 Yuan Cao
Yuan Cao Lin-Wei Zhao4†
Lin-Wei Zhao4†- 1Department of Neurology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou University, Zhengzhou, China
- 2NHC Key Laboratory of Prevention and Treatment of Cerebrovascular Diseases, Zhengzhou, China
- 3Clinical Systems Biology Laboratories, Translation Medicine Center, The First Affiliated Hospital of Zhengzhou University, Zhengzhou University, Zhengzhou, China
- 4Department of Cardiology, People’s Hospital of Zhengzhou University, Henan Provincial People’s Hospital, Zhengzhou University Central China Fuwai Hospital, Zhengzhou, China
Alzheimer’s disease (AD) is increasingly recognized as being intertwined with the dysregulation of lipid metabolism. Lipids are a significant class of nutrients vital to all organisms, playing crucial roles in cellular structure, energy storage, and signaling. Alterations in the levels of various lipids in AD brains and dysregulation of lipid pathways and transportation have been implicated in AD pathogenesis. Clinically, evidence for a high-fat diet firmly links disrupted lipid metabolism to the pathogenesis and progression of AD, although contradictory findings warrant further exploration. In view of the significance of various lipids in brain physiology, the discovery of complex and diverse mechanisms that connect lipid metabolism with AD-related pathophysiology will bring new hope for patients with AD, underscoring the importance of lipid metabolism in AD pathophysiology, and promising targets for therapeutic intervention. Specifically, cholesterol, sphingolipids, and fatty acids have been shown to influence amyloid-beta (Aβ) accumulation and tau hyperphosphorylation, which are hallmarks of AD pathology. Recent studies have highlighted the potential therapeutic targets within lipid metabolism, such as enhancing apolipoprotein E lipidation, activating liver X receptors and retinoid X receptors, and modulating peroxisome proliferator-activated receptors. Ongoing clinical trials are investigating the efficacy of these strategies, including the use of ketogenic diets, statin therapy, and novel compounds like NE3107. The implications of these findings suggest that targeting lipid metabolism could offer new avenues for the treatment and management of AD. By concentrating on alterations in lipid metabolism within the central nervous system and their contribution to AD development, this review aims to shed light on novel research directions and treatment approaches for combating AD, offering hope for the development of more effective management strategies.
1 Introduction
Dementia is a worldwide emergency that affects countries with aging demographics and communities at large, with around 10 million new cases emerging globally each year (Prince et al., 2015; Anstey et al., 2017). Alzheimer’s disease (AD), the predominant type of dementia, is characterized by accumulation of amyloid plaques and neurofibrillary tangles (NFTs) in the brain. AD not only impairs the functionality of the elderly, but also imposes significant physical, psychological, and economic strain on families and society. Despite the severe and enduring consequences of AD, existing treatments fail to deliver effective relief or halt disease (Blanco-Silvente et al., 2018). Consequently, there is a pressing need for innovative AD treatments.
Exploring the mechanisms underlying the pathogenesis of AD could lead to the development of novel therapeutic approaches. Interestingly, the presence of ‘adipose inclusions’ or ‘lipoid granules’ in the brains of patients with AD has been noted as an additional neuropathological hallmark, alongside the well-known amyloid plaques and NFTs, first identified by Alois Alzheimer in 1907. Initially, this third marker was largely overlooked (Foley, 2010). However, subsequent research has consistently indicated that lipid droplets (LDs), the organelles responsible for storing neutral lipids, tend to accumulate in the brains of patients with AD (Nguyen et al., 2017; Gómez-Ramos and Asunción Morán, 2007).
Recent clinical studies have underscored the significance of lipid metabolism in AD development. Research has shown that high-fat diet (HFD) correlates with the onset of AD pathologies and cognitive deficits. This is through the activation of neuronal C/EBPβ/AEP signaling and the induction of insulin resistance, alongside inflammatory and stress responses, as observed in rodent models (Liu et al., 2022; Kothari et al., 2017). Additional research has shown that targeting pro-neuroinflammation and deficits in neurogenesis could mitigate the cognitive decline caused by HFD, suggesting that lipid metabolism is crucial for neuropathological changes in AD (Wang Q. et al., 2018). In contrast to these findings, studies on the ketogenic diet, a high-fat, low-carbohydrate regimen, highlight its neuroprotective benefits, including improved mitochondrial function and reduced inflammation and apoptosis (Rusek et al., 2019). The ability of this diet to compensate for impaired glucose metabolism by providing alternative energy sources such as ketones has sparked interest in its potential as an AD treatment (Broom et al., 2019). Furthermore, the glycation of apolipoprotein E (ApoE), which hinders the transport of essential lipids to the brain and leads to lipid shortage, may contribute to the progression of AD in its later stages. Collectively, this evidence firmly links disrupted lipid metabolism to the pathogenesis and progression of AD; however, contradictory findings warrant further exploration.
Lipids are essential nutrients for all living organisms, and play a critical role in brain physiology. The brain is the second most lipid-rich organ after the adipose tissue, comprising 10–12% of its fresh weight and over 50% of its dry weight (Naudí et al., 2015; Han, 2007). The pivotal role of various lipids in brain function has led to extensive research to uncover the intricate relationships between lipid metabolism and AD pathology, offering new avenues for treatment. Therapies targeting lipid metabolism have shown varying degrees of success against AD. This review provides clinical evidence for altered lipid metabolism in AD, explores the mechanisms linking lipid metabolism with AD etiology, and assesses the potential of targeting lipid metabolism in AD therapeutics.
2 Lipid metabolism in the central nervous system
Lipids, a crucial type of macrobiomolecule, are vital for providing energy for cellular metabolism, maintaining cellular and organelle structures, and acting as signaling molecules within the central nervous system (CNS). Throughout the first 20 years of life, the levels of cerebral lipids increase, only starting to diminish after reaching 50 years of age (Naudí et al., 2015). Brain lipids predominantly fall into one of the following groups: phospholipids, sphingolipids, fatty acids (FAs), and sterols, with phospholipids accounting for approximately 50% of the brain’s total lipid content (Figure 1; Sastry, 1985). Additionally, the transport and distribution of lipids are critical for the ability of the CNS to sustain balance and support neuronal function, processes that depend significantly on apolipoproteins. Thus, understanding lipid and apolipoprotein functions is the key to understanding the fundamentals of fat metabolism (Table 1).
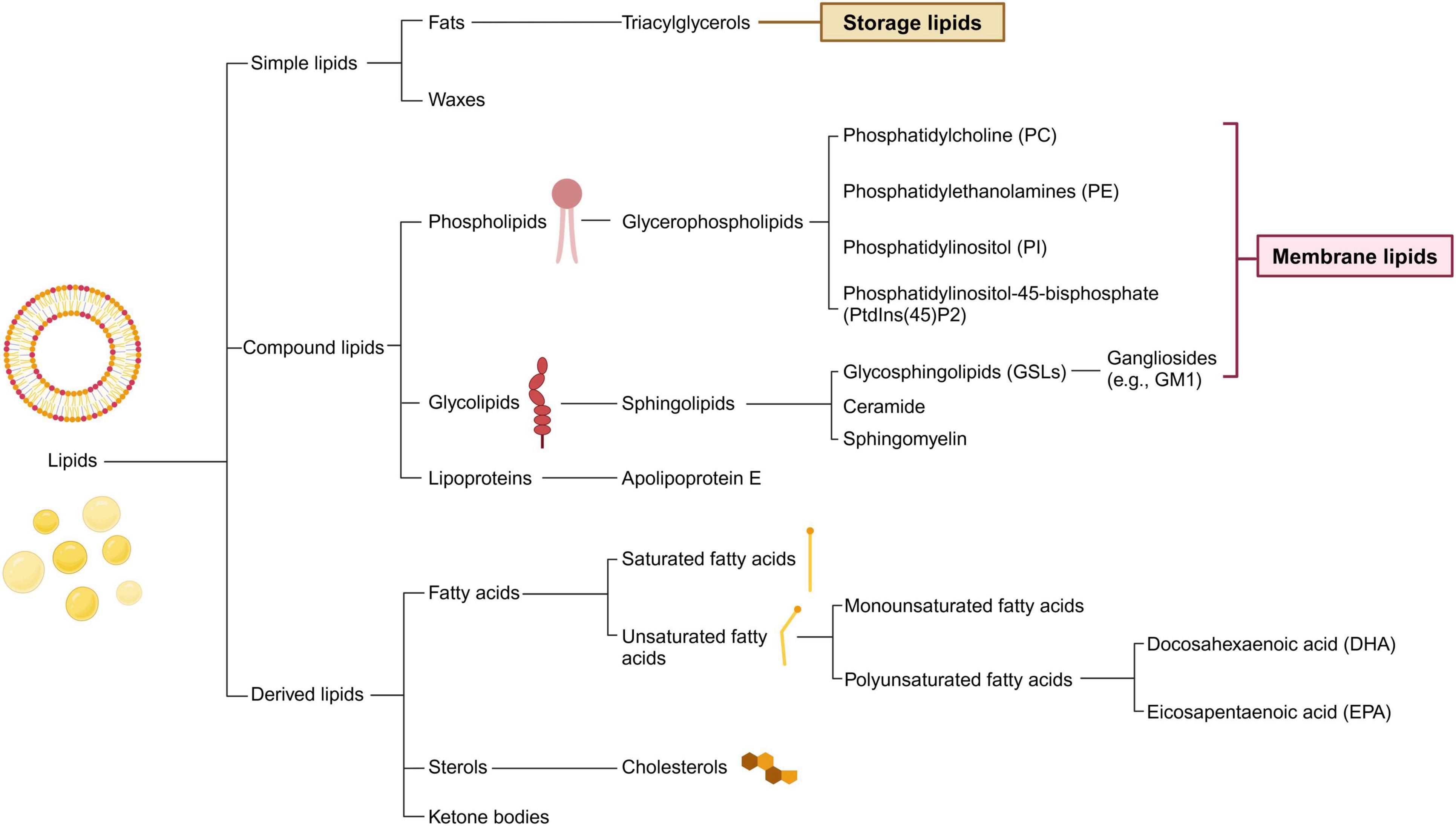
Figure 1. Classification of lipids and its main compounds in central nervous system. Lipids are categorized into three fundamental types: simple, compound, and derived. Within the central nervous system, the prevalent lipid species differ from those in other tissues. The flowchart illustrates common brain lipids, which are instrumental in the metabolic interchange between neurons and glial cells. Simple lipids mainly serve as storage molecules and encompass fats and waxes. Compound lipids, including phospholipids and glycolipids, are essential components of cell membranes and may contain additional functional groups. Derived lipids comprise entities such as fatty acids and sterols.
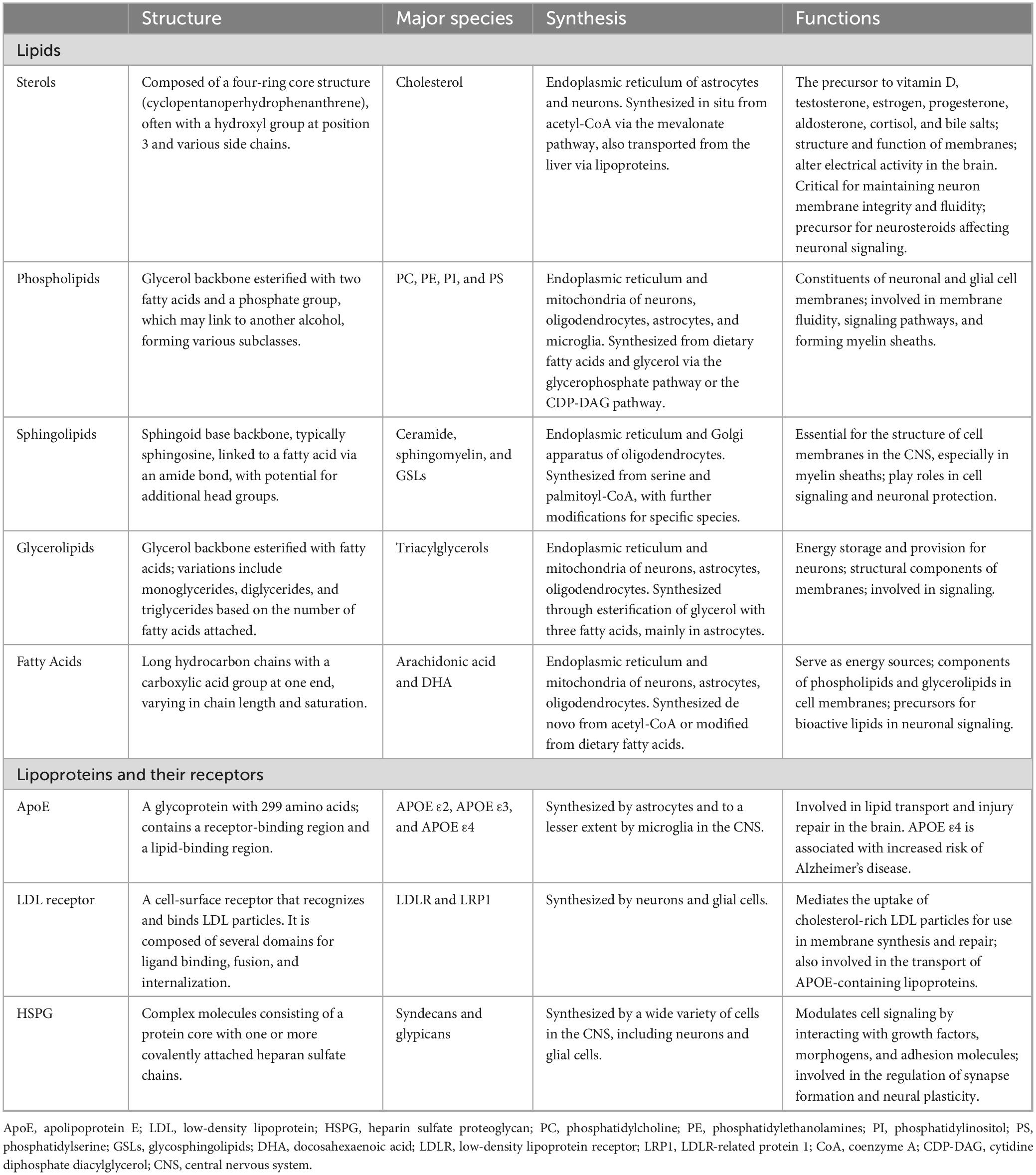
Table 1. Major lipid species and apolipoprotein in the central nervous system.
2.1 Lipids
2.1.1 Cholesterols
The brain, which is rich in cholesterol, contains approximately 25% of the body’s total cholesterol, which is primarily unesterified, in addition to minor amounts of desmosterol and cholesteryl ester, distinguishing it from other tissues (Dietschy and Turley, 2001; Dietschy and Turley, 2004). Cholesterol in the CNS is divided into two main pools: one found in the myelin sheaths, accounting for 70%–80% of the brain’s cholesterol, and the other in the neuronal plasma membranes, sourced from astrocytes via cholesterol-rich lipoproteins, including ApoE (Dietschy and Turley, 2004; Orth and Bellosta, 2012; Zhang and Liu, 2015). This distribution is crucial for axonal insulation, neuronal morphology, and synaptic connectivity. The brain’s cholesterol metabolism, which is vital for its function, operates independently from the rest of the body because of the blood-brain barrier (BBB), and disruptions in cholesterol homeostasis are linked to neurodegenerative diseases and cognitive decline in the elderly (Morell and Jurevics, 1996).
Cholesterol synthesis and regulation occur in the endoplasmic reticulum of astrocytes and neurons, and are managed by mechanisms contrastingly independent of and dependent on the Golgi apparatus (DeGrella and Simoni, 1982; Heino et al., 2000). This synthesis is regulated by sterol regulatory element-binding proteins and liver X receptors (LXRs), which adjust cholesterol levels through feedback loops that control its production and eflux, particularly through ATP-binding cassette (ABC) transporters (Vance et al., 1994; Barber and Raben, 2019; Moutinho and Landreth, 2017). In neurons, excess cholesterol is managed by efflux to ApoE-containing particles, maintaining cellular health and facilitating functions, such as synapse and dendrite formation, axonal guidance, and neurotransmission (Kim et al., 2008). Because cholesterol is water-insoluble, very little unbound cholesterol is detected in the cytosol, and most cholesterol exists in protein-bound forms, such as ApoE-containing cholesterol particles in the cytosol (Zhang and Liu, 2015). The role of cholesterol in neuronal physiology underscores its importance in both developmental and adult neurofunction, with its depletion leading to significant neural impairments (Linetti et al., 2010; Liu et al., 2010; Liu et al., 2007).
2.1.2 Phospholipid
The predominant phospholipids in the CNS are glycerophospholipids (GPs), including phosphatidylcholine (PC) and phosphatidylethanolamines (PE), which are crucial for the cell membrane structure (Figure 2), each comprising two fatty acyl tails and a phosphate polar head on a glycerol backbone.
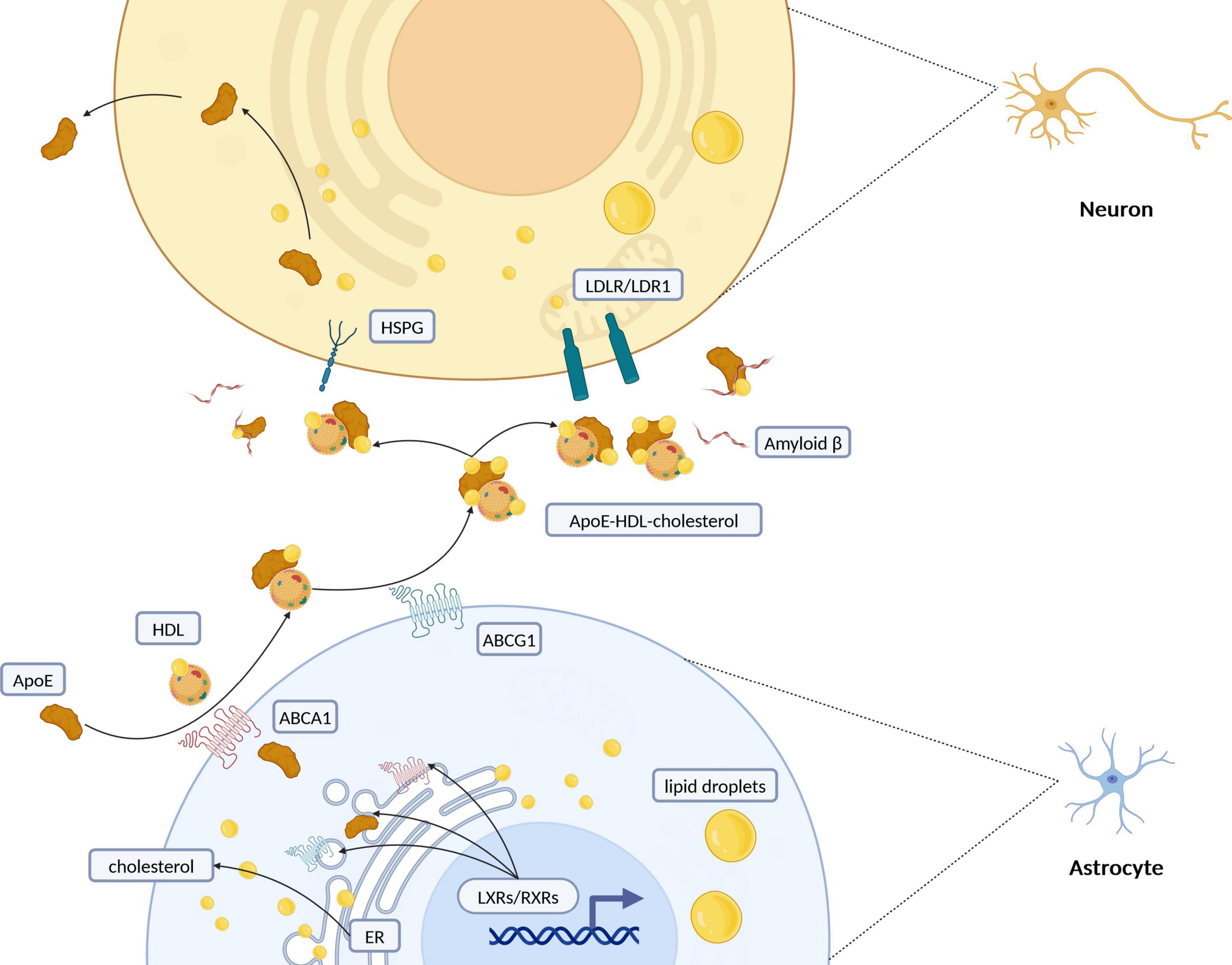
Figure 2. Schematic representation of intercellular cholesterol transportation between astrocytes and neurons. Cholesterol synthesis and regulation primarily take place in the endoplasmic reticulum (ER) of astrocytes. Once synthesized, cholesterol is packaged into lipid droplets and transported predominantly from glial cells to neurons. Its efflux is managed by the formation of ApoE-containing HDL-like particles, facilitated especially by ATP-binding cassette (ABC) transporters such as ABCA1 and ABCG1. Retinoid X receptors (RXRs) and liver X receptors (LXRs), part of the nuclear receptor superfamily, regulate cholesterol levels through feedback mechanisms that control its synthesis and efflux, particularly via ABC transporters. Cholesterol is redistributed to neurons through interactions with low-density lipoprotein receptors (LDLR/LDLR1) or heparan sulfate proteoglycans (HSPG). Additionally, the interaction of lipoprotein particles with amyloid beta (Aβ) peptides suggests a complex interplay between cholesterol metabolism and the pathogenesis of Alzheimer’s disease. ApoE, apolipoprotein E; HDL, high-density lipoprotein; ER, endoplasmic reticulum; RXRs, retinoid X receptors; LXRs, liver X Receptors; LDLR, low-density lipoprotein receptor; LRP1, LDLR-related protein 1; HSPG, heparin sulfate proteoglycan. Created with BioRender.com.
Studies on mild cognitive impairment (MCI) and AD subjects’ post-mortem brains have identified reduced glycerophospholipid levels in regions such as the hippocampus and cortex, which are areas susceptible to AD (Guan et al., 1999; Nitsch et al., 1992; Stokes and Hawthorne, 1987; Pettegrew et al., 2001). Research has revealed alterations in phospholipid composition and metabolism in AD, including changes in phospholipid-metabolizing enzymes (Stokes and Hawthorne, 1987; Garner, 2010). Phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2), which is vital for plasma membrane functions, has been linked to γ-secretase activity and amyloid beta (Aβ) 42 levels, inversely affecting AD progression (Landman et al., 2006). The hydrolysis of PtdIns(4,5)P2 by phospholipase C (PLC) increases intracellular Ca2+, implicating familial AD mutations and suggesting that PLC inhibition could reduce Aβ42 secretion and Ca2+ entry (Landman et al., 2006; Yoo et al., 2000). Furthermore, phospholipase D (PLD), particularly PLD1, plays a role in amyloidogenesis by affecting PC metabolism, influencing the cell surface delivery of amyloid precursor protein (APP) and presenilin-1 (PS1) and modulating PS1 activity, both of which are critical processes in AD development (Garner, 2010; Cai et al., 2006a; Cai et al., 2006b).
2.1.3 Sphingolipids
Sphingolipids, including ceramide and sphingomyelin, and glycosphingolipids (GSLs), such as gangliosides, are crucial for the composition of lipid rafts alongside cholesterol. Ceramides play a pivotal role in sphingolipid metabolism, acting as foundational elements for creating sphingomyelin and complex GSLs by attaching to phosphocholine or sugars. These components are not only abundant in the brain, but are also essential for the structure of neuronal membranes, with gangliosides being significant for their high sialic acid content.
Studies have identified significant changes in sphingolipid levels at the onset of AD, with further results showing that increased ceramide levels that may contribute to neuronal death through oxidative stress and decreased sphingomyelin levels, implicating these lipids in the metabolism of APP and suggesting a complex role in AD progression (He et al., 2010; Castro et al., 2009; Söderberg et al., 1992). Further investigation into gangliosides reveals their significant presence in membrane rafts, affecting Aβ aggregation (Yu et al., 2011; Ariga et al., 2008). The interaction between specific gangliosides, such as GM1 gangliosidosis (GM1), and Aβ alters Aβ’s structure, facilitating the formation of amyloid fibrils and plaques associated with AD. Moreover, the manipulation of ganglioside levels, particularly through genetic modifications, has been shown to affect Aβ deposition in both the brain parenchyma and vascular tissues, illustrating the nuanced role gangliosides play in AD pathology and their potential impact on immune responses and plaque maintenance (Ariga et al., 2008; Matsuzaki, 2020; Kracun et al., 1990; Kracun et al., 1991).
2.1.4 Fatty acids
Short- and medium-chain FAs are transported to the brain from peripheral sources, whereas the synthesis of long- and very-long-chain FAs occurs predominantly within the brain, starting with acetyl-CoA (Tracey et al., 2018). Moreover, FAs in the brain can also be produced through the hydrolysis of phospholipids, a process catalyzed by phospholipase A2, and possibly through the mechanism of lipophagy (Du et al., 2009; Singh and Cuervo, 2012). Astrocytes are the primary cell type involved in FAs degradation; however, microglia also contribute to this process, playing a role in determining the microglial phenotype through FA metabolism (Eraso-Pichot et al., 2018; Fecher et al., 2019; Edmond et al., 1987).
FAs are categorized as saturated, trans-, monounsaturated (MUFAs), or polyunsaturated (PUFAs), depending on their bond structures, with PUFAs being particularly significant. Despite the brain’s ability to produce saturated FAs, it relies on external sources of PUFAs acquired from peripheral blood through passive diffusion or ATP-dependent transporters (Dabravolski et al., 2021). FAs traverse the BBB either by dissociating from albumin carriers and entering endothelial cells without ATP or through transport proteins such as fatty acid transport proteins (FATP1-6), fatty acid translocase (FAT/CD36), intracellular fatty acid-binding proteins (FABP1-9), plasma membrane fatty acid binding protein (FABPpm), and caveolin-1 (Mitchell et al., 2011). These mechanisms are crucial for regulating FA metabolism in the brain. FAs are fundamental for synthesizing lipid classes and excluding cholesterol, and they play a pivotal role in forming phospholipids and sphingolipids. Notably, PUFAs, such as docosahexaenoic acid (DHA) and arachidonic acid, are vital for modulating synaptic plasticity and neurotransmission, underscoring their importance in the CNS (Bazinet and Layé, 2014; Liu et al., 2024).
2.2 Lipoproteins and its receptors
Mature neurons have a high demand for cholesterol, and while they can synthesize it under physiological conditions, additional ApoE-associated cholesterol is required (Pfrieger and Ungerer, 2011; Nieweg et al., 2009). ApoE, a 34 kDa glycoprotein consisting of 299 amino acids, is primarily synthesized by astrocytes within the CNS and plays a crucial role in lipid homeostasis (Mahley, 1988; Xu et al., 2006; Kang et al., 2018). With three common alleles—ε2, ε3, ε4—the isoforms of ApoE exhibit varied efficiencies in cholesterol and phospholipid binding/transporting due to structural differences. Notably, ApoE2 demonstrates superior cholesterol efflux capabilities compared to ApoE3 and ApoE4 (Minagawa et al., 2009; Wahrle et al., 2007; Hara et al., 2003).
In the brain, cholesterol transport predominantly occurs from glial cells to neurons, with ApoE acting as the main transporter by enabling cholesterol efflux from astrocytes and uptake by neurons (Figure 3A) (Boyles et al., 1989; Michikawa et al., 2000). Once ApoE has been secreted from the cells, the cell surface ABC family of transporters, ABCA1 (Nieweg et al., 2009) and ABCG1 (Karten et al., 2006), transfer cholesterol and phospholipids to nascent ApoE to form high-density lipoprotein (HDL)-like particles (Horiuchi et al., 2019). ApoE is critical in redistributing cholesterol and other lipids to neurons through binding to cell-surface ApoE receptors. The CNS’s lipid metabolism intricately involves the low-density lipoprotein receptor (LDLR) family, with numerous receptors like LDLR, LDLR-related protein 1 (LRP1), and others playing pivotal roles in the uptake of ApoE-containing lipoprotein particles (Herz, 2009; Pottier et al., 2012; Pitas et al., 1987). The distribution of these receptors—LRP1’s prevalence in neurons versus LDLR’s in glial cells—highlights the specialized functions and interactions within the brain’s lipid homeostasis framework (Rebeck et al., 1993). LRP1, known for its rapid endocytic rates, demonstrates significant transport capacity for ApoE, underscoring the dynamic interplay between ApoE isoforms, their lipidation profiles, and receptor affinities (Li et al., 2001; Fagan et al., 1996). Additionally, ApoE-enriched lipoproteins are captured by binding to heparin sulfate proteoglycan (HSPG), brought into the environment of the LRP, and then transferred to the LRP for internalization by the cells or form a complex that is internalized (Huang and Mahley, 2014). The efficacy of these processes is influenced by the lipidation extent of ApoE particles, with variations observed across different ApoE isoforms, significantly affecting lipid metabolism and receptor interaction capacities (Qi et al., 2021; Zhao et al., 2017; Lanfranco et al., 2020).
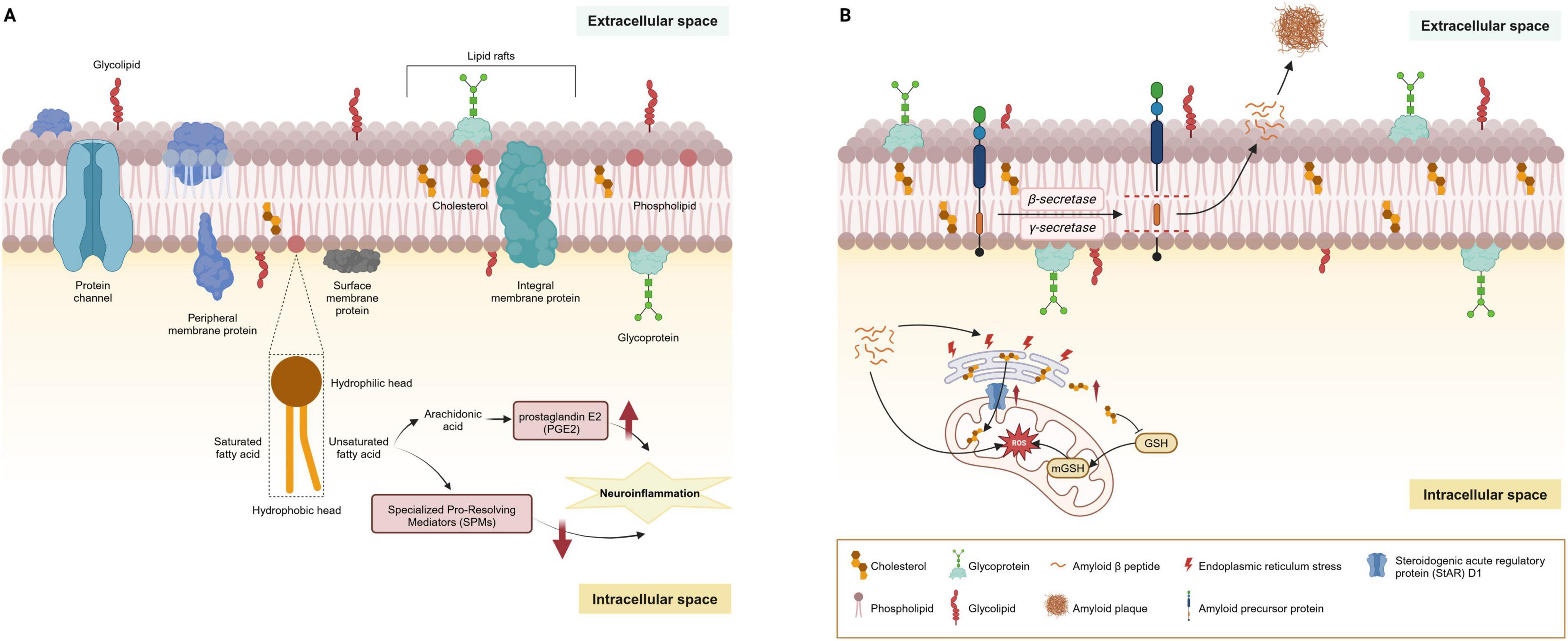
Figure 3. Main lipids of cell membranes and their dysregulation in AD. (A) Lipids are the primary constituents of cell membranes in the central nervous system. Under physiological conditions, the cell membrane is principally composed of phospholipids arranged in a bilayer. These phospholipids consist of a hydrophilic head and a hydrophobic tail; the tail may be comprised of saturated or unsaturated fatty acids, varying according to the membrane’s function and location. Additionally, cholesterol and sphingolipids are interspersed throughout the membrane, playing a significant role in the formation of lipid rafts. Glycoproteins and glycolipids are embedded in the membrane’s extracellular surface, contributing to cellular recognition and signaling. (B) Lipid rafts are crucial in the amyloidogenic processing of APP, where the enrichment of cholesterol and sphingolipids facilitates the anchoring of AD-related proteins such as β-site APP cleaving enzyme 1 (BACE1) and γ-secretase. The production of Aβ peptides begins with APP embedded in the cell membrane. This protein is sequentially cleaved by β-secretase and γ-secretase enzymes, releasing Aβ peptides into the extracellular space. Over time, these peptides can aggregate to form amyloid plaques. MAMs, rich in cholesterol and sphingomyelin, structurally mirror lipid rafts and are critical for lipid synthesis and trafficking. The steroidogenic acute regulatory protein (StAR) D1 is essential for mitochondrial cholesterol trafficking and is located at the MAM interface between the ER and mitochondria. ER stress induced by amyloid-β may contribute to the upregulation of StARD1. Accumulation of cholesterol in mitochondria disrupts the physical properties of the membrane and impairs the transport of GSH into mitochondria, undermining the mitochondrial antioxidant defense and leading to subsequent oxidative stress. AD, Alzheimer’s disease; AbPP, amyloid-β precursor protein; APP, amyloid precursor protein; MAMs, mitochondria-associated membranes; ER, endoplasmic reticulum.
3 Lipid metabolism changes in AD
Lipid metabolism undergoes significant changes across the lifespan, and these changes are critical in the context of AD. For instance, cholesterol levels in the brain are known to increase with age, which can exacerbate Aβ accumulation and plaque formation, key features of AD. Moreover, the reduction in the efficiency of lipid transport mechanisms with age can lead to lipid imbalances and contribute to neurodegeneration. Sex differences also play a pivotal role in lipid metabolism and AD risk. Women generally have higher HDL cholesterol levels and differences in fatty acid composition compared to men. These differences can influence the susceptibility to and progression of AD. Estrogen, which has neuroprotective properties and modulates lipid metabolism, declines significantly in postmenopausal women, potentially increasing their risk of developing AD. Our review focuses on the intricate relationships between lipid metabolism and AD pathology. However, we acknowledge the necessity to highlight how age and sex differences contribute to these processes.
3.1 Cholesterol metabolism
Cholesterol levels are notably altered. Previous studies have shown that low cholesteryl esters and free cholesterol levels are associated with increased amyloid production (Abad-Rodriguez et al., 2004). However, studies in cell cultures have shown that cholesterol deficiency can increase the risk of damage from excitotoxicity (Chou et al., 2003), affect neurosteroid production (Brinton, 2013), and alter cell membrane properties. Rodent studies have shown that HFD correlates with the onset of AD pathologies and cognitive deficits through the activation of neuronal C/EBPβ/AEP signaling (Liu et al., 2022). Despite these findings, studies have generally shown that cholesterol levels in the brain and blood of AD patients are higher than those in healthy individuals, correlating with disease severity (Cutler et al., 2004; Heverin et al., 2004; Popp et al., 2013; Liu et al., 2020). Additionally, this increase has been observed in the plaque cores of AD-affected brains (Mori et al., 2001).
3.1.1 Cholesterol and ApoE
Mounting evidence suggests that cholesterol levels regulate the amyloidogenic pathway (Di Paolo and Kim, 2011; Puglielli et al., 2001). Suppression of cholesterol synthesis in astrocytes substantially reduces amyloid and tau pathology in a mouse model of AD (Wang et al., 2021). Initial investigations into AD’s lipid-related mechanisms of AD have revealed disrupted cholesterol movement from astrocytes to neurons. ApoE serves as the primary cholesterol conduit, also playing a role in binding and removing Aβ peptides. Furthermore, the ApoE4 allele, a genetic variant of ApoE, is associated with AD risk, and is linked to changes in cholesterol and sphingolipids, highlighting its critical role in AD pathogenesis and lipid homeostasis (as explained in detail below) (Lane and Farlow, 2005; Bandaru et al., 2009).
3.1.2 Cholesterol and lipid rafts
Lipid rafts, characterized by their dynamic nature within cell membranes, are critical for signal transduction, cell adhesion, and sorting of lipids and proteins. These structures primarily consist of sphingolipids, cholesterol, and saturated FAs, with a lower content of PUFAs. These structures are pivotal in the amyloidogenic processing of AβPP, where the enrichment of cholesterol and sphingolipids facilitates the anchoring of AD-related proteins such as β-site APP cleaving enzyme 1 (BACE1) and γ-secretase (Figure 3B). Research has underscored the role of cholesterol in lipid rafts in bringing APP and BACE1 closer, promoting rapid endocytosis, with the activities of BACE1 and γ-secretase also being influenced by cholesterol levels, indicating the significant impact of cholesterol metabolism on AD development (Allen et al., 2007; Nickels et al., 2019; Fabelo et al., 2014).
Furthermore, alterations in the composition of lipid rafts, particularly the lipid makeup, are crucial for the progression of neurodegenerative disorders such as AD. Lipid rafts serve as essential platforms for signaling pathways, and their disruption can lead to abnormal protein distribution and aggregation, thus contributing to neurodegeneration. Key components of AD pathology, including β and γ-secretase, AβPP, and Aβ oligomers, are localized within lipid rafts, exacerbating toxicity and disrupting signaling. The interaction between Aβ and ApoE in the lipid rafts is also vital for neurofibrillary tangle formation, highlighting the importance of these membrane domains in AD pathogenesis, as well as the potential of targeting cholesterol synthesis in astrocytes as a therapeutic approach to mitigate amyloid and tau pathology (Wang et al., 2021; Yin et al., 2019; Kojro et al., 2001; Lee et al., 1998; Wahrle et al., 2002; Oshima et al., 2001; Kawarabayashi et al., 2004; Riddell et al., 2001).
3.1.3 Cholesterol and membrane fluidity
Cholesterol deficiency could alter cell membrane fluidity modifying their physicochemical properties. The composition of the acyl chains in membrane lipids dictates membrane fluidity, with straight and saturated acyl chains packed closely together. Integral membrane proteins such as AβPP, as well as β-and γ-secretases, rely heavily on the lipid levels within the membrane for their functionality. Cholesterol homeostasis has been highlighted as a key factor affecting amyloid production, with studies having shown its significant influence (Fabiani and Antollini, 2019; Hartmann et al., 1994). The process of AβPP cleavage by secretases is closely tied to the membrane’s physicochemical state, with increased fluidity enhancing α-secretase activity (thus preventing Aβ peptide formation) and decreased fluidity boosting γ-secretase activity, leading to amyloidogenic processing of AβPP, and an increase in longer Aβ forms (Zhang et al., 2012; Niu et al., 2018; Tan and Gleeson, 2019; Verdier et al., 2004). Additionally, cholesterol is involved in forming amyloid pores triggered by various Aβ peptides (Aβ 1–42, Aβ 22–35, Aβ 25–35) (Di Scala et al., 2013; Hashemi et al., 2022; Kandel et al., 2019).
3.1.4 Cholesterol and acetylcholine receptors
The interaction of Aβ with cell membranes disrupts the structure and fluidity of the lipid microenvironment, impacting the functionality of membrane receptors, notably the α7 nicotinic acetylcholine receptor (α7 nAChR) (Small et al., 1996). Cholesterol, the principal lipid, plays a crucial role in determining the function, stability, and supramolecular organization of nAChRs within the plasma membrane. Consequently, Aβ’s detrimental effects on membrane lipids lead to a dysfunction in nAChR activity, contributing to cognitive decline (Small et al., 1996). In the CNS, neuronal nAChR, particularly α7nAChR, is closely associated with lipid rafts, and is essential for their formation and maintenance (Brusés et al., 2001; Head et al., 2014). Evidence suggests that reducing cholesterol levels decrease the presence of α7AChRs on the cell surface (Peña et al., 2011; Borroni and Barrantes, 2011), while inhibiting cholesterol synthesis alters the desensitization kinetics of α7 nAChRs without affecting their current amplitude, thereby enhancing cholinergic function and, subsequently, cognition and memory in animal models (Hernandez et al., 2010).
3.1.5 Cholesterol metabolism and Aβ production
Cholesterol plays a crucial role in maintaining membrane integrity and fluidity, which are essential for the proper functioning of membrane-bound enzymes involved in Aβ production. Elevated cholesterol levels in neuronal membranes have been shown to increase the cleavage of APP by β-secretase and γ-secretase, leading to higher Aβ production (Di Paolo and Kim, 2011). Additionally, cholesterol-rich lipid rafts serve as platforms for the colocalization of APP and secretases, thereby facilitating the amyloidogenic pathway (Allen et al., 2007).
3.1.6 Cholesterol and tau pathology
Cholesterol accumulation in neurons can disrupt cellular signaling pathways, leading to tau hyperphosphorylation. Elevated cholesterol levels enhance the activity of kinases such as glycogen synthase kinase-3β (GSK-3β) and cyclin-dependent kinase 5 (CDK5), which are known to phosphorylate tau (Puglielli et al., 2001). Moreover, cholesterol-induced oxidative stress can activate stress kinases, further contributing to tau pathology (Huang et al., 2001).
3.2 Phospholipids metabolism
Phospholipids play a pivotal role in the activities of γ-secretase and APP processing proteins (Do Carmo et al., 2024). Investigations into AD patient-derived cell cultures have uncovered shifts in various phospholipid types, such as PC, PE, and phosphatidylinositol (PI), along with enzymes, such as PLC and PLD, responsible for phospholipid metabolism (Di Paolo and Kim, 2011). Studies in animal models have indicated that changes in phospholipid composition and metabolism are linked to the pathogenesis of AD, with alterations in key enzymes affecting amyloidogenesis (Garner, 2010; Cai et al., 2006b). GPs, crucial structural components of cellular membranes, influence membrane fluidity and have been found to be dysregulated in AD, showing a widespread reduction across PC, PE, and PI species (Bennett et al., 2013; Kosicek and Hecimovic, 2013; Wong et al., 2017). Moreover, changes in plasmalogen PE levels in AD brains could act as potential lipid biomarkers, with a significant decrease in white matter and an increase in gray matter developing as AD progresses (Han et al., 2001). PtdIns(4,5)P2, a key derivative of PI and a substrate for PLC, sees reduced turnover upon PLC inhibition, which subsequently lowers Aβ42 secretion (Landman et al., 2006). Conversely, inhibiting PLC activity adversely affects the activity of non-amyloidogenic γ-secretase (Rossner, 2004), casting doubt on PLC signaling’s role in mitigating amyloidogenesis in AD. Collectively, the correlation between phospholipid metabolism and APP processing is further underscored by the accelerated intracellular Aβ accumulation and memory issues stemming from a phospholipid transfer protein deficiency (Tong et al., 2015).
3.3 Sphingolipid metabolism
Research on cell cultures has shown that Aβ42 and Aβ40 can reduce sphingomyelin and cholesterol levels by activating neutral sphingomyelinases and inhibiting hydroxymethylglutaryl-CoA reductase activity (Grimm et al., 2005). In rodent models, increased ceramide levels contribute to neuronal death through oxidative stress, implicating sphingolipid metabolism in AD progression (He et al., 2010). Analyses of AD patient brains have shown elevated levels of acid sphingomyelinase and acid ceramidase, leading to decreased sphingomyelin levels and increased ceramide production, which in turn reduces γ-secretase activity and Aβ secretion (He et al., 2010; Grimm et al., 2005; Di Paolo and Kim, 2011). This interaction showcases the close relationship between Aβ generation and the metabolism of cholesterol and sphingolipids. Interestingly, Aβ42 and Aβ40 can further diminish sphingomyelin and cholesterol levels by activating neutral sphingomyelinases and inhibiting hydroxymethylglutaryl-CoA reductase activity, respectively, suggesting a reciprocal regulatory mechanism (Grimm et al., 2005). Moreover, fibrillar Aβ triggers sphingomyelinase activity and ceramide production through ROS-dependent pathways, potentially creating a feedback loop that amplifies ceramide accumulation and worsens AD pathology (Grimm et al., 2005; Jana and Pahan, 2004; Malaplate-Armand et al., 2006).
Mitochondria-associated membranes (MAMs), critical for lipid synthesis and trafficking, mirror lipid rafts structurally and function as a nexus for lipid and Aβ metabolism within neurons (Figure 3B; Sezgin et al., 2017; Vance, 2014; Giorgi et al., 2015). MAMs are rich in cholesterol and sphingomyelin, which are house-keeping proteins essential for phospholipid exchange, calcium signaling, and redox regulation. Studies have revealed an enrichment of β- and γ-secretases at MAMs, linking the elevated β-secretase cleaved APP product (C99) to diminished mitochondrial function, spurred by increased sphingomyelin hydrolysis and ceramide levels (Pera et al., 2017; Area-Gomez et al., 2009; Area-Gomez et al., 2018). Ceramides play a role in stabilizing BACE1, extending its half-life, and thus elevating the Aβ production rates (Puglielli et al., 2003). GSLs are also implicated in amyloid fibril formation, with the GM1 specifically binding to Aβ. These GM1-Aβ complexes, identified in early-stage AD patient brains, are linked to the presence of Aβ oligomers in the cerebrospinal fluid (CSF), underlining the significant impact of sphingolipids on APP processing, Aβ generation, and amyloid accumulation (Hong et al., 2014). This interplay between sphingolipids and Aβ suggests a critical role for these lipids in the molecular mechanisms underlying AD progression.
3.4 Fatty acids metabolism
The cortex of individuals with AD reveals increased FAs synthase protein levels and elevated free fatty acids (FFAs), which disrupt mitochondrial function and energy metabolism (Ates et al., 2020; Thangavel et al., 2017; Schönfeld and Reiser, 2017; Li L. O. et al., 2010). These compounds are neurotoxic and disrupt mitochondrial function and energy metabolism. Research has shown that the levels of FFAs and even-chain saturated FAs are higher in the CSF of patients with AD, and that these FFAs promote the assembly of amyloid and tau filaments in vitro. Conversely, levels of unsaturated FAs, including ω-3 PUFAs, DHA, and MUFAs, are lower in the AD brain and plasma; (Fonteh et al., 2014; Cunnane et al., 2012; Snowden et al., 2017; Belkouch et al., 2016; Fonteh et al., 2020). The lipid raft profiles in AD brains further reflect lipid imbalances, showcasing reduced ω-3 PUFAs and MUFAs, particularly oleic acid, and a lower unsaturation index of phospholipid acyl chains, indicating alterations in lipid homeostasis from the early stages of AD (Martín et al., 2010; Marin et al., 2016; Fabelo et al., 2014).
Imbalances in fatty acid metabolism, particularly an increase in saturated fatty acids (SFAs) and a decrease in PUFAs, can exacerbate Aβ toxicity. SFAs are known to increase the rigidity of cell membranes, promoting Aβ aggregation, while PUFAs, such as DHA, have been shown to inhibit Aβ fibrillogenesis and enhance its clearance (Bazinet and Layé, 2014).
The alteration of fatty acid composition in neuronal membranes can influence tau phosphorylation and aggregation. For instance, the depletion of DHA has been associated with increased tau hyperphosphorylation through the activation of GSK-3β and CDK5 (Rankin et al., 2007). Conversely, supplementation with omega-3 FAs has been shown to reduce tau pathology in animal models of AD (Morris et al., 2003).
Studies in vitro have highlighted that ApoE4 impedes FA sequestration in LDs, leading to increased FFA levels and neuroenergetic and synaptic disturbances (Qi et al., 2021). The degradation of FFAs, particularly those which are oxidized, is vital for brain health, with neurons depending on glial cells, mainly astrocytes, for the clearance and degradation of excessive FFAs in LDs through oxidative processes like β-oxidation within mitochondria and peroxisomes (Schönfeld and Reiser, 2017; Ioannou et al., 2019; Qi et al., 2021).
LDs serve as an initial defense against the lipotoxicity caused by FFAs or cholesterol, trapping them as neutral triacylglycerols (TAGs) and cholesterol esters (CEs) (Nguyen et al., 2017). The accumulation of LDs in AD corresponds to their role in the presence of peroxidised FAs and susceptible PUFAs (Zhang et al., 2021). Research in rodent models has indicated that LDs accumulation in ependymal cells preludes amyloid and tau pathology, thereby impacting neural stem cell proliferation (Hamilton et al., 2015). Furthermore, in vitro evidence has highlighted that ApoE4 impedes FA sequestration in LDs, leading to increased FFA levels, and neuroenergetic and synaptic disturbances (Qi et al., 2021). During oxidative stress, peroxidized FAs move from neurons to astrocytes through ApoE-mediated processes, and ApoE4 negatively affects this transfer (Ioannou et al., 2019; Liu L. et al., 2017). Studies have revealed that while ApoE3 promotes astrocytic clearance of neuronal LDs, ApoE4 does not, underscoring the varying influence of ApoE isoforms on lipid management. Additionally, AD risk genes, such as ABCA1, ABCA7, and PICALM, play roles in glial LDs formation (Moulton et al., 2021), suggesting that impaired lipid clearance and transport contribute to lipid imbalance and diminished neurotrophic support in AD and aging.
3.5 Lipid droplets and AD pathogenesis
LDs are dynamic organelles involved in the storage and metabolism of lipids and have been increasingly recognized for their roles in cellular homeostasis and AD processes. Recent studies have highlighted several functional roles of LDs in the context of AD.
LDs store excess FAs and cholesterol, preventing lipotoxicity and maintaining cellular lipid homeostasis. This detoxification role is crucial in the AD brain, where lipid metabolism is disrupted. Studies have shown that LDs trap harmful substances as neutral TAGs and CEs to mitigate damage (Nguyen et al., 2017).
LDs provide a reservoir of lipids that can be mobilized for energy production, particularly in conditions of metabolic stress. The dysregulation of this function in AD could contribute to the observed energy deficits in affected neurons. Evidence from rodent models indicates that LDs accumulation in ependymal cells precedes amyloid and tau pathology, impacting neural stem cell proliferation and highlighting the potential link between LDs and neurogenesis (Hamilton et al., 2015).
LDs are involved in the cellular response to oxidative stress by sequestering peroxidized lipids, thus protecting cells from oxidative damage. In AD, impaired LD function due to ApoE4 can exacerbate oxidative stress and neuronal injury. Clinical studies have highlighted that LDs in neurons and glial cells are associated with the presence of peroxidized FAs and susceptible PUFAs (Zhang et al., 2021).
LDs interact with immune cells in the brain, influencing the inflammatory response. Changes in LD dynamics can affect neuroinflammation, a key feature of AD pathology. Research has shown that LDs play a role in the presence of peroxidised FAs and susceptible PUFAs, and ApoE-mediated processes are crucial for the transfer of these FAs. However, the ApoE4 isoform negatively affects this transfer, leading to inefficient lipid management and increased neuronal damage (Ioannou et al., 2019).
3.6 Link between lipids and mitochondria in AD
The link between lipid metabolism and mitochondrial function is crucial in understanding the pathogenesis of AD. Mitochondria are vital for cellular energy production and lipid metabolism, and their dysfunction is a hallmark of AD. Several studies have demonstrated how lipid imbalances can affect mitochondrial function, contributing to neurodegeneration in AD. MAMs are specialized subdomains of the endoplasmic reticulum (ER) that physically and functionally interact with mitochondria. MAMs are critical for lipid synthesis and trafficking, calcium signaling, and apoptosis regulation. In AD, alterations in MAMs contribute to disrupted lipid metabolism and mitochondrial dysfunction. Studies have shown that β- and γ-secretases, involved in APP processing enriched in MAMs, link lipid metabolism directly to amyloidogenic pathways (Area-Gomez et al., 2018).
Elevated levels of certain lipids, such as ceramides and cholesterol, have been shown to impair mitochondrial function. Ceramides, in particular, can induce mitochondrial apoptosis pathways, leading to neuronal death. Increased ceramide levels have been observed in AD brains, correlating with mitochondrial dysfunction and neurodegeneration (He et al., 2010).
FAs are metabolized in mitochondria through β-oxidation, producing energy. However, imbalances in FA metabolism can lead to mitochondrial stress and dysfunction. In AD, dysregulated FA metabolism results in the accumulation of toxic lipid intermediates, which impair mitochondrial respiration and increase oxidative stress. Studies have found that AD patients exhibit altered levels of free fatty acids (FFAs) and their metabolic by-products, which are neurotoxic and disrupt mitochondrial function (Ates et al., 2020).
Mitochondrial dysfunction in AD is often accompanied by increased oxidative stress and lipid peroxidation. Oxidative damage to lipids within mitochondrial membranes can further impair mitochondrial function, creating a vicious cycle of mitochondrial dysfunction and lipid peroxidation. This oxidative stress is exacerbated in AD, leading to progressive neuronal damage (Song et al., 2021).
3.7 Lipids and vascular contributions to dementia
Atherosclerosis, characterized by the buildup of cholesterol-rich plaques in the arterial walls, is a major vascular condition linked to lipid metabolism (Mourino-Alvarez et al., 2024). It leads to reduced blood flow and increased risk of cerebrovascular events such as strokes, which are significant risk factors for cognitive decline and dementia. Studies have shown that high LDL cholesterol and low HDL cholesterol levels are associated with increased risk of atherosclerosis and subsequent cognitive impairment (Poliakova and Wellington, 2023). However, controversial results from older adults showed that very high HDL-C levels were associated with an increased risk of cognitive impairment, indicating that the very high levels of HDL-C may change its protective role in maintaining brain health (Huang et al., 2024).
Cerebral Small Vessel Disease (CSVD) encompasses a range of pathological processes affecting small blood vessels in the brain, including arteriolosclerosis, microinfarcts, and microbleeds. Dyslipidemia, particularly elevated triglycerides and low HDL cholesterol, has been implicated in the development of CSVD. CSVD is a common cause of vascular cognitive impairment and can exacerbate AD pathology by promoting amyloid deposition and tau pathology (Hong et al., 2024).
3.8 BBB dysfunction
The BBB plays an essential role in separating the brain from and connecting the brain to peripheral circulation by forming an interface for the transcytotic exchange of substances, including lipids (Segarra et al., 2021). BBB dysfunction manifests in the early stages of AD, irrespective of changes in Aβ or tau biomarkers (Montagne et al., 2017; Nation et al., 2019). In another study focusing on the effect of the peripheral lipid profile, obesity-associated high circulating saturated FAs led to elevated CSF levels of palmitate and increased BBB permeability, and high brain levels of palmitate further elicited neuroinflammation and impaired synaptic and cognitive function (Rhea et al., 2017; Ouyang et al., 2014; Melo et al., 2020). Mfsd2a, a phospholipid flippase, and transporters expressed by BBB endothelial cells suppress transcytosis and maintain BBB integrity (Ben-Zvi et al., 2014). It has further been discovered that, as a major DHA transporter across the BBB, Mfsd2a increases DHA-containing phospholipids in CNS endothelial cells, which inhibits caveolae-mediated transcytosis and helps to maintain BBB integrity (Andreone et al., 2017).
3.9 ApoE4
3.9.1 ApoE4 impairs Aβ accumulation and clearance
The role of ApoE role in AD pathology is multifaceted, with evidence showing its co-deposition with Aβ in amyloid plaques (Namba et al., 1991). Post-mortem studies have further identified an increase in Aβ plaque deposition and an earlier onset of amyloid pathology in ApoE4 carriers (Kok et al., 2009; Schmechel et al., 1993), with apoE4 specifically accelerating early amyloid seeding due to its higher affinity for Aβ compared to other isoforms. Research involving AD mouse models and human-induced pluripotent stem cells has demonstrated that the apoE4 isoform not only increases Aβ load in the brain interstitial fluid, but also shows distinctive cellular behaviors, with neurons exhibiting increased synapse numbers and Aβ42 secretion, while astrocytes display reduced Aβ uptake and increased cholesterol accumulation, highlighting the isoform’s unique impact on amyloid pathology (Liu C. C. et al., 2017; Huynh et al., 2017; Lin et al., 2018).
Furthermore, while ApoE plays a critical role in Aβ clearance through mechanisms like receptor-mediated clearance and proteolytic degradation, apoE4’s effectiveness in these processes is lower than that of apoE2 and apoE3. The inefficiency of apoE4 in lipid transport extends to compromised proteolytic degradation of Aβ, leading to slower Aβ clearance. This is evidenced by in vivo studies showing differential clearance rates of ApoE-Aβ complexes by the very low density lipoproteins receptor and LRP1 on the BBB, with apoE4 complexes clearing more slowly. Additionally, ApoE4 microglia-like cells show altered morphologies and reduced Aβ phagocytosis. The interaction between ApoE and Aβ, particularly the influence of ApoE lipidation on this process, sheds light on how disruptions in lipid homeostasis may contribute to AD progression, with delipidated apoE4 particularly prone to accelerating fibril formation (Lin et al., 2018; Wildsmith et al., 2013; Sanan et al., 1994; Pillot et al., 1999).
3.9.2 ApoE4 stimulates tau phosphorylation
Research has shown that fragments of apoE4 lacking parts of their carboxyl terminus can penetrate the cytosol, leading to neurotoxic effects (Pillot et al., 1999; Harris et al., 2003; Huang et al., 2001). These fragments specifically target components of the cytoskeleton such as tau and neurofilaments, thus altering their functions. In transgenic mice, these truncated ApoE4 fragments have been observed to promote tau phosphorylation and the formation of intracellular NFTs, similar to those observed in AD (Brecht et al., 2004; Harris et al., 2003). Remarkably, the removal of tau from mice offers protection against neurotoxic effects induced by these ApoE4 fragments (Andrews-Zwilling et al., 2010). Furthermore, studies in humans have identified that ApoE4 can independently increase the levels of phosphorylated tau in the CSF without the involvement of Aβ (Cruchaga et al., 2013).
3.9.3 ApoE4 fragments impair mitochondrial function
Mitochondrial dysfunction in AD is influenced by the ApoE genotype, with the effects being notably more pronounced in ApoE4 carriers than in ApoE3 (Ghosh et al., 1999; Hirai et al., 2001; Kamino et al., 2000; Gibson et al., 2000). ApoE4’s association with reduced cerebral glucose metabolism has been observed in both AD patients and non-demented individuals of the same age, a phenomenon that precedes cognitive symptoms by decades and likely occurs before extensive Aβ accumulation (Hirono et al., 2002; Mosconi et al., 2004; Reiman et al., 2004; Reiman et al., 2005; Small, 2001; Small et al., 2004; Scarmeas et al., 2005). This suggests that apoE4-related mitochondrial dysfunction may be an early event in the pathogenesis of AD. Additionally, apoE4 fragments specifically interact with neuronal mitochondria, exacerbating mitochondrial dysfunction, and contributing to neurotoxicity (Chang et al., 2005). The detrimental impact of ApoE on mitochondrial dynamics is isoform-dependent, with cultured neuronal cells showing significantly reduced mitochondrial mobility in the presence of apoE4 fragments, followed by apoE4, and to a lesser extent, apoE3 (Brodbeck et al., 2011). Furthermore, in transgenic mice with neuron-specific expression of ApoE4, the axonal transport of mitochondria is impaired, highlighting the detrimental role of apoE4 in neuronal health and function (Tesseur et al., 2000).
3.10 Brain-derived extracellular vesicles (BDEVs)
Extracellular vesicles (EVs) are released by all the cells, and contain a subset of their parental cells, including lipids. One prior in-depth lipidomic analysis revealed that the lipid composition of brain-derived EVs (BDEVs) in the human frontal cortex is altered in AD (Su et al., 2021). AD BDEVs had significantly altered glycerophospholipid and sphingolipid levels, specifically increased PS lipids, decreased polyunsaturated fatty acyl-containing lipids, and altered amide-linked acyl chain content in sphingomyelin and ceramide lipids relative to the controls. PS lipids constitute a substantial proportion of the EV membrane and have been proposed to play a role in facilitating EVs uptake by recipient cells (Kastelowitz and Yin, 2014; Laulagnier et al., 2004; Matsumura et al., 2019; Record et al., 2014; Record et al., 2018; Sharma et al., 2017; Wei et al., 2016). The most prominent alteration was a two-fold decrease in lipid species containing anti-inflammatory/pro-resolving DHA.
Notably, BDEVs can be considered as ideal lipid peripheral biomarkers because they can readily cross the BBB (Kanninen et al., 2016; Skoumalová et al., 2011). Future studies should assess PE lipids and polyunsaturated fatty acyl-containing lipids in peripherally sourced BDEVs, alongside other clinical measures (CSF and neuroimaging assessments), to determine whether BDEVs in the periphery can predict the progression from mild MCI to AD.
3.11 Genetic risk factors involved in lipid metabolism
Among the key risk genes for sporadic late-onset AD, APOE, TREM2, APOJ, PICALM, SREBP-2, ABCA1, and ABCA7 are involved in lipid trafficking and metabolism (Table 2; von Maydell et al., 2024). Similarly, genes linked to familial AD, including APP, PSEN1, and PSEN2, influence lipid metabolism (Table 2; Karch and Goate, 2015). The APOE4 allele is identified as the most potent genetic risk factor for AD, enhancing the disease’s likelihood by 4 to 14 times depending on the presence of one or two alleles, respectively, when compared to the more common ε3 isoform. This risk alteration is attributed to a combination of the gain of toxic effects and the loss of protective functions seen with APOE4, whereas the ε2 isoform (APOE2) is associated with a reduced risk (Liu et al., 2013; Corder et al., 1994; Reiman et al., 2020). Additionally, TREM2, primarily located on microglial surfaces, plays a crucial role in phagocytosis and the inflammatory response, with loss-of-function mutations resulting in early onset dementia, including Nasu-Hakola disease (Jay et al., 2017; Ulland and Colonna, 2018; Kleinberger et al., 2014; Paloneva et al., 2002). Recent studies have underscored TREM2’s involvement in myelin debris clearance and remyelination, highlighting how TREM2 deficiency leads to disrupted lipid metabolism and transport (Gouna et al., 2021; Nugent et al., 2020; Li et al., 2022). APOJ (Clusterin) and PICALM have also been recognized for their roles in AD risk, with APOJ implicated in Aβ aggregation and clearance and PICALM in lipid particle internalization and transport (Harold et al., 2009; Lambert et al., 2009; Zhao et al., 2015). SREBP-2, regulating cholesterol synthesis and has been linked to AD progression (Chan et al., 2018). These findings highlight the complex interactions between lipid-related genes and AD pathology, and further emphasize the significance of lipid metabolism and trafficking in disease development and progression (Wang Y. et al., 2015; Yeh et al., 2016; Kober and Brett, 2017; Nuutinen et al., 2009). ABCA1 and ABCA7 belong to the ABC transporter family, showing 54% sequence similarity (Aikawa et al., 2018). ABCA1 plays a crucial role in lipid efflux, specifically of cholesterol and phospholipids, by attaching them to lipid-free lipoproteins, notably ApoE, which serves as the main lipidation substrate in the brain (Nordestgaard et al., 2015; Wahrle et al., 2004). Mutations leading to ABCA1 dysfunction are linked to reduced plasma ApoE levels and increased risk of AD (Nordestgaard et al., 2015). Similarly, ABCA7 participates in the transport of cholesterol and phospholipids (Abe-Dohmae et al., 2004). Human genetic and epigenetic studies have identified various SNPs, gene variants, alternative splicing events, and methylation events in the ABCA7 gene, all of which are associated with functional impairment. These alterations in ABCA7 have been implicated in disrupted lipid and Aβ metabolism, ultimately contributing to an increased likelihood of AD development (Aikawa et al., 2018; De Roeck et al., 2019). This study identified differentially expressed lipid metabolism-related genes (DELMRGs) in AD, emphasizing the role of these genes in disease progression and potential therapeutic targets (Zeng et al., 2023).
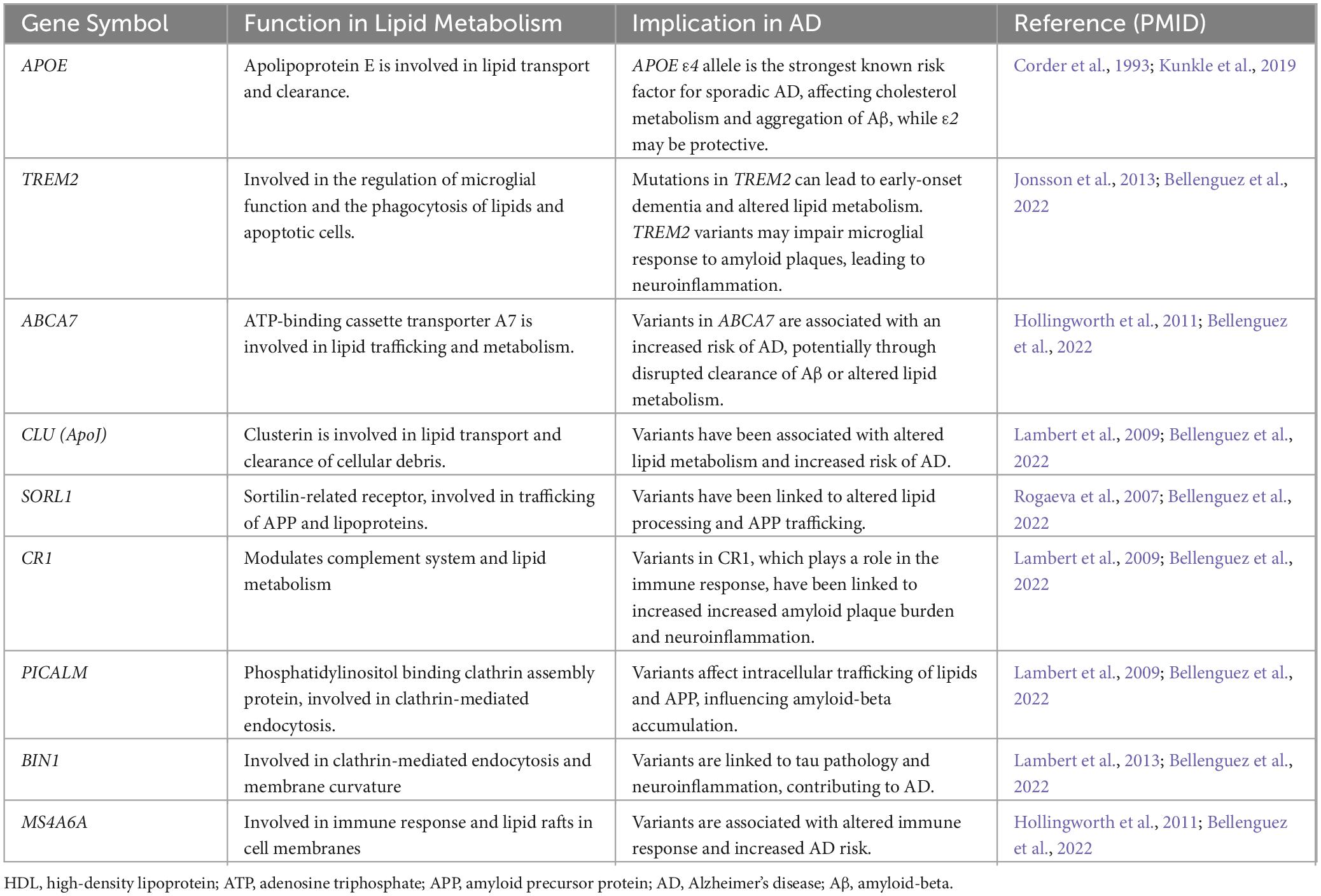
Table 2. Confirmed Alzheimer’s disease risk gene from large cohort genetic studies.
3.12 Immunomodulatory properties of lipid metabolism
Lipid metabolism directly influences neuroinflammation through the production of specialized pro-resolution mediators (SPMs) such as resolvins, lipoxins, protectins, and maresins, derived from PUFAs (ω-3 PUFAs, like DHA and EPA) (Basil and Levy, 2016). SPMs play crucial roles in dampening inflammation and promoting homeostasis (Emre et al., 2020; Zhu et al., 2016). In AD, neuroinflammation is a critical aspect of the pathogenesis and progression, with decreased levels of multiple SPMs observed in human AD brains, underlying the challenge of overcoming persistent inflammation. Notably, there was a positive relationship between CSF levels of SPMs (LXA4 and RvD1) and cognitive performance in both healthy and AD subjects. Experimental treatments involving SPM supplementation have been shown to counteract declines in hippocampal SPM levels, diminish inflammatory cytokines, reduce glial activation, and mitigate Aβ and tau pathology in AD mouse models (Weiner et al., 2015; Kantarci et al., 2018; Dunn et al., 2015).
Furthermore, the interplay between lipid metabolism and neuroinflammation extends to the expression of anti-inflammatory genes triggered by lipid-sensing nuclear receptors such as LXRs and PPARs (Kang and Rivest, 2012; Heneka et al., 2011). Pro-inflammatory prostaglandins, especially prostaglandin E2 (PGE2), synthesized from arachidonic acid by cyclooxygenase enzymes (COX-1 and COX-2) and prostaglandin E synthases, are found at increased concentrations in the early stages of AD, and their presence has been linked to intensified neuroinflammation, oxidative stress, and memory deficits in rodent models (Ricciotti and FitzGerald, 2011). Targeting PGE2 receptors can alleviate these adverse effects via microglial-driven mechanisms. In addition, PGE2 has been shown to impair microglial function and mitochondrial energy production via the EP2 receptor (Montine et al., 1999; Combrinck et al., 2006; Shi et al., 2012; Liang et al., 2005; Johansson et al., 2015; Minhas et al., 2021). Finally, the role of lipid rafts in glial cells, which are rich in cholesterol and sphingolipids, suggests a pivotal role in initiating inflammatory responses, where elevated cholesterol levels contribute to the aggregation of inflammatory proteins and increase inflammation (Miller et al., 2020).
4 Treatment of AD by targeting lipid metabolism
4.1 ApoE lipidation
For ApoE to carry out its essential roles, including lipid/cholesterol transport, synapse regeneration, immune modulation, and Aβ clearance/degradation, it must be secreted and adequately lipidated (Kanekiyo et al., 2014; Hanson et al., 2013). However, ApoE4 exhibits poor lipidation, and is less abundantly expressed than ApoE2 and ApoE3 (Kanekiyo et al., 2014; Wang L. et al., 2015; Heinsinger et al., 2016). This shortfall in ApoE4 lipidation indicates that enhancing ApoE lipidation may be a promising therapeutic strategy for treating AD and other neurological conditions (Table 3).
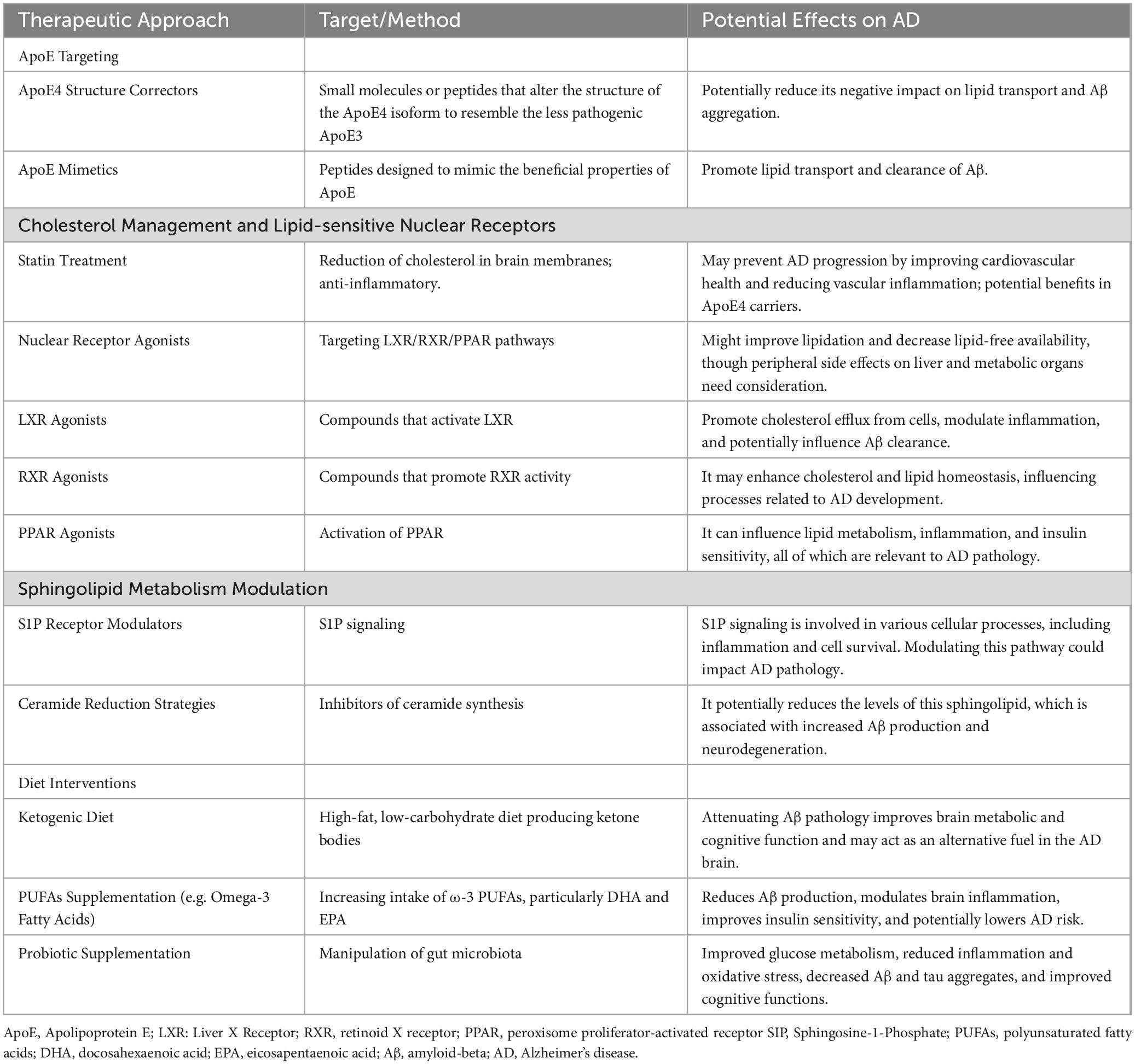
Table 3. Therapies of Alzheimer’s disease by targeting lipid metabolism.
4.1.1 Small molecules that enhance ABCA1-mediated ApoE4 lipidation
Targeting ApoE lipidation has emerged as a potential strategy for mitigating AD symptoms. The ABCA1 transporter plays a crucial role in this process by transferring cholesterol to apolipoproteins, including apoE, thus regulating lipidation in the brain (Koldamova et al., 2014). Enhancing ABCA1 transporter activity can be achieved through micro-RNA manipulation, notably by inhibiting miR-33, a brain-pervasive microRNA that influences Aβ levels via ABCA1. Inhibition of miR-33 in the brain leads to increased ABCA1 and ApoE lipidation and facilitates the degradation of extracellular Aβ, simultaneously decreasing Aβ secretion in the cortex of APP/PS1 mice (Kim et al., 2015). Furthermore, the activation of ABCA1 can also be accomplished through the application of therapeutic peptides, such as CS-6253, which is derived from the C-terminal domain of ApoE. This peptide can oligomerize ABCA1, boost its activity, reverse cognitive deficits linked to ApoE4, and reduce both tau hyperphosphorylation and Aβ buildup in hippocampal neurons (Boehm-Cagan et al., 2016; Handattu et al., 2013). Another promising peptide, Ac-hE18A-NH2, counteracts the adverse effects of Aβ on ApoE secretion and, when administered to mice, enhances cognition, reduces amyloid plaques, and diminishes glial activation while increasing CNS levels of ApoE (Handattu et al., 2013). Peptide 4F, which mimics HDL function, significantly boosts ApoE secretion and lipidation in glial cells without causing cytotoxicity (Chernick et al., 2018). While these interventions primarily focus on Aβ pathology, their impact on lipid metabolism and trafficking warrants further exploration.
4.1.2 ApoE4 structure correctors
Efforts to counteract the ApoE4-associated neuropathology have led to the development of structural correctors that transform ApoE4 into a more ApoE3-like conformation, enhancing lipidation and improving phospholipid and cholesterol transport (Mahley, 2016). The compound PH002 has been identified to mitigate ApoE4 toxicity in human iPSC-derived neurons, evidenced by a reduction in toxic ApoE4 fragments, an increase in GABAergic neurons and GAD67 levels, alongside a decrease in hyperphosphorylated tau and Aβ production (Wang C. et al., 2018). Additionally, compounds such as GIND-25 and GIND105 have been designed to adjust ApoE4 towards ApoE3-like lipid-binding characteristics, positively affecting neuronal cultures (Chen et al., 2011; Chen et al., 2012; Brodbeck et al., 2011). Another approach, anti-ApoE4 immunotherapy, uses antibodies targeting non-lipidated ApoE4 to reduce its toxicity. The anti-human ApoE antibody HAE-4, which identifies non-lipidated ApoE3 and ApoE4, has shown promise in reducing Aβ deposition in APP/PS1–21/ApoE4 mice, whether administered centrally or peripherally (Liao et al., 2018).
Enhancing ApoE lipidation through the activation of ABC transporters, such as ABCA1, has shown promise in preclinical studies. Further strategies include silencing of ARF6 with siRNAs to boost ABCA1 activity, as ApoE4 astrocytes express more ARF6 than ApoE3, leading to decreased ABCA1 expression on the membrane and increased degradation (Rawat et al., 2019). Advancements in genetic therapies, such as using AAV-APOE2 to express the protective APOE2 allele in APOE4 homozygotes, aim to prevent or reverse disease progression by enhancing ApoE lipidation, increasing ApoE-associated cholesterol, and reducing Aβ levels. This approach has been effective in mouse models overexpressing mutant APP or APP/PS1/APOE4 transgenes, offering a glimpse into potential therapies for altering APOE function and addressing AD pathology (Xu et al., 2015).
4.2 Lipid-sensitive nuclear receptors
Retinoid X receptors (RXRs), LXRs, and PPARs are all members of the nuclear receptor superfamily, and act as key regulators of lipid balance within the body and brain. They activate the genes that play critical roles in lipid metabolism (Table 3; Ma and Nelson, 2019).
4.2.1 LXRs and RXRs agonists
LXRs, including LXRα and LXRβ, and RXRs, play a crucial role in lipid homeostasis by forming heterodimers and activating genes essential for lipid metabolism, with LXRβ being the dominant form in the brain, significantly higher than LXRα expression (Ma and Nelson, 2019; Gofflot et al., 2007). LXRs act as cholesterol sensors, promoting the expression of genes responsible for cholesterol efflux, such as ApoE, and can be activated alongside RXRs by their respective ligands to stimulate the transcription of ABCA1, ABCG1, and APOE. This activation pathway is explored in therapies aiming to correct ApoE4 structure, enhance ApoE lipidation, and facilitate Aβ clearance through strategies such as gene therapy and immunotherapy, showing promise in reversing memory deficits in AD mouse models (Olivares et al., 2015; Zelcer and Tontonoz, 2006; Liang et al., 2004; Cramer et al., 2012; Boehm-Cagan and Michaelson, 2014). LXR agonists, including T0901317 and GW3965, have been shown to increase ApoE lipidation and improve microglial association with Aβ plaques, enhancing Aβ clearance and mitigating cognitive impairments, though they also bring unwanted side effects on liver health and triglyceride production by inducing lipogenesis-related genes (Koldamova et al., 2005; Namjoshi et al., 2013; Schultz et al., 2000; Jalil et al., 2019; Cummings et al., 2016). RXRs can dimerize with themselves or with PPARs and retinoic acid receptors (RARs) in response to natural ligands, such as retinoic acid and DHA. RXRs agonists such as bexarotene and LG100268, have been found to further promote LXRs and PPAR-γ dimerization and activation, thus stimulating ApoE lipidation and ABCA1 activity, improving cognitive deficits in AD mouse models. Additionally, specific flavonoids from Sophora tonkinensis (SPF1 and SPF2) with selective RXRs agonistic activity enhanced ABCA1 upregulation and promoted ApoE lipidation. These findings suggest a potential for RXRs and LXRs agonists in AD therapy, although the therapeutic development of synthetic LXRs agonists is hampered by their peripheral side effects, highlighting the need for strategies that balance efficacy with safety (Corona et al., 2016; Boehm-Cagan and Michaelson, 2014; Tai et al., 2014; Wang et al., 2019b; Wang et al., 2019a).
4.2.2 PPARγ
PPARγ stands out as the most explored PPAR target in AD research, with early studies highlighting the role of PPARγ agonists in combatting AD pathologies through anti-inflammatory actions. These include inhibiting pro-inflammatory transcription factors such as NFκB and STATs, showcasing the deep ties between lipid metabolism and the inflammatory response (Daynes and Jones, 2002; DiBattista et al., 2016). In animal studies, PPARγ agonists have been shown to mitigate Aβ pathology and enhance cognitive functions by influencing both Aβ production and clearance (Sastre et al., 2003; Pedersen et al., 2006; Mandrekar-Colucci et al., 2012; Yamanaka et al., 2012; O’Reilly and Lynch, 2012; Toledo and Inestrosa, 2010; Yu et al., 2015). However, larger human trials with PPARγ agonists, such as pioglitazone and rosiglitazone, have yielded negative outcomes (Burns et al., 2021; Gold et al., 2010; Watson et al., 2005; Tzimopoulou et al., 2010; Harrington et al., 2011), a discrepancy that might stem from these agonists’ limited ability to penetrate the BBB. This suggests that next-generation PPARγ agonists with enhanced BBB permeability could offer more effective AD interventions by targeting lipid dysregulation more efficiently.
Conversely, PPARα plays a crucial role in regulating genes crucial for fatty acid oxidation, including those for carnitine palmitoyl transferases and acyl-CoA oxidase, predominantly in astrocytes within the brain (Reddy and Hashimoto, 2001; Zhang et al., 2014; Basu-Modak et al., 1999). In the context of AD, PPARα contributes to amyloid metabolism regulation by promoting the non-amyloidogenic α-secretase activity and suppressing amyloidogenic β-secretase, offering a potential therapeutic angle for AD. Indeed, the PPARα agonist Wy-14643 has been found to alleviate AD-like pathologies and memory impairments in APP/PS1 mice, indicating its potential for therapeutic applications in AD (Zhang et al., 2015; Corbett et al., 2015; Inestrosa et al., 2013; Luo et al., 2020).
4.3 Other therapies affecting lipid metabolism
4.3.1 High-cholesterol diet
Research has indicated that high-cholesterol diets activate astrocytes and amplify the detrimental effects of Aβ on nAChR subunits, leading to cognitive impairments associated with learning and memory (Luo et al., 2020). Elevated cholesterol levels may further impede α-secretase activity in the metabolic processing of APP and reduce the expression of α7nAChR, with AD patients showing a significant decline in these receptors’ expression (Xiu et al., 2006). Although numerous animal studies have linked HFD to impaired brain insulin signaling (Clegg et al., 2011; Carvalheira et al., 2003), few human studies have directly investigated the impact of diet on brain insulin levels. One study found that a diet high in fats and glycemic index lowers CSF insulin levels, aligning with patterns seen in AD, whereas a diet low in these components raised insulin levels in individuals with MCI to those of healthy subjects, with dietary changes in CSF insulin also correlating with shifts in CSF Aβ42 levels (Bayer-Carter et al., 2011). Systematic reviews of dietary interventions focusing on increased intake of PUFAs, nuts, and plant-based foods while reducing saturated fats, animal proteins, and refined sugars have shown that adherence to such diets is associated with improved peripheral insulin sensitivity and a reduced risk of cognitive decline and AD (Solfrizzi et al., 2017; van den Brink et al., 2019; Pistollato et al., 2018).
Epidemiological studies have suggested that a higher intake of ω-3 PUFAs, particularly DHA and EPA, is associated with a reduced risk of AD, whereas a lower intake increases this risk (Morris et al., 2003; Zhang et al., 2016). DHA and EPA are believed to diminish Aβ production and augment its degradation, modulating the brain’s inflammatory response to Aβ, and acting as activators of RXRs and PPARs (de Urquiza et al., 2000; Forman et al., 1997; Casali et al., 2015). The supplementation of ω-3 FA in patients with MCI has been shown to enhance the macrophage levels of specialized pro-resolving mediators (SPMs) like RvD1, increase Aβ phagocytosis, and lower cytokine expression, suggesting that the resolution of neuroinflammation via SPMs could mediate some of the beneficial effects of ω-3 FA (Fiala et al., 2015).
Ketogenic diets (KDs), high in fat and low in carbohydrates, produce ketone bodies such as β-hydroxybutyrate and acetoacetate, which serve as alternative fuel sources for the brain, particularly in AD, where glucose metabolism is compromised early in the disease’s development (Martin et al., 2006; Mosconi et al., 2008; Cunnane et al., 2011). KDs have shown promise in attenuating Aβ pathology and enhancing metabolic and cognitive functions in animal models of aging and AD, with clinical trials in early-stage AD patients demonstrating cognitive improvements (Cunnane et al., 2011; Newman et al., 2017; Kimoto et al., 2017; Henderson and Poirier, 2011; Henderson et al., 2009; Kashiwaya et al., 2000). Moreover, ketone bodies may promote neuronal growth and survival and potentially upregulate mitochondrial biogenesis, thus improving oxidative phosphorylation and ATP production in the brain (Kashiwaya et al., 2000; Bough et al., 2006). However, the efficacy of KDs against AD may vary based on the individual metabolic status and lipid profiles, highlighting the need for further research to determine the long-term applicability of KDs in AD patient care.
4.3.2 Statins
Statin therapy is known to decrease cholesterol levels in the brain membranes, and further exhibits anti-inflammatory, antioxidant, and neuroprotective properties. Although preclinical, retrospective/prospective, and observational studies have suggested that statins may offer protective benefits against AD development, randomized clinical trials have not confirmed the beneficial effects of statins in patients (Dai et al., 2021; Geifman et al., 2017; Li G. et al., 2010; Sano et al., 2011; Feldman et al., 2010; Li et al., 2017). The potential protective mechanism of statins is thought to be derived largely from lowering circulating cholesterol levels, which positively affects the cardiovascular and cerebrovascular systems and diminishes vascular inflammation, indirectly impacting AD risk (Schultz et al., 2018). Additionally, the influence of statins in reducing the risk of AD may also be related to their effects on vascular dementia, which is frequently mistaken for AD. Recent research has suggested that statin therapy could be particularly advantageous for individuals with the ApoE4 genotype, aligning with the role of ApoE4 in disrupting both the central and systemic cholesterol balance. Beyond cholesterol regulation, statins have been proposed to mitigate neuroinflammatory processes that contribute to neurodegeneration (Geifman et al., 2017; Dagliati et al., 2021; McFarland et al., 2018).
4.3.3 Gut microbiota modulation
Probiotics have recently been recognized as an effective and safe method for altering the composition of the gut microbiota and enhancing overall health through mechanisms that are still being explored. In particular, chronic dietary supplementation with a multi-strain probiotic formulation, SLAB51, showed promising results in 3xTg-AD mice. This treatment adjusted the gut microbiota, improved glucose metabolism, reduced inflammatory and oxidative states, partially restored impaired neuronal proteolysis, and diminished Aβ and tau aggregates, leading to enhanced cognitive functions and a delay in AD progression (Bonfili et al., 2017; Bonfili et al., 2018; Bonfili et al., 2020). Probiotic administration has also been found to suppress cholesterol biosynthesis in mice with AD via pathways involving sterol regulatory element-binding protein 1c and LXRs, along with increased brain expression of cholesterol 24S-hydroxylase. These findings suggest that modulating the microbiota with probiotics can beneficially alter the lipid composition in AD mice, highlighting arachidonic acid as a key metabolite linking changes in the lipid profile induced by probiotics to insulin sensitivity and inflammation.
Short-chain fatty acids (SCFAs), which include acetate, butyrate, and propionate, are produced by colonic bacteria through the anaerobic fermentation of dietary fibers and undigested carbohydrates, and range from one to six carbon atoms in length (Tan et al., 2014). These SCFAs can cross the BBB or influence the brain via the gut-brain axis, with butyrate offering neuroprotective benefits as a histone deacetylase inhibitor, affecting G protein-coupled receptor signaling pathways, and exhibiting anti-inflammatory effects (Braniste et al., 2014). Butyrate has further been shown to enhance hippocampal histone acetylation and elevate the expression of genes related to learning (Bonfili et al., 2022). In AD mouse models, sodium butyrate treatment improved learning and memory functions, reduced amyloid plaque buildup, and restored dendritic spine density in the hippocampus (Govindarajan et al., 2011; Ricobaraza et al., 2012). Additionally, acetate has been observed to modulate microglial activity and decrease BBB permeability (Bienenstock et al., 2015), and is reduced in Drosophila models of AD (Kong et al., 2018), suggesting a multifaceted role for SCFAs in AD management and progression.
Taken together, summarizing the clinical trials targeting lipid metabolism in MCI or AD in a table will enhance the clarity and accessibility of this information (Table 4).
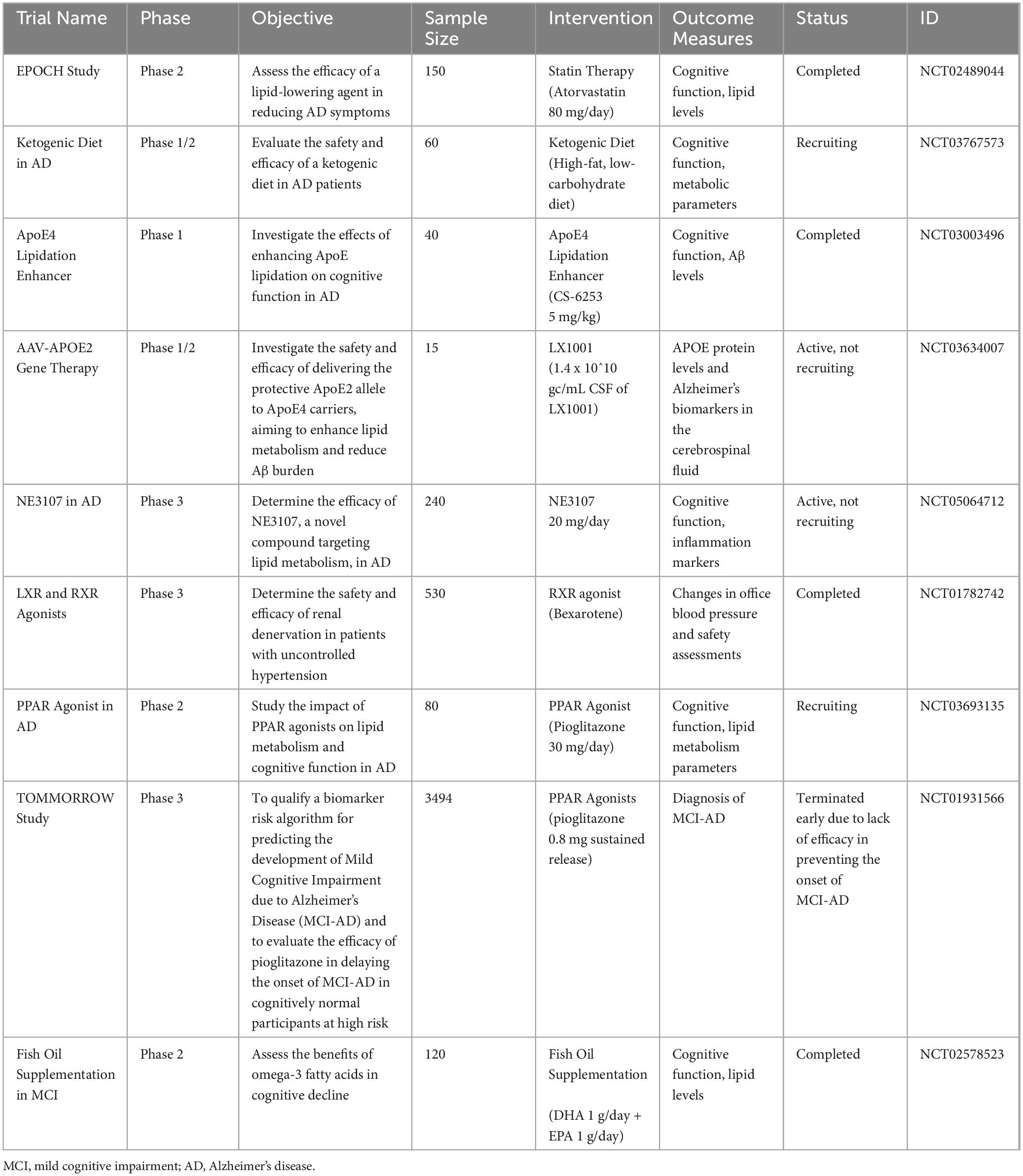
Table 4. Summarizes several clinical trials targeting lipid metabolism in MCI or AD.
5 Summary
Extensive clinical research has shown significant changes in lipid composition and metabolism to be at the core of AD development and progression, with alterations often appearing at disease onset. This evidence points to lipid imbalance as a potential early trigger of AD, likely through its interaction with amyloid buildup, synapse formation, and energy metabolism, underscoring the need for timely lipid-focused intervention strategies.
As AD advances, lipid imbalances not only persist but also intensify, potentially driving a harmful cycle that exacerbates neuroinflammation, energy failure, and oxidative damage, thus accelerating neuronal loss and cognitive decline. This outlook suggests that dysregulated lipid metabolism plays a central role in bridging the gap between reduced cellular energy and ongoing brain inflammation, which are the key features of AD. Moreover, the complex interplay between lipid transport, Aβ, and ApoE underscores the potential of lipid metabolism as a critical junction for both direct and indirect pathways influencing AD pathology.
Insights from preclinical successes and shortcomings of clinical trials targeting lipid metabolism in AD have highlighted the influence of genetic factors, particularly the ApoE genotype, on treatment outcomes. Treatments aimed at modulating lipid metabolism appear to be more promising for AD prevention, especially among individuals carrying the ApoE4 allele, who constitute a significant proportion of both the general and AD-affected populations. Future therapeutic developments must consider the need to target the brain while minimizing side effects on peripheral organs, such as the liver. The collective findings of decades of research emphasize the importance of tailoring lipid metabolism interventions to individual risk profiles, including genetic and lifestyle factors, to effectively combat AD. Overall, adapting therapeutic approaches to address the evolving landscape of lipid metabolism across all stages of AD could enhance treatment efficacy.
Author contributions
YC: Writing – original draft. L-WZ: Writing – original draft. Z-XC: Data curation, Writing – original draft. S-HL: Writing – review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82301501); the Henan Medical Science and Technique Foundation (LHGJ20230180); the National Natural Science Foundation of China (82301345); and the Henan Medical Science and Technique Foundation (LHGJ20230311).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abad-Rodriguez, J., Ledesma, M. D., Craessaerts, K., Perga, S., Medina, M., Delacourte, A., et al. (2004). Neuronal membrane cholesterol loss enhances amyloid peptide generation. J. Cell Biol. 167, 953–960. doi: 10.1083/jcb.200404149
Abe-Dohmae, S., Ikeda, Y., Matsuo, M., Hayashi, M., Okuhira, K., Ueda, K., et al. (2004). Human ABCA7 supports apolipoprotein-mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J. Biol. Chem. 279, 604–611. doi: 10.1074/jbc.M309888200
Aikawa, T., Holm, M. L., and Kanekiyo, T. (2018). ABCA7 and pathogenic pathways of Alzheimers disease. Brain Sci. 8:27. doi: 10.3390/brainsci8020027
Allen, J. A., Halverson-Tamboli, R. A., and Rasenick, M. M. (2007). Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 8, 128–140. doi: 10.1038/nrn2059
Andreone, B. J., Chow, B. W., Tata, A., Lacoste, B., Ben-Zvi, A., Bullock, K., et al. (2017). Blood-brain barrier permeability is regulated by lipid transport-dependent suppression of caveolae-mediated transcytosis. Neuron 94:581–594.e5. doi: 10.1016/j.neuron.2017.03.043
Andrews-Zwilling, Y., Bien-Ly, N., Xu, Q., Li, G., Bernardo, A., Yoon, S. Y., et al. (2010). Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci. 30, 13707–13717. doi: 10.1523/jneurosci.4040-10.2010
Anstey, K. J., Peters, R., Clare, L., Lautenschlager, N. T., Dodge, H. H., Barnes, D. E., et al. (2017). Joining forces to prevent dementia: The international research network on dementia prevention (IRNDP). Int. Psychogeriatr. 29, 1757–1760. doi: 10.1017/s1041610217001685
Area-Gomez, E., de Groof, A. J., Boldogh, I., Bird, T. D., Gibson, G. E., Koehler, C. M., et al. (2009). Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 175, 1810–1816. doi: 10.2353/ajpath.2009.090219
Area-Gomez, E., de Groof, A., Bonilla, E., Montesinos, J., Tanji, K., Boldogh, I., et al. (2018). A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 9:335. doi: 10.1038/s41419-017-0215-0
Ariga, T., McDonald, M. P., and Yu, R. K. (2008). Role of ganglioside metabolism in the pathogenesis of Alzheimers disease–a review. J. Lipid Res. 49, 1157–1175. doi: 10.1194/jlr.R800007-JLR200
Ates, G., Goldberg, J., Currais, A., and Maher, P. (2020). CMS121, a fatty acid synthase inhibitor, protects against excess lipid peroxidation and inflammation and alleviates cognitive loss in a transgenic mouse model of Alzheimers disease. Redox Biol. 36:101648. doi: 10.1016/j.redox.2020.101648
Bandaru, V. V., Troncoso, J., Wheeler, D., Pletnikova, O., Wang, J., Conant, K., et al. (2009). ApoE4 disrupts sterol and sphingolipid metabolism in Alzheimers but not normal brain. Neurobiol. Aging 30, 591–599. doi: 10.1016/j.neurobiolaging.2007.07.024
Barber, C. N., and Raben, D. M. (2019). Lipid metabolism crosstalk in the brain: Glia and neurons. Front. Cell. Neurosci. 13:212. doi: 10.3389/fncel.2019.00212
Basil, M. C., and Levy, B. D. (2016). Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 16, 51–67. doi: 10.1038/nri.2015.4
Basu-Modak, S., Braissant, O., Escher, P., Desvergne, B., Honegger, P., and Wahli, W. (1999). Peroxisome proliferator-activated receptor beta regulates acyl-CoA synthetase 2 in reaggregated rat brain cell cultures. J. Biol. Chem. 274, 35881–35888. doi: 10.1074/jbc.274.50.35881
Bayer-Carter, J. L., Green, P. S., Montine, T. J., VanFossen, B., Baker, L. D., Watson, G. S., et al. (2011). Diet intervention and cerebrospinal fluid biomarkers in amnestic mild cognitive impairment. Arch. Neurol. 68, 743–752. doi: 10.1001/archneurol.2011.125
Bazinet, R. P., and Layé, S. (2014). Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 15, 771–785. doi: 10.1038/nrn3820
Belkouch, M., Hachem, M., Elgot, A., Lo Van, A., Picq, M., Guichardant, M., et al. (2016). The pleiotropic effects of omega-3 docosahexaenoic acid on the hallmarks of Alzheimers disease. J. Nutr. Biochem. 38, 1–11. doi: 10.1016/j.jnutbio.2016.03.002
Bellenguez, C., Küçükali, F., Jansen, I. E., Kleineidam, L., Moreno-Grau, S., Amin, N., et al. (2022). New insights on the genetic etiology of Alzheimers and related dementia. Nat. Genet. 54, 412–424.
Bennett, S. A., Valenzuela, N., Xu, H., Franko, B., Fai, S., and Figeys, D. (2013). Using neurolipidomics to identify phospholipid mediators of synaptic (dys)function in Alzheimers Disease. Front. Physiol. 4:168. doi: 10.3389/fphys.2013.00168
Ben-Zvi, A., Lacoste, B., Kur, E., Andreone, B. J., Mayshar, Y., Yan, H., et al. (2014). Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 509, 507–511. doi: 10.1038/nature13324
Bienenstock, J., Kunze, W., and Forsythe, P. (2015). Microbiota and the gut-brain axis. Nutr. Rev. 73, 28–31. doi: 10.1093/nutrit/nuv019
Blanco-Silvente, L., Capellà, D., Garre-Olmo, J., Vilalta-Franch, J., and Castells, X. (2018). Predictors of discontinuation, efficacy, and safety of memantine treatment for Alzheimers disease: Meta-analysis and meta-regression of 18 randomized clinical trials involving 5004 patients. BMC Geriatr. 18:168. doi: 10.1186/s12877-018-0857-5
Boehm-Cagan, A., and Michaelson, D. M. (2014). Reversal of apoE4-driven brain pathology and behavioral deficits by bexarotene. J. Neurosci. 34, 7293–7301. doi: 10.1523/jneurosci.5198-13.2014
Boehm-Cagan, A., Bar, R., Liraz, O., Bielicki, J. K., Johansson, J. O., and Michaelson, D. M. (2016). ABCA1 agonist reverses the ApoE4-driven cognitive and brain pathologies. J. Alzheimers Dis. 54, 1219–1233. doi: 10.3233/jad-160467
Bonfili, L., Cecarini, V., Berardi, S., Scarpona, S., Suchodolski, J. S., Nasuti, C., et al. (2017). Microbiota modulation counteracts Alzheimers disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci. Rep. 7:2426. doi: 10.1038/s41598-017-02587-2
Bonfili, L., Cecarini, V., Cuccioloni, M., Angeletti, M., Berardi, S., Scarpona, S., et al. (2018). SLAB51 probiotic formulation activates SIRT1 pathway promoting antioxidant and neuroprotective effects in an AD mouse model. Mol. Neurobiol. 55, 7987–8000. doi: 10.1007/s12035-018-0973-4
Bonfili, L., Cecarini, V., Gogoi, O., Berardi, S., Scarpona, S., Angeletti, M., et al. (2020). Gut microbiota manipulation through probiotics oral administration restores glucose homeostasis in a mouse model of Alzheimers disease. Neurobiol. Aging 87, 35–43. doi: 10.1016/j.neurobiolaging.2019.11.004
Bonfili, L., Cuccioloni, M., Gong, C., Cecarini, V., Spina, M., Zheng, Y., et al. (2022). Gut microbiota modulation in Alzheimers disease: Focus on lipid metabolism. Clin. Nutr. 41, 698–708. doi: 10.1016/j.clnu.2022.01.025
Borroni, V., and Barrantes, F. J. (2011). Cholesterol modulates the rate and mechanism of acetylcholine receptor internalization. J. Biol. Chem. 286, 17122–17132. doi: 10.1074/jbc.M110.211870
Bough, K. J., Wetherington, J., Hassel, B., Pare, J. F., Gawryluk, J. W., Greene, J. G., et al. (2006). Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 60, 223–235. doi: 10.1002/ana.20899
Boyles, J. K., Zoellner, C. D., Anderson, L. J., Kosik, L. M., Pitas, R. E., Weisgraber, K. H., et al. (1989). A role for apolipoprotein E, apolipoprotein A-I, and low density lipoprotein receptors in cholesterol transport during regeneration and remyelination of the rat sciatic nerve. J. Clin. Invest. 83, 1015–1031. doi: 10.1172/jci113943
Braniste, V., Al-Asmakh, M., Kowal, C., Anuar, F., Abbaspour, A., Tóth, M., et al. (2014). The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 6:263ra158. doi: 10.1126/scitranslmed.3009759
Brecht, W. J., Harris, F. M., Chang, S., Tesseur, I., Yu, G. Q., Xu, Q., et al. (2004). Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci. 24, 2527–2534. doi: 10.1523/jneurosci.4315-03.2004
Brinton, R. D. (2013). Neurosteroids as regenerative agents in the brain: Therapeutic implications. Nat. Rev. Endocrinol. 9, 241–250. doi: 10.1038/nrendo.2013.31
Brodbeck, J., McGuire, J., Liu, Z., Meyer-Franke, A., Balestra, M. E., Jeong, D. E., et al. (2011). Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J. Biol. Chem. 286, 17217–17226. doi: 10.1074/jbc.M110.217380
Broom, G. M., Shaw, I. C., and Rucklidge, J. J. (2019). The ketogenic diet as a potential treatment and prevention strategy for Alzheimers disease. Nutrition 60, 118–121. doi: 10.1016/j.nut.2018.10.003
Brusés, J. L., Chauvet, N., and Rutishauser, U. (2001). Membrane lipid rafts are necessary for the maintenance of the (alpha)7 nicotinic acetylcholine receptor in somatic spines of ciliary neurons. J. Neurosci. 21, 504–512. doi: 10.1523/jneurosci.21-02-00504.2001
Burns, D. K., Alexander, R. C., Welsh-Bohmer, K. A., Culp, M., Chiang, C., ONeil, J., et al. (2021). Safety and efficacy of pioglitazone for the delay of cognitive impairment in people at risk of Alzheimers disease (TOMMORROW): A prognostic biomarker study and a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 20, 537–547. doi: 10.1016/s1474-4422(21)00043-0
Cai, D., Netzer, W. J., Zhong, M., Lin, Y., Du, G., Frohman, M., et al. (2006a). Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc. Natl. Acad. Sci. U.S.A. 103, 1941–1946. doi: 10.1073/pnas.0510708103
Cai, D., Zhong, M., Wang, R., Netzer, W. J., Shields, D., Zheng, H., et al. (2006b). Phospholipase D1 corrects impaired betaAPP trafficking and neurite outgrowth in familial Alzheimers disease-linked presenilin-1 mutant neurons. Proc. Natl. Acad. Sci. U.S.A. 103, 1936–1940. doi: 10.1073/pnas.0510710103
Carvalheira, J. B., Ribeiro, E. B., Araújo, E. P., Guimarães, R. B., Telles, M. M., Torsoni, M., et al. (2003). Selective impairment of insulin signalling in the hypothalamus of obese Zucker rats. Diabetologia 46, 1629–1640. doi: 10.1007/s00125-003-1246-x
Casali, B. T., Corona, A. W., Mariani, M. M., Karlo, J. C., Ghosal, K., and Landreth, G. E. (2015). Omega-3 fatty acids augment the actions of nuclear receptor agonists in a mouse model of Alzheimers disease. J. Neurosci. 35, 9173–9181. doi: 10.1523/jneurosci.1000-15.2015
Castro, B. M., Silva, L. C., Fedorov, A., de Almeida, R. F., and Prieto, M. (2009). Cholesterol-rich fluid membranes solubilize ceramide domains: Implications for the structure and dynamics of mammalian intracellular and plasma membranes. J. Biol. Chem. 284, 22978–22987. doi: 10.1074/jbc.M109.026567
Chan, D., Shafto, M., Kievit, R., Matthews, F., Spink, M., Valenzuela, M., et al. (2018). Lifestyle activities in mid-life contribute to cognitive reserve in late-life, independent of education, occupation, and late-life activities. Neurobiol. Aging 70, 180–183. doi: 10.1016/j.neurobiolaging.2018.06.012
Chang, S., Ma, T., Miranda, R. D., Balestra, M. E., Mahley, R. W., and Huang, Y. (2005). Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 102, 18694–18699. doi: 10.1073/pnas.0508254102
Chen, H. K., Ji, Z. S., Dodson, S. E., Miranda, R. D., Rosenblum, C. I., Reynolds, I. J., et al. (2011). Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J. Biol. Chem. 286, 5215–5221. doi: 10.1074/jbc.M110.151084
Chen, H. K., Liu, Z., Meyer-Franke, A., Brodbeck, J., Miranda, R. D., McGuire, J. G., et al. (2012). Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J. Biol. Chem. 287, 5253–5266. doi: 10.1074/jbc.M111.276162
Chernick, D., Ortiz-Valle, S., Jeong, A., Swaminathan, S. K., Kandimalla, K. K., Rebeck, G. W., et al. (2018). High-density lipoprotein mimetic peptide 4F mitigates amyloid-β-induced inhibition of apolipoprotein E secretion and lipidation in primary astrocytes and microglia. J. Neurochem. 147, 647–662. doi: 10.1111/jnc.14554
Chou, Y. C., Lin, S. B., Tsai, L. H., Tsai, H. I., and Lin, C. M. (2003). Cholesterol deficiency increases the vulnerability of hippocampal glia in primary culture to glutamate-induced excitotoxicity. Neurochem. Int. 43, 197–209. doi: 10.1016/s0197-0186(03)00003-2
Clegg, D. J., Gotoh, K., Kemp, C., Wortman, M. D., Benoit, S. C., Brown, L. M., et al. (2011). Consumption of a high-fat diet induces central insulin resistance independent of adiposity. Physiol. Behav. 103, 10–16. doi: 10.1016/j.physbeh.2011.01.010
Combrinck, M., Williams, J., De Berardinis, M. A., Warden, D., Puopolo, M., Smith, A. D., et al. (2006). Levels of CSF prostaglandin E2, cognitive decline, and survival in Alzheimers disease. J. Neurol. Neurosurg. Psychiatry 77, 85–88. doi: 10.1136/jnnp.2005.063131
Corbett, G. T., Gonzalez, F. J., and Pahan, K. (2015). Activation of peroxisome proliferator-activated receptor α stimulates ADAM10-mediated proteolysis of APP. Proc. Natl. Acad. Sci. U.S.A. 112, 8445–8450. doi: 10.1073/pnas.1504890112
Corder, E. H., Saunders, A. M., Risch, N. J., Strittmatter, W. J., Schmechel, D. E., and Gaskell, P. C. Jr., et al. (1994). Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 7, 180–184. doi: 10.1038/ng0694-180
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D., Gaskell, P., Small, G., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimers disease in late onset families. Science 261, 921–923.
Corona, A. W., Kodoma, N., Casali, B. T., and Landreth, G. E. (2016). ABCA1 is necessary for bexarotene-mediated clearance of soluble amyloid beta from the hippocampus of APP/PS1 mice. J. Neuroimmune Pharmacol. 11, 61–72. doi: 10.1007/s11481-015-9627-8
Cramer, P. E., Cirrito, J. R., Wesson, D. W., Lee, C. Y., Karlo, J. C., Zinn, A. E., et al. (2012). ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506. doi: 10.1126/science.1217697
Cruchaga, C., Kauwe, J. S., Harari, O., Jin, S. C., Cai, Y., Karch, C. M., et al. (2013). GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimers disease. Neuron 78, 256–268. doi: 10.1016/j.neuron.2013.02.026
Cummings, J. L., Zhong, K., Kinney, J. W., Heaney, C., Moll-Tudla, J., Joshi, A., et al. (2016). Double-blind, placebo-controlled, proof-of-concept trial of bexarotene Xin moderate Alzheimers disease. Alzheimers Res. Ther. 8:4. doi: 10.1186/s13195-016-0173-2
Cunnane, S. C., Schneider, J. A., Tangney, C., Tremblay-Mercier, J., Fortier, M., Bennett, D. A., et al. (2012). Plasma and brain fatty acid profiles in mild cognitive impairment and Alzheimers disease. J. Alzheimers Dis. 29, 691–697. doi: 10.3233/jad-2012-110629
Cunnane, S., Nugent, S., Roy, M., Courchesne-Loyer, A., Croteau, E., Tremblay, S., et al. (2011). Brain fuel metabolism, aging, and Alzheimers disease. Nutrition 27, 3–20. doi: 10.1016/j.nut.2010.07.021
Cutler, R. G., Kelly, J., Storie, K., Pedersen, W. A., Tammara, A., Hatanpaa, K., et al. (2004). Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimers disease. Proc. Natl. Acad. Sci. U.S.A. 101, 2070–2075. doi: 10.1073/pnas.0305799101
Dabravolski, S. A., Bezsonov, E. E., and Orekhov, A. N. (2021). The role of mitochondria dysfunction and hepatic senescence in NAFLD development and progression. Biomed. Pharmacother. 142:112041. doi: 10.1016/j.biopha.2021.112041
Dagliati, A., Peek, N., Brinton, R. D., and Geifman, N. (2021). Sex and APOE genotype differences related to statin use in the aging population. Alzheimers Dement. 7:e12156. doi: 10.1002/trc2.12156
Dai, L., Zou, L., Meng, L., Qiang, G., Yan, M., and Zhang, Z. (2021). Cholesterol metabolism in neurodegenerative diseases: Molecular mechanisms and therapeutic targets. Mol. Neurobiol. 58, 2183–2201. doi: 10.1007/s12035-020-02232-6
Daynes, R. A., and Jones, D. C. (2002). Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2, 748–759. doi: 10.1038/nri912
De Roeck, A., Van Broeckhoven, C., and Sleegers, K. (2019). The role of ABCA7 in Alzheimers disease: Evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 138, 201–220. doi: 10.1007/s00401-019-01994-1
de Urquiza, A. M., Liu, S., Sjöberg, M., Zetterström, R. H., Griffiths, W., Sjövall, J., et al. (2000). Docosahexaenoic acid, a ligand for the retinoid X receptor in mouse brain. Science 290, 2140–2144. doi: 10.1126/science.290.5499.2140
DeGrella, R. F., and Simoni, R. D. (1982). Intracellular transport of cholesterol to the plasma membrane. J. Biol. Chem. 257, 14256–14262.
Di Paolo, G., and Kim, T. W. (2011). Linking lipids to Alzheimers disease: Cholesterol and beyond. Nat. Rev. Neurosci. 12, 284–296. doi: 10.1038/nrn3012
Di Scala, C., Yahi, N., Lelièvre, C., Garmy, N., Chahinian, H., and Fantini, J. (2013). Biochemical identification of a linear cholesterol-binding domain within Alzheimers β amyloid peptide. ACS Chem. Neurosci. 4, 509–517. doi: 10.1021/cn300203a
DiBattista, A. M., Dumanis, S. B., Newman, J., and Rebeck, G. W. (2016). Identification and modification of amyloid-independent phenotypes of APOE4 mice. Exp. Neurol. 280, 97–105. doi: 10.1016/j.expneurol.2016.04.014
Dietschy, J. M., and Turley, S. D. (2001). Cholesterol metabolism in the brain. Curr. Opin. Lipidol. 12, 105–112. doi: 10.1097/00041433-200104000-00003
Dietschy, J. M., and Turley, S. D. (2004). Thematic review series: Brain lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 45, 1375–1397. doi: 10.1194/jlr.R400004-JLR200
Do Carmo, S., Kautzmann, M. I., Bhattacharjee, S., Jun, B., Steinberg, C., Emmerson, J. T., et al. (2024). Differential effect of an evolving amyloid and tau pathology on brain phospholipids and bioactive lipid mediators in rat models of Alzheimer-like pathology. J. Neuroinflamm. 21:185. doi: 10.1186/s12974-024-03184-7
Du, L., Hickey, R. W., Bayir, H., Watkins, S. C., Tyurin, V. A., Guo, F., et al. (2009). Starving neurons show sex difference in autophagy. J. Biol. Chem. 284, 2383–2396. doi: 10.1074/jbc.M804396200
Dunn, H. C., Ager, R. R., Baglietto-Vargas, D., Cheng, D., Kitazawa, M., Cribbs, D. H., et al. (2015). Restoration of lipoxin A4 signaling reduces Alzheimers disease-like pathology in the 3xTg-AD mouse model. J. Alzheimers Dis. 43, 893–903. doi: 10.3233/jad-141335
Edmond, J., Robbins, R. A., Bergstrom, J. D., Cole, R. A., and de Vellis, J. (1987). Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J. Neurosci. Res. 18, 551–561. doi: 10.1002/jnr.490180407
Emre, C., Hjorth, E., Bharani, K., Carroll, S., Granholm, A. C., and Schultzberg, M. (2020). Receptors for pro-resolving mediators are increased in Alzheimers disease brain. Brain Pathol. 30, 614–640. doi: 10.1111/bpa.12812
Eraso-Pichot, A., Brasó-Vives, M., Golbano, A., Menacho, C., Claro, E., Galea, E., et al. (2018). GSEA of mouse and human mitochondriomes reveals fatty acid oxidation in astrocytes. Glia 66, 1724–1735. doi: 10.1002/glia.23330
Fabelo, N., Martín, V., Marín, R., Moreno, D., Ferrer, I., and Díaz, M. (2014). Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimers disease and facilitates APP/BACE1 interactions. Neurobiol. Aging 35, 1801–1812. doi: 10.1016/j.neurobiolaging.2014.02.005
Fabiani, C., and Antollini, S. S. (2019). Alzheimers disease as a membrane disorder: Spatial cross-talk among beta-amyloid peptides, nicotinic acetylcholine receptors and lipid rafts. Front. Cell. Neurosci. 13:309. doi: 10.3389/fncel.2019.00309
Fagan, A. M., Bu, G., Sun, Y., Daugherty, A., and Holtzman, D. M. (1996). Apolipoprotein E-containing high density lipoprotein promotes neurite outgrowth and is a ligand for the low density lipoprotein receptor-related protein. J. Biol. Chem. 271, 30121–30125. doi: 10.1074/jbc.271.47.30121
Fecher, C., Trovò, L., Müller, S. A., Snaidero, N., Wettmarshausen, J., Heink, S., et al. (2019). Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 22, 1731–1742. doi: 10.1038/s41593-019-0479-z
Feldman, H. H., Doody, R. S., Kivipelto, M., Sparks, D. L., Waters, D. D., Jones, R. W., et al. (2010). Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology 74, 956–964. doi: 10.1212/WNL.0b013e3181d6476a
Fiala, M., Halder, R. C., Sagong, B., Ross, O., Sayre, J., Porter, V., et al. (2015). ω-3 Supplementation increases amyloid-β phagocytosis and resolvin D1 in patients with minor cognitive impairment. FASEB J. 29, 2681–2689. doi: 10.1096/fj.14-264218
Foley, P. (2010). Lipids in Alzheimers disease: A century-old story. Biochim. Biophys. Acta 1801, 750–753. doi: 10.1016/j.bbalip.2010.05.004
Fonteh, A. N., Cipolla, M., Chiang, A. J., Edminster, S. P., Arakaki, X., and Harrington, M. G. (2020). Polyunsaturated fatty acid composition of cerebrospinal fluid fractions shows their contribution to cognitive resilience of a pre-symptomatic Alzheimers disease cohort. Front. Physiol. 11:83. doi: 10.3389/fphys.2020.00083
Fonteh, A. N., Cipolla, M., Chiang, J., Arakaki, X., and Harrington, M. G. (2014). Human cerebrospinal fluid fatty acid levels differ between supernatant fluid and brain-derived nanoparticle fractions, and are altered in Alzheimers disease. PLoS One 9:e100519. doi: 10.1371/journal.pone.0100519
Forman, B. M., Chen, J., and Evans, R. M. (1997). Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc. Natl. Acad. Sci. U.S.A. 94, 4312–4317. doi: 10.1073/pnas.94.9.4312
Garner, B. (2010). Lipids and Alzheimers disease. Biochim. Biophys. Acta 1801, 747–749. doi: 10.1016/j.bbalip.2010.06.003
Geifman, N., Brinton, R. D., Kennedy, R. E., Schneider, L. S., and Butte, A. J. (2017). Evidence for benefit of statins to modify cognitive decline and risk in Alzheimers disease. Alzheimers Res. Ther. 9:10. doi: 10.1186/s13195-017-0237-y
Ghosh, S. S., Swerdlow, R. H., Miller, S. W., Sheeman, B., Parker, W. D. Jr., and Davis, R. E. (1999). Use of cytoplasmic hybrid cell lines for elucidating the role of mitochondrial dysfunction in Alzheimers disease and Parkinsons disease. Ann. N. Y. Acad. Sci. 893, 176–191. doi: 10.1111/j.1749-6632.1999.tb07825.x
Gibson, G. E., Haroutunian, V., Zhang, H., Park, L. C., Shi, Q., Lesser, M., et al. (2000). Mitochondrial damage in Alzheimers disease varies with apolipoprotein E genotype. Ann. Neurol. 48, 297–303.
Giorgi, C., Missiroli, S., Patergnani, S., Duszynski, J., Wieckowski, M. R., and Pinton, P. (2015). Mitochondria-associated membranes: Composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal. 22, 995–1019. doi: 10.1089/ars.2014.6223
Gofflot, F., Chartoire, N., Vasseur, L., Heikkinen, S., Dembele, D., Le Merrer, J., et al. (2007). Systematic gene expression mapping clusters nuclear receptors according to their function in the brain. Cell 131, 405–418. doi: 10.1016/j.cell.2007.09.012
Gold, M., Alderton, C., Zvartau-Hind, M., Egginton, S., Saunders, A. M., Irizarry, M., et al. (2010). Rosiglitazone monotherapy in mild-to-moderate Alzheimers disease: Results from a randomized, double-blind, placebo-controlled phase III study. Dement. Geriatr. Cogn. Disord. 30, 131–146. doi: 10.1159/000318845
Gómez-Ramos, P., and Asunción Morán, M. (2007). Ultrastructural localization of intraneuronal Abeta-peptide in Alzheimer disease brains. J. Alzheimers Dis. 11, 53–59. doi: 10.3233/jad-2007-11109
Gouna, G., Klose, C., Bosch-Queralt, M., Liu, L., Gokce, O., Schifferer, M., et al. (2021). TREM2-dependent lipid droplet biogenesis in phagocytes is required for remyelination. J. Exp. Med. 218:227. doi: 10.1084/jem.20210227
Govindarajan, N., Agis-Balboa, R. C., Walter, J., Sananbenesi, F., and Fischer, A. (2011). Sodium butyrate improves memory function in an Alzheimers disease mouse model when administered at an advanced stage of disease progression. J. Alzheimers Dis. 26, 187–197. doi: 10.3233/jad-2011-110080
Grimm, M. O., Grimm, H. S., Pätzold, A. J., Zinser, E. G., Halonen, R., Duering, M., et al. (2005). Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat. Cell Biol. 7, 1118–1123. doi: 10.1038/ncb1313
Guan, Z., Wang, Y., Cairns, N. J., Lantos, P. L., Dallner, G., and Sindelar, P. J. (1999). Decrease and structural modifications of phosphatidylethanolamine plasmalogen in the brain with Alzheimer disease. J. Neuropathol. Exp. Neurol. 58, 740–747. doi: 10.1097/00005072-199907000-00008
Hamilton, L. K., Dufresne, M., Joppé, S. E., Petryszyn, S., Aumont, A., Calon, F., et al. (2015). Aberrant lipid metabolism in the forebrain niche suppresses adult neural stem cell proliferation in an animal model of Alzheimers disease. Cell Stem Cell 17, 397–411. doi: 10.1016/j.stem.2015.08.001
Han, X. (2007). Neurolipidomics: Challenges and developments. Front. Biosci. 12:2601–2615. doi: 10.2741/2258
Han, X., Holtzman, D. M., and McKeel, D. W. Jr. (2001). Plasmalogen deficiency in early Alzheimers disease subjects and in animal models: Molecular characterization using electrospray ionization mass spectrometry. J. Neurochem. 77, 1168–1180. doi: 10.1046/j.1471-4159.2001.00332.x
Handattu, S. P., Monroe, C. E., Nayyar, G., Palgunachari, M. N., Kadish, I., van Groen, T., et al. (2013). In vivo and in vitro effects of an apolipoprotein e mimetic peptide on amyloid-β pathology. J. Alzheimers Dis. 36, 335–347. doi: 10.3233/jad-122377
Hanson, A. J., Bayer-Carter, J. L., Green, P. S., Montine, T. J., Wilkinson, C. W., Baker, L. D., et al. (2013). Effect of apolipoprotein E genotype and diet on apolipoprotein E lipidation and amyloid peptides: Randomized clinical trial. JAMA Neurol. 70, 972–980. doi: 10.1001/jamaneurol.2013.396
Hara, M., Matsushima, T., Satoh, H., Iso-o, N., Noto, H., Togo, M., et al. (2003). Isoform-dependent cholesterol efflux from macrophages by apolipoprotein E is modulated by cell surface proteoglycans. Arterioscler. Thromb. Vasc. Biol. 23, 269–274. doi: 10.1161/01.atv.0000054199.78458.4b
Harold, D., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Hamshere, M. L., et al. (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimers disease. Nat. Genet. 41, 1088–1093. doi: 10.1038/ng.440
Harrington, C., Sawchak, S., Chiang, C., Davies, J., Donovan, C., Saunders, A. M., et al. (2011). Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimers disease: Two phase 3 studies. Curr. Alzheimer Res. 8, 592–606. doi: 10.2174/156720511796391935
Harris, F. M., Brecht, W. J., Xu, Q., Tesseur, I., Kekonius, L., Wyss-Coray, T., et al. (2003). Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimers disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 100, 10966–10971. doi: 10.1073/pnas.1434398100
Hartmann, H., Eckert, A., and Müller, W. E. (1994). Apolipoprotein E and cholesterol affect neuronal calcium signalling: The possible relationship to beta-amyloid neurotoxicity. Biochem. Biophys. Res. Commun. 200, 1185–1192. doi: 10.1006/bbrc.1994.1576
Hashemi, M., Banerjee, S., and Lyubchenko, Y. L. (2022). Free cholesterol accelerates Aβ self-assembly on membranes at physiological concentration. Int. J. Mol. Sci. 23:2803. doi: 10.3390/ijms23052803
He, X., Huang, Y., Li, B., Gong, C. X., and Schuchman, E. H. (2010). Deregulation of sphingolipid metabolism in Alzheimers disease. Neurobiol. Aging. 31, 398–408. doi: 10.1016/j.neurobiolaging.2008.05.010
Head, B. P., Patel, H. H., and Insel, P. A. (2014). Interaction of membrane/lipid rafts with the cytoskeleton: Impact on signaling and function: Membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim. Biophys. Acta 1838, 532–545. doi: 10.1016/j.bbamem.2013.07.018
Heino, S., Lusa, S., Somerharju, P., Ehnholm, C., Olkkonen, V. M., and Ikonen, E. (2000). Dissecting the role of the golgi complex and lipid rafts in biosynthetic transport of cholesterol to the cell surface. Proc. Natl. Acad. Sci. U.S.A. 97, 8375–8380. doi: 10.1073/pnas.140218797
Heinsinger, N. M., Gachechiladze, M. A., and Rebeck, G. W. (2016). Apolipoprotein E genotype affects size of ApoE complexes in cerebrospinal fluid. J. Neuropathol. Exp. Neurol. 75, 918–924. doi: 10.1093/jnen/nlw067
Henderson, S. T., and Poirier, J. (2011). Pharmacogenetic analysis of the effects of polymorphisms in APOE, IDE and IL1B on a ketone body based therapeutic on cognition in mild to moderate Alzheimers disease; a randomized, double-blind, placebo-controlled study. BMC Med. Genet. 12:137. doi: 10.1186/1471-2350-12-137
Henderson, S. T., Vogel, J. L., Barr, L. J., Garvin, F., Jones, J. J., and Costantini, L. C. (2009). Study of the ketogenic agent AC-1202 in mild to moderate Alzheimers disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 6:31. doi: 10.1186/1743-7075-6-31
Heneka, M. T., Reyes-Irisarri, E., Hüll, M., and Kummer, M. P. (2011). Impact and therapeutic potential of PPARs in Alzheimers disease. Curr. Neuropharmacol. 9, 643–650. doi: 10.2174/157015911798376325
Hernandez, C. M., Kayed, R., Zheng, H., Sweatt, J. D., and Dineley, K. T. (2010). Loss of alpha7 nicotinic receptors enhances beta-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimers disease. J. Neurosci. 30, 2442–2453. doi: 10.1523/jneurosci.5038-09.2010
Herz, J. (2009). Apolipoprotein E receptors in the nervous system. Curr. Opin. Lipidol. 20, 190–196. doi: 10.1097/MOL.0b013e32832d3a10
Heverin, M., Bogdanovic, N., Lütjohann, D., Bayer, T., Pikuleva, I., Bretillon, L., et al. (2004). Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimers disease. J. Lipid Res. 45, 186–193. doi: 10.1194/jlr.M300320-JLR200
Hirai, K., Aliev, G., Nunomura, A., Fujioka, H., Russell, R. L., Atwood, C. S., et al. (2001). Mitochondrial abnormalities in Alzheimers disease. J. Neurosci. 21, 3017–3023. doi: 10.1523/jneurosci.21-09-03017.2001
Hirono, N., Hashimoto, M., Yasuda, M., Ishii, K., Sakamoto, S., Kazui, H., et al. (2002). The effect of APOE epsilon4 allele on cerebral glucose metabolism in AD is a function of age at onset. Neurology 58, 743–750. doi: 10.1212/wnl.58.5.743
Hollingworth, P., Harold, D., Sims, R., Gerrish, A., Lambert, J., Carrasquillo, M., et al. (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimers disease. Nat. Genet. 43, 429–435.
Hong, H., Hong, L., Luo, X., Zeng, Q., Li, K., Wang, S., et al. (2024). The relationship between amyloid pathology, cerebral small vessel disease, glymphatic dysfunction, and cognition: A study based on Alzheimers disease continuum participants. Alzheimers Res. Ther. 16:43. doi: 10.1186/s13195-024-01407-w
Hong, S., Ostaszewski, B. L., Yang, T., OMalley, T. T., Jin, M., Yanagisawa, K., et al. (2014). Soluble Aβ oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 82, 308–319. doi: 10.1016/j.neuron.2014.02.027
Horiuchi, Y., Ohkawa, R., Lai, S. J., Yamazaki, A., Ikoma, H., Yano, K., et al. (2019). Characterization of the cholesterol efflux of apolipoprotein E-containing high-density lipoprotein in THP-1 cells. Biol. Chem. 400, 209–218. doi: 10.1515/hsz-2018-0284
Huang, H., Yang, B., Yu, R., Ouyang, W., Tong, J., and Le, Y. (2024). Very high high-density lipoprotein cholesterol may be associated with higher risk of cognitive impairment in older adults. Nutr. J. 23:79. doi: 10.1186/s12937-024-00983-9
Huang, Y., and Mahley, R. W. (2014). Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimers diseases. Neurobiol. Dis. 72, 3–12. doi: 10.1016/j.nbd.2014.08.025
Huang, Y., Liu, X. Q., Wyss-Coray, T., Brecht, W. J., Sanan, D. A., and Mahley, R. W. (2001). Apolipoprotein E fragments present in Alzheimers disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc. Natl. Acad. Sci. U.S.A. 98, 8838–8843. doi: 10.1073/pnas.151254698
Huynh, T. V., Liao, F., Francis, C. M., Robinson, G. O., Serrano, J. R., Jiang, H., et al. (2017). Age-dependent effects of apoE reduction using antisense oligonucleotides in a model of β-amyloidosis. Neuron 96:1013–1023.e4. doi: 10.1016/j.neuron.2017.11.014
Inestrosa, N. C., Carvajal, F. J., Zolezzi, J. M., Tapia-Rojas, C., Serrano, F., Karmelic, D., et al. (2013). Peroxisome proliferators reduce spatial memory impairment, synaptic failure, and neurodegeneration in brains of a double transgenic mice model of Alzheimers disease. J. Alzheimers Dis. 33, 941–959. doi: 10.3233/jad-2012-120397
Ioannou, M. S., Jackson, J., Sheu, S. H., Chang, C. L., Weigel, A. V., Liu, H., et al. (2019). Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell 177:1522–1535.e14. doi: 10.1016/j.cell.2019.04.001
Jalil, A., Bourgeois, T., Ménégaut, L., Lagrost, L., Thomas, C., and Masson, D. (2019). Revisiting the role of LXRs in PUFA metabolism and phospholipid homeostasis. Int. J. Mol. Sci. 20:3787. doi: 10.3390/ijms20153787
Jana, A., and Pahan, K. (2004). Fibrillar amyloid-beta peptides kill human primary neurons via NADPH oxidase-mediated activation of neutral sphingomyelinase. Implications for Alzheimers disease. J. Biol. Chem. 279, 51451–51459. doi: 10.1074/jbc.M404635200
Jay, T. R., von Saucken, V. E., and Landreth, G. E. (2017). TREM2 in neurodegenerative diseases. Mol. Neurodegener. 12:56. doi: 10.1186/s13024-017-0197-5
Johansson, J. U., Woodling, N. S., Wang, Q., Panchal, M., Liang, X., Trueba-Saiz, A., et al. (2015). Prostaglandin signaling suppresses beneficial microglial function in Alzheimers disease models. J. Clin. Invest. 125, 350–364. doi: 10.1172/jci77487
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimers disease. N. Engl. J. Med. 368, 107–116.
Kamino, K., Nagasaka, K., Imagawa, M., Yamamoto, H., Yoneda, H., Ueki, A., et al. (2000). Deficiency in mitochondrial aldehyde dehydrogenase increases the risk for late-onset Alzheimers disease in the Japanese population. Biochem. Biophys. Res. Commun. 273, 192–196. doi: 10.1006/bbrc.2000.2923
Kandel, N., Matos, J. O., and Tatulian, S. A. (2019). Structure of amyloid β(25-35) in lipid environment and cholesterol-dependent membrane pore formation. Sci. Rep. 9:2689. doi: 10.1038/s41598-019-38749-7
Kanekiyo, T., Xu, H., and Bu, G. (2014). ApoE and Aβ in Alzheimers disease: Accidental encounters or partners? Neuron 81, 740–754. doi: 10.1016/j.neuron.2014.01.045
Kang, J., and Rivest, S. (2012). Lipid metabolism and neuroinflammation in Alzheimers disease: A role for liver X receptors. Endocr. Rev. 33, 715–746. doi: 10.1210/er.2011-1049
Kang, S. S., Ebbert, M. T. W., Baker, K. E., Cook, C., Wang, X., Sens, J. P., et al. (2018). Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J. Exp. Med. 215, 2235–2245. doi: 10.1084/jem.20180653
Kanninen, K. M., Bister, N., Koistinaho, J., and Malm, T. (2016). Exosomes as new diagnostic tools in CNS diseases. Biochim. Biophys. Acta 1862, 403–410. doi: 10.1016/j.bbadis.2015.09.020
Kantarci, A., Aytan, N., Palaska, I., Stephens, D., Crabtree, L., Benincasa, C., et al. (2018). Combined administration of resolvin E1 and lipoxin A4 resolves inflammation in a murine model of Alzheimers disease. Exp. Neurol. 300, 111–120. doi: 10.1016/j.expneurol.2017.11.005
Karch, C. M., and Goate, A. M. (2015). Alzheimers disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. doi: 10.1016/j.biopsych.2014.05.006
Karten, B., Campenot, R. B., Vance, D. E., and Vance, J. E. (2006). Expression of ABCG1, but not ABCA1, correlates with cholesterol release by cerebellar astroglia. J. Biol. Chem. 281, 4049–4057. doi: 10.1074/jbc.M508915200
Kashiwaya, Y., Takeshima, T., Mori, N., Nakashima, K., Clarke, K., and Veech, R. L. (2000). D-beta-hydroxybutyrate protects neurons in models of Alzheimers and Parkinsons disease. Proc. Natl. Acad. Sci. U.S.A. 97, 5440–5444. doi: 10.1073/pnas.97.10.5440
Kastelowitz, N., and Yin, H. (2014). Exosomes and microvesicles: Identification and targeting by particle size and lipid chemical probes. Chembiochem 15, 923–928. doi: 10.1002/cbic.201400043
Kawarabayashi, T., Shoji, M., Younkin, L. H., Wen-Lang, L., Dickson, D. W., Murakami, T., et al. (2004). Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimers disease. J. Neurosci. 24, 3801–3809. doi: 10.1523/jneurosci.5543-03.2004
Kim, J., Yoon, H., Horie, T., Burchett, J. M., Restivo, J. L., Rotllan, N., et al. (2015). microRNA-33 regulates ApoE lipidation and amyloid-β metabolism in the brain. J. Neurosci. 35, 14717–14726. doi: 10.1523/jneurosci.2053-15.2015
Kim, W. S., Weickert, C. S., and Garner, B. (2008). Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J. Neurochem. 104, 1145–1166. doi: 10.1111/j.1471-4159.2007.05099.x
Kimoto, A., Ohnuma, T., Toda, A., Takebayashi, Y., Higashiyama, R., Tagata, Y., et al. (2017). Medium-chain triglycerides given in the early stage of mild-to-moderate Alzheimers disease enhance memory function. Psychogeriatrics 17, 520–521. doi: 10.1111/psyg.12257
Kleinberger, G., Yamanishi, Y., Suárez-Calvet, M., Czirr, E., Lohmann, E., Cuyvers, E., et al. (2014). TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 6:243ra86. doi: 10.1126/scitranslmed.3009093
Kober, D. L., and Brett, T. J. (2017). TREM2-ligand interactions in health and disease. J. Mol. Biol. 429, 1607–1629. doi: 10.1016/j.jmb.2017.04.004
Kojro, E., Gimpl, G., Lammich, S., Marz, W., and Fahrenholz, F. (2001). Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc. Natl. Acad. Sci. U.S.A. 98, 5815–5820. doi: 10.1073/pnas.081612998
Kok, E., Haikonen, S., Luoto, T., Huhtala, H., Goebeler, S., Haapasalo, H., et al. (2009). Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann. Neurol. 65, 650–657. doi: 10.1002/ana.21696
Koldamova, R. P., Lefterov, I. M., Staufenbiel, M., Wolfe, D., Huang, S., Glorioso, J. C., et al. (2005). The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimers disease. J. Biol. Chem. 280, 4079–4088. doi: 10.1074/jbc.M411420200
Koldamova, R., Fitz, N. F., and Lefterov, I. (2014). ATP-binding cassette transporter A1: From metabolism to neurodegeneration. Neurobiol. Dis. 72, 13–21. doi: 10.1016/j.nbd.2014.05.007
Kong, Y., Jiang, B., and Luo, X. (2018). Gut microbiota influences Alzheimers disease pathogenesis by regulating acetate in Drosophila model. Future Microbiol. 13, 1117–1128. doi: 10.2217/fmb-2018-0185
Kosicek, M., and Hecimovic, S. (2013). Phospholipids and Alzheimers disease: Alterations, mechanisms and potential biomarkers. Int. J. Mol. Sci. 14, 1310–1322. doi: 10.3390/ijms14011310
Kothari, V., Luo, Y., Tornabene, T., ONeill, A. M., Greene, M. W., Geetha, T., et al. (2017). High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 499–508. doi: 10.1016/j.bbadis.2016.10.006
Kracun, I., Kalanj, S., Cosovic, C., and Talan-Hranilovic, J. (1990). Brain gangliosides in Alzheimers disease. J. Hirnforsch. 31, 789–793.
Kracun, I., Rosner, H., Drnovsek, V., Heffer-Lauc, M., Cosović, C., and Lauc, G. (1991). Human brain gangliosides in development, aging and disease. Int. J. Dev. Biol. 35, 289–295.
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J., Damotte, V., Naj, A., et al. (2019). Genetic meta-analysis of diagnosed Alzheimers disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430.
Lambert, J. C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimers disease. Nat. Genet. 41, 1094–1099. doi: 10.1038/ng.439
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimers disease. Nat. Genet. 45, 1452–1458.
Landman, N., Jeong, S. Y., Shin, S. Y., Voronov, S. V., Serban, G., Kang, M. S., et al. (2006). Presenilin mutations linked to familial Alzheimers disease cause an imbalance in phosphatidylinositol 4,5-bisphosphate metabolism. Proc. Natl. Acad. Sci. U.S.A. 103, 19524–19529. doi: 10.1073/pnas.0604954103
Lane, R. M., and Farlow, M. R. (2005). Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimers disease. J. Lipid Res. 46, 949–968. doi: 10.1194/jlr.M400486-JLR200
Lanfranco, M. F., Ng, C. A., and Rebeck, G. W. (2020). ApoE lipidation as a therapeutic target in Alzheimers disease. Int. J. Mol. Sci. 21:6336. doi: 10.3390/ijms21176336
Laulagnier, K., Motta, C., Hamdi, S., Roy, S., Fauvelle, F., Pageaux, J. F., et al. (2004). Mast cell- and dendritic cell-derived exosomes display a specific lipid composition and an unusual membrane organization. Biochem. J. 380, 161–171. doi: 10.1042/bj20031594
Lee, S. J., Liyanage, U., Bickel, P. E., Xia, W., Lansbury, P. T. Jr., and Kosik, K. S. (1998). A detergent-insoluble membrane compartment contains A beta in vivo. Nat. Med. 4, 730–734. doi: 10.1038/nm0698-730
Li, G., Mayer, C. L., Morelli, D., Millard, S. P., Raskind, W. H., Petrie, E. C., et al. (2017). Effect of simvastatin on CSF Alzheimer disease biomarkers in cognitively normal adults. Neurology 89, 1251–1255. doi: 10.1212/wnl.0000000000004392
Li, L. O., Klett, E. L., and Coleman, R. A. (2010). Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim. Biophys. Acta 1801, 246–251. doi: 10.1016/j.bbalip.2009.09.024
Li, G., Shofer, J. B., Rhew, I. C., Kukull, W. A., Peskind, E. R., McCormick, W., et al. (2010). Age-varying association between statin use and incident Alzheimers disease. J. Am. Geriatr. Soc. 58, 1311–1317. doi: 10.1111/j.1532-5415.2010.02906.x
Li, R. Y., Qin, Q., Yang, H. C., Wang, Y. Y., Mi, Y. X., Yin, Y. S., et al. (2022). TREM2 in the pathogenesis of AD: A lipid metabolism regulator and potential metabolic therapeutic target. Mol. Neurodegener. 17:40. doi: 10.1186/s13024-022-00542-y
Li, Y., Lu, W., Marzolo, M. P., and Bu, G. (2001). Differential functions of members of the low density lipoprotein receptor family suggested by their distinct endocytosis rates. J. Biol. Chem. 276, 18000–18006. doi: 10.1074/jbc.M101589200
Liang, X., Wang, Q., Hand, T., Wu, L., Breyer, R. M., Montine, T. J., et al. (2005). Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimers disease. J. Neurosci. 25, 10180–10187. doi: 10.1523/jneurosci.3591-05.2005
Liang, Y., Lin, S., Beyer, T. P., Zhang, Y., Wu, X., Bales, K. R., et al. (2004). A liver X receptor and retinoid X receptor heterodimer mediates apolipoprotein E expression, secretion and cholesterol homeostasis in astrocytes. J. Neurochem. 88, 623–634. doi: 10.1111/j.1471-4159.2004.02183.x
Liao, F., Li, A., Xiong, M., Bien-Ly, N., Jiang, H., Zhang, Y., et al. (2018). Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Invest. 128, 2144–2155. doi: 10.1172/jci96429
Lin, Y. T., Seo, J., Gao, F., Feldman, H. M., Wen, H. L., Penney, J., et al. (2018). APOE4 causes widespread molecular and cellular alterations associated with Alzheimers disease phenotypes in human iPSC-derived brain cell types. Neuron 98:1141–1154.e7. doi: 10.1016/j.neuron.2018.05.008
Linetti, A., Fratangeli, A., Taverna, E., Valnegri, P., Francolini, M., Cappello, V., et al. (2010). Cholesterol reduction impairs exocytosis of synaptic vesicles. J. Cell Sci. 123, 595–605. doi: 10.1242/jcs.060681
Liu, C. C., Liu, C. C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Liu, L., MacKenzie, K. R., Putluri, N., Maletić-Savatić, M., and Bellen, H. J. (2017). The glia-neuron lactate shuttle and elevated ros promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab. 26:719–737.e6. doi: 10.1016/j.cmet.2017.08.024
Liu, C. C., Zhao, N., Fu, Y., Wang, N., Linares, C., Tsai, C. W., et al. (2017). ApoE4 accelerates early seeding of amyloid pathology. Neuron 96:1024–1032.e3. doi: 10.1016/j.neuron.2017.11.013
Liu, L., Xu, J., Huang, X., Wang, Y., Ma, X., Wang, X., et al. (2024). DHA dietary intervention caused different hippocampal lipid and protein profile in ApoE-/- and C57BL/6J mice. Biomed. Pharmacother. 177:117088. doi: 10.1016/j.biopha.2024.117088
Liu, P., Wang, Z. H., Kang, S. S., Liu, X., Xia, Y., Chan, C. B., et al. (2022). High-fat diet-induced diabetes couples to Alzheimers disease through inflammation-activated C/EBPβ/AEP pathway. Mol. Psychiatry 27, 3396–3409. doi: 10.1038/s41380-022-01600-z
Liu, Q., Trotter, J., Zhang, J., Peters, M. M., Cheng, H., Bao, J., et al. (2010). Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J. Neurosci. 30, 17068–17078. doi: 10.1523/jneurosci.4067-10.2010
Liu, Q., Zerbinatti, C. V., Zhang, J., Hoe, H. S., Wang, B., Cole, S. L., et al. (2007). Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 56, 66–78. doi: 10.1016/j.neuron.2007.08.008
Liu, Y., Zhong, X., Shen, J., Jiao, L., Tong, J., Zhao, W., et al. (2020). Elevated serum TC and LDL-C levels in Alzheimers disease and mild cognitive impairment: A meta-analysis study. Brain Res. 1727:146554. doi: 10.1016/j.brainres.2019.146554
Luo, R., Su, L. Y., Li, G., Yang, J., Liu, Q., Yang, L. X., et al. (2020). Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 16, 52–69. doi: 10.1080/15548627.2019.1596488
Ma, L., and Nelson, E. R. (2019). Oxysterols and nuclear receptors. Mol. Cell Endocrinol. 484, 42–51. doi: 10.1016/j.mce.2019.01.016
Mahley, R. W. (1988). Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 240, 622–630. doi: 10.1126/science.3283935
Mahley, R. W. (2016). Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 94, 739–746. doi: 10.1007/s00109-016-1427-y
Malaplate-Armand, C., Florent-Béchard, S., Youssef, I., Koziel, V., Sponne, I., Kriem, B., et al. (2006). Soluble oligomers of amyloid-beta peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol. Dis. 23, 178–189. doi: 10.1016/j.nbd.2006.02.010
Mandrekar-Colucci, S., Karlo, J. C., and Landreth, G. E. (2012). Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimers disease. J. Neurosci. 32, 10117–10128. doi: 10.1523/jneurosci.5268-11.2012
Marin, R., Fabelo, N., Fernández-Echevarría, C., Canerina-Amaro, A., Rodríguez-Barreto, D., Quinto-Alemany, D., et al. (2016). Lipid raft alterations in aged-associated neuropathologies. Curr. Alzheimer Res. 13, 973–984. doi: 10.2174/1567205013666160314150017
Martin, P. M., Gopal, E., Ananth, S., Zhuang, L., Itagaki, S., Prasad, B. M., et al. (2006). Identity of SMCT1 (SLC5A8) as a neuron-specific Na+-coupled transporter for active uptake of L-lactate and ketone bodies in the brain. J. Neurochem. 98, 279–288. doi: 10.1111/j.1471-4159.2006.03878.x
Martín, V., Fabelo, N., Santpere, G., Puig, B., Marín, R., Ferrer, I., et al. (2010). Lipid alterations in lipid rafts from Alzheimers disease human brain cortex. J. Alzheimers Dis. 19, 489–502. doi: 10.3233/jad-2010-1242
Matsumura, S., Minamisawa, T., Suga, K., Kishita, H., Akagi, T., Ichiki, T., et al. (2019). Subtypes of tumour cell-derived small extracellular vesicles having differently externalized phosphatidylserine. J. Extracell. Vesicles 8:1579541. doi: 10.1080/20013078.2019.1579541
Matsuzaki, K. (2020). Aβ-ganglioside interactions in the pathogenesis of Alzheimers disease. Biochim. Biophys. Acta Biomembr. 1862:183233. doi: 10.1016/j.bbamem.2020.183233
McFarland, A. J., Davey, A. K., McDermott, C. M., Grant, G. D., Lewohl, J., and Anoopkumar-Dukie, S. (2018). Differences in statin associated neuroprotection corresponds with either decreased production of IL-1β or TNF-α in an in vitro model of neuroinflammation-induced neurodegeneration. Toxicol. Appl. Pharmacol. 344, 56–73. doi: 10.1016/j.taap.2018.03.005
Melo, H. M., Seixas, da Silva, G. D. S., SantAna, M. R., Teixeira, C. V. L., Clarke, J. R., et al. (2020). Palmitate is increased in the cerebrospinal fluid of humans with obesity and induces memory impairment in mice via pro-inflammatory TNF-α. Cell Rep. 30:2180–2194.e8. doi: 10.1016/j.celrep.2020.01.072
Michikawa, M., Fan, Q. W., Isobe, I., and Yanagisawa, K. (2000). Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J. Neurochem. 74, 1008–1016. doi: 10.1046/j.1471-4159.2000.0741008.x
Miller, Y. I., Navia-Pelaez, J. M., Corr, M., and Yaksh, T. L. (2020). Lipid rafts in glial cells: Role in neuroinflammation and pain processing. J. Lipid Res. 61, 655–666. doi: 10.1194/jlr.TR119000468
Minagawa, H., Gong, J. S., Jung, C. G., Watanabe, A., Lund-Katz, S., Phillips, M. C., et al. (2009). Mechanism underlying apolipoprotein E (ApoE) isoform-dependent lipid efflux from neural cells in culture. J. Neurosci. Res. 87, 2498–2508. doi: 10.1002/jnr.22073
Minhas, P. S., Latif-Hernandez, A., McReynolds, M. R., Durairaj, A. S., Wang, Q., Rubin, A., et al. (2021). Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature 590, 122–128. doi: 10.1038/s41586-020-03160-0
Mitchell, R. W., On, N. H., Del Bigio, M. R., Miller, D. W., and Hatch, G. M. (2011). Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J. Neurochem. 117, 735–746. doi: 10.1111/j.1471-4159.2011.07245.x
Montagne, A., Zhao, Z., and Zlokovic, B. V. (2017). Alzheimers disease: A matter of blood-brain barrier dysfunction? J. Exp. Med. 214, 3151–3169. doi: 10.1084/jem.20171406
Montine, T. J., Sidell, K. R., Crews, B. C., Markesbery, W. R., Marnett, L. J., and Roberts, L. J. II, et al. (1999). Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology 53, 1495–1498. doi: 10.1212/wnl.53.7.1495
Morell, P., and Jurevics, H. (1996). Origin of cholesterol in myelin. Neurochem. Res. 21, 463–470. doi: 10.1007/bf02527711
Mori, T., Paris, D., Town, T., Rojiani, A. M., Sparks, D. L., Delledonne, A., et al. (2001). Cholesterol accumulates in senile plaques of Alzheimer disease patients and in transgenic APP(SW) mice. J. Neuropathol. Exp. Neurol. 60, 778–785. doi: 10.1093/jnen/60.8.778
Morris, M. C., Evans, D. A., Bienias, J. L., Tangney, C. C., Bennett, D. A., Wilson, R. S., et al. (2003). Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch. Neurol. 60, 940–946. doi: 10.1001/archneur.60.7.940
Mosconi, L., Pupi, A., and De Leon, M. J. (2008). Brain glucose hypometabolism and oxidative stress in preclinical Alzheimers disease. Ann. N. Y. Acad. Sci. 1147, 180–195. doi: 10.1196/annals.1427.007
Mosconi, L., Sorbi, S., Nacmias, B., De Cristofaro, M. T., Fayyaz, M., Bracco, L., et al. (2004). Age and ApoE genotype interaction in Alzheimers disease: An FDG-PET study. Psychiatry Res. 130, 141–151. doi: 10.1016/j.pscychresns.2003.12.005
Moulton, M. J., Barish, S., Ralhan, I., Chang, J., Goodman, L. D., Harland, J. G., et al. (2021). Neuronal ROS-induced glial lipid droplet formation is altered by loss of Alzheimers disease-associated genes. Proc. Natl. Acad. Sci. U.S.A. 118:e2112095118. doi: 10.1073/pnas.2112095118
Mourino-Alvarez, L., Juarez-Alia, C., Sastre-Oliva, T., Perales-Sánchez, I., Hernandez-Fernandez, G., Chicano-Galvez, E., et al. (2024). Dysregulation of lipid metabolism serves as a link between Alzheimers and cardiovascular disease, as witnessed in a cross-sectional study. Aging Dis. doi: 10.14336/ad.2024.0434 [Epub ahead of print].
Moutinho, M., and Landreth, G. E. (2017). Therapeutic potential of nuclear receptor agonists in Alzheimers disease. J. Lipid Res. 58, 1937–1949. doi: 10.1194/jlr.R075556
Namba, Y., Tomonaga, M., Kawasaki, H., Otomo, E., and Ikeda, K. (1991). Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimers disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 541, 163–166. doi: 10.1016/0006-8993(91)91092-f
Namjoshi, D. R., Martin, G., Donkin, J., Wilkinson, A., Stukas, S., Fan, J., et al. (2013). The liver X receptor agonist GW3965 improves recovery from mild repetitive traumatic brain injury in mice partly through apolipoprotein E. PLoS One 8:e53529. doi: 10.1371/journal.pone.0053529
Nation, D. A., Sweeney, M. D., Montagne, A., Sagare, A. P., DOrazio, L. M., Pachicano, M., et al. (2019). Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 25, 270–276. doi: 10.1038/s41591-018-0297-y
Naudí, A., Cabré, R., Jové, M., Ayala, V., Gonzalo, H., Portero-Otín, M., et al. (2015). Lipidomics of human brain aging and Alzheimers disease pathology. Int. Rev. Neurobiol. 122, 133–189. doi: 10.1016/bs.irn.2015.05.008
Newman, J. C., Covarrubias, A. J., Zhao, M., Yu, X., Gut, P., Ng, C. P., et al. (2017). Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab. 26:547–557.e8. doi: 10.1016/j.cmet.2017.08.004
Nguyen, T. B., Louie, S. M., Daniele, J. R., Tran, Q., Dillin, A., Zoncu, R., et al. (2017). DGAT1-dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy. Dev Cell 42:9–21.e5. doi: 10.1016/j.devcel.2017.06.003
Nickels, J. D., Smith, M. D., Alsop, R. J., Himbert, S., Yahya, A., Cordner, D., et al. (2019). Lipid rafts: Buffers of cell membrane physical properties. J. Phys. Chem. B 123, 2050–2056. doi: 10.1021/acs.jpcb.8b12126
Nieweg, K., Schaller, H., and Pfrieger, F. W. (2009). Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J. Neurochem. 109, 125–134. doi: 10.1111/j.1471-4159.2009.05917.x
Nitsch, R. M., Blusztajn, J. K., Pittas, A. G., Slack, B. E., Growdon, J. H., and Wurtman, R. J. (1992). Evidence for a membrane defect in Alzheimer disease brain. Proc. Natl. Acad. Sci. U.S.A. 89, 1671–1675. doi: 10.1073/pnas.89.5.1671
Niu, Z., Zhang, Z., Zhao, W., and Yang, J. (2018). Interactions between amyloid β peptide and lipid membranes. Biochim. Biophys. Acta Biomembr. 1860, 1663–1669. doi: 10.1016/j.bbamem.2018.04.004
Nordestgaard, L. T., Tybjærg-Hansen, A., Nordestgaard, B. G., and Frikke-Schmidt, R. (2015). Loss-of-function mutation in ABCA1 and risk of Alzheimers disease and cerebrovascular disease. Alzheimers Dement. 11, 1430–1438. doi: 10.1016/j.jalz.2015.04.006
Nugent, A. A., Lin, K., van Lengerich, B., Lianoglou, S., Przybyla, L., Davis, S. S., et al. (2020). TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105:837-854.e9. doi: 10.1016/j.neuron.2019.12.007
Nuutinen, T., Suuronen, T., Kauppinen, A., and Salminen, A. (2009). Clusterin: A forgotten player in Alzheimers disease. Brain Res. Rev. 61, 89–104. doi: 10.1016/j.brainresrev.2009.05.007
Olivares, A. M., Moreno-Ramos, O. A., and Haider, N. B. (2015). Role of nuclear receptors in central nervous system development and associated diseases. J. Exp. Neurosci. 9, 93–121. doi: 10.4137/jen.s25480
O’Reilly, J. A., and Lynch, M. (2012). Rosiglitazone improves spatial memory and decreases insoluble Aβ(1-42) in APP/PS1 mice. J. Neuroimmune Pharmacol. 7, 140–144. doi: 10.1007/s11481-011-9282-7
Orth, M., and Bellosta, S. (2012). Cholesterol: Its regulation and role in central nervous system disorders. Cholesterol 2012:292598. doi: 10.1155/2012/292598
Oshima, N., Morishima-Kawashima, M., Yamaguchi, H., Yoshimura, M., Sugihara, S., Khan, K., et al. (2001). Accumulation of amyloid beta-protein in the low-density membrane domain accurately reflects the extent of beta-amyloid deposition in the brain. Am. J. Pathol. 158, 2209–2218. doi: 10.1016/s0002-9440(10)64693-7
Ouyang, S., Hsuchou, H., Kastin, A. J., Wang, Y., Yu, C., and Pan, W. (2014). Diet-induced obesity suppresses expression of many proteins at the blood-brain barrier. J. Cereb. Blood Flow Metab. 34, 43–51. doi: 10.1038/jcbfm.2013.166
Paloneva, J., Manninen, T., Christman, G., Hovanes, K., Mandelin, J., Adolfsson, R., et al. (2002). Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656–662. doi: 10.1086/342259
Pedersen, W. A., McMillan, P. J., Kulstad, J. J., Leverenz, J. B., Craft, S., and Haynatzki, G. R. (2006). Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp. Neurol. 199, 265–273. doi: 10.1016/j.expneurol.2006.01.018
Peña, V. B., Bonini, I. C., Antollini, S. S., Kobayashi, T., and Barrantes, F. J. (2011). alpha 7-type acetylcholine receptor localization and its modulation by nicotine and cholesterol in vascular endothelial cells. J. Cell Biochem. 112, 3276–3288. doi: 10.1002/jcb.23254
Pera, M., Larrea, D., Guardia-Laguarta, C., Montesinos, J., Velasco, K. R., Agrawal, R. R., et al. (2017). Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. Embo J. 36, 3356–3371. doi: 10.15252/embj.201796797
Pettegrew, J. W., Panchalingam, K., Hamilton, R. L., and McClure, R. J. (2001). Brain membrane phospholipid alterations in Alzheimers disease. Neurochem. Res. 26, 771–782. doi: 10.1023/a:1011603916962
Pfrieger, F. W., and Ungerer, N. (2011). Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 50, 357–371. doi: 10.1016/j.plipres.2011.06.002
Pillot, T., Goethals, M., Najib, J., Labeur, C., Lins, L., Chambaz, J., et al. (1999). Beta-amyloid peptide interacts specifically with the carboxy-terminal domain of human apolipoprotein E: Relevance to Alzheimers disease. J. Neurochem. 72, 230–237. doi: 10.1046/j.1471-4159.1999.0720230.x
Pistollato, F., Iglesias, R. C., Ruiz, R., Aparicio, S., Crespo, J., Lopez, L. D., et al. (2018). Nutritional patterns associated with the maintenance of neurocognitive functions and the risk of dementia and Alzheimers disease: A focus on human studies. Pharmacol. Res. 131, 32–43. doi: 10.1016/j.phrs.2018.03.012
Pitas, R. E., Boyles, J. K., Lee, S. H., Foss, D., and Mahley, R. W. (1987). Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim. Biophys. Acta 917, 148–161. doi: 10.1016/0005-2760(87)90295-5
Poliakova, T., and Wellington, C. L. (2023). Roles of peripheral lipoproteins and cholesteryl ester transfer protein in the vascular contributions to cognitive impairment and dementia. Mol. Neurodegener. 18:86. doi: 10.1186/s13024-023-00671-y
Popp, J., Meichsner, S., Kölsch, H., Lewczuk, P., Maier, W., Kornhuber, J., et al. (2013). Cerebral and extracerebral cholesterol metabolism and CSF markers of Alzheimers disease. Biochem. Pharmacol. 86, 37–42. doi: 10.1016/j.bcp.2012.12.007
Pottier, C., Hannequin, D., Coutant, S., Rovelet-Lecrux, A., Wallon, D., Rousseau, S., et al. (2012). High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol. Psychiatry 17, 875–879. doi: 10.1038/mp.2012.15
Prince, M., Wimo, A., Guerchet, M., Ali, G.-C., Wu, Y.-T., and Prina, M. (2015). World Alzheimer report 2015. The global impact of dementia. An analysis of prevalence, incidence, cost and trends. London: Alzheimers Disease International.
Puglielli, L., Ellis, B. C., Saunders, A. J., and Kovacs, D. M. (2003). Ceramide stabilizes beta-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid beta-peptide biogenesis. J. Biol. Chem. 278, 19777–19783. doi: 10.1074/jbc.M300466200
Puglielli, L., Konopka, G., Pack-Chung, E., Ingano, L. A., Berezovska, O., Hyman, B. T., et al. (2001). Acyl-coenzyme A: Cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat. Cell Biol. 3, 905–912. doi: 10.1038/ncb1001-905
Qi, G., Mi, Y., Shi, X., Gu, H., Brinton, R. D., and Yin, F. (2021). ApoE4 impairs neuron-astrocyte coupling of fatty acid metabolism. Cell Rep. 34:108572. doi: 10.1016/j.celrep.2020.108572
Rankin, C. A., Sun, Q., and Gamblin, T. C. (2007). Tau phosphorylation by GSK-3β promotes tangle-like filament morphology. Mol. Neurodegener. 2:12. doi: 10.1186/1750-1326-2-12
Rawat, V., Wang, S., Sima, J., Bar, R., Liraz, O., Gundimeda, U., et al. (2019). ApoE4 alters ABCA1 membrane trafficking in astrocytes. J. Neurosci. 39, 9611–9622. doi: 10.1523/jneurosci.1400-19.2019
Rebeck, G. W., Reiter, J. S., Strickland, D. K., and Hyman, B. T. (1993). Apolipoprotein E in sporadic Alzheimers disease: Allelic variation and receptor interactions. Neuron 11, 575–580. doi: 10.1016/0896-6273(93)90070-8
Record, M., Carayon, K., Poirot, M., and Silvente-Poirot, S. (2014). Exosomes as new vesicular lipid transporters involved in cell-cell communication and various pathophysiologies. Biochim. Biophys. Acta 1841, 108–120. doi: 10.1016/j.bbalip.2013.10.004
Record, M., Silvente-Poirot, S., Poirot, M., and Wakelam, M. J. O. (2018). Extracellular vesicles: Lipids as key components of their biogenesis and functions. J. Lipid Res. 59, 1316–1324. doi: 10.1194/jlr.E086173
Reddy, J. K., and Hashimoto, T. (2001). Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: An adaptive metabolic system. Annu. Rev. Nutr. 21, 193–230. doi: 10.1146/annurev.nutr.21.1.193
Reiman, E. M., Arboleda-Velasquez, J. F., Quiroz, Y. T., Huentelman, M. J., Beach, T. G., Caselli, R. J., et al. (2020). Exceptionally low likelihood of Alzheimers dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat. Commun. 11:667. doi: 10.1038/s41467-019-14279-8
Reiman, E. M., Chen, K., Alexander, G. E., Caselli, R. J., Bandy, D., Osborne, D., et al. (2004). Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimers dementia. Proc. Natl. Acad. Sci. U.S.A. 101, 284–289. doi: 10.1073/pnas.2635903100
Reiman, E. M., Chen, K., Alexander, G. E., Caselli, R. J., Bandy, D., Osborne, D., et al. (2005). Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism. Proc. Natl. Acad. Sci. U.S.A. 102, 8299–8302. doi: 10.1073/pnas.0500579102
Rhea, E. M., Salameh, T. S., Logsdon, A. F., Hanson, A. J., Erickson, M. A., and Banks, W. A. (2017). Blood-brain barriers in obesity. AAPS J. 19, 921–930. doi: 10.1208/s12248-017-0079-3
Ricciotti, E., and FitzGerald, G. A. (2011). Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 31, 986–1000. doi: 10.1161/atvbaha.110.207449
Ricobaraza, A., Cuadrado-Tejedor, M., Marco, S., Pérez-Otaño, I., and García-Osta, A. (2012). Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 22, 1040–1050. doi: 10.1002/hipo.20883
Riddell, D. R., Christie, G., Hussain, I., and Dingwall, C. (2001). Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 11, 1288–1293. doi: 10.1016/s0960-9822(01)00394-3
Rogaeva, E., Meng, Y., Lee, J. H., Gu, Y., Kawarai, T., Zou, F., et al. (2007). The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 39, 168–177.
Rossner, S. (2004). New players in old amyloid precursor protein-processing pathways. Int. J. Dev. Neurosci. 22, 467–474. doi: 10.1016/j.ijdevneu.2004.07.004
Rusek, M., Pluta, R., Ułamek-Kozioł, M., and Czuczwar, S. J. (2019). Ketogenic diet in Alzheimers disease. Int. J. Mol. Sci. 20:3892. doi: 10.3390/ijms20163892
Sanan, D. A., Weisgraber, K. H., Russell, S. J., Mahley, R. W., Huang, D., Saunders, A., et al. (1994). Apolipoprotein E associates with beta amyloid peptide of Alzheimers disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J. Clin. Invest. 94, 860–869. doi: 10.1172/jci117407
Sano, M., Bell, K. L., Galasko, D., Galvin, J. E., Thomas, R. G., van Dyck, C. H., et al. (2011). A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology 77, 556–563. doi: 10.1212/WNL.0b013e318228bf11
Sastre, M., Dewachter, I., Landreth, G. E., Willson, T. M., Klockgether, T., van Leuven, F., et al. (2003). Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J. Neurosci. 23, 9796–9804. doi: 10.1523/jneurosci.23-30-09796.2003
Sastry, P. S. (1985). Lipids of nervous tissue: Composition and metabolism. Prog. Lipid Res. 24, 69–176. doi: 10.1016/0163-7827(85)90011-6
Scarmeas, N., Habeck, C. G., Hilton, J., Anderson, K. E., Flynn, J., Park, A., et al. (2005). APOE related alterations in cerebral activation even at college age. J. Neurol. Neurosurg. Psychiatry 76, 1440–1444. doi: 10.1136/jnnp.2004.053645
Schmechel, D. E., Saunders, A. M., Strittmatter, W. J., Crain, B. J., Hulette, C. M., Joo, S. H., et al. (1993). Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 9649–9653. doi: 10.1073/pnas.90.20.9649
Schönfeld, P., and Reiser, G. (2017). Brain energy metabolism spurns fatty acids as fuel due to their inherent mitotoxicity and potential capacity to unleash neurodegeneration. Neurochem. Int. 109, 68–77. doi: 10.1016/j.neuint.2017.03.018
Schultz, B. G., Patten, D. K., and Berlau, D. J. (2018). The role of statins in both cognitive impairment and protection against dementia: A tale of two mechanisms. Transl. Neurodegener. 7:5. doi: 10.1186/s40035-018-0110-3
Schultz, J. R., Tu, H., Luk, A., Repa, J. J., Medina, J. C., Li, L., et al. (2000). Role of LXRs in control of lipogenesis. Genes Dev. 14, 2831–2838. doi: 10.1101/gad.850400
Segarra, M., Aburto, M. R., and Acker-Palmer, A. (2021). Blood-brain barrier dynamics to maintain brain homeostasis. Trends Neurosci. 44, 393–405. doi: 10.1016/j.tins.2020.12.002
Sezgin, E., Levental, I., Mayor, S., and Eggeling, C. (2017). The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 18, 361–374. doi: 10.1038/nrm.2017.16
Sharma, R., Huang, X., Brekken, R. A., and Schroit, A. J. (2017). Detection of phosphatidylserine-positive exosomes for the diagnosis of early-stage malignancies. Br. J. Cancer 117, 545–552. doi: 10.1038/bjc.2017.183
Shi, J., Wang, Q., Johansson, J. U., Liang, X., Woodling, N. S., Priyam, P., et al. (2012). Inflammatory prostaglandin E2 signaling in a mouse model of Alzheimer disease. Ann. Neurol. 72, 788–798. doi: 10.1002/ana.23677
Singh, R., and Cuervo, A. M. (2012). Lipophagy: Connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012:282041. doi: 10.1155/2012/282041
Skoumalová, A., Ivica, J., Santorová, P., Topinková, E., and Wilhelm, J. (2011). The lipid peroxidation products as possible markers of Alzheimers disease in blood. Exp. Gerontol. 46, 38–42. doi: 10.1016/j.exger.2010.09.015
Small, D. H., Michaelson, S., and Sberna, G. (1996). Non-classical actions of cholinesterases: Role in cellular differentiation, tumorigenesis and Alzheimers disease. Neurochem. Int. 28, 453–483. doi: 10.1016/0197-0186(95)00099-2
Small, S. A. (2001). Age-related memory decline: Current concepts and future directions. Arch. Neurol. 58, 360–364. doi: 10.1001/archneur.58.3.360
Small, S. A., Chawla, M. K., Buonocore, M., Rapp, P. R., and Barnes, C. A. (2004). Imaging correlates of brain function in monkeys and rats isolates a hippocampal subregion differentially vulnerable to aging. Proc. Natl. Acad. Sci. U.S.A. 101, 7181–7186. doi: 10.1073/pnas.0400285101
Snowden, S. G., Ebshiana, A. A., Hye, A., An, Y., Pletnikova, O., OBrien, R., et al. (2017). Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: A nontargeted metabolomic study. PLoS Med. 14:e1002266. doi: 10.1371/journal.pmed.1002266
Söderberg, M., Edlund, C., Alafuzoff, I., Kristensson, K., and Dallner, G. (1992). Lipid composition in different regions of the brain in Alzheimers disease/senile dementia of Alzheimers type. J. Neurochem. 59, 1646–1653. doi: 10.1111/j.1471-4159.1992.tb10994.x
Solfrizzi, V., Custodero, C., Lozupone, M., Imbimbo, B. P., Valiani, V., Agosti, P., et al. (2017). Relationships of dietary patterns, foods, and micro- and macronutrients with alzheimers disease and late-life cognitive disorders: A systematic review. J. Alzheimers Dis. 59, 815–849. doi: 10.3233/jad-170248
Song, T., Song, X., Zhu, C., Patrick, R., Skurla, M., Santangelo, I., et al. (2021). Mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic alterations in the progression of Alzheimers disease: A meta-analysis of in vivo magnetic resonance spectroscopy studies. Ageing Res. Rev. 72:101503. doi: 10.1016/j.arr.2021.101503
Stokes, C. E., and Hawthorne, J. N. (1987). Reduced phosphoinositide concentrations in anterior temporal cortex of Alzheimer-diseased brains. J. Neurochem. 48, 1018–1021. doi: 10.1111/j.1471-4159.1987.tb05619.x
Su, H., Rustam, Y. H., Masters, C. L., Makalic, E., McLean, C. A., Hill, A. F., et al. (2021). Characterization of brain-derived extracellular vesicle lipids in Alzheimers disease. J. Extracell. Vesicles 10:e12089. doi: 10.1002/jev2.12089
Tai, L. M., Koster, K. P., Luo, J., Lee, S. H., Wang, Y. T., Collins, N. C., et al. (2014). Amyloid-β pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J. Biol. Chem. 289, 30538–30555. doi: 10.1074/jbc.M114.600833
Tan, J. Z. A., and Gleeson, P. A. (2019). The role of membrane trafficking in the processing of amyloid precursor protein and production of amyloid peptides in Alzheimers disease. Biochim. Biophys. Acta Biomembr. 1861, 697–712. doi: 10.1016/j.bbamem.2018.11.013
Tan, J., McKenzie, C., Potamitis, M., Thorburn, A. N., Mackay, C. R., and Macia, L. (2014). The role of short-chain fatty acids in health and disease. Adv. Immunol. 121, 91–119. doi: 10.1016/b978-0-12-800100-4.00003-9
Tesseur, I., Van Dorpe, J., Bruynseels, K., Bronfman, F., Sciot, R., Van Lommel, A., et al. (2000). Prominent axonopathy and disruption of axonal transport in transgenic mice expressing human apolipoprotein E4 in neurons of brain and spinal cord. Am. J. Pathol. 157, 1495–1510. doi: 10.1016/s0002-9440(10)64788-8
Thangavel, R., Kempuraj, D., Zaheer, S., Raikwar, S., Ahmed, M. E., Selvakumar, G. P., et al. (2017). Glia maturation factor and mitochondrial uncoupling proteins 2 and 4 expression in the temporal cortex of Alzheimers disease brain. Front. Aging Neurosci. 9:150. doi: 10.3389/fnagi.2017.00150
Toledo, E. M., and Inestrosa, N. C. (2010). Activation of WNT signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimers disease. Mol. Psychiatry 27:228. doi: 10.1038/mp.2009.72
Tong, Y., Sun, Y., Tian, X., Zhou, T., Wang, H., Zhang, T., et al. (2015). Phospholipid transfer protein (PLTP) deficiency accelerates memory dysfunction through altering amyloid precursor protein (APP) processing in a mouse model of Alzheimers disease. Hum. Mol. Genet. 24, 5388–5403. doi: 10.1093/hmg/ddv262
Tracey, T. J., Steyn, F. J., Wolvetang, E. J., and Ngo, S. T. (2018). Neuronal lipid metabolism: Multiple pathways driving functional outcomes in health and disease. Front. Mol. Neurosci. 11:10. doi: 10.3389/fnmol.2018.00010
Tzimopoulou, S., Cunningham, V. J., Nichols, T. E., Searle, G., Bird, N. P., Mistry, P., et al. (2010). A multi-center randomized proof-of-concept clinical trial applying [18F]FDG-PET for evaluation of metabolic therapy with rosiglitazone XR in mild to moderate Alzheimers disease. J. Alzheimers Dis. 22, 1241–1256. doi: 10.3233/jad-2010-100939
Ulland, T. K., and Colonna, M. (2018). TREM2 - a key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 14, 667–675. doi: 10.1038/s41582-018-0072-1
van den Brink, A. C., Brouwer-Brolsma, E. M., Berendsen, A. A. M., and van de Rest, O. (2019). The mediterranean, dietary approaches to stop hypertension (DASH), and mediterranean-DASH intervention for neurodegenerative delay (MIND) diets are associated with less cognitive decline and a lower risk of Alzheimers disease-a review. Adv. Nutr. 10, 1040–1065. doi: 10.1093/advances/nmz054
Vance, J. E. (2014). MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 1841, 595–609. doi: 10.1016/j.bbalip.2013.11.014
Vance, J. E., Pan, D., Campenot, R. B., Bussière, M., and Vance, D. E. (1994). Evidence that the major membrane lipids, except cholesterol, are made in axons of cultured rat sympathetic neurons. J. Neurochem. 62, 329–337. doi: 10.1046/j.1471-4159.1994.62010329.x
Verdier, Y., Zarándi, M., and Penke, B. (2004). Amyloid beta-peptide interactions with neuronal and glial cell plasma membrane: Binding sites and implications for Alzheimers disease. J. Pept. Sci. 10, 229–248. doi: 10.1002/psc.573
von Maydell, D., Wright, S., Bonner, J. M., Staab, C., Spitaleri, A., Liu, L., et al. (2024). Single-cell atlas of ABCA7 loss-of-function reveals impaired neuronal respiration via choline-dependent lipid imbalances. bioRxiv [Preprint]. doi: 10.1101/2023.09.05.556135
Wahrle, S. E., Jiang, H., Parsadanian, M., Legleiter, J., Han, X., Fryer, J. D., et al. (2004). ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem. 279, 40987–40993. doi: 10.1074/jbc.M407963200
Wahrle, S. E., Shah, A. R., Fagan, A. M., Smemo, S., Kauwe, J. S., Grupe, A., et al. (2007). Apolipoprotein E levels in cerebrospinal fluid and the effects of ABCA1 polymorphisms. Mol. Neurodegener. 2:7. doi: 10.1186/1750-1326-2-7
Wahrle, S., Das, P., Nyborg, A. C., McLendon, C., Shoji, M., Kawarabayashi, T., et al. (2002). Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 9, 11–23. doi: 10.1006/nbdi.2001.0470
Wang, Q., Yuan, J., Yu, Z., Lin, L., Jiang, Y., Cao, Z., et al. (2018). FGF21 attenuates high-fat diet-induced cognitive impairment via metabolic regulation and anti-inflammation of obese mice. Mol. Neurobiol. 55, 4702–4717. doi: 10.1007/s12035-017-0663-7
Wang, C., Najm, R., Xu, Q., Jeong, D. E., Walker, D., Balestra, M. E., et al. (2018). Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 24, 647–657. doi: 10.1038/s41591-018-0004-z
Wang, H., Kulas, J. A., Wang, C., Holtzman, D. M., Ferris, H. A., and Hansen, S. B. (2021). Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc. Natl. Acad. Sci. U.S.A. 118:e2102191118. doi: 10.1073/pnas.2102191118
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimers disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Wang, L., Cheng, S., Yin, Z., Xu, C., Lu, S., Hou, J., et al. (2015). Conditional inactivation of Akt three isoforms causes tau hyperphosphorylation in the brain. Mol. Neurodegener. 10:33. doi: 10.1186/s13024-015-0030-y
Wang, W., Nakashima, K. I., Hirai, T., and Inoue, M. (2019b). Neuroprotective effect of naturally occurring RXR agonists isolated from Sophora tonkinensis Gagnep. on amyloid-β-induced cytotoxicity in PC12 cells. J. Nat. Med. 73, 154–162. doi: 10.1007/s11418-018-1257-z
Wang, W., Nakashima, K. I., Hirai, T., and Inoue, M. (2019a). Anti-inflammatory effects of naturally occurring retinoid X receptor agonists isolated from Sophora tonkinensis Gagnep. via retinoid X receptor/liver X receptor heterodimers. J. Nat. Med. 73, 419–430. doi: 10.1007/s11418-018-01277-1
Watson, G. S., Cholerton, B. A., Reger, M. A., Baker, L. D., Plymate, S. R., Asthana, S., et al. (2005). Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatry 13, 950–958. doi: 10.1176/appi.ajgp.13.11.950
Wei, X., Liu, C., Wang, H., Wang, L., Xiao, F., Guo, Z., et al. (2016). Surface Phosphatidylserine is responsible for the internalization on microvesicles derived from hypoxia-induced human bone marrow mesenchymal stem cells into human endothelial cells. PLoS One 11:e0147360. doi: 10.1371/journal.pone.0147360
Weiner, M. W., Veitch, D. P., Aisen, P. S., Beckett, L. A., Cairns, N. J., Cedarbaum, J., et al. (2015). 2014 update of the Alzheimers disease neuroimaging initiative: A review of papers published since its inception. Alzheimers Dement. 11:e120. doi: 10.1016/j.jalz.2014.11.001
Wildsmith, K. R., Holley, M., Savage, J. C., Skerrett, R., and Landreth, G. E. (2013). Evidence for impaired amyloid β clearance in Alzheimers disease. Alzheimers Res. Ther. 5, 33. doi: 10.1186/alzrt187
Wong, M. W., Braidy, N., Poljak, A., Pickford, R., Thambisetty, M., and Sachdev, P. S. (2017). Dysregulation of lipids in Alzheimers disease and their role as potential biomarkers. Alzheimers Dement. 13, 810–827. doi: 10.1016/j.jalz.2017.01.008
Xiu, J., Nordberg, A., Qi, X., and Guan, Z. Z. (2006). Influence of cholesterol and lovastatin on alpha-form of secreted amyloid precursor protein and expression of alpha7 nicotinic receptor on astrocytes. Neurochem. Int. 49, 459–465. doi: 10.1016/j.neuint.2006.03.007
Xu, Q., Bernardo, A., Walker, D., Kanegawa, T., Mahley, R. W., and Huang, Y. (2006). Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 26, 4985–4994. doi: 10.1523/jneurosci.5476-05.2006
Xu, Z., Xiao, N., Chen, Y., Huang, H., Marshall, C., Gao, J., et al. (2015). Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits. Mol. Neurodegener. 10:58. doi: 10.1186/s13024-015-0056-1
Yamanaka, M., Ishikawa, T., Griep, A., Axt, D., Kummer, M. P., and Heneka, M. T. (2012). PPARγ/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J. Neurosci. 32, 17321–17331. doi: 10.1523/jneurosci.1569-12.2012
Yeh, F. L., Wang, Y., Tom, I., Gonzalez, L. C., and Sheng, M. (2016). TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 91, 328–340. doi: 10.1016/j.neuron.2016.06.015
Yin, Y., Le, S. C., Hsu, A. L., Borgnia, M. J., Yang, H., and Lee, S. Y. (2019). Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 363:eaav9334. doi: 10.1126/science.aav9334
Yoo, A. S., Cheng, I., Chung, S., Grenfell, T. Z., Lee, H., Pack-Chung, E., et al. (2000). Presenilin-mediated modulation of capacitative calcium entry. Neuron 27, 561–572. doi: 10.1016/s0896-6273(00)00066-0
Yu, R. K., Tsai, Y. T., Ariga, T., and Yanagisawa, M. (2011). Structures, biosynthesis, and functions of gangliosides–an overview. J. Oleo Sci. 60, 537–544. doi: 10.5650/jos.60.537
Yu, Y., Li, X., Blanchard, J., Li, Y., Iqbal, K., Liu, F., et al. (2015). Insulin sensitizers improve learning and attenuate tau hyperphosphorylation and neuroinflammation in 3xTg-AD mice. J. Neural Transm. 122, 593–606. doi: 10.1007/s00702-014-1294-z
Zelcer, N., and Tontonoz, P. (2006). Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Invest. 116, 607–614. doi: 10.1172/jci27883
Zeng, Y., Cao, S., Li, N., Tang, J., and Lin, G. (2023). Identification of key lipid metabolism-related genes in Alzheimers disease. Lipids Health Dis. 22:155. doi: 10.1186/s12944-023-01918-9
Zhang, H., Gao, Y., Qiao, P. F., Zhao, F. L., and Yan, Y. (2015). PPAR-α agonist regulates amyloid-β generation via inhibiting BACE-1 activity in human neuroblastoma SH-SY5Y cells transfected with APPswe gene. Mol. Cell Biochem. 408, 37–46. doi: 10.1007/s11010-015-2480-5
Zhang, H., Ma, Q., Zhang, Y. W., and Xu, H. (2012). Proteolytic processing of Alzheimers β-amyloid precursor protein. J. Neurochem. 120, 9–21. doi: 10.1111/j.1471-4159.2011.07519.x
Zhang, J., and Liu, Q. (2015). Cholesterol metabolism and homeostasis in the brain. Protein Cell 6, 254–264. doi: 10.1007/s13238-014-0131-3
Zhang, J., Hu, Y., Wang, Y., Fu, L., Xu, X., Li, C., et al. (2021). mmBCFA C17iso ensures endoplasmic reticulum integrity for lipid droplet growth. J. Cell Biol. 220:2122. doi: 10.1083/jcb.202102122
Zhang, Y., Chen, J., Qiu, J., Li, Y., Wang, J., and Jiao, J. (2016). Intakes of fish and polyunsaturated fatty acids and mild-to-severe cognitive impairment risks: A dose-response meta-analysis of 21 cohort studies. Am. J. Clin. Nutr. 103, 330–340. doi: 10.3945/ajcn.115.124081
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., OKeeffe, S., et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. doi: 10.1523/jneurosci.1860-14.2014
Zhao, J., Davis, M. D., Martens, Y. A., Shinohara, M., Graff-Radford, N. R., Younkin, S. G., et al. (2017). APOE ε4/ε4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum. Mol. Genet. 26, 2690–2700. doi: 10.1093/hmg/ddx155
Zhao, Z., Sagare, A. P., Ma, Q., Halliday, M. R., Kong, P., Kisler, K., et al. (2015). Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat. Neurosci. 18, 978–987. doi: 10.1038/nn.4025
Keywords: lipid, lipid metabolism, Alzheimer’s disease, therapy strategies, diet
Citation: Cao Y, Zhao L-W, Chen Z-X and Li S-H (2024) New insights in lipid metabolism: potential therapeutic targets for the treatment of Alzheimer’s disease. Front. Neurosci. 18:1430465. doi: 10.3389/fnins.2024.1430465
Received: 10 May 2024; Accepted: 14 August 2024;
Published: 11 September 2024.
Edited by:
Francesco Panza, University of Bari Aldo Moro, ItalyReviewed by:
Takahisa Kanekiyo, Mayo Clinic Florida, United StatesMohd Moin Khan, Harvard Medical School, United States
Copyright © 2024 Cao, Zhao, Chen and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shao-Hua Li, MTgwMzk2NzMxOTdAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship