


- 1Department of Biological Sciences, Korea Advanced Institute of Science and Technology, Daejeon, South Korea
- 2Center for Synaptic Brain Dysfunctions, Institute for Basic Science, Daejeon, South Korea
Autism spectrum disorder (ASD) is a group of developmental disabilities characterized by impairments in social interaction and communication and restricted and repetitive interests/behaviors. Advances in human genomics have identified a large number of genetic variations associated with ASD. These associations are being rapidly verified by a growing number of studies using a variety of approaches, including mouse genetics. These studies have also identified key mechanisms underlying the pathogenesis of ASD, many of which involve synaptic dysfunctions, and have investigated novel, mechanism-based therapeutic strategies. This review will try to integrate these three key aspects of ASD research: human genetics, animal models, and potential treatments. Continued efforts in this direction should ultimately reveal core mechanisms that account for a larger fraction of ASD cases and identify neural mechanisms associated with specific ASD symptoms, providing important clues to efficient ASD treatment.
Introduction to Autism Spectrum Disorder
Autism spectrum disorder (ASD) is a group of developmental disabilities characterized by abnormal social interaction and communication, and stereotyped behaviors with restricted interest. Autism was first reported by Kanner (1943) with a clinical description of 11 children showing “extreme aloneness from the very beginning of life, not responding to anything that comes to them from the outside world.” He proposed the behavioral combination of autism, obsessiveness, stereotypy, and echolalia as childhood schizophrenia. However, until the 1980s, ASD was not accepted as an individual developmental disorder with a biological origin. In the early 1980s, studies demonstrated the high heritability of ASD and its association with other genetic syndromes (Gillberg and Wahlstrom, 1985; Wahlstrom et al., 1986), providing compelling evidence for a genetic etiology of ASD and fueling the conceptualization of autism as a distinct neurodevelopmental disorder. From the definition of “childhood or early-onset schizophrenia” put forward by Kanner, autism was renamed “infantile autism” in 1980, “autism disorder” in 1987 and, more recently, “autism” or the umbrella term “ASD”.
Diagnosis
Currently, ASD is included in the diagnostic category of a neurodevelopmental disorders in the Diagnostic and Statistical Manual of Mental Disorders V (Grzadzinski et al., 2013). The diagnosis of autism is mainly based on the presence of two major aforementioned symptoms: social-communication deficits, and restricted and repetitive interests/behaviors (Grzadzinski et al., 2013). These symptoms must be shown from early childhood of individuals with ASD. But autism is also associated with various comorbidities, including sensory and motor abnormalities, sleep disturbance, epilepsy, attention deficit/hyperactivity disorder (ADHD)-like hyperactivity, intellectual disability, and mood disorders such as anxiety and aggression (Goldstein and Schwebach, 2004; Simonoff et al., 2008; Geschwind, 2009). Some monogenic syndromes including fragile X syndrome and Rett syndrome also have autistic features, while we should be cautious to directly interpret the disorders as autism since the major symptoms for these syndromes are intellectual disabilities.
Prevalence
An early study conducted in the UK in 1966 reported a prevalence rate of autism of 4.5 in 10,000 children (Lotter, 1966). The estimated prevalence increased to 19 in 10,000 American children in 1992 and rose steeply to 1 in 150 in 2002 (Autism et al., 2007) and 1 in 110 in 2006 (Autism et al., 2009) (see also data from the US Centers for Disease Control and Prevention [CDC]). The currently accepted prevalence of ASD, based on consistent reports of ASD prevalence by multiple sources in different populations, is ~1% worldwide, placing this disorder as one of the most common pervasive developmental disorders and elevating public concerns.
Genetics
On the basis of numerous studies that have been undertaken to elucidate the pathogenic mechanisms underlying ASD, it is widely accepted that ASD is a disorder with strong genetic components. In support of this notion, the concordance rates for autism reach up to 90% in monozygotic twins and 10% in dizygotic twins (Rutter, 2000; Folstein and Rosen-Sheidley, 2001; Veenstra-Vanderweele et al., 2003).
However, autism is an etiologically heterogeneous disorder in that no single genetic mutation accounts for more than 1–2% of ASD cases (Abrahams and Geschwind, 2008). Thus far, linkage and candidate-gene analyses, genome-wide association studies (GWAS), and assessments of chromosomal variations have uncovered a wide range of genes with predisposing mutations and polymorphisms associated with ASD (International Molecular Genetic Study of Autism, 1998, 2001; Abrahams and Geschwind, 2008; Glessner et al., 2009; Ma et al., 2009; Wang et al., 2009; Weiss et al., 2009; Anney et al., 2010; Pinto et al., 2010; Devlin and Scherer, 2012; Moreno-De-Luca et al., 2013) (see Tables 1, 2 for examples). Moreover, recent advancements in exome sequencing and next-generation sequencing have enabled the discovery of an overwhelming number of de novo mutations that confer a risk for ASD (Iossifov et al., 2012; Neale et al., 2012; O'Roak et al., 2012a,b; Sanders et al., 2012). These mutations include rare mutations or copy number variations in synaptic proteins such as Shanks/ProSAPs (Durand et al., 2007; Berkel et al., 2010; Sato et al., 2012) and neuroligins (Jamain et al., 2003).
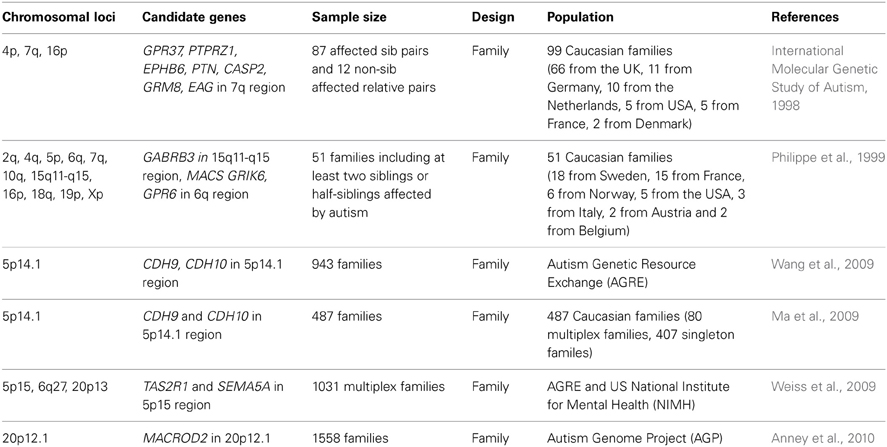
Table 1. Examples of ASD-associated chromosomal loci and candidate genes from GWAS.
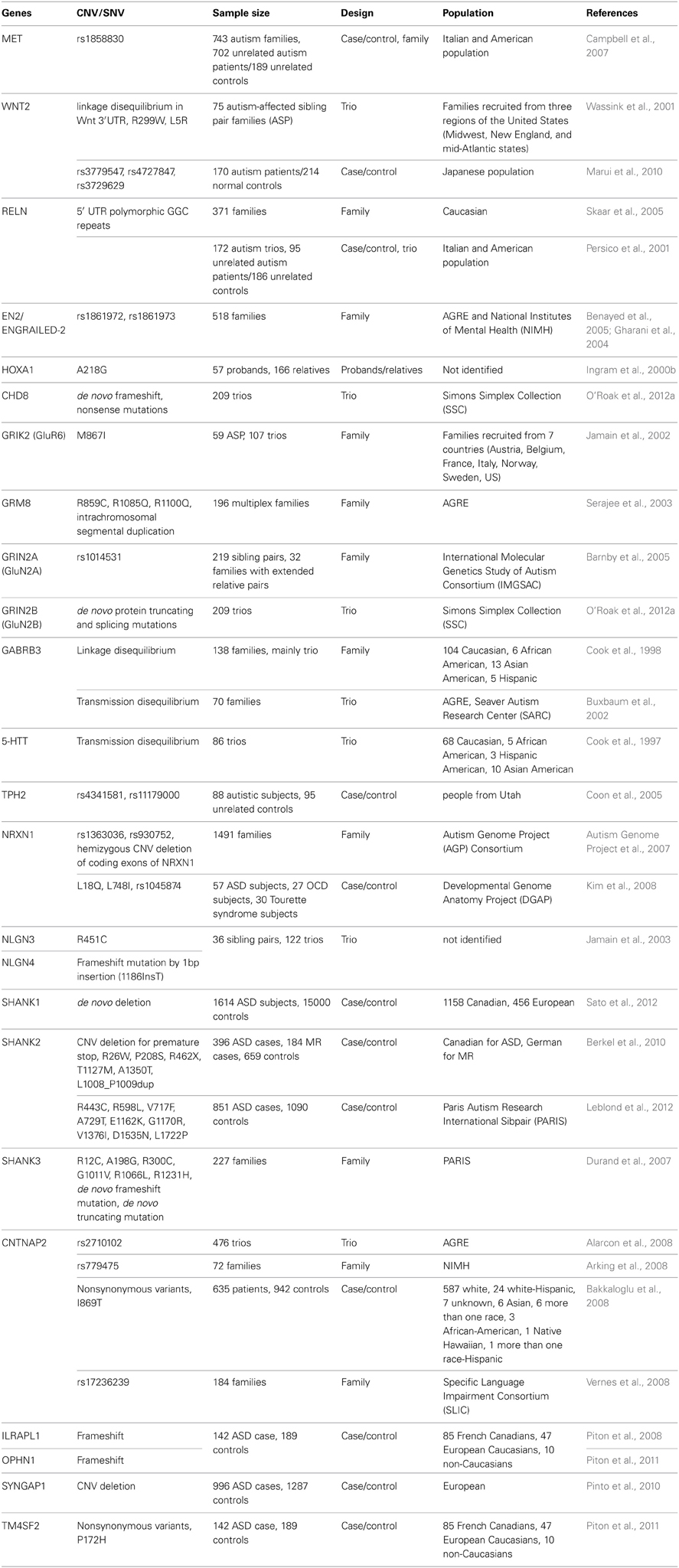
Table 2. Examples of ASD-associated human genetic variations.
However, how these mutations lead to ASD phenotypes is poorly understood. In addition, many ASD-related genes are also associated with other neuropsychiatric disorders. For example, IL1RAPL1 and OPHN1 are associated with X chromosome-linked intellectual disability (Billuart et al., 1998a; Carrie et al., 1999). Additional examples include schizophrenia for RELN, GluR6, GRIN2A, GRIN2B, and CNTNAP2 (Bah et al., 2004; Friedman et al., 2008; Shifman et al., 2008; Demontis et al., 2011), childhood absence epilepsy for GABRB3 (Feucht et al., 1999), ADHD and depression for 5-HTT (Manor et al., 2001; Caspi et al., 2003), and major depression for TPH2 (Zill et al., 2004). Dissecting the neural mechanisms underlying diverse symptoms/disorders caused by single genetic defects is one of the key directions for neuropsychiatric research.
Animal Models for ASD
Animal models of human diseases need to satisfy three major criteria; face validity, construct validity, and predictive validity. Animal models for ASD should display behavioral abnormalities, including impaired sociability, impaired social communication, and repetitive and restricted behaviors (face validity). These models should share analogous genetic or anatomical impairments with humans (construct validity), and show similar responses to the medications used to treat ASD in humans (predictive validity).
Dedicated efforts of many behavioral neuroscientists including Jacqueline Crawley led to the establishment of several well-known assays for rat/mouse models of ASD (Silverman et al., 2010b). Examples include 3-chambered test to assess sociability and social novelty recognition of rodents, ultrasonic vocalization (USV) test to measure the communication patterns of rodents, T-maze test for restricted interests, and home cage behavior or marble burying assay for repetitive behaviors. Through these assays, many genetic and non-genetic animal models of ASD have been characterized and used to identify the etiology of ASD and develop novel treatments (see Tables 3–6 for four different groups of ASD models).
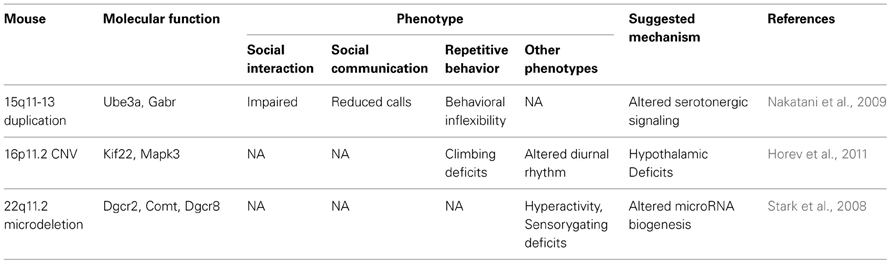
Table 3. ASD models with chromosomal abnormality.
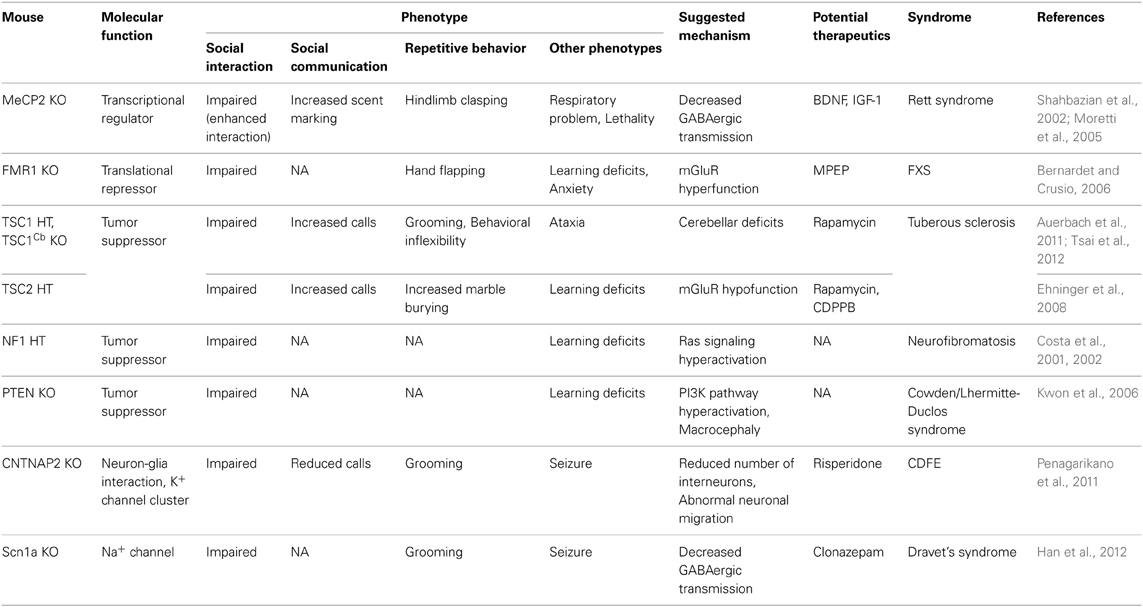
Table 4. Syndromic ASD models.
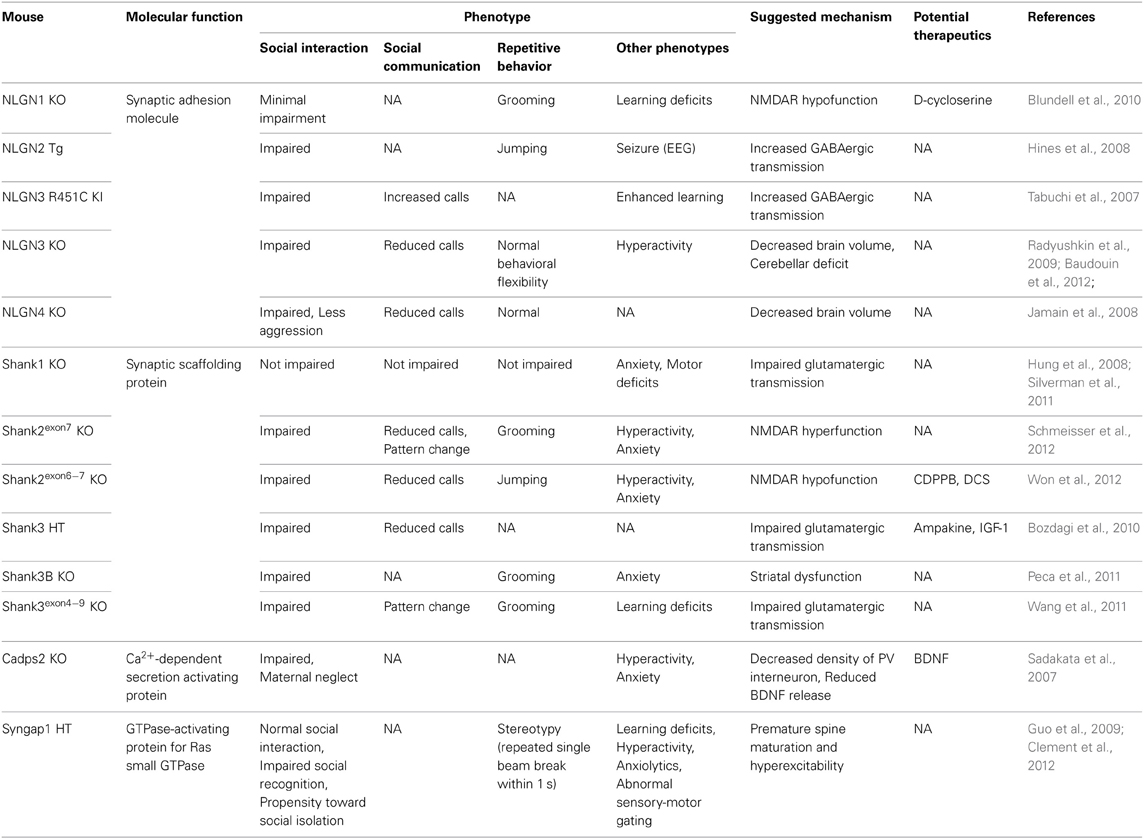
Table 5. Synaptopathy ASD models.
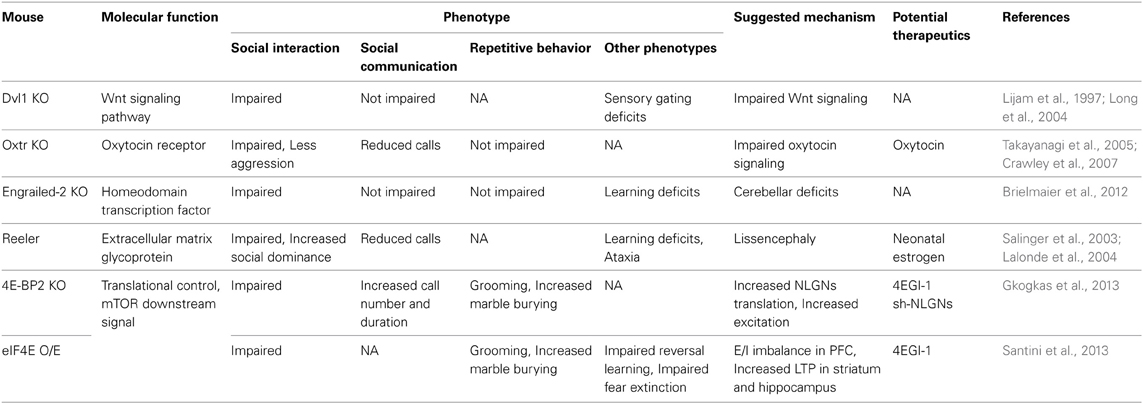
Table 6. Non-synaptopathy ASD models.
Although animal models are useful for exploring ASD mechanisms and testing novel interventions, we should be cautious in interpreting the results from animal models of ASD because what we are observing in animals are behavioral features that look similar to some of the ASD symptoms in humans. This notion partly stems from the fact that the brains of humans and rodents are fundamentally different. For instance, there are small but significant differences in gene expression patterns in the cerebral cortex in different species (Zeng et al., 2012), suggesting that the same cell types in different species may have different functions. Moreover, the size, structural complexity, and neural connectivity of the human brain are much greater than those in rodent brains. These functional and anatomical differences between species may create difficulties in translating the ASD-related mechanisms identified in model organisms into human applications. However, some fundamental aspects of the neural mechanisms identified in animal models such as alterations in synaptic transmission, excitation-inhibition balance, and neuronal excitability might be conserved across species and translatable. In addition, given that stem cell technologies are rapidly improving, it is becoming easier for the changes observed in rodent neurons to be compared with those in human neurons derived from individuals with neuropsychiatric disorders (Brennand et al., 2011).
Potential Mechanisms Underlying ASD
Mechanisms underlying autism have been extensively studied using various approaches. Neuroanatomical studies have reported macrocephaly and abnormal neuronal connectivity in autistic individuals, while genetics studies using mouse models have implicated a variety of neuronal proteins in the development of ASD. More recently, defects in a number of synaptic proteins have been suggested to cause ASD via alterations in synaptic structure/function and neural circuits, suggesting that “synaptopathy” is an important component of ASD.
Neuroanatomical Abnormalities
A change frequently observed in the brains of individuals with ASD is the overgrowth of the brain termed macrocephaly, which is observed in ~20% of autistic children (Bolton et al., 2001; Courchesne, 2002; Courchesne et al., 2003, 2007; Fombonne et al., 1999; Hazlett et al., 2005). Aberrations in cytoarchitectural organization in autistic brains are observed during early brain development in regions including the frontal lobe, parieto-temporal lobe, cerebellum, and subcortical limbic structures (Fombonne et al., 1999; Bolton et al., 2001; Courchesne, 2002; Courchesne et al., 2003, 2007; Hazlett et al., 2005).
The cerebellum is a strong candidate for anatomic abnormalities in autism (Courchesne, 1997, 2002). Magnetic resonance imaging (MRI) studies have found hypoplasia of the cerebellar vermis and hemispheres, and autopsy studies have reported a reduction in the number of cerebellar Purkinje cells. In line with these anatomical changes, cerebellar activation is significantly reduced during selective attention tasks (Allen and Courchesne, 2003), whereas it is enhanced during a simple motor task (Allen et al., 2004). Although the putative role of the cerebellum in ASD has been restricted to sensory and motor dysfunctions, it is becoming increasingly clear that the cerebellum is associated with the core symptoms of autism.
In support of this notion, selective deletion of Tsc1 (tuberous sclerosis 1) in cerebellar Purkinje cells is sufficient to cause all core autism-like behaviors in mice in association with reduced excitability in Purkinje cells (see also Table 4 for summary of syndromic ASD models) (Tsai et al., 2012). In addition, mice lacking the neuroligin-3 gene (Nlgn3−/− mice), another autism model with an Nlgn3 deletion identified in autistic patients, show occluded metabotropic glutamatergic receptor (mGluR)-dependent long-term depression (LTD) at synapses between parallel fibers and Purkinje cells in association with motor coordination deficits (see also Table 5 for summary of synaptopathy ASD models) (Baudouin et al., 2012). Both synaptic and behavioral perturbations are rescued by Purkinje cell-specific re-expression of Nlgn-3 in juvenile mice, suggesting the interesting possibility that altered neural circuits can be corrected after completion of development.
The cerebral cortex is another brain region frequently affected in ASD. Abnormal enlargement or hyperplasia of the cerebral cortex has been reported in MRI studies on young children with ASD (Sparks et al., 2002; Herbert et al., 2003). Because frontal and temporal lobes are important for higher brain functions including social functioning and language development, these anatomical anomalies are likely to underlie the pathophysiology of autism.
The amygdala and hippocampus are subcortical brain regions associated with ASD (Aylward et al., 1999; Schumann et al., 2004; Schumann and Amaral, 2006). Some studies have reported that the autistic amygdala exhibits early enlargement, whereas others have reported a reduction in neuron numbers and amygdala volume. Increases and decreases in the volume of hippocampus are also associated with ASD.
Aberrant connectivity is an emerging theory to account for anatomical abnormalities in autism. Neuroimaging techniques, such as diffusion tensor imaging (DTI) and functional MRI (fMRI), have suggested that ASD involves abrogation of white matter tracts in brain regions associated with social cognition, such as the prefrontal cortex, anterior cingulate cortex, and superior temporal regions (Barnea-Goraly et al., 2004; Minshew and Williams, 2007). Alterations in connectivity across diverse brain regions associated with language, working memory, and social cognition have also been linked to autism. In general, it appears that autism subjects display local over-connectivity and long-range or inter-regional under-connectivity (Herbert et al., 2003, 2004; Baron-Cohen and Belmonte, 2005; Herbert, 2005; Just et al., 2007).
Potential ASD-related neural circuitries have also been proposed based on animal studies. Shank3b−/− mice, which exhibit autistic-like behaviors, have striatal dysfunctions (Table 5) (Peca et al., 2011). In addition, a shift in the balance between excitation and inhibition (E-I balance) toward excitation in the mouse medial prefrontal cortex (mPFC) induced by optogenetic stimulation causes sociability impairments (Yizhar et al., 2011). These results suggest that the striatum and mPFC are components of ASD-related neural circuits.
Although various neuroanatomical defects are observed in autistic brains, a direct linkage between neuroanatomical anomalies and behavioral symptoms of ASD remains to be elucidated. Uncovering the detailed circuitries underlying autistic behaviors would help us understand higher cognitive functions, such as language and sociability.
Extracellular Factors
It has been found that growth factors and neurotrophic factors are associated with ASD. Genetic and protein expression studies have shown that MET, a transmembrane receptor for hepatocyte growth factor (HGF) with tyrosine kinase activity, is associated with ASD. Genetic variations including rs1858830 in the promoter region that abrogate MET transcription are associated with ASD in Italian and American families and case/control studies, and the levels of MET mRNA and protein are reduced in the cortex of autistic patients (Campbell et al., 2007, 2006). However, this association between rs1858830 and ASD failed to replicate in another study (Sousa et al., 2009). By binding to MET, HGF acts as a neurotrophic factor for neurons to influence neurite outgrowth and dendritic morphology (Figure 1) (Powell et al., 2001, 2003; Sun et al., 2002; Gutierrez et al., 2004), implicating abnormal neuronal structures in ASD pathology.
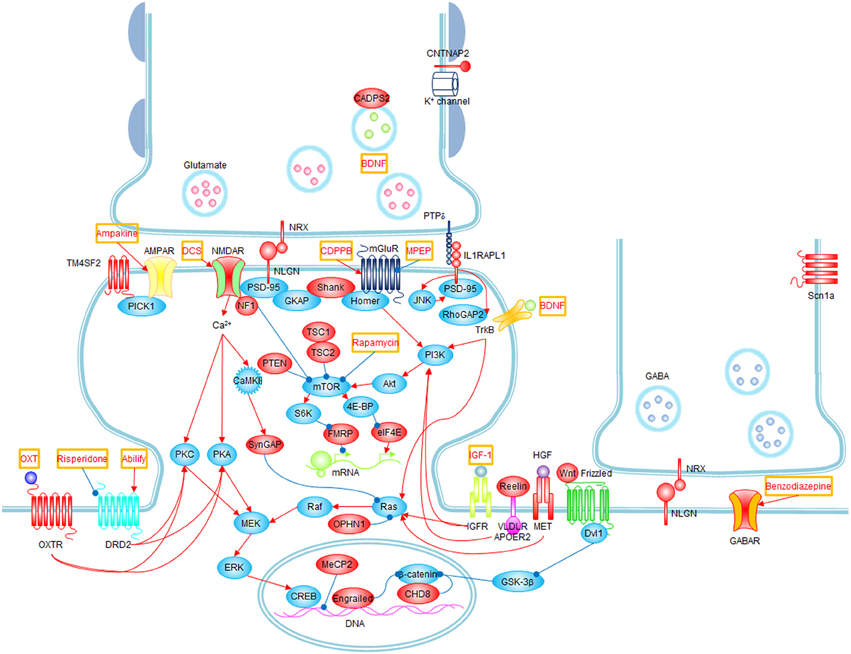
Figure 1. Signaling pathways and possible treatments associated with ASD. Molecules whose mutations or polymorphisms are associated with ASD are indicated in red. Stimulations and inhibitions are indicated by red and blue arrows, respectively. Possible treatments and their target molecules are indicated by red texts in orange boxes. SynGAP1, which directly interacts with PSD-95, could not be placed next to PSD-95 for simplicity.
WNT2 is a secreted growth factor that has been linked to ASD. Acting through the canonical Wnt pathway, WNT2 triggers a signal transduction cascade mediated by Dishevelled (Dvl1). WNT2 is a critical regulator of multiple biological functions, including embryonic development, cellular differentiation, and cell-polarity generation. It also regulates neuronal migration, axon guidance, and dendrite branching (Figure 1) (Logan and Nusse, 2004). Multiple lines of evidence have implicated the WNT2 locus in ASD: the WNT2 gene is located at the autism-susceptibility chromosomal locus 7q31 (Vincent et al., 2000; Warburton et al., 2000), and single nucleotide polymorphisms (SNP; rs3779547, rs4727847, and rs3729629, in a case/control study in a Japanese population) and several WNT2 locus variants (R299W and L5R, in autism-affected sibling pair [ASP] and trio families) are associated with autism (Wassink et al., 2001; Marui et al., 2010), although a subsequent study in Han Chinese trios failed to replicate the SNP association with ASD (Chien et al., 2011). While the majority of Wnt2−/− mice are lethal (Goss et al., 2009), null mutants of Dvl1 show deficits in nest building and home-cage huddling (see also Table 6 for summary of non-synaptopathy ASD models) (Lijam et al., 1997; Long et al., 2004). Moreover, the Wnt signaling pathway is associated with and is regulated by chromodomain-helicase-DNA-binding protein 8 (CHD8; Figure 1), de novo mutations of which are repeatedly detected in autistic patients (Neale et al., 2012; O'Roak et al., 2012b; Sanders et al., 2012).
Brain-derived neurotrophic factor (BDNF) is associated with ASD. BDNF is a member of the neurotrophin family of growth factors that supports neurogenesis, axodendritic growth, neuronal/synaptic differentiation, and brain dysfunctions (Figure 1) (Huang and Reichardt, 2001; Martinowich et al., 2007). Elevated levels of BDNF were reproducibly found in the sera of Japanese and American autistic individuals (Connolly et al., 2006; Miyazaki et al., 2004). Another clue comes from calcium-dependent secretion activator 2 (CADPS2), a calcium binding protein in the presynaptic nerve terminal that interacts with and regulates exocytosis of BDNF-containing dense-core vesicles (Figure 1) (Cisternas et al., 2003). CADPS2, located at the autism-susceptibility locus on chromosome 7q31, is abnormally spliced in autism patients, and Cadps2−/− mice exhibit social interaction deficits, including maternal neglect (Table 5) (Sadakata et al., 2007). Hence, although it is unclear how BDNF contributes to autism pathogenesis, evidence for its role in ASD is becoming clear.
Reelin is also involved in autism. Reelin is a large secreted extracellular matrix glycoprotein that acts as a serine protease for the extracellular matrix, a function that is essential for neuronal migration, cortical patterning, and brain development (Figure 1) (Forster et al., 2006). The RELN gene is located in an autism susceptibility locus on chromosome 7q22, and triplet GGC repeats in 5′ untranslated regions (5′UTR) in the RELN gene have been associated with autism in a Caucasian population (Persico et al., 2001; Skaar et al., 2005) (Table 2). Expression levels of Reelin are decreased in postmortem autism brains (Fatemi et al., 2005). Reelin has also been implicated in pathogenesis of various neuropsychiatric disorders, including schizophrenia, bipolar disorder, lissencephaly, and epilepsy (Fatemi, 2001). Reeler mice, with a 150-kb deletion of the Reln gene, exhibit deficits in motor coordination, increased social dominance, and learning and memory impairments (Table 6) (Salinger et al., 2003; Lalonde et al., 2004).
Transcription Factors
Syndromic forms of ASD frequently involve transcription factors. This is likely because defective transcription factors have significant influences on many genes and their downstream molecules, affecting diverse neuronal functions.
MeCP2 (X-linked gene methyl CpG binding protein 2) is one of the best examples. It is a member of a large family of methyl-CpG binding domain (MBD) proteins that selectively binds to methylated DNA and represses gene transcription (Figure 1) (Bienvenu and Chelly, 2006). Its downstream targets encompass ASD-related genes such as BDNF and CDKL5. Mutations in MeCP2 are the major cause of Rett syndrome, a progressive neurodevelopmental disorder with autistic features (Amir et al., 1999; Bienvenu and Chelly, 2006; Chahrour and Zoghbi, 2007). Mecp2-null mice, an animal model for Rett syndrome, recapitulate most symptomatic traits of Rett syndrome such as respiratory dysfunction, forelimb and hindlimb clasping stereotypy, motor dysfunction, tremor, hypoactivity, anxiety, cognitive impairments, and altered sociability (Table 4) (Shahbazian et al., 2002; Moretti et al., 2005).
Engrailed-2 is a homeodomain transcription factor associated with ASD. Engrailed-2 is involved in a diverse range of biological processes from embryological development and segmental polarity to brain development and axon guidance (Figure 1) (Brunet et al., 2005; Joyner, 1996). The Engrailed-2 gene on human chromosome 7q36 is in the autism susceptibility locus, and an association between two intronic SNPs rs1861972 and rs1861973 at Engrailed-2 locus and ASD has been repeatedly identified in 518 ASD families (Gharani et al., 2004; Benayed et al., 2005) (Table 2). However, these SNPs were not found to be associated with ASD in Han Chinese trios (Wang et al., 2008). This association between Engrailed-2 and ASD was further confirmed by animal model studies, which showed Engrailed-2 null mice display social dysfunction and cognitive impairments (Table 6) (Brielmaier et al., 2012). Because Engrailed-2 is expressed upon activation of WNT2-Dvl1 signaling, it appears that the WNT2-Dvl1-Engrailed-2 pathway, which regulates neuronal migration and axonal guidance, may significantly contribute to ASD pathogenesis via neuroanatomical abnormalities. In addition, a base substitution (A218G) mutant of HOXA1, another homeobox gene, was reported in autistic individuals (Ingram et al., 2000b), indicating the importance of homeobox genes in normal brain function and ASD.
Excitatory and Inhibitory Imbalance
Mutations identified in important synaptic molecules including neuroligins (Jamain et al., 2003), neurexin (Autism Genome Project et al., 2007; Kim et al., 2008) and Shank (Durand et al., 2007; Berkel et al., 2010; Sato et al., 2012) in autistic subjects have prompted investigations into exploring the roles of synaptic dysfunctions in ASD pathogenesis. This “synaptopathy” model of autism has provided much insight into the field (Table 5).
Defects in synaptic proteins would lead to defective transmissions at excitatory and inhibitory synapses, disrupting the E-I balance in postsynaptic neurons, a key mechanism implicated in ASD. In line with this, ASD has been genetically associated with diverse glutamate receptors, including the kainite receptor subunit GluR6 (M867I in the intracytoplasmic C-terminal region of GluR6) (Jamain et al., 2002), the metabotropic glutamate receptor 8 (GRM8) (R859C, R1085Q, R1100Q, and intrachromosomal segmental duplication) (Serajee et al., 2003), and the N-methyl-D-aspartic acid receptor (NMDAR) subunit GluN2A (rs1014531) (Barnby et al., 2005), and GluN2B (de novo protein truncating and splice mutations) (O'Roak et al., 2012a,b) (Table 2). Decreased levels of glutamine and abnormal levels of glutamate were observed in the plasma of autistic children (Rolf et al., 1993; Moreno-Fuenmayor et al., 1996). In addition, neuropathological studies of postmortem autism brains show perturbations in the glutamate neurotransmitter system (Purcell et al., 2001).
Abnormal GABAergic system is also proposed as a potential mechanism for ASD. Reduced expression levels in a rate-limiting enzyme for GABA synthesis, glutamic acid decarboxylase (GAD), and GABA receptors with altered subunit composition were observed in autistic brains (Fatemi et al., 2002, 2010). Furthermore, linkage disequilibrium and transmission disequilibrium between GABRB3, a gene encoding the β3 subunit of GABAα receptors, with Angelman syndrome and autism has been reported (Cook et al., 1998; Bass et al., 2000; Buxbaum et al., 2002) (Table 2).
The serotonergic system would also play a role in ASD pathogenesis by modulating the E-I balance. Serotonin levels in blood or urine are increased in subjects with autism (Cook and Leventhal, 1996; Burgess et al., 2006), and various genes in the serotonin system are linked to autism. Among them are genes encoding the serotonin transporter 5-HTT (transmission disequilibrium at the 5-HTT locus in 86 autism trios) (Cook et al., 1997), and a rate-limiting enzyme for serotonin synthesis TPH2 (two intronic SNPs rs4341581 and rs11179000 at introns 1 and 4, respectively, have been associated with autism) (Coon et al., 2005) (Table 2).
Neurexins and neuroligins are synaptic cell adhesion molecules enriched at pre- and post-synaptic membranes, respectively (Figure 1) (Craig and Kang, 2007; Sudhof, 2008). Specific interactions between neurexins and neuroligins regulate various aspects of both excitatory and inhibitory synaptic development and function, affecting the E-I balance in postsynaptic neurons. Many mutations in genes encoding neurexins (including hemizygous CNV deletions and missense mutations) and neuroligins (e.g., R451C for NLGN3 and a frameshift insertion mutation for NLGN4) have been associated with ASD, intellectual disability, and schizophrenia (Jamain et al., 2003; Laumonnier et al., 2004; Autism Genome Project et al., 2007; Kim et al., 2008; Walsh et al., 2008) (Table 2). Neuroligin3 knockin mice with the R451C mutation found in autistic patients recapitulate autistic features including moderately impaired sociability (Table 5) (Tabuchi et al., 2007). Notably, inhibitory transmission was enhanced in the cortical regions of the mutant brains of these mutant mice, suggesting that disrupted E-I balance may contribute to ASD.
SHANK family genes encode scaffolding proteins enriched in the postsynaptic density (PSD), a postsynaptic membrane specialization composed of multi-synaptic protein complexes (Figure 1) (Sheng and Kim, 2000). The Shank family contains three known members, Shank1, Shank2 and Shank3, also known as ProSAP3, ProSAP1, and ProSAP2, respectively. The idea that Shanks are involved in the etiology of ASD firstly emerged from Phelan-McDermid syndrome (PMS) or 22q13 deletion syndrome, a neurodevelopmental disorder caused by a microdeletion on chromosome 22 (Boeckers et al., 2002; Wilson et al., 2003; Phelan and McDermid, 2012). The association between SHANK and ASD became evident by identifying numerous mutations including de novo frameshift, truncating, and missense mutations on SHANK3 locus in autistic individuals (Durand et al., 2007) (Table 2). Mutations in SHANK2 and SHANK1 including de novo CNV deletions and missense mutations in Canadian and European populations were also identified in individuals with ASD and intellectual disability (Berkel et al., 2010; Leblond et al., 2012; Sato et al., 2012).
Multiple lines of transgenic mice with Shank mutations found in human patients have been reported. Shank3 heterozygous mice show sociability deficits and reductions in miniature excitatory postsynaptic currents (mEPSC) amplitude and basal synaptic transmission (Bozdagi et al., 2010); mice with deletion of exon 4–9 of Shank3 are socially impaired and exhibit alterations in dendritic spine morphology and activity-dependent surface expression of AMPARs (Wang et al., 2011); Shank1−/− mice display reduced basal synaptic transmission in the hippocampal CA1 region and reduced motor function and anxiety-like behavior, although they show normal sociability (Hung et al., 2008; Silverman et al., 2011); mice expressing Shank2-R462X in hippocampal CA1 neurons exhibit cognitive dysfunction accompanied by reduced mEPSC amplitude and changes in neuronal morphologies (Table 5) (Berkel et al., 2012).
CNTNAP2, a neuronal transmembrane protein, is a member of the neurexin family localized at juxtaparanodes of myelinated axons. Here, CNTNAP2 regulates neuron-glia interactions and potassium channel clustering in myelinated axons (Figure 1) (Poliak et al., 1999). Several SNPs (e.g., rs2710102, rs7794745, rs17236239) and nonsynonymous variants (e.g., I867T) in CNTNAP2 locus were found to be associated with ASD, language impairment, and cortical dysplasia-focal epilepsy syndrome in humans (Alarcon et al., 2008; Arking et al., 2008; Bakkaloglu et al., 2008; Vernes et al., 2008) (Table 2). In a case-control association study in Spanish autistic patients and controls, however, CNTNAP2 SNPs rs2710102 and rs7794745 did not associate with ASD (Toma et al., 2013). Cntnap2−/− mice recapitulate all three core symptoms of autism, and display abnormal neuronal migration, reduced number of GABAergic interneurons, and abnormal neuronal synchronization (Table 4) (Penagarikano et al., 2011). Excessive grooming and hyperactivity in these mice were restored by the treatment of the antipsychotic risperidone (Table 4), suggesting the possibility of therapeutic intervention for certain symptoms of autism.
SynGAP is a GTPase-activating protein for the Ras small GTPase. SynGAP directly interacts with PSD-95, and negatively regulates the Ras-MAPK signaling pathway, excitatory synapse development, and synaptic transmission and plasticity (Figure 1) (Chen et al., 1998; Kim et al., 1998). In humans, de novo mutations of SYNGAP1 have been associated with intellectual disability and autism (Hamdan et al., 2011). In addition, a genetic case/control study in European populations associates a rare de novo copy number variation in SYNGAP1 with ASD (Pinto et al., 2010). Syngap1 heterozygous mice show schizophrenia-like phenotypes including hyperactivity, impaired sensory-motor gating, impaired social memory and fear conditioning, and preference to social isolation (Guo et al., 2009) (Table 5). In a more recent study, Syngap1 heterozygous mice showed premature dendritic spine development together with enhanced hippocampal excitability and abnormal behaviors, suggesting that over-paced excitatory synaptic development during a critical time window of postnatal brain development causes intellectual disability and ASD (Clement et al., 2012).
Several genes associated with X chromosome-linked intellectual disability (XLID) and synaptic regulations have been associated with ASD. One of them is interleukin 1 receptor accessory protein-like 1 (IL1RAPL1) that encodes a synaptic transmembrane protein (Carrie et al., 1999). Recently, a systematic sequencing screen of X chromosomes of ASD-affected individuals has identified a de novo frameshift mutation in IL1RAPL1 (Piton et al., 2008). IL1RAPL1 plays an important role in the formation and stabilization of excitatory synapses by recruiting the scaffolding protein PSD-95 to excitatory postsynaptic sites through the JNK signaling pathway (Figure 1) (Pavlowsky et al., 2010). In addition, IL1RAPL1 induces the presynaptic differentiation through its trans-synaptic interaction with protein tyrosine phosphatase δ (PTPδ) (Figure 1) (Valnegri et al., 2011b; Yoshida et al., 2011). This interaction between IL1RAPL1 and PTPδ recruits RhoGAP2 to the excitatory synapses and induces dendritic spine formation (Valnegri et al., 2011b). Interestingly, IL1RAPL1 regulates the development of inhibitory circuits in the cerebellum, an ASD-related brain region, and disrupts the excitatory and inhibitory balance, as determined by a study using Il1rapl1−/− mice (Gambino et al., 2009). These results suggest that IL1RAPL1 is involved in the regulation of excitatory synaptic development and the balance between excitatory and inhibitory synaptic inputs.
Another XLID gene related with ASD is OLIGOPHRENIN-1 (OPHN1), which encodes a GTPase-activating protein that inhibits Rac, Cdc42, and RhoA small GTPases. Since the initial report of the association of a truncation mutation of OPHN1 with XLID (Billuart et al., 1998a,b), additional studies have associated nonsynonymous rare missense variants in OPHN1 with ASD (e.g., H705R) and schizophrenia (e.g., M461V) (Piton et al., 2011). OPHN1 regulates dendritic spine morphogenesis through the RhoA signaling pathway (Govek et al., 2004) and activity-dependent synaptic stabilization of AMPA receptors (Nadif Kasri et al., 2009). OPHN1 also interacts with the transcription repressor Rev-erba to regulate expression of circadian oscillators (Valnegri et al., 2011a). Importantly, Ophn1−/− mice show immature spine morphology, impaired spatial memory and social behavior, and hyperactivity (Khelfaoui et al., 2007). These results suggest that OPHN1 regulates excitatory synaptic development and function.
TM4SF2 or tetraspanin 7 (TSPAN7), another X-linked gene which encodes a membrane protein which belongs to transmembrane 4 superfamily (TM4SF), plays important roles in the cell proliferation, activation, growth, adhesion, and migration (Maecker et al., 1997). TM4SF proteins form a complex with integrin, which regulates cell motility and migration by modulating the actin cytoskeleton (Berditchevski and Odintsova, 1999). A balanced translocation and mutations (a nonsense mutation and a P172H missense mutation) of TM4SF2 was firstly discovered in the individuals with XLID (Zemni et al., 2000). In subsequent studies, the P172H missense mutation was found in individuals with XLID (Maranduba et al., 2004) and ASD (Piton et al., 2011). A microduplication in the locus of TM4SF2 was revealed, but this duplication was also present in unaffected controls, suggesting that it may be a neutral polymorphism (Cai et al., 2008). In neurons, TM4SF2 regulates excitatory synaptic development and AMPA receptor trafficking by binding to the synaptic PDZ protein PICK1 (Figure 1) (Bassani et al., 2012).
Synaptic Signaling
Disrupted synaptic signaling may be a key determinant of ASD. Components in mGluR- or NMDAR-dependent signaling cascades have recently been implicated in ASD.
Neurofibromin 1 (NF1), tuberous sclerosis complex (TSC1/TSC2), and phosphatase and tensin homolog (PTEN) are genes associated with neurological diseases with common autistic symptoms including neurofibromatosis (Rasmussen and Friedman, 2000), tuberous sclerosis (van Slegtenhorst et al., 1997), and Cowden/Lhermitte-Duclos syndrome (Pilarski and Eng, 2004). They are tumor suppressors sharing a common function; they negatively regulate the mammalian target of rapamycin (mTOR) signaling pathway. Although Tsc1 null mice are embryonically lethal (Wilson et al., 2005), mutant mice with loss of Tsc1 in cerebellar Purkinje cells display autistic-like behaviors (Tsai et al., 2012), and Tsc2 heterozygote mice exhibit abnormal social communication (Young et al., 2010); Nf1 mutant mice show aberrant social transmission of food preference and deficits in hippocampus-dependent learning (Costa et al., 2001, 2002); Pten deficient mice show altered social interaction and macrocephaly with hyperactivation of mTOR pathway (Table 4) (Kwon et al., 2006).
Signaling molecules in the downstream of mTOR in the mTOR pathway play crucial roles in ASD pathogenesis. Upon phosphorylation by mTORC1, 4E-BP proteins are detached from eIF4E to promote eIF4E-dependent protein translation (Figure 1) (Richter and Sonenberg, 2005). A SNP at eIF4E promoter region which increases its promotor activity was found in autism patients (Neves-Pereira et al., 2009). Implications of mTOR downstream signaling in ASD were demonstrated as 4E-BP2 knockout mice and eIF4E overexpression mice display autistic-like behaviors. 4E-BP2 knockout mice show enhanced translational control of neuroligins and increased excitatory transmission in the hippocampus (Table 6) (Gkogkas et al., 2013), while eIF4E overexpressing transgenic mice show impaired excitatory/inhibitory balance in the mPFC and increased LTD in the hippocampus and striatum (Table 6) (Santini et al., 2013). Autistic features of these mutant mice were ameliorated by 4EGI-1 infusion, which inhibits the eIF4E–eIF4G interaction.
Fragile X syndrome is the most common cause of intellectual disability and autism. It is mostly caused by the expansion of CGG trinucleotide repeats in the promoter region of the FMR1 gene, which enhances the methylation of the promoter and represses generation of FMR1-encoded protein (FMRP), which binds to target mRNAs and regulates their translation and transport of mRNA into dendrites and synapses (Figure 1) (Bassell and Warren, 2008). In the absence of FMRP, target mRNA translation becomes excessive and uncontrolled, leading to an aberrant activity-dependent protein synthesis. Fmr1 mutant mice show enhanced protein synthesis-dependent mGluR-mediated LTD and dendritic spine elongation, together with cognitive deficits, social anxiety and impaired social interaction (Table 4) (Bernardet and Crusio, 2006). Interestingly, target molecules of FMRP include Shank3, GluN2A, mTOR, TSC2, NF1, neuroligin2, and neurexin1 (Darnell et al., 2011), which are associated with ASD pathogenesis.
It should be noted that the ASD-related signaling molecules mentioned above are also associated with NMDAR and mGluR signaling pathways. NMDARs and mGluRs play critical roles in the regulation of synaptic function and plasticity at excitatory synapses. NF1 interacts with the NMDAR complex and regulates GluN2A phosphorylation (Figure 1) (Husi et al., 2000). FMRP and TSC have profound effects on mGluR-dependent LTD and protein synthesis, which are upregulated in Fmr1−/y mice, while downregulated in Tsc2+/− mice (Auerbach et al., 2011). FMRP is also in the downstream of mGluR signaling (Figure 1) (Bassell and Warren, 2008).
Defects in NMDAR function and associated signaling are also observed in nonsyndromic ASD models with Shank mutations. Shank proteins are physically connected to both NMDARs and mGluRs, suggesting that Shank may regulate signaling pathways downstream of NMDAR or mGluR activation, and the functional interaction between the two receptors (Figure 1). Shank2−/− mice with the deletion of exons 6 and 7 display autistic-like behaviors and reductions in NMDAR function and associated signaling, without affecting mGluR-dependent LTD (Won et al., 2012), while Shank2−/− mice with exon 7 deletion show similar behavioral abnormalities with NMDAR hyperfunction (Table 5) (Schmeisser et al., 2012). Although how similar exon deletions in Shank2 lead to comparable behavioral abnormalities but different changes in NMDAR function remains to be determined, these results point to that Shank2 is an important regulator of NMDAR function, and that NMDAR function and NMDAR-associated signaling are associated with ASD.
Neuroimmune Response
The implication of the immune system on autism was initially proposed in 1976 based on that some autistic children do not have detectable Rubella titers in spite of previous vaccination (Stubbs, 1976). Levels of serum IgG and autoantibodies to neuronal and glial molecules were elevated in autistic patients (Singh et al., 1997; Croonenberghs et al., 2002), proposing involvement of autoimmune responses in autism. In addition, plasma or cerebrospinal fluid (CSF) levels of pro-inflammatory cytokines and chemokines including MCP-1, IL-6, IL-12, IFN-γ and TGFβ1 were increased in autistic individuals (Ashwood and Van de Water, 2004; Ashwood et al., 2006).
Astrocytes and microglia are two glial cell types important for immune responses in the brain as well as regulation of neuronal functions and homeostasis (Fields and Stevens-Graham, 2002). Postmortem analyses demonstrated abnormal glial activation and neuroinflammatory responses in autistic brains (Vargas et al., 2005). Transcriptome analysis of autistic postmortem brain tissues has also revealed upregulation in the expression of genes belonging to immune and inflammatory networks (Voineagu et al., 2011). Reactive astrocytes were also detected in Cntnap2−/− brains, a well-established autism model (Penagarikano et al., 2011). These results clearly suggest the association between neuroimmune defects with ASD, although further details remain to be determined.
Non-Genetic Models of ASD
Although we have thus far described genetic factors underlying ASD, environmental factors also have strong influences on ASD. Epidemiologic studies suggest that maternal exposure to stress, viral or bacterial infection, thalidomide, and valproic acid can increase the risk for ASD in offspring (Grabrucker, 2012).
Maternal immune activation (MIA) induced by poly(I:C), the synthetic doublestrand RNA polyriboinosinic-polyribocytidilic acid, in pregnant mice leads to the development of core ASD-like phenotypes in the offspring, including impaired sociability, decreased USV, and increased repetitive behaviors (Malkova et al., 2012). MIA by lipopolysaccharide (LPS) treatment during pregnancy can also induces ASD-like phenotypes in rodent offspring, including impaired social interaction (Hava et al., 2006; Kirsten et al., 2010) and reduced USV (Baharnoori et al., 2012). IL-6 is thought to play a critical role in this process, as IL-6 knockout mice do not show poly(I:C) induced social deficits (Smith et al., 2007), and IL-6 levels are significantly elevated in the cerebellum of autistic subjects (Wei et al., 2011). Although further details remain to be determined, the underlying mechanisms may include IL6-dependent regulation of excitatory and inhibitory synaptic transmission and neuroprotection (Sallmann et al., 2000; Biber et al., 2008; Dugan et al., 2009).
Prenatal exposure to teratogens can increase the risk for ASD in animals, as in humans. Thalidomide (THAL) and valproic acid (VPA) cause rat offspring to display brain morphological abnormalities observed in ASD, including altered cerebellar structures and reduced number of cranial motor neurons (Rodier et al., 1997; Ingram et al., 2000a). Behaviorally, VPA-exposed rats show decreases in prepulse inhibition, stereotypy, and social behaviors (Schneider and Przewlocki, 2005). VPA-exposed rats display elevated serotonin levels and abnormal serotonergic neurons (Anderson et al., 1990; Narita et al., 2002; Miyazaki et al., 2005), decreased parvalbumin-positive interneurons in the neocortex (Gogolla et al., 2009), and elevated NMDA receptor levels and enhanced LTP (Rinaldi et al., 2007), suggesting that these mechanisms may contribute to the development of ASD-like phenotypes.
Potential Treatments for ASD
Currently, only two medicines have been approved for ASD by US FDA; risperidone (Risperdal®) and aripiprazole (Abilify®), which act as dopamine/5-HT receptor antagonists (McPheeters et al., 2011). These drugs are useful for correcting irritability and stereotypy, but not sociability defects. Recently, a number of candidate ASD medications for treating social abnormalities have been suggested (Figure 1).
mGluR Positive Allosteric Modulators
mGluR1 and mGluR5 are group I mGluRs that are postsynaptically expressed in broad brain regions, including the cerebral cortex, striatum, hippocampus, nucleus accumbens, and inferior colliculus (Testa et al., 1995). Upon activation, group I mGluRs enhance calcium release from intracellular stores resulting in neuronal depolarization, augmentation of neuronal excitability, and activation of intracellular signaling cascades such as PKA, PKC, MAPK, ERK, and CREB (Niswender and Conn, 2010). mGluR5 is physically linked to NMDARs via Homer-Shank/ProSAP-GKAP/SAPAP-PSD-95 interactions (Naisbitt et al., 1999; Tu et al., 1999), and is functionally coupled to NMDARs via aforementioned signaling molecules including PKC (Niswender and Conn, 2010). Through these structural and biochemical interactions, mGluR5 activation is thought to potentiate NMDAR function (Awad et al., 2000; Attucci et al., 2001; Mannaioni et al., 2001; Pisani et al., 2001; Alagarsamy et al., 2002; Rosenbrock et al., 2010).
Positive allosteric modulators of mGluR5 receptors were first developed to alleviate symptoms of schizophrenia (Gregory et al., 2011). Although antipsychotics are available for positive symptoms of schizophrenia, such as hallucinations, no medications are currently available for negative symptoms or cognitive impairments. Two main hypotheses have been proposed for schizophrenia: dopaminergic hyperactivity and NMDA hypofunction. Dopaminergic hyperactivity can be treated by dopamine receptor-antagonistic antipsychotics such as risperidone, but NMDA hypofunction is difficult to modulate given the expected side effects of enhancing NMDAR functions.
Therefore, the concept of augmenting NMDAR signaling via mGluR potentiation was proposed to improve negative symptoms of schizophrenia (Uslaner et al., 2009; Stefani and Moghaddam, 2010). mGluR positive allosteric modulators increase the function of NMDAR only when they are occupied by the endogenous ligand glutamate (Figure 1). mGluR positive allosteric modulators have significant advantages over the conventional mGluR agonist, (RS)-3,5-dihydroxyphenylglycine (DHPG). While DHPG has poor specificity toward particular mGluR subtypes, mGluR positive allosteric modulators offer high subtype specificity. Some positive allosteric modulators have high brain blood barrier penetrance, which enables the systemic administration of the drugs. Furthermore, whereas direct mGluR agonists cause rapid receptor desensitization, mGluR positive allosteric modulators potentiate mGluR function with minimal desensitization, because they bind to an allosteric site on the receptor distinct from the orthosteric glutamate binding site. These properties of positive allosteric modulators are predicted to minimize their excitotoxicity and enable high-dose administrations.
A large number of mGluR5 allosteric modulators have been developed (Williams and Lindsley, 2005; Gregory et al., 2011). Of these, CDPPB, ADX47273, MPPA, and VU0092273 readily cross the blood-brain barrier, and CDPPB, particular, has been examined in various behavioral assays and model animals. In CHO (Chinese hamster ovary) cells expressing human mGluR5, CDPPB treatment was shown to enhance mGluR5 activity in a concentration-dependent manner (Kinney et al., 2005). Behaviorally, CDPPB alleviates prepulse inhibition and hyperactivity produced by amphetamine, suggesting that CDPPB could be a potential antipsychotic agent.
Because NMDARs play an essential role in learning and memory, indirect potentiation of NMDARs by mGluR5 positive allosteric modulators may facilitate synaptic plasticity and learning and memory. Indeed, CDPPB and ADX47273 enhance the performance of wild-type mice in the Morris water maze test, a hippocampus-dependent learning and memory paradigm (Ayala et al., 2009). In addition, VU-29 and ADX47273 potentiate two forms of NMDAR-dependent synaptic plasticity—LTP and LTD—in the CA1 region of the hippocampus (Ayala et al., 2009). DFB-treated rats make fewer errors in the Y-maze spatial alternation task (Balschun et al., 2006), and CDPPB and ADX47273 enhance performance in novel object recognition and five-choice serial reaction time tasks (Liu et al., 2008; Uslaner et al., 2009). CDPPB not only improves learning and memory performance of wild-type mice, but also reverses cognitive dysfunction and behavioral inflexibility induced by the NMDAR antagonist MK-801 (Uslaner et al., 2009; Stefani and Moghaddam, 2010). These results suggest that mGluR5 positive allosteric modulators have the potential to improve cognitive impairments associated with brain disorders including schizophrenia and autism.
Indeed, CDPPB has recently shown promise as a potential treatment for ASD. In a study using Tsc2+/− mice, a mouse model of tuberous sclerosis characterized by intellectual disability and autism, Mark Bear and colleagues showed that cognitive impairments observed in these mice could be alleviated by CDPPB administration (Table 4) (Auerbach et al., 2011). In addition, social deficits of Shank2−/− mice are rescued by CDPPB treatment (Won et al., 2012), implicating hypofunction of mGluRs and NMDARs in social impairment, and suggesting mGluR positive allosteric modulators as novel therapeutics for the treatment of social deficits (Table 5). More recently, CDPPB was shown to reverse defects in social novelty recognition induced by neonatal phencyclidine treatment (Clifton et al., 2012).
D-Cycloserine
Although it is well established that NMDARs critically regulate normal brain functions, the excitotoxicity and poor bioavailability of direct NMDAR agonists have hampered attempts to control brain activity by modulating NMDARs (Quartermain et al., 1994). D-cycloserine is a high-affinity partial agonist of NMDA-coupled, strychnine-insensitive glycine receptors (Figure 1) (Hood et al., 1989). Similar to glycine, D-cycloserine also binds to the glycine site of NMDARs as a partial agonist, potentiating NMDARs by increasing the frequency of channel opening. In addition, because NMDARs are not maximally potentiated by endogenous glycine, there is room for D-cycloserine to further potentiate NMDARs. These properties enable D-cycloserine to act as a positive modulator of NMDARs.
D-cycloserine is a viable drug candidate because it is a partial agonist, displaying efficacy of 40-50% relative to glycine, and has low toxicity and decent bioavailability (Hood et al., 1989). Although the brain penetrance of D-cycloserine is not high, it can nonetheless infiltrate the blood-brain barrier, exerting a peak effect 1 h after intraperitoneal administration (Peterson, 1992). D-cycloserine shows dose-dependent elimination (higher elimination rates with lower doses) and a half-life of 7–15 h in humans and 23 min in mice (lwainsky, 1988; Wlaz et al., 1994).
When glycine levels are low, D-cycloserine amplifies the activity of the NMDAR complex and enhances synaptic plasticity and cognitive function. D-cycloserine alleviates senescence-associated behavioral defects (Flood et al., 1992) and facilitates memory acquisition, consolidation, and retrieval (Quartermain et al., 1994). While low doses (10–20 mg kg−1) of D-cycloserine have cognition-enhancing effects (Monahan et al., 1989; Flood et al., 1992; Schuster and Schmidt, 1992; Sirvio et al., 1992; Quartermain et al., 1994), higher doses (>100 mg kg−1) exert anticonvulsant effects in tonic convulsion models (Peterson, 1992; Peterson and Schwade, 1993).
Putative effects of D-cycloserine on ASD have been suggested by previous studies. Mice with a neuroligin1 (Nlgn1) deficiency exhibit abnormally increased grooming behavior, and this behavioral anomaly is reversed by D-cycloserine treatment (Table 5) (Blundell et al., 2010). Low-dose D-cycloserine alleviates negative symptoms of schizophrenia-affected individuals (Goff et al., 1999), and reduces social withdrawal and increases social responsiveness in autistic patients (Posey et al., 2004). Moreover, D-cycloserine partially rescues social deficits of Shank2−/− mice, supporting the role of NMDAR functionality in autism (Table 5) (Won et al., 2012).
Benzodiazepines
Recently, benzodiazepines were suggested as putative therapeutic agents for Dravet's syndrome, which is a developmental disorder with myoclonic infantile seizure, ADHD-like inattention and hyperactivity, motor impairment, sleep disorder, anxiety-like behaviors, cognitive defects, autism-like social dysfunction, and restricted interests. Mice heterozygous for a deletion of the α-subunit of the type 1 voltage-gated sodium channel (Scn1α+/− mice), an animal model for Dravet's syndrome, recapitulate most features of the disorder, including epilepsy, ataxia, sleep disorder, anxiety-like behaviors, hippocampus-dependent learning impairments, sociability deficits, and excessive repetitive grooming behaviors (Table 4) (Yu et al., 2006; Kalume et al., 2007; Han et al., 2012). In Scn1α+/− mouse brains, expression of the voltage-gated sodium channel type-1 (Nav1.1) is decreased in GABAergic interneurons, and GABAergic transmission onto postsynaptic neurons was reduced. This would cause a shift in the balance between excitation and inhibition in postsynaptic neurons toward excitation, which may be corrected by stimulating GABA receptors in these neurons. Indeed, it was shown that both behavioral abnormalities and aberrant GABAergic transmission are rescued by low-dose administration of clonazepam (Table 4) (Han et al., 2012).
Clonazepam, a type of benzodiazepine, is a positive allosteric modulator of GABAA receptors that exerts sedative, hypnotic, anxiolytic, anticonvulsant, and muscle relaxing effects (Figure 1) (Rudolph and Knoflach, 2011). Similar to the action of mGluR positive allosteric modulators, clonazepam potentiates GABA signaling only when GABAA receptors are bound by their endogenous ligand, GABA. Therefore, these results indicate that normalization of disrupted E-I balance may be a novel and promising strategy for treating symptoms of ASD.
mGluR Negative Allosteric Modulators
The potential of mGluR of negative allosteric modulation as a therapeutic strategy in ASD was first proposed based on studies in Fmr1−/y mice, an animal model for fragile X syndrome (Bakker et al., 1994). The enhanced mGluR5-dependent LTD and protein synthesis observed in Fmr1−/y mice provided a conceptual framework for the mGluR theory of fragile X pathogenesis (Bear et al., 2004; Bassell and Warren, 2008). Synaptic protein synthesis is stimulated by local mRNA translation, a process that depends on group I mGluR activation. FMRP, encoded by the Fmr1 gene, is a repressor of mRNA translation; thus, mGluR-mediated protein synthesis could be enhanced in the absence of FMRP. Therefore, attempts have been made to correct fragile X syndrome by suppressing abnormally enhanced mGluR5-dependent synaptic plasticity and protein synthesis.
Two approaches have been used to normalize behavioral and neuronal deficits of Fmr1−/y mice: genetic crossbreeding with Tsc2+/− mice, which exhibit suppressed mGluR activity, and acute administration of an mGluR antagonist (Auerbach et al., 2011). Administering the mGluR5 antagonist 2-methyl-6-(phenylethynyl) pyridine hydrochloride (MPEP) to Fmr1−/y mice normalizes defective phenotypes, including cognitive deficits, perturbed mGluR-dependent LTD and protein synthesis, and excessive filopodia-like long and thin spines (Figure 1, Table 4) (Yan et al., 2005; de Vrij et al., 2008). In line with this, mGluR negative allosteric modulators are now in clinical trials for fragile X syndrome patients (Krueger and Bear, 2011).
The therapeutic potential of mGluR antagonists in ASD has also been suggested. Repetitive grooming behaviors in BTBR and valproic acid (VPA) mouse models of autism are significantly alleviated by MPEP treatment (Silverman et al., 2010a; Mehta et al., 2011). Impairments in social interaction of BTBR mice are also ameliorated by MPEP administration (Silverman et al., 2010a). GRN-529, a selective negative allosteric modulator of mGluR5 developed by Pfizer, was shown to fully rescue excessive repetitive grooming behavior and social dysfunction in BTBR mice and jumping stereotypy in C58/J mice (Silverman et al., 2012). These findings suggest that mGluR negative allosteric modulators have novel therapeutic potential in autism, in addition to fragile X syndrome.
NMDAR Antagonists
NMDAR antagonists including amantadine and its close analogue memantine are now in clinical trials for autistic patients (Nightingale, 2012; Spooren et al., 2012). Amantadine and memantine are non-competitive antagonists for NMDARs with multiple clinical uses (Chen et al., 1992; Blanpied et al., 2005). Memantine is currently being used for Alzheimer's disease, while it is also useful for viral infection and Parkinson's disease. Because both drugs are weak NMDAR antagonists with moderate affinity, prolonged receptor blockade during treatment is unlikely to cause significant side effects.
In a double-blind, placebo controlled study, amantadine-treated group show significant improvements in hyperactivity and inappropriate speech (King et al., 2001). Memantine is also effective in improving language and social behavior and clinical global impressions (CGI) scale in autistic patients (Chez et al., 2007; Erickson et al., 2007; Niederhofer, 2007).
With regard to mechanisms of memantine and amantadine underlying the treatment of ASD remains, both medications are highly likely to exert their therapeutic effects by suppressing NMDAR function and modulating excitotoxicity in autistic subjects. However, care should be taken because other possibilities exist. For instance, memantine treatment promotes excitatory synapse formation and maturation and cell adhesion properties of cerebellar granule cells (CGCs) of Fmr1 knockout mice (Wei et al., 2012). In addition, memantine exerts neuroprotective activities by promoting glia-derived neurotrophic factor (GDNF) release and preventing migroglial inflammatory responses (Wu et al., 2009). Memantine can also act as a non-competitive antagonist for 5-HT receptors (Rammes et al., 2001) and nicotinic acetylcholine receptors (Aracava et al., 2005), while it functions as an agonist for D2 dopamine receptors (Seeman et al., 2008).
IGF-1
A new approach for alleviating phenotypic traits of ASD in animal models has come from research on Rett syndrome. Rett syndrome is an X-linked neurological disorder caused by mutations in the MeCP2 gene. MeCP2 is a transcriptional repressor and activator, which binds widely across the genome and influence a large number of genes (Chahrour et al., 2008). One of the best characterized targets of MeCP2 is BDNF, a neurotrophic factor that regulates neuronal development and synaptic plasticity (Figure 1) (Greenberg et al., 2009). Bdnf conditional knockout mice show features analogous to Rett syndrome, including smaller brain size and hindlimb-clasping behavior (Chang et al., 2006). Mice with double knockout of Bdnf and MeCP2 show earlier onset of Rett-like symptoms, whereas overexpression of Bdnf in MeCP2 knockout mice delays the onset and relieves the electrophysiological defects of MeCP2 mutants. Moreover, restoring Bdnf expression through ampakine administration alleviates respiratory problems of MeCP2 mutant mice (Ogier et al., 2007). Although BDNF appears to have significant effects in Rett syndrome model animals, it poorly penetrates the blood-brain barrier, limiting its therapeutic application.
Another growth factor associated with Rett syndrome is insulin-like growth factor 1 (IGF-1) (Figure 1). IGF-1 is a polypeptide hormone with structural similarity to insulin. While it has a profound effect on overall cell growth, it also plays an important role in regulating neuronal functions by promoting axonal outgrowth (Ozdinler and Macklis, 2006), neuro- and synaptogenesis (O'Kusky et al., 2000), and activity-dependent cortical plasticity (Tropea et al., 2006). IGF-1 binds to IGF-binding proteins (IGFBP1–6), resulting in extension of the half-life of IGF-1 (Hwa et al., 1999). Upon binding to its cognate receptor, IGF-1 activates Ras-MAPK and PI3K-Akt pathways (Fernandez and Torres-Aleman, 2012), signaling cascades that are also activated by BDNF.
Because IGF-1 crosses the blood-brain barrier, it may be a viable alternative to BDNF as a therapeutic agent for Rett syndrome. Indeed, IGF-1 and IGFBP have been implicated in Rett syndrome and autism: IGFBP3 levels are abnormally elevated in MeCP2 mutant mice and Rett syndrome patients (Itoh et al., 2007), and the concentration of IGF-1 in CSF is reduced in autistic individuals (Riikonen et al., 2006). The therapeutic utility of IGF-1 in Rett syndrome was originally suggested by Mriganka Sur and coworkers, who reported that lethality, hypoactivity, and respiratory problems of MeCP2-null mice are partially rescued by IGF-1 treatment in association with normalization of impaired spine density, synaptic transmission, and cortical plasticity (Table 4) (Tropea et al., 2009). IGF-1 also reverses the reduction in excitatory synapse number and density of neurons derived from Rett patients (Marchetto et al., 2010).
Rapamycin
Rapamycin is an immunosuppressant originally identified as an antifungal agent in isolates from Streptomyces hygroscopicus (Sehgal et al., 1975; Vezina et al., 1975; Baker et al., 1978; Singh et al., 1979). Rapamycin strongly binds to FK506-binding protein (FKBP); this complex then binds and inhibits mTOR, a serine/threonine kinase implicated in transcription, cytoskeleton dynamics, ubiquitin-dependent protein degradation, autophagy, and membrane trafficking (Figure 1) (Dennis et al., 1999). mTOR signaling has profound effects on neuronal cells in addition to cancer cells (Busaidy et al., 2012), immune cells (Araki et al., 2011), and cells that regulate lifespan (Powers et al., 2006; Harrison et al., 2009). In the nervous system, mTOR regulates axon guidance, dendrite arborization, synaptogenesis, and synaptic plasticity (Troca-Marin et al., 2012).
Perturbations in mTOR signaling have significant impacts on normal brain functions. Patients with Alzheimer's disease and Drosophila tauopathy models show enhanced mTOR signaling in the brain (Li et al., 2005; Khurana et al., 2006). Hyperactivation of the Akt-mTOR pathway is observed in hippocampal neurons of Ts1Cje mice, which models Down syndrome (Troca-Marin et al., 2011). Animal models and patients of Parkinson's disease exhibit enhanced levels of REDD1, which inhibits mTOR activity (Malagelada et al., 2006). mTOR is observed in inclusion bodies from Huntington's disease patients and corresponding mouse models (Ravikumar et al., 2004). Importantly, rapamycin treatment alleviates several pathogenic traits observed in in vivo and in vitro models of Alzheimer's disease (Khurana et al., 2006; Harrison et al., 2009), Parkinson's disease (Pan et al., 2009; Tain et al., 2009), and polyglutamine diseases (Ravikumar et al., 2004; Berger et al., 2006; Pandey et al., 2007).
The therapeutic utility of rapamycin in ASD was suggested in 2008 based on studies in Tsc2+/− mice (Ehninger et al., 2008). The mTOR pathway is associated with TSC because TSC1 and TSC2 are upstream inhibitory regulators of mTOR activity (Han and Sahin, 2011). In this study, the learning and memory deficits, lethality, aberrant brain overgrowth, and altered synaptic plasticity of Tsc2+/− mice were ameliorated by acute treatment with rapamycin (Table 4). The social dysfunction and behavioral inflexibility of Purkinje cell-specific Tsc1 mutant mice were also improved by rapamycin (Tsai et al., 2012), further suggesting that rapamycin may be useful in reversing core symptoms of autism.
Oxytocin
Oxytocin is a nine amino acid neuropeptide hormone synthesized by magnocellular neurons in paraventricular and supraoptic nuclei of the hypothalamus and secreted from the posterior pituitary gland into the circulation (Figure 1). Oxytocin acts through oxytocin receptors (OXTRs), which are abundantly expressed in the amygdala, hippocampus, and hypothalamus (Gould and Zingg, 2003). Oxytocin is associated with various social behaviors including affiliation, maternity, aggression, and pair bonding (Lee et al., 2009; Caldwell, 2012; Feldman, 2012). Given the prominence of oxytocin in the regulation of social behavior, the association of oxytocin with autism pathogenesis has been extensively examined.
Several SNPs of OXTRs are associated with ASD (Wu et al., 2005; Jacob et al., 2007; Yrigollen et al., 2008; Liu et al., 2010). Oxtr knockout mice display autistic-like behaviors; they emit fewer USVs upon social isolation, show defects in social recognition and discrimination, and are less aggressive (Table 6) (Takayanagi et al., 2005; Crawley et al., 2007). Supporting the pharmacotherapeutic potential of oxytocin, nasal administration of oxytocin improves social interactions and communications (Andari et al., 2010; Kosaka et al., 2012), reduces repetitive behaviors (Hollander et al., 2003), and enhances social cognition (Hollander et al., 2007) in autism-affected individuals.
Perspectives
Homeostatic Mechanisms Underlying ASD
Tuberous sclerosis and fragile X syndrome are disorders with common symptoms including intellectual disabilities, seizures, and autism. While their genetic determinants are different (TSC1/TSC2 for tuberous sclerosis and FMR1 for fragile X syndrome), their gene products both regulate protein synthesis in neurons (Bassell and Warren, 2008; Ehninger et al., 2009). Interestingly, animal models of tuberous sclerosis (Tsc2+/− mice) and fragile X syndrome (Fmr1−/y mice) display abnormal protein synthesis in opposite directions (Auerbach et al., 2011).
Tsc2+/− mice exhibit diminished mGluR-dependent LTD and protein synthesis in the hippocampus, whereas Fmr1−/y mice show excessive mGluR-dependent LTD and protein synthesis. Consistent with this, cognitive impairments of the two animal models are corrected by drugs that modulate mGluR5 in the opposite manner (CDPPB for Tsc2+/− mice and MPEP for Fmr1−/y mice). In addition, crossbreeding of these two mouse lines rescues behavioral impairments and synaptic dysfunctions. These results strongly suggest that mGluR5-mediated synaptic plasticity and protein synthesis in the normal range is important and that deviation in either direction from a normal range can cause brain dysfunctions that yield similar behavioral manifestations.
Another such example comes from two mouse models with different mutations in the same gene. Shank2−/− mouse lines lacking exons 6 and 7 (Won et al., 2012) or exon 7 only (Schmeisser et al., 2012), both of which mimic mutations found in humans (Berkel et al., 2010), display similar autistic-like behaviors, but NMDAR function in their brains shows opposite changes: NMDAR hypofunction with exons 6 and 7 deletion and NMDAR hyperfunction with exon 7 deletion. Although further details remain to be explored, this is another example suggesting that NMDAR function in a normal range is important, and that deviations in either directions can lead to similar behavioral abnormalities. Therefore, individuals with mutations in the same gene may have to be carefully diagnosed, for example by high-through sequencing, in order to receive proper treatment.
Core Mechanisms Underlying ASD
Given the diverse genetic variations underlying the development of ASD, one obvious challenge in understanding how ASD develops is the wide range of mechanisms associated with it. This diversity poses a serious additional problem in treating ASD: a single medication is likely to cover only a small fraction of individuals with ASD, or a limited spectrum of ASD symptoms.
A related well-known example is the selective effect of risperidone. Risperidone, a dopamine antagonist, is an antipsychotic mainly used to treat schizophrenia and bipolar disorder, and it is currently one of the few FDA-approved medications for autism. The drug mainly ameliorates irritability, hyperactivity, and repetitive and restricted behaviors, but is largely ineffective against social withdrawal and language deficits of autistic individuals (McPheeters et al., 2011). Similarly, risperidone rescues repetitive grooming and hyperactivity, but not social deficits, in Cntnap2−/− mice (Penagarikano et al., 2011). Another example is the demonstration that CDPPB rescues only social interaction in Shank2-deficient mice but fails to rescue impaired pup retrieval, repetitive jumping, hyperactivity, and anxiety-like behavior (Won et al., 2012). The fact that some medications reverse only selective symptoms/phenotypes of ASD, however, may provide an opportunity to further explore detailed mechanisms underlying particular aspects of ASD etiology. This would, in principle, allow us to dissect and study synaptic or circuit mechanisms that are specifically associated with certain aspects of ASD, such as impaired social interaction, impaired social communication, repetitive behavior, restricted interests, intellectual disability, anxiety, and hyperactivity.
A possible solution to the apparent diversity of ASD-related mechanisms is to identify “core” mechanisms that cover a large fraction of genetic variations, or a broader spectrum of ASD symptoms. The concept of core mechanisms is based on the assumption that a fraction of ASD-related proteins may act together and converge on a common pathway. A possible core mechanism could be excitatory synaptic transmission. Excitatory synaptic development can be regulated by a number of factors including synaptic adhesion molecules, synaptic scaffolding proteins, and actin-regulatory proteins. In addition, excitatory synaptic transmission, which is mainly mediated by AMPAR receptors, can be determined by the regulators of the synaptic trafficking and stabilization of AMPA receptors, and regulated by the signaling pathways in the downstream of NMDA receptors, mGluRs, and monoamine receptors. Another core mechanism could be the E-I balance, which is determined by the relative amounts of excitatory and inhibitory synaptic transmissions, and, together with the excitability of postsynaptic neurons, determines firing patterns of postsynaptic neurons and, subsequently, network activities across the brain. Establishing these core mechanisms, if any, would require rigorous and time-consuming verifications using a range of approaches, including mouse genetics, electrophysiology, and behavior.
Integrating Three Aspects of ASD Research: Human Genetics, Mouse Models, and Potential Treatments
An important starting point for ASD research using mouse models would be to select best possible genetic variations that can provide us decent insights into the underlying mechanisms and potential treatments. Luckily, a large number of ASD-related papers are being published each year (i.e., ~2500 papers in 2012 when “autism” was used as a search key word in PubMed). These publications, which use diverse genetic and genomic approaches and often large size samples, have identified overlapping genes and mutations, which are likely to have greater influences on the development of ASD. Characterization of transgenic mouse lines that carry these frequent genetic variations would help us efficiently obtain ASD mechanisms with a greater impact.
The synaptic and circuit mechanisms derived from ASD mouse model researches would provide clues to the ways to rescue synaptic/circuit phenotypes and ASD-like behaviors in mice. Given that there is no FDA-approved treatment for social deficits in ASD as of now, these rescue results will only be useful in supporting that the candidate mechanisms are indeed causing the ASD-like phenotypes in mice. Importantly, however, some of these mechanism-based rescues may serve as the basis for clinical trials. Eventually, some of the clinically verified medications may return to basic ASD research and be used to identify additional ASD mouse models with similar or novel underlying mechanisms, which will help us understand a bigger picture, where many synaptic and circuit mechanisms act together and converge into more comprehensive mechanisms.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the Institute for Basic Science (IBS).
References
Abrahams, B. S., and Geschwind, D. H. (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 9, 341–355. doi: 10.1038/nrg2346
Alagarsamy, S., Rouse, S. T., Junge, C., Hubert, G. W., Gutman, D., Smith, Y., et al. (2002). NMDA-induced phosphorylation and regulation of mGluR5. Pharmacol. Biochem. Behav. 73, 299–306. doi: 10.1016/S0091-3057(02)00826-2
Alarcon, M., Abrahams, B. S., Stone, J. L., Duvall, J. A., Perederiy, J. V., Bomar, J. M., et al. (2008). Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 82, 150–159. doi: 10.1016/j.ajhg.2007.09.005
Allen, G., and Courchesne, E. (2003). Differential effects of developmental cerebellar abnormality on cognitive and motor functions in the cerebellum: an fMRI study of autism. Am. J. Psychiatry 160, 262–273. doi: 10.1176/appi.ajp.160.2.262
Allen, G., Muller, R. A., and Courchesne, E. (2004). Cerebellar function in autism: functional magnetic resonance image activation during a simple motor task. Biol. Psychiatry 56, 269–278. doi: 10.1016/j.biopsych.2004.06.005
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Andari, E., Duhamel, J. R., Zalla, T., Herbrecht, E., Leboyer, M., and Sirigu, A. (2010). Promoting social behavior with oxytocin in high-functioning autism spectrum disorders. Proc. Natl. Acad. Sci. U.S.A. 107, 4389–4394. doi: 10.1073/pnas.0910249107
Anderson, G. M., Horne, W. C., Chatterjee, D., and Cohen, D. J. (1990). The hyperserotonemia of autism. Ann. N.Y. Acad. Sci. 600, 331–340. discussion: 341–332. doi: 10.1111/j.1749-6632.1990.tb16893.x
Anney, R., Klei, L., Pinto, D., Regan, R., Conroy, J., Magalhaes, T. R., et al. (2010). A genome-wide scan for common alleles affecting risk for autism. Hum. Mol. Genet. 19, 4072–4082. doi: 10.1093/hmg/ddq307
Aracava, Y., Pereira, E. F., Maelicke, A., and Albuquerque, E. X. (2005). Memantine blocks alpha7* nicotinic acetylcholine receptors more potently than n-methyl-D-aspartate receptors in rat hippocampal neurons. J. Pharmacol. Exp. Ther. 312, 1195–1205. doi: 10.1124/jpet.104.077172
Araki, K., Ellebedy, A. H., and Ahmed, R. (2011). TOR in the immune system. Curr. Opin. Cell Biol. 23, 707–715. doi: 10.1016/j.ceb.2011.08.006
Arking, D. E., Cutler, D. J., Brune, C. W., Teslovich, T. M., West, K., Ikeda, M., et al. (2008). A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 82, 160–164. doi: 10.1016/j.ajhg.2007.09.015
Ashwood, P., and Van de Water, J. (2004). A review of autism and the immune response. Clin. Dev. Immunol. 11, 165–174. doi: 10.1080/10446670410001722096
Ashwood, P., Wills, S., and Van de Water, J. (2006). The immune response in autism: a new frontier for autism research. J. Leukoc. Biol. 80, 1–15. doi: 10.1189/jlb.1205707
Attucci, S., Carla, V., Mannaioni, G., and Moroni, F. (2001). Activation of type 5 metabotropic glutamate receptors enhances NMDA responses in mice cortical wedges. Br. J. Pharmacol. 132, 799–806. doi: 10.1038/sj.bjp.0703904
Auerbach, B. D., Osterweil, E. K., and Bear, M. F. (2011). Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68. doi: 10.1038/nature10658
Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators; Centers for Disease Control and Prevention. (2007). Prevalence of autism spectrum disorders–autism and developmental disabilities monitoring network, 14 sites, United States. MMWR Surveill. Summ. 56, 12–28.
Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators; Centers for Disease Control and Prevention. (2009). Prevalence of autism spectrum disorders - Autism and developmental disabilities monitoring network, United States, 2006. MMWR Surveill. Summ. 58, 1–20.
Autism Genome Project, C., Szatmari, P., Paterson, A. D., Zwaigenbaum, L., Roberts, W., Brian, J., et al. (2007). Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 39, 319–328. doi: 10.1038/ng1985
Awad, H., Hubert, G. W., Smith, Y., Levey, A. I., and Conn, P. J. (2000). Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. J. Neurosci. 20, 7871–7879.
Ayala, J. E., Chen, Y., Banko, J. L., Sheffler, D. J., Williams, R., Telk, A. N., et al. (2009). mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology 34, 2057–2071. doi: 10.1038/npp.2009.30
Aylward, E. H., Minshew, N. J., Goldstein, G., Honeycutt, N. A., Augustine, A. M., Yates, K. O., et al. (1999). MRI volumes of amygdala and hippocampus in non-mentally retarded autistic adolescents and adults. Neurology 53, 2145–2150. doi: 10.1212/WNL.53.9.2145
Bah, J., Quach, H., Ebstein, R. P., Segman, R. H., Melke, J., Jamain, S., et al. (2004). Maternal transmission disequilibrium of the glutamate receptor GRIK2 in schizophrenia. Neuroreport 15, 1987–1991. doi: 10.1097/00001756-200408260-00031
Baharnoori, M., Bhardwaj, S. K., and Srivastava, L. K. (2012). Neonatal behavioral changes in rats with gestational exposure to lipopolysaccharide: a prenatal infection model for developmental neuropsychiatric disorders. Schizophr. Bull. 38, 444–456. doi: 10.1093/schbul/sbq098
Baker, H., Sidorowicz, A., Sehgal, S. N., and Vezina, C. (1978). Rapamycin (AY-22,989), a new antifungal antibiotic. III. In vitro and in vivo evaluation. J. Antibiot. 31, 539–545. doi: 10.7164/antibiotics.31.539
Bakkaloglu, B., O'Roak, B. J., Louvi, A., Gupta, A. R., Abelson, J. F., Morgan, T. M., et al. (2008). Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 82, 165–173. doi: 10.1016/j.ajhg.2007.09.017
Bakker, C. E., Verheij, C., Willemsen, R., Helm, R.v.d., Oerlemans, F., Vermey, M., et al. (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78, 23–33.
Balschun, D., Zuschratter, W., and Wetzel, W. (2006). Allosteric enhancement of metabotropic glutamate receptor 5 function promotes spatial memory. Neuroscience 142, 691–702. doi: 10.1016/j.neuroscience.2006.06.043
Barnby, G., Abbott, A., Sykes, N., Morris, A., Weeks, D. E., Mott, R., et al. (2005). Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. Am. J. Hum. Genet. 76, 950–966. doi: 10.1086/430454
Barnea-Goraly, N., Kwon, H., Menon, V., Eliez, S., Lotspeich, L., and Reiss, A. L. (2004). White matter structure in autism: preliminary evidence from diffusion tensor imaging. Biol. Psychiatry 55, 323–326. doi: 10.1016/j.biopsych.2003.10.022
Baron-Cohen, S., and Belmonte, M. K. (2005). Autism: a window onto the development of the social and the analytic brain. Annu. Rev. Neurosci. 28, 109–126. doi: 10.1146/annurev.neuro.27.070203.144137
Bass, M. P., Menold, M. M., Wolpert, C. M., Donnelly, S. L., Ravan, S. A., Hauser, E. R., et al. (2000). Genetic studies in autistic disorder and chromosome 15. Neurogenetics 2, 219–226. doi: 10.1007/s100489900081
Bassani, S., Cingolani, L. A., Valnegri, P., Folci, A., Zapata, J., Gianfelice, A., et al. (2012). The X-linked intellectual disability protein TSPAN7 regulates excitatory synapse development and AMPAR trafficking. Neuron 73, 1143–1158. doi: 10.1016/j.neuron.2012.01.021
Bassell, G. J., and Warren, S. T. (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214. doi: 10.1016/j.neuron.2008.10.004
Baudouin, S. J., Gaudias, J., Gerharz, S., Hatstatt, L., Zhou, K., Punnakkal, P., et al. (2012). Shared synaptic pathophysiology in syndromic and nonsyndromic rodent models of autism. Science 338, 128–132. doi: 10.1126/science.1224159
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. doi: 10.1016/j.tins.2004.04.009
Benayed, R., Gharani, N., Rossman, I., Mancuso, V., Lazar, G., Kamdar, S., et al. (2005). Support for the homeobox transcription factor gene ENGRAILED 2 as an autism spectrum disorder susceptibility locus. Am. J. Hum. Genet. 77, 851–868. doi: 10.1086/497705
Berditchevski, F., and Odintsova, E. (1999). Characterization of integrin-tetraspanin adhesion complexes: role of tetraspanins in integrin signaling. J. Cell Biol. 146, 477–492. doi: 10.1083/jcb.146.2.477
Berger, Z., Ravikumar, B., Menzies, F. M., Oroz, L. G., Underwood, B. R., Pangalos, M. N., et al. (2006). Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 15, 433–442. doi: 10.1093/hmg/ddi458
Berkel, S., Marshall, C. R., Weiss, B., Howe, J., Roeth, R., Moog, U., et al. (2010). Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 42, 489–491. doi: 10.1038/ng.589
Berkel, S., Tang, W., Trevino, M., Vogt, M., Obenhaus, H. A., Gass, P., et al. (2012). Inherited and de novo SHANK2 variants associated with autism spectrum disorder impair neuronal morphogenesis and physiology. Hum. Mol. Genet. 21, 344–357. doi: 10.1093/hmg/ddr470
Bernardet, M., and Crusio, W. E. (2006). Fmr1 KO mice as a possible model of autistic features. ScientificWorldJournal 6, 1164–1176. doi: 10.1100/tsw.2006.220
Biber, K., Pinto-Duarte, A., Wittendorp, M. C., Dolga, A. M., Fernandes, C. C., Von Frijtag Drabbe Kunzel, J., et al. (2008). Interleukin-6 upregulates neuronal adenosine A1 receptors: implications for neuromodulation and neuroprotection. Neuropsychopharmacology 33, 2237–2250.
Bienvenu, T., and Chelly, J. (2006). Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat. Rev. Genet. 7, 415–426. doi: 10.1038/nrg1878
Billuart, P., Bienvenu, T., Ronce, N., des Portes, V., Vinet, M. C., Zemni, R., et al. (1998a). Oligophrenin 1 encodes a rho-GAP protein involved in X-linked mental retardation. Pathol. Biol. 46, 678.
Billuart, P., Bienvenu, T., Ronce, N., des Portes, V., Vinet, M. C., Zemni, R., et al. (1998b). Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature 392, 923–926.
Blanpied, T. A., Clarke, R. J., and Johnson, J. W. (2005). Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J. Neurosci. 25, 3312–3322. doi: 10.1523/JNEUROSCI.4262-04.2005
Blundell, J., Blaiss, C. A., Etherton, M. R., Espinosa, F., Tabuchi, K., Walz, C., et al. (2010). Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. 30, 2115–2129. doi: 10.1523/JNEUROSCI.4517-09.2010
Boeckers, T. M., Bockmann, J., Kreutz, M. R., and Gundelfinger, E. D. (2002). ProSAP/Shank proteins - a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J. Neurochem. 81, 903–910. doi: 10.1046/j.1471-4159.2002.00931.x
Bolton, P. F., Roobol, M., Allsopp, L., and Pickles, A. (2001). Association between idiopathic infantile macrocephaly and autism spectrum disorders. Lancet 358, 726–727. doi: 10.1016/S0140-6736(01)05903-7
Bozdagi, O., Sakurai, T., Papapetrou, D., Wang, X., Dickstein, D. L., Takahashi, N., et al. (2010). Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 1, 15. doi: 10.1186/2040-2392-1-15
Brennand, K. J., Simone, A., Jou, J., Gelboin-Burkhart, C., Tran, N., Sangar, S., et al. (2011). Modelling schizophrenia using human induced pluripotent stem cells. Nature 473, 221–225. doi: 10.1038/nature09915
Brielmaier, J., Matteson, P. G., Silverman, J. L., Senerth, J. M., Kelly, S., Genestine, M., et al. (2012). Autism-relevant social abnormalities and cognitive deficits in engrailed-2 knockout mice. PLoS ONE 7:e40914. doi: 10.1371/journal.pone.0040914
Brunet, I., Weinl, C., Piper, M., Trembleau, A., Volovitch, M., Harris, W., et al. (2005). The transcription factor Engrailed-2 guides retinal axons. Nature 438, 94–98. doi: 10.1038/nature04110
Burgess, N. K., Sweeten, T. L., McMahon, W. M., and Fujinami, R. S. (2006). Hyperserotoninemia and altered immunity in autism. J. Autism Dev. Disord. 36, 697–704. doi: 10.1007/s10803-006-0100-7
Busaidy, N. L., Farooki, A., Dowlati, A., Perentesis, J. P., Dancey, J. E., Doyle, L. A., et al. (2012). Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J. Clin. Oncol. 30, 2919–2928. doi: 10.1200/JCO.2011.39.7356
Buxbaum, J. D., Silverman, J. M., Smith, C. J., Greenberg, D. A., Kilifarski, M., Reichert, J., et al. (2002). Association between a GABRB3 polymorphism and autism. Mol. Psychiatry 7, 311–316. doi: 10.1038/sj.mp.4001011
Cai, G., Edelmann, L., Goldsmith, J. E., Cohen, N., Nakamine, A., Reichert, J. G., et al. (2008). Multiplex ligation-dependent probe amplification for genetic screening in autism spectrum disorders: efficient identification of known microduplications and identification of a novel microduplication in ASMT. BMC Med. Genomics 1:50. doi: 10.1186/1755-8794-1-50
Caldwell, H. K. (2012). Neurobiology of sociability. Adv. Exp. Med. Biol. 739, 187–205. doi: 10.1007/978-1-4614-1704-0_12
Campbell, D. B., D'Oronzio, R., Garbett, K., Ebert, P. J., Mirnics, K., Levitt, P., et al. (2007). Disruption of cerebral cortex MET signaling in autism spectrum disorder. Ann. Neurol. 62, 243–250. doi: 10.1002/ana.21180
Campbell, D. B., Sutcliffe, J. S., Ebert, P. J., Militerni, R., Bravaccio, C., Trillo, S., et al. (2006). A genetic variant that disrupts MET transcription is associated with autism. Proc. Natl. Acad. Sci. U.S.A. 103, 16834–16839. doi: 10.1073/pnas.0605296103
Carrie, A., Jun, L., Bienvenu, T., Vinet, M. C., McDonell, N., Couvert, P., et al. (1999). A new member of the IL-1 receptor family highly expressed in hippocampus and involved in X-linked mental retardation. Nat. Genet. 23, 25–31. doi: 10.1038/12623
Caspi, A., Sugden, K., Moffitt, T. E., Taylor, A., Craig, I. W., Harrington, H., et al. (2003). Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301, 386–389. doi: 10.1126/science.1083968
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437. doi: 10.1016/j.neuron.2007.10.001
Chang, Q., Khare, G., Dani, V., Nelson, S., and Jaenisch, R. (2006). The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49, 341–348. doi: 10.1016/j.neuron.2005.12.027
Chen, H. J., Rojas-Soto, M., Oguni, A., and Kennedy, M. B. (1998). A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron 20, 895–904. doi: 10.1016/S0896-6273(00)80471-7
Chen, H. S., Pellegrini, J. W., Aggarwal, S. K., Lei, S. Z., Warach, S., Jensen, F. E., et al. (1992). Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J. Neurosci. 12, 4427–4436.
Chez, M. G., Burton, Q., Dowling, T., Chang, M., Khanna, P., and Kramer, C. (2007). Memantine as adjunctive therapy in children diagnosed with autistic spectrum disorders: an observation of initial clinical response and maintenance tolerability. J. Child Neurol. 22, 574–579. doi: 10.1177/0883073807302611
Chien, Y. L., Wu, Y. Y., Chiu, Y. N., Liu, S. K., Tsai, W. C., Lin, P. I., et al. (2011). Association study of the CNS patterning genes and autism in Han Chinese in Taiwan. Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 1512–1517. doi: 10.1016/j.pnpbp.2011.04.010
Cisternas, F. A., Vincent, J. B., Scherer, S. W., and Ray, P. N. (2003). Cloning and characterization of human CADPS and CADPS2, new members of the Ca2+-dependent activator for secretion protein family. Genomics 81, 279–291. doi: 10.1016/S0888-7543(02)00040-X
Clement, J. P., Aceti, M., Creson, T. K., Ozkan, E. D., Shi, Y., Reish, N. J., et al. (2012). Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151, 709–723. doi: 10.1016/j.cell.2012.08.045
Clifton, N. E., Morisot, N., Girardon, S., Millan, M. J., and Loiseau, F. (2012). Enhancement of social novelty discrimination by positive allosteric modulators at metabotropic glutamate 5 receptors: adolescent administration prevents adult-onset deficits induced by neonatal treatment with phencyclidine. Psychopharmacology 225, 579–594. doi: 10.1007/s00213-012-2845-3
Connolly, A. M., Chez, M., Streif, E. M., Keeling, R. M., Golumbek, P. T., Kwon, J. M., et al. (2006). Brain-derived neurotrophic factor and autoantibodies to neural antigens in sera of children with autistic spectrum disorders, Landau-Kleffner syndrome, and epilepsy. Biol. Psychiatry 59, 354–363. doi: 10.1016/j.biopsych.2005.07.004
Cook, E. H., Jr., Courchesne, R., Lord, C., Cox, N. J., Yan, S., Lincoln, A., et al. (1997). Evidence of linkage between the serotonin transporter and autistic disorder. Mol. Psychiatry 2, 247–250.
Cook, E. H. Jr., Courchesne, R. Y., Cox, N. J., Lord, C., Gonen, D., Guter, S. J., et al. (1998). Linkage-disequilibrium mapping of autistic disorder, with 15q11-13 markers. Am. J. Hum. Genet. 62, 1077–1083.
Cook, E. H., and Leventhal, B. L. (1996). The serotonin system in autism. Curr. Opin. Pediatr. 8, 348–354. doi: 10.1097/00008480-199608000-00008
Coon, H., Dunn, D., Lainhart, J., Miller, J., Hamil, C., Battaglia, A., et al. (2005). Possible association between autism and variants in the brain-expressed tryptophan hydroxylase gene (TPH2). Am. J. Med. Genet. B Neuropsychiatr. Genet. 135B, 42–46.
Costa, R. M., Federov, N. B., Kogan, J. H., Murphy, G. G., Stern, J., Ohno, M., et al. (2002). Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 415, 526–530. doi: 10.1038/nature711
Costa, R. M., Yang, T., Huynh, D. P., Pulst, S. M., Viskochil, D. H., Silva, A. J., et al. (2001). Learning deficits, but normal development and tumor predisposition, in mice lacking exon 23a of Nf1. Nat. Genet. 27, 399–405. doi: 10.1038/86898
Courchesne, E. (1997). Brainstem, cerebellar and limbic neuroanatomical abnormalities in autism. Curr. Opin. Neurobiol. 7, 269–278. doi: 10.1016/S0959-4388(97)80016-5
Courchesne, E. (2002). Abnormal early brain development in autism. Mol. Psychiatry 7(Suppl. 2), S21–S23.
Courchesne, E., Carper, R., and Akshoomoff, N. (2003). Evidence of brain overgrowth in the first year of life in autism. JAMA 290, 337–344. doi: 10.1001/jama.290.3.337
Courchesne, E., Pierce, K., Schumann, C. M., Redcay, E., Buckwalter, J. A., Kennedy, D. P., et al. (2007). Mapping early brain development in autism. Neuron 56, 399–413. doi: 10.1016/j.neuron.2007.10.016
Craig, A. M., and Kang, Y. (2007). Neurexin-neuroligin signaling in synapse development. Curr. Opin. Neurobiol. 17, 43–52. doi: 10.1016/j.conb.2007.01.011
Crawley, J. N., Chen, T., Puri, A., Washburn, R., Sullivan, T. L., Hill, J. M., et al. (2007). Social approach behaviors in oxytocin knockout mice: comparison of two independent lines tested in different laboratory environments. Neuropeptides 41, 145–163. doi: 10.1016/j.npep.2007.02.002
Croonenberghs, J., Wauters, A., Devreese, K., Verkerk, R., Scharpe, S., Bosmans, E., et al. (2002). Increased serum albumin, gamma globulin, immunoglobulin IgG, and IgG2 and IgG4 in autism. Psychol. Med. 32, 1457–1463. doi: 10.1017/S0033291702006037
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
de Vrij, F. M., Levenga, J., van der Linde, H. C., Koekkoek, S. K., De Zeeuw, C. I., Nelson, D. L., et al. (2008). Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol. Dis. 31, 127–132. doi: 10.1016/j.nbd.2008.04.002
Demontis, D., Nyegaard, M., Buttenschon, H. N., Hedemand, A., Pedersen, C. B., Grove, J., et al. (2011). Association of GRIN1 and GRIN2A-D with schizophrenia and genetic interaction with maternal herpes simplex virus-2 infection affecting disease risk. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156B, 913–922.
Dennis, P. B., Fumagalli, S., and Thomas, G. (1999). Target of rapamycin (TOR): balancing the opposing forces of protein synthesis and degradation. Curr. Opin. Genet. Dev. 9, 49–54. doi: 10.1016/S0959-437X(99)80007-0
Devlin, B., and Scherer, S. W. (2012). Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 22, 229–237. doi: 10.1016/j.gde.2012.03.002
Dugan, L. L., Ali, S. S., Shekhtman, G., Roberts, A. J., Lucero, J., Quick, K. L., et al. (2009). IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS ONE 4:e5518. doi: 10.1371/journal.pone.0005518
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Ehninger, D., de Vries, P. J., and Silva, A. J. (2009). From mTOR to cognition: molecular and cellular mechanisms of cognitive impairments in tuberous sclerosis. JIDR 53, 838–851. doi: 10.1111/j.1365-2788.2009.01208.x
Ehninger, D., Han, S., Shilyansky, C., Zhou, Y., Li, W., Kwiatkowski, D. J., et al. (2008). Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat. Med. 14, 843–848. doi: 10.1038/nm1788
Erickson, C. A., Posey, D. J., Stigler, K. A., Mullett, J., Katschke, A. R., and McDougle, C. J. (2007). A retrospective study of memantine in children and adolescents with pervasive developmental disorders. Psychopharmacology 191, 141–147. doi: 10.1007/s00213-006-0518-9
Fatemi, S. H. (2001). Reelin mutations in mouse and man: from reeler mouse to schizophrenia, mood disorders, autism and lissencephaly. Mol. Psychiatry 6, 129–133. doi: 10.1038/sj.mp.4000129
Fatemi, S. H., Halt, A. R., Stary, J. M., Kanodia, R., Schulz, S. C., and Realmuto, G. R. (2002). Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol. Psychiatry 52, 805–810. doi: 10.1016/S0006-3223(02)01430-0
Fatemi, S. H., Reutiman, T. J., Folsom, T. D., Rooney, R. J., Patel, D. H., and Thuras, P. D. (2010). mRNA and protein levels for GABAAalpha4, alpha5, beta1 and GABABR1 receptors are altered in brains from subjects with autism. J. Autism Dev. Disord. 40, 743–750. doi: 10.1007/s10803-009-0924-z
Fatemi, S. H., Snow, A. V., Stary, J. M., Araghi-Niknam, M., Reutiman, T. J., Lee, S., et al. (2005). Reelin signaling is impaired in autism. Biol. Psychiatry 57, 777–787. doi: 10.1016/j.biopsych.2004.12.018
Feldman, R. (2012). Oxytocin and social affiliation in humans. Horm. Behav. 61, 380–391. doi: 10.1016/j.yhbeh.2012.01.008
Fernandez, A. M., and Torres-Aleman, I. (2012). The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 13, 225–239. doi: 10.1038/nrn3209
Feucht, M., Fuchs, K., Pichlbauer, E., Hornik, K., Scharfetter, J., Goessler, R., et al. (1999). Possible association between childhood absence epilepsy and the gene encoding GABRB3. Biol. Psychiatry 46, 997–1002. doi: 10.1016/S0006-3223(99)00039-6
Fields, R. D., and Stevens-Graham, B. (2002). New insights into neuron-glia communication. Science 298, 556–562. doi: 10.1126/science.298.5593.556
Flood, J. F., Morley, J. E., and Lanthorn, T. H. (1992). Effect on memory processing by D-cycloserine, an agonist of the NMDA/glycine receptor. Eur. J. Pharmacol. 221, 249–254. doi: 10.1016/0014-2999(92)90709-D
Folstein, S. E., and Rosen-Sheidley, B. (2001). Genetics of autism: complex aetiology for a heterogeneous disorder. Nat. Rev. Genet. 2, 943–955. doi: 10.1038/35103559
Fombonne, E., Roge, B., Claverie, J., Courty, S., and Fremolle, J. (1999). Microcephaly and macrocephaly in autism. J. Autism Dev. Disord. 29, 113–119. doi: 10.1023/A:1023036509476
Forster, E., Jossin, Y., Zhao, S., Chai, X., Frotscher, M., and Goffinet, A. M. (2006). Recent progress in understanding the role of Reelin in radial neuronal migration, with specific emphasis on the dentate gyrus. Eur. J. Neurosci. 23, 901–909. doi: 10.1111/j.1460-9568.2006.04612.x
Friedman, J. I., Vrijenhoek, T., Markx, S., Janssen, I. M., van der Vliet, W. A., Faas, B. H., et al. (2008). CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol. Psychiatry 13, 261–266. doi: 10.1038/sj.mp.4002049
Gambino, F., Kneib, M., Pavlowsky, A., Skala, H., Heitz, S., Vitale, N., et al. (2009). IL1RAPL1 controls inhibitory networks during cerebellar development in mice. Eur. J. Neurosci. 30, 1476–1486. doi: 10.1111/j.1460-9568.2009.06975.x
Geschwind, D. H. (2009). Advances in autism. Annu. Rev. Med. 60, 367–380. doi: 10.1146/annurev.med.60.053107.121225
Gharani, N., Benayed, R., Mancuso, V., Brzustowicz, L. M., and Millonig, J. H. (2004). Association of the homeobox transcription factor, ENGRAILED 2, 3, with autism spectrum disorder. Mol. Psychiatry 9, 474–484. doi: 10.1038/sj.mp.4001498
Gillberg, C., and Wahlstrom, J. (1985). Chromosome abnormalities in infantile autism and other childhood psychoses: a population study of 66 cases. Dev. Med. Child Neurol. 27, 293–304. doi: 10.1111/j.1469-8749.1985.tb04539.x
Gkogkas, C. G., Khoutorsky, A., Ran, I., Rampakakis, E., Nevarko, T., Weatherill, D. B., et al. (2013). Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377. doi: 10.1038/nature11628
Glessner, J. T., Wang, K., Cai, G., Korvatska, O., Kim, C. E., Wood, S., et al. (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. doi: 10.1038/nature07953
Goff, D. C., Tsai, G., Levitt, J., Amico, E., Manoach, D., Schoenfeld, D. A., et al. (1999). A placebo-controlled trial of D-cycloserine added to conventional neuroleptics in patients with schizophrenia. Arch. Gen. Psychiatry 56, 21–27. doi: 10.1001/archpsyc.56.1.21
Gogolla, N., Leblanc, J. J., Quast, K. B., Sudhof, T. C., Fagiolini, M., and Hensch, T. K. (2009). Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J. Neurodev. Disord. 1, 172–181. doi: 10.1007/s11689-009-9023-x
Goldstein, S., and Schwebach, A. J. (2004). The comorbidity of pervasive developmental disorder and attention deficit hyperactivity disorder: results of a retrospective chart review. J. Autism Dev. Disord. 34, 329–339. doi: 10.1023/B:JADD.0000029554.46570.68
Goss, A. M., Tian, Y., Tsukiyama, T., Cohen, E. D., Zhou, D., Lu, M. M., et al. (2009). Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev. Cell 17, 290–298. doi: 10.1016/j.devcel.2009.06.005
Gould, B. R., and Zingg, H. H. (2003). Mapping oxytocin receptor gene expression in the mouse brain and mammary gland using an oxytocin receptor-LacZ reporter mouse. Neuroscience 122, 155–167. doi: 10.1016/S0306-4522(03)00283-5
Govek, E. E., Newey, S. E., Akerman, C. J., Cross, J. R., Van der Veken, L., and Van Aelst, L. (2004). The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat. Neurosci. 7, 364–372. doi: 10.1038/nn1210
Grabrucker, A. M. (2012). Environmental factors in autism. Front. Psychiatry 3:118. doi: 10.3389/fpsyt.2012.00118.
Greenberg, M. E., Xu, B., Lu, B., and Hempstead, B. L. (2009). New insights in the biology of BDNF synthesis and release: implications in CNS function. J. Neurosci. 29, 12764–12767.
Gregory, K. J., Dong, E. N., Meiler, J., and Conn, P. J. (2011). Allosteric modulation of metabotropic glutamate receptors: structural insights and therapeutic potential. Neuropharmacology 60, 66–81. doi: 10.1016/j.neuropharm.2010.07.007
Grzadzinski, R., Huerta, M., and Lord, C. (2013). DSM-5 and autism spectrum disorders (ASDs): an opportunity for identifying ASD subtypes. Mol. Autism 4, 12. doi: 10.1186/2040-2392-4-12
Guo, X., Hamilton, P. J., Reish, N. J., Sweatt, J. D., Miller, C. A., and Rumbaugh, G. (2009). Reduced expression of the NMDA receptor-interacting protein SynGAP causes behavioral abnormalities that model symptoms of Schizophrenia. Neuropsychopharmacology 34, 1659–1672. doi: 10.1038/npp.2008.223
Gutierrez, H., Dolcet, X., Tolcos, M., and Davies, A. (2004). HGF regulates the development of cortical pyramidal dendrites. Development 131, 3717–3726. doi: 10.1242/dev.01209
Hamdan, F. F., Daoud, H., Piton, A., Gauthier, J., Dobrzeniecka, S., Krebs, M. O., et al. (2011). De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol. Psychiatry 69, 898–901. doi: 10.1016/j.biopsych.2010.11.015
Han, J. M., and Sahin, M. (2011). TSC1/TSC2 signaling in the CNS. FEBS Lett. 585, 973–980. doi: 10.1016/j.febslet.2011.02.001
Han, S., Tai, C., Westenbroek, R. E., Yu, F. H., Cheah, C. S., Potter, G. B., et al. (2012). Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390. doi: 10.1038/nature11356
Harrison, D. E., Strong, R., Sharp, Z. D., Nelson, J. F., Astle, C. M., Flurkey, K., et al. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395.
Hava, G., Vered, L., Yael, M., Mordechai, H., and Mahoud, H. (2006). Alterations in behavior in adult offspring mice following maternal inflammation during pregnancy. Dev. Psychobiol. 48, 162–168. doi: 10.1002/dev.20116
Hazlett, H. C., Poe, M., Gerig, G., Smith, R. G., Provenzale, J., Ross, A., et al. (2005). Magnetic resonance imaging and head circumference study of brain size in autism: birth through age 2 years. Arch. Gen. Psychiatry 62, 1366–1376. doi: 10.1001/archpsyc.62.12.1366
Herbert, M. R. (2005). Large brains in autism: the challenge of pervasive abnormality. Neuroscientist 11, 417–440. doi: 10.1177/0091270005278866
Herbert, M. R., Ziegler, D. A., Deutsch, C. K., O'Brien, L. M., Lange, N., Bakardjiev, A., et al., Jr. (2003). Dissociations of cerebral cortex, subcortical and cerebral white matter volumes in autistic boys. Brain 126, 1182–1192. doi: 10.1093/brain/awg110
Herbert, M. R., Ziegler, D. A., Makris, N., Filipek, P. A., Kemper, T. L., Normandin, J. J., et al. (2004). Localization of white matter volume increase in autism and developmental language disorder. Ann. Neurol. 55, 530–540. doi: 10.1002/ana.20032
Hines, R. M., Wu, L., Hines, D. J., Steenland, H., Mansour, S., Dahlhaus, R., et al. (2008). Synaptic imbalance, stereotypies, and impaired social interactions in mice with altered neuroligin 2 expression. J. Neurosci. 28, 6055–6067. doi: 10.1523/JNEUROSCI.0032-08.2008
Hollander, E., Bartz, J., Chaplin, W., Phillips, A., Sumner, J., Soorya, L., et al. (2007). Oxytocin increases retention of social cognition in autism. Biol. Psychiatry 61, 498–503. doi: 10.1016/j.biopsych.2006.05.030
Hollander, E., Novotny, S., Hanratty, M., Yaffe, R., DeCaria, C. M., Aronowitz, B. R., et al. (2003). Oxytocin infusion reduces repetitive behaviors in adults with autistic and Asperger's disorders. Neuropsychopharmacology 28, 193–198. doi: 10.1038/sj.npp.1300021
Hood, W. F., Compton, R. P., and Monahan, J. B. (1989). D-cycloserine: a ligand for the N-methyl-D-aspartate coupled glycine receptor has partial agonist characteristics. Neurosci. Lett. 98, 91–95. doi: 10.1016/0304-3940(89)90379-0
Horev, G., Ellegood, J., Lerch, J. P., Son, Y. E., Muthuswamy, L., Vogel, H., et al. (2011). Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl. Acad. Sci. U.S.A. 108, 17076–17081. doi: 10.1073/pnas.1114042108
Huang, E. J., and Reichardt, L. F. (2001). Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736. doi: 10.1146/annurev.neuro.24.1.677
Hung, A. Y., Futai, K., Sala, C., Valtschanoff, J. G., Ryu, J., Woodworth, M. A., et al. (2008). Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J. Neurosci. 28, 1697–1708. doi: 10.1523/JNEUROSCI.3032-07.2008
Husi, H., Ward, M. A., Choudhary, J. S., Blackstock, W. P., and Grant, S. G. (2000). Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 3, 661–669. doi: 10.1038/76615
Hwa, V., Oh, Y., and Rosenfeld, R. G. (1999). The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr. Rev. 20, 761–787. doi: 10.1210/er.20.6.761
Ingram, J. L., Peckham, S. M., Tisdale, B., and Rodier, P. M. (2000a). Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neurotoxicol. Teratol. 22, 319–324.
Ingram, J. L., Stodgell, C. J., Hyman, S. L., Figlewicz, D. A., Weitkamp, L. R., and Rodier, P. M. (2000b). Discovery of allelic variants of HOXA1 and HOXB1: genetic susceptibility to autism spectrum disorders. Teratology 62, 393–405.
International Molecular Genetic Study of Autism, C. (1998). A full genome screen for autism with evidence for linkage to a region on chromosome 7q. International Molecular Genetic Study of Autism Consortium. Hum. Mol. Genet. 7, 571–578.
International Molecular Genetic Study of Autism, C. (2001). A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am. J. Hum. Genet. 69, 570–581.
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. doi: 10.1016/j.neuron.2012.04.009
Itoh, M., Ide, S., Takashima, S., Kudo, S., Nomura, Y., Segawa, M., et al. (2007). Methyl CpG-binding protein 2 (a mutation of which causes Rett syndrome) directly regulates insulin-like growth factor binding protein 3 in mouse and human brains. J. Neuropathol. Exp. Neurol. 66, 117–123. doi: 10.1097/nen.0b013e3180302078
Jacob, S., Brune, C. W., Carter, C. S., Leventhal, B. L., Lord, C., and Cook, E. H. Jr. (2007). Association of the oxytocin receptor gene (OXTR) in Caucasian children and adolescents with autism. Neurosci. Lett. 417, 6–9. doi: 10.1016/j.neulet.2007.02.001
Jamain, S., Betancur, C., Quach, H., Philippe, A., Fellous, M., Giros, B., et al. (2002). Linkage and association of the glutamate receptor 6 gene with autism. Mol. Psychiatry. 7, 302–310. doi: 10.1038/sj.mp.4000979
Jamain, S., Quach, H., Betancur, C., Rastam, M., Colineaux, C., Gillberg, I. C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. doi: 10.1038/ng1136
Jamain, S., Radyushkin, K., Hammerschmidt, K., Granon, S., Boretius, S., Varoqueaux, F., et al. (2008). Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc. Natl. Acad. Sci. U.S.A. 105, 1710–1715. doi: 10.1073/pnas.0711555105
Joyner, A. L. (1996). Engrailed, Wnt and Pax genes regulate midbrain–hindbrain development. TIG 12, 15–20. doi: 10.1016/0168-9525(96)81383-7
Just, M. A., Cherkassky, V. L., Keller, T. A., Kana, R. K., and Minshew, N. J. (2007). Functional and anatomical cortical underconnectivity in autism: evidence from an FMRI study of an executive function task and corpus callosum morphometry. Cereb. cortex 17, 951–961. doi: 10.1093/cercor/bhl006
Kalume, F., Yu, F. H., Westenbroek, R. E., Scheuer, T., and Catterall, W. A. (2007). Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J. Neurosci. 27, 11065–11074.
Khelfaoui, M., Denis, C., van Galen, E., de Bock, F., Schmitt, A., Houbron, C., et al. (2007). Loss of X-linked mental retardation gene oligophrenin1 in mice impairs spatial memory and leads to ventricular enlargement and dendritic spine immaturity. J. Neurosci. 27, 9439–9450. doi: 10.1523/JNEUROSCI.2029-07.2007
Khurana, V., Lu, Y., Steinhilb, M. L., Oldham, S., Shulman, J. M., and Feany, M. B. (2006). TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr. Biol. 16, 230–241. doi: 10.1016/j.cub.2005.12.042
Kim, H. G., Kishikawa, S., Higgins, A. W., Seong, I. S., Donovan, D. J., Shen, Y., et al. (2008). Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 82, 199–207. doi: 10.1016/j.ajhg.2007.09.011
Kim, J. H., Liao, D., Lau, L. F., and Huganir, R. L. (1998). SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron 20, 683–691. doi: 10.1016/S0896-6273(00)81008-9
King, B. H., Wright, D. M., Handen, B. L., Sikich, L., Zimmerman, A. W., McMahon, W., et al., Jr. (2001). Double-blind, placebo-controlled study of amantadine hydrochloride in the treatment of children with autistic disorder. J. Am. Acad. Child Adolesc. Psychiatry 40, 658–665. doi: 10.1097/00004583-200106000-00010
Kinney, G. G., O'Brien, J. A., Lemaire, W., Burno, M., Bickel, D. J., Clements, M. K., et al. (2005). A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J. Pharmacol. Exp. Ther. 313, 199–206. doi: 10.1124/jpet.104.079244
Kirsten, T. B., Taricano, M., Maiorka, P. C., Palermo-Neto, J., and Bernardi, M. M. (2010). Prenatal lipopolysaccharide reduces social behavior in male offspring. Neuroimmunomodulation 17, 240–251. doi: 10.1159/000290040
Kosaka, H., Munesue, T., Ishitobi, M., Asano, M., Omori, M., Sato, M., et al. (2012). Long-term oxytocin administration improves social behaviors in a girl with autistic disorder. BMC Psychiatry 12:110. doi: 10.1186/1471-244X-12-110
Krueger, D. D., and Bear, M. F. (2011). Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annu. Rev. Med. 62, 411–429. doi: 10.1146/annurev-med-061109-134644
Kwon, C. H., Luikart, B. W., Powell, C. M., Zhou, J., Matheny, S. A., Zhang, W., et al. (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388. doi: 10.1016/j.neuron.2006.03.023
Lalonde, R., Hayzoun, K., Derer, M., Mariani, J., and Strazielle, C. (2004). Neurobehavioral evaluation of Reln-rl-orl mutant mice and correlations with cytochrome oxidase activity. Neurosci. Res. 49, 297–305. doi: 10.1016/j.neures.2004.03.012
Laumonnier, F., Bonnet-Brilhault, F., Gomot, M., Blanc, R., David, A., Moizard, M. P., et al. (2004). X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 74, 552–557. doi: 10.1086/382137
Leblond, C. S., Heinrich, J., Delorme, R., Proepper, C., Betancur, C., Huguet, G., et al. (2012). Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 8:e1002521. doi: 10.1371/journal.pgen.1002521
Lee, H. J., Macbeth, A. H., Pagani, J. H., and Young, W. S., 3rd. (2009). Oxytocin: the great facilitator of life. Prog. Neurobiol. 88, 127–151.
Li, X., Alafuzoff, I., Soininen, H., Winblad, B., and Pei, J. J. (2005). Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer's disease brain. FEBS J. 272, 4211–4220. doi: 10.1111/j.1742-4658.2005.04833.x
Lijam, N., Paylor, R., McDonald, M. P., Crawley, J. N., Deng, C. X., Herrup, K., et al. (1997). Social interaction and sensorimotor gating abnormalities in mice lacking Dvl1. Cell 90, 895–905. doi: 10.1016/S0092-8674(00)80354-2
Liu, F., Grauer, S., Kelley, C., Navarra, R., Graf, R., Zhang, G., et al. (2008). ADX47273 [S-(4-fluoro-phenyl)-{3-[3-(4-fluoro-phenyl)-[1,2,4]-oxadiazol-5-yl]-piperidin-1- yl}-methanone]: a novel metabotropic glutamate receptor 5-selective positive allosteric modulator with preclinical antipsychotic-like and procognitive activities. J. Pharmacol. Exp. Ther. 327, 827–839. doi: 10.1124/jpet.108.136580
Liu, X., Kawamura, Y., Shimada, T., Otowa, T., Koishi, S., Sugiyama, T., et al. (2010). Association of the oxytocin receptor (OXTR) gene polymorphisms with autism spectrum disorder (ASD) in the Japanese population. J. Hum. Genet. 55, 137–141. doi: 10.1038/jhg.2009.140
Logan, C. Y., and Nusse, R. (2004). The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810. doi: 10.1146/annurev.cellbio.20.010403.113126
Long, J. M., LaPorte, P., Paylor, R., and Wynshaw-Boris, A. (2004). Expanded characterization of the social interaction abnormalities in mice lacking Dvl1. Genes Brain Behav. 3, 51–62. doi: 10.1046/j.1601-183x.2003.00045.x
Lotter, V. (1966). Epidemiology of autistic conditions in young children: I. Prevalence. Soc. Psychiatry 1, 124–137. doi: 10.1007/BF00584048
lwainsky, H. (1988). Mode of action, biotransformation and pharmacokinetics of antituberculosis drugs in animals and man, in: antituberculosis Drugs. Handb. Exp. Pharmacol. 84, 399.
Ma, D., Salyakina, D., Jaworski, J. M., Konidari, I., Whitehead, P. L., Andersen, A. N., et al. (2009). A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Ann. Hum. Genet. 73, 263–273. doi: 10.1111/j.1469-1809.2009.00523.x
Maecker, H. T., Todd, S. C., and Levy, S. (1997). The tetraspanin superfamily: molecular facilitators. FASEB J. 11, 428–442.
Malagelada, C., Ryu, E. J., Biswas, S. C., Jackson-Lewis, V., and Greene, L. A. (2006). RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson's disease by a mechanism involving mammalian target of rapamycin inactivation. J. Neurosci. 26, 9996–10005. doi: 10.1523/JNEUROSCI.3292-06.2006
Malkova, N. V., Yu, C. Z., Hsiao, E. Y., Moore, M. J., and Patterson, P. H. (2012). Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav. Immun. 26, 607–616. doi: 10.1016/j.bbi.2012.01.011
Mannaioni, G., Marino, M. J., Valenti, O., Traynelis, S. F., and Conn, P. J. (2001). Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J. Neurosci. 21, 5925–5934.
Manor, I., Eisenberg, J., Tyano, S., Sever, Y., Cohen, H., Ebstein, R. P., et al. (2001). Family-based association study of the serotonin transporter promoter region polymorphism (5-HTTLPR) in attention deficit hyperactivity disorder. Am. J. Med. Genet. 105, 91–95.
Maranduba, C. M., Sa Moreira, E., Muller Orabona, G., Pavanello, R. C., Vianna-Morgante, A. M., and Passos-Bueno, M. R. (2004). Does the P172H mutation at the TM4SF2 gene cause X-linked mental retardation? Am. J. Med. Genet. A 124A, 413–415.
Marchetto, M. C., Carromeu, C., Acab, A., Yu, D., Yeo, G. W., Mu, Y., et al. (2010). A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539. doi: 10.1016/j.cell.2010.10.016
Martinowich, K., Manji, H., and Lu, B. (2007). New insights into BDNF function in depression and anxiety. Nat. Neurosci. 10, 1089–1093. doi: 10.1038/nn1971
Marui, T., Funatogawa, I., Koishi, S., Yamamoto, K., Matsumoto, H., Hashimoto, O., et al. (2010). Association between autism and variants in the wingless-type MMTV integration site family member 2 (WNT2) gene. Int. J. Neuropsychopharmacol. 13, 443–449. doi: 10.1017/S1461145709990903
McPheeters, M. L., Warren, Z., Sathe, N., Bruzek, J. L., Krishnaswami, S., Jerome, R. N., et al. (2011). A systematic review of medical treatments for children with autism spectrum disorders. Pediatrics 127, e1312–e1321.
Mehta, M. V., Gandal, M. J., and Siegel, S. J. (2011). mGluR5-antagonist mediated reversal of elevated stereotyped, repetitive behaviors in the VPA model of autism. PLoS ONE 6:e26077. doi: 10.1371/journal.pone.0026077
Minshew, N. J., and Williams, D. L. (2007). The new neurobiology of autism: cortex, connectivity, and neuronal organization. Arch. Neurol. 64, 945–950. doi: 10.1001/archneur.64.7.945
Miyazaki, K., Narita, N., and Narita, M. (2005). Maternal administration of thalidomide or valproic acid causes abnormal serotonergic neurons in the offspring: implication for pathogenesis of autism. Int. J. Dev. Neurosci. 23, 287–297. doi: 10.1016/j.ijdevneu.2004.05.004
Miyazaki, K., Narita, N., Sakuta, R., Miyahara, T., Naruse, H., Okado, N., et al. (2004). Serum neurotrophin concentrations in autism and mental retardation: a pilot study. Brain Dev. 26, 292–295. doi: 10.1016/S0387-7604(03)00168-2
Monahan, J. B., Handelmann, G. E., Hood, W. F., and Cordi, A. A. (1989). D-cycloserine, a positive modulator of the N-methyl-D-aspartate receptor, enhances performance of learning tasks in rats. Pharmacol. Biochem. Behav. 34, 649–653. doi: 10.1016/0091-3057(89)90571-6
Moreno-De-Luca, A., Myers, S. M., Challman, T. D., Moreno-De-Luca, D., Evans, D. W., and Ledbetter, D. H. (2013). Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol. 12, 406–414. doi: 10.1016/S1474-4422(13)70011-5
Moreno-Fuenmayor, H., Borjas, L., Arrieta, A., Valera, V., and Socorro-Candanoza, L. (1996). Plasma excitatory amino acids in autism. Invest. Clin. 37, 113–128.
Moretti, P., Bouwknecht, J. A., Teague, R., Paylor, R., and Zoghbi, H. Y. (2005). Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum. Mol. Genet. 14, 205–220. doi: 10.1093/hmg/ddi016
Nadif Kasri, N., Nakano-Kobayashi, A., Malinow, R., Li, B., and Van Aelst, L. (2009). The Rho-linked mental retardation protein oligophrenin-1 controls synapse maturation and plasticity by stabilizing AMPA receptors. Genes Dev. 23, 1289–1302. doi: 10.1101/gad.1783809
Naisbitt, S., Kim, E., Tu, J. C., Xiao, B., Sala, C., Valtschanoff, J., et al. (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23, 569–582. doi: 10.1016/S0896-6273(00)80809-0
Nakatani, J., Tamada, K., Hatanaka, F., Ise, S., Ohta, H., Inoue, K., et al. (2009). Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell 137, 1235–1246. doi: 10.1016/j.cell.2009.04.024
Narita, N., Kato, M., Tazoe, M., Miyazaki, K., Narita, M., and Okado, N. (2002). Increased monoamine concentration in the brain and blood of fetal thalidomide- and valproic acid-exposed rat: putative animal models for autism. Pediatr. Res. 52, 576–579.
Neale, B. M., Kou, Y., Liu, L., Ma'Ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. doi: 10.1038/nature11011
Neves-Pereira, M., Muller, B., Massie, D., Williams, J. H., O'Brien, P. C., Hughes, A., et al. (2009). Deregulation of EIF4E: a novel mechanism for autism. J. Med. Genet. 46, 759–765. doi: 10.1136/jmg.2009.066852
Niederhofer, H. (2007). Glutamate antagonists seem to be slightly effective in psychopharmacologic treatment of autism. J. Clin. Psychopharmacol. 27, 317–318. doi: 10.1097/01.jcp.0000270082.30500.69
Nightingale, S. (2012). Autism spectrum disorders. Nat. Rev. Drug Discov. 11, 745–746. doi: 10.1038/nrd3771
Niswender, C. M., and Conn, P. J. (2010). Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322. doi: 10.1146/annurev.pharmtox.011008.145533
O'Kusky, J. R., Ye, P., and D'Ercole, A. J. (2000). Insulin-like growth factor-I promotes neurogenesis and synaptogenesis in the hippocampal dentate gyrus during postnatal development. J. Neurosci. 20, 8435–8442.
O'Roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G., et al. (2012a). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622.
O'Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012b). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250.
Ogier, M., Wang, H., Hong, E., Wang, Q., Greenberg, M. E., and Katz, D. M. (2007). Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J. Neurosci. 27, 10912–10917. doi: 10.1523/JNEUROSCI.1869-07.2007
Ozdinler, P. H., and Macklis, J. D. (2006). IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat. Neurosci. 9, 1371–1381. doi: 10.1038/nn1789
Pan, T., Rawal, P., Wu, Y., Xie, W., Jankovic, J., and Le, W. (2009). Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 164, 541–551. doi: 10.1016/j.neuroscience.2009.08.014
Pandey, U. B., Nie, Z., Batlevi, Y., McCray, B. A., Ritson, G. P., Nedelsky, N. B., et al. (2007). HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863. doi: 10.1038/nature05853
Pavlowsky, A., Gianfelice, A., Pallotto, M., Zanchi, A., Vara, H., Khelfaoui, M., et al. (2010). A postsynaptic signaling pathway that may account for the cognitive defect due to IL1RAPL1 mutation. Curr. Biol. 20, 103–115. doi: 10.1016/j.cub.2009.12.030
Peca, J., Feliciano, C., Ting, J. T., Wang, W., Wells, M. F., Venkatraman, T. N., et al. (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. doi: 10.1038/nature09965
Penagarikano, O., Abrahams, B. S., Herman, E. I., Winden, K. D., Gdalyahu, A., Dong, H., et al. (2011). Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246. doi: 10.1016/j.cell.2011.08.040
Persico, A. M., D'Agruma, L., Maiorano, N., Totaro, A., Militerni, R., Bravaccio, C., et al. (2001). Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol. Psychiatry. 6, 150–159. doi: 10.1038/sj.mp.4000850
Peterson, S. L. (1992). 7-Chlorokynurenic acid antagonizes the anticonvulsant activity of D-cycloserine in maximal electroshock seizures. Epilepsy Res. 13, 73–81. doi: 10.1016/0920-1211(92)90009-I
Peterson, S. L., and Schwade, N. D. (1993). The anticonvulsant activity of D-cycloserine is specific for tonic convulsions. Epilepsy Res. 15, 141–148. doi: 10.1016/0920-1211(93)90094-N
Phelan, K., and McDermid, H. E. (2012). The 22q13.3 Deletion Syndrome (Phelan-McDermid Syndrome). Mol. Syndromol. 2, 186–201.
Philippe, A., Martinez, M., Guilloud-Bataille, M., Gillberg, C., Rastam, M., Sponheim, E., et al. (1999). Genome-wide scan for autism susceptibility genes. Paris Autism Research International Sibpair Study. Hum. Mol. Genet. 8, 805–812. doi: 10.1093/hmg/8.5.805
Pilarski, R., and Eng, C. (2004). Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J. Med. Genet. 41, 323–326. doi: 10.1136/jmg.2004.018036
Pinto, D., Pagnamenta, A. T., Klei, L., Anney, R., Merico, D., Regan, R., et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372.
Pisani, A., Gubellini, P., Bonsi, P., Conquet, F., Picconi, B., Centonze, D., Bernardi, G., and Calabresi, P. (2001). Metabotropic glutamate receptor 5 mediates the potentiation of N-methyl-D-aspartate responses in medium spiny striatal neurons. Neuroscience 106, 579–587. doi: 10.1016/S0306-4522(01)00297-4
Piton, A., Gauthier, J., Hamdan, F. F., Lafreniere, R. G., Yang, Y., Henrion, E., et al. (2011). Systematic resequencing of X-chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol. Psychiatry 16, 867–880. doi: 10.1038/mp.2010.54
Piton, A., Michaud, J. L., Peng, H., Aradhya, S., Gauthier, J., Mottron, L., et al. (2008). Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum. Mol. Genet. 17, 3965–3974. doi: 10.1093/hmg/ddn300
Poliak, S., Gollan, L., Martinez, R., Custer, A., Einheber, S., Salzer, J. L., Trimmer, J. S., Shrager, P., and Peles, E. (1999). Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 24, 1037–1047. doi: 10.1016/S0896-6273(00)81049-1
Posey, D. J., Kem, D. L., Swiezy, N. B., Sweeten, T. L., Wiegand, R. E., and McDougle, C. J. (2004). A pilot study of D-cycloserine in subjects with autistic disorder. Am. J. Psychiatry 161, 2115–2117. doi: 10.1176/appi.ajp.161.11.2115
Powell, E. M., Mars, W. M., and Levitt, P. (2001). Hepatocyte growth factor/scatter factor is a motogen for interneurons migrating from the ventral to dorsal telencephalon. Neuron 30, 79–89. doi: 10.1016/S0896-6273(01)00264-1
Powell, E. M., Muhlfriedel, S., Bolz, J., and Levitt, P. (2003). Differential regulation of thalamic and cortical axonal growth by hepatocyte growth factor/scatter factor. Dev. Neurosci. 25, 197–206. doi: 10.1159/000072268
Powers, R. W. 3rd., Kaeberlein, M., Caldwell, S. D., Kennedy, B. K., and Fields, S. (2006). Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 20, 174–184. doi: 10.1101/gad.1381406
Purcell, A. E., Jeon, O. H., Zimmerman, A. W., Blue, M. E., and Pevsner, J. (2001). Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 57, 1618–1628. doi: 10.1212/WNL.57.9.1618
Quartermain, D., Mower, J., Rafferty, M. F., Herting, R. L., and Lanthorn, T. H. (1994). Acute but not chronic activation of the NMDA-coupled glycine receptor with D-cycloserine facilitates learning and retention. Eur. J. Pharmacol. 257, 7–12. doi: 10.1016/0014-2999(94)90687-4
Radyushkin, K., Hammerschmidt, K., Boretius, S., Varoqueaux, F., El-Kordi, A., Ronnenberg, A., et al. (2009). Neuroligin-3-deficient mice: model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav. 8, 416–425. doi: 10.1111/j.1601-183X.2009.00487.x
Rammes, G., Rupprecht, R., Ferrari, U., Zieglgansberger, W., and Parsons, C. G. (2001). The N-methyl-D-aspartate receptor channel blockers memantine, MRZ 2/579 and other amino-alkyl-cyclohexanes antagonise 5-HT(3) receptor currents in cultured HEK-293 and N1E-115 cell systems in a non-competitive manner. Neurosci. Lett. 306, 81–84. doi: 10.1016/S0304-3940(01)01872-9
Rasmussen, S. A., and Friedman, J. M. (2000). NF1 gene and neurofibromatosis 1. Am. J. Epidemiol. 151, 33–40. doi: 10.1093/oxfordjournals.aje.a010118
Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595. doi: 10.1038/ng1362
Richter, J. D., and Sonenberg, N. (2005). Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433, 477–480. doi: 10.1038/nature03205
Riikonen, R., Makkonen, I., Vanhala, R., Turpeinen, U., Kuikka, J., and Kokki, H. (2006). Cerebrospinal fluid insulin-like growth factors IGF-1 and IGF-2 in infantile autism. Dev. Med. Child Neurol. 48, 751–755. doi: 10.1017/S0012162206001605
Rinaldi, T., Kulangara, K., Antoniello, K., and Markram, H. (2007). Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc. Natl. Acad. Sci. U.S.A. 104, 13501–13506. doi: 10.1073/pnas.0704391104
Rodier, P. M., Ingram, J. L., Tisdale, B., and Croog, V. J. (1997). Linking etiologies in humans and animal models: studies of autism. Reprod. Toxicol. 11, 417–422. doi: 10.1016/S0890-6238(97)80001-U
Rolf, L. H., Haarmann, F. Y., Grotemeyer, K. H., and Kehrer, H. (1993). Serotonin and amino acid content in platelets of autistic children. Acta Psychiatr. Scand. 87, 312–316. doi: 10.1111/j.1600-0447.1993.tb03378.x
Rosenbrock, H., Kramer, G., Hobson, S., Koros, E., Grundl, M., Grauert, M., et al. (2010). Functional interaction of metabotropic glutamate receptor 5 and NMDA-receptor by a metabotropic glutamate receptor 5 positive allosteric modulator. Eur. J. Pharmacol. 639, 40–46. doi: 10.1016/j.ejphar.2010.02.057
Rudolph, U., and Knoflach, F. (2011). Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat. Rev. Drug Discov. 10, 685–697. doi: 10.1038/nrd3502
Rutter, M. (2000). Genetic studies of autism: from the 1970s into the millennium. J. Abnorm. Child Psychol. 28, 3–14. doi: 10.1023/A:1005113900068
Sadakata, T., Washida, M., Iwayama, Y., Shoji, S., Sato, Y., Ohkura, T., et al. (2007). Autistic-like phenotypes in Cadps2-knockout mice and aberrant CADPS2 splicing in autistic patients. J. Clin. Invest. 117, 931–943. doi: 10.1172/JCI29031
Salinger, W. L., Ladrow, P., and Wheeler, C. (2003). Behavioral phenotype of the reeler mutant mouse: effects of RELN gene dosage and social isolation. Behav. Neurosci. 117, 1257–1275. doi: 10.1037/0735-7044.117.6.1257
Sallmann, S., Juttler, E., Prinz, S., Petersen, N., Knopf, U., Weiser, T., et al. (2000). Induction of interleukin-6 by depolarization of neurons. J. Neurosci. 20, 8637–8642.
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. doi: 10.1038/nature10945
Santini, E., Huynh, T. N., MacAskill, A. F., Carter, A. G., Pierre, P., Ruggero, D., et al. (2013). Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 493, 411–415. doi: 10.1038/nature11782
Sato, D., Lionel, A. C., Leblond, C. S., Prasad, A., Pinto, D., Walker, S., et al. (2012). SHANK1 Deletions in Males with Autism Spectrum Disorder. Am. J. Hum. Genet. 90, 879–887. doi: 10.1016/j.ajhg.2012.03.017
Schmeisser, M. J., Ey, E., Wegener, S., Bockmann, J., Stempel, A. V., Kuebler, A., et al. (2012). Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486, 256–260.
Schneider, T., and Przewlocki, R. (2005). Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology 30, 80–89. doi: 10.1038/sj.npp.1300518
Schumann, C. M., and Amaral, D. G. (2006). Stereological analysis of amygdala neuron number in autism. J. Neurosci. 26, 7674–7679. doi: 10.1523/JNEUROSCI.1285-06.2006
Schumann, C. M., Hamstra, J., Goodlin-Jones, B. L., Lotspeich, L. J., Kwon, H., Buonocore, M. H., et al. (2004). The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. J. Neurosci. 24, 6392–6401. doi: 10.1523/JNEUROSCI.1297-04.2004
Schuster, G. M., and Schmidt, W. J. (1992). D-cycloserine reverses the working memory impairment of hippocampal-lesioned rats in a spatial learning task. Eur. J. Pharmacol. 224, 97–98. doi: 10.1016/0014-2999(92)94825-G
Seeman, P., Caruso, C., and Lasaga, M. (2008). Memantine agonist action at dopamine D2High receptors. Synapse 62, 149–153. doi: 10.1002/syn.20472
Sehgal, S. N., Baker, H., and Vezina, C. (1975). Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J. Antibiot. 28, 727–732. doi: 10.7164/antibiotics.28.727
Serajee, F. J., Zhong, H., Nabi, R., and Huq, A. H. (2003). The metabotropic glutamate receptor 8 gene at 7q31: partial duplication and possible association with autism. J. Med. Genet. 40:e42.
Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., et al. (2002). Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35, 243–254. doi: 10.1016/S0896-6273(02)00768-7
Sheng, M., and Kim, E. (2000). The Shank family of scaffold proteins. J. Cell Sci. 113(Pt 11), 1851–1856.
Shifman, S., Johannesson, M., Bronstein, M., Chen, S. X., Collier, D. A., Craddock, N. J., et al. (2008). Genome-wide association identifies a common variant in the reelin gene that increases the risk of schizophrenia only in women. PLoS Genet. 4:e28. doi: 10.1371/journal.pgen.0040028
Silverman, J. L., Smith, D. G., Rizzo, S. J., Karras, M. N., Turner, S. M., Tolu, S. S., et al. (2012). Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci. Transl. Med. 4:131ra51. doi: 10.1126/scitranslmed.3003501
Silverman, J. L., Tolu, S. S., Barkan, C. L., and Crawley, J. N. (2010a). Repetitive self-grooming behavior in the BTBR mouse model of autism is blocked by the mGluR5 antagonist MPEP. Neuropsychopharmacology 35, 976–989.
Silverman, J. L., Yang, M., Lord, C., and Crawley, J. N. (2010b). Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 11, 490–502.
Silverman, J. L., Turner, S. M., Barkan, C. L., Tolu, S. S., Saxena, R., Hung, A. Y., et al. (2011). Sociability and motor functions in Shank1 mutant mice. Brain Res. 1380, 120–137. doi: 10.1016/j.brainres.2010.09.026
Simonoff, E., Pickles, A., Charman, T., Chandler, S., Loucas, T., and Baird, G. (2008). Psychiatric disorders in children with autism spectrum disorders: prevalence, comorbidity, and associated factors in a population-derived sample. J. Am. Acad. Child Adolesc. Psychiatry 47, 921–929. doi: 10.1097/CHI.0b013e318179964f
Singh, K., Sun, S., and Vezina, C. (1979). Rapamycin (AY-22,989), a new antifungal antibiotic. IV. Mechanism of action. J. Antibiot. 32, 630–645. doi: 10.7164/antibiotics.32.630
Singh, V. K., Warren, R., Averett, R., and Ghaziuddin, M. (1997). Circulating autoantibodies to neuronal and glial filament proteins in autism. Pediatr. Neurol. 17, 88–90. doi: 10.1016/S0887-8994(97)00045-3
Sirvio, J., Ekonsalo, T., Riekkinen, P. Jr., Lahtinen, H., and Riekkinen, P. Sr. (1992). D-cycloserine, a modulator of the N-methyl-D-aspartate receptor, improves spatial learning in rats treated with muscarinic antagonist. Neurosci. Lett. 146, 215–218. doi: 10.1016/0304-3940(92)90081-H
Skaar, D. A., Shao, Y., Haines, J. L., Stenger, J. E., Jaworski, J., Martin, E. R., et al. (2005). Analysis of the RELN gene as a genetic risk factor for autism. Mol. Psychiatry 10, 563–571. doi: 10.1038/sj.mp.4001614
Smith, S. E., Li, J., Garbett, K., Mirnics, K., and Patterson, P. H. (2007). Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 27, 10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007
Sousa, I., Clark, T. G., Toma, C., Kobayashi, K., Choma, M., Holt, R., et al. (2009). MET and autism susceptibility: family and case-control studies. Eur. J. Hum. Genet. 17, 749–758. doi: 10.1038/ejhg.2008.215
Sparks, B. F., Friedman, S. D., Shaw, D. W., Aylward, E. H., Echelard, D., Artru, A. A., et al. (2002). Brain structural abnormalities in young children with autism spectrum disorder. Neurology 59, 184–192. doi: 10.1212/WNL.59.2.184
Spooren, W., Lindemann, L., Ghosh, A., and Santarelli, L. (2012). Synapse dysfunction in autism: a molecular medicine approach to drug discovery in neurodevelopmental disorders. Trends Pharmacol. Sci. 33, 669–684. doi: 10.1016/j.tips.2012.09.004
Stark, K. L., Xu, B., Bagchi, A., Lai, W. S., Liu, H., Hsu, R., et al. (2008). Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 40, 751–760. doi: 10.1038/ng.138
Stefani, M. R., and Moghaddam, B. (2010). Activation of type 5 metabotropic glutamate receptors attenuates deficits in cognitive flexibility induced by NMDA receptor blockade. Eur. J. Pharmacol. 639, 26–32. doi: 10.1016/j.ejphar.2010.01.028
Stubbs, E. G. (1976). Autistic children exhibit undetectable hemagglutination-inhibition antibody titers despite previous rubella vaccination. J. Autism Child. Schizophr. 6, 269–274. doi: 10.1007/BF01543467
Sudhof, T. C. (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911. doi: 10.1038/nature07456
Sun, W., Funakoshi, H., and Nakamura, T. (2002). Localization and functional role of hepatocyte growth factor (HGF) and its receptor c-met in the rat developing cerebral cortex. Brain Res. Mol. Brain Res. 103, 36–48. doi: 10.1016/S0169-328X(02)00168-7
Tabuchi, K., Blundell, J., Etherton, M. R., Hammer, R. E., Liu, X., Powell, C. M., et al. (2007). A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 318, 71–76. doi: 10.1126/science.1146221
Tain, L. S., Mortiboys, H., Tao, R. N., Ziviani, E., Bandmann, O., and Whitworth, A. J. (2009). Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat. Neurosci. 12, 1129–1135. doi: 10.1038/nn.2372
Takayanagi, Y., Yoshida, M., Bielsky, I. F., Ross, H. E., Kawamata, M., Onaka, T., et al. (2005). Pervasive social deficits, but normal parturition, in oxytocin receptor-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 102, 16096–16101. doi: 10.1073/pnas.0505312102
Testa, C. M., Standaert, D. G., Landwehrmeyer, G. B., Penney, J. B., Jr., and Young, A. B. (1995). Differential expression of mGluR5 metabotropic glutamate receptor mRNA by rat striatal neurons. J. Comp. Neurol. 354, 241–252. doi: 10.1002/cne.903540207
Toma, C., Hervas, A., Torrico, B., Balmana, N., Salgado, M., Maristany, M., et al. (2013). Analysis of two language-related genes in autism: a case-control association study of FOXP2 and CNTNAP2. Psychiatr. Genet. 23, 82–85. doi: 10.1097/YPG.0b013e32835d6fc6
Troca-Marin, J. A., Alves-Sampaio, A., and Montesinos, M. L. (2011). An increase in basal BDNF provokes hyperactivation of the Akt-mammalian target of rapamycin pathway and deregulation of local dendritic translation in a mouse model of Down's syndrome. J. Neurosci. 31, 9445–9455. doi: 10.1523/JNEUROSCI.0011-11.2011
Troca-Marin, J. A., Alves-Sampaio, A., and Montesinos, M. L. (2012). Deregulated mTOR-mediated translation in intellectual disability. Prog. Neurobiol. 96, 268–282. doi: 10.1016/j.pneurobio.2012.01.005
Tropea, D., Giacometti, E., Wilson, N. R., Beard, C., McCurry, C., Fu, D. D., et al. (2009). Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 106, 2029–2034. doi: 10.1073/pnas.0812394106
Tropea, D., Kreiman, G., Lyckman, A., Mukherjee, S., Yu, H., Horng, S., et al. (2006). Gene expression changes and molecular pathways mediating activity-dependent plasticity in visual cortex. Nat. Neurosci. 9, 660–668. doi: 10.1038/nn1689
Tsai, P. T., Hull, C., Chu, Y., Greene-Colozzi, E., Sadowski, A. R., Leech, J. M., et al. (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 488, 647–651. doi: 10.1038/nature11310
Tu, J. C., Xiao, B., Naisbitt, S., Yuan, J. P., Petralia, R. S., Brakeman, P., et al. (1999). Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 23, 583–592. doi: 10.1016/S0896-6273(00)80810-7
Uslaner, J. M., Parmentier-Batteur, S., Flick, R. B., Surles, N. O., Lam, J. S., McNaughton, C. H., et al. (2009). Dose-dependent effect of CDPPB, the mGluR5 positive allosteric modulator, on recognition memory is associated with GluR1 and CREB phosphorylation in the prefrontal cortex and hippocampus. Neuropharmacology. 57, 531–538. doi: 10.1016/j.neuropharm.2009.07.022
Valnegri, P., Khelfaoui, M., Dorseuil, O., Bassani, S., Lagneaux, C., Gianfelice, A., et al. (2011a). A circadian clock in hippocampus is regulated by interaction between oligophrenin-1 and Rev-erbalpha. Nat. Neurosci. 14, 1293–1301.
Valnegri, P., Montrasio, C., Brambilla, D., Ko, J., Passafaro, M., and Sala, C. (2011b). The X-linked intellectual disability protein IL1RAPL1 regulates excitatory synapse formation by binding PTPdelta and RhoGAP2. Hum. Mol. Genet. 20, 4797–4809.
van Slegtenhorst, M., de Hoogt, R., Hermans, C., Nellist, M., Janssen, B., Verhoef, S., et al. (1997). Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277, 805–808. doi: 10.1126/science.277.5327.805
Vargas, D. L., Nascimbene, C., Krishnan, C., Zimmerman, A. W., and Pardo, C. A. (2005). Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 57, 67–81. doi: 10.1002/ana.20315
Veenstra-Vanderweele, J., Cook, E. Jr., and Lombroso, P. J. (2003). Genetics of childhood disorders: XLVI. Autism, part 5: genetics of autism. J. Am. Acad. Child Adolesc. Psychiatry 42, 116–118. doi: 10.1097/00004583-200301000-00018
Vernes, S. C., Newbury, D. F., Abrahams, B. S., Winchester, L., Nicod, J., Groszer, M., et al. (2008). A functional genetic link between distinct developmental language disorders. N. Engl. J. Med. 359, 2337–2345. doi: 10.1056/NEJMoa0802828
Vezina, C., Kudelski, A., and Sehgal, S. N. (1975). Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 28, 721–726. doi: 10.7164/antibiotics.28.721
Vincent, J. B., Herbrick, J. A., Gurling, H. M., Bolton, P. F., Roberts, W., and Scherer, S. W. (2000). Identification of a novel gene on chromosome 7q31 that is interrupted by a translocation breakpoint in an autistic individual. Am. J. Hum. Genet. 67, 510–514. doi: 10.1086/303005
Voineagu, I., Wang, X., Johnston, P., Lowe, J. K., Tian, Y., Horvath, S., et al. (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. doi: 10.1038/nature10110
Wahlstrom, J., Gillberg, C., Gustavson, K. H., and Holmgren, G. (1986). Infantile autism and the fragile X. A Swedish multicenter study. Am. J. Med. Genet. 23, 403–408. doi: 10.1002/ajmg.1320230132
Walsh, T., McClellan, J. M., McCarthy, S. E., Addington, A. M., Pierce, S. B., Cooper, G. M., et al. (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543. doi: 10.1126/science.1155174
Wang, K., Zhang, H., Ma, D., Bucan, M., Glessner, J. T., Abrahams, B. S., et al. (2009). Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459, 528–533. doi: 10.1038/nature07999
Wang, L., Jia, M., Yue, W., Tang, F., Qu, M., Ruan, Y., et al. (2008). Association of the ENGRAILED 2 (EN2) gene with autism in Chinese Han population. Am. J. Med. Genet. Part B Neuropsychiat. Genet. 147B, 434–438.
Wang, X., McCoy, P. A., Rodriguiz, R. M., Pan, Y., Je, H. S., Roberts, A. C., et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20, 3093–3108. doi: 10.1093/hmg/ddr212
Warburton, P., Baird, G., Chen, W., Morris, K., Jacobs, B. W., Hodgson, S., et al. (2000). Support for linkage of autism and specific language impairment to 7q3 from two chromosome rearrangements involving band 7q31. Am. J. Med. Genet. 96, 228–234.
Wassink, T. H., Piven, J., Vieland, V. J., Huang, J., Swiderski, R. E., Pietila, J., et al. (2001). Evidence supporting WNT2 as an autism susceptibility gene. Am. J. Med. Genet. 105, 406–413. doi: 10.1002/ajmg.1401
Wei, H., Dobkin, C., Sheikh, A. M., Malik, M., Brown, W. T., and Li, X. (2012). The therapeutic effect of memantine through the stimulation of synapse formation and dendritic spine maturation in autism and fragile X syndrome. PLoS ONE 7:e36981. doi: 10.1371/journal.pone.0036981
Wei, H., Zou, H., Sheikh, A. M., Malik, M., Dobkin, C., Brown, W. T., et al. (2011). IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J. Neuroinflamm. 8, 52. doi: 10.1186/1742-2094-8-52
Weiss, L. A., Arking, D. E., Gene Discovery Project of Johns Hopkins and the Autism Consortium, Daly, M. J., and Chakravarti, A. (2009). A genome-wide linkage and association scan reveals novel loci for autism. Nature 461, 802–808. doi: 10.1038/nature08490
Williams, D. L. Jr., and Lindsley, C. W. (2005). Discovery of positive allosteric modulators of metabotropic glutamate receptor subtype 5 (mGluR5). Curr. Top. Med. Chem. 5, 825–846. doi: 10.2174/1568026054750290
Wilson, C., Idziaszczyk, S., Parry, L., Guy, C., Griffiths, D. F., Lazda, E., et al. (2005). A mouse model of tuberous sclerosis 1 showing background specific early post-natal mortality and metastatic renal cell carcinoma. Hum. Mol. Genet. 14, 1839–1850. doi: 10.1093/hmg/ddi190
Wilson, H. L., Wong, A. C., Shaw, S. R., Tse, W. Y., Stapleton, G. A., Phelan, M. C., et al. (2003). Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J. Med. Genet. 40, 575–584. doi: 10.1136/jmg.40.8.575
Wlaz, P., Baran, H., and Loscher, W. (1994). Effect of the glycine/NMDA receptor partial agonist, D-cycloserine, on seizure threshold and some pharmacodynamic effects of MK-801 in mice. Eur. J. Pharmacol. 257, 217–225. doi: 10.1016/0014-2999(94)90132-5
Won, H., Lee, H.-R., Gee, H. Y., Mah, W., Kim, J.-I., Lee, J., et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265. doi: 10.1038/nature11208
Wu, H. M., Tzeng, N. S., Qian, L., Wei, S. J., Hu, X., Chen, S. H., et al. (2009). Novel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology 34, 2344–2357.
Wu, S., Jia, M., Ruan, Y., Liu, J., Guo, Y., Shuang, M., et al. (2005). Positive association of the oxytocin receptor gene (OXTR) with autism in the Chinese Han population. Biol. Psychiatry 58, 74–77. doi: 10.1016/j.biopsych.2005.03.013
Yan, Q. J., Rammal, M., Tranfaglia, M., and Bauchwitz, R. P. (2005). Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49, 1053–1066. doi: 10.1016/j.neuropharm.2005.06.004
Yizhar, O., Fenno, L. E., Prigge, M., Schneider, F., Davidson, T. J., O'Shea, D. J., et al. (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178. doi: 10.1038/nature10360
Yoshida, T., Yasumura, M., Uemura, T., Lee, S. J., Ra, M., Taguchi, R., et al. (2011). IL-1 receptor accessory protein-like 1 associated with mental retardation and autism mediates synapse formation by trans-synaptic interaction with protein tyrosine phosphatase delta. J. Neurosci. 31, 13485–13499. doi: 10.1523/JNEUROSCI.2136-11.2011
Young, D. M., Schenk, A. K., Yang, S. B., Jan, Y. N., and Jan, L. Y. (2010). Altered ultrasonic vocalizations in a tuberous sclerosis mouse model of autism. Proc. Natl. Acad. Sci. U.S.A. 107, 11074–11079. doi: 10.1073/pnas.1005620107
Yrigollen, C. M., Han, S. S., Kochetkova, A., Babitz, T., Chang, J. T., Volkmar, F. R., et al. (2008). Genes controlling affiliative behavior as candidate genes for autism. Biol. Psychiatry 63, 911–916. doi: 10.1016/j.biopsych.2007.11.015
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., et al. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149. doi: 10.1038/nn1754
Zemni, R., Bienvenu, T., Vinet, M. C., Sefiani, A., Carrie, A., Billuart, P., et al. (2000). A new gene involved in X-linked mental retardation identified by analysis of an X;2 balanced translocation. Nat. Genet. 24, 167–170. doi: 10.1038/72829
Zeng, H., Shen, E. H., Hohmann, J. G., Oh, S. W., Bernard, A., Royall, J. J., et al. (2012). Large-scale cellular-resolution gene profiling in human neocortex reveals species-specific molecular signatures. Cell 149, 483–496. doi: 10.1016/j.cell.2012.02.052
Keywords: autism spectrum disorder, therapeutics, genetics, animal model, synapse, synaptopathy
Citation: Won H, Mah W and Kim E (2013) Autism spectrum disorder causes, mechanisms, and treatments: focus on neuronal synapses. Front. Mol. Neurosci. 6:19. doi: 10.3389/fnmol.2013.00019
Received: 03 April 2013; Accepted: 16 July 2013;
Published online: 05 August 2013.
Edited by:
Nicola Maggio, The Chaim Sheba Medical Center, IsraelReviewed by:
Carlo Sala, CNR Institute of Neuroscience, ItalyLior Greenbaum, Hadassah Medical Center, Israel
Copyright © 2013 Won, Mah and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eunjoon Kim, Center for Synaptic Brain Dysfunctions, Institute for Basic Science, and Department of Biological Sciences, Korea Advanced Institute of Science and Technology, Kuseong-dong, Yuseong-ku, Daejeon 305-701, South Korea e-mail:a2ltZUBrYWlzdC5hYy5rcg==