
 Gabriel Zimmerman2,3‡
Gabriel Zimmerman2,3‡



- 1Departments of Physiology and Neurobiology, Zlotowski Center of Neuroscience, Ben-Gurion University of the Negev, Beer-Sheva, Israel
- 2Interdisciplinary Center for Neural Computation, The Hebrew University of Jerusalem, Jerusalem, Israel
- 3The Edmond and Lily Safra Center of Brain Sciences, The Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem, Israel
Recent studies show a key role of brain inflammation in epilepsy. However, the mechanisms controlling brain immune response are only partly understood. In the periphery, acetylcholine (ACh) release by the vagus nerve restrains inflammation by inhibiting the activation of leukocytes. Recent reports suggested a similar anti-inflammatory effect for ACh in the brain. Since brain cholinergic dysfunctions are documented in epileptic animals, we explored changes in brain cholinergic gene expression and associated immune response during pilocarpine-induced epileptogenesis. Levels of acetylcholinesterase (AChE) and inflammatory markers were measured using real-time RT-PCR, in-situ hybridization and immunostaining in wild type (WT) and transgenic mice over-expressing the “synaptic” splice variant AChE-S (TgS). One month following pilocarpine, mice were video-monitored for spontaneous seizures. To test directly the effect of ACh on the brain's innate immune response, cytokines expression levels were measured in acute brain slices treated with cholinergic agents. We report a robust up-regulation of AChE as early as 48 h following pilocarpine-induced status epilepticus (SE). AChE was expressed in hippocampal neurons, microglia, and endothelial cells but rarely in astrocytes. TgS mice overexpressing AChE showed constitutive increased microglial activation, elevated levels of pro-inflammatory cytokines 48 h after SE and accelerated epileptogenesis compared to their WT counterparts. Finally we show a direct, muscarine-receptor dependant, nicotine-receptor independent anti-inflammatory effect of ACh in brain slices maintained ex vivo. Our work demonstrates for the first time, that ACh directly suppresses brain innate immune response and that AChE up-regulation after SE is associated with enhanced immune response, facilitating the epileptogenic process. Our results highlight the cholinergic system as a potential new target for the prevention of seizures and epilepsy.
Introduction
Accumulating experimental evidence indicates that brain immune response and inflammatory mediators decrease the threshold for individual seizures and contributes to the process of epileptogenesis (for review see: Vezzani and Granata, 2005; Rijkers et al., 2009; Vezzani et al., 2011). Therefore, understanding the mechanisms controlling the extent and duration of the brain's inflammatory response to status epilepticus (SE) and injury may promote the understanding of epileptogenesis and highlight new therapeutic targets for the prevention and treatment of epilepsy.
Peripheral inflammation is controlled, amongst others, by the cholinergic anti-inflammatory pathway which involves inhibition of innate immune responses by ACh released from the vagus nerve. This mechanism is dependent on the alpha7 subunit of the nicotinic acetylcholine receptor (α7nAChR), which inhibits NF-kappaB nuclear translocation and suppresses cytokine release by monocytes and macrophages (Tracey, 2002, 2009). Centrally acting inhibitors of the ACh hydrolyzing enzyme, acetylcholinesterase (AChE), were shown to enhance the peripheral cholinergic anti-inflammatory pathway via central muscarinic receptors (Pavlov et al., 2006, 2009). Recent reports point toward a cholinergic anti-inflammatory mechanism also in the central nervous system (CNS). In cultured microglia, ACh pre-treatment inhibited lipopolysaccharide (LPS)-induced tumor necrosis factor alpha (TNFα) release via α7nAChR (Shytle et al., 2004). Accordingly, AChE inhibition reduced microglial production of TNFα in a hypoxia model (Wang et al., 2010). AChE inhibitors have also been shown to reduce glial activation and inflammatory cytokine production in experimental autoimmune encephalomyelitis (Nizri et al., 2008) and in a cerebral hypoperfusion model in the rat (Wang et al., 2010).
We recently reported cholinergic dysfunction and up-regulation of AChE in temporal lobe structures of SE-experienced epileptic rats (Zimmerman et al., 2008). These results raised the hypothesis that transcriptionally controlled cholinergic imbalance may have a role in brain immune response during epileptogenesis. We thus used the pilocarpine model of epilepsy to study AChE expression and the role of consequent cholinergic imbalance on brain immune response and epileptogenesis.
Materials and Methods
All animal experiments were approved by the Committee for Ethics in Animal experimentation of the Faculty of Health Sciences, Ben-Gurion University of the Negev. Wild type (WT) FVB/N (Harlan Laboratories, Jerusalem), and transgenic mice over-expressing human AChE-S (TgS) (Beeri et al., 1995) were maintained on a 12 h light/dark cycle and had access to food and water ad libitum. All chemicals were purchased from Sigma unless otherwise mentioned.
Pilocarpine Treatment
The well-established pilocapine model for temporal lobe epilepsy was induced (Cavalheiro et al., 1996; Shibley and Smith, 2002; Chen et al., 2005). Pilocarpine (290–340 mg/kg i.p.) was injected to adult (8–16 weeks old, 26–34 g BW) male mice 15 min after pre-treatment with methyl-scopolamine (1.5 mg/kg i.p.). Animals were observed for 2 h after pilocarpine injection, and seizures were evaluated according to a modified version of the Racine scale (Racine, 1972; Borges et al., 2003). SE was defined by continuous seizure activity consisting of stages 3.5–5 seizures, five taken as the onset. Forty minutes from onset, SE was interrupted by injection of diazepam (4 mg/kg i.p.). Control mice were treated with the same protocol, except that saline was injected instead of pilocarpine, followed, 1 h later, by diazepam.
Epilepsy was diagnosed in the presence of at least one spontaneous stage five seizure observed during a 48 h period of video-monitoring, 1–3 months after SE (in 11 WT mice). For the comparison of spontaneous seizures frequency between strains, WT (eight mice) and TgS (12 mice) were simultaneously video-monitored 1 month following SE.
Processing of Tissue Samples
Forty-eight hours after SE, animals were deeply anesthetized with isofluorane and decapitated. For enzymatic activity and RT-PCR, brains were quickly removed and the hippocampus was dissected and frozen in liquid nitrogen. All tissues were stored fresh at −80°C. For native protein extraction, frozen tissue was thawed and homogenized on ice with 200 μl homogenization buffer (0.01 M sodium phosphate pH 7.4, 1% triton, 4°C). Lysates were pre-cleared by centrifugation at 17,000 rpm at 4°C for 30 min, and the total protein concentration of the supernatant was determined using Bradford solution (Bio-Rad, Israel) (Bradford, 1976). Bovine serum albumin (BSA) was used as a standard. The supernatants were kept on ice until the cholinesterase assay was performed within 2 h after homogenization. For histological staining, hemispheres were either fixed in 4% paraformaldehyde (PFA) in phosphate buffer saline (PBS) for 48 h and paraffin-embedded, or horizontally sliced to 400 μm sections, fixed in 4% PFA in phosphate buffer (PB) (0.1 M Na2HPO4, 0.02 M NaH2PO4, pH 7.4) and kept in 4°C until re-slicing. Slices were immersed in 30% sucrose overnight and then re-sliced to 30 μm sections in a freezing microtome (Leica Microsystems, Wetzlar, Germany).
Karnovsky-Root Staining in Non-Denaturing Electrophoresis Gel and Brain Sections
Electrophoresis of active AChE was performed in non-denaturing conditions. Fifty milligrams protein from each supernatant was loaded on 7.5% vertical polyacrylamide gel at 4°C for 1 h (Bon et al., 1988). Migration was performed in the presence of 0.25% Triton X-100; AChE activity was revealed as reported by Karnovsky and Roots (Karnovsky and Roots, 1964; Kaufer et al., 1998). Bands intensity was quantified using a homemade MatLab script. AChE activity staining was performed in 200 μm fresh horizontal sections using the same staining solution (Zimmerman et al., 2008). Images were taken with a stereomicroscope (Zeiss, Lumar.V12 and an AxiCam MRc5).
Real-Time RT-PCR, In-situ Hybridization and Immunostaining
Real-time RT-PCR was performed using a standard protocol as previously described (Zimmerman et al., 2008). Total RNA was extracted using Absolutely RNA Miniprep Kit (Stratagene, CA, USA), and reverse transcribed (300 ng) using Verso™ cDNA Kit (Thermo, MA, USA). RT-PCR was performed using ABsolute™ QPCR SYBR® Green ROX (ABgene, UK) in a 7900 HT Sequence Detection System (Applied Biosystems, CA, USA). Quantification was assessed at the logarithmic phase of the PCR reaction. The PCR annealing temperature was 60°C for all primer pairs (Table 1). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a house-keeping gene.
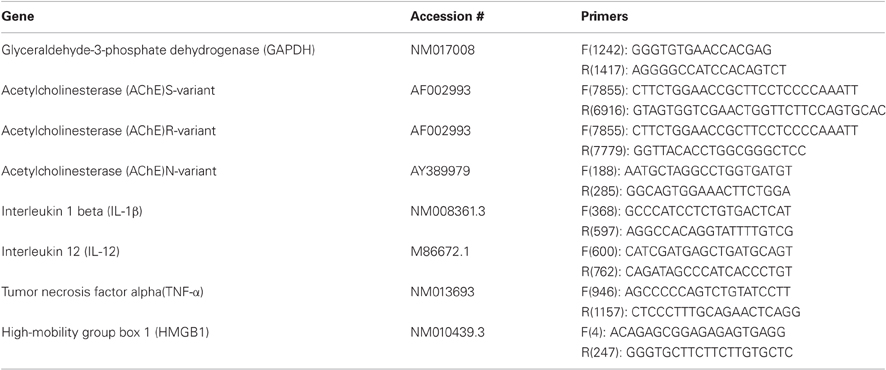
Table 1. Primer pairs.
For double labeling histological experiments we combined in-situ hybridization (FISH) with immunofluorescence (IF). Stainings were done on seven μm-thick paraffin-embedded sagittal sections or 30 μm-thick freezing microtome horizontal sections. Prior to staining, 30 μm sections were washed three times in PB and mounted on SuperFrost slides (Thermo, MA, USA). FISH was performed as previously described (Berson et al., 2008) with slight modifications: for antigen retrieval, slides were soaked in 0.01 M citrate buffer pH six, heated in the micro-wave for 15 min, washed in double-distilled water and with PBT. Hybridization mix [including 50-mer 5′-biotinylated, 2-O-methylprotected complementary RNA probes (Table 2) 10 μg/ml] was applied for 10 min in 60°C and then for 90 min in 52°C (90 min and overnight respectively for 30 μm sections). After washing with TBST buffer (25 mM Tris–HCl pH 7.5, 136 mM NaCl, 2.7 mM KCl, 0.05% Tween-20), slides were incubated in blocking solution for 30 min. Primary antibodies (Table 3) diluted in the blocking solution were incubated (90 min) at room temperature (30 μm sections—overnight in 4°C). After three rinses in TBST, sections were incubated in a mix of streptavidin-conjugated Cy2 and Cy3 or Cy5 conjugated goat IgG (Jackson Immunoresearch, 1:100 dilution) in TBST for 1 h at room temperature. Slides were then washed with TBST, and mounted with ImmunoMount (Thermo, MA, USA). Slides stained for Iba1 were incubated in PBS with 0.5% Triton X-100 for 1 h, before blocking.

Table 2. Complementary RNA probes.
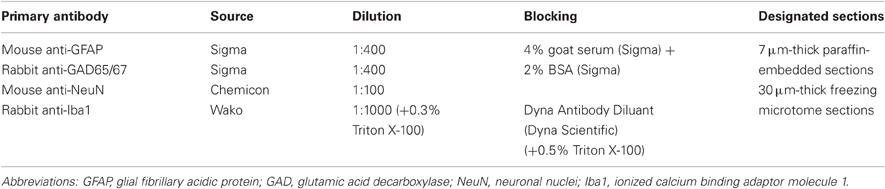
Table 3. Dilution, blocking, and designated sections for all primary antibodies.
Acute Slices Preparation and Drug Application
Preparation and maintenance of brain slices ex-vivo was performed using standard procedures as previously reported (Zimmerman et al., 2008). In short, mice (2–4 month old) were deeply anesthetized; brains quickly removed and 400 μm horizontal slices were obtained (NVSLM1—motorized advance vibroslice, WPI, USA). Slices were maintained in a standard interface chamber at 36 ± 1°C and were superfused with artificial cerebrospinal fluid (ACSF) contained (in mM): NaCl, 129; NaHCO3, 21; NaH2PO4, 1.25; MgSO4, 1.8; CaCl2, 1.6; KCl, 3; glucose, 10; pH 7.4. Treated slices were incubated simultaneously with matched controls (in separate chambers). Drugs were added to the bathing solution after 30 min incubation with ACSF for 4 h. Slices were then transferred to liquid nitrogen for RT-PCR. Drugs included carbamylcholine chloride (CCh, 50 μM), acetylcholine (ACh 10 or 50 μM), physostigmine (1 μM), Atropine (1 μM), Mecamylamine (50 μM), α-bungarotoxin (100 μM) and tetrodotoxine (TTX, 1 μM).
Microscopy and Image Acquisition
All fluorescent images were obtained using XYZ scanning with an Olympus FluoView FV1000 confocal microscope (Olympus, Hamburg, Germany) or with a Nikon inverted TI microscope (Nikon, Japan) and a cooled 14 bit CCD camera (Coolsnap HQ2, Photometrics, Tucson AZ). For quantification, Six to eight images (×20) from non-overlapping hippocampal fields were taken from each slice (three slices per animal, two to three animals per group) using the same acquisition parameters. Analysis was done using a homemade MatLab script: intensity values for each image were normalized to the mean. A constant cut-off value was set to establish a threshold defining background (“black”) and signal (“white”) pixels. The total number of white pixels (stained area) for each binary image was summed and that value was later used for statistical analysis.
Statistical Analysis
The non-parametric Mann–Whitney test, One-Way ANOVA, Chi-square (χ2), and two-tailed independent student t-test were applied (SPSS; SPSS Inc., Chicago, USA) as mentioned in the text. P < 0.05 was considered significant. Data is expressed as the mean ± SEM.
Results
SE was achieved in 75.7% of mice injected with pilocarpine (n = 53 out of 70) with a mortality rate of 22.6%. In WT mice, 48 h video-monitoring at 3 months after SE revealed a high percentage (n = 9 out of 11, 82%) of epileptic mice (≥1 spontaneous unprovoked stage 5 seizure).
AChE is Up-Regulated During Epileptogenesis
As previous studies showed AChE upregulation in chronic epileptic animals, we studied AChE expression 48 h after SE to challenge the hypothesis that cholinergic imbalance occurs early during the epileptogenesis process. All known alternatively spliced AChE mRNA transcripts, including AChE-S, AChE-R, and N-AChE were found to be significantly up-regulated in the hippocampus of treated mice compared to saline-injected controls (Figure 1A). Karnovsky–Root staining confirmed increased AChE activity in hippocampal homogenates (Figure 1B) as well as in sections obtained from SE-experienced mice (Figure 1C). Within the hippocampus, AChE activity was most prominent in the stratum molecular of the dentate gyrus, the stratum oriens of the CA1 and CA3 regions, the subiculum and the entorhinal cortex (Figure 1C).
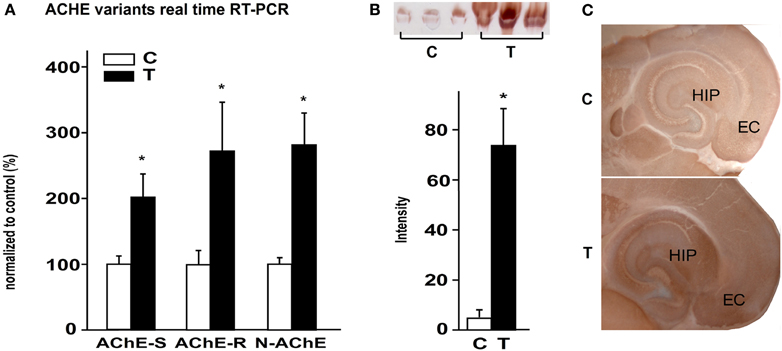
Figure 1. AChE up-regulation 48 h post SE. (A) expression levels of AChE-S, AChE-R, and N-AChE 48 h after SE, as revealed by real-time RT-PCR (p = 0.05, 0.053, and 0.025 respectively; n = 5 controls, n = 3 or 4 treated; mean ± SEM; Mann–Whitney test). (B) Top: Karnovsky–Roots staining on non-denatured activity gel of control and 48 h after SE samples. Bottom: gel quantification (p = 0.05; n = 3; mean ± SEM; Mann–Whitney test). (C) Karnovsky–Roots staining of 200 μm horizontal brain sections from control and 48 h post SE mice. C, control; T, treated.
To identify specific cell populations expressing AChE mRNA following SE, we combined AChE FISH with immunostaining against specific cell-type markers. FISH showed intensified AChE expression in neurons within all regions of the hippocampus, which was most prominent in the pyramidal cell layer of all CA sub-regions (Figures 2A,B,G). In addition, ACHE mRNA was co-localized with GAD positive inhibitory interneurons in all hippocampal sub-regions (Figure 2H). We further searched for AChE expression in glia cells, known to be activated early following SE and suggested to play a role in epileptogenesis (Borges et al., 2003; Turrin and Rivest, 2004; Ivens et al., 2007; Pitkanen et al., 2007; David et al., 2009). GFAP immunostaning indeed indicated a strong astroglial activation after SE (Figures 2C,D); However, while AChE mRNA was observed along blood vessels, probably in endothelial cells, co-labeling experiments failed to reveal significant ACHE mRNA expression within GFAP-positive astrocytes (Figures 2I,J). Conversely, in SE-exposed mice we found a robust increase in immunolabeling for the microglia marker, Iba1 (Figures 2E,F; p < 0.001; control: n = 47, treated: n = 70 fields from three sections per animal, 2/3 animals per group; independent t-test), co-labeled with a strong FISH signal for AChE mRNA (Figures 2K,L). In addition, Iba1 positive cells following SE presented perikaryal hypertrophy and processes retraction (Figure 2L), a clear morphological pattern for their activation (Rappold et al., 2006).
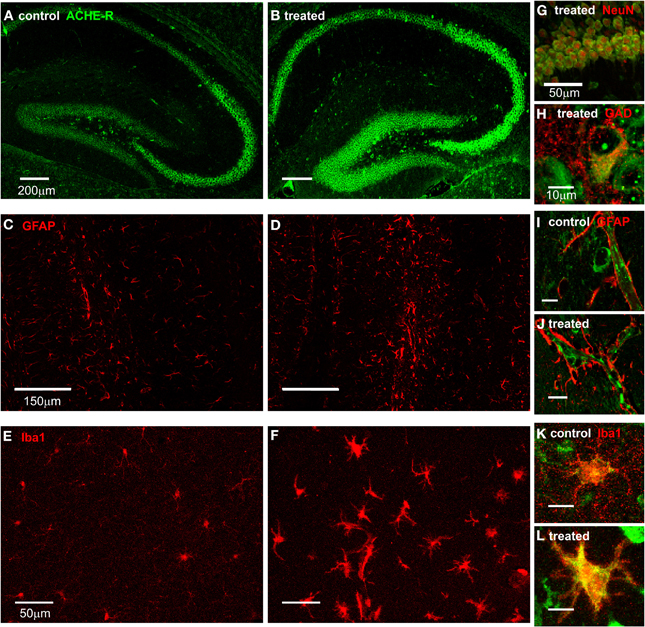
Figure 2. AChE expression in the hippocampus. (A,B) FISH for AChE-R in the hippocampus of control (A) and 48 h after SE (B) mice. (C,D) GFAP staining in the hippocampus of control (C) and 48 h post SE (D) mice. (E,F) Iba1 staining in the hippocampus of control (E) and 48 h post SE (F) mice. (G) Double labeling of AChE (green) and NeuN (red) in CA1 area of a treated animal. (H) Double labeling of AChE (green) and GAD65/67 (red) in the hilus of a treated animal. (I,J) Double labeling of AChE (green) and GFAP (red) in hippocampus of control (I) and treated (J) mice (scale 10 μm). Note the clear positive AChE staining in the blood vessel and the endothelial cells but not in the end fit of GFAP-positive astrocytes surrounding it. (K,L) Double labeling of AChE (green) and Iba1 (red) in hippocampus of control (K) and treated (L) mice (scale 10 μm).
Brain AChE Levels and Local Immune Response
The rapid up-regulation and expression pattern of AChE after SE together with the known cholinergic control of immune response in the periphery led us to challenge the hypothesis that the observed cholinergic imbalance is sufficient to induce a local innate immune response within the brain. We thus tested the immune response in transgenic mice with a constitutive over-expression of the human synaptic variant of acetylcholinesterase (AChE-S) (TgS) before and 48 h following SE. In TgS mice, showing 50% more AChE activity than WT animals (Beeri et al., 1995; and Figure 3A), immunolabeling against Iba1 was significantly increased compared to WT (p = 0.046; WT: n = 47, TgS: n = 70 fields from three sections per animal, 2/3 animals per group; independent t-test). This increase was associated with upregulation of mRNA for high mobility group box 1 (HMGB1)—a pro-inflammatory cytokine-like mediator expressed in activated microglia (Lotze and Tracey, 2005; Kim et al., 2006; Hayakawa et al., 2008) (Figure 3F; p = 0.014). Forty-eight hours following SE both WT and TgS mice showed higher labeling of Iba1 compared to their matched controls (Figure 3B); However, treated TgS mice showed significantly higher (30%) labeling compared to that observed in treated WTs (17%) (p = 0.043; control: n = 47, treated: n = 71 fields from three sections per animal, 2/3 animals per group; independent t-test). In addition, up-regulation of IL-1β and IL-12 was significantly higher in TgS mice compared to WT (Figures 3C,D). SE-induced up-regulation of TNFα mRNA was similar in both WT and TgS mice (Figure 3E). HMGB1 was also upregulated (p = 0.027) in WT exposed to SE and matched the already high levels in non-treated TgS (Figure 3F).
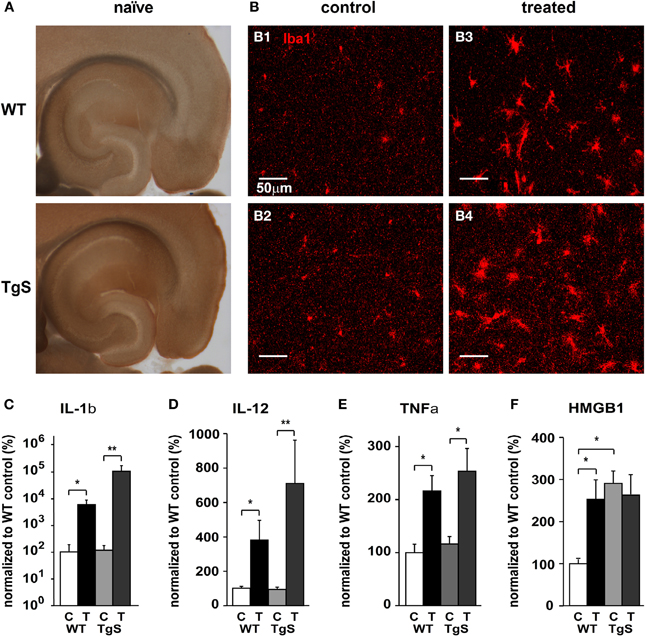
Figure 3. Compared microglia status in WT and TgS mice. (A) Karnovsky–Roots staining of 200 μm horizontal brain sections from naïve WT and TgS mice. (B) Iba1 staining in the hippocampus of control WT (B1), control TgS (B2), treated WT (B3), and treated TgS (B4) mice. (C–F) Real-time RT-PCR for key inflammatory cytokines. (C) IL-1β mRNA relative levels (*p = 0.014; **p = 0.004). (D) IL-12 mRNA relative levels (*p = 0.014; **p = 0.004). (E) TNFα mRNA relative levels. WT: p = 0.027, TgS: p = 0.054. (F) HMGB1 mRNA relative levels. p < 0.05 (see text). (WT: n = 5 controls, n = 4 treated, TgS: n = 6 controls/treated. In F; TgS control: n = 4; C, control; T, treated; Statistical analysis was done using the non-parametric Mann–Whitney test, mean ± SEM.)
TgS Mice Over-Expressing AChE Show Accelerated Epileptogenesis
To test whether the observed increase in immune response in treated TgS mice is associated with altered epileptogenesis (Vezzani et al., 2008), we video-monitored WT and TgS mice for 48 h one month after SE. Importantly, no differences in the acute response to pilocarpine (rate of SE, delay to SE, seizure phenotypes, response to diazepam or survival) were observed between WT and TgS mice (Figures 4A–C). In contrast, video-monitoring at 1 month uncovered a significantly higher rate of TgS mice presenting spontaneous seizures compared to WT mice (Figure 4D). These results support the notion that AChE over-expression is associated with enhanced immune response and facilitated epileptogenesis.
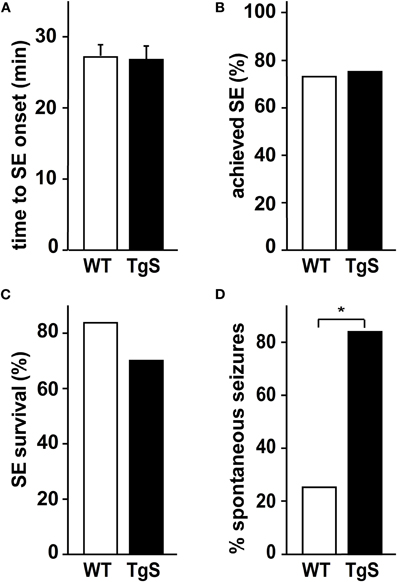
Figure 4. SE and spontaneous seizures in TgS and WT mice. (A) Time (in min) was measured from pilocarpine injection to onset of SE. (B) Percentage of mice achieving SE. (C) Percentage of mice surviving 40 min of SE (total number of injected animals: WT: n = 33, TgS: n = 44). (D) Percentage of mice presenting spontaneous seizures 1 month after SE (*p = 0.019, Chi-square test; number of monitored animals: WT: n = 8, TgS: n = 12).
Cholinergic Activation is Anti-Inflammatory in Hippocampal Slices
Our results suggested that the rapid up-regulation of AChE following SE is associated with enhanced local immune response. To explore to what extent this effect is associated with reduced cholinergic control over the brain innate immune response, we studied the immune response in acute brain slices maintained ex-vitro 4 h following the slicing procedure. We hypothesized that the injury associated with the slicing procedure would activate a significant inflammatory response (Lucas et al., 2006) which could be suppressed by cholinergic stimuli. Indeed, cholinergic activation by 50 μM CCh reduced mRNA levels of IL1β and TNFα (Figure 5A). Similar results were found with low levels of the physiological agonist, ACh (10 μM), in the presence of the AChE inhibitor physostigmine (1 μM, Figure 5A). The addition of TTX did not prevent CCh action on the immune response, suggesting it is not dependent on neuronal firing (Figure 5B). Importantly, while the non-specific muscarinic receptor antagonist atropine blocked the cholinergic-suppression of inflammatory response, the non-specific nicotinic receptor antagonist, mecamylamine (50 μM) or the α7 nicotinic receptor antagonist α-bungarotoxin (100 nM) did not (Figure 5B and data not shown).
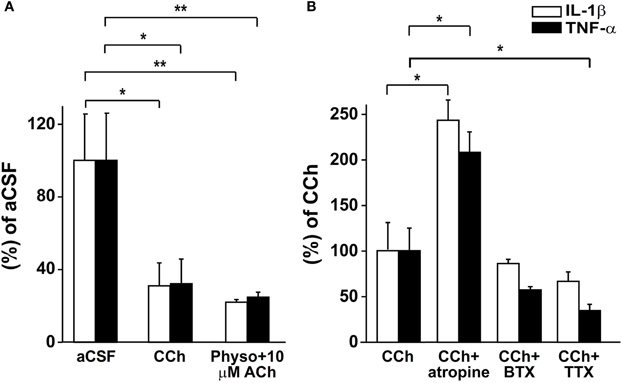
Figure 5. Real-time RT-PCR for IL-1β and TNF-α in acute slices. (A) mRNA relative levels in slices treated with aCSF, 50 μM CCh (*p = 0.025; n = 6) or 1 μM physostigmine with 10 μM ACh (**p = 0.006; n = 5–6). (B) mRNA relative levels in slices treated with 50 μM CCh (n = 5) and 50 μM CCh with 1 μM atropine (*p = 0.028; n = 5), 100 nM BTX (n = 5) or 1 μM TTX (*p = 0.027; n = 4). Statistical analysis was done using the non-parametric Mann–Whitney test, mean ± SEM. Physo, physostigmine; BTX, bungarotoxine.
Discussion
In this study we offer experimental evidence indicating that (1) cholinergic imbalance due to upregulation of AChE within the hippocampus is associated with a local immune response; (2) such imbalance and the associated inflammation following SE is associated with epileptogenesis; and (3) ACh acts directly as a modulator of brain innate immune response and thereby affect epileptogenesis.
AChE is the fastest enzyme in mammalian cells and as the enzyme in charge of executing ACh breakdown it is a key component of cholinergic signaling (Meshorer and Soreq, 2006). AChE levels are controlled via transcriptional and post-transcriptional mechanisms and are dramatically increased under toxicological, physical and psychological stress (Kaufer et al., 1998; Shohami et al., 2000; Meshorer et al., 2002). This fast and robust increase in AChE levels in response to neuronal activation has been suggested to act as an homeostatic control to reduce neuronal excitability (Meshorer et al., 2005b) and on the long-run this response was reported to be associated with enhanced network excitability (Meshorer et al., 2002; Zimmerman et al., 2008). These previous findings raised the hypothesis that AChE will be similarly up-regulated following SE in the pilocarpine model of epilepsy. Indeed, 48 h following SE we recorded increased AChE mRNA and protein levels in SE-experienced mice. Unlike milder and more specific stress manipulations (e.g., psychological) where upregulation was reported to include the readthrough alternatively spliced AChE-R variant (Kaufer et al., 1998; Meshorer et al., 2002), SE was associated with a significant increase in mRNA levels of all three alternatively spliced transcripts (Figure 1). In this study we used the muscarinic agonist, pilocarpine, to induce SE and thus it is possible that AChE upregulation represents a specific response to the activation of muscarinic receptors. While we cannot entirely exclude this possibility, it seems unlikely in light of a similar up-regulation observed in the brains from chronic epileptic rats following kainic acid-induced SE (Zimmerman et al., 2008). The presence of c-fos binding sites in the promoter of the gene encoding AChE (Meshorer et al., 2004) further suggest that its upregulation is part of the transcriptional response to excess neuronal activation. AChE transcripts were found in both principle and inhibitory interneurons, supporting its role in controlling the hyperexcitable network. In addition, following SE high expression levels of AChE mRNA were also found in morphologically identified endothelial cells, suggesting it may have a role in vascular changes observed following SE, specifically the robust increase in blood-brain barrier (BBB) permeability (van Vliet et al., 2007; Weissberg et al., 2011). This notion is supported by reports on compromised BBB in transgenic mice over-expressing AChE (Meshorer et al., 2005a). The activation of the immune system, and specifically HMGB1 upregulation in these transgenic animals (Figure 3) may also underlie BBB opening (Zhang et al., 2011). AChE transcripts were not found in GFAP positive astrocytes, despite their robust activation during SE. These results are consistent with a previous report showing AChE activity in different lines of neuronal and non-neuronal cells except for astrocytes (Thullbery et al., 2005), although mixed astroglia cells were reported to upregulate the readthrough ACHE transcript following oxidative stress in culture (Bond et al., 2006). High expression levels of ACHE transcripts were found in Iba1 positive microglia following SE and to much less extent in control animals. These results together with the well-described brain immune activation following SE (review by Vezzani et al., 2011) raised the hypothesis that AChE upregulation in microglia may have a role in modulating the innate immune response. This hypothesis is supported by previous studies showing that in microglial cell cultures AChE induces changes in cell morphology as well as iNOS upregulation (von Bernhardi et al., 2003). That the AChE effect is likely to be associated with cholinergic signaling is suggested by the observation that ACh inhibits LPS-induced TNFα release in microglia cultures, an effect attenuated by α-bungarotoxin (Shytle et al., 2004). To test directly the role of AChE overexpression on microglia functions we studied transgenic mice with CNS limited, constitutive up-regulation of synaptic AChE-S (Sternfeld et al., 2000). Indeed, the increase in AChE activity led to increased immunostaining of the microglial marker Iba1 and significant upregulation of mRNA levels for HMGB1 (Figure 3), a newly defined cytokine that can be passively released from necrotic cells or actively released from immune activated cells (Wang et al., 1999; Abraham et al., 2000). Our finding showing that AChE over expression in transgenic animals facilitated epileptogenesis are consistent with recent reports showing a proconvulsant effect of HMGB1 in kainic acid treated mice (Maroso et al., 2010). It is worth noticing that TgS mice showed a robust increase in IL-1β expression (18-fold higher than WT mice) 48 h following SE, consistent with the well-described epileptogenic effects of IL-1β (Vezzani et al., 1999; Friedman and Dingledine, 2011). Facilitated epileptogenesis in TgS mice could not be solely attributed to enhanced seizure activity due to increased network sensitivity to ACh (Meshorer et al., 2002) since the latency and duration of SE in response to pilocarpine were similar in WT mice. This strengthens the notion that AChE upregulation following SE is a response to excess neuronal activation rather than a direct effect of pilocarpine. We cannot rule out at this stage the contribution of other epileptogenic mechanisms including vascular changes (i.e., enhanced BBB permeability) (Marchi et al., 2007; Friedman et al., 2009) or changes in peripheral immune response (Fabene et al., 2008; Marchi et al., 2009). When monitoring WTs for 1–3 month, 82% showed spontaneous seizures, while TgS were not monitored at such a late time point. Therefore, it is feasible that the rate of epileptogenesis and not the end result are different between the strains.
The most direct evidence for cholinergic control over the innate immune response is given in the acute slices experiments demonstrating that adding ACh or CCh to the bathing solution suppressed mRNA levels of IL-1β and TNF-α. In accordance with these results, previous studies demonstrated anti-inflammatory effect for ACh and AChE inhibitors in glial cultures (Shytle et al., 2004; Wang et al., 2010) and for AChE inhibitors in animals (Nizri et al., 2008; Wang et al., 2008; Wang et al., 2010). It is important to note that exposure to centrally acting potent AChE inhibitors induces SE and neuroinflammation that may promote epileptogenesis (Dhote et al., 2007, 2012; De Araujo et al., 2010). Thus, it seems clear that AChE inhibition per se, perhaps due to enhanced neuronal excitability is usually associated with increased brain inflammation and the transient increase in ACh levels are not sufficient to block completely the brain innate immune processes associated with SE and chronic recurrent seizures (Pernot et al., 2009). Unlike previous studies (mostly in the peripheral nervous system) (Tracey, 2002, 2007; Shytle et al., 2004; Wang et al., 2008), our findings point to muscarinic dependant activation as the underlying pathway. Although muscarinic receptors in the CNS were shown to control the peripheral nicotinic pathway, hinting to complex interactions between the involved pathways (Pavlov et al., 2006, 2009), these interactions could not explain the observations in the ex-vivo slice preparation. Since recent studies did show that cytokines act as pro-convulsants (Vezzani et al., 2011), their upregulation may also be attributed to a positive feedback enhancement mechanism. However, it is noteworthy that the sodium channel blocker, TTX had no effect on cholinergic-suppression of cytokine production suggesting that this cholinergic effect is independent of neuronal firing.
In summary, our study suggests for the first time, a role for activity-dependant cholinergic imbalance in brain immune response, epileptogenesis and seizures and may offer a new basis for understanding the role of AChE-inhibitors in the treatment of Alzheimer's disease and similar neurodegenerative disorders. Future studies are warranted to delineate the contribution of cholinergic signaling to brain immune response in neurological and neurodegenerative disorders where immune response has been reported (Amor et al., 2010) as well as its potential as a novel target in the treatment of uncontrolled brain immune response.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Sonderforschungsbereich TR3, the Israel Science Foundation (566/07), The German-Israel Foundation (GIF 1066-124.1/2008) and the National Institute for Neurological Disorders and Stroke (1RO1N5066005).
References
Abraham, E., Arcaroli, J., Carmody, A., Wang, H., and Tracey, K. J. (2000). HMG-1 as a mediator of acute lung inflammation. J. Immunol. 165, 2950–2954.
Amor, S., Puentes, F., Baker, D., and van der Valk, P. (2010). Inflammation in neurodegenerative diseases. Immunology 129, 154–169.
Beeri, R., Andres, C., Lev-Lehman, E., Timberg, R., Huberman, T., Shani, M., and Soreq, H. (1995). Transgenic expression of human acetylcholinesterase induces progressive cognitive deterioration in mice. Curr. Biol. 5, 1063–1071.
Berson, A., Knobloch, M., Hanan, M., Diamant, S., Sharoni, M., Schuppli, D., Geyer, B. C., Ravid, R., Mor, T. S., Nitsch, R. M., and Soreq, H. (2008). Changes in readthrough acetylcholinesterase expression modulate amyloid-beta pathology. Brain 131, 109–119.
Bond, C. E., Patel, P., Crouch, L., Tetlow, N., Day, T., Abu-Hayyeh, S., Williamson, C., and Greenfield, S. A. (2006). Astroglia up-regulate transcription and secretion of ‘readthrough’ acetylcholinesterase following oxidative stress. Eur. J. Neurosci. 24, 381–386.
Bon, S., Toutant, J. P., Meflah, K., and Massoulie, J. (1988). Amphiphilic and nonamphiphilic forms of Torpedo cholinesterases: I. Solubility and aggregation properties. J. Neurochem. 51, 776–785.
Borges, K., Gearing, M., McDermott, D. L., Smith, A. B., Almonte, A. G., Wainer, B. H., and Dingledine, R. (2003). Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp. Neurol. 182, 21–34.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254.
Cavalheiro, E. A., Santos, N. F., and Priel, M. R. (1996). The pilocarpine model of epilepsy in mice. Epilepsia 37, 1015–1019.
Chen, J., Larionov, S., Pitsch, J., Hoerold, N., Ullmann, C., Elger, C. E., Schramm, J., and Becker, A. J. (2005). Expression analysis of metabotropic glutamate receptors I and III in mouse strains with different susceptibility to experimental temporal lobe epilepsy. Neurosci. Lett. 375, 192–197.
David, Y., Cacheaux, L. P., Ivens, S., Lapilover, E., Heinemann, U., Kaufer, D., and Friedman, A. (2009). Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J. Neurosci. 29, 10588–10599.
De Araujo Furtado, M., Lumley, L. A., Robison, C., Tong, L. C., Lichtenstein, S., and Yourick, D. L. (2010). Spontaneous recurrent seizures after status epilepticus induced by soman in Sprague–Dawley rats. Epilepsia 51, 1503–1510.
Dhote, F., Carpentier, P., Barbier, L., Peinnequin, A., Baille, V., Pernot, F., Testylier, G., Beaup, C., Foquin, A., and Dorandeu, F. (2012). Combinations of ketamine and atropine are neuroprotective and reduce neuroinflammation after a toxic status epilepticus in mice. Toxicol. Appl. Pharmacol. 259, 195–209.
Dhote, F., Peinnequin, A., Carpentier, P., Baille, V., Delacour, C., Foquin, A., Lallement, G., and Dorandeu, F. (2007). Prolonged inflammatory gene response following soman-induced seizures in mice. Toxicology 238, 166–176.
Fabene, P. F., Navarro, M. G., Martinello, M., Rossi, B., Merigo, F., Ottoboni, L., Bach, S., Angiari, S., Benati, D., Chakir, A., Zanetti, L., Schio, F., Osculati, A., Marzola, P., Nicolato, E., Homeister, J. W., Xia, L., Lowe, J. B., McEver, R. P., Osculati, F., Sbarbati, A., Butcher, E. C., and Constantin, G. (2008). A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat. Med. 14, 1377–1383.
Friedman, A., and Dingledine, R. (2011). Molecular cascades that mediate the influence of inflammation on epilepsy. Epilepsia 52 (Suppl. 3), 33–39.
Friedman, A., Kaufer, D., and Heinemann, U. (2009). Blood-brain barrier breakdown-inducing astrocytic transformation: novel targets for the prevention of epilepsy. Epilepsy Res. 85, 142–149.
Hayakawa, K., Mishima, K., Nozako, M., Hazekawa, M., Mishima, S., Fujioka, M., Orito, K., Egashira, N., Iwasaki, K., and Fujiwara, M. (2008). Delayed treatment with minocycline ameliorates neurologic impairment through activated microglia expressing a high-mobility group box1-inhibiting mechanism. Stroke 39, 951–958.
Ivens, S., Kaufer, D., Flores, L. P., Bechmann, I., Zumsteg, D., Tomkins, O., Seiffert, E., Heinemann, U., and Friedman, A. (2007). TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 130, 535–547.
Karnovsky, M. J., and Roots, L. (1964). A “direct-coloring” thiocholine method for cholinesterases. J. Histochem. Cytochem. 12, 219–221.
Kaufer, D., Friedman, A., Seidman, S., and Soreq, H. (1998). Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature 393, 373–377.
Kim, J. B., Sig, C. J., Yu, Y. M., Nam, K., Piao, C. S., Kim, S. W., Lee, M. H., Han, P. L., Park, J. S., and Lee, J. K. (2006). HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 26, 6413–6421.
Lotze, M. T., and Tracey, K. J. (2005). High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 5, 331–342.
Lucas, S. M., Rothwell, N. J., and Gibson, R. M. (2006). The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 147 (Suppl. 1), S232–S240.
Marchi, N., Angelov, L., Masaryk, T., Fazio, V., Granata, T., Hernandez, N., Hallene, K., Diglaw, T., Franic, L., Najm, I., and Janigro, D. (2007). Seizure-promoting effect of blood-brain barrier disruption. Epilepsia 48, 732–742.
Marchi, N., Fan, Q., Ghosh, C., Fazio, V., Bertolini, F., Betto, G., Batra, A., Carlton, E., Najm, I., Granata, T., and Janigro, D. (2009). Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol. Dis. 33, 171–181.
Maroso, M., Balosso, S., Ravizza, T., Liu, J., Aronica, E., Iyer, A. M., Rossetti, C., Molteni, M., Casalgrandi, M., Manfredi, A. A., Bianchi, M. E., and Vezzani, A. (2010). Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 16, 413–419.
Meshorer, E., Biton, I. E., Ben-Shaul, Y., Ben-Ari, S., Assaf, Y., Soreq, H., and Cohen, Y. (2005a). Chronic cholinergic imbalances promote brain diffusion and transport abnormalities. FASEB J. 19, 910–922.
Meshorer, E., Bryk, B., Toiber, D., Cohen, J., Podoly, E., Dori, A., and Soreq, H. (2005b). SC35 promotes sustainable stress-induced alternative splicing of neuronal acetylcholinesterase mRNA. Mol. Psychiatry 10, 985–997.
Meshorer, E., Erb, C., Gazit, R., Pavlovsky, L., Kaufer, D., Friedman, A., Glick, D., Ben Arie, N., and Soreq, H. (2002). Alternative splicing and neuritic mRNA translocation under long-term neuronal hypersensitivity. Science 295, 508–512.
Meshorer, E., and Soreq, H. (2006). Virtues and woes of AChE alternative splicing in stress-related neuropathologies. Trends Neurosci. 29, 216–224.
Meshorer, E., Toiber, D., Zurel, D., Sahly, I., Dori, A., Cagnano, E., Schreiber, L., Grisaru, D., Tronche, F., and Soreq, H. (2004). Combinatorial complexity of 5' alternative acetylcholinesterase transcripts and protein products. J. Biol. Chem. 279, 29740–29751.
Nizri, E., Irony-Tur-Sinai, M., Faranesh, N., Lavon, I., Lavi, E., Weinstock, M., and Brenner, T. (2008). Suppression of neuroinflammation and immunomodulation by the acetylcholinesterase inhibitor rivastigmine. J. Neuroimmunol. 203, 12–22.
Pavlov, V. A., Ochani, M., Gallowitsch-Puerta, M., Ochani, K., Huston, J. M., Czura, C. J., Al-Abed, Y., and Tracey, K. J. (2006). Central muscarinic cholinergic regulation of the systemic inflammatory response during endotoxemia. Proc. Natl. Acad. Sci. U.S.A. 103, 5219–5223.
Pavlov, V. A., Parrish, W. R., Rosas-Ballina, M., Ochani, M., Puerta, M., Ochani, K., Chavan, S., Al-Abed, Y., and Tracey, K. J. (2009). Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav. Immun. 23, 41–45.
Pernot, F., Carpentier, P., Baille, V., Testylier, G., Beaup, C., Foquin, A., Filliat, P., Liscia, P., Coutan, M., Pierard, C., Beracochea, D., and Dorandeu, F. (2009). Intrahippocampal cholinesterase inhibition induces epileptogenesis in mice without evidence of neurodegenerative events. Neuroscience 162, 1351–1365.
Pitkanen, A., Kharatishvili, I., Karhunen, H., Lukasiuk, K., Immonen, R., Nairismägi, J., Gröhn, O., and Nissinen, J. (2007). Epileptogenesis in experimental models. Epilepsia 48, 13–20.
Racine, R. J. (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281–294.
Rappold, P. M., Lynd-Balta, E., and Joseph, S. A. (2006). P2X7 receptor immunoreactive profile confined to resting and activated microglia in the epileptic brain. Brain Res. 1089, 171–178.
Rijkers, K., Majoie, H. J., Hoogland, G., Kenis, G., De, B. M., and Vles, J. S. (2009). The role of interleukin-1 in seizures and epilepsy: a critical review. Exp. Neurol. 216, 258–271.
Shibley, H., and Smith, B. N. (2002). Pilocarpine-induced status epilepticus results in mossy fiber sprouting and spontaneous seizures in C57BL/6 and CD-1 mice. Epilepsy Res. 49, 109–120.
Shohami, E., Kaufer, D., Chen, Y., Seidman, S., Cohen, O., Ginzberg, D., Melamed-Book, N., Yirmiya, R., and Soreq, H. (2000). Antisense prevention of neuronal damages following head injury in mice. J. Mol. Med. 78, 228–236.
Shytle, R. D., Mori, T., Townsend, K., Vendrame, M., Sun, N., Zeng, J., Ehrhart, J., Silver, A. A., Sanberg, P. R., and Tan, J. (2004). Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J. Neurochem. 89, 337–343.
Sternfeld, M., Shoham, S., Klein, O., Flores-Flores, C., Evron, T., Idelson, G. H., Kitsberg, D., Patrick, J. W., and Soreq, H. (2000). Excess “read-through” acetylcholinesterase attenuates but the “synaptic” variant intensifies neurodeterioration correlates. Proc. Natl. Acad. Sci. U.S.A. 97, 8647–8652.
Thullbery, M. D., Cox, H. D., Schule, T., Thompson, C. M., and George, K. M. (2005). Differential localization of acetylcholinesterase in neuronal and non-neuronal cells. J. Cell. Biochem. 96, 599–610.
Tracey, K. J. (2007). Physiology and immunology of the cholinergic antiinflammatory pathway. J. Clin. Invest. 117, 289–296.
Turrin, N. P., and Rivest, S. (2004). Innate immune reaction in response to seizures: implications for the neuropathology associated with epilepsy. Neurobiol. Dis. 16, 321–334.
van Vliet, E. A., da Costa, A. S., Redeker, S., van Schaik, R., Aronica, E., and Gorter, J. A. (2007). Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 130, 521–534.
Vezzani, A., Balosso, S., and Ravizza, T. (2008). The role of cytokines in the pathophysiology of epilepsy. Brain Behav. Immun. 22, 797–803.
Vezzani, A., Conti, M., De, L. A., Ravizza, T., Moneta, D., Marchesi, F., and De Simoni, M. G. (1999). Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J. Neurosci. 19, 5054–5065.
Vezzani, A., French, J., Bartfai, T., and Baram, T. Z. (2011). The role of inflammation in epilepsy. Nat. Rev. Neurol. 7, 31–40.
Vezzani, A., and Granata, T. (2005). Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia 46, 1724–1743.
von Bernhardi, R., Ramirez, G., De Ferrari, G. V., and Inestrosa, N. C. (2003). Acetylcholinesterase induces the expression of the beta-amyloid precursor protein in glia and activates glial cells in culture. Neurobiol. Dis. 14, 447–457.
Wang, H., Bloom, O., Zhang, M., Vishnubhakat, J. M., Ombrellino, M., Che, J., Frazier, A., Yang, H., Ivanova, S., Borovikova, L., Manogue, K. R., Faist, E., Abraham, E., Andersson, J., Andersson, U., Molina, P. E., Abumrad, N. N., Sama, A., and Tracey, K. J. (1999). HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251.
Wang, J., Zhang, H. Y., and Tang, X. C. (2010). Huperzine a improves chronic inflammation and cognitive decline in rats with cerebral hypoperfusion. J. Neurosci. Res. 88, 807–815.
Wang, Z. F., Wang, J., Zhang, H. Y., and Tang, X. C. (2008). Huperzine A exhibits anti-inflammatory and neuroprotective effects in a rat model of transient focal cerebral ischemia. J. Neurochem. 106, 1594–1603.
Weissberg, I., Reichert, A., Heinemann, U., and Friedman, A. (2011). Blood-brain barrier dysfunction in epileptogenesis of the temporal lobe. Epilepsy Res. Treat. doi: 10.1155/2011/143908
Zhang, J., Takahashi, H. K., Liu, K., Wake, H., Liu, R., Maruo, T., Date, I., Yoshino, T., Ohtsuka, A., Mori, S., and Nishibori, M. (2011). Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke 42, 1420–1428.
Keywords: acetylcholinesterase, epileptogenesis, hippocampus, inflammation, status epilepticus
Citation: Gnatek Y, Zimmerman G, Goll Y, Najami N, Soreq H and Friedman A (2012) Acetylcholinesterase loosens the brain's cholinergic anti-inflammatory response and promotes epileptogenesis. Front. Mol. Neurosci. 5:66. doi: 10.3389/fnmol.2012.00066
Received: 30 January 2012; Paper pending published: 13 February 2012;
Accepted: 07 May 2012; Published online: 18 May 2012.
Edited by:
Karl Tsim, The Hong Kong University of Science and Technology, ChinaReviewed by:
Oleg Krishtal, Ukrainian Academy of Sciences, UkraineFabien Pernot, Rhenovia Pharma, France
Copyright: © 2012 Gnatek, Zimmerman, Goll, Najami, Soreq and Friedman. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Alon Friedman, Department of physiology, Laboratory for Experimental Neuroscience, Ben-Gurion University, Beer-Sheva, 84105 Israel. e-mail: alonf@bgu.ac.il
†Present Address: Yehudit Gnatek, Sheba Medical Center, Pediatric Stem Cell Research Institute, Tel HaShomer, Israel.
Naim Najami, Department of Biology, The Academic Arab College of Education, Haifa, Israel.
‡ These authors contributed equally to this work.