
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mol. Neurosci. , 24 August 2011
Sec. Molecular Signalling and Pathways
Volume 4 - 2011 | https://doi.org/10.3389/fnmol.2011.00014
This article is part of the Research Topic The tumor and the embryo: oxygen and oxygen dependent pathways in development View all 6 articles
 Anja Bondke Persson1* and Ivo R. Buschmann2
Anja Bondke Persson1* and Ivo R. Buschmann2
Vascular growth forms the first functional organ system during development, and continues into adult life, wherein it is often associated with disease states. Genetically determined vasculogenesis produces a primary vascular plexus during ontogenesis. Angiogenesis, occurring, e.g., in response to metabolic stress within hypoxic tissues, enhances tissue capillarization. Arteriogenesis denotes the adaptive outgrowth of pre-existent collateral arteries to bypass arterial stenoses in response to hemodynamic changes. It has been debated whether vasculogenesis occurs in the adult, and whether or not circulating progenitor cells structurally contribute to vessel regeneration. Secondly, the major determinants of vascular growth – genetic predisposition, metabolic factors (hypoxia), and hemodynamics – cannot be assigned in a mutually exclusive fashion to vasculogenesis, angiogenesis, and arteriogenesis, respectively; rather, mechanisms overlap. Lastly, all three mechanisms of vessel growth seem to contribute to physiological embryogenesis as well as adult adaptive vascularization as occurs in tumors or to circumvent arterial stenosis. Thus, much conceptual and terminological confusion has been created, while therapies targeting neovascularization have yielded promising results in the lab, but failed randomized studies when taken to the bedside. Therefore, this review article aims at providing an exact definition of the mechanisms of vascular growth and their contribution to embryonic development as well as adult adaptive revascularization. We have been looking for potential reasons for why clinical trials have failed, how vitally the application of appropriate methods of measuring and assessment influences study outcomes, and how relevant, e.g., results gained in models of vascular occlusive disease may be for antineoplastic strategies, advocating a reverse bedside-to-bench approach, which may hopefully yield successful approaches to therapeutically targeting vascular growth.
In vertebrate organogenesis, the blood vessels constitute the first organ system that arises and reaches a functional state (Risau and Flamme, 1995); a prerequisite for the growth of complex organisms which develop an increasing need for oxygen and nutrients and the removal of metabolic waste products. Mice failing to establish a functioning circulatory system by E10 die shortly thereafter (Semenza, 2007b).
Vascular growth continues into adult life, either in the form of cyclic processes such as decidual arteriogenesis, or in response to acute or chronic stress, as occurs in atherosclerotic vascular disease, chronic inflammation or the hypoxic environment of growing malignancies, or in response to physical training (Hänze et al., 2007). Most major causes of disease in humans involve changes in tissue vascularization and oxygen availability (Semenza, 2007b).
The recent decades have seen an evolution of our understanding of the (patho)physiology that underlies vascular growth, providing a rationale for intense efforts toward either its therapeutic stimulation or inhibition. This requires a thorough understanding of the underlying mechanisms.
In this systematic review article we discuss key mechanisms of vasculogenesis, angiogenesis, and arteriogenesis, the three major processes of vascular growth. We thereby set out to provide a basis for understanding current therapeutic strategies which target the vascular system.
The formation of the earliest blood vessels (around embryonic day E18 in humans, E6.5 in mice) occurs through vasculogenesis in all mammals. Vasculogenesis comprises mesoderm formation, the establishment of the endothelial cell (EC) lineage as so-called angioblasts, their organization into cord-like vascular structures, lumen formation, and the organization of early vessels into vascular networks (Figure 1A).
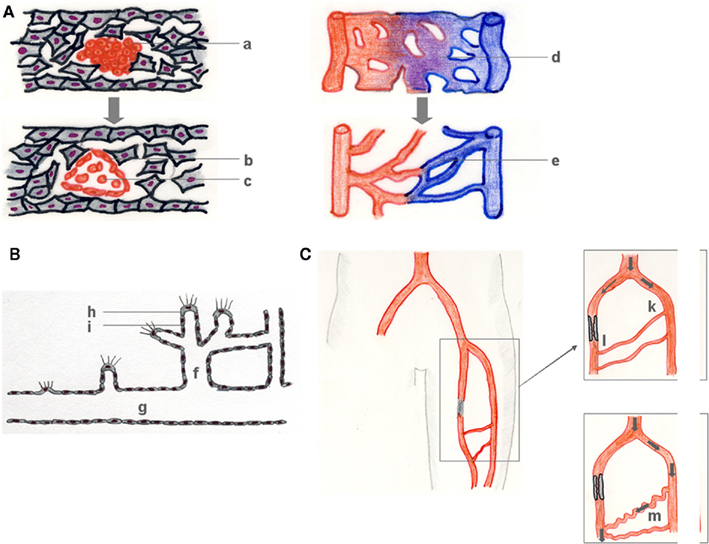
Figure 1. Vasculogenesis, angiogenesis, and arteriogenesis. During vasculogenesis (A), (hem-) angioblast precursors form blood islands (a), which subsequently differentiate into primary ECs (b), and early blood cells (c). The resulting primary vascular plexus (d) matures into a secondary vessel network (e). Angiogenesis (B) denotes the growth of capillaries, mostly in response to a lack in sufficient tissue supply with oxygen and nutrients that is signaled by a growth factor gradient (B). Capillary sprouts (f) grow from pre-existent vessels (g) in the direction of a growth factor gradient (e.g., VEGF) that is sensed by a tip cell (h) and followed by a vascular stalk (i) that subsequently lumenizes. Arteriogenesis describes the outgrowth of pre-existent arteriolar collaterals which then bypass an arterial stenosis (C). Atherosclerotic occlusion of a major conductance artery induces a pressure gradient between the collateral stem region (k) (approx. 100 mmHg) and the reentry zone (l) (approx. 25 mmHg), followed by a redistribution of perfusion, an increase in collateral flow and a subsequent outgrowth of the collateral arteriole of typical corkscrew-like morphology (m).
During gastrulation, the three germ layers ectoderm, mesoderm, and endoderm are formed, of which the mesoderm gives rise to the vascular system as well as the primordial blood cells (Risau and Flamme, 1995). Mesodermal differentiation to ECs occurs first within the extraembryonic visceral mesoderm of the yolk sac (splanchnopleuric mesoderm) around developmental days E6–7 in mice (reviewed in Jin and Patterson, 2009), and shortly afterward within the embryo (somatopleuric mesoderm) at the one-somite stage. Additional, atypical regions of EC formation have been described in avian as well as mammalian embryos, such as placental (Demir et al., 2007) or allantoic tissues (Caprioli et al., 2001). EC differentiation, both in the yolk sac and within the embryo proper, occurs in form of blood islands or – islets, cellular aggregates whose peripheral cells are destined to become EC precursors, while their centers form blood cells (Risau and Flamme, 1995; Carmeliet, 2000; Figure 1A).
Already in the late nineteenth century, His (1900) described as “angioblasts” the mesenchymal cells that later form blood vessels. Murray (1932) renamed these cells “hemangioblasts,” as he assumed that ECs and blood cells would always develop from the same progenitor cell.
Indeed, in the extraembryonic visceral mesoderm of the yolk sac, endothelial, and hematopoietic progenitors are closely related (Sabin, 1917; Choi et al., 1998; Eichmann et al., 2000). Genuine hemangioblasts have been isolated from both mouse and zebrafish embryos and give rise to both vascular ECs as well as hematopoietic cells (Huber et al., 2004; Vogeli et al., 2006).
However, the blood islands forming within the intraembryonic mesoderm and the earlier extraembryonic vasculogenic aggregates of the yolk sac differ. While extraembryonic hemangioblasts are (at least) bipotential progenitors, angioblast differentiation within the embryo is not coupled as closely to the development of the blood cells (Risau and Flamme, 1995). Rather, a hemogenic EC intermediate, i.e., an EC of hematopoietic potential, later generates hematopoietic cells (Lancrin et al., 2009).
Recent results suggest an even more complex picture. ECs share a progenitor cell with a number of different cell populations which give rise to the cardiovascular system, not only blood cells, but also cardiomyocytes, and vascular smooth muscle cells (SMCs; Jin and Patterson, 2009). Transdifferentiation seems possible between these organ-specific mesodermal progenitors (Song and Tuan, 2004; Mudumana et al., 2008).
Angioblast differentiation from mesodermal cells and the formation of immature blood vessels from angioblasts have together been defined as vasculogenesis (Risau and Flamme, 1995), followed by lumen formation and network maturation (Figure 1A).
Precise mechanisms of lumen formation have been elucidated for epithelia, but remain somewhat ambiguous for vascular endothelia. The peripheral cells of the blood islands, destined to become ECs, flatten and proceed to surround primitive liquid-filled spaces, which fuse, forming the first lumina in form of angioblastic cords/strands. Once a lumen has been generated, angioblastic cords originating from blood islands in spatial proximity associate to form a primary embryonic vascular network (Hirakow and Hiruma, 1981). Migratory angioblasts (Flamme et al., 1997; Risau, 1997) assemble along the posterior axis to form the precursors of the main body vasculature, i.e., the dorsal aorta, cardinal veins, and primary endocardial tubes (Flamme et al., 1997).
Thus, vasculogenesis leads to the formation of the earliest primary vascular network, which consists of the precursors of major vessels and the honeycomb-like plexus which connects them (Yancopoulos et al., 2000).
Known molecular pathways of vasculogenesis constantly increase in number and complexity, and are beyond the scope of this article. In short, molecular signals from neighboring tissues interact with the mesoderm during vasculogenesis, examples of which are Hedgehog protein originating in the hypochord and VEGF from the ventral somites (Jin and Patterson, 2009). If endo- and ectodermal signals are critical, is still debated. Fibroblast growth factors (FGFs), transforming-growth factor β family members (TGFβ-like factors), and VEGF-A may induce mesoderm formation and differentiation (Green et al., 1992; Isaacs et al., 1992; Duan et al., 2003). However, Risau and Flamme (1995) collected ample evidence of the mesoderm alone being capable of vascular differentiation.
With the emergence of the angioblast the molecular picture becomes more clear, with leading roles taken on by (a) the Angpt/Tie and (b) the VEGF signaling pathways.
The VEGF family comprises seven different molecules (VEGF1 through VEGF7, PIGF), which act via the tyrosine kinases VEGFR1 (Flt1), VEGFR2 (Flk1, KDR), VEGFR3 (Flt4), and their coreceptors Neuropilin-1 and -2. Embryonic defects in VEGF signaling lead to antenatal lethality due to early and fundamental defects in endothelial and hematopoietic development (Shalaby et al., 1995). Current candidates for the superordinate regulation of VEGF signaling during vasculogenesis are HoxB5 and FoxH1 (Jin and Patterson, 2009).
Angiopoietins 1–3, via their Tie-2 receptor, regulate the late vasculogenic processes, vascular network development, and vessel maturation (Schnurch and Risau, 1993).
It is debated whether vasculogenesis occurs in postnatal life (Luttun and Carmeliet, 2003; Péault, 2010). On the one hand, cells sharing molecular characteristics of embryonic hemangioblasts, namely CD34/VEGF-2R coexpression, have been detected in the adult circulation (Pelosi et al., 2002). Factors such as VEGF, GM-CSF, bFGF, and IGF-1 mediate their mobilization and/or differentiation (reviewed in Carmeliet, 2000). Other authors, however, assign vasculogenesis to a restricted time period in early embryogenesis (Hänze et al., 2007; Semenza, 2007b) and state that the use of the term “vasculogenesis” for adult blood vessel development, hypothetically associated with circulating vascular progenitor cells, was ”an inappropriate use of this term based on its natural definition” (Ferguson et al., 2005).
Although environmental factors modify the vasculogenic program in the embryo, genetic predetermination governs vasculogenesis.
Angiogenesis denotes capillary growth either by sprouting from or by splitting of pre-existing vascular structures (intussusception) (Risau, 1997).
Sprouting angiogenesis is initiated by a proteolytic degradation of the extracellular matrix (ECM) by EC in response to proangiogenic factors such as VEGF released in their proximity, e.g., in response to local hypoxia. ECs proliferate and migrate toward gradients of proangiogenic factors, form lumina, and vessels are stabilized, mature, and acquire tissue-specific properties (Risau, 1997; Carmeliet, 2000; Fischer et al., 2006; Figure 1B).
While sprouting angiogenesis relies on EC mitosis and migration, intussusceptive growth multiplies vessels by splitting their lumina by the insertion of tissue pillars (Patan, 2000). Intussusception subsumes a series of processes, extensively reviewed in (Djonov and Makanya, 2005). Vessel maturation is comparable to the events observed in sprouting angiogenesis, which this chapter mainly focuses on.
During development, angiogenesis establishes a functional circulation in primarily avascular tissues. While vasculogenesis forms the primary vascular plexus in endo- and mesoderm-derived organs, ectoderm-derived tissues such as the brain are vascularized by angiogenesis (Pardanaud et al., 1989).
Physiological angiogenesis is a highly ordered and tightly controlled process critically dependent (a) on a controlled interaction of the proliferating endothelium with its environment and (b) orchestrated by an interplay of cytokines and growth factors (Chung et al., 2010).
Sprouting angiogenesis is initiated by vascular leakage and local degradation of the basement membrane (BM) of the mother vessel. A preceding NO-mediated local increase in vascular diameter has especially been described for pathological angiogenesis (Carmeliet, 2000). Leaking plasma proteins then form a scaffold for migrating ECs in a PECAM-1/VE-Cadherin/Src kinase-mediated process (Eliceiri et al., 1999). VEGF leads to vasodilation and increased permeability.
The angiopoietin Angpt-1, through interaction with its specific receptor Tie-2, counteracts vasodilation to prevent excess leakage. Its physiological antagonist Angpt-2 destabilizes the matrix, with a subsequent partial degradation of the BM and surrounding ECM as well as proteolytic activation of angiogenic growth factors (Carmeliet, 2000). Angpt-2 activated MMPs and proteolytic enzymes liberate matrix-bound pro- and antiangiogenic mediators in an orchestrated sequence, and regulate the biological activities of factors such as VEGF by proteolytic cleavage (Fischer et al., 2006).
Next, some ECs acquire an activated phenotype, which permits mitosis and migration. The “initial decision of an EC to become angiogenic” (Carmeliet, 2000) potentially involves factors such as p54, the vascular endothelial zinc finger-1 and the Hox and GATA families (Carmeliet, 2000). In the current biomedical literature, no uniform definition of an activated endothelium can be found. The term is used to refer to, e.g., an upregulation of adhesion molecules in response to inflammatory mediators or hemodynamic forces or the acquisition of an angiogenic phenotype, which is what we refer to herein.
Angiogenesis is induced by a pioneering “tip cell” which increases BM and ECM degradation by membrane-type 1 MMP expression and secretion (Figure 1C). Thereby, more ECs loose contact to the BM and interact with exposed tissue collagen, which induces a reorganization of the actin cytoskeleton and sprout formation (sprouting morphogenesis; Chung et al., 2010). Ausprunk and Folkman (1977) described mitosis and migration in angiogenesis, with migration predominating at the tip of the growing capillary and mitosis occurring several cells proximal to the tip. Selection of tip cells vs. stalk cells depends on intracellular VEGFR1/VEGFR2 ratios, with ECs dynamically competing for the tip cell position (Jakobsson et al., 2010).
Leaked plasma proteins serve as a matrix for EC migration. ECs interact with the matrix in an integrin-mediated process. Sprouting ECs are guided by gradients of angiogenic factors such as VEGFs/VEGFR2. VEGF isoforms fulfill different functions during angiogenesis: context-specifically, they increase vascular permeability, aid ECM degradation and pericyte recruitment and induce EC sprouting, proliferation, migration, lumen formation, and EC survival (for a detailed review, see Chung et al., 2010 or Ferrara et al., 2003). Hypoxia drives angiogenesis through intracellular stabilization of the hypoxia-inducible factor (HIF-1) and subsequent induction of VEGF expression.
The HIFs are transcription factors (HIF-1, -2 and -3) which are induced when partial pressures of tissue oxygen decrease to values below 6%/40 mmHg (Semenza, 2007a). HIFs are heterodimers of a regulatory α subunit and a constitutive β subunit (Wang and Semenza, 1995). Cellular HIF levels are regulated in a complex manner, involving prolyl-4-hydroxylases and asparaginyl hydroxylases, phosphorylation and sumoylation (Hänze et al., 2007; Scholz and Kirschner, 2011). Transcriptional activation of HIF-1 occurs downstream of protein-kinase C mediated signaling pathways, in response to stimuli such as inflammatory mediators or angiotensin II (Hänze et al., 2007). HIF-1α translation is enhanced downstream of the PI3K/Akt pathway, while post-transcriptional control of HIF-1α synthesis may be relevant during vascular growth and remodeling (Gorospe et al., 2011).
Hypoxia-inducible factor-1 assumes a central role in oxygen-dependent vascularization via binding to the hypoxia-responsive cis elements [5′(A/G)CGTG-3′] within the promoter regions (Rey and Semenza, 2010) of genes associated with vessel growth and remodeling. HIF-1 is essential for proper embryonic vascular development, regulating angiogenic growth factors such as VEGF, PLGF, and the Angiopoietins and their coordinated activation (Semenza, 2007b).
The FGFs family and their tyrosine kinase receptors (FGFR) stimulate angiogenesis. FGFs 1, 2, 4, and 5 target ECs and exert pro angiogenic activities in vivo, directly or through the recruitment of proangiogenic cells (Fischer et al., 2006).
Angpt-1/Angpt-2 mediate the balance between vascular quiescence and proliferation during the sprouting/migration phase of angiogenesis. Local Angpt-2 expression, together with VEGF, appears to be critical for EC reentry into the cell cycle. Angpt-1 is mitogenic and chemotactic for sprouting ECs, while Angpt-2 is not (Witzenbichler et al., 1998; Yancopoulos et al., 2000).
Targeted tip cell migration is aided by PIGF via VEGFR1 (Autiero et al., 2003; Fischer et al., 2006). Disruption of this pathway causes defects in the stress response toward hypoxia in the adult, as in ischemia, inflammation or malignancies (Carmeliet et al., 2001).
Many other molecules are pro- or antiangiogenic, often context- and/or tissue-specifically (Semenza, 2007b), and reach beyond the scope of this paper. Examples are integrins and their receptors, tissue-specific growth factors, erythropoietin, and interleukins (Carmeliet, 2003; Kertesz et al., 2004; Fischer et al., 2006; Sciesielski et al., 2010).
Leukocytes affect angiogenesis. Monocytes/macrophages function as cellular regulators of angiogenesis, recruited by chemokines (Bernardini et al., 2003), VEGF-A or PIGF to sites of active angiogenesis (Barleon et al., 1996) to exert subpopulation-specific effects (Carmeliet, 2003).
Both vasculogenesis and angiogenesis lead to chord-like EC alignments (Zeeb et al., 2010), from which vascular networks are formed by fusion, anastomosis, and lumen formation. Although large amounts of in vitro data have been compiled, functional models of lumenization have only recently been suggested: apparently, angiogenic sprouts form lumina by cell or cord hollowing (Iruela-Arispe and Davis, 2009). Either, intracellular vacuoles associate, or ECs form cylinders, which subsequently create an internal, extracellular lumen.
Until recently, angiogenesis was thought to proceed via cell hollowing, generating seamless tubes (Iruela-Arispe and Davis, 2009). Since Sabin (1917) had described coalescing vacuoles, modern techniques, especially in cell and tissue culture, led to the discovery of EC lumen formation by a pinocytosis-resembling process (depending on integrin and cdc42/Rac1 downstream of integrin–ECM signaling interactions; Bayless and Davis, 2002), which could analogously be observed in vivo in the zebrafish (Kamei et al., 2006). However, lumen formation seems to differ in health and disease, sprouting and lumen formation occur either at the same time or in sequence, and pathways appear to be context-specific. Vacuolation-based lumenization has, for example, especially been observed for isolated ECs deprived of cell–cell contacts (Iruela-Arispe and Davis, 2009). Recent data have been tipping the scales in favor of the extracellular lumen model. A closer look at zebrafish vasculogenesis revealed that ECs form a multicellular tube (Blum et al., 2008). VEGF is necessary for lumen formation (Iruela-Arispe and Davis, 2009). Angpt-1 increases vessel diameter, while Thrombospondin-1 and tubedown-1 reduce it (Fischer et al., 2006). Once functional vessels have been generated, mechanical factors displace genetic pre-specifications in determining lumen diameter (Jones et al., 2006).
Angiogenic vascular morphogenesis enters its resolution phase when the activated, proteolytic phenotype of the invading tip cell is “turned off” by EC-pericyte contacts. Tip cell MMP activity is downregulated and replaced by an activation of its inhibitors TIMP-2 and -3 (Saunders et al., 2006). An involvement of Dll4–Notch1 in restricting tip cell formation in response to VEGF-A has been described (Hellström et al., 2007). A downregulation of major angiogenic factors such as VEGF, and a rise in local levels of PDGF, Angpt-1, and TGFβ1, occur (Chung et al., 2010).
Mature blood vessels are made of ECs and mural cells (pericytes, SMCs) within a matrix. Following early angiogenesis, no mural cells are present, and homotypic tight and adherens junctions resist blood flow (Iruela-Arispe and Davis, 2009). When mural cells are recruited and a BM, elastic laminae, and surrounding matrices are generated (Jain, 2003; “vessel maturation”), the newly generated vessel stabilizes. Vessels then specialize to fulfill the requirements of their host tissue by EC differentiation (Carmeliet, 2000; Jain, 2003). Not only the individual vessel, but vascular networks acquire their functionality through patterning events such as branching, remodelling and pruning (“network maturation”) (Jain, 2003).
To tighten and stabilize the vessel (a) mural cells are recruited and (b) matrix and laminae are formed.
PDGF (-BB), via PDGF-Rβ1 on mural cells, regulates pericyte, and SMC recruitment (Lindblom et al., 2003). Similarly, Angpt-1 on mural cells interacts with Tie-2 on ECs. Both are involved in the tightening of the EC layer, conferring leakage-resistance. SIP-1/EDG-1 interactions mediate mural cell recruitment and matrix production (Kluk and Hla, 2002). The latter is also stimulated by the TGFβ/Alk-5 pathway (Jain, 2003).
Network maturation processes occur concomitantly or sequentially. Pruning, the removal of superfluous vascular branches, is mediated by tissue oxygen tension (Chung et al., 2010). Directional branching or secondary outgrowth can be guided by mechanical forces transmitted by the ECM (Krishnan et al., 2008) via integrin-dependent pathways (Jain, 2003; Chung et al., 2010). The ECM generates extracellular growth factor gradients for branching, in which proteases (e.g., MMPs) and their inhibitors (e.g., TIMPs, ADAM) play a major role by cleaving and activating angiogenic factors (VEGF, FGF, angiostatin; Jain, 2003; van Hinsbergh and Koolwijk, 2008). Growth factors relevant for neuronal growth share roles in angiogenic network maturation, e.g., the neuropilins (Neufeld et al., 2002), ephrins, or the Dll4/Notch pathway (Djokovic et al., 2010). Other network remodeling factors are VEGF/VEGFR3, the Ets family of transcription factors and gridlock in zebrafish (Carmeliet, 2000).
Vessel specialization involves the development of organ-specific EC characteristics and network-related processes such as arteriovenous (AV) fate determination. Most processes are context-specific, such as the VEGF-mediated EC fenestration in endocrine glands (Jain, 2003) or the GFAP/angiotensinogen interaction in the development of the blood–brain barrier (Carmeliet, 2000). During the development of the arterial system, vessels are “muscularized” (Carmeliet, 2000; “arterialization”): pericytes/SMCs grow alongside the nascent vessel, or differentiate in situ from mesenchymal precursors.
Some authors refer to this process as arteriogenesis. This may lead to confusion, as “arteriogenesis” had been coined by W. Risau, W. Schaper, and R. Munoz-Chapuli (Schaper et al., 1999) for the postnatal development of collateral conductance arteries from arteriolar networks. Arteriogenesis, in this review, will only be used in the latter sense.
When a functional, local vascular network has developed, tissue oxygenation provides a negative feedback loop for angiogenesis.
Unlike vasculogenesis, angiogenesis is not only well-established in the adult organism (Risau, 1997), but numerically the main mechanism through which new vessels arise (Carmeliet, 2000). Adult angiogenesis resembles embryonic angiogenesis (Risau, 1997), both are facilitated by reductions in oxygen tension, but adult angiogenesis requires additional events: cell-cycle progression, remodeling of cell adhesions and junctions, induction of proteolysis, and neutralization of angiogenesis inhibitors (Carmeliet, 2000) differ between angiogenesis seen in the embryo and in the adult. Egginton and Gerritsen (2003) have systematically reviewed the events that specify embryonic angiogenesis: ECs are more mitotic, early angiogenic sprouts contain a subpopulation of EC precursors that display an invasive migratory behavior, intercellular junctions are readily formed, and nascent embryonic capillaries are characterized by wide lumina and fenestrated endothelia.
Deregulated angiogenesis has a major pathophysiological impact. Cancer, atherosclerosis, infectious diseases, psoriasis, and diabetes involve changes in tissue capillarization with significant consequences on morbidity and mortality. Therapeutic attempts are being made to (a) induce angiogenesis, mostly through the targeted application of proangiogenic growth factors, and (b) to apply angiogenesis inhibitors.
Angiogenesis occurs during embryonic development and in the adult organism. In contrast to embryonic vasculogenesis, and although angiogenesis is co-determined by genetic factors, it is a change in tissue oxygenation that induces angiogenesis, mainly through its master regulator, VEGF.
Angiogenesis is initiated by hypoxia, and leads to a capillary vascular network. Arteriogenesis, in contrast, is the outgrowth of a mature collateral arteriole upon stenosis or occlusion of a major conductance artery (Figure 1D), driven by hemodynamic factors such as an increase in stretch and/or fluid shear stress (FSS) on ECs (Schaper and Ito, 1996; Carmeliet, 2000).
Stenosis or occlusion of a conductance vessel trigger arteriogenesis (Figure 1D): with an arterial occlusion, a pressure gradient develops over interconnecting collaterals between the pre- (high pressure areal) and postocclusive (low pressure areal) regions. Flow through these collateral vessels increases and deforms ECs, raising FSS on the endothelium, which functionally and morphologically activates it. Shear patterns need to be pulsatile to prompt an arteriogenic response and support arterial identity through molecular factors such as Gja5 (connexin 40; Buschmann et al., 2010).
An activated endothelial phenotype results from increases in flow, morphologically characterized by EC swelling and ultrastructural protrusions (Schaper et al., 1976, 1988; Bondke et al., 2008), correlated with an increased cellular metabolism, changes in protein expression, and the activation of transient receptor potential cation channels, which affect cellular osmotic control and volume regulation, and upon chronic stimulation, EC and SMC proliferation (Bondke et al., 2008; Troidl et al., 2009) reviewed in (van Royen et al., 2010).
Changes in glycocalix structure prompt EC–leukocyte interactions (Grundmann et al., 2009).
Shear-activated ECs attract mononuclear cells (Bergmann et al., 2006) by expression of MCP-1 and ICAM-1, which invade the vessel wall and facilitate its proteolytic remodeling (Hillmeister et al., 2008).
Whether or not endothelial progenitor cells (EPCs) contribute structurally to postnatal vascular growth is unclear (Shintani et al., 2001; Ziegelhoeffer et al., 2004). Probably, their function is in line with macrophage function in arteriogenesis, i.e., the provision of local factors that facilitate vascular remodeling (Grundmann et al., 2007a; Patenaude et al., 2010).
By ECM digestion and remodeling, monocytes enable SMC dedifferentiation to a synthetic, proliferative phenotype, SMC migration, proliferation, and division. The media of the growing vessel undergoes a dramatic phenotypic change. Elastic laminae become digested, and SMC proliferation leads to an outgrowth of the growing collateral to about 50-fold increases in tissue volume (Scholz et al., 2000).
Arteriogenesis has been investigated meticulously since the 1970s, owing at large to the work of W. Schaper and his students from Bad Nauheim, while recent research on the molecular pathways of arteriogenesis sheds new light on the process.
Endothelial cells sense elevated FSS levels by mechanosensory complexes upstream of integrin signaling (Tzima et al., 2005). Transcription factors activator protein 1 (AP-1), early growth response protein 1 (Egr-1), and other DNA binding proteins are activated via the Rho- and MEK/Erk-1/-2 pathways, while actin-binding proteins, integrins, and connexins assume important functions (reviewed in Schaper, 2009). Rho controls the expression ICAM-1 and MCP-1. Integrins act as upstream effectors of Rho (Deindl, 2007). Carp, differentially expressed during arteriogenesis, may function as an upstream regulator of Egr-1 (Deindl, 2007). As is true for other processes of vascular growth, the identified factors are specific for species and tissues.
However, a number of unifying principles apply to arteriogenic processes (Schaper, 2009).
First, collateral conductance vessels develop from pre-existent arterioles. Arteriogenesis does not increase the number of collaterals. Detection by imaging yields ambiguous results when vessels grow from below detection level during the observation period to an above-threshold size (Limbourg et al., 2009; Schaper, 2009). Moreover, assessment of collateralization may be easier in the periphery than in the myocardium due to spatial limitations and close anatomical relations in the latter.
Second, arteriogenesis occurs outside the ischemic area at risk. In peripheral arterial disease (PAD) in human patients, femoral artery occlusion leads to ischemia of the lower leg, while collaterals develop in parallel to the stenosis (Yang et al., 2008; van Royen et al., 2010). Stenosis of one of the vessels supplying the Circle of Willis in the brain leads to a redistribution of blood flow and subsequent collateral enlargement outside the ischemic area (Busch et al., 2003). In the myocardium, the ischemic area and the growing collateral lie close together, however, the stem of collaterals, “the most important single factor in determining the extent and distribution of myocardial infarction” develops outside the ischemic area (Fulton, 1964). Growing collaterals conduct arterial blood and are thus sufficiently supplied with oxygen (van Royen et al., 2010): Hypoxia-inducible gene expression is not relevant for collateral growth (Ito et al., 1997; Deindl et al., 2001). VEGF expression occurs (Toyota et al., 2005; Schaper, 2009), however, independent of the angiogenic HIF–VEGF-axis, while hypoxia impairs monocyte migration (Grimshaw and Balkwill, 2001). In the zebrafish, where sufficient oxygenation can be maintained by diffusion alone, adaptive arteriogenesis occurs in response to changes in flow (Gray et al., 2007).
Third, arteriogenesis is initiated by changes in blood flow. The vascular wall is constantly subjected to changes in stretch, cyclic mechanical strain, and shear stress, with shear stress primarily affecting the EC layer, while stretch acts on all layers of the vessel wall (Lehoux and Tedgui, 2003). The steep pressure gradient between the pre- and poststenotic areas is bridged by collateral vessels, in which the sudden increase in flow leads to significantly rising shear rates. This brings about the structural and functional changes in EC morphology and function that initiate arteriogenesis (Schaper, 2009).
Angiogenesis is terminated when local tissue oxygenation is restored (Chung et al., 2010). A similar negative feedback control mechanism applies to arteriogenesis: in adaptive arteriogenesis, the restoration of conductance by collateral growth ceases when FSS normalizes, thereby achieving a restoration of blood flow capacity of 30–40% (Schaper, 2009; Figure 1C). This “adaptive arteriogenesis” leads to a species-dependent 2- to 20-fold enlargement of collateral arterioles (Schaper and Ito, 1996; Schaper, 2009).
Collateral vessel development varies significantly between individuals (Rehman, 2008). Arteriogenesis may eliminate the signs and symptoms of a severe vascular occlusion such as a complete occlusion of a main coronary artery (Schaper, 1971; Grundmann et al., 2007a), but may also remain ineffective due to individual differences, e.g., in the expression of antiarteriogenic genes (Schirmer et al., 2008). Arterial occlusions often occur in patients who (a) have one or more cardiovascular risk factors or (b) regularly take cardiovascular drugs, both of which influence arteriogenic capacity. Hypercholesterolemia, hypertension, tobacco use, hyperglycemia, obesity, and advanced age all impair collateral development (reviewed in de Groot et al., 2009). Risk factor reduction by medication may counteract these effects, however, antiarteriogenic effects of some antiplatelet agents (Hoefer et al., 2005; Duelsner et al., 2011) are due to their anti-inflammatory side effects.
Adaptive arteriogenesis occurs in the adult organism, and can be stimulated to overcome its physiological restrictions to induce or enhance arteriogenesis in patients with vascular occlusive disease (therapeutic arteriogenesis). Two therapeutic focus points have emerged: the augmentation of monocyte migration and subsequent vascular remodeling through the application of growth factors, and the sustained elevation of FSS as the mechanical stimulus for the induction of collateral growth.
The critical role of monocytes and pro-inflammatory cytokines (Buschmann et al., 2003) for adaptive arteriogenesis yields a number of candidates for its therapeutic stimulation. Proarteriogenic cytokines aim at a prolongation of monocyte life span, augmenting monocyte migration into the vessel wall and subsequent SMC proliferation. Concern has been expressed for most of this substances, as their effects seem to overlap with atherosclerotic disease mechanisms (Janus phenomenon; Epstein et al., 2004; Grundmann et al., 2007a). A detailed analysis of the mechanisms involved is most important for the safe and efficient pharmacological and/or hemodynamic augmentation of arteriogenesis.
Substances that have been proarteriogenic in preclinical studies are MCP-1, the colony-stimulating factors GM-CSF and G-CSF, TGFβ1, FGFs and, to a certain degree, VEGF. MCP-1 enhances collateral conductance in PAD, but showed proarteriogenic side effects (van Royen et al., 2003). G-CSF and GM-CSF are attractive pharmacological targets, as safety and dosage data in human subjects are available from their use in the treatment of leucopenia. GM-CSF promotes arteriogenesis in the brain (Schneeloch et al., 2004; Love, 2003), the myocardial vasculature (Seiler et al., 2001), and the periphery (Grundmann et al., 2006). G-CSF has yielded similarly promising first results in the brain (Sugiyama et al., 2011) and heart (Deindl et al., 2006), respectively. TGFβ1 augments collateral growth in animal models of PAD and coronary stenosis, even when applied locally (Grundmann et al., 2007b). However, a deregulation of the TGFβ cascade is implied in the progression of atherosclerosis, necessitating more research. FGF-2 stimulates arteriogenesis in the heart and periphery (Grundmann et al., 2007a). In addition, there are some conflicting data on the role of exogenous VEGF in promoting arteriogenesis in the heart (van Royen et al., 2010).
Collateral growth can be stimulated by enhancing and/or sustaining elevated FSS levels on the endothelium. Arteriovenous (AV) shunts lead to a sustained FSS level, wherein collateral growth and increasing collateral conductance generate a positive feedback mechanism. High shunt flows can overcome the physiological restrictions of arteriogenesis, leading to a normalization and overcompensation of maximal conductance (Pipp et al., 2004; Eitenmüller et al., 2006). Besides advancing our understanding of the basic mechanisms of arteriogenesis and of establishing benchmarks for therapies, the results from shunt models translate into practical application. For decades, mechanisms to non-invasively increase FSS have been known, namely the beneficial effects of physical training on patients with atherosclerotic diseases, probably even before Heberden (1772) reported an alleviation of exercise-induced angina pectoris by sawing wood each day. During adaptive collateral growth the restoration of flow brought about by the growing collateral reduces FSS and prematurely terminates arteriogenesis. Exercise training maintains elevated FSS level, thus augmenting collateral proliferation (reviewed in Bondke Persson et al., 2011). External counterpulsation is currently under evaluation for the clinical use in patients who are unable to undergo the necessary long and intense training intervals, with promising first results (Buschmann et al., 2009; Thakkar et al., 2010).
Unfortunately, therapeutic neovascularization has not found its way to large-scale clinical application (Grundmann et al., 2007a; Rehman, 2008). This is probably due to a number of reasons:
First, individual differences seem to be decisive. In a large study population, Wustmann et al. (2003) found 20–25% of patients with functional collaterals able to prevent all signs of myocardial ischemia during coronary occlusion, indicating a complex genetic predisposition that acts in favor of adaptive collateral growth, while it fails in others.
Second, adequate endpoints and sufficient methods of assessment are critical. If, for example, Laser Doppler flow measurements alone are utilized, they will not provide a thorough characterization of the extremity’s angioarchitecture, as superficial vessels contribute overproportionally to the results.
Other obstacles include, e.g., the difficulty to blind studies with growth factor administration due to the side effects of the medication (van Royen et al., 2010). Therefore, intense further research seems necessary before therapeutic neovascularization will be applicable in the clinical setting.
Arteriogenesis counteracts the narrowing of an adult conductance artery. In contrast to the genetic program for vasculogenesis, and changes in tissue oxygenation for angiogenic processes, mechanical factors, mainly changes in blood flow, regulate arteriogenesis.
The growth of blood vessels is an integral part of embryonic development. In adult life, it is often related to pathologic processes, such as the vascularization of neoplastic tissue or the organism’s attempt at compensating for an arterial stenosis (Figure 2). Three major growth processes, vasculogenesis, angiogenesis, and arteriogenesis, and their main determinants, i.e., genetic predisposition, oxygen tension, and hemodynamic forces, have been introduced. Vascular growth processes in vivo – within the embryo, in growing collaterals or in tumors – usually comprise more than one of these mechanisms. Thus, an interplay of genetics, hemodynamics, and oxygen tension determines vessel identity, morphology, and function during ontogenesis and in adult life.
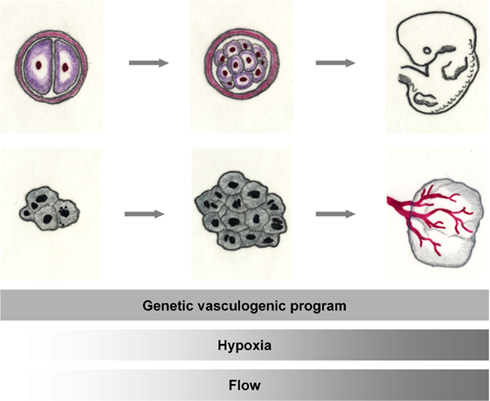
Figure 2. The tumor and the embryo. Rapid cell division leads to exponential tissue growth both during the growth of the mammalian embryo as well as in malignant tissues. Basic mechanisms of vascular growth occur both during normal development and tumor growth: vessel growth is promoted by the expression of angiogenic factors. When adequate oxygenation can no longer be warranted by diffusion, hypoxia, in addition, activates the angiogenic cascade. With the onset of flow, hemodynamics shape the developing vasculature.
Genetic hardwiring controls embryonic vessel formation, identity, and positioning (Jones et al., 2006). However, the significant interindividual differences in, e.g., the ability to form collaterals in response to environmental factors in adult life indicates a role of genetic predetermination also later in life.
The physical environment modifies vessel morphology and function, examples of which are the hypoxic induction of angiogenesis and the initiation of arteriogenesis by hemodynamic factors. The picture is rendered more complex by the fact that although one environmental factor has essentially been associated with one growth mechanism, they overlap: tissue oxygen tension is essentially associated with angiogenesis, but influences both vasculogenesis and arteriogenesis (Semenza, 2007b), and hemodynamic factors such as flow determine embryonic vessel identity (Jones et al., 2006), branching morphogenesis of angiogenic sprouts (Carmeliet, 2000), and arteriogenic EC activation (Bondke et al., 2008; Troidl et al., 2009).
A thorough understanding of how these events occur and how they are controlled is a critical prerequisite for therapeutical interventions that aim at (neo-)vascularization.
During embryogenesis, vasculogenesis and primary remodeling processes give rise to a vascular plexus, in which arteriovenous identity is established and primary maturation takes place. Second, the embryonic vascular network is optimized to hemodynamic and functional efficiency (Jones et al., 2006; Rey and Semenza, 2010).
During the initial phase, vasculogenesis predominates, tightly controlled by genetic determination (Jones et al., 2006; Semenza, 2007b; Jin and Patterson, 2009). This phase is dominated by cellular differentiation and decreasing plasticity (Jin and Patterson, 2009). A primary vascular plexus is formed rather stereotypically (Semenza, 2007b). The VEGF family and the angiopoietins assume a leading role, others are the Wnt and Delta/Notch pathways, the ephrins, growth factors such as TGFβ and FGF as well as a number of transcription factors (reviewed in Jin and Patterson, 2009).
Factors guiding vascular development are shared with the nervous system (Carmeliet and Tessier-Lavigne, 2005). Tip cells are functional equivalents to the axonal growth cone, following repulsive or attractive signals. VEGF and the netrin receptor unc-5 homolog b (UNC5B) are prominent examples of such guidance cues, while Dll4/Notch signaling is in control of tip cell differentiation (Le Noble et al., 2008). The genetic regulation of AV identity before the onset of blood flow is determined by signaling molecules such as Neuropilin-1 and Notch1 for arteries and Neuropilin-2 and Ephrin B4 for veins.
Environmental influences do not significantly determine initial vascular plexus generation, but hemodynamics (Lucitti et al., 2007) and tissue oxygenation (Ramírez-Bergeron et al., 2004; Semenza, 2007b) modulate early vasculogenesis (Jin and Patterson, 2009). The role of HIF in earliest vascularization is still debated: HIF-1α knockout mice are lethal due to early vascular remodeling defects, while mice embryos subjected to constant hypoxia develop normally (Lucitti et al., 2007). On the other hand, factors critically involved in early vasculogenesis such as VEGF and TGFβ are major HIF-1α target genes (Jin and Patterson, 2009), and deletion of the HIF-beta subunit (ARNT) results in defective hematopoiesis and the inhibition of both vasculogenesis and angiogenesis (Ramírez-Bergeron et al., 2006).
As soon as the heart starts pumping blood through the primary circulation, flow contributes to the initial remodeling processes that affect embryonic vessels. In this phase, the primary vascular plexus is remodeled to a secondary one (Patan, 2000). Cardiac failure in mice during development leads to severe early remodeling defects, and flow-induced eNOS expression seems to control secondary plexus generation (Lucitti et al., 2007). Vasculogenesis is supplemented or replaced by hypoxia-induced angiogenesis. The capillary network that forms within the individual tissues is much more individual-specific then the primary vascular plexus (Semenza, 2007b), indicating environmental influences.
AV determination is a model process in which environmental factors determine vascular form and function before a genetically determined background. Feedback signals from vascular function (pressure, flow, metabolic state of the tissue) modify the genetically determined outline after the onset of blood flow. Without flow, the expression of specific AV markers in the yolk sac vasculature is not sufficient to induce differentiation, while perfusion with venous blood transforms an early artery into a vein (Le Noble et al., 2008). Local tissue hypoxia suppresses the arterial markers Delta-like 4 (Dll4) and Ephrin B2 (Claxton and Fruttiger, 2005).
In this regard, the embryonic development of pre-existent collateral arteries remains elusive. Differentiation and maturation of the arterial system during development occurs in a proximal-to-distal direction, forming the arterial tree (Schaper and Schaper, 2004). Collateral arteries, however, are devoid of a clear proximal-to-distal axis. It has been shown that collaterals continue to develop in postnatal life, by arterialization of a capillary connection of two arteriolar endings (Price et al., 1994). It is tempting to presume a similar mechanism for the embryonic period, but it has until now not been demonstrated. However, as inhibition of artery formation occurs when a maturing artery approaches the irrigation domain of a neighboring branch (Durán et al., 1998), the disinhibition of these not yet known signaling pathways may contain clues to how pre-existent collaterals develop and why they differ between individuals (Schaper and Schaper, 2004).
Until the onset of circulation, vascular development is genetically determined. Environmental factors such as hemodynamics and oxygen tension then contribute significantly to the remodeling events that shape the individual-specific, secondary vascular plexus.
Atherosclerosis, a systemic disease of the vascular system, leads to progressive narrowing and occlusion of conductance arteries, a major cause of morbidity and mortality worldwide. Arterial occlusion prompts an adaptive response of the organisms to compensate for perfusion deficits. While acute changes in flow and tissue perfusion are counteracted by changes in vascular tone, chronic stenoses lead to adaptive vascular growth. Downstream of the stenosis, reduced perfusion leads to ischemic hypoxia. HIF-1 is upregulated, VEGF and other angiogenic factors are secreted, inducing a capillary proliferation within or in the border zone of the ischemic tissue (Wagner et al., 2002; Semenza, 2007b). However, stenosis of a conduit artery cannot be compensated for by capillary growth: according to the law of Hagen and Poiseuille, the number of capillaries needed to compensate for the lost conductance capacity would leave no more room for the non-vascular tissue that is to be supplied (Scholz et al., 2001).
Arteriogenesis occurs upstream of and in time with ischemia-induced angiogenesis (Figure 3). In different animal models, arteriogenesis was the most efficient mechanism to increase blood flow to an ischemic region (Scholz et al., 2002). Therapies aiming at the restoration of arterial blood supply must therefore target the proximal conduit vessel (Carmeliet and Tessier-Lavigne, 2005).
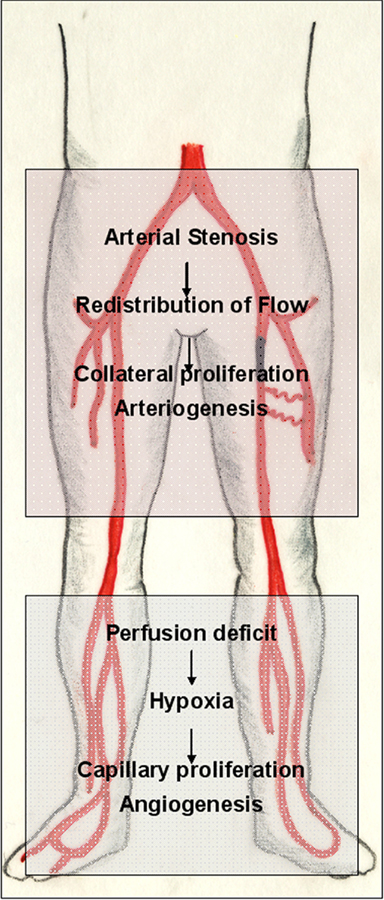
Figure 3. Adaptive vascular growth in peripheral arterial disease (PAD). The pathophysiology of PAD is a suitable example of the interplay of different mechanisms partaking in adaptive vascular growth. Arterial inflow is usually compromised by a proximal (iliac or femoropopliteal) stenosis of one of the arteries that supply the lower extremity. Ischemia endangers and damages the distal (infrapopliteal) region. Hypoxia in the lower leg triggers an angiogenic response in the border zone of the ischemic tissue, which, however, cannot compensate for the loss of pulsatile arterial inflow. If sufficient collateral arterioles are present to circumvent the proximal stenosis, however, an increase in flow triggers their outgrowth, developing a biological bypass through arteriogenesis.
Arteriogenesis, as a homeostatic mechanism, is switched off when FSS normalizes, as does angiogenesis when tissue oxygen tensions have reached physiologic values (Semenza, 2007a). This may occur before perfusion is restored sufficiently. Therefore, therapeutic arteriogenesis aims at an augmentation of the naturally occurring adaptive response.
Arteriogenesis is not related primarily to changes in oxygen tension (Deindl et al., 2001). It affects vessels that are continuously perfused with oxygenated arterial blood (van Royen et al., 2010) upstream of the ischemic area at risk. Interestingly, however, experimental administration of a constitutively active form of HIF-1α positively influenced angiogenesis as well as arteriogenesis in a hindlimb ischemia model (Patel et al., 2005). HIF-1α polymorphisms are associated with individual differences in coronary collateralization (Resar et al., 2005) and the role of VEGF in coronary arteriogenesis is still debated (van Royen et al., 2010).
Interestingly, one of the properties of angiogenic cytokines is the mobilization and recruitment of heterogeneous bone-marrow derived cells (Grunewald et al., 2006). This observation prompted the assumption that vasculogenesis would be a part of the postnatal adaptive response to ischemia. However, vasculogenesis as such is not regulated by oxygen gradients (Semenza, 2007b), and the cells detected in the postnatal circulation and recruited to sites of vascular remodeling are often inflammatory in nature (Semenza, 2007b) and not incorporated into growing collaterals, but rather support vessel wall remodeling, as monocytes do (Deindl and Schaper, 2005). Angiogenic cytokines might contribute to their recruitment and proarteriogenic action.
While angiogenesis inhibitors have made it to clinical application, the administration of individual proangiogenic factors alone or in combination has so far not yielded results that would justify their use in a larger patient population (Semenza, 2007b). Unfortunately, the induction of arteriogenesis through growth factor administration, although more than promising in animal models, has also not yet reached the clinics. Reasons for that might lie in the multifactorially impaired response to ischemia in patients that calls for multifactorial therapies, the necessary individual risk factor management and differences in genetic background (Semenza, 2007b) as well as the sensitivity of assessment methods that had been available so far for the use in human subjects (van Royen et al., 2010). Considering the biophysics of oxygenated blood flow in case of an arterial stenosis, a focus on the restoration of superordinate arterial inflow is critical, which should then be followed by an attenuation of the distribution within the ischemic area.
Further research will be necessary to elucidate the related pathophysiological mechanisms, while special attention must be paid to appropriate assessment techniques and endpoints.
In vascular occlusive disease, angiogenesis occurs within the ischemic tissues in an attempt to compensate for the lack in tissue oxygen. Arteriogenesis, leading to the positive outward remodeling of collateral arteries upstream of the ischemic tissue, is capable of restoring arterial inflow and must thus be at the focus of therapeutic approaches.
The size and aggressiveness of solid malignancies depends on their vascularization. Vascular growth in tumors shares features of physiological angiogenesis during development (Figure 2). Tissues grow until the delivery of oxygen and nutrients by diffusion is no longer possible. Then, hypoxia initiates the angiogenic cascade, ensuring oxygen and nutrient supply. During development, the restoration of tissue oxygen tension shuts down the angiogenic cascade, and vessels mature to meet specific demands of their host tissue. Malignant growth, however, is accompanied by an almost unlimited, uncoordinated proliferation of vascular tissues (Chung et al., 2010), which leads to blood vessels that differ from their physiological counterparts (Figure 4).
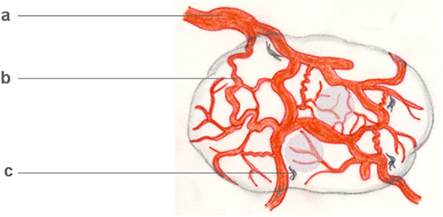
Figure 4. Tumor arteriogenesis/angiogenesis. Malignant neoplasms often critically depend on the development of an autonomous vascular system, which differs from physiological vascular tissue. Tumor vessels are devoid of a conventional arterial–venous hierarchy, unevenly distributed, often irregularly enlarged (a), leaky and often insufficient. Even in vascularized tumors, hypoxic regions develop (shaded areas). Interestingly, however, some tumor vessels share features of growing collateral arterioles in arteriogenesis, examples being their tortuosity (b) and the presence of perivascular macrophages (c). On a cellular and subcellular level, basement membrane degradation, SMC dedifferentiation, and growth factor profiles further indicate tumor arteriogenesis.
Current research efforts have focused on the distinguishing characteristics of tumor vessels, aiming at targeted therapies (Nagy et al., 2009). Tumor blood vessels do not exhibit the conventional hierarchy of the vascular system. They are distributed unevenly, are tortuous and leaky and comprise AV shunts without an obvious feeding function. Interestingly, flow directions may vary. Despite the continuous growth of the tumor vasculature, tumors tend to outgrow their blood supply. Vessel density is generally higher at the interface with the host organism, all of these factors indicating inefficient growth, which is confirmed by the lower overall blood flow in tumors when compared to normal tissues.
The formation of tumor blood vessels roughly follows the three physiological mechanisms of vascular growth discussed above, however, significant aberrations occur.
Vasculogenesis denotes the de novo generation of blood vessels from immature progenitor cells, an issue still debated for the adult organism. EPCs have been observed in the adult bloodstream, in conjunction with growing malignancies, and have even been suggested to be surrogate markers of cancer progression and chemotherapeutic efficacy (Patenaude et al., 2010). While the existence of bone-marrow derived EPC that structurally incorporate into the tumor vasculature is indicated by a number of high-quality studies, the relevance of this phenomenon for cancer progression remains controversial.
In addition, (sub-)populations of cancer cells are apparently able to aid tumor vascularization through a mechanisms called “vasculogenic mimicry,” namely the physical drilling of blood-conducting tunnels (Maniotis et al., 1999), which are primarily devoid of ECs, but mimic EC behavior to such an extent, that cell lines derived from these tissues have mistakenly been considered to be of endothelial origin (Moldovan et al., 2000). In later stages, endothelialization takes place (Ribatti and Vacca, 2009). Vasculogenic mimicry occurs in different cancer types and stages, and is currently met with much interest as a mechanisms that determines therapeutic sensitivity and invasiveness.
Tumor angiogenesis has been a therapeutic target since the suggestion of antiangiogenic chemotherapy 40 years ago (Folkman, 1971). When the growing tumor reaches a critical dimension, hypoxia upregulates mediators such as the HIFs, which then transcriptionally activate the proangiogenic cascade via, e.g., the VEGF family and its co-players. Moreover, uncoordinated secretion of proangiogenic factors occurs, driven by oncogenes, by tumor suppressor inactivation etc. (Dvorak, 2003). VEGF-A, which assumes a central role in tumor angiogenesis (Nagy et al., 2009; Chung et al., 2010), has, together with its receptor, until now been at the center of attention. However, drugs targeting VEGF-A are costly, have serious adverse effects, and have until now only been shown to be effective as part of an aggressive combined therapeutic regimen (Jain et al., 2006).
Searching the extensive biomedical databases for “tumor arteriogenesis” still yields only a handful of peer-reviewed publications. However, if one has a closer look at tumor vascularization, features of collateral growth, as described for adaptive arteriogenesis, seem to fabricate the feeding vessels of growing malignancies. It appears that in the hunt for innovative cancer therapies, tumor arteriogenesis has been somewhat neglected, but may be of great value as a therapeutic target.
Tumor vessels are tortuous and enlarged (Nagy et al., 2009; Chung et al., 2010). In this, they bear a striking resemblance to growing collateral arteries, which proliferate in response to an immediate pressure gradient. Thus, tumor vessels assume aspects of a growing collateral which are so characteristic that they have been defined as arteriogenesis’ distinguishing features (Schaper and Schaper, 2004).
Capillarization, especially of highly vascularized tumors, leads to a decrease in vascular resistance over the feeding vessel, followed by an increase in blood flow, which increases FSS on its endothelium, as occurs in growing collaterals of a vascular stenosis. Indeed, “tumor arteriogenesis” has been postulated (Yu and Rak, 2003), and the enlargement of feeding tumor vessels with increasing tumor capillarization has been observed (Folkman, 1997). The ultrastructure of growing tumor vessels strikingly resembles that of growing collateral arteries. Mother/feeding vessel formation requires BM degradation, pericyte detachment, and vascular enlargement (Nagy et al., 2009). These processes similarly occur in growing collateral arterioles of the extremities and the heart (Schaper, 1971), although in adaptive arteriogenesis, the process is, unsurprisingly, ordered and controlled: the thin-walled collaterals of the first phase of collateral development are subsequently stabilized and matured. Tumor vessels may remain in a disordered state (Nagy et al., 2009). Transient SMC dedifferentiation has been observed in tumor vessels as well as in adaptive collateral growth. The resulting feeder arteries and draining veins are often endowed with a complete, multilayered pericyte/SMC coat, have lost VEGFR2, no longer depend on VEGF-A and survive indefinitely without VEGF-A (Nagy et al., 2009). This may explain the fact that anti-VEGF/VEGFR damage tumors at early stages of development, but not at later stages when their vessels contain mature, SMC-coated vessels (Bergers et al., 2003). An inhibition of PDGF, however, blocked the further growth of late-to-end-stage tumors, and was, in combination with VEGF inhibitors, more effective against all stages of experimentally induced pancreatic carcinoma (Bergers et al., 2003). Although PDGF has been detected in the pericyte layer of tumor feeding vessels, the exact mechanism of action is not clear. PDGF may play a role in the attraction of progenitor cells to growing tumor vessels, thereby promoting their growth, and that this may be where the PDGF-inhibition proves effective. However, taking into account that (a) the structural contribution of progenitor cells to adult vascular proliferation remains elusive (Patenaude et al., 2010) and that (b) PDGF mediates collateral proliferation in adaptive arteriogenesis (Wu et al., 2010), an alternative explanation for its effects on solid tumor vascularization would be the inhibition of tumor arteriogenesis that complements antiangiogenic treatment by VEGF-inhibition.
Macrophages assume a critical role within the tumor environment. Macrophagocytic cells are educated by the tumor environment as tumor-associated macrophages (TAMs), to be able to promote tumor invasion and metastasis by facilitating cell motility, matrix degradation and tumor vessel growth (Pollard, 2004). Thus, they co-determine prognosis and therapeutic results. The pivotal role of macrophages in angiogenesis has been described (Carmeliet, 2003), in tumor microenvironments, they produce and release VEGF and an array of proangiogenic cytokines and trophic factors (Pollard, 2004). They can assume a critical role in vascular mimicry, namely the drilling and endothelialization of vascular tunnels into the tumor tissue (Vacca and Ribatti, 2011).
However, arteriogenesis is the one mechanism of vascular growth which critically depends on macrophage function (Schaper et al., 1976; Schaper and Schaper, 2004; Bergmann et al., 2006). VEGF, abundantly expressed in tumor tissues, stimulates arteriogenesis by activating monocytic cells and stimulating their endothelial adhesion and transmigration (Heil et al., 2000). Given that tumor arteriogenesis has been underestimated, research efforts to elucidate the contribution of arteriogenesis to tumor vascularization and the cellular mediators involved therein will hopefully be an important step in the therapeutic approach against one major scourge of humanity.
Targeting the tumor vasculature is a critical step in cancer chemotherapy, but current results are limited and side effects are significant. A thorough look at the individual contribution of the different vascular growth processes to tumor vascularization (angiogenesis, arteriogenesis) may allow for more targeted approaches.
Vascular growth fabricates the first working organ system during organogenesis, and continues throughout life. Vasculogenesis, angiogenesis, and arteriogenesis are the three major processes through which vessels grow, each one occurring in response to a principal stimulus of genetic or environmental origin: vasculogenesis is determined genetically, decreased oxygen levels initiate angiogenesis, and increased flow gives rise to arteriogenic growth. All three growth processes contribute to embryonic growth and postnatal adaptive vascular proliferation, in response to, e.g., the stenotic narrowing of an artery or the expansion of a malignant neoplasm, making vascular growth processes a tempting therapeutic target. However, benchmarks raised in the lab have until now not been met by the results achieved at the bedside. It may prove helpful to go back to the lab to accurately distinguish between vasculogenesis, angiogenesis, and arteriogenesis contributing to the disease in question. When we overcome the borders of the individual biomedical disciplines, to, e.g., apply the current knowledge about arteriogenesis to re-examine how tumor vessels grow, we may significantly advance today’s research results as well as tomorrow’s health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ausprunk, D. H., and Folkman, J. (1977). Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc. Res. 14, 53–65.
Autiero, M., Waltenberger, J., Communi, D., Kranz, A., Moons, L., Lambrechts, D., Kroll, J., Plaisance, S., De Mol, M., Bono, F., Kliche, S., Fellbrich, G., Ballmer-Hofer, K., Maglione, D., Mayr-Beyrle, U., Dewerchin, M., Dombrowski, S., Stanimirovic, D., Van Hummelen, P., Dehio, C., Hicklin, D. J., Persico, G., Herbert, J. M., Communi, D., Shibuya, M., Collen, D., Conway, E. M., and Carmeliet, P. (2003). Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat. Med. 9, 936–943.
Barleon, B., Sozzani, S., Zhou, D., Weich, H. A., Mantovani, A., and Marmé, D. (1996). Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 87, 3336–3343.
Bayless, K. J., and Davis, G. E. (2002). The Cdc42 and Rac1 GTPases are required for capillary lumen formation in three-dimensional extracellular matrices. J. Cell. Sci. 115(Pt 6), 1123–1136.
Bergers, G., Song, S., Meyer-Morse, N., Bergsland, E., and Hanahan, D. (2003). Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J. Clin. Invest. 111, 1287–1295.
Bergmann, C. E., Hoefer, I. E., Meder, B., Roth, H., van Royen, N., Breit, S. M., Jost, M. M., Aharinejad, S., Hartmann, S., and Buschmann, I. R. (2006). Arteriogenesis depends on circulating monocytes and macrophage accumulation and is severely depressed in op/op mice. J. Leukoc. Biol. 80, 59–65.
Bernardini, G., Ribatti, D., Spinetti, G., Morbidelli, L., Ziche, M., Santoni, A., Capogrossi, M. C., and Napolitano, M. (2003). Analysis of the role of chemokines in angiogenesis. J. Immunol. Methods 273, 83–101.
Blum, Y., Belting, H. G., Ellertsdottir, E., Herwig, L., Lüders, F., and Affolter, M. (2008). Complex cell rearrangements during intersegmental vessel sprouting and vessel fusion in the zebrafish embryo. Dev. Biol. 316, 312–322.
Bondke, A., Hillmeister, P., Duelsner, A., Gruber, C., Buschmann, I. R., and Lehmann, K. (2008). Early-phase cerebral arteriogenesis: evidence of endothelial activation, vascular proliferation and local inflammation. FASEB J. 22, 1147.13.
Bondke Persson, A., Buschmann, E. E., Lindhorst, R., Troidl, K., Langhoff, R., Schulte, K. L., and Buschmann, I. (2011). Therapeutic arteriogenesis in peripheral arterial disease: combining intervention and passive training. VASA 40, 177–187.
Busch, H. J., Buschmann, I. R., Mies, G., Bode, C., and Hossmann, K. A. (2003). Arteriogenesis in hypoperfused rat brain. J. Cereb. Blood Flow Metab. 5, 621–628.
Buschmann, E. E., Utz, W., Pagonas, N., Schulz-Menger, J., Busjahn, A., Monti, J., Maerz, W., le Noble, F., Thierfelder, L., Dietz, R., Klauss, V., Gross, M., Buschmann, I. R., and Arteriogenesis Network (Art. Net.). (2009). Improvement of fractional flow reserve and collateral flow by treatment with external counterpulsation (Art.Net.-2 Trial). Eur. J. Clin. Invest. 39, 866–875.
Buschmann, I., Heil, M., Jost, M., and Schaper, W. (2003). Influence of inflammatory cytokines on arteriogenesis. Microcirculation 10, 371–379.
Buschmann, I., Pries, A., Styp-Rekowska, B., Hillmeister, P., Loufrani, L., Henrion, D., Shi, Y., Duelsner, A., Hoefer, I., Gatzke, N., Wang, H., Lehmann, K., Ulm, L., Ritter, Z., Hauff, P., Hlushchuk, R., Djonov, V., van Veen, T., and le Noble, F. (2010). Pulsatile shear and Gja5 modulate arterial identity and remodeling events during flow-driven arteriogenesis. Development 137, 2187–2196.
Caprioli, A., Minko, K., Drevon, C., Eichmann, A., Dieterlen-Lievre, F., and Jaffredo, T. (2001). Hemangioblast commitment in the avian allantois: cellular and molecular aspects. Dev. Biol. 238, 64–78.
Carmeliet, P., Moons, L., Luttun, A., Vincenti, V., Compernolle, V., De Mol, M., Wu, Y., Bono, F., Devy, L., Beck, H., Scholz, D., Acker, T., DiPalma, T., Dewerchin, M., Noel, A., Stalmans, I., Barra, A., Blacher, S., Vandendriessche, T., Ponten, A., Eriksson, U., Plate, K. H., Foidart, J. M., Schaper, W., Charnock-Jones, D. S., Hicklin, D. J., Herbert, J. M., Collen, D., and Persico, M. G. (2001). Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 7, 575–583.
Carmeliet, P., and Tessier-Lavigne, M. (2005). Common mechanisms of nerve and blood vessel wiring. Nature 436, 193–200.
Choi, K., Kennedy, M., Kazarov, A., Papadimitriou, J. C., and Keller, G. (1998). A common precursor for hematopoietic and endothelial cells. Development 125, 725–732.
Chung, A. S., Lee, J., and Ferrara, N. (2010). Targeting the tumour vasculature: insights from physiological angiogenesis. Nat. Rev. Cancer 10, 505–514.
Claxton, S., and Fruttiger, M. (2005). Oxygen modifies artery differentiation and network morphogenesis in the retinal vasculature. Dev. Dyn. 233, 822–828.
de Groot, D., Pasterkamp, G., and Hoefer, I. E. (2009). Cardiovascular risk factors and collateral artery formation. Eur. J. Clin. Invest. 39, 1036–1047.
Deindl, E. (2007). Arteriogenesis: a focus on signal transduction cascades and transcription factors. Thromb. Haemost. 98, 940–943.
Deindl, E., Buschmann, I., Hoefer, I. E., Podzuweit, T., Boengler, K., Vogel, S., van Royen, N., Fernandez, B., and Schaper, W. (2001). Role of ischemia and of hypoxia-inducible genes in arteriogenesis after femoral artery occlusion in the rabbit. Circ. Res. 89, 779–786.
Deindl, E., Zaruba, M. M., Brunner, S., Huber, B., Mehl, U., Assmann, G., Hoefer, I. E., Mueller-Hoecker, J., and Franz, W. M. (2006). G-CSF administration after myocardial infarction in mice attenuates late ischemic cardiomyopathy by enhanced arteriogenesis. FASEB J. 20, 956–958.
Demir, R., Seval, Y., and Huppertz, B. (2007). Vasculogenesis and angiogenesis in the early human placenta. Acta Histochem. 109, 257–265.
Djokovic, D., Trindade, A., Gigante, J., Badenes, M., Silva, L., Liu, R., Li, X., Gong, M., Krasnoperov, V., Gill, P. S., and Duarte, A. (2010). Combination of Dll4/Notch and Ephrin-B2/EphB4 targeted therapy is highly effective in disrupting tumor angiogenesis. BMC Cancer 10, 641. doi: 10.1186/1471-2407-10-641
Djonov, V., and Makanya, A. N. (2005). “New insights into intussusceptive angiogenesis,” in Mechanisms of Angiogenesis, eds M. Clauss and G. Breier (Berlin: Birkhäuser Verlag), 17–34.
Duan, L. J., Nagy, A., and Fong, G. H. (2003). Gastrulation and angiogenesis, not endothelial specification, is sensitive to partial deficiency in vascular endothelial growth factor-a in mice. Biol. Reprod. 69, 1852–1858.
Duelsner, A., Gatzke, N., Glaser, J., et al. (2011). Acetylsalicylic acid, but not clopidogrel, inhibits therapeutically induced cerebral arteriogenesis in the hypoperfused rat brain. J. Cereb. Blood Flow Metab. (in print).
Durán, A. C., Arqué, J. M., Sans-Coma, V., Fernández, B., and de Vega, N. G. (1998). Severe congenital stenosis of the left coronary artery ostium and its possible pathogenesis according to current knowledge on coronary artery development. Cardiovasc. Pathol. 7, 261–266.
Dvorak, H. F. (2003). Rous-Whipple Award Lecture: how tumors make bad blood vessels and stroma. Am. J. Pathol. 162, 1747–1757.
Egginton, S., and Gerritsen, M. (2003). Lumen formation: in vivo versus in vitro observations. Microcirculation 10, 45–61.
Eichmann, A., Corbel, C., Pardanaud, L., Bréant, C., Moyon, D., and Yuan, L. (2000). Hemangioblastic precursors in the avian embryo. Curr. Top Microbiol. Immunol. 251, 83–90.
Eitenmüller, I., Volger, O., Kluge, A., Troidl, K., Barancik, M., Cai, W. J., Heil, M., Pipp, F., Fischer, S., Horrevoets, A. J., Schmitz-Rixen, T., and Schaper, W. (2006). The range of adaptation by collateral vessels after femoral artery occlusion. Circ. Res. 99, 656–662.
Eliceiri, B. P., Paul, R., Schwartzberg, P. L., Hood, J. D., Leng, J., and Cheresh, D. A. (1999). Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol. Cell 4, 915–924.
Epstein, S. E., Stabile, E., Kinnaird, T., Lee, C. W., Clavijo, L., and Burnett, M. S. (2004). Janus phenomenon: the interrelated tradeoffs inherent in therapies designed to enhance collateral formation and those designed to inhibit atherogenesis. Circulation 109, 2826–2831.
Ferguson, J. E. III, Kelley, R. W., and Patterson, C. (2005). Mechanisms of endothelial differentiation in embryonic vasculogenesis. Arterioscler. Thromb. Vasc. Biol. 25, 2246–2254.
Ferrara, N., Gerber, H. P., and LeCouter, J. (2003). The biology of VEGF and its receptors. Nat. Med. 9, 669–676.
Fischer, C., Schneider, M., and Carmeliet, P. (2006). Principles and therapeutic implications of angiogenesis, vasculogenesis and arteriogenesis. Handb. Exp. Pharmacol. 176(Pt 2), 157–212.
Flamme, I., Frölich, T., and Risau, W. (1997). Molecular mechanisms of vasculogenesis and embryonic angiogenesis. J. Cell. Physiol. 173, 206–210.
Folkman, J. (1997). “Tumor angiogenesis,” in Cancer Medicine, eds J. F. Holland, R. C. Bast, D. L. Morton, E. Frei, D. W. Kufe and R. R. Weichselbaum (Baltimore, MD: Williams & Wilkins), 181–204.
Gorospe, M., Tominaga, K., Wu, X., Fähling, M., and Ivan, M. (2011). Post-transcriptional control of the hypoxic response by RNA-binding proteins and microRNAs. Front. Mol. Neurosci. 4:7. doi: 10.3389/fnmol.2011.00007
Gray, C., Packham, I. M., Wurmser, F., Eastley, N. C., Hellewell, P. G., Ingham, P. W., Crossman, D. C., and Chico, T. J. (2007). Ischemia is not required for arteriogenesis in zebrafish embryos. Arterioscler. Thromb. Vasc. Biol. 10, 2135–2141.
Green, J. B. A., New, H. V., and Smith, J. C. (1992). Responses of embryonic Xenopus cells to activin and IGI are separated by multiple dose thresholds and correspond to distinct axes of the mesoderm. Cell 71, 731–739.
Grimshaw, M. J., and Balkwill, F. R. (2001). Inhibition of monocyte and macrophage chemotaxis by hypoxia and inflammation – a potential mechanism. Eur. J. Immunol. 31, 480–489.
Grundmann, S., Hoefer, I., Ulusans, S., Bode, C., Oesterle, S., Tijssen, J. G., Piek, J. J., Buschmann, I., and van Royen, N. (2006). Granulocyte-macrophage colony-stimulating factor stimulates arteriogenesis in a pig model of peripheral artery disease using clinically applicable infusion pumps. J. Vasc. Surg. 43, 1263–1269.
Grundmann, S., Piek, J. J., Pasterkamp, G., and Hoefer, I. E. (2007a). Arteriogenesis: basic mechanisms and therapeutic stimulation. Eur. J. Clin. Invest. 37, 755–766.
Grundmann, S., van Royen, N., Pasterkamp, G., Gonzalez, N., Tijsma, E. J., Piek, J. J., and Hoefer, I. E. (2007b). A new intra-arterial delivery platform for pro-arteriogenic compounds to stimulate col- lateral artery growth via transforming growth factor-beta1 release. J. Am. Coll. Cardiol. 50, 351–358.
Grundmann, S., Schirmer, S. H., Hekking, L. H., Post, J. A., Ionita, M. G., de Groot, D., van Royen, N., van den Berg, B., Vink, H., Moser, M., Bode, C., de Kleijn, D., Pasterkamp, G., Piek, J. J., and Hoefer, I. E. (2009). Endothelial glycocalyx dimensions are reduced in growing collateral arteries and modulate leucocyte adhesion in arteriogenesis. J. Cell. Mol. Med. 13, 3463–1374.
Grunewald, M., Avraham, I., Dor, Y., Bachar-Lustig, E., Itin, A., Jung, S., Chimenti, S., Landsman, L., Abramovitch, R., and Keshet, E. (2006). VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell 124, 175–189.
Hänze, J., Weissmann, N., Grimminger, F., Seeger, W., and Rose, F. (2007). Cellular and molecular mechanisms of hypoxia-inducible factor driven vascular remodeling. Thromb. Haemost. 97, 774–787.
Heberden, W. (1772). Some account of a disorder of the breast. Med. Trans. R. Coll. Physicians Lond. 2, 59–67.
Heil, M., Clauss, M., Suzuki, K., Buschmann, I. R., Willuweit, A., Fischer, S., and Schaper, W. (2000). Vascular endothelial growth factor (VEGF) stimulates monocyte migration through endothelial monolayers via increased integrin expression. Eur. J. Cell Biol. 79, 850–857.
Hellström, M., Phng, L. K., Hofmann, J. J., Wallgard, E., Coultas, L., Lindblom, P., Alva, J., Nilsson, A. K., Karlsson, L., Gaiano, N., Yoon, K., Rossant, J., Iruela-Arispe, M. L., Kalén, M., Gerhardt, H., and Betsholtz, C. (2007). Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445, 776–780.
Hillmeister, P., Lehmann, K. E., Bondke, A., Witt, H., Duelsner, A., Gruber, C., Busch, H. J., Jankowski, J., Ruiz-Noppinger, P., Hossmann, K. A., and Buschmann, I. R. (2008). Induction of cerebral arteriogenesis leads to early-phase expression of protease inhibitors in growing collaterals of the brain. J. Cereb. Blood Flow Metab. 28, 1811–1823.
Hirakow, R., and Hiruma, T. (1981). Scanning electron microscopic study on the development of primitive blood vessels in chick embryos at the early somite-stage. Anat. Embryol. 163, 299–306.
His, W. (1900). Lecithoblast und Angioblast der Wirbeltiere. Abhandl. Math-Phys. Ges. Wiss. 26, 171–328.
Hoefer, I. E., Grundmann, S., Schirmer, S., van Royen, N., Meder, B., Bode, C., Piek, J. J., and Buschmann, I. R. (2005). Aspirin, but not clopidogrel, reduces collateral conductance in a rabbit model of femoral artery occlusion. J. Am. Coll. Cardiol. 46, 994–1001.
Huber, T. L., Kouskoff, V., Fehling, H. J., Palis, J., and Keller, G. (2004). Haemangioblast commitment is initiated in the primitive streak of the mouse embryo. Nature 432, 625–630.
Iruela-Arispe, M. L., and Davis, G. E. (2009). Cellular and molecular mechanisms of vascular lumen formation. Dev. Cell 16, 222–231.
Isaacs, H. V., Tannahill, O., and Slack, M. (1992). Expression of a novel FGF in the Xenopus embryo. A new candidate-inducing factor for mesoderm formation and anteroposterior specification. Development 14, 711–720.
Ito, W. D., Arras, M., Scholz, D., Winkler, B., Htun, P., and Schaper, W. (1997). Angiogenesis but not collateral growth is associated with ischemia after femoral artery occlusion. Am. J. Physiol. 273(3 Pt 2), H1255–H1265.
Jain, R. K., Duda, D. G., Clark, J. W., and Loeffler, J. S. (2006). Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat. Clin. Pract. Oncol. 3, 24–40.
Jakobsson, L., Franco, C. A., Bentley, K., Collins, R. T., Ponsioen, B., Aspalter, I. M., Rosewell, I., Busse, M., Thurston, G., Medvinsky, A., Schulte-Merker, S., and Gerhardt, H. (2010). Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 12, 943–953.
Jin, S. W., and Patterson, C. (2009). The opening act: vasculogenesis and the origins of circulation. Arterioscler. Thromb. Vasc. Biol. 29, 623–629.
Jones, E. A., le Noble, F., and Eichmann, A. (2006). What determines blood vessel structure? Genetic prespecification vs. hemodynamics. Physiology (Bethesda) 21, 388–395.
Kamei, M., Saunders, W. B., Bayless, K. J., Dye, L., Davis, G. E., and Weinstein, B. M. (2006). Endothelial tubes assemble from intracellular vacuoles in vivo. Nature 442, 453–456.
Kertesz, N., Wu, J., Chen, T. H., Sucov, H. M., and Wu, H. (2004). The role of erythropoietin in regulating angiogenesis. Dev. Biol. 276, 101–110.
Kluk, M. J., and Hla, T. (2002). Signaling of sphingosine-1-phosphate via the S1P/EDG-family of G-protein-coupled receptors. Biochim. Biophys. Acta 1582, 72–80.
Krishnan, L., Underwood, C. J., Maas, S., Ellis, B. J., Kode, T. C., Hoying, J. B., and Weiss, J. A. (2008). Effect of mechanical boundary conditions on orientation of angiogenic microvessels. Cardiovasc. Res. 78, 324–332.
Lancrin, C., Sroczynska, P., Stephenson, C., Allen, T., Kouskoff, V., and Lacaud, G. (2009). The haemangioblast generates haematopoietic cells through a haemogenic endothelium stage. Nature 457, 892–897.
Le Noble, F., Klein, C., Tintu, A., Pries, A., and Buschmann, I. (2008). Neural guidance molecules, tip cells, and mechanical factors in vascular development. Cardiovasc. Res. 78, 232–241.
Lehoux, S., and Tedgui, A. (2003). Cellular mechanics and gene expression in blood vessels. J. Biomech. 36, 631–643.
Limbourg, A., Korff, T., Napp, L. C., Schaper, W., Drexler, H., and Limbourg, F. P. (2009). Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nat. Protoc. 12, 1737–1746.
Lindblom, P., Gerhardt, H., Liebner, S., Abramsson, A., Enge, M., Hellstrom, M., Backstrom, G., Fredriksson, S., Landegren, U., Nystrom, H. C., Bergstrom, G., Dejana, E., Ostman, A., Lindahl, P., and Betsholtz, C. (2003). Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 17, 1835–1840.
Love, R. (2003). GM-CSF induced arteriogenesis: a potential treatment for stroke? Lancet Neurol. 2, 458.
Lucitti, J. L., Jones, E. A., Huang, C., Chen, J., Fraser, S. E., and Dickinson, M. E. (2007). Vascular remodeling of the mouse yolk sac requires hemodynamic force. Development 134, 3317–3326.
Luttun, A., and Carmeliet, P. (2003). De novo vasculogenesis in the heart. Cardiovasc. Res. 58, 378–389.
Maniotis, A. J., Folberg, R., Hess, A., Seftor, E. A., Gardner, L. M., Pe’er, J., Trent, J. M., Meltzer, P. S., and Hendrix, M. J. (1999). Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am. J. Pathol. 155, 739–752.
Moldovan, N. I., Goldschmidt-Clermont, P. J., Parker-Thornburg, J., Shapiro, S. D., and Kolattukudy, P. E. (2000). Contribution of monocytes/macrophages to compensatory neovascularization: the drilling of metalloelastase-positive tunnels in ischemic myocardium. Circ. Res. 87, 378–384.
Mudumana, S. P., Hentschel, D., Liu, Y., Vasilyev, A., and Drummond, I. A. (2008). Odd skipped related-1 reveals a novel role for endoderm in regulating kidney versus vascular cell fate. Development 135, 3355–3367.
Murray, P. D. F. (1932). The development in vitro of the blood of the early chick embryo. Proc. Biol. Sci. 111, 497–521.
Nagy, J. A., Chang, S. H., Dvorak, A. M., and Dvorak, H. F. (2009). Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 100, 865–869.
Neufeld, G., Cohen, T., Shraga, N., Lange, T., Kessler, O., and Herzog, Y. (2002). The neuropilins: multifunctional semaphorin and VEGF receptors that modulate axon guidance and angiogenesis. Trends Cardiovasc. Med. 12, 13–19.
Pardanaud, L., Yassine, F., and Dieterlen-Lievre, F. (1989). Relationship between vasculogenesis, angiogenesis and haemopoiesis during avian ontogeny. Development 105, 473–485.
Patan, S. (2000). Vasculogenesis and angiogenesis as mechanisms of vascular network formation, growth and remodeling. J. Neurooncol. 50, 1–15.
Patel, T. H., Kimura, H., Weiss, C. R., Semenza, G. L., and Hofmann, L. V. (2005). Constitutively active HIF-1alpha improves perfusion and arterial remodeling in an endovascular model of limb ischemia. Cardiovasc. Res. 68, 144–154.
Patenaude, A., Parker, J., and Karsan, A. (2010). Involvement of endothelial progenitor cells in tumor vascularization. Microvasc. Res. 79, 217–223.
Pelosi, E., Valtieri, M., Coppola, S., Botta, R., Gabbianelli, M., Lulli, V., Marziali, G., Masella, B., Müller, R., Sgadari, C., Testa, U., Bonanno, G., and Peschle, C. (2002). Identification of the hemangioblast in postnatal life. Blood 100, 3203–3208.
Pipp, F., Boehm, S., Cai, W. J., Adili, F., Ziegler, B., Karanovic, G., Ritter, R., Balzer, J., Scheler, C., Schaper, W., and Schmitz-Rixen, T. (2004). Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arterioscler. Thromb. Vasc. Biol. 24, 1664–1668.
Pollard, J. W. (2004). Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 4, 71–78.
Price, R. J., Owens, G. K., and Skalak, T. C. (1994). Immunohistochemical identification of arteriolar development using markers of smooth muscle differentiation. Evidence that capillary arterialization proceeds from terminal arterioles. Circ. Res. 75, 520–527.
Ramírez-Bergeron, D. L., Runge, A., Adelman, D. M., Gohil, M., and Simon, M. C. (2006). HIF-dependent hematopoietic factors regulate the development of the embryonic vasculature. Dev. Cell 11, 81–92.
Ramírez-Bergeron, D. L., Runge, A., Dahl, K. D., Fehling, H. J., Keller, G., and Simon, M. C. (2004). Hypoxia affects mesoderm and enhances hemangioblast specification during early development. Development 131, 4623–4634.
Rehman, J. (2008). An inconvenient truth: recognizing individual differences in arteriogenesis. Circ. Res. 102, 1146–1147.
Resar, J. R., Roguin, A., Voner, J., Nasir, K., Hennebry, T. A., Miller, J. M., Ingersoll, R., Kasch, L. M., and Semenza, G. L. (2005). Hypoxia-inducible factor 1alpha polymorphism and coronary collaterals in patients with ischemic heart disease. Chest 128, 787–791.
Rey, S., and Semenza, G. L. (2010). Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc. Res. 86, 236–242.
Ribatti, D., and Vacca, A. (2009). The role of monocytes-macrophages in vasculogenesis in multiple myeloma. Leukemia 23, 1535–1536.
Sabin, F. (1917). Preliminary note on the differentiation of angioblasts and the method by which they produce blood-vessels, blood-plasma and red blood-cells as seen in the living chick (1917). J. Hematother. Stem Cell Res. 11, 5–7.
Saunders, W. B., Bohnsack, B. L., Faske, J. B., Anthis, N. J., Bayless, K. J., Hirschi, K. K., and Davis, G. E. (2006). Coregulation of vascular tube stabilization by endothelial cell TIMP-2 and pericyte TIMP-3. J. Cell Biol. 175, 179–191.
Schaper, J., König, R., Franz, D., and Schaper, W. (1976). The endothelial surface of growing coronary collateral arteries. Intimal margination and diapedesis of monocytes. A combined SEM and TEM study. Virchows Arch. A Pathol. Anat. Histol. 370, 193–205.
Schaper, W., Görge, G., Winkler, B., and Schaper, J. (1988). The collateral circulation of the heart. Prog. Cardiovasc. Dis. 31, 57–77.
Schaper, W., and Ito, W. D. (1996). Molecular mechanisms of coronary collateral vessel growth. Circ. Res. 79, 911–919.
Schaper, W., Piek, J. J., Munoz-Chapuli, R., Wolf, C., and Ito, W. (1999). “Collateral circulation of the heart,” in Angiogenesis and Cardiovascular Disease, eds J. A. Ware and M. Simons (Oxford: Oxford University Press), 159–198.
Schirmer, S. H., Fledderus, J. O., Bot, P. T., Moerland, P. D., Hoefer, I. E., Baan, J. Jr., Henriques, J. P., van der Schaaf, R. J., Vis, M. M., Horrevoets, A. J., Piek, J. J., and van Royen, N. (2008). Interferon-beta signaling is enhanced in patients with insufficient coronary collateral artery development and inhibits arteriogenesis in mice. Circ. Res. 102, 1286–1294.
Schneeloch, E., Mies, G., Busch, H. J., Buschmann, I. R., and Hossmann, K. A. (2004). Granulocyte-macrophage colony-stimulating factor-induced arteriogenesis reduces energy failure in hemodynamic stroke. Proc. Natl. Acad. Sci. U.S.A. 101, 12730–12735.
Schnurch, H., and Risau, W. (1993). Expression of tie-2, a member of a novel family of receptor tyrosine kinases, in the endothelial cell lineage. Development 119, 957–968.
Scholz, D., Cai, W. J., and Schaper, W. (2001). Arteriogenesis, a new concept of vascular adaptation in occlusive disease. Angiogenesis 4, 247–257.
Scholz, D., Ito, W., Fleming, I., Deindl, E., Sauer, A., Wiesnet, M., Busse, R., Schaper, J., and Schaper, W. (2000). Ultrastructure and molecular histology of rabbit hind-limb collateral artery growth (arteriogenesis). Virchows Arch. 436, 257–270.
Scholz, D., Ziegelhoeffer, T., Helisch, A., Wagner, S., Friedrich, C., Podzuweit, T., and Schaper, W. (2002). Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J. Mol. Cell. Cardiol. 34, 775–787.
Scholz, H., and Kirschner, K. M. (2011). Oxygen-dependent gene expression in development and cancer: lessons learned from the Wilms’ tumor gene, WT1. Front. Mol. Neurosci. 4:4. doi: 10.3389/fnmol.2011.00004
Sciesielski, L. K., Kirschner, K. M., Scholz, H., and Persson, A. B. (2010). Wilms’ tumor protein Wt1 regulates the Interleukin-10 (IL-10) gene. FEBS Lett. 584, 4665–4671.
Seiler, C., Pohl, T., Wustmann, K., Hutter, D., Nicolet, P. A., Windecker, S., Eberli, F. R., and Meier, B. (2001). Promotion of collateral growth by granulocyte-macrophage colony-stimulating factor in patients with coronary artery disease: a randomized, double-blind, placebo-controlled study. Circulation 104, 2012–2017.
Semenza, G. L. (2007a). Regulation of tissue perfusion in mammals by hypoxia-inducible factor 1. Exp. Physiol. 92, 988–991.
Semenza, G. L. (2007b). Vasculogenesis, angiogenesis, and arteriogenesis: mechanisms of blood vessel formation and remodeling. J. Cell Biochem. 102, 840–847.
Shalaby, F., Rossant, J., Yamaguchi, T. P., Gertsenstein, M., Wu, X. F., Breitman, M. L., and Schuh, A. C. (1995). Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376, 62–66.
Shintani, S., Murohara, T., Ikeda, H., Ueno, T., Sasaki, K., Duan, J., and Imaizumi, T. (2001). Augmentation of postnatal neovascularization with autologous bone marrow transplantation. Circulation 103, 897–903.
Song, L., and Tuan, R. S. (2004). Transdifferentiation potential of human mesenchymal stem cells derived from bone marrow. FASEB J. 18, 980–982.
Sugiyama, Y., Yagita, Y., Oyama, N., Terasaki, Y., Omura-Matsuoka, E., Sasaki, T., and Kitagawa, K. (2011). Granulocyte colony-stimulating factor enhances arteriogenesis and ameliorates cerebral damage in a mouse model of ischemic stroke. Stroke 42, 770–775.
Thakkar, B. V., Hirsch, A. T., Satran, D., Bart, B. A., Barsness, G., McCullough, P. A., Kennard, E. D., Kelsey, S. F., and Henry, T. D. (2010). The efficacy and safety of enhanced external counterpulsation in patients with peripheral arterial disease. Vasc. Med. 15, 15–20.
Toyota, E., Warltier, D. C., Brock, T., Ritman, E., Kolz, C., O’Malley, P., Rocic, P., Focardi, M., and Chilian, W. M. (2005). Vascular endothelial growth factor is required for coronary collateral growth in the rat. Circulation 112, 2108–2113.
Troidl, C., Troidl, K., Schierling, W., Cai, W. J., Nef, H., Möllmann, H., Kostin, S., Schimanski, S., Hammer, L., Elsässer, A., Schmitz-Rixen, T., and Schaper, W. (2009). Trpv4 induces collateral vessel growth during regeneration of the arterial circulation. J. Cell. Mol. Med. 13, 2613–2621.
Tzima, E., Irani-Tehrani, M., Kiosses, W. B., Dejana, E., Schultz, D. A., Engelhardt, B., Cao, G., DeLisser, H., and Schwartz, M. A. (2005). A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437, 426–431.
Vacca, A., and Ribatti, D. (2011). Angiogenesis and vasculogenesis in multiple myeloma: role of inflammatory cells. Recent Results Cancer Res. 183, 87–95.
van Hinsbergh, V. W., and Koolwijk, P. (2008). Endothelial sprouting and angiogenesis: matrix metalloproteinases in the lead. Cardiovasc. Res. 78, 203–212.
van Royen, N., Hoefer, I., Böttinger, M., Hua, J., Grundmann, S., Voskuil, M., Bode, C., Schaper, W., Buschmann, I., and Piek, J. J. (2003). Local monocyte chemoattractant protein-1 therapy increases collateral artery formation in apolipoprotein E-deficient mice but induces systemic monocytic CD11b expression, neointimal formation, and plaque progression. Circ. Res. 92, 218–225.
van Royen, N., Piek, J. J., Schaper, W., and Fulton, W. F. (2010). A critical review of clinical arteriogenesis research. J. Am. Coll. Cardiol. 55, 17–25.
Vogeli, K. M., Jin, S. W., Martin, G. R., and Stainier, D. Y. (2006). A common progenitor for haematopoietic and endothelial lineages in the zebrafish gastrula. Nature 443, 337–339.
Wagner, K. D., Wagner, N., Bondke, A., Nafz, B., Flemming, B., Theres, H., and Scholz, H. (2002). The Wilms’ tumor suppressor Wt1 is expressed in the coronary vasculature after myocardial infarction. FASEB J. 16, 1117–1119.
Wang, G. L., and Semenza, G. L. (1995). Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 270, 1230–1237.
Witzenbichler, B., Maisonpierre, P. C., Jones, P., Yancopoulos, G. D., and Isner, J. M. (1998). Chemotactic properties of angiopoietin-1 and -2, ligands for the endothelial-specific receptor tyrosine kinase Tie-2. J. Biol. Chem. 273, 18514–18521.
Wu, S., Wu, X., Zhu, W., Cai, W. J., Schaper, J., and Schaper, W. (2010). Immunohistochemical study of the growth factors, aFGF, bFGF, PDGF-AB, VEGF-A and its receptor (Flk-1) during arteriogenesis. Mol. Cell. Biochem. 343, 223–229.
Wustmann, K., Zbinden, S., Windecker, S., Meier, B., and Seiler, C. (2003). Is there functional collateral flow during vascular occlusion in angiographically normal coronary arteries? Circulation 107, 2213–2220.
Yancopoulos, G. D., Davis, S., Gale, N. W., Rudge, J. S., Wiegand, S. J., and Holash, J. (2000). Vascular-specific growth factors and blood vessel formation. Nature 407, 242–248.
Yang, H. T., Prior, B. M., Lloyd, P. G., Taylor, J. C., Li, Z., Laughlin, M. H., and Terjung, R. L. (2008). Training-induced vascular adaptations to ischemic muscle. J. Physiol. Pharmacol. 59(Suppl. 7), 57–70.
Yu, J. L., and Rak, J. W. (2003). Host microenvironment in breast cancer development: inflammatory and immune cells in tumour angiogenesis and arteriogenesis. Breast Cancer Res. 5, 83–88.
Zeeb, M., Strilic, B., and Lammert, E. (2010). Resolving cell-cell junctions: lumen formation in blood vessels. Curr. Opin. Cell Biol. 22, 626–632.
Keywords: vascular growth, vasculogenesis, angiogenesis, arteriogenesis
Citation: Persson AB and Buschmann IR (2011) Vascular growth in health and disease. Front. Mol. Neurosci. 4:14. doi: 10.3389/fnmol.2011.00014
Received: 10 June 2011; Accepted: 18 July 2011;
Published online: 24 August 2011.
Edited by:
Michael Fähling, Charité – Universitätsmedizin Berlin, GermanyReviewed by:
Carlo Sala, National Research Council Institute of Neuroscience, ItalyCopyright: © 2011 Persson and Buschmann. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Anja Bondke Persson, Institut für Vegetative Physiologie, Charité Universitaetsmedizin Berlin, Hessische Strasse 3-4, 10115 Berlin, Germany. e-mail:YW5qYS5ib25ka2VAY2hhcml0ZS5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.