 Nobuyuki Takei
Nobuyuki Takei Hiroyuki Nawa
Hiroyuki Nawa
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 23 April 2014
Sec. Neuroplasticity and Development
Volume 7 - 2014 | https://doi.org/10.3389/fnmol.2014.00028
This article is part of the Research Topic The Role of PI3K/Pten-mTOR Signalling in Neuronal Form and Function View all 11 articles
Target of rapamycin (TOR) was first identified in yeast as a target molecule of rapamycin, an anti-fugal and immunosuppressant macrolide compound. In mammals, its orthologue is called mammalian TOR (mTOR). mTOR is a serine/threonine kinase that converges different extracellular stimuli, such as nutrients and growth factors, and diverges into several biochemical reactions, including translation, autophagy, transcription, and lipid synthesis among others. These biochemical reactions govern cell growth and cause cells to attain an anabolic state. Thus, the disruption of mTOR signaling is implicated in a wide array of diseases such as cancer, diabetes, and obesity. In the central nervous system, the mTOR signaling cascade is activated by nutrients, neurotrophic factors, and neurotransmitters that enhances protein (and possibly lipid) synthesis and suppresses autophagy. These processes contribute to normal neuronal growth by promoting their differentiation, neurite elongation and branching, and synaptic formation during development. Therefore, disruption of mTOR signaling may cause neuronal degeneration and abnormal neural development. While reduced mTOR signaling is associated with neurodegeneration, excess activation of mTOR signaling causes abnormal development of neurons and glia, leading to brain malformation. In this review, we first introduce the current state of molecular knowledge of mTOR complexes and signaling in general. We then describe mTOR activation in neurons, which leads to translational enhancement, and finally discuss the link between mTOR and normal/abnormal neuronal growth during development.
Mammalian target of rapamycin (mTOR) controls growth and metabolism by activating anabolic processes and suppressing catabolic processes. Dysregulation of mTOR signaling induces various disorders including cancer, diabetes, obesity, cardiovascular disease, inflammation, and neurodevelopmental and neurodegenerative disorders. Several inhibitors of mTOR have been developed and are now clinically approved. Recently, remarkable progress has been made in understanding the molecular nature of mTOR signaling and its biological significance. Here we overview the signaling network of mTOR cascades with special emphasis on translational control. We also focus on the normal and abnormal growth of neurons regulated by mTOR complex 1 (mTORC1).
Mammalian target of rapamycin is a 289-kDa serine/threonine protein kinase that is, as indicated by its name, a target molecule of the immunosuppressant rapamycin. Rapamycin is an anti-fungal macrolide compound isolated from soil bacterium, Streptomyces hygroscopicus, in Rapa Nui (Easter Island; Vézina et al., 1975). The 12-kDa FK506-binding protein (FKBP12) was first identified as a rapamycin-binding protein (Heitman et al., 1991a; Schreiber, 1991). However, yeast genetic screening revealed that FKBP12 was not a functional TOR because disruption of FRP1 (FKBP12 coding gene in the budding yeast Saccharomyces cerevisiae) did not cause growth inhibition/toxicity. This suggested that FKBP12 was likely a co-factor in executing the rapamycin action. Screening of rapamycin-resistant yeast mutants led to the identification of genes TOR1 and TOR2 as molecular targets of rapamycin-FKBP12 complex and functional targets of rapamycin (Heitman et al., 1991b). Subsequently, four groups identified rapamycin targets in mammalian cells: mTOR, FRAP (FKBP12 and rapamycin associated protein), RAFT1 (rapamycin and FKBP12 target 1), and RAPT1. Although recent database nomenclature defined the abbreviation MTOR as the “mechanistic TOR” in mammals, many researchers still use the name mTOR (Hall, 2013).
Whereas yeast expresses two TOR molecules, TOR1 and TOR2, only one TOR homolog, mTOR, exists in mammals. mTOR nucleates two distinct complexes called mTORC 1 and 2 with several interacting proteins; these complexes have different functions. For example, mTORC1 governs cell growth, metabolism and cell cycle, whereas mTORC2 controls cell survival and cytoskeleton organization (see review by Laplante and Sabatini, 2012). The composition of mTORC1 and mTORC2 is shown in Figure 1 and detailed below. The main characteristic component of mTORC1 is raptor (Hara et al., 2002; Kim et al., 2002). Raptor is a scaffold protein that regulates complex assembly in addition to substrate recognition, that is to say, it determines downstream signaling of mTORC1. Likewise, rictor is a crucial binding partner of mTOR to make it function as mTORC2 (Jacinto et al., 2004; Sarbassov et al., 2004). In fact, as discussed below, mTORC1 and mTORC2 functions are impaired by the knockout/knockdown of raptor or rictor, respectively. The mammalian lethal with sec13 protein 8 (mLST8) is thought to be a requisite for mTORCs activities and mSin1 is thought to be a scaffold for mTORC2. In addition, deptor and PRAS40 are inhibitor molecules for mTORCs. Several other molecules are reported to be involved in these complexes but their roles are still unclear. Rapamycin, in binding with FKBP-12, interacts with mTOR on its FKBP12-rapamycin binding (FRB) domain and inhibits mTORC1 activity. Many, but not all (Kang et al., 2013), of the substrates and biological processes controlled under mTORC1 are rapamycin-sensitive. mTORC2 activity, in contrast, has been thought to be rapamycin-insensitive. Recently structural analysis of mTOR gives us an important insight into the mechanism of rapamycin action (Yang et al., 2013). The work predicts that FKBP12-rapamycin complex bound FRB domain may come close to mLST8 thus reducing the access of the substrates to active site of mTOR. These structural properties of mTOR may contribute to the preferences of substrates and/or phosphorylation sites by rapamycin inhibition (Choo and Blenis, 2009; Kang et al., 2013). In case of mTORC2, its components may already be bound proximately to FRB domain thus inhibits FRB. This notion is supported by the two findings. Prolonged treatment with rapamycin affects mTORC2 activity (Sarbassov et al., 2006). Rapamycin does not liberate a pre-existing rictor-mTORC but inhibits the interaction of newly synthesized rictor with mTOR. Another finding is that micromolar order of rapamycin inhibits mTORC2 activity independently of FKBP12 (Shor et al., 2008). High dose of rapamycin may allows free-rapamycin that can bind FRB domain and thus inhibits mTORC2. These findings advance our knowledge about the mechanism of the inhibitory action of rapamycin.
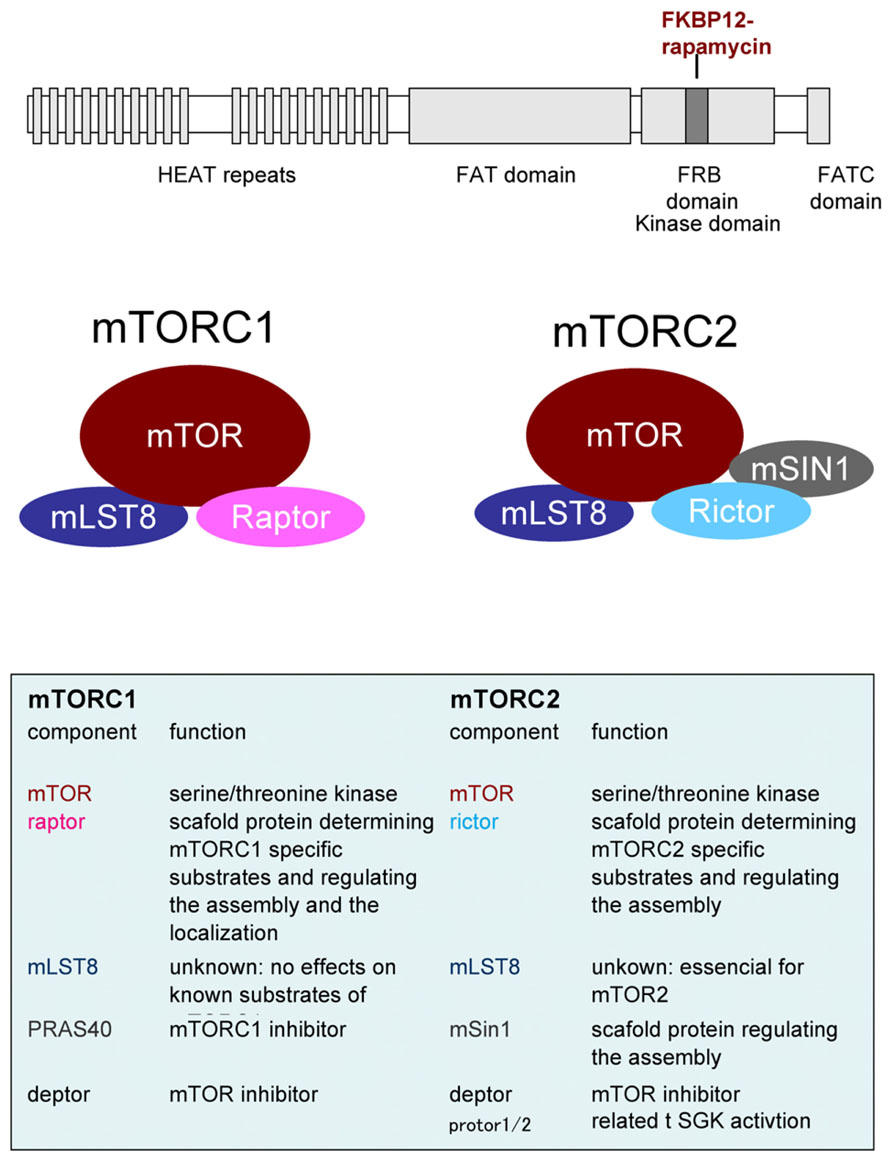
FIGURE 1. Upper panel. Domain structure of mTOR. HEAT: Huntington Elongation Factor 3 PR65/A TOR, FAT: FRAP ATM TTRAP, FRB: FKBP12-Rapamycin Binding. Middle and lower panel: Components of mTOR complexs.
In contrast to the dearth of knowledge about the signaling pathway of mTORC2, the upstream and downstream pathways of mTORC1 are much better understood. mTORC1 integrates several intracellular and extracellular cues, and transduces divergent downstream events. Growth factors and amino acids are the best-characterized extracellular stimuli that activate mTORC1. In addition, cellular energy status, oxygen/hypoxia and stressors regulate mTORC1 activity. In the central nervous system (CNS), neurotransmitters, neuromodulators, and hormones are reported to activate mTORC1.
Growth factors bind to and activate receptor tyrosine kinases (RTKs). The RTK to Akt (aka PKB) pathway has been well investigated so far. Then, tuberous sclerosis complex 1 (TSC1; aka hamartin) and TSC2 (aka tuberin) have been identified as links between Akt and mTOR (Gao et al., 2002; Inoki et al., 2002; Tee et al., 2002). Akt phosphorylates TSC2 and causes it to dissociate from TSC1. Dissociated TSC2 is captured by 14-3-3 and is prevented to form the complex (Li et al., 2002; Nellist et al., 2002; Shumway et al., 2003). Phosphorylated and liberated TSC2 is degraded by ubiquitination (Chong-Kopera et al., 2006; Hu et al., 2008), although the role of 14-3-3 in this process is unclear. The TSC1/2 plays a key role in the activation of mTOR. TSC1/2, together with a third component TBC1D7 (Nakashima et al., 2007) functions as a GTPase-activating protein (GAP) for small G-protein Ras homology enriched in brain (Rheb; Inoki et al., 2003a; Dibble et al., 2012). GTP-bound Rheb, the active form, directly binds to mTORC1 and activates its kinase activity (Long et al., 2005). The GAP accelerates GTP hydrolysis, which inactivates Rheb into its GDP-bound form. In steady state, without growth factor stimulation, unphosphorylated TSC2 maintains a heteromeric interaction with TSC1, and this complex is a suppressor of mTRC1. Until now, the guanine nucleotide exchange factor (GEF) that promotes GDP to GTP conversion of Rheb had not been identified. In addition to Akt, mitogen-activated protein kinase (MAPK), and Rsk inhibit TSC2 thus activating mTORC1. As discussed below, the mutations that causes loss of function of TSCs result in mTORC1 over activation and cause brain diseases.
Tuberous sclerosis complex 2 directly receives many upstream signals by its phosphorylation. The phosphorylation of TSC2 induces both activation and inactivation of mTORC1 depending on the phospho-acceptor amino acid residues. For example, phosphorylation at Thr1462 of TSC2 by Akt (Inoki et al., 2002; Manning et al., 2002) and Ser664 by MAPK (aka Erk; Ma et al., 2007) inhibits TSC1/2 activity thus activating mTORC1, whereas Ser1345 phosphorylation by AMP-activated protein kinase (AMPK) enhances TSC1/2 activity (Inoki et al., 2003b). GSK3β (glycogen synthase kinase) also phosphorylates TSC2 at Ser 1337 and Ser1341 after priming phosphorylation at Ser1345 by AMPK and activates TSC1/2, thus inhibiting mTORC1 (Choo et al., 2006; Inoki et al., 2006).
Insulin is the most-characterized growth factor that activates mTORCs through the PI3K/Akt/TSC/Rheb pathway (Avruch et al., 2006). Indeed, insulin and insulin-like growth factor 1 (IGF1) enhance mRNA translation in neurons possibly through mTORC1 (Quevedo et al., 2002; Lee et al., 2005; Chenal et al., 2008). Among several growth factors tested, brain-derived neurotrophic factor (BDNF), the most prominent neurotrophic factor in the CNS (Lewin and Barde, 1996; Nawa and Takei, 2001), has been shown to activate mTORC1 signaling and enhance novel protein synthesis in cortical neurons (Takei et al., 2001, 2004). Interestingly, the effects of BDNF on translation were stronger than that of insulin likely because BDNF activates both Akt and MAPK, whereas insulin activates only Akt in neurons. This makes sense because TSC2 is phosphorylated by both Akt and MAPK. In addition to BDNF and insulin, several growth factors/RTKs (Kim et al., 2008a; Yao et al., 2013) and guidance molecules such as Eph, Slit, and Reelin (detailed in the axonal growth section) have been reported to activate mTORC1 signaling in neuronal cells.
A list of activators of mTORC1 in neurons is shown in Table 1. For example, G-protein coupled receptors (GPCRs) such as μ-opioid (Polakiewicz et al., 1998), metabotropic glutamate (Hou and Klann, 2004; Page et al., 2006), and cannabinoid (Puighermanal et al., 2009) receptors activate mTORC1 in neurons. Because GPCRs are known to transduce signals to Akt and/or MAPK, mTORC1 activation triggered by these ligands seems to inhibit TSC2. Other neurotransmitters were reported to possibly activate mTORC1 (meaning that the outputs are sensitive to rapamycin), such as serotonin (5-HT; Casadio et al., 1999; Carroll et al., 2004, 2006), may utilize this pathway.
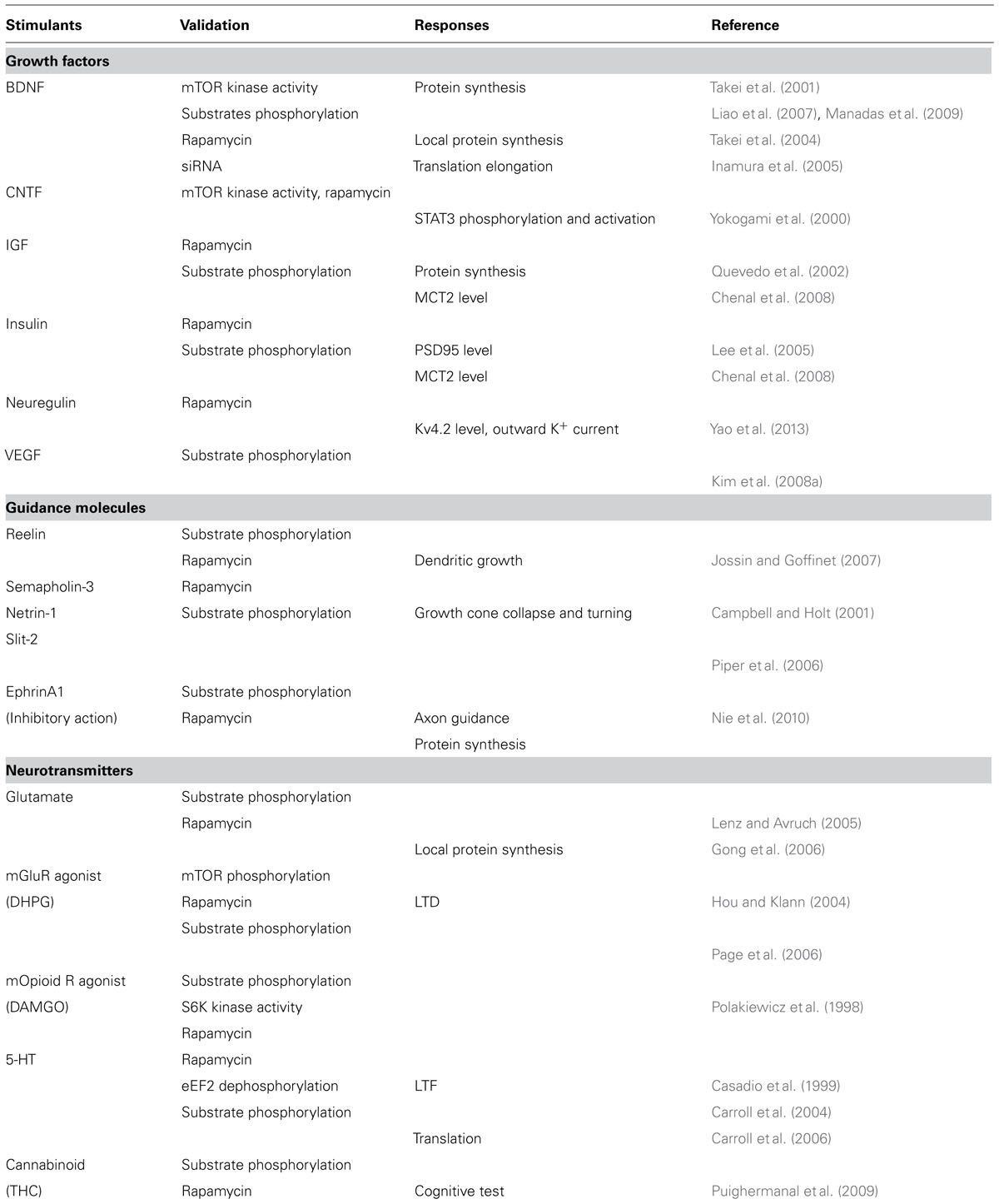
TABLE 1. Stimulants of mTOR in CNS neurons.
Neural activity-dependent regulation is another characteristic feature of mTORC1 activation in the CNS. For instance, the paradigms of synaptic plasticity such as long-term potentiation (LTP) and long-term depression (LTD) often induce mTORC1 activation. mTORC1 activation associates with spatial learning and fear conditioning as well. The direct activator molecules driving these plasticity-related or behavioral paradigms are likely the growth factors or neurotransmitters mentioned above. Indeed, the involvement of BDNF in LTP and metabotropic glutamate receptors (mGluRs) in LTD is well known. Interestingly, Rheb, an upstream activator of mTORC1, was originally found as a growth factor- and an activity-dependent transcript in the brain so that it named “enriched in brain” (Yamagata et al., 1994). This mechanism may also participate in mTORC1 activation by neural activity. The increase of blood flow during learning, as revealed by blood oxygenation level-dependent functional magnetic resonance imaging (BOLD fMRI) may supply enough nutrients such as glucose and amino acids. As mentioned below, these nutrients are other stimulants of mTORC1. More detailed discussions on mTORC1 in synaptic plasticity can be found in other reviews in this issue.
Yeast TOR activity is controlled by nutrients. Similarly, amino acids, especially leucine, are other essential extracellular stimuli that activate mTORC1 (Hara et al., 1998) in mammalian cells. Amino acids are not only the building blocks of proteins or intermediates of metabolism but are also evolutionarily conserved inter-cellular signaling molecules in both eukaryotes and prokaryotes. For example, glutamate and GABA are important neurotransmitters in the CNS. Recent reports have answered the long-lasting question, why and how leucine activates mTORC1. The analysis of raptor binding partner (Sancak et al., 2008) and siRNA screening of small GTPases (Kim et al., 2008b) revealed the link between Rag family small G-proteins (RagA–D) and mTORC1. RagA or RagB (~98% sequence similarity) forms a heterodimer with RagC or RagD (~87% sequence similarity). The active conformation is a heterodimer of RagA/B·GTP and RagC/D·GDP. When amino acid availability is sufficient, this complex directly binds to raptor and recruits mTORC1 to the lysosome with “Ragulator (consisting with three proteins, MP1/P14/P18),” which is a GEF for RagA/B. This makes mTORC1 close to and bind to Rheb, an activator of mTORC1 on the lysosomal membrane. A surprising molecule now comes into play – leucyl-tRNA synthetase (LeuRS) – both in mammalian cells (Han et al., 2012) and in yeast (Bonfils et al., 2012). LeuRS is an aminoacyl-tRNA synthetase that loads leucine to tRNA with high fidelity. LeuRS binds to RagD and acts as a GAP. Thus, RagD stays in a GDP-bound form, which is necessary for mTORC1 activation. This may be the mechanism that cells sense leucine and activates mTORC1. However, there are still unsolved questions (Segev and Hay, 2012), such as why among the 20 aminoacyl-tRNA synthetases only LeuRS works in the mTORC1 system. Because other amino acids also activate mTORC1, additional players must be involved.
mTORC1 converges nutrients and growth factor signals, and there is cross-talk between them. Reports suggest that amino acid(s) sufficiency is essential for the insulin-induced activation of mTORC1 in several cell lines (Hara et al., 1998; Campbell et al., 1999). Leucine activates mTORC1 in neurons after influx through system L amino acid transporter (LAT), consisting of LAT1 or LAT2 and 4F2hc (CD98; Ishizuka et al., 2008). Uptake of arginine by cationic amino acid transporters CAT1 and CAT3 has also been reported to activate mTORC1 in neurons (Huang et al., 2007). However, contrary to the above-mentioned essential role of amino acids, starvation of amino acids is less effective on BDNF-induced mTORC1 activation in neurons (Ishizuka et al., 2013). This discrepancy is unclear; however, cell-type specific amino acid contents or autophagy mechanisms may influence the system responses.
mTORC1 signaling in neurons seems to be more sensitive to energy status. For instance, glucose starvation increases the ratio of AMP/ATP. Increased AMP activates AMPK (Witters et al., 2006). AMPK directly phosphorylates TSC2 and enhances TSC1/2 activity, leading to the inhibition of mTORC1 (Inoki et al., 2003b, 2006). Another target to inhibit mTORC1 by AMPK is raptor. Raptor is directly phosphorylated by AMPK at Ser722/Ser792 and this leads to the inhibition of mTORC1 activity (Gwinn et al., 2008). In neurons, AMPK activation completely abrogated growth factor-induced mTORC1 activation and protein synthesis, with TSC2 and raptor phosphorylation (Ishizuka et al., 2013). AMPK is phosphorylated and activated by LKB1 (Woods et al., 2003) and calcium/calmodulin-dependent protein kinase kinase (CaMKK; Hurley et al., 2005). Because CaMKK activity is dependent on calcium, both ionotropic and metabotropic receptor ligands (neurotransmitters) may participate in mTORC1 signaling through the Ca2+-CaMKK-AMPK pathway in neurons.
Many substrates lead to diverse cellular responses downstream of mTOR. p70 ribosomal protein S6 kinase (p70S6K) and eukaryotic initiation factor 4E (eIF4E)-binding protein (4EBP) are the best-characterized substrates for mTORC1 that regulate translation. In contrast, ULK1 and ATG13 phosphorylation suppress autophagy. PRAS40, an inhibitory component of mTORC1, is a substrate of mTORC1 itself (Oshiro et al., 2007). All these mTORC1 substrates contain TOR signaling (TOS) motif. TOS motif is recognized by raptor and utilized for substrates recruitment to mTORC1 for optimal phosphorylation (Schalm and Blenis, 2002; Nojima et al., 2003). As mTORC2 substrates, Akt mediates a variety of actions including mTORC1 activation and the inhibition of apoptosis, among others. Protein kinase C (PKC) also mediates a myriad of cellular responses, and serum- and glucocorticoid-induced protein kinase (SGK) affects transcription through FoxO. Recent phosphoproteomic analyses revealed many mTOR substrates that may link to the known cellular responses such as ribosome biogenesis, mitochondrial biogenesis, metabolism, mRNA splicing etc. The flow sheet of mTORCs cascades is summarized in Figure 2. The physiological roles of these events in neural functions are less understood. In contrast, translational control mediated by mTORC1 is well studied in the aspect of neural development, plasticity, and diseases. Thus we review this process in detail below.
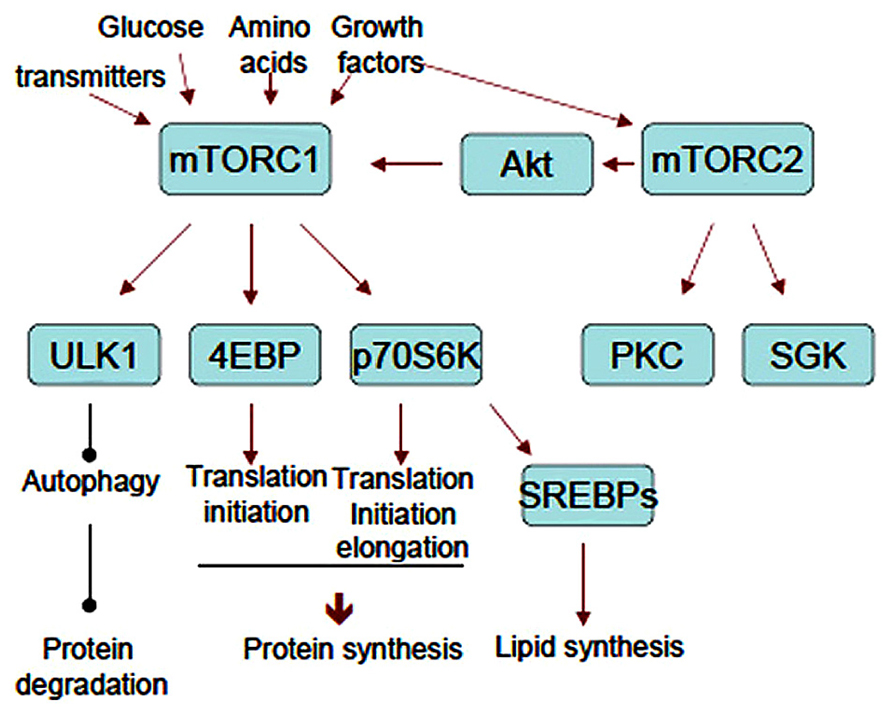
FIGURE 2. The flow sheet of upstream and downstream of mTORCs. Representative substrates and cellular responses mentioned in the text are shown. Note that mTORC2 activates mTORC1 through Akt.
Upon activation, mTOR phosphorylates p70S6K at Thr389 and 4EBPs [there are three 4EBPs (4EBP1–3), and 4EBP2 is a major isoform in neurons] at Thr37/46 and Ser65 directly. These substrates bind to the scaffold protein raptor by a TOS motif (Schalm and Blenis, 2002; Nojima et al., 2003) and are phosphorylated by the kinase mTOR. Phosphorylation of 4EBPs liberates eIF4E and allows eIF4E to bind to eIF4G and form an eIF4F complex together with eIF4A, an RNA helicase. eIF4E recognizes the 7-methylguanosine 5-triphosphate cap structure of 5′-UTR and poly(A)-binding protein (PABP) binds to the poly(A) tail of mRNAs so that the eIF4F complex makes mRNA circular. Indeed, the circular form of mRNA is thought to stabilize it and facilitate translation. Phosphorylation and activation of p70S6K by mTORC1 induced by insulin facilitates the association of eIF3 (a large molecular complex consisting of 13 subunits) with eIF4G (Holz et al., 2005; Harris et al., 2006). The process is thought to be important for recruiting the 40S ribosome to the mRNA-eIF4F complex. eIF4B is phosphorylated at Ser422 by Akt directly (van Gorp et al., 2009) and by Ser406 and Ser422 in MEK and mTOR/p79S6K-dependent manner (Raught et al., 2004; Shahbazian et al., 2006). Phosphorylated eIF4B enhances eIF4A helicase activity, suggesting that mTORC2 also participates in translational control. eIF4G phosphorylation at Ser1108, Ser1148, and Ser1192 is reportedly rapamycin-sensitive (Raught et al., 2000) (Figure 3, Upper panel). In addition, p70S6K phosphorylates eukaryotic elongation factor 2 kinase (eEF2K) and suppresses its activity (Wang et al., 2001), This causes the downregulation of eEF2 phosphorylation thus induces its activation (Figure 3, Lower panel).
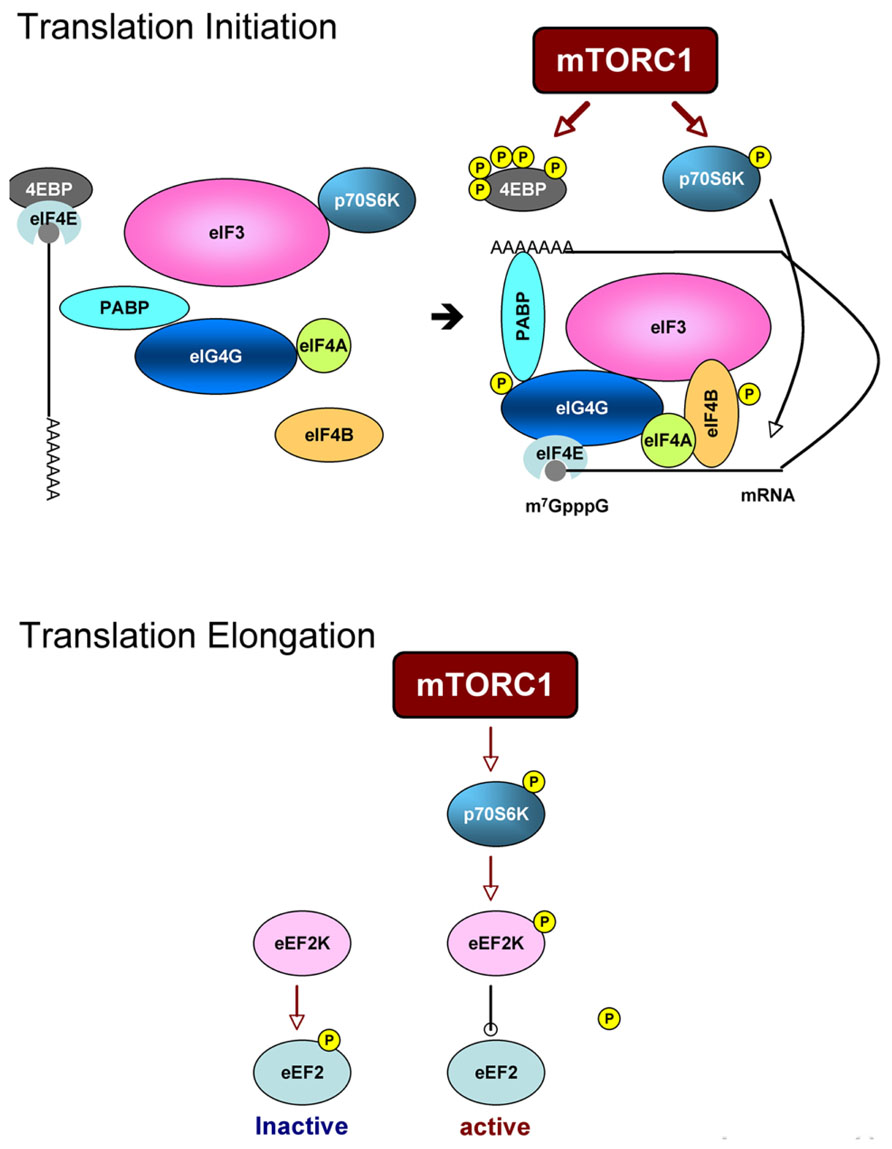
FIGURE 3. Scheme of translation processes that are regulated by mTORC1. Upper panel: translation initiation. mTORC1 directly phosphorylates 4EBP and liberates eIF4E. eIF4E with mRNA then binds to eIF4G to form eIF4F complex. Phosphorylation of eIF4G and eIF4B is mTORC1-dependent. Assembly of eIF3 subunits and eIF4G is also thought to be mTORC1-dependent. Lower panel: translation elongation. p70S6K downstream of mTORC1 phosphorylates eEF2K and suppresses its activity to phosphorylate eEF2. Non-phosphorylated form of eEF2 is an active form thus enhances elongation process.
In neurons, BDNF (Takei et al., 2001, 2004; Liao et al., 2007; Manadas et al., 2009), insulin (Lee et al., 2005; Chenal et al., 2008), and IGF1 (Quevedo et al., 2002; Chenal et al., 2008) have been shown to enhance translation by activating initiation processes through mTORC1 signaling. In addition, BDNF enhances translation elongation processes through mTORC1-dependent downregulation of eEF2 phosphorylation (Inamura et al., 2005). Enhancement of translation by dephosphorylation of eEF2 is also induced by 5-HT (Carroll et al., 2004). A neuron-specific feature of mTORC1-dependent translational control is its spatial property. For example, the mTORC1 system is localized in dendrites as well as in cell bodies (Takei et al., 2004). BDNF (Takei et al., 2004) and transmitters (Casadio et al., 1999; Carroll et al., 2004; Gong et al., 2006) have been shown to induce “local” activation of mTORC1 and translation (see review in this issue).
In summary, mTORC1 (and possibly mTORC2) modulates translational processes by the following: mTOR 1) phosphorylates 4EBP and induces cap-dependent translation; 2) phosphorylates p70S6K and allows eIF3 to bind eIF4G; 3) induces eIF4B phosphorylation through p70S6K and Akt; and 4) activates eEF2 through p-70S6K/eEF2K. Therefore, mTOR regulates translation both at initiation and elongation steps.
A recent report suggested that mTORC1 specifically controls the translation of a certain subset of mRNAs (Thoreen et al., 2012). eIF4E preferentially binds to mRNAs that harbor 5′-terminal oligopyrimidine tract (5′-TOP) or related sequences. These mRNAs encode proteins of translation machinery such as ribosomal proteins and translation factors. It has been postulated that p70S6K/S6 regulates translation of 5′-TOP mRNAs. However, although these events occur in parallel, genetic and biochemical analysis have refuted a direct relation between them (Shima et al., 1998; Ruvinsky and Meyuhas, 2006). This new insight can explain the direct interaction of mTORC1 and 5′-TOP mRNA translation. Indeed, acute signal(s) that activates mTORC1 enhances the production of proteins necessary for translation machinery and may prepare the succeeding and continuous activation of translation that leads to cellular growth. The authors concluded that 4EBP and eIF4G were the master regulators of mTORC1-dependent translation. However, further research must be conducted in postmitotic neurons, because the elongation process was the rate limiting step for translation in neurons (Takei et al., 2009).
Many reports have suggested that mTORC1-dependent translational control is crucial in the development and plasticity of neurons. However, in the majority of the studies, conclusions are based on pharmacological approaches using inhibitors. Rapamycin, the most commonly used tool for analyzing mTORC1 signals, does not effectively inhibit all mTORC1-dependent translation (Kang et al., 2013). An ATP-competitive mTOR inhibitor such as Torin1 both inhibits rapamycin-resistant mTORC1 reactions and mTORC2 activity. Chronic rapamycin also inhibits mTORC2. The effectiveness is somewhat cell-type specific, and the efficacy of rapamycin on mTORC2 in neurons has not yet been verified. In any case, we suggest caution when making conclusions using simple pharmacological approaches. Although it is technically challenging, monitoring the actual translation processes together with analysis of translation machinery as well as mTORC1 signaling is desired in combined with certain biological/physiological responses.
In addition to protein synthesis, mTORC1 also enhances lipid biosynthesis (see review by Laplante and Sabatini, 2009). mTORC1 has been reported to activate SREBP (sterol regulatory element binding protein) 1 and 2, transcription factors responsible for the expression of many lipid metabolic enzymes (Düvel et al., 2010). SREBPs are cleaved and the processed forms are translocated to the nuclei and promote gene transcription. Transcriptome and metabolome analysis have shown that SREBP-dependent lipid biosynthesis is rapamycin-sensitive (Düvel et al., 2010). Rapamycin inhibited lipid biosynthesis and the expression of lipid synthetic enzymes including acetyl-CoA carboxylase (Brown et al., 2007), fatty acid synthase (Peng et al., 2002), and HMG-CoA reductase (Yamauchi et al., 2011). Fatty acid oxidation (Brown et al., 2007) and cholesterol synthesis (Yamauchi et al., 2011) have also been shown to be rapamycin-sensitive. A recent report showed that mTORC1-mediated activation of SREBP1 and 2 was mediated by p70S6K (Düvel et al., 2010). Deletion of p70S6K induces the small body and small cell phenotype but does not affect translation; thus, the phenotype may be the result of inhibition of lipid synthesis, rather than protein synthesis. Because lipids are necessary to form plasma and organelle membrane component, as well as energy storage source and intracellular signaling, there is no doubt that lipid synthesis is essential to cell growth. Indeed, SREBP1 knockdown, as well as rapamycin treatment, can reduce cell size (Porstmann et al., 2008). Thus, both protein and lipid synthesis controlled by mTORC1 must be necessary for cell size regulation.
In the CNS, BDNF, the most potent activator of mTORC1 in neurons, has been shown to enhance cholesterol synthesis (Suzuki et al., 2007), as well as protein synthesis. Thus, mTORC1 must contribute to BDNF-mediated neuronal cell growth and dendritic arborization (e.g., McAllister et al., 1995, see review by Lewin and Barde, 1996). Cancer cells that exhibit uncontrolled growth factor signaling often show activation of SREBPs and lipogenesis. Enough (or excess) lipids required for membrane synthesis are a critical process of cancer progression including migration and invasion along with membrane expansion. These cellular responses give us insight into the characteristics of neuritis extension and spine formation during development and synaptic plasticity. Thus, BDNF-mediated neuronal growth may be dependent, at least in part, on de novo lipid synthesis.
mTOR plays a pivotal role in growth, proliferation, and migration of every cell; thus, it is thought to be essential for the development of an organism. Indeed, a knockout (KO) mouse study showed that mTOR was indispensable for normal development and viability (Murakami et al., 2004). The first genetic evidence that mTOR plays an important role in brain development came from an ethyl-nitroso-urea-induced mouse mutation screening (Hentges et al., 2001). This mutant, named flat-top, was a loss of function mutant of mTOR by missplicing; it showed a defect in telencephalon formation and died in mid-gestation. The milder phenotype compared to full KO may result from the incomplete loss of mTOR function. Indeed, the mutant still had about 10% p70S6K activity compared to the wild-type mouse.
Complete ablation of mTORC components in mice resulted in embryonic lethality (Guertin et al., 2006; Shiota et al., 2006). In addition to mice lacking mTOR itself, raptor KO mice died very early in development, while mLST8 or rictor-null mice died at embryonic day 10.5. Therefore, a conditional KO (CKO) strategy was applied for analyzing the function of mTORCs in brain. Elimination of mTORC1 function in brain was achieved by crossing raptor-floxed mice and nestin-Cre mice (Cloëtta et al., 2013). Nestin is a marker of neural progenitors. Although the raptor-CKO mice died soon after birth, they showed microcephaly via reductions in cell size and cell number. In addition, gliogenesis was affected in parallel with the downregulation of STAT3 phosphorylation at Ser727. This is plausible because STAT3 is pivotal for gliogenesis (Bonni et al., 1997) and is phosphorylated by mTORC1 (Yokogami et al., 2000).
The conditional deletion of rictor in several tissues shows a relatively mild phenotype compared with that of raptor. Mice with brain-specific deletion of rictor by crossing nestin-Cre mice have been shown to survive but with smaller brain size, like raptor-KO mice (Thomanetz et al., 2013). Brain weight of this rictor-KO mouse was about 60% (nestin-Cre) of the wild-type mouse brain. Total dendritic lengths and soma sizes of pyramidal neurons of hippocampus and Purkinje neurons of cerebellum were reduced. Purkinje cell-specific deletion of rictor via an L7/Pcp-2-cre mouse showed impairments in synaptic function and morphology of these neurons, which are correlated with the ataxia-like phenotype of this mouse (Thomanetz et al., 2013). Another report employed Emx1 promoter to drive Cre (Carson et al., 2013). Emx1 expresses only in dorsal neural progenitor cells that generate excitatory neurons and astrocytes in the dorsal cortex. The Emx1-Cre:rictor CKO mice also showed a small brain but to a lesser extent (about 80% of the wild-type mice), because the rictor-deficient cell population was more limited than that in the nesti-Cre:rictor mice. Neurons in cortical layers II–IV of the Emx1-Cre:rictor conditional mice were smaller than that of wild-type mice. These results implicate mTORC2 in the progression of neuronal size and morphology during brain development. To put it simply, because mTORC2 activates Akt, it will cause mTORC1 activation. If mTORC2 is inhibited, mTORC1 activity may be downregulated. However, mTORC1 activity looked normal in this rictor CKO mouse. In addition, in other cell types in other organs, for example muscle cells, mTORC2 inactivation has little effect on cell size compared to the mTORC1 inactivation (Bentzinger et al., 2008). The precise mechanism of how mTORC2 regulates cell size is still unresolved.
Detailed morphological analysis of mTOR on neurite formation comes from a culture study. Specifically, transfection of constitutively active or dominant negative forms of PI3K, Akt, and Ras revealed that PI3K-Akt increased the size of the soma and dendrites (Jaworski et al., 2005). Ras combined with PI3K-Akt enhanced the complexity of dendrites of hippocampal neurons (Kumar et al., 2005). Similarly, knockdown of phosphatase and tensin homolog (PTEN), the phosphatase for Akt, by siRNA induced arborization of hippocampal dendrites (Jaworski et al., 2005). The dendritic growth induced by these manipulations was antagonized by chronic rapamycin treatment, siRNA-mediated knockdown of mTOR and p70S6K, and overexpression of phosphorylation-defective mutant 4EBP (Jaworski et al., 2005). These results indicate that mTOR, especially mTORC1, play pivotal roles in dendritic growth/maturation. Mice with neuron-specific deletion of PTEN showed macrocephaly and hypertrophy of neurons with enhanced mTORC1 signaling (Kwon et al., 2006). Enhanced mTORC1 signaling and dendritic arborization by extracellular stimuli like BDNF (Jaworski et al., 2005) and Reelin (Jossin and Goffinet, 2007) are both inhibited by rapamycin. In addition to mTORC1, mTORC2 is implicated in the dendritic growth of hippocampal neurons. The dendritic arbor was disturbed not only by raptor but also by rictor small hairpin (sh) RNA, as well as by mTOR inhibitor Ku-006379A, which inhibits both mTORC1 and 2.
Axon guidance during development is regulated by balanced chemotactic cues by attractive and repulsive molecules. Semaphorin-3 and netrin-1 have been shown to induce growth cone collapse and repulsive turning in Xenopus retinal neurons. The response was abrogated by rapamycin, as well as by protein synthesis inhibitors, cycloheximide and anisomycin. Phosphorylation of 4EBP was observed in growth cones in response to semaphorin-3 and netrin-1 (Campbell and Holt, 2001). Slit2 has been shown to act on growth cone collapse and 4EBP phosphorylation similarly, but at later stage, in a rapamycin-sensitive manner (Piper et al., 2006). TSC2 haploinsufficiency in mice (TSC-/+) caused aberrant retinogenicular projection, suggesting the disruption of axon guidance. Since axon guidance of this tract is known to depend on ephrin-Eph signaling, the effect of ephrinA on mTOR pathway was investigated. In contrast to the case with semaphorin-3, the ephrinA-EphA signal that induced growth cone collapse suppressed the MAPK-TSC2-mTORC1 cascade and inhibited novel protein synthesis. TSC deficiency and constitutively active Rheb expression have been reported to counteract the actions of ephrinA-EphA (Nie et al., 2010). These results indicate that mTORC1-mediated translational control of growth cones plays pivotal roles in axon guidance during development.
These studies reveal the universal and specific roles of mTORCs in neurons. mTORC1 activation induces protein and lipid synthesis so that it increases cellular mass with expansion of plasma membrane. Local protein (and possibly lipid) synthesis mediated by mTORC1 may participate in the extension of an axon and dendrites of a neuron. mTORC2 may be the a putative facilitator of growth cone motility, including neurite pathfinding and elongation because it is known to affect actin dynamics. Although the precise mechanisms are still not fully understood, regulated and coordinated activities of mTORC1 and 2 must be necessary for normal development of neurons and a brain.
mTOR has been implicated as a cause of various diseases. Among them, most famous and directly related to mTOR disease is TSC. As mentioned previously, this disease is caused by the genetic mutation (including nonsense, missense, insertion, and deletion) of TSC1 or TSC2, the genes encoding TSC1 (hamartin) or TSC2 (tuberin), respectively. TSC was first described in late 19th century and the mutations have been identified in TSC1 (9q34) and TSC2 (16p13.3) in the 1990s. In 2002, several groups reported that TSC1/2 complex is a suppressor of mTOR. TSC1/2 complex is a GAP for Rheb so it inhibits mTORC1.
Tuberous sclerosis complex is an autosomal dominant, multisystem disorder that affects brain, lung, heart, skin, and kidney. The neurological symptoms are intractable epilepsy, autism, and mental retardation. Pathological features are cortical tubers, subependymal nodules (SENs), and subependymal giant cell tumors (SGCTs) and subependymal giant cell astrocytomas (SEGAs). The characteristic features are abnormal cell proliferation and growth (see review Crino et al., 2006; Crino, 2013). Because TSC is caused by a loss-of-function of TSC1 or TSC2, mTORC1 must be constitutively active in these cells. Indeed, highly phosphorylated p70S6K (Thr389), S6 (Ser235/236), and 4EBP (Thr37/46) have been observed immunohistochemically in pathologically abnormal, enlarged cells that may be of progenitor cell origin (Tsai et al., 2012; Prabowo et al., 2013). Although the direct interaction of TSC1/2 and mTORC2 is still unclear, phosphorylation of mTORC2 substrates PKCa at Ser657, Akt at Ser473, and SGK1 at Ser422 have also been observed in the fetal TSC brain (Tsai et al., 2012). The relationship between the activation of mTORC1 and cell (over)growth observed in human disease corroborates the experimental findings. In fact, the rapamycin analogue (called “Rapalog”) everolimus (RAD-001) is clinically approved for the medication of TSC (http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm317490.html).
There have been many reports about the KO of TSC1 or TSC2 genes. Homozygotes of these gene deficiency are lethal, and heterozygotes do not have tubers, although the mice show some abnormalities. Several CKO mice carrying biallelic deletion in certain cells have been generated, and they mimic some symptoms and pathology of human TSC. This may be because biallelic mutation is necessary to induce severe morphological alteration such as tubers or giant astrocytomas. In fact, so-called “two-hit” mechanisms of germline mutation and somatic mutation have been proposed for human tubers of TSC patients (Crino et al., 2010), although another report argued that the second hit was rare (Qin et al., 2010). The two-hit mechanism has been evaluated with homozygous deletion of TSC2 only in radial glia, and heterozygous deletion in all other cells by crossing Tsc2+/-, Tsc2flox, and GFAP-Cre mice (Way et al., 2009). The mice showed cytomegaly, defects in lamination, astrogliosis, and hypomyelination. mTORC1 activation was confirmed by S6 phosphorylation. Another approach of double hit of TSC1 gene by in utero electroporation has been reported. The mice were made by crossing TSC1+/- mice and TSC1flox mice. TSC1 in the cells of certain brain regions were biallelically deleted via in utero electroporation of Cre plasmid. The offspring showed tuber-like lesions and cortical hyperexcitability (Feliciano et al., 2011). Other conditional homozygotic deletion of either TSC1 or 2 represented some of the features of TSC (Magri et al., 2011). All the animal models indicate that biallelic deletion (or dysfunction) is necessary to induce TSC pathology. We have learned a lot about mTOR biology from TSC including the signaling mechanism of mTORCs; however, there are still many unsolved questions like the above-mentioned two-hit hypothesis. Further comprehensive genetic analysis (not limited in TSC genes) in the TSC brain may address the pathological obscurity of genotype-phenotype interaction.
There are two diseases with neurological symptoms and brain pathology similar to TSC – hemimegalencephaly and focal cortical dysplasia. Genetic mutations of these diseases have not been identified until now. Recently, somatic mutations of Akt, PI3K, and mTOR were reported in the hemimegalencephaly brain (Lee et al., 2012; Poduri et al., 2012). Two of the cases are a trisomy of 1q that contains Akt3, and one is an activating mutation of Akt3 (G49A; Poduri et al., 2012). This mutation encompasses the amino acid substitution E17K in a coding region, which converts Akt to a constitutively active form. Another group found same somatic mutation of Akt3 in one case, and constitutively active PI3K mutations (G1633A, E545K) in four cases. In addition, an mTOR somatic mutation at C4448T that causes C1483Y substitution was observed in hemimegalencephaly brain (Lee et al., 2012). In contrast to some mTOR active mutations developed in laboratories in mammalian cell (Urano et al., 2007; Ohne et al., 2008; Hardt et al., 2011), whether C1483Y mutation makes mTOR active or not is unknown. This is the first report of mTOR mutation in neurological disease so that the validation of activity is awaited. Dysmorphic cells in the brain of focal cortical dysplasia type IIB patient showed hyper-phosphorylation of S6 (Baybis et al., 2004; Miyata et al., 2004). Although genetic analysis forthcoming, it is likely that there may be somatic mutations of unknown genes in mTORC1 signaling pathways that are responsible for cellular abnormality in focal cortical dysplasia.
In this review, we summarize the updated molecular interaction of mTOR signaling. We focus on the growth and size regulation of neurons during development as a biological output of mTOR signaling. Diseases that are picked are limited to this aspect. TSC is caused by the mutation of TSC1 or 2 that are regulators of mTORC1. TSC and other pathologically related disease, hemimegalencephaly and forcal cortical dysplasia are characteristics of enlarged and dysmorphic neurons and glia. These are diseases of dysregulated cell growth.
Although mTOR governs various processes, we describes much on the translational control and a bit about lipid biogenesis, because these two processes seem to be related with cell growth regulation directly. In addition, mTOR-mediated translational control is most (or almost only) studied in the field of neuroscience as a mechanism behind the neural plasticity, learning and memory.
Pathology of TSC has given us a hit of link between mTORC1 activity and size control in the brain. Overactivation of mTORC1 (and potentially mTORC2) induces enlargement and dysmorphism of neurons and glia (or their progenitors). On the other hand, it has been well known that fetal and neonatal malnutrition causes reduced brain size (Morgane et al., 1993). Insufficient intake of proteins and carbohydrates causes reduced amino acid and glucose. Because these nutrients are essential factors to induce mTORC1 function, it is likely that mTORC1 inhibition is related to dysgenesis of a brain. Beside these unusual situations, size differences are found in neurons. One characteristic feature of neurons is its high heterogeneity in function and morphology. Soma sizes vary from about 10 (cerebellar granule neurons) to 100 (Betz cell) micrometers in diameter. The cell size is thought to be determined genetically (and/or epigenetically). The concept includes timing and levels of expression of nutrients transporters and growth factor receptors that may affect mTORC1 activity. Although mTOR signaling components are ubiquitously expressed, rheb, for example, is an activity-inducible molecule in neurons. It suggests that neural activity may enhance mTORC1 through the upregulation of rheb level.
mTOR is undoubtedly a master regulator of cell growth from yeast to human. However, how mTOR controls cell size is still not clear enough. Enhanced protein and lipid synthesis, and inhibition of protein degradation controlled by mTORC1 surely increase cellular mass that includes cell volume, axon elongation, and dendrite arborization. Conceptual scheme is shown in Figure 4. It must be clarify whether novel protein and/or lipid synthesis are necessary and sufficient for cell growth in the brain. Further studies will reveal the correspondence to mTOR downstream signaling pathway to phenotype (cell growth). It will shed light on the biology of cell size and may also contribute to the drug discovery for the TSC and related diseases more selective than rapamycin and rapalogs.
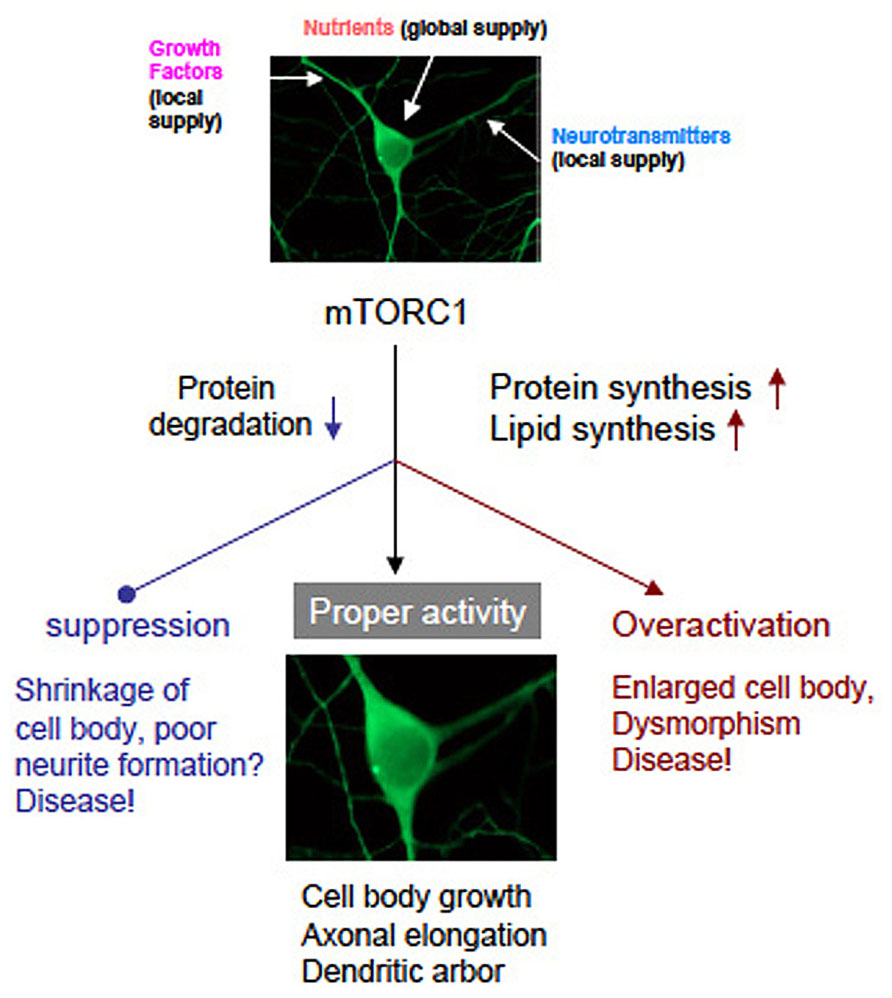
FIGURE 4. A graphic of hypothetical neuronal development governed by mTORC1. Neurons receive nutrients globally and growth factors/transmitters locally. Both inputs coordinately activate mTORC1 that leads normal neuronal development. Suppression or overactivation of mTORC1 result dysregulation of neuronal morphology and function. (note that photographs of a neuron was image processed).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by the Grant-in-Aid for Scientific Research C (JSPS) and the Grant-in-Aid for Scientific Research on Innovative Areas (MEXT).
Avruch, J., Hara, K., Lin, Y., Liu, M., Long, X., Ortiz-Vega, S., et al. (2006). Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene 25, 6361–6372. doi: 10.1038/sj.onc.1209882
Baybis, M., Yu, J., Lee, A., Golden, J. A., Weiner, H., McKhann, G. II, et al. (2004). mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann. Neurol. 56, 478–487. doi: 10.1002/ana.20211
Bentzinger, C. F., Romanino, K., Cloëtta, D., Lin, S., Mascarenhas, J. B., Oliveri, F., et al. (2008). Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 8, 411–424. doi: 10.1016/j.cmet.2008.10.002
Bonfils, G., Jaquenoud, M., Bontron, S., Ostrowicz, C., Ungermann, C., and De Virgilio, C. (2012). Leucyl-tRNA synthetase controls TORC1 via the EGO complex. Mol. Cell 46, 105–110. doi: 10.1016/j.molcel.2012.02.009
Bonni, A., Sun, Y., Nadal-Vicens, M., Bhatt, A., Frank, D. A., Rozovsky, I., et al. (1997). Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 278, 477–483. doi: 10.1126/science.278.5337.477
Brown, N. F., Stefanovic-Racic, M., Sipula, I. J., and Perdomo, G. (2007). The mammalian target of rapamycin regulates lipid metabolism in primary cultures of rat hepatocytes. Metabolism 56, 1500–1507. doi: 10.1016/j.metabol.2007.06.016
Campbell, D. S., and Holt, C. E. (2001). Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron 32, 1013–1026. doi: 10.1016/S0896-6273(01)00551-7
Campbell, L. E., Wang, X., and Proud, C. G. (1999). Nutrients differentially regulate multiple translation factors and their control by insulin. Biochem. J. 344, 433–441. doi: 10.1042/0264-6021:3440433
Carroll, M., Dyer, J., and Sossin, W. S. (2006). Serotonin increases phosphorylation of synaptic 4EBP through TOR, but eukaryotic initiation factor 4E levels do not limit somatic cap-dependent translation in aplysia neurons. Mol. Cell. Biol. 26, 8586–8598. doi: 10.1128/MCB.00955-06
Carroll, M., Warren, O., Fan, X., and Sossin, W. S. (2004). 5-HT stimulates eEF2 dephosphorylation in a rapamycin-sensitive manner in Aplysia neurites. J. Neurochem. 90, 1464–1476. doi: 10.1111/j.1471-4159.2004.02634.x
Carson, R. P., Fu, C., Winzenburger, P., and Ess, K. C. (2013). Deletion of Rictor in neural progenitor cells reveals contributions of mTORC2 signaling to tuberous sclerosis complex. Hum. Mol. Genet. 22, 140–152. doi: 10.1093/hmg/dds414
Casadio, A., Martin, K. C., Giustetto, M., Zhu, H., Chen, M., Bartsch, D., et al. (1999). A transient, neuron-wide form of CREB-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis. Cell 99, 221–237. doi: 10.1016/S0092-8674(00)81653-0
Chenal, J., Pierre, K., and Pellerin, L. (2008). Insulin and IGF-1 enhance the expression of the neuronal monocarboxylate transporter MCT2 by translational activation via stimulation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin pathway. Eur. J. Neurosci. 27, 53–65. doi: 10.1111/j.1460-9568.2007.05981.x
Choo, A. Y., and Blenis, J. (2009). Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle 8, 567–572. doi: 10.4161/cc.8.4.7659
Chong-Kopera, H., Inoki, K., Li, Y., Zhu, T., Garcia-Gonzalo, F. R., Rosa, J. L., et al. (2006). TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligase. J. Biol. Chem. 281, 8313–8316. doi: 10.1074/jbc.C500451200
Choo, A. Y., Roux, P. P., and Blenis, J. (2006). Mind the GAP: Wnt steps onto the mTORC1 train. Cell 126, 834–836. doi: 10.1016/j.cell.2006.08.025
Cloëtta, D., Thomanetz, V., Baranek, C., Lustenberger, R. M., Lin, S., Oliveri, F., et al. (2013). Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J. Neurosci. 33, 7799–7810. doi: 10.1523/JNEUROSCI.3294-12.2013
Crino, P. B. (2013). Evolving neurobiology of tuberous sclerosis complex. Acta Neuropathol. 125, 317–332. doi: 10.1007/s00401-013-1085-x
Crino, P. B., Aronica, E., Baltuch, G., and Nathanson, K. L. (2010). Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology 74, 1716–1723. doi: 10.1212/WNL.0b013e3181e04325
Crino, P. B., Nathanson, K. L., and Henske, E. P. (2006). The tuberous sclerosis complex. N. Engl. J. Med. 355, 1345–1356. doi: 10.1056/NEJMra055323
Dibble, C. C., Elis, W., Menon, S., Qin, W., Klekota, J., Asara, J. M., et al. (2012). TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 47, 535–546. doi: 10.1016/j.molcel.2012.06.009
Düvel, K., Yecies, J. L., Menon, S., Raman, P., Lipovsky, A. I., Souza, A. L., et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183. doi: 10.1016/j.molcel.2010.06.022
Feliciano, D. M., Su, T., Lopez, J., Platel, J. C., and Bordey, A. (2011). Single-cell Tsc1 knockout during corticogenesis generates tuber-like lesions and reduces seizure threshold in mice. J. Clin. Invest. 121, 1596–1607. doi: 10.1172/JCI44909
Gao, X., Zhang, Y., Arrazola, P., Hino, O., Kobayashi, T., Yeung, R. S., et al. (2002). Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat. Cell Biol. 4, 699–704. doi: 10.1038/ncb847
Gong, R., Park, C. S., Abbassi, N. R., and Tang, S. J. (2006). Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J. Biol. Chem. 281, 18802–18815. doi: 10.1074/jbc.M512524200
Guertin, D. A., Stevens, D. M., Thoreen, C. C., Burds, A. A., Kalaany, N. Y., Moffat, J., et al. (2006). Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Dev. Cell 11, 859–871. doi: 10.1016/j.devcel.2006.10.007
Gwinn, D. M., Shackelford, D. B., Egan, D. F., Mihaylova, M. M., Mery, A., Vasquez, D. S., et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. doi: 10.1016/j.molcel.2008.03.003
Hall, M. N. (2013). On mTOR nomenclature. Biochem. Soc. Trans. 41, 887–888. doi: 10.1042/BST20130092
Han, J. M., Jeong, S. J., Park, M. C., Kim, G., Kwon, N. H., Kim, H. K., et al. (2012). Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 149, 410–424. doi: 10.1016/j.cell.2012.02.044
Hara, K., Maruki, Y., Long, X., Yoshino, K., Oshiro, N., Hidayat, S., et al. (2002). Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177–189. doi: 10.1016/S0092-8674(02)00833-4
Hara, K., Yonezawa, K., Weng, Q. P., Kozlowski, M. T., Belham, C., and Avruch, J. (1998). Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 273, 14484–14494. doi: 10.1074/jbc.273.23.14484
Hardt, M., Chantaravisoot, N., and Tamanoi, F. (2011). Activating mutations of TOR (target of rapamycin). Genes Cells 16, 141–151. doi: 10.1111/j.1365-2443.2010.01482.x
Harris, T. E., Chi, A., Shabanowitz, J., Hunt, D. F., Rhoads, R. E., and Lawrence, J. C. Jr. (2006). mTOR-dependent stimulation of the association of eIF4G and eIF3 by insulin. EMBO J. 25, 1659–1668. doi: 10.1038/sj.emboj.7601047
Heitman, J., Movva, N. R., Hiestand, P. C., and Hall, M. N. (1991a). FK 506-binding protein proline rotamase is a target for the immunosuppressive agent FK 506 in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 88, 1948–1952. doi: 10.1073/pnas.88.5.1948
Heitman, J., Movva, N. R., and Hall, M. N. (1991b). Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253, 905–909. doi: 10.1126/science.1715094
Hentges, K. E., Sirry, B., Gingeras, A. C., Sarbassov, D., Sonenberg, N., Sabatini, D., et al. (2001). FRAP/mTOR is required for proliferation and patterning during embryonic development in the mouse. Proc. Natl. Acad. Sci. U.S.A. 98, 13796–13801. doi: 10.1073/pnas.241184198
Holz, M. K., Ballif, B. A., Gygi, S. P., and Blenis, J. (2005). mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123, 569–580. doi: 10.1016/j.cell.2005.10.024
Hou, L., and Klann, E. (2004). Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 24, 6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004
Hu, J., Zacharek, S., He, Y. J., Lee, H., Shumway, S., Duronio, R. J., et al. (2008). WD40 protein FBW5 promotes ubiquitination of tumor suppressor TSC2 by DDB1-CUL4-ROC1 ligase. Genes Dev. 22, 866–871. doi: 10.1101/gad.1624008
Huang, Y., Kang, B. N., Tian, J., Liu, Y., Luo, H. R., Hester, L., et al. (2007). The cationic amino acid transporters CAT1 and CAT3 mediate NMDA receptor activation-dependent changes in elaboration of neuronal processes via the mammalian target of rapamycin mTOR pathway. J. Neurosci. 27, 449–458. doi: 10.1523/JNEUROSCI.4489-06.2007
Hurley, R. L., Anderson, K. A., Franzone, J. M., Kemp, B. E., Means, A. R., and Witters, L. A. (2005). The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 280, 29060–29066. doi: 10.1074/jbc.M503824200
Inamura, N., Nawa, H., and Takei, N. (2005). Enhancement of translation elongation in neurons by brain-derived neurotrophic factor: implications for mammalian target of rapamycin signaling. J. Neurochem. 95, 1438–1445. doi: 10.1111/j.1471-4159.2005.03466.x
Inoki, K., Li, Y., Xu, T., and Guan, K. L. (2003a). Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834. doi: 10.1101/gad.1110003
Inoki, K., Zhu, T., and Guan, K. L. (2003b). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. doi: 10.1016/S0092-8674(03)00929-2
Inoki, K., Li, Y., Zhu, T., Wu, J., and Guan, K. L. (2002). TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648–657. doi: 10.1038/ncb839
Inoki, K., Ouyang, H., Zhu, T., Lindvall, C., Wang, Y., Zhang, X., et al. (2006). TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 126, 955–968. doi: 10.1016/j.cell.2006.06.055
Ishizuka, Y., Kakiya, N., Nawa, H., and Takei, N. (2008). Leucine induces phosphorylation and activation of p70S6K in cortical neurons via the system L amino acid transporter. J. Neurochem. 106, 934–942. doi: 10.1111/j.1471-4159.2008.05438.x
Ishizuka, Y., Kakiya, N., Witters, L. A., Oshiro, N., Shirao, T., Nawa, H., et al. (2013). AMP-activated protein kinase counteracts brain-derived neurotrophic factor-induced mammalian target of rapamycin complex 1 signaling in neurons. J. Neurochem. 127, 66–77. doi: 10.1111/jnc.12362
Jacinto, E., Loewith, R., Schmidt, A., Lin, S., Rüegg, M. A., Hall, A., et al. (2004). Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 6, 1122–1128. doi: 10.1038/ncb1183
Jaworski, J., Spangler, S., Seeburg, D. P., Hoogenraad, C. C., and Sheng, M. (2005). Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J. Neurosci. 25, 11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005
Jossin, Y., and Goffinet, A. M. (2007). Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol. Cell. Biol. 27, 7113–7124. doi: 10.1128/MCB.00928-07
Kang, S. A., Pacold, M. E., Cervantes, C. L., Lim, D., Lou, H. J., Ottina, K., et al. (2013). mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 341, 12365–12366. doi: 10.1126/science.1236566
Kim, B. W., Choi, M., Kim, Y. S., Park, H., Lee, H. R., Yun, C. O., et al. (2008a). Vascular endothelial growth factor (VEGF) signaling regulates hippocampal neurons by elevation of intracellular calcium and activation of calcium/calmodulin protein kinase II and mammalian target of rapamycin. Cell. Signal. 20, 714–725. doi: 10.1016/j.cellsig.2007.12.009
Kim, E., Goraksha-Hicks, P., Li, L., Neufeld, T. P., and Guan, K. L. (2008b). Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 10, 935–945. doi: 10.1038/ncb1753
Kim, D. H., Sarbassov, D. D., Ali, S. M., King, J. E., Latek, R. R., Erdjument-Bromage, H., et al. (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175. doi: 10.1016/S0092-8674(02)00808-5
Kumar, V., Zhang, M. X., Swank, M. W., Kunz, J., and Wu, G. Y. (2005). Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J. Neurosci. 25, 11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005
Kwon, C. H., Luikart, B. W., Powell, C. M., Zhou, J., Matheny, S. A., Zhang, W., et al. (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388. doi: 10.1016/j.neuron.2006.03.023
Laplante, M., and Sabatini, D. M. (2009). An emerging role of mTOR in lipid biosynthesis. Curr. Biol. 19, R1046–R1052. doi: 10.1016/j.cub.2009.09.058
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Lee, C. C., Huang, C. C., Wu, M. Y., and Hsu, K. S. (2005). Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J. Biol. Chem. 280, 18543–18550. doi: 10.1074/jbc.M414112200
Lee, J. H., Huynh, M., Silhavy, J. L., Kim, S., Dixon-Salazar, T., Heiberg, A., et al. (2012). De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet. 44, 941–945. doi: 10.1038/ng.2329
Lenz, G., and Avruch, J. (2005). Glutamatergic regulation of the p70S6 kinase in primary mouse neurons. J. Biol. Chem. 280, 38121–38124. doi: 10.1074/jbc.C500363200
Lewin, G. R., and Barde, Y. A. (1996). Physiology of the neurotrophins. Annu. Rev. Neurosci. 19, 289–317. doi: 10.1146/annurev.ne.19.030196.001445
Li, Y., Inoki, K., Yeung, R., and Guan, K. L. (2002). Regulation of TSC2 by 14-3-3 binding. J. Biol. Chem. 277, 44593–44596. doi: 10.1074/jbc.C200510200
Liao, L., Pilotte, J., Xu, T., Wong, C. C., Edelman, G. M., Vanderklish, P., et al. (2007). BDNF induces widespread changes in synaptic protein content and up-regulates components of the translation machinery: an analysis using high-throughput proteomics. J. Proteome Res. 6, 1059–1071. doi: 10.1021/pr060358f
Long, X., Lin, Y., Ortiz-Vega, S., Yonezawa, K., and Avruch, J. (2005). Rheb binds and regulates the mTOR kinase. Curr. Biol. 15, 702–713. doi: 10.1016/j.cub.2005.02.053
Ma, L., Teruya-Feldstein, J., Bonner, P., Bernardi, R., Franz, D. N., Witte, D., et al. (2007). Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res. 67, 7106–7112. doi: 10.1158/0008-5472.CAN-06-4798
Magri, L., Cambiaghi, M., Cominelli, M., Alfaro-Cervello, C., Cursi, M., Pala, M., et al. (2011). Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell 9, 447–462. doi: 10.1016/j.stem.2011.09.008
Manadas, B., Santos, A. R., Szabadfi, K., Gomes, J. R., Garbis, S. D., Fountoulakis, M., et al. (2009). BDNF-induced changes in the expression of the translation machinery in hippocampal neurons: protein levels and dendritic mRNA. J. Proteome Res. 8, 4536–4552. doi: 10.1021/pr900366x
Manning, B. D., Tee, A. R., Logsdon, M. N., Blenis, J., and Cantley, L. C. (2002). Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 10, 151–162. doi: 10.1016/S1097-2765(02)00568-3
McAllister, A. K., Lo, D. C., and Katz, L. C. (1995). Neurotrophins regulate dendritic growth in developing visual cortex. Neuron 15, 791–803. doi: 10.1016/0896-6273(95)90171-X
Miyata, H., Chiang, A. C., and Vinters, H. V. (2004). Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann. Neurol. 56, 510–519. doi: 10.1002/ana.20234
Morgane, P. J., Austin-LaFrance, R., Bronzino, J., Tonkiss, J., Díaz-Cintra, S., Cintra, L., et al. (1993). Prenatal malnutrition and development of the brain. Neurosci. Biobehav. Rev. 17, 91–128. doi: 10.1016/S0149-7634(05)80234-9
Murakami, M., Ichisaka, T., Maeda, M., Oshiro, N., Hara, K., Edenhofer, F., et al. (2004). mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol. Cell. Biol. 24, 6710–6718. doi: 10.1128/MCB.24.15.6710-6718.2004
Nakashima, A., Yoshino, K., Miyamoto, T., Eguchi, S., Oshiro, N., Kikkawa, U., et al. (2007). Identification of TBC7 having TBC domain as a novel binding protein to TSC1-TSC2 complex. Biochem. Biophys. Res. Commun. 361, 218–223. doi: 10.1016/j.bbrc.2007.07.011
Nawa, H., and Takei, N. (2001). BDNF as an anterophin; a novel neurotrophic relationship between brain neurons. Trends Neurosci. 24, 683–684. doi: 10.1016/S0166-2236(00)01955-X
Nellist, M., Goedbloed, M. A., de Winter, C., Verhaaf, B., Jankie, A., Reuser, A. J., et al. (2002). Identification and characterization of the interaction between tuberin and 14-3-3ϖ. J. Biol. Chem. 277, 39417–39424. doi: 10.1074/jbc.M204802200
Nie, D., Di Nardo, A., Han, J. M., Baharanyi, H., Kramvis, I., Huynh, T., et al. (2010). Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat. Neurosci. 13, 163–167. doi: 10.1038/nn.2477
Nojima, H., Tokunaga, C., Eguchi, S., Oshiro, N., Hidayat, S., Yoshino, K., et al. (2003). The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem. 278, 15461–15464. doi: 10.1074/jbc.C200665200
Ohne, Y., Takahara, T., Hatakeyama, R., Matsuzaki, T., Noda, M., Mizushima, N., et al. (2008). Isolation of hyperactive mutants of mammalian target of rapamycin. J. Biol. Chem. 283, 31861–31870. doi: 10.1074/jbc.M801546200
Oshiro, N., Takahashi, R., Yoshino, K., Tanimura, K., Nakashima, A., Eguchi, S., et al. (2007). The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J. Biol. Chem. 282, 20329–20339. doi: 10.1074/jbc.M702636200
Page, G., Khidir, F. A., Pain, S., Barrier, L., Fauconneau, B., Guillard, O., et al. (2006). Group I metabotropic glutamate receptors activate the p70S6 kinase via both mammalian target of rapamycin (mTOR) and extracellular signal-regulated kinase (ERK 1/2) signaling pathways in rat striatal and hippocampal synaptoneurosomes. Neurochem. Int. 49, 413–421. doi: 10.1016/j.neuint.2006.01.020
Peng, T., Golub, T. R., and Sabatini, D. M. (2002). The immunosuppressant rapamycin mimics a starvation-like signal distinct from amino acid and glucose deprivation. Mol. Cell. Biol. 22, 5575–5584. doi: 10.1128/MCB.22.15.5575-5584.2002
Piper, M., Anderson, R., Dwivedy, A., Weinl, C., van Horck, F., Leung, K. M., et al. (2006). Signaling mechanisms underlying Slit2-induced collapse of Xenopus retinal growth cones. Neuron 49, 215–228. doi: 10.1016/j.neuron.2005.12.008
Poduri, A., Evrony, G. D., Cai, X., Elhosary, P. C., Beroukhim, R., Lehtinen, M. K., et al. (2012). Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 74, 41–48. doi: 10.1016/j.neuron.2012.03.010
Polakiewicz, R. D., Schieferl, S. M., Gingras, A. C., Sonenberg, N., and Comb, M. J. (1998). mu-Opioid receptor activates signaling pathways implicated in cell survival and translational control. J. Biol. Chem. 273, 23534–23541. doi: 10.1074/jbc.273.36.23534
Porstmann, T., Santos, C. R., Griffiths, B., Cully, M., Wu, M., Leevers, S., et al. (2008). SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 8, 224–236. doi: 10.1016/j.cmet.2008.07.007
Prabowo, A. S., Anink, J. J., Lammens, M., Nellist, M., van den Ouweland, A. M., Adle-Biassette, H., et al. (2013). Fetal brain lesions in tuberous sclerosis complex: TORC1 activation and inflammation. Brain Pathol. 23, 45–59. doi: 10.1111/j.1750-3639.2012.00616.x
Puighermanal, E., Marsicano, G., Busquets-Garcia, A., Lutz, B., Maldonado, R., and Ozaita, A. (2009). Cannabinoid modulation of hippocampal long-term memory is mediated by mTOR signaling. Nat. Neurosci. 12, 1152–1158. doi: 10.1038/nn.2369
Qin, W., Chan, J. A., Vinters, H. V., Mathern, G. W., Franz, D. N., Taillon, B. E., et al. (2010). Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second-hit mutations in these genes are rare events. Brain Pathol. 20, 1096–1105. doi: 10.1111/j.1750-3639.2010.00416.x
Quevedo, C., Salinas, M., and Alcázar, A. (2002). Regulation of cap-dependent translation by insulin-like growth factor-1 in neuronal cells. Biochem. Biophys. Res. Commun. 291, 560–566. doi: 10.1006/bbrc.2002.6479
Raught, B., Gingras, A. C., Gygi, S. P., Imataka, H., Morino, S., Gradi, A., et al. (2000). Serum-stimulated, rapamycin-sensitive phosphorylation sites in the eukaryotic translation initiation factor 4GI. EMBO J. 19, 434–444. doi: 10.1093/emboj/19.3.434
Raught, B., Peiretti, F., Gingras, A. C., Livingstone, M., Shahbazian, D., Mayeur, G. L., et al. (2004). Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 23, 1761–1769. doi: 10.1038/sj.emboj.7600193
Ruvinsky, I., and Meyuhas, O. (2006). Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem. Sci. 31, 342–348. doi: 10.1016/j.tibs.2006.04.003
Sancak, Y., Peterson, T. R., Shaul, Y. D., Lindquist, R. A., Thoreen, C. C., Bar-Peled, L., et al. (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501. doi: 10.1126/science.1157535
Sarbassov, D. D., Ali, S. M., Kim, D. H., Guertin, D. A., Latek, R. R., Erdjument-Bromage, H., et al. (2004). Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302. doi: 10.1016/j.cub.2004.06.054
Sarbassov, D. D., Ali, S. M., Sengupta, S., Sheen, J. H., Hsu, P. P., Bagley, A. F., et al. (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168. doi: 10.1016/j.molcel.2006.03.029
Schalm, S. S., and Blenis, J. (2002). Identification of a conserved motif required for mTOR signaling. Curr. Biol. 12, 632–639. doi: 10.1016/S0960-9822(02)00762-5
Schreiber, S. L. (1991). Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science 251, 283–287. doi: 10.1126/science.1702904
Segev, N., and Hay, N. (2012). Hijacking leucyl-tRNA synthetase for amino acid-dependent regulation of TORC1. Mol. Cell 46, 4–6. doi: 10.1016/j.molcel.2012.03.028
Shahbazian, D., Roux, P. P., Mieulet, V., Cohen, M. S., Raught, B., Taunton, J., et al. (2006). The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 25, 2781–2791. doi: 10.1038/sj.emboj.7601166
Shima, H., Pende, M., Chen, Y., Fumagalli, S., Thomas, G., and Kozma, S. C. (1998). Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 17, 6649–6659. doi: 10.1093/emboj/17.22.6649
Shiota, C., Woo, J. T., Lindner, J., Shelton, K. D., and Magnuson, M. A. (2006). Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev. Cell 11, 583–589. doi: 10.1016/j.devcel.2006.08.013
Shor, B., Zhang, W. G., Toral-Barza, L., Lucas, J., Abraham, R. T., Gibbons, J. J., et al. (2008). A new pharmacologic action of CCI-779 involves FKBP12-independent inhibition of mTOR kinase activity and profound repression of global protein synthesis. Cancer Res. 68, 2934–2943. doi: 10.1158/0008-5472.CAN-07-6487
Shumway, S. D., Li, Y., and Xiong, Y. (2003). 14-3-3β binds to and negatively regulates the tuberous sclerosis complex 2 (TSC2) tumor suppressor gene product, tuberin. J. Biol. Chem. 278, 2089–2092. doi: 10.1074/jbc.C200499200
Suzuki, S., Kiyosue, K., Hazama, S., Ogura, A., Kashihara, M., Hara, T., et al. (2007). Brain-derived neurotrophic factor regulates cholesterol metabolism for synapse development. J. Neurosci. 27, 6417–6427. doi: 10.1523/JNEUROSCI.0690-07.2007
Takei, N., Inamura, N., Kawamura, M., Namba, H., Hara, K., Yonezawa, K., et al. (2004). Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J. Neurosci. 24, 9760–9769. doi: 10.1523/JNEUROSCI.1427-04.2004
Takei, N., Kawamura, M., Hara, K., Yonezawa, K., and Nawa, H. (2001). Brain-derived neurotrophic factor enhances neuronal translation by activating multiple initiation processes: comparison with the effects of insulin. J. Biol. Chem. 276, 42818–42825. doi: 10.1074/jbc.M103237200
Takei, N., Kawamura, M., Ishizuka, Y., Kakiya, N., Inamura, N., Namba, H., et al. (2009). Brain-derived neurotrophic factor enhances the basal rate of protein synthesis by increasing active eukaryotic elongation factor 2 levels and promoting translation elongation in cortical neurons. J. Biol. Chem. 284, 26340–26348. doi: 10.1074/jbc.M109.023010
Tee, A. R., Fingar, D. C., Manning, B. D., Kwiatkowski, D. J., Cantley, L. C., and Blenis, J. (2002). Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. U.S.A. 99, 13571–13576. doi: 10.1073/pnas.202476899
Thomanetz, V., Angliker, N., Cloëtta, D., Lustenberger, R. M., Schweighauser, M., Oliveri, F., et al. (2013). Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol. 201, 293–308. doi: 10.1083/jcb.201205030
Thoreen, C. C., Chantranupong, L., Keys, H. R., Wang, T., Gray, N. S., and Sabatini, D. M. (2012). A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113. doi: 10.1038/nature11083
Tsai, V., Parker, W. E., Orlova, K. A., Baybis, M., Chi, A. W., Berg, B. D., et al. (2012). Fetal brain mTOR signaling activation in tuberous sclerosis complex. Cereb. Cortex 24, 315–327. doi: 10.1093/cercor/bhs310
Urano, J., Sato, T., Matsuo, T., Otsubo, Y., Yamamoto, M., and Tamanoi, F. (2007). Point mutations in TOR confer Rheb-independent growth in fission yeast and nutrient-independent mammalian TOR signaling in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 104, 3514–3519. doi: 10.1073/pnas.0608510104
van Gorp, A. G., van der Vos, K. E., Brenkman, A. B., Bremer, A., van den Broek, N., Zwartkruis, F., et al. (2009). AGC kinases regulate phosphorylation and activation of eukaryotic translation initiation factor 4B. Oncogene 28, 95–106. doi: 10.1038/onc.2008.367
Vézina, C., Kudelski, A., and Sehgal, S. N. (1975). Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. (Tokyo) 28, 721–726. doi: 10.7164/antibiotics.28.721
Wang, X., Li, W., Williams, M., Terada, N., Alessi, D. R., and Proud, C. G. (2001). Regulation of elongation factor 2 kinase by p90RSK1 and p70 S6 kinase. EMBO J. 20, 4370–4379. doi: 10.1093/emboj/20.16.4370
Way, S. W., McKenna, J. III, Mietzsch, U., Reith, R. M., Wu, H. C., and Gambello, M. J. (2009). Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Hum. Mol. Genet. 18, 1252–1265. doi: 10.1093/hmg/ddp025
Witters, L. A., Kemp, B. E., and Means, A. R. (2006). Chutes and Ladders: the search for protein kinases that act on AMPK. Trends Biochem. Sci. 31, 13–16. doi: 10.1016/j.tibs.2005.11.009
Woods, A., Johnstone, S. R., Dickerson, K., Leiper, F. C., Fryer, L. G., Neumann, D., et al. (2003). LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13, 2004–2008. doi: 10.1016/j.cub.2003.10.031
Yamagata, K., Sanders, L. K., Kaufmann, W. E., Yee, W., Barnes, C. A., Nathans, D., et al. (1994). rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J. Biol. Chem. 269, 16333–16339.
Yamauchi, Y., Furukawa, K., Hamamura, K., and Furukawa, K. (2011). Positive feedback loop between PI3K-Akt-mTORC1 signaling and the lipogenic pathway boosts Akt signaling: induction of the lipogenic pathway by a melanoma antigen. Cancer Res. 71, 4989–4997. doi: 10.1158/0008-5472.CAN-10-4108
Yang, H., Rudge, D. G., Koos, J. D., Vaidialingam, B., Yang, H. J., and Pavletich, N. P. (2013). mTOR kinase structure, mechanism and regulation. Nature 497, 217–223. doi: 10.1038/nature12122
Yao, J. J., Sun, J., Zhao, Q. R., Wang, C. Y., and Mei, Y. A. (2013). Neuregulin-1/ErbB4 signaling regulates Kv4.2-mediated transient outward K+ current through the Akt/mTOR pathway. Am. J. Physiol. Cell Physiol. 305, C197–C206. doi: 10.1152/ajpcell.00041.2013
Keywords: mTORC1 signaling, translational control, protein synthesis, BDNF, CNS neurons, amino acids, TSC/mTOR, brain malformation
Citation: Takei N and Nawa H (2014) mTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci. 7:28. doi: 10.3389/fnmol.2014.00028
Received: 10 January 2014; Accepted: 30 March 2014;
Published online: 23 April 2014.
Edited by:
Bryan Weston Luikart, Geisel School of Medicine at Dartmouth, USAReviewed by:
Leonid Tarassishin, Albert Einstein College of Medicine, USACopyright © 2014 Takei and Nawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nobuyuki Takei, Department of Molecular Neurobiology, Brain Research Institute, Niigata University, Asahimachi, Niigata 951-8585, Japan e-mail:bm9idGFrQGJyaS5uaWlnYXRhLXUuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.