



- 1 Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA, USA
- 2 Department of Cell Biology, Harvard Medical School, Boston, MA, USA
- 3 Division of Biology, California Institute of Technology, Pasadena, CA, USA
- 4 Max Planck Institute for Brain Research, Frankfurt am Main, Germany
Proteasome-mediated proteolysis is important for synaptic plasticity, neuronal development, protein quality control, and many other processes in neurons. To define proteasome composition in brain, we affinity purified 26S proteasomes from cytosolic and synaptic compartments of the rat cortex. Using tandem mass spectrometry, we identified the standard 26S subunits and a set of 28 proteasome-interacting proteins that associated substoichiometrically and may serve as regulators or cofactors. This set differed from those in other tissues and we also found several proteins that associated only with either the cytosolic or the synaptic proteasome. The latter included the ubiquitin-binding factor TAX1BP1 and synaptic vesicle protein SNAP-25. Native gel electrophoresis revealed a higher proportion of doubly-capped 26S proteasome (19S-20S-19S) in the cortex than in the liver or kidney. To investigate the interplay between proteasome regulation and synaptic plasticity, we exposed cultured neurons to glutamate receptor agonist NMDA. Within 4 h, this agent caused a prolonged decrease in the activity of the ubiquitin-proteasome system as shown by disassembly of 26S proteasomes, decrease in ubiquitin-protein conjugates, and dissociation of the ubiquitin ligases UBE3A (E6-AP) and HUWE1 from the proteasome. Surprisingly, the regulatory 19S particles were rapidly degraded by proteasomal, not lysosomal degradation, and the dissociated E3 enzymes also degraded. Thus the content of proteasomes and their set of associated proteins can be altered by neuronal activity, in a manner likely to influence synaptic plasticity and learning.
Introduction
In eukaryotic cells, most intracellular proteins are degraded by the ubiquitin-proteasome system (UPS). In this pathway, polyubiquitylation of a protein marks it for degradation by the 26S proteasome (Hershko and Ciechanover, 1998 ). In neurons, the UPS modulates the function and plasticity of synapses in normal and diseased states (DiAntonio and Hicke, 2004 ; Yi and Ehlers, 2007 ; Tai and Schuman, 2008 ). The UPS has been shown to regulate presynaptic vesicle release (Willeumier et al., 2006 ; Yao et al., 2007 ) and the restructuring of the postsynaptic density (PSD) (Ehlers, 2003 ; Bingol and Schuman, 2004 ). In fact, the inhibition of proteasome activity can impair synaptic plasticity (Fonseca et al., 2006 ; Hou et al., 2006 ; Karpova et al., 2006 ), and certain types of learning in animals (Lopez-Salon et al., 2001 ; Lee et al., 2008 ). Another critical function of the UPS is to protect neurons by clearing damaged and misfolded proteins (Goldberg, 2003 ). Intraneuronal aggregates of misfolded proteins are the hallmarks of many neurodegenerative disorders, such as α-synuclein in Parkinson’s disease and hyperphosphorylated tau in Alzheimer’s disease. Frequently, these aggregates contain ubiquitylated proteins and proteasomes, and it has often been suggested that impairment of the UPS underlies neurodegeneration (Sherman and Goldberg, 2001 ; Rubinsztein, 2006 ; Bedford et al., 2008b ).
There is growing evidence that ubiquitylation is not the only site of regulation of this pathway and that proteasome composition, and presumably function, can be regulated in multiple ways (Glickman and Raveh, 2005 ; Schmidt et al., 2005 ). The proteolytic component of the proteasome is the 20S core particle, which may be associated with several different types of activating particles in addition to the 19S regulatory particle, which binds to ubiquitylated proteins (Rechsteiner and Hill, 2005 ). In the 26S proteasome, a single 20S particle may associate with one or two 19S particles to form a singly- or doubly-capped 26S proteasome. Several studies have shown that neuron-specific mechanisms of 26S proteasome regulation may underlie synaptic plasticity (DiAntonio and Hicke, 2004 ; Bingol and Schuman, 2005 ; Yi and Ehlers, 2007 ). A striking example is the rapid redistribution of 26S proteasomes into dendritic spines following neuronal stimulation (Bingol and Schuman, 2006 ). Inside the synapse, proteasome-mediated proteolysis can regulate the internalization of glutamate receptors (Patrick et al., 2003 ) as well as the abundance of many PSD proteins (Ehlers, 2003 ; Bingol and Schuman, 2004 ).
It is also likely that proteasome regulation occurs through changes in the levels of proteasome-interacting proteins. In mammals, USP14 and Rpn13/UCH37, both deubiquitylating enzymes or DUBs, influence substrate degradation through their interaction with the 26S proteasome (Koulich et al., 2008 ). The spontaneous mutation of Usp14 in mice leads to synaptic ubiquitin defects and altered short-term synaptic plasticity (Wilson et al., 2002 ; Chen et al., 2009 ). In addition to 33 different integral subunits, mass-spectrometric analysis of gently-purified proteasomes from non-neuronal tissues and cells has revealed many proteins that interact with 26S proteasomes (Wang and Huang, 2008 ; Besche et al., 2009 ; Scanlon et al., 2009 ). However, their specific functions and mode of regulation are generally unclear.
In particular, there are no published studies about what proteins interact substoichiometrically with 26S proteasomes in the brain and how they are regulated. Recently, Besche et al. (2009) have described an affinity purification method that allows gentle, rapid isolation of 26S proteasomes together with associated proteins. This approach, which is based upon the high affinity of 26S proteasomes for the ubiquitin-like (UBL) domain of Rad23B (Schauber et al., 1998 ), has revealed various new proteasome-associated proteins, among them several ubiquitin ligases and DUBs. In this study, we have utilized this newly developed method for purifying 26S proteasomes from the adult rat cortex. We specifically isolated 26S proteasomes from biochemically purified neuronal fractions and compared the interacting proteins found in synaptic and cytosolic fractions by tandem mass spectrometry. We found that the 26S proteasome-interacting proteins differ between the synapse and the cytosol, and even more so between the cortex and other tissues. Moreover, neuronal activity was found to alter the functional capacity of the UPS. Following the exposure of neurons to N-methyl-D-aspartate (NMDA), we identified several novel mechanisms that down-regulate the UPS, including the disassembly of 26S proteasomes, the degradation of proteasome-associated ubiquitin ligases (E3s), and the degradation of 19S regulatory particles in a proteasome-dependent manner.
Materials and Methods
Reagents
Antibodies against the following antigens were purchased commercially: α7 (PW8110), Rpt1 (PW8825), UBE3A (PW0535), ubiquitin conjugates (UG9510), PI31 (PW9710) from Biomol/Enzo (Plymouth Meeting, PA, USA); ECM29 (PA3-035), USP14 (MA1-57109), PA28α (PA1-960) from Affinity Bioreagents/Thermo (Rockford, IL, USA); parkin (MAB5512), ubiquitin monomer (MAB1510) from Chemicon/Millipore (Billerica, MA, USA); PSMD9 (sc-10670), 14-3-3γ (sc-731) from Santa Cruz Biotechnology (Santa Cruz, CA, USA); HUWE1 (A300-486A) from Bethyl (Montgomery, TX, USA); tubulin β3 (T8860) from Sigma (St Louis, MO, USA); KCMF1 (15-288-21213) from Genway (San Diego, CA, USA); drebrin (ab12350) and GRASP-1 (ab30576) from Abcam (Cambridge, MA, USA). Nonidet P-40 (NP-40) was purchased from BDH (Poole, England). Protease inhibitor cocktail (complete, EDTA-free) was from Roche (Indianapolis, IN, USA). Phosphatase inhibitor (cocktail 2), NMDA, and chloroquine were from Sigma. Epoxomicin and clasto-lactacystin β-lactone were from Enzo. Tetrodotoxin (TTX), (S)-3,5-dihydroxyphenylglycine (DHPG), 2-amino-5-phosphonopentanoic acid (APV), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and concanamycin A were from Tocris (Ellisville, MO, USA).
Recombinant Protein Purification
GST-UBL and His10-UIM plasmids and their expression in bacteria have been previously described (Besche et al., 2009 ). pDEST-15-HHR23BUBL plasmid was transformed into BL21AI (Invitrogen) according to manufacturer’s instructions. Cells were grown at 37°C with ampicillin to an OD600 of 0.5 and induced for 3 h with 1 g/L L-arabinose. Cell pellet from 1 L culture was resuspended in 100 mL ice-cold phosphate buffered saline (PBS, 10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mL KCl, 137 mM NaCl, pH 7.4) with 2 mM DTT, 10 mM MgCl2, 0.2 mg/mL lysozyme and protease inhibitors (complete, Roche). The entire purification procedure was carried out at 4°C. Cells were lysed by sonication, and the cell debris was removed by centrifugation at 20,000 × g for 30 min. The supernatant was passed through 0.2-μm filters and supplemented with 0.2% Triton X-100, followed by incubation with 20 mL of glutathione sepharose for 2 h. The resin was transferred to a gravity-flow column, and washed several times in PBS with 0.2% Triton X-100. GST-UBL was eluted by adding 50 mL of elution buffer (75 mM Tris pH 8.8, 150 mM NaCl, 40 mM reduced glutathione), and the collected fractions were combined and dialyzed against buffer B (25 mM HEPES pH 7.5, 10% glycerol, 5 mM MgCl2 1 mM DTT). Small aliquots of 10 mg/mL GST-UBL were frozen in liquid nitrogen and stored at −80°C. The final yield from 1 L culture was ∼100 mg.
pET26b-S5aUIM2 plasmid was transformed into BL21 (DE3) (Promega) according to manufacturer’s instructions. Cells were grown at 37°C with kanamycin to an OD600 of 0.8, followed by induction with 0.5 mM IPTG for 2 h at 30°C. Pellets from 1-L culture (∼20 mg of His10-UIM) were resuspended in 30 mL B-PER extraction reagent (Pierce/Thermo) supplemented with 3 mM 2-mercaptoethanol and DNase I (Sigma). After 15 min of extraction at 25°C with mixing, the lysate was cooled to 4°C and centrifuged at 20,000 × g for 20 min. The supernatant was passed through 0.2-μm filters, supplemented with 10 mM imidazole, and incubated with 10 mL of Ni-NTA for 1 h. The resin was transferred to a gravity-flow column and washed first with a 1:1 mixture of B-PER and wash buffer (50 mM Tris pH 7.5, 40 mM imidazole, 300 mM NaCl), and then just wash buffer. Twenty milliliters of elution buffer (50 mM Tris pH 7.5, 250 mM imidazole, 300 mM NaCl) was added, and collected fractions containing His10-UIM were combined and dialyzed against buffer B with 40 mM KCl. Protein concentration was compared to bovine serum albumin standards (Pierce/Thermo) using two gel-staining methods: Coomassie blue and E-Zinc stain (Pierce/Thermo). Small aliquots of 2 mg/mL His10-UIM were frozen in liquid nitrogen and stored at −80°C.
Synaptosome Isolation
Adult rat cortices rapidly frozen in liquid nitrogen and stored at −80°C were used for isolating synaptosomes, based on standard protocols (Gordon-Weeks, 1987 ) with buffer modifications to stabilize proteasomes. The procedure is schematically represented in Figure 1 . Briefly, cortices from four rats were homogenized in a motor-driven Potter-Elvehjem homogenizer in 30 mL buffer A (20 mM HEPES, 0.32 M sucrose, 5 mM MgCl2, 2 mM ATP, 2 mM DTT, protease and phosphatase inhibitors, pH 7.2). The homogenate was centrifuged at 1000 × g for 5 min, and the supernatant was collected as S1 (if cloudy, centrifuged one more time). S1 was centrifuged at 12,000 × g for 20 min, and the pellet (P12) was resuspended in buffer A and centrifuged again at 12,000 × g for 20 min. The synaptosome pellet (P12’) was resuspended in 11 mL buffer A with 1% NP-40 using a Potter-Elvehjem homogenizer to disrupt the vesicles and extract proteins. This was followed by centrifugation at 18,000 × g for 10 min, and then at 100,000 × g for 1 h to sediment clathrin complexes and debris. The supernatant (S100) was collected as synaptosomal extract. To collect the cytosolic extract, supernatant S12 was centrifuged at 100,000 × g to remove microsomes. The supernatant was collected as cytosolic extract and supplemented with 1% NP-40. Synaptosomal protein yield from four cortices was ∼50 mg.
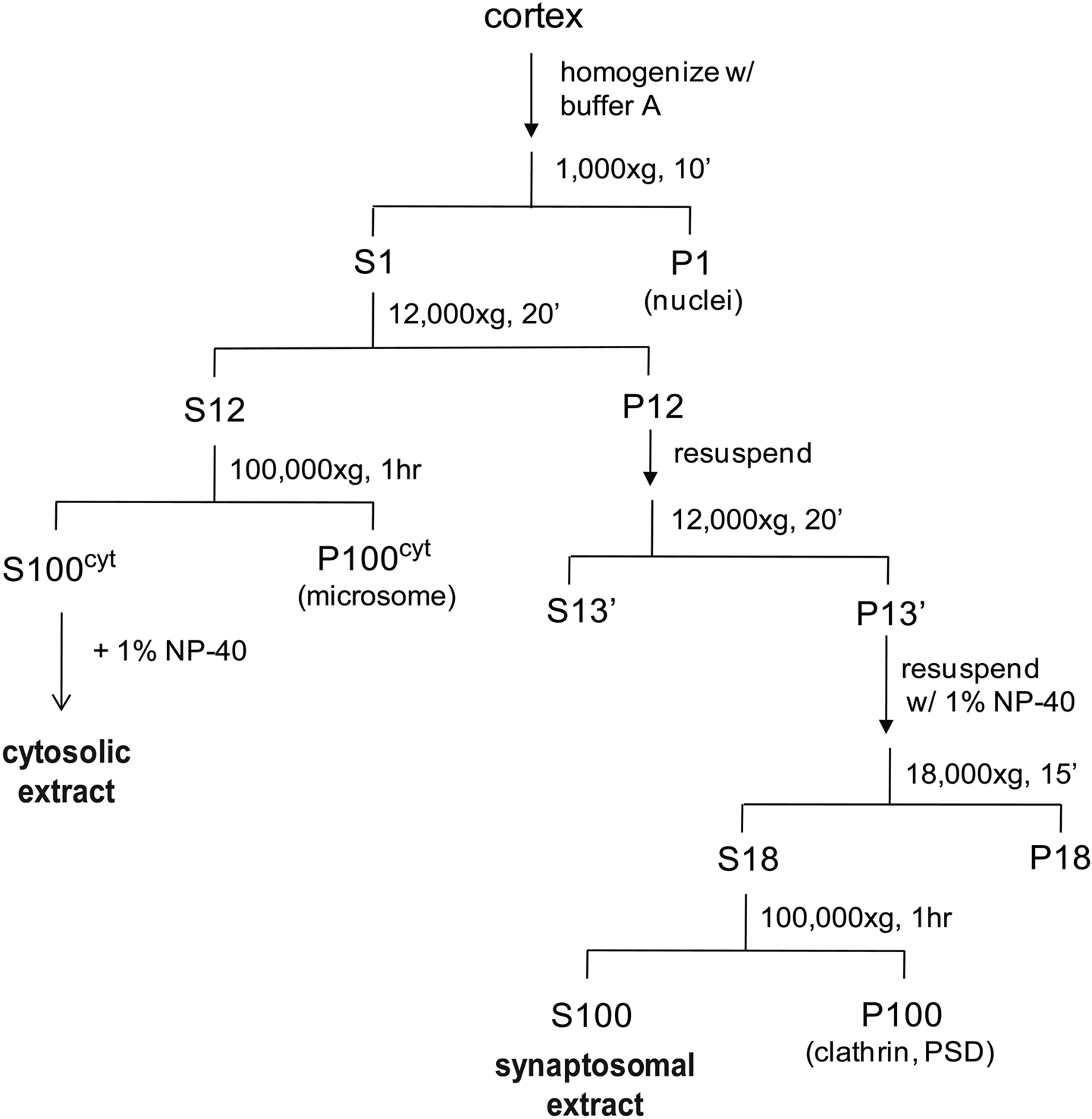
Figure 1. A schematic showing the preparation of synaptosomal and cytosolic extracts from rat cortices. We include an additional centrifugation step (1 h, 100,000 × g) after solubilization of synaptosomes with NP-40 and before incubation with the UBL-domain. Solubilization leads to binding of an unrelated clathrin complex to the UBL matrix, which complicates the mass spectrometric analysis (data not shown). The respective centrifugation step removes the bulk of this clathrin complex while the proteasome remains fully soluble.
Proteasome Purification
The purification of 26S proteasomes was conducted at 4°C. First, cytosolic or synaptosomal extracts (20 mg total protein) were incubated for 2 h with 1 mg GST-UBL and 200 μL glutathione sepharose 4B. After transferring to a gravity-glow column, resins were washed with 20 mL buffer A plus 0.5% NP-40, followed by 30 mL of buffer B plus 1 mM ATP and 5 mM MgCl2. Proteasomes were eluted by a 20-min incubation with 250 μL His-UIM (2 mg/mL, with 2 mM ATP), and repeated one more time. The combined eluate was incubated with 120 μ Ni-NTA for 20 min, and passed through 0.22-μm spin-filters (ultrafree MC, Millipore). The filtrate contained purified 26S proteasomes. For control samples, 0.7 mg GST was substituted for GST-UBL in the purification procedure.
To deplete the cell extract of 26S proteasomes, GST-UBL was added to cell extracts (50 μg/1 mg of total protein) and incubated with glutathione sepharose. To sediment proteasomes, cell extracts were pre-cleared at 100,000 × g for 1 h, followed by centrifugation at 100,000 × g for 6 h. Pellets were redissolved in 50 mM Tris pH 7.5, 6 M urea, and 1% SDS.
Tissues and Neuronal Cultures
Cortex, liver and kidney were dissected from 4- to 6-weeks old male Sprague-Dawley rats. Dissociated hippocampal neurons were prepared and maintained as previously described (Aakalu et al., 2001 ). Briefly, hippocampi from postnatal day 0 to 2 rats were dissected out and dissociated by papain and plated at a density of 70,000 cells/cm2 onto poly-D-lysine-coated 60-mm culture dishes (Falcon). Cultures were maintained in Neurobasal A medium containing B-27 and Glutamax supplements (Invitrogen) at 37°C for 21–28 days before use. Tissue extracts were prepared in buffer A (20 mM HEPES, 0.32 M sucrose, 5 mM MgCl2, 2 mM ATP, 2 mM DTT, protease and phosphatase inhibitors, pH 7.2) with 0.2% NP-40, using motor-driven Potter-Elvehjem homogenizers. Extracts of cultured neurons were prepared in buffer A with 0.5% NP-40, using glass dounce homogenizers. Extracts were cleared by centrifugation at 18,000 × g for 15 min to remove nuclei and cell debris, and protein concentrations were measured by Coomassie Plus protein assay (Pierce).
Native Gel Electrophoresis
To resolve proteasomes in cell extracts, 2–5% gradient native gels were prepared using the mini-protean 3 system and acrylamide/Bis (37.5:1) from Bio-Rad. The gel formulation was modified from published protocols for discontinuous native gels (Dohmen et al., 2005 ) based on Tris-borate-EDTA buffer (TBE, 90 mM Tris base, 80 mM boric acid, 0.1 mM EDTA, pH 8.3). Each 1.5 mm, 10-well gradient gel was made by mixing 3.3 mL of the upper gel solution (70 mM Tris pH 6.8, 2% acrylamide, 0.06% APS, 0.12% TEMED) and 6.3 mL of lower gel solution (1X TBE, 5% acrylamide, 3.5% sucrose, 1 mM DTT, 1 mM ATP, 5 mM MgCl2, 0.05% APS, 0.1% TEMED). Samples were mixed with 4X loading buffer (100 mM Tris pH 8.0, 20% glycerol) just before loading. Electrophoresis was carried out at 6–10°C, in TBE running buffer (with 0.5 mM DTT, 0.5 mM ATP and 2 mM MgCl2). The applied voltage was gradually increased with time (30 V for 30 min, 35 V for 1 h, 50 V for 1 h, 65 V for 2–4 h). To resolve isolated 26S proteasomes, 1.5 mm, 10-well 3–8% Tri-acetate gels (Invitrogen) were used. Gel loading buffer and running buffer were the same as above. Electrophoresis was carried out at 4°C and 150 V for 4 h. For immunoblotting, proteins in native gels were transferred to PVDF membranes using Bio-Rad mini-protean 3 transfer system (buffer contains 25 mM Tris base, 192 mM glycine, 0.1% SDS) for 4 h at 70 V (current limit: 350 mA) in a 4°C room.
Immunoblotting and Silver Staining
PVDF membranes with transferred proteins were blocked with 5% non-fat milk in 50 mM Tris pH 7.5, 150 mM NaCl, and 0.05% Tween-20. Primary antibodies and HRP-conjugated secondary antibodies were applied in blocking reagent, followed by standard chemiluminescence detection. Band intensities on scanned films were quantified by densitometry using the gel analysis function in ImageJ software. Silver staining was performed with SilverSNAP kit II (Pierce/Thermo).
Proteasome Activity Assay
26S proteasome activity in cell extracts was measured by a proteasome activity kit (Biomol/Enzo) and a fluorometer, based on the hydrolysis of Suc-LLVY-AMC, a fluorogenic substrate for chymotrypsin-like peptidase activity. Native gels containing 26S proteasomes were incubated with reaction buffer (50 mM Tris pH 7.5, 10 mM MgCl2, 1 mM ATP, 1 mM DTT, 50 μM Suc-LLVY-AMC) for 30 min at 37°C. Fluorescent bands around 26S proteasomes were visualized and quantified by standard gel-imaging systems for DNA staining by ethidium bromide.
Mass Spectrometry
Purified cytosolic and synaptic proteasome samples were digested with trypsin and the generated peptides were subjected to nanoscale-microcapillary reversed phase liquid chromatography tandem mass spectrometry (LC-MS/MS) using an in-house packed C18 125 μm I.D. capillary column and a hybrid linear ion trap/FT-ICR mass spectrometer (LTQ FT, Thermo Electron, Bremen, Germany) as described previously (Haas et al., 2006 ). Using the SEQUEST algorithm, MS/MS data were searched against a concatenated target-decoy database created based on the rat IPI protein database including sequences of known contaminant proteins. We determined an overall protein false-discovery rate of 1% for the ∼100 proteins that made it above the unbiased filter thresholds (Elias and Gygi, 2007 ). The maximum number of incorrectly assigned proteins in the final list of <50 protein is therefore 1. We believe that this false-discovery rate is hardly matched by other medium-to-large scale analysis methods currently available. Data in Tables 1 and 2 are compiled from two independent experiments using four rats each.
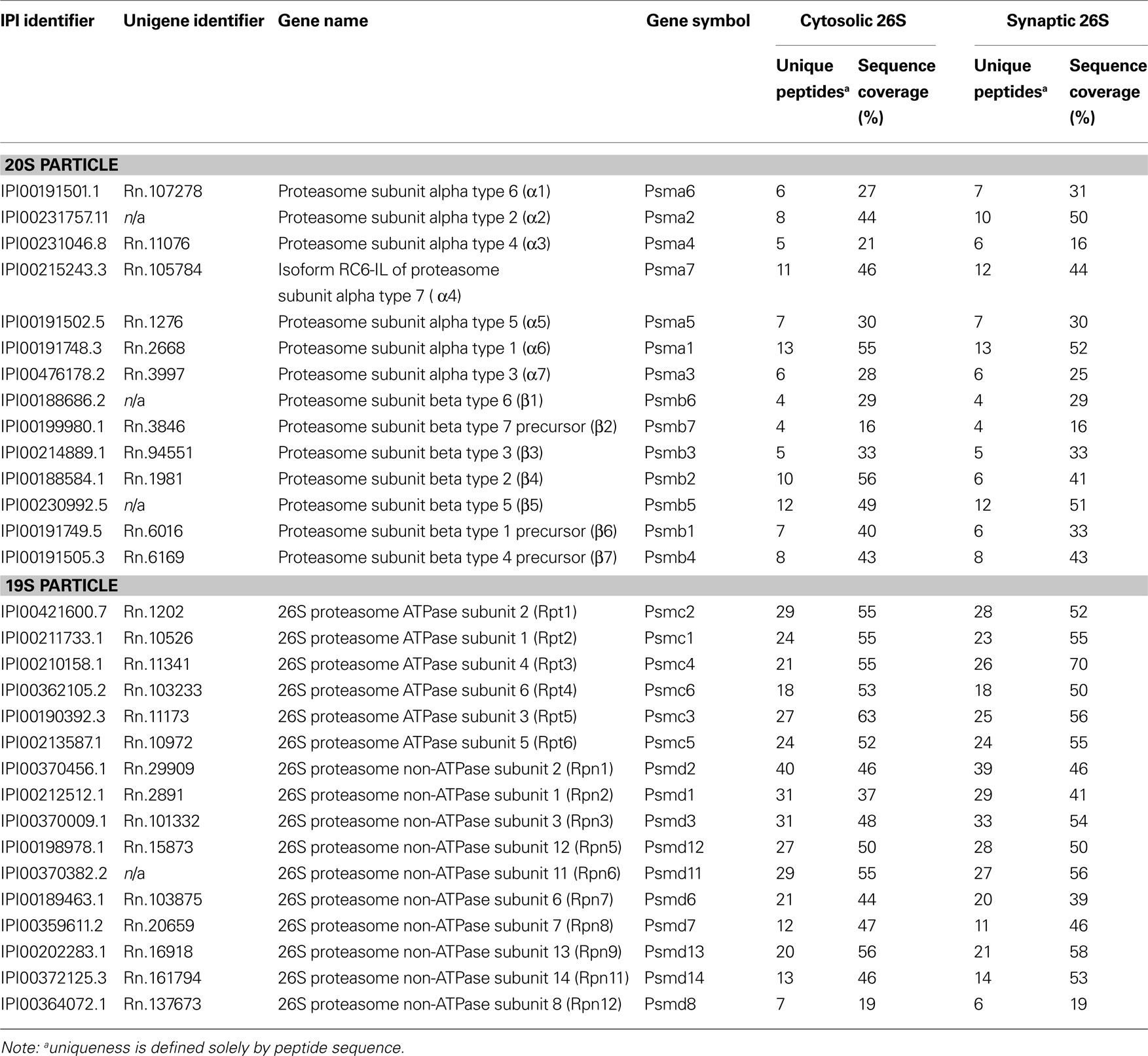
Table 1. 26S proteasome subunits identified by mass spectrometry.
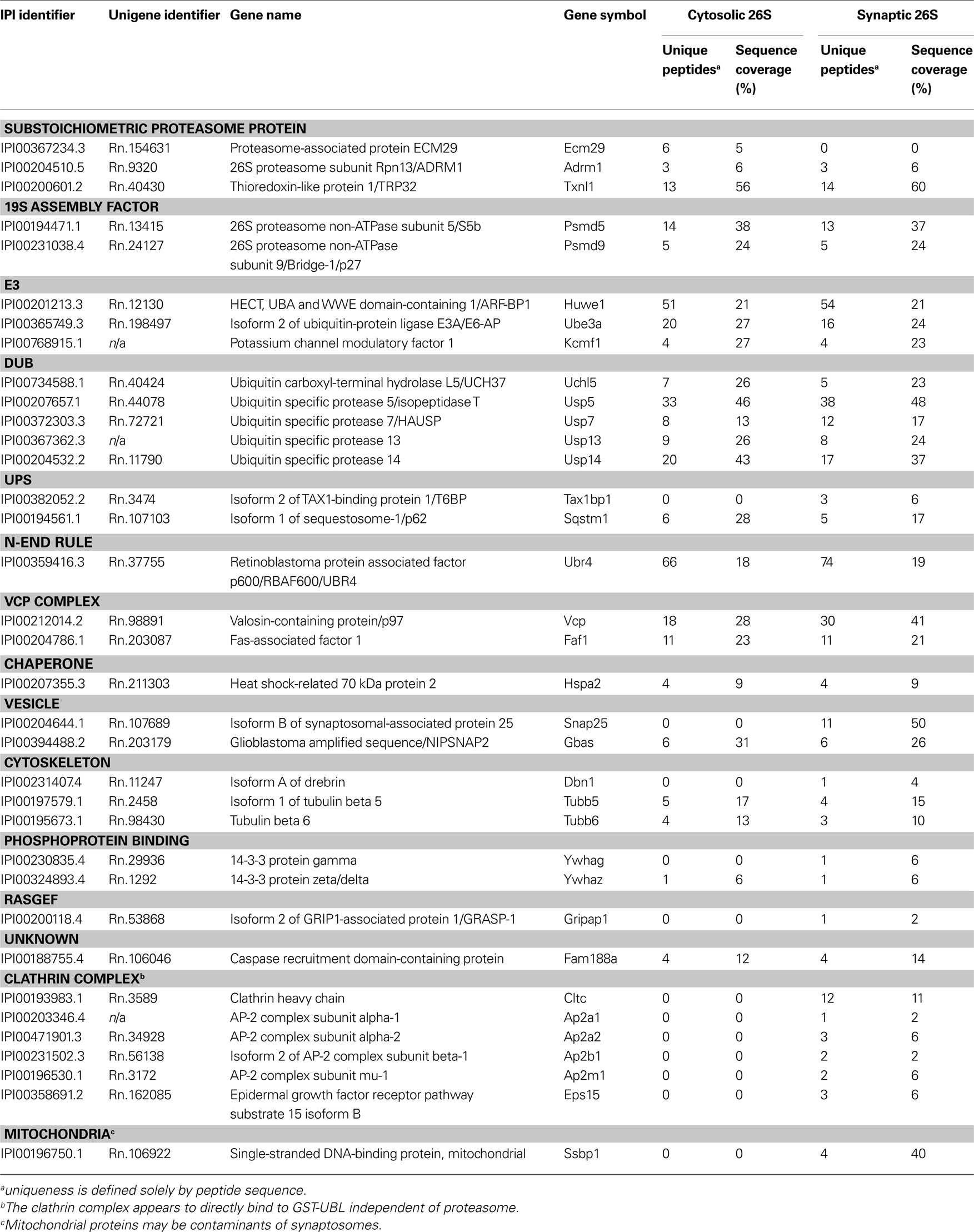
Table 2. Mass spectrometric identification of proteins that co-purify with cytosolic and synaptic 26S proteasomes.
Results
The Distribution of Proteasome Species in the Cortex
Since very little is known about the proteasome content of the brain, we first investigated what types of proteasomes may exist in the rat cortex. Using native gel electrophoresis followed by immunoblotting against 19S and 20S subunits, we found doubly-capped 26S, singly-capped 26S, and 20S proteasome in rat cortical extracts (Figure 2 A). Their relative abundance in cortex, kidney, liver and Hela cells is quantified and summarized in Figure 2 B. Interestingly, the level of doubly-capped 26S proteasome was higher in the cortex than in other cell types (p < 0.01 by Tukey’s test). Additionally, we detected free 19S particles in these cell types, as well as a smaller complex containing the 19S subunit Rpt1 (Figure 2 A). We characterized this complex from liver extracts by tandem mass spectrometry (LC-MS/MS) and found S5b, Rpn1, Rpt1 and Rpt2. This complex has been recently shown to be an intermediate in 19S assembly (Funakoshi et al., 2009 ; Kaneko et al., 2009 ; Le Tallec et al., 2009 ; Park et al., 2009 ; Roelofs et al., 2009 ; Saeki et al., 2009 ).
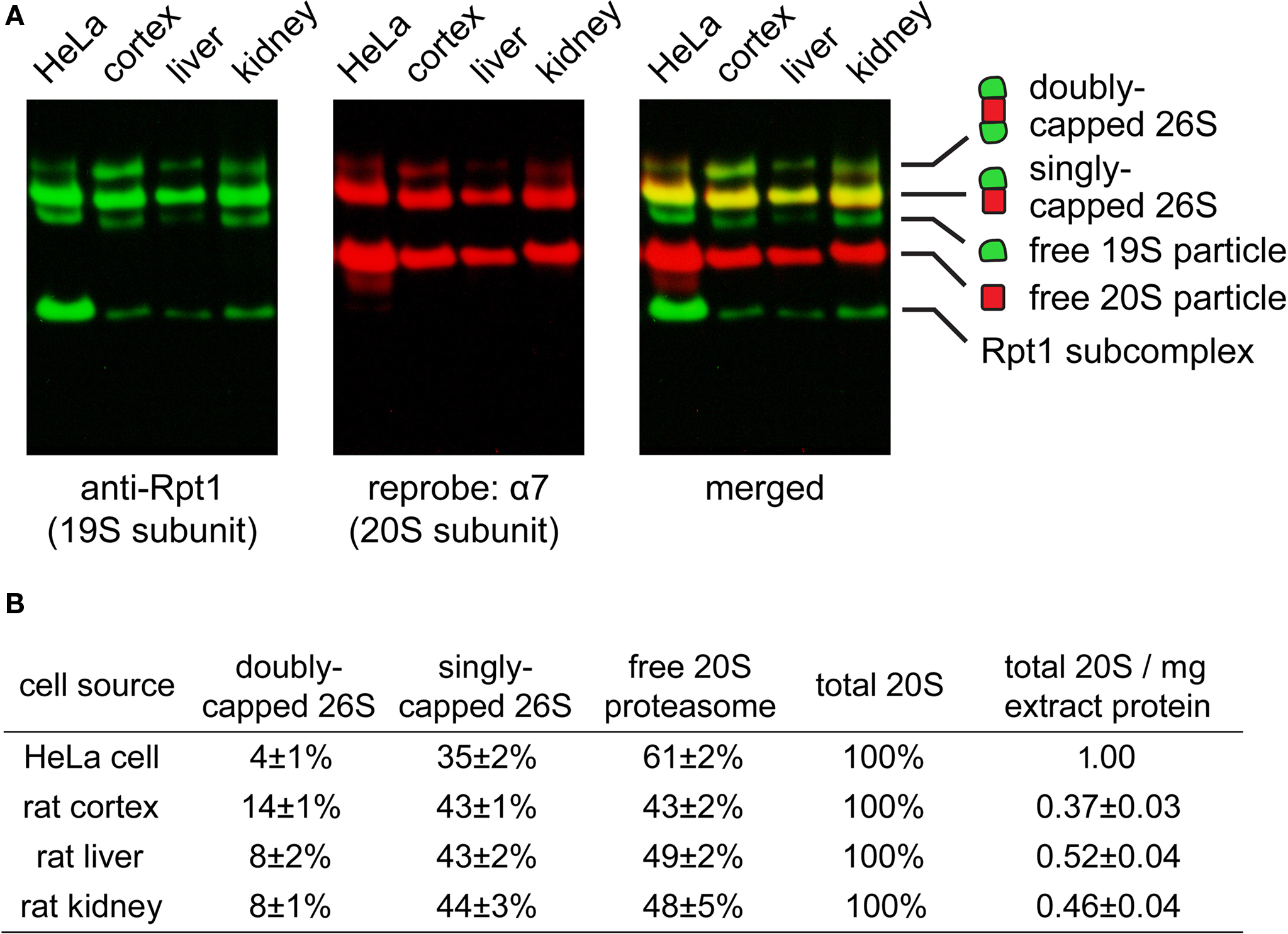
Figure 2. (A) Proteasome species found in different cell types. Extracts of HeLa cell (30 μg), rat cortex (60 μg), rat liver (40 μg) and rat kidney (50 μg) were resolved by 2–5% gradient native gel, immunoblotted against 19S subunit Rpt1, and reprobed against 20S subunit α7. Protein bands were pseudo-colored and merged to illustrate the presence of proteasomes and related species (from top to bottom): doubly-capped 26S proteasome, singly-capped 26S proteasome, free 19S particle and free 20S particle. A fast-migrating band corresponding to a subcomplex of the 19S containing Rpt1 protein was also observed. (B) The distribution of different proteasome species in (A), quantified by densitometry (mean ± SEM, n = 6). The experiment was performed independently six times with tissues from three animals. By one-way ANOVA and Turkey’s multiple comparison test, doubly-capped 26S levels in cortex are found to be significantly higher (p < 0.01) than those in liver, kidney or HeLa cells.
Proteasome Integrity after Subcellular Fractionation
Next, we investigated if proteasomes in different subcellular compartments have different properties by comparing cytosolic and synaptic 26S proteasomes. Slight modifications of standard protocols for isolating cytosolic and synaptosomal fractions (Figure 1 ) were sufficient to preserve proteasome integrity, as assessed by native gel electrophoresis (Figure 3 A) and enzymatic assays (Figure 3 C). When cytosolic and synaptosomal extracts were incubated with GST-UBL and glutathione sepharose, the flow-through fraction was depleted of 19S-subunit Rpt1 protein (Figure 3 B). Thus GST-UBL affinity capture provides an efficient method to purify the entire population of 26S proteasomes without apparent bias towards a specific subset. By contrast, there was a high amount of α7 subunit remaining in the flow-through (Figure 3 B), which was consistent with the abundance of free 20S particles (which lack affinity for ubiquitin or UBL-proteins), as detected in the native gel (Figure 3 A). Also, when GST-alone was used, instead of GST-UBL, no proteasome or other complexes were detectable (Figure 4 A).
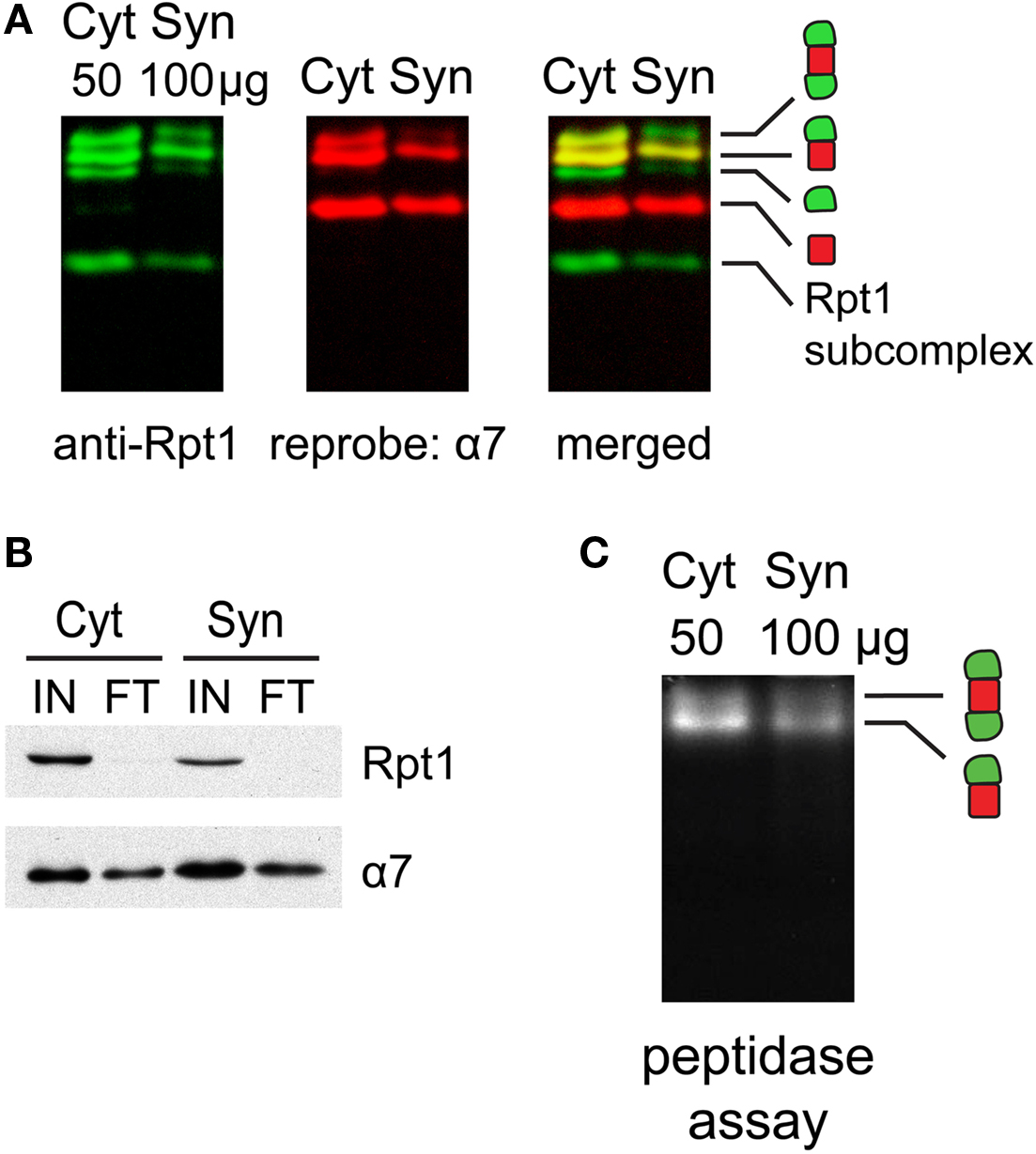
Figure 3. (A) Proteasome integrity after subcellular fractionation. Cytosolic (Cyt) and synaptic (Syn) extracts prepared from rat cortices were resolved by 2–5% gradient native gel, immunoblotted against 19S subunit Rpt1, and reprobed for 20S subunit α7. Singly-, doubly-capped 26S, 20S proteasomes and free 19S were detected in both fractions. (B) Efficacy of 26S proteasome isolation by UBL affinity chromatography. Cytosolic (Cyt) and synaptic (Syn) extracts were incubated with GST-UBL and glutathione sepharose. The input (IN) and flow-through (FT) materials were analyzed by SDS-PAGE and immunoblotted against Rpt1 and α7. The depletion of Rpt1 from the flow-through indicated that most 26S proteasomes and 19S particles remained intact during subcellular fractionation and were captured with high efficiency. The high remaining level of α7 subunit in the flow-through is consistent with the abundance of free 20S particles detected in (A). (C) Cytosolic and synaptosomal extracts contain active 26S proteasomes. Cytosolic (Cyt) and synaptic (Syn) extracts isolated from rat cortices were resolved by 2–5% gradient native gel, followed by incubation with Suc-LLVY-AMC, a fluorogenic proteasome substrate. Active 26S proteasomes appear as fluorescent bands, corresponding to doubly- and singly-capped 26S proteasomes.
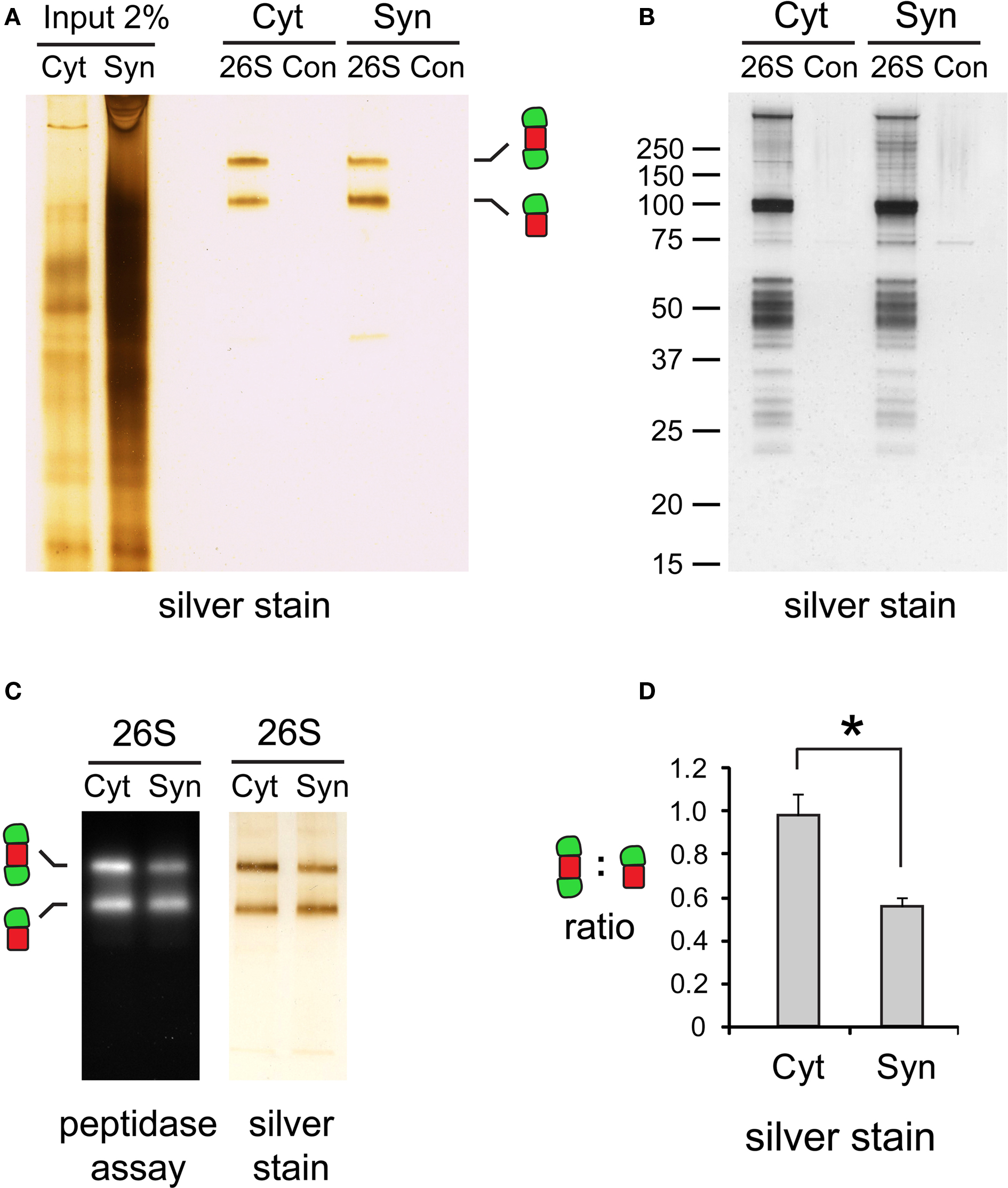
Figure 4. (A) Purity of isolated 26S proteasomes. Cytosolic (Cyt) and synaptic (Syn) 26S proteasomes were isolated using GST-UBL/His-UIM. In control (Con) experiments, GST was added instead of GST-UBL. Samples were resolved by 3–8% gradient native gel and silver-stained. (B) The same samples in (A) analyzed by SDS-PAGE, followed by silver staining. The control experiments showed only a few non-specific protein bands. (C) Activity of isolated proteasomes. Chymotrypsin-like peptidase activity of isolated 26S proteasomes resolved by 3–8% native gel was assayed with Suc-LLVY-AMC. (D) Differences in capped proteasome ratios. Silver-stained 26S proteasome bands in (C) were quantified by densitometry. The ratio of doubly- over singly-capped 26S was significantly higher in the cytosol (Cyt) than the synaptosome (Syn) (mean ± SEM, n = 4, *p < 0.05 by paired t-test).
Cytosolic and synaptic 26S proteasomes bound to the GST-UBL matrix were recovered by elution with His-UIM. When resolved by native gel electrophoresis and silver stained, each 26S sample showed only two bands, corresponding to doubly- and singly-capped 26S proteasomes (Figure 4 A), both of which remain enzymatically active (Figure 4 C). Interestingly, the ratio of doubly- to singly-capped 26S was significantly higher in the cytosol than in the synapse (Figure 4 D). By sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and silver staining, the band patterns of the cytosolic and synaptic 26S proteasomes showed some differences, and very few proteins were found in GST controls (Figure 4 B). Taken together, our data demonstrate that the distribution of proteasome species varies between different cells and tissues, as well as between different subcellular compartments of the brain.
The GST-UBL capture method offers several advantages over proteasome-purification strategies based on tandem affinity purification (TAP) tags (Krogan et al., 2004 ; Guerrero et al., 2006 ; Wang et al., 2007 ). TAP tag requires genetic manipulation and the tag may affect proteasome properties, while GST-UBL can efficiently capture endogenous proteasomes from almost any biological source. Instead of tandem affinity chromatography, a one-step elution using His-UIM yields functionally intact, highly-purified proteasomes which can be used for further enzymatic characterizations.
Identifying Proteasome-Interacting Proteins by Mass Spectrometry
Because the 26S proteasomes isolated from the cytosol and the synaptosome were highly purified, it was possible to determine their compositions and the nature of the associated proteins by trypsin digestion and LC-MS/MS. These proteasome purifications were carried out in the absence of salt to maximize the content of proteasome-associated proteins (Besche et al., 2009 ). Proteins bound to the GST-UBL sample that matched proteins bound by GST-alone were non-specific contaminants that interacted with the matrix and were not analyzed further. The remaining identified proteins represented either stoichiometric proteasome subunits (Table 1 ) or 26S proteasome-interacting proteins (Table 2 ). There were no discernible differences in proteasome subunits between the cytosol and synaptosome, and no immunoproteasome subunits were detected. Among the ∼30 interacting proteins identified, one protein ECM29 was restricted to cytosolic proteasomes, and five proteins (TAX1BP1, SNAP-25, drebrin, GRASP-1, 14-3-3γ) were found only in synaptic proteasomes. Those interactors shared between the two proteasome populations include several ubiquitin ligases (E3s), DUBs, molecular chaperones, and components of the UPS.
During purification, the majority of ubiquitylated substrates associated with 26S proteasomes were removed because they became bound to His-UIM proteins on Ni-NTA resins as previously reported (Besche et al., 2009 ). Consequently, neither ubiquitin nor ubiquitin conjugates were detected in purified 26S samples. Proteins listed in Table 2 , therefore, most likely represent true proteasome-interacting proteins, not transiently-associated proteasome substrates. Some of these proteins have previously also been reported to interact with proteasomes from non-neuronal cells (Table 3 ); some appear to be brain-specific, such as the ubiquitin-binding factor TAX1BP1, the E3 enzyme KCMF1, and the synaptic vesicle protein SNAP-25.
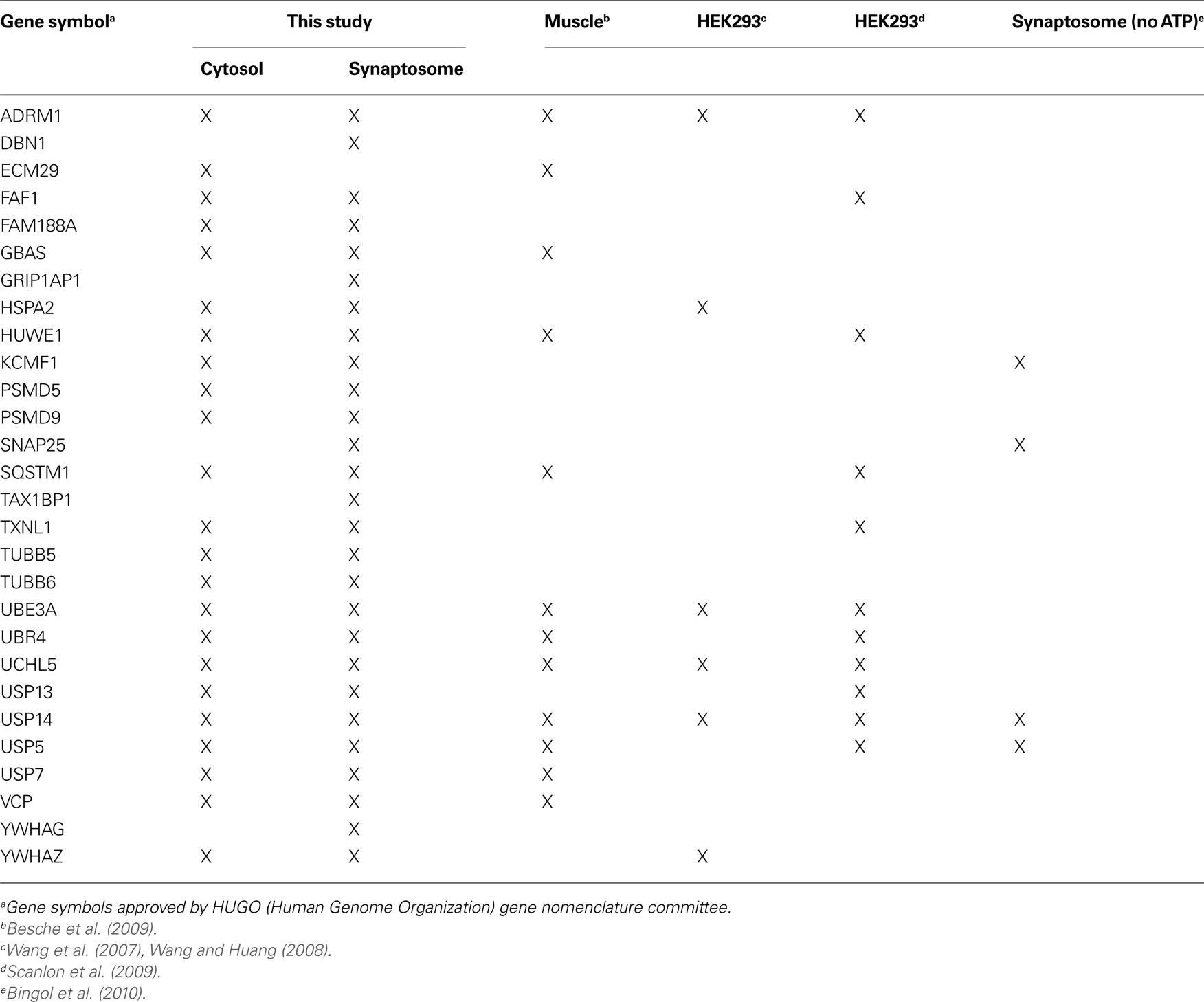
Table 3. Comparison of proteasome composition from different mammalian cells determined by proteomic approaches.
Validation of Proteasome-Interactions
A few proteins (such as drebrin and GRASP-1) listed in Table 2 were identified only by a single matching peptide, and we wanted to validate that they were bona fide interactors using an independent approach. We were able to confirm their presence in purified proteasome preparations by Western blotting using specific antibodies (Figure 5 A). In addition, we utilized a co-sedimentation assay to independently assess (in the absence of GST-UBL) if proteins listed in Table 2 are indeed associated with proteasomes. When cytosolic and synaptic extracts were subjected to centrifugation at 100,000 × g for 6 h, 20S and 26S proteasomes were largely found in the pellet fraction (Figure 5 C). Under this condition, true proteasome-interacting proteins should also sediment with the proteasome in the pellet, and a total of seven putative interacting proteins from Table 2 were tested. These included three ubiquitin ligases, HUWE1 (HECT, UBA and WWE domain containing 1), UBE3A (E6-associated protein, E6-AP), and KCMF1 (potassium channel modulatory factor 1); USP14 (ubiquitin-specific protease 14, a DUB); PSMD9, a recently identified 19S assembly factor (Funakoshi et al., 2009 ; Kaneko et al., 2009 ; Saeki et al., 2009 ); ECM29, the only proteasome protein detected exclusively in the cytosol; and 14-3-3γ, which binds phosphoproteins and was found only in synaptic proteasomes. All seven proteins co-sedimented with proteasomes as expected, and their subcellular distribution agrees very well with the mass spectrometry results (Figure 5 C). This validates our purification and identification strategy.
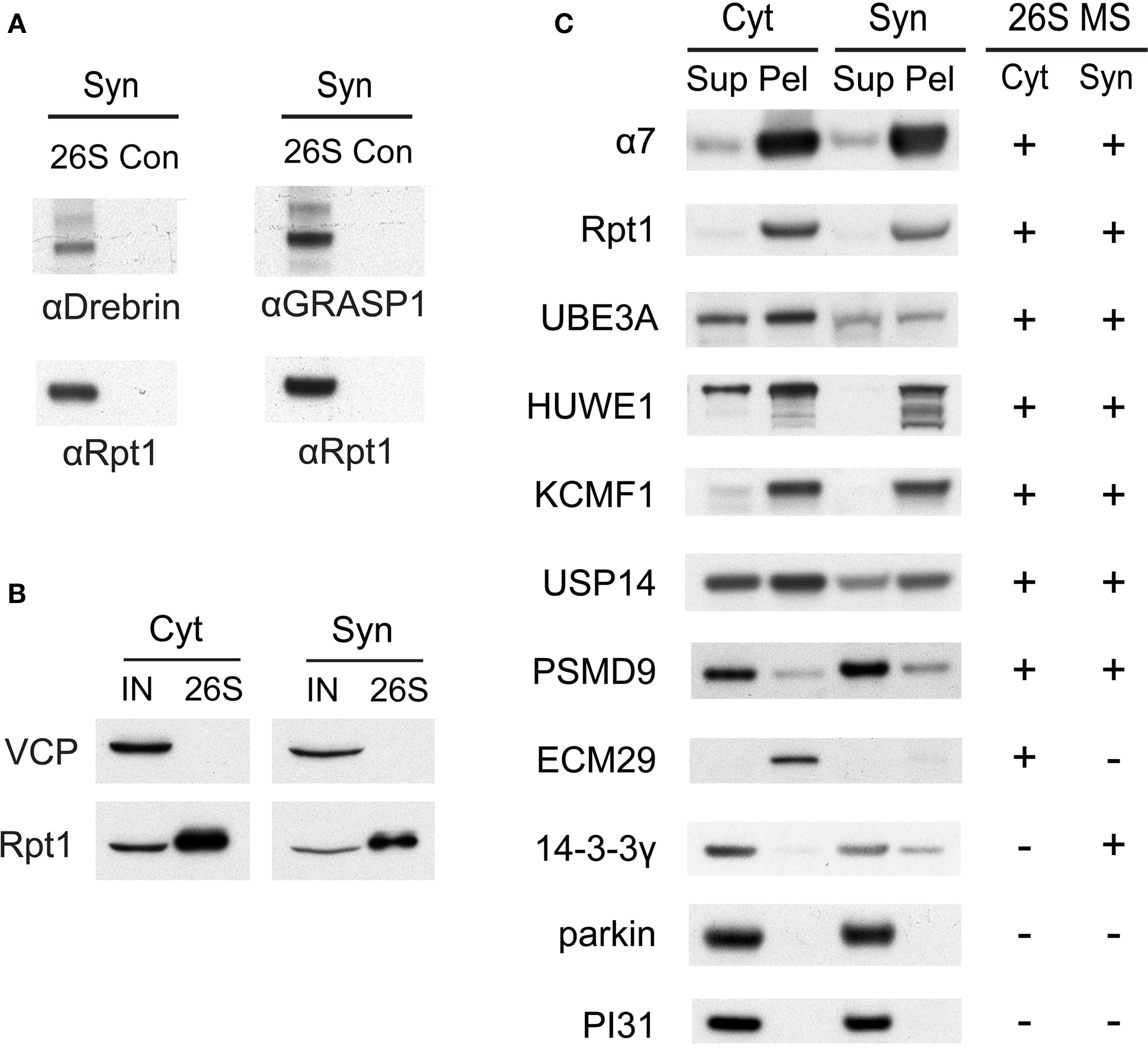
Figure 5. (A) Verification of drebrin and GRASP-1 as interactors of synaptic proteasomes. Both proteins were initially detected in synaptic proteasomes by mass spectrometry, based on a single matching peptide (Table 2 ). These specific interactions are confirmed by SDS-PAGE and Western blotting. Synaptosome (Syn)-derived proteasomes (26S) and control (Con) purifications using only GST are probed with specific antibodies against drebrin, GRASP-1, and 19S subunit Rpt1. (B) The VCP complex does not interact appreciably with GST-UBL. 26S proteasomes from the cytosolic (Cyt) and synaptic (Syn) extracts were purified by the GST-UBL/His-UIM method. The input (IN) material and purified 26S proteasomes were immunoblotted against VCP and Rpt1. The amount of VCP captured by the GST-UBL matrix was undetectable by Western blot in three independent experiments. (C) Verification of proteasome-interacting proteins by co-sedimentation. Cytosolic (Cyt) and synaptosomal (Syn) extracts were subjected to centrifugation at 100,000 × g for 6 h. The supernatant (Sup) and pellet (Pel) materials were loaded at 1:1 ratio and resolved by SDS-PAGE. The sedimentation of proteasomes was confirmed by the high levels of 20S subunit α7 and 19S subunit Rpt1 in the pellet fraction. Proteins identified by mass spectrometry in 26S proteasomes (26S MS) from the cytosol and the synaptosome are indicated by the positive sign. Agreement between mass spectrometry data and co-sedimentation data was observed for all seven 26S-interacting proteins (UBE3A, HUWE1, KCMF1, USP14, PSMD9, ECM29, 14-3-3γ) probed by specific antibodies (see Materials and Methods). Parkin and PI31 did not show appreciable association with brain proteasomes. Results shown here are representative of three experiments.
In our preliminary experiments, it was observed that the clathrin complex can bind to GST-UBL independently of the proteasome in purifications from synaptosomes and microsomes (data not shown). In this study, ultracentrifugation for 1 h at 100,000 × g before the UBL-purification efficiently removed most of the clathrin complex from solubilized synaptosomal proteasomes; however, traces were still detectable by LC-MS/MS. Thus, we do not consider clathrin and related vesicular proteins as 26S proteasome interactors (Table 2 ), although our data cannot formally rule out the possibility of such interactions. The VCP/p97 complex, which is also a key component of the UPS, has been previously observed to bind proteasomes in both muscle and yeast (Verma et al., 2000 ; Besche et al., 2009 ). However, it also directly binds to the UBL-domain in complex with associated cofactors in muscle extracts (Besche et al. 2009 ). We failed to detect by Western blotting any appreciable interaction between VCP and the preparation of GST-UBL used in this study (Figure 5 B). The relatively low levels of VCP and one of its associated factors detected by mass spectrometry in purified 26S proteasomes are thus likely a result of VCP-proteasome interaction in brain, although we cannot formally rule out the potential interaction between VCP and the GST-UBL matrix.
It is also noteworthy that we could not detect by mass spectrometry parkin (an E3 mutated in autosomal-recessive juvenile Parkinsonism) and PI31 (which may function as an inhibitor of the 20S proteasome), which have been reported to interact with proteasomes in non-neuronal cells (McCutchen-Maloney et al., 2000 ; Sakata et al., 2003 ). Also, neither of these proteins co-sedimented with proteasomes (Figure 5 C). These findings further illustrate the specificity of our purification strategy and the differences between proteasome interactors in different cell types.
NMDA Treatment Suppresses Neuronal UPS Activity
Several forms of synaptic plasticity have been shown to require activity of the UPS (Fonseca et al., 2006 ; Hou et al., 2006 ; Karpova et al., 2006 ), and hence there is a growing interest in understanding how neuronal activity regulates different aspects of the UPS (Ehlers, 2003 ; Patrick et al., 2003 ; DiAntonio and Hicke, 2004 ; Bingol and Schuman, 2006 ; Bingol et al., 2010 ). However, the regulation of proteasomes by synaptic activity is hindered by a lack of basic knowledge about neuronal proteasomes. The biochemical characterization of brain proteasomes described above now provides a basis for investigating the interplay between proteasomes and synaptic plasticity. To induce long-term synaptic plasticity, we exposed cultured hippocampal neurons to 20 μM NMDA for 3 min. NMDA has been shown to induce rapid internalization of glutamate receptors (Li et al., 2004 ; Delgado et al., 2007 ), resulting in depressed neurotransmission that lasts for hours, which is also called “chemical long-term depression” (chemical LTD) (Lee et al., 1998 ). This is a well-established protocol to investigate synaptic plasticity in cultured hippocampal neurons and hippocampal slices (Lee et al., 1998 ; Li et al., 2004 ; Lu and Ziff, 2005 ; Delgado et al., 2007 ; Davidson et al., 2009 ).
Following the exposure of neurons to NMDA, we observed a long-lasting reduction in 26S proteasome activity assayed with fluorogenic peptide substrates (Figures 6 A,B). The decrease was statistically significant compared to untreated neurons at 4 h post-NMDA treatment. A decrease in 26S proteasome activity should lead to the accumulation of ubiquitin-protein conjugates, as occurs upon treatment with proteasome inhibitors, provided there were no global changes in the rates of ubiquitylation as well as deubiquitylation. Therefore, we measured the levels of ubiquitin-protein conjugates in neurons following NMDA treatment. Surprisingly, the content of ubiquitin-protein conjugates was significantly reduced 4 h after NMDA exposure, while levels of free (monomeric) ubiquitin remained unaffected (Figures 6 C,D).
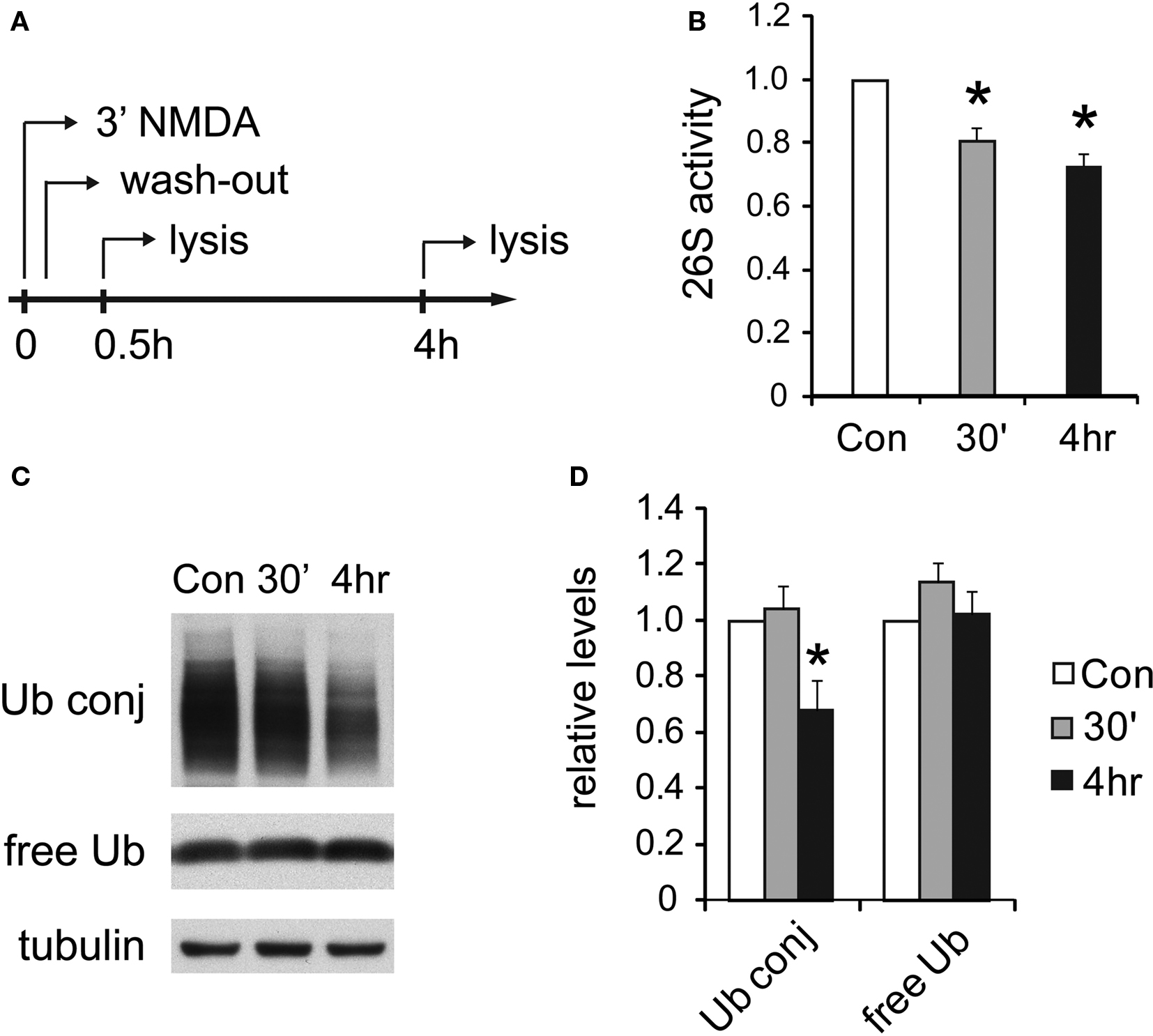
Figure 6. UPS activity is reduced after NMDA treatment. (A) Cultured hippocampal neurons were treated with 20 μM NMDA for 3 min, a standard procedure for inducing chemical LTD. Whole-cell lysates were prepared at 30 min or 4 h post-treatment. (B) Decreases in proteasome activity after NMDA exposure. 26S proteasome activities in neuronal lysates were assayed with Suc-LLVY-AMC (see Materials and Methods). NMDA-treated samples (30’ and 4 h) are normalized to the untreated control (Con) (mean ± SEM, n = 5, *p < 0.05 by paired t-test). (C) Ubiquitin conjugates decrease after NMDA exposure. Neuronal lysates resolved by SDS-PAGE were blotted using antibodies against ubiquitin-protein conjugates (Ub conj) and free monomeric ubiquitin (free Ub). Tubulin served as the loading control. (D) Signals in (C) quantified by densitometry (n = 5, *p < 0.05 by paired t-test).
The similar decrease in 26S proteasome activity and ubiquitin-conjugate levels 4 h after NMDA treatment strongly implies that protein ubiquitylation also decreased and suggests a coordinated reduction in the rate of proteolysis mediated by the ubiquitin-proteasome pathway during NMDA-induced plasticity. These results are consistent with earlier reports showing that activity-blockade with TTX leads to decreased proteasome activity (Djakovic et al., 2009 ) and lower ubiquitin-conjugate levels (Ehlers, 2003 ).
NMDA Treatment Causes 26S Disassembly and E3 Dissociation
We next investigated the mechanisms responsible for the long-lasting reduction in UPS activity. In yeast, during stationary phase when proteolysis by the UPS appears to be down-regulated, 26S proteasomes gradually disassemble into 19S and 20S particles (Bajorek et al., 2003 ). Hence, we assessed if similar changes occur in neurons by native gel analysis. As expected, there was a shift from 26S proteasomes to 20S proteasomes 4 h after NMDA exposure, indicating the disassembly of 26S proteasomes (Figures 7 A,C).
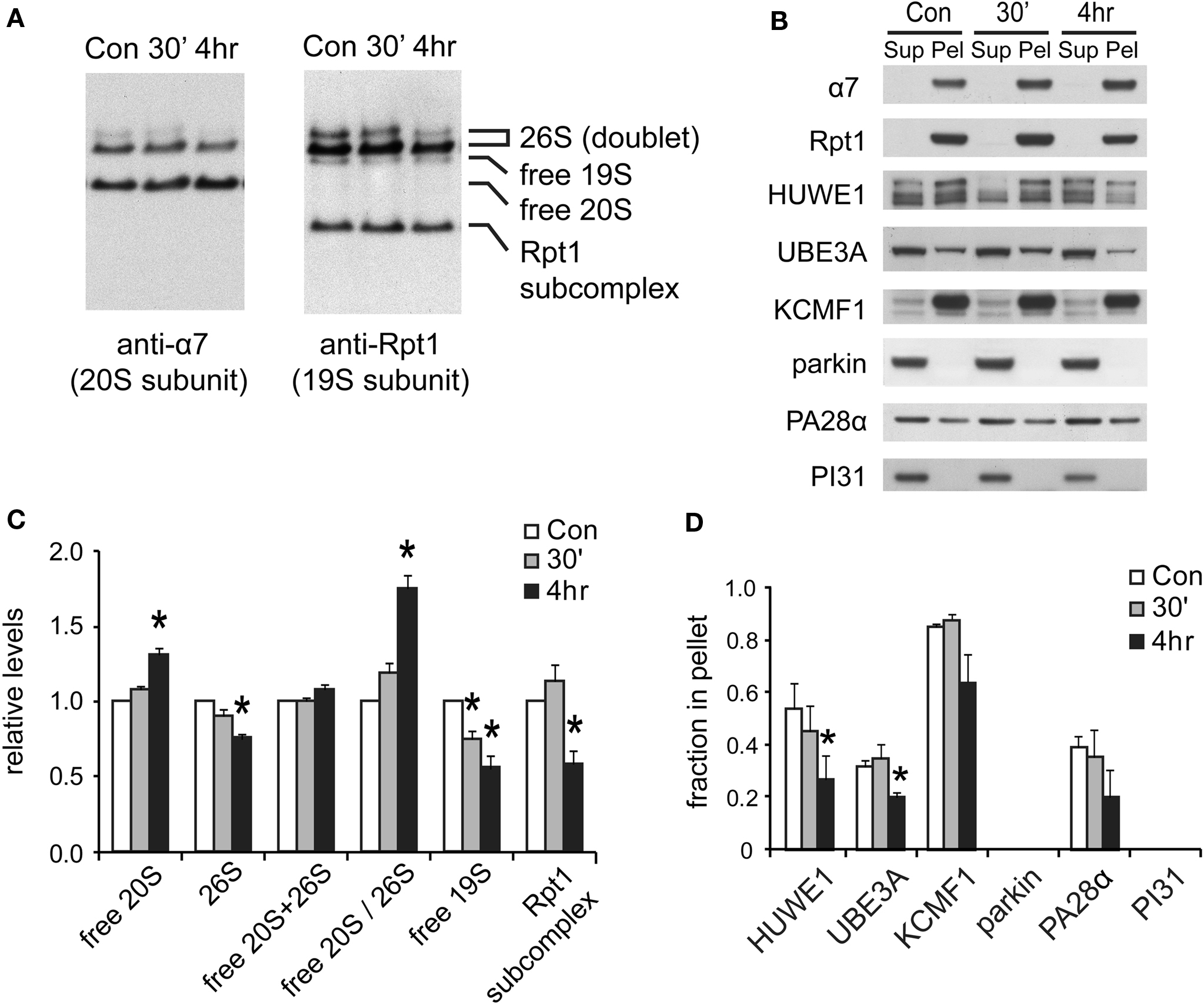
Figure 7. Changes in proteasome complexes after NMDA exposure. (A) The disassembly of 26S proteasomes. Neuronal lysates collected at 30’ and 4 h post-NMDA (20 μM, 3 min) were resolved by 2–5% gradient native gel and immunoblotted against 20S subunit α7 and 19S subunit Rpt1. (B) Changes in proteasome-associated proteins. Lysates from (A) were subjected to ultracentrifugation to sediment proteasomes. Equal amounts of supernatant (Sup) and pellet (Pel) materials were analyzed by SDS-PAGE. The sedimentation property of proteasome-interacting proteins (HUWE1, UBE3A, KCMF1, PA28α) was examined by Western blotting. Parkin and PI31 were not detected in the pellet. (C) Quantification of changes in proteasome distributions in (A) by densitometry (n = 6, mean ± SEM, *p < 0.05 by paired t-test). (D) The graph represents the sedimentation data in (B). The protein level in the pellet is divided by the total level (supernatant + pellet), and the results are plotted (n = 4, *p < 0.05 by paired t-test).
The above findings suggest that the reduction in 26S levels after NMDA exposure in neurons is coordinated with reduced protein ubiquitylation (Figures 6 C,D). One possible mechanism is that some of the cell’s many ubiquitin ligases may be down-regulated following NMDA treatment. We therefore focused on the three proteasome-bound E3s identified earlier in this study. The degree of association between the E3s and the proteasome was determined by their co-sedimentation properties (at 100,000 × g for 6 h). Two of these E3s, UBE3A and HUWE1, were found to be largely dissociated from proteasomes at 4 h after NMDA exposure, while the ligase KCMF1 was not significantly affected (Figure 7 D). The loss of specific ligases from the proteasome may potentially decrease proteasome activity and alter substrate specificity (Kraut et al., 2007 ). It is noteworthy that UBE3A (E6-AP) was the first E3 to be implicated in synaptic plasticity (Jiang et al., 1998 ), and it can target synaptic protein Arc for degradation to control glutamate receptor endocytosis (Greer et al., 2010 ). Our data implies that UBE3A may provide a mechanistic link between NMDA receptor signaling and proteasome activity.
Degradation of 19S Particles and Proteasome-Bound E3S
What is the fate of the 19S particles dissociated from 26S proteasomes? In yeast, during stationary phase, free 19S particles appear to remain intact and available for reuse when growth is reinitiated (Bajorek et al., 2003 ). However, in neurons during NMDA-induced plasticity, we detected a simultaneous decrease in free 19S particles (Figure 7 C), suggesting the further disassembly or degradation of the 19S. Therefore, we investigated if the subunits of the disassembled 19S existed as free proteins in the cytosol or were rapidly degraded. Total 19S subunit levels in the lysate were found to decrease significantly (Figures 8 A,B), and no free subunits were detectable (Figure 7 B). Altogether, these data suggest that 19S particles released from 26S proteasomes were degraded within 4 h of NMDA treatment.
In addition, we investigated the fate of the E3s that dissociated from the proteasome. Like the 19S particle, UBE3A and HUWE1 were degraded by 4 h after NMDA exposure, as indicated by lower protein levels than untreated controls (Figures 8 C,D). On the other hand, the E3 KCMF1 did not show either significant degradation or dissociation from the proteasome (Figures 7 D and 8 D).
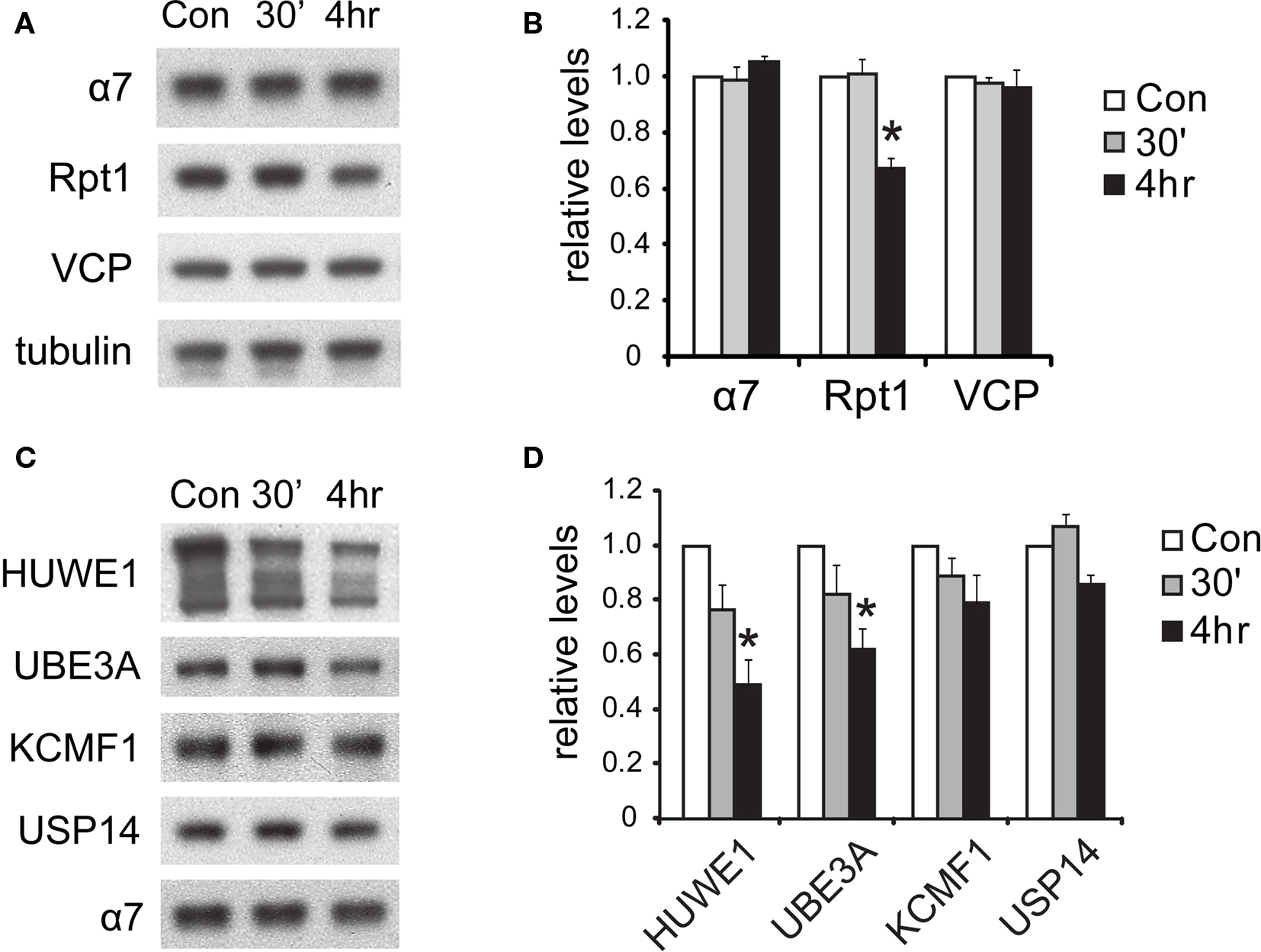
Figure 8. Degradation of proteasome components after NMDA exposure. (A) The degradation of 19S subunits. Neuronal lysates collected at 30’ and 4 h post-NMDA were resolved by SDS-PAGE and probed for total levels of 20S subunit α7, 19S subunit Rpt1, and VCP (an abundant UPS protein). Tubulin served as the loading control. (B) Quantification of the results in (A) by densitometry (n = 6, mean ± SEM, *p < 0.05 by paired t-test). (C) Degradation of proteasome-interacting E3s. Neuronal lysates from (A) were immunoblotted against E3s (HUWE1, UBE3A, KCMF1) and DUBs (USP14) that interact with proteasomes. (D) The graph represents protein levels in (C) normalized to α7 levels (n = 5, *p < 0.05 by paired t-test).
In contrast to acute NMDA treatment, which is often associated with a depression of synaptic transmission, other pharmacological manipulations that suppress neuronal activities do not elicit the degradation of 19S particles (Figure 9 ). These manipulations included adding DHPG to induce metabotropic glutamate receptor (mGluR)-dependent LTD, applying TTX to block action potentials, and TTX + APV + CNQX to block both action potentials and miniature synaptic events These results suggest that only NMDA receptor signaling may be specifically required for the degradation of 19S particles, in addition to reduced synaptic activity.
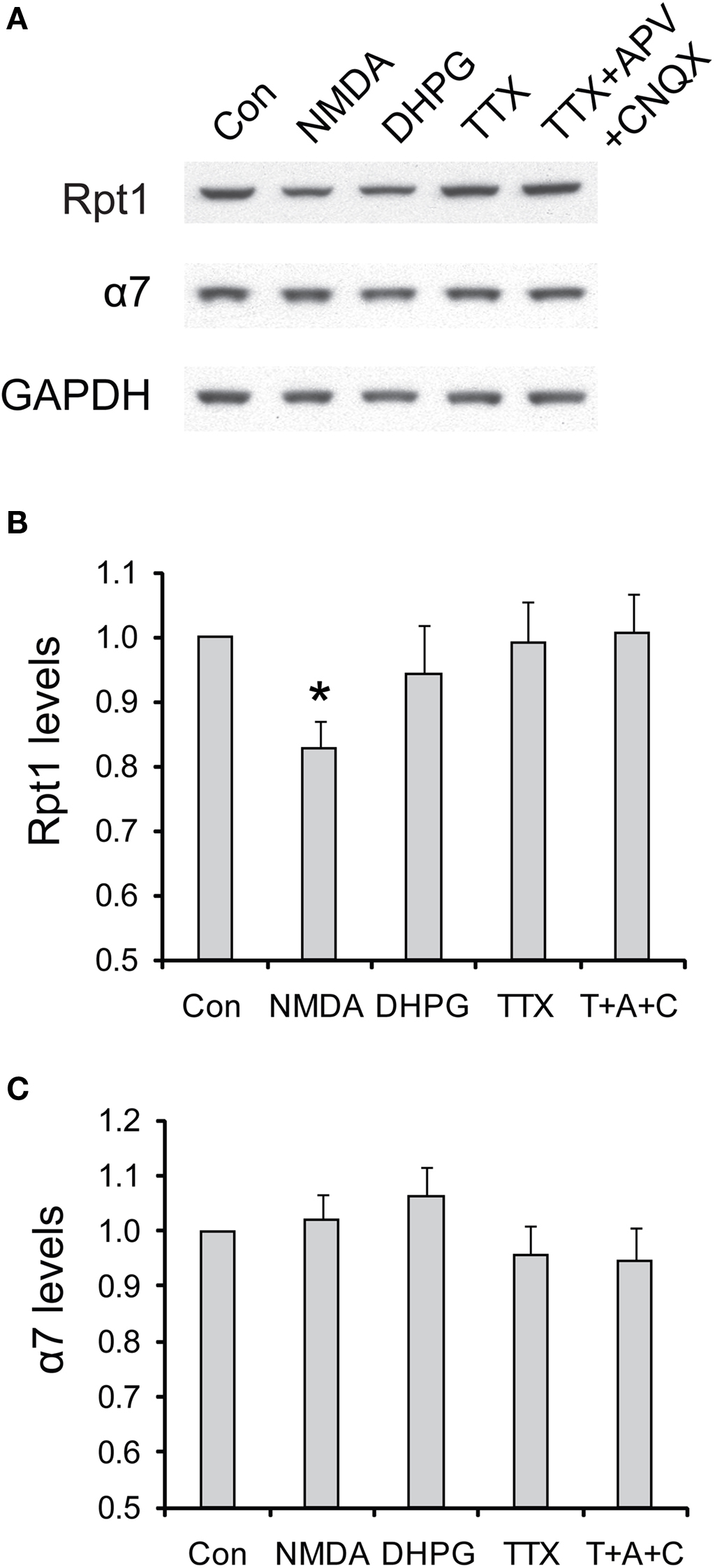
Figure 9. The degradation of 19S particles is specific to NMDA treatment. (A) Cultured hippocampal neurons were briefly treated with NMDA (20 μM, 3 min) or DHPG (100 μM, 10 min), or persistently treated with TTX (2 μM) or TTX + CNQX + APV (2, 40, and 50 μM, respectively). Only the vehicle was added in control experiments (Con). Neuronal lysates were collected at 4 h after drug addition, and analyzed by SDS-PAGE and Western blot for the levels of 19S subunit Rpt1 and 20S subunit α7. GAPDH served as loading control. (B,C) represent the results in (A) quantified by densitometry. Rpt1 and α7 levels (mean ± SEM) were normalized to the control. Statistical significance (*p < 0.05) was determined by paired t-test against the control (n = 8 for NMDA and DHPG, n = 4 for TTX and TTX + APV + CNQX).
19S Particles are Degraded by a Proteasome-Dependent Mechanism
Although the proteasome is the major site for protein degradation in the cell and comprises about 2% of cell proteins, surprisingly little is known about the catabolism of the proteasome itself. A prior study suggested that 20S proteasomes undergo a slow turnover in the liver via lysosomal digestion (Cuervo et al., 1995 ). To our knowledge, no study has examined the catabolic fate of 19S particles. Thus, we tested if in response to NMDA treatment they are degraded by the proteasome or the lysosomal system.
We tested many different combinations of proteasome and lysosome inhibitors and examined their effects on the degradation of 19S particles induced by NMDA (Figure 10 ). Proteasome inhibitors including MG132 and epoxomicin + lactacytsin were the most effective in preventing the NMDA-induced degradation of the 19S. On the other hand, the lysosome inhibitors chloroquine and concanamycin A, which prevent lysosomal acidification, showed no effects on 19S degradation.
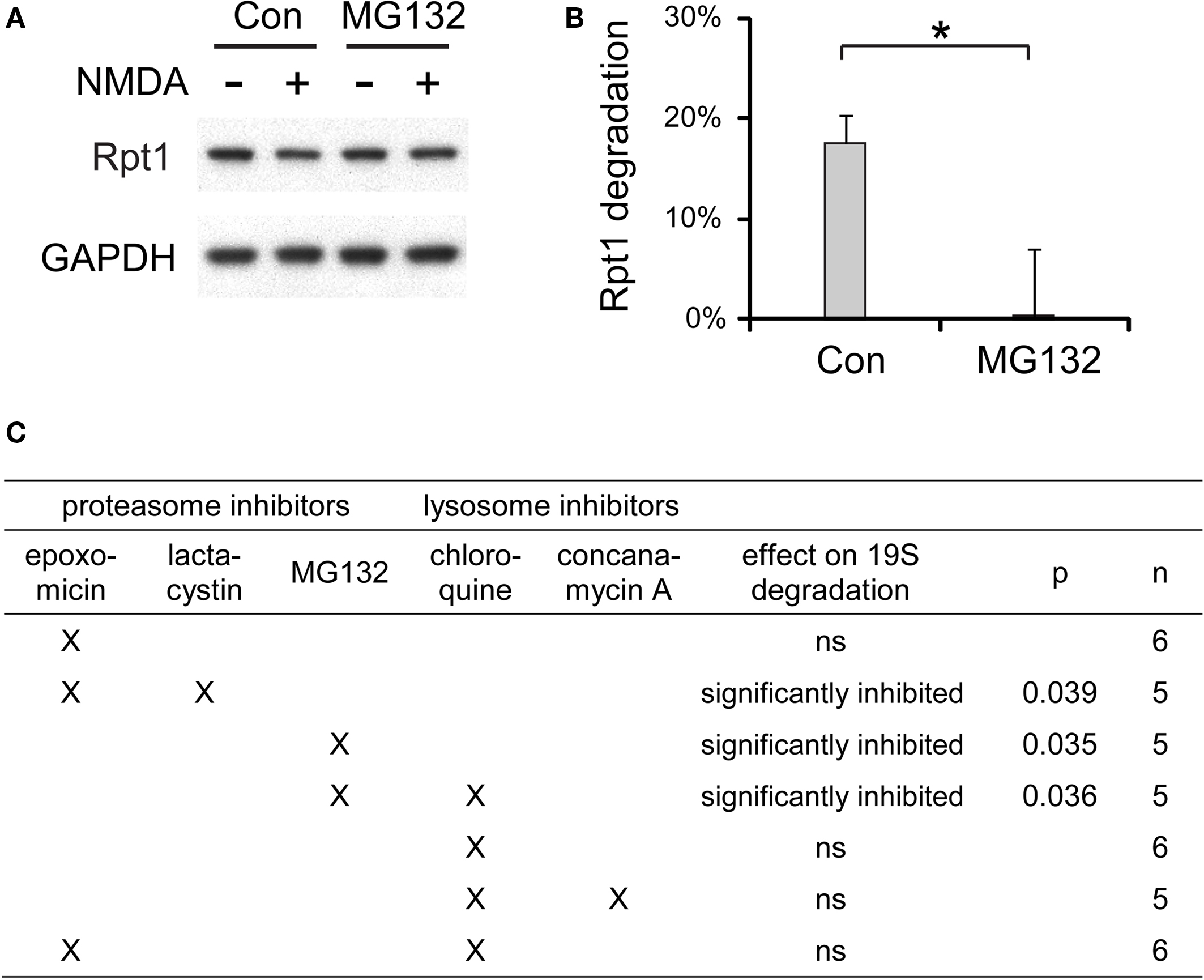
Figure 10. The degradation of 19S particles is proteaosme-dependent. (A) Proteasome inhibitor MG132 (40 μM) was added to cultured hippocampal neurons 30 min before NMDA stimulation (20 μM, 3 min). After NMDA washed-out, MG132 was applied again for 4 h before neuronal lysates were collected. In control (Con) experiments, neurons were only treated with NMDA. Rpt1 levels were determined by SDS-PAGE and immunoblotting. GAPDH served as loading control. (B) 19S degradation induced by NMDA in (A) was quantified by densitometry (n = 5, mean ± SEM). Statistical significance (*p < 0.05) was determined by t-test assuming unequal variance. (C) The same experiments as in (A) and (B), performed with different combinations of proteasome inhibitors (2 μM epoxomicin; 10 μM lactacystin; 40 μM MG132) and lysosome inhibitors (150 μM chloroquine; 100 nM concanamycin A). Statistical significance was determined using t-test (p-value is listed, or ns for not significant, p > 0.05).
In summary, our data indicate that the reduction in UPS activity after NMDA exposure occurs through several mechanisms, including a reduction of 26S proteasome levels and activities, dissociation of 19S particles from the 26S and their subsequent degradation, decrease of protein–ubiquitin conjugates, and degradation of the E3 enzymes released from the proteasome. The degradation of 19S particles occurs via proteasomal and not lysosomal mechanisms.
Discussion
Cytosolic and Synaptic Proteasomes have Different Compositions
In this study, we isolated and characterized cytosolic and synaptic 26S proteasomes from the rat cortex. One unexpected observation was that the ratio of doubly- to singly-capped 26S proteasomes was higher in the cytosol than in the synapse. Both forms can degrade ubiquitylated proteins (Kriegenburg et al., 2008 ), but the functional differences between doubly- and singly-capped 26S have not been established. For example, it is unknown whether they differ in rates of degradation or in their substrate affinity or specificity. Singly-capped 26S has an exposed 20S α-ring that allows it to interact with additional regulators such as PA28 or PA200 (Rechsteiner and Hill, 2005 ). These interacting complexes are likely to dissociate during native gel electrophoresis. Consequently, the singly-capped species observed in our study may in fact represent hybrid 26S (e.g. 19S-20S-PA28) in vivo. Unfortunately, the functional roles of these factors on the proteasome are also not well understood. They may facilitate degradation of specific substrates, e.g. PA28γ has been reported to promote the ubiquitin-independent degradation of the transcriptional coactivator SRC-3 (Li et al., 2006 ). Thus, it is possible that some singly-capped 26S proteasomes in association with such cofactors represent more specialized particles that serve distinct functions in different subcellular locations, such as the synapse.
The 28 26S-associated proteins we found in rat cortex differ considerably from those identified by the same approach in rat muscle (Besche et al., 2009 ). Surprisingly, only 12 are identical, even though myocytes are also postmitotic. Protein degradation however, serves certain very different roles in neurons and skeletal muscle, which represents the major amino acid reservoir in mammals. Unlike proteins in neurons, muscle proteins are mobilized through accelerated proteolysis in fasting or other stressful conditions (Mitch and Goldberg, 1996 ). Also, the protein composition in skeletal muscles and neurons are very different, which may necessitate the involvement of different factors to assist proteasome-mediated degradation. Whether further differences in the spectrum of proteasome-associated proteins exist between different brain regions remains to be explored.
A surprising finding is that ECM29, which has been reported to stabilize the association of 19S and 20S particles (Leggett et al., 2002 ; Kleijnen et al., 2007 ), is only present in the cytosolic 26S but not the synaptic 26S. No factor is known that promotes formation of doubly-capped 26S, but our data suggest ECM29 as a possible candidate.
By mass spectrometry, we identified several proteasome-interacting proteins unique to synaptic 26S proteasomes (14-3-3γ, TAX1BP1, drebrin, SNAP-25), which may modulate proteolysis in a synapse-specific manner. 14-3-3γ, which binds many phosphoproteins in the cytosol (Bridges and Moorhead, 2004 ), may coordinate proteasome function with the protein phosphorylation events that regulate synaptic transmission (Malenka and Bear, 2004 ). TAX1BP1 is a ubiquitin-binding protein (Iha et al., 2008 ; Shembade et al., 2008 ) that may promote substrate-proteasome association at the synapse. It will be interesting to examine which synaptic proteins interact with TAX1BP1. Previous studies suggested that proteasomes are sequestered in dendritic spines by interacting with the actin cytoskeleton (Bingol and Schuman, 2006 ). Our data suggests that the interaction may be partially mediated by drebrin, an important actin-binding protein in the spine (Takahashi et al., 2009 ). Moreover, proteasome-mediated proteolysis has been shown to regulate synaptic vesicle release in several ways (Willeumier et al., 2006 ; Yao et al., 2007 ). The interaction between synaptic proteasomes and SNAP-25, a key synaptic vesicle protein (DeBello et al., 1995 ; Montecucco et al., 2005 ), may provide a mechanism for the proteolytic control of vesicle dynamics.
The various features of the proteasomes in the brain raise many obvious fundamental questions that unfortunately cannot now be answered due to the limits of our knowledge about the UPS. For example, as noted above, the functional differences (if any) between singly-capped and doubly-capped 26S is presently unknown, nor is it clear whether free 20S proteasomes play any significant role in overall intracellular proteolysis by themselves (i.e. when not part of the 26S complex) or in the breakdown of specific proteins although an important role in eliminating inherently structureless proteins has been proposed (Tsvetkov et al., 2009 ). Another fundamental issue raised by the present study is the role of the proteasome-associated ubiquitin ligases. Neurons, like other cells, contain many, perhaps hundreds of E3s, and the functional significance of the localization of UBE3A, HUWE1 and KCMF1 on the proteasome is a major mystery. Presumably, the presence of a ubiquitylation system on the proteasome allows more efficient destruction of a substrate than if the substrate is free in the cytosol (or nucleus or synapse) and has to diffuse or be delivered to the 26S for degradation. Interestingly, UBE3A has recently been implicated in degrading proteins important in synaptic plasticity (Greer et al., 2010 ). Alternatively, the presence of these E3s on the 26S may indicate a direct role in proteasome function, e.g. in remodeling proteasome-bound ubiquitin chains or enhancing processivity, as has been suggested for the E3, Hul5, which is associated with yeast 26S particle (Crosas et al., 2006 ). Finally, interpretation of the significance of this spectrum of proteasome-interacting proteins, and differences between synaptic and cytosolic proteasomes, will be facilitated when we learn more about the proteins associated with the 26S in other mammalian tissues. Similar systematic analyses of these proteasomes have not yet been carried out.
Neuronal Activity Influences Proteasome Degradation and Interacting E3s
Recent studies have shown that neuronal activity influences the activity of UPS in dendrites and synapses (Bingol and Schuman, 2006 ; Djakovic et al., 2009 ). Our findings further demonstrate that the global UPS activity of neurons can also be affected by neuronal activity. Neurons exposed to NMDA showed decreases in the levels of ubiquitin conjugates in whole-cell lysates, which may result from decreased ubiquitylation or increased conjugate degradation. However, after NMDA treatment, the levels of 26S proteasomes decreased due to the degradation of 19S particles. One possible interpretation is that neurons reduce 26S proteasome levels when there are fewer substrates to degrade. We found that 19S degradation is catalyzed by proteasomes and not lysosomes. Although the detailed mechanism is not clear yet, it is likely that the 19S particle dissociates into individual subunits, which are then degraded by the proteasome. Presumably, this NMDA-induced destruction of proteasomes is a mechanism for suppressing overall proteolysis. The proteasome is generally assumed to be a long-lived cell constituent, although the few prior studies (Cuervo et al., 1995 ) have only investigated turnover of 20S particles. In the NMDA-treated neurons, about a third of the 26S particles and up to 50% of the free 19S were lost in four hours without any concomitant loss of the 20S particle. To our knowledge, this is the first study to observe selective degradation of the 19S particle and the detailed mechanisms will be important to understand in future studies.
The selective degradation of the 19S also resulted in the apparent shift from 26S to 20S proteasomes, which cannot degrade ubiquitylated proteins. The role(s) of free 20S in the cell is not clear since the entry of substrates into its proteolytic chamber is restricted by a gate formed by the N-terminal residues of the α-subunits (Smith et al., 2007 ; Tanaka, 2009 ). In vitro this gate is continually opening spontaneously and peptides or unfolded proteins can enter the 20S, though at a much lower rate than in the assembled 26S. Degradation of unfolded or inherently structureless proteins by the 20S has often been proposed to occur in vivo (e.g. after oxidative damage, or with aging) (Poppek and Grune, 2006 ; Tsvetkov et al., 2009 ), but clear evidence for such a role in vivo is lacking. On the contrary, there is strong evidence that most unfolded or oxidant-damaged proteins are degraded by a ubiquitin-dependent mechanism requiring the 26S proteasome and VCP/p97 complexes (Medicherla and Goldberg, 2008 ). Whether having additional 20S proteasomes actually enhances proteolytic functions in neurons is another important issue for further study raised by these data.
Proteasomes and Synaptic Function
Several studies have shown that modifying synaptic strength requires the proteasomal degradation of synaptic proteins (Colledge et al., 2003 ; Ehlers, 2003 ; Bingol and Schuman, 2004 ; Guo and Wang, 2007 ). In particular, proteasome-mediated proteolysis is required for the endocytosis of glutamate receptors (Colledge et al., 2003 ; Patrick et al., 2003 ), after neurons are exposed to high concentrations of glutamate or NMDA (Ehlers, 2000 ; Lee et al., 2004 ). Hence, the proteins interacting with synaptic proteasomes identified in this study are likely to be important for synaptic regulation.
We found three E3s (KCMF1, HUWE1, and UBE3A) and five DUBs (USP5, USP7, USP13, USP14, and UCH37) in association with synaptic proteasomes, which may help proteasomes function more efficiently, help determine specificity for certain types of conjugates, or insure the rapid elimination of ubiquitin chains released from the substrate (since free ubiquitin chains can inhibit proteasomal degradation in vitro) (Finley, 2009 ). The proteasome-associated DUBs presumably function together with the Rpn11 subunit (a different type of DUB) to catalyze the disassembly of the ubiquitin chain and the recycling of free ubiquitin (Koulich et al., 2008 ). In particular, removal of Ubp6 (USP14 homolog) from yeast proteasomes leads to a failure to efficiently remove the ubiquitin chain from substrates and the degradation of some of the attached ubiquitins (Hanna and Finley, 2007 ). USP14/Ubp6 also has an important regulatory role in controlling proteasome function (Finley, 2009 ), for example, together with the ATPases, it regulates gate opening and substrate entry in the 20S (Peth et al., 2009 ). In fact, there are mice with spontaneous mutations in USP14, called ataxia (axJ) mice, which develop synaptic dysfunction (Wilson et al., 2002 ; Anderson et al., 2005 ). Our identification of USP14 on synaptic proteasomes supports the model that reduced synaptic levels of monomeric ubiquitin and ubiquitin conjugates observed in axJ mice (Chen et al., 2009 ) may be due to altered proteasomes that degrade the substrate-linked ubiquitin and fail to recycle it.
Since hundreds of E3s encoded in the mammalian genome appear to function without apparent association with the proteasome (Semple, 2003 ), it will be important to learn what is special about the three E3s we identified on synaptic proteasomes. Curiously, two of them have been previously linked to genetic disorders associated with mental retardation in humans. UBE3A mutations cause Angelman syndrome (Lalande and Calciano, 2007 ), and knockout mice lacking this E3 show defects in synapse morphology, glutamate receptor endocytosis, and LTP (Jiang et al., 1998 ; Dindot et al., 2008 ; Greer et al., 2010 ). HUWE1 mutations are found in X-linked mental retardation (Froyen et al., 2008 ). Since many mental retardation genes appear to be involved in synaptic dysfunction (Humeau et al., 2009 ), our data imply that UBE3A and HUWE1 mutations may cause synaptic defects by altering synaptic proteasome function. Interestingly, UBE3A and HUWE1 dissociate from 26S proteasomes and become degraded after neurons are exposed to NMDA. This simultaneous degradation of 19S particles and these bound E3s represents a novel mechanism of UPS regulation in mammalian cells. It will be important to determine what synaptic protein substrates are normally targeted for degradation by these two E3s, and which are presumably stabilized after NMDA treatment.
Proteasomes and Neuronal Maintenance
Unlike yeast and other mitotic cells, neurons cannot dispose of damaged proteins by cell division (Aguilaniu et al., 2003 ). Hence, neuronal proteasomes and their interacting proteins must play important roles in clearing abnormal proteins.
The hallmarks of many neurodegenerative disorders are neuronal aggregates of misfolded proteins, which often contain ubiquitylated proteins and proteasomes (Ross and Poirier, 2004 ; Rubinsztein, 2006 ). It is clearly important to identify the factors that help deliver ubiquitylated proteins to proteasomes in neurons, because they may be critical in preventing aggregate formation (Chung et al., 2001 ; Bedford et al., 2008b ; Tai and Schuman, 2008 ). It is now clear that many ubiquitin conjugates (e.g. misfolded proteins degraded by the ER-associated degradation pathway or oxidatively damaged proteins in yeast) bind first to the VCP/p97 ATPase complex and then are delivered to the 26S complex bound to UBL-proteins (e.g. Rad23 or ubiquilin) (Madura, 2004 ; Medicherla and Goldberg, 2008 ; Nakatsukasa et al., 2008 ; Finley, 2009 ). Our data show that p62 and the VCP ATPase complex, both of which bind ubiquitylated proteins and assist degradation (Dreveny et al., 2004 ; Seibenhener et al., 2004 ), are associated with neuronal proteasomes. It is attractive to conclude that p62 and VCP/p97 function together with UBL-proteins to handle various ubiquitylated substrates at neuronal 26S proteasomes.
Interestingly, genetic studies have shown that the disruption of p62 (Ramesh Babu et al., 2008 ), VCP (Watts et al., 2004 ; Neumann et al., 2007 ), and 19S particle (Bedford et al., 2008a ) in the brain leads to ubiquitylated inclusions of tau, TAR-DNA binding protein (TDP-43), and α-synuclein, respectively. In fact, these three types of inclusions are associated with over 80% of sporadic dementias in humans (Cummings, 2004 ; Bugiani, 2007 ), which include Alzheimer’s disease (tau), dementia with Lewy bodies (α-synuclein), and certain types of frontotemporal dementias (tau or TDP-43). Therefore, protein aggregates found in sporadic dementias may reflect the impairment of different aspects of 26S proteasome function or substrate delivery to it. Severe 26S dysfunction may also contribute to the co-morbidity observed in some dementia cases, characterized by multiple aggregate types (for example, α-synuclein plus TDP-43) (Nakashima-Yasuda et al., 2007 ; Kovacs et al., 2008 ). Testing these hypotheses in humans or animals has proven difficult (Lansbury and Lashuel, 2006 ) in the past due to difficulties in isolation of 26S proteasome in its native forms. The affinity purification methods used here should greatly facilitate such studies of 26S proteasome function in neurodegenerative disorders, studies of how neuronal activity regulates 26S proteasome properties, or the reported progressive decline of proteasome activity with aging (Dahlmann, 2007 ; Tonoki et al., 2009 ).
In summary, we found that neuronal proteasomes have a large set of interacting proteins that differs from the set of interacting proteins in muscle (Besche et al., 2009 ) or yeast (Verma et al., 2000 ). The proteasome complex varies in different subcellular localizations and changes its content markedly with neuronal activity. Surprisingly, brief activation of NMDA-type glutamate receptors leads to a reduced capacity of the ubiquitin proteasome pathway through rapid disassembly and degradation of the 26S components. This newly discovered level of complexity should be taken into consideration in efforts to understand the molecular mechanisms of synaptic plasticity, the selective degradation of misfolded brain proteins, and the failure to clear such proteins in neurodegenerative diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Steven Gygi and Dr. Wilhelm Haas at Harvard Medical School for their generous support in mass spectrometric analysis. Dr. Sonja Hess and Geoffrey Smith at Caltech Protein Exploration Lab also assisted in preliminary mass spectrometric experiments. We thank members of the Schuman lab and the Goldberg lab for helpful suggestions. Supported by Howard Hughes Medical Institute (Erin M. Schuman) and California Tobacco-Related Disease Research Program (Hwan-Ching Tai).
References
Aakalu, G., Smith, W. B., Nguyen, N., Jiang, C., and Schuman, E. M. (2001). Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron 30, 489–502.
Aguilaniu, H., Gustafsson, L., Rigoulet, M., and Nystrom, T. (2003). Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 299, 1751–1753.
Anderson, C., Crimmins, S., Wilson, J. A., Korbel, G. A., Ploegh, H. L., and Wilson, S. M. (2005). Loss of Usp14 results in reduced levels of ubiquitin in ataxia mice. J. Neurochem. 95, 724–731.
Bajorek, M., Finley, D., and Glickman, M. H. (2003). Proteasome disassembly and downregulation is correlated with viability during stationary phase. Curr. Biol. 13, 1140–1144.
Bedford, L., Hay, D., Devoy, A., Paine, S., Powe, D. G., Seth, R., Gray, T., Topham, I., Fone, K., Rezvani, N., Mee, M., Soane, T., Layfield, R., Sheppard, P. W., Ebendal, T., Usoskin, D., Lowe, J., and Mayer, R. J. (2008a). Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J. Neurosci. 28, 8189–8198.
Bedford, L., Hay, D., Paine, S., Rezvani, N., Mee, M., Lowe, J., and Mayer, R. J. (2008b). Is malfunction of the ubiquitin proteasome system the primary cause of alpha-synucleinopathies and other chronic human neurodegenerative disease? Biochim. Biophys. Acta 1782, 683–690.
Besche, H., Haas, W., Gygi, S., and Goldberg, A. (2009). Isolation of mammalian 26S proteasomes and p97/VCP complexes using the ubiquitin-like domain from HHR23B reveals novel proteasome-associated proteins. Biochemistry 48, 2538–2549.
Bingol, B., and Schuman, E. M. (2004). A proteasome-sensitive connection between PSD-95 and GluR1 endocytosis. Neuropharmacology 47, 755–763.
Bingol, B., and Schuman, E. M. (2005). Synaptic protein degradation by the ubiquitin proteasome system. Curr. Opin. Neurobiol. 15, 536–541.
Bingol, B., and Schuman, E. M. (2006). Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature 441, 1144–1148.
Bingol, B., Wang, C. F., Arnott, D., Cheng, D., Peng, J., and Sheng, M. (2010). Autophosphorylated CaMKIIα acts as a scaffold to recruit proteasomes to dendritic spines. Cell 140, 567–578.
Bridges, D., and Moorhead, G. B. (2004). 14-3-3 proteins: a number of functions for a numbered protein. Sci. STKE 2004, re10.
Bugiani, O. (2007). The many ways to frontotemporal degeneration and beyond. Neurol. Sci. 28, 241–244.
Chen, P. C., Qin, L. N., Li, X. M., Walters, B. J., Wilson, J. A., Mei, L., and Wilson, S. M. (2009). The proteasome-associated deubiquitinating enzyme Usp14 is essential for the maintenance of synaptic ubiquitin levels and the development of neuromuscular junctions. J. Neurosci. 29, 10909–10919.
Chung, K. K., Dawson, V. L., and Dawson, T. M. (2001). The role of the ubiquitin-proteasomal pathway in Parkinson’s disease and other neurodegenerative disorders. Trends Neurosci. 24, S7–S14.
Colledge, M., Snyder, E. M., Crozier, R. A., Soderling, J. A., Jin, Y. T., Langeberg, L. K., Lu, H., Bear, M. F., and Scott, J. D. (2003). Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron 40, 595–607.
Crosas, B., Hanna, J., Kirkpatrick, D. S., Zhang, D. P., Tone, Y., Hathaway, N. A., Buecker, C., Leggett, D. S., Schmidt, M., King, R. W., Gygi, S. P., and Finley, D. (2006). Ubiquitin chains are remodeled at the proteasome by opposing ubiquitin ligase and deubiquitinating activities. Cell 127, 1401–1413.
Cuervo, A. M., Palmer, A., Rivett, A. J., and Knecht, E. (1995). Degradation of proteasomes by lysosomes in rat liver. Eur. J. Biochem. 227, 792–800.
Cummings, J. L. (2004). Dementia with Lewy bodies: Molecular pathogenesis and implications for classification. J. Geriatr. Psychiatry Neurol. 17, 112–119.
Davidson, H. T., Xiao, J., Dai, R., and Bergson, C. (2009). Calcyon is necessary for activity-dependent AMPA receptor internalization and LTD in CA1 neurons of hippocampus. Eur. J. Neurosci. 29, 42–54.
DeBello, W. M., O’Connor, V., Dresbach, T., Whiteheart, S. W., Wang, S. S., Schweizer, F. E., Betz, H., Rothman, J. E., and Augustine, G. J. (1995). SNAP-mediated protein-protein interactions essential for neurotransmitter release. Nature 373, 626–630.
Delgado, J. Y., Coba, M., Anderson, C. N., Thompson, K. R., Gray, E. E., Heusner, C. L., Martin, K. C., Grant, S. G., and O’Dell, T. J. (2007). NMDA receptor activation dephosphorylates AMPA receptor glutamate receptor 1 subunits at threonine 840. J. Neurosci. 27, 13210–13221.
DiAntonio, A., and Hicke, L. (2004). Ubiquitin-dependent regulation of the synapse. Annu. Rev. Neurosci. 27, 223–246.
Dindot, S. V., Antalffy, B. A., Bhattacharjee, M. B., and Beaudet, A. L. (2008). The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 17, 111–118.
Djakovic, S. N., Schwarz, L. A., Barylko, B., Demartino, G. N., and Patrick, G. N. (2009). Regulation of the proteasome by neuronal activity and CAMKII. J. Biol. Chem. 284, 26655–26665.
Dohmen, R. J., London, M. K., Glanemann, C., and Ramos, P. C. (2005). “Assays for proteasome assembly and maturation,” in Methods in Molecular Biology, Vol. 301, Ubiquitin-Proteasome Protocols. eds C. Patterson and D. M. Cyr (Totowa, NJ: Humana), 243–254.
Dreveny, I., Pye, V. E., Beuron, F., Briggs, L. C., Isaacson, R. L., Matthews, S. J., McKeown, C., Yuan, X., Zhang, X., and Freemont, P. S. (2004). p97 and close encounters of every kind: a brief review. Biochem. Soc. Trans. 32, 715–720.
Ehlers, M. D. (2000). Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28, 511–525.
Ehlers, M. D. (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 6, 231–242.
Elias, J. E., and Gygi, S. P. (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214.
Finley, D. (2009). Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513.
Fonseca, R., Vabulas, R. M., Hartl, F. U., Bonhoeffer, T., and Nagerl, U. V. (2006). A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron 52, 239–245.
Froyen, G., Corbett, M., Vandewalle, J., Jarvela, I., Lawrence, O., Meldrum, C., Bauters, M., Govaerts, K., Vandeleur, L., Van Esch, H., Chelly, J., Sanlaville, D., van Bokhoven, H., Ropers, H. H., Laumonnier, F., Ranieri, E., Schwartz, C. E., Abidi, F., Tarpey, P. S., Futreal, P. A., Whibley, A., Raymond, F. L., Stratton, M. R., Fryns, J. P., Scott, R., Peippo, M., Sipponen, M., Partington, M., Mowat, D., Field, M., Hackett, A., Marynen, P., Turner, G., and Gecz, J. (2008). Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am. J. Hum. Genet. 82, 432–443.
Funakoshi, M., Tomko, R. J. Jr., Kobayashi, H., and Hochstrasser, M. (2009). Multiple assembly chaperones govern biogenesis of the proteasome regulatory particle base. Cell 137, 887–899.
Goldberg, A. L. (2003). Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899.
Gordon-Weeks, P. R. (1987). “Isolation of synaptosomes, growth cones and their subcellular components,” in Neurochemistry: A Practical Approach. eds A. J. Turner and H. S. Bachelard (Oxford: IRL), 1–26.
Greer, P. L., Hanayama, R., Bloodgood, B. L., Mardinly, A. R., Lipton, D. M., Flavell, S. W., Kim, T. K., Griffith, E. C., Waldon, Z., Maehr, R. Ploegh, H. L., Chowdhury, S., Worley, P. F., Steen, J., and Greenberg, M. E. (2010). The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 140, 704–716.
Guerrero, C., Tagwerker, C., Kaiser, P., and Huang, L. (2006). An integrated mass spectrometry-based proteomic approach: quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol. Cell Proteomics 5, 366–378.
Guo, L., and Wang, Y. (2007). Glutamate stimulates glutamate receptor interacting protein 1 degradation by ubiquitin-proteasome system to regulate surface expression of GluR2. Neuroscience 145, 100–109.
Haas, W., Faherty, B. K., Gerber, S. A., Elias, J. E., Beausoleil, S. A., Bakalarski, C. E., Li, X., Villen, J., and Gygi, S. P. (2006). Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol. Cell Proteomics 5, 1326–1337.
Hou, L., Antion, M. D., Hu, D., Spencer, C. M., Paylor, R., and Klann, E. (2006). Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51, 441–454.
Humeau, Y., Gambino, F., Chelly, J., and Vitale, N. (2009). X-linked mental retardation: focus on synaptic function and plasticity. J. Neurochem. 109, 1–14.
Iha, H., Peloponese, J. M., Verstrepen, L., Zapart, G., Ikeda, F., Smith, C. D., Starost, M. F., Yedavalli, V., Heyninck, K., Dikic, I., Beyaert, R., and Jeang, K. T. (2008). Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. EMBO J. 27, 629–641.
Jiang, Y. H., Armstrong, D., Albrecht, U., Atkins, C. M., Noebels, J. L., Eichele, G., Sweatt, J. D., and Beaudet, A. L. (1998). Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21, 799–811.
Kaneko, T., Hamazaki, J., Iemura, S., Sasaki, K., Furuyama, K., Natsume, T., Tanaka, K., and Murata, S. (2009). Assembly pathway of the Mammalian proteasome base subcomplex is mediated by multiple specific chaperones. Cell 137, 914–925.
Karpova, A., Mikhaylova, M., Thomas, U., Knopfel, T., and Behnisch, T. (2006). Involvement of protein synthesis and degradation in long-term potentiation of Schaffer collateral CA1 synapses. J. Neurosci. 26, 4949–4955.
Kleijnen, M. F., Roelofs, J., Park, S., Hathaway, N. A., Glickman, M., King, R. W., and Finley, D. (2007). Stability of the proteasome can be regulated allosterically through engagement of its proteolytic active sites. Nat. Struct. Mol. Biol. 14, 1180–1188.
Koulich, E., Li, X., and DeMartino, G. N. (2008). Relative structural and functional roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Mol. Biol. Cell 19, 1072–1082.
Kovacs, G. G., Alafuzoff, I., Al-Sarraj, S., Arzberger, T., Bogdanovic, N., Capellari, S., Ferrer, I., Gelpi, E., Kovari, V., Kretzschmar, H., Nagy, Z., Parchi, P., Seilhean, D., Soininen, H., Troakes, C., and Budka, H. (2008). Mixed brain pathologies in dementia: the BrainNet Europe consortium experience. Dement. Geriatr. Cogn. Disord. 26, 343–350.
Kraut, D. A., Prakash, S., and Matouschek, A. (2007). To degrade or release: ubiquitin-chain remodeling. Trends Cell Biol. 17, 419–421.
Kriegenburg, F., Seeger, M., Saeki, Y., Tanaka, K., Lauridsen, A. M., Hartmann-Petersen, R., and Hendil, K. B. (2008). Mammalian 26S proteasomes remain intact during protein degradation. Cell 135, 355–365.
Krogan, N. J., Lam, M. H., Fillingham, J., Keogh, M. C., Gebbia, M., Li, J., Datta, N., Cagney, G., Buratowski, S., Emili, A., and Greenblatt, J. F. (2004). Proteasome involvement in the repair of DNA double-strand breaks. Mol. Cell. 16, 1027–1034.
Lalande, M., and Calciano, M. A. (2007). Molecular epigenetics of Angelman syndrome. Cell. Mol. Life Sci. 64, 947–960.
Lansbury, P. T., and Lashuel, H. A. (2006). A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature 443, 774–779.
Lee, H. K., Kameyama, K., Huganir, R. L., and Bear, M. F. (1998). NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21, 1151–1162.
Lee, S. H., Choi, J. H., Lee, N., Lee, H. R., Kim, J. I., Yu, N. K., Choi, S. L., Kim, H., and Kaang, B. K. (2008). Synaptic protein degradation underlies destabilization of retrieved fear memory. Science 319, 1253–1256.
Lee, S. H., Simonetta, A., and Sheng, M. (2004). Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron 43, 221–236.
Leggett, D. S., Hanna, J., Borodovsky, A., Crosas, B., Schmidt, M., Baker, R. T., Walz, T., Ploegh, H., and Finley, D. (2002). Multiple associated proteins regulate proteasome structure and function. Mol. Cell 10, 495–507.
Le Tallec, B., Barrault, M. B., Guerois, R., Carre, T., and Peyroche, A. (2009). Hsm3/S5b participates in the assembly pathway of the 19S regulatory particle of the proteasome. Mol. Cell 33, 389–399.
Li, R., Dozmorov, M., Hellberg, F., Tian, Y., Jilderos, B., and Wigstrom, H. (2004). Characterization of NMDA induced depression in rat hippocampus: involvement of AMPA and NMDA receptors. Neurosci. Lett. 357, 87–90.
Li, X., Lonard, D. M., Jung, S. Y., Malovannaya, A., Feng, Q., Qin, J., Tsai, S. Y., Tsai, M. J., and O’Malley, B. W. (2006). The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell 124, 381–392.
Lopez-Salon, M., Alonso, M., Vianna, M. R. M., Viola, H., Souza, T. M. E., Izquierdo, I., Pasquini, J. M., and Medina, J. H. (2001). The ubiquitin-proteasome cascade is required for mammalian long-term memory formation. Eur. J. Neurosci. 14, 1820–1826.
Lu, W., and Ziff, E. B. (2005). PICK1 interacts with ABP/GRIP to regulate AMPA receptor trafficking. Neuron 47, 407–421.
Madura, K. (2004). Rad23 and Rpn10: perennial wallflowers join the melee. Trends Biochem. Sci. 29, 637–640.
McCutchen-Maloney, S. L., Matsuda, K., Shimbara, N., Binns, D. D., Tanaka, K., Slaughter, C. A., and DeMartino, G. N. (2000). cDNA cloning, expression, and functional characterization of PI31, a proline-rich inhibitor of the proteasome. J. Biol. Chem. 275, 18557–18565.
Medicherla, B., and Goldberg, A. L. (2008). Heat shock and oxygen radicals stimulate ubiquitin-dependent degradation mainly of newly synthesized proteins. J. Cell Biol. 182, 663–673.
Mitch, W. E., and Goldberg, A. L. (1996). Mechanisms of muscle wasting. The role of the ubiquitin-proteasome pathway. N. Engl. J. Med. 335, 1897–1905.
Montecucco, C., Schiavo, G., and Pantano, S. (2005). SNARE complexes and neuroexocytosis: how many, how close? Trends Biochem. Sci. 30, 367–372.
Nakashima-Yasuda, H., Uryu, K., Robinson, J., Xie, S. X., Hurtig, H., Duda, J. E., Arnold, S. E., Siderowf, A., Grossman, M., Leverenz, J. B., Woltjer, R., Lopez, O. L., Hamilton, R., Tsuang, D. W., Galasko, D., Masliah, E., Kaye, J., Clark, C. M., Montine, T. J., Lee, V. M., and Trojanowski, J. Q. (2007). Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 114, 221–229.
Nakatsukasa, K., Huyer, G., Michaelis, S., and Brodsky, J. L. (2008). Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell 132, 101–112.
Neumann, M., Mackenzie, I. R., Cairns, N. J., Boyer, P. J., Markesbery, W. R., Smith, C. D., Taylor, J. P., Kretzschmar, H. A., Kimonis, V. E., and Forman, M. S. (2007). TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J. Neuropathol. Exp. Neurol. 66, 152–157.
Park, S., Roelofs, J., Kim, W., Robert, J., Schmidt, M., Gygi, S. P., and Finley, D. (2009). Hexameric assembly of the proteasomal ATPases is templated through their C termini. Nature 459, 866–870.
Patrick, G. N., Bingol, B., Weld, H. A., and Schuman, E. M. (2003). Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Curr. Biol. 13, 2073–2081.
Peth, A., Besche, H. C., and Goldberg, A. L. (2009). Ubiquitinated proteins activate the proteasome by binding to Usp14/Ubp6, which causes 20S gate opening. Mol. Cell 36, 794–804.
Poppek, D., and Grune, T. (2006). Proteasomal defense of oxidative protein modifications. Antioxid. Redox Signal. 8, 173–184.
Ramesh Babu, J., Lamar Seibenhener, M., Peng, J., Strom, A. L., Kemppainen, R., Cox, N., Zhu, H., Wooten, M. C., Diaz-Meco, M. T., Moscat, J., and Wooten, M. W. (2008). Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J. Neurochem. 106, 107–120.
Rechsteiner, M., and Hill, C. P. (2005). Mobilizing the proteolytic machine: cell biological roles of proteasome activators and inhibitors. Trends Cell Biol. 15, 27–33.
Roelofs, J., Park, S., Haas, W., Tian, G., McAllister, F. E., Huo, Y., Lee, B.-H., Zhang, F., Shi, Y., Gygi, S. P., and Finley, D. (2009). Chaperone-mediated pathway of proteasome regulatory particle assembly. Nature 459, 861–865.
Ross, C. A., and Poirier, M. A. (2004). Protein aggregation and neurodegenerative disease. Nat. Med. 10, S10–S17.
Rubinsztein, D. C. (2006). The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786.
Saeki, Y., Toh, E. A., Kudo, T., Kawamura, H., and Tanaka, K. (2009). Multiple Proteasome-interacting proteins assist the assembly of the yeast 19S regulatory particle. Cell 137, 900–913.
Sakata, E., Yamaguchi, Y., Kurimoto, E., Kikuchi, J., Yokoyama, S., Yamada, S., Kawahara, H., Yokosawa, H., Hattori, N., Mizuno, Y., Tanaka, K., and Kato, K. (2003). Parkin binds the Rpn10 subunit of 26S proteasomes through its ubiquitin-like domain. EMBO Rep. 4, 301–306.
Scanlon, T. C., Gottlieb, B., Durcan, T. M., Fon, E. A., Beitel, L. K., and Trifiro, M. A. (2009). Isolation of human proteasomes and putative proteasome-interacting proteins using a novel affinity chromatography method. Exp. Cell Res. 315, 176–189.
Schauber, C., Chen, L., Tongaonkar, P., Vega, I., Lambertson, D., Potts, W., and Madura, K. (1998). Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature 391, 715–718.
Schmidt, M., Hanna, J., Elsasser, S., and Finley, D. (2005). Proteasome-associated proteins: regulation of a proteolytic machine. Biol. Chem. 386, 725–737.
Seibenhener, M. L., Babu, J. R., Geetha, T., Wong, H. C., Krishna, N. R., and Wooten, M. W. (2004). Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 24, 8055–8068.
Semple, C. A. M. (2003). The comparative proteomics of ubiquitination in mouse. Genome Res. 13, 1389–1394.
Shembade, N., Harhaj, N. S., Parvatiyar, K., Copeland, N. G., Jenkins, N. A., Matesic, L. E., and Harhaj, E. W. (2008). The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat. Immunol. 9, 254–262.
Sherman, M. Y., and Goldberg, A. L. (2001). Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29, 15–32.
Smith, D. M., Chang, S. C., Park, S., Finley, D., Cheng, Y., and Goldberg, A. L. (2007). Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Mol. Cell 27, 731–744.
Tai, H. C., and Schuman, E. M. (2008). Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 9, 826–838.
Takahashi, H., Yamazaki, H., Hanamura, K., Sekino, Y., and Shirao, T. (2009). Activity of the AMPA receptor regulates drebrin stabilization in dendritic spine morphogenesis. J. Cell. Sci. 122, 1211–1219.
Tanaka, K. (2009). The proteasome: overview of structure and functions. Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci. 85, 12–36.
Tonoki, A., Kuranaga, E., Tomioka, T., Hamazaki, J., Murata, S., Tanaka, K., and Miura, M. (2009). Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol. Cell. Biol. 29, 1095–1106.
Tsvetkov, P., Reuven, N., and Shaul, Y. (2009). The nanny model for IDPs. Nat. Chem. Biol. 5, 778–781.
Verma, R., Chen, S., Feldman, R., Schieltz, D., Yates, J., Dohmen, T., and Deshaies, R. J. (2000). Proteasomal proteomics: Identification of nucleotide-sensitive proteasome-interacting proteins by mass spectrometric analysis of affinity-purified proteasomes. Mol. Biol. Cell 11, 3425–3439.
Wang, X. R., Chen, C. F., Baker, P. R., Chen, P. L., Kaiser, P., and Huang, L. (2007). Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry 46, 3553–3565.
Wang, X. R., and Huang, L. (2008). Identifying dynamic interactors of protein complexes by quantitative mass spectrometry. Mol. Cell Proteomics 7, 46–57.
Watts, G. D., Wymer, J., Kovach, M. J., Mehta, S. G., Mumm, S., Darvish, D., Pestronk, A., Whyte, M. P., and Kimonis, V. E. (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381.
Willeumier, K., Pulst, S. M., and Schweizer, F. E. (2006). Proteasome inhibition triggers activity-dependent increase in the size of the recycling vesicle pool in cultured hippocampal neurons. J. Neurosci. 26, 11333–11341.
Wilson, S. M., Bhattacharyya, B., Rachel, R. A., Coppola, V., Tessarollo, L., Householder, D. B., Fletcher, C. F., Miller, R. J., Copeland, N. G., and Jenkins, N. A. (2002). Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat. Genet. 32, 420–425.
Yao, I., Takagi, H., Ageta, H., Kahyo, T., Sato, S., Hatanaka, K., Fukuda, Y., Chiba, T., Morone, N., Yuasa, S. Inokuchi, K., Ohtsuka, T., MacGregor, G. R., Tanaka, K., and Setou, M. (2007). SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell 130, 943–957.
Keywords: proteasome, proteasome-interacting protein, ubiquitin ligase, synaptic plasticity, UBE3A, NMDA
Citation: Tai H-C, Besche H, Goldberg AL and Schuman EM (2010) Characterization of the brain 26S proteasome and its interacting proteins. Front. Mol. Neurosci. 3:12. doi: 10.3389/fnmol.2010.00012
Received: 04 December 2009;
Paper pending published: 26 February 2010;
Accepted: 21 April 2010;
Published online: 21 May 2010
Edited by:
Seth G. N. Grant, Wellcome Trust Sanger Institute, UKReviewed by:
Mark Collins, Wellcome Trust Sanger Institute, UKCopyright: © 2010 Tai, Besche, Goldberg and Schuman. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Erin M. Schuman, Caltech MC114-96, 1200 E. California Blvd., Pasadena, CA 91125, USA; Max Planck Institute for Brain Research, Max-von-Laue Strasse 3, 60438 Frankfurt am Main, Germany e-mail: schumane@caltech.edu; schumane@brain.mpg.de