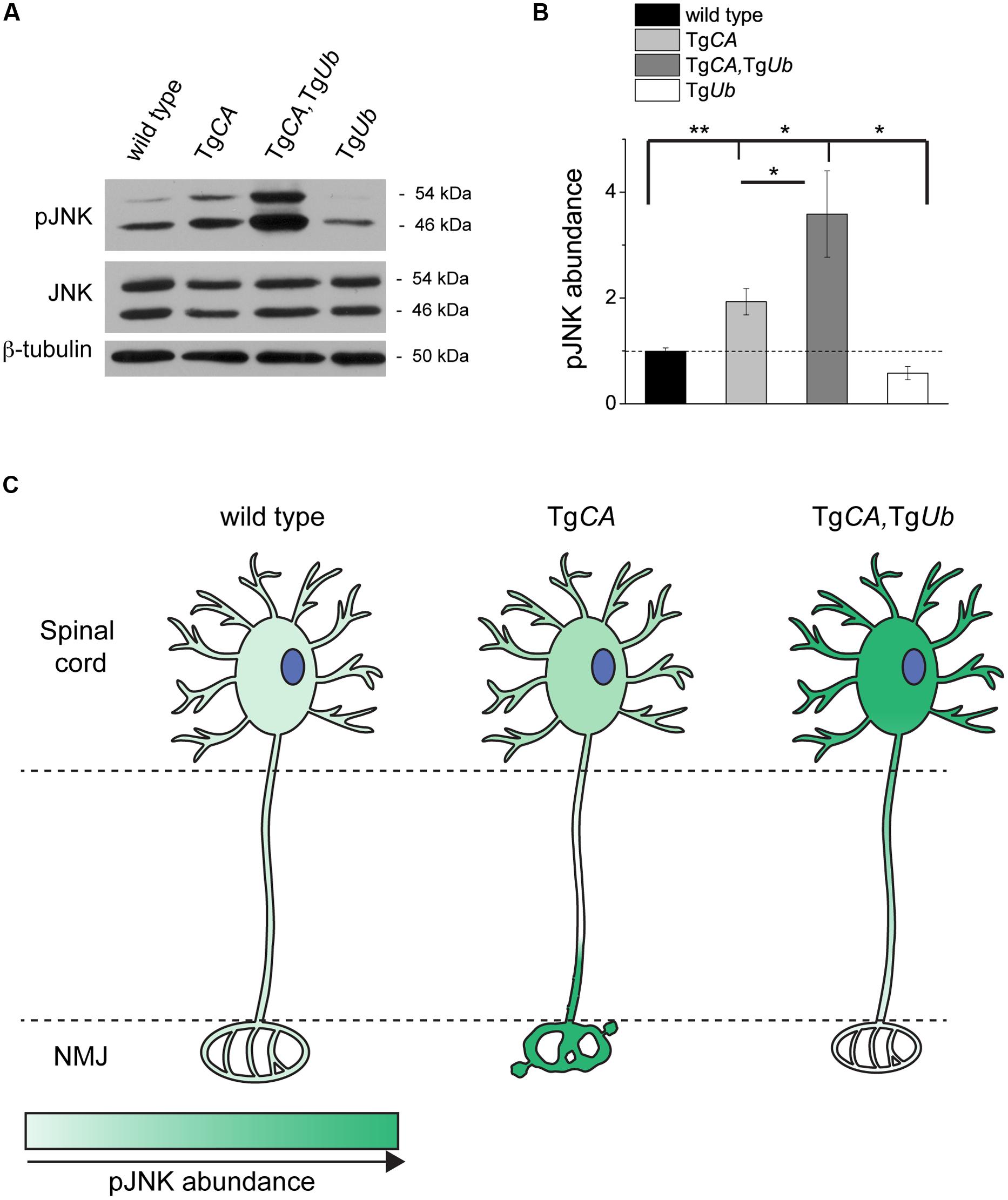
FIGURE 7. Ubiquitin overexpression does not reduce pJNK abundance in TgCA spinal cords. (A) Representative immunoblots of pJNK and total Jun N-terminal kinase (JNK) from spinal cords of 4 weeks-old wild type, TgCA, TgCA,TgUb, and TgUb mice. β-tubulin was used as a loading control. (B) Quantitation of pJNK levels in 4-weeks-old mice normalized to wt and total JNK abundance [F(3,36) = 9.532; p < 0.0001, one-way ANOVA]. Symbols represent unpaired t-tests corrected for multiple comparisons with a Bonferroni adjustment, where ∗p < 0.05, ∗∗p < 0.01. (C) Schematic representation of the levels of pJNK abundance observed in the spinal cords, distal axons of motor neurons, and NMJs of 4- to 5-weeks-old mice of the genotypes indicated, where darker green indicates greater pJNK abundance.
Three main conclusions can be drawn from these studies. First, overexpression of ubiquitin increased the abundance of ubiquitin conjugates in the spinal cord (Figure 4), demonstrating that free ubiquitin is a limiting factor in some ubiquitin-dependent processes in the nervous system. Second, USP14’s ubiquitin hydrolase activity plays a significant role in regulating the distribution of free and conjugated ubiquitin pools in neurons (Figure 1). The increased abundance of ubiquitin conjugates, and not depletion of the free ubiquitin pool, directly correlated with impaired motor function when USP14’s ubiquitin hydrolase activity was inhibited (Figures 2 and 3). Third, both ubiquitin levels and the catalytic activity of USP14 impact the abundance of pJNK at the NMJ and in the spinal cord (Figures 6 and 7) and, in turn, the levels of pJNK at the NMJ impact the structure of the presynaptic terminal.
Neuronal overexpression of ubiquitin led to increased ubiquitin conjugates in a dose-dependent manner in two transgenic founder lines, TgUb and TgUb-H, with low and high levels of ubiquitin overexpression, respectively (Figure 4A). These ubiquitin conjugates had dose-dependent effects on motor function and muscle development in wild type mice (Figure 4). Mild ubiquitin overexpression led to increased muscle development and reduced expression of the fetal γ subunit of the muscle AChR, which is correlated with enhanced NMJ synaptic transmission (Chen et al., 2009, 2011; Vaden et al., 2015), in TgUb mice compared to controls (Figures 4B–D). In contrast, more dramatic ubiquitin overexpression reduced muscle development, increased AChR-γ abundance, and hindered motor performance in the TgUb-H mice compared to controls (Figures 4B–D). Moreover, we have previously reported that the TgUb-H mice develop adult-onset motor endplate disease (Hallengren et al., 2013). Together, these data highlight the importance of maintaining ubiquitin levels within a relatively narrow window.
Ubiquitin-specific protease 14 is an important regulator of ubiquitin homeostasis. We found that increasing the level of wild type USP14 in the nervous system resulted in an increase in the free ubiquitin pool and decreased levels of ubiquitin conjugates as compared to controls. Because both the axJ mice and TgCA mice exhibit severe motor endplate disease, we were surprised to find differences in ubiquitin homeostasis between these two mouse lines. Loss of USP14 in the axJ mice significantly reduced the abundance of both free and conjugated ubiquitin. In contrast, expression of catalytically inactive USP14CA in the TgCA mice led to a 40% increase in ubiquitin conjugates and a 25% reduction of free ubiquitin. Further studies will be required to determine if these effects of USP14 on ubiquitin pools are due to changes in ubiquitin-dependent degradation by the proteasome. These differences in ubiquitin homeostasis in the spinal cords of the axJ and TgCA mice, along with the dose-dependent impact of increased ubiquitin conjugates on motor function and muscle development, may provide a framework for understanding the opposite effects of ubiquitin overexpression in the axJ and TgCA mice. One possibility for these differences is that USP14CA may have an increased affinity for its substrates compared to USP14. Binding of USP14CA may slow their eventual deubiquitination by another DUB and result in prolonged ubiquitin-dependent signaling. Alternatively, increased affinity of USP14CA to ubiquitin conjugates may block their proteasomal degradation and lead to protein aggregation that could affect synaptic function.
In the axJ, TgUb mice, the levels of ubiquitin conjugates and free ubiquitin are slightly elevated over what is normally observed in wild type mice (Chen et al., 2011). We have now shown that a modest increase in ubiquitin conjugates and free ubiquitin in the TgUb mice correlated with increased muscle development and motor function, even when wild type USP14 was present (Figure 4). In contrast, the levels of ubiquitin conjugates in the spinal cords of the TgCA, TgUb mice were nearly twofold what is observed in wild type mice (Figures 2A,B), and reduced muscle development and mobility were observed in the TgCA,TgUb mice compared to both wild type and TgCA mice. Our studies of the TgUb-H mice indicated that a twofold increase in ubiquitin conjugates resulted in deficits in motor function and muscle development even when USP14’s catalytic activity was intact (Figure 4). Together, these data open the possibility that the restoration of motor neuron function in axJTgUb mice is indirect and can be attributed to increased ubiquitin conjugates, while suggesting that ubiquitin overexpression aggravates the accumulation of ubiquitin conjugates caused by loss of USP14’s DUB activity in TgCA mice to exaggerate the existing phenotype (Figures 2 and 3). Since both increases and decreases in ubiquitin pools are associated with deleterious effects in cells and animals models (Parag et al., 1987; Ferguson et al., 1990; Osaka et al., 2003; Anderson et al., 2005; Hirsch et al., 2006) the ubiquitin depletion observed in the absence of functional USP14 may contribute to other neurological deficits observed in the TgCA and axJ mice.
However, when considered together with our previous work demonstrating a role for pJNK in the NMJ pathology observed in the TgCA mice (Vaden et al., 2015), this study suggests that ubiquitin complementation corrects these structural deficits directly, by reducing pJNK-positive pathology (Figures 5 and 6). While we have not examined pJNK abundance at the NMJs of axJ mice, the increased JNK activation in axJ spinal cords suggests that the mechanism underlying NMJ pathology caused by loss of USP14 is the same as that caused by loss of USP14’s DUB activity (Vaden et al., 2015). Given the reduction of pJNK at TgCA,TgUb NMJs compared to TgCA NMJs (Figure 6), we were surprised by the dramatic increase in pJNK levels in the spinal cords of the TgCA,TgUb mice (Figures 7A,B). However, we have recently reported that loss of USP14’s DUB activity leads to enhanced K63-linked ubiquitination of MLK3 (Vaden et al., 2015), which drives it to dimerize, autophosphorylate, and activate its immediate downstream targets, MKK4/7, which, in turn, activate JNK (Humphrey et al., 2013). It is therefore possible that the increased JNK activation that we observed in TgCA,TgUb spinal cords compared to TgCA spinal cords (Figures 7A,B) results from increased activation of MLK3.
The means by which ubiquitin complementation reduced pJNK-positive pathology at the NMJ remains unclear. One possibility is that the increased ubiquitin in TgCA,TgUb mice stimulates the degradation of pJNK or its upstream kinases at the NMJ or, alternatively, that ubiquitin contributes to the activation of phosphatases that act on pJNK. Both of these explanations are consistent with the reduction of pJNK abundance in TgUb spinal cords compared to wild type, but not with the increase in pJNK levels over both wild type and TgCA levels in the spinal cords of the TgCA,TgUb mice (Figures 7A,B). In contrast, the well-documented retrograde axonal transport of activated JNK (Cavalli et al., 2005; Lindwall and Kanje, 2005; Shin et al., 2012; Drerup and Nechiporuk, 2013) and the pJNK-positive swellings at TgCA NMJs (Figure 6) are all consistent with a deficit in the retrograde transport of pJNK out of axon terminals in the TgCA mice. The lack of pJNK-positive pathology in the nerve terminals (Figure 6) of the TgCA,TgUb mice, combined with the robust elevation of pJNK abundance in TgCA,TgUb spinal cords (Figures 7A,B), the site of motor neuron cell bodies, suggest that ubiquitin may stimulate the retrograde axonal transport of pJNK (Figure 7C). Although a role for ubiquitin in retrograde axonal transport has not been demonstrated directly, it was recently reported that altered ubiquitin homeostasis contributes to the deficits in TrkB retrograde transport caused by the exposure of cultured neurons to Aβ oligomers (Poon et al., 2013).
Finally, our findings also suggest that the functional deficits observed in the TgCA and axJ mice do not arise because of altered NMJ structure. This is consistent with our previous report that acute inhibition of USP14 at the NMJs of wild type mice causes deficits in synaptic transmission that are similar to what is observed in the TgCA and axJ mice, while intramuscular injections of the same inhibitor given over the course of a week do not cause NMJ pathology (Vaden et al., 2015). Together, these findings may indicate that USP14 regulates synapse structure and function through distinct pathways. The same is true of the ubiquitin ligase highwire, which regulates the structure and function of the Drosophila NMJ through separate pathways (Collins et al., 2006).
Scott Wilson is a paid consultant for Progenera Inc. The collection, analysis, and interpretation of the data presented in this manuscript were not influenced by my relationship with Progenra Inc.
This work was supported by grants to UAB from the Howard Hughes Medical Institute through the Med into Grad Initiative (JV), the Civitan Research Center, the Evelyn F. McKnight Brain Institute, and the National Institutes of Health (R01 NS047533 and R21 NS074456 to SW).
TgCA (transgene expressing catalytically inactive USP14, generated by mutating the active site cysteine to an alanine residue, expressed under the neuronal Thy1.2 promoter; also refers to mice expressing this transgene); TgUb (transgene expressing ubiquitin under the Thy1.2 promoter; also refers to mice expressing this transgene); TgUb-H (a transgenic founder line with more robust ubiquitin overexpression than TgUb); TgCA,TgUb (mice expressing both TgCA and TgUb).
Anderson, C., Crimmins, S., Wilson, J. A., Korbel, G. A., Ploegh, H. L., and Wilson, S. M. (2005). Loss of Usp14 results in reduced levels of ubiquitin in ataxia mice. J. Neurochem. 95, 724–731. doi: 10.1111/j.1471-4159.2005.03409.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baker, R. T., and Board, P. G. (1987). The human ubiquitin gene family: structure of a gene and pseudogenes from the Ub B subfamily. Nucleic Acids Res. 15, 443–463. doi: 10.1093/nar/15.2.443
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baker, R. T., and Board, P. G. (1991). The human ubiquitin-52 amino acid fusion protein gene shares several structural features with mammalian ribosomal protein genes. Nucleic Acids Res. 19, 1035–1040. doi: 10.1093/nar/19.5.1035
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cavalli, V., Kujala, P., Klumperman, J., and Goldstein, L. S. (2005). Sunday Driver links axonal transport to damage signaling. J. Cell Biol. 168, 775–787. doi: 10.1083/jcb.200410136
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, P. C., Bhattacharyya, B. J., Hanna, J., Minkel, H., Wilson, J. A., Finley, D.,et al. (2011). Ubiquitin homeostasis is critical for synaptic development and function. J. Neurosci. 31, 17505–17513. doi: 10.1523/JNEUROSCI.2922-11.2011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, P. C., Qin, L. N., Li, X. M., Walters, B. J., Wilson, J. A., Mei, L.,et al. (2009). The proteasome-associated deubiquitinating enzyme Usp14 is essential for the maintenance of synaptic ubiquitin levels and the development of neuromuscular junctions. J. Neurosci. 29, 10909–10919. doi: 10.1523/jneurosci.2635-09.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Collins, C. A., Wairkar, Y. P., Johnson, S. L., and Diantonio, A. (2006). Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron 51, 57–69. doi: 10.1016/j.neuron.2006.05.026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Crimmins, S., Jin, Y., Wheeler, C., Huffman, A. K., Chapman, C., Dobrunz, L. E.,et al. (2006). Transgenic rescue of ataxia mice with neuronal-specific expression of ubiquitin-specific protease 14. J. Neurosci. 26, 11423–11431. doi: 10.1523/JNEUROSCI.3600-06.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Drerup, C. M., and Nechiporuk, A. V. (2013). JNK-interacting protein 3 mediates the retrograde transport of activated c-Jun N-terminal kinase and lysosomes. PLoS Genet. 9:e1003303. doi: 10.1371/journal.pgen.1003303
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Etter, P. D., Narayanan, R., Navratilova, Z., Patel, C., Bohmann, D., Jasper, H.,et al. (2005). Synaptic and genomic responses to JNK and AP-1 signaling in Drosophila neurons. BMC Neurosci. 6:39. doi: 10.1186/1471-2202-6-39
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ferguson, D. L., Guikema, J. A., and Paulsen, G. M. (1990). Ubiquitin pool modulation and protein degradation in wheat roots during high temperature stress. Plant Physiol. 92, 740–746. doi: 10.1104/pp.92.3.740
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Finley, D., Bartel, B., and Varshavsky, A. (1989). The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature 338, 394–401. doi: 10.1038/338394a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hallengren, J., Chen, P. C., and Wilson, S. M. (2013). Neuronal ubiquitin homeostasis. Cell Biochem. Biophys. 67, 67–73. doi: 10.1007/s12013-013-9634-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425–479. doi: 10.1146/annurev.biochem.67.1.425
Hirsch, C., Gauss, R., and Sommer, T. (2006). Coping with stress: cellular relaxation techniques. Trends Cell Biol. 16, 657–663. doi: 10.1016/j.tcb.2006.10.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Humphrey, R. K., Yu, S. M., Bellary, A., Gonuguntla, S., Yebra, M., and Jhala, U. S. (2013). Lysine 63-linked ubiquitination modulates mixed lineage kinase-3 interaction with JIP1 scaffold protein in cytokine-induced pancreatic beta cell death. J. Biol. Chem. 288, 2428–2440. doi: 10.1074/jbc.M112.425884
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jara, J. H., Frank, D. D., and Ozdinler, P. H. (2013). Could dysregulation of UPS be a common underlying mechanism for cancer and neurodegeneration? Lessons from UCHL1. Cell Biochem. Biophys. 67, 45–53. doi: 10.1007/s12013-013-9631-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lindwall, C., and Kanje, M. (2005). Retrograde axonal transport of JNK signaling molecules influence injury induced nuclear changes in p-c-Jun and ATF3 in adult rat sensory neurons. Mol. Cell. Neurosci. 29, 269–282. doi: 10.1016/j.mcn.2005.03.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lowe, J., Blanchard, A., Morrell, K., Lennox, G., Reynolds, L., Billett, M.,et al. (1988). Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson’s disease, Pick’s disease, and Alzheimer’s disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J. Pathol. 155, 9–15. doi: 10.1002/path.1711550105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lund, P. K., Moats-Staats, B. M., Simmons, J. G., Hoyt, E., D’ercole, A. J., Martin, F.,et al. (1985). Nucleotide sequence analysis of a cDNA encoding human ubiquitin reveals that ubiquitin is synthesized as a precursor. J. Biol. Chem. 260, 7609–7613. doi: 10.1002/path.1711550105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Osaka, H., Wang, Y. L., Takada, K., Takizawa, S., Setsuie, R., Li, H.,et al. (2003). Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum. Mol. Genet. 12, 1945–1958. doi: 10.1093/hmg/ddg211
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Parag, H. A., Raboy, B., and Kulka, R. G. (1987). Effect of heat shock on protein degradation in mammalian cells: involvement of the ubiquitin system. EMBO J. 6, 55–61.
Perry, G., Friedman, R., Shaw, G., and Chau, V. (1987). Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc. Natl. Acad. Sci. U.S.A. 84, 3033–3036. doi: 10.1073/pnas.84.9.3033
Poon, W. W., Carlos, A. J., Aguilar, B. L., Berchtold, N. C., Kawano, C. K., Zograbyan, V.,et al. (2013). beta-Amyloid (Abeta) oligomers impair brain-derived neurotrophic factor retrograde trafficking by down-regulating ubiquitin C-terminal hydrolase, UCH-L1. J. Biol. Chem. 288, 16937–16948. doi: 10.1074/jbc.M113.463711
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Redman, K. L., and Rechsteiner, M. (1989). Identification of the long ubiquitin extension as ribosomal protein S27a. Nature 338, 438–440. doi: 10.1038/338438a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ryu, K. Y., Garza, J. C., Lu, X. Y., Barsh, G. S., and Kopito, R. R. (2008). Hypothalamic neurodegeneration and adult-onset obesity in mice lacking the Ubb polyubiquitin gene. Proc. Natl. Acad. Sci. U.S.A. 105, 4016–4021. doi: 10.1073/pnas.0800096105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schmukle, A. C., and Walczak, H. (2012). No one can whistle a symphony alone - how different ubiquitin linkages cooperate to orchestrate NF-kappaB activity. J. Cell Sci. 125, 549–559. doi: 10.1242/jcs.091793
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shin, J. E., Cho, Y., Beirowski, B., Milbrandt, J., Cavalli, V., and Diantonio, A. (2012). Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron 74, 1015–1022. doi: 10.1016/j.neuron.2012.04.028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tanno, H., and Komada, M. (2013). The ubiquitin code and its decoding machinery in the endocytic pathway. J. Biochem. 153, 497–504. doi: 10.1093/jb/mvt028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vaden, J. H., Bhattacharyya, B. J., Chen, P. C., Watson, J. A., Marshall, A. G., Phillips, S. E.,et al. (2015). Ubiquitin-specific protease 14 regulates c-Jun N-terminal kinase signaling at the neuromuscular junction. Mol. Neurodegener. 10:3. doi: 10.1186/1750-1326-10-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Walters, B. J., Hallengren, J. J., Theile, C. S., Ploegh, H. L., Wilson, S. M., and Dobrunz, L. E. (2014). A catalytic independent function of the deubiquitinating enzyme USP14 regulates hippocampal synaptic short-term plasticity and vesicle number. J. Physiol. 592, 571–586. doi: 10.1113/jphysiol.2013.266015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wiborg, O., Pedersen, M. S., Wind, A., Berglund, L. E., Marcker, K. A., and Vuust, J. (1985). The human ubiquitin multigene family: some genes contain multiple directly repeated ubiquitin coding sequences. EMBO J. 4, 755–759.
Yang, W. L., Wu, C. Y., Wu, J., and Lin, H. K. (2010). Regulation of Akt signaling activation by ubiquitination. Cell Cycle 9, 487–497. doi: 10.4161/cc.9.3.10508
Zhou, A. Y., Shen, R. R., Kim, E., Lock, Y. J., Xu, M., Chen, Z. J.,et al. (2013). IKKepsilon-mediated tumorigenesis requires K63-linked polyubiquitination by a cIAP1/cIAP2/TRAF2 E3 ubiquitin ligase complex. Cell Rep. 3, 724–733. doi: 10.1016/j.celrep.2013.01.031
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: neuromuscular junction, ubiquitin, USP14, pJNK, proteasomes, motor neuron and deubiquitinating enzyme
Citation: Vaden JH, Watson JA, Howard AD, Chen P-C, Wilson JA and Wilson SM (2015) Distinct effects of ubiquitin overexpression on NMJ structure and motor performance in mice expressing catalytically inactive USP14. Front. Mol. Neurosci. 8:11. doi: 10.3389/fnmol.2015.00011
Received: 17 February 2015; Accepted: 06 April 2015;
Published online: 23 April 2015
Edited by:
Ashok Hegde, Wake Forest School of Medicine, USAReviewed by:
Izhak Michaelevski, Tel Aviv University, IsraelCopyright © 2015 Vaden, Watson, Howard, Chen, Wilson and Wilson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Scott M. Wilson, Evelyn F. McKnight Brain Institute, Department of Neurobiology and Civitan International Research Center, University of Alabama at Birmingham, 1825 University Boulevard, Shelby 914, Birmingham, AL 35294, USAbGl2dnkwMUB1YWIuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.