 Marcel Brügel
Marcel Brügel Ann-Sophie Kiesel
Ann-Sophie Kiesel Tobias B. Haack
Tobias B. Haack Susana Peralta
Susana Peralta- Institute of Medical Genetics and Applied Genomics, University of Tübingen, Tübingen, Germany
Pathogenic variants in the ATAD3 gene cluster have been associated with different neurodevelopmental disorders showing clinical symptoms like global developmental delay, muscular hypotonia, cardiomyopathy, congenital cataracts, and cerebellar atrophy. ATAD3A encodes for a mitochondrial ATPase whose function is unclear and has been considered one of the five most common nuclear genes associated with mitochondrial diseases in childhood. However, the mechanism causing ATAD3-associated disorders is still unknown. In vivo models have been used to identify ATAD3 function. Here we summarize the features of mouse models with ATAD3 loss of function and Drosophila models overexpressing pathogenic ATAD3 variants. We discuss how these models have contributed to our understanding of ATAD3 function and the pathomechanism of the ATAD3-associated disease.
Introduction
ATPase family AAA domain-containing protein 3 (ATAD3) is a mitochondrial membrane protein from the family of ATPases associated with diverse cellular activities and conserved in metazoans. ATAD3 gene absence results in embryonic lethality in worms (Hoffmann et al., 2009), flies (Gilquin et al., 2010; Harel et al., 2016), and mice (Goller et al., 2013; Peralta et al., 2018), indicating that it plays a role in early developmental stages and may be essential for proper mitochondrial function. In hominids, ATAD3 has been duplicated twice to form an array of three paralog genes organized in tandem close to the telomere in chromosome 1p: ATAD3A, ATAD3B, and ATAD3C, whereas other species, such as fruit flies and mice, harbor only one gene (Li et al., 2014). ATAD3B differs from the ancestral paralog ATAD3A by having a C-terminal extension of 62 amino acids, which is caused by a mutation in the original stop codon, while ATAD3C seems to be a truncated gene, missing the first 70 amino acids of the protein (Li and Rousseau, 2012; Merle et al., 2012).
ATAD3A has been described within mitochondria as spanning both mitochondrial membranes with its C terminus facing the matrix and the N-terminal region in the outer membrane (Gilquin et al., 2010; Baudier, 2018). The N-terminal domain comprises two transmembrane domains (TM1 and TM2), two coiled-coil domains (Cc1 and Cc2) important for protein–protein interactions and ATAD3A oligomerization, and a proline-rich domain (PR) of unknown function (Hubstenberger et al., 2010). The C-terminal region of ATAD3A contains an ATPase domain in the mitochondrial matrix with two conserved Walker A and Walker B motifs for ATP binding and ATPase activity (Figure 1). As a member of the AAA+ ATPase family, ATAD3 is predicted to form hexameric ring structures (Frickey and Lupas, 2004).
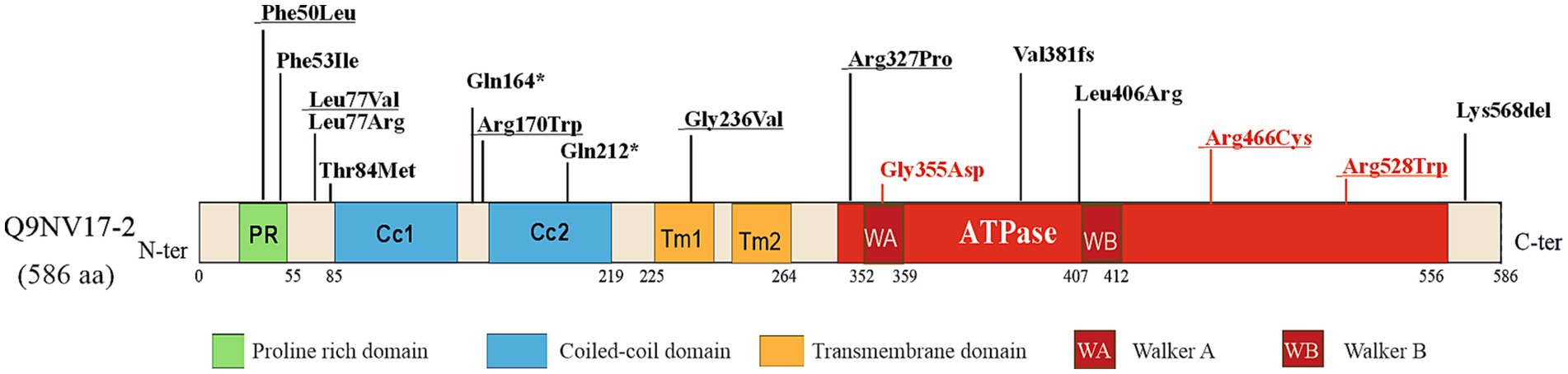
Figure 1. Single Nucleotide Pathogenic variants of human ATAD3A. Schematic representation of the human isoform 2 ATAD3A protein labeled with the main functional domains (NM_001170535.2, Q9NV17–2). PR stands for the proline-rich-domain and CC for the coiled-coil domain. The transmembrane (TM) domains are shown in orange, the ATPase domain in red, and the Walker A motif and Walker B motifs are denoted with WA and WB, respectively. The recessive and biallelic variants are marked by black arrows, while dominant variants are marked by red arrows. The variants that have been overexpressed in Drosophila and have been described in Table 1 are underlined.
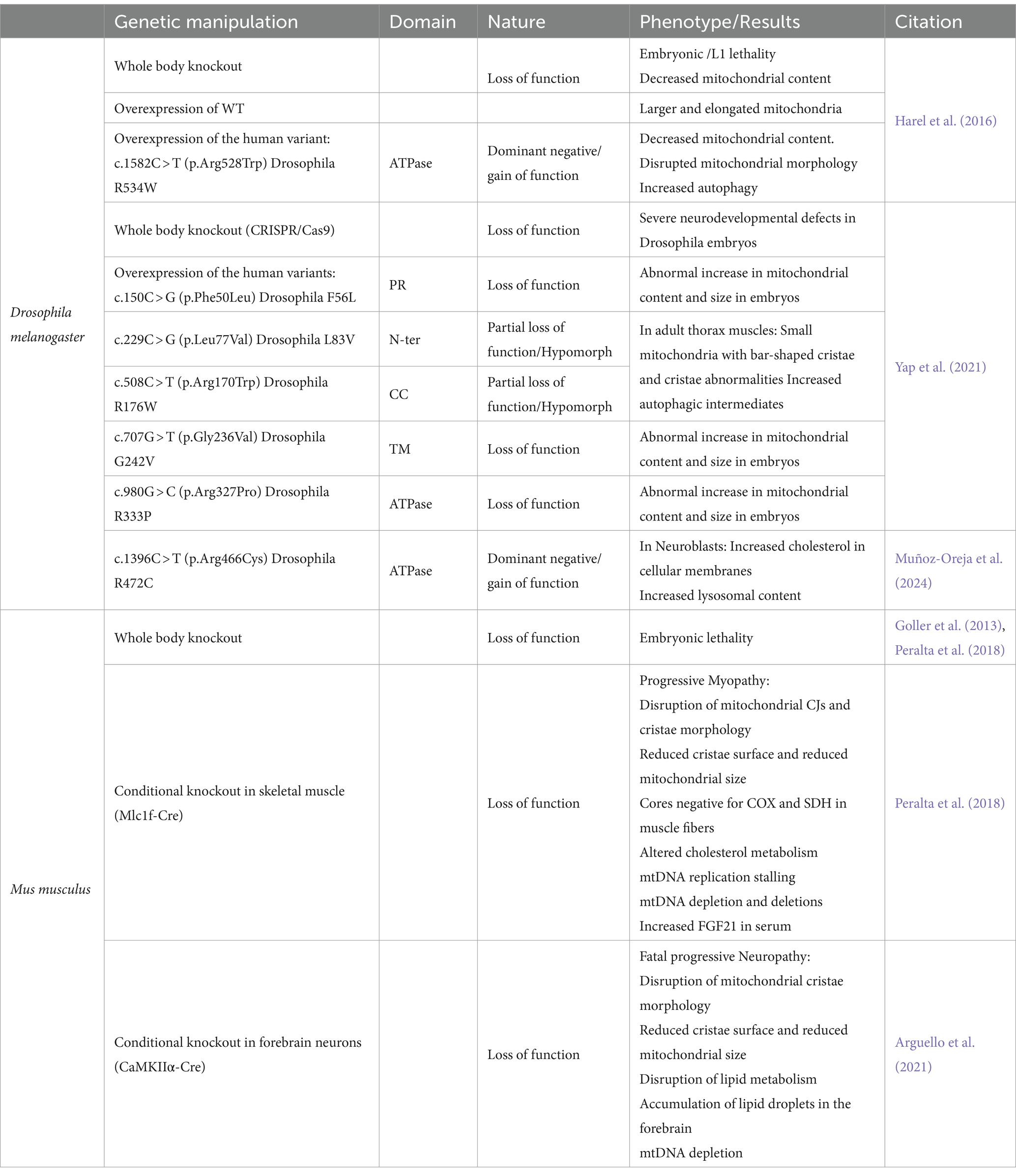
Table 1. Drosophila and mouse models developed to study the role of ATAD3.
Functionally, ATAD3A has been associated with several roles within mitochondria including regulation of the inner membrane structure, protein assembly (Peralta et al., 2018; Arguello et al., 2021), mitochondrial DNA (mtDNA) nucleoid organization (He et al., 2007; Gerhold et al., 2015; Desai et al., 2017), cholesterol trafficking, and lipid metabolism (Issop et al., 2015) among others (van den Ecker et al., 2015; Fang et al., 2010; Lang et al., 2020; Jin et al., 2018). However, the primary role of the protein remains unknown.
ATAD3A variants and their associated diseases
ATAD3A is considered one of the five most common nuclear genes associated with mitochondrial diseases in childhood (Frazier et al., 2021). Due to the extensive sequence homology among the paralogs ATAD3A, ATAD3B, and ATAD3C, the 1p36.33 region is prone to non-allelic homologous recombination (NAHR), resulting in copy number variants (CNVs). Disease-causing mutations in ATAD3A include duplications and deletions among the ATAD3 paralogs (Harel et al., 2016; Desai et al., 2017; Peeters-Scholte et al., 2017; Gunning et al., 2020; Frazier et al., 2021; Ebihara et al., 2022; Tawfik et al., 2023) and single nucleotide variants (SNVs) in the highly expressed ATAD3A gene (Cooper et al., 2017; Peralta et al., 2019; Dorison et al., 2020; Hanes et al., 2020; Muñoz-Oreja et al., 2024; Al Madhoun et al., 2019; Chen et al., 2023). The allelic spectrum of ATAD3A-associated diseases includes null, hypomorphic, and antimorphic alleles and the variants can be inherited and de novo. The SNVs reported to date in ATAD3A are depicted in Figure 1.
In 2016, pathogenic variants in ATAD3A were first associated with Harel-Yoon syndrome (MIM:617183), characterized by global developmental delay and muscular hypotonia, along with other features such as hypertrophic cardiomyopathy, optic atrophy, congenital cataracts, and peripheral neuropathy (Harel et al., 2016). Since then, ATAD3A variants have been associated with different neurodevelopmental disorders. The dominantly inherited heterozygous variant c.1064G > A (p.Gly355Asp) in the Walker A domain of ATAD3A was associated with hereditary spastic paraplegia and axonal neuropathy (Cooper et al., 2017). The heterozygous c.1396.C > T (p.Arg466Cys) missense variant, involved in the ATP hydrolysis, produces a form of neurological syndrome associated with optic atrophy (Muñoz-Oreja et al., 2024). Bi-allelic deletions of ATAD3 via NAHR and compound heterozygous variants (some combinations of deletions/truncating mutations with missense mutations) have been associated with neonatal lethal pontocerebellar hypoplasia, hypotonia, and respiratory insufficiency syndrome (PHRINL, MIM618810; Desai et al., 2017; Peeters-Scholte et al., 2017; Skopkova et al., 2023). Moreover, a dominant 68 kilobase (kb) de novo duplication in the ATAD3 locus was reported in 22 patients from 21 families, associated with a severe multisystemic disorder characterized by neonatal respiratory insufficiency, hypotonia, and cardiomyopathy, resulting in death in the first weeks of life (MIM:618815; Gunning et al., 2020; Frazier et al., 2021). These duplications generate an extra copy of ATAD3B and an in-frame ATAD3A/C fusion gene that forms a stable chimeric ATAD3A/C protein disrupting regular ATAD3 oligomerization. The extra copy of ATAD3B is not thought to play a role in the phenotype as healthy individuals and patients with distinct phenotypes were found to have benign duplications affecting only ATAD3B.
Drosophila models overexpressing pathogenic ATAD3 variants
To functionally evaluate the potential pathogenicity of the ATAD3A variants several transgenic flies have been generated using the UAS-Gal4 system for tissue-specific expression (Table 1). The Drosophila melanogaster ortholog to the human ATAD3A is called Belphegor (bor), hereafter referred to as “Drosophila ATAD3” or “dATAD3.”
In 2016, Harel et al. studied the recurrent heterozygous de novo variant c.1582C > T (p.Arg528Trp), located in the ATPase domain of the protein (equivalent in Drosophila, p.R534W; Figure 1; Harel et al., 2016). This variant was found in five unrelated families associated with global developmental delay, axonal neuropathy, and hypertrophic cardiomyopathy. They demonstrated that ubiquitous (tub-Gal4 and Ubi-Gal4), pan-neuronal (n-syb-Gal4), and motoneuronal (D42-Gal4) expression of UAS-dAtad3R534W resulted in complete embryonic lethality with no viable adult flies. Muscle-specific expression (C57-Gal4) led to approximately 90% lethality. In contrast, overexpression of the wild-type allele UAS-dAtad3WT with the same Gal4 drivers consistently produced viable flies with no phenotype. On a cellular level, the p.R534W mutation induced a significant reduction of mitochondria in the ventral nerve cord, axons, and synaptic boutons, suggesting increased mitophagy. Transmission electron microscopy (TEM) showed that muscle tissue contained very few and small mitochondria with highly aberrant cristae and a substantial increase in autophagic intermediates. Overexpression of dAtad3WT, however, resulted in the opposite phenotype, with large, elongated mitochondria. The authors proposed that ATAD3 may promote mitochondrial fusion or inhibit fission, while the mutation likely inhibits fusion and/or promotes fission.
In 2021, Yap et al. investigated five ATAD3A missense variants inherited in trans to loss-of-function (LOF) alleles and associated with distinct neurological phenotypes (Yap et al., 2021). The variants included c.150C > G (p.Phe50Leu), c.229C > G (p.Leu77Val), in the N-terminal of ATAD3A, c.508C > T (p.Arg170Trp) located between the coiled-coil domains; c.707G > T (p.Gly236Val) in the transmembrane domain, and c.980G > C (p.Arg327Pro) in the ATPase domain (Figure 1). The equivalent variants in Drosophila are dAtad3F56L, dAtad3L83K, dAtad3R176W, dAtad3G242V and dAtad3R333P (Table 1). Using CRISPR/Cas9-mediated genome editing, the researchers integrated a gene cassette into the first intron of dAtad3 generating a LOF allele. All flies harboring this LOF allele and a null allele (PBac{PB}dAtad3ac05496), resulted in a functional knockout. The lethality was rescued by UAS-dAtad3WT expression using a pan-neuronal Gal4 driver (elavC155-Gal4), confirming the severe LOF nature of the introduced cassette. The expression of dAtad3L83V and dAtad3R176W variants also rescued the developmental lethality caused by ATAD3A loss, suggesting that they are partial LOF alleles. The other 3 variants, dAtad3F56L, dAtad3G242V, and dAtad3R333P failed to rescue lethality indicating that they are severe LOF alleles.
Further characterization of the dAtad3L83V and dAtad3R176W variants showed a decreased lifespan, and locomotion and flight defects. TEM analysis showed both mutations caused small mitochondria with cristae abnormalities and increased autophagic intermediates. Confocal images showed increased expression of the autophagic marker p62 in adult thorax muscles expressing dAtad3L83V and dAtad3R176W. The authors concluded that ATAD3A function is required for the homeostasis of mitochondrial dynamics and mitophagy. One possible mechanism proposed was through increased interaction with Drp1, as the coiled-coil domain was shown to interact with Drp1 promoting mitochondrial fission (Zhao et al., 2019). Moreover, this study revealed the functional importance of the N-terminal, coiled-coil, and transmembrane domains of ATAD3A.
Recently, Muñoz-Oreja et al. investigated the heterozygous p.Arg466Cys variant (p.R472C in Drosophila), affecting a conserved arginine finger crucial for ATP hydrolysis (Figure 1). Transgenic flies with UAS-dAtad3R472C were created and crossed with various tissue-specific Gal4 drivers (Muñoz-Oreja et al., 2024). Ubiquitous expression resulted in complete lethality, likely through a dominant-negative mechanism. Expression in nervous and muscle tissue (Atad3-Gal4) or neurons (elavC155-Gal4) was lethal as well. However, expression driven by eyeless-Gal4 (ey-Gal4), which is limited to the eyes and part of the brain, resulted only in partial lethality (65%), with the surviving flies exhibiting abnormal or missing eyes. Moreover, using the neuroblast-specific inscuteable-Gal4 (insc-Gal4) driver or the late-onset eye and neuronal driver glass multiple reporter-Gal4 (GMR-Gal4), viable flies expressing the mutant variant dAtad3R472C were produced similarly to controls. Therefore, the Arg466Cys variant is highly deleterious unless expression is highly restricted. Expression of the p.Arg466Cys variant in neuroblasts led to the formation of membrane-bound cholesterol aggregates and increased lysosomal content. The cholesterol aggregates, detected by the reporter mKate-D4, co-localized with the lysosomal marker LAMP-GFP, suggesting that this excess of cholesterol is targeted to the lysosomal pathway. In agreement with the results obtained in Drosophila, patient-derived fibroblasts also exhibited membrane-embedded cholesterol aggregation in the form of membrane whorls and increased lysosomal content. Interestingly, flies expressing the p.Arg466Cys variant under the ey-Gal4 driver showed higher dependency on dietary cholesterol. A diet with reduced cholesterol significantly decreased the number of viable adults, and, by contrast, cholesterol supplementation on the diet enhanced viability. Based on these results, the authors propose a model where defective ATAD3 results in a mitochondrial cholesterol deficit that is attenuated by increasing the cytosolic cholesterol levels. This increased cholesterol would be a cellular compensatory response that leads to an aberrant aggregation in membranes that can cascade to lysosomal insufficiency contributing to the pathomechanism of the disease.
Mouse models of ATAD3 loss of function
In mice, the ubiquitous disruption of ATAD3 was embryonic lethal (Peralta et al., 2018; Goller et al., 2013). To understand the in vivo function of ATAD3 in mammals we generated two different LOF mouse models: the ATAD3 skeletal muscle deficient mice (Peralta et al., 2018) and ATAD3-neuron deficient mice (Arguello et al., 2021). The main features observed in the animal models are summarized in Table 1.
ATAD3 skeletal muscle knockout
ATAD3 skeletal muscle-deficient mice (ATAD3 muscle KO) were obtained by crossing the ATAD3 floxed with Mlc1f-Cre transgenic mice, expressing Cre recombinase under the myosin light chain 1 (Mlc1) promoter (Bothe et al., 2000). The lack of ATAD3 in skeletal muscle induced a progressive myopathy with an onset between 2 and 3 months of age, characterized by motor-impaired coordination and weakness, developing into muscle wasting, and reduced fiber size (Peralta et al., 2018). Despite the dramatically reduced muscle tonus, ATAD3 muscle KO mice did not show reduced survival. This fact suggests that skeletal muscle mitochondria are either able to compensate for the resulting functional consequences, or that in skeletal muscle, ATAD3 is not essential for general survival.
The first phenotype detected by TEM in the muscles of 2-month-old ATAD3 KO mice was a disruption of the inner mitochondrial membrane (IMM) structure. The cristae, formed by the inner membrane, lost the cristae junctions (CJs), and the cristae surface per mitochondria was decreased (Table 1). This was accompanied by a reduction in high molecular weight mitochondrial contact site and cristae organizing system (MICOS) complexes. As degeneration of the cristae increased over time, in the muscles from 5-month-old animals, the CJs and the lamellar structure of the cristae were mostly absent with predominant circular structures. In addition, ATAD3 KO muscles had reduced mitochondrial size. These findings indicated that ATAD3 is required for the integrity of mitochondrial cristae in skeletal muscle.
Furthermore, the lack of muscular ATAD3 resulted in mtDNA replication stalling (indicated by the accumulation of replication intermediates), causing progressive mtDNA depletion and deletions in the KO muscle. Indeed, the levels of the myokine FGF21, a biomarker for mtDNA-related myopathies (Lehtonen et al., 2016), were increased in ATAD3 KO muscle compared to controls. This result suggested that mtDNA replication might be coupled with cristae organization.
Lipidomic studies showed that ATAD3 KO muscles had decreased levels of cholesterol esters (CEs) synthesized in the ER (generally containing short saturated or monounsaturated acyl chains) and increased levels of CEs obtained from the diet (generally containing longer and more unsaturated acyl chains). A 30% decrease in the ratio of total CEs versus free cholesterol (unesterified cholesterol) was detected in the KO muscles of 5-month-old mice when most of the mitochondria had disrupted cristae. These results indicated that cholesterol is internalized but does not reach the mitochondria, hinting at a cholesterol-trafficking defect. As cholesterol-rich membrane structures are important for tethering mtDNA nucleoids to the inner mitochondrial membranes (Gerhold et al., 2015), these results link ATAD3 to cholesterol-dependent cristae organization and mtDNA maintenance.
Interestingly, in the immunohistochemistry staining the muscle fibers of the KO tissues presented pale cores that were negative for both COX (cytochrome oxidase) and SDH (succinate dehydrogenase) activities, indicating reduced mitochondrial mass in focal areas. This suggests that ATAD3 has a key role in the preservation of the mitochondrial network in muscle. Oxidative phosphorylation (OXPHOS) activity within muscle fibers in the KO mice was not severely affected. Only the complexes known to be dependent on cristae structure, such as complex V, and supercomplexes containing complex I and III were reduced in the KO muscles. These results demonstrate that ATAD3 does not have a significant role in mitochondrial translation, as previously suggested (He et al., 2012) and that it is not crucial for OXPHOS assembly. ATAD3 KO skeletal muscles presented increased PGC-1α and SDHA levels, probably as a result of the induction of mitochondrial biogenesis as a compensatory mechanism.
Altogether, these results indicated a critical early role of ATAD3 in regulating IMM structure, leading to secondary defects in cholesterol homeostasis, mtDNA replication, and OXPHOS levels in muscle tissue.
ATAD3 neuron knockout
ATAD3-neuron deficient mice (ATAD3 neuron KO) were obtained by crossing the ATAD3 floxed mice with CaMKIIα transgenic mice, expressing Cre recombinase under the calcium/calmodulin-dependent protein kinase II (CaMKIIα) promoter. The CaMKIIα gene is expressed predominantly in the cortex and hippocampus neurons (Dragatsis and Zeitlin, 2000). The lack of ATAD3 in forebrain neurons resulted in a fatal progressive encephalopathy with an onset at 5 months old (Arguello et al., 2021). The ATAD3 neuron KO mice showed symptoms such as impaired motor coordination and disrupted stereotypical rodent behavior that worsened with time. Contrary to what was observed in the muscle model, ablation of ATAD3 in neurons resulted in premature death, indicating that ATAD3 function in neurons is essential for survival.
Coinciding with the muscle model, the first phenotype detected in the ATAD3 neuron KO model was a disrupted cristae morphology in the hippocampus region of pre-symptomatic KO mice of 2 months old. Quantification analysis of the TEM images demonstrated that ATAD3 KO neurons had reduced mitochondrial size and reduced cristae surface per mitochondria. The mtDNA levels were similar in ATAD3 neuron KO and control tissues of 3 months old (pre-symptomatic stage). However, 2 months later, mtDNA levels were decreased in ATAD3 neuron KO cortical and hippocampal areas. Neuronal cell death and decreased OXPHOS levels were detected only in 5-month-old tissues, concurring with the mtDNA depletion.
Metabolomics and lipidomic analysis performed in ATAD3 neuron KO mice at the pre-symptomatic stage revealed altered pathways related to the transport of lipids along mitochondria membranes, for example in the carnitine shuttle pathway, which transports long fatty acid chains through the IMM. Also, several precursors of the cardiolipin synthesis pathway, the main fatty acyl moiety in mitochondria, and phosphatidylcholine, one of the most abundant phospholipids in both mitochondrial membranes, were decreased.
Overall, these results indicated a role of ATAD3 in the preservation of the cristae morphology and mitochondrial lipid metabolism in neurons. This results over the months in mtDNA depletion and neuron cell death.
Discussion and concluding remarks
Animal models for ATAD3/Atad3 dysfunction have become indispensable for studying ATAD3 functions in vivo and effectively recapitulate many features observed in patients’ cells with ATAD3 variants. In the last years, several advanced techniques have been used to study these features.
High-resolution approaches like TEM have provided critical insights into mitochondrial cristae structure. Both animal models and patient cells show disrupted mitochondrial cristae morphology (Peralta et al., 2019; Dorison et al., 2020) and mitochondrial fragmentation (Cooper et al., 2017). This aligns with ATAD3A’s interaction with other proteins like PROHIBITIN and LETM1, that are essential for maintaining cristae morphology (Arguello et al., 2021; Antonicka et al., 2020). Also, in human embryonic kidney (HEK) cells, ATAD3A showed a remarkably regular distribution across the mitochondrial membrane, a typical characteristic of scaffolding proteins (Arguello et al., 2021).
Furthermore, fluorescence microscopy plays a crucial role in the analysis of cholesterol metabolism. In patient cells harboring ATAD3 gene cluster deletions, or duplication resulting in the formation of an ATAD3A/C fusion protein, elevated levels of unesterified cholesterol have been identified by filipin staining (Desai et al., 2017; Gunning et al., 2020). The animal models for ATAD3 dysfunction also reflect patient findings in terms of altered lipid metabolism. In 2024, a study using the Drosophila model introduced the novel application of the cholesterol reporter mKate-D4, which enabled the detection of membrane-bound cholesterol in vivo (Muñoz-Oreja et al., 2024). This novel approach facilitates to detect this difficult to study area of the cell metabolism, the cholesterol metabolism, which has been previously implicated in the disease. The researchers demonstrated that Atad3 dysfunction leads to a compensatory increase in cellular cholesterol, resulting in its abnormal aggregation in membranes that can cascade into lysosomal insufficiency, which may contribute to the pathomechanism of the disease.
Aberrant organization of mtDNA was observed in ATAD3 patients’ fibroblasts by immunocytochemistry staining (Desai et al., 2017; Gunning et al., 2020; Muñoz-Oreja et al., 2024), but has not been studied in animal models. However, in mouse models, mtDNA depletion occurs after cristae disorganization, suggesting that ATAD3 affects mtDNA maintenance in vivo by stabilizing the mitochondrial cristae morphology (Peralta et al., 2018; Arguello et al., 2021).
As previously described, ATAD3 LOF in skeletal muscle tissue of mice resulted in a milder phenotype than the neuron model. This result highlights the existence of different compensatory mechanisms to counteract ATAD3 dysfunction in different tissues. Indeed, Frazier et al. also indicated this tissue specificity, where complex I activity was more profoundly reduced in cardiac tissue than in skeletal muscle or fibroblasts from ATAD3-deficient patients (Frazier et al., 2021).
However, species-specific variations observed in the ATAD3A gene structure present a potential limitation in the direct application of findings from animal models to human disease. To illustrate, consider the case of the NAHR-mediated duplication syndrome, which is characterized by pontocerebellar hypoplasia, seizures, and respiratory insufficiency (Gunning et al., 2020; Frazier et al., 2021). On a molecular level, the duplications typically result in a stable chimeric ATAD3A/C protein harboring 29 missense changes, including the previously referenced p.Arg466Cys variant, which is postulated to be a significant contributor to pathogenicity. However, the p.Arg466Cys variant itself is associated with a milder phenotype including syndromic dominant optic atrophy with neurological involvement (Muñoz-Oreja et al., 2024). This suggests that the variant is only partially responsible for the phenotype observed in the duplication syndrome. Nevertheless, ubiquitous expression of the Arg466Cys variant in Drosophila proved to be lethal. Thus, the animal model, which lacks the ATAD3B and ATAD3C genes, is unable to fully elucidate the underlying pathomechanisms in this case.
Overall, the results obtained from the Drosophila and mouse models, along with other in vitro studies, have yielded valuable insights into the function of ATAD3A. In summary, it has been demonstrated that ATAD3A LOF or CNVs in the ATAD3 locus result in mitochondrial dysfunction, due to the protein’s role in the structural organization of mitochondrial membranes and its impact on essential processes such as mtDNA maintenance and cholesterol metabolism. Nevertheless, the precise function of ATAD3A and the cellular mechanisms underlying ATAD3-associated disorders remain unclear and require further research. The integration of genomic, transcriptomic, proteomic, lipidomic, and metabolomic data, coupled with the application of novel advanced technologies, is a promising avenue for advancing our understanding of pathomechanisms and may also facilitate the identification of potential therapeutic targets.
Author contributions
MB: Writing – original draft. A-SK: Writing – original draft. TH: Writing – review & editing. SP: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by University of Tübingen, Faculty of Medicine, “Doctoral College program” to A-SK and TH and SP.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Al Madhoun, A., Alnaser, F., Melhem, M., Nizam, R., Al-Dabbous, T., and Al-Mulla, F. (2019). Ketogenic diet attenuates cerebellar atrophy progression in a subject with a biallelic variant at the Atad3A locus. Appl. Clin. Genet. 12, 79–86. doi: 10.2147/TACG.S194204
Antonicka, H., Lin, Z. Y., Janer, A., Aaltonen, M. J., Weraarpachai, W., Gingras, A. C., et al. (2020). A high-density human mitochondrial proximity interaction network. Cell Metab. 32, 479–497.e9. doi: 10.1016/j.cmet.2020.07.017
Arguello, T., Peralta, S., Antonicka, H., Gaidosh, G., Diaz, F., Tu, Y.-T., et al. (2021). Atad3A has a scaffolding role regulating mitochondria inner membrane structure and protein assembly. Cell Rep. 37:110139. doi: 10.1016/j.celrep.2021.110139
Baudier, J. (2018). Atad3 proteins: brokers of a mitochondria-endoplasmic reticulum connection in mammalian cells. Biol. Rev. Camb. Philos. Soc. 93, 827–844. doi: 10.1111/brv.12373
Bothe, G. W., Haspel, J. A., Smith, C. L., Wiener, H. H., and Burden, S. J. (2000). Selective expression of Cre recombinase in skeletal muscle fibers. Genesis 26, 165–166. doi: 10.1002/(SICI)1526-968X(200002)26:2<165:AID-GENE22>3.0.CO;2-F
Chen, L., Li, Y., Zambidis, A., and Papadopoulos, V. (2023). Atad3A: a key regulator of mitochondria-associated diseases. Int. J. Mol. Sci. 24:12511. doi: 10.3390/ijms241512511
Cooper, H. M., Yang, Y., Ylikallio, E., Khairullin, R., Woldegebriel, R., Lin, K. L., et al. (2017). Atpase-deficient mitochondrial inner membrane protein Atad3A disturbs mitochondrial dynamics in dominant hereditary spastic paraplegia. Hum. Mol. Genet. 26, 1432–1443. doi: 10.1093/hmg/ddx042
Desai, R., Frazier, A. E., Durigon, R., Patel, H., Jones, A. W., Dalla Rosa, I., et al. (2017). Atad3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial Dna and cholesterol metabolism. Brain 140, 1595–1610. doi: 10.1093/brain/awx094
Dorison, N., Gaignard, P., Bayot, A., Gelot, A., Becker, P. H., Fourati, S., et al. (2020). Mitochondrial dysfunction caused by novel Atad3A mutations. Mol. Genet. Metab. 131, 107–113. doi: 10.1016/j.ymgme.2020.09.002
Dragatsis, I., and Zeitlin, S. (2000). Camkiialpha-Cre transgene expression and recombination patterns in the mouse brain. Genesis 26, 133–135. doi: 10.1002/(SICI)1526-968X(200002)26:2<133:AID-GENE10>3.0.CO;2-V
Ebihara, T., Nagatomo, T., Sugiyama, Y., Tsuruoka, T., Osone, Y., Shimura, M., et al. (2022). Severe spinal cord hypoplasia due to a novel Atad3A compound heterozygous deletion. Mol Genet Metab Rep 33:100912. doi: 10.1016/j.ymgmr.2022.100912
Fang, H. Y., Chang, C. L., Hsu, S. H., Huang, C. Y., Chiang, S. F., Chiou, S. H., et al. (2010). Atpase family Aaa domain-containing 3A is a novel anti-apoptotic factor in lung adenocarcinoma cells. J. Cell Sci. 123, 1171–1180. doi: 10.1242/jcs.062034
Frazier, A. E., Compton, A. G., Kishita, Y., Hock, D. H., Welch, A. E., Amarasekera, S. S. C., et al. (2021). Fatal perinatal mitochondrial cardiac failure caused by recurrent de novo duplications in the Atad3 locus. Med (N Y) 2, 49–73. doi: 10.1016/j.medj.2020.06.004
Frickey, T., and Lupas, A. N. (2004). Phylogenetic analysis of Aaa proteins. J. Struct. Biol. 146, 2–10. doi: 10.1016/j.jsb.2003.11.020
Gerhold, J. M., Cansiz-Arda, Ş., Lõhmus, M., Engberg, O., Reyes, A., Van Rennes, H., et al. (2015). Human mitochondrial Dna-protein complexes attach to a cholesterol-rich membrane structure. Sci. Rep. 5:15292. doi: 10.1038/srep15292
Gilquin, B., Taillebourg, E., Cherradi, N., Hubstenberger, A., Gay, O., Merle, N., et al. (2010). The Aaa+ Atpase Atad3A controls mitochondrial dynamics at the interface of the inner and outer membranes. Mol. Cell. Biol. 30, 1984–1996. doi: 10.1128/MCB.00007-10
Goller, T., Seibold, U. K., Kremmer, E., Voos, W., and Kolanus, W. (2013). Atad3 function is essential for early post-implantation development in the mouse. PLoS One 8:e54799. doi: 10.1371/journal.pone.0054799
Gunning, A. C., Strucinska, K., Muñoz Oreja, M., Parrish, A., Caswell, R., Stals, K. L., et al. (2020). Recurrent De novo Nahr reciprocal duplications in the Atad3 gene cluster cause a Neurogenetic trait with perturbed cholesterol and mitochondrial metabolism. Am. J. Hum. Genet. 106, 272–279. doi: 10.1016/j.ajhg.2020.01.007
Hanes, I., Mcmillan, H. J., Ito, Y., Kernohan, K. D., Lazier, J., Lines, M. A., et al. (2020). A splice variant in Atad3A expands the clinical and genetic spectrum of Harel-Yoon syndrome. Neurol Genet 6:e452. doi: 10.1212/NXG.0000000000000452
Harel, T., Yoon, W. H., Garone, C., Gu, S., Coban-Akdemir, Z., Eldomery, M. K., et al. (2016). Recurrent De novo and Biallelic variation of Atad3A, encoding a mitochondrial membrane protein, results in distinct neurological syndromes. Am. J. Hum. Genet. 99, 831–845. doi: 10.1016/j.ajhg.2016.08.007
He, J., Cooper, H. M., Reyes, A., Di Re, M., Sembongi, H., Litwin, T. R., et al. (2012). Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 40, 6109–6121. doi: 10.1093/nar/gks266
He, J., Mao, C. C., Reyes, A., Sembongi, H., Di Re, M., Granycome, C., et al. (2007). The Aaa+ protein Atad3 has displacement loop binding properties and is involved in mitochondrial nucleoid organization. J. Cell Biol. 176, 141–146. doi: 10.1083/jcb.200609158
Hoffmann, M., Bellance, N., Rossignol, R., Koopman, W. J., Willems, P. H., Mayatepek, E., et al. (2009). C. elegans Atad-3 is essential for mitochondrial activity and development. PLoS One 4:e7644. doi: 10.1371/journal.pone.0007644
Hubstenberger, A., Merle, N., Charton, R., Brandolin, G., and Rousseau, D. (2010). Topological analysis of Atad3A insertion in purified human mitochondria. J. Bioenerg. Biomembr. 42, 143–150. doi: 10.1007/s10863-010-9269-8
Issop, L., Fan, J., Lee, S., Rone, M. B., Basu, K., Mui, J., et al. (2015). Mitochondria-associated membrane formation in hormone-stimulated Leydig cell steroidogenesis: role of Atad3. Endocrinology 156, 334–345. doi: 10.1210/en.2014-1503
Jin, G., Xu, C., Zhang, X., Long, J., Rezaeian, A. H., Liu, C., et al. (2018). Atad3a suppresses Pink1-dependent mitophagy to maintain homeostasis of hematopoietic progenitor cells. Nat. Immunol. 19, 29–40. doi: 10.1038/s41590-017-0002-1
Lang, L., Loveless, R., and Teng, Y. (2020). Emerging links between control of mitochondrial protein Atad3A and Cancer. Int. J. Mol. Sci. 21:7917. doi: 10.3390/ijms21217917
Lehtonen, J. M., Forsström, S., Bottani, E., Viscomi, C., Baris, O. R., Isoniemi, H., et al. (2016). Fgf21 is a biomarker for mitochondrial translation and mtdna maintenance disorders. Neurology 87, 2290–2299. doi: 10.1212/WNL.0000000000003374
Li, S., Lamarche, F., Charton, R., Delphin, C., Gires, O., Hubstenberger, A., et al. (2014). Expression analysis of Atad3 isoforms in rodent and human cell lines and tissues. Gene 535, 60–69. doi: 10.1016/j.gene.2013.10.062
Li, S., and Rousseau, D. (2012). Atad3, a vital membrane bound mitochondrial Atpase involved in tumor progression. J. Bioenerg. Biomembr. 44, 189–197. doi: 10.1007/s10863-012-9424-5
Merle, N., Féraud, O., Gilquin, B., Hubstenberger, A., Kieffer-Jacquinot, S., Assard, N., et al. (2012). Atad3B is a human embryonic stem cell specific mitochondrial protein, re-expressed in cancer cells, that functions as dominant negative for the ubiquitous Atad3A. Mitochondrion 12, 441–448. doi: 10.1016/j.mito.2012.05.005
Muñoz-Oreja, M., Sandoval, A., Bruland, O., Perez-Rodriguez, D., Fernandez-Pelayo, U., De Arbina, A. L., et al. (2024). Elevated cholesterol in Atad3 mutants is a compensatory mechanism that leads to membrane cholesterol aggregation. Brain. 147, 1899–1913. doi: 10.1093/brain/awae018
Peeters-Scholte, C. M. P. C. D., Adama Van Scheltema, P. N., Klumper, F. J. C. M., Everwijn, S. M. P., Koopmans, M., Hoffer, M. J. V., et al. (2017). Genotype-phenotype correlation in Atad3A deletions: not just of scientific relevance. Brain 140:e66. doi: 10.1093/brain/awx239
Peralta, S., Goffart, S., Williams, S. L., Diaz, F., Garcia, S., Nissanka, N., et al. (2018). Atad3 controls mitochondrial cristae structure in mouse muscle, influencing mtdna replication and cholesterol levels. J. Cell Sci. 131:217075. doi: 10.1242/jcs.217075
Peralta, S., González-Quintana, A., Ybarra, M., Delmiro, A., Pérez-Pérez, R., Docampo, J., et al. (2019). Novel Atad3A recessive mutation associated to fatal cerebellar hypoplasia with multiorgan involvement and mitochondrial structural abnormalities. Mol. Genet. Metab. 128, 452–462. doi: 10.1016/j.ymgme.2019.10.012
Skopkova, M., Stufkova, H., Rambani, V., Stranecky, V., Brennerova, K., Kolnikova, M., et al. (2023). Atad3A-related pontocerebellar hypoplasia: new patients and insights into phenotypic variability. Orphanet J. Rare Dis. 18:92. doi: 10.1186/s13023-023-02689-3
Tawfik, C. A., Zaitoun, R., and Farag, A. A. (2023). Harel Yoon syndrome: a novel mutation in Atad3A gene and expansion of the clinical spectrum. Ophthalmic Genet. 44, 226–233. doi: 10.1080/13816810.2023.2183223
Van Den Ecker, D., Hoffmann, M., Müting, G., Maglioni, S., Herebian, D., Mayatepek, E., et al. (2015). Caenorhabditis elegans Atad-3 modulates mitochondrial iron and heme homeostasis. Biochem. Biophys. Res. Commun. 467, 389–394. doi: 10.1016/j.bbrc.2015.09.143
Yap, Z. Y., Park, Y. H., Wortmann, S. B., Gunning, A. C., Ezer, S., Lee, S., et al. (2021). Functional interpretation of Atad3A variants in neuro-mitochondrial phenotypes. Genome Med. 13:55. doi: 10.1186/s13073-021-00873-3
Keywords: ATAD3 , cholesterol, mtDNA depletion and deletion, neurodegeneration, animal model, mitochondrial disease
Citation: Brügel M, Kiesel A-S, Haack TB and Peralta S (2024) Mutations in mitochondrial ATAD3 gene and disease, lessons from in vivo models. Front. Neurosci. 18:1496142. doi: 10.3389/fnins.2024.1496142
Edited by:
Federica De Lazzari, University of Cambridge, United KingdomReviewed by:
Adam Gunning, Royal Devon & Exeter NHS Foundation Trust, United KingdomMikel Muñoz, Biodonostia Health Research Institute (IIS Biodonostia), Spain
Copyright © 2024 Brügel, Kiesel, Haack and Peralta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susana Peralta, c3VzYW5hLnBlcmFsdGEtbW9yYWxlc0BtZWQudW5pLXR1ZWJpbmdlbi5kZQ==
†These authors have contributed equally to this work