 Tabitha R. F. Green1†
Tabitha R. F. Green1† Sean D. Carey2,3†Grant Mannino1John A. Craig2
Sean D. Carey2,3†Grant Mannino1John A. Craig2 Rachel K. Rowe1
Rachel K. Rowe1 Mark R. Zielinski2,3*
Mark R. Zielinski2,3*- 1Department of Integrative Physiology, University of Colorado Boulder, Boulder, CO, United States
- 2Veterans Affairs (VA) Boston Healthcare System, West Roxbury, MA, United States
- 3Department of Psychiatry, Harvard Medical School, West Roxbury, MA, United States
Traumatic brain injury (TBI) can induce dysregulation of sleep. Sleep disturbances include hypersomnia and hyposomnia, sleep fragmentation, difficulty falling asleep, and altered electroencephalograms. TBI results in inflammation and altered hemodynamics, such as changes in blood brain barrier permeability and cerebral blood flow. Both inflammation and altered hemodynamics, which are known sleep regulators, contribute to sleep impairments post-TBI. TBIs are heterogenous in cause and biomechanics, which leads to different molecular and symptomatic outcomes. Animal models of TBI have been developed to model the heterogeneity of TBIs observed in the clinic. This review discusses the intricate relationship between sleep, inflammation, and hemodynamics in pre-clinical rodent models of TBI.
1 TBI as a disease process
Traumatic brain injury (TBI) is a major cause of morbidity and mortality worldwide. TBI is caused by a mechanical insult to the brain that results in motor, cognitive, affective, and behavioral symptoms. Although TBI is a rapid onset condition, a complex disease process ensues and can progress chronically. TBI causes a robust inflammatory response triggered by tissue damage. Initially, inflammation can be beneficial and allow restoration and clean-up of damaged tissues. However, inflammation often progresses chronically post-TBI and creates a unique and progressive disease environment causing symptoms to evolve over time. Sleep is a biological system that is tightly coupled with inflammation due to the crossover in signaling molecules that are involved in both inflammatory and sleep processes. Furthermore, brain hemodynamics alter with sleep–wake state, which can become dysregulated post-TBI (Figure 1; Townsend et al., 1973; Madsen et al., 1991a,b; Bouma et al., 1992; Braun et al., 1997; Soustiel et al., 2005; Bangash et al., 2008; Salehi et al., 2017).
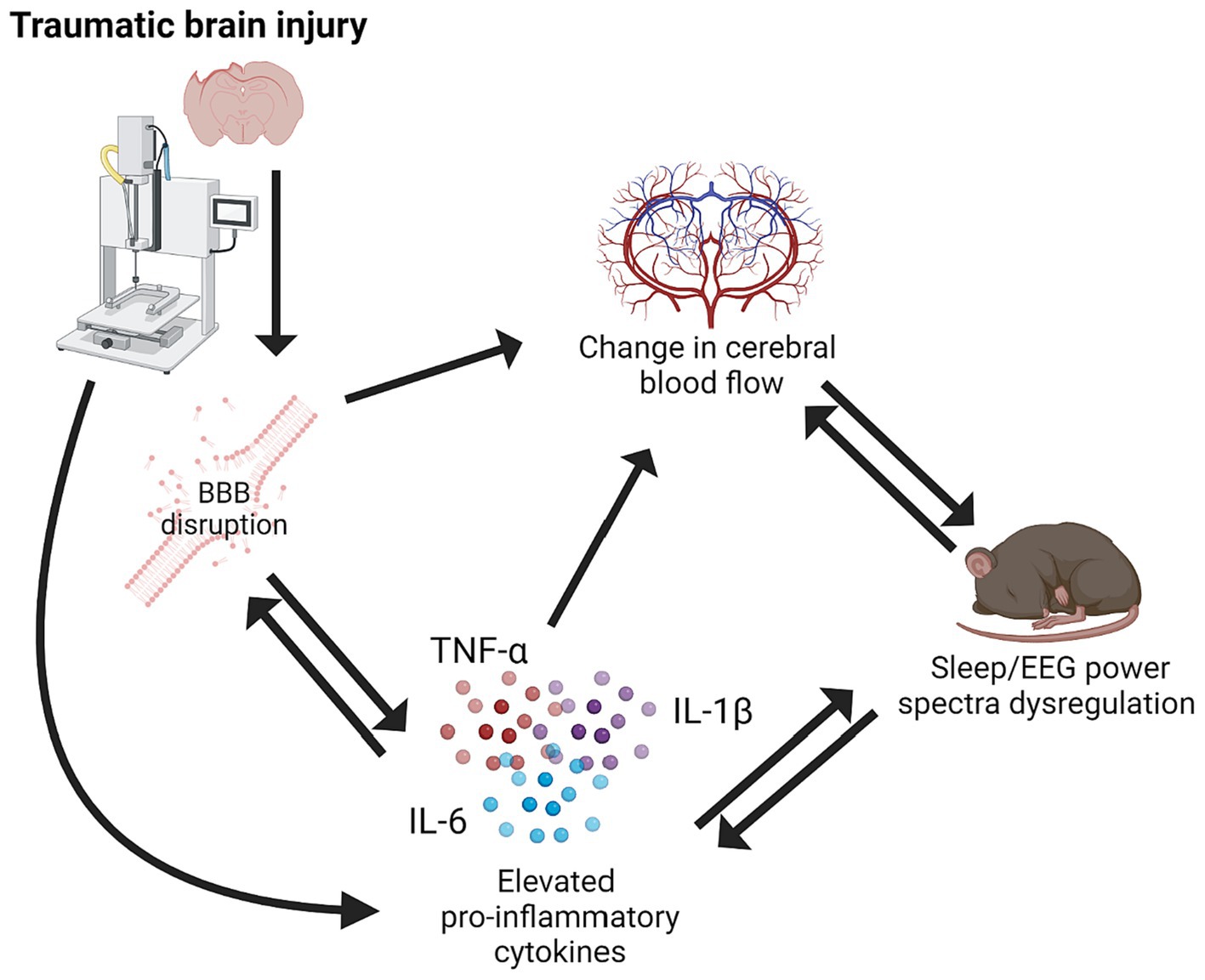
Figure 1. Schematic of relationship between traumatic brain injury, pro-inflammatory cytokines, BBB disruption, cerebral blood flow, and sleep/EEG power spectra dysregulation. Created using BioRender.com.
TBI can be broadly classified into three categories based on injury modality: diffuse, focal, and blast. These injury modalities are not mutually exclusive. Diffuse TBI is caused by the head or neck forcefully colliding with an object or surface. Common causes of diffuse TBI are falls, domestic violence, contact/collision sports, and motor vehicle accidents. In contrast, focal TBI results from a penetrating head wound, and common causes include gunshot wounds, skull fractures, and shrapnel injuries. Blast TBI results from shock waves, often associated with combat related explosions (Bass et al., 2012). Shock waves are transmitted through the brain causing direct movement and disruption of brain tissue (Säljö et al., 2008; Kovacs et al., 2014). Furthermore, shock waves cause displacement and oscillations of blood and other fluids in vessels throughout the body, which is a second, but simultaneous, cause of blast-induced TBI (Long et al., 2009; Kovacs et al., 2014).
A hallmark symptom of diffuse TBI is diffuse axonal injury. Diffuse axonal injury, characterized by damage to white matter tracts and axons, is present in all severities of diffuse TBI (Johnson et al., 2013). There are many pathological features of diffuse axonal injury that contribute to the acute and chronic symptoms associated with the condition, for example structural axonal damage, disrupted axonal transport, and axonal swelling (Johnson et al., 2013). Diffuse axonal injury can progress to Wallerian degeneration, a type of neurodegeneration associated with physical damage to the neuron (Johnson et al., 2013; Koliatsos and Alexandris, 2019). Diffuse TBI does not exhibit immediate cell death; however, neuronal cell death does occur over time because of secondary inflammatory processes (Lifshitz et al., 2007, 2016). In contrast, focal TBI is characterized by overt tissue disruption and cell death at the site of injury as early as 4–6 h post-TBI (Flygt et al., 2016; Díaz et al., 2023).
2 Pre-clinical models of TBI
TBI is often studied preclinically using animal models (Figure 2). Most commonly, rodent models are used, however, there are many other established models including ferrets, pigs, drosophila, cats, and rabbits (Ma et al., 2019). Diffuse TBI can be modeled using a closed head injury technique. Closed head TBI is often modeled using a weight drop technique. The weight drop injury (WDI) TBI model involves a gravity falling weight onto the head of a rodent where weight position and displacement of the force can be altered. Variations in WDI models include the Feeney model that causes a direct impact to the dura or the Marmarou model that induces a diffuse impact due to a plate placed above the skull (Feeney et al., 1981; Foda and Marmarou, 1994). The size of the object and the speed and height from which it is dropped change the severity and biomechanics of the weight drop generated injury (Bodnar et al., 2019). A controlled cortical impact (CCI) device can be used to deliver an impact to the intact skull causing acceleration/deceleration of the brain. Closed head piston driven methods can be performed with or without a rodent helmet and on either an intact scalp or exposed skull. For an in-depth review of closed head injury models refer to Bodnar et al. (2019). Another method to induce a diffuse TBI is midline fluid percussion injury (mFPI). To induce an mFPI, a craniectomy is performed on the sagittal suture between bregma and lambda markings on the skull. The fluid impulse is delivered to the intact dura through an injury hub assembly attached to the skull (Lifshitz et al., 2016). The forces applied to the brain are dissipated through the tissue and cause pathology at multiple sites, determined by the structural properties of the skull (Beitchman et al., 2021).
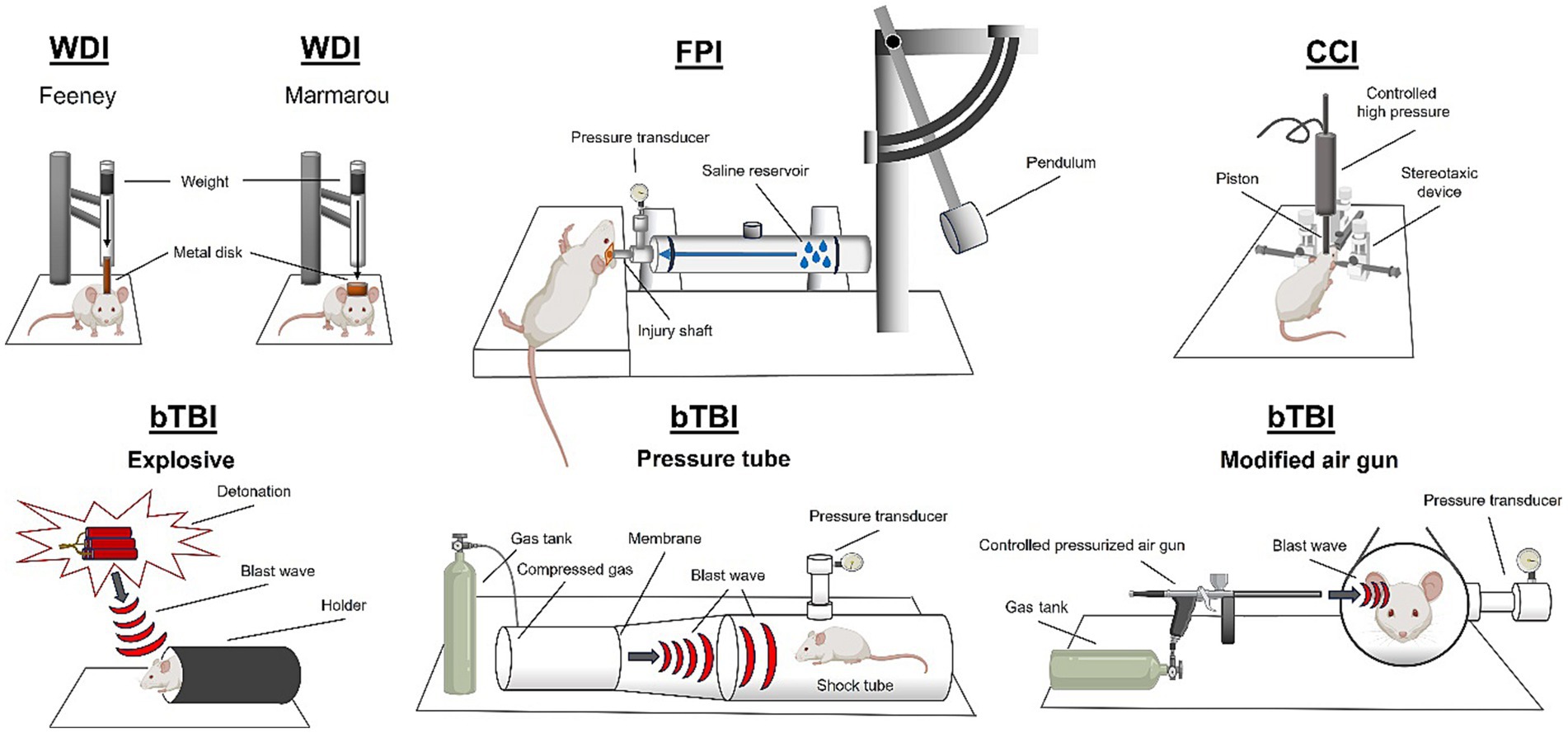
Figure 2. Rodent models of traumatic brain injury. Weight drop injury (WDI), fluid percussion injury (FPI), controlled cortical impact (CCI), and blast traumatic brain injury (bTBI). Created using BioRender.com.
Focal TBI is most studied using the CCI method. CCI was designed to administer focal TBIs to Ferrets in 1988 (Lighthall, 1988), but since has been adapted to many species (primate, mouse, swine; Kobeissy, 2015). Most commonly, CCI is used in mice and rats and involves craniectomy surgery to remove a small portion of the parietal bone. After removal of the skull, a pneumatic (or electromagnetic) piston is driven into the brain tissue at the desired depth and velocity causing tissue disruption. Lateral fluid percussion injury (LFPI) results in focal brain damage at the site of the impact, and also causes diffuse pathology throughout the brain. LFPI requires a craniectomy in the parietal bone, and the assembly of an injury hub onto the skull. Using the fluid percussion device, a fluid impulse is delivered directly to the brain through the injury hub, onto the intact dura, which causes a focal lesion to the brain, as well as applying rotational forces to the brain that results in widespread diffuse pathology (Alder et al., 2011). The craniectomy site can be adjusted to change the location of the focal lesion (i.e., frontal bone; Ouyang et al., 2018).
To induce pathology associated with a blast, models of blast TBI (bTBI) use peak overpressure, and number of exposures as metrics (Skotak et al., 2019). Explosive bTBI methods can be modeled in rodents using an open field explosive device detonation which results in both primary and secondary blast pathology (Kovacs et al., 2014). To study the primary blast mechanism in the absence of secondary shockwave reverberations in the rat, blast tubes have been developed (Säljö et al., 2000; Risling et al., 2011). Blast tubes have also been invented to study blast injury in swine (Bauman et al., 2009). bTBI can also be induced in rodents using a pressure tube, in which build-up of compressed gas is used to rupture a breakable membrane which causes a shockwave to travel down the tube (Long et al., 2009; Reneer et al., 2011). Blast injury with secondary shrapnel penetration can be modeled using a blast gun (Gamboa et al., 2021). Additionally, blast injury can be modeled using a modified air gun to administer focal injury via directed pressurized air (Heldt et al., 2014).
3 Sleep dysregulation after TBI
Sleep disturbances occur in 30–84% of patients post-TBI (Paredes et al., 2021). TBIs can induce profound sleep dysregulation, including hypersomnia and hyposomnia, sleep fragmentation, sleepiness, difficulty falling asleep, and altered electroencephalograms (EEGs; Sandsmark et al., 2017; Aoun et al., 2019; Green et al., 2020; Rowe and Griesbach, 2022). In the clinic, patients report both acute and chronic sleep disturbances, and these occur following mild, moderate, and severe TBI (Verma et al., 2007; Viola-Saltzman and Watson, 2012; Wickwire et al., 2016; Mollayeva et al., 2017). Rodent models of TBI are used to investigate sleep disturbances. Experimental TBI results in acute and chronic sleep disturbances, and these sleep disturbances occur following both mild and moderate TBI (Table 1). It is important to note that there are multiple factors that can influence sleep disturbances including the location of the injury, severity, depth of impact, diffuse or focal, pressure of blast, and brain area of injury.
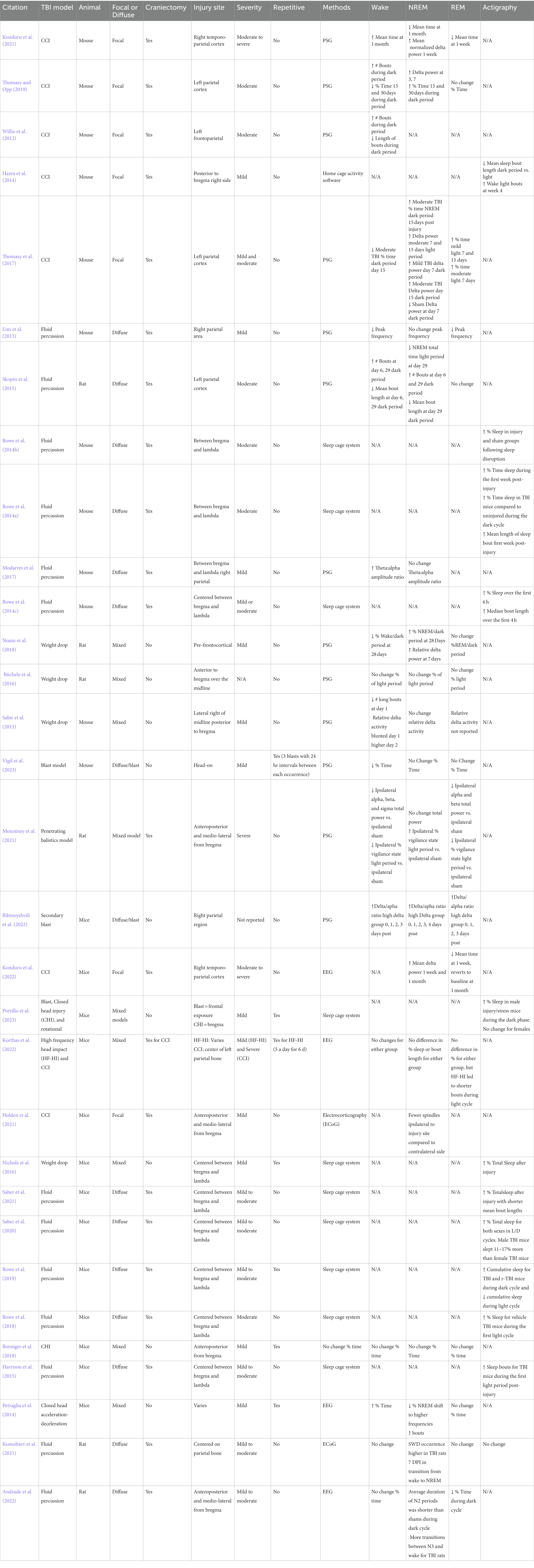
Table 1. Summary of key sleep and EEG findings of studies involving rodent TBI models.
Sleep disturbances can exacerbate pathology after a TBI and lead to longer recovery times in patients, thus, treating sleep disturbances is of high priority to increase the quality of life of TBI survivors (Wiseman-Hakes et al., 2013; Theadom et al., 2016; Sandsmark et al., 2017; Morse and Garner, 2018). Unfortunately, there is a lack of effective long lasting sleep treatments for TBI (Wickwire, 2020). While the exact mechanisms responsible for sleep disturbances caused by TBI are complex and unknown, the contribution of pro-inflammatory cytokines and alteration of neurovascular hemodynamics is discussed in this review (Pop and Badaut, 2011; Viola-Saltzman and Watson, 2012; Lok et al., 2015; Sandsmark et al., 2017; Aoun et al., 2019; Green et al., 2020; Rowe and Griesbach, 2022).
4 Models of TBI and sleep disturbances
Sleep has been studied post-TBI in a range of animal models (Table 1). Here, we discuss sleep post-TBI after diffuse, focal, and blast injuries. Most research on sleep post-TBI focuses on the acute period post-injury and details the post-traumatic sleep response (Rowe et al., 2014c, 2019). Few studies assess sleep at chronic post-injury time points. This is an important area for investigation since individuals with TBI report sleep dysregulation occurring 6 months or longer following TBI (Baumann et al., 2007).
4.1 Sleep after diffuse and mixed-model TBI
It is well established that diffuse TBI causes an acute increase in sleep, known as a period of ‘post-traumatic sleep’. There is a robust increase (over 50%) in time spent asleep in the first 6 h post-injury in mice (Rowe et al., 2014c). This period of post-traumatic increase in sleep occurs regardless of injury severity or time of day the injury occurs (Rowe et al., 2014c). This increase in sleep extends to the first week after mFPI, where brain-injured mice slept more than sham controls (Saber et al., 2020). Additionally, evidence suggests that sex differences in sleep responses to TBI occur with male mice sleeping more than female mice acutely after a TBI (Saber et al., 2020). While chronic sleep disturbances post-TBI are often reported in the clinic, enhanced sleep from mFPI was not sustained chronically in the mouse (Rowe et al., 2014a). After LFPI, a mixed model of diffuse and focal TBI, acute (6 days post-TBI) and chronic (29 days post-TBI) sleep disturbances were observed (Skopin et al., 2015), although there were no differences in sleep architecture at the midpoint (19 days post-TBI).
One study found that 6 days post-LFPI mice had increased bouts of NREM sleep (Skopin et al., 2015). Differences in sleep responses to TBI in rodents are shown to occur mostly during the dark period, which might be due to ceiling effects from their increased sleep during the light period. Nevertheless, increases in EEG delta power after weight drop are observed 7 days post-injury (Noain et al., 2018). Decreased long bouts of wakefulness during the dark period 1 day after TBI and increased EEG delta activity during the second day after weight drop are reported (Sabir et al., 2015). Increased bouts of NREM sleep following LFPI have also been observed during the dark period in rats (Skopin et al., 2015).
4.2 Sleep after focal TBI
After CCI, mice have identifiable gliosis and changes in sleep and EEG delta power (Willie et al., 2012; Hazra et al., 2014; Thomasy et al., 2017; Thomasy and Opp, 2019; Konduru et al., 2021, 2022). Another study found that EEG delta power during NREM sleep decreased by 55% after CCI at 7 days post-injury, and theta power (5.5–8.5 Hz) increased 59% during REM sleep. However, the same study found that there were no differences in time spent awake, in REM, or in NREM sleep after CCI at 7 days post-injury (Korthas et al., 2022). At 1–2 months post-TBI, CCI mice had greater sleep efficiency and altered sleep architecture compared to sham controls (Konduru et al., 2021). CCI mice also exhibited increased epileptiform activity acutely at week 1 or chronically at 1, 2, or 3 months, which may further cause sleep disruptions (Konduru et al., 2021).
4.3 Sleep after blast TBI
Hypersomnia and reduced EEG gamma power during a slow-wave-sleep state post-injury was reported in one bTBI study (Vigil et al., 2023). However, the polysomnography (PSG) surgery occurred post-TBI so proper baseline normalization of EEG signals was not done and sleep was assessed 24 h after the injury where inflammation and surgical procedures could affect sleep responses (Beuckmann et al., 2019). Another bTBI and PSG study in rats reported 24 h post-injury hypersomnia effects and did not show persistent effects or determine hallmarks of bTBI sequalae (Mountney et al., 2021). A third study found hypersomnia and EEG delta/theta ratios but only assessed within 1 week post-injury (Bibineyshvili et al., 2022).
5 TBI-induced mechanisms that dysregulate sleep
5.1 Pro-inflammatory cytokines and sleep
Cytokines are small cell-signaling molecules that have multiple functions including immune, inflammatory, vasoregulatory, and sleep regulatory functions. Many pro-inflammatory cytokines can alter sleep, electroencephalogram (EEG) power spectra, and induce sleepiness, and cytokines with anti-inflammatory properties tend to attenuate sleep responses to sleep promoting stimuli (Zielinski and Gibbons, 2022). Interleukin-1 beta (IL-1β), tumor necrosis factor-alpha (TNF-α), and IL-6 are the most well characterized pro-inflammatory cytokines that regulate sleep (Kinoshita et al., 2002; Krueger, 2008; Krueger et al., 2011). These cytokines are largely enhanced in sleep regulatory brain areas, such as the cortex, brainstem, and basal forebrain, and enhance non-rapid eye movement (NREM) sleep and EEG delta power (0.5–4 Hz frequency) after stimulation by sleep loss, disease, or injury (Krueger, 2008; Krueger et al., 2011). Pro-inflammatory cytokines can also alter sleep architecture, for example, they result in suppressed or delayed rapid eye movement (REM; Krueger et al., 2011; Zielinski and Gibbons, 2022).
Inflammation can promote sleep or result in sleep fragmentation depending on the stimuli severity or dose, stimuli duration, or timing post-stimuli (Zielinski and Gibbons, 2022). IL-1β, TNF-α, and IL-6 are increased in the circulation after sleep loss (Krueger, 2008). Application of IL-1β or TNF-α directly to the brain enhances NREM sleep (Dickstein et al., 1999; Baker et al., 2005). Inhibiting these cytokines or their receptors with transgenic mice, pharmacologically, or with small interfering RNA reduces sleep responses to sleep promoting stimuli (Krueger, 2008). Moreover, nucleotide-binding domain and leucine-rich repeat protein-3 (i.e., NLRP3) inflammasomes, which are sensing protein complexes that induce the somnogenic molecules IL-1β and IL-18 and are a critical regulator or NREM sleep and EEG delta power, are activated in the brain from sleep loss, pathogens, altered metabolism, and following a TBI (Zielinski et al., 2017; Carey et al., 2023). This further implicates the role of pro-inflammatory cytokines in TBI-induced sleep dysregulation. Although speculative, it is likely that sleep dysregulation following TBI is, in part, caused by both activation and modulation of cytokines and their receptors, and the reciprocating changes in these molecules caused by altering sleep amounts and sleep fragmentation.
There is a temporal association between the acute increase in sleep observed after diffuse TBI, and levels of pro-inflammatory cytokines (Rowe et al., 2014c). Consistent with diffuse TBI models, cytokines IL-1β and TNF-α are elevated hours after injury in CCI and LFPI models of focal TBI which can potentially contribute to sleep dysregulation (Dalgard et al., 2012; Perez-Polo et al., 2013). The literature reports that bTBI animal models can increase pro-inflammatory somnogenic cytokines in the brain, such as IL-1α, IL-1β, and IL-6 that could contribute to sleep dysregulation (Heyburn et al., 2023). Additionally, patients with TBI have elevated serum levels of pro-inflammatory cytokines (Morganti-Kossman et al., 1997; Morganti-Kossmann et al., 2002), including IL-1β (Tasçı et al., 2003). These findings are consistent with studies of increased sleepiness after TBI in humans and level of TBI severity with IL-1β potentially increasing sleepiness after injury (Watson et al., 2007). Cellular and molecular mechanisms other than those involving inflammation can also contribute to the sleep effects, such as loss of hypocretin neurons post-TBI (Thomasy and Opp, 2019), changes in hypocretin ligands and receptors (Korthas et al., 2022), and changes in circadian gene expression (e.g., Clock, Per1, Per 2, Bmal, Cry1, Cry2; Boone et al., 2012; Govindarajulu et al., 2023).
5.2 The blood–brain-barrier and cerebral blood flow post-TBI
The mechanical force of TBI can cause disruption to the blood–brain-barrier (BBB) upon impact (Cash and Theus, 2020). However, secondary BBB damage can occur due to ongoing inflammatory processes including pro-inflammatory cytokine signaling (Figure 1; Cash and Theus, 2020). Both focal and diffuse brain injuries disrupt the BBB, but the type of damage differs between brain injury models (Chodobski et al., 2011). For example, CCI and LFPI cause immediate shearing of blood vessels upon impact. Diffuse and closed head injury models also cause some bleeding upon impact; however, this is not due to the penetration of a metal piston or fluid impulse, this is due to compression of the brain from acceleration/deceleration forces. Blast TBI causes widespread disruption of the BBB, consisting of many microbleeds (Kawoos et al., 2021). One study found that in 8/10 brain regions tested there was significant bleeding within 15 min of blast induced TBI in rats (Logsdon et al., 2018). Mechanistically, the diffuse BBB disruption caused by blast TBI is due to disruption of tight junctions from loss of transmembrane and adhesion molecules (Hue et al., 2013; Heyburn et al., 2019). Furthermore, increased permeability of the BBB has been observed chronically post-injury, regardless of severity (Andrews et al., 2016). Damage to cerebral vasculature can cause long term alteration in cerebral blood flow (CBF; Golding, 2002; Monson et al., 2019). Reduced CBF can worsen patient outcomes and prolong secondary inflammation post-TBI (Jenkins et al., 1989; Robertson et al., 1992). After TBI, hyperpermeability of the cerebral vasculature can cause edema which increases intracranial pressure and decreases cerebral perfusion pressure (Alluri et al., 2018).
5.3 Cerebral blood flow and sleep post-TBI
CBF is intrinsically related to changes in neuronal activity and functions to support the rapid need for glucose and oxygen from changing brain demands (Zheng et al., 2016). An interaction between neuronal activity and CBF is referred to as neurovascular coupling and it is tightly regulated (Iadecola, 2017). Altered CBF often accompanies TBI as early as hours after injury in the very acute post-injury phase with decreases observed in patients (Bouma et al., 1992; Soustiel et al., 2005; Salehi et al., 2017), although an increase in CBF is reported in day 1–5 post-injury and is associated with better outcome (Kelly et al., 1997). Intriguingly, CBF is also reported to be reduced at 6 and 12 months post-injury in TBI survivors compared to healthy controls (Gaggi et al., 2023). There are physiologic, neurologic, and immunologic changes that mediate changes in neurovascular hemodynamics to affect CBF and link alterations in sleep. Specifically, cytokines such as TNF-α and IL-1β can modulate CBF (Vila and Salaices, 2005). For example, chronic intracerebroventricular infusion of IL-1β reduces CBF in rodent models suggesting that chronic IL-1β might be involved in the TBI hypoperfusion (Maher et al., 2003).
5.4 CBF and sleep
Evidence indicates that CBF and sleep have a dynamic relationship, which alludes to the potential effects on this relationship from TBI since both CBF and sleep are altered by TBI (Viola-Saltzman and Watson, 2012; Salehi et al., 2017). Broadly, global decreases in CBF during NREM compared to wake are observed (Townsend et al., 1973; Madsen et al., 1991a,b; Braun et al., 1997; Bangash et al., 2008). A further decrease in CBF during NREM was found in humans as well as parallel findings in measurements of cerebral metabolic rate (Townsend et al., 1973; Madsen et al., 1991b; Braun et al., 1997; Bangash et al., 2008). Recent reports demonstrate both vascular diameter and hemoglobin concentrations increase in NREM and REM compared to awake brain states in mice (Turner et al., 2020). Typically, blood flow increases to areas of the brain by vessel dilation, although reductions in microvascular pressure and myogenic mechanisms can also modulate CBF (Cipolla, 2009). Consequently, it is plausible that pro-inflammatory cytokines, which can induce vasodilation, can affect hemodynamics, in part, from the modulation of cytokines from TBI (Vila and Salaices, 2005). Nevertheless, in both humans and rodents, blood flow changes during sleep states and during sleep state transitions, suggesting that altered hemodynamics from TBI could influence sleep or vice versa (Zielinski and Gibbons, 2022).
6 Treating TBI-induced sleep disturbances
Sleep disturbances are experienced by TBI survivors; however, treating sleep disturbances has proven challenging. Therapeutics are typically administered to mitigate the dominant symptom or treat a specific sleep–wake disorder. Clinical studies support the potential use of pharmacological compounds that promote wakefulness, such as modafinil, to treat excessive daytime sleepiness in TBI patients. Wake-promoting agents have shown some success in ameliorating excessive sleepiness after TBI (Kaiser et al., 2010; Menn et al., 2014), while stimulants have been less studied in the clinical setting. In a preclinical model of TBI, modafinil attenuates neuroinflammation and exerts neuroprotective effects (Ozturk et al., 2021). These anti-inflammatory actions may underlie the effectiveness of modafinil in treating excessive daytime sleepiness after a brain injury.
Treating the underlying pathology, as opposed to the dominant symptom, has shown efficacy in preclinical TBI models. Novel compounds that target cytokines have been administered to reduce inflammation and subsequently prevent TBI-induced sleep disturbances in the mouse (Rowe et al., 2018; Apostol et al., 2022). Importantly, when acute TBI-induced sleep is reduced with a pharmacological intervention, mice have improved functional outcomes (Rowe et al., 2018; Apostol et al., 2022). This highlights the importance of treating sleep disturbances with the goal of improving functional recovery and quality of life.
Other novel treatments for TBI-induced sleep disturbances include light therapy, dietary supplementation, and cognitive-behavioral therapy. Dietary supplementation with branched chain amino acids (BCAAs) improves wakefulness and cognition in a rodent model of TBI (Lim et al., 2013; Elkind et al., 2015). BCAA supplementation also improved insomnia severity and sleep measures determined by actigraphy, in the chronic phase of recovery from TBI in a cohort of veterans (Elliott et al., 2022). In a mechanistic study to investigate the action of BCAAs on TBI-induced sleep disturbances it was identified that BCAAs restore excitatory glutamate within presynaptic terminals on wake-promoting orexin neurons (Elliott et al., 2018).
Survivors of TBI experience medical, psychological, and mental health comorbidities during the long-term recovery process that complicate treatment strategies. Comorbidities associated with TBI include pain, headaches, and endocrine dysfunction, which further exacerbate sleep disturbances (Rowe and Griesbach, 2022). It should be noted that treatment of these comorbidities can include medications that interfere with healthy sleep. There is a critical gap in the current literature around combination therapies to treat TBI-induced sleep disturbances and comorbid symptoms. Further research is needed to elucidate effective treatment strategies for TBI survivors. A combination of therapies that include targeted pharmacological intervention, dietary supplementation, light therapy, and behavioral therapies should be explored.
7 Conclusion
Sleep is dysregulated post-TBI regardless of severity, and injury biomechanics which is consistent with reports in humans. There are diverse changes in sleep architecture, inflammation, and hemodynamics after TBI. Many of these differences are driven by the biomechanics of the injury model such as diffuse, focal, or blast injury, and by the brain regions injured. Understanding the complex interactions between the biological systems affected by TBI is essential in progressing both research and patient treatments.
Author contributions
TG: Writing – original draft, Writing – review & editing. SC: Writing – original draft, Writing – review & editing. GM: Writing – original draft, Writing – review & editing. JC: Writing – original draft, Writing – review & editing. RR: Writing – original draft, Writing – review & editing. MZ: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Preparation of the report was supported by the Department of Veterans Affairs grant I21RX003722 (MZ) and 5I01BX005379 (MZ) and Congressionally Directed Medical Research Programs Department of Defense grant W81XWH-22-1-0383 (RR).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alder, J., Fujioka, W., Lifshitz, J., Crockett, D. P., and Thakker-Varia, S. (2011). Lateral fluid percussion: model of traumatic brain injury in mice. J. visualized experiments: JoVE. 54, 1–6. doi: 10.3791/3063-v
Alluri, H., Shaji, C. A., Davis, M. L., and Tharakan, B. (2018). A mouse controlled cortical impact model of traumatic brain injury for studying blood-brain barrier dysfunctions. Methods Mol. Biol. 1717, 37–52. doi: 10.1007/978-1-4939-7526-6_4
Andrade, P., Lara-Valderrábano, L., Manninen, E., Ciszek, R., Tapiala, J., Ndode-Ekane, X. E., et al. (2022). Seizure susceptibility and sleep disturbance as biomarkers of Epileptogenesis after experimental TBI. Biomedicine 10:1138. doi: 10.3390/biomedicines10051138
Andrews, A. M., Lutton, E. M., Merkel, S. F., Razmpour, R., and Ramirez, S. H. (2016). Mechanical injury induces brain endothelial-derived microvesicle release: implications for cerebral vascular injury during traumatic brain injury. Front. Cell. Neurosci. 10:43. doi: 10.3389/fncel.2016.00043
Aoun, R., Rawal, H., Attarian, H., and Sahni, A. (2019). Impact of traumatic brain injury on sleep: an overview. Nat Sci Sleep. 11, 131–140. doi: 10.2147/NSS.S182158
Apostol, C. R., Bernard, K., Tanguturi, P., Molnar, G., Bartlett, M. J., Szabo, L., et al. (2022). Design and synthesis of brain penetrant Glycopeptide analogues of PACAP with neuroprotective potential for traumatic brain injury and parkinsonism. Front Drug Discov (Lausanne). 1:1. doi: 10.3389/fddsv.2021.818003
Baker, F. C., Shah, S., Stewart, D., Angara, C., Gong, H., Szymusiak, R., et al. (2005). Interleukin 1beta enhances non-rapid eye movement sleep and increases c-Fos protein expression in the median preoptic nucleus of the hypothalamus. Am. J. Phys. Regul. Integr. Comp. Phys. 288, R998–r1005. doi: 10.1152/ajpregu.00615.2004
Bangash, M. F., Xie, A., Skatrud, J. B., Reichmuth, K. J., Barczi, S. R., and Morgan, B. J. (2008). Cerebrovascular response to arousal from NREM and REM sleep. Sleep 31, 321–327. doi: 10.1093/sleep/31.3.321
Bass, C. R., Panzer, M. B., Rafaels, K. A., Wood, G., Shridharani, J., and Capehart, B. (2012). Brain injuries from blast. Ann. Biomed. Eng. 40, 185–202. doi: 10.1007/s10439-011-0424-0
Bauman, R. A., Ling, G., Tong, L., Januszkiewicz, A., Agoston, D., Delanerolle, N., et al. (2009). An introductory characterization of a combat-casualty-care relevant swine model of closed head injury resulting from exposure to explosive blast. J. Neurotrauma 26, 841–860. doi: 10.1089/neu.2008.0898
Baumann, C. R., Werth, E., Stocker, R., Ludwig, S., and Bassetti, C. L. (2007). Sleep-wake disturbances 6 months after traumatic brain injury: a prospective study. Brain J. Neurol. 130, 1873–1883. doi: 10.1093/brain/awm109
Beitchman, J., Lifshitz, J., Harris, N., Currier-Thomas, T., Lafrenaye, A., Hanell, A., et al. (2021). Spatial distribution of neuropathology and Neuroinflammation elucidate the biomechanics of fluid percussion injury. Neurotrauma Reports. 2, 59–75. doi: 10.1089/neur.2020.0046
Beuckmann, C. T., Ueno, T., Nakagawa, M., Suzuki, M., and Akasofu, S. (2019). Preclinical in vivo characterization of lemborexant (E2006), a novel dual orexin receptor antagonist for sleep/wake regulation. Sleep 42, 1–14. doi: 10.1093/sleep/zsz076
Bibineyshvili, Y., Schiff, N. D., and Calderon, D. P. (2022). Dexmedetomidine-mediated sleep phase modulation ameliorates motor and cognitive performance in a chronic blast-injured mouse model. Front. Neurol. 13, 1–18. doi: 10.3389/fneur.2022.1040975
Bodnar, C. N., Roberts, K. N., Higgins, E. K., and Bachstetter, A. D. (2019). A systematic review of closed head injury models of mild traumatic brain injury in mice and rats. J. Neurotrauma 36, 1683–1706. doi: 10.1089/neu.2018.6127
Boone, D. R., Sell, S. L., Micci, M. A., Crookshanks, J. M., Parsley, M., Uchida, T., et al. (2012). Traumatic brain injury-induced dysregulation of the circadian clock. PLoS One 7:e46204. doi: 10.1371/journal.pone.0046204
Borniger, J. C., Ungerleider, K., Zhang, N., Karelina, K., Magalang, U. J., and Weil, Z. M. (2018). Repetitive brain injury of juvenile mice impairs environmental enrichment-induced modulation of REM sleep in adulthood. Neuroscience 375, 74–83. doi: 10.1016/j.neuroscience.2018.01.064
Bouma, G. J., Muizelaar, J. P., Stringer, W. A., Choi, S. C., Fatouros, P., and Young, H. F. (1992). Ultra-early evaluation of regional cerebral blood flow in severely head-injured patients using xenon-enhanced computerized tomography. J. Neurosurg. 77, 360–368. doi: 10.3171/jns.1992.77.3.0360
Braun, A. R., Balkin, T. J., Wesenten, N. J., Carson, R. E., Varga, M., Baldwin, P., et al. (1997). Regional cerebral blood flow throughout the sleep-wake cycle. An H2(15)O PET study. Brain 120, 1173–1197. doi: 10.1093/brain/120.7.1173
Büchele, F., Morawska, M. M., Schreglmann, S. R., Penner, M., Muser, M., Baumann, C. R., et al. (2016). Novel rat model of weight drop-induced closed diffuse traumatic brain injury compatible with electrophysiological recordings of vigilance states. J. Neurotrauma 33, 1171–1180. doi: 10.1089/neu.2015.4001
Carey, S., Gibbons, A., Prabhala, R., Kaplan, G., and Zielinski, M. (2023). 0155 persistent activation of TXNIP and NLRP3 Inflammasomes in cortical glia and neurons after mild traumatic brain injury. Sleep 46, A69–A70. doi: 10.1093/sleep/zsad077.0155
Cash, A., and Theus, M. H. (2020). Mechanisms of blood-brain barrier dysfunction in traumatic brain injury. Int. J. Mol. Sci. 21, 1–27. doi: 10.3390/ijms21093344
Chodobski, A., Zink, B. J., and Szmydynger-Chodobska, J. (2011). Blood-brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2, 492–516. doi: 10.1007/s12975-011-0125-x
Cipolla, M. J. The cerebral circulation. San Rafael (CA): Morgan & Claypool Life Sciences; (2009). 1, 1–59.
Dalgard, C. L., Cole, J. T., Kean, W. S., Lucky, J. J., Sukumar, G., McMullen, D. C., et al. (2012). The cytokine temporal profile in rat cortex after controlled cortical impact. Front. Mol. Neurosci. 5:6. doi: 10.3389/fnmol.2012.00006
Díaz, M. M., Tsenkina, Y., Arizanovska, D., Mehlen, P., and Liebl, D. J. (2023). DCC/netrin-1 regulates cell death in oligodendrocytes after brain injury. Cell Death Differ. 30, 397–406. doi: 10.1038/s41418-022-01091-z
Dickstein, J. B., Moldofsky, H., Lue, F. A., and Hay, J. B. (1999). Intracerebroventricular injection of TNF-alpha promotes sleep and is recovered in cervical lymph. Am. J. Phys. 276, R1018–R1022. doi: 10.1152/ajpregu.1999.276.4.R1018
Elkind, J. A., Lim, M. M., Johnson, B. N., Palmer, C. P., Putnam, B. J., Kirschen, M. P., et al. (2015). Efficacy, dosage, and duration of action of branched chain amino acid therapy for traumatic brain injury. Front. Neurol. 6:73. doi: 10.3389/fneur.2015.00073
Elliott, J. E., De Luche, S. E., Churchill, M. J., Moore, C., Cohen, A. S., Meshul, C. K., et al. (2018). Dietary therapy restores glutamatergic input to orexin/hypocretin neurons after traumatic brain injury in mice. Sleep 41, 1–13. doi: 10.1093/sleep/zsx212
Elliott, J. E., Keil, A. T., Mithani, S., Gill, J. M., O'Neil, M. E., Cohen, A. S., et al. (2022). Dietary supplementation with branched chain amino acids to improve sleep in veterans with traumatic brain injury: a randomized double-blind placebo-controlled pilot and feasibility trial. Front. Syst. Neurosci. 16:854874. doi: 10.3389/fnsys.2022.854874
Feeney, D. M., Boyeson, M. G., Linn, R. T., Murray, H. M., and Dail, W. G. (1981). Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res. 211, 67–77. doi: 10.1016/0006-8993(81)90067-6
Flygt, J., Gumucio, A., Ingelsson, M., Skoglund, K., Holm, J., Alafuzoff, I., et al. (2016). Human traumatic brain injury results in oligodendrocyte death and increases the number of oligodendrocyte progenitor cells. J. Neuropathol. Exp. Neurol. 75, 503–515. doi: 10.1093/jnen/nlw025
Foda, M. A., and Marmarou, A. (1994). A new model of diffuse brain injury in rats. Part II: Morpholog. characterization. J Neurosurg. 80, 301–313. doi: 10.3171/jns.1994.80.2.0301
Gaggi, N. L., Ware, J. B., Dolui, S., Brennan, D., Torrellas, J., Wang, Z., et al. (2023). Temporal dynamics of cerebral blood flow during the first year after moderate-severe traumatic brain injury: a longitudinal perfusion MRI study. Neuroimage Clin. 37:103344. doi: 10.1016/j.nicl.2023.103344
Gamboa, J., Horvath, J., Simon, A., Islam, M. S., Gao, S., Perk, D., et al. (2021). Secondary-blast injury in rodents produces cognitive sequelae and distinct motor recovery trajectories. Brain Res. 1755:147275. doi: 10.1016/j.brainres.2020.147275
Golding, E. M. (2002). Sequelae following traumatic brain injury. The cerebrovascular perspective. Brain Res. Brain Res. Rev. 38, 377–388. doi: 10.1016/S0165-0173(02)00141-8
Govindarajulu, M., Patel, M. Y., Krishnan, J., Long, J. B., and Arun, P. (2023). Blast exposure induces acute alterations in circadian clock genes in the hypothalamus and pineal gland in rats: an exploratory study. Neurotrauma Reports. 4, 586–597. doi: 10.1089/neur.2023.0048
Green, T. R. F., Ortiz, J. B., Wonnacott, S., Williams, R. J., and Rowe, R. K. The Bidirectional Relationship Between Sleep and Inflammation Links Traumatic Brain Injury and Alzheimer's Disease. Front. Neurosci. 14:894. doi: 10.3389/fnins.2020.00894
Harrison, J. L., Rowe, R. K., Ellis, T. W., Yee, N. S., O’Hara, B. F., Adelson, P. D., et al. (2015). Resolvins AT-D1 and E1 differentially impact functional outcome, post-traumatic sleep, and microglial activation following diffuse brain injury in the mouse. Brain Behav. Immun. 47, 131–140. doi: 10.1016/j.bbi.2015.01.001
Hazra, A., Macolino, C., Elliott, M. B., and Chin, J. (2014). Delayed thalamic astrocytosis and disrupted sleep-wake patterns in a preclinical model of traumatic brain injury. J. Neurosci. Res. 92, 1434–1445. doi: 10.1002/jnr.23430
Heldt, S., Elberger, A., Deng, Y., Guley, N., Del Mar, N., Rogers, J., et al. (2014). A novel closed-head model of mild traumatic brain injury caused by primary overpressure blast to the cranium produces sustained emotional deficits in mice. Front. Neurol. 5, 1–14. doi: 10.3389/fneur.2014.00002
Heyburn, L., Abutarboush, R., Goodrich, S., Urioste, R., Batuure, A., Statz, J., et al. (2019). Repeated low-level blast overpressure leads to endovascular disruption and alterations in TDP-43 and Piezo2 in a rat model of blast TBI. Front. Neurol. 10:766. doi: 10.3389/fneur.2019.00766
Heyburn, L., Batuure, A., Wilder, D., Long, J., and Sajja, V. S. (2023). Neuroinflammation profiling of brain cytokines following repeated blast exposure. Int. J. Mol. Sci. 24:12564. doi: 10.3390/ijms241612564
Holden, S. S., Grandi, F. C., Aboubakr, O., Higashikubo, B., Cho, F. S., Chang, A. H., et al. (2021). Complement factor C1q mediates sleep spindle loss and epileptic spikes after mild brain injury. Science 373, 1–9. doi: 10.1126/science.abj2685
Hue, C. D., Cao, S., Haider, S. F., Vo, K. V., Effgen, G. B., Vogel, E., et al. (2013). Blood-brain barrier dysfunction after primary blast injury in vitro. J. Neurotrauma 30, 1652–1663. doi: 10.1089/neu.2012.2773
Iadecola, C. (2017). The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron 96, 17–42. doi: 10.1016/j.neuron.2017.07.030
Jenkins, L. W., Moszynski, K., Lyeth, B. G., Lewelt, W., DeWitt, D. S., Allen, A., et al. (1989). Increased vulnerability of the mildly traumatized rat brain to cerebral ischemia: the use of controlled secondary ischemia as a research tool to identify common or different mechanisms contributing to mechanical and ischemic brain injury. Brain Res. 477, 211–224. doi: 10.1016/0006-8993(89)91409-1
Johnson, V. E., Stewart, W., and Smith, D. H. (2013). Axonal pathology in traumatic brain injury. Exp. Neurol. 246, 35–43. doi: 10.1016/j.expneurol.2012.01.013
Kaiser, P. R., Valko, P. O., Werth, E., Thomann, J., Meier, J., Stocker, R., et al. (2010). Modafinil ameliorates excessive daytime sleepiness after traumatic brain injury. Neurology 75, 1780–1785. doi: 10.1212/WNL.0b013e3181fd62a2
Kawoos, U., Abutarboush, R., Gu, M., Chen, Y., Statz, J. K., Goodrich, S. Y., et al. (2021). Blast-induced temporal alterations in blood-brain barrier properties in a rodent model. Sci. Rep. 11:5906. doi: 10.1038/s41598-021-84730-8
Kelly, D. F., Martin, N. A., Kordestani, R., Counelis, G., Hovda, D. A., Bergsneider, M., et al. (1997). Cerebral blood flow as a predictor of outcome following traumatic brain injury. J. Neurosurg. 86, 633–641. doi: 10.3171/jns.1997.86.4.0633
Kinoshita, K., Chatzipanteli, I. K., Vitarbo, E., Truettner, J. S., and Alonso OFDietrich, W. D. (2002). Interleukin-1beta messenger ribonucleic acid and protein levels after fluid-percussion brain injury in rats: importance of injury severity and brain temperature. Neurosurgery 51, 195–203. doi: 10.1097/00006123-200207000-00027
Kobeissy, F. H. Brain Neurotrauma: Molecular, neuropsychological, and rehabilitation aspects. (2015).
Koliatsos, V. E., and Alexandris, A. S. (2019). Wallerian degeneration as a therapeutic target in traumatic brain injury. Curr. Opin. Neurol. 32, 786–795. doi: 10.1097/WCO.0000000000000763
Komoltsev, I. G., Frankevich, S. O., Shirobokova, N. I., Volkova, A. A., Levshina, I. P., Novikova, M. R., et al. (2021). Differential early effects of traumatic brain injury on spike-wave discharges in Sprague-Dawley rats. Neurosci. Res. 166, 42–54. doi: 10.1016/j.neures.2020.05.005
Konduru, S. R., Isaacson, J. R., Lasky, D. J., Zhou, Z., Rao, R. K., Vattem, S. S., et al. (2022). Dual orexin antagonist normalized sleep homeostatic drive, enhanced GABAergic inhibition, and suppressed seizures after traumatic brain injury. Sleep 45, 1–9. doi: 10.1093/sleep/zsac238
Konduru, S. S., Wallace, E. P., Pfammatter, J. A., Rodrigues, P. V., Jones, M. V., and Maganti, R. K. (2021). Sleep-wake characteristics in a mouse model of severe traumatic brain injury: relation to posttraumatic epilepsy. Epilepsia Open. 6, 181–194. doi: 10.1002/epi4.12462
Korthas, H. T., Main, B. S., Harvey, A. C., Buenaventura, R. G., Wicker, E., Forcelli, P. A., et al. (2022). The effect of traumatic brain injury on sleep architecture and circadian rhythms in mice-a comparison of high-frequency head impact and controlled cortical injury. Biology (Basel). 11, 1–18. doi: 10.3390/biology11071031
Kovacs, S. K., Leonessa, F., and Ling, G. S. (2014). Blast TBI models, neuropathology, and implications for seizure risk. Front. Neurol. 5:47. doi: 10.3389/fneur.2014.00047
Krueger, J. M. (2008). The role of cytokines in sleep regulation. Curr. Pharm. Des. 14, 3408–3416. doi: 10.2174/138161208786549281
Krueger, J. M., Majde, J. A., and Rector, D. M. (2011). “Chapter 15 - cytokines in immune function and sleep regulation” in Handbook of clinical neurology. eds. P. Montagna and S. Chokroverty (Netherlands: Elsevier), 229–240.
Lifshitz, J., Kelley, B. J., and Povlishock, J. T. (2007). Perisomatic thalamic axotomy after diffuse traumatic brain injury is associated with atrophy rather than cell death. J. Neuropathol. Exp. Neurol. 66, 218–229. doi: 10.1097/01.jnen.0000248558.75950.4d
Lifshitz, J., Rowe, R. K., Griffiths, D. R., Evilsizor, M. N., Thomas, T. C., Adelson, P. D., et al. (2016). Clinical relevance of midline fluid percussion brain injury: acute deficits, chronic morbidities and the utility of biomarkers. Brain injury 30, 1293–1301. doi: 10.1080/02699052.2016.1193628
Lighthall, J. W. (1988). Controlled cortical impact: a new experimental brain injury model. J. Neurotrauma 5, 1–15. doi: 10.1089/neu.1988.5.1
Lim, M. M., Elkind, J., Xiong, G., Galante, R., Zhu, J., Zhang, L., et al. (2013). Dietary therapy mitigates persistent wake deficits caused by mild traumatic brain injury. Sci. Transl. Med. 5:215ra173. doi: 10.1126/scitranslmed.3007092
Logsdon, A. F., Meabon, J. S., Cline, M. M., Bullock, K. M., Raskind, M. A., Peskind, E. R., et al. (2018). Blast exposure elicits blood-brain barrier disruption and repair mediated by tight junction integrity and nitric oxide dependent processes. Sci. Rep. 8:11344. doi: 10.1038/s41598-018-29341-6
Lok, J., Wang, X.-S., Xing, C.-H., Maki, T.-K., Wu, L.-M., Guo, S.-Z., et al. (2015). Targeting the neurovascular unit in brain trauma. CNS Neurosci. Ther. 21, 304–308. doi: 10.1111/cns.12359
Long, J. B., Bentley, T. L., Wessner, K. A., Cerone, C., Sweeney, S., and Bauman, R. A. (2009). Blast overpressure in rats: recreating a battlefield injury in the laboratory. J. Neurotrauma 26, 827–840. doi: 10.1089/neu.2008.0748
Ma, X., Aravind, A., Pfister, B. J., Chandra, N., and Haorah, J. (2019). Animal models of traumatic brain injury and assessment of injury severity. Mol. Neurobiol. 56, 5332–5345. doi: 10.1007/s12035-018-1454-5
Madsen, P. L., Holm, S., Vorstrup, S., Friberg, L., Lassen, N. A., and Wildschiødtz, G. (1991a). Human regional cerebral blood flow during rapid-eye-movement sleep. J. Cereb. Blood Flow Metab. 11, 502–507. doi: 10.1038/jcbfm.1991.94
Madsen, P. L., Schmidt, J. F., Wildschiodtz, G., Friberg, L., Holm, S., Vorstrup, S., et al. (1991b). Cerebral O2 metabolism and cerebral blood flow in humans during deep and rapid-eye-movement sleep. J. Appl. Physiol. 70, 2597–2601. doi: 10.1152/jappl.1991.70.6.2597
Maher, C. O., Anderson, R. E., Martin, H. S., McClelland, R. L., and Meyer, F. B. (2003). Interleukin-1beta and adverse effects on cerebral blood flow during long-term global hypoperfusion. J. Neurosurg. 99, 907–912. doi: 10.3171/jns.2003.99.5.0907
Menn, S. J., Yang, R., and Lankford, A. (2014). Armodafinil for the treatment of excessive sleepiness associated with mild or moderate closed traumatic brain injury: a 12-week, randomized, double-blind study followed by a 12-month open-label extension. J. Clin. Sleep Med.: JCSM: Official Pub. American Academy of Sleep Med. 10, 1181–1191. doi: 10.5664/jcsm.4196
Modarres, M. H., Kuzma, N. N., Kretzmer, T., Pack, A. I., and Lim, M. M. (2017). EEG slow waves in traumatic brain injury: convergent findings in mouse and man. Neurobiol. Sleep and Circadian Rhythms. 2, 59–70. doi: 10.1016/j.nbscr.2016.06.001
Mollayeva, T., D'Souza, A., and Mollayeva, S. (2017). Sleep and psychiatric disorders in persons with mild traumatic brain injury. Curr. Psychiatry Rep. 19:47. doi: 10.1007/s11920-017-0800-z
Monson, K. L., Converse, M. I., and Manley, G. T. (2019). Cerebral blood vessel damage in traumatic brain injury. Clin. Biomech. (Bristol, Avon) 64, 98–113. doi: 10.1016/j.clinbiomech.2018.02.011
Morganti-Kossman, M. C., Lenzlinger, P. M., Hans, V., Stahel, P., Csuka, E., Ammann, E., et al. (1997). Production of cytokines following brain injury: beneficial and deleterious for the damaged tissue. Mol. Psychiatry 2, 133–136. doi: 10.1038/sj.mp.4000227
Morganti-Kossmann, M. C., Rancan, M., Stahel, P. F., and Kossmann, T. (2002). Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr. Opin. Crit. Care 8, 101–105. doi: 10.1097/00075198-200204000-00002
Morse, A. M., and Garner, D. R. (2018). Traumatic brain injury, sleep disorders, and psychiatric disorders: an Underrecognized relationship. Med Sci (Basel). 6, 1–16. doi: 10.3390/medsci6010015
Mountney, A., Blaze, J., Wang, Z., Umali, M., Flerlage, W. J., Dougherty, J., et al. (2021). Penetrating ballistic brain injury produces acute alterations in sleep and circadian-related genes in the rodent cortex: a preliminary study. Front. Neurol. 12, 1–15. doi: 10.3389/fneur.2021.745330
Nichols, J. N., Deshane, A. S., Niedzielko, T. L., Smith, C. D., and Floyd, C. L. (2016). Greater neurobehavioral deficits occur in adult mice after repeated, as compared to single, mild traumatic brain injury (mTBI). Behav. Brain Res. 298, 111–124. doi: 10.1016/j.bbr.2015.10.052
Noain, D., Büchele, F., Schreglmann, S. R., Valko, P. O., Gavrilov, Y. V., Morawska, M. M., et al. (2018). Increased sleep need and reduction of Tuberomammillary histamine neurons after rodent traumatic brain injury. J. Neurotrauma 35, 85–93. doi: 10.1089/neu.2017.5067
Ouyang, W., Wu, W., Fan, Z., Wang, J., Pan, H., and Yang, W. (2018). Modified device for fluid percussion injury in rodents. J. Neurosci. Res. 96, 1412–1429. doi: 10.1002/jnr.24261
Ozturk, Y., Bozkurt, I., Guvenc, Y., Kepoglu, U., Cingirt, M., Gulbahar, O., et al. (2021). Modafinil attenuates the neuroinflammatory response after experimental traumatic brain injury. J. Neurosurg. Sci, 15, 498–506. doi: 10.23736/S0390-5616.21.053820
Paredes, I., Navarro, B., and Lagares, A. (2021). Sleep disorders in traumatic brain injury. Neurocirugia (Astur: Engl Ed). 32, 178–187. doi: 10.1016/j.neucir.2020.09.001
Perez-Polo, J. R., Rea, H. C., Johnson, K. M., Parsley, M. A., Unabia, G. C., Xu, G., et al. (2013). Inflammatory consequences in a rodent model of mild traumatic brain injury. J. Neurotrauma 30, 727–740. doi: 10.1089/neu.2012.2650
Petraglia, A. L., Plog, B. A., Dayawansa, S., Chen, M., Dashnaw, M. L., Czerniecka, K., et al. (2014). The Spectrum of neurobehavioral sequelae after repetitive mild traumatic brain injury: a novel mouse model of chronic traumatic encephalopathy. J. Neurotrauma 31, 1211–1224. doi: 10.1089/neu.2013.3255
Pop, V., and Badaut, J. (2011). A neurovascular perspective for long-term changes after brain trauma. Transl. Stroke Res. 2, 533–545. doi: 10.1007/s12975-011-0126-9
Portillo, E., Zi, X., Kim, Y., Tucker, L. B., Fu, A., Miller, L. A., et al. (2023). Persistent hypersomnia following repetitive mild experimental traumatic brain injury: roles of chronic stress and sex differences. J. Neurosci. Res. 101, 843–865. doi: 10.1002/jnr.25165
Reneer, D. V., Hisel, R. D., Hoffman, J. M., Kryscio, R. J., Lusk, B. T., and Geddes, J. W. (2011). A multi-mode shock tube for investigation of blast-induced traumatic brain injury. J. Neurotrauma 28, 95–104. doi: 10.1089/neu.2010.1513
Risling, M., Plantman, S., Angeria, M., Rostami, E., Bellander, B. M., Kirkegaard, M., et al. (2011). Mechanisms of blast induced brain injuries, experimental studies in rats. NeuroImage 54, S89–S97. doi: 10.1016/j.neuroimage.2010.05.031
Robertson, C. S., Contant, C. F., Gokaslan, Z. L., Narayan, R. K., and Grossman, R. G. (1992). Cerebral blood flow, arteriovenous oxygen difference, and outcome in head injured patients. J. Neurol. Neurosurg. Psychiatry 55, 594–603. doi: 10.1136/jnnp.55.7.594
Rowe, R. K., and Griesbach, G. S. (2022). Immune-endocrine interactions in the pathophysiology of sleep-wake disturbances following traumatic brain injury: a narrative review. Brain Res. Bull. 185, 117–128. doi: 10.1016/j.brainresbull.2022.04.017
Rowe, R. K., Harrison, J. L., Morrison, H. W., Subbian, V., Murphy, S. M., and Lifshitz, J. (2019). Acute post-traumatic sleep may define vulnerability to a second traumatic brain injury in mice. J. Neurotrauma 36, 1318–1334. doi: 10.1089/neu.2018.5980
Rowe, R. K., Harrison, J. L., O'Hara, B. F., and Lifshitz, J. (2014a). Diffuse brain injury does not affect chronic sleep patterns in the mouse. Brain injury 28, 504–510. doi: 10.3109/02699052.2014.888768
Rowe, R. K., Harrison, J. L., O'Hara, B. F., and Lifshitz, J. (2014b). Recovery of neurological function despite immediate sleep disruption following diffuse brain injury in the mouse: clinical relevance to medically untreated concussion. Sleep 37, 743–752. doi: 10.5665/sleep.3582
Rowe, R. K., Harrison, J. L., Zhang, H., Bachstetter, A. D., Hesson, D. P., O'Hara, B. F., et al. (2018). Novel TNF receptor-1 inhibitors identified as potential therapeutic candidates for traumatic brain injury. J. Neuroinflammation 15:154. doi: 10.1186/s12974-018-1200-y
Rowe, R. K., Striz, M., Bachstetter, A. D., Van Eldik, L. J., Donohue, K. D., O'Hara, B. F., et al. (2014c). Diffuse brain injury induces acute post-traumatic sleep. PLoS One 9:e82507. doi: 10.1371/journal.pone.0082507
Saber, M., Giordano, K. R., Hur, Y., Ortiz, J. B., Morrison, H., Godbout, J. P., et al. (2020). Acute peripheral inflammation and post-traumatic sleep differ between sexes after experimental diffuse brain injury. Eur. J. Neurosci. 52, 2791–2814. doi: 10.1111/ejn.14611
Saber, M., Murphy, S. M., Cho, Y., Lifshitz, J., and Rowe, R. K. (2021). Experimental diffuse brain injury and a model of Alzheimer's disease exhibit disease-specific changes in sleep and incongruous peripheral inflammation. J. Neurosci. Res. 99, 1136–1160. doi: 10.1002/jnr.24771
Sabir, M., Gaudreault, P.-O., Freyburger, M., Massart, R., Blanchet-Cohen, A., Jaber, M., et al. (2015). Impact of traumatic brain injury on sleep structure, electrocorticographic activity and transcriptome in mice. Brain Behav. Immun. 47, 118–130. doi: 10.1016/j.bbi.2014.12.023
Salehi, A., Zhang, J. H., and Obenaus, A. (2017). Response of the cerebral vasculature following traumatic brain injury. J. Cereb. Blood Flow Metab. 37, 2320–2339. doi: 10.1177/0271678X17701460
Säljö, A., Arrhén, F., Bolouri, H., Mayorga, M., and Hamberger, A. (2008). Neuropathology and pressure in the pig brain resulting from low-impulse noise exposure. J. Neurotrauma 25, 1397–1406. doi: 10.1089/neu.2008.0602
Säljö, A., Bao, F., Haglid, K. G., and Hansson, H. A. (2000). Blast exposure causes redistribution of phosphorylated neurofilament subunits in neurons of the adult rat brain. J. Neurotrauma 17, 719–726. doi: 10.1089/089771500415454
Sandsmark, D. K., Elliott, J. E., and Lim, M. M. (2017). Sleep-wake disturbances after traumatic brain injury: synthesis of human and animal studies. Sleep 40, 1–18. doi: 10.1093/sleep/zsx044
Skopin, M. D., Kabadi, S. V., Viechweg, S. S., Mong, J. A., and Faden, A. I. (2015). Chronic decrease in wakefulness and disruption of sleep-wake behavior after experimental traumatic brain injury. J. Neurotrauma 32, 289–296. doi: 10.1089/neu.2014.3664
Skotak, M., Townsend, M. T., Ramarao, K. V., and Chandra, N. (2019). A comprehensive review of experimental rodent models of repeated blast TBI. Front. Neurol. 10:1015. doi: 10.3389/fneur.2019.01015
Soustiel, J. F., Glenn, T. C., Shik, V., Boscardin, J., Mahamid, E., and Zaaroor, M. (2005). Monitoring of cerebral blood flow and metabolism in traumatic brain injury. J. Neurotrauma 22, 955–965. doi: 10.1089/neu.2005.22.955
Tasçı, A., Okay, Ö., Gezici, A. R., Ergün, R., and Ergüngör, F. (2003). Prognostic value of interleukin-1 beta levels after acute brain injury. Neurol. Res. 25, 871–874. doi: 10.1179/016164103771953998
Theadom, A., Starkey, N., Jones, K., Cropley, M., Parmar, P., Barker-Collo, S., et al. (2016). Sleep difficulties and their impact on recovery following mild traumatic brain injury in children. Brain injury 30, 1243–1248. doi: 10.1080/02699052.2016.1183171
Thomasy, H. E., Febinger, H. Y., Ringgold, K. M., Gemma, C., and Opp, M. R. (2017). Hypocretinergic and cholinergic contributions to sleep-wake disturbances in a mouse model of traumatic brain injury. Neurobiol. Sleep and Circadian Rhythms. 2, 71–84. doi: 10.1016/j.nbscr.2016.03.001
Thomasy, H. E., and Opp, M. R. (2019). Hypocretin mediates sleep and wake disturbances in a mouse model of traumatic brain injury. J. Neurotrauma 36, 802–814. doi: 10.1089/neu.2018.5810
Townsend, R. E., Prinz, P. N., and Obrist, W. D. (1973). Human cerebral blood flow during sleep and waking. J. Appl. Physiol. 35, 620–625. doi: 10.1152/jappl.1973.35.5.620
Turner, K. L., Gheres, K. W., Proctor, E. A., and Drew, P. J. (2020). Neurovascular coupling and bilateral connectivity during NREM and REM sleep. elife 9:e62071. doi: 10.7554/eLife.62071
Verma, A., Anand, V., and Verma, N. P. (2007). Sleep disorders in chronic traumatic brain injury. J. Clin. Sleep Med.: JCSM: Official Pub. American Academy of Sleep Med. 3, 357–362. doi: 10.5664/jcsm.26856
Vigil, F. A., Belchior, H., Bugay, V., Bazaldua, I. I., Stoja, A., Dantas, D. C., et al. (2023). Acute treatment with the M-channel (K(v)7, KCNQ) opener Retigabine reduces the Long-term effects of repetitive blast traumatic brain injuries. Neurotherapeutics 20, 853–869. doi: 10.1007/s13311-023-01361-9
Vila, E., and Salaices, M. (2005). Cytokines and vascular reactivity in resistance arteries. Am. J. Phys. Heart Circ. Phys. 288, H1016–H1021. doi: 10.1152/ajpheart.00779.2004
Viola-Saltzman, M., and Watson, N. F. (2012). Traumatic brain injury and sleep disorders. Neurol. Clin. 30, 1299–1312. doi: 10.1016/j.ncl.2012.08.008
Watson, N. F., Dikmen, S., Machamer, J., Doherty, M., and Temkin, N. (2007). Hypersomnia following traumatic brain injury. J. Clin. Sleep Med. 3, 363–368. doi: 10.5664/jcsm.26857
Wickwire, E. M. (2020). Why sleep matters after traumatic brain injury. J. Clin. Sleep Med. 16, 5–6. doi: 10.5664/jcsm.8872
Wickwire, E. M., Williams, S. G., Roth, T., Capaldi, V. F., Jaffe, M., Moline, M., et al. (2016). Sleep, sleep disorders, and mild traumatic brain injury. What we know and what we need to know: findings from a National Working Group. Neurotherapeutics: J. American Society for Experimental NeuroTherapeutics. 13, 403–417. doi: 10.1007/s13311-016-0429-3
Willie, J. T., Lim, M. M., Bennett, R. E., Azarion, A. A., Schwetye, K. E., and Brody, D. L. (2012). Controlled cortical impact traumatic brain injury acutely disrupts wakefulness and extracellular orexin dynamics as determined by intracerebral microdialysis in mice. J. Neurotrauma 29, 1908–1921. doi: 10.1089/neu.2012.2404
Wiseman-Hakes, C., Murray, B., Moineddin, R., Rochon, E., Cullen, N., Gargaro, J., et al. (2013). Evaluating the impact of treatment for sleep/wake disorders on recovery of cognition and communication in adults with chronic TBI. Brain Inj. 27, 1364–1376. doi: 10.3109/02699052.2013.823663
Zheng, H., Wang, R., and Qu, J. (2016). Effect of different glucose supply conditions on neuronal energy metabolism. Cogn. Neurodyn. 10, 563–571. doi: 10.1007/s11571-016-9401-5
Zielinski, M. R., Gerashchenko, D., Karpova, S. A., Konanki, V., McCarley, R. W., Sutterwala, F. S., et al. (2017). The NLRP3 inflammasome modulates sleep and NREM sleep delta power induced by spontaneous wakefulness, sleep deprivation and lipopolysaccharide. Brain Behav. Immun. 62, 137–150. doi: 10.1016/j.bbi.2017.01.012
Keywords: sleep, hemodynamics, cerebral blood flow, neuroinflammation, TBI
Citation: Green TRF, Carey SD, Mannino G, Craig JA, Rowe RK and Zielinski MR (2024) Sleep, inflammation, and hemodynamics in rodent models of traumatic brain injury. Front. Neurosci. 18:1361014. doi: 10.3389/fnins.2024.1361014
Edited by:
Kazuhiko Kume, University of Tsukuba, JapanReviewed by:
Rama Maganti, University of Wisconsin-Madison, United StatesCopyright © 2024 Green, Carey, Mannino, Craig, Rowe and Zielinski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark R. Zielinski, TWFya19aaWVsaW5za2lAaG1zLmhhcnZhcmQuZWR1
†These authors have contributed equally to this work