 Maria E. Figueiredo-Pereira
Maria E. Figueiredo-Pereira Patricia Rockwell
Patricia Rockwell Thomas Schmidt-Glenewinkel
Thomas Schmidt-Glenewinkel Peter Serrano
Peter Serrano
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 13 January 2015
Sec. Molecular Signalling and Pathways
Volume 7 - 2014 | https://doi.org/10.3389/fnmol.2014.00104
This article is part of the Research Topic Ubiquitin and the Brain: Roles of Proteolysis in the Normal and Abnormal Nervous System View all 19 articles
The immune response of the CNS is a defense mechanism activated upon injury to initiate repair mechanisms while chronic over-activation of the CNS immune system (termed neuroinflammation) may exacerbate injury. The latter is implicated in a variety of neurological and neurodegenerative disorders such as Alzheimer and Parkinson diseases, amyotrophic lateral sclerosis, multiple sclerosis, traumatic brain injury, HIV dementia, and prion diseases. Cyclooxygenases (COX-1 and COX-2), which are key enzymes in the conversion of arachidonic acid into bioactive prostanoids, play a central role in the inflammatory cascade. J2 prostaglandins are endogenous toxic products of cyclooxygenases, and because their levels are significantly increased upon brain injury, they are actively involved in neuronal dysfunction induced by pro-inflammatory stimuli. In this review, we highlight the mechanisms by which J2 prostaglandins (1) exert their actions, (2) potentially contribute to the transition from acute to chronic inflammation and to the spreading of neuropathology, (3) disturb the ubiquitin-proteasome pathway and mitochondrial function, and (4) contribute to neurodegenerative disorders such as Alzheimer and Parkinson diseases, and amyotrophic lateral sclerosis, as well as stroke, traumatic brain injury (TBI), and demyelination in Krabbe disease. We conclude by discussing the therapeutic potential of targeting the J2 prostaglandin pathway to prevent/delay neurodegeneration associated with neuroinflammation. In this context, we suggest a shift from the traditional view that cyclooxygenases are the most appropriate targets to treat neuroinflammation, to the notion that J2 prostaglandin pathways and other neurotoxic prostaglandins downstream from cyclooxygenases, would offer significant benefits as more effective therapeutic targets to treat chronic neurodegenerative diseases, while minimizing adverse side effects.
Chronic neuroinflammation is recognized as a primary mechanism involved in the pathogenesis of a variety of neurodegenerative disorders including Alzheimer, Parkinson, and Huntington diseases as well as amyotrophic lateral sclerosis (Wyss-Coray and Mucke, 2002; Liu and Hong, 2003; Glass et al., 2010; Herrup, 2010). Neuroinflammation is an active process detectable in the earliest stages of these diseases (Zagol-Ikapitte et al., 2005; Liang et al., 2007; Yoshiyama et al., 2007). The neurotoxicity associated with inflammation makes it a potential risk factor in their pathogenesis. Characterizing the self-perpetuating cycle of inflammatory processes involving microglia and astrocytes in the brain that drives the slow progression of neurodegeneration could be critical for preventing/arresting these devastating disorders (Schwab and McGeer, 2008; Herrup, 2010).
Major players in inflammation are the cyclooxygenases COX-1 and COX-2 (Figure 1), which function as homodimers and are key enzymes in the biosynthesis of prostaglandins (Smyth et al., 2009). Although the brain expresses COX-1 and COX-2 under normal physiological conditions, it is clear that cyclooxygenases are implicated in neurodegeneration (Liang et al., 2007; Bartels and Leenders, 2010). COX-1 is generally viewed as being the homeostatic isoform, but studies suggest that it is actively involved in some forms of brain injury (Choi et al., 2009; Aid and Bosetti, 2011). The expression and activity of COX-2 are largely responsive to adverse stimuli, such as inflammation and physiologic imbalances (Yamagata et al., 1993). COX-2 activity is markedly induced in a range of neurodegenerative disorders subsequently leading to neuronal injury (Feng et al., 2003; Klivenyi et al., 2003; Teismann et al., 2003). COX-2 up-regulation following CNS injury is not restricted to neurons since COX-2 induction is also apparent in glia (Consilvio et al., 2004). Although many studies support the notion that COX-2 is involved in neurodegeneration, its contribution to the neurodegenerative process remains poorly defined. Inhibiting cyclooxygenases with non-steroidal anti-inflammatory drugs (NSAIDs) is being explored as a therapeutic strategy to mitigate chronic inflammation and prevent the onset/progression of neuropathology (Klegeris et al., 2007; Vlad et al., 2008). However, the effectiveness of NSAIDs could be counterproductive by blocking the generation of all prostaglandin products of cyclooxygenases (Figure 1).
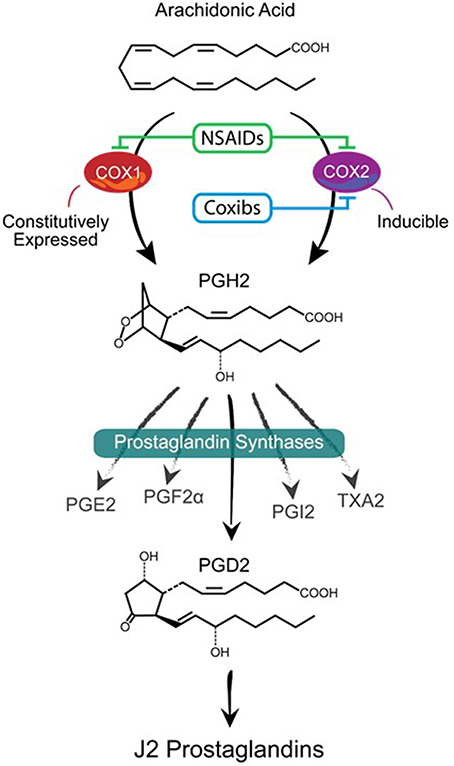
Figure 1. Prostanoid biosynthetic pathway. Arachidonic acid is converted via a two-step process (cyclooxygenation and hydroperoxidation) by cyclooxygenase enzymes COX-1 or COX-2 into the unstable prostaglandin PGH2. COX-1 is constitutively expressed while COX-2 is mostly an inducible enzyme that is upregulated under stress conditions. Non-steroidal anti-inflammatory drugs (NSAIDs) block the activities of both enzymes while Coxibs are selective COX-2 inhibitors. PGH2 is then converted to prostanoid products (PGE2, PGF2α, PGD2, PGI2, and TXA2) by specific prostaglandin synthases that differ in their cell type distribution. Of these products, PGD2 is highly unstable (estimated brain half-life of 1.1 min) resulting in the non-enzymatic formation of J2 prostaglandins.
Current animal and cell models of neurodegenerative diseases fail to address how prostaglandins redirect cellular events to promote neurodegeneration. This is a crucial gap since some prostaglandins are neuroprotective and others neurotoxic (Lucin and Wyss-Coray, 2009; Iadecola and Gorelick, 2005). Since prostaglandins act as potent local regulators of physiologic and pathologic pathways linked to CNS inflammation, elucidating the prostaglandin-dependent pathologic pathways will have a major impact on blocking neurotoxicity linked to chronic neuroinflammation with fewer undesirable side effects, and could lead to preventing/delaying neurodegeneration.
Prostaglandins (PGs) are a family of 20-carbon unsaturated fatty acids produced via the cyclooxygenase pathway in response to numerous extrinsic and intrinsic stimuli (Figure 2). The initial step in prostaglandin synthesis involves the hydrolysis of membrane sn-2 glycerophospholipids (phosphatidylcholine, phosphatidylethanolamine, and phosphatidylinositol) by phospholipase A2 (PLA2 group IVA) to release arachidonic acid (Tassoni et al., 2008; Smyth et al., 2009; Astudillo et al., 2012). PLA2 is activated by increased calcium levels and phosphorylation. This event leads to the translocation of PLA2 from the cytoplasm to intracellular membranes including the endoplasmic reticulum and nuclear envelope, to allow its access to arachidonic acid-containing phospholipid substrates (Shimizu et al., 2008).
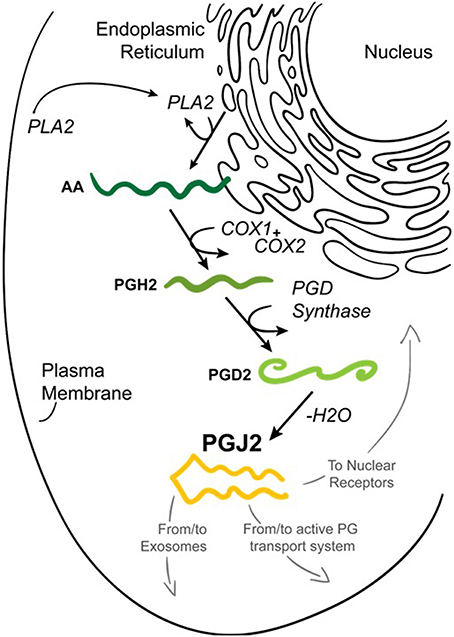
Figure 2. Formation of prostaglandin J2 (PGJ2). Upon cell activation by mechanical trauma, cytokines, growth factors or other stressful stimuli, phospholipase A2 (PLA2) is recruited from the cytoplasm to intracellular membranes (nucleus or endoplasmic reticulum) to catalyze the hydrolysis of membrane sn-2 glycerophospholipids releasing arachidonic acid (AA, dark green). AA is converted by COX-1 or COX-2 to PGH2 (medium green) which is then converted to PGD2 (light green) by PGD synthase. PGD2 undergoes a non-enzymatic dehydration (–H2O) to biologically active PGJ2 (yellow). PGJ2 can be localized to exosomes, to transport systems or to nuclear receptors to mediate its function.
Cyclooxygenases, which are bifunctional enzymes inserted into the ER and nuclear membranes, will then catalyze the cyclooxygenation of arachidonic acid to PGG2 followed by hydroperoxidation of PGG2 to PGH2 (Kulkarni et al., 2000; Smith et al., 2000; Simmons et al., 2004). PGH2 diffuses from the ER lumen through its membrane to the cytoplasm to be converted to more polar prostanoids via synthases localized on the cytoplasmic face of the ER (Schuster, 2002). The coupling of PGH2 synthesis with the respective downstream synthase enzymes that produce the different types of prostaglandins is intricately orchestrated in a tissue and/or cell specific manner (Funk, 2001).
J2 prostaglandins (Figure 3) are derived from PGD2, which is the most abundant prostaglandin in the brain (Ogorochi et al., 1984; Hertting and Seregi, 1989; Uchida and Shibata, 2008; Ricciotti and FitzGerald, 2011), and the one that changes the most under pathological conditions (Liang et al., 2005). PGD2 is produced by two distinct prostaglandin D2 synthases (PGDS), which carry out the isomerization of PGH2 to PGD2: (i) the hematopoietic enzyme (H-PGDS) and (ii) the lipocalin enzyme (L-PGDS) (Urade and Hayaishi, 2000; Urade and Eguchi, 2002). H-PGDS is a cytosolic protein found abundantly in mast cells, antigen presenting cells, and T helper type 2 (Th2) cells (Kanaoka and Urade, 2003). L-PGDS is localized in the CNS, heart and male genital organs (Urade and Eguchi, 2002). L-PGDS is one of the most abundant CSF proteins produced in the brain (Kanekiyo et al., 2007), representing 3% of total CSF protein (Xu and Venge, 2000). Secreted L-PGDS in the CSF has a dual function: it increases CSF-PGD2 levels (Scher and Pillinger, 2005) and also acts as a lipophilic-ligand carrier (Urade and Hayaishi, 2000), being a major endogenous Aβ-chaperone in the brain (Kanekiyo et al., 2007).
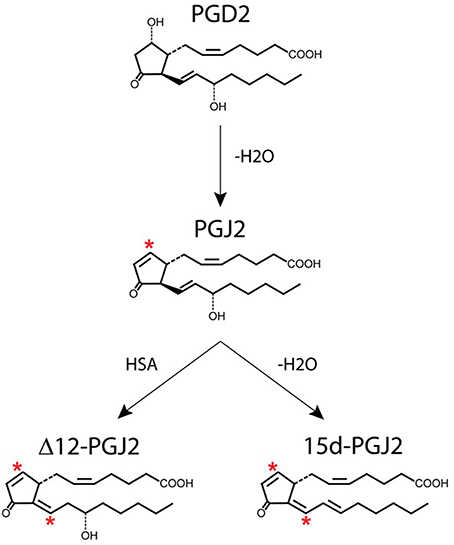
Figure 3. Generation of J2 prostaglandins. PGJ2 is generated by non-enzymatic dehydration of PGD2. The J2 metabolites Δ12-PGJ2 and 15d-PGJ2 are formed from PGJ2 either by reactions catalyzed by human serum albumin (HSA) or by dehydration (–H2O), respectively. Asterisks indicate α,β-unsaturated carbonyl groups.
PGD2 is unstable and readily undergoes in vivo and in vitro non-enzymatic dehydration to generate the biologically active cyclopentenone J2 prostaglandins (Figure 3), which include PGJ2, Δ12-PGJ2, and 15-deoxy-Δ12,14-PGJ2 (15d-PGJ2) (Shibata et al., 2002; Uchida and Shibata, 2008; Gilroy, 2010). The half-life of PGD2 in the brain was estimated to be 1.1 min and in the blood 0.9 min (Suzuki et al., 1986).
PGJ2 and its metabolites are not stored in tissues or cells and their production increases with diverse stimuli. Prostaglandins are largely produced in the brain by activated microglia, reactive astrocytes and neurons. During CNS inflammation, these cells make large quantities of prostaglandins such as PGE2 and PGD2 (Liu et al., 2003) as well as J2 prostaglandins (Bernardo et al., 2003). For example, LPS-activated microglia in culture, produced ~3 ng/ml media of 15d-PGJ2 upon 72 h, and ~2 ng/ml of PGD2 upon 24 h (Bernardo et al., 2003). J2 prostaglandins have been detected in vivo in human body fluids (Hirata et al., 1988), human atherosclerotic plaques (Shibata et al., 2002) and tissues of patients with sporadic ALS (Kondo et al., 2002; Zhang et al., 2010). In addition, a range of studies showed that J2 prostaglandins are generated in vivo upon various conditions related to brain injury (see below).
Prostaglandins are present in body fluids in the pico to nanomolar range reaching low micromolar levels at local sites of acute inflammation (Offenbacher et al., 1986; Hertting and Seregi, 1989). For example, in human airways PGD2 rose in 9 min to an average of 150-fold in five patients in response to an allergen (Murray et al., 1986). Moreover, exosomes, which are extracellular bioactive vesicles released from multivesicular bodies that mediate intercellular signaling (Subra et al., 2010), were found to contain a large panel of free fatty acids, including arachidonic acid and its derivatives, such as PGE2 and PGJ2 (Subra et al., 2010). In fact, the levels of these prostaglandins within exosomes was determined to be in the micromolar range, thus at concentrations capable of triggering prostaglandin-dependent biological effects (Subra et al., 2010).
J2 prostaglandins (Table 1) are bioactive cyclopentenone prostaglandins produced in vivo during inflammation (Rajakariar et al., 2007). Like their precursor, J2 prostaglandins can be considered some of the most abundant prostaglandins in the brain (Katura et al., 2010). For example, plasma levels of 15d-PGJ2 increased 12-fold and 23-fold in patients following acute stroke or with vascular risk factors and atherothrombotic infarcts, respectively (Blanco et al., 2005). In rodents, stroke (cerebral ischemia) and traumatic brain injury (TBI) elevate PGJ2 levels in the brain to concentrations similar to those shown to be neurotoxic in vitro (Kunz et al., 2002; Hickey et al., 2007; Liu et al., 2013a,b,c; Shaik et al., 2014). Accordingly, the in vivo concentration of free PGJ2 in the brain upon stroke and TBI, increases from almost undetectable to the 100 nM range (Liu et al., 2011, 2013a). These levels represent average brain concentrations, but it is predicted that local cellular and intracellular concentrations of J2 prostaglandins are much higher (Liu et al., 2013b). It is also clear that this is an underestimation of the overall J2 prostaglandin levels in vivo, as they bind covalently to proteins (see below), and therefore reported levels of free J2 prostaglandins do not represent their total amounts.
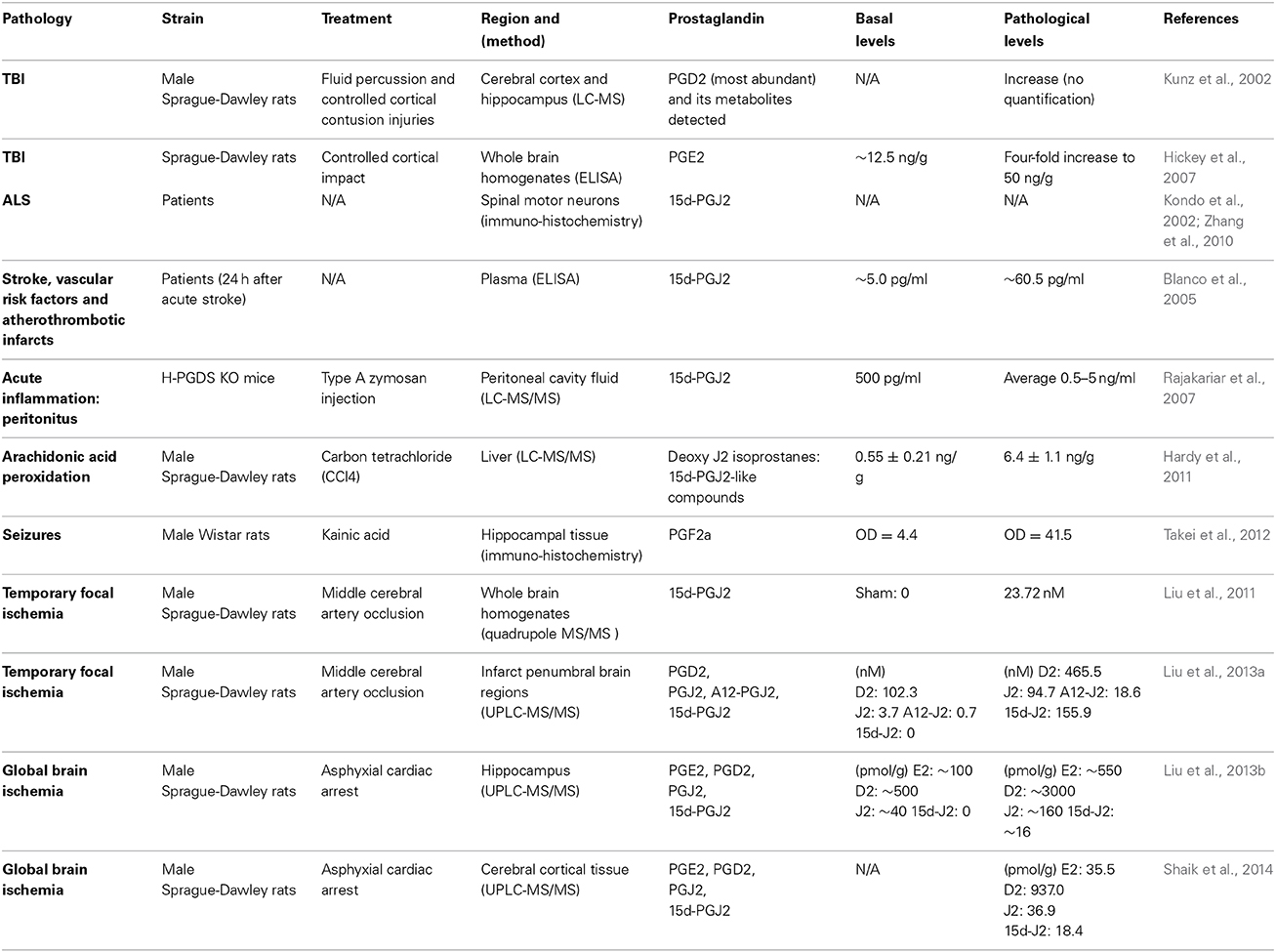
Table 1. In vivo levels of J2 prostaglandins.
Despite their lipid nature, prostaglandins are charged anions thus have low intrinsic permeability across the plasma membrane, but they cross it twice (Figure 4): once following their synthesis as they are released from the cytoplasm into the extracellular environment (efflux), and then again when they undergo reuptake into the cytoplasm (influx), a process that mimics neurotransmitter reuptake (Schuster, 1998; Chan et al., 2002; Chi et al., 2014). Prostaglandin efflux is mediated (a) by diffusion driven by pH and the membrane potential, and (b) by the action of transporters such as multidrug resistance-associated proteins (MRPs) and prostaglandin transporters (PGTs) (Schuster, 2002; Ohkura et al., 2014). Prostaglandin influx is mediated by PGTs as well (Schuster, 2002; Chi et al., 2006). Overall, prostaglandin transport requires further investigation. A well-characterized PGT belongs to the family of the 12-transmembrane organic anion transporting polypeptides (OATPs), and mediates the influx of prostaglandins only (Chi et al., 2006; Chi and Schuster, 2010). PGT mediated influx is a required step for prostaglandin metabolism (Nomura et al., 2004), which occurs intracellularly. PGD2 is a substrate for PGT (Itoh et al., 1996) and its metabolism to PGJ2 highly likely involves PGT mediated uptake. Interestingly, this PGT is a lactate/prostaglandin exchanger in which prostaglandin influx varies with lactate levels, so that cells engaged in glycolysis thus producing high levels of lactate, are energetically poised to uptake prostaglandins via this PGT (Chan et al., 2002; Banu et al., 2003). This PGT is present in many tissues including the brain (Kanai et al., 1995; Lu et al., 1996).
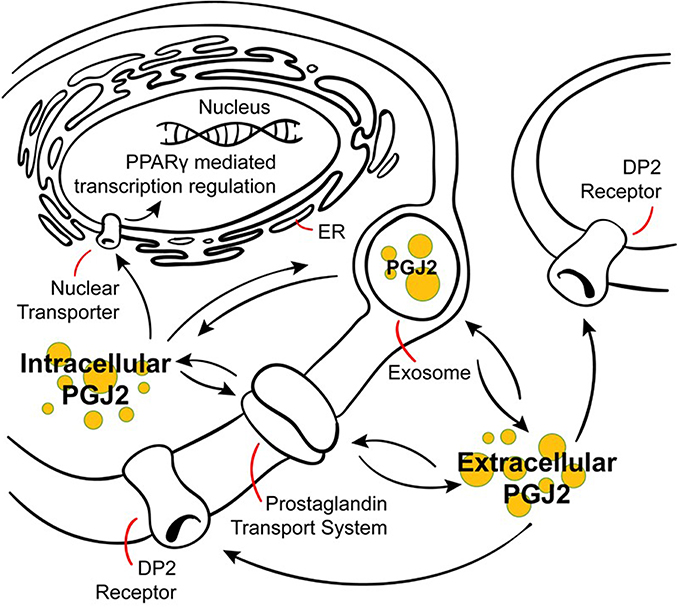
Figure 4. Modes of action of J2 prostaglandins. PGJ2 and its metabolites exit the cell via diffusion or poorly defined transporters, and can enter cells or the nucleus via active transporters at the plasma or nuclear membranes. PGJ2 and its metabolites exert their actions by different mechanisms. They can bind to the DP2 receptor on the plasma membrane or to the peroxisomal proliferator activator receptor (PPARγ) at the nuclear membrane. Trafficking of PGJ2 and its metabolites in and out of cells can also occur via exosomes.
Prostaglandins also can be transferred from cell to cell via exosomes, which are extracellular bioactive vesicles released from multivesicular bodies that mediate intercellular signaling (Subra et al., 2010). As such, exosomes are considered intercellular “signalosomes” as they carry arachidonic acid, phospholipases (A2 and D2), COX-1 and 2, and a whole set of prostaglandins, including PGD2, E2, and J2 (Subra et al., 2010). In fact, the concentrations of these prostaglandins within exosomes is in the micromolar range, thus at concentrations capable of triggering prostaglandin-dependent biological effects (Subra et al., 2010). Moreover, exosome internalization by neighboring cells is considered a mechanism for prostaglandins to reach their intracellular targets (Subra et al., 2010). Exosomes are released from and taken up by neurons in a synaptic activity-dependent manner that is also regulated by calcium (Lachenal et al., 2011; Perez-Gonzalez et al., 2012; Chivet et al., 2013; Morel et al., 2013). Furthermore, exosomes have recently been considered to be propagation vehicles for spreading of toxic proteins (Bellingham et al., 2012) as well as prostaglandins, such as neurotoxic PGJ2, and could thus play a significant role in the spread of pathology in a variety of neurodegenerative disorders (Schneider and Simons, 2013).
As they are unstable, prostaglandins exert their effects near their sites of synthesis thus acting as autocrine or paracrine ligands (Scher and Pillinger, 2005). The efflux of newly synthesized prostaglandins mediates their biological actions through their cell surface receptors, and prostaglandin influx from the extracellular milieu mediates their action through specific nuclear receptors or their inactivation (Banu et al., 2003). Prostaglandin inactivation in the cytoplasm is carried out by the enzyme NAD(+)-linked 15 hydroxyprostaglandin dehydrogenase (15-PGDH); its expression and that of COX-2 are reciprocally regulated in cancer, thus both enzymes control the cellular levels of prostaglandins by opposing means (Tai et al., 2006; Tai, 2011).
The effects of J2 prostaglandins are mediated by at least three different means.
In general, the prostaglandin GPCRs are present not only at the plasma membrane (Ricciotti and FitzGerald, 2011) but also at the nuclear membrane, thus providing for intracrine (intracellular) signaling (Zhu et al., 2006). PGD2, the precursor of PGJ2, binds to the DP1 and DP2 receptors (Urade and Eguchi, 2002). While the activation of the DP1 receptor is coupled to the G protein Gs, resulting in increased cAMP levels, activation of the DP2 receptor and its coupling to Gi decreases cAMP levels, and increases intracellular calcium (Hata and Breyer, 2004). J2 prostaglandins bind to DP1 and DP2, however they have a higher affinity for DP2 (as much as 100-fold) and bind to it with an affinity similar to PGD2, i.e., in the nanomolar range (Monneret et al., 2002; Pettipher et al., 2007). DP2 activation was shown to potentiate neuronal injury in hippocampal neuronal cultures and organotypic slices, while DP1 activation is neuroprotective (Liang et al., 2005, 2007).
15d-PGJ2 and Δ12-PGJ2 are endogenous ligands for the nuclear peroxisomal proliferator activator receptor (PPARγ), to which they bind with high affinity (Gilroy, 2010; Paulitschke et al., 2012), although this remains controversial (Ricciotti and FitzGerald, 2011). PPARγ plays a major role in the regulation of adipogenesis, glucose homeostasis, cellular differentiation, apoptosis and inflammation (Qi et al., 2010). PPARγ agonists promote neuroprotection in models of stroke, AD, HD, PD, MS, and spinal cord injury, via anti-inflammatory or antioxidant-dependent mechanisms (Kapadia et al., 2008; Kiaei, 2008).
J2 prostaglandins also act through PPARγ-independent mechanisms including activation of the MAPK and JNK pathways (Wilmer et al., 2001; Li et al., 2004a), stabilization of the transcription factor Nrf2 via its interaction with Keap1 (Itoh et al., 2004; Kaspar et al., 2009; Haskew-Layton et al., 2013), and inhibition of the NFκB pathway (Rossi et al., 2000; Straus et al., 2000). This may account for the different effects of 15d-PGJ2 and other PPAR ligands (Scher and Pillinger, 2005).
J2 prostaglandins are unique among the prostaglandin family in that they have α,β-unsaturated carbonyl groups (asterisks, Figure 5), promoting Michael addition reactions with free sulfhydryl groups of cysteines in glutathione and cellular proteins (Straus and Glass, 2001). These cyclopentenone PGs covalently modify several proteins, including the p50 subunit of NFκB, which may explain its anti-inflammatory effects (Cernuda-Morollon et al., 2001). They also modify thioredoxin reductase, an enzyme that protects against oxidative damage (Moos et al., 2003) and activate Ras, a small GTPase oncogene known to activate Erk signaling pathways (Oliva et al., 2003).
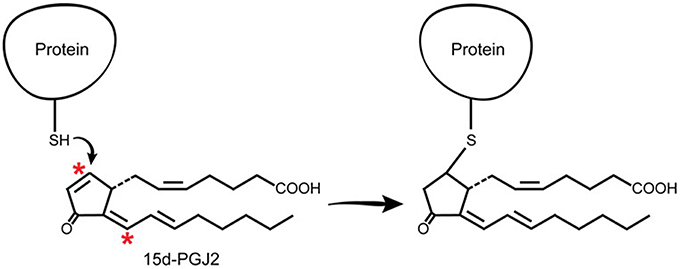
Figure 5. J2 prostaglandins interact directly with cellular proteins. J2 prostaglandins (shown for 15d-PGJ2) covalently modify selective proteins through Michael addition. Their α,β-unsaturated carbonyl groups (asterisks) react with free sulfhydryls (SH) in cysteines on glutathione and cellular proteins.
Electrophile binding to key protein cysteine(s) by endogenous compounds such as PGJ2 is regarded as playing an important role in determining whether neurons will live or die (Satoh and Lipton, 2007). In fact, with neuronal cultures, we established that PGJ2 was by far the most neurotoxic of four prostaglandins tested, including PGA1, D2, E2, and J2, with PGE2 being the least neurotoxic of the four under the conditions tested (Li et al., 2004b). The response to J2 prostaglandins is different from that of agonists that do not form covalent adducts with proteins. A slow steady stream-like release of J2 prostaglandins as a result of chronic neuroinflammation could be cumulative, leading overtime to accumulation of covalent PGJ2-protein adducts until they reach a toxic threshold. Thus, covalent protein modification in the brain by highly reactive electrophiles such as J2 prostaglandins, represents a novel pathologic post-translational change (Higdon et al., 2012b) and could play a critical role in progressive neurodegeneration.
The mechanisms underlying the transition from acute to chronic inflammation are poorly understood. One hypothesis supported by animal studies is that in addition to their role in mediating acute inflammation, prostaglandins also function in the transition and maintenance of chronic inflammation, culminating in long-lasting effects (Aoki and Narumiya, 2012). Prostaglandins accomplish this by amplifying cytokine signaling, up-regulating COX-2 (Figure 6), inducing chemokines, and recruiting inflammatory cells such as macrophages (Aoki and Narumiya, 2012). We and others demonstrated that J2 prostaglandins induce COX-2 up-regulation in cancer cells (Kim et al., 2008; Kitz et al., 2011) and neuronal cells (Li et al., 2004a) and this event can be driven by MAPK activation (Li et al., 2004a; Kitz et al., 2011) or Akt/AP-1 activation (Kim et al., 2008).
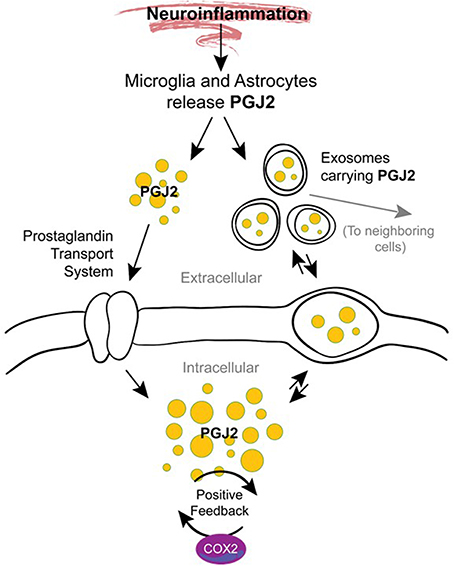
Figure 6. Potential mechanisms by which J2 prostaglandins promote neurodegeneration. During neuroinflammation PGJ2 (and its metabolites) are released from activated microglia and astrocytes. Free or exosome enclosed PGJ2 mediates the spread of neurodamage within the brain via intercellular uptake. PGJ2 also increases the levels of COX-2, thus activating a positive feedback loop that could mediate the transition from acute to chronic inflammation.
It is also possible that J2 prostaglandins mediate the spread of neurodamage within the brain via exosomes (Figure 6), as discussed above under “Modes of action of J2 prostaglandins.” This is important because exosomes were recently considered propagation vehicles for spreading neuropathology in a range of neurodegenerative diseases including AD, PD, and HD (Schneider and Simons, 2013). In vitro studies demonstrate that exosomes are released from neurons in a synaptic activity-dependent manner regulated by calcium (Lachenal et al., 2011). This later aspect of the regulation of the exosome-release by calcium could be particularly relevant to J2 prostaglandins. They are potent agonists (EC50 of ~10 nM) of the DP2 receptor (Monneret et al., 2002), which signals through elevation of intracellular calcium and reduction in intracellular cAMP (Pettipher et al., 2007), thus J2 prostaglandins could induce exosome-release.
These findings support the notion that targeting prostaglandin signaling for therapeutics represents a highly innovative strategy to prevent/block chronic neuroinflammation and disease progression.
COX-2 neurotoxicity seems to be mediated by PGD2 but not by PGE2 (Liang et al., 2005). PGD2 is the most abundant prostaglandin in the brain (Abdel-Halim et al., 1977; Narumiya et al., 1982; Hertting and Seregi, 1989). For example, in young rats (16–18 post-natal) subjected to a 12-min asphyxial cardiac arrest, the brain levels of PGE2 assessed by UPLC–MS/MS were ~35.5 pmol/g of tissue, while those of PGD2 were at least 26 fold higher, reaching ~937 pmol/g of tissue (Shaik et al., 2014). PGD2 elicits its cytotoxicity via its bioactive metabolites J2 prostaglandins (Liu et al., 2013b). In contrast to other reviews (Musiek et al., 2005; Uchida and Shibata, 2008; Scher and Pillinger, 2009; Surh et al., 2011; Oeste and Perez-Sala, 2014), our review addresses in detail the effects of J2 prostaglandins on two targets that play key roles in the neurodegenerative process, namely, the ubiquitin-proteasome pathway and mitochondrial function (Figure 7).
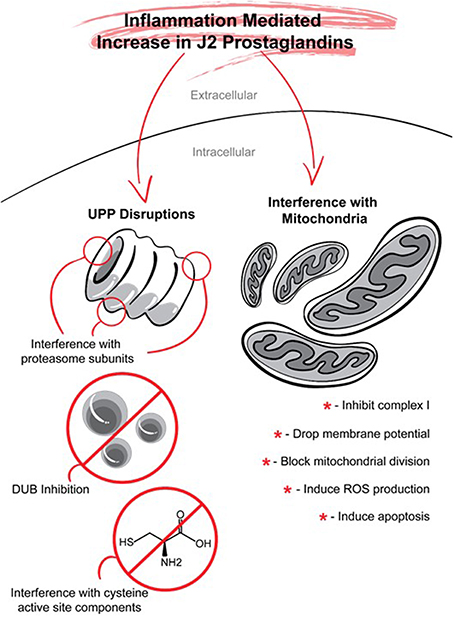
Figure 7. J2 prostaglandins target the ubiquitin proteasome pathway (UPP) and mitochondria. J2 prostaglandins affect the UPP by: (1) impairing the 26S proteasome by inducing oxidation of proteasome subunits, or promoting its disassembly, (2) inhibiting de-ubiquitinating enzymes (DUBs), and (3) covalently modifying specific active site cysteines on UPP components such as E1 activating enzymes, E2 conjugating enzymes, and some E3 ligases. J2 prostaglandins can also inhibit mitochondrial function by: (1) inhibiting NADH-ubiquinone reductase in complex I, (2) reducing membrane potential, (3) blocking fission, and (4) inducing the generation of reactive oxygen species (ROS) and apoptosis.
It is well-established that in neuronal cells J2 prostaglandins trigger the accumulation/aggregation of ubiquitinated proteins (see for example Li et al., 2004b; Ogburn and Figueiredo-Pereira, 2006; Liu et al., 2011, 2013a). This protein accumulation/aggregation can be mediated by at least two mechanisms shown to be affected by J2 prostaglandins.
We and others demonstrated that J2 prostaglandins impair the 26S proteasome (Shibata et al., 2003; Ishii and Uchida, 2004; Wang et al., 2006; Koharudin et al., 2010). In neuronal cells, these prostaglandins induce the oxidation of at least one proteasome subunit, i.e., S6 ATPase (Rpt5), which seems to be one of the proteasome subunits most vulnerable to protein carbonylation (Ishii et al., 2005). PGJ2 also promotes dissociation of the 20S core particle from the 19S regulatory particle (Wang et al., 2006), resembling the effects of agents that induce oxidative stress (Aiken et al., 2011). The effect of J2 prostaglandins on the proteasome are attributed to their electrophilic properties, since a 15d-PGJ2 analog that lacks the double bond in the cyclopentenone ring failed to inhibit the proteasome (Shibata et al., 2003). Furthermore, 15d-PGJ2/proteasome conjugates were detected in neuronal cells treated with biotinylated 15d-PGJ2 (Shibata et al., 2003).
These data indicate that one of the effects of inflammation mediated by J2 prostaglandin is proteasome inhibition. Conversely, proteasome inhibition suppresses inflammation (Bi et al., 2012). It was recently shown that the mechanisms by which the antibiotic rifampicin suppresses microglia activation upon LPS-treatment, is by downregulating the Rpt1 (MSS1) proteasome subunit (Bi et al., 2012). Likewise, downregulation of the proteasome subunit Rpn9 (PSMD13) by siRNA suppresses microglial activation and diminishes the production of inflammation associated factors such as nitric oxide synthase, nitric oxide, cyclooxygenase-2, and prostaglandin E2 (Bi et al., 2014).
A close relation between proteasome and inflammation is further supported by the finding that cyclooxygenases 1 and 2 are turned over by the 26S proteasome (Rockwell et al., 2000; Yazaki et al., 2012) via the endoplasmic reticulum-associated degradation (ERAD) pathway (Mbonye et al., 2006, 2008). Proteasomal-mediated turnover of COX-2 is regulated by the G protein-coupled receptor prostaglandin E1 (EP1 for PGE2) independently of receptor activation (Haddad et al., 2012). EP1 is present both at the plasma membrane and at the inner and outer membrane of the nuclear envelop (Bhattacharya et al., 1998). EP1 may act as a scaffold for an E3 ligase that ubiquitinates COX-2 (Haddad et al., 2012). In addition to the cyclooxygenases, prostaglandin synthases are also turned over by the proteasome. As such, increases in intracellular calcium result in the rapid proteasomal-mediated degradation of the PGD2 prostaglandin synthase H-PGDS in human megakaryocytic cells (Yazaki et al., 2012). Degradation of other prostaglandin synthases by the proteasome remains to be investigated.
The UPP and autophagy play a critical role in protein quality control and thus have attracted special attention for drug development (Edelmann et al., 2011). In regard to the UPP, which is the focus of this review, the initial effort was directed toward proteasome inhibitors, although not particularly for neurological conditions (Ristic et al., 2014). However, proteasome inhibition can be quite unspecific and thus may lead to a range of undesirable side effects. De-ubiquitinating enzymes known as DUBs are relevant UPP targets upstream of the proteasome and are the focus of recent attention for drug development. Several DUBs have been targeted for cancer treatment (Crosas, 2014), and one for neurological conditions (USP14) (Lee et al., 2010). At least one hundred (if not more) DUBs from five different gene families are present in humans, and four of these families are thiol proteases and one is a metalloprotease (Eletr and Wilkinson, 2014). These DUBs carry-out different functions, some of which involve processing newly translated ubiquitin (Ub) to provide monomers for conjugation and chain formation, or trimming mono-Ub from the distal end of a poly-Ub chain, or disassembling poly-Ub chains, or removing poly-Ub chains from their substrates (Clague et al., 2013). All together these functions provide a means for DUBs to regulate “where, when, and why” ubiquitinated substrates are degraded by the 26S proteasome (Ristic et al., 2014).
We and others showed that J2 prostaglandins inhibit some of the thiol DUBs including UCH-L1 and UCH-L3 (Mullally et al., 2001; Li et al., 2004b; Liu et al., 2011). In particular, UCH-L1 is predicted to be one of the most abundant proteins in the brain reaching 1–2% of the total protein content (Wilkinson et al., 1992), and its activity is highly diminished by J2 prostaglandins (Li et al., 2004b; Koharudin et al., 2010). These prostaglandins trigger UCH-L1 unfolding and aggregation by forming a covalent adduct with a single thiol group on Cys 152 (Koharudin et al., 2010). UCH-L1 plays an important role in aging and neurodegenerative disorders such as AD and PD, thus its alterations by J2 prostaglandins are highly relevant to the neurodegenerative process as discussed below under “neurodegenerative disorders” and reviewed in (Ristic et al., 2014).
Other components of the UPP, such as E1 activating enzymes, E2 conjugating enzymes, and some E3 ligases also have active site cysteines (Metzger et al., 2014). Whether J2 prostaglandins affect these and/or other thiol DUBs remains to be investigated. Protein modification by J2 prostaglandins seems to be highly selective where only a specific set of cysteine residues are vulnerable to covalent modification and this event occurs independently of protein thiol content (Higdon et al., 2012a; Vasil'ev et al., 2014). Of the several hundred cellular proteins with potentially reactive thiols, only ~10% form covalent adducts with J2 prostaglandins, and are considered members of the “electrophile-responsive proteome” (Ceaser et al., 2004). Although J2 prostaglandins interact with selective targets, they can covalently modify a wide variety of intracellular proteins. Proteomic approaches established that J2 prostaglandins covalently bind to specific sites within the plasma membrane, nuclear and cytosolic subcellular fractions (Yamamoto et al., 2011). At least eleven plasma membrane proteins were identified as binding biotinylated 15d-PGJ2 and they were distributed into three functional groups: glycolytic enzymes, molecular chaperones, and cytoskeletal proteins (Yamamoto et al., 2011). Furthermore, J2 prostaglandin binding alters protein catalysis, binding, structural function, and transport (Marcone and Fitzgerald, 2013; Oeste and Perez-Sala, 2014).
J2 prostaglandins inhibit mitochondrial function leading to oxidative stress and apoptosis (Kondo et al., 2001; Lee et al., 2009; Paulitschke et al., 2012). These prostaglandins inhibit the activity of the enzyme NADH-ubiquinone reductase from mitochondrial respiratory complex I, most likely by adduct formation (Martinez et al., 2005). J2 prostaglandins also induce the generation of reactive oxygen species (ROS), trigger a drop in membrane potential (Pignatelli et al., 2005; Gutierrez et al., 2006), interact with the cytoskeleton (Stamatakis et al., 2006) and block mitochondrial division through covalent modification of dynamin-related protein 1 (Drp1) which regulates mitochondrial fission (Mishra et al., 2010). A mitochondrial targeted analog of 15d-PGJ2 (mito-15d-PGJ2) was more efficient than 15d-PGJ2 at inducing apoptosis, was less potent at up-regulating Keap1-dependent antioxidant expression of HO-1 and GSH, and caused profound defects in mitochondrial bioenergetics and mitochondrial membrane depolarization (Diers et al., 2010). This interesting approach that involved specifically targeting 15d-PGJ2 to mitochondria by conjugation to a lipophilic cation, demonstrates the feasibility of manipulating its biological effects (Diers et al., 2010).
UPP and mitochondrial function are inherently connected, and since J2 prostaglandins affect both processes we will discuss this interaction. On the one hand the UPP degrades various mitochondrial proteins contributing to mitochondrial quality control (Taylor and Rutter, 2011). Mitochondrial proteins that are UPP substrates include (a) damaged and/or misfolded nuclear encoded proteins that are destined for import into mitochondria, (b) defective proteins at the outer mitochondrial membrane (OMM) extracted by p97 and delivered to the proteasome, and (c) non-OMM proteins, although the mechanistic details of how these proteins in the inner compartments retrotranslocate to the OMM remains poorly defined (Shanbhag et al., 2012). In addition, both the ubiquitin ligase parkin (Narendra and Youle, 2011) and the de-ubiquitinating enzyme USP30 (Bingol et al., 2014) seem to regulate mitochondrial turnover via mitophagy. The PINK1-Parkin pathway proposed to promote mitophagy remains controversial in neurons (Exner et al., 2012).
On the other hand, mitochondria provide ATP for protein ubiquitination and for 26S proteasome function (Livnat-Levanon and Glickman, 2011). E1 activity, the first step of the ubiquitination cascade, requires ATP for formation of a thiol ester adduct with ubiquitin (Haas et al., 1982; Schulman and Harper, 2009). If E1 activity is impaired all protein ubiquitination should be diminished. Moreover, the degradation of proteins by the 26S proteasome is highly dependent on ATP binding and hydrolysis (Liu et al., 2006).
It is postulated that in neurons even a modest restriction of ATP production by mitochondria far outweighs the negligible effects of ROS, although the underlying mechanisms are not clearly understood (Nicholls, 2008). In a recent study with neurons (Huang et al., 2013), we demonstrated that low ATP levels caused by mitochondrial dysfunction, correlated with impairment of the UPP: there was a decline in E1 and 26S proteasome activities with a concomitant rise in 20S proteasomes. This decline in UPP function occurred upon acute and long-term mitochondrial impairment. Notably, upon energy depletion, calpain activation led to the selective cleavage of Rpn10, a 26S proteasome subunit, without affecting other proteasome subunits tested. Rpn10 cleavage combined with ATP depletion, contributed to the decline in 26S proteasome function. We postulated that upon mitochondrial dysfunction, ATP-depletion and calpain activation contribute to the demise of protein turnover by the UPP in favor of unregulated and energy-independent protein degradation by 20S proteasomes. This adaptive response to energy deficiency may be suitable for short-term periods to promote degradation of randomly unfolded oxidized proteins. However, if chronic it can lead to neurodegeneration, as regulated protein degradation by the UPP is essential for neuronal survival (Huang et al., 2013).
Overall, the role of J2 prostaglandins in inflammation is complex (Harris et al., 2002; Wall et al., 2012). On the one hand, they have emerged as key anti-inflammatory agents as they inhibit the production of pro-inflammatory mediators such as iNOS, TNFα, and IL1β, suppress microglia and astrocyte activation and induce apoptosis (Eucker et al., 2004; Giri et al., 2004; Mrak and Landreth, 2004). On the other hand, J2 prostaglandins are pro-inflammatory agents. They stimulate the production of pro-inflammatory mediators such as IL8 and activate MAPK (Meade et al., 1999; Zhang et al., 2001). Furthermore, 15d-PGJ2 seems to play a role in the regulation of human autoimmune diseases and to inhibit inflammation in models of arthritis, ischemia-reperfusion injury, inflammatory bowel disease, lupus nephritis, and AD (Scher and Pillinger, 2009).
J2 prostaglandins also display both protective and destructive effects. Their biological activities include antiviral and antitumoral effects, modulation of the heat shock response, induction of oxidative stress, and apoptosis (Uchida and Shibata, 2008), as well as up-regulation of the death receptor 5 (DR5), sensitization to TRAIL-induced cytotoxicity, and caspase 8 activation (Kondo et al., 2002; Nakata et al., 2006; Su et al., 2008; Metcalfe et al., 2012). Although their anti-proliferative and pro-apoptotic effects are most frequently described, J2 prostaglandins also induce the proliferation of different forms of cancer cells when used at nanomolar to low micromolar concentrations (Oliva et al., 2003).
The conflicting effects of J2 prostaglandins most likely depend on their intracellular targets and downstream pathways which can be dose- and cell-type dependent (Servidei et al., 2004). J2 prostaglandins may exert some of their anti- or pro-inflammatory as well as anti- or pro-survival effects through PPARγ-dependent mechanisms. However, these prostaglandins also exert their actions through PPARγ-independent pathways as discussed above under “modes of action of J2 prostaglandins.” As such, it is known that specific PPARγ ligands, such as pioglitazone, do not replicate all of the effects attributed to J2 prostaglandins. Identifying the mechanism by which J2 prostaglandins exert their neurotoxic effects could lead to new strategies to prevent and/or delay neurodegeneration linked to inflammation.
Chronic neuroinflammation is recognized as a primary mechanism involved in neurodegenerative diseases such as AD, PD, and ALS (Liu and Hong, 2003; Glass et al., 2010; Cudaback et al., 2014; Mosher and Wyss-Coray, 2014). Moreover, UPP and mitochondrial dysfunction as interdependent cellular events (Livnat-Levanon and Glickman, 2011; Taylor and Rutter, 2011) are both impaired in many neurodegenerative disorders (Lin and Beal, 2006; Paul, 2008). However, the underlying mechanisms that bring about and/or maintain these malfunctions are unknown. A self-perpetuating cycle of inflammatory processes involving brain immune cells (microglia and astrocytes) could drive the slow progression of the neurodegenerative process, leading to dysfunction in protein degradation by the UPP and ATP production by mitochondria. Preventing/arresting this self-perpetuating inflammatory cycle is a very promising neuroprotective strategy for these disorders (Lima et al., 2012). We will focus on a review of studies showing that J2 prostaglandins are associated with AD, PD, and ALS, as well as stroke, TBI and Krabbe disease (Table 2).
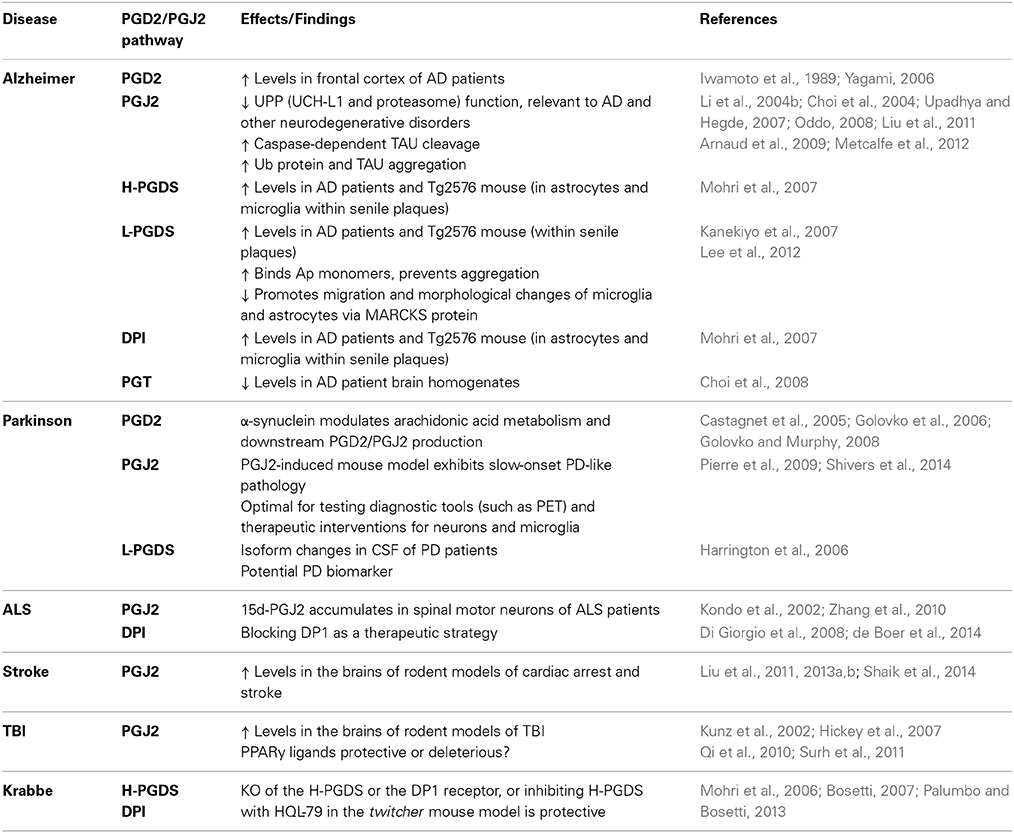
Table 2. J2 postaglandins and neurodegenerative disorders.
PGD2 levels were found to be significantly increased in the frontal cortex of AD patients compared to age matched controls (Iwamoto et al., 1989; Yagami, 2006). In AD patients and in Tg2576 mice, a well-established AD model (Hsiao et al., 1996), the levels of the PGD2 synthase H-PGDS and the PGD2 receptor DP1 were found to be selectively up-regulated in microglia and astrocytes within senile plaques (Mohri et al., 2007). These results support the notion that PGD2 acts as a mediator of plaque associated inflammation in the AD brain and they could also explain the pharmacologic mechanisms underlying the favorable response of patients with AD to non-steroidal anti-inflammatory drugs (Mohri et al., 2007).
The other PGD2 synthase L-PGDS, which is one of the most abundant CSF proteins produced in the brain, was localized in amyloid plaques in both AD patients and Tg2576 mice (Kanekiyo et al., 2007). Secreted L-PGDS in the CSF has a dual function: it increases CSF-PGD2 levels (Scher and Pillinger, 2005) and also acts as a lipophilic-ligand carrier (Urade and Hayaishi, 2000). L-PGDS was found to bind Aβ monomers and prevent Aβ aggregation, suggesting that L-PGDS is a major Aβ chaperone and disruption of this function could be related to the onset and progression of AD (Kanekiyo et al., 2007).
L-PGDS and PGD2 also promote migration and morphological changes of microglia and astrocytes that resemble those exhibited under reactive gliosis (Lee et al., 2012). This L-PGDS function is mediated by its interaction with myristoylated alanine-rich protein kinase C substrate (MARCKS), which in turn activates the AKT/Rho/JNK pathway (Lee et al., 2012). MARCKS is a plasma membrane resident abundant in the nervous system, and that depending on its phosphorylation by PKC acts as an actin cross-linker to regulate cellular adhesion and spreading, migration, proliferation, and fusion through its interaction with the cytoskeleton (Arbuzova et al., 2002). MARCKS also plays a role in the maintenance of dendritic spines and contributes to PKC-dependent morphological plasticity in hippocampal neurons (Calabrese and Halpain, 2005). Together these results support an essential role for L-PGDS in the regulation of glial cell migration and morphology, and perhaps neuronal plasticity, within the CNS (Lee et al., 2012).
The transport of prostaglandins across cell and organelle membranes involves, among others, a prostaglandin transporter (PGT) (Kanai et al., 1995). Immunohistochemical and immunofluorescent analyses of this PGT in human brains showed its localization in neurons, microglia, and astrocytes in all brain tissues assessed (Choi et al., 2008). In addition, PGT levels were lower in AD than in age-matched control brain homogenates, suggesting that prostaglandins might not be cleared at the normal rate in AD brains (Choi et al., 2008).
In relation to J2 prostaglandins, the finding that they impair the UPP is highly relevant to AD (Selkoe, 2004; Shaw et al., 2007). Defective proteasome activity is linked to the early phase of AD characterized by synaptic dysfunction, as well as to late AD stages linked to accumulation and aggregation of ubiquitinated (Ub)-proteins in both senile plaques and neurofibrillary tangles (Upadhya and Hegde, 2007; Oddo, 2008). Moreover, J2 prostaglandins inhibit the de-ubiquitinating enzyme UCH-L1 (Li et al., 2004b; Liu et al., 2011), which is down-regulated in AD brains; UCH-L1 down-regulation is inversely proportional to the number of neurofibrillary tangles (Choi et al., 2004).
Besides the effects on the UPP, we showed in rat primary cerebral cortical cultures that PGJ2 induced accumulation of Ub-proteins, caspase-activation, TAU cleavage at Asp421, and neuritic dystrophy (Arnaud et al., 2009; Metcalfe et al., 2012). TAU cleavage at Asp421 was identified as an early event in AD tangle pathology (Gamblin et al., 2003; Rissman et al., 2004; de Calignon et al., 2010). In summary, J2 prostaglandins mimic many pathological processes observed in AD.
Up-regulation of PGD2 or J2 prostaglandins in PD patients has not been addressed yet, but there is ample evidence linking PGD2, the precursor of PGJ2, to PD. Changes in PGD2 levels occurring in PD brains will lead to parallel changes in the highly reactive cyclopentenone J2 prostaglandins because PGD2 is unstable and is spontaneously metabolized to J2 prostaglandins. It was calculated that the half-life of PGD2 in the brain is 1.1 min (Suzuki et al., 1986).
Significant changes in L-PGDS isoforms were detected in the CSF of at least 20 idiopathic PD patients compared to 100 controls (Harrington et al., 2006). These alterations reflected up/down regulation of L-PGDS isoforms likely (a) to represent pathology at the cellular level expected to impact prostaglandin production, and (b) to correlate with disease symptoms (Harrington et al., 2006). It was speculated that these altered isoforms could be candidate diagnostic PD biomarkers and may have predictive value (Harrington et al., 2006).
A number of studies suggest that α-synuclein plays a role in brain fatty acid metabolism including arachidonic acid, through modulation of ER-localized acyl-CoA synthetase activity (Castagnet et al., 2005; Golovko et al., 2006). Acyl-CoA synthetase is an enzyme that converts fatty acids to acyl-coA for subsequent beta oxidation. In α-synuclein KO mice, exogenous addition of wild-type mouse or human α-synuclein restored acyl-CoA synthetase activity, while mutant (A30P, E46K, and A53T) forms of α-synuclein did not (Golovko et al., 2006). In addition, the levels of several prostaglandins in brains following a 30 s global ischemia were compared in wild type vs. α-synuclein KO mice (Golovko and Murphy, 2008). Among all prostaglandins assayed (E2, D2, F2α, TxB2, and 6-ketoF1α) PGD2 showed the greatest increase (two-fold) in the α-synuclein KO mice relative to the wild type. The levels of PGD2 in brains of α-synuclein KO mice reached ~35 ng/g following the 30 s global ischemia. Under normal physiological conditions, α-synuclein ablation had no effect. Together these studies suggest that α-synuclein could play a role in brain inflammatory responses through modulation of arachidonic acid metabolism and downstream PGD2/PGJ2 production.
As far as we know, we established the first model of inflammation in which the endogenous highly reactive product of inflammation PGJ2, induces PD pathology convincingly (Pierre et al., 2009). Microinfusion of PGJ2 into the brainstem and striatum of adult FVB male mice led to a dose-dependent reduction in the number of dopaminergic (TH+) neurons in the substantia nigra pars compacta, with little damage to local GABAergic interneurons, and to the appearance of Lewy-like bodies and activated glial cells. PGJ2-treatment resulted in a PD-like phenotype exhibiting gait disturbance and impaired balance. More recently we showed that this PGJ2-induced mouse model that mimics in part chronic inflammation, exhibits slow-onset PD-like pathology (Shivers et al., 2014). In this mouse model, microglia activation was evaluated in vivo by PET with [11C](R)PK11195 (Banati, 2002) to provide a regional estimation of brain inflammation. We also demonstrated that PACAP27, a peptide that increases intracellular cAMP levels (Moody et al., 2011), reduced dopaminergic neuronal loss and motor deficits induced by PGJ2, without preventing microglia activation. The latter could be problematic in that persistent microglia activation can exert long-term deleterious effects on neurons and behavior. In conclusion, this PGJ2-induced mouse model is optimal for testing diagnostic tools such as PET, which is a powerful technique to quantitatively assess neuroinflammation in vivo (Stoessl, 2014), as well as therapies designed to target the integrated signaling across neurons and microglia, to fully benefit patients with PD.
There is evidence supporting the involvement of J2 prostaglandins in ALS. As such, 15d-PGJ2 was shown to accumulate in spinal motor neurons of patients with amyotrophic lateral sclerosis (ALS) (Kondo et al., 2002; Zhang et al., 2010). Moreover, astrocytes from mice carrying the SOD1G93A mutation were shown to be toxic to stem cell-derived human motor neurons but not to interneurons (Di Giorgio et al., 2008). The astrocyte induced neurotoxicity was mediated by up-regulation of PGD2 signaling, and was prevented by MK05524, an antagonist for the PGD2 receptor DP1 (Di Giorgio et al., 2008). In a more recent study, an in vivo genetic approach validated the importance of this DP1-mediated mechanism for neuronal degeneration. As such, genetic ablation of DP1 in SOD1G93A mice extended their life span, decreased microglial activation, and reduced motor neuron loss (de Boer et al., 2014). These results suggest that blocking DP1 may be a therapeutic strategy in ALS (Di Giorgio et al., 2008; de Boer et al., 2014).
Stroke and silent brain infarcts are high risk factors for dementia and neurodegenerative diseases such as AD (Vermeer et al., 2003) and PD (Becker et al., 2010; Rodriguez-Grande et al., 2013). The cyclooxygenase pathway was considered to be a valuable therapeutic target for stroke, however while COX-2 inhibitors are able to diminish injury in stroke models, they also produce an unbalance in prostanoid synthesis that promotes damaging vascular effects (Iadecola and Gorelick, 2005). For this reason, new therapeutic strategies targeting the factors that mediate the damage downstream from COX-2 may offer stroke patients powerful new tools to ameliorate brain damage and improve their functional outcome (Iadecola and Gorelick, 2005). Some of these factors could be J2 prostaglandins, as their levels in the brain are highly elevated in rodent models of cardiac arrest and stroke (Liu et al., 2011, 2013a,c; Shaik et al., 2014). Due to their inhibitory effects on the UPP, J2 prostaglandins could play an important role in brain ischemia brought about by cardiac arrest or stroke, as UPP impairment is highly relevant to these conditions (Caldeira et al., 2014). Thus, therapeutic strategies targeting the deleterious effects of J2 prostaglandins could offer great promise.
TBI is another neurological condition associated with J2 prostaglandins. In rodents, TBI elevates J2 prostaglandin levels in the brain to concentrations similar to those shown to be neurotoxic in vitro (Kunz et al., 2002; Hickey et al., 2007). TBI initiates an inflammatory cascade that leads to acute pathologic processes as well as long-term neuronal damage (Ziebell and Morganti-Kossmann, 2010). The nuclear receptor PPARγ, for which 15d-PGJ2 is an endogenous ligand, is considered a major anti-inflammatory and neuroprotective target for treating patients with TBI. However, PPARγ activation can also trigger apoptosis (Qi et al., 2010). These opposing effects seem to be related to the level of PPARγ agonists produced (Clay et al., 1999; Na and Surh, 2003). Low PPARγ agonist levels exert neuroprotective and anti-inflammatory effects that include down-regulation of inflammatory responses, reduction of oxidative stress, inhibition of apoptosis, and promotion of neurogenesis, while high levels induced apoptosis (Qi et al., 2010). The regulatory mechanisms and signaling cascades underlying the opposing PPARγ effects require further elucidation (Qi et al., 2010).
This disease is associated with demyelination, for which the twitcher mouse is an authentic animal model (Duchen et al., 1980; Kobayashi et al., 1980). In this mouse model, myelination proceeds normally up to post-natal day 30, when demyelination initiates due to oligodendrocyte apoptosis accompanied by microglia activation and astroglyosis (Mohri et al., 2006). Remarkably, a blockade of the PGD2 signaling cascade in the twitcher mouse via knock-out of the PGD2 synthase H-PGDS or the PGD2 receptor DP1, or treating these mice with the H-PGDS inhibitor HQL-79 [4-benzhydryloxy-1-[3-(1H-tetrazol-5-yl)-propyl]piperidine], suppressed astroglyosis, demyelination, twitching and spasticity. These results support the notion that PGD2 and perhaps its metabolites, are key to the pathological demyelination occurring in the twitcher mouse, and the neuroprotective potential of manipulating the PGD2-signaling to overcome demyelination in Krabbe disease (Mohri et al., 2006; Bosetti, 2007).
While inflammation can be beneficial, failure to adequately control its abatement when the injurious agent is neutralized, is increasingly believed to be one of the major causes of chronic inflammation (Gilroy, 2010). The therapeutic potential of the prostanoid pathway is supported by the clinical efficacy of NSAIDs, although they present risky side effects as current NSAIDs non-selectively inhibit the overall synthesis of all prostaglandins (Table 3).
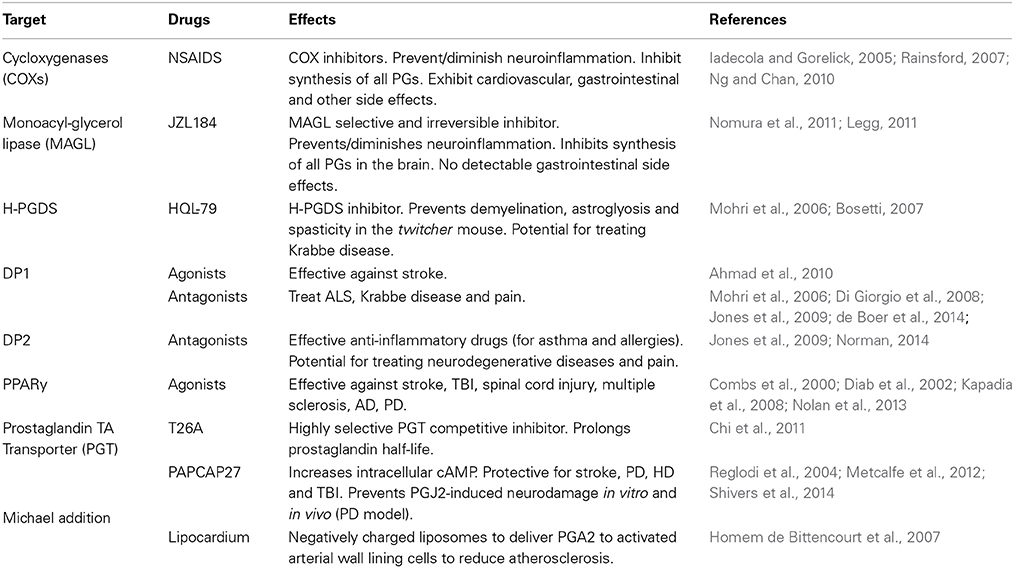
Table 3. Potential J2 prostaglandin therapeutic targets.
A more recent approach of diminishing prostaglandin-induced neuroinflammation is to block the activity of the enzyme monoacylglycerol lipase (MAGL) (Table 3). MAGL, which hydrolyzes endocannabinoids, can also regulate arachidonic acid release in the brain but not in the gastrointestinal tract, that culminates in the generation of neuroinflammatory prostaglandins (Nomura et al., 2011; Piro et al., 2012). A novel MAGL selective and irreversible inhibitor (JZL184, which is 4-nitrophenyl-4-[bis(1,3-benzodioxol-5-yl)(hydroxy)methyl]piperidine-1-carboxylate) has shown promising results for blocking the neuroinflammation in the brain associated with PD and other neurodegenerative disorders (Legg, 2011; Nomura et al., 2011).
Additional specific targets in the prostaglandin pathways, such as prostaglandin synthases, prostaglandin transporters, and prostaglandin receptors have also emerged as drug targets (Table 3). For example, recently a highly selective competitive inhibitor of the prostaglandin transporter PGT was found to prolong PGE2 half-life, in vitro and in vivo (Chi et al., 2011). It is expected that further delineation of the prostaglandin pathway will yield novel beneficial therapeutics in the years to come (Smyth et al., 2009).
As discussed throughout this review, further knowledge on the neurotoxic mechanisms mediated by J2 prostaglandins and their contribution to the progression and longevity/resolution of the inflammatory response are needed to develop novel and more effective neuroprotective therapeutic strategies to attenuate inflammation. J2 prostaglandins represent attractive therapeutic targets because of their important roles in the development and resolution of inflammation. In particular, J2 prostaglandin-dependent therapeutics should target mechanisms of action that include receptor activation and Michael addition.
It is critical to fully understand the dual roles of these prostaglandins as pro- and anti-inflammatory agents. The opposing effects of PGD2/J2 prostaglandins on inflammation are reflected by the responses induced by activation of the DP1 or DP2 receptors, for which a range of antagonists are undergoing clinical evaluation (Pettipher et al., 2007; Sandig et al., 2007; Ricciotti and FitzGerald, 2011). Some of these potential drugs can reach the cerebrospinal fluid upon oral administration (Pettipher et al., 2007). While DP1 agonists seem to alleviate brain damage upon stroke (Ahmad et al., 2010), DP1 antagonists are being proposed to treat disorders such as ALS (de Boer et al., 2014) and Krabbe disease (Mohri et al., 2006). DP2 antagonists are highly relevant to treating allergies and asthma (Norman, 2014). The effective anti-inflammatory properties of these drugs may be relevant to blocking inflammation associated with neurodegeneration and pain (Jones et al., 2009).
PPARγ agonists seem to protect neurons not only following acute CNS injury including stroke, spinal cord injury and TBI after which massive inflammation plays a detrimental role, but also in neurodegenerative conditions including multiple sclerosis (Diab et al., 2002; Chaudhuri, 2013), AD (Combs et al., 2000) and PD (Kapadia et al., 2008). While 15d-PGJ2 is thought to be the endogenous ligand for PPARγ, the thiazolidinediones (TZDs) are used as potent exogenous agonists exerting their neuroprotective effects via prevention of microglial activation, inflammatory cytokine and chemokine expression, and promoting the anti-oxidant mechanisms in the injured CNS (Kapadia et al., 2008). More recent evidence suggests that the nuclear receptor (NR) superfamily of transcription factors including Nuclear receptor-related factor1 (Nurr1), PPARs, retinoic acid and glucocorticoid receptors show promise as therapeutic targets for PD. Since they are known to regulate an array of inflammatory mediators, it is postulated that modulating Nurr1 expression or NR receptor activation, including PPARs, via agonists would protect against dopaminergic neuronal death induced by inflammation (Nolan et al., 2013).
Overall, the effectiveness of J2 prostaglandin receptor agonists/antagonists on the treatment of neurodegenerative conditions needs to be carefully investigated as these receptors may act sequentially to initiate and sustain disease states, and/or play complementary roles. Potentially, a combination of these receptor agonists/antagonists could prove to be a very promising therapeutic approach (Jones et al., 2009). Whether these drugs can be applied to preventing chronic long-term neuroinflammation remains to be explored.
The best characterized mechanism of action of J2 prostaglandins is the covalent modification of proteins at cysteine residues through Michael addition, which is attributed to their electrophilic nature (Oeste and Perez-Sala, 2014). In this regard, proteomic approaches used in the past few years have provided much needed knowledge about the pharmacological actions and signaling mechanisms of J2 prostaglandins (Oeste and Perez-Sala, 2014). Detailed investigation of the protein targets directly affected by these lipid mediators of inflammation, is critical to the development of more specific and effective therapeutic approached against the deleterious effects of neuroinflammation. Identification of these protein targets will provide important clues on the pathways modulated by J2 prostaglandins and the mechanisms underlying their beneficial or deleterious effects.
As we discussed above, two of these pathways that are affected by J2 prostaglandins and that are highly relevant to neurodegeneration are the UPP and mitochondrial function. We recently investigated in vitro (Metcalfe et al., 2012) and in vivo (Shivers et al., 2014) a potential therapeutic approach to overcome J2 prostaglandin neurotoxicity, based on elevating intracellular cAMP with PACAP27 (pituitary adenylate cyclase-activating polypeptide). PACAP27 is a potent neuroprotective lipophilic peptide in different models of neuronal injury such as stroke, PD, HD, TBI, retinal degeneration, and others, where it exhibits anti-apoptotic, anti-inflammatory and anti-oxidant effects (Reglodi et al., 2004; Atlasz et al., 2010; Ohtaki et al., 2010; Dejda et al., 2011; Mao et al., 2012). While we confirmed the anti-apoptotic effects of PACAP27 in our studies, it was clear that PACAP27 as tested and by itself was not sufficient to overcome all of the neurodamaging effects of PGJ2 (Metcalfe et al., 2012; Shivers et al., 2014). Ideal therapeutic interventions against J2 prostaglandins may require a combinatorial approach to effectively prevent the pleiotropic effects of these highly reactive endogenous mediators of inflammation.
In contrast, negatively charged cyclopentenone prostaglandin-based liposomes (LipoCardium) were developed to specifically deliver prostaglandins, in this case PGA2, to injured arterial wall cells of atherosclerotic mice (Homem de Bittencourt et al., 2007). Anti-inflammatory, anti-proliferative, anti-cholesterogenic and cytoprotective effects were obtained with LipoCardium. This strategy opens up new avenues for specific prostaglandin delivery to humans, including a chronic slow delivery.
In conclusion, much remains to be discovered about the biology of the J2 prostaglandins, to prevent their neurotoxic effects and possibly “trick” ongoing inflammation into resolution. Further investigations may provide important clues “to bring us into a new era of inflammation research, which, if approached with creativity and persistence, might provide numerous benefits for those suffering from inflammation-mediated diseases” (Gilroy, 2010).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Please note that this review is not intended to be comprehensive and we apologize to the authors whose work is not mentioned. We would like to thank Mr. Omer Goodovich for the illustrations. This work was supported by National Institutes of Health (NIH) [CTSC GRANT UL1-RR024996 (pilot award to Maria E. Figueiredo-Pereira, co-investigator and another pilot award to Thomas Schmidt-Glenewinkel), NIGMS 1SC3GM086323 to Thomas Schmidt-Glenewinkel, 5R24DA012136-13 (pilot award to Peter Serrano)]; PSC-CUNY Research Award to Patricia Rockwell and Maria E. Figueiredo-Pereira.
Abdel-Halim, M. S., Hamberg, M., Sjoquist, B., and Anggard, E. (1977). Identification of prostaglandin D2 as a major prostaglandin in homogenates of rat brain. Prostaglandins 14, 633–643. doi: 10.1016/0090-6980(77)90190-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ahmad, A. S., Ahmad, M., Maruyama, T., Narumiya, S., and Dore, S. (2010). Prostaglandin D2 DP1 receptor is beneficial in ischemic stroke and in acute exicitotoxicity in young and old mice. Age (Dordr.) 32, 271–282. doi: 10.1007/s11357-010-9135-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aid, S., and Bosetti, F. (2011). Targeting cyclooxygenases-1 and -2 in neuroinflammation: therapeutic implications. Biochimie 93, 46–51. doi: 10.1016/j.biochi.2010.09.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aiken, C. T., Kaake, R. M., Wang, X., and Huang, L. (2011). Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell Proteomics 10, R110. doi: 10.1074/mcp.M110.006924
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aoki, T., and Narumiya, S. (2012). Prostaglandins and chronic inflammation. Trends Pharmacol. Sci. 33, 304–311. doi: 10.1016/j.tips.2012.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arbuzova, A., Schmitz, A. A., and Vergeres, G. (2002). Cross-talk unfolded: MARCKS proteins. Biochem. J. 362, 1–12. doi: 10.1042/0264-6021:3620001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arnaud, L. T., Myeku, N., and Figueiredo-Pereira, M. E. (2009). Proteasome-caspase-cathepsin sequence leading to tau pathology induced by prostaglandin J2 in neuronal cells. J. Neurochem. 110, 328–342. doi: 10.1111/j.1471-4159.2009.06142.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Astudillo, A. M., Balgoma, D., Balboa, M. A., and Balsinde, J. (2012). Dynamics of arachidonic acid mobilization by inflammatory cells. Biochim. Biophys. Acta 1821, 249–256. doi: 10.1016/j.bbalip.2011.11.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Atlasz, T., Szabadfi, K., Kiss, P., Tamas, A., Toth, G., Reglodi, D., et al. (2010). Evaluation of the protective effects of PACAP with cell-specific markers in ischemia-induced retinal degeneration. Brain Res. Bull. 81, 497–504. doi: 10.1016/j.brainresbull.2009.09.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Banati, R. B. (2002). Visualising microglial activation in vivo. Glia 40, 206–217. doi: 10.1002/glia.10144
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Banu, S. K., Arosh, J. A., Chapdelaine, P., and Fortier, M. A. (2003). Molecular cloning and spatio-temporal expression of the prostaglandin transporter: a basis for the action of prostaglandins in the bovine reproductive system. Proc. Natl. Acad. Sci. U.S.A. 100, 11747–11752. doi: 10.1073/pnas.1833330100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bartels, A. L., and Leenders, K. L. (2010). Cyclooxygenase and neuroinflammation in Parkinson's disease neurodegeneration. Curr. Neuropharmacol. 8, 62–68. doi: 10.2174/157015910790909485
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Becker, C., Jick, S. S., and Meier, C. R. (2010). Risk of stroke in patients with idiopathic Parkinson disease. Parkinsonism. Relat. Disord. 16, 31–35. doi: 10.1016/j.parkreldis.2009.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bellingham, S. A., Guo, B. B., Coleman, B. M., and Hill, A. F. (2012). Exosomes: vehicles for the transfer of toxic proteins associated with neurodegenerative diseases? Front. Physiol. 3:124. doi: 10.3389/fphys.2012.00124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bernardo, A., Ajmone-Cat, M. A., Levi, G., and Minghetti, L. (2003). 15-Deoxy-delta12,14-prostaglandin J2 regulates the functional state and the survival of microglial cells through multiple molecular mechanisms. J. Neurochem. 87, 742–751. doi: 10.1046/j.1471-4159.2003.02045.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bhattacharya, M., Peri, K. G., Almazan, G., Ribeiro-da-Silva, A., Shichi, H., Durocher, Y., et al. (1998). Nuclear localization of prostaglandin E2 receptors. Proc. Natl. Acad. Sci. U.S.A. 95, 15792–15797. doi: 10.1073/pnas.95.26.15792
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bi, W., Jing, X., Zhu, L., Liang, Y., Liu, J., Yang, L., et al. (2012). Inhibition of 26S protease regulatory subunit 7 (MSS1) suppresses neuroinflammation. PLoS ONE 7:e36142. doi: 10.1371/journal.pone.0036142
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bi, W., Zhu, L., Zeng, Z., Jing, X., Liang, Y., Guo, L., et al. (2014). Investigations into the role of 26S proteasome non-ATPase regulatory subunit 13 in neuroinflammation. Neuroimmunomodulation 21, 331–337. doi: 10.1159/000357811
Bingol, B., Tea, J. S., Phu, L., Reichelt, M., Bakalarski, C. E., Song, Q., et al. (2014). The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370–375. doi: 10.1038/nature13418
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blanco, M., Moro, M. A., Davalos, A., Leira, R., Castellanos, M., Serena, J., et al. (2005). Increased plasma levels of 15-deoxyDelta prostaglandin J2 are associated with good outcome in acute atherothrombotic ischemic stroke. Stroke 36, 1189–1194. doi: 10.1161/01.STR.0000166054.55993.e5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bosetti, F. (2007). Arachidonic acid metabolism in brain physiology and pathology: lessons from genetically altered mouse models. J. Neurochem. 102, 577–586. doi: 10.1111/j.1471-4159.2007.04558.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Calabrese, B., and Halpain, S. (2005). Essential role for the PKC target MARCKS in maintaining dendritic spine morphology. Neuron 48, 77–90. doi: 10.1016/j.neuron.2005.08.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caldeira, M. V., Salazar, I. L., Curcio, M., Canzoniero, L. M., and Duarte, C. B. (2014). Role of the ubiquitin-proteasome system in brain ischemia: friend or foe? Prog. Neurobiol. 112, 50–69. doi: 10.1016/j.pneurobio.2013.10.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Castagnet, P. I., Golovko, M. Y., Barcelo-Coblijn, G. C., Nussbaum, R. L., and Murphy, E. J. (2005). Fatty acid incorporation is decreased in astrocytes cultured from alpha-synuclein gene-ablated mice. J. Neurochem. 94, 839–849. doi: 10.1111/j.1471-4159.2005.03247.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ceaser, E. K., Moellering, D. R., Shiva, S., Ramachandran, A., Landar, A., Venkartraman, A., et al. (2004). Mechanisms of signal transduction mediated by oxidized lipids: the role of the electrophile-responsive proteome. Biochem. Soc. Trans. 32, 151–155. doi: 10.1042/BST0320151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cernuda-Morollon, E., Pineda-Molina, E., Canada, F. J., and Perez-Sala, D. (2001). 15-Deoxy-Delta 12,14-prostaglandin J2 inhibition of NF-kappaB-DNA binding through covalent modification of the p50 subunit. J. Biol. Chem. 276, 35530–35536. doi: 10.1074/jbc.M104518200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chan, B. S., Endo, S., Kanai, N., and Schuster, V. L. (2002). Identification of lactate as a driving force for prostanoid transport by prostaglandin transporter PGT. Am. J. Physiol. Renal. Physiol. 282, F1097–F1102. doi: 10.1152/ajprenal.00151.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chaudhuri, A. (2013). Multiple sclerosis is primarily a neurodegenerative disease. J. Neural Transm. 120, 1463–1466. doi: 10.1007/s00702-013-1080-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chi, Y., Khersonsky, S. M., Chang, Y. T., and Schuster, V. L. (2006). Identification of a new class of prostaglandin transporter inhibitors and characterization of their biological effects on prostaglandin E2 transport. J. Pharmacol. Exp. Ther. 316, 1346–1350. doi: 10.1124/jpet.105.091975
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chi, Y., Min, J., Jasmin, J. F., Lisanti, M. P., Chang, Y. T., and Schuster, V. L. (2011). Development of a high-affinity inhibitor of the prostaglandin transporter. J. Pharmacol. Exp. Ther. 339, 633–641. doi: 10.1124/jpet.111.181354
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chi, Y., and Schuster, V. L. (2010). The prostaglandin transporter PGT transports PGH(2). Biochem. Biophys. Res. Commun. 395, 168–172. doi: 10.1016/j.bbrc.2010.03.108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chi, Y., Suadicani, S., and Schuster, V. L. (2014). Regulation of prostaglandin EP1 and EP4 receptor signaling by carrier-meditaed ligand reuptake. Pharma Res. Per. 2:e00051. doi: 10.1002/prp2.51
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chivet, M., Javalet, C., Hemming, F., Pernet-Gallay, K., Laulagnier, K., Fraboulet, S., et al. (2013). Exosomes as a novel way of interneuronal communication. Biochem. Soc. Trans. 41, 241–244. doi: 10.1042/BST20120266
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choi, J., Levey, A. I., Weintraub, S. T., Rees, H. D., Gearing, M., Chin, L. S., et al. (2004). Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson's and Alzheimer's diseases. J. Biol. Chem. 279, 13256–13264. doi: 10.1074/jbc.M314124200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choi, K., Zhuang, H., Crain, B., and Dore, S. (2008). Expression and localization of prostaglandin transporter in Alzheimer disease brains and age-matched controls. J. Neuroimmunol. 195, 81–87. doi: 10.1016/j.jneuroim.2008.01.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choi, S. H., Aid, S., and Bosetti, F. (2009). The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: implications for translational research. Trends Pharmacol. Sci. 30, 174–181. doi: 10.1016/j.tips.2009.01.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clague, M. J., Barsukov, I., Coulson, J. M., Liu, H., Rigden, D. J., and Urbe, S. (2013). Deubiquitylases from genes to organism. Physiol. Rev. 93, 1289–1315. doi: 10.1152/physrev.00002.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clay, C. E., Namen, A. M., Atsumi, G., Willingham, M. C., High, K. P., Kute, T. E., et al. (1999). Influence of J series prostaglandins on apoptosis and tumorigenesis of breast cancer cells. Carcinogenesis 20, 1905–1911. doi: 10.1093/carcin/20.10.1905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Combs, C. K., Johnson, D. E., Karlo, J. C., Cannady, S. B., and Landreth, G. E. (2000). Inflammatory mechanisms in Alzheimer's disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J. Neurosci. 20, 558–567.
Consilvio, C., Vincent, A. M., and Feldman, E. L. (2004). Neuroinflammation, COX-2, and ALS–a dual role? Exp. Neurol. 187, 1–10. doi: 10.1016/j.expneurol.2003.12.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crosas, B. (2014). Deubiquitinating enzyme inhibitors and their potential in cancer therapy. Curr. Cancer Drug Targets 14, 506–516. doi: 10.2174/1568009614666140725090620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cudaback, E., Jorstad, N. L., Yang, Y., Montine, T. J., and Keene, C. D. (2014). Therapeutic implications of the prostaglandin pathway in Alzheimer's disease. Biochem. Pharmacol. 88, 565–572. doi: 10.1016/j.bcp.2013.12.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Boer, A. S., Koszka, K., Kiskinis, E., Suzuki, N., Davis-Dusenbery, B. N., and Eggan, K. (2014). Genetic validation of a therapeutic target in a mouse model of ALS. Sci. Transl. Med. 6, 248ra104. doi: 10.1126/scitranslmed.3009351
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Calignon, A., Fox, L. M., Pitstick, R., Carlson, G. A., Bacskai, B. J., Spires-Jones, T. L., et al. (2010). Caspase activation precedes and leads to tangles. Nature 464, 1201–1204. doi: 10.1038/nature08890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dejda, A., Seaborn, T., Bourgault, S., Touzani, O., Fournier, A., Vaudry, H., et al. (2011). PACAP and a novel stable analog protect rat brain from ischemia: insight into the mechanisms of action. Peptides 32, 1207–1216. doi: 10.1016/j.peptides.2011.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Diab, A., Deng, C., Smith, J. D., Hussain, R. Z., Phanavanh, B., Lovett-Racke, A. E., et al. (2002). Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-Delta(12,14)-prostaglandin J(2) ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 168, 2508–2515. doi: 10.4049/jimmunol.168.5.2508
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Diers, A. R., Higdon, A. N., Ricart, K. C., Johnson, M. S., Agarwal, A., Kalyanaraman, B., et al. (2010). Mitochondrial targeting of the electrophilic lipid 15-deoxy-Delta12,14-prostaglandin J2 increases apoptotic efficacy via redox cell signalling mechanisms. Biochem. J. 426, 31–41. doi: 10.1042/BJ20091293
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Di Giorgio, F. P., Boulting, G. L., Bobrowicz, S., and Eggan, K. C. (2008). Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 3, 637–648. doi: 10.1016/j.stem.2008.09.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Duchen, L. W., Eicher, E. M., Jacobs, J. M., Scaravilli, F., and Teixeira, F. (1980). Hereditary leucodystrophy in the mouse: the new mutant twitcher. Brain 103, 695–710. doi: 10.1093/brain/103.3.695
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edelmann, M. J., Nicholson, B., and Kessler, B. M. (2011). Pharmacological targets in the ubiquitin system offer new ways of treating cancer, neurodegenerative disorders and infectious diseases. Expert Rev. Mol. Med. 13, e35. doi: 10.1017/S1462399411002031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eletr, Z. M., and Wilkinson, K. D. (2014). Regulation of proteolysis by human deubiquitinating enzymes. Biochim. Biophys. Acta 1843, 114–128. doi: 10.1016/j.bbamcr.2013.06.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eucker, J., Bangeroth, K., Zavrski, I., Krebbel, H., Zang, C., Heider, U., et al. (2004). Ligands of peroxisome proliferator-activated receptor gamma induce apoptosis in multiple myeloma. Anticancer Drugs 15, 955–960. doi: 10.1097/00001813-200411000-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Exner, N., Lutz, A. K., Haass, C., and Winklhofer, K. F. (2012). Mitochondrial dysfunction in Parkinson's disease: molecular mechanisms and pathophysiological consequences. EMBO J. 31, 3038–3062. doi: 10.1038/emboj.2012.170
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feng, Z., Li, D., Fung, P. C., Pei, Z., Ramsden, D. B., and Ho, S. L. (2003). COX-2-deficient mice are less prone to MPTP-neurotoxicity than wild-type mice. Neuroreport 14, 1927–1929. doi: 10.1097/00001756-200310270-00009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Funk, C. D. (2001). Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294, 1871–1875. doi: 10.1126/science.294.5548.1871
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gamblin, T. C., Chen, F., Zambrano, A., Abraha, A., Lagalwar, S., Guillozet, A. L., et al. (2003). Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 100, 10032–10037. doi: 10.1073/pnas.1630428100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gilroy, D. W. (2010). Eicosanoids and the endogenous control of acute inflammatory resolution. Int. J. Biochem. Cell Biol. 42, 524–528. doi: 10.1016/j.biocel.2009.12.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Giri, S., Rattan, R., Singh, A. K., and Singh, I. (2004). The 15-deoxy-delta12,14-prostaglandin J2 inhibits the inflammatory response in primary rat astrocytes via down-regulating multiple steps in phosphatidylinositol 3-kinase-Akt-NF-kappaB-p300 pathway independent of peroxisome proliferator-activated receptor gamma. J. Immunol. 173, 5196–5208. doi: 10.4049/jimmunol.173.8.5196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934. doi: 10.1016/j.cell.2010.02.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Golovko, M. Y., and Murphy, E. J. (2008). Brain prostaglandin formation is increased by alpha-synuclein gene-ablation during global ischemia. Neurosci. Lett. 432, 243–247. doi: 10.1016/j.neulet.2007.12.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Golovko, M. Y., Rosenberger, T. A., Faergeman, N. J., Feddersen, S., Cole, N. B., Pribill, I., et al. (2006). Acyl-CoA synthetase activity links wild-type but not mutant alpha-synuclein to brain arachidonate metabolism. Biochemistry 45, 6956–6966. doi: 10.1021/bi0600289
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gutierrez, J., Ballinger, S. W., Darley-Usmar, V. M., and Landar, A. (2006). Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circ. Res. 99, 924–932. doi: 10.1161/01.RES.0000248212.86638.e9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haas, A. L., Warms, J. V., Hershko, A., and Rose, I. A. (1982). Ubiquitin-activating enzyme. Mechanism and role in protein-ubiquitin conjugation. J. Biol. Chem. 257, 2543–2548.
Haddad, A., Flint-Ashtamker, G., Minzel, W., Sood, R., Rimon, G., and Barki-Harrington, L. (2012). Prostaglandin EP1 receptor down-regulates expression of cyclooxygenase-2 by facilitating its proteasomal degradation. J. Biol. Chem. 287, 17214–17223. doi: 10.1074/jbc.M111.304220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hardy, K. D., Cox, B. E., Milne, G. L., Yin, H., and Roberts, L. J. (2011). Nonenzymatic free radical-catalyzed generation of 15-deoxy-Delta(12,14)-prostaglandin J(2)-like compounds (deoxy-J(2)-isoprostanes) in vivo. J. Lipid Res. 52, 113–124. doi: 10.1194/jlr.M010264
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harrington, M. G., Fonteh, A. N., Biringer, R. G., AF, R. H., and Cowan, R. P. (2006). Prostaglandin D synthase isoforms from cerebrospinal fluid vary with brain pathology. Dis. Markers 22, 73–81. doi: 10.1155/2006/241817
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harris, S. G., Padilla, J., Koumas, L., Ray, D., and Phipps, R. P. (2002). Prostaglandins as modulators of immunity. Trends Immunol. 23, 144–150. doi: 10.1016/S1471-4906(01)02154-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haskew-Layton, R. E., Payappilly, J. B., Xu, H., Bennett, S. A., and Ratan, R. R. (2013). 15-Deoxy-Delta12,14-prostaglandin J2 (15d-PGJ2) protects neurons from oxidative death via an Nrf2 astrocyte-specific mechanism independent of PPARgamma. J. Neurochem. 124, 536–547. doi: 10.1111/jnc.12107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hata, A. N., and Breyer, R. M. (2004). Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol. Ther. 103, 147–166. doi: 10.1016/j.pharmthera.2004.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Herrup, K. (2010). Reimagining Alzheimer's disease–an age-based hypothesis. J. Neurosci. 30, 16755–16762. doi: 10.1523/JNEUROSCI.4521-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hertting, G., and Seregi, A. (1989). Formation and function of eicosanoids in the central nervous system. Ann. N. Y. Acad. Sci. 559, 84–99. doi: 10.1111/j.1749-6632.1989.tb22600.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hickey, R. W., Adelson, P. D., Johnnides, M. J., Davis, D. S., Yu, Z., Rose, M. E., et al. (2007). Cyclooxygenase-2 activity following traumatic brain injury in the developing rat. Pediatr. Res. 62, 271–276. doi: 10.1203/PDR.0b013e3180db2902
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Higdon, A., Diers, A. R., Oh, J. Y., Landar, A., and Darley–Usmar, V. M. (2012a). Cell signalling by reactive lipid species: new concepts and molecular mechanisms. Biochem. J. 442, 453–464.
Higdon, A. N., Landar, A., Barnes, S., and Darley-Usmar, V. M. (2012b). The electrophile responsive proteome: integrating proteomics and lipidomics with cellular function. Antioxid. Redox. Signal. 17, 1580–1589. doi: 10.1089/ars.2012.4523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hirata, Y., Hayashi, H., Ito, S., Kikawa, Y., Ishibashi, M., Sudo, M., et al. (1988). Occurrence of 9-deoxy-delta 9,delta 12-13,14-dihydroprostaglandin D2 in human urine. J. Biol. Chem. 263, 16619–16625.
Homem de Bittencourt, P. I. Jr., Lagranha, D. J., Maslinkiewicz, A., Senna, S. M., Tavares, A. M., Baldissera, L. P., et al. (2007). LipoCardium: endothelium-directed cyclopentenone prostaglandin-based liposome formulation that completely reverses atherosclerotic lesions. Atherosclerosis 193, 245–258. doi: 10.1016/j.atherosclerosis.2006.08.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hsiao, K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S., et al. (1996). Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice [see comments]. Science 274, 99–102. doi: 10.1126/science.274.5284.99
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, Q., Wang, H., Perry, S. W., and Figueiredo-Pereira, M. E. (2013). Negative regulation of 26S proteasome stability via calpain-mediated cleavage of Rpn10 subunit upon mitochondrial dysfunction in neurons. J. Biol. Chem. 288, 12161–12174. doi: 10.1074/jbc.M113.464552
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iadecola, C., and Gorelick, P. B. (2005). The Janus face of cyclooxygenase-2 in ischemic stroke: shifting toward downstream targets. Stroke 36, 182–185. doi: 10.1161/01.STR.0000153797.33611.d8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ishii, T., Sakurai, T., Usami, H., and Uchida, K. (2005). Oxidative modification of proteasome: identification of an oxidation-sensitive subunit in 26 s proteasome. Biochemistry 44, 13893–13901. doi: 10.1021/bi051336u
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ishii, T., and Uchida, K. (2004). Induction of reversible cysteine-targeted protein oxidation by an endogenous electrophile 15-deoxy-delta12,14-prostaglandin J2. Chem. Res. Toxicol. 17, 1313–1322. doi: 10.1021/tx049860+
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Itoh, K., Mochizuki, M., Ishii, Y., Ishii, T., Shibata, T., Kawamoto, Y., et al. (2004). Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Delta(12,14)-prostaglandin j(2). Mol. Cell Biol. 24, 36–45. doi: 10.1128/MCB.24.1.36-45.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Itoh, S., Lu, R., Bao, Y., Morrow, J. D., Roberts, L. J., and Schuster, V. L. (1996). Structural determinants of substrates for the prostaglandin transporter PGT. Mol. Pharmacol. 50, 738–742.
Iwamoto, N., Kobayashi, K., and Kosaka, K. (1989). The formation of prostaglandins in the postmortem cerebral cortex of Alzheimer-type dementia patients. J. Neurol. 236, 80–84. doi: 10.1007/BF00314401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jones, R. L., Giembycz, M. A., and Woodward, D. F. (2009). Prostanoid receptor antagonists: development strategies and therapeutic applications. Br. J. Pharmacol. 158, 104–145. doi: 10.1111/j.1476-5381.2009.00317.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kanai, N., Lu, R., Satriano, J. A., Bao, Y., Wolkoff, A. W., and Schuster, V. L. (1995). Identification and characterization of a prostaglandin transporter. Science 268, 866–869. doi: 10.1126/science.7754369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kanaoka, Y., and Urade, Y. (2003). Hematopoietic prostaglandin D synthase. Prostaglandins Leukot. Essent. Fatty Acids 69, 163–167. doi: 10.1016/S0952-3278(03)00077-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kanekiyo, T., Ban, T., Aritake, K., Huang, Z. L., Qu, W. M., Okazaki, I., et al. (2007). Lipocalin-type prostaglandin D synthase/beta-trace is a major amyloid beta-chaperone in human cerebrospinal fluid. Proc. Natl. Acad. Sci. U.S.A. 104, 6412–6417. doi: 10.1073/pnas.0701585104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kapadia, R., Yi, J. H., and Vemuganti, R. (2008). Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front. Biosci. 13:1813–1826. doi: 10.2741/2802
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaspar, J. W., Niture, S. K., and Jaiswal, A. K. (2009). Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 47, 1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Katura, T., Moriya, T., and Nakahata, N. (2010). 15-Deoxy-delta 12,14-prostaglandin J2 biphasically regulates the proliferation of mouse hippocampal neural progenitor cells by modulating the redox state. Mol. Pharmacol. 77, 601–611. doi: 10.1124/mol.109.061010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kiaei, M. (2008). Peroxisome proliferator-activated receptor-gamma in amyotrophic lateral sclerosis and Huntington's disease. PPAR. Res. 2008, 418765. doi: 10.1155/2008/418765
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, E. H., Na, H. K., Kim, D. H., Park, S. A., Kim, H. N., Song, N. Y., et al. (2008). 15-Deoxy-Delta12,14-prostaglandin J2 induces COX-2 expression through Akt-driven AP-1 activation in human breast cancer cells: a potential role of ROS. Carcinogenesis 29, 688–695. doi: 10.1093/carcin/bgm299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kitz, K., Windischhofer, W., Leis, H. J., Huber, E., Kollroser, M., and Malle, E. (2011). 15-Deoxy-Delta12,14-prostaglandin J2 induces Cox-2 expression in human osteosarcoma cells through MAPK and EGFR activation involving reactive oxygen species. Free Radic. Biol. Med. 50, 854–865. doi: 10.1016/j.freeradbiomed.2010.12.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klegeris, A., McGeer, E. G., and McGeer, P. L. (2007). Therapeutic approaches to inflammation in neurodegenerative disease. Curr. Opin. Neurol. 20, 351–357. doi: 10.1097/WCO.0b013e3280adc943
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klivenyi, P., Gardian, G., Calingasan, N. Y., Yang, L., and Beal, M. F. (2003). Additive neuroprotective effects of creatine and a cyclooxygenase 2 inhibitor against dopamine depletion in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson's disease. J. Mol. Neurosci. 21, 191–198. doi: 10.1385/JMN:21:3:191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kobayashi, T., Yamanaka, T., Jacobs, J. M., Teixeira, F., and Suzuki, K. (1980). The Twitcher mouse: an enzymatically authentic model of human globoid cell leukodystrophy (Krabbe disease). Brain Res. 202, 479–483. doi: 10.1016/0006-8993(80)90159-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koharudin, L. M., Liu, H., Di, M. R., Kodali, R. B., Graham, S. H., and Gronenborn, A. M. (2010). Cyclopentenone prostaglandin-induced unfolding and aggregation of the Parkinson disease-associated UCH-L1. Proc. Natl. Acad. Sci. U.S.A. 107, 6835–6840. doi: 10.1073/pnas.1002295107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kondo, M., Oya-Ito, T., Kumagai, T., Osawa, T., and Uchida, K. (2001). Cyclopentenone prostaglandins as potential inducers of intracellular oxidative stress. J. Biol. Chem. 276, 12076–12083. doi: 10.1074/jbc.M009630200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kondo, M., Shibata, T., Kumagai, T., Osawa, T., Shibata, N., Kobayashi, M., et al. (2002). 15-Deoxy-Delta(12,14)-prostaglandin J(2): the endogenous electrophile that induces neuronal apoptosis. Proc. Natl. Acad. Sci. U.S.A. 99, 7367–7372. doi: 10.1073/pnas.112212599
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kulkarni, S. K., Jain, N. K., and Singh, A. (2000). Cyclooxygenase isoenzymes and newer therapeutic potential for selective COX-2 inhibitors. Methods Find. Exp. Clin. Pharmacol. 22, 291–298. doi: 10.1358/mf.2000.22.5.796648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kunz, T., Marklund, N., Hillered, L., and Oliw, E. H. (2002). Cyclooxygenase-2, prostaglandin synthases, and prostaglandin H2 metabolism in traumatic brain injury in the rat. J. Neurotrauma 19, 1051–1064. doi: 10.1089/089771502760341965
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lachenal, G., Pernet-Gallay, K., Chivet, M., Hemming, F. J., Belly, A., Bodon, G., et al. (2011). Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell Neurosci. 46, 409–418. doi: 10.1016/j.mcn.2010.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, B. H., Lee, M. J., Park, S., Oh, D. C., Elsasser, S., Chen, P. C., et al. (2010). Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467, 179–184. doi: 10.1038/nature09299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, D. R., Kwon, C. H., Park, J. Y., Kim, Y. K., and Woo, J. S. (2009). 15-Deoxy-Delta(12,14)-prostaglandin J(2) induces mitochondrial-dependent apoptosis through inhibition of PKA/NF-kappaB in renal proximal epithelial cells. Toxicology 258, 17–24. doi: 10.1016/j.tox.2009.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, S., Jang, E., Kim, J. H., Kim, J. H., Lee, W. H., and Suk, K. (2012). Lipocalin-type prostaglandin D2 synthase protein regulates glial cell migration and morphology through myristoylated alanine-rich C-kinase substrate: prostaglandin D2-independent effects. J. Biol. Chem. 287, 9414–9428. doi: 10.1074/jbc.M111.330662
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Legg, K. (2011). Neurodegenerative diseases: an alternative path to reduce neuroinflammation. Nat. Rev. Drug Discov. 10:901. doi: 10.1038/nrd3607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Z., Jansen, M., Ogburn, K., Salvatierra, L., Hunter, L., Mathew, S., et al. (2004a). Neurotoxic prostaglandin J2 enhances cyclooxygenase-2 expression in neuronal cells through the p38MAPK pathway: a death wish? J. Neurosci. Res. 78, 824–836. doi: 10.1002/jnr.20346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Z., Melandri, F., Berdo, I., Jansen, M., Hunter, L., Wright, S., et al. (2004b). Delta12-Prostaglandin J2 inhibits the ubiquitin hydrolase UCH-L1 and elicits ubiquitin-protein aggregation without proteasome inhibition. Biochem. Biophys. Res. Commun. 319, 1171–1180. doi: 10.1016/j.bbrc.2004.05.098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liang, X., Wu, L., Hand, T., and Andreasson, K. (2005). Prostaglandin D2 mediates neuronal protection via the DP1 receptor. J. Neurochem. 92, 477–486. doi: 10.1111/j.1471-4159.2004.02870.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liang, X., Wu, L., Wang, Q., Hand, T., Bilak, M., McCullough, L., et al. (2007). Function of COX-2 and prostaglandins in neurological disease. J. Mol. Neurosci. 33, 94–99. doi: 10.1007/s12031-007-0058-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lima, I. V., Bastos, L. F., Limborco-Filho, M., Fiebich, B. L., and de Oliveira, A. C. (2012). Role of prostaglandins in neuroinflammatory and neurodegenerative diseases. Mediators Inflamm. 2012:946813. doi: 10.1155/2012/946813
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, B., Gao, H. M., and Hong, J. S. (2003). Parkinson's disease and exposure to infectious agents and pesticides and the occurrence of brain injuries: role of neuroinflammation. Environ. Health Perspect. 111, 1065–1073. doi: 10.1289/ehp.6361
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, B., and Hong, J. S. (2003). Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 304, 1–7. doi: 10.1124/jpet.102.035048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, C. W., Li, X., Thompson, D., Wooding, K., Chang, T. L., Tang, Z., et al. (2006). ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome. Mol. Cell 24, 39–50. doi: 10.1016/j.molcel.2006.08.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, H., Li, W., Ahmad, M., Miller, T. M., Rose, M. E., Poloyac, S. M., et al. (2011). Modification of ubiquitin-C-terminal hydrolase-L1 by cyclopentenone prostaglandins exacerbates hypoxic injury. Neurobiol. Dis. 41, 318–328. doi: 10.1016/j.nbd.2010.09.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, H., Li, W., Ahmad, M., Rose, M. E., Miller, T. M., Yu, M., et al. (2013a). Increased generation of cyclopentenone prostaglandins after brain ischemia and their role in aggregation of ubiquitinated proteins in neurons. Neurotox. Res. 24, 191–204. doi: 10.1007/s12640-013-9377-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, H., Li, W., Rose, M. E., Pascoe, J. L., Miller, T. M., Ahmad, M., et al. (2013b). Prostaglandin D toxicity in primary neurons is mediated through its bioactive cyclopentenone metabolites. Neurotoxicology 39C, 35–44. doi: 10.1016/j.neuro.2013.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, H., Rose, M. E., Miller, T. M., Li, W., Shinde, S. N., Pickrell, A. M., et al. (2013c). COX2-derived primary and cyclopentenone prostaglandins are increased after asphyxial cardiac arrest. Brain Res. 1519, 71–77. doi: 10.1016/j.brainres.2013.04.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Livnat-Levanon, N., and Glickman, M. H. (2011). Ubiquitin-proteasome system and mitochondria – reciprocity. Biochim. Biophys. Acta 1809, 80–87. doi: 10.1016/j.bbagrm.2010.07.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, R., Kanai, N., Bao, Y., and Schuster, V. L. (1996). Cloning, in vitro expression, and tissue distribution of a human prostaglandin transporter cDNA(hPGT). J. Clin. Invest. 98, 1142–1149. doi: 10.1172/JCI118897
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lucin, K. M., and Wyss-Coray, T. (2009). Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 64, 110–122. doi: 10.1016/j.neuron.2009.08.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mao, S. S., Hua, R., Zhao, X. P., Qin, X., Sun, Z. Q., Zhang, Y., et al. (2012). Exogenous administration of PACAP alleviates traumatic brain injury in rats through a mechanism involving the TLR4/MyD88/NF-kappaB pathway. J. Neurotrauma 29, 1941–1959. doi: 10.1089/neu.2011.2244
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marcone, S., and Fitzgerald, D. J. (2013). Proteomic identification of the candidate target proteins of 15-deoxy-delta12,14-prostaglandin J2. Proteomics 13, 2135–2139. doi: 10.1002/pmic.201200289
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martinez, B., Perez-Castillo, A., and Santos, A. (2005). The mitochondrial respiratory complex I is a target for 15-deoxy-delta12,14-prostaglandin J2 action. J. Lipid Res. 46, 736–743. doi: 10.1194/jlr.M400392-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mbonye, U. R., Wada, M., Rieke, C. J., Tang, H. Y., DeWitt, D. L., and Smith, W. L. (2006). The 19-amino acid cassette of cyclooxygenase-2 mediates entry of the protein into the endoplasmic reticulum-associated degradation system. J. Biol. Chem. 281, 35770–35778. doi: 10.1074/jbc.M608281200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mbonye, U. R., Yuan, C., Harris, C. E., Sidhu, R. S., Song, I., Arakawa, T., et al. (2008). Two distinct pathways for cyclooxygenase-2 protein degradation. J. Biol. Chem. doi: 10.1074/jbc.M710137200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meade, E. A., McIntyre, T. M., Zimmerman, G. A., and Prescott, S. M. (1999). Peroxisome proliferators enhance cyclooxygenase-2 expression in epithelial cells. J. Biol. Chem. 274, 8328–8334. doi: 10.1074/jbc.274.12.8328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Metcalfe, M. J., Huang, Q., and Figueiredo-Pereira, M. E. (2012). Coordination between proteasome impairment and caspase activation leading to TAU pathology: neuroprotection by cAMP. Cell Death Dis. 3, e326. doi: 10.1038/cddis.2012.70
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Metzger, M. B., Pruneda, J. N., Klevit, R. E., and Weissman, A. M. (2014). RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta 1843, 47–60. doi: 10.1016/j.bbamcr.2013.05.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mishra, N., Kar, R., Singha, P. K., Venkatachalam, M. A., McEwen, D. G., and Saikumar, P. (2010). Inhibition of mitochondrial division through covalent modification of Drp1 protein by 15 deoxy-Delta(12,14)-prostaglandin J2. Biochem. Biophys. Res. Commun. 395, 17–24. doi: 10.1016/j.bbrc.2010.03.093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mohri, I., Kadoyama, K., Kanekiyo, T., Sato, Y., Kagitani-Shimono, K., Saito, Y., et al. (2007). Hematopoietic prostaglandin D synthase and DP1 receptor are selectively upregulated in microglia and astrocytes within senile plaques from human patients and in a mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol. 66, 469–480. doi: 10.1097/01.jnen.0000240472.43038.27
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mohri, I., Taniike, M., Taniguchi, H., Kanekiyo, T., Aritake, K., Inui, T., et al. (2006). Prostaglandin D2-mediated microglia/astrocyte interaction enhances astrogliosis and demyelination in twitcher. J. Neurosci. 26, 4383–4393. doi: 10.1523/JNEUROSCI.4531-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Monneret, G., Li, H., Vasilescu, J., Rokach, J., and Powell, W. S. (2002). 15-Deoxy-delta 12,14-prostaglandins D2 and J2 are potent activators of human eosinophils. J. Immunol. 168, 3563–3569. doi: 10.4049/jimmunol.168.7.3563
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moody, T. W., Ito, T., Osefo, N., and Jensen, R. T. (2011). VIP and PACAP: recent insights into their functions/roles in physiology and disease from molecular and genetic studies. Curr. Opin. Endocrinol. Diabetes Obes. 18, 61–67. doi: 10.1097/MED.0b013e328342568a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moos, P. J., Edes, K., Cassidy, P., Massuda, E., and Fitzpatrick, F. A. (2003). Electrophilic prostaglandins and lipid aldehydes repress redox-sensitive transcription factors p53 and hypoxia-inducible factor by impairing the selenoprotein thioredoxin reductase. J. Biol. Chem. 278, 745–750. doi: 10.1074/jbc.M211134200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morel, L., Regan, M., Higashimori, H., Ng, S. K., Esau, C., Vidensky, S., et al. (2013). Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J. Biol. Chem. 288, 7105–7116. doi: 10.1074/jbc.M112.410944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mosher, K. I., and Wyss-Coray, T. (2014). Microglial dysfunction in brain aging and Alzheimer's disease. Biochem. Pharmacol. 88, 594–604. doi: 10.1016/j.bcp.2014.01.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mrak, R. E., and Landreth, G. E. (2004). PPARgamma, neuroinflammation, and disease. J. Neuroinflammation 1:5. doi: 10.1186/1742-2094-1-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mullally, J. E., Moos, P. J., Edes, K., and Fitzpatrick, F. A. (2001). Cyclopentenone prostaglandins of the J series inhibit the ubiquitin isopeptidase activity of the proteasome pathway. J. Biol. Chem. 276, 30366–30373. doi: 10.1074/jbc.M102198200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murray, J. J., Tonnel, A. B., Brash, A. R., Roberts, L. J., Gosset, P., Workman, R., et al. (1986). Release of prostaglandin D2 into human airways during acute antigen challenge. N. Engl. J. Med. 315, 800–804. doi: 10.1056/NEJM198609253151304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Musiek, E. S., Milne, G. L., McLaughlin, B., and Morrow, J. D. (2005). Cyclopentenone eicosanoids as mediators of neurodegeneration: a pathogenic mechanism of oxidative stress-mediated and cyclooxygenase-mediated neurotoxicity. Brain Pathol. 15, 149–158. doi: 10.1111/j.1750-3639.2005.tb00512.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Na, H. K., and Surh, Y. J. (2003). Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands as bifunctional regulators of cell proliferation. Biochem. Pharmacol. 66, 1381–1391. doi: 10.1016/S0006-2952(03)00488-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nakata, S., Yoshida, T., Shiraishi, T., Horinaka, M., Kouhara, J., Wakada, M., et al. (2006). 15-Deoxy-Delta12,14-prostaglandin J(2) induces death receptor 5 expression through mRNA stabilization independently of PPARgamma and potentiates TRAIL-induced apoptosis. Mol. Cancer Ther. 5, 1827–1835. doi: 10.1158/1535-7163.MCT-06-0023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Narendra, D. P., and Youle, R. J. (2011). Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid. Redox. Signal. 14, 1929–1938. doi: 10.1089/ars.2010.3799
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Narumiya, S., Ogorochi, T., Nakao, K., and Hayaishi, O. (1982). Prostaglandin D2 in rat brain, spinal cord and pituitary: basal level and regional distribution. Life Sci. 31, 2093–2103. doi: 10.1016/0024-3205(82)90101-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ng, S. C., and Chan, F. K. (2010). NSAID-induced gastrointestinal and cardiovascular injury. Curr. Opin. Gastroenterol. 26, 611–617. doi: 10.1097/MOG.0b013e32833e91eb
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicholls, D. G. (2008). Oxidative stress and energy crises in neuronal dysfunction. Ann. N. Y. Acad. Sci. 1147, 53–60. doi: 10.1196/annals.1427.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nolan, Y. M., Sullivan, A. M., and Toulouse, A. (2013). Parkinson's disease in the nuclear age of neuroinflammation. Trends Mol. Med. 19, 187–196. doi: 10.1016/j.molmed.2012.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nomura, D. K., Morrison, B. E., Blankman, J. L., Long, J. Z., Kinsey, S. G., Marcondes, M. C., et al. (2011). Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 334, 809–813. doi: 10.1126/science.1209200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nomura, T., Lu, R., Pucci, M. L., and Schuster, V. L. (2004). The two-step model of prostaglandin signal termination: in vitro reconstitution with the prostaglandin transporter and prostaglandin 15 dehydrogenase. Mol. Pharmacol. 65, 973–978. doi: 10.1124/mol.65.4.973
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Norman, P. (2014). Update on the status of DP2 receptor antagonists; from proof of concept through clinical failures to promising new drugs. Expert Opin. Investig. Drugs 23, 55–66. doi: 10.1517/13543784.2013.839658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oddo, S. (2008). The ubiquitin-proteasome system in Alzheimer's disease. J. Cell Mol. Med. 12, 363–373. doi: 10.1111/j.1582-4934.2008.00276.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oeste, C. L., and Perez-Sala, D. (2014). Modification of cysteine residues by cyclopentenone prostaglandins: interplay with redox regulation of protein function. Mass Spectrom. Rev. 33, 110–125. doi: 10.1002/mas.21383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Offenbacher, S., Odle, B. M., and Van Dyke, T. E. (1986). The use of crevicular fluid prostaglandin E2 levels as a predictor of periodontal attachment loss. J. Periodontal. Res. 21, 101–112. doi: 10.1111/j.1600-0765.1986.tb01443.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ogburn, K. D., and Figueiredo-Pereira, M. E. (2006). Cytoskeleton/endoplasmic reticulum collapse induced by prostaglandin J2 parallels centrosomal deposition of ubiquitinated protein aggregates. J. Biol. Chem. 281, 23274–23284. doi: 10.1074/jbc.M600635200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ogorochi, T., Narumiya, S., Mizuno, N., Yamashita, K., Miyazaki, H., and Hayaishi, O. (1984). Regional distribution of prostaglandins D2, E2, and F2 alpha and related enzymes in postmortem human brain. J. Neurochem. 43, 71–82. doi: 10.1111/j.1471-4159.1984.tb06680.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ohkura, N., Shigetani, Y., Yoshiba, N., Yoshiba, K., and Okiji, T. (2014). Prostaglandin transporting protein-mediated prostaglandin E2 transport in lipopolysaccharide-inflamed rat dental pulp. J. Endod. 40, 1112–1117. doi: 10.1016/j.joen.2013.12.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ohtaki, H., Satoh, A., Nakamachi, T., Yofu, S., Dohi, K., Mori, H., et al. (2010). Regulation of oxidative stress by pituitary adenylate cyclase-activating polypeptide (PACAP) mediated by PACAP receptor. J. Mol. Neurosci. 42, 397–403. doi: 10.1007/s12031-010-9350-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oliva, J. L., Perez-Sala, D., Castrillo, A., Martinez, N., Canada, F. J., Bosca, L., et al. (2003). The cyclopentenone 15-deoxy-delta 12,14-prostaglandin J2 binds to and activates H-Ras. Proc. Natl. Acad. Sci. U.S.A. 100, 4772–4777. doi: 10.1073/pnas.0735842100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Palumbo, S., and Bosetti, F. (2013). Alterations of brain eicosanoid synthetic pathway in multiple sclerosis and in animal models of demyelination: role of cyclooxygenase-2. Prostaglandins Leukot. Essent. Fatty Acids 89, 273–278. doi: 10.1016/j.plefa.2013.08.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paul, S. (2008). Dysfunction of the ubiquitin-proteasome system in multiple disease conditions: therapeutic approaches. Bioessays 30, 1172–1184. doi: 10.1002/bies.20852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paulitschke, V., Gruber, S., Hofstatter, E., Haudek-Prinz, V., Klepeisz, P., Schicher, N., et al. (2012). Proteome analysis identified the PPARgamma ligand 15d-PGJ2 as a novel drug inhibiting melanoma progression and interfering with tumor-stroma interaction. PLoS ONE 7:e46103. doi: 10.1371/journal.pone.0046103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Perez-Gonzalez, R., Gauthier, S. A., Kumar, A., and Levy, E. (2012). The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J. Biol. Chem. 287, 43108–43115. doi: 10.1074/jbc.M112.404467
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pettipher, R., Hansel, T. T., and Armer, R. (2007). Antagonism of the prostaglandin D2 receptors DP1 and CRTH2 as an approach to treat allergic diseases. Nat. Rev. Drug Discov. 6, 313–325. doi: 10.1038/nrd2266
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pierre, S. R., Lemmens, M. A., and Figueiredo-Pereira, M. E. (2009). Subchronic infusion of the product of inflammation prostaglandin J2 models sporadic Parkinson's disease in mice. J. Neuroinflammation 6:18. doi: 10.1186/1742-2094-6-18
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pignatelli, M., Sanchez-Rodriguez, J., Santos, A., and Perez-Castillo, A. (2005). 15-Deoxy-{Delta}-12,14-prostaglandin J2 induces programmed cell death of breast cancer cells by a pleiotropic mechanism. Carcinogenesis 26, 81–92. doi: 10.1093/carcin/bgh308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Piro, J. R., Benjamin, D. I., Duerr, J. M., Pi, Y., Gonzales, C., Wood, K. M., et al. (2012). A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer's disease. Cell Rep. 1, 617–623. doi: 10.1016/j.celrep.2012.05.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qi, L., Jacob, A., Wang, P., and Wu, R. (2010). Peroxisome proliferator activated receptor-gamma and traumatic brain injury. Int. J. Clin. Exp. Med. 3, 283–292.
Rainsford, K. D. (2007). Anti-inflammatory drugs in the 21st century. Subcell. Biochem. 42, 3–27. doi: 10.1007/1-4020-5688-5_1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rajakariar, R., Hilliard, M., Lawrence, T., Trivedi, S., Colville-Nash, P., Bellingan, G., et al. (2007). Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15-deoxyDelta12 14 PGJ2. Proc. Natl. Acad. Sci. U.S.A. 104, 20979–20984. doi: 10.1073/pnas.0707394104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reglodi, D., Lubics, A., Tamas, A., Szalontay, L., and Lengvari, I. (2004). Pituitary adenylate cyclase activating polypeptide protects dopaminergic neurons and improves behavioral deficits in a rat model of Parkinson's disease. Behav. Brain Res. 151, 303–312. doi: 10.1016/j.bbr.2003.09.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ricciotti, E., and FitzGerald, G. A. (2011). Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 31, 986–1000. doi: 10.1161/ATVBAHA.110.207449
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rissman, R. A., Poon, W. W., Blurton-Jones, M., Oddo, S., Torp, R., Vitek, M. P., et al. (2004). Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J. Clin. Invest. 114, 121–130. doi: 10.1172/JCI200420640
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ristic, G., Tsou, W. L., and Todi, S. V. (2014). An optimal ubiquitin-proteasome pathway in the nervous system: the role of deubiquitinating enzymes. Front. Mol. Neurosci. 7:72. doi: 10.3389/fnmol.2014.00072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rockwell, P., Yuan, H. M., Magnusson, R., and Figueiredo-Pereira, M. E. (2000). Proteasome inhibition in neuronal cells induces a proinflammatory response manifested by upregulation of cyclooxygenase-2, its accumulation as ubiquitin conjugates, and production of the prostaglandin PGE2. Arch. Biochem. Biophys. 374, 325–333. doi: 10.1006/abbi.1999.1646
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rodriguez-Grande, B., Blackabey, V., Gittens, B., Pinteaux, E., and Denes, A. (2013). Loss of substance P and inflammation precede delayed neurodegeneration in the substantia nigra after cerebral ischemia. Brain Behav. Immun. 29, 51–61. doi: 10.1016/j.bbi.2012.11.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rossi, A., Kapahi, P., Natoli, G., Takahashi, T., Chen, Y., Karin, M., et al. (2000). Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature 403, 103–108. doi: 10.1038/47520
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sandig, H., Pease, J. E., and Sabroe, I. (2007). Contrary prostaglandins: the opposing roles of PGD2 and its metabolites in leukocyte function. J. Leukoc. Biol. 81, 372–382. doi: 10.1189/jlb.0706424
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Satoh, T., and Lipton, S. A. (2007). Redox regulation of neuronal survival mediated by electrophilic compounds. Trends Neurosci. 30, 37–45. doi: 10.1016/j.tins.2006.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scher, J. U., and Pillinger, M. H. (2005). 15d-PGJ2: the anti-inflammatory prostaglandin? Clin. Immunol. 114, 100–109. doi: 10.1016/j.clim.2004.09.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scher, J. U., and Pillinger, M. H. (2009). The anti-inflammatory effects of prostaglandins. J. Investig. Med. 57, 703–708. doi: 10.231/JIM.0b013e31819aaa76
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schneider, A., and Simons, M. (2013). Exosomes: vesicular carriers for intercellular communication in neurodegenerative disorders. Cell Tissue Res. 352, 33–47. doi: 10.1007/s00441-012-1428-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schulman, B. A., and Harper, J. W. (2009). Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 10, 319–331. doi: 10.1038/nrm2673
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schuster, V. L. (1998). Molecular mechanisms of prostaglandin transport. Annu. Rev. Physiol. 60, 221–242. doi: 10.1146/annurev.physiol.60.1.221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schuster, V. L. (2002). Prostaglandin transport. Prostaglandins Other Lipid Mediat. 68–69, 633–647. doi: 10.1016/S0090-6980(02)00061-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schwab, C., and McGeer, P. L. (2008). Inflammatory aspects of Alzheimer disease and other neurodegenerative disorders. J. Alzheimers. Dis. 13, 359–369.
Selkoe, D. J. (2004). Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat. Cell Biol. 6, 1054–1061. doi: 10.1038/ncb1104-1054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Servidei, T., Morosetti, R., Ferlini, C., Cusano, G., Scambia, G., Mastrangelo, R., et al. (2004). The cellular response to PPARgamma ligands is related to the phenotype of neuroblastoma cell lines. Oncol. Res. 14, 345–354.
Shaik, J. S., Miller, T. M., Graham, S. H., Manole, M. D., and Poloyac, S. M. (2014). Rapid and simultaneous quantitation of prostanoids by UPLC-MS/MS in rat brain. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 945–946, 207–216. doi: 10.1016/j.jchromb.2013.11.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shanbhag, R., Shi, G., Rujiviphat, J., and McQuibban, G. A. (2012). The emerging role of proteolysis in mitochondrial quality control and the etiology of Parkinson's disease. Parkinsons Dis. 2012, 382175. doi: 10.1155/2012/382175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shaw, L. M., Korecka, M., Clark, C. M., Lee, V. M., and Trojanowski, J. Q. (2007). Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nat. Rev. Drug Discov. 6, 295–303. doi: 10.1038/nrd2176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shibata, T., Kondo, M., Osawa, T., Shibata, N., Kobayashi, M., and Uchida, K. (2002). 15-deoxy-delta 12,14-prostaglandin J2. A prostaglandin D2 metabolite generated during inflammatory processes. J. Biol. Chem. 277, 10459–10466. doi: 10.1074/jbc.M110314200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shibata, T., Yamada, T., Kondo, M., Tanahashi, N., Tanaka, K., Nakamura, H., et al. (2003). An endogenous electrophile that modulates the regulatory mechanism of protein turnover: inhibitory effects of 15-deoxy-delta(12,14)-prostaglandin J2 on proteasome. Biochemistry 42, 13960–13968. doi: 10.1021/bi035215a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shimizu, M., Nakamura, H., Hirabayashi, T., Suganami, A., Tamura, Y., and Murayama, T. (2008). Ser515 phosphorylation-independent regulation of cytosolic phospholipase A2alpha (cPLA2alpha) by calmodulin-dependent protein kinase: possible interaction with catalytic domain A of cPLA2alpha. Cell Signal. 20, 815–824. doi: 10.1016/j.cellsig.2007.12.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shivers, K. Y., Nikolopoulou, A., Machlovi, S. I., Vallabhajosula, S., and Figueiredo-Pereira, M. E. (2014). PACAP27 prevents Parkinson-like neuronal loss and motor deficits but not microglia activation induced by prostaglandin J2. Biochim. Biophys. Acta 1842, 1707–1719. doi: 10.1016/j.bbadis.2014.06.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Simmons, D. L., Botting, R. M., and Hla, T. (2004). Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 56, 387–437. doi: 10.1124/pr.56.3.3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smith, W. L., DeWitt, D. L., and Garavito, R. M. (2000). Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 69, 145–182. doi: 10.1146/annurev.biochem.69.1.145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smyth, E. M., Grosser, T., Wang, M., Yu, Y., and FitzGerald, G. A. (2009). Prostanoids in health and disease. J. Lipid Res. 50(Suppl.), S423–S428. doi: 10.1194/jlr.R800094-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stamatakis, K., Sanchez-Gomez, F. J., and Perez-Sala, D. (2006). Identification of novel protein targets for modification by 15-deoxy-Delta12,14-prostaglandin J2 in mesangial cells reveals multiple interactions with the cytoskeleton. J. Am. Soc. Nephrol. 17, 89–98. doi: 10.1681/ASN.2005030329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stoessl, A. J. (2014). Developments in neuroimaging: positron emission tomography. Parkinsonism Relat. Disord. 20(Suppl. 1), S180–S183. doi: 10.1016/S1353-8020(13)70042-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Straus, D. S., and Glass, C. K. (2001). Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med. Res. Rev. 21, 185–210. doi: 10.1002/med.1006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Straus, D. S., Pascual, G., Li, M., Welch, J. S., Ricote, M., Hsiang, C. H., et al. (2000). 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 97, 4844–4849. doi: 10.1073/pnas.97.9.4844
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Su, R. Y., Chi, K. H., Huang, D. Y., Tai, M. H., and Lin, W. W. (2008). 15-deoxy-Delta12,14-prostaglandin J2 up-regulates death receptor 5 gene expression in HCT116 cells: involvement of reactive oxygen species and C/EBP homologous transcription factor gene transcription. Mol. Cancer Ther. 7, 3429–3440. doi: 10.1158/1535-7163.MCT-08-0498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Subra, C., Grand, D., Laulagnier, K., Stella, A., Lambeau, G., Paillasse, M., et al. (2010). Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J. Lipid Res. 51, 2105–2120. doi: 10.1194/jlr.M003657
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Surh, Y. J., Na, H. K., Park, J. M., Lee, H. N., Kim, W., Yoon, I. S., et al. (2011). 15-Deoxy-Delta(1)(2),(1)(4)-prostaglandin J(2), an electrophilic lipid mediator of anti-inflammatory and pro-resolving signaling. Biochem. Pharmacol. 82, 1335–1351. doi: 10.1016/j.bcp.2011.07.100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suzuki, F., Hayashi, H., and Hayaishi, O. (1986). Transport of prostaglandin D2 into brain. Brain Res. 385, 321–328. doi: 10.1016/0006-8993(86)91079-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tai, H. H. (2011). Prostaglandin catabolic enzymes as tumor suppressors. Cancer Metastasis Rev. 30, 409–417. doi: 10.1007/s10555-011-9314-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tai, H. H., Cho, H., Tong, M., and Ding, Y. (2006). NAD+-linked 15-hydroxyprostaglandin dehydrogenase: structure and biological functions. Curr. Pharm. Des. 12, 955–962. doi: 10.2174/138161206776055958
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takei, S., Hasegawa-Ishii, S., Uekawa, A., Chiba, Y., Umegaki, H., Hosokawa, M., et al. (2012). Immunohistochemical demonstration of increased prostaglandin F2α levels in the rat hippocampus following kainic acid-induced seizures. Neuroscience 218, 295–304. doi: 10.1016/j.neuroscience.2012.05.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tassoni, D., Kaur, G., Weisinger, R. S., and Sinclair, A. J. (2008). The role of eicosanoids in the brain. Asia Pac. J. Clin. Nutr. 17(Suppl. 1), 220–228.
Taylor, E. B., and Rutter, J. (2011). Mitochondrial quality control by the ubiquitin-proteasome system. Biochem. Soc. Trans. 39, 1509–1513. doi: 10.1042/BST0391509
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Teismann, P., Tieu, K., Choi, D. K., Wu, D. C., Naini, A., Hunot, S., et al. (2003). Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 100, 5473–5478. doi: 10.1073/pnas.0837397100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uchida, K., and Shibata, T. (2008). 15-Deoxy-Delta(12,14)-prostaglandin J2: an electrophilic trigger of cellular responses. Chem. Res. Toxicol. 21, 138–144. doi: 10.1021/tx700177j
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Upadhya, S. C., and Hegde, A. N. (2007). Role of the ubiquitin proteasome system in Alzheimer's disease. BMC Biochem. 8(Suppl. 1):S12. doi: 10.1186/1471-2091-8-S1-S12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Urade, Y., and Eguchi, N. (2002). Lipocalin-type and hematopoietic prostaglandin D synthases as a novel example of functional convergence. Prostaglandins Other Lipid Mediat. 68–69, 375–382. doi: 10.1016/S0090-6980(02)00042-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Urade, Y., and Hayaishi, O. (2000). Prostaglandin D synthase: structure and function. Vitam. Horm. 58, 89–120. doi: 10.1016/S0083-6729(00)58022-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vasil'ev, Y. V., Tzeng, S. C., Huang, L., and Maier, C. S. (2014). Protein modifications by electrophilic lipoxidation products: adduct formation, chemical strategies and tandem mass spectrometry for their detection and identification. Mass Spectrom. Rev. 33, 157–182. doi: 10.1002/mas.21389
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vermeer, S. E., Prins, N. D., den, H. T., Hofman, A., Koudstaal, P. J., and Breteler, M. M. (2003). Silent brain infarcts and the risk of dementia and cognitive decline. N. Engl. J. Med. 348, 1215–1222. doi: 10.1056/NEJMoa022066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vlad, S. C., Miller, D. R., Kowall, N. W., and Felson, D. T. (2008). Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70, 1672–1677. doi: 10.1212/01.wnl.0000311269.57716.63
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wall, S. B., Oh, J. Y., Diers, A. R., and Landar, A. (2012). Oxidative modification of proteins: an emerging mechanism of cell signaling. Front. Physiol. 3:369. doi: 10.3389/fphys.2012.00369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, Z., Aris, V. M., Ogburn, K. D., Soteropoulos, P., and Figueiredo-Pereira, M. E. (2006). Prostaglandin J2 alters pro-survival and pro-death gene expression patterns and 26 S proteasome assembly in human neuroblastoma cells. J. Biol. Chem. 281, 21377–21386. doi: 10.1074/jbc.M601201200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wilkinson, K. D., Deshpande, S., and Larsen, C. N. (1992). Comparisons of neuronal (PGP 9.5) and non-neuronal ubiquitin C-terminal hydrolases. Biochem. Soc. Trans. 20, 631–637.
Wilmer, W. A., Dixon, C., Lu, L., Hilbelink, T., and Rovin, B. H. (2001). A cyclopentenone prostaglandin activates mesangial MAP kinase independently of PPARgamma. Biochem. Biophys. Res. Commun. 281, 57–62. doi: 10.1006/bbrc.2001.4301
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wyss-Coray, T., and Mucke, L. (2002). Inflammation in neurodegenerative disease–a double-edged sword. Neuron 35, 419–432. doi: 10.1016/S0896-6273(02)00794-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, S., and Venge, P. (2000). Lipocalins as biochemical markers of disease. Biochim. Biophys. Acta 1482, 298–307. doi: 10.1016/S0167-4838(00)00163-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yagami, T. (2006). Cerebral arachidonate cascade in dementia: Alzheimer's disease and vascular dementia. Curr. Neuropharmacol. 4, 87–100. doi: 10.2174/157015906775203011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamagata, K., Andreasson, K. I., Kaufmann, W. E., Barnes, C. A., and Worley, P. F. (1993). Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron 11, 371–386. doi: 10.1016/0896-6273(93)90192-T
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamamoto, Y., Takase, K., Kishino, J., Fujita, M., Okamura, N., Sakaeda, T., et al. (2011). Proteomic identification of protein targets for 15-deoxy-Delta(12,14)-prostaglandin J2 in neuronal plasma membrane. PLoS ONE 6:e17552. doi: 10.1371/journal.pone.0017552
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yazaki, M., Kashiwagi, K., Aritake, K., Urade, Y., and Fujimori, K. (2012). Rapid degradation of cyclooxygenase-1 and hematopoietic prostaglandin D synthase through ubiquitin-proteasome system in response to intracellular calcium level. Mol. Biol. Cell 23, 12–21. doi: 10.1091/mbc.E11-07-0623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yoshiyama, Y., Higuchi, M., Zhang, B., Huang, S. M., Iwata, N., Saido, T. C., et al. (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. doi: 10.1016/j.neuron.2007.01.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zagol-Ikapitte, I., Masterson, T. S., Amarnath, V., Montine, T. J., Andreasson, K. I., Boutaud, O., et al. (2005). Prostaglandin H(2)-derived adducts of proteins correlate with Alzheimer's disease severity. J. Neurochem. 94, 1140–1145. doi: 10.1111/j.1471-4159.2005.03264.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, H. X., Tanji, K., Yoshida, H., Hayakari, M., Shibata, T., Mori, F., et al. (2010). Alteration of biochemical and pathological properties of TDP-43 protein by a lipid mediator, 15-deoxy-Delta(12,14)-prostaglandin J(2). Exp. Neurol. 222, 296–303. doi: 10.1016/j.expneurol.2010.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, X., Wang, J. M., Gong, W. H., Mukaida, N., and Young, H. A. (2001). Differential regulation of chemokine gene expression by 15-deoxy-delta 12,14 prostaglandin J2. J. Immunol. 166, 7104–7111. doi: 10.4049/jimmunol.166.12.7104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhu, T., Gobeil, F., Vazquez-Tello, A., Leduc, M., Rihakova, L., Bossolasco, M., et al. (2006). Intracrine signaling through lipid mediators and their cognate nuclear G-protein-coupled receptors: a paradigm based on PGE2, PAF, and LPA1 receptors. Can. J. Physiol. Pharmacol. 84, 377–391. doi: 10.1139/y05-147
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ziebell, J. M., and Morganti-Kossmann, M. C. (2010). Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 7, 22–30. doi: 10.1016/j.nurt.2009.10.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: J2 prostaglandins, neuroinflammation, UPP, mitochondria, neurodegeneration
Citation: Figueiredo-Pereira ME, Rockwell P, Schmidt-Glenewinkel T and Serrano P (2015) Neuroinflammation and J2 prostaglandins: linking impairment of the ubiquitin-proteasome pathway and mitochondria to neurodegeneration. Front. Mol. Neurosci. 7:104. doi: 10.3389/fnmol.2014.00104
Received: 03 November 2014; Accepted: 17 December 2014;
Published online: 13 January 2015.
Edited by:
Ashok Hegde, Wake Forest School of Medicine, USAReviewed by:
Björn Spittau, Albert-Ludwigs-University Freiburg, GermanyCopyright © 2015 Figueiredo-Pereira, Rockwell, Schmidt-Glenewinkel and Serrano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria E. Figueiredo-Pereira, Department of Biological Sciences, Hunter College, The Graduate School and University Center, City University of New York, 695 Park Avenue, Room 827N, New York, NY 10065, USA e-mail:cGVyZWlyYUBnZW5lY3RyLmh1bnRlci5jdW55LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.