 Seyed Mehrdad Savar1,2
Seyed Mehrdad Savar1,2 Bin Ma1,3
Bin Ma1,3 Eugene Hone4
Eugene Hone4 Farzana Jahan1,5Shaun Markovic1,2
Farzana Jahan1,5Shaun Markovic1,2 Steve Pedrini4,6Soudabeh Shemehsavar1,5
Steve Pedrini4,6Soudabeh Shemehsavar1,5 Vandhana Easwaran1,2
Vandhana Easwaran1,2 Kevin Taddei4,6
Kevin Taddei4,6 Samantha Gardener4,6Jasmeer P. Chhatwal7
Samantha Gardener4,6Jasmeer P. Chhatwal7 Ellis S. van Etten8
Ellis S. van Etten8 Matthias J. P. van Osch9
Matthias J. P. van Osch9 Daniel Clarke6,10Anastazija Gnjec6,10
Daniel Clarke6,10Anastazija Gnjec6,10 Mark A. van Buchem9
Mark A. van Buchem9 Marieke J. H. Wermer8,11Graeme J. Hankey12,13Steven M. Greenberg14*
Marieke J. H. Wermer8,11Graeme J. Hankey12,13Steven M. Greenberg14* Ralph N. Martins1,4,6,15*
Ralph N. Martins1,4,6,15* Hamid R. Sohrabi1,2,4,6,15*
Hamid R. Sohrabi1,2,4,6,15*- 1Centre for Healthy Ageing, Health Future Institute, Murdoch University, Perth, WA, Australia
- 2School of Psychology, Murdoch University, Murdoch, WA, Australia
- 3School of Medical, Molecular and Forensic Sciences, Murdoch University, Murdoch, WA, Australia
- 4School of Medical and Health Sciences, Sarich Neuroscience Research Institute, Edith Cowan University, Nedlands, WA, Australia
- 5College of Science, Technology, Engineering and Mathematics, Murdoch University, Perth, WA, Australia
- 6Australian Alzheimer's Research Foundation, Nedlands, WA, Australia
- 7Massachusetts General Hospital, Harvard Medical School, Boston, MA, United States
- 8Department of Neurology, Leiden University Medical Center, Leiden, Netherlands
- 9Department of Radiology, Leiden University Medical Center, Leiden, Netherlands
- 10Sir Charles Gairdner Hospital, Perth Neurology Center, Nedlands, WA, Australia
- 11Department of Neurology, University Medical Center Groningen, Groningen, Netherlands
- 12Medical School, The University of Western Australia, Crawley, WA, Australia
- 13Perron Institute for Neurological and Translational Science, Nedlands, WA, Australia
- 14Haemorrhagic Stroke Research Program, Massachusetts General Hospital Stroke Research Center, Boston, MA, United States
- 15Macquarie Medical School, Macquarie University, North Ryde, NSW, Australia
Cerebral amyloid angiopathy (CAA) is a type of cerebrovascular disorder characterised by the accumulation of amyloid within the leptomeninges and small/medium-sized cerebral blood vessels. Typically, cerebral haemorrhages are one of the first clinical manifestations of CAA, posing a considerable challenge to the timely diagnosis of CAA as the bleedings only occur during the later disease stages. Fluid biomarkers may change prior to imaging biomarkers, and therefore, they could be the future of CAA diagnosis. Additionally, they can be used as primary outcome markers in prospective clinical trials. Among fluid biomarkers, blood-based biomarkers offer a distinct advantage over cerebrospinal fluid biomarkers as they do not require a procedure as invasive as a lumbar puncture. This article aimed to provide an overview of the present clinical data concerning fluid biomarkers associated with CAA and point out the direction of future studies. Among all the biomarkers discussed, amyloid β, neurofilament light chain, matrix metalloproteinases, complement 3, uric acid, and lactadherin demonstrated the most promising evidence. However, the field of fluid biomarkers for CAA is an under-researched area, and in most cases, there are only one or two studies on each of the biomarkers mentioned in this review. Additionally, a small sample size is a common limitation of the discussed studies. Hence, it is hard to reach a solid conclusion on the clinical significance of each biomarker at different stages of the disease or in various subpopulations of CAA. In order to overcome this issue, larger longitudinal and multicentered studies are needed.
1 Introduction
1.1 Cerebral amyloid angiopathy: pathogenesis, clinical manifestations, and epidemiology
Proteins that are considered amyloidogenic can undergo conformational changes, leading to the formation of β-sheet aggregates. The aggregated amyloidogenic proteins make insoluble fibrils called amyloid fibrils, which are involvedin a number of neurodegenerative diseases, including Alzheimer’s disease (AD), in which amyloid β (Aβ) is the amyloidogenic protein (Chiti and Dobson, 2006; Harrison et al., 2007).
Another condition that is caused by Aβ is cerebral amyloid angiopathy (CAA). CAA is the result of Aβ deposition in leptomeningeal and cortical blood vessel walls, resulting in haemorrhages and cognitive decline. The Aβ deposition usually involves small to medium-sized vessels, with or without the involvement of capillaries, namely CAA type I and CAA type II, respectively (Thal et al., 2002; Gatti et al., 2020; Greenberg et al., 2020). Moderate to severe CAA can be seen in about 6% of cognitively normal older adults and in about 50% of patients with AD (Jakel et al., 2022). According to the Religious Orders Study, the prevalence of CAA pathology in autopsied brains is 84.9%, indicating how commonly CAA occurs (Arvanitakis et al., 2011). While sporadic CAA (sCAA) is the more widespread form of the disease, there are also rare hereditary forms of CAA that typically arise as a consequence of a mutation occurring in the amyloid precursor protein (APP) (Jakel et al., 2022). More recently, two other conditions have been added to the CAA spectrum, namely CAA-related inflammation (CAA-ri) and iatrogenic CAA (i-CAA). CAA-ri is the result of an excessive inflammatory response to vascular Aβ deposition, typically manifesting as a subacute cognitive decline. i-CAA is a condition caused by the prion-like transmission of Aß from cadaveric dural grafts (cadaveric dura), cadaveric human growth hormone, or contaminated surgical instruments (Kaushik et al., 2023; Storti et al., 2023).
In both sCAA and hereditary CAA, the clinical presence of lobar microbleeds and intracerebral haemorrhage (ICH) is noteworthy (Biffi, 2022). More than half the patients with lobar ICH have CAA pathology (Jakel et al., 2022). The global incidence of ICH in 2020 was 3.41 million, with Oceania having the highest age-standardised mortality rate (Tsao et al., 2023). ICH and different types of dementias are among the top 20 causes of years of life lost to premature mortality and death in the United States (Tsao et al., 2023). The lobar microbleeds associated with CAA may have a greater impact on health-related quality of life than deep microbleeds related to hypertension (Tang et al., 2011).
1.2 Diagnosis, follow-up, and treatment of cerebral amyloid angiopathy
A definite diagnosis of CAA is only possible through postmortem examination of the brain (Charidimou et al., 2022). There have been some advances in finding neuroimaging biomarkers for diagnosing and tracking CAA, such as using the small vessel disease (SVD) score, which is a score ranging from zero to six and based on the presence of lobar cerebral microbleeds, cortical superficial siderosis (cSS), white matter hyperintensities, and centrum semiovale-enlarged perivascular spaces (CSO-EPVS) in MRI (Charidimou et al., 2016; Valenti et al., 2017). Another instance is using positron emission tomography (PET) agent Pittsburgh compound B (PiB) as a measure of amyloid load to track the disease progression in hereditary CAA (Schultz et al., 2019). In addition to PiB, the clinical application of florbetapir and florbetaben, which are 18F-labeled amyloid PET ligands, have also been studied in CAA (Charidimou et al., 2018; Jang et al., 2019). Cerebral haemorrhages and other imaging markers including presence of superficial siderosis, increased perivascular spaces and white matter lesions in a multispot pattern, are employed as diagnostic markers for CAA in the Boston criteria 2.0. These criteria provide the chance to diagnose probable and possible CAA via magnetic resonance imaging (MRI) and clinical data without the need for histopathologic analysis of the brain tissue (Charidimou et al., 2022). The Edinburgh criteria use subarachnoid haemorrhage and finger-like projections observed in computed tomography (CT) and APOE4 genotype to determine the probability of CAA, which can be used in patients with large bleeds if MRI is not available (Rodrigues et al., 2018; Sembill et al., 2022). That said, the Edinburgh criteria may have low sensitivity in small-sized lobar ICH (van Etten et al., 2020).
As for the management of CAA, currently, there are no treatments available for CAA, except for CAA-ri, in which anti-inflammatory and immunomodulatory medications have shown a reduction in inflammation and a better clinical outcome (Regenhardt et al., 2020).
1.3 Aim and search strategy
According to the previous section, the current diagnostic criteria are mainly based on neuroimaging and do not incorporate any fluid biomarkers. However, as seen in AD, cerebrospinal fluid (CSF) biomarkers can be used to detect the disease earlier than PET imaging (Schindler et al., 2018). Furthermore, cerebral haemorrhage is believed to happen at the final stages of the disease (Jakel et al., 2022; Koemans et al., 2023). Therefore, there is a need for biomarkers that reflect the CAA pathology and can be used for diagnostic or prognostic purposes at earlier stages. Studying hereditary CAA provides an opportunity to research CAA before any symptoms. That said, due to the variety of APP mutations and the small number of patients with hereditary CAA, studying these populations has its own limitations (Biffi, 2022).
Another limitation of the current diagnostic criteria could be the availability of resources and expertise, as CAA may be underdiagnosed in rural areas compared to urban areas (Nagaraja and Patel, 2021). Having a proper blood biomarker for CAA could probably be beneficial in that regard since a blood test is relatively cheaper (Padala and Newhouse, 2023).
This article aims to review fluid biomarkers of CAA that have been studied in clinical settings so far and discuss their clinical application and potential for future studies by looking into the body of literature supporting the usage of each biomarker in CAA and, if available, their respective sensitivity and specificity. The search strategy was based on using a combination of the Medical Subject Heading (MeSH) term “Cerebral Amyloid Angiopathy” and other relevant MeSH terms, including “Cerebral Amyloid Angiopathy/blood” and “Cerebral Amyloid Angiopathy/cerebrospinal fluid” in the PubMed database. The next search strategy was to search for “Cerebral Amyloid Angiopathy” and “Biomarker” within the title and abstract of the articles. The output of the two search strategies on August 2, 2023, was 68 and 105 articles, respectively. The references of the obtained results were also screened for relevant articles. A complementary search was implemented on Google Scholar, searching for other potential biomarkers known to the authors to enhance the coverage of the review. Preclinical investigations, conference abstracts, presentations, and research papers that were not in English were excluded. In order to present a clear overview and comparison between the included studies, the sample size, sample characteristics, methods, and main findings were recorded.
2 Amyloid beta
The transmembrane protein APP can undergo an amyloidogenic pathway by being metabolised via β-secretase and γ-secretase enzymes into Aβ peptides, which could have 37 to 49 amino acid residues (Chen et al., 2017). Based on the framework proposed by Koemans et al., CAA initially begins with cerebrovascular amyloid deposition (Koemans et al., 2023). Several investigations have explored the viability of Aβ, a crucial component in the definition of CAA, as a potential CSF and plasma biomarker for this condition. Each of the studies has subtle differences from the others, and therefore, a summary of their study design and population is also provided for a better understanding of the role of Aβ as a biomarker for CAA.
A study by Verbeek and colleagues in 2009 with 17 CAA patients (Mean age ± Standard deviation (SD): 62.8 ± 11.9), 72 AD patients (Mean age ± SD: 69.4 ± 8.3), and 58 control subjects (Mean age ± SD: 61.0 ± 8.7) investigated whether CSF Aβ could differentiate between healthy controls and the two clinical groups. This study included the CAA patients according to the original Boston criteria instead of the modified or 2.0 version. The CAA group consisted of both sporadic (15 patients) and hereditary (2 patients) CAA. The results indicated that the CSF levels of Aβ42 were decreased in CAA and AD compared to healthy controls (CAA 42.4% of the controls; AD 51.6% of control values). However, the decrease in CSF Aβ40 was only significant for CAA patients compared to AD and controls. The ratio of Aβ40 to Aβ42 was elevated in AD (182% of control) and CAA (192% of control), but it could not differentiate the two conditions (Verbeek et al., 2009).
A similar study recruited CAA patients using the modified Boston criteria (possible and probable) but on an older cohort (mean age of 78 compared to 62.8) (Renard et al., 2012). CSF Aβ40 and Aβ42 levels in this study were lower in the CAA group compared to the control group. However, the difference was only significant for Aβ42 (Area under the receiver operating characteristic curve (AUC) = 0.75). Similar to the previous research, CSF Aβ40 was significantly lower in CAA patients compared to AD (AUC = 0.795). Additionally, no significant difference was observed between CAA patients with lobar haemorrhage and superficial siderosis with regard to the studied CSF biomarkers (Renard et al., 2012).
A small study with 10 CAA patients (mean age ± SD: 68.6 ± 3), 20 AD patients (mean age ± SD: 62.5 ± 4.1), and ten healthy controls (mean age ± SD: 62.2 ± 5.4) concluded that the median CSF concentrations of Aβ38, Aβ40, and Aβ42 were significantly lower in CAA patients compared to the other two groups. In contrast, AD and the control group had a similar CSF profile regarding the aforementioned biomarkers (Banerjee et al., 2020).
Examining the shared findings among the three investigations mentioned above, it appears that CSF levels of Aβ40 could act as a specific marker for CAA. In fact, this is in line with post-mortem analysis of the human brain tissue, where Aβ40 deposition was more profound in CAA than in AD (Gkanatsiou et al., 2019). That said, those studies did not reject the idea of using Aβ42, which reflects amyloid plaque formation in AD pathology, for differentiating CAA from controls (Gkanatsiou et al., 2019).
One of the largest studies examining the variations in CSF-biomarkers between CAA and AD patients was conducted using a retrospective cohort in two French centres with 63 probable CAA patients (mean age ± SD: 72.6 ± 7.4), 27 AD patients (mean age ± SD: 64.1 ± 6.3), and 21 controls (mean age ± SD: 65.8 ± 9.6). CAA patients recruited in this study had three different CSF profiles. About half the patients (32 patients out of 63) had a CSF profile similar to AD, about a third of them (22 patients out of 63) had isolated decreased Aβ42 levels, and the rest (9 patients out of 63) had normal Aβ42 levels. Aβ40 levels were significantly higher in CAA patients with normal Aβ42 CSF profiles compared to participants with decreased Aβ42 levels. The number of cerebral haemorrhages, including ICH and microbleeds, and the presence of superficial siderosis did not differ between the groups. Additionally, Aβ40 and Aβ42 levels were not correlated with the number of microbleeds detected by MRI. Critically, while Aβ40 levels were not significantly different between AD and controls, they were able to distinguish CAA patients from the other comparison groups, confirming the previous findings (Grangeon et al., 2022a).
In the largest study to date, conducted on 372 participants (67 with CAA, 76 with probable AD, 75 with mild cognitive impairment (MCI) due to AD, 76 with MCI, and 78 healthy controls), Sembill and colleagues reported that CSF levels of both Aβ40 and Aβ42 were lower in CAA compared to healthy controls. However, unlike Aβ40, Aβ42 was comparable in CAA, MCI due to AD, and AD patients. The authors suggested that both Aβ40 and Aβ42 could be used for diagnostic purposes as they could properly distinguish CAA patients from controls (Aβ40: AUC = 0.83, 95%CI (0.76–0.89), p < 0.001; Aβ42: AUC = 0.82, 95%CI (0.75–0.88), p < 0.001) (Sembill et al., 2023).
Aβ levels have also been investigated in CAA-related inflammation (CAA-ri). It has been reported that Aβ40 levels in the CSF are negatively correlated with amyloid load measured via PET scan when using pons as a reference. There was also a negative, non-significant correlation between CSF Aβ40 levels and the number of microbleeds in MRI (Renard et al., 2018). When comparing CAA-ri to non-inflammatory CAA, CSF Aβ42 levels are lower in CAA-ri (373.3 pg./mL vs. 490.8 pg./mL, p = 0.05) (Grangeon et al., 2022b). On the contrary, no significant difference has been reported when comparing CSF Aβ40 and Aβ42 in patients with acute CAA-ri (48 patients with a median age of 77 [71–84] years) and CAA patients with AD (48 patients with a median age of 71 [59–82] years) (Sakai et al., 2023). The similarity of CSF Aβ40 and Aβ42 in CAA-ri and CAA + AD patients suggests that these two biomarkers may not be useful for distinguishing the two populations, yet they can reflect the response to anti-inflammatory treatment in CAA-ri (Sakai et al., 2023).
An important sub-population of hereditary CAA is the Dutch-type CAA (D-CAA). Research has shown that Aβ38, Aβ40, Aβ42, and Aβ43 levels are correlated with each other, and they are lower in both sCAA patients and D-CAA patients compared to controls (De Kort et al., 2023b). However, sCAA and D-CAA patients did not follow the same pattern when it came to SVD burden, as all Aβ levels in sCAA patients were correlated with SVD score, while in D-CAA patients there was only a correlation between Aβ42 and SVD score. Furthermore, except for Aβ43, all the other peptides were significantly lower in sCAA compared to AD (De Kort et al., 2023b).
In a 2017 study, 10 symptomatic D-CAA patients (mean age ± SD: 55 ± 6) were compared to 5 presymptomatic D-CAA patients (mean age ± SD: 36 ± 13) and their age-matched healthy controls regarding their CSF profile. Aβ40 and Aβ42 levels were lower in symptomatic and presymptomatic D-CAA patients compared to their respective controls. Furthermore, presymptomatic individuals reported higher decreases in Aβ42 than Aβ40 at least a decade before the first symptoms of CAA (van Etten et al., 2017). Notably, CSF Aβ40 and Aβ42 levels did not correlate with the number of ICHs quantified via MRI, yet Aβ40 was correlated with the number of microbleeds and white matter hyperintensity volume in an age-adjusted analysis (van Etten et al., 2017).
Bornebroek et al. (2003) compared plasma levels of Aβ40 and Aβ42 in seven asymptomatic D-CAA mutation carriers (mean age: 51 years) to 13 non-carriers (mean age: 57 years). The authors found that Aβ40 was the only amyloid-based biomarker significantly lower in D-CAA plasma samples compared to healthy controls (Bornebroek et al., 2003). In line with this result, it is hypothesised that plasma levels of Aβ40 may initially decrease and then rise in later haemorrhagic stages of CAA (Muir et al., 2023).
Analysing plasma levels of Aβ40 and Aβ42 via the Single Molecule Array (SIMOA) platform in 9 presymptomatic D-CAA mutation carriers (mean age ± SD: 44.11 ± 4.31) and 8 D-CAA mutation non-carriers (mean age ± SD: 43.5 ± 6.57) from the Dominantly Inherited Alzheimer Network (DIAN) study demonstrated promising results using plasma samples, rather than CSF. Both plasma Aβ40 and Aβ42 levels were significantly lower in mutation carriers than controls (Chatterjee et al., 2021b). Similar results were seen in comparing symptomatic and asymptomatic D-CAA patients to controls, where plasma levels of Aβ38, Aβ40, and Aβ42 were all significantly decreased in addition to demonstrating strong inter-correlations suggesting that a panel of these biomarkers could reflect disease progression (Verbeek et al., 2021).
A recent publication with a larger sample size (54 D-CAA patients, 61 sCAA patients) than the aforementioned studies (Bornebroek et al., 2003; Verbeek et al., 2021; Chatterjee et al., 2021b) has provided a more comprehensive understanding of the differences in plasma Aβ expression between hereditary and sporadic CAA. Specifically, plasma levels of Aβ38, Aβ40, and Aβ42 were not significantly lower in presymptomatic D-CAA patients than in age-matched controls. Similar observations were made in the case of sCAA patients and their respective control groups. Notably, a substantial decrease in plasma concentrations of Aβ42 was only evident in symptomatic individuals with D-CAA. Consequently, the outcomes of this investigation propose the potential utility of plasma Aβ42 levels as a promising biomarker for identifying individuals with symptomatic D-CAA (De Kort et al., 2023a). In addition, plasma levels of Aβ in patients with D-CAA were correlated with specific imaging findings such as the Fazekas score and categorised number of lobar cerebral microbleeds but not the microbleed count. Yet, the plasma Aβ levels did not have a correlation with imaging markers in sCAA patients (De Kort et al., 2023a).
Employing a comprehensive panel of both CSF and plasma Aβ might yield a precise distinction in identifying sCAA, D-CAA, AD, and cognitively normal individuals. However, this supposition necessitates further validation through investigations involving larger cohorts. CSF levels of Aβ37, Aβ38, Aβ40, Aβ42, and Aβ43 were lower in CAA patients compared to AD and controls in almost every study. In contrast, measuring Aβ levels in the blood seems to have limited applications, although reduced plasma levels of Aβ42 may be specific to the pathological progression of D-CAA (De Kort et al., 2023a). Furthermore, studies have shown that plasma and CSF levels of Aβ are not correlated in CAA patients (De Kort et al., 2023a). Hence, the dynamics between plasma and CSF Aβ is an area requiring further investigation.
As shown in Table 1, the reported sensitivity and specificity of amyloid and tau as diagnostic biomarkers for CAA varies substantially across different research studies. This may be due to the small sample sizes, the difference in the mean ages of the participants, and the difference in the populations of interest (D-CAA and sCAA).
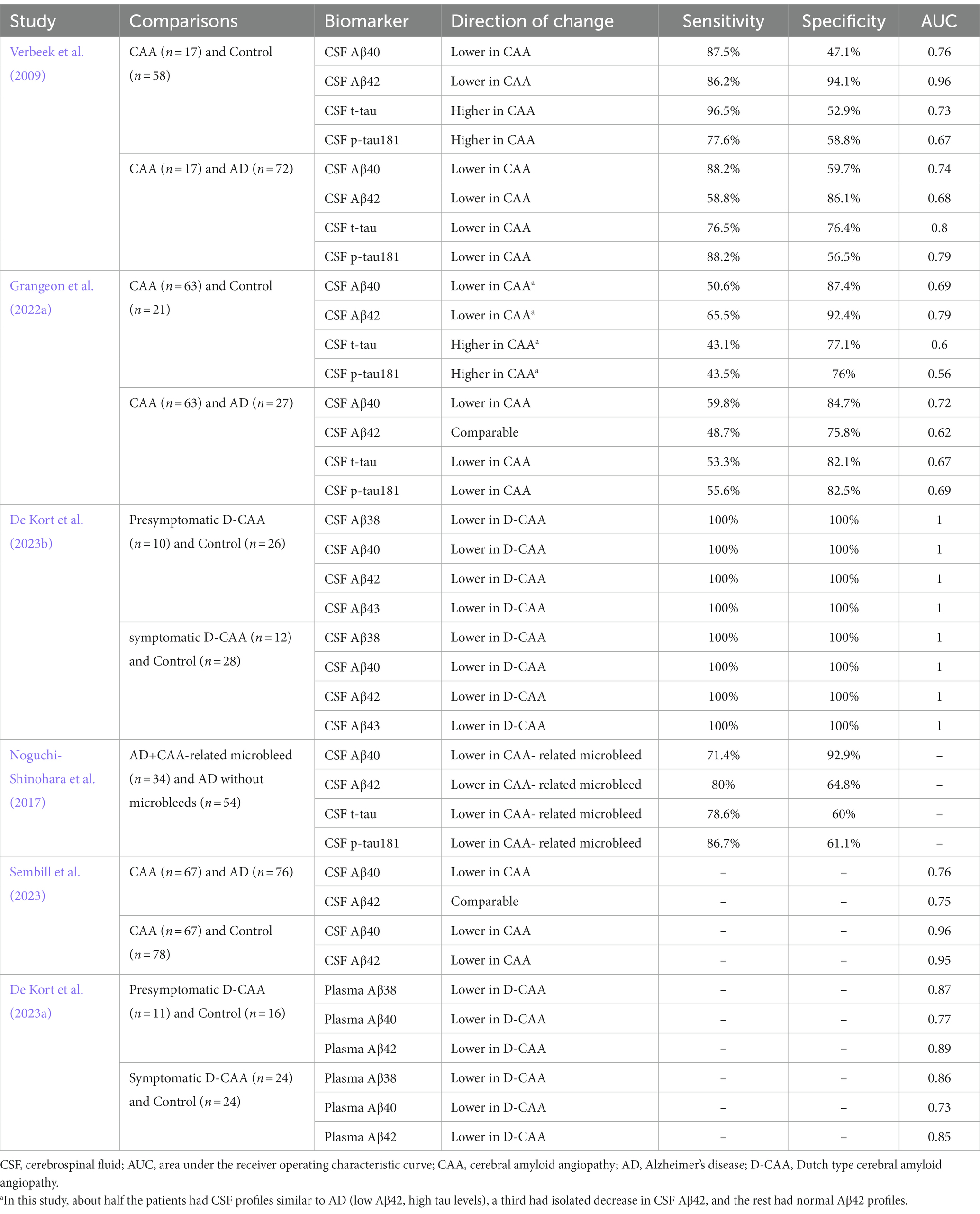
Table 1. Sensitivity and specificity of amyloid β and tau in cerebral amyloid angiopathy studies.
In addition to relying on single biomarkers, a combination of individual biomarkers could also be considered to optimise sensitivity and specificity levels further. For instance, one study proposed an index comprising of Aβ40 and phosphorylated tau (p-tau) 181 (reg = 0.00013164×Aβ40 + 0.0313×p-tau181) that differentiated CAA patients from the control group the best (Sensitivity = 84.6%, specificity = 76.8%, AUC =0.861) (Renard et al., 2012). Finding the optimum tool for each population requires further investigation.
A frequently measured combination of biomarkers is using the Aβ42/40 ratio, as it could reflect brain amyloidosis using amyloid PET scans in cognitively normal individuals (Schindler et al., 2019). This ratio is hypothesised to account for interindividual differences in both overall Aβ production and CSF dynamics in AD (Blennow and Zetterberg, 2018). In a population-based study with 712 participants, a lower plasma Aβ42/Aβ40 ratio was associated with the level of amyloidosis in individuals with cortical microbleeds (McCarter et al., 2022). Yet, this should be considered with caution as a recent publication on a small CAA cohort (n = 12) did not show such discrimination using Aβ42/Aβ40 (Jang et al., 2021). As mentioned before, some studies suggested a decrease in both CSF Aβ40 and Aβ42 in CAA, and that may be the reason for these contradicting results for the Aβ42/Aβ40 ratio as a biomarker for CAA.
The most recent meta-analysis available on the significance of Aβ40, Aβ42, total tau (t-tau), and p-tau181 in CAA is a study from early 2022 (Margraf et al., 2022). The results of this meta-analysis on four studies concluded that a CSF ratio of Aβ42/Aβ40 can accurately differentiate CAA and control groups. Still, the study pointed out that the Aβ40, Aβ42, t-tau, and p-tau181 could not tell AD and CAA apart, and other biomarkers should be used in that regard (Margraf et al., 2022). That said, with more recent publications available, the results of an updated meta-analysis may differ from the aforementioned study.
3 Tau
Tau protein, along with Aβ42, is one of the core CSF biomarkers for AD (Molinuevo et al., 2018). A conformational change in tau protein could result in the production of highly toxic monomers prone to aggregation, forming oligomers and less toxic neurofibrillary tangles. The toxic tau monomer and oligomers could cause oxidative stress and synaptic dysfunction (Niewiadomska et al., 2021). Furthermore, tau may have a critical role in mediating Aβ-induced neurodegeneration as no signs of neurodegeneration are observed in tau-depleted neurons that are exposed to Aβ (Rapoport et al., 2002). The same observation has been reported in older adults, where an increase in the severity of CAA and burden of amyloid plaques was associated with higher tau burden and cognitive decline (Rabin et al., 2022). Therefore, most of the studies that measured Aβ as a biomarker for CAA also measured the p-tau181 and t-tau levels.
Discrepancies exist in the outcomes concerning the discriminatory capacity of t-tau and p-tau181 in distinguishing between CAA, AD, and healthy control subjects. Some studies have reported that there is a significant difference between the three groups regarding CSF t-tau levels, where AD patients exhibited the highest levels, while healthy individuals demonstrated the lowest t-tau levels (Verbeek et al., 2009; Renard et al., 2012). Other studies report that CSF t-tau levels follow the same order among the three groups, but the difference between CAA patients and the control group is not significant (Banerjee et al., 2020; Grangeon et al., 2022a).
In a study on AD patients with or without microbleeds, it was observed that CSF levels of p-tau181 were markedly diminished in individuals with cortical microbleeds. Notably, all AD patients exhibiting microbleeds in this study were concurrently diagnosed with CAA (Noguchi-Shinohara et al., 2017). Similar to t-tau, some studies have reported a statistically significant difference between CSF p-tau181 levels in CAA patients compared to controls, whereas other investigations have not confirmed such a distinction (Verbeek et al., 2009; Renard et al., 2012).
Looking at the D-CAA population in particular, it has been reported that CSF t-tau and p-tau181 levels are not significantly different between presymptomatic patients and controls. This observation remains consistent when comparing presymptomatic and symptomatic D-CAA participants. Notably, among symptomatic D-CAA patients, solely p-tau181 levels were reduced in comparison to control subjects (van Etten et al., 2017).
To sum up these findings, tau appears to be involved in limited cases of CAA, as it did not show promising results in the presymptomatic D-CAA population. A study has shown that memory impairment in probable CAA could suggest a tau pathology where there is an elevated tau-PET retention (Schoemaker et al., 2021). Hence, measuring the CSF levels of p-tau181 might be most informative in CAA patients with AD pathology. As for t-tau, larger studies are needed to resolve the current discrepancy in the literature.
4 Neuroinflammation and oxidative stress biomarkers
4.1 Matrix metalloproteinases
A reason for the Aβ deposition (and thus, reduced concentrations of Aβ seen in CSF) may stem from disruptions within enzymatic or non-enzymatic pathways that typically facilitate the clearance of Aβ (Gatti et al., 2020). One group of enzymes responsible for Aβ clearance are the matrix metalloproteinases (MMPs). MMP-2 and (to a lesser extent) MMP-9 have a proteolytic effect on Aβ in vitro, producing soluble fragments (Hernandez-Guillamon et al., 2015). In contrast to this function, MMP-2 and MMP-9 can disrupt the tight junctions of the blood–brain barrier (BBB). The immune cells can then enter the brain and produce cytokines that promote MMP-9 expression, leading to more damage to the BBB and cerebral haemorrhage (Gireud-Goss et al., 2021). In fact, MMPs could activate proinflammatory pathways in various neurodegenerative diseases (Brkic et al., 2015). The in vivo inhibition of MMP-9 can enhance Aβ clearance across the BBB (Shackleton et al., 2019). A study by Vervuurt and colleagues published in 2023 measured the CSF levels of MMP-2, MMP-9, and MMP-14 in sporadic and D-CAA patients. They also measured the tissue inhibitors of metalloproteinases (TIMP) levels. When comparing sCAA patients (n = 28, median age: 72.3 (65.7–77.1) years) with controls (n = 40, median age: 64.2 (56.1–69.8) years), the only notable difference was observed for MMP-2/TIMP-2 and MMP-14/TIMP-2 ratios. Furthermore, the symptomatic D-CAA patients (n = 12, median age 58.5 (52.8–65.8) years) had lower MMP-14/TIMP-1 and MMP-14/TIMP-2 ratios than the presymptomatic D-CAA patients (n = 11, median age: 38.0 (31.0–52.0) years) and controls (n = 28, median age: 60.3 (51.6–65.9) years). The symptomatic D-CAA patients also had lower MMP-14 levels than healthy controls (Vervuurt et al., 2023).
The serum levels of MMPs have also been examined in CAA-related ICH. It was observed that the levels of MMP-2 were notably diminished in CAA-related ICH, whereas MMP-9 levels exhibited significant elevation in comparison to the control group (Xia et al., 2021). Moreover, there was an association between serum MMP-3 levels and lobar cerebral microbleed count. Regarding their connection with neuroimaging and cognitive biomarkers, the serum concentrations of MMPs displayed correlations that did not reach statistical significance (Xia et al., 2021). Two studies have shown that MMPs could be a promising biomarker for CAA in CSF and serum. Nevertheless, it is imperative to acknowledge that quantifying the levels of MMPs in CSF or serum is not the same as measuring their enzymatic activity. This common limitation is applicable to both of the aforementioned investigations (Xia et al., 2021; Vervuurt et al., 2023). Additionally, MMPs may not be a specific biomarker for CAA, as they could play a role in other conditions such as AD, Parkinson’s disease (PD), multiple sclerosis, stroke, and meningitis (Rosenberg, 2009). As mentioned before, MMPs have a dual function, where they could be beneficial in Aβ clearance or harmful in promoting neuroinflammation (Rosenberg, 2009). Hence, their temporal changes during the course of the disease need further investigation.
4.2 Transforming growth factor-β1
Preclinical evidence suggests that the cytokine transforming growth factor β1 (TGFβ1), which is produced by astrocytes, is involved in vascular deposition of Aβ in CAA (Wyss-Coray et al., 1997). Howe and colleagues have reported that a blood-based panel of MMPs, TGFβs, and a protein associated with vascular basement membrane called fibronectin could accurately differentiate CAA-related ICH from hypertensive ICH (AUC = 0.81) (Howe et al., 2018). However, there is conflicting evidence suggesting that TGF-β1 cannot differentiate subjects with CAA-related cerebral haemorrhage, hypertension-related cerebral haemorrhage, and controls (Greenberg et al., 2000). These contradictions can be explained by looking into the dual function of TGFβ1. According to the literature, TGFβ1 has synaptogenic and neuroprotective effects and reduces parenchymal Aβ deposition, and yet, it may promote vascular amyloid deposition (Diniz et al., 2019; Kapoor and Chinnathambi, 2023). Furthermore, TGFβ1 might not be a proper biomarker at early stages of AD and autosomal dominant AD (Flanders et al., 1995). Therefore, it is conceivable that TGFβ1 might exhibit restricted utility as a biomarker for CAA at different stages of the disease. More investigations are needed to assess the validity of this hypothesis.
4.3 Apolipoproteins
Apolipoprotein D (Apo D) is a glycoprotein that assumes a role in numerous neurological disorders characterised by heightened oxidative stress, such as AD, stroke, and PD (Muffat and Walker, 2010). The association of Apo D and CAA pathology has been established. However, the application of Apo D as a biomarker for CAA is not a straightforward matter, as Apo D levels significantly vary between the two genders and different age groups (Kuiperij et al., 2020).
In addition to Apo D, Apo E and Apo J are two other proteins that highlight the involvement of lipid metabolism in CAA. A study of 60 patients with lobar ICH tried to find the connection between imaging biomarkers of CAA with Apo D and Apo E. Lower levels of Apo E and Apo J in low-density lipoproteins (LDL) fractions of the blood samples showed an association with the cSS extent and CSO-EPVS, respectively (Bonaterra-Pastra et al., 2023).
Apo E ε4 is a well-known genetic risk factor for AD and CAA, affecting Aβ conformation and clearance (Holtzman, 2001). Furthermore, the APO E ε2 allele is associated with an increased risk of CAA-related ICH (Camacho et al., 2019). As such, apolipoproteins may be a key component in CAA pathology, and measuring their dynamic changes during the course of the disease is an area worth exploring.
4.4 Complement 3
Complement 3 (C3), responsible for inflammation and blood–brain barrier (BBB) disruption, has been investigated in CAA patients. The microglia, activated in response to the accumulation of Aβ, alongside perivascular macrophages, can produce a number of cytokines, including C3. C3b, which is a fragment of C3, then attaches to Aβ and the complement receptor three on the microglia, forming a complex that can migrate to the vessel walls. High levels of C3 in blood samples of CAA patients indicate that this proposed pathway may have a substantial role in CAA pathology (Saito et al., 2022). The C3b can also tag synapses for elimination by microglial phagocytosis, causing neurodegeneration in cases such as AD. There is clinical evidence of C3 being involved in a number of neurological diseases, including AD, traumatic brain injury, Huntington’s disease, and amyotrophic lateral sclerosis (ALS) (Dalakas et al., 2020).
In a retrospective cohort, serum samples of 16 CAA patients with MCI (mean age ± SD: 76.6 ± 4.9 years) were compared to 39 non-CAA patients with MCI (mean age ± SD: 76.1 ± 7.5 years). The outcomes of this investigation have indicated that elevated C3 levels possess the potential to act as an independent predictor for CAA (Saito et al., 2022). That said, a single study is not enough to draw a solid conclusion. Furthermore, the role of C3 as a diagnostic biomarker in cognitively unimpaired CAA patients also remains to be investigated in the future.
4.5 Lactadherin
Lactadherin or milk fat globule-EGF factor 8 (MFG-E8) is a glycoprotein involved in various roles, including tissue regeneration, anti-inflammation, and modulation of neurogenesis (Cheyuo et al., 2019). Lactadherin is also associated with CAA Aβ pathology and, therefore, is proposed as a biomarker for CAA. The lactadherin CSF levels are reported to be lower in CAA patients compared to AD patients and healthy controls (Marazuela et al., 2021). Unlike the CSF levels of lactadherin, its serum levels could not differentiate between AD, CAA, and healthy controls (Marazuela et al., 2021). Lactadherin, known for its anti-inflammatory properties within the CNS, looks like a very promising biomarker for CAA as it was correlated with CSF levels of Aβ 40 and Aβ 42 and could differentiate CAA from AD and controls. The exact role of this protein in CAA remains to be clarified through forthcoming investigations (Marazuela et al., 2021).
4.6 Uric acid
Uric acid (UA) is another interesting biomarker candidate. UA is involved in anti-oxidative as well as pro-oxidative pathways, depending on its serum level. UA acts as an anti-oxidant at normal levels but shows cytotoxic effects at higher levels (Mijailovic et al., 2022). It is evident, from studying animal models, that a lower level of UA in the blood is associated with higher amyloid deposition in blood vessels and higher Aβ40 expression. Additionally, a higher level of UA reduced the risk of cerebral haemorrhage by preserving the integrity of blood vessels (Wang et al., 2023).
Accordingly, in a study by Hu and colleagues, 82 CAA patients (according to the modified Boston criteria) with ICH (mean age ± SD: 68.4 ± 8.1 years) were compared to 82 healthy controls (mean age ± SD: 68.3 ± 8.6 years) with regard to their serum UA levels. This study demonstrated a pattern for the UA levels, where probable CAA patients (193 ± 57) had a lower UA concentration compared to possible CAA patients (233 ± 78), while the possible CAA patients, in turn, had lower concentrations compared to healthy controls (309 ± 60). The UA levels, however, were not indicative of the region of haemorrhagic lesions (temporooccipital lesions, frontoparietal lesions, multiple lesions) or the severity of neurologic impairment, measured via the National Institute of Health Stroke Scale and the Glasgow Coma Scale (Hu et al., 2014).
4.7 Platelet-derived growth factor receptor-Β
Pericytes are vital cells situated between neurons, astrocytes, and endothelial cells that have many roles, including regulation of the BBB integrity, capillary hemodynamic responses, neuroinflammation, and toxic metabolite clearance (Sweeney et al., 2016). One of the surface antigens expressed by pericytes is platelet-derived growth factor receptor-β (PDGFR-β). In neurodegenerative diseases such as AD, CSF levels of PDGFRβ may rise due to the malfunction or destruction of pericytes (Salmina et al., 2019).
Hence, PDGFRβ has also been investigated as a novel CSF biomarker, potentially capable of differentiating CAA, AD and healthy controls. However, the results were not promising for CAA since this biomarker only differentiated controls from amnestic MCI and AD patients with an AD-positive biomarker profile (De Kort et al., 2022).
High levels of CSF PDGFRβ may be associated with BBB disruption and neuroinflammation. However, it could not act as a biomarker for CAA and did not reflect the pathological stages of AD. That said, it is hard to draw a conclusion from this limited data and more studies are needed to find the exact role of PDGFRβ in aging (De Kort et al., 2022; Cicognola et al., 2023).
4.8 Neuroleukin
The accumulation of Aβ also promotes the production of reactive oxygen species and neuroinflammation (Carrano et al., 2011). In vitro study of human brain pericytes has shown that Aβ exposure can induce the expression of the neuroleukin (NLK) gene (Rensink et al., 2004). CSF levels of NLK could differentiate amnestic MCI and AD from controls. Conversely, this biomarker proved ineffective in distinguishing between individuals with CAA and the control group (De Kort et al., 2021). Although NLK is expected to promote neuroprotective effects in neurodegenerative disease, CSF levels of NLK did not show promising results as a biomarker for diagnosis or disease progression in PD either (Santaella et al., 2020). That said, it is hard to completely disregard NLK as a biomarker for CAA or a specific subpopulation of CAA, given the limited data available to date.
4.9 Iron and other metals
An excessive quantity of metals, particularly iron, is associated with the initiation of oxidative stress and the aggregation of Aβ (Carocci et al., 2018). CSF levels of iron, cobalt, ferritin, manganese, and nickel have been explored in CAA. When comparing CSF iron levels, CAA patients had higher levels than AD patients and healthy controls. Ferritin only showed a significant difference in a comparison when the data was not age-adjusted. However, ferritin was significantly correlated with CSF levels of Aβ42. As for the other metals, only CSF levels of nickel were significantly higher in CAA compared to AD patients in an age-adjusted comparison (Banerjee et al., 2022). In a study on patients referred from a memory clinic, no associations were discovered between haemorrhagic markers in MRI and CSF iron levels (Shams et al., 2020).
The aggregation of Aβ is interconnected with the generation of reactive oxygen species and the induction of neuroinflammatory processes (Carrano et al., 2011). Iron can also increase APP expression, leading to more Aβ aggregation and oxidative stress, and the oxidative stress continues the cycle by increasing Aβ aggregation (Uranga and Salvador, 2018). This mechanism, along with the study on metallomics in CAA, suggests that iron may play a role in CAA pathology (Banerjee et al., 2022).
5 Neuronal damage biomarkers
In addition to AD-related neurodegeneration in CAA, ischemic or haemorrhagic insults to the brain can also contribute to neuronal damage and brain atrophy (Smith, 2018). The non-haemorrhagic lesions can be observed via neuroimaging about two decades after the initial Aβ deposition within the cerebral vessel walls, which eventually leads to haemorrhagic injuries after another 10 years (Koemans et al., 2023).
Neurofilament light chain (NfL), a specific biomarker for axonal injury, has shown promising results in predicting ICH recurrence in CAA (AUC = 0.84, 95% CI: 0.74–0.93) (Cheng et al., 2020). Serum NfL level is significantly higher in CAA-related ICH compared to controls and is correlated with disease severity (Cheng et al., 2020). The CSF levels of NfL are higher in CAA patients compared to controls, and among those, the amyloid-PET positive CAA patients showed higher levels than the amyloid-PET negative ones (Banerjee et al., 2020). CAA-ri is another condition in which NfL levels in the CSF can be helpful in differentiating between cognitively normal controls and CAA-ri patients (Plotzker et al., 2021). Additionally, the inflammation present in CAA leads to neuronal injury, where NfL could act as a biomarker for it (Banerjee et al., 2020; Cheng et al., 2020; Plotzker et al., 2021).
Serum and CSF levels of NfL are correlated in a variety of neurodegenerative diseases, including Guillain-Barré syndrome (GBS), ALS, and AD, where the CSF levels are about 100 times higher than the serum (Gaiottino et al., 2013). The utilisation of NfL for a differential diagnosis of CAA from the aforementioned diseases appears to exhibit a lack of specificity. That said, the temporal changes in NfL levels could potentially serve as a promising biomarker for tracking disease progression in CAA, something that needs to be explored further.
Glial fibrillary acidic protein (GFAP), an intermediate filament protein produced by astrocytes, faces an increase in production following neuronal damage. In fact, this biomarker is currently used to assess the severity of traumatic brain injuries (TBIs) (Abdelhak et al., 2022). This plasma biomarker is correlated with the number and density of amyloid plaques in AD. However, until now, GFAP plasma levels have failed to reflect vascular deposition of Aβ (Benedet et al., 2022). Nevertheless, the data available on GFAP, especially in CSF, as a biomarker for CAA is limited, making it challenging to derive a definitive conclusion from the current data.
6 Other biomarkers
6.1 Metabolites
Metabolomics, or the comprehensive evaluation of endogenous metabolites, is based on measuring the gross alterations of biochemical hemostasis within the body fluids, including blood, thereby offering valuable insights into the pathogenesis of various diseases (Serkova et al., 2011).
One of the most extensive studies on blood-based biomarkers for CAA was published in 2021. This study compared 275 metabolites in 9 presymptomatic D-CAA mutation carriers (mean age ± SD: 44.1 ± 4.3 years) to 8 controls (mean age ± SD: 43.5 ± 6.6 years) from the same pedigree. A total number of 22 metabolites from 6 different subgroups, including acylcarnitines, amino acids, biogenic amines, hexoses, phospholipids, and sphingolipids, were found to have different plasma levels between the D-CAA mutation carriers and non-carriers. However, after correction for multiple comparisons, only spermidine levels remained significantly different between the two studied groups (Chatterjee et al., 2021a). A similar study on a larger cohort is recommended to enhance statistical power for finding other relevant metabolites. Spermidine is a metabolite with anti-inflammation and anti-oxidative properties. This polyamine is an autophagy inducer in microglia and astrocytes. Spermidine has been studied in animal models of various brain disorders, including traumatic brain injury, Parkinson’s disease, and memory deficits (Ghosh et al., 2020). In the AD mice model (APP/PS1 mice), Spermidine successfully inhibited the Aβ degradation and the neuroinflammation caused by microglia (Freitag et al., 2022).
6.2 Autoantibodies
A dysregulation in B-cell tolerance checkpoints may result in the formation of plasma cells that can enter the brain and produce autoantibodies. These autoantibodies can play a role in neurological and neurodegenerative diseases (Pruss, 2021).
Anti-amyloid β autoantibodies have been studied in CAA-ri as a diagnostic tool. The CSF levels of anti-amyloid β autoantibodies are increased in CAA-ri, and the levels decrease after remission (Piazza et al., 2013; Carmona-Iragui et al., 2016). Studies conducted by other research groups and with larger datasets are needed to validate these findings. The use case of anti-amyloid β autoantibodies as a biomarker for CAA seems to be limited to CAA-ri as the levels come back to the baseline after the acute inflammatory phase. Yet, this biomarker could perform as well as a PiB-PET scan in CAA-ri (Carmona-Iragui et al., 2016). Given the existence of several autoantibodies that are linked with AD, it is conceivable that numerous autoantibodies may also still remain undiscovered within the context of CAA (San Segundo-Acosta et al., 2021; DeMarshall et al., 2022).
6.3 MicroRNAs
An additional group of potential biomarkers with considerable promise for investigation in clinical contexts are microRNAs (miRNAs). These miRNAs, which are non-coding RNAs, hold the potential to participate in the pathological mechanisms of both CAA and AD. While miRNAs could be detected in body fluids such as blood, saliva, and CSF, identifying an optimal subset of miRNAs could be challenging due to the existence of over 2,500 identified miRNA variants (Weldon Furr et al., 2019).
To date, two microRNAs, namely miR-582-3p and miR-892b, associated with APP messenger ribonucleic acid (mRNA) translation, have been identified from blood samples of CAA patients (Nicolas et al., 2016).
Using microRNAs as biomarkers for CAA is an under-studied area that holds great potential in discovering novel biomarkers with high specificity for CAA.
6.4 Synaptic proteins
As discussed before, the tau protein is responsible for oxidative stress and synaptic dysfunction in AD (Niewiadomska et al., 2021). To assess synaptic dysfunction in CAA, van den Berg and colleagues conducted a study in 2023, where they quantified the concentrations of 15 synaptic proteins within the CSF of patients with CAA, AD, and cognitively un-impaired controls. The levels of all assessed synaptic proteins were found to be comparable between CAA patients and the control group, with the exception of Neuronal Pentraxin 2 (NPTX2). In contrast, AD patients had elevated levels in 12 out of the 15 investigated proteins compared to controls. This significant observation underscores the potential utility of synaptic proteins as biomarkers for distinguishing between AD and CAA (AUC = 0.987) (van den Berg et al., 2023). Furthermore, this data suggests that CAA may not affect synaptic function as much as AD.
7 Limitations and future directions
With the advent of new methods such as SIMOA, it is now possible to simultaneously detect multiple biomarkers, from proteins to small molecules, with high sensitivity and specificity (Wang and Walt, 2020). Using a blood-based biomarker instead of a CSF biomarker could markedly benefit patients as it would require a less invasive procedure. However, as seen in the case of lactadherin, a good CSF biomarker does not necessarily translate to a good blood-based biomarker. Similarly, results from a plasma analysis may not be arbitrarily extrapolated to a CSF analysis and making decisions on the viability of using a potential fluid biomarker that is present in both CSF and blood, requires measurements and establishment of firm correlation of such a biomarker in both fluids.
Thus far, some promising biomarkers for AD, like NLK (in CSF), GFAP (in plasma), tau (in CSF), and synaptic proteins (in CSF), have failed to provide convincing results for their utility in distinguishing CAA from cognitively normal controls, highlighting the differing pathology of CAA and AD. That said, confirmatory studies should be considered due to the limited data available on these biomarkers.
Despite this difference, and although AD and CAA are two different diseases with different aetiologies, the two disorders share many similarities (Greenberg et al., 2020). In AD, the disease progression does not occur in a linear manner. Each contributing pathological process, such as neuroinflammation, has a distinct pattern during the course of the disease. Hence, a different panel of biomarkers may become relevant at each stage of AD (Fan et al., 2017). For example, neuroinflammation and microglial activation during prodromal stages of AD may be a neural defence mechanism, given studies have shown that microglial activation is negatively associated with plasma NfL levels, signifying neuronal injury (Parbo et al., 2020). We hypothesise that this may be the case for CAA as well. Some of the discrepancies between the discussed studies may arise from the differences in the mean ages of the participant in these studies, and thus the disease stage of the patients recruited in them. Therefore, longitudinal studies on CAA patients could present a clearer picture of how each biomarker changes during the course of the disease.
Discovering the similarities and differences between D-CAA and sCAA regarding their respective pathological pathway is another crucial step towards finding a reliable biomarker for CAA. The generalisability of findings in presymptomatic D-CAA patients to sCAA patients requires further investigations (Chatterjee et al., 2021b).
Investigating the inflammatory mechanisms inherent to the progression of CAA needs more attention, as there are not enough data available on the interconnection of all the different pathways. Investigations on inflammatory biomarkers could shed light on the pathophysiology of CAA and may have utility in monitoring disease progression, but probably not for diagnostic purposes as they are not specific to CAA. Additionally, apart from the biomarkers discussed in this paper, there remains a lot to be discovered within the field of biomarkers for CAA. Fibrinogen, which is correlated with Aβ levels in AD patients, might be associated with BBB disruption and brain haemorrhage in CAA patients (Fan et al., 2020; Kozberg et al., 2022). Plasma and platelet levels of phosphatidylcholines have preclinical evidence to be used as a diagnostic biomarker for CAA (Foidl et al., 2020). Neutrophil to leukocyte ratio and the systemic immune-inflammation index (SII) could act as prognostic biomarkers in ICH and potentially CAA-related ICH (Liang et al., 2023; Pereira et al., 2023). A number of highly specific proteins, including norrin and collagen alpha-2, have been identified in post-mortem analysis of brain tissue of patients with CAA type I and translating those results into fluid-based measurements in living CAA patients could potentially result in finding a novel and specific biomarker for CAA (Hondius et al., 2018).
A common limitation of biomarker studies on CAA is the small number of enrolled patients. For instance, Banerjee and Werr’ng’s study on the CSF metallomics profile only had 10 CAA patients (Banerjee et al., 2022). This study provided a novel approach to finding a biomarker in CAA, but without better-powered, more comprehensive studies over a longer period of time, no clear conclusions on the validity of metallomics (or other biomarkers of interest) can be drawn. Recruitment of CAA patients from a single center may be feasible for a small study, but a larger study may require a multicentre design (Banerjee and Werring, 2020). A multicentre design brings researchers from different centres and backgrounds together, providing the opportunity to enhance the study design and protocol. Additionally, due to sampling from a more diverse population, the results of a multicentre study are more generalisable to the whole population compared to a single-center study, and smaller effects with enough statistical power can be detected in the large multicentre studies (Chung et al., 2010).
Another factor that needs to be considered concerning the limitations of the studies presented here is the concomitant diseases within the enrolled populations. For instance, in the study on Apo-D, AD patients with concomitant CAA were not identified, and it would be difficult to know if the observed results were related to CAA alone (Kuiperij et al., 2020).
8 Conclusion
This review has conducted a comprehensive analysis of the major fluid biomarkers that have been investigated in relation to all subtypes of CAA in a clinical setting (Table 2). Some of the biomarkers discussed in this review and their interactions are illustrated in Figure 1, providing an overview of the ongoing pathophysiological pathways identified in CAA.
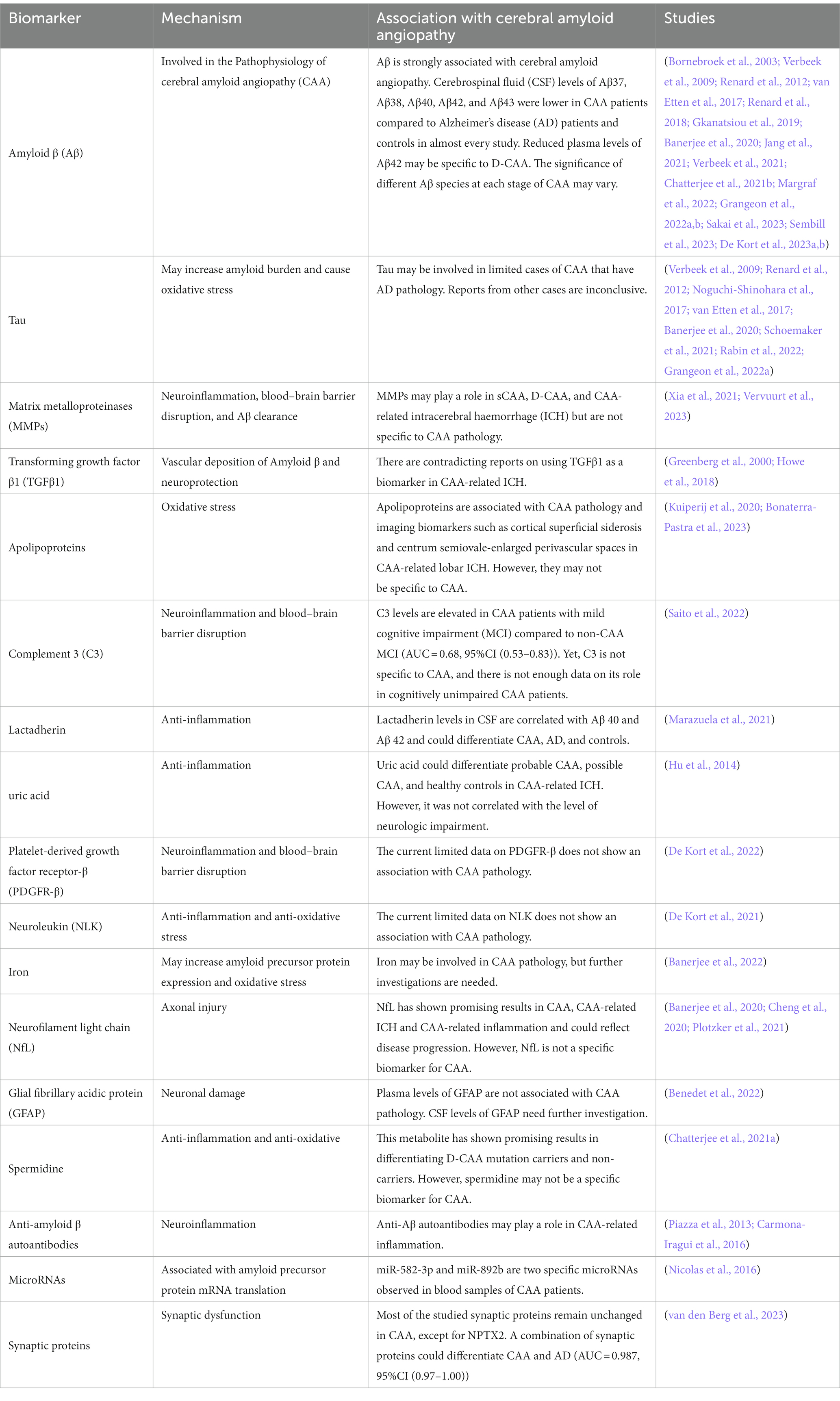
Table 2. A summary of fluid biomarkers of cerebral amyloid angiopathy.
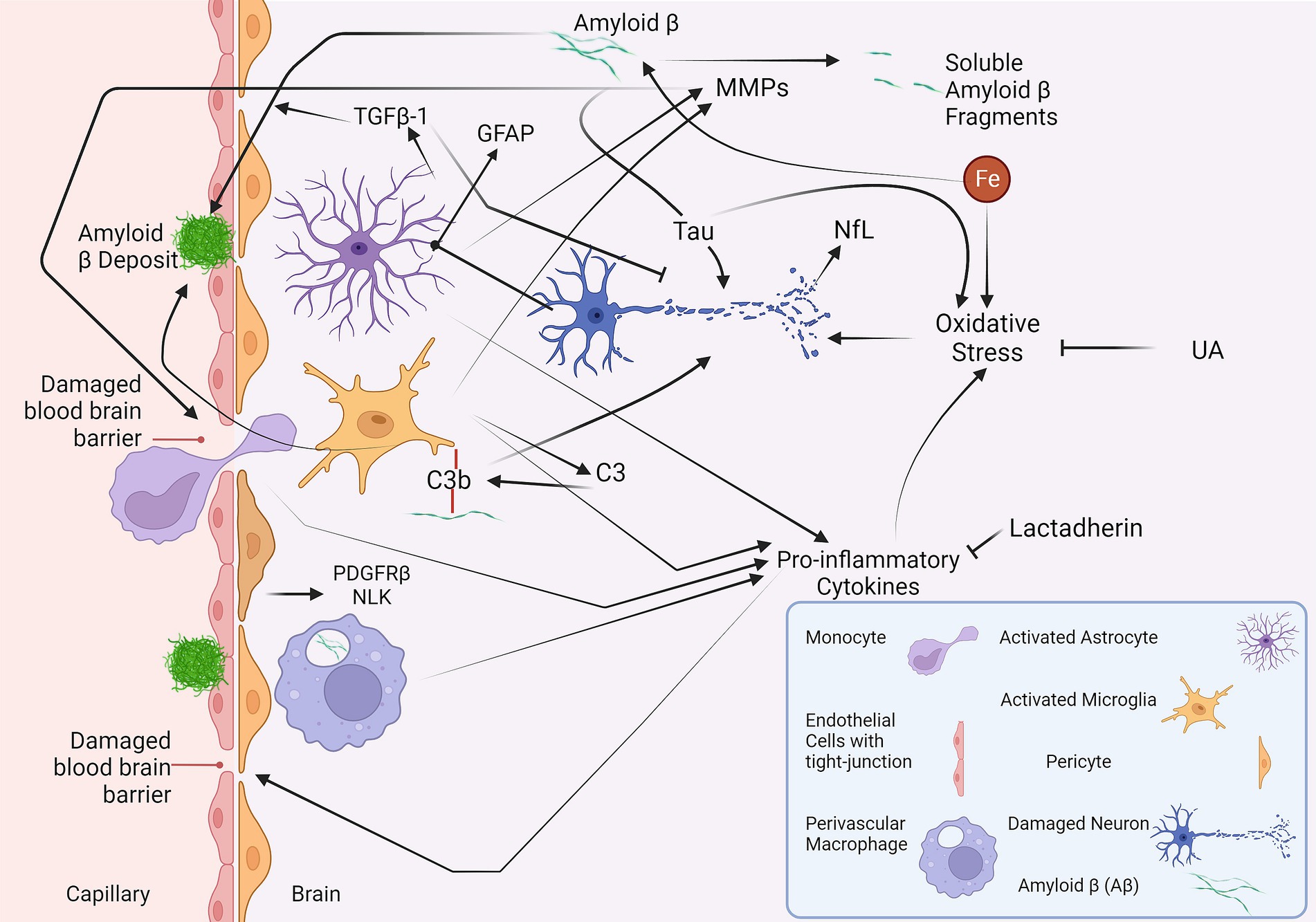
Figure 1. A summary of the fluid biomarkers of cerebral amyloid angiopathy (CAA). The vascular deposition of Aβ leads to CAA (Greenberg et al., 2020). Iron can promote the production of Aβ, whereas MMPs can break down Aβ into soluble fragments (Hernandez-Guillamon et al., 2015; Uranga and Salvador, 2018). The MMPs could also play a harmful role in disrupting the blood–brain barrier (Gireud-Goss et al., 2021). Aβ may also contribute to neurodegeneration with the aid of tau protein (Rapoport et al., 2002). The oxidative stress induced by iron, pro-inflammatory cytokines, and tau results in neuronal damage (Teleanu et al., 2022). In response to this damage, production of GFAP can be induced in activated astrocytes (Abdelhak et al., 2022). NfL is another neuronal damage marker that could be utilised in CAA (Banerjee et al., 2020; Cheng et al., 2020; Plotzker et al., 2021). TGFβ-1, another protein produced by astrocytes, has a dual function and is involved in vascular deposition and clearance of Aβ and, at the same time, has neuroprotective effects (Diniz et al., 2019; Kapoor and Chinnathambi, 2023). C3, produced by the microglia, may play a role in the vascular deposition of Aβ and neurodegeneration (Saito et al., 2022). NLK and PDGFRβ, released by pericytes, are proposed to increase in response to Aβ and damage to pericytes, respectively (De Kort et al., 2021, 2022). Yet, neither NLK nor PDGFRβ has shown significant changes in CAA. Lactadherin and UA have anti-inflammatory and anti-oxidative features, respectively (Cheyuo et al., 2019; Mijailovic et al., 2022). However, their levels are decreased in CAA (Hu et al., 2014; Marazuela et al., 2021), preventing them from mitigating the neuroinflammation and oxidative stress present in CAA. Aβ, amyloid β; C3, complement 3; GFAP, glial fibrillary acidic protein; MMP, matrix metalloproteinase; NFL, neurofilament light chain; NLK, neuroleukin; PDGFRβ, platelet-derived growth factor receptor-β; TGFβ, transforming growth factor; UA, uric acid.
Many biomarkers (e.g., Aβ, UA, C3, MMPs, lactadherin, and NfL) have shown promising evidence in their ability to diagnose and provide a prognosis of disease progression. However, biomarkers that were related to pericytes (NLK and PDGFRβ) did not show promising results, and the results for tau and TGFβ were inconclusive. Additionally, some biomarkers, such as GFAP, autoantibodies, microRNAs, and metabolites, are under-researched and require more investigation. In fact, the current body of evidence for biomarkers of CAA needs an enhancement in both quality and quantity by conducting confirmatory and exploratory studies. Finding a suitable CSF biomarker for CAA reduces the need for special equipment for the diagnosis and follow-up of the patients. A blood-based biomarker has an additional advantage since it requires a less invasive procedure than a lumbar puncture.
Given the differences and similarities between sporadic CAA, hereditary CAA, and AD, it seems that a panel of fluid biomarkers is needed for a differential diagnosis instead of a single fluid biomarker. Additionally, these biomarkers could add to the accuracy of existing diagnostic criteria that are mostly based on imaging biomarkers. Further investigations are needed to identify the optimal combination of biomarkers with high sensitivity and specificity at each stage of the disease. Identifying more sensitive biomarker panels with better clinical utility will help us better characterise CAA and, thus, may help inform future therapeutic avenues. In addition, a reliable biomarker that could reflect the disease progression may be utilised to monitor treatment response effectively and thus help validate new treatment approaches.
Author contributions
SMS: Conceptualization, Writing – original draft, Writing – review & editing. BM: Conceptualization, Supervision, Writing – review & editing, Project administration. EH: Supervision, Writing – review & editing, Conceptualization. FJ: Writing – review & editing, Supervision. SM: Writing – review & editing. SP: Writing – review & editing. SoS: Writing – review & editing. VE: Writing – review & editing. KT: Writing – review & editing. SaG: Writing – review & editing. JC: Writing – review & editing. EE: Writing – review & editing. MO: Writing – review & editing. DC: Writing – review & editing. AG: Writing – review & editing. MB: Writing – review & editing. MW: Writing – review & editing. GH: Writing – review & editing. StG: Conceptualization, Writing – review & editing. RM: Conceptualization, Supervision, Writing – review & editing. HS: Conceptualization, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The details for the sources of funding are disclosed in the “Acknowledgments” and “Conflict of interest” sections.
Acknowledgments
Figure 1 was adapted from “Damaged Blood Brain Barrier Background,” by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates (Publication and Licensing Rights Agreement number: TD268Q3IZO). We would like to thank Murdoch University, Western Australia and the Australian Alzheimer’s Research Foundation (AARF) for their support. SMS and VE are supported by the International Tuition Fee Scholarships from Murdoch University and Stipend Scholarships from the AARF supported by the funding from the TRACK Dutch-CAA Study Consortium.
Conflict of interest
The authors declare that while some of their CAA research projects have been supported by commercial funding from Biogen Inc. and Alnylam Pharmaceuticals, this specific manuscript did not receive any funding and the funders of our research were not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdelhak, A., Foschi, M., Abu-Rumeileh, S., Yue, J. K., D'anna, L., Huss, A., et al. (2022). Blood Gfap as an emerging biomarker in brain and spinal cord disorders. Nat. Rev. Neurol. 18, 158–172. doi: 10.1038/s41582-021-00616-3
Arvanitakis, Z., Leurgans, S. E., Wang, Z., Wilson, R. S., Bennett, D. A., and Schneider, J. A. (2011). Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann. Neurol. 69, 320–327. doi: 10.1002/ana.22112
Banerjee, G., Ambler, G., Keshavan, A., Paterson, R. W., Foiani, M. S., Toombs, J., et al. (2020). Cerebrospinal fluid biomarkers in cerebral amyloid Angiopathy. J. Alzheimers Dis. 74, 1189–1201. doi: 10.3233/JAD-191254
Banerjee, G., Forsgard, N., Ambler, G., Keshavan, A., Paterson, R. W., Foiani, M. S., et al. (2022). Cerebrospinal fluid metallomics in cerebral amyloid angiopathy: an exploratory analysis. J. Neurol. 269, 1470–1475. doi: 10.1007/s00415-021-10711-6
Banerjee, G., and Werring, D. J. (2020). Feasibility of clinical trial recruitment for cerebral amyloid angiopathy: a specialist single Centre experience. J. Neurol. Sci. 409:116580. doi: 10.1016/j.jns.2019.116580
Benedet, A. L., Smirnov, D. S., Ashton, N. J., Hiniker, A. O., Simrén, J., Rodriguez, J. L., et al. (2022). Neuropathological validation of plasma Gfap. Alzheimers Dement. 18:e066008. doi: 10.1002/alz.066008
Biffi, A. (2022). Main features of hereditary cerebral amyloid angiopathies: a systematic review. Cereb. Circ. Cogn. Behav. 3:100124. doi: 10.1016/j.cccb.2022.100124
Blennow, K., and Zetterberg, H. (2018). Biomarkers for Alzheimer's disease: current status and prospects for the future. J. Intern. Med. 284, 643–663. doi: 10.1111/joim.12816
Bonaterra-Pastra, A., Benitez, S., Pancorbo, O., Rodriguez-Luna, D., Vert, C., Rovira, A., et al. (2023). Association of candidate genetic variants and circulating levels of ApoE/ApoJ with common neuroimaging features of cerebral amyloid angiopathy. Front. Aging Neurosci. 15:1134399. doi: 10.3389/fnagi.2023.1134399
Bornebroek, M., De Jonghe, C., Haan, J., Kumar-Singh, S., Younkin, S., Roos, R., et al. (2003). Hereditary cerebral hemorrhage with amyloidosis Dutch type (Abetapp 693): decreased plasma amyloid-beta 42 concentration. Neurobiol. Dis. 14, 619–623. doi: 10.1016/j.nbd.2003.08.019
Brkic, M., Balusu, S., Libert, C., and Vandenbroucke, R. E. (2015). Friends or foes: matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediat. Inflamm. 2015:620581. doi: 10.1155/2015/620581
Camacho, J., Moline, T., Bonaterra-Pastra, A., Ramon, Y. C. S., Martinez-Saez, E., and Hernandez-Guillamon, M. (2019). Brain ApoA-I, ApoJ and ApoE Immunodetection in cerebral amyloid Angiopathy. Front. Neurol. 10:187. doi: 10.3389/fneur.2019.00187
Carmona-Iragui, M., Fernández-Arcos, A., Alcolea, D., Piazza, F., Morenas-Rodriguez, E., Antón-Aguirre, S., et al. (2016). Cerebrospinal fluid anti-amyloid-β autoantibodies and amyloid pet in cerebral amyloid angiopathy-related inflammation. J. Alzheimers Dis. 50, 1–7. doi: 10.3233/JAD-150614
Carocci, A., Catalano, A., Sinicropi, M. S., and Genchi, G. (2018). Oxidative stress and neurodegeneration: the involvement of iron. Biometals 31, 715–735. doi: 10.1007/s10534-018-0126-2
Carrano, A., Hoozemans, J. J., Van Der Vies, S. M., Rozemuller, A. J., Van Horssen, J., and De Vries, H. E. (2011). Amyloid beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid. Redox Signal. 15, 1167–1178. doi: 10.1089/ars.2011.3895
Charidimou, A., Boulouis, G., Frosch, M. P., Baron, J. C., Pasi, M., Albucher, J. F., et al. (2022). The Boston criteria version 2.0 for cerebral amyloid angiopathy: a multicentre, retrospective, Mri-neuropathology diagnostic accuracy study. Lancet Neurol. 21, 714–725. doi: 10.1016/S1474-4422(22)00208-3
Charidimou, A., Farid, K., Tsai, H. H., Tsai, L. K., Yen, R. F., and Baron, J. C. (2018). Amyloid-pet burden and regional distribution in cerebral amyloid angiopathy: a systematic review and meta-analysis of biomarker performance. J. Neurol. Neurosurg. Psychiatry 89, 410–417. doi: 10.1136/jnnp-2017-316851
Charidimou, A., Martinez-Ramirez, S., Reijmer, Y. D., Oliveira-Filho, J., Lauer, A., Roongpiboonsopit, D., et al. (2016). Total magnetic resonance imaging burden of small vessel disease in cerebral amyloid angiopathy: an imaging-pathologic study of concept validation. JAMA Neurol. 73, 994–1001. doi: 10.1001/jamaneurol.2016.0832
Chatterjee, P., Fagan, A. M., Xiong, C., Mckay, M., Bhatnagar, A., Wu, Y., et al. (2021a). Presymptomatic Dutch-type hereditary cerebral amyloid angiopathy-related blood metabolite alterations. J. Alzheimers Dis. 79, 895–903. doi: 10.3233/JAD-201267
Chatterjee, P., Tegg, M., Pedrini, S., Fagan, A. M., Xiong, C., Singh, A. K., et al. (2021b). Plasma amyloid-Beta levels in a pre-symptomatic Dutch-type hereditary cerebral amyloid angiopathy pedigree: a cross-sectional and longitudinal investigation. Int. J. Mol. Sci. 22:2931. doi: 10.3390/ijms22062931
Chen, G. F., Xu, T. H., Yan, Y., Zhou, Y. R., Jiang, Y., Melcher, K., et al. (2017). Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38, 1205–1235. doi: 10.1038/aps.2017.28
Cheng, X., Su, Y., Wang, Q., Gao, F., Ye, X., Wang, Y., et al. (2020). Neurofilament light chain predicts risk of recurrence in cerebral amyloid angiopathy-related intracerebral hemorrhage. Aging (Albany NY) 12, 23727–23738. doi: 10.18632/aging.103927
Cheyuo, C., Aziz, M., and Wang, P. (2019). Neurogenesis in neurodegenerative diseases: role of Mfg-E8. Front. Neurosci. 13:569. doi: 10.3389/fnins.2019.00569
Chiti, F., and Dobson, C. M. (2006). Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366. doi: 10.1146/annurev.biochem.75.101304.123901
Chung, K. C., and Song, J. W.Group, W. S (2010). A guide to organizing a multicenter clinical trial. Plast. Reconstr. Surg. 126, 515–523. doi: 10.1097/PRS.0b013e3181df64fa
Cicognola, C., Mattsson-Carlgren, N., Van Westen, D., Zetterberg, H., Blennow, K., Palmqvist, S., et al. (2023). Associations of Csf Pdgfrbeta with aging, blood-brain barrier damage, Neuroinflammation, and Alzheimer disease pathologic changes. Neurology 101, e30–e39. doi: 10.1212/WNL.0000000000207358
Dalakas, M. C., Alexopoulos, H., and Spaeth, P. J. (2020). Complement in neurological disorders and emerging complement-targeted therapeutics. Nat. Rev. Neurol. 16, 601–617. doi: 10.1038/s41582-020-0400-0
De Kort, A. M., Kuiperij, H. B., Alcolea, D., Kersten, I., Versleijen, A. A. M., Greenberg, S. M., et al. (2021). Cerebrospinal fluid levels of the neurotrophic factor neuroleukin are increased in early Alzheimer's disease, but not in cerebral amyloid angiopathy. Alzheimers Res. Ther. 13:160. doi: 10.1186/s13195-021-00899-0
De Kort, A. M., Kuiperij, H. B., Jäkel, L., Kersten, I., Rasing, I., Van Etten, E. S., et al. (2023a). Plasma amyloid beta 42 is a biomarker for patients with hereditary, but not sporadic, cerebral amyloid angiopathy. Alzheimers Res. Ther. 15:102. doi: 10.1186/s13195-023-01245-2
De Kort, A. M., Kuiperij, H. B., Kersten, I., Versleijen, A. A. M., Schreuder, F., Van Nostrand, W. E., et al. (2022). Normal cerebrospinal fluid concentrations of Pdgfrbeta in patients with cerebral amyloid angiopathy and Alzheimer's disease. Alzheimers Dement. 18, 1788–1796. doi: 10.1002/alz.12506
De Kort, A. M., Kuiperij, H. B., Marques, T. M., Jakel, L., Van Den Berg, E., Kersten, I., et al. (2023b). Decreased cerebrospinal fluid amyloid beta 38, 40, 42, and 43 levels in sporadic and hereditary cerebral amyloid angiopathy. Ann. Neurol. 93, 1173–1186. doi: 10.1002/ana.26610
DeMarshall, C., Viviano, J., Emrani, S., Godsey, G., Sarkar, A., Belinka, B., et al. (2022). Preclinical detection and monitoring of Alzheimer’s disease using a multi-disease diagnostic platform employing autoantibodies as blood-based biomarkers. Alzheimers Dement. 18:e066955. doi: 10.1002/alz.066955
Diniz, L. P., Matias, I., Siqueira, M., Stipursky, J., and Gomes, F. C. A. (2019). Astrocytes and the Tgf-beta1 pathway in the healthy and diseased brain: a double-edged sword. Mol. Neurobiol. 56, 4653–4679. doi: 10.1007/s12035-018-1396-y
Fan, Z., Brooks, D. J., Okello, A., and Edison, P. (2017). An early and late peak in microglial activation in Alzheimer's disease trajectory. Brain 140, 792–803. doi: 10.1093/brain/aww349
Fan, D. Y., Sun, H. L., Sun, P. Y., Jian, J. M., Li, W. W., Shen, Y. Y., et al. (2020). The correlations between plasma fibrinogen with amyloid-Beta and tau levels in patients with Alzheimer's disease. Front. Neurosci. 14:625844. doi: 10.3389/fnins.2020.625844
Flanders, K. C., Lippa, C. F., Smith, T. W., Pollen, D. A., and Sporn, M. B. (1995). Altered expression of transforming growth factor-beta in Alzheimer's disease. Neurology 45, 1561–1569. doi: 10.1212/WNL.45.8.1561
Foidl, B. M., Oberacher, H., Marksteiner, J., and Humpel, C. (2020). Platelet and plasma phosphatidylcholines as biomarkers to diagnose cerebral amyloid angiopathy. Front. Neurol. 11:359. doi: 10.3389/fneur.2020.00359
Freitag, K., Sterczyk, N., Wendlinger, S., Obermayer, B., Schulz, J., Farztdinov, V., et al. (2022). Spermidine reduces neuroinflammation and soluble amyloid beta in an Alzheimer's disease mouse model. J. Neuroinflammation 19:172. doi: 10.1186/s12974-022-02534-7
Gaiottino, J., Norgren, N., Dobson, R., Topping, J., Nissim, A., Malaspina, A., et al. (2013). Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS One 8:e75091. doi: 10.1371/journal.pone.0075091
Gatti, L., Tinelli, F., Scelzo, E., Arioli, F., Di Fede, G., Obici, L., et al. (2020). Understanding the pathophysiology of cerebral amyloid angiopathy. Int. J. Mol. Sci. 21:3435. doi: 10.3390/ijms21103435
Ghosh, I., Sankhe, R., Mudgal, J., Arora, D., and Nampoothiri, M. (2020). Spermidine, an autophagy inducer, as a therapeutic strategy in neurological disorders. Neuropeptides 83:102083. doi: 10.1016/j.npep.2020.102083
Gireud-Goss, M., Mack, A. F., Mccullough, L. D., and Urayama, A. (2021). Cerebral amyloid Angiopathy and blood-brain barrier dysfunction. Neuroscientist 27, 668–684. doi: 10.1177/1073858420954811
Gkanatsiou, E., Portelius, E., Toomey, C. E., Blennow, K., Zetterberg, H., Lashley, T., et al. (2019). A distinct brain beta amyloid signature in cerebral amyloid angiopathy compared to Alzheimer's disease. Neurosci. Lett. 701, 125–131. doi: 10.1016/j.neulet.2019.02.033
Grangeon, L., Paquet, C., Guey, S., Zarea, A., Martinaud, O., Rotharmel, M., et al. (2022a). Cerebrospinal fluid profile of tau, phosphorylated tau, Aβ42, and Aβ40 in probable cerebral amyloid Angiopathy. J. Alzheimers Dis. 87, 791–802. doi: 10.3233/JAD-215208
Grangeon, L., Quesney, G., Verdalle-Cazes, M., Coulette, S., Renard, D., Wacongne, A., et al. (2022b). Different clinical outcomes between cerebral amyloid angiopathy-related inflammation and non-inflammatory form. J. Neurol. 269, 4972–4984. doi: 10.1007/s00415-022-11145-4
Greenberg, S. M., Bacskai, B. J., Hernandez-Guillamon, M., Pruzin, J., Sperling, R., and Van Veluw, S. J. (2020). Cerebral amyloid angiopathy and Alzheimer disease – one peptide, two pathways. Nat. Rev. Neurol. 16, 30–42. doi: 10.1038/s41582-019-0281-2
Greenberg, S. M., Cho, H. S., O'Donnell, H. C., Rosand, J., Segal, A. Z., Younkin, L. H., et al. (2000). Plasma beta-amyloid peptide, transforming growth factor-beta 1, and risk for cerebral amyloid angiopathy. Ann. N. Y. Acad. Sci. 903, 144–149. doi: 10.1111/j.1749-6632.2000.tb06361.x
Harrison, R. S., Sharpe, P. C., Singh, Y., and Fairlie, D. P. (2007). Amyloid peptides and proteins in review. Rev. Physiol. Biochem. Pharmacol. 159, 1–77. doi: 10.1007/112_2007_0701
Hernandez-Guillamon, M., Mawhirt, S., Blais, S., Montaner, J., Neubert, T. A., Rostagno, A., et al. (2015). Sequential amyloid-beta degradation by the matrix metalloproteases Mmp-2 and Mmp-9. J. Biol. Chem. 290, 15078–15091. doi: 10.1074/jbc.M114.610931
Holtzman, D. M. (2001). Role of apoe/Abeta interactions in the pathogenesis of Alzheimer's disease and cerebral amyloid angiopathy. J. Mol. Neurosci. 17, 147–155. doi: 10.1385/JMN:17:2:147
Hondius, D. C., Eigenhuis, K. N., Morrema, T. H. J., Van Der Schors, R. C., Van Nierop, P., Bugiani, M., et al. (2018). Proteomics analysis identifies new markers associated with capillary cerebral amyloid angiopathy in Alzheimer's disease. Acta Neuropathol. Commun. 6:46. doi: 10.1186/s40478-018-0540-2
Howe, M. D., Zhu, L., Sansing, L. H., Gonzales, N. R., Mccullough, L. D., and Edwards, N. J. (2018). Serum markers of blood-brain barrier remodeling and fibrosis as predictors of etiology and Clinicoradiologic outcome in intracerebral hemorrhage. Front. Neurol. 9:746. doi: 10.3389/fneur.2018.00746
Hu, Q., Liu, A., Huang, M., Cheng, L., Kang, H., Xu, F., et al. (2014). Lower serum uric acid levels in cerebral amyloid angiopathy: a pilot study. Neurol. Sci. 35, 1035–1039. doi: 10.1007/s10072-014-1634-7
Jakel, L., De Kort, A. M., Klijn, C. J. M., Schreuder, F., and Verbeek, M. M. (2022). Prevalence of cerebral amyloid angiopathy: a systematic review and meta-analysis. Alzheimers Dement. 18, 10–28. doi: 10.1002/alz.12366
Jang, H., Jang, Y. K., Kim, H. J., Werring, D. J., Lee, J. S., Choe, Y. S., et al. (2019). Clinical significance of amyloid beta positivity in patients with probable cerebral amyloid angiopathy markers. Eur. J. Nucl. Med. Mol. Imaging 46, 1287–1298. doi: 10.1007/s00259-019-04314-7
Jang, H., Kim, J. S., Lee, H. J., Kim, C. H., Na, D. L., Kim, H. J., et al. (2021). Performance of the plasma Abeta42/Abeta40 ratio, measured with a novel Hplc-Ms/Ms method, as a biomarker of amyloid pet status in a Dpuk-Korean cohort. Alzheimers Res. Ther. 13:179. doi: 10.1186/s13195-021-00911-7
Kapoor, M., and Chinnathambi, S. (2023). Tgf-beta1 signalling in Alzheimer's pathology and cytoskeletal reorganization: a specialized tau perspective. J. Neuroinflammation 20:72. doi: 10.1186/s12974-023-02751-8
Kaushik, K., Van Etten, E. S., Siegerink, B., Kappelle, L. J., Lemstra, A. W., Schreuder, F., et al. (2023). Iatrogenic cerebral amyloid Angiopathy post neurosurgery: frequency, clinical profile, radiological features, and outcome. Stroke 54, 1214–1223. doi: 10.1161/STROKEAHA.122.041690
Koemans, E. A., Chhatwal, J. P., Van Veluw, S. J., Van Etten, E. S., Van Osch, M. J. P., Van Walderveen, M. A. A., et al. (2023). Progression of cerebral amyloid angiopathy: a pathophysiological framework. Lancet Neurol. 22, 632–642. doi: 10.1016/S1474-4422(23)00114-X
Kozberg, M. G., Yi, I., Freeze, W. M., Auger, C. A., Scherlek, A. A., Greenberg, S. M., et al. (2022). Blood-brain barrier leakage and perivascular inflammation in cerebral amyloid angiopathy. Brain Commun. 4:fcac245. doi: 10.1093/braincomms/fcac245
Kuiperij, H. B., Hondius, D. C., Kersten, I., Versleijen, A. A. M., Rozemuller, A. J. M., Greenberg, S. M., et al. (2020). Apolipoprotein D: a potential biomarker for cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 46, 431–440. doi: 10.1111/nan.12595
Liang, Z., Liu, H., Xue, L., Ma, B., Yang, L. Z., Liang, Q. L., et al. (2023). A retrospective study about association of dynamic systemic immune-inflammation index (Sii) with 180-day functional outcome after basal ganglia intracerebral hemorrhage. Heliyon 9:e16937. doi: 10.1016/j.heliyon.2023.e16937
Marazuela, P., Solé, M., Bonaterra-Pastra, A., Pizarro, J., Camacho, J., Martínez-Sáez, E., et al. (2021). Mfg-E8 (Lactadherin): a novel marker associated with cerebral amyloid angiopathy. Acta Neuropathol. Commun. 9:154. doi: 10.1186/s40478-021-01257-9
Margraf, N. G., Jensen-Kondering, U., Weiler, C., Leypoldt, F., Maetzler, W., Philippen, S., et al. (2022). Cerebrospinal fluid biomarkers in cerebral amyloid Angiopathy: new data and quantitative Meta-analysis. Front. Aging Neurosci. 14:783996. doi: 10.3389/fnagi.2022.783996
Mccarter, S. J., Lesnick, T. G., Lowe, V. J., Rabinstein, A. A., Przybelski, S. A., Algeciras-Schimnich, A., et al. (2022). Association between plasma biomarkers of amyloid, tau, and neurodegeneration with cerebral microbleeds. J. Alzheimers Dis. 87, 1–11. doi: 10.3233/JAD-220158
Mijailovic, N. R., Vesic, K., and Borovcanin, M. M. (2022). The influence of serum uric acid on the brain and cognitive dysfunction. Front. Psych. 13:828476. doi: 10.3389/fpsyt.2022.828476
Molinuevo, J. L., Ayton, S., Batrla, R., Bednar, M. M., Bittner, T., Cummings, J., et al. (2018). Current state of Alzheimer's fluid biomarkers. Acta Neuropathol. 136, 821–853. doi: 10.1007/s00401-018-1932-x
Muffat, J., and Walker, D. W. (2010). Apolipoprotein D: an overview of its role in aging and age-related diseases. Cell Cycle 9, 269–273. doi: 10.4161/cc.9.2.10433
Muir, R. T., Ismail, Z., Black, S. E., and Smith, E. E. (2023). Comparative methods for quantifying plasma biomarkers in Alzheimer's disease: implications for the next frontier in cerebral amyloid angiopathy diagnostics. Alzheimers Dement. 1–23. doi: 10.1002/alz.13510
Nagaraja, N., and Patel, U. K. (2021). Disparities in diagnosis of cerebral amyloid angiopathy based on hospital characteristics. J. Clin. Neurosci. 89, 39–42. doi: 10.1016/j.jocn.2021.04.021
Nicolas, G., Wallon, D., Goupil, C., Richard, A. C., Pottier, C., Dorval, V., et al. (2016). Mutation in the 3'untranslated region of app as a genetic determinant of cerebral amyloid angiopathy. Eur. J. Hum. Genet. 24, 92–98. doi: 10.1038/ejhg.2015.61
Niewiadomska, G., Niewiadomski, W., Steczkowska, M., and Gasiorowska, A. (2021). Tau oligomers neurotoxicity. Life (Basel) 11:28. doi: 10.3390/life11010028
Noguchi-Shinohara, M., Komatsu, J., Samuraki, M., Matsunari, I., Ikeda, T., Sakai, K., et al. (2017). Cerebral amyloid Angiopathy-related microbleeds and cerebrospinal fluid biomarkers in Alzheimer's disease. J. Alzheimers Dis. 55, 905–913. doi: 10.3233/JAD-160651
Padala, S. P., and Newhouse, P. A. (2023). Blood-based biomarkers in Alzheimer's disease: a mini-review. Metab. Brain Dis. 38, 185–193. doi: 10.1007/s11011-022-01114-1
Parbo, P., Madsen, L. S., Ismail, R., Zetterberg, H., Blennow, K., Eskildsen, S. F., et al. (2020). Low plasma neurofilament light levels associated with raised cortical microglial activation suggest inflammation acts to protect prodromal Alzheimer's disease. Alzheimers Res. Ther. 12:3. doi: 10.1186/s13195-019-0574-0
Pereira, M., Batista, R., Marreiros, A., and Nzwalo, H. (2023). Neutrophil-to-leukocyte ratio and admission glycemia as predictors of short-term death in very old elderlies with lobar intracerebral hemorrhage. Brain Circ. 9, 94–98. doi: 10.4103/bc.bc_5_23
Piazza, F., Greenberg, S. M., Savoiardo, M., Gardinetti, M., Chiapparini, L., Raicher, I., et al. (2013). Anti-amyloid β autoantibodies in cerebral amyloid angiopathy-related inflammation: implications for amyloid-modifying therapies. Ann. Neurol. 73, 449–458. doi: 10.1002/ana.23857
Plotzker, A. S., Henson, R. L., Fagan, A. M., Morris, J. C., and Day, G. S. (2021). Clinical and Paraclinical measures associated with outcome in cerebral amyloid Angiopathy with related inflammation. J. Alzheimers Dis. 80, 133–142. doi: 10.3233/JAD-201299
Pruss, H. (2021). Autoantibodies in neurological disease. Nat. Rev. Immunol. 21, 798–813. doi: 10.1038/s41577-021-00543-w
Rabin, J. S., Nichols, E., La Joie, R., Casaletto, K. B., Palta, P., Dams-O'connor, K., et al. (2022). Cerebral amyloid angiopathy interacts with neuritic amyloid plaques to promote tau and cognitive decline. Brain 145, 2823–2833. doi: 10.1093/brain/awac178
Rapoport, M., Dawson, H. N., Binder, L. I., Vitek, M. P., and Ferreira, A. (2002). Tau is essential to beta -amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. U. S. A. 99, 6364–6369. doi: 10.1073/pnas.092136199
Regenhardt, R. W., Thon, J. M., Das, A. S., Thon, O. R., Charidimou, A., Viswanathan, A., et al. (2020). Association between immunosuppressive treatment and outcomes of cerebral amyloid Angiopathy-related inflammation. JAMA Neurol. 77, 1261–1269. doi: 10.1001/jamaneurol.2020.1782
Renard, D., Castelnovo, G., Wacongne, A., Le Floch, A., Thouvenot, E., Mas, J., et al. (2012). Interest of Csf biomarker analysis in possible cerebral amyloid angiopathy cases defined by the modified Boston criteria. J. Neurol. 259, 2429–2433. doi: 10.1007/s00415-012-6520-8
Renard, D., Collombier, L., Demattei, C., Wacongne, A., Charif, M., Ayrignac, X., et al. (2018). Cerebrospinal fluid, Mri, and Florbetaben-pet in cerebral amyloid Angiopathy-related inflammation. J. Alzheimers Dis. 61, 1107–1117. doi: 10.3233/JAD-170843
Rensink, A. A., Otte-Holler, I., Ten Donkelaar, H. J., De Waal, R. M., Kremer, B., and Verbeek, M. M. (2004). Differential gene expression in human brain pericytes induced by amyloid-beta protein. Neuropathol. Appl. Neurobiol. 30, 279–291. doi: 10.1111/j.1365-2990.2004.00536.x
Rodrigues, M. A., Samarasekera, N., Lerpiniere, C., Humphreys, C., Mccarron, M. O., White, P. M., et al. (2018). The Edinburgh Ct and genetic diagnostic criteria for lobar intracerebral haemorrhage associated with cerebral amyloid angiopathy: model development and diagnostic test accuracy study. Lancet Neurol. 17, 232–240. doi: 10.1016/S1474-4422(18)30006-1
Rosenberg, G. A. (2009). Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 8, 205–216. doi: 10.1016/S1474-4422(09)70016-X
Saito, S., Yamashiro, T., Yamauchi, M., Yamamoto, Y., Noguchi, M., Tomita, T., et al. (2022). Complement 3 is a potential biomarker for cerebral amyloid Angiopathy. J. Alzheimers Dis. 89, 381–387. doi: 10.3233/JAD-220494
Sakai, K., Noguchi-Shinohara, M., Tanaka, H., Ikeda, T., Hamaguchi, T., Kakita, A., et al. (2023). Cerebrospinal fluid biomarkers and amyloid-β elimination from the brain in cerebral amyloid Angiopathy-related inflammation. J. Alzheimers Dis. 91, 1173–1183. doi: 10.3233/JAD-220838
Salmina, A. B., Komleva, Y. K., Lopatina, O. L., and Birbrair, A. (2019). Pericytes in Alzheimer's disease: novel clues to cerebral amyloid Angiopathy pathogenesis. Adv. Exp. Med. Biol. 1147, 147–166. doi: 10.1007/978-3-030-16908-4_7
San Segundo-Acosta, P., Montero-Calle, A., Jernbom-Falk, A., Alonso-Navarro, M., Pin, E., Andersson, E., et al. (2021). Multiomics profiling of Alzheimer's disease serum for the identification of autoantibody biomarkers. J. Proteome Res. 20, 5115–5130. doi: 10.1021/acs.jproteome.1c00630
Santaella, A., Kuiperij, H. B., Van Rumund, A., Esselink, R. A. J., Van Gool, A. J., Bloem, B. R., et al. (2020). Cerebrospinal fluid monocyte chemoattractant protein 1 correlates with progression of Parkinson's disease. NPJ Parkinsons Dis. 6:21. doi: 10.1038/s41531-020-00124-z
Schindler, S. E., Bollinger, J. G., Ovod, V., Mawuenyega, K. G., Li, Y., Gordon, B. A., et al. (2019). High-precision plasma beta-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 93, e1647–e1659. doi: 10.1212/WNL.0000000000008081
Schindler, S. E., Gray, J. D., Gordon, B. A., Xiong, C., Batrla-Utermann, R., Quan, M., et al. (2018). Cerebrospinal fluid biomarkers measured by Elecsys assays compared to amyloid imaging. Alzheimers Dement. 14, 1460–1469. doi: 10.1016/j.jalz.2018.01.013
Schoemaker, D., Charidimou, A., Zanon Zotin, M. C., Raposo, N., Johnson, K. A., Sanchez, J. S., et al. (2021). Association of Memory Impairment with Concomitant tau Pathology in patients with cerebral amyloid Angiopathy. Neurology 96, e1975–e1986. doi: 10.1212/WNL.0000000000011745
Schultz, A. P., Kloet, R. W., Sohrabi, H. R., van der Weerd, L., van Rooden, S., Wermer, M. J. H., et al. (2019). Amyloid imaging of dutch-type hereditary cerebral amyloid angiopathy carriers. Ann. Neurol. 86, 616–625. doi: 10.1002/ana.25560
Sembill, J. A., Knott, M., Xu, M., Roeder, S. S., Hagen, M., Sprugel, M. I., et al. (2022). Simplified Edinburgh Ct criteria for identification of lobar intracerebral hemorrhage associated with cerebral amyloid Angiopathy. Neurology 98, e1997–e2004. doi: 10.1212/WNL.0000000000200261
Sembill, J. A., Lusse, C., Linnerbauer, M., Sprugel, M. I., Mrochen, A., Knott, M., et al. (2023). Cerebrospinal fluid biomarkers for cerebral amyloid angiopathy. Brain Commun. 5:fcad159. doi: 10.1093/braincomms/fcad159
Serkova, N. J., Standiford, T. J., and Stringer, K. A. (2011). The emerging field of quantitative blood metabolomics for biomarker discovery in critical illnesses. Am. J. Respir. Crit. Care Med. 184, 647–655. doi: 10.1164/rccm.201103-0474CI
Shackleton, B., Ringland, C., Abdullah, L., Mullan, M., Crawford, F., and Bachmeier, C. (2019). Influence of matrix metallopeptidase 9 on Beta-amyloid elimination across the blood-brain barrier. Mol. Neurobiol. 56, 8296–8305. doi: 10.1007/s12035-019-01672-z
Shams, M., Martola, J., Charidimou, A., Granberg, T., Ferreira, D., Westman, E., et al. (2020). Cerebrospinal fluid metals and the association with cerebral small vessel disease. J. Alzheimers Dis. 78, 1229–1236. doi: 10.3233/JAD-200656
Smith, E. E. (2018). Cerebral amyloid angiopathy as a cause of neurodegeneration. J. Neurochem. 144, 651–658. doi: 10.1111/jnc.14157
Storti, B., Gabriel, M. M., Sennfalt, S., Canavero, I., Rifino, N., Gatti, L., et al. (2023). Rare forms of cerebral amyloid angiopathy: pathogenesis, biological and clinical features of Caa-ri and icaa. Front. Neurosci. 17:1219025. doi: 10.3389/fnins.2023.1219025
Sweeney, M. D., Ayyadurai, S., and Zlokovic, B. V. (2016). Pericytes of the neurovascular unit: key functions and signaling pathways. Nat. Neurosci. 19, 771–783. doi: 10.1038/nn.4288
Tang, W. K., Chen, Y. K., Lu, J., Ahuja, A. T., Chu, W. C., Mok, V. C., et al. (2011). Cerebral microbleeds and quality of life in acute ischemic stroke. Neurol. Sci. 32, 449–454. doi: 10.1007/s10072-011-0571-y
Teleanu, D. M., Niculescu, A. G., Lungu, I., Radu, C. I., Vladacenco, O., Roza, E., et al. (2022). An overview of oxidative stress, neuroinflammation, and neurodegenerative diseases. Int. J. Mol. Sci. 23:5938. doi: 10.3390/ijms23115938
Thal, D. R., Ghebremedhin, E., Rub, U., Yamaguchi, H., Del Tredici, K., and Braak, H. (2002). Two types of sporadic cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 61, 282–293. doi: 10.1093/jnen/61.3.282
Tsao, C. W., Aday, A. W., Almarzooq, Z. I., Anderson, C. A. M., Arora, P., Avery, C. L., et al. (2023). Heart disease and stroke Statistics-2023 update: a report from the American Heart Association. Circulation 147, e93–e621. doi: 10.1161/CIR.0000000000001123
Uranga, R. M., and Salvador, G. A. (2018). Unraveling the burden of Iron in neurodegeneration: intersections with amyloid beta peptide pathology. Oxid. Med. Cell. Longev. 2018:2850341. doi: 10.1155/2018/2850341
Valenti, R., Reijmer, Y. D., Charidimou, A., Boulouis, G., Martinez, S. R., Xiong, L., et al. (2017). Total small vessel disease burden and brain network efficiency in cerebral amyloid angiopathy. J. Neurol. Sci. 382, 10–12. doi: 10.1016/j.jns.2017.09.015
Van Den Berg, E., Nilsson, J., Kersten, I., Brinkmalm, G., De Kort, A. M., Klijn, C. J. M., et al. (2023). Cerebrospinal fluid panel of synaptic proteins in cerebral amyloid Angiopathy and Alzheimer's disease. J. Alzheimers Dis. 92, 467–475. doi: 10.3233/JAD-220977
Van Etten, E. S., Kaushik, K., Van Zwet, E. W., Voigt, S., Van Walderveen, M. A. A., Van Buchem, M. A., et al. (2020). Sensitivity of the Edinburgh criteria for lobar intracerebral hemorrhage in hereditary cerebral amyloid Angiopathy. Stroke 51, 3608–3612. doi: 10.1161/STROKEAHA.120.031264
Van Etten, E. S., Verbeek, M. M., Van Der Grond, J., Zielman, R., Van Rooden, S., Van Zwet, E. W., et al. (2017). β-Amyloid in Csf: biomarker for preclinical cerebral amyloid angiopathy. Neurology 88, 169–176. doi: 10.1212/WNL.0000000000003486
Verbeek, M. M., De Kort, A. M., Wermer, M. J., Marques, T. M., Kersten, I., Schreuder, F. H., et al. (2021). Amyloid-βeta peptides in Csf and plasma discriminate cerebral amyloid angiopathy from controls. Alzheimers Dement. 17:e053858. doi: 10.1002/alz.053858
Verbeek, M. M., Kremer, B. P., Rikkert, M. O., Van Domburg, P. H., Skehan, M. E., and Greenberg, S. M. (2009). Cerebrospinal fluid amyloid beta(40) is decreased in cerebral amyloid angiopathy. Ann. Neurol. 66, 245–249. doi: 10.1002/ana.21694
Vervuurt, M., De Kort, A. M., Jakel, L., Kersten, I., Abdo, W. F., Schreuder, F., et al. (2023). Decreased ratios of matrix metalloproteinases to tissue-type inhibitors in cerebrospinal fluid in sporadic and hereditary cerebral amyloid angiopathy. Alzheimers Res. Ther. 15:26. doi: 10.1186/s13195-023-01171-3
Wang, X., and Walt, D. R. (2020). Simultaneous detection of small molecules, proteins and micrornas using single molecule arrays. Chem. Sci. 11, 7896–7903. doi: 10.1039/D0SC02552F
Wang, D., Wang, Y., Xu, D., Zhou, G., and He, S. (2023). Relationship between uric acid and cerebral amyloid angiopathy. Int. J. Neurosci. 133, 222–231. doi: 10.1080/00207454.2021.1903001
Weldon Furr, J., Morales-Scheihing, D., Manwani, B., Lee, J., and Mccullough, L. D. (2019). Cerebral amyloid Angiopathy, Alzheimer's disease and Microrna: mirna as diagnostic biomarkers and potential therapeutic targets. NeuroMolecular Med. 21, 369–390. doi: 10.1007/s12017-019-08568-0
Wyss-Coray, T., Masliah, E., Mallory, M., Mcconlogue, L., Johnson-Wood, K., Lin, C., et al. (1997). Amyloidogenic role of cytokine Tgf-beta1 in transgenic mice and in Alzheimer's disease. Nature 389, 603–606. doi: 10.1038/39321
Keywords: cerebral amyloid angiopathy, amyloid beta, familial cerebral amyloid angiopathy, differential diagnosis, fluid biomarkers, surrogate endpoints
Citation: Savar SM, Ma B, Hone E, Jahan F, Markovic S, Pedrini S, Shemehsavar S, Easwaran V, Taddei K, Gardener S, Chhatwal JP, van Etten ES, van Osch MJP, Clarke D, Gnjec A, van Buchem MA, Wermer MJH, Hankey GJ, Greenberg SM, Martins RN and Sohrabi HR (2024) Fluid biomarkers in cerebral amyloid angiopathy. Front. Neurosci. 18:1347320. doi: 10.3389/fnins.2024.1347320
Edited by:
Scott Miners, University of Bristol, United KingdomReviewed by:
Siranjeevi Nagaraj, Université libre de Bruxelles, BelgiumRobbie Fisher, University of Bristol, United Kingdom
Copyright © 2024 Savar, Ma, Hone, Jahan, Markovic, Pedrini, Shemehsavar, Easwaran, Taddei, Gardener, Chhatwal, van Etten, van Osch, Clarke, Gnjec, van Buchem, Wermer, Hankey, Greenberg, Martins and Sohrabi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hamid R. Sohrabi, aGFtaWQuc29ocmFiaUBtdXJkb2NoLmVkdS5hdQ==; Ralph N. Martins, cmFscGgubWFydGluc0BtcS5lZHUuYXU=; Steven M. Greenberg, c2dyZWVuYmVyZ0BtZ2guaGFydmFyZC5lZHU=