 David Bradford
David Bradford Kathleen E. Rodgers
Kathleen E. Rodgers
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci., 22 May 2024
Sec. Neurodegeneration
Volume 18 - 2024 | https://doi.org/10.3389/fnins.2024.1401706
Amyotrophic lateral sclerosis (ALS) continues to pose a significant challenge due to the disease complexity and heterogeneous manifestations. Despite recent drug approvals, there remains a critical need for the development of more effective therapies. This review explores the underlying mechanisms involved; including neuroinflammation, glutamate mediated excitotoxicity, mitochondrial dysfunction, and hypermetabolism, and how researchers are trying to develop novel drugs to target these pathways. While progress has been made, the unmet need of ALS patients highlights the urgency for continued research and resource allocation in the pursuit of effective treatments.
Amyotrophic lateral sclerosis (ALS) is a rare, progressive neurodegenerative disease leading to a rapid loss of motor neurons, leading to complete paralysis and death. Classified as an orphan disease, the prevalence for ALS is as high as 9.9 per 100,000 people, equating to about 31,843 people nationally (Mehta P. et al., 2023). Globally, this number increased to 222,801 in 2015, with this number expected to increase by 69% by 2040, highlighting the importance in understanding this devastating disease (Arthur et al., 2016). First identified in 1869 by Jean-Martin Charcot, ALS remained relatively unknown until 1941 when Lou Gehrig was diagnosed, and the term Lou Gehrig’s Disease was coined (The ALS Association, 2024a). It then took another 50 years for the first genetic risk factor, SOD1 mutation, to be discovered (Rosen et al., 1993).
Beginning from symptomatic onset, the average life expectancy is between 2 to 5 years, with about half of people dying within 3 years (Štětkářová and Ehler, 2021; The ALS Association, 2024b). A challenge with treating this disease lays with the difficulties around diagnosing ALS. It can take up to 14 months between the first clinic visit to an accurate diagnosis, with some groups reporting a delay of up to 24 months at the high end (Chiò, 1999; Segura et al., 2023). This delay in diagnosis is detrimental to the patient’s overall treatment, causing a heavy burden to both the patient’s mental health and more importantly, drastically delaying targeted treatment. Unlike other diseases, ALS is difficult to diagnosis due to a number of confounding factors. Clinicians undergo a “rule-out” approach when diagnosing a patient. In fact, over half of patients receive a different diagnosis prior to the correct ALS diagnosis, with the four most common alternative diagnosis being neuropathy (28%), spinal disease (18%), vascular disease (11%), or another neurodegenerative disease (11%) (Paganoni et al., 2014). This difficulty in diagnosing is in part due to the heterogeneity of the disease. ALS can mimic a variety of different neurological and motor diseases. The symptoms a patient has comes from where the disease first manifests. The two main classifications are bulbar-onset or limb-onset. Bulbar-onset first arise in the head and neck, affecting speech and swallowing. Limb-onset first affect the muscles in the periphery, primarily the hands and feet (Masrori and Van Damme, 2020). To further complicate things, the majority of ALS cases are sporadic in nature, making up 90% of cases, where the remaining 10% are familial, having a genetic component (Brotman et al., 2023). Understanding the complexity of this neurodegenerative disease may serve as a starting point to stratify this heterogeneous population to better develop targeted therapies and find potential biomarkers for earlier diagnosis.
To date there are over 50 genes implicated in the development of ALS affecting a variety of neuronal processes (Mejzini et al., 2019). The four most common mutations seen in fALS patients lay within mutations in the SOD1, C9orf72, TARDBP, and FUS genes, however there are multiple other genes with a definitive or likely causative link to ALS (Boylan, 2015). In addition to the previously mentioned genes with a definitive link to ALS, kinesin family member 5A (KIF5A), is also linked (Nicolas et al., 2018). KIF5, expressed in motor neurons, is responsible for cargo binding to adaptor proteins (Kanai et al., 2000). In ALS, there is a loss of function, disrupting axonal transport leading to neurodegeneration (Millecamps and Julien, 2013). In addition to these genetic mutations with a definitive link to ALS development, over 30 additional genes have been associated with a “likely causative” link. These include mutations in genes linked to the formation of toxic aggregates (ANXA11, DCTN1, EWSR1, MATR3, NEFH, PRPH, TAF15, TIA1, UBQLN2, and VAPB) or affecting mitochondrial function (CHCHD10, CHMP2B, SIGMAR1, SQSTM1, and VCP) (Smukowski et al., 2022). The wide variety of genes contributing to the development of ALS highlights the complexity of this disease and contributes to the challenges with treating this disease.
The superoxide dismutase 1 (SOD1) gene has been a major focus of ALS research since it was first identified in 1993, making it the first genetic risk factor for familial ALS (fALS). SOD1 encodes an antioxidant enzyme reducing oxidative stress by converting superoxide radicals into less reactive peroxide and oxygen (McCord and Fridovich, 1969). Despite being linked to a relatively small proportion of ALS cases (around 8–23% of all fALS cases and up to about 5% of all sporadic forms), the SOD1 mutations have provided crucial insights in the understanding of ALS pathology (Müller et al., 2022).
To date, over 200 unique mutations in the SOD1 gene have been identified (Gagliardi et al., 2023). These mutations typically exhibit an autosomal dominant inheritance pattern, where only a single mutated copy of the gene is sufficient to increase susceptibility to ALS (Boylan, 2015; Feneberg et al., 2020). In the context of disease progression, mutated SOD1 works in a gain-of-function manner, evident by a myriad of studies knocking down or entirely knocking out SOD1 and seeing either a slowing in disease progression or no ALS like phenotype, respectfully (Reaume et al., 1996; Sau et al., 2007; Bunton-Stasyshyn et al., 2015). Largely cytosolic, SOD1 is also found within the mitochondrial inner membrane space, making these areas affected (Sturtz et al., 2001). The gain-of-function is presumably believed to a result of increased aggregation, destabilization of dimers, and oligomerization of the mutant SOD1 protein (Pansarasa et al., 2018). The formation of insoluble aggregates is also believed to go on and either be neurotoxic in nature or block normal intracellular signaling and cellular function (McAlary et al., 2016; Pansarasa et al., 2018). These aggregates can then go on and affect neighboring cells, leading to the progressive nature seen in ALS pathology. While there is clearly a role SOD1 plays in ALS, the question of their exact role and how these aggregates interact in cellular transmission remains areas of active investigation. Answering the questions around SOD1 mutations could be a future therapeutic avenue for future drug development.
The C9orf72 gene, located on the short arm of chromosome 9, gained prominence in ALS research due to its association with a hexanucleotide repeat expansion (GGGGCC), making it the most prevalent genetic mutation in both familial and sporadic forms of ALS. This repeat expansion is seen in upwards to 50% of all fALS cases and 10% of sALS (Umoh et al., 2016). First identified in 2011 as a link between ALS and Frontotemporal dementia (FTD), it is believed these repeats work again in a gain-of-function fashion (DeJesus-Hernandez et al., 2011; Renton et al., 2011). The normal function of C9orf72 is still not fully understood, however it is thought to play a role in various cellular processes, including vesicle trafficking and autophagy (Sellier et al., 2016; Smeyers et al., 2021).
These repeats are often present in healthy individuals, but evidence shows that deleterious events may begin when the hexanucleotide repeat expansions reaches over 30 times (Zhang et al., 2015). In fact, most ALS patients with the C9orf72 mutation have expansions that range from hundreds to thousands of repeats, far exceeding what is seen in healthy populations (Mejzini et al., 2019). The exact molecular mechanism underlying the toxicity that leads to ALS pathology remains up for debate and not completely understood. It is theorized that the G rich regions have a tendency to form highly stable quadruplex structures (G-quadruplex). These G-quadruplexes can go on and interfere with various cellular processes, interfering and improperly interacting with intracellular proteins, affecting telomere stability and RNA transcription, splicing, translation, and transport (Fratta et al., 2012; Endoh and Sugimoto, 2016). Patients with the C9orf72 mutation often exhibit a variety of symptoms, including both motor and cognitive impairments, as evident by the overlap with FTD. Further research may provide insights into the toxic mechanisms these aggregates play, and may pave the way for targeted interventions that not only help alleviate ALS pathology, but also relieve FTD symptoms.
Another genetic mutation leading to the formation of ALS lies within the TARDBP gene, which encodes the TDP43 protein (Jo et al., 2020). Since its causative link was first discovered in 2008, more than 60 TARDBP mutations have been identified (Chen et al., 2021). In fact, an overwhelming number of ALS cases exhibit TDP-43 proteopathy. Notably, 97% of all ALS cases have TDP-43 aggregates present, regardless of disease classification (sALS vs. fALS), making this a pathological hallmark of ALS (Swarup et al., 2011; Ling et al., 2013).
TDP-43 is a versatile protein, involved in various cellular functions relating to RNA metabolism (Prasad et al., 2019). Under normal conditions, TDP-43 is primarily localized within the nucleus, contributing to the regulation of RNA processes and repressing the cryptic exon inclusion during RNA splicing (Jo et al., 2020; Ma et al., 2022). However, in ALS patients with TDP-43 proteopathy, there is a loss of functional TDP-43 within the nucleus and an increase in cytoplasmic levels (Arai et al., 2006). This loss of nuclear localization is a consequence from post-translational modifications, primarily through phosphorylation and ubiquitination (Neumann et al., 2006; Inukai et al., 2008). The accumulation of TDP-43 aggregates in the cytoplasm disrupts normal cellular processes, decreasing autophagy (by affecting TFEB [transcription factor EB] localization), increasing stress granule formation, and increases mitochondrial dysfunction (Liu-Yesucevitz et al., 2010; Stribl et al., 2014; Xia et al., 2016).
In addition to forming toxic aggregates, the loss of nuclear TDP-43 leads to a loss in RNA splicing regulation, often including cryptic exons leading to the inclusion of frameshifts or stop codons (Torres et al., 2018). In 2019, two independent groups found that the loss of functional TDP-43 leads to the inclusion of a cryptic exon within the STMN2 gene, which encodes a microtubule-associated protein (stathmin-2) critical for axonal growth and neuronal repair (Klim et al., 2019; Melamed et al., 2019; Mehta P. R. et al., 2023). Other cryptic exons can be found implanted within genes due to the loss of TDP-43. For instance, these exons have been found in ATG4B (autophagy related 4B cysteine peptidase), inhibiting autophagy. The inclusion of cryptic exons have been primarily found within regions implicated in ALS, including the motor cortex and spinal cords of ALS patients (Ling et al., 2015; Torres et al., 2018). The role of TDP-43 seems to be two-fold, acting as both a gain-of-function (through their ability to form toxic aggregates) and a loss-of-function (evident by their mismanagement of RNA splicing). Even with this, the exact mechanisms through which TDP-43 aggregates contribute to neurodegeneration are actively under investigation with many groups looking at stabilizing their functions to alleviate ALS like symptoms. Inhibiting the formation of TDP-43 aggregates and promoting cytoplasmic clearance may be a promising target to treat ALS, not just in those with a TARDBP mutation.
FUS (Fused in Sarcoma) is a gene that encodes an RNA-binding protein crucial for various cellular processes, including transcription, translation, DNA repair, and the maintenance of genomic stability (Lagier-Tourenne et al., 2010; Chen et al., 2019). Mutations within the FUS gene are heavily linked to the development of ALS, especially in juvenile cases, where the FUS mutation is the most frequent genetic cause (Vance et al., 2009; Zou et al., 2016). The majority of disease-causing mutations are located at the C-terminal region, disrupting the nuclear localization signals (Nakaya and Maragkakis, 2018). These signals are responsible for mediating the transport of proteins from the cytoplasm into the nucleus (Lu et al., 2021). Instead of residing in the nucleus, the FUS is mislocalized into the cytoplasm, leading again to a gain of function phenotype, leading to neuronal toxicity and ALS pathology (Qiu et al., 2014). Like the other mutations mentioned, the specific mechanism of how FUS mutations lead to ALS is an ongoing area of research. However, it is clear that FUS mutations add to the complexity of this disease, further highlighting the heterogeneity present in ALS.
Outside of the genetic mutations, being a male is the strongest risk factor for developing ALS. Men have a 1.5 times greater risk of developing ALS than women. This is likely due to early testosterone exposure possibly driving the disease pathogenesis (Mehta et al., 2014). This trend continues up until menopause, where after menopause, the incidence rates between men and women equal out (Oskarsson et al., 2015). Even with this increase in prevalence, only one mutation is linked to the X chromosome, with the others acting in an autosomal dominant fashion (Benarroch et al., 2023). Therefore, this risk factor could also be tied to an increase in exposure to other risk factors described below.
Outside of the genetic risk factors, there are no definitive risk factors that are known to cause the development of ALS. However, many groups have investigated whether certain environmental factors lead to a higher incidence of disease. It is believed that military experience has an increased incidence compared to non-military personnel (Beard and Kamel, 2015). This could also be true due to exposure of other neurotoxic agents like lead, pesticides, and smoking (Armon, 2009; Malek et al., 2012; Wang et al., 2014; Nieh et al., 2021). Another explanation for this trend in military personnel is with their overall physical fitness. Having a low body mass index is also associated with an increased risk of ALS, however, the causative link remains unknown (Mattsson et al., 2012). While these trends exist in certain cohorts, it should be emphasized that these are not definitive, with further research being needed to find the causative link, if there is one.
Since its discovery, ALS has been long time viewed as an isolated muscle disease; however, sentiment within the field has been changing as this disease has systemic effects not isolated to voluntary muscles (Silani et al., 2017). Regardless of the disease classification, there are shared mechanisms contributing the neurodegeneration. While the exact mutation leading to the onset of the disease vary; neuronal hyperexcitability, neuroinflammation, and mitochondrial dysfunction are seen throughout disease progression.
Although not unique to neurodegenerative diseases, neuroinflammation and immune activation are seen throughout the disease course of ALS. Neuroinflammation, led by microglial and astrocyte activation, T-lymphocyte infiltration, and the overproduction of proinflammatory cytokines are observed before symptomatic onset, suggesting a link between the immune response and ALS progression (Komine and Yamanaka, 2015). The balance between the protective vs. toxic role of immune activation remains elusive. Microglia act as the first line of defense within the CNS, surveying the microenvironment existing in two states; resting or activated (Cherry et al., 2014). Once activated, microglia can either enter one of two opposing pathways, the classical (M1) phenotype or alternative (M2) phenotype (Franco and Fernández-Suárez, 2015; Thompson and Tsirka, 2017).
In the presence of an injury, microglia become classically activated; exhibiting a proinflammatory and cytotoxic phenotype (Orihuela et al., 2016). These microglia produce inflammatory cytokines and chemokines, including but not limited to tumor necrosis factor alpha (TNFα), interleukin (IL)-1b, IL-6, IL-12, and increased oxidative stress (Villalta et al., 2009; Colonna and Butovsky, 2017). TNFα has been shown to have a direct effect on neuronal excitability both in-vivo and within hippocampal slices from animals with peripheral inflammation (Riazi et al., 2008; Galic et al., 2012). Other proinflammatory cytokines and chemokines, like the interleukins, also lead to neuronal death via the NF-kB signaling pathway, ultimately leading to apoptosis and neuronal loss (Wang et al., 2015; Singh and Singh, 2020). Increasing oxidative stress and excessive formation of reactive oxygen species (ROS) exacerbate the neurodegenerative process in a feed-forward pathway. ROS, released from damaged neurons or glial cells, can bind to glutamate receptors, thus elevating synaptic glutamate furthering neuronal excitability. This causes intracellular calcium to increase, causing mitochondrial dysfunction and directly causing more ROS production (Heath and Shaw, 2002; Hemerková and Vališ, 2021).
Another integral glial cell involved in neuroinflammation are astrocytes. Like microglia, astrocytes can become polarized, acting in either a neuroprotective role or facilitating neurodegeneration and releasing neurotoxins (Li et al., 2019). Cytotoxic astrocytes upregulate proinflammatory cytokines (like IL-1 and TNFα), which promotes neuronal inflammation. These astrocytes also upregulate glutamate and ATP secretion (Orellana et al., 2011; Liddelow and Barres, 2017). This increase in glutamate release is also paired with a downregulation in excitatory amino acid transporter 2 (EAAT2) expression further promoting another neurodegenerative pathway, glutamate mediated excitability (Gong et al., 2000). These studies highlight the detrimental effects elevated proinflammatory agents has on organism as a whole, and not just locally where they are produced.
Glutamate-mediated excitotoxicity has been widely observed within ALS. So much so that the first FDA approved medication for ALS, Riluzole, targets this phenomenon (Doble, 1996). Under normal physiological conditions, glutamate release and clearance are highly regulated. So much so that within CNS, 1% of the total brain proteins are the glial glutamate transporter, EAAT2 (Danbolt, 2001; Foran and Trotti, 2009). This highlights the importance of quickly clearing out released glutamate and preventing glutamate from spilling over to neighboring synapses.
Alterations in glutamate homeostasis in ALS pathology has been widely established, but the cause remains unknown. As alluded to earlier, ALS patients demonstrate both an increase in glutamate release as well as a decrease in glutamate uptake, primarily due to the loss of the astrocytic glutamate transporter, EAAT2 (Rothstein et al., 1992; Ferrarese et al., 2001). Incorrect splicing due to abnormal EAAT2 mRNA were found in ALS patients (Lin et al., 1998). This decrease in clearance leads to an accumulation of glutamate, leading to neurotoxic levels and further pathogenesis. The levels of glutamate are elevated in both the cerebrospinal fluid (CSF) and plasma, indicating compensatory mechanisms are unable to adapt, leading to systemic stress on the patient.
The accumulation of damaged mitochondria is a prevalent feature in age-related neurodegeneration, and a common feature in ALS (Kim et al., 2019). As previously mentioned, neuronal inflammation and glutamate mediated excitotoxicity contribute to the development of mitochondrial dysfunction. Mitochondria are the main site for ATP synthesis and metabolism and their function is essential, especially in energy-demanding organs like the brain and muscle (Balasubramanian, 2021; Zhao et al., 2022). Therefore, mitochondrial dysfunction is heavily linked to ALS pathogenesis and disease progression. Prolonged elevated intracellular calcium overloads the mitochondria, resulting in ROS production, oxidative stress and eventually apoptosis (Kawamata and Manfredi, 2010).
In addition to calcium buffering, mitochondrial dysfunction leads to axonal transport dysfunction (Genin et al., 2023). ALS patients have mitochondrial with abnormal morphology, which includes increased cristae and mitochondrial swelling (Sasaki and Iwata, 2007). These alterations are all believed to affect ATP production and mitochondrial respiration. ALS mitochondria exhibit decreased activity of the electron transport chain (ETC), with defects seen across most aspects, including complex I, II, IV as well as certain mitochondrial enzymes, including citrate synthase and cytochrome c oxidase (Borthwick et al., 1999; Wiedemann et al., 2002; Vandoorne et al., 2018). Impaired cellular respiration and ATP production also contribute to a phenomenon typically seen in ALS patients, hypermetabolism.
Hypermetabolism is a state of exhibiting an elevated resting energy expenditure (REE) and has been seen in upwards between 55–60% of ALS patients (Bouteloup et al., 2009; D'Amico et al., 2021). Other groups have found that the REE is increased by 10% on average in ALS patients when compared to that of healthy populations (Desport et al., 2001). In fact, ALS patients are recommended to consume more calories than their calculated need (Kasarskis et al., 1996; Ngo et al., 2014). It is not surprising that metabolic dysfunction would be implicated in ALS considering the brain and muscles are two of the most metabolically active tissue groups in the body (Wang et al., 2010).
Neurons and muscles, along with other tissues, utilize glucose as their main source of energy (Mergenthaler et al., 2013). Glucose is metabolized into pyruvate, via glycolysis, which can go on to produce more ATP by the TCA cycle. Under normal conditions, the energy required for homeostasis is met, promoting proper synaptic function and axonal transport in the neuron, two processes heavily reliant on glycolysis (Zala et al., 2013; Rangaraju et al., 2014). In both ALS patients and in SOD1 symptomatic mice, decreased levels of pyruvate have been observed, indicating an impairment of glucose utilization and the glycolytic pathway (Tefera et al., 2019, 2021).
With defects in the glycolytic pathway, the body must get energy from another source in order to meet the increased energy demand. One way the body does this is through b-oxidation, where lipids and fatty acids are utilized rather than glucose. While this process is normally fine short term, prolonged β-oxidation results in the formation of ROS and cellular stress (Zhang et al., 2010). When agents are added to promote glycolysis and the entry of pyruvate into the Krebs cycle, like dichloroacetate, to improve glycolytic capacity, there is a delay in muscle denervation and atrophy (Palamiuc et al., 2015). This switch from glycolysis to b-oxidation is shown to be cytotoxic in neurons and muscles, contributing to muscle denervation and disease progression.
It is important to note that these mechanisms leading to neurodegeneration and ALS disease progression should not be viewed as mutually exclusive, but rather a highly dynamic and reactive process potentially exacerbating one another (Figure 1). This phenomenon may be responsible for the failure in drug development and the limited benefit in the currently approved treatment options available for patients.
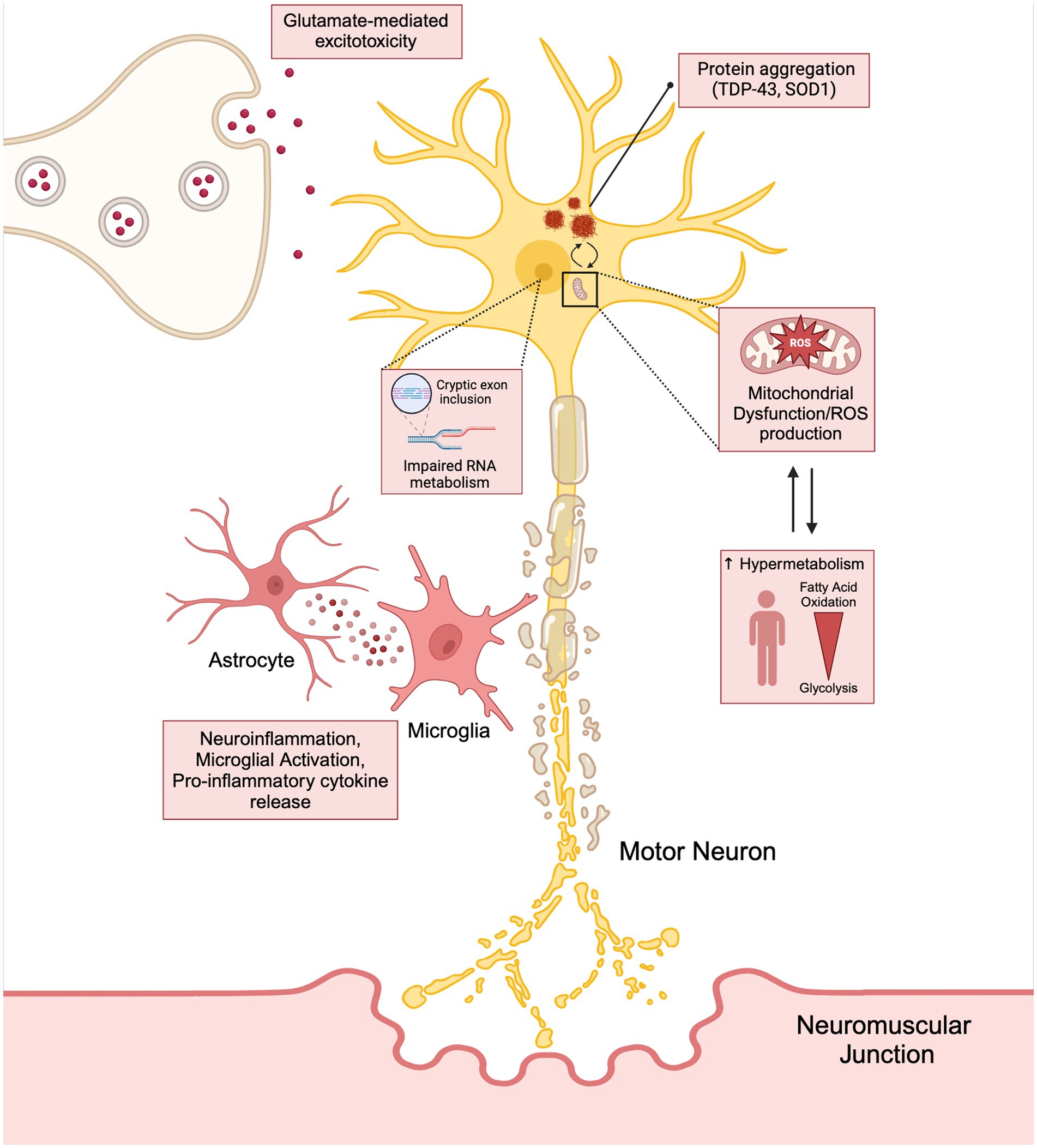
Figure 1. The interconnection between the hallmarks of ALS (neuroinflammation, glutamate mediated excitotoxicity, and mitochondrial dysfunction) have on stressing the motor neuron and how these cytotoxic pathways lead to disease progression and hypermetabolism. Created with BioRender.com.
Drug development is a long and complex process, often taking up to 15 years and costing over a billion dollars (Hughes et al., 2011). With the unmet need for viable new therapeutics, drug repurposing may be a more time and cost effect strategy. The utilization of network-based approaches, like SAveRunner, GEO, and STRING databases allow for the identification of possible targets with theoretical feasibility in ALS (Fiscon et al., 2021; Sunildutt et al., 2024). These methods have facilitated research in drugs with known safety profiles, speeding up clinical trial opportunities.
Many drugs undergoing clinical trials for ALS involve repurposed drugs that are already FDA approved for another indication. These include drugs previously approved for cancer treatments (Bosutinib and Mastinib), HIV treatments (Dolutegravir, Abacavir, and Lamivudine), and rheumatoid arthritis (Baricitinib) (Greig and Deeks, 2015; Isfort et al., 2018; Urits et al., 2020; Ketabforoush et al., 2023). Additional agents being investigated preclinically include autophagy inducers (Rapamycin), hormone antagonists (Tamoxifen and Imatinib), Alkylating agents (Cisplatin and Carboplatin), and immunomodulating drugs (Thalidomide and Lenalidomide) (Mandrioli et al., 2023; Potenza et al., 2024).
In recent years, there has been a push for accelerated approvals and drug development for rare diseases. The National Center for Advancing Translational Sciences (NCATS) plans to grow and improve the experimental drug pipeline for rare diseases (Mullard, 2023). Since the first drug was approved in 1995, three others have been approved, with two of those coming within the last two years (Saitoh and Takahashi, 2020; Center for Drug Evaluation and Research, 2023a,b). Even with these new treatment options for patients, there is still a desperate need to develop more effective therapies. This fact is especially evident by the recent determination of Relyvrio. Approved by the FDA in 2022, Relyvrio was taken off the market due to failed phase-III results, where there was no change in the disease progression in patients with or without treatment (Bryson, 2024). This recent withdrawal highlights the complexity and difficulties surrounding the development of effective treatment.
Currently, there are 35 clinical trials in phase I, II, or III that are recruiting on clinicaltrials.gov. Of these 35 trials, 29 are in either phase 1 or 2, with the primary focus of those drugs broadly targeting the immune system and neuroinflammation. Other targets involve either targeting neurons directly through the integrated stress response and oxidative stress, trying to mitigate symptoms and serving as comfort care, or looking at diagnostic approaches through imaging for screening and faster disease identification.
One way the ALS research community has tried to streamline clinical research is through the formation of the HEALY ALS Platform. According to the HEALEY ALS Platform Trial Research Partners,1 this master protocol platform first began enrolling patients in 2020, with the goal being to reduce the cost, speed up enrollment, and encourage more patient participation. This is accomplished by testing multiple treatments simultaneously, rather than one at a time. Currently, the HEALY Platform (Clinical Trials: NCT04297683) has drugs under Phase II/III with others in the pipeline. Below is a breakdown outlining the current on-going clinical trials with their broad mechanism we identified. These mechanisms are classified between targeting the immune system/neuroinflammation, the neuron/neuroprotective, symptoms/comfort care, or a diagnostic tool. It is important to reiterate that these pathways crosstalk and the true mechanism of the drug can be argued to be in multiple categories (Figure 2; Table 1).
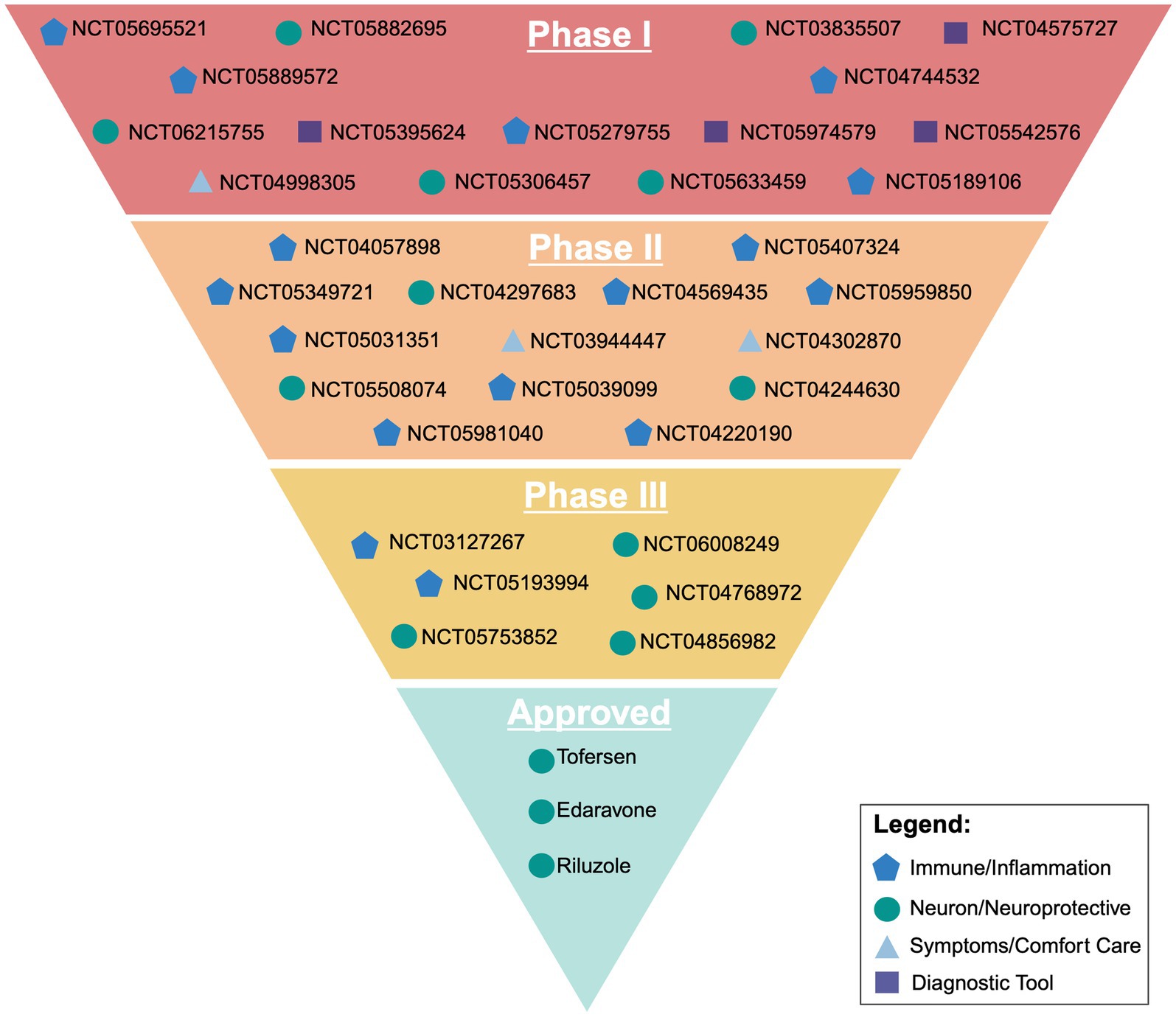
Figure 2. Visual representation of current ongoing clinical trials and their broad target of action outlined in the sponsors clinicaltrials.gov entry. Created with BioRender.com.
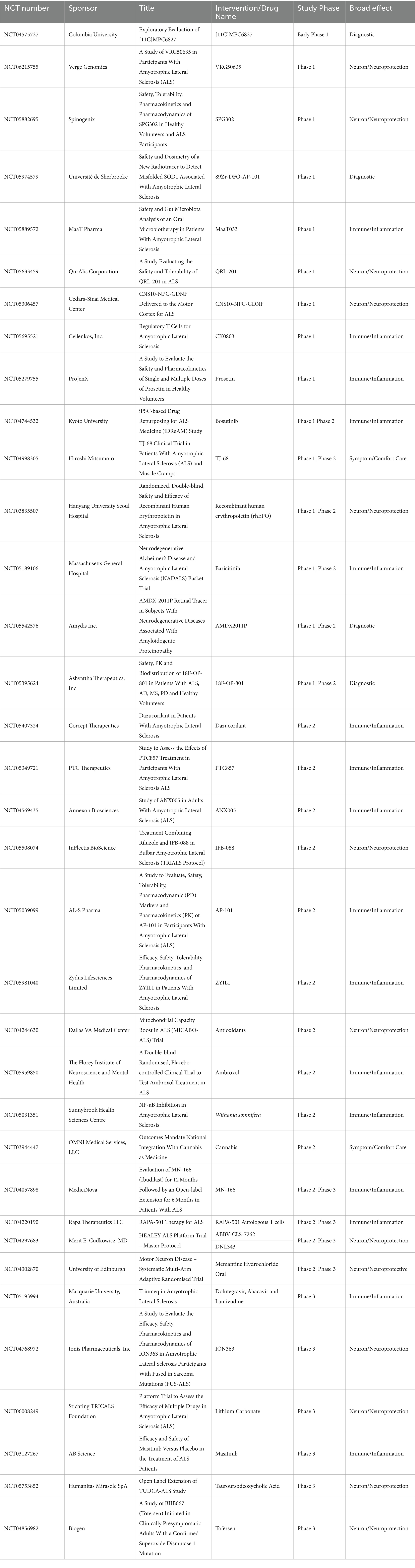
Table 1. Table of all active clinical trials for ALS listed on clinicaltrials.gov.
Despite recent advancements in ALS research, significant gaps persist in our understanding of the disease’s pathophysiology, hindering the development of targeted therapies. While precision medicine has made strides, like with the development of Tofersen for SOD1 mutant patients, the fundamental triggers initiating ALS pathology remain elusive. Common neurodegenerative disease pathways involving mitochondrial dysfunction, neuroinflammation, and glutamate-mediated cytotoxicity have been identified, however, the primary driving force being disease progression remains elusive.
One critical aspect that demands attention is the need for a more robust diagnostic pipeline. The prolonged delay in ALS diagnosis, which in some cases can take up to 14 months, poses substantial challenges for patient outcomes. An improved and expedited diagnostic process would allow for earlier treatment intervention, potentially leading to better survival outcomes and allowing patients to maintain their independence longer. A comprehensive understanding of the heterogeneity within ALS, both genetic and non-genetic factors, is essential for tailoring treatment to the individual patient.
Another significant hurdle lies in drug development. Targeting a single mechanism has proven to be insufficient, with the majority of clinical trials failing to meet primary endpoints. This may be attributed to the potential circumvention of certain pathways, enabling neurodegeneration to persist. While a universal approach would be ideal, acknowledging ALS as a heterogeneous disease suggests the need for a more nuanced approach. Stratifying patients based on their underlying pathology and genetic mutations may lead to more efficacious clinical trial designs.
The emergence of the HEALY ALS Platform spotlights the efforts made to streamline clinical research and accelerating drug development. However, the dynamic and reactive nature of ALS pathology, as highlighted in this review, underscores the complexity that may impede the success of monotherapeutic approaches. Collaborative efforts should be directed toward understanding the intricacies present and identifying synergistic interventions. Ultimately, ALS remains a challenging neurodegenerative disease to understand. Despite the continued progress, the factors contributing to disease progression needs continued research and collaboration. By addressing the gaps in our knowledge and optimizing both therapeutic development and diagnostic tools, there is hope for improving outcomes and the quality of life for individuals living with ALS.
DB: Writing – original draft, Writing – review & editing. KR: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Institute of Health (T32-AG061897) and the Department of Defense (W81XWH-19-1-0471 and W81XWH-22-1-0074).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi: 10.1016/j.bbrc.2006.10.093
Armon, C. (2009). Smoking may be considered an established risk factor for sporadic ALS. Neurology 73, 1693–1698. doi: 10.1212/WNL.0b013e3181c1df48
Arthur, K. C., Calvo, A., Price, T. R., Geiger, J. T., Chiò, A., and Traynor, B. J. (2016). Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 7:12408. doi: 10.1038/ncomms12408
Balasubramanian, V. (2021). Brain power. Proc. Natl. Acad. Sci. USA 118. doi: 10.1073/pnas.2107022118
Beard, J. D., and Kamel, F. (2015). Military service, deployments, and exposures in relation to amyotrophic lateral sclerosis etiology and survival. Epidemiol. Rev. 37, 55–70. doi: 10.1093/epirev/mxu001
Benarroch, L., Bonne, G., Rivier, F., and Hamroun, D. (2023). The 2023 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 33, 76–117. doi: 10.1016/j.nmd.2022.12.002
Borthwick, G. M., Johnson, M. A., Ince, P. G., Shaw, P. J., and Turnbull, D. M. (1999). Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann. Neurol. 46, 787–790. doi: 10.1002/1531-8249(199911)46:5<787::AID-ANA17>3.0.CO;2-8
Bouteloup, C., Desport, J. C., Clavelou, P., Guy, N., Derumeaux-Burel, H., Ferrier, A., et al. (2009). Hypermetabolism in ALS patients: an early and persistent phenomenon. J. Neurol. 256, 1236–1242. doi: 10.1007/s00415-009-5100-z
Boylan, K. (2015). Familial amyotrophic lateral sclerosis. Neurol. Clin. 33, 807–830. doi: 10.1016/j.ncl.2015.07.001
Brotman, R. G., Moreno-Escobar, M. C., Joseph, J., and Pawar, G. (2023). Amyotrophic lateral sclerosis. Available at: https://www.ncbi.nlm.nih.gov/books/NBK556151/
Bryson, S. (2024). Relyvrio withdrawn from US, Canada after phase 3 trial results. Available at: https://alsnewstoday.com/news/relyvrio-withdrawn-from-us-canada-after-phase-3-trial-results/
Bunton-Stasyshyn, R. K., Saccon, R. A., Fratta, P., and Fisher, E. M. C. (2015). SOD1 function and its implications for amyotrophic lateral sclerosis pathology: new and renascent themes. Neuroscientist 21, 519–529. doi: 10.1177/1073858414561795
Center for Drug Evaluation and Research . (2023a). Drug trials snapshots: Relyvrio. Available at: https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trials-snapshots-relyvrio
Center for Drug Evaluation and Research . (2023b). FDA approves treatment of amyotrophic lateral sclerosis associated with a mutation in the SOD1 gene. Available at: https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-treatment-amyotrophic-lateral-sclerosis-associated-mutation-sod1-gene.
Chen, C., Ding, X., Akram, N., Xue, S., and Luo, S.-Z. (2019). Fused in sarcoma: properties, self-assembly and correlation with neurodegenerative diseases. Molecules 24. doi: 10.3390/molecules24081622
Chen, S., Zhou, R. L., Zhang, W., Che, C. H., Feng, S. Y., Huang, H. P., et al. (2021). Novel TARDBP missense mutation caused familial amyotrophic lateral sclerosis with frontotemporal dementia and parkinsonism. Neurobiol. Aging 107, 168–173. doi: 10.1016/j.neurobiolaging.2021.05.017
Cherry, J. D., Olschowka, J. A., and O’Banion, M. K. (2014). Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J. Neuroinflammation 11:98. doi: 10.1186/1742-2094-11-98
Chiò, A. (1999). ISIS survey: an international study on the diagnostic process and its implications in amyotrophic lateral sclerosis. J. Neurol. 246:III1. doi: 10.1007/BF03161081
Colonna, M., and Butovsky, O. (2017). Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 35, 441–468. doi: 10.1146/annurev-immunol-051116-052358
D'Amico, E., Grosso, G., Nieves, J. W., Zanghì, A., Factor-Litvak, P., and Mitsumoto, H. (2021). Metabolic abnormalities, dietary risk factors and nutritional Management in Amyotrophic Lateral Sclerosis. Nutrients 13:2273. doi: 10.3390/nu13072273
Danbolt, N. C. (2001). Glutamate uptake. Prog. Neurobiol. 65, 1–105. doi: 10.1016/S0301-0082(00)00067-8
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Desport, J. C., Preux, P. M., Magy, L., Boirie, Y., Vallat, J. M., Beaufrère, B., et al. (2001). Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am. J. Clin. Nutr. 74, 328–334. doi: 10.1093/ajcn/74.3.328
Doble, A. (1996). The pharmacology and mechanism of action of riluzole. Neurology 47, S233–S241. doi: 10.1212/WNL.47.6_Suppl_4.233S
Endoh, T., and Sugimoto, N. (2016). Mechanical insights into ribosomal progression overcoming RNA G-quadruplex from periodical translation suppression in cells. Sci. Rep. 6:22719. doi: 10.1038/srep22719
Feneberg, E., Turner, M. R., Ansorge, O., and Talbot, K. (2020). Amyotrophic lateral sclerosis with a heterozygous D91A SOD1 variant and classical ALS-TDP neuropathology. Neurology 95, 595–596. doi: 10.1212/WNL.0000000000010587
Ferrarese, C., Sala, G., Riva, R., Begni, B., Zoia, C., Tremolizzo, L., et al. (2001). Decreased platelet glutamate uptake in patients with amyotrophic lateral sclerosis. Neurology 56, 270–272. doi: 10.1212/WNL.56.2.270
Fiscon, G., Conte, F., Amadio, S., Volonté, C., and Paci, P. (2021). Drug repurposing: a network-based approach to amyotrophic lateral sclerosis. Neurotherapeutics 18, 1678–1691. doi: 10.1007/s13311-021-01064-z
Foran, E., and Trotti, D. (2009). Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid. Redox Signal. 11, 1587–1602. doi: 10.1089/ars.2009.2444
Franco, R., and Fernández-Suárez, D. (2015). Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 131, 65–86. doi: 10.1016/j.pneurobio.2015.05.003
Fratta, P., Mizielinska, S., Nicoll, A. J., Zloh, M., Fisher, E. M. C., Parkinson, G., et al. (2012). C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci. Rep. 2:1016. doi: 10.1038/srep01016
Gagliardi, D., Ripellino, P., Meneri, M., del Bo, R., Antognozzi, S., Comi, G. P., et al. (2023). Clinical and molecular features of patients with amyotrophic lateral sclerosis and SOD1 mutations: a monocentric study. Front. Neurol. 14:1169689. doi: 10.3389/fneur.2023.1169689
Galic, M. A., Riazi, K., and Pittman, Q. J. (2012). Cytokines and brain excitability. Front. Neuroendocrinol. 33, 116–125. doi: 10.1016/j.yfrne.2011.12.002
Genin, E. C., Abou-Ali, M., and Paquis-Flucklinger, V. (2023). Mitochondria, a key target in amyotrophic lateral sclerosis pathogenesis. Genes 14:1981. doi: 10.3390/genes14111981
Gong, Y. H., Parsadanian, A. S., Andreeva, A., Snider, W. D., and Elliott, J. L. (2000). Restricted expression of G86R cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J. Neurosci. 20, 660–665. doi: 10.1523/JNEUROSCI.20-02-00660.2000
Greig, S. L., and Deeks, E. D. (2015). Abacavir/dolutegravir/lamivudine single-tablet regimen: a review of its use in HIV-1 infection. Drugs 75, 503–514. doi: 10.1007/s40265-015-0361-6
Heath, P. R., and Shaw, P. J. (2002). Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve 26, 438–458. doi: 10.1002/mus.10186
Hemerková, P., and Vališ, M. (2021). Role of oxidative stress in the pathogenesis of amyotrophic lateral sclerosis: antioxidant Metalloenzymes and therapeutic strategies. Biomol. Ther. 11:437. doi: 10.3390/biom11030437
Hughes, J. P., Rees, S., Kalindjian, S. B., and Philpott, K. L. (2011). Principles of early drug discovery. Br. J. Pharmacol. 162, 1239–1249. doi: 10.1111/j.1476-5381.2010.01127.x
Inukai, Y., Nonaka, T., Arai, T., Yoshida, M., Hashizume, Y., Beach, T. G., et al. (2008). Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett. 582, 2899–2904. doi: 10.1016/j.febslet.2008.07.027
Isfort, S., Crysandt, M., Gezer, D., Koschmieder, S., Brümmendorf, T. H., and Wolf, D. (2018). Bosutinib: a potent second-generation tyrosine kinase inhibitor. Recent Results Cancer Res. 212, 87–108. doi: 10.1007/978-3-319-91439-8_4
Jo, M., Lee, S., Jeon, Y. M., Kim, S., Kwon, Y., and Kim, H. J. (2020). The role of TDP-43 propagation in neurodegenerative diseases: integrating insights from clinical and experimental studies. Exp. Mol. Med. 52, 1652–1662. doi: 10.1038/s12276-020-00513-7
Kanai, Y., Okada, Y., Tanaka, Y., Harada, A., Terada, S., and Hirokawa, N. (2000). KIF5C, a novel neuronal kinesin enriched in motor neurons. J. Neurosci. 20, 6374–6384. doi: 10.1523/JNEUROSCI.20-17-06374.2000
Kasarskis, E. J., Berryman, S., Vanderleest, J. G., Schneider, A. R., and McClain, C. (1996). Nutritional status of patients with amyotrophic lateral sclerosis: relation to the proximity of death. Am. J. Clin. Nutr. 63, 130–137. doi: 10.1093/ajcn/63.1.130
Kawamata, H., and Manfredi, G. (2010). Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech. Ageing Dev. 131, 517–526. doi: 10.1016/j.mad.2010.05.003
Ketabforoush, A., Chegini, R., Barati, S., Tahmasebi, F., Moghisseh, B., and Joghataei, M. T. (2023). Masitinib: the promising actor in the next season of the amyotrophic lateral sclerosis treatment series. Biomed. Pharmacother. 160:114378. doi: 10.1016/j.biopha.2023.114378
Kim, Y., Park, J., and Choi, Y. K. (2019). The role of astrocytes in the central nervous system focused on BK Channel and Heme oxygenase metabolites: a review. Antioxidants 8:121. doi: 10.3390/antiox8050121
Klim, J. R., Williams, L. A., Limone, F., Guerra San Juan, I., Davis-Dusenbery, B. N., Mordes, D. A., et al. (2019). ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 22, 167–179. doi: 10.1038/s41593-018-0300-4
Komine, O., and Yamanaka, K. (2015). Neuroinflammation in motor neuron disease. Nagoya J. Med. Sci. 77, 537–549,
Lagier-Tourenne, C., Polymenidou, M., and Cleveland, D. W. (2010). TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 19, R46–R64. doi: 10.1093/hmg/ddq137
Li, K., Li, J., Zheng, J., and Qin, S. (2019). Reactive astrocytes in neurodegenerative diseases. Aging Dis. 10, 664–675. doi: 10.14336/AD.2018.0720
Liddelow, S. A., and Barres, B. A. (2017). Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967. doi: 10.1016/j.immuni.2017.06.006
Lin, C. L., Bristol, L. A., Jin, L., Dykes-Hoberg, M., Crawford, T., Clawson, L., et al. (1998). Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 20, 589–602. doi: 10.1016/S0896-6273(00)80997-6
Ling, J. P., Pletnikova, O., Troncoso, J. C., and Wong, P. C. (2015). TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655. doi: 10.1126/science.aab0983
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Liu-Yesucevitz, L., Bilgutay, A., Zhang, Y. J., Vanderwyde, T., Citro, A., Mehta, T., et al. (2010). Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5:e13250. doi: 10.1371/journal.pone.0013250
Lu, J., Wu, T., Zhang, B., Liu, S., Song, W., Qiao, J., et al. (2021). Types of nuclear localization signals and mechanisms of protein import into the nucleus. Cell Commun. Signal. 19:60. doi: 10.1186/s12964-021-00741-y
Ma, X. R., Prudencio, M., Koike, Y., Vatsavayai, S. C., Kim, G., Harbinski, F., et al. (2022). TDP-43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature 603, 124–130. doi: 10.1038/s41586-022-04424-7
Malek, A. M., Barchowsky, A., Bowser, R., Youk, A., and Talbott, E. O. (2012). Pesticide exposure as a risk factor for amyotrophic lateral sclerosis: a meta-analysis of epidemiological studies: pesticide exposure as a risk factor for ALS. Environ. Res. 117, 112–119. doi: 10.1016/j.envres.2012.06.007
Mandrioli, J., D’Amico, R., Zucchi, E., de Biasi, S., Banchelli, F., Martinelli, I., et al. (2023). Randomized, double-blind, placebo-controlled trial of rapamycin in amyotrophic lateral sclerosis. Nat. Commun. 14:4970. doi: 10.1038/s41467-023-40734-8
Masrori, P., and Van Damme, P. (2020). Amyotrophic lateral sclerosis: a clinical review. Eur. J. Neurol. 27, 1918–1929. doi: 10.1111/ene.14393
Mattsson, P., Lönnstedt, I., Nygren, I., and Askmark, H. (2012). Physical fitness, but not muscle strength, is a risk factor for death in amyotrophic lateral sclerosis at an early age. J. Neurol. Neurosurg. Psychiatry. 83, 390–394. doi: 10.1136/jnnp.2010.218982
McAlary, L., Aquilina, J. A., and Yerbury, J. J. (2016). Susceptibility of mutant SOD1 to form a destabilized monomer predicts cellular aggregation and toxicity but not in vitro aggregation propensity. Front. Neurosci. 10:499. doi: 10.3389/fnins.2016.00499
McCord, J. M., and Fridovich, I. (1969). Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244, 6049–6055. doi: 10.1016/S0021-9258(18)63504-5
Mehta, P., Antao, V., Kaye, W., Sanchez, M., Williamson, D., Bryan, L., et al. (2014). Prevalence of amyotrophic lateral sclerosis – United States, 2010-2011. MMWR Suppl. 63, 1–14
Mehta, P. R., Brown, A. L., Ward, M. E., and Fratta, P. (2023). The era of cryptic exons: implications for ALS-FTD. Mol. Neurodegener. 18:16. doi: 10.1186/s13024-023-00608-5
Mehta, P., Raymond, J., Punjani, R., Han, M., Larson, T., Kaye, W., et al. (2023). Prevalence of amyotrophic lateral sclerosis in the United States using established and novel methodologies, 2017. Amyotroph Lateral Scler Frontotemporal Degener 24, 108–116. doi: 10.1080/21678421.2022.2059380
Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., and Akkari, P. A. (2019). ALS genetics, mechanisms, and therapeutics: where are we now? Front. Neurosci. 13:1310. doi: 10.3389/fnins.2019.01310
Melamed, Z.'e., López-Erauskin, J., Baughn, M. W., Zhang, O., Drenner, K., Sun, Y., et al. (2019). Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 22, 180–190. doi: 10.1038/s41593-018-0293-z
Mergenthaler, P., Lindauer, U., Dienel, G. A., and Meisel, A. (2013). Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 36, 587–597. doi: 10.1016/j.tins.2013.07.001
Millecamps, S., and Julien, J.-P. (2013). Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 14, 161–176. doi: 10.1038/nrn3380
Mullard, A. (2023). NCATS's plan to grow the rare disease pipeline. Nat. Rev. Drug Discov. 22, 440–441. doi: 10.1038/d41573-023-00082-0
Müller, K., Oh, K. W., Nordin, A., Panthi, S., Kim, S. H., Nordin, F., et al. (2022). De novo mutations in SOD1 are a cause of ALS. J. Neurol. Neurosurg. Psychiatry 93, 201–206. doi: 10.1136/jnnp-2021-327520
Nakaya, T., and Maragkakis, M. (2018). Amyotrophic lateral sclerosis associated FUS mutation shortens mitochondria and induces neurotoxicity. Sci. Rep. 8:15575. doi: 10.1038/s41598-018-33964-0
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Ngo, S. T., Steyn, F. J., and McCombe, P. A. (2014). Body mass index and dietary intervention: implications for prognosis of amyotrophic lateral sclerosis. J. Neurol. Sci. 340, 5–12. doi: 10.1016/j.jns.2014.02.035
Nicolas, A., Kenna, K. P., Renton, A. E., Ticozzi, N., Faghri, F., Chia, R., et al. (2018). Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1267–1288. doi: 10.1016/j.neuron.2018.02.027
Nieh, C., Mancuso, J. D., Powell, T. M., Welsh, M. M., Gackstetter, G. D., and Hooper, T. I. (2021). Cigarette smoking patterns among U.S. military service members before and after separation from the military. PLoS One 16:e0257539. doi: 10.1371/journal.pone.0257539
Orellana, J. A., Froger, N., Ezan, P., Jiang, J. X., Bennett, M. V. L., Naus, C. C., et al. (2011). ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J. Neurochem. 118, 826–840. doi: 10.1111/j.1471-4159.2011.07210.x
Orihuela, R., McPherson, C. A., and Harry, G. J. (2016). Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 173, 649–665. doi: 10.1111/bph.13139
Oskarsson, B., Horton, D. K., and Mitsumoto, H. (2015). Potential environmental factors in amyotrophic lateral sclerosis. Neurol. Clin. 33, 877–888. doi: 10.1016/j.ncl.2015.07.009
Paganoni, S., Macklin, E. A., Lee, A., Murphy, A., Chang, J., Zipf, A., et al. (2014). Diagnostic timelines and delays in diagnosing amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler Frontotemporal Degener 15, 453–456. doi: 10.3109/21678421.2014.903974
Palamiuc, L., Schlagowski, A., Ngo, S. T., Vernay, A., Dirrig-Grosch, S., Henriques, A., et al. (2015). A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 7, 526–546. doi: 10.15252/emmm.201404433
Pansarasa, O., Bordoni, M., Diamanti, L., Sproviero, D., Gagliardi, S., and Cereda, C. (2018). SOD1 in amyotrophic lateral sclerosis: “ambivalent” behavior connected to the disease. Int. J. Mol. Sci. 19:1345. doi: 10.3390/ijms19051345
Potenza, R. L., Armida, M., and Popoli, P. (2024). Can some anticancer drugs be repurposed to treat amyotrophic lateral sclerosis? A brief narrative review. Int. J. Mol. Sci. 25:1751. doi: 10.3390/ijms25031751
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A., and Patel, B. K. (2019). Molecular mechanisms of TDP-43 Misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 12:25. doi: 10.3389/fnmol.2019.00025
Qiu, H., Lee, S., Shang, Y., Wang, W. Y., Au, K. F., Kamiya, S., et al. (2014). ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Invest. 124, 981–999. doi: 10.1172/JCI72723
Rangaraju, V., Calloway, N., and Ryan, T. A. (2014). Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835. doi: 10.1016/j.cell.2013.12.042
Reaume, A. G., Elliott, J. L., Hoffman, E. K., Kowall, N. W., Ferrante, R. J., Siwek, D. R., et al. (1996). Motor neurons in cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 13, 43–47. doi: 10.1038/ng0596-43
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Riazi, K., Galic, M. A., Kuzmiski, J. B., Ho, W., Sharkey, K. A., and Pittman, Q. J. (2008). Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc. Natl. Acad. Sci. USA 105, 17151–17156. doi: 10.1073/pnas.0806682105
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rothstein, J. D., Martin, L. J., and Kuncl, R. W. (1992). Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 326, 1464–1468. doi: 10.1056/NEJM199205283262204
Saitoh, Y., and Takahashi, Y. (2020). Riluzole for the treatment of amyotrophic lateral sclerosis. Neurodegener. Dis. Manag. 10, 343–355. doi: 10.2217/nmt-2020-0033
Sasaki, S., and Iwata, M. (2007). Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 66, 10–16. doi: 10.1097/nen.0b013e31802c396b
Sau, D., de Biasi, S., Vitellaro-Zuccarello, L., Riso, P., Guarnieri, S., Porrini, M., et al. (2007). Mutation of SOD1 in ALS: a gain of a loss of function. Hum. Mol. Genet. 16, 1604–1618. doi: 10.1093/hmg/ddm110
Segura, T., Medrano, I. H., Collazo, S., Maté, C., Sguera, C., del Rio-Bermudez, C., et al. (2023). Symptoms timeline and outcomes in amyotrophic lateral sclerosis using artificial intelligence. Sci. Rep. 13:702. doi: 10.1038/s41598-023-27863-2
Sellier, C., Campanari, M. L., Julie Corbier, C., Gaucherot, A., Kolb-Cheynel, I., Oulad-Abdelghani, M., et al. (2016). Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 35, 1276–1297. doi: 10.15252/embj.201593350
Silani, V., Ludolph, A., and Fornai, F. (2017). The emerging picture of ALS: a multisystem, not only a “motor neuron disease”. Arch. Ital. Biol. 155, 99–109. doi: 10.12871/00039829201741
Singh, S., and Singh, T. G. (2020). Role of nuclear factor kappa B (NF-κB) Signalling in neurodegenerative diseases: an mechanistic approach. Curr. Neuropharmacol. 18, 918–935. doi: 10.2174/1570159X18666200207120949
Smeyers, J., Banchi, E. G., and Latouche, M. (2021). C9ORF72: what it is, what it does, and why it matters. Front. Cell. Neurosci. 15:661447. doi: 10.3389/fncel.2021.661447
Smukowski, S. N., Maioli, H., Latimer, C. S., Bird, T. D., Jayadev, S., and Valdmanis, P. N. (2022). Progress in amyotrophic lateral sclerosis gene discovery. Neurol. Genet. 8:e669. doi: 10.1212/NXG.0000000000000669
Štětkářová, I., and Ehler, E. (2021). Diagnostics of amyotrophic lateral sclerosis: up to date. Diagnostics (Basel) 11:231. doi: 10.3390/diagnostics11020231
Stribl, C., Samara, A., Trümbach, D., Peis, R., Neumann, M., Fuchs, H., et al. (2014). Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43. J. Biol. Chem. 289, 10769–10784. doi: 10.1074/jbc.M113.515940
Sturtz, L. A., Diekert, K., Jensen, L. T., Lill, R., and Culotta, V. C. (2001). A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J. Biol. Chem. 276, 38084–38089. doi: 10.1074/jbc.M105296200
Sunildutt, N., Ahmed, F., Salih, A. R. C., Lim, J. H., and Choi, K. H. (2024). Integrating transcriptomic and structural insights: revealing drug repurposing opportunities for sporadic ALS. ACS Omega 9, 3793–3806. doi: 10.1021/acsomega.3c07296
Swarup, V., Phaneuf, D., Bareil, C., Robertson, J., Rouleau, G. A., Kriz, J., et al. (2011). Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain 134, 2610–2626. doi: 10.1093/brain/awr159
Tefera, T. W., Bartlett, K., Tran, S. S., Hodson, M. P., and Borges, K. (2019). Impaired pentose phosphate pathway in the spinal cord of the hSOD1 (G93A) mouse model of amyotrophic lateral sclerosis. Mol. Neurobiol. 56, 5844–5855. doi: 10.1007/s12035-019-1485-6
Tefera, T. W., Steyn, F. J., Ngo, S. T., and Borges, K. (2021). CNS glucose metabolism in amyotrophic lateral sclerosis: a therapeutic target? Cell Biosci. 11:14. doi: 10.1186/s13578-020-00511-2
The ALS Association . (2024a). Lou Gehrig and the history of ALS. Available at: https://www.als.org/understanding-als/lou-gehrig
The ALS Association . (2024b). Understanding ALS. Available at: https://www.als.org/understanding-als
Thompson, K. K., and Tsirka, S. E. (2017). The diverse roles of microglia in the neurodegenerative aspects of central nervous system (CNS) autoimmunity. Int. J. Mol. Sci. 18. doi: 10.3390/ijms18030504
Torres, P., Ramírez-Núñez, O., Romero-Guevara, R., Barés, G., Granado-Serrano, A. B., Ayala, V., et al. (2018). Cryptic exon splicing function of TARDBP interacts with autophagy in nervous tissue. Autophagy 14, 1398–1403. doi: 10.1080/15548627.2018.1474311
Umoh, M. E., Fournier, C., Li, Y., Polak, M., Shaw, L., Landers, J. E., et al. (2016). Comparative analysis of C9orf72 and sporadic disease in an ALS clinic population. Neurology 87, 1024–1030. doi: 10.1212/WNL.0000000000003067
Urits, I., Israel, J., Hakobyan, H., Yusin, G., Lassiter, G., Fackler, N., et al. (2020). Baricitinib for the treatment of rheumatoid arthritis. Reumatologia 58, 407–415. doi: 10.5114/reum.2020.102006
Vance, C., Rogelj, B., Hortobágyi, T., de Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Vandoorne, T., De Bock, K., and Van Den Bosch, L. (2018). Energy metabolism in ALS: an underappreciated opportunity? Acta Neuropathol. 135, 489–509. doi: 10.1007/s00401-018-1835-x
Villalta, S. A., Nguyen, H. X., Deng, B., Gotoh, T., and Tidball, J. G. (2009). Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 18, 482–496. doi: 10.1093/hmg/ddn376
Wang, M. D., Gomes, J., Cashman, N. R., Little, J., and Krewski, D. (2014). A meta-analysis of observational studies of the association between chronic occupational exposure to lead and amyotrophic lateral sclerosis. J. Occup. Environ. Med. 56, 1235–1242. doi: 10.1097/JOM.0000000000000323
Wang, W. Y., Tan, M.-S., Yu, J.-T., and Tan, L. (2015). Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease. Ann. Transl. Med 3:136. doi: 10.3978/j.issn.2305-5839.2015.03.49
Wang, Z., Ying, Z., Bosy-Westphal, A., Zhang, J., Schautz, B., Later, W., et al. (2010). Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am. J. Clin. Nutr. 92, 1369–1377. doi: 10.3945/ajcn.2010.29885
Wiedemann, F. R., Manfredi, G., Mawrin, C., Beal, M. F., and Schon, E. A. (2002). Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 80, 616–625. doi: 10.1046/j.0022-3042.2001.00731.x
Xia, Q., Wang, H., Hao, Z., Fu, C., Hu, Q., Gao, F., et al. (2016). TDP‐43 loss of function increasesTFEBactivity and blocks autophagosome–lysosome fusion. EMBO J. 35, 121–142. doi: 10.15252/embj.201591998
Zala, D., Hinckelmann, M. V., Yu, H., Lyra da Cunha, M. M., Liot, G., Cordelières, F. P., et al. (2013). Vesicular glycolysis provides on-board energy for fast axonal transport. Cell 152, 479–491. doi: 10.1016/j.cell.2012.12.029
Zhang, K., Donnelly, C. J., Haeusler, A. R., Grima, J. C., Machamer, J. B., Steinwald, P., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61. doi: 10.1038/nature14973
Zhang, L., Keung, W., Samokhvalov, V., Wang, W., and Lopaschuk, G. D. (2010). Role of fatty acid uptake and fatty acid beta-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim. Biophys. Acta 1801, 1–22. doi: 10.1016/j.bbalip.2009.09.014
Zhao, J., Wang, X., Huo, Z., Chen, Y., Liu, J., Zhao, Z., et al. (2022). The impact of mitochondrial dysfunction in amyotrophic lateral sclerosis. Cells 11:2049. doi: 10.3390/cells11132049
Keywords: amyotrophic lateral sclerosis, inflammation, excitotoxicity, metabolism, neurodegenerative disease
Citation: Bradford D and Rodgers KE (2024) Advancements and challenges in amyotrophic lateral sclerosis. Front. Neurosci. 18:1401706. doi: 10.3389/fnins.2024.1401706
Edited by:
Cassie S. Mitchell, Georgia Institute of Technology, United StatesReviewed by:
Francesco Liguori, Santa Lucia Foundation (IRCCS), ItalyCopyright © 2024 Bradford and Rodgers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathleen E. Rodgers, a3JvZGdlcnNAYXJpem9uYS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.