 Francesca Fontana1†
Francesca Fontana1† Michela A. Denti
Michela A. Denti- 1Laboratory of RNA Biology and Biotechnology, Centre for Integrative Biology, University of Trento, Trento, Italy
- 2CNR, Institute of Neuroscience, Padua, Italy
Frontotemporal dementia (FTD) is a neurodegenerative disorder characterized by degeneration of the fronto temporal lobes and abnormal protein inclusions. It exhibits a broad clinicopathological spectrum and has been linked to mutations in seven different genes. We will provide a picture, which connects the products of these genes, albeit diverse in nature and function, in a network. Despite the paucity of information available for some of these genes, we believe that RNA processing and post-transcriptional regulation of gene expression might constitute a common theme in the network. Recent studies have unraveled the role of mutations affecting the functions of RNA binding proteins and regulation of microRNAs. This review will combine all the recent findings on genes involved in the pathogenesis of FTD, highlighting the importance of a common network of interactions in order to study and decipher the heterogeneous clinical manifestations associated with FTD. This approach could be helpful for the research of potential therapeutic strategies.
Frontotemporal Dementia
Despite 90% of the human genome being transcribed to RNA, only 1.2% of genomic sequence is protein-coding, indicating that a huge proportion of non-coding RNAs (ncRNAs) are likely to participate in a number of physiological processes in cell types, including neurons (Lander et al., 2001; Birney et al., 2007; Wilhelm et al., 2008; Clark et al., 2011). The transcribed precursors of messenger RNAs (pre-mRNA) undergo splicing, such that the non-coding introns are removed and exons are combined variably to produce an RNA that would code for protein (Pandit et al., 2008). The pre-mRNAs undergoes alternative splicing producing mature messenger RNAs (mRNAs) which are then expressed in specific tissues and cell types in different stages of development. These mRNAs then associate with the ribosomal machinery to be translated into proteins in the cytoplasm. Non-coding RNAs (among which microRNAs and long non-coding RNAs), might regulate the translation of specific mRNAs, thereby representing a post-transcriptional mechanism exerting a fine-tuned control in the production of specific proteins.
microRNAs (miRNAs) are a group of small non-coding RNAs of 21–22 nt with important regulatory roles on the post-transcriptional expression of target mRNAs (Bartel, 2009; Ghildiyal and Zamore, 2009). MiRNAs are generating from longer transcripts of different lengths called primary transcripts (pri-miRNAs), usually transcribed by RNA polymerase II, from intragenic or intergenic DNA regions (Lee et al., 2004; Garzon et al., 2010). The pri-miRNAs are processed in the nucleus by the micro-processor complex, formed by an RNase III enzyme, Drosha, and its cofactor DiGeorge syndrome critical region in gene eight termed (DGCR8) (Lee et al., 2003). The process lead to the production of small hairpin structure of 70–100 nt called precursor miRNAs (pre-miRNAs). Pre-miRNAs are exported to the cytoplasm through Exportin 5 (Kim, 2004), where they are further processed by an RNase III nuclease, Dicer to produce RNA duplex (Bernstein et al., 2001; Grishok et al., 2001; Hutvagner et al., 2001). One strand is loaded on the RNA-Induced Silencing Complex (RISC) and associated with Argonaute-2 (Ago2) to interact with the target mRNA. The miRNA-RISC complex induces mRNA downregulation through two different ways: mRNA cleavage in case of perfect complementarity between miRNA and target mRNA or translation inhibition if there is an imperfect binding (Wahid et al., 2010) (Figure 1). In case of perfect complementarity, Ago2 is the protein involved in the cleavage of the target mRNA in humans (Liu et al., 2004). However, in animals, translational repression is the most frequent way of action for miRNAs (Huntzinger and Izaurralde, 2011; Pasquinelli, 2012), although the exact process is still unknown since is not clear if the repression occur at the initiation step or during the translation process (Wahid et al., 2010). Even the mechanisms for target regulation played by miRNAs are still unclear, the target mRNA could be repressed by the promotion of deadenylation, sequestration of miRNAs and target by stress granules and P-Bodies (Valencia-Sanchez et al., 2006), disruption of translation initiation or protein degradation caused by RISC after translation (Tang et al., 2008).
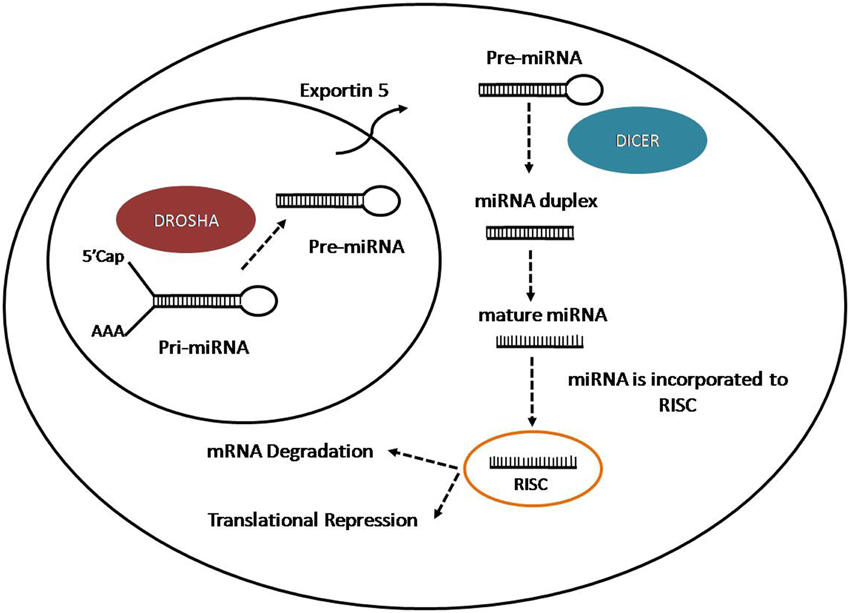
Figure 1. Representation of miRNA biogenesis.
The enormous content of non-coding RNA (ncRNA) in the cell intrigues its role and function in the cells. LncRNAs are defined as transcripts longer than 200 nucleotides and lacking an appreciable open reading frame (usually less than 100 amino acids). They may be transcribed by RNA polymerase II (RNA Pol II) or RNA Pol III, and may undergo splicing or comprise of a single exon. In contrast to small ncRNAs, lncRNAs tend to be poorly conserved evolutionarily and regulate gene expression by diverse mechanisms that are not entirely understood. As a functionally diverse macromolecule, the biological roles of lncRNAs cannot be determined solely from their nucleotide sequence, secondary structures, or genomic locations (Ng et al., 2013).
Recent work has begun to elucidate the roles of some lncRNAs, such as architectural function in nuclear paraspeckles (Sunwoo et al., 2009; Souquere et al., 2010), transcriptional co-regulators (Feng et al., 2006; Bond et al., 2009), and as endogenous competing RNAs (ceRNAs) (Cesana et al., 2011; Tay et al., 2011). LncRNA expression is abundant in cells of the CNS (Mehler and Mattick, 2007; Mercer et al., 2008) and recent studies have suggested that lncRNAs play crucial roles in spatial-temporal control of gene expression in brain development (Mercer et al., 2008). They have also known to be involved in brain development, neural differentiation and maintenance, synaptic plasticity, cognitive function and memory, and in aging and neurodegenerative disorders (Wu et al., 2013b).
Though different mechanisms may play a role in causing neurodegenerative disorders, recent studies show increasing evidence of abnormalties in RNA processes, highlighting the possible putative role of RNA in neurodegeneration. An mRNA gain-of-toxic-function has been proposed for some neurodegenerative diseases (Osborne and Thornton, 2006; O'Rourke and Swanson, 2009; Todd and Paulson, 2010) whereas other neurodegenerative disorders are driven through altered or lost non-coding RNA, RNA splicing and RNA binding activities (Gallo et al., 2005; Cooper et al., 2009; Lagier-Tourenne et al., 2010).
Fronto temporal lobar degeneration (FTLD) is the most common cause of dementia after Alzheimer's disease. The clinicopathological spectrum of FTLD includes frontal and temporal variants of frontotemporal dementia (FTD), primary progressive aphasia, semantic dementia, Cortico-basal degeneration (CBD), progressive supranuclear palsy (PSP), progressive subcortical gliosis (PSG) and FTD with motor neuron disease (FTD–MND) (Bugiani, 2007). Moreover, despite Amyotrophic Lateral Sclerosis (ALS) and FTD being two different neurodegenerative disorders, they often share genetic, neuropathological and clinical characteristics; therefore they are considered part of the same spectrum of diseases (Ling et al., 2013). Frontotemporal dementia symptoms can also be present along with disabling muscle weakness and osteolytic bone lesions, in IBMPFD1 (Inclusion body myopathy with early-onset Paget disease with or without Frontotemporal dementia 1).
It is estimated that one in seven people in the US might develop a neurodegenerative disorder in their lifetime, with dementia being one of the leading causes of death in US (Thies and Bleiler, 2011). Though this broad spectrum of disorders has been studied based on protein aggregation and research has been focusing on protein functions and alterations, emerging avenues in research unravels the role of RNA and RNA processing in contributing to neurodegeneration (Belzil et al., 2013).
To date, FTD has been linked to mutations in seven different genes (TARDBP, FUS, MAPT, GRN, VCP, CHMP2B, C9ORF72).
Findings that showed the presence of ubiquitinated protein TDP-43 in sporadic cases of ALS with FTD further linked these two diseases (Arai et al., 2006; Neumann et al., 2006). Following these findings, mutations in the gene coding for the RNA binding protein TDP-43 were discovered in ALS cases (Kabashi et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008) and FTD cases (Borroni et al., 2009; Kovacs et al., 2009).
With the broadening knowledge on the impact of impaired RNA binding proteins in mediating the disease process, mutations in the fused in sarcoma/translocated in liposarcoma (FUS/TLS) gene were found to account for an additional 5% of familial ALS and few rare cases of FTD (Kwiatkowski et al., 2009; Vance et al., 2009).
TDP-43 and FUS share similar structural and functional properties with a likely role in multiple steps of RNA processing and they are both linked to RNA metabolism. The pathological accumulation of these proteins is observed in over 90% ALS and 50% FTD patients. These studies also highlight that errors in RNA processing might be enough to initiate the disease process.
MAPT mutations were observed in several FTD families with abnormally phosphorylated tau proteins being isolated from neuroectoderm cells of patients. Mutations present in the C terminal repeat domains lead to the inhability of abnormal tau protein to bind microtubules, thus leading to its instability and accumulation and causing neuronal degeneration (Bugiani, 2007). FTD with tau inclusions was characterized as a tauopathy and dubbed FTLD-tau.
However, a different class of patients were found to have had accumulated ubiquitin and ubiquitin-associated proteins (FTLD-U). Co-localization of abnormal proteins with ubiquitin in the nucleus and perikaryon of neuronal cells, indicated the involvement of proteasome dysfunction in the pathology. Analysis of significant genes on chromosome 17, close to the MAPT locus, led to the discovery of mutations in GRN (Baker et al., 2006; Cruts et al., 2006). GRN is known to be involved in the cell cycle control and motility.
Studies on an ALS/FTD-affected Scandinavian family (Morita et al., 2006) and on IBMPFD1 families suggested the possible role of mutations in chromosome 9 in FTD. The disorder was associated to mutations in VCP, encoding the valosin-containing protein essential for ubiquitin-mediated protein degradation (Watts et al., 2004; Johnson et al., 2010).
Other FTLD mutations are located on chromosome 3 (FTD-3), in the CHMP2B gene, which encodes for a protein involved in degradation of surface receptor proteins and formation of endocytic multivesicular bodies (Skibinski et al., 2005).
Another link between ALS and FTD are the large intronic hexanucleotide repeat expansions in the C9ORF72 gene located on chromosome 9 found in ALS, FTD, or ALS/FTD cases (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Gijselinck et al., 2012).
This review will focus on the single genes known to have implications in FTD and their altered functions in the diseased state. The ultimate aim is to explorepossible functional connections between these seven diverse genes and describe a network in which a possible common thread might be represented through RNA mediated processes.
TARDBP (TDP 43)
Human TDP-43 was discovered in 1995 in a screen for transcriptional repressors of the trans-active response (TAR) DNA binding element of the HIV-1 virus, and thus the gene is named TARDNA Binding Protein (TARDBP) (Ou et al., 1995). TARDBP is composed of six exons and maps on chromosome 1p36.22.
The protein TARDBP produces is being labeled as TDP-43 due to its molecular weight of 43 KDa (Neumann et al., 2006). TARDBP is ubiquitously expressed in various human tissues (Table 1) including brain and spinal cord (Wang et al., 2008a). To date, 34 different TDP-43 mutations have been discovered in 131 different FTD and ALS families (Cruts et al., 2012). Pathogenic mutations observed in TDP-43 are highlighted in Table 2.
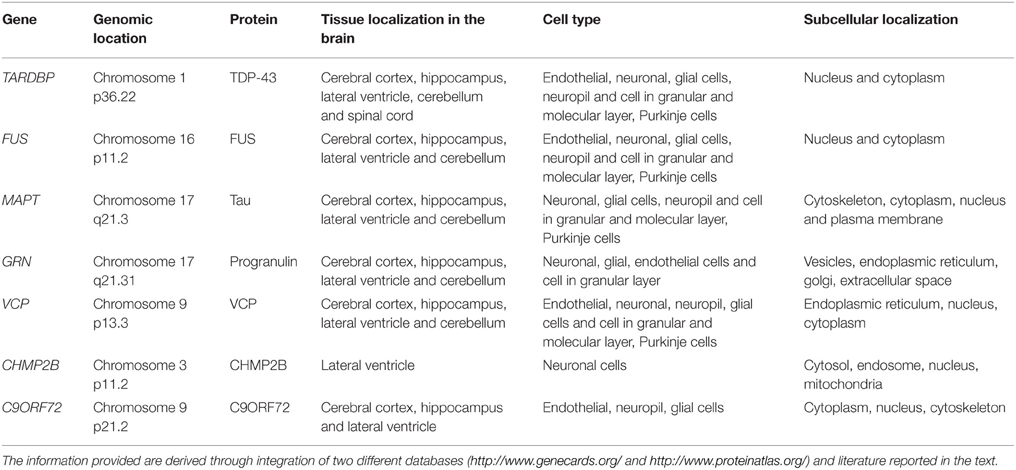
Table 1. Protein localisation of different genes associated to FTD.
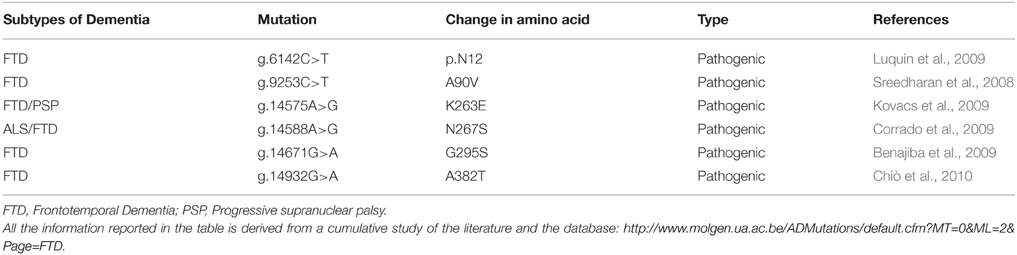
Table 2. List of mutations in TARDBP and their characteristic phenotypes.
Structure
TDP-43 is a 414 amino acids protein (Figure 2A) containing two RNA recognition motifs (RRMs), a glycine-rich low sequence complexity prion-like domain (Wang et al., 2013). A nuclear localization signal motif (NLS) and a nuclear export signal motif (NES) allow TDP-43 to shuttle between the nucleus and the cytosol (Buratti and Baralle, 2001).
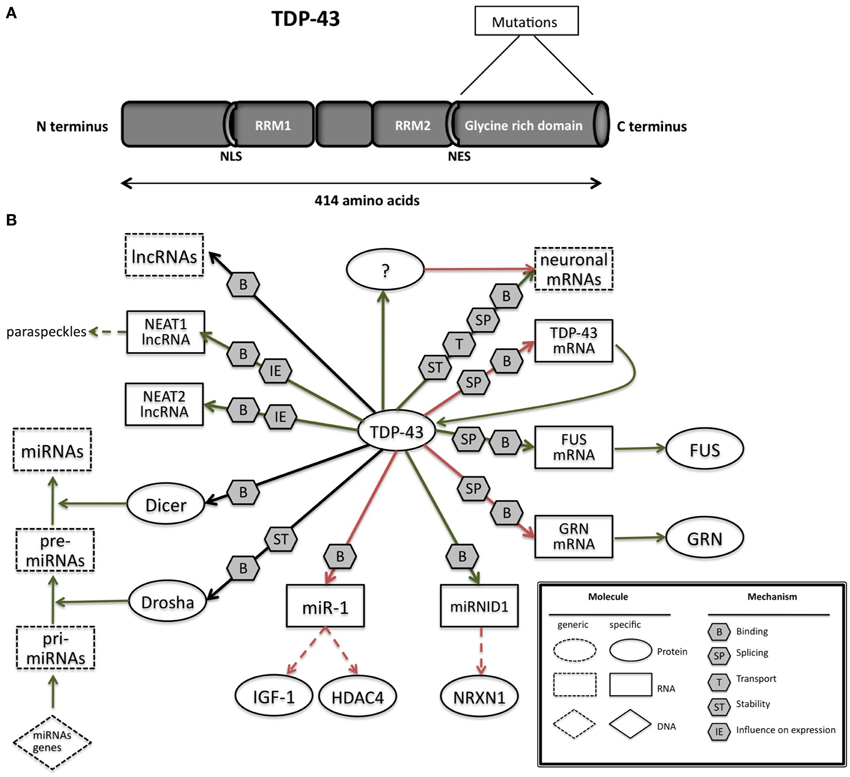
Figure 2. (A) TAR DNA-binding protein 43 (TDP-43) contains two RNA-recognition motifs (RRM1 and RRM2), a carboxy-terminal glycine-rich domain, a bipartite nuclear localization signal (NLS), and a nuclear export signal (NES). Mutations are mainly located in the glycine-rich domain. (B) The network of interactions of TDP-43 with proteins and RNAs. Green arrows indicate binding interactions or processes that result in activation or increased expression. Red arrows indicate binding interactions or processes that result in inhibition of activity or reduced expression. Black arrows indicate binding interactions or processes whose result can be either positive or negative. Dashed arrows indicated indirect processes. Symbols as in Legend. lncRNAs, long non-coding RNAs; IGF-1, insulin-like growth factor 1; HDAC4, histone deacetylase 4; NRXN1, neurexin 1; TDP-43, TAR DNA binding protein; FUS, fused in sarcoma; GRN, progranulin; NEAT1, nuclear-enriched autosomal transcript 1; NEAT2, nuclear-enriched autosomal transcript 1.
Localization and Function
Though TDP-43 expression is seen in the nucleus with low cytosolic localization (Ayala et al., 2005), there is a significant cytosolic TDP-43 expression especially in large motor neurons where TDP-43 has an additional role in mRNA transport as a neuronal activity responsive factor in dendrites thus promoting dendritic branching (Wang et al., 2008a; Barmada et al., 2010; Kabashi et al., 2010).
TDP-43 was found to be accumulated in cytoplasmic stress granules due to oxidative stress (Colombrita et al., 2009). Stress granules are aggregations, formed after cell insults such as oxidative stress or heat shock that temporarily store non-translating mRNAs, small ribosome subunits, RNA-binding proteins and translation initiation factors (Buchan and Parker, 2009). Formation of stress granules protects the cells, allowing a translational block and initiation of repair processes (Anderson and Kedersha, 2008).
Upregulation of nuclear TDP-43 has also been shown to provide protection to primary neurons against glutamate induced excitotoxicity (Zheng et al., 2012). These findings also suggest that TDP-43 regulates synaptic plasticity by governing the transport and splicing of synaptic mRNAs. In a recent review, Belzil and co-authors postulate that altered TDP-43 could lead to impaired hippocampal plasticity and render neurons more vulnerable to cellular stressors (Belzil et al., 2013).
TDP-43 is highly conserved from human to C. elegans, both in the RNA binding motifs and in the carboxy-terminal portion (Ayala et al., 2005). In situ hybridization studies showed that TDP-43 is expressed very early in the brain and spinal cord of zebrafish (Shankaran et al., 2008) suggesting that it plays an important role in nervous system development.
Implications of RNA in Pathogenesis
Many studies have linked TDP-43 to neurodegenerative disorders, including ALS and FTLD (Neumann et al., 2006; Lagier-Tourenne et al., 2010; Lee et al., 2012). Janssens and Van Broeckhoven (2013) have highlighted the increasing evidence of role of impaired RNA metabolism in TDP-43-driven neurodegeneration.
TARDBP primary transcript undergoes alternative splicing to produce eleven different mRNAs including the one encoding TDP-43. Seven of these are shorter transcripts which are generated through the seven different splicing reactions within exon 6 of TARDBP pre-mRNA using a combination of four different 5′ donor sites and four different 3′ acceptor sites (Wang et al., 2004a).
In few ALS cases a smaller TDP-43 isoform (~28 KDa) was observed additionally to the 43 kDa isoform, lacking exon 3 and a significant portion of exon 6-encoded amino acids (Strong et al., 2007). This smaller isoform lacks the carboxy-terminal portion of the protein and is thought to be associated with disease pathology (Neumann et al., 2006).
Converging lines of evidence in research suggest that TDP-43 regulates RNA in various ways (Figure 2B; Lee et al., 2012). The RRM1 domain of TDP-43 is critical for its binding to single-stranded RNA (Ou et al., 1995; Buratti and Baralle, 2001; Wang et al., 2004a; Ayala et al., 2005). TDP-43 preferentially binds UG repeats, but is also found to be associated with non-UG repeat sequences (Buratti and Baralle, 2001; Ayala et al., 2005; Polymenidou et al., 2011; Tollervey et al., 2011).
Pathological TDP-43 aggregates are ubiquitinated and phosphorylated. Under normal conditions, these forms are not readily detectable in brain tissues, thus making them disease-specific. Over-expression of full-length TDP-43 in a variety of transgenic animal models lead to the presence of phosphorylated TDP-43 aggregates similar to ALS and FTD cases (Wegorzewska et al., 2009; Shan et al., 2010; Stallings et al., 2010; Wils et al., 2010; Xu et al., 2010a). The phosphorylated form has a longer half-life than the non-phosphorylated form thus leading to accumulation of phosphorylated proteins. Despite the progress toward describing the full spectrum of TDP-43 pathology in human neurodegenerative diseases, the fundamental question of whether TDP-43 dysfunction mediates neuro-degeneration through gain of toxic function or a loss of normal function remains unanswered (Lee et al., 2012).
Upon depletion of TDP-43 from adult mouse brain with antisense oligonucleotides, levels of 601 mRNAs, including FUS, GRN and other transcripts involved in neurodegeneration, were altered, along with 965 varied splicing events. RNAs depleted by the reduction of TDP-43 were coded by genes with long introns (Polymenidou et al., 2011).
In-vivo searches for TDP-43 RNA targets in mouse (Polymenidou et al., 2011), human brain (Tollervey et al., 2011), rat cortical neurons (Sephton et al., 2011), a mouse NSC-34 cell line (Colombrita et al., 2012), and a human neuroblastoma cell line (Xiao et al., 2011) revealed that there are more than 6000 RNA targets which constitutes to about 30% of total transcriptome. TDP-43 was found to preferentially bind to introns (including deep intronic sites), 3′ untranslated regions (UTRs), and non-coding RNAs (Polymenidou et al., 2011; Tollervey et al., 2011), indicating a multifaceted role in RNA maturation. TDP-43 can influence splice site selection by binding to exon-intron junctions and intronic regions, mRNA stability and transport by binding on 3′UTRs. A substantial amount of mRNAs regulated by TDP-43 at splicing levels were involved in neuronal development or in neurological diseases (Tollervey et al., 2011). Additional data show that when TDP-43 is reduced the levels of several other mRNAs increase. As the affected mRNAs include more than 300 mRNAs without TDP-43 binding sites, these observation point toward an indirect mechanism (Polymenidou et al., 2011) of modulation.
i-CLIP experiments have also shown that TDP-43 binds to long ncRNAs (lncRNAs), including nuclear-enriched autosomal transcript 1 (NEAT1) and metastasis-associated lung adenocarcinoma transcript 1 (MALAT1, also called NEAT 2) (Tollervey et al., 2011). Expression of both lncRNAs is elevated in FTD patients with TDP-43 inclusions, thus correlating with their increased association with TDP-43 (Tollervey et al., 2011).
The binding of TDP-43 to small (<200 base) ncRNAs and miRNAs remains largely unexplored. However, the association of TDP-43 with Drosha microprocessor (Ling et al., 2010) and Dicer complexes (Freibaum et al., 2011; Kawahara and Mieda-Sato, 2012) provides a suggestive role of TDP-43 involvement in miRNA biogenesis. Indeed, let-7b miRNA is downregulated, whereas miR-663 is upregulated upon reduction of TDP-43 (Buratti et al., 2010). Di Carlo and collegues demonstrated that TDP-43 directly interacts with Drosha and controls its stability at different levels. Moreover, TDP-43 is also involved in the Drosha substrate recognition as in the regulation mediated by Drosha of Neurogenin 2, an important and master gene in neurogenesis (Di Carlo et al., 2013).
Fan et al. (2014) have performed CLIP-seq analysis to examine the small RNAs (pri-miRNAs, miRNAs, and piRNAs) bound to TDP-43 and found that a novel miRNA (miR-NID1), which is processed from the intron five of human neurexin 1 gene (NRXN1), interacts with TDP-43 and represses expression of NRXN1. Neurexins are cellular proteins that function as cell adhesion molecules and receptors in the vertebrate nervous system, involved in synaptic development including calcium signaling, heterogeneous cell-to-cell adhesion and synaptogenesis (Craig et al., 2006; Bottos et al., 2011) Disruptions or mutations of NRXN1 have been reported to associate with autistic spectrum disorder (ASD), mental retardation, and schizophrenia (Reichelt et al., 2012).
Recent studies by King and colleagues identified a physical interaction between TDP-43 and miR-1 family which is known to be involved in smooth muscle gene repression in heart and an opposing myogenic differentiation (King et al., 2014). TDP-43 overexpression in skeletal muscle led to decrease of miR-1 and increased protein levels of the miR-1 family targets, IGF-1 and HDAC4. These results demonstrate that TDP-43 could influence miRNA regulation through a physical interaction by limiting their bioavailability for RISC loading and offer a mechanism by which mature miRNAs can be differentially regulated.
The expression of TDP-43 is tightly autoregulated through a complex interplay between transcription, splicing, and 3′ end processing (Avendaño-Vázquez et al., 2012): TDP-43 over-expression in humans and mice leads to activation of a 3′ UTR intron which results in excision of proximal polyA site (PAS) which in turn activates a cryptic PAS and prevents TDP-43 expression through a nuclear retention mechanism.
The above mentioned studies have highlighted that TDP-43 is linked to various mRNAs and non-coding RNAs, in a neuronal context wherein it mediates effects through splicing or interaction with Drosha and Dicer complexes. It is also involved in its autoregulation mediated at the RNA level.
Additionally, TDP 43 is known to interact with MATR3, a DNA RNA binding protein. Their interaction was confirmed to be RNA based. Mutations in this gene have been linked to cases of ALS. The authors further report that the phenotype observed in patients with MATR3 was a combination of those observed in cases of ALS and myopathy. Clinical symptoms were similar to patients with VCP mutations (Johnson et al., 2014).
FUS
FUS, (fused in sarcoma, also called TLS: translocated in liposarcoma) belongs to the TET family of RNA binding proteins involved in many different cellular processes (Bertolotti et al., 1996; Law et al., 2006; Tan and Manley, 2009). FUS, located on chromosome 16 at locus p11.2, encodes a multifunctional protein able to bind and interact with single stranded RNA and double stranded DNA, participating in different aspects of RNA metabolism (Shelkovnikova et al., 2014).
Structure
FUS is characterized by different domains (Figure 3A): a N-terminal domain with transcriptional activating properties mainly composed of glutamine, glycine, serine, and tyrosine residues (Law et al., 2006), a glycine rich region, a RNA binding domain, and a highly conserved C-terminus capable of binding DNA, RNA and splicing factors (Law et al., 2006).
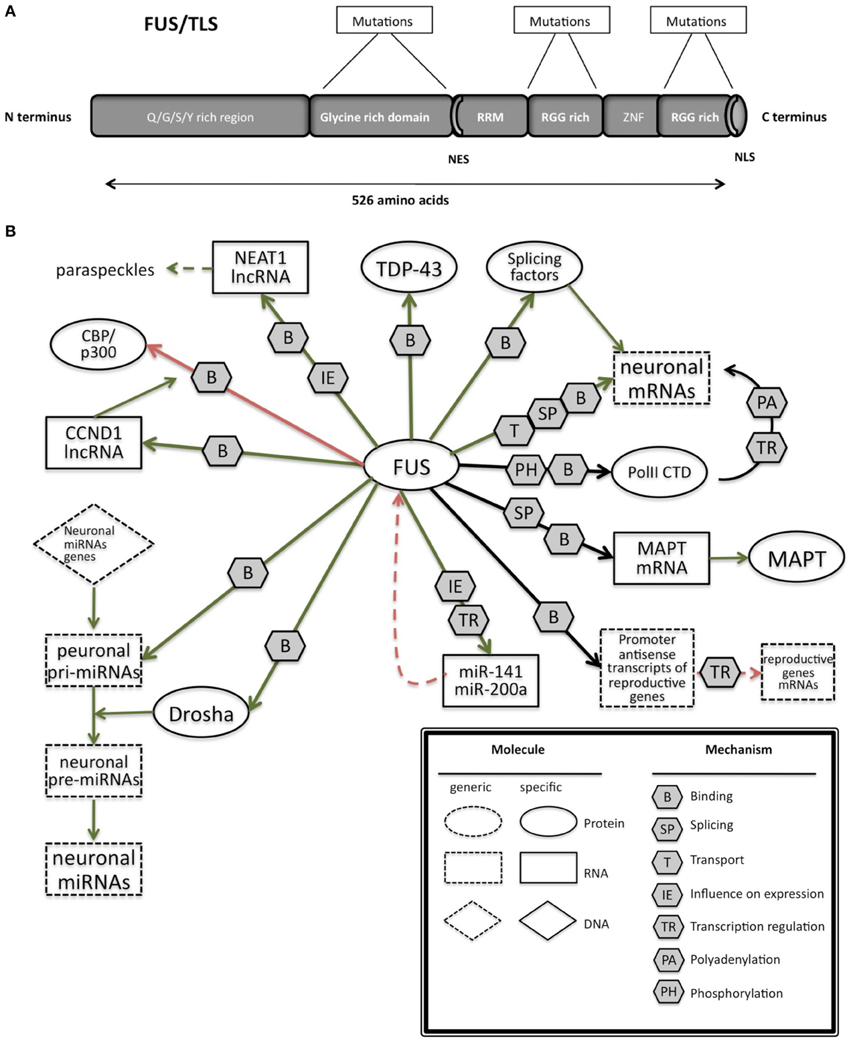
Figure 3. (A) Schematic representation of the functional domains in FUS/TLS. FUS contains a N-terminal part enriched in glutamine, glycine, serine and tyrosine residues (QGSY region), a glycine-rich region, a nuclear export signal (NES), an RNA recognition motif (RRM), repeats of arginine, glycine, glycine (RGG), a zinc finger motif (ZNF), and a C-terminal nuclear localization signal (NLS). Most of the mutation are localized in the glycine rich region and in the last 17 amino acids of the NLS part. (B) The network of interactions of FUS with proteins and RNAs. Green arrows indicate binding interactions or processes that result in activation or increased expression. Red arrows indicate binding interactions or processes that result in inhibition of activity or reduced expression. Black arrows indicate binding interactions or processes whose result can be either positive or negative. Dashed arrows indicated indirect processes. Symbols as in Legend. lncRNAs, long non-coding RNAs; IGF-1, insulin-like growth factor 1; HDAC4, histone deacetylase 4; NRXN1, neurexin 1; TDP-43, TAR DNA binding protein; FUS, fused in sarcoma; MAPT, microtubule-associated protein tau; NEAT1, nuclear-enriched autosomal transcript 1; CCND1, G1/S-specific cyclin-D1; CBP, CREB-binding protein; p300, Histone acetyltransferase p300; PolII CTD, Carboxy-terminal Domain of the RNA polymerase II.
Localization and Function
FUS is mainly localized in the nucleus (Colombrita et al., 2009; Van Blitterswijk and Landers, 2010; Kawahara and Mieda-Sato, 2012) but it is also actively implicated in other cellular processes that occur in the cytoplasm such as mRNA transport, mRNA stability and translation (Buratti and Baralle, 2010; Colombrita et al., 2011). Indeed FUS was reported to shuttle between the nucleus and the cytoplasm, exporting to the cytoplasm spliced mRNAs in ribonucleoprotein complexes (Zinszner et al., 1997). Particularly, upon stimulation in hippocampal neurons FUS was reported to accumulate in the spines of mature dendrites, where local translation occurred (Fujii and Takumi, 2005). FUS immunoreactivity was also observed in dendritic spines in mature primary cultures and in adult hippocampus in situ (Belly et al., 2005; Table 1).
The C-terminal part of FUS encodes for a non-classic nuclear localization signal (Figure 3A; Iko et al., 2004) that is necessary for nuclear import, as it was demonstrated through the generation of deletion mutant lacking 13 amino acids in the C-terminal part of FUS(Dormann et al., 2010).
Several papers reported that mutations and aberrations of FUS are linked to the pathogenesis of frontotemporal degeneration (FTD) as well as familial and sporadic ALS (Kwiatkowski et al., 2009; Vance et al., 2009), as reported in Table 3. Moreover, FUS accumulates in inclusions in the cytoplasm of autopsied spinal cords and brains of sporadic and familial ALS and FTD. FUS inclusions are not only observed in presence of FUS mutations, as they were found in patients with different or unknown genetic defects such as sporadic ALS, ALS/dementia or FTLD (with or without progranulin mutations), FUS or TDP43 mutation-linked familial ALS, SOD1-negative familial ALS. These inclusions were also positive for TDP43/ubiquitin and p62 (Deng et al., 2010).
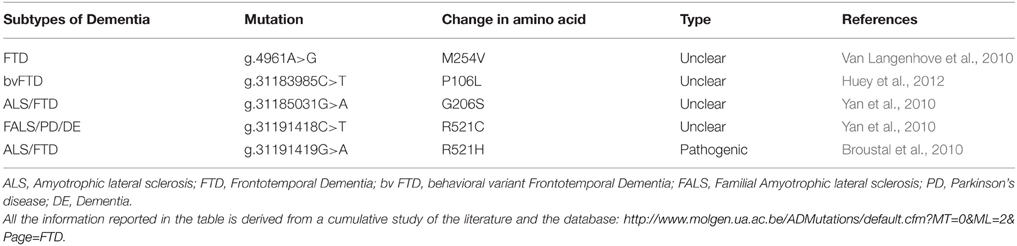
Table 3. List of mutations in FUS and their characteristic phenotypes.
ALS/FTD patients show mutations mainly in the Glycine rich region and C-terminal part (Lagier-Tourenne et al., 2010). The mechanism underlying the pathogenesis of FUS mutations was related to FUS nucleus/cytoplasmic imbalance since ALS mutations increase its localization in the cytoplasm, observed through immunostaining of FUS in postmortem ALS brain samples (Kwiatkowski et al., 2009), or through the analysis in neuroblastoma cell lines of the subcellular localization of recombinant mutant FUS fused either to green fluorescent protein (GFP) (Kwiatkowski et al., 2009; Morlando et al., 2012), an N-terminal hemagglutinin (HA) tag (Vance et al., 2009), a C-terminal V5-His tag, or an N-terminal myc tag in HeLa (Ito et al., 2011).
Both the loss of FUS nuclear function and the potential gain of toxic effect by FUS in the cytoplasm could explain pathogenesis (Shelkovnikova et al., 2014).
Very few studies so far reported FTD cases associated with FUS mutations. The first analysis of FUS in FTD patients showed a novel missense mutation in the glycine-rich region of FUS, predicted to be pathogenic by in silico analysis (Van Langenhove et al., 2010). Subsequently another study found novel missense mutations in patients with familial ALS with features of frontotemporal dementia (FALS/FTD) and one with familial ALS with parkinsonism and dementia (FALS/PD/DE) (Yan et al., 2010). Recently, another study found two novel heterozygous missense mutations in FUS in patients with behavioral variant FTD (bvFTD), however the pathogenicity of these mutations needs to be further investigated in other screening (Huey et al., 2012).
FUS has been reported to co-localize with TDP-43 in nuclear complexes (Kim et al., 2010b; Ling et al., 2010) and in larger cytoplasmic complexes (Kim et al., 2010b). Purified FUS has also been reported to interact with purified His-tagged TDP-43 in vitro in an RNA-independent manner, associated to the C-terminal region of TDP-43 (Kim et al., 2010b). These ubiquitously expressed binding proteins seem to have similar and complementary functions.
Only the mutant form of FUS was found in stress granules in reponse to translational arrest (Bosco et al., 2010). FUS and TDP-43 were observed to co-localize in cytoplasmic aggregations of ALS/FTLD-affected neurons (Da Cruz and Cleveland, 2011). Dormann and colleagues found stress granule markers such as PABP-1 and eIF4G co-deposited with FUS inclusions in sections of post-mortem brain and spinal cord tissue from familial ALS-FUS and sporadic FTDLD-U. On the contrary, TDP inclusions did not show any co-localization with stress granules proteins in HeLa transiently transfected with the mutated form of FUS, after heat shock for 1 h (Dormann et al., 2010). Another study reported that ubiquitin-positive inclusions in frozen post-mortem tissue from FTLD-TDP patients were not stained with anti-FUS antibodies (Neumann et al., 2009b), therefore FUS and TDP-43 are not always found in the same inclusions or aggregates.
The relation between FUS and TDP-43 is reported as a delicate equilibrium, where small alteration on their relative quantity and presence in nucleus/cytoplasm could very likely cause serious problem over a long period (Colombrita et al., 2012), which might be an accumulation of events due to an alteration of their targetome.
Implications of RNA in Pathogenesis
FUS is involved in pre-mRNA splicing (Figure 3B), by interacting with splicing factors such as SRm160, PTB, and serine/arginine rich proteins (SR proteins) (Yang et al., 1998; Meissner et al., 2003). In addition the recent sequencing approaches applied to clarify the function and identify the targets of FUS reinforced its fundamental role in splicing (Colombrita et al., 2012) by revealing its binding to intronic sequences or to splice site acceptors.
Similarly to many other splicing factors, FUS can bind the C-terminal domain of RNA polymerase II and prevent the premature hyperphosphorylation of Ser2 in the C-terminal domain of RNA polymerase II. Moreover the lack of FUS leads to an accumulation of RNA polymerase II at the transcription start site with a shift toward abundance of mRNA isoforms with early polyadenylation (Schwartz et al., 2012).
FUS can bind to the promoter antisense strand transcript of some genes such as Ptprn2, Xrn1, Gak, or Glt1d1 and this interaction downregulates the transcription of the coding sense strand, but this effect seems to be specific for some genes enriched with GO terms connected to the reproductive process(Ishigaki et al., 2012).
As FUS was shown to regulate RNA polymerase II at many more gene promoters than the genes reported for splicing defects, its role on transcription could be a separated function in addition to the regulation on splicing (Schwartz et al., 2012). However, a small proportion of FUS target regions is localized in exonic sequences and in the 3′UTRs (Hoell et al., 2011), suggesting another potential role, such as the transport of mRNAs or the control of mRNA stability and translation (Fujii et al., 2005; Fujii and Takumi, 2005). A model was suggested, in which FUS is released from actin filaments, when cytoskeletal organization collapses, becoming free to be linked to the mRNA that is transported to the local translational machinery in the spines (Fujii and Takumi, 2005).
Recent techniques, like HITS-CLIP or RIP-CHIP were also used to identify FUS binding motif, but all the studies lead to the common assumption that FUS binds to specific secondary structures on its RNA targets and a primary sequences analysis is not sufficient (Colombrita et al., 2012; Ishigaki et al., 2012).
Interestingly, silencing of FUS was reported to alter splicing events in genes, such as MAPT, that have an important neuronal function (Ishigaki et al., 2012). This finding leads an unexpected connection between these two genes, both involved in the pathogenesis of FTD. In particular, FUS was shown to help the skipping of MAPT exon 10 in primary cortical neurons (Ishigaki et al., 2012). The alternative splicing of MAPT exon 10 is known to have a causative role in FTD as discussed later (MAPT paragraph).
FUS is also involved in microRNA biogenesis (Morlando et al., 2012), specifically interacting with pri-miRNAs and Drosha, and helping the recruitment of Drosha for the correct miRNA processing in neuronal cells. Several miRNAs like miR-9, miR-132, and miR-125b whose biogenesis is controlled by FUSare important for neuronal functions, neuronal differentiation, and synaptogenesis (Morlando et al., 2012). Additionally miR-9 and miR-132 have also been shown to control neurite extension and branching through downregulation of Foxp2 (Forkhead box protein P2) (Clovis et al., 2012) Moreover this role of FUS seems to be prominent in neuronal cells compared to non-neuronal cells, such as HeLa cells, in which the proportion of miRNAs affected by FUS knockdown was lower. Indeed the mutations known to induce a cytoplasmic delocalization of FUS would impede its nuclear role as pri-miRNA processor. Though the balance of nuclear and cytoplasmic FUS seems necessary, the sole role of nuclear FUS should not be neglected and further investigations would be needed to clarify its biological function within this cell compartment. Recently, the same laboratory demonstrated the presence of a regulatory loop in which FUS can enhance the expression of miR-141 and miR-200a, which in turn regulate FUS, through a binding on its 3′UTR. This pathway seems to be affected in the presence of one mutation found in two ALS patients (Dini Modigliani et al., 2014).
FUS is also reported to bind lncRNAs. The binding to lncRNA CCND1 induces an allosteric change in FUS, thus in turn permits its interaction with CBP/p300. As FUS represses CBP/p300-mediated transcription by inhibiting their histone acetyltransferase (HAT) functions (Wang et al., 2008a), in the presence of ncRNA CCDN1, CBP/p300-mediated transcription is repressed.
The nuclear-enriched abundant transcript 1 (NEAT1) produces two types of lncRNAs from the same promoter NEAT1_1 and NEAT1_2 (Nishimoto et al., 2013). FUS was shown to bind NEAT1_2, known to assemble and organize the core proteins of paraspeckles (Wang et al., 2008a; Hoell et al., 2011; Lagier-Tourenne et al., 2012), which represent a storage for the rapid release of RNAs during stress condition or a nuclear retention of long hyperedited transcripts (Prasanth et al., 2005; Chen and Carmichael, 2009). According to observations and data obtained from cultured cells, transgenic mice and human post-mortem tissue, paraspeckles represents an important protective cell mechanism during stress conditions (Nakagawa et al., 2011; Nakagawa and Hirose, 2012; Shelkovnikova et al., 2014).
Paraspeckels are present in almost all the cultured cells (Fox and Lamond, 2010), but in normal tissues are found only in cells that contain high levels of NEAT1_2 RNA and coherently, in neurons where NEAT1 is express at low levels, paraspeckles are not observed (Nakagawa et al., 2011).
The presence of FUS in paraspeckles was confirmed in different cell lines by three studies (Naganuma et al., 2012; Nishimoto et al., 2013; Shelkovnikova et al., 2014) Moreover, NEAT1 was shown through PAR-CLIP to be a target of both WT and mutant FUS (Hoell et al., 2011).
Paraspeckles are found in spinal motoneurons of patients at early stage of ALS. The possibility that aging induces an increase in the level of NEAT1_2 was ruled out due to the fact that human control cases were older that ALS cases of an average of 10 years. However, the process that induces an up-regulation of NEAT1_2 lncRNA during the early phases of ALS is still unknown (Nishimoto et al., 2013). Overall FUS seems to play a key role on the regulation of RNA at different levels, acting on transcription, splicing, transport, and stability of mRNA with a particular function in microRNA biogenesis and interaction with non-coding RNAs.
MAPT (Tau)
MAPT (microtubule associated protein) encodes for protein Tau and is located on chromosome 17q21.3. The gene, which is 150 kb-long, contains 16 exons, out of which 11 are expressed in CNS (Wolfe, 2012).
Structure
The protein consists of a projection domain, including an acidic and a proline-rich region, which interacts with cytoskeletal elements (Figure 4A). The N-terminal part is involved in signal transduction pathways by interacting with proteins such as PLC-γ and Src-kinases. The C-terminal part, referred to as the microtubule binding domain, regulates the rate of microtubules polymerization and is involved in binding with functional proteins such as protein phosphatase 2A (PP2A) or presenilin 1 (PS1) (Luna-Muñoz et al., 2013).
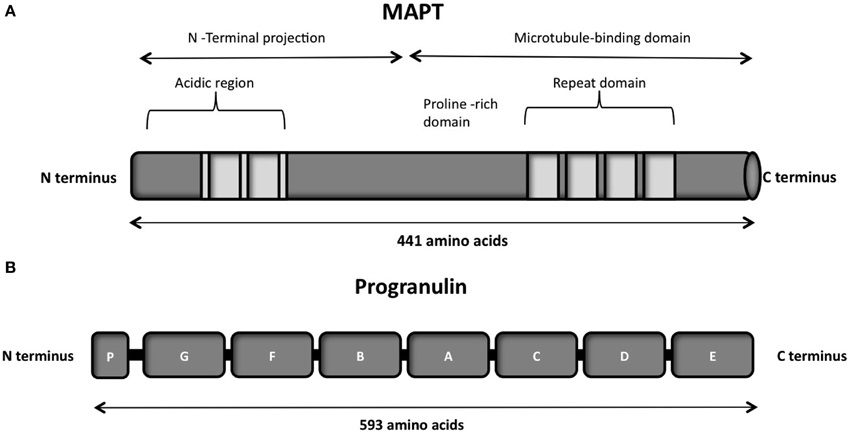
Figure 4. (A) Schematic representation of the functional domains of the largest tau isoform (441 amino acids 2N4R isoform). The N-terminal projection domain, including an acidic and a proline-rich region, interacts with cytoskeletal elements. The N-terminal part is also involved in signal transduction pathways by interacting with proteins such as PLC-γ and Src-kinases. The C-terminal part, also known as the microtubule-binding domain, regulates the rate of microtubules polymerization and is involved in binding with proteins such as protein phosphatase 2A (PP2A) or presenilin 1(PS1). (B) Schematic representation of the progranulin structure encoded by the major human transcript containing all the 13 exons. The lettered boxes on the progranulin scheme represent the individual granulin domains containing cystein-rich motifs.
Localization and Function
Tau is a microtubule-associated protein which is found in abundance in the axons of Central nervous system (CNS) and Peripheral nervous system (PNS) (Binder et al., 1985; Couchie et al., 1992; Table 1). It is also observed in astrocytes and oligodendrocytes in the CNS. The tau pre-mRNA undergoes alternative splicing at exons 2, 3, and 10 to give six different possible isoforms. Inclusion of exon 10 generates 4-repeat or 4R tau, while exclusion forms 3-repeat or 3R tau. In neurons this ratio is controlled throughout development, emphasizing the importance of this balance for neuronal functions.
Implications of RNA in Pathogenesis
In FTD populations, MAPT mutation frequency ranges from 8 to 50%. To date, 44 different MAPT mutations, either mis-sense or splice mutations or both, have been discovered in 138 different families (Cruts et al., 2012). The list of pathogenic mutations observed in MAPT are reported in Table 4). Most missense mutations alter ability of tau to bind to microtubules, thus leading to the formation of inclusion in neurons and glia, called neurofibrillary tangles (NFT) (Lee et al., 2005).
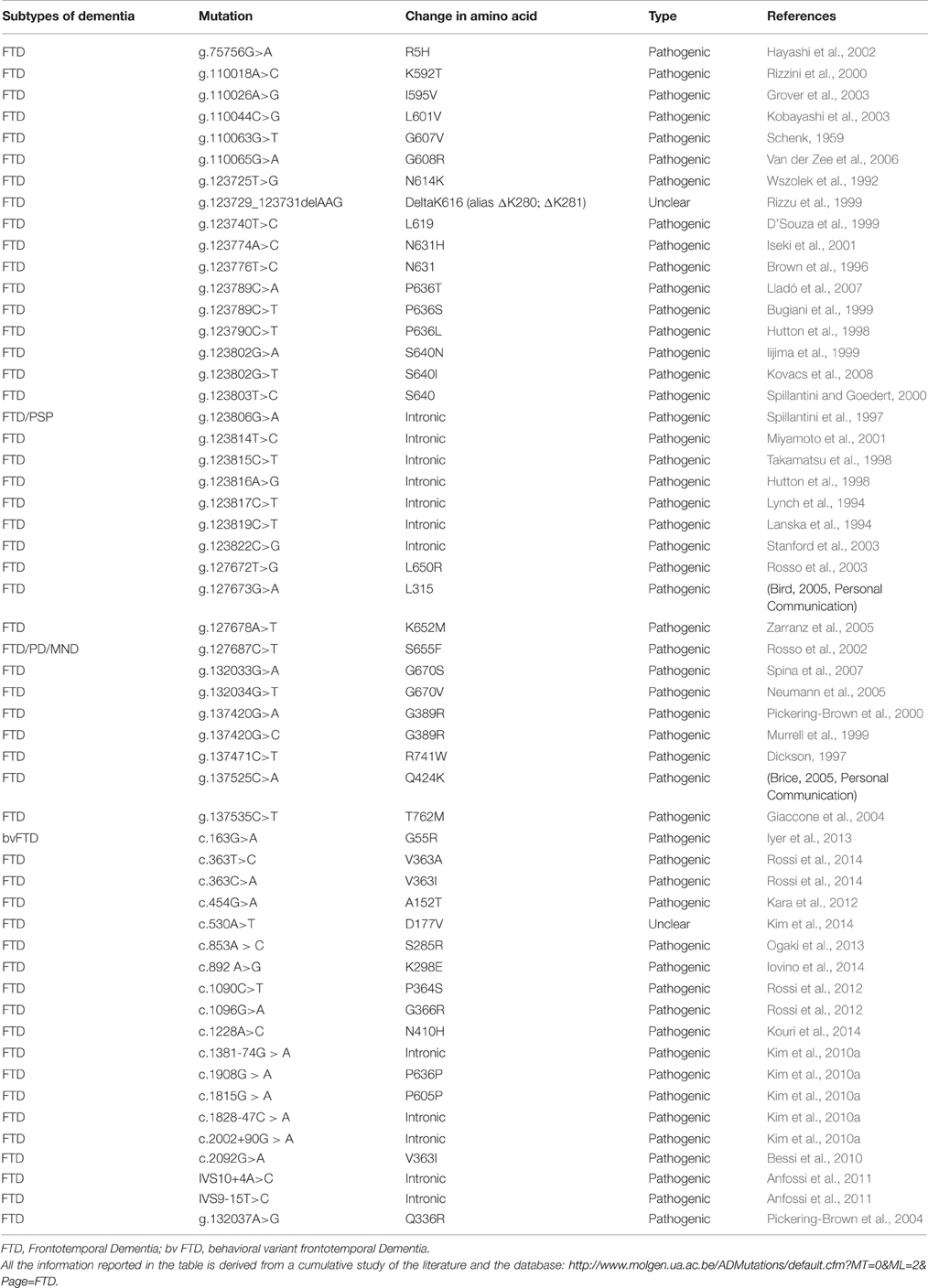
Table 4. List of mutations in MAPT and their characteristic phenotypes.
About half of the mutations in MAPT, however, are associated with alteration of splicing of exon 10 and increase the ratio of 4R to 3R. The mutations near exon 10 5′splice site enhance inclusion of exon 10 either by altering the linear cis-splicing elements or by destabilizing a stem-loop structure at the exon-intron junction (D'Souza et al., 1999; Grover et al., 1999; Spillantini and Goedert, 2013). This stem-loop arises as a result of the self complementarity among bases in this region and has a putative role in masking the 5′ splice site Mutations that disrupt the stem-loop structure make the 5′ splice site accesible to splicing factors, leading to inclusion of exon 10 (Wolfe, 2012).
Though mutations lead to alteration of splicing at the mRNA level, their primary effect becomes pathogenic through changes in the protein level in about half of the cases (Goedert and Jakes, 2005).
The human MAPT 3′UTR, as well as that of rodents, contains two Polyadenylation Signals (PAS) in tandem and can undergo alternative polyadenylation (APA) to produce transcripts of approximately 2 or 6 kb, namely the short and long transcript variants (Poorkaj et al., 2001). Dickson and colleagues investigated the role of human MAPT 3′-UTR in regulating tau expression (Dickson et al., 2013). They observed that the two MAPT 3′UTR variants are differentially regulated and influence both mRNA stability and protein expression levels. The same authors have reported that miR-34a can bind the human MAPT 3′-UTR long form and reduce tau levels, whereas inhibition of endogenous miR-34 family members leads to increased tau levels, leading to a hypothesis that up-regulation of miR-34 observed during neuronal differentiation could be a compensatory mechanism to decrease the expression of tau aggregates. Recent work (Wu et al., 2013a) also confirms the finding that MAPT is regulated by miRNA 34c-5p and miRNA 34c-3p, which bind to its 3′UTR.
Additionally, work by Zovoilis and colleagues have suggested that miR-34c could be a marker for the onset of cognitive disturbances linked to Alzheimers disease and they also indicate that targeting miR-34c could thus be a suitable therapy (Zovoilis et al., 2011).
Studies also reported that miR-34 regulates apoptosis by blocking the SIRT1 gene (Hermeking, 2010) and astrocytic apoptosis has been observed as an early event in FTLD conditions (Broe et al., 2004). These findings suggest that miRNAs might be involved in FTD through apoptotic mechanisms.
Tau is known to spread through synaptic and non-synaptic mechanisms (Medina and Avila, 2014) and its accumulation is thought to be mediated through spreading of the protein from cell to cell. Tau has been reported to be secreted unconventionally in naked form (Chai et al., 2012) or associated to exosomes (Saman et al., 2012) and/or other membrane vesicles (Simón et al., 2012). This method of elimination of tau has been suggested as a response mechanism to inhibit tau secretion and toxicity. Recent reports have suggested that tau is released into culture medium from neuroblastoma cells, tau-expressing non-neuronal cells, induced pluripotent stem cell-derived human neurons, and mouse primary neurons (Kim et al., 2010a; Shi et al., 2012). This has also been observed in the brain interstitial fluid of both wild-type and P301S tau-expressing mice in micro-dialysis studies (Yamada et al., 2011). Clinico-pathological studies underline the tau pathology progression from entorhinal cortex through the hippocampus and into limbic system (Arriagada et al., 1992). Recent in vivo studies in tauopathy transgenic mouse models have also highlighted the spreading of tau pathology through a trans-synaptic mechanism in anatomically connected neuronal networks (De Calignon et al., 2012; Liu et al., 2012). Apart from these, intracerebral inoculation of synthetic tau fibrils induced NFT (Neuro fibrillary tangles) like inclusions that propagated from injected sites to other connected regions of brain (Iba et al., 2013).
Current hypotheses also include that pathological progression of improperly folded of tau could be transferred between neuronal cells via a prion-like seeding mechanism which might lead to neurodegeneration.
The major implication observed upon mutations which lead to splice defects highlights the relevance of regulation at RNA level which decides the fate of onset of neurodegeneration. The regulation of MAPT mediated through miRNAs further indicates the role of non-coding RNAs in determining tau protein levels.
GRN (Progranulin)
GRN is located on the long arm of chromosome 17 at the locus q21.31 which is present at a distance of 1.7 Mb from MAPT (Baker et al., 2006; Cruts et al., 2006). GRN encodes for a 593 aa precursor protein of 68.5 kDa called progranulin.
Structure
Progranulin can be N-glycosylated at five potential sites and secreted as a mature protein of 88 kDa (Chen-Plotkin et al., 2010; Songsrirote et al., 2010). The protein is formed by 7.5 cysteine-rich granulin domains, separated through linker sequences that contain disulfide bridges (He and Bateman, 2003), as represented in Figure 4B. This characteristic structure can be cleaved at the intra-linker spacer sequences to produce seven non-identical granulins that contain cysteine-rich motifs. Different proteases can cleave progranulin, such as matrix metalloproteinase-14 (Butler et al., 2008), elastase (Zhu et al., 2002), proteinase 3, and neutrophil elastase (NE) at the pericellular microenvironment of the neutrophil cell surface (Kessenbrock et al., 2008). The full-length progranulin, once secreted, is protected from cleavage by the high-density lipoprotein (HDL)/Apolipoprotein A-I complex (Okura et al., 2010) and the secretory leukocyte protease inhibitor (SLPI) (Zhu et al., 2002).
Localization and Function
Progranulin is present in many tissues, is highly expressed in immune system cells (Daniel et al., 2000) and in a medium level in the brain (Bhandari et al., 1996; Ahmed et al., 2007), where it is highly expressed in specific populations of neuronal cells, such as cortical neurons, and granule cells of the hippocampus (Daniel et al., 2000; Table 1). The subcellular location of progranulin seems to be the endoplasmic reticulum (ER) and Golgi, where it is particular abundant in mouse cortical neurons and mouse microglia (Almeida et al., 2011). Progranulin is implicated in a wide range of biological processes such as embryogenesis (Díaz-Cueto et al., 2000; Daniel et al., 2003; Bateman and Bennett, 2009), cell survival and cell growth (Plowman et al., 1992; He and Bateman, 1999), inflammation and wound repair (Zhu et al., 2002; He et al., 2003; Kessenbrock et al., 2008; Yin et al., 2010), transcriptional repression (Hoque et al., 2003, 2010) and several reports suggest its role in neuronal development (Van Damme et al., 2008). Interestingly, progranulin and the proteolytically cleaved granulins can have coherent functions, such as in the regulation of neurite outgrowth (Van Damme et al., 2008), or they can have contrasting roles, such as in inflammation processes (He and Bateman, 2003).
To date, 69 different GRN mutations have been discovered in 231 families (Cruts et al., 2012). A list of detailed pathogenic mutations are reported in Table 5. The GRN mutations frequency range from 1 to 11.7% in FTD patients, but the frequency rises to 12–25% in familial FTD (Cruts et al., 2006; Gass et al., 2006; Huey et al., 2006; Bronner et al., 2007; Borroni et al., 2008). There are different types of GRN mutations, the majority are classified as non-sense, frameshift, and splice site mutations that cause a premature stop codons (Baker et al., 2006; Cruts et al., 2006). However, the pathogenic variants include also missense mutations with a partial decrease of progranulin and a loss of its function (Mukherjee et al., 2006, 2008; Shankaran et al., 2008; Wang et al., 2010). Silent and intronic mutation with unknown pathology can also occur. Generally the pathogenic GRN mutations lead to a decreased GRN expression due to a non-sense-mediated mRNA decay, resulting in a GRN haploinsufficiency inherited in an autosomal dominant manner (Baker et al., 2006; Cruts et al., 2006; Gass et al., 2006; Cruts and Van Broeckhoven, 2008).
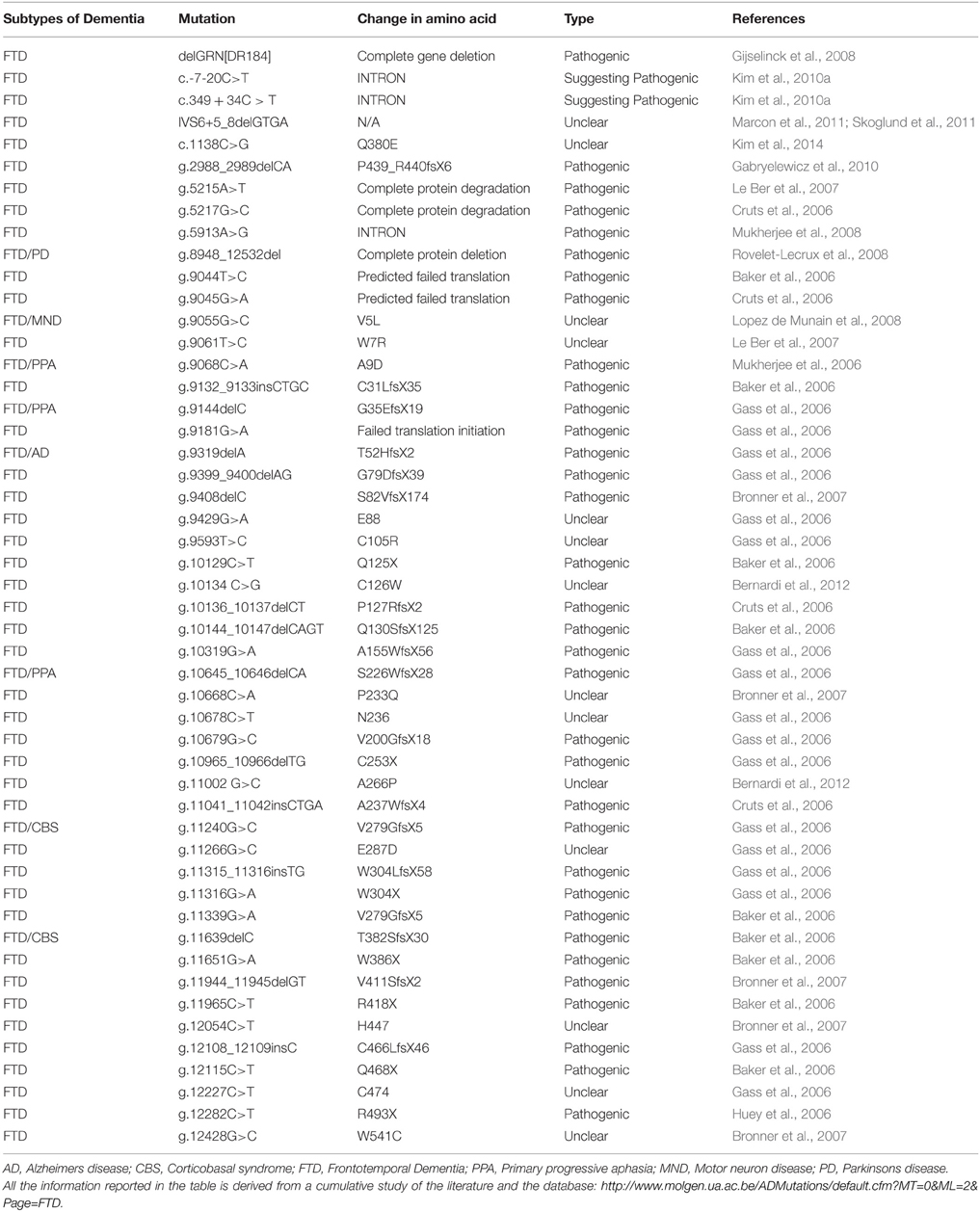
Table 5. List of mutations in GRN and their characteristic phenotypes.
Indeed progranulin levels, measured in either the serum or cerebrospinal fluid (CSF) of patients with GRN mutations are ~30–50% of normal (Van Damme et al., 2008). Moreover, a decreased progranulin level can be also detected in plasma of GRN mutations patients (Finch et al., 2009) and a reduced GRN mRNA level can be observed in patient whole blood samples through microarray experiments (Coppola et al., 2008). In contrast an increased level of GRN mRNA was observed in the frontal cortex from post-mortem brain samples of FTD patients with GRN mutations, as compared to FTD patients without GRN mutations (Chen-Plotkin et al., 2010). The higher level of GRN transcripts could be due to the robust microglia infiltrations, observed in the brain tissues of GRN mutation patients. Indeed microglia shows high level of GRN expression.
Implications of RNA in Pathogenesis
Most of the patients with FTLD-U show GRN mutations with presence of TDP-43 ubiquitin positive inclusions, hence bearing the term FTLD-TDP (Mackenzie et al., 2006, 2010; Sampathu et al., 2006). The relation between TDP-43 and progranulin is not fully understood, however several recent studies indicate that TDP-43 controls the expression of progranulin by binding to GRN mRNA. On a study in which TDP-43 targets were identified through a RIP-chip analysis, it is shown that TDP-43 has a post-transcriptional regulation on GRN and VEGFA (Vascular endothelial growth factor A) (Colombrita et al., 2012).
As previously mentioned, TDP-43 was shown to specifically bind GRN 3′UTR controlling GRN mRNA stability and the final quantity of progranulin protein (Polymenidou et al., 2011; Colombrita et al., 2012). Moreover a knock-down of TDP-43 in mice showed an increase in the amount of GRN mRNA level (Polymenidou et al., 2011; Colombrita et al., 2012). Depletion of TDP-43 also led to altered splicing of sortilin, the putative progranulin receptor (Polymenidou et al., 2011). The relation between GRN and TDP-43 was also demonstrated in vitro: cells that were treated with siRNA against GRN for 72 h, showed a caspase-dependent cleavage of TDP-43 into fragments (Zhang et al., 2007); whereas primary neuronal cultures upon knowckdown of GRN showed a re-localization of TDP-43 in the cytoplasm (Guo et al., 2010).
Through genetic association analysis, a common genetic variation localized on the 3′UTR of GRN (rs5848) was shown to represent a genetic risk factor for FTD (Rademakers et al., 2008). Progranulin levels in brain extracts from rs5848 TT homozygous FTD patients were lower than in CC carriers, as observed through western blot analyses, ELISA, and immunohistochemistry. A stronger binding of miR-659 in the 3′UTR of GRN was shown in the presence of the rs5848 T variant, and might explain the reduced progranulin levels.
It is reported that miR-107 is downregulated in presence of Alzheimer's disease at early stage (Wang et al., 2008b). Another study demonstrated through a RIP-Chip analysis performed in human H4 neuroglioma cells that the open reading frame of GRN mRNA contains many recognizing sequences elements for miR-107 (Wang et al., 2010), showing implications of miR-107 deregulation in neurodegenerative diseases. In particular miR-107 regulation of GRN seems to be relevant to glucose metabolism in cultured H4 neuroglioma cells. Previous analysis identified miR-107 as one of the microRNAs that increase their expression with glucose supplementation in cell culture medium (Tang et al., 2009). Wang and colleagues reported that glucose metabolic pathway may recruit miR-107 to regulate GRN expression. Another microRNAs that was found significantly down-regulated in brains of Alzheimer's disease patients is miR-29b, that beloged to the miR-29a/b-1 cluster (Hébert et al., 2008). Interestingly progranulin can also be regulated by miR-29b through a binding in the 3′UTR of GRN mRNA (Jiao et al., 2010). It would be useful to know if these microRNAs deregulation can contribute to the pathogenesis of dementia. So far different microRNAs seem to be important for the control of progranulin along with the role played by TDP-43 on the stability of GRN mRNA and its expression.
VCP
The VCP (Valosin-containging protein) gene is located on chromosome 9p13.3. It also called p97 or CDC48, consists of 17 coding exons.
Structure
The VCP protein is composed of four domains vital for its proper functioning, namely the N, D1 D2 and C-terminal domains (Figure 5B; DeLaBarre et al., 2006; Pye et al., 2007). The VCP N domain is encoded by exons 1, 2, 3, 4 and 5, while the D1 and D2 domains are encoded by exons 6, 7, 8, 9, 10 and 12, 13, 14, respectively. There are two linker domains in the protein: the N-D1 linker and the flexible D1-D2 linker.
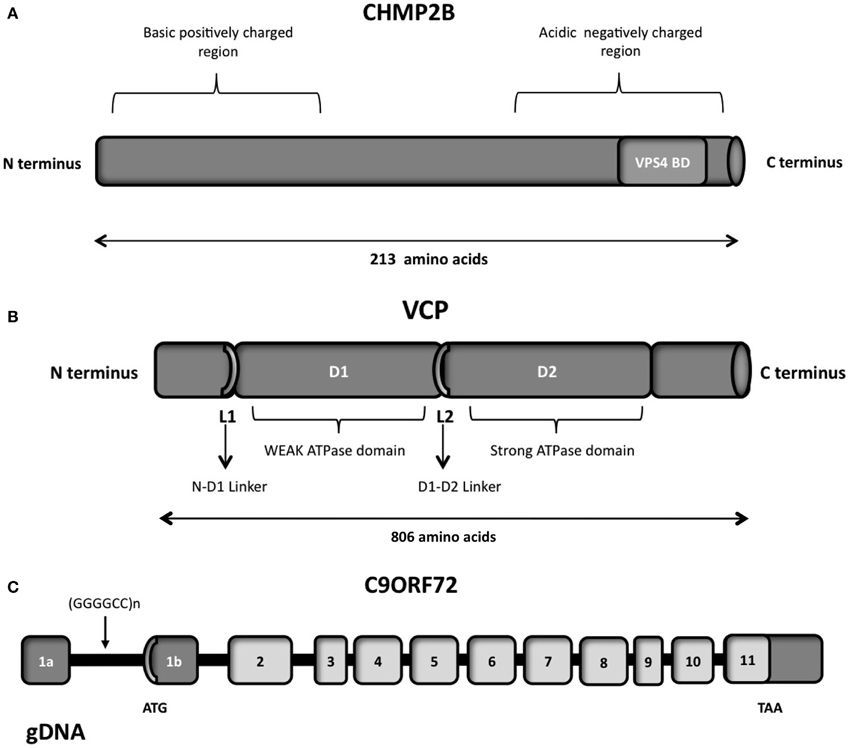
Figure 5. (A) Schematic representation of the CHMP2B which contains an acidic negatively charged C-terminal domain and a basic positively charged N-terminal domain, a predicted coiled-coil structure (14-51 aa) and a conserved Snf7 domain (16-178 aa). The autoinhibitory structure formed in the cytosol due to the C- and N- terminal part interactions is reverted through the binding of VSP4 on the VPS4 binding domain (VPS4 BD), localized on the C-terminal part. (B) Schematic representation of the six functional domains of the VCP protein: the N-terminal domain, the weak ATPase domain (D1), the major ATPase domain (D2), the N-D1 linker domain, the flexible D1–D2 linker domain and the C-terminal domain. (C) Overview of the genomic structure of the C9ORF72 gene, with white boxes representing the coding exons and gray boxes representing the non-coding exons. The position of the hexanucleotide repeat (GGGGCC), the start codon (ATG), and the stop codon (TAA) are indicated in the scheme.
VCP is a member of the AAA-ATPase gene superfamily (ATPase Associated with diverse cellular Activities) (Woodman, 2003; Wang et al., 2004b), and is one of the most abundant cytosolic proteins (Table 1) conserved throughout in mammals. The complete protein contains 806 amino acids. The N domain of VCP is responsible for the cofactor and ubiquitin binding function (Wang et al., 2004b). While the D1 domain mediates oligomerization-independent nucleotide binding, the D2 domain confers most of the ATPase activity (Wang et al., 2004b).
Localization and Function
This protein functions as a molecular chaperone in various distinct cellular processes including ubiquitin-dependent protein degradation, stress responses, programmed cell death, nuclear envelope reconstruction, and Golgi and endoplasmic reticulum (ER) assembly (Guinto et al., 2007).
VCP is known to be involved in protein aggregation/quality control of mitochondria and cell proliferation (Hayashi, 2013) and is vital for retro-translocation of misfolded proteins from Endoplasmic reticulum to cytoplasm (Kimonis et al., 2008). Mutation and depletion studies of VCP have provided evidence of accumulation of poly-ubiquitinated proteins (Dai and Li, 2001). Mutations in this gene may suggest the disruption of normal protein degradation pathway in the disease. This could be facilitated through the disruption of binding between the VCP and protein adaptors.
The expression of mutant VCP in myoblastic cell lines is associated with increased ubiquitin conjugated proteins (Weihl et al., 2006). Studies on overexpression of mutant VCP protein in transgenic mice implicated an age-dependent muscle weakness and Ubiquitin-positive inclusions and accumulation of high molecular weight protein aggregates (Weihl et al., 2007).
VCP functions as a homohexamer (Zhang et al., 2000; Rouiller et al., 2002) by binding to multiple proteins associated with Ubiquitin proteasome system (UPS). The VCP complex binds to polyubiquitin chains and unbounds ubiquitinated proteins from their binding partners thereby facilitating transport to the UPS.
Implications of RNA in Pathogenesis
To date, 18 different VCP mutations have been discovered in 48 different families, which include FTLD that is associated with ALS, inclusion body myopathy, and Paget disease (Cruts et al., 2012). Table 6 high-lights the list of pathogenic mutations observed so far. The association of inclusion body myopathy and FTD was established by Kovach et al. (2001).
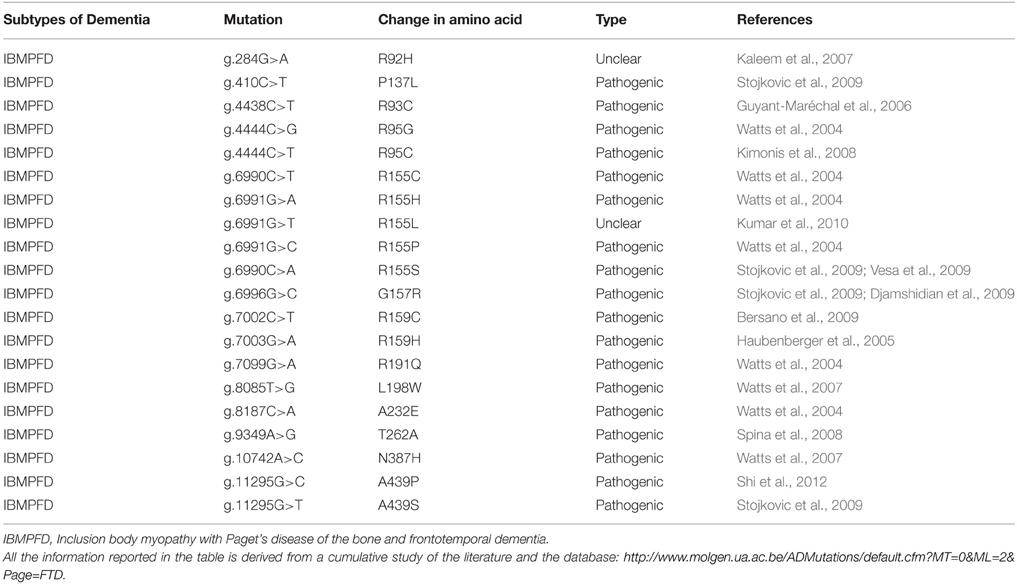
Table 6. List of mutations in VCP and their characteristic phenotypes.
A recent work by Jacquin et al. (2013) has showed R155H (464 G>A) mutation of the VCP gene in a French family, led to the Inclusion body myopathy with Paget's disease of the bone and frontotemporal dementia (IBMPFD), with a psychiatric onset of FTD.
The expression of IMBPFD-associated VCP mutations in skeletal muscle cells reduced UNC-45 (a molecular chaperone involved in myosin assembly) degradation that is linked to severe myofibril disorganization in myotubules. This study suggests a possible mechanism for the selective vulnerability of skeletal muscle in IBMPFD; however, the implication for the pathogenesis of FTD still remains unknown. Studies on a VCP-mutant transgenic mouse have shown TDP-43 and ubiquitin positive accumulations (Custer et al., 2010) suggesting a possible interplay between these proteins. IBMPFD is known to have TDP-43 aggregation with VCP mutations (Nalbandian et al., 2011). Ju et al. (2009) have established a link between VCP and autophagosomes wherein the loss of VCP leads to accumulation of autophagosomes, thus establishing a possible cause of aggregation of proteins such as TDP-43.
VCP has been detected in a few inclusions of neurodegenerative diseases such as senile plaques in Alzheimer's disease, Lewy bodies in Parkinson's disease, neuronal intranuclear inclusions in CAG/polyglutamine diseases and ubiquitin-positive inclusions in ALS (Hirabayashi et al., 2001; Mizuno et al., 2003; Ishigaki et al., 2004).
Bartolome and colleagues have performed analyses in fibroblasts derived from patients with three different pathogenic VCP mutations, VCP-deficient cells, mouse cortical primary neurons and astrocytes, to conclude that reduction of VCP led to uncoupling of mitochondria and increased oxygen consumption and a subsequent decrease in ATP of cells leading to cellular toxicity and neuronal death (Bartolome et al., 2013).
VCP has been recently involved in clearance of mRNP granules (Buchan et al., 2013), thereby unraveling a new mechanism in clearance of RNPs from the cell. This might indicate why VCP mutations lead to accumulation of stress granule constituents or cytoplasmic inclusions. mRNP granules assemble to form stress granules as a consequence of their aggregation (Erickson and Lykke-Andersen, 2011). Wang et al. (2015) have shown a direct interaction between VCP and FUS. VCP being a key regulator of protein degradation, DNA interaction, and mitochondrial activity, its direct interaction with FUS is intriguing. Although there is no evidence which shows a direct interaction or implication of VCP mutations on RNA, its association with TDP-43 and FUS, both of which are RNA binding proteins may suggest their unraveled interactions in RNA metabolism.
CHMP2B
CHMP2B (Charged multivesicular body protein) is encoded by a gene located on chromosome 3 and is a component of the endosomal sorting complex required for transport-III (ESCRT-III complex).
Structure
CHMP2B is a protein of 213 amino acids, with an acid C-terminal domain and basic N-terminal domain (Figure 5A), and a predicted coiled-coil structure (Skibinski et al., 2005). The negatively charged C-terminal domain interacts with the positively charged N-terminal part, creating a closed and autoinhibitory structure in the cytosol (Whitley et al., 2003; Shim et al., 2007). CHMP2B requires therefore an activation process performed by VPS4, which binds to its C-terminal domain. Indeed the C-terminal part of CHMP2B contains a binding domain for VPS4. VPS4 is a member of the AAA-ATPase family and it has a role in catalyzing the dissociation of ESCRT complexes from endosomes (Katzmann et al., 2002). The ATPase activity of VPS4 is important for endosomal sorting (Katzmann et al., 2002; Obita et al., 2007). Mutations localized in the VSP4-binding region impair the function of CHMP2B, preventing the formation of protrusions from the cell (Bodon et al., 2011).
Localization and Function
Northern-Blot analysis demonstrated that CHMP2B is expressed in all the major parts of the brain, including the temporal and frontal lobes (Table 1). Moreover through in situ hybridization of mouse brain, an enhanced expression of CHMP2B in the hippocampus, frontal and temporal lobes, and cerebellum was shown (Skibinski et al., 2005). The endosomal-sorting complex required for transport (ESCRT) is a protein complex involved in the endocytosed protein trafficking from endosome to lysosomes, playing an important role for sorting of proteins and for their efficient lysosomal degradation (Urwin et al., 2010). Moreover ESCRT complexes have a relevant role at the plasma membrane, during cytokinesis (Carlton and Martin-Serrano, 2007; Elia and Sougrat, 2011), budding of some enveloped viruses (Usami et al., 2009; Martin-Serrano and Neil, 2011), autophagy and transcriptional regulation (Roxrud et al., 2010; Schmidt and Teis, 2012). Endosomal trafficking is a key process for the internalization and transport of neuronal growth factors, secreted growth factors, signaling molecules (Bronfman et al., 2007). A dysfunction in this process could lead to an altered cell-signaling and aberrant communication between cells.
Implications of RNA in Pathogenesis
In particular ESCRT dysfunction is associated with neurodegeneration, indeed mutation in CHMP2B have been reported in frontotemporal dementia linked to chromosome 3 (FTD-3) (Urwin et al., 2010). FTD-3 has an onset of 48–67 years and is an autosomal dominant dementia with TDP-43 negative FTLD-U, ubiquitin positive inclusions (Urwin et al., 2010).
As reported in Table 7, several missense mutations are connected with FTD-3 (Skibinski et al., 2005), with familial or sporadic cases of ALS (Parkinson et al., 2006; Urwin et al., 2010), familial frontotemporal lobar degeneration (FTLD) (Ghanim et al., 2010) or CBD (Van der Zee et al., 2008), however only few studies analyzed their pathogenic features in cultured neurons.
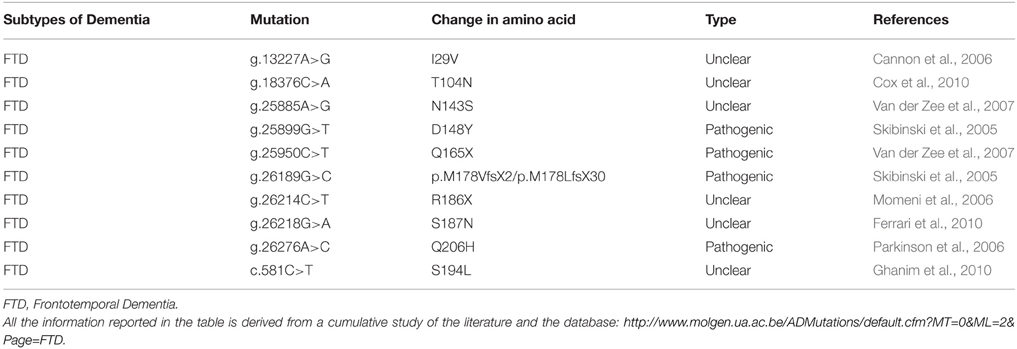
Table 7. List of mutations in CHMP2B and their characteristic phenotypes.
A point mutation has been identified in the 5′ acceptor site of exon 6, causing the production of two abnormal transcripts: CHMP2Bintron5 with retention of intron 5 and CHMP2BΔ10 that has a 10 bp deletion and a sequence frameshift due to the use of a criptic splice site in exon 6 (Skibinski et al., 2005). Both proteins lack 36 amino acids in the C-terminal part with the subsequent absence of VPS4-binding domain and an accumulation of CHMP2B on the endosomal membrane (Skibinski et al., 2005; Urwin et al., 2010). This accumulation suggests that the binding of mutated proteins to the endosomes prevents the recruitment of the correct proteins necessary for the fusion with lysosomes (Metcalf and Isaacs, 2010; Urwin et al., 2010). Indeed large and abnormal endosomal structures are observed in post-mortem brain tissues, in patient fibroblast cultures and in case of overexpression of CHMP2B mutant in PC12 and human neuroblastoma cell lines (Skibinski et al., 2005; Van der Zee et al., 2008; Urwin et al., 2010). Moreover CHMP2B seems to act through a gain of function mechanism in the presence of mutations, since only the transgenic mice expressing CHMP2Bintron5 show similar neuropathology features observed in FTD-3 patients, whereas the knockout mice with the inactivation of CHMP2B do not show any pathological characteristics (Ghazi-Noori et al., 2012).
Another non-sense mutation replaces a glutamine residue with a stop codon, creating a more severe C-terminal truncation (Van der Zee et al., 2008).
Since CHMP2B is involved in the endosomal trafficking of signal molecules, it could be interesting and possibly relevant for the pathology to check if an altered endosomal process can affect the function of other proteins involved in FTLD, such as progranulin, as is it also suggested by Urwin et al. (2010).
C9ORF72
Structure
Large expansions of the non-coding GGGGCC repeat in C9ORF72 (Chromosome 9 open reading frame 72) were recently demonstrated to cause ALS and FTD (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Indeed 20–80% of familial and 5–15% of sporadic ALS and FTD in North American and European patients show this hexanucleotide expansion with a range of 700–1600 repeats, whereas the normal population carries less than 30 repeats (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Smith et al., 2012). Pathogenic mutations reported in C9ORF72 are listed in Table 8. C9ORF72 is localized on chromosome 9 and is composed of 12 exons, with two alternate non-coding first exons (Figure 5C, exon 1a and 1b) (DeJesus-Hernandez et al., 2011). Specifically, the polymorphic hexanucleotides repeat was identified between the two alternatively spliced non-coding exons, through a sequencing approach (DeJesus-Hernandez et al., 2011).

Table 8. List of mutations in C9ORF72 and their characteristic phenotypes.
Depending on alternative transcription initiation, the GGGGCC repeat can be located on the promoter of transcriptional variant 1 or in the intron 1 of transcriptional variants 2 and 3. Variant 2 results from splicing of exons 1a and exons 2–5 whereas variant 3 is composed of exon 1a and exons 2–11.
Localization and Function
Expression array data showed wild type C9ORF72 RNA present in different CNS tissues, including spinal cord and higher expression in the cerebellum (Renton et al., 2011; Table 1).
The protein encoded by C9ORF72 is still uncharacterized and with unknown function even if it is well-conserved across species (DeJesus-Hernandez et al., 2011).
Recently, Farg et al. (2014) demonstrated for the first time that the endogenous C9ORF72 protein has a function in the regulation of intracellular trafficking processes in the endosomal and autophagy-lysosomal compartments in neuronal cell lines. Therefore, they reported the normal cellular function of C9ORF72 that is essential to understand its role in FTD/ALS.
In particular, they found co-localization in neuronal cell lines and primary cortical neurons of C9ORF72 with four Rab proteins, which are involved in endosomal trafficking. In motor neurons, they found 70% of colocalization with Rab7, which is a fundamental protein implicated in the biogenesis of lysosomes, the transport of endosomes and the maturation of autophagosomes (Gutierrez et al., 2004). A similar mechanism of interaction and recruitment of Rab7 was also described for CHMP2B by Urwin and collaborators. In CHMP2B mutant cells, an impaired recruitment of Rab7 onto endosomes was observed with a decreased fusion with lysosomes and a delayed degradation (Urwin et al., 2010).
C9ORF72 protein was also found to colocalize with lysosomes, ubiquilin-2 and autophagosomes, involved in autophagy (Farg et al., 2014). Interestingly, the ability of C9ORF72 to interact with hnRNPs and induce not yet characterized nuclear aggregates and stress granules, could link the C9ORF72 protein with RNA metabolism processes (Farg et al., 2014).
Implications of RNA in Pathogenesis
Immunocytochemistry analysis on human fibloblasts and mouse motor neuron NSC-34 cell line revealed a predominant nuclear localization of C9ORF72 protein (Renton et al., 2011). Immunohistochemical analysis showed C9ORF72 expression in neurons and in FTD- and ALS-affected regions with a predominant cytoplasmic staining and a synaptic localization, but the quantitative mRNA analysis demonstrated that the repeat expansion reduces C9ORF72 transcript variant 1 expression in lymphoblast cell lines of expanded repeats carriers and in frontal cortex samples from unrelated FTLD-TDP patients carrying expanded repeats (DeJesus-Hernandez et al., 2011). The hexanucleotide repetitions present in the C9ORF72 transcript can form G-tetrad units, called G-quartets, where G bases are rearranged in a cyclic pattern with eight hydrogen bonds (Fratta et al., 2012). The presence of RNA G-quadruplexes has been found in different organisms and has been observed in vitro and in vivo (Kikin et al., 2008; Xu et al., 2010b). Transcripts are enriched in RNA G-quadruplexes structures in the 5′UTR, 3′UTR and in the first exon (Eddy and Maizels, 2008; Huppert et al., 2008). Recently a molecular mechanism was described by which the DNA and RNA G-quadruplexes in C9ORF72 create structures that promote the formation of RNA/DNA hybrids (R-loops) (Haeusler et al., 2014). The pathological mechanism involvingC9ORF72 gene or its function are not clear, even if several studies showed a decrease in the mRNA levels of some C9ORF72 variants in ALS, which suggests a loss-of-function mechanism (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Gijselinck et al., 2012; Mori et al., 2013b). Moreover, the aberrant transcripts containing the hexanucleotide repeats can accumulate and form structures in the nucleus called RNA foci, which may produce neurodegeneration through a toxic effect (DeJesus-Hernandez et al., 2011). These transcripts can be aberrantly expressed through repeat-associated non-ATG (RAN) translation (Mori et al., 2013b). Several groups reported that the RAN translation of the hexanucleotide repeats produces poly(GP), poly(GA) and poly(GR) proteins, since this type of translation, without an initiation codon, can have all the possible reading frames (Ash et al., 2013; Mori et al., 2013b) and RNA can be also bidirectionally transcribed (Gendron et al., 2013). These RAN proteins can form inclusions in neurons and are considered a hallmark of the disease (Ash et al., 2013; Mori et al., 2013b). The neuronal inclusions can be detected through antibodies that recognize putative GGGGCC repeat RAN-translated peptides, therefore this type of immunoreactivity can be use as a potential biomarker for the disease (Ash et al., 2013).
It is also reported that RNA foci may sequester important RNA binding proteins, causing an alteration inside the cell and a subsequent dysfunction of RNA targets, in a process similar to the formation of RNA foci in myotonic dystrophy type 1 (DM1) (Lee et al., 2013; Mori et al., 2013a; Reddy et al., 2013; Xu et al., 2013). Specifically, one study demonstrated that hnRNP-H is sequestrated by RNA foci, reducing its available amount and its splicing efficiency on different target transcripts (Lee et al., 2013). A recent paper by Gendron et al. (2013) contains detailed descriptions of the proteins found to be sequestered on RNA foci in in vitro studies.
The presence of both sense and antisense RNA foci in frontotemporal dementia with the presence of C9ORF72 repeats (C9FTLD), was demonstrated in patients, specifically in the frontal cortex, motor cortex, hippocampus, cerebellum, and spinal cord (Gendron et al., 2013; Lagier-Tourenne et al., 2013; Mizielinska et al., 2013; Zu et al., 2013). RNA foci were identified in neurons and with lower frequency in astrocytes, microglia, and oligodendrocytes; the highest concentration of foci was found in the frontal cortex region, compared to cerebellum and hippocampus (Mizielinska et al., 2013). However, another work reported that foci are localized with higher frequency in the cerebellum (Lee et al., 2013). Despite this inconsistency, the major part of RNA foci is localized at the very edge of the nucleus, but the explanation for this localization is still unknown (Mizielinska et al., 2013). The cellular toxicity associated with the longer hexanucleotide repeats and the presence of RNA foci was demonstrated using neuroblastoma cells and zebrafish embryos (Lee et al., 2013). One patient, who was homozygous for the C9ORF72 hexanucleotide repeats, showed a higher proportion of sense and antisence foci with an early onset of FTD and severe pathological characteristics, compared to the heterozygous case (Mizielinska et al., 2013). A recent study found a mechanism for the disease in which the DNA and RNA–DNA structures formed in the repeat regions, alter the RNA transcription, with a result of transcriptional pausing and abortion. The accumulation of abortive transcripts with hexanucleotides repeats, creates G-quadruplexes, and hairpins structures with a binding of essential proteins, leading to nuclear stress, and further defects (Haeusler et al., 2014).
TDP-43 and FUS, two FTD related proteins previously decribed, are structurally related to the hnRNPs that are found to bind C9ORF72 RNA foci (Lee et al., 2013; Mori et al., 2013b), however FUS and TDP-43 do not colocalize with C9ORF72 RNA foci in cells, patient motor neuron cultures or in spinal motor neurons from patients (Lagier-Tourenne et al., 2013; Lee et al., 2013; Sareen et al., 2013). Since TDP-43 is capable to bind through its C-terminal region the hnRNP proteins (Buratti et al., 2005), the accumulation of these ribonucleoprotein on the RNA foci could indirectly influence TDP-43 function, creating a possible link of interaction between these two factors involved in FTD and ALS (Gendron et al., 2013). Indeed most of the cases with C9ORF72 expansion show TDP-43 inclusions (FTLD-TDP) (DeJesus-Hernandez et al., 2011; Lagier-Tourenne et al., 2013; Mackenzie et al., 2014) with some exception, such as a case in UK with C9ORF72 repeats with FTLD-tau pathology (Snowden et al., 2012). It was reported that the plasma and CSF level of phosphorylated TDP-43 is significantly higher in patients with FTD carrying C9ORF72 expansion or GRN mutations compared to other FTD patients or healthy controls (Suárez-Calvet et al., 2014). This finding creates another possible link of interaction or regulation between TDP-43 and C9ORF72 that needs further analysis.
Discussion
In this review we describe the different genes involved in FTD, focusing on their possible interactions, in order to identify a common network of their combined regulations. We created this network focusing on the RNA aspect, an emerging and crucial molecule that plays critical and fundamental functions in the cells. Recently, research has increased its focus onthe role of RNA in neurodegeneration (Renoux and Todd, 2012). We believe that the RNA mediated regulation plays a key role in the unique integration of all the known genes involved in FTD.
In this picture (Figure 6) FUS and TDP-43 RNA binding proteins are at the core of the network, since they often are associated factors that share similar features, with sometimes different but complementary roles (Colombrita et al., 2012). They interact with RNA in three main roles: as RNA binding proteins participating on the different aspects of mRNA processing (Bosco et al., 2010; Colombrita et al., 2011), as regulators of microRNAs processing, and as regulators of lncRNAs. FUS and TDP-43 were both found in aggregates in ALS/FTLD affected neurons (Da Cruz and Cleveland, 2011), nuclear complexes and in cytoplasmic RNPs (Kim et al., 2010b). TDP-43 appears to be the main regulator of this network, being able to interact with FUS pre-mRNA and regulate its splicing, and auto-regulate its own pre-mRNA, causing a reduction of its own expression (Polymenidou et al., 2011).
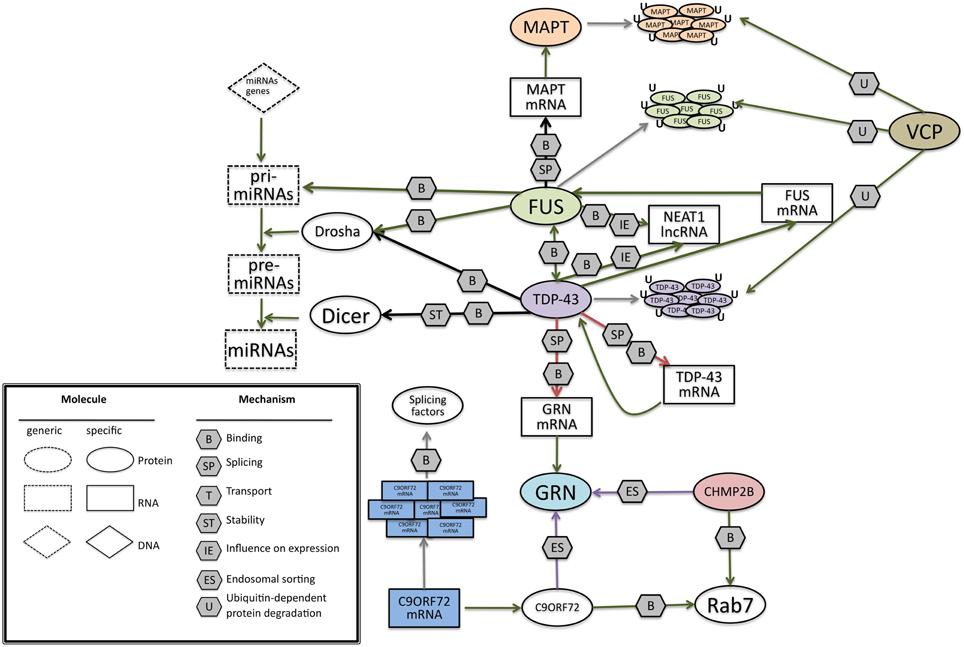
Figure 6. A possible network of interactions between proteins and RNAs, at the basis of FrontoTemporal Dementia. Green arrows indicate binding interactions or processes that result in activation or increased expression. Red arrows indicate binding interactions or processes that result in inhibition of activity or reduced expression. Black arrows indicate binding interactions or processes whose result can be either positive or negative. Purple arrows indicate binding interactions or processes which are hypotetical. Symbols as in Legend. lncRNAs, long non-coding RNA; TDP-43, TAR DNA binding protein; FUS, Fused in Sarcoma; GRN, progranulin; MAPT, Microtubule-Associated Protein Tau; VCP, Valosin Containing Protein; C9ORF72. CHMP2B, Charged multivesicular body protein 2b; Rab7, Ras-related protein 7.
TDP-43 can also bind GRN pre-mRNA, negatively controlling its splicing and, accordingly, knock-down of TDP-43 was shown to increase the amount of GRN mRNA level (Polymenidou et al., 2011; Colombrita et al., 2012). In the presence of GRN mutations, TDP-43 regulation can be altered, causing the formation of inclusions containing TDP-43 (Mackenzie et al., 2006, 2010; Sampathu et al., 2006). Though TDP-43 aggregation is a typical hallmark of many other neurodegenerative disorders, such as Alzheimer's disease, Guam Parkinsonism dementia complex, and Lewy body disease (Dickson, 2008), its impact on FTD in influencing the regulation of the network should not be underestimated.
On the other side, FUS acts as a splicing regulator of MAPT mRNA, indeed it was demonstrated that silencing of FUS alters the splicing of MAPT. In particular, FUS helps the skipping of exon 10 in primary cortical neurons (Ishigaki et al., 2012). The presence and the absence of exon 10 in MAPT gene has a fundamental role in the regulation of a delicate balance in the ratio of 4 or 3 repeats that can lead to FTD.
For the recently identified C9ORF72 gene, large expansions of the non-coding GGGGCC repeat correlate with pathogenesis, making an RNA gain-of function mechanism possible. Indeed the aberrant C9ORF72 transcripts accumulate in nuclear RNA foci and sequester several RNA-binding proteins, including some splicing factors. However, other possible pathogenetic mechanisms are under scrutiny for C9ORF72, including impaired transcription of the expanded gene or repeat-associated non-ATG (RAN) translation of expanded transcripts.
In this scenario, two FTD genes code for proteins that fit in the picture not for their relation to RNA, but for their role in protein degradation.
VCP, taking part in the ubiquitin-proteasome pathway and protein turnover (Zhang et al., 2000; Rouiller et al., 2002), could be involved in the degradation of protein inclusions present in different forms of FTD. TDP-43 inclusions were found in the presence of a VCP mutation (Neumann et al., 2009a). A direct interaction between VCP and FUS has been observed suggesting a possible convergence in their functions (Wang et al., 2015).
CHMP2B regulates protein trafficking between endosomes and lysosomes and is involved in the protein degradation pathway through lysosomes (Urwin et al., 2010). Therefore, CHMP2B could be relevant for the internalization and transport of neuronal growth factors or signaling molecules such as progranulin.
Recently, a function for the C9ORF72 protein was uncovered, in the regulation of intracellular trafficking processes in the endosomal and autophagy-lysosomal compartments (Farg et al., 2014), providing an additional link to VCP and CHMP2B proteins.
During the preparation of this review a recent study performed by Van der Zee and colleagues have demonstrated ananalysis on a European cohort of 1808 FTLD patients revealing mutationsin SQSTM1 (Sequestosome 1) or p62. The p62 protein is a stress-responsiveubiquitin-binding protein, which is shown to have a role in degradation ofpolyubiquitinated proteins via the proteasome pathway or autophagicprocesses (Van der Zee et al., 2014). This gene was known to be associated with ALS and found as a rare mutation with a frequency of 1–3% in both ALS and FTLD cases. This further intrigued its possible role in pathogenicity with a common patho-mechanism. p62 is present in neuronal and glial ubiquitin-positive inclusions in different tauopathies and synucleinopathies (Van der Zee et al., 2014). The meta-analysis performed by Van der Zee and colleagues revealed that rare mutations clustering in the UBA domain of SQSTM1 may influence disease susceptibility by doubling the risk for FTLD. Further, histopathology analysis of autopsied brain of SQSTM1 mutation carriers demonstrated a widespread of neuronal and glial phospho-TDP-43 pathology. Therefore, this study opens up another target gene SQSTM1, which is known to have implications in FTLD/ALS and additionally associated with TDP-43. Despite further work being needed, to unravel and confirm the details of the proposed network, we foresee that the construction of a picture of the interactions between proteins and RNAs at the basis of the FTD pathology will be of invaluable importance, not only to comprehend the pathogenetic mechanisms but also to develop new and more effective therapeutical approaches. Through the network analysis proposed in this review, it can be foreseen that more genes can be linked to FTD and their roles will possibly fall in to two main categories: regulation of gene expression through RNA or protein degradation. Additionally it could be predicted that novel genes related to FTD in future will be possibly a part of the proposed network.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a Futuro in Ricerca-Italian Ministry of Education, University and Research Grant RBFR-0895DC “Mechanisms of post-transcriptional regulation of gene expression in dementias.”
References
Ahmed, Z., Mackenzie, I. R., Hutton, M. L., and Dickson, D. W. (2007). Progranulin in frontotemporal lobar degeneration and neuroinflammation. J. Neuroinflammation 4:7. doi: 10.1186/1742-2094-4-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Almeida, S., Zhou, L., and Gao, F.-B. (2011). Progranulin, a glycoprotein deficient in frontotemporal dementia, is a novel substrate of several protein disulfide isomerase family proteins. PLoS ONE 6:e26454. doi: 10.1371/journal.pone.0026454
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Anderson, P., and Kedersha, N. (2008). Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 33, 141–150. doi: 10.1016/j.tibs.2007.12.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Anfossi, M., Vuono, R., Maletta, R., Virdee, K., Mirabelli, M., Colao, R., et al. (2011). Compound heterozygosity of 2 novel MAPT mutations in frontotemporal dementia. Neurobiol. Aging 32, 757.e1-757.e11. doi: 10.1016/j.neurobiolaging.2010.12.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi: 10.1016/j.bbrc.2006.10.093
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Arriagada, P. V., Marzloff, K., and Hyman, B. T. (1992). Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer's disease. Neurology 42, 1681–1688. doi: 10.1212/WNL.42.9.1681
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ash, P. E., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W.-L., Dejesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Avendaño-Vázquez, S. E., Dhir, A., Bembich, S., Buratti, E., Proudfoot, N., and Baralle, F. (2012). Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 26, 1679–1684. doi: 10.1101/gad.194829.112
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ayala, Y. M., Pantano, S., D'Ambrogio, A., Buratti, E., Brindisi, A., Marchetti, C., et al. (2005). Human, Drosophila, and C.elegans TDP43: nucleic acid binding properties and splicing regulatory function. J. Mol. Biol. 348, 575–588. doi: 10.1016/j.jmb.2005.02.038
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baker, M., Mackenzie, I. R., Pickering-Brown, S. M., Gass, J., Rademakers, R., Lindholm, C., et al. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. doi: 10.1038/nature05016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barmada, S. J., Skibinski, G., Korb, E., Rao, E. J., and Wu, J. Y. (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial ALS 30. J. Neurosci. 30, 639–649. doi: 10.1523/JNEUROSCI.4988-09.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. doi: 10.1016/j.cell.2009.01.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bartolome, F., Wu, H.-C., Burchell, V. S., Preza, E., Wray, S., Mahoney, C. J., et al. (2013). Pathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levels. Neuron 78, 57–64. doi: 10.1016/j.neuron.2013.02.028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bateman, A., and Bennett, H. P. (2009). The granulin gene family: from cancer to dementia. Bioessays 31, 1245–1254. doi: 10.1002/bies.200900086
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Belly, A., Moreau-Gachelin, F., Sadoul, R., and Goldberg, Y. (2005). Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci. Lett. 379, 152–157. doi: 10.1016/j.neulet.2004.12.071
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Belzil, V. V., Gendron, T. F., and Petrucelli, L. (2013). RNA-mediated toxicity in neurodegenerative disease. Mol. Cell. Neurosci. 56, 406–419. doi: 10.1016/j.mcn.2012.12.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Benajiba, L., Le Ber, I., Camuzat, A., Lacoste, M., Thomas-Anterion, C., Couratier, P., et al. (2009). TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann. Neurol. 65, 470–473. doi: 10.1002/ana.21612
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bernardi, L., Frangipane, F., Smirne, N., Colao, R., Puccio, G., Curcio, S. M., et al. (2012). Epidemiology and genetics of frontotemporal dementia: a door-to-door survey in southern Italy. Neurobiol. Aging 33, 2948.e1-2948.e10. doi: 10.1016/j.neurobiolaging.2012.06.017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bernstein, E., Caudy, A. A., Hammond, S. M., and Hannon, G. J. (2001). Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409, 363–366. doi: 10.1038/35053110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bersano, A., Del Bo, R., Lamperti, C., Ghezzi, S., Fagiolari, G., Fortunato, F., et al. (2009). Inclusion body myopathy and frontotemporal dementia caused by a novel VCP mutation. Neurobiol. Aging 30, 752–758. doi: 10.1016/j.neurobiolaging.2007.08.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bertolotti, A., Lutz, Y., and Heard, D. (1996). hTAF (II) 68, a novel RNA/ssDNA-binding protein with homology to the pro-oncoproteins TLS/FUS and EWS is associated with both TFIID and RNA polymerase II. EMBO J. 15, 5022–5031.
Bessi, V., Bagnoli, S., Nacmias, B., Tedde, A., Sorbi, S., and Bracco, L. (2010). Semantic dementia associated with mutation V363I in the tau gene. J. Neurol. Sci. 296, 112–114. doi: 10.1016/j.jns.2010.06.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bhandari, V., Daniel, R., Lim, P. S., and Bateman, A. (1996). Structural and functional analysis of a promoter of the human granulin/epithelin gene. Biochem. J. 319(Pt 2), 441–447.
Binder, L. I., Frankfurter, A., and Rebhun, L. I. (1985). The distribution of tau in the mammalian central nervous system. J. Cell Biol. 101, 1371–1378. doi: 10.1083/jcb.101.4.1371
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Birney, E., Stamatoyannopoulos, J. A., Dutta, A., Guigó, R., Gingeras, T. R., and Margulies, E. H. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816. doi: 10.1038/nature05874
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bodon, G., Chassefeyre, R., Pernet-Gallay, K., Martinelli, N., Effantin, G., Hulsik, D. L., et al. (2011). Charged multivesicular body protein 2B (CHMP2B) of the endosomal sorting complex required for transport-III (ESCRT-III) polymerizes into helical structures deforming the plasma membrane. J. Biol. Chem. 286, 40276–40286. doi: 10.1074/jbc.M111.283671
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bond, A. M., Vangompel, M. J., Sametsky, E. A., Clark, M. F., Savage, J. C., Disterhoft, J. F., et al. (2009). Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat. Neurosci. 12, 1020–1027. doi: 10.1038/nn.2371
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Borroni, B., Archetti, S., Alberici, A., Agosti, C., Gennarelli, M., Bigni, B., et al. (2008). Progranulin genetic variations in frontotemporal lobar degeneration: evidence for low mutation frequency in an Italian clinical series. Neurogenetics 9, 197–205. doi: 10.1007/s10048-008-0127-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Borroni, B., Bonvicini, C., Alberici, A., Buratti, E., Agosti, C., Archetti, S., et al. (2009). Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat. 30, E974–E983. doi: 10.1002/humu.21100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bosco, D. A., Lemay, N., Ko, H. K., Zhou, H., Burke, C., Kwiatkowski, T. J., et al. (2010). Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 19, 4160–4175. doi: 10.1093/hmg/ddq335
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bottos, A., Rissone, A., Bussolino, F., and Arese, M. (2011). Neurexins and neuroligins: synapses look out of the nervous system. Cell. Mol. Life Sci. 68, 2655–2666. doi: 10.1007/s00018-011-0664-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Broe, M., Kril, J., and Halliday, G. M. (2004). Astrocytic degeneration relates to the severity of disease in frontotemporal dementia. Brain 127, 2214–2220. doi: 10.1093/brain/awh250
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bronfman, F. C., Escudero, C. A., and Weis, J. K. A. (2007). Endosomal transport of neurotrophins: roles in signaling and neurodegenerative diseases. Dev. Neurobiol. 67, 1183–1203. doi: 10.1002/dneu.20513
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bronner, I. F., Rizzu, P., Seelaar, H., van Mil, S. E., Anar, B., Azmani, A., et al. (2007). Progranulin mutations in Dutch familial frontotemporal lobar degeneration. Eur. J. Hum. Genet. 15, 369–374. doi: 10.1038/sj.ejhg.5201772
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Broustal, O., Camuzat, A., Guillot-Noël, L., Guy, N., Millecamps, S., Deffond, D., et al. (2010). FUS mutations in frontotemporal lobar degeneration with amyotrophic lateral sclerosis. J. Alzheimers Dis. 22, 765–769.
Brown, J., Lantos, P. L., Roques, P., Fidani, L., and Rossor, M. N. (1996). Familial dementia with swollen achromatic neurons and corticobasal inclusion bodies: a clinical and pathological study. J. Neurol. Sci. 135, 21–30. doi: 10.1016/0022-510X(95)00236-U
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buchan, J. R., Kolaitis, R.-M., Taylor, J. P., and Parker, R. (2013). Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153, 1461–1474. doi: 10.1016/j.cell.2013.05.037
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buchan, J. R., and Parker, R. (2009). Eukaryotic stress granules: the ins and outs of translation. Mol. Cell. 36, 932–941. doi: 10.1016/j.molcel.2009.11.020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bugiani, O. (2007). The many ways to frontotemporal degeneration and beyond. Neurol. Sci. 28, 241–244. doi: 10.1007/s10072-007-0829-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bugiani, O., Murrell, J. R., Giaccone, G., Hasegawa, M., Ghigo, G., Tabaton, M., et al. (1999). Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J. Neuropathol. Exp. Neurol. 58, 667–677. doi: 10.1097/00005072-199906000-00011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buratti, E., and Baralle, F. E. (2001). Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem. 276, 36337–36343. doi: 10.1074/jbc.M104236200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buratti, E., and Baralle, F. E. (2010). The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 7, 420–429. doi: 10.4161/rna.7.4.12205
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buratti, E., Brindisi, A., Giombi, M., Tisminetzky, S., Ayala, Y. M., and Baralle, F. E. (2005). TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 280, 37572–37584. doi: 10.1074/jbc.M505557200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buratti, E., De Conti, L., Stuani, C., Romano, M., Baralle, M., and Baralle, F. (2010). Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J. 277, 2268–2281. doi: 10.1111/j.1742-4658.2010.07643.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Butler, G. S., Dean, R. A., Tam, E. M., and Overall, C. M. (2008). Pharmacoproteomics of a metalloproteinase hydroxamate inhibitor in breast cancer cells: dynamics of membrane type 1 matrix metalloproteinase-mediated membrane protein shedding. Mol. Cell. Biol. 28, 4896–4914. doi: 10.1128/MCB.01775-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cannon, A., Baker, M., Boeve, B., Josephs, K., Knopman, D., Petersen, R., et al. (2006). CHMP2B mutations are not a common cause of frontotemporal lobar degeneration. Neurosci. Lett. 398, 83–84. doi: 10.1016/j.neulet.2005.12.056
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Carlton, J. G., and Martin-Serrano, J. (2007). Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Science 316, 1908–1912. doi: 10.1126/science.1143422
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cesana, M., Cacchiarelli, D., Legnini, I., Santini, T., Sthandier, O., Chinappi, M., et al. (2011). A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147, 358–369. doi: 10.1016/j.cell.2011.09.028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chai, X., Dage, J. L., and Citron, M. (2012). Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol. Dis. 48, 356–366. doi: 10.1016/j.nbd.2012.05.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, L., and Carmichael, G. (2009). Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Mol. Cell 35, 467–478. doi: 10.1016/j.molcel.2009.06.027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen-Plotkin, A. S., Xiao, J., Geser, F., Martinez-Lage, M., Grossman, M., Unger, T., et al. (2010). Brain progranulin expression in GRN-associated frontotemporal lobar degeneration. Acta Neuropathol. 119, 111–122. doi: 10.1007/s00401-009-0576-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chiò, A., Calvo, A., Moglia, C., Restagno, G., Ossola, I., Brunetti, M., et al. (2010). Amyotrophic lateral sclerosis-frontotemporal lobar dementia in 3 families with p.Ala382Thr TARDBP mutations. Arch. Neurol. 67, 1002–1009. doi: 10.1001/archneurol.2010.173
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clark, M. B., Amaral, P. P., Schlesinger, F. J., Dinger, M. E., Taft, R. J., Rinn, J. L., et al. (2011). The reality of pervasive transcription. PLoS Biol. 9:e1000625; discussion e1001102. doi: 10.1371/journal.pbio.1000625
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clovis, Y. M., Enard, W., Marinaro, F., Huttner, W. B., and De Pietri Tonelli, D. (2012). Convergent repression of Foxp2 3′UTR by miR-9 and miR-132 in embryonic mouse neocortex: implications for radial migration of neurons. Development 139, 3332–3342. doi: 10.1242/dev.078063
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Colombrita, C., Onesto, E., Megiorni, F., Pizzuti, A., Baralle, F. E., Buratti, E., et al. (2012). TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J. Biol. Chem. 287, 15635–15647. doi: 10.1074/jbc.M111.333450
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Colombrita, C., Onesto, E., Tiloca, C., Ticozzi, N., Silani, V., and Ratti, A. (2011). RNA-binding proteins and RNA metabolism: a new scenario in the pathogenesis of Amyotrophic lateral sclerosis. Arch. Ital. Biol. 149, 83–99. doi: 10.4449/aib.v149i1.1261
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Colombrita, C., Zennaro, E., Fallini, C., Weber, M., Sommacal, A., Buratti, E., et al. (2009). TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 111, 1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cooper, T. A., Wan, L., and Dreyfuss, G. (2009). RNA and disease. Cell 136, 777–793. doi: 10.1016/j.cell.2009.02.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Coppola, G., Karydas, A., Rademakers, R., Wang, Q., Baker, M., Hutton, M., et al. (2008). Gene expression study on peripheral blood identifies progranulin mutations. Ann. Neurol. 64, 92–96. doi: 10.1002/ana.21397
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Corrado, L., Ratti, A., Gellera, C., Buratti, E., Castellotti, B., Carlomagno, Y., et al. (2009). High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum. Mutat. 30, 688–694. doi: 10.1002/humu.20950
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Couchie, D., Mavilia, C., Georgieff, I. S., Liem, R. K., Shelanski, M. L., and Nunez, J. (1992). Primary structure of high molecular weight tau present in the peripheral nervous system. Proc. Natl. Acad. Sci. U.S.A. 89, 4378–4381. doi: 10.1073/pnas.89.10.4378
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cox, L. E., Ferraiuolo, L., Goodall, E. F., Heath, P. R., Higginbottom, A., Mortiboys, H., et al. (2010). Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 5:e9872. doi: 10.1371/journal.pone.0009872
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Craig, A., Graf, E., and Linhoff, M. (2006). How to build a central synapse: clues from cell culture. Trends Neurosci. 29, 8–20. doi: 10.1016/j.tins.2005.11.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cruts, M., Gijselinck, I., Van der Zee, J., Engelborghs, S., Wils, H., Pirici, D., et al. (2006). Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924. doi: 10.1038/nature05017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cruts, M., Theuns, J., and Van Broeckhoven, C. (2012). Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 33, 1340–1344. doi: 10.1002/humu.22117
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cruts, M., and Van Broeckhoven, C. (2008). Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet. 24, 186–194. doi: 10.1016/j.tig.2008.01.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Custer, S. K., Neumann, M., Lu, H., Wright, A. C., and Taylor, J. P. (2010). Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum. Mol. Genet. 19, 1741–1755. doi: 10.1093/hmg/ddq050
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
D'Souza, I., Poorkaj, P., Hong, M., Nochlin, D., Lee, V. M., Bird, T. D., et al. (1999). Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc. Natl. Acad. Sci. U.S.A. 96, 5598–5603. doi: 10.1073/pnas.96.10.5598
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Da Cruz, S., and Cleveland, D. W. (2011). Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 21, 904–919. doi: 10.1016/j.conb.2011.05.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dai, R. M., and Li, C. C. (2001). Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat. Cell Biol. 3, 740–744. doi: 10.1038/35087056
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Daniel, R., Daniels, E., He, Z., and Bateman, A. (2003). Progranulin (acrogranin/PC cell-derived growth factor/granulin-epithelin precursor) is expressed in the placenta, epidermis, microvasculature, and brain during murine development. Dev. Dyn. 227, 593–599. doi: 10.1002/dvdy.10341
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Daniel, R., He, Z., Carmichael, K. P., Halper, J., and Bateman, A. (2000). Cellular localization of gene expression for progranulin. J. Histochem. Cytochem. 48, 999–1009. doi: 10.1177/002215540004800713
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Calignon, A., Polydoro, M., Suárez-Calvet, M., William, C., Adamowicz, D. H., Kopeikina, K. J., et al. (2012). Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73, 685–697. doi: 10.1016/j.neuron.2011.11.033
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
DeLaBarre, B., Christianson, J. C., Kopito, R. R., and Brunger, A. T. (2006). Central pore residues mediate the p97/VCP activity required for ERAD. Mol. Cell 22, 451–462. doi: 10.1016/j.molcel.2006.03.036
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deng, H.-X., Zhai, H., Bigio, E. H., Yan, J., Fecto, F., Ajroud, K., et al. (2010). FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 67, 739–748. doi: 10.1002/ana.22051
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Díaz-Cueto, L., Stein, P., Jacobs, A., Schultz, R. M., and Gerton, G. L. (2000). Modulation of mouse preimplantation embryo development by acrogranin (epithelin/granulin precursor). Dev. Biol. 217, 406–418. doi: 10.1006/dbio.1999.9564
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Di Carlo, V., Grossi, E., Laneve, P., Morlando, M., Dini Modigliani, S., Ballarino, M., et al. (2013). TDP-43 regulates the microprocessor complex activity during in vitro neuronal differentiation. Mol. Neurobiol. 48, 952–963. doi: 10.1007/s12035-013-8564-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dickson, D. W. (1997). Neurodegenerative diseases with cytoskeletal pathology: a biochemical classification. Ann. Neurol. 42, 541–544. doi: 10.1002/ana.410420403
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dickson, D. W. (2008). TDP-43 immunoreactivity in neurodegenerative disorders: disease versus mechanism specificity. Acta Neuropathol. 115, 147–149. doi: 10.1007/s00401-007-0323-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dickson, J. R., Kruse, C., Montagna, D. R., Finsen, B., and Wolfe, M. S. (2013). Alternative polyadenylation and miR-34 family members regulate tau expression. J. Neurochem. 127, 739–749. doi: 10.1111/jnc.12437
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dini Modigliani, S., Morlando, M., Errichelli, L., Sabatelli, M., and Bozzoni, I. (2014). An ALS-associated mutation in the FUS 3′-UTR disrupts a microRNA-FUS regulatory circuitry. Nat. Commun. 5:4335. doi: 10.1038/ncomms5335
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Djamshidian, A., Schaefer, J., Haubenberger, D., Stogmann, E., Zimprich, F., Auff, E., et al. (2009). A novel mutation in the VCP gene (G157R) in a German family with inclusion-body myopathy with Paget disease of bone and frontotemporal dementia. Muscle Nerve 39, 389–391. doi: 10.1002/mus.21225
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dormann, D., Rodde, R., Edbauer, D., Bentmann, E., Fischer, I., Hruscha, A., et al. (2010). ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. EMBO J. 29, 2841–2857. doi: 10.1038/emboj.2010.143
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eddy, J., and Maizels, N. (2008). Conserved elements with potential to form polymorphic G-quadruplex structures in the first intron of human genes. Nucleic Acids Res. 36, 1321–1333. doi: 10.1093/nar/gkm1138
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Elia, N., and Sougrat, R. (2011). Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc Natl Acad Sci U.S.A. 108, 4846–4851. doi: 10.1073/pnas.1102714108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Erickson, S. L., and Lykke-Andersen, J. (2011). Cytoplasmic mRNP granules at a glance. J. Cell Sci. 1, 293–297. doi: 10.1242/jcs.072140
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fan, Z., Chen, X., and Chen, R. (2014). Transcriptome-wide analysis of TDP-43 binding small RNAs identifies miR-NID1 (miR-8485), a novel miRNA that represses NRXN1 expression. Genomics 103, 76–82. doi: 10.1016/j.ygeno.2013.06.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farg, M. A., Sundaramoorthy, V., Sultana, M. J., Yang, S., Atkinson, R. A. K., Levina, V., et al. (2014). C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 23, 3579–3595. doi: 10.1093/hmg/ddu068
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Feng, J., Bi, C., Clark, B. S., Mady, R., Shah, P., and Kohtz, J. D. (2006). The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. 20, 1470–1484. doi: 10.1101/gad.1416106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ferrari, R., Kapogiannis, D., Huey, E. D., Grafman, J., Hardy, J., and Momeni, P. (2010). Novel missense mutation in charged multivesicular body protein 2B in a patient with frontotemporal dementia. Alzheimer Dis. Assoc. Disord. 24, 397–401. doi: 10.1097/WAD.0b013e3181df20c7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Finch, N., Baker, M., Crook, R., Swanson, K., Kuntz, K., Surtees, R., et al. (2009). Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 132, 583–591. doi: 10.1093/brain/awn352
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fox, A. H., and Lamond, A. I. (2010). Paraspeckles. Cold Spring Harb. Perspect. Biol. 2:a000687. doi: 10.1101/cshperspect.a000687
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fratta, P., Mizielinska, S., Nicoll, A. J., Zloh, M., Fisher, E. M. C., Parkinson, G., et al. (2012). C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci. Rep. 2:1016. doi: 10.1038/srep01016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Freibaum, B. D., Chitta, R., High, A. A., and Taylor, J. P. (2011). Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 9, 1104–1120. doi: 10.1021/pr901076y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fujii, R., Okabe, S., Urushido, T., Inoue, K., Yoshimura, A., Tachibana, T., et al. (2005). The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr. Biol. 15, 587–593. doi: 10.1016/j.cub.2005.01.058
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fujii, R., and Takumi, T. (2005). TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J. Cell Sci. 118, 5755–5765. doi: 10.1242/jcs.02692
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gabryelewicz, T., Masellis, M., Berdynski, M., Bilbao, J. M., Rogaeva, E., St George-Hyslop, P., et al. (2010). Intra-familial clinical heterogeneity due to FTLD-U with TDP-43 proteinopathy caused by a novel deletion in progranulin gene (PGRN). J. Alzheimer Dis. 22, 1123–1133. doi: 10.3233/JAD-2010-101413
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gallo, J.-M., Jin, P., Thornton, C. A., Lin, H., Robertson, J., D'Souza, I., et al. (2005). The role of RNA and RNA processing in neurodegeneration. J. Neurosci. 25, 10372–10375. doi: 10.1523/JNEUROSCI.3453-05.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Garzon, R., Marcucci, G., and Croce, C. (2010). Targeting microRNAs in cancer: rationale, strategies and challenges. Nat. Rev. Drug Discov. 9, 775–789. doi: 10.1038/nrd3179
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gass, J., Cannon, A., Mackenzie, I. R., Boeve, B., Baker, M., Adamson, J., et al. (2006). Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet. 15, 2988–3001. doi: 10.1093/hmg/ddl241
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gendron, T. F., Bieniek, K. F., Zhang, Y.-J., Jansen-West, K., Ash, P. E., Caulfield, T., et al. (2013). Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126, 829–844. doi: 10.1007/s00401-013-1192-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ghanim, M., Guillot-Noel, L., Pasquier, F., Jornea, L., Deramecourt, V., Dubois, B., et al. (2010). CHMP2B mutations are rare in French families with frontotemporal lobar degeneration. J. Neurol. 257, 2032–2036. doi: 10.1007/s00415-010-5655-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ghazi-Noori, S., Froud, K. E., Mizielinska, S., Powell, C., Smidak, M., Fernandez de Marco, M., et al. (2012). Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain 135, 819–832 doi: 10.1093/brain/aws006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ghildiyal, M., and Zamore, P. D. (2009). Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 10, 94–108. doi: 10.1038/nrg2504
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Giaccone, G., Rossi, G., Di Fede, G., Marcon, G., Farina, L., Sacco, L., et al. (2004). Familial frontotemporal dementia is associated with the novel tau mutation T427M. Neurobiol. Aging 25, 449–450. doi: 10.1016/S0197-4580(04)81481-7
Gijselinck, I., Van der Zee, J., Engelborghs, S., Goossens, D., Peeters, K., Mattheijssens, M., et al. (2008). Progranulin locus deletion in frontotemporal dementia. Hum. Mutat. 29, 53–58 doi: 10.1002/humu.20651
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gijselinck, I., Van Langenhove, T., van der Zee, J., Sleegers, K., Philtjens, S., Kleinberger, G., et al. (2012). A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 11, 54–65. doi: 10.1016/S1474-4422(11)70261-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goedert, M., and Jakes, R. (2005). Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta. 1739, 240–250. doi: 10.1016/j.bbadis.2004.08.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grishok, A., Pasquinelli, A. E., Conte, D., Li, N., Parrish, S., Ha, I., et al. (2001). Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 106, 23–34 doi: 10.1016/S0092-8674(01)00431-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grover, A., England, E., Baker, M., Sahara, N., Adamson, J., Granger, B., et al. (2003). A novel tau mutation in exon 9 (1260V) causes a four-repeat tauopathy. Exp. Neurol. 184, 131–140. doi: 10.1016/S0014-4886(03)00393-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grover, A., Houlden, H., Baker, M., Adamson, J., Lewis, J., Prihar, G., et al. (1999). J. Biol. Chem. 274, 15134–15143. doi: 10.1074/jbc.274.21.15134
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guinto, J. B., Ritson, G. P., Taylor, J. P., and Forman, M. S. (2007). Valosin-containing protein and the pathogenesis of frontotemporal dementia associated with inclusion body myopathy. Acta Neuropathol. 114, 55–61. doi: 10.1007/s00401-007-0224-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guo, A., Tapia, L., Bamji, S. X., Cynader, M. S., and Jia, W. (2010). Progranulin deficiency leads to enhanced cell vulnerability and TDP-43 translocation in primary neuronal cultures. Brain Res. 1366, 1–8. doi: 10.1016/j.brainres.2010.09.099
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gutierrez, M. G., Munafo, D. B., Beron, W., and Colombo, M. I. (2004). Rab7 is requie for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 117(Pt 13), 2687–2697. doi: 10.1242/jcs.01114
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guyant-Maréchal, L., Laquerrière, A., Duyckaerts, C., Dumanchin, C., Bou, J., Dugny, F., et al. (2006). Valosin-containing protein gene mutations: clinical and neuropathologic features. Neurology 67, 644–651. doi: 10.1212/01.wnl.0000225184.14578.d3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Haeusler, A. R., Donnelly, C. J., Periz, G., Simko, E. A. J., Shaw, P. G., Kim, M.-S., et al. (2014). C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200. doi: 10.1038/nature13124
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Haubenberger, D., Bittner, R. E., Rauch-Shorny, S., Zimprich, F., Mannhalter, C., Wagner, L., et al. (2005). Inclusion body myopathy and Paget disease is linked to a novel mutation in the VCP gene. Neurology 65, 1304–1305 doi: 10.1212/01.wnl.0000180407.15369.92
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hayashi, S., Toyoshima, Y., Hasegawa, M., Umeda, Y., Wakabayashi, K., Tokiguchi, S., et al. (2002). Late-onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann. Neurol. 51, 525–530. doi: 10.1002/ana.10163
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hayashi, Y. K. (2013). Inclusion body myopathy with Paget's disease of bone and frontotemporal dementia. Rinsho Shinkeigaku 53, 947–950. doi: 10.5692/clinicalneurol.53.947
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
He, Z., and Bateman, A. (1999). Progranulin gene expression regulates epithelial cell growth and promotes tumor growth in vivo. Cancer Res. 3222–3229.
He, Z., and Bateman, A. (2003). Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J. Mol. Med. (Berl). 81, 600–612. doi: 10.1007/s00109-003-0474-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
He, Z., Ong, C. H. P., Halper, J., and Bateman, A. (2003). Progranulin is a mediator of the wound response. Nat. Med. 9, 225–229. doi: 10.1038/nm816
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hébert, S. S., Horré, K., Nicolaï, L., Papadopoulou, A. S., Mandemakers, W., Silahtaroglu, A. N., et al. (2008). Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. U.S.A. 105, 6415–1520. doi: 10.1073/pnas.0710263105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hermeking, H. (2010). The miR-34 family in cancer and apoptosis. Cell Death Differ. 2, 193–199. doi: 10.1038/cdd.2009.56
Hirabayashi, M., Inoue, K., Tanaka, K., Nakadate, K., Ohsawa, Y., Kamei, Y., et al. (2001). VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ. 8, 977–984. doi: 10.1038/sj.cdd.4400907
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hoell, J. I., Larsson, E., Runge, S., Nusbaum, J. D., Duggimpudi, S., Farazi, T., et al. (2011). RNA targets of wild-type and mutant FET family proteins. Nat. Struct. Mol. Biol. 18, 1428–1431. doi: 10.1038/nsmb.2163
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hoque, M., Mathews, M. B., and Pe'ery, T. (2010). Progranulin (granulin/epithelin precursor) and its constituent granulin repeats repress transcription from cellular promoters. J. Cell. Physiol. 223, 224–233. doi: 10.1002/jcp.22031
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hoque, M., Young, T., and Lee, C. (2003). The growth factor granulin interacts with cyclin T1 and modulates P-TEFb-dependent transcription. Cell. Biol. 23, 1688–1702.
Huey, E. D., Ferrari, R., Moreno, J. H., Jensen, C., Morris, C. M., Potocnik, F., et al. (2012). FUS and TDP43 genetic variability in FTD and CBS. Neurobiol. Aging 33, 1016.e9–1016.e17. doi: 10.1016/j.neurobiolaging.2011.08.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huey, E. D., Grafman, J., Wassermann, E. M., Pietrini, P., Tierney, M. C., Ghetti, B., et al. (2006). Characteristics of frontotemporal dementia patients with a progranulin mutation. Ann. Neurol. 60, 374–380. doi: 10.1002/ana.20969
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huntzinger, E., and Izaurralde, E. (2011). Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat. Rev. Genet. 12, 99–110. doi: 10.1038/nrg2936
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huppert, J. L., Bugaut, A., Kumari, S., and Balasubramanian, S. (2008). G-quadruplexes: the beginning and end of UTRs. Nucleic Acids Res. 36, 6260–6268. doi: 10.1093/nar/gkn511
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., et al. (1998). Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. doi: 10.1038/31508
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hutvagner, G., McLachlan, J., Pasquinelli, A. E., Balint, E., Tuschl, T., and Zamore, P. D. (2001). A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293, 834–838. doi: 10.1126/science.1062961
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iba, M., Guo, J. L., Mcbride, J. D., Zhang, B., Trojanowski, J. Q., and Lee, V. M. (2013). Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like Tauopathy J. Neurosci. 33, 1024–1037. doi: 10.1523/JNEUROSCI.2642-12.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iijima, M., Tabira, T., Poorkaj, P., Schellenberg, G. D., Trojanowski, J. Q., Lee, V. M., et al. (1999). A distinct familial presenile dementia with a novel missense mutation in the tau gene. Neuroreport 10, 497–501. doi: 10.1097/00001756-199902250-00010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iko, Y., Kodama, T. S., Kasai, N., Oyama, T., Morita, E. H., Muto, T., et al. (2004). Domain architectures and characterization of an RNA-binding protein, TLS. J. Biol. Chem. 279, 44834–44840. doi: 10.1074/jbc.M408552200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iovino, M., Pfisterer, U., Holton, J. L., Lashley, T., Swingler, R. J., Calo, L., et al. (2014). The novel MAPT mutation K,98E: mechanisms of mutant tau toxicity, brain pathology and tau expression in induced fibroblast-derived neurons. Acta Neuropathol. 127, 283–295. doi: 10.1007/s00401-013-1219-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iseki, E., Matsumura, T., Marui, W., Hino, H., Odawara, T., Sugiyama, N., et al. (2001). Familial frontotemporal dementia and parkinsonism with a novel N296H mutation in exon 10 of the tau gene and a widespread tau accumulation in the glial cells. Acta Neuropathol. 102, 285–292.
Ishigaki, S., Hishikawa, N., Niwa, J., Iemura, S., Natsume, T., Hori, S., et al. (2004). Physical and functional interaction between Dorfin and Valosin-containing protein that are colocalized in ubiquitylated inclusions in neurodegenerative disorders. J. Biol. Chem. 279, 51376–51385. doi: 10.1074/jbc.M406683200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ishigaki, S., Masuda, A., Fujioka, Y., Iguchi, Y., Katsuno, M., Shibata, A., et al. (2012). Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci. Rep. 2, 529. doi: 10.1038/srep00529
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ito, D., Seki, M., Tsunoda, Y., Uchiyama, H., and Suzuki, N. (2011). Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann. Neurol. 69, 152–162. doi: 10.1002/ana.22246
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iyer, A., Lapointe, N. E., Zielke, K., Berdynski, M., Guzman, E., Barczak, A., et al. (2013). A novel MAPT mutation, G55R, in a frontotemporal dementia patient leads to altered Tau function. PLoS ONE 8:e76409. doi: 10.1371/journal.pone.0076409
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jacquin, A., Rouaud, O., Soichot, P., Bejot, Y., Dygai-Cochet, I., Sarazin, M., et al. (2013). Psychiatric presentation of frontotemporal dementia associated with inclusion body myopathy due to the VCP mutation (R155H) in a French family. Case Rep. Neurol. 5, 187–194. doi: 10.1159/000356481
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Janssens, J., and Van Broeckhoven, C. (2013). Pathological mechanisms underlying TDP-43 driven neurodegeneration in FTLD-ALS spectrum disorders. Hum. Mol. Genet. 22, R77–R87. doi: 10.1093/hmg/ddt349
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jiao, J., Herl, L. D., Farese, R. V., and Gao, F.-B. (2010). MicroRNA-29b regulates the expression level of human progranulin, a secreted glycoprotein implicated in frontotemporal dementia. PLoS ONE 5:e10551. doi: 10.1371/journal.pone.0010551
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Johnson, J. O., Mandrioli, J., Benatar, M., Van Deerlin, V. M., Trojanowski, J. Q., Mora, G., et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. doi: 10.1016/j.neuron.2010.11.036
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Johnson, J. O., Pioro, E. P., Boehringer, A., Chia, R., Feit, H., Renton, A. E., et al. (2014). Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 17, 664–666. doi: 10.1038/nn.3688
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ju, J.-S., Fuentealba, R. A., Miller, S. E., Jackson, E., Piwnica-Worms, D., Baloh, R. H., et al. (2009). Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 187, 875–888. doi: 10.1083/jcb.200908115
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kabashi, E., Lin, L., Tradewell, M. L., Dion, P. A., Bercier, V., Bourgouin, P., et al. (2010). Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 19, 671–683. doi: 10.1093/hmg/ddp534
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., McConkey, B. J., Vande Velde, C., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi: 10.1038/ng.132
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kaleem, M., Zhao, A., Hamshere, M., and Myers, A. J. (2007). Identification of a novel valosin-containing protein polymorphism in late-onset Alzheimer's disease. Neurodegener Dis. 4, 376–381. doi: 10.1159/000105158
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kara, E., Ling, H., Pittman, A. M., Shaw, K., de Silva, R., Simone, R., et al. (2012). The MAPT p.A152T variant is a risk factor associated with tauopathies with atypical clinical and neuropathological features. Neurobiol. Aging. 33, 2231.e7-2231.e14. doi: 10.1016/j.neurobiolaging.2012.04.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Katzmann, D. J., Odorizzi, G., and Emr, S. D. (2002). Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 3, 893–905. doi: 10.1038/nrm973
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kawahara, Y., and Mieda-Sato, A. (2012). TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. U.S.A. 109, 3347–3352. doi: 10.1073/pnas.1112427109
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kessenbrock, K., Fröhlich, L., Sixt, M., Lämmermann, T., Pfister, H., Bateman, A., et al. (2008). Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J. Clin. Invest. 118, 2438–2447. doi: 10.1172/JCI34694
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kikin, O., Zappala, Z., D'Antonio, L., and Bagga, P. S. (2008). GRSDB2 and GRS_UTRdb: databases of quadruplex forming G-rich sequences in pre-mRNAs and mRNAs. Nucleic Acids Res. 36, D141-D8.(Database issue):D141-8. doi: 10.1093/nar/gkm982
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, E.-J., Kwon, J. C., Park, K. H., Park, K.-W., Lee, J.-H., Choi, S. H., et al. (2014). Clinical and genetic analysis of MAPT, GRN, and C9orf72 genes in Korean patients with frontotemporal dementia. Neurobiol. Aging. 35, 1213.e13–1213.e17. doi: 10.1016/j.neurobiolaging.2013.11.033
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, H.-J., Jeon, B. S., Yun, J. Y., Seong, M.-W., Park, S. S., and Lee, J.-Y. (2010a). Screening for MAPT and PGRN mutations in Korean patients with PSP/CBS/FTD. Parkinsonism Relat. Disord. 16, 305–306. doi: 10.1016/j.parkreldis.2010.01.004
Kim, S. H., Shanware, N. P., Bowler, M. J., and Tibbetts, R. S. (2010b). Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. J. Biol. Chem. 285, 34097–34105. doi: 10.1074/jbc.M110.154831
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, V. N. (2004). MicroRNA precursors in motion: exportin-5 mediates their nuclear export. Trends Cell Biol. 14, 156–159. doi: 10.1016/j.tcb.2004.02.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kimonis, V. E., Mehta, S. G., Fulchiero, E. C., Thomasova, D., Boycott, K., Neilan, E. G., et al. (2008). Clinical studies in familial VCP myopathy associated with paget disease of bone and frontotemporal dementia. Am. J. Med. Genet. A. 28, 745–757. doi: 10.1002/ajmg.a.31862
King, I., Yartseva, V., Salas, D., Kumar, A., Heidersbach, A., Ando, D. M., et al. (2014). The RNA binding protein TDP-43 selectively disrupts MicroRNA-1/206 incorporation into the RNA-induced silencing complex. J. Biol. Chem. 289, 14263–14271. doi: 10.1074/jbc.M114.561902
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kobayashi, T., Ota, S., Tanaka, K., Ito, Y., Hasegawa, M., Umeda, Y., et al. (2003). Novel L266V mutation of the tau gene causes frontotemporal dementia with a unique tau pathology. Ann. Neurol. 53, 133–137. doi: 10.1002/ana.10447
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kouri, N., Carlomagno, Y., Baker, M., Liesinger, A. M., Caselli, R. J., Wszolek, Z. K., et al. (2014). Novel mutation in MAPT exon 13 (p.N410H) causes corticobasal degeneration. Acta Neuropathol. 127, 271–282. doi: 10.1007/s00401-013-1193-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kovach, M. J., Waggoner, B., Leal, S. M., Gelber, D., Khardori, R., Levenstien, M., et al. (2001). Clinical delineation and localization to chromosome 9p13.3-p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol. Genet. Metab. 74, 458–475. doi: 10.1006/mgme.2001.3256
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kovacs, G. G., Murrell, J. R., Horvath, S., Haraszti, L., Majtenyi, K., Molnar, M. J., et al. (2009). TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov. Disord. 24, 1843–1847. doi: 10.1002/mds.22697
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kovacs, G. G., Pittman, A., Revesz, T., Luk, C., Lees, A., Kiss, E., et al. (2008). MAPT S305I mutation: implications for argyrophilic grain disease. Acta Neuropathol. 116, 103–118. doi: 10.1007/s00401-007-0322-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kumar, K. R., Needham, M., Mina, K., Davis, M., Brewer, J., Staples, C., et al. (2010). Two Australian families with inclusion-body myopathy, Paget's disease of bone and frontotemporal dementia: novel clinical and genetic findings. Neuromuscul. Disord. 20, 330–334. doi: 10.1016/j.nmd.2010.03.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kwiatkowski, T. J., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lagier-Tourenne, C., Baughn, M., Rigo, F., Sun, S., Liu, P., Li, H.-R., et al. (2013). Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. U.S.A. 110, E4530–E4539. doi: 10.1073/pnas.1318835110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lagier-Tourenne, C., Polymenidou, M., and Cleveland, D. W. (2010). TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 19, R46–R64. doi: 10.1093/hmg/ddq137
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lagier-Tourenne, C., Polymenidou, M., Hutt, K. R., Vu, A. Q., Baughn, M., Huelga, S. C., et al. (2012). Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 15, 1488–1497. doi: 10.1038/nn.3230
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. doi: 10.1038/35057062
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lanska, D. J., Currier, R. D., Cohen, M., Gambetti, P., Smith, E. E., Bebin, J., et al. (1994). Familial progressive subcortical gliosis. Neurology 44, 1633–1643. doi: 10.1212/WNL.44.9.1633
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Law, W. J., Cann, K. L., and Hicks, G. G. (2006). TLS, EWS and TAF15: a model for transcriptional integration of gene expression. Brief. Funct. Genomic. Proteomic. 5, 8–14. doi: 10.1093/bfgp/ell015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Le Ber, I., Van der Zee, J., Hannequin, D., Gijselinck, I., Campion, D., Puel, M., et al. (2007). Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum. Mutat. 28, 846–855. doi: 10.1002/humu.20520
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, E. B., Lee, V. M.-Y., and Trojanowski, J. Q. (2012). Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 13, 38–50. doi: 10.1038/nrn3121
Lee, H.-G., Perry, G., Moreira, P. I., Garrett, M. R., Liu, Q., Zhu, X., et al. (2005). Tau phosphorylation in Alzheimer's disease: pathogen or protector? Trends Mol. Med. 11, 164–169. doi: 10.1016/j.molmed.2005.02.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, Y., Ahn, C., Han, J., Choi, H., Kim, J., Yim, J., et al. (2003). The nuclear RNase III Drosha initiates microRNA processing. Nature 425, 415–419. doi: 10.1038/nature01957
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, Y.-B., Chen, H.-J., Peres, J. N., Gomez-Deza, J., Attig, J., Stalekar, M., et al. (2013). Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 5, 1178–1186. doi: 10.1016/j.celrep.2013.10.049
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, Y., Kim, M., Han, J., Yeom, K.-H., Lee, S., Baek, S. H., et al. (2004). MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23, 4051–4060. doi: 10.1038/sj.emboj.7600385
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ling, S. C., Albuquerque, C. P., Han, J. S., Lagier-Tourenne, C., Tokunaga, S., Zhou, H., et al. (2010). ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc. Natl. Acad. Sci. U.S.A. 107, 13318–13323. doi: 10.1073/pnas.1008227107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ling, S.-C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, J., Carmell, M., and Rivas, F. (2004). Argonaute2 is the catalytic engine of mammalian RNAi. Science 305, 1437–1441. doi: 10.1126/science.1102513
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, L., Drouet, V., Wu, J. W., Witter, M. P., Small, S. A., Clelland, C., et al. (2012). Trans-synaptic spread of tau pathology in vivo. PLoS ONE 7:e31302. doi: 10.1371/journal.pone.0031302
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lladó, A., Ezquerra, M., Sánchez-Valle, R., Rami, L., Tolosa, E., and Molinuevo, J. L. (2007). A novel MAPT mutation (P301T) associated with familial frontotemporal dementia. Eur. J. Neurol. 14, e9–e10. doi: 10.1111/j.1468-1331.2007.01763.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lopez de Munain, A., Alzualde, A., Gorostidi, A., Otaequi, D., Ruiz-Martinez, J., Indakoetxea, B., et al. (2008). Mutations in progranulin gene: clinical, pathological, and ribonucleic acid expression findings. Biol. Psychiatry 63, 946–952. doi: 10.1016/j.biopsych.2007.08.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luna-Muñoz, J., Harrington, C. R., Wischik, C. M., Flores-Rodriìguez, P., Avila, J., Zamudio, S. R., et al. (2013). “Phosphorylation of tau protein associated as a protective mechanism in the presence of toxic, C-terminally truncated tau in Alzheimer's disease,” in Understanding Alzheimer's Disease, ed I. Zerr (InTech). doi: 10.5772/54228
Luquin, N., Yu, B., Saunderson, R. B., Trent, R. J., and Pamphlett, R. (2009). Genetic variants in the promoter of TARDBP in sporadic amyotrophic lateral sclerosis. Neuromuscul Disord. 19, 696–700. doi: 10.1016/j.nmd.2009.07.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lynch, T., Sano, M., Marder, K. S., Bell, K. L., Foster, N. L., Defendini, R. F., et al. (1994). Clinical characteristics of a family with chromosome 17-linked disinhibition-dementia-parkinsonism-amyotrophy complex. Neurology 44, 1878–1884. doi: 10.1212/WNL.44.10.1878
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mackenzie, I. R., Baker, M., Pickering-Brown, S., Hsiung, G.-Y. R., Lindholm, C., Dwosh, E., et al. (2006). The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 129, 3081–3090. doi: 10.1093/brain/awl271
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mackenzie, I. R. A, Frick, P., and Neumann, M. (2014). The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol. 127, 347–357. doi: 10.1007/s00401-013-1232-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mackenzie, I. R., Rademakers, R., and Neumann, M. (2010). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9, 995–1007. doi: 10.1016/S1474-4422(10)70195-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marcon, G., Rossi, G., Giaccone, G., Giovagnoli, A. R., Piccoli, E., Zanini, S., et al. (2011). Variability of the clinical phenotype in an Italian family with dementia associated with an intronic deletion in the GRN gene. J. Alzheimers Dis. 22, 1123–1133. doi: 10.3233/JAD-2011-110332
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Martin-Serrano, J., and Neil, S. J. D. (2011). Host factors involved in retroviral budding and release. Nat. Rev. Microbiol. 9, 519–531. doi: 10.1038/nrmicro2596
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Medina, M., and Avila, J. (2014). The role of extracellular Tau in the spreading of neurofibrillary pathology. Front. Cell. Neurosci. 8:113. doi: 10.3389/fncel.2014.00113
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mehler, M. F., and Mattick, J. S. (2007). Noncoding RNAs and RNA editing in brain development, functional diversification, and neurological disease. Physiol. Rev. 87, 799–823. doi: 10.1152/physrev.00036.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meissner, M., Lopato, S., Gotzmann, J., Sauermann, G., and Barta, A. (2003). Proto-oncoprotein tls/fus is associated to the nuclear matrix and complexed with splicing factors ptb, srm160, and sr proteins. Exp. Cell Res. 283, 184–195. doi: 10.1016/S0014-4827(02)00046-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mercer, T. R., Dinger, M. E., Sunkin, S. M., Mehler, M. F., and Mattick, J. S. (2008). Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. U.S.A. 105, 716–721. doi: 10.1073/pnas.0706729105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Metcalf, D., and Isaacs, A. M. (2010). The role of ESCRT proteins in fusion events involving lysosomes, endosomes and autophagosomes. Biochem. Soc. Trans. 38, 1469–1473. doi: 10.1042/BST0381469
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miyamoto, K., Kowalska, A., Hasegawa, M., Tabira, T., Takahashi, K., Araki, W., et al. (2001). Familial frontotemporal dementia and parkinsonism with a novel mutation at an intron 10+11-splice site in the tau gene. Ann. Neurol. 50, 117–120. doi: 10.1002/ana.1083
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mizielinska, S., Lashley, T., Norona, F. E., Clayton, E. L., Ridler, C. E., Fratta, P., et al. (2013). C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 126, 845–857. doi: 10.1007/s00401-013-1200-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mizuno, Y., Hori, S., Kakizuka, A., and Okamoto, K. (2003). Vacuole-creating protein in neurodegenerative diseases in humans. Neurosci. Lett. 343, 77–80. doi: 10.1016/S0304-3940(03)00280-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Momeni, P., Rogaeva, E., Van Deerlin, V., Yuan, W., Grafman, J., Tierney, M., et al. (2006). Genetic variability in CHMP2B and frontotemporal dementia. Neurodegener. Dis. 3, 129–133. doi: 10.1159/000094771
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mori, K., Lammich, S., Mackenzie, I. R., Forné, I., Zilow, S., Kretzschmar, H., et al. (2013a). hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 125, 413–423. doi: 10.1007/s00401-013-1088-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mori, K., Weng, S.-M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013b). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science (New York, NY), 339, 1335–1338. doi: 10.1126/science.1232927
Morita, M., Al-Chalabi, A., Andersen, P. M., Hosler, B., Sapp, P., Englund, E., et al. (2006). A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology 66, 839–844. doi: 10.1212/01.wnl.0000200048.53766.b4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Morlando, M., Dini Modigliani, S., Torrelli, G., Rosa, A., Di Carlo, V., Caffarelli, E., et al. (2012). FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. 31, 4502–4510. doi: 10.1038/emboj.2012.319
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mukherjee, O., Pastor, P., Cairns, N. J., Chakraverty, S., Kauve, J. S., Shears, S., et al. (2006). HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, taunegative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann. Neurol. 60, 314–322. doi: 10.1002/ana.20963
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mukherjee, O., Wang, J., Gitcho, M., Chakraverty, S., Taylor-Reinwald, L., Shears, S., et al. (2008). Molecular characterization of novel progranulin (GRN) mutations in frontotemporal dementia. Hum. Mutat. 29, 512–521. doi: 10.1002/humu.20681
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murrell, J. R., Spillantini, M. G., Zolo, P., Guazzelli, M., Smith, M. J., Hasegawa, M., et al. (1999). Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J. Neuropathol. Exp. Neurol. 58, 1207–1226. doi: 10.1097/00005072-199912000-00002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Naganuma, T., Nakagawa, S., Tanigawa, A., Sasaki, Y. F., Goshima, N., and Hirose, T. (2012). Alternative 3′-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. EMBO J. 31, 4020–4034. doi: 10.1038/emboj.2012.251
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakagawa, S., and Hirose, T. (2012). Paraspeckle nuclear bodies–useful uselessness? Cell. Mol. Life Sci. 69, 3027–3036. doi: 10.1007/s00018-012-0973-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakagawa, S., Naganuma, T., Shioi, G., and Hirose, T. (2011). Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J. Cell Biol. 193, 31–39. doi: 10.1083/jcb.201011110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nalbandian, A., Donkervoort, S., Dec, E., Badadani, M., Katheria, V., Rana, P., et al. (2011). The multiple faces of valosin-containing protein-associated diseases: inclusion body myopathy with Paget's disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J. Mol. Neurosci. 45, 522–531. doi: 10.1007/s12031-011-9627-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Neumann, M., Diekmann, S., Bertsch, U., Vanmassenhove, B., Bogerts, B., and Kretzschmar, H. A. (2005). Novel G335V mutation in the tau gene associated with early onset familial frontotemporal dementia. Neurogenetics 6, 91–95. doi: 10.1007/s10048-005-0210-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Neumann, M., Kwong, L. K., Lee, E. B., Kremmer, E., Flatley, A., Xu, Y., et al. (2009a). Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 117, 137–149. doi: 10.1007/s00401-008-0477-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Neumann, M., Rademakers, R., Roeber, S., Baker, M., Kretzschmar, H. A., and Mackenzie, I. R. A. (2009b). A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132(Pt 11), 2922–2931. doi: 10.1093/brain/awp214
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (New York, NY) 314, 130–133. doi: 10.1126/science.1134108
Ng, S. Y., Lin, L., Soh, B. S., and Stanton, L. W. (2013). Long noncoding RNAs in development and disease of the central nervous system. Trends Genet. 29, 461–468. doi: 10.1016/j.tig.2013.03.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nishimoto, Y., Nakagawa, S., Hirose, T., Okano, H. J., Takao, M., Shibata, S., et al. (2013). The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol. Brain 6, 31. doi: 10.1186/1756-6606-6-31
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Obita, T., Saksena, S., Ghazi-Tabatabai, S., Gill, D. J., Perisic, O., Emr, S. D., et al. (2007). Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature 449, 735–739. doi: 10.1038/nature06171
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
O'Rourke, J. R., and Swanson, M. S. (2009). Mechanisms of RNA-mediated disease. J. Biol. Chem. 284, 7419–7423. doi: 10.1074/jbc.R800025200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ogaki, K., Li, Y., Takanashi, M., Ishikawa, K.-I., Kobayashi, T., Nonaka, T., et al. (2013). Analyses of the MAPT, PGRN, and C9orf72 mutations in Japanese patients with FTLD, PSP, and CBS. Parkinsonism Relat. Disord. 19, 15–20. doi: 10.1016/j.parkreldis.2012.06.019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Okura, H., Yamashita, S., Ohama, T., Saga, A., Yamamoto-Kakuta, A., Hamada, Y., et al. (2010). HDL/apolipoprotein A-I binds to macrophage-derived progranulin and suppresses its conversion into proinflammatory granulins. J. Atheroscler. Thromb. 17, 568–577. doi: 10.5551/jat.3921
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Osborne, R. J., and Thornton, C., a. (2006). RNA-dominant diseases. Hum. Mol. Genet. 15 Spec No(2), R162–R9. doi: 10.1093/hmg/ddl181
Ou, S. H., Wu, F., Harrich, D., García-Martínez, L. F., and Gaynor, R. B. (1995). Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 69, 3584–3596.
Pandit, S., Wang, D., and Fu, X.-D. (2008). Functional integration of transcriptional and RNA processing machineries. Curr. Opin. Cell Biol. 20, 260–265. doi: 10.1016/j.ceb.2008.03.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Parkinson, N., Ince, P. G., Smith, M. O., Highley, R., Skibinski, G., Andersen, P. M., et al. (2006). ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67, 1074–1077. doi: 10.1212/01.wnl.0000231510.89311.8b
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pasquinelli, A. E. (2012). MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 13, 271–282. doi: 10.1038/nrg3162
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pickering-Brown, S., Baker, M., Yen, S. H., Liu, W. K., Hasegawa, M., Cairns, N., et al. (2000). Pick's disease is associated with mutations in the tau gene. Ann. Neurol. 48, 859–867. doi: 10.1002/1531-8249(200012)48:6<859::AID-ANA6>3.0.CO;2-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pickering-Brown, S. M., Baker, M., Nonaka, T., Ikeda, K., Sharma, S., Mackenzie, J., et al. (2004). Frontotemporal dementia with Pick-type histology associated with Q336R mutation in the tau gene. Brain 127(Pt. 6), 1415–1426. doi: 10.1093/brain/awh147
Plowman, G. D., Green, J. M., Neubauer, M. G., Buckley, S. D., Mcdonald, V. L., Todaro, G. J., et al. (1992). The epithelin precursor encodes two proteins with opposing activities on epithelial cell growth. J. Biol. Chem. 267, 13073–13078.
Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R., Huelga, S. C., Moran, J., Liang, T. Y., et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468. doi: 10.1038/nn.2779
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Poorkaj, P., Kas, A., D'Souza, I., Zhou, Y., Pham, Q., Stone, M., et al. (2001). A genomic sequence analysis of the mouse and human microtubule-associated protein tau. Mamm. Genome. 12, 700–712. doi: 10.1007/s00335-001-2044-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Prasanth, K. V., Prasanth, S. G., Xuan, Z., Hearn, S., Freier, S. M., Bennett, C. F., et al. (2005). Regulating gene expression through RNA nuclear retention. Cell 123, 249–263. doi: 10.1016/j.cell.2005.08.033
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pye, V. E., Beuron, F., Keetch, C. A, McKeown, C., Robinson, C. V., Meyer, H. H., et al. (2007). Structural insights into the p97-Ufd1-Npl4 complex. Proc. Natl. Acad. Sci. U.S.A. 104, 467–472. doi: 10.1073/pnas.0603408104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rademakers, R., Eriksen, J. L., Baker, M., Robinson, T., Ahmed, Z., Lincoln, S. J., et al. (2008). Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum. Mol. Genet. 17, 3631–3642. doi: 10.1093/hmg/ddn257
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reddy, K., Zamiri, B., Stanley, S. Y. R., Macgregor, R. B., and Pearson, C. E. (2013). The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J. Biol. Chem. 288, 9860–9866. doi: 10.1074/jbc.C113.452532
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reichelt, A. C., Rodgers, R. J., and Clapcote, S. J. (2012). The role of neurexins in schizophrenia and autistic spectrum disorder. Neuropharmacology 62, 1519–1526. doi: 10.1016/j.neuropharm.2011.01.024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Renoux, A., and Todd, P. (2012). Neurodegeneration the RNA way. Prog. Neurobiol. 97, 173–189. doi: 10.1016/j.pneurobio.2011.10.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rizzini, C., Goedert, M., Hodges, J. R., Smith, M. J., Jakes, R., Hills, R., et al. (2000). Tau gene mutation K257T causes a tauopathy similar to Pick's disease. J. Neuropathol. Exp. Neurol. 59, 990–1001.
Rizzu, P., van Swieten, J. C., Joosse, M., Hasegawa, M., Stevens, M., Tibben, A., et al. (1999). High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am. J. Human Genet. 64, 414–421. doi: 10.1086/302256
Rossi, G., Bastone, A., Piccoli, E., Mazzoleni, G., Morbin, M., Uggetti, A., et al. (2012). New mutations in MAPT gene causing frontotemporal lobar degeneration: biochemical and structural characterization. Neurobiol. Aging 33, 834.e1–834.e6. doi: 10.1016/j.neurobiolaging.2011.08.008
Rossi, G., Bastone, A., Piccoli, E., Morbin, M., Mazzoleni, G., Fugnanesi, V., et al. (2014). Different mutations at V363 MAPT codon are associated with atypical clinical phenotypes and show unusual structural and functional features. Neurobiol. Aging 35, 408–417. doi: 10.1016/j.neurobiolaging.2013.08.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rosso, S. M., Donker, K. L., Baks, T., Joosse, M., de Koning, I., Pijnenburg, Y., et al. (2003). Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain 126, 2016–2022. doi: 10.1093/brain/awg204
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rosso, S. M., van Herpen, E., Deelen, W., Kamphorst, W., Severijnen, L. A., Willemsen, R., et al. (2002). A novel tau mutation, S320F, causes a tauopathy with inclusions similar to those in Pick's disease. Ann. Neurol. 51, 373–376. doi: 10.1002/ana.10140
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rouiller, I., DeLaBarre, B., May, A. P., Weis, W. I., Brunger, A. T., Milligan, R., et al. (2002). Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nat. Struct. Biol. 9, 950–957 doi: 10.1038/nsb872
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rovelet-Lecrux, A., Deramecourt, V., Legallic, S., Maurage, C. A., Le Ber, I., Brice, A., et al. (2008). Deletion of the progranulin gene in patients with frontotemporal lobar degeneration or Parkinson disease. Neurobiol. Dis. 31, 41–45. doi: 10.1016/j.nbd.2008.03.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roxrud, I., Stenmark, H., and Malerød, L. (2010). ESCRT and Co. Biol. Cell, 102, 293–318. doi: 10.1042/BC20090161
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Saman, S., Kim, W., Raya, M., Visnick, Y., Miro, S., Jackson, B., et al. (2012). Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849. doi: 10.1074/jbc.M111.277061
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sampathu, D. M., Neumann, M., Kwong, L. K., Chou, T. T., Micsenyi, M., Truax, A., et al. (2006). Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am. J. Pathol. 169, 1343–1352. doi: 10.2353/ajpath.2006.060438
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sareen, D., O'Rourke, J. G., Meera, P., Muhammad, A. K., Grant, S., Simpkinson, M., et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5, 208ra149. doi: 10.1126/scitranslmed.3007529
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schenk, V. W. (1959). Re-examination of a family with Pick's disease. Ann. Hum. Genet. 23, 325–333. doi: 10.1111/j.1469-1809.1959.tb01476.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schmidt, O., and Teis, D. (2012). The ESCRT machinery. Curr. Biol. 22, R116–R120. doi: 10.1016/j.cub.2012.01.028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schwartz, J. C., Ebmeier, C. C., Podell, E. R., Heimiller, J., Taatjes, D. J., and Cech, T. R. (2012). FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev. 26, 2690–2695. doi: 10.1101/gad.204602.112
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sephton, C. F., Cenik, C., Kucukural, A., Dammer, E. B., Cenik, B., Han, Y., et al. (2011). Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J. Biol. Chem. 286, 1204–1215. doi: 10.1074/jbc.M110.190884
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shan, X., Chiang, P.-M., Price, D. L., and Wong, P. C. (2010). Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 107, 16325–16330. doi: 10.1073/pnas.1003459107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shankaran, S. S., Capell, A., Hruscha, A. T., Fellerer, K., Neumann, M., Schmid, B., et al. (2008). Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J. Biol. Chem. 283, 1744–1753. doi: 10.1074/jbc.M705115200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shelkovnikova, T. A., Robinson, H. K., Troakes, C., Ninkina, N., and Buchman, V. L. (2014). Compromised paraspeckle formation as a pathogenic factor in FUSopathies. Hum. Mol. Genet. 23, 2298–2312. doi: 10.1093/hmg/ddt622
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shi, Z., Hayashi, Y. K., Mitsuhashi, S., Goto, K., Kaneda, D., Choi, Y.-C., et al. (2012). Characterization of the Asian myopathy patients with VCP mutations. Eur. J. Neurol. 19, 501–509. doi: 10.1111/j.1468-1331.2011.03575.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shim, S., Kimpler, L. A., and Hanson, P. I. (2007). Structure/function analysis of four core ESCRT-III proteins reveals common regulatory role for extreme C-terminal domain. Traffic 8, 1068–1079. doi: 10.1111/j.1600-0854.2007.00584.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Simón, D., Garcia-Garcia, E., Royo, F., Falcon-Perez, J. M., and Avila, J. (2012). Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett. 586, 47–54. doi: 10.1016/j.febslet.2011.11.022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Skibinski, G., Parkinson, N. J., Brown, J. M., Chakrabarti, L., Lloyd, S. L., Hummerich, H., et al. (2005). Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37, 806–808. doi: 10.1038/ng1609
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Skoglund, L., Matsui, T., Freeman, S. H., Wallin, A., Blom, E. S., Frosch, M. P., et al. (2011). Novel progranulin mutation detected in 2 patients with FTLD. Alzheimer Dis. Assoc. Disord. 25, 173–178. doi: 10.1097/WAD.0b013e3181fbc22c
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Smith, B. N., Newhouse, S., Shatunov, A., Vance, C., Topp, S., Johnson, L., et al. (2012). The C9ORF72 expansion mutation is a common cause of ALS+/-FTD in Europe and has a single founder. Eur. J. Hum. Genet. 21, 102–108. doi: 10.1038/ejhg.2012.98
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Snowden, J. S., Rollinson, S., Thompson, J. C., Harris, J. M., Stopford, C. L., Richardson, A. M. T., et al. (2012). Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 135(Pt 3), 693–708. doi: 10.1093/brain/awr355
Songsrirote, K., Li, Z., Ashford, D., Bateman, A., and Thomas-Oates, J. (2010). Development and application of mass spectrometric methods for the analysis of progranulin N-glycosylation. J. Proteomics 73, 1479–1490. doi: 10.1016/j.jprot.2010.02.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Souquere, S., Beauclair, G., Harper, F., Fox, A., and Pierron, G. (2010). Highly ordered spatial organization of the structural long noncoding NEAT1 RNAs within paraspeckle nuclear bodies. Mol. Biol. Cell 21, 4020–4027. doi: 10.1091/mbc.E10-08-0690
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spillantini, M. G., and Goedert, M. (2000). Tau mutations in familial frontotemporal dementia. Brain 123, 857–859. doi: 10.1093/brain/123.5.857
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spillantini, M. G., and Goedert, M. (2013). Tau pathology and neurodegeneration. Lancet Neurol. 12, 609–622. doi: 10.1016/S1474-4422(13)70090-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spillantini, M. G., Goedert, M., Crowther, R. A., Murrell, J. R., Farlow, M. R., and Ghetti, B. (1997). Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc. Natl. Acad. Sci. U.S.A. 94, 4113–4118. doi: 10.1073/pnas.94.8.4113
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spina, S., Murrell, J. R., Yoshida, H., Ghetti, B., Bermingham, N., Sweeney, B., et al. (2007). The novel Tau mutation G335S: clinical, neuropathological and molecular characterization. Acta Neuropathol. 113, 461–470. doi: 10.1007/s00401-006-0182-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spina, S., Murrell, J. R., Vidal, R., and Ghetti, B. (2008). Neuropathologic and genetic characterization of frontotemporal lobar degeneration with Ubiquitin- and/or Tdp-43-positive inclusions: a large series. Alzheimer's Dementia 4(Supp. 2), T431. doi: 10.1016/j.jalz.2008.05.1280
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stallings, N. R., Puttaparthi, K., Luther, C. M., Burns, D. K., and Elliott, E. J. (2010). Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol. Dis. 40, 404–414. doi: 10.1016/j.nbd.2010.06.017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stanford, P. M., Shepherd, C. E., Halliday, G. M., Brooks, W. S., Schofield, P. W., Brodaty, H., et al. (2003). Mutations in the tau gene that cause an increase in three repeat tau and frontotemporal dementia. Brain 126, 814–826 doi: 10.1093/brain/awg090
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stojkovic, T., Hammouda, E. H., Richard, P., Munain, A. L., De Ruiz-Martinez, J., Camaño, P., et al. (2009). Clinical outcome in 19 French and Spanish patients with valosin-containing protein myopathy associated with Paget's disease of bone and frontotemporal dementia. Neuromuscul. Disord. 21, 316–323. doi: 10.1016/j.nmd.2009.02.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Strong, M. J., Volkening, K., Hammond, R., Yang, W., Strong, W., Leystra-Lantz, C., et al. (2007). TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol. Cell. Neurosci. 35, 320–327. doi: 10.1016/j.mcn.2007.03.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Suárez-Calvet, M., Dols-Icardo, O., Lladó, A., Sánchez-Valle, R., Hernández, I., Amer, G., et al. (2014). Plasma phosphorylated TDP-43 levels are elevated in patients with frontotemporal dementia carrying a C9orf72 repeat expansion or a GRN mutation. J. Neurol. Neurosurg. Psychiatr. 85, 684–691. doi: 10.1136/jnnp-2013-305972
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sunwoo, H., Dinger, M. E., Wilusz, J. E., Amaral, P. P., Mattick, J. S., and Spector, D. L. (2009). MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 19, 347–359. doi: 10.1101/gr.087775.108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Takamatsu, J., Kondo, A., Ikegami, K., Kimura, T., Fujii, H., Mitsuyama, Y., et al. (1998). Selective expression of Ser 199/202 phosphorylated tau in a case of frontotemporal dementia. Dement. Geriatr. Cogn. Disord. 9, 82–89. doi: 10.1159/000017028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tan, A. Y., and Manley, J. L. (2009). The TET family of proteins: functions and roles in disease. J. Mol. Cell Biol. 1, 82–92. doi: 10.1093/jmcb/mjp025
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, G., Tang, X., Mendu, V., Tang, X., Jia, X., Chen, Q.-J., et al. (2008). The art of microRNA: various strategies leading to gene silencing via an ancient pathway. Biochim. Biophys. Acta 1779, 655–662. doi: 10.1016/j.bbagrm.2008.06.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, X., Muniappan, L., Tang, G., and Özcan, S. (2009). Identification of glucose-regulated miRNAs from pancreatic β cells reveals a role for miR-30d in insulin transcription. RNA 15, 287–293. doi: 10.1261/rna.1211209
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tay, Y., Kats, L., Salmena, L., Weiss, D., Tan, S. M., Ala, U., et al. (2011). Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell 147, 344–357. doi: 10.1016/j.cell.2011.09.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thies, W., and Bleiler, L. (2011). 2011 Alzheimer's disease facts and figures. Alzheimers Dement. 7, 208–244. doi: 10.1016/j.jalz.2011.02.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Todd, P. K., and Paulson, H. L. (2010). RNA-mediated neurodegeneration in repeat expansion disorders. Ann. Neurol. 67, 291–300. doi: 10.1002/ana.21948
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tollervey, J. R., Curk, T., Rogelj, B., Briese, M., Cereda, M., Kayikci, M., et al. (2011). Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458. doi: 10.1038/nn.2778
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Urwin, H., Authier, A., Nielsen, J. E., Metcalf, D., Powell, C., Froud, K., et al. (2010). Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum. Mol. Genet. 19, 2228–2238. doi: 10.1093/hmg/ddq100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Usami, Y., Popov, S., Popova, E., Inoue, M., Weissenhorn, W., and G Göttlinger, H. (2009). The ESCRT pathway and HIV-1 budding. Biochem. Soc. Trans. 37(Pt 1), 181–184. doi: 10.1042/BST0370181
Valencia-Sanchez, M. A., Liu, J., Hannon, G. J., and Parker, R. (2006). Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 20, 515–524. doi: 10.1101/gad.1399806
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van Blitterswijk, M., and Landers, J. E. (2010). RNA processing pathways in amyotrophic lateral sclerosis. Neurogenetics 11, 275–290. doi: 10.1007/s10048-010-0239-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van Damme, P., Van Hoecke, A., Lambrechts, D., Vanacker, P., Bogaert, E., van Swieten, J., et al. (2008). Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J. Cell Biol. 181, 37–41. doi: 10.1083/jcb.200712039
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van Deerlin, V. M., Leverenz, J. B., Bekris, L. M., Bird, T. D., Yuan, W., Elman, L. B., et al. (2008). TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 7, 409–416. doi: 10.1016/S1474-4422(08)70071-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van der Zee, J., Gijselinck, I., Dillen, L., Van Langenhove, T., Theuns, J., Engelborghs, S., et al. (2013). A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum. Mutat. 34, 363–373. doi: 10.1002/humu.22244
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van der Zee, J., Rademakers, R., Engelborghs, S., Gijselinck, I., Bogaerts, V., Vandenberghe, R., et al. (2006). A Belgian ancestral haplotype harbours a highly prevalent mutation for 17q21-linked tau-negative FTLD. Brain 129(Pt 4), 841–852. doi: 10.1093/brain/awl029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van der Zee, J., Le Ber, I., Maurer-Stroh, S., Engelborghs, S., Gijselinck, I., Camuzat, A., et al. (2007). Mutations other than null mutations producing a pathogenic loss of progranulin in frontotemporal dementia. Hum. Mutat. 28, 416. doi: 10.1002/humu.9484
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van der Zee, J., Urwin, H., Engelborghs, S., Bruyland, M., Vandenberghe, R., Dermaut, B., et al. (2008). CHMP2B C-truncating mutations in frontotemporal lobar degeneration are associated with an aberrant endosomal phenotype in vitro. Hum. Mol. Genet. 17, 313–322. doi: 10.1093/hmg/ddm309
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van der Zee, J., Van Langenhove, T., Kovacs, G. G., Dillen, L., Deschamps, W., Engelborghs, S., et al. (2014). Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol. 128, 397–410. doi: 10.1007/s00401-014-1298-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van Langenhove, T., van der Zee, J., Sleegers, K., Engelborghs, S., Vandenberghe, R., Gijselinck, I., et al. (2010). Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 74, 366–371. doi: 10.1212/WNL.0b013e3181ccc732
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vesa, J., Su, H., Watts, G. D., Krause, S., Walter, M. C., Martin, B., et al. (2009). Valosin containing protein associated inclusion body myopathy: abnormal vacuolization, autophagy and cell fusion in myoblasts. Neuromuscul. Disord. 19, 766–772. doi: 10.1016/j.nmd.2009.08.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wahid, F., Shehzad, A., Khan, T., and Kim, Y. Y. (2010). MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim. Biophys. Acta 1803, 1231–1243. doi: 10.1016/j.bbamcr.2010.06.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, H.-Y., Wang, I.-F., Bose, J., and Shen, C.-K. J. (2004a). Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics 83, 130–139. doi: 10.1016/S0888-7543(03)00214-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Q., Song, C., and Li, C.-C. H. (2004b). Molecular perspectives on p97-VCP: progress in understanding its structure and diverse biological functions. J. Struct. Biol. 146, 44–57. doi: 10.1016/j.jsb.2003.11.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, T., Jiang, X., Chen, G., and Xu, J. (2015). Interaction of amyotrophic lateral sclerosis/frontotemporal lobar degeneration-associated fused-in-sarcoma with proteins involved in metabolic and protein degradation pathways. Neurobiol. Aging 36, 527–535. doi: 10.1016/j.neurobiolaging.2014.07.044
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, W. X., Rajeev, B. W., Stromberg, A. J., Ren, N., Tang, G., Huang, Q., et al. (2008b). The expression of microRNA miR-107 decreases arly in Alzheimer's disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 28, 1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, W.-X., Wilfred, B. R., Madathil, S. K., Tang, G., Hu, Y., Dimayuga, J., et al. (2010). miR-107 regulates granulin/progranulin with implications for traumatic brain injury and neurodegenerative disease. Am. J. Pathol. 177, 334–345. doi: 10.2353/ajpath.2010.091202
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, X., Arai, S., Song, X., Reichart, D., Du, K., Pascual, G., et al. (2008a). Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454, 126–130. doi: 10.1038/nature06992
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Y. T., Kuo, P. H., Chiang, C. H., Liang, J. R., Chen, Y. R., Wang, S., et al. (2013). The Truncated C-terminal RNA recognition motif of TDP-43 protein plays a key role in forming proteinaceous aggregates. J. Biol. Chem. 288, 9049–9057. doi: 10.1074/jbc.M112.438564
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Watts, G. D. J., Thomasova, D., Ramdeen, S. K., Fulchiero, E. C., Mehta, S. G., Drachman, D. A., et al. (2007). Novel VCP mutations in inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Clin. Genet. 72, 420–426. doi: 10.1111/j.1399-0004.2007.00887.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Watts, G. D. J., Wymer, J., Kovach, M. J., Mehta, S. G., Mumm, S., Darvish, D., et al. (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381. doi: 10.1038/ng1332
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wegorzewska, I., Bell, S., Cairns, N. J., Miller, T. M., and Baloh, R. H. (2009). TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. U.S.A. 106, 18809–18814. doi: 10.1073/pnas.0908767106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weihl, C. C., Dalal, S., Pestronk, A., and Hanson, P. I. (2006). Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum. Mol. Genet. 15, 189–199. doi: 10.1093/hmg/ddi426
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weihl, C. C., Miller, S. E., Hanson, P. I., and Pestronk, A. (2007). Transgenic expression of inclusion body myopathy associated mutant p97/VCP causes weakness and ubiquitinated protein inclusions in mice. Hum. Mol. Genet. 16, 919–928. doi: 10.1093/hmg/ddm037
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Whitley, P., Reaves, B. J., Hashimoto, M., Riley, A. M., Potter, B. V. L., and Holman, G. D. (2003). Identification of mammalian Vps24p as an effector of phosphatidylinositol 3,5-bisphosphate-dependent endosome compartmentalization. J. Biol. Chem. 278, 38786–38795. doi: 10.1074/jbc.M306864200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wilhelm, B. T., Marguerat, S., Watt, S., Schubert, F., Wood, V., Goodhead, I., et al. (2008). Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 453, 1239–1243. doi: 10.1038/nature07002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wils, H., Kleinberger, G., Janssens, J., Pereson, S., Joris, G., Cuijt, I., et al. (2010). TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. U.S.A. 107, 3858–3863. doi: 10.1073/pnas.0912417107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wolfe, M. S. (2012). The role of tau in neurodegenerative diseases and its potential as a therapeutic target. Scientifica 2012:796024. doi: 10.6064/2012/796024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Woodman, P. G. (2003). P97, a protein coping with multiple identities. J. Cell Sci. 116(Pt 21), 4283–4290. doi: 10.1242/jcs.00817
Wszolek, Z. K., Pfeiffer, R. F., Bhatt, M. H., Schelper, R. L., Cordes, M., Snow, B. J., et al. (1992). Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann. Neurol. 32, 312–320. doi: 10.1002/ana.410320303
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, H., Huang, M., Lu, M., Zhu, W., Shu, Y., Cao, P., et al. (2013a). Regulation of microtubule-associated protein tau (MAPT) by miR-34c-5p determines the chemosensitivity of gastric cancer to paclitaxel. Cancer Chemother. Pharmacol. 71, 1159–1171. doi: 10.1007/s00280-013-2108-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, P., Zuo, X., Deng, H., Liu, X., Liu, L., and Ji, A. (2013b). Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases. Brain Res. Bull. 97, 69–80. doi: 10.1016/j.brainresbull.2013.06.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xiao, S., Sanelli, T., Dib, S., Sheps, D., Findlater, J., Bilbao, J., et al. (2011). RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol. Cell. Neurosci. 47, 167–180. doi: 10.1016/j.mcn.2011.02.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, Y. F., Gendron, T. F., Zhang, Y. J., Lin, W. L., D'Alton, S., Sheng, H., et al. (2010a). Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 30, 10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, Y., Suzuki, Y., Ito, K., and Komiyama, M. (2010b). Telomeric repeat-containing RNA structure in living cells. Proc. Natl. Acad. Sci. U.S.A. 107, 14579–14584. doi: 10.1073/pnas.1001177107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, Z., Poidevin, M., Li, X., and Li, Y. (2013). Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 7778–7783. doi: 10.1073/pnas.1219643110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamada, S., Inoue, Y., Suga, A., and Iwazaki, M. (2011). Surgical risk of vessel injury: an unusual anatomical variant of the right medial basal segmental pulmonary artery. Gen. Thorac. Cardiovasc. Surg. 59, 301–303. doi: 10.1007/s11748-010-0655-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yan, J., Deng, H.-X., Siddique, N., Fecto, F., Chen, W., Yang, Y., et al. (2010). Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 75, 807–814. doi: 10.1212/WNL.0b013e3181f07e0c
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, L., Embree, L. J., Tsai, S., and Hickstein, D. D. (1998). Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J. Biol. Chem. 273, 27761–27764. doi: 10.1074/jbc.273.43.27761
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yin, F., Banerjee, R., Thomas, B., Zhou, P., Qian, L., Jia, T., et al. (2010). Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J. Exp. Med. 207, 117–128. doi: 10.1084/jem.20091568
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zarranz, J. J., Ferrer, I., Lezcano, E., Forcadas, M. I., Eizaguirre, B., Atares, B., et al. (2005). A novel mutation (K317M) in the MAPT gene causes FTDP and motor neuron disease. Neurology 64, 1578–1585. doi: 10.1212/01.WNL.0000160116.65034.12
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, H., Wang, Q., and Kajino Kiichi, G. M. I. (2000). VCP, a weak ATPase involved in multiple cellular events, interacts physically with BRCA1 in the nucleus of living cells. DNA Cell Biol. 19, 253–263. doi: 10.1089/10445490050021168
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, Y.-J., Xu, Y., Dickey, C. A., Buratti, E., Baralle, F., Bailey, R., et al. (2007). Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J. Neurosci. 27, 10530–10534. doi: 10.1523/JNEUROSCI.3421-07.2007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zheng, M., Liao, M., Cui, T., Tian, H., Fan, D. S., and Wan, Q. (2012). Regulation of nuclear TDP-43 by NR2A-containing NMDA receptors and PTEN. J. Cell Sci. 125(Pt 6), 1556–1567. doi: 10.1242/jcs.095729
Zhu, J., Nathan, C., Jin, W., Sim, D., Ashcroft, G. S., Wahl, S. M., et al. (2002). Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell 111, 867–878. doi: 10.1016/S0092-8674(02)01141-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zinszner, H., Sok, J., Immanuel, D., Yin, Y., and Ron, D. (1997). TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell Sci. 110(Pt 1), 1741–1750.
Zovoilis, A., Agbemenyah, H. Y., Agis-Balboa, R. C., Stilling, R. M., Edbauer, D., Rao, P., et al. (2011). microRNA-34c is a novel target to treat dementias. EMBO J. 30, 4299–4308. doi: 10.1038/emboj.2011.327
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zu, T., Liu, Y., and Bañez-Coronel, M. (2013). RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. U.S.A. 110, E4968–E4977. doi: 10.1073/pnas.1315438110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: FTD, TDP-43, FUS, progranulin, tau, CHMP2B. C9ORF72
Citation: Fontana F, Siva K and Denti MA (2015) A network of RNA and protein interactions in Fronto Temporal Dementia. Front. Mol. Neurosci. 8:9. doi: 10.3389/fnmol.2015.00009
Received: 28 December 2014; Accepted: 25 February 2015;
Published: 19 March 2015.
Edited by:
Nicola Maggio, The Chaim Sheba Medical Center, IsraelReviewed by:
Mark R. Cookson, National Institutes of Health, USADavide De Pietri Tonelli, Fondazione Istituto Italiano di Tecnologia, Italy
Copyright © 2015 Fontana, Siva and Denti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michela A. Denti, Centre for Integrative Biology, University of Trento, Via Sommarive 9, 38123 Trento, ItalyZGVudGlAc2NpZW5jZS51bml0bi5pdA==
†These authors have contributed equally to this work.