 Zihao Yang1,2†
Zihao Yang1,2† Kai Yao
Kai Yao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 17 January 2024
Sec. Visual Neuroscience
Volume 18 - 2024 | https://doi.org/10.3389/fnins.2024.1265630
This article is part of the Research Topic Insights in Visual Neuroscience: 2023 View all 8 articles
Color blindness is a retinal disease that mainly manifests as a color vision disorder, characterized by achromatopsia, red-green color blindness, and blue-yellow color blindness. With the development of technology and progress in theory, extensive research has been conducted on the genetic basis of color blindness, and various approaches have been explored for its treatment. This article aims to provide a comprehensive review of recent advances in understanding the pathological mechanism, clinical symptoms, and treatment options for color blindness. Additionally, we discuss the various treatment approaches that have been developed to address color blindness, including gene therapy, pharmacological interventions, and visual aids. Furthermore, we highlight the promising results from clinical trials of these treatments, as well as the ongoing challenges that must be addressed to achieve effective and long-lasting therapeutic outcomes. Overall, this review provides valuable insights into the current state of research on color blindness, with the intention of informing further investigation and development of effective treatments for this disease.
Vision is one of the most important sources of information for human beings. The human visual system relies on two types of photoreceptors located in the retina (Figure 1A), namely rods and cones. The human retina contains around 90 to 120 million rod cells, while only 4 to 6 million cone cells. Rod cells are located in all areas except the central fovea, while cone cells are clustered in the central fovea. Among them, the rod cells are responsible for night vision, that is, black and white vision. They are highly sensitive and can reliably receive the signal of a single photon. However, this high sensitivity makes rods susceptible to saturation; thus, for example, human rod monochromats (persons whose retina lacks functional cones) are light-blinded even at low diurnal levels of illumination. The cones are responsible for daytime vision and color vision. And the sensitivity of cones is 100 to 1,000 times less than that of rods. The main cause of the differing sensitivity of rods and cones is the ability of cones to terminate their photo response faster. This ensures that the cone does not saturate and provides useful vision at the maximum intensity available in nature (Astakhova et al., 2015). When cone function is impaired, it can result in color blindness.
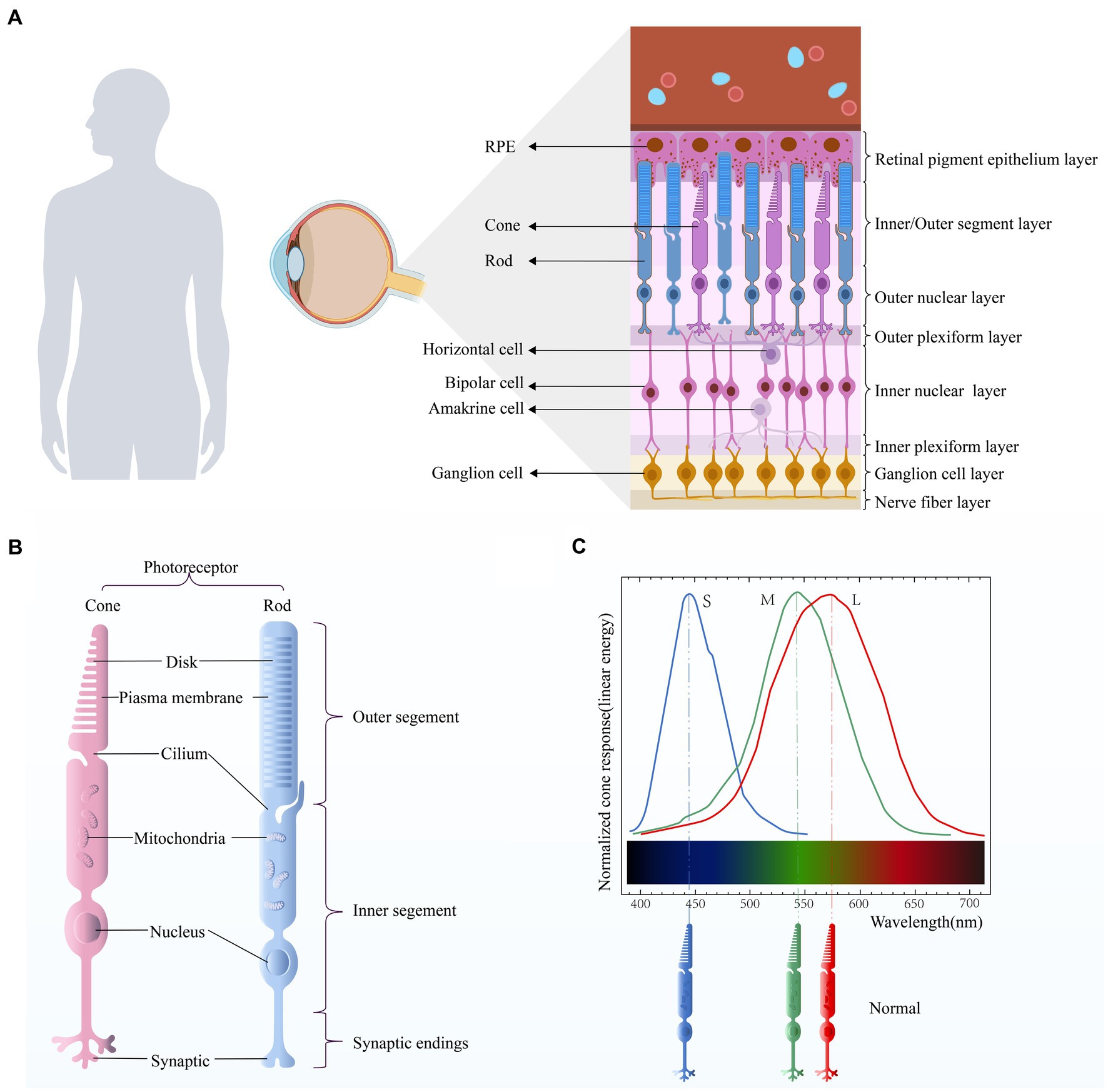
Figure 1. (A) Retinal stratification. The retina is mainly composed of retinal pigment epithelial (RPE) cells, cone cells, rod cells, horizontal cells, bipolar cells, amacrine cells, and ganglion cells. (B) Photoreceptor structure. There are two types of photoreceptors in the human retina, namely the cone and the rod. Their structures are similar and can be roughly divided into three parts: outer segment, inner segment, and synaptic ending. (C) Optic cone cell absorption spectra. Different types of cone cells have different peak sensitivities.
Color blindness, a type of cone dysfunction syndrome (CDS), is a color vision disease that includes both congenital and acquired forms. Patients with this disease are unable to distinguish one or more colors in the natural spectrum. Congenital color vision disease is an inherited retinal disease (IRD) that is passed down through chromosomes, affecting both males and females but showing a greater prevalence in men. Acquired color vision diseases have various causes, including ocular and neurological disorders, some metabolic disorders, drug toxicity, and exposure to certain solvents (Swanson and Cohen, 2003). Furthermore, research indicates that prolonged exposure to alcohol during adolescence and young adulthood can result in visual deficits and impair color vision (Brasil et al., 2015).
The origins of our understanding of color blindness can be traced to a 1,602 account by a Chinese medical scientist. Coincidentally, British physicist and chemist Robert Boyle also described a case of “visual impairment” in an article in 1688. Boyle wrote that the maid sometimes “wanted to gather violets,” but “she knelt where the violets grew, and could not distinguish weeds and flowers by color, only by shape or feeling” (Simunovic, 2010). About 100 years later, the British chemist and physicist Dalton also discovered color blindness as a result of his own and his brother’s deficiencies and published “On Color Blindness” in 1793 (Emery, 1988). In honor of his achievements, congenital red-green color blindness is also known as Daltonism.
With the development of modern scientific understanding, including genetics, there has been an improved understanding of color blindness. Congenital color blindness is divided into Achromatopsia (ACHM), red-green colorblindness, and blue-yellow colorblindness.
Achromatopsia is an autosomal recessive genetic disorder of the retina. This disease affects three types of cone cells in the retina, resulting in complete cone cell dysfunction, poor vision, complete loss of color vision, leaving only the ability to differentiate between light and dark, often accompanied by photophobia, nystagmus, and other phenomena.
Red-green color blindness (anerythrochloropsia), a recessive disease carried on the X chromosome, is divided into protanopia (red blindness) and deuteranopia (green blindness). This condition is the most prevalent type of color vision disorder, constituting around 95% of all color vision disorders (Salih et al., 2021) and is the most common single locus genetic disorder (Mancuso et al., 2009). Patients are unable to distinguish between red and green.
Blue-yellow color blindness (tritanopia), an autosomal dominant disorder, is characterized by the inability to recognize blue and yellow, although the patient can recognize the red and green spectrum (Weitz et al., 1992a).
In addition to these diseases with severely impaired color vision, CDS also encompasses some diseases of lesser severity, such as color weakness, oligocone trichromacy (OT) and bradyopsia (Aboshiha et al., 2016). The boundary between color blindness and color weakness is not very clear, and some patients are close to colorblindness when the light is dim. Color weakness includes panchromatic weakness, red-green color weakness and blue-yellow color weakness.
Patients with panchromatic weakness display a reduced level of color vision impairment in contrast to those with achromatopsia, without any associated abnormal vision or complications. While the ability to perceive colors under bright lighting conditions is preserved, distinguishing between them becomes challenging in low-light settings.
The red-green color weakness (dyserythrochloropsia), including the red color weakness (protanomalia) and the green color weakness (deuteranomalia), affects the patient’s ability to distinguish red and green. In dim lighting, the patient is close to red-green color blindness, while in bright lighting, the patient is close to normal. The prevalence rate of protanomalia is approximately 1.08%, while the prevalence rate of deuteranomalia is approximately 4.63% (Salih et al., 2020).
The blue color weakness (tritanomalia), similar to the red-green color weakness, and the ability to distinguish blue is poor. The patient is close to blue-yellow color blindness when the light is dark, and close to normal when the light is bright. The prevalence rate is about 0.2% (Salih et al., 2020).
Currently, the rate of color blindness is approximately 5% for male sand 0.7% for females. The frequency of color blindness gene carriers is estimated to be 8.98%. Among them, the prevalence of total color blindness is estimated to be 1 in 30,000 (Kohl and Hamel, 2013), the prevalence of red blindness is approximately 1.01%, the prevalence of green blindness is approximately 1.27%, and the prevalence of blue-yellow blindness is approximately 0.2% (Salih et al., 2020).
OT is characterized by severe impairment of cone function on ERG assessment coupled with normal or near-normal color discrimination. It was first described in 1973 by Van Lith, who reported a boy that, despite his poor vision and reduced photopic ERG responses, had nearly normal color vision. This was hypothesized to be due to a low number of normal functioning cones (from the Greek oligos for ‘few’), which retained their normal distribution proportions between the three cone types, hence preserving trichromatic vision.
Bradyopsia is a rare condition that affects vision. The term “bradyopsia” is from the Greek words for slow vision. In affected individuals, the eyes adapt more slowly than usual to changing light conditions. Bradyopsia was first reported in 1991 in four Dutch patients, who demonstrated an abnormally long interval of suppression in their ERG amplitude responses to the second of a pair of bright stimuli flashes. This was postulated to be due to a deficit in the normally fast regeneration of the visual pathway signaling processes. The term bradyopsia (Greek for slow vision) was devised in 2004 to describe this stationary retinal phenotype, wherein affected patients had difficulty in adapting to sudden changes in cone-mediated luminance levels and difficulty in seeing moving objects (Aboshiha et al., 2016).
It has been suggested that people with color vision disorders have relevant advantages in nighttime vision tasks (Chapanis, 1947). Yet this assumption clearly does not stand up to close scrutiny (Simunovic et al., 2001). Color vision disorders not only affects the patient in daily life, such as the inability to recognize traffic lights and an inability to see the colorful world, but also has a negative impact on the patient’s employment, making them unable to engage in aviation, navigation, engineering, and military fields of work (Holroyd and Hall, 1997). Therefore, it is necessary to diagnose color vision disorders as early as possible (Hathibelagal, 2022).
In order to rescue the vision of patients with color vision disorder, it is an urgent task to solve the pathological mechanism of the color vision disorder and find a correct and effective treatment. This paper briefly summarizes the progress in related fields.
Patients with achromatopsia have complete loss of cone function, loss of color vision and poor vision (<0.1), accompanied by nystagmus, photophobia (Simunovic and Moore, 1998) and eccentric fixation (Sundaram et al., 2014). Hyperopia is common (Wissinger et al., 2001; Kelly et al., 2003) and there are extensive reports of refractive errors (Haegerstrom-Portnoy et al., 1996). However, the rod cells of patients with achromatopsia have normal function and a faster dark adaptation ability than in normal subjects. Fundoscopy examination is normal in most cases, but there are exceptions (Goodman et al., 1963; Khan et al., 2007), and findings of fundus pigmentation, small macular granularity, and macular defects have also been reported (Khan et al., 2007).
As genetic technology continues to advance, our comprehension of achromatopsia deepens. Furthermore, newly emerging technologies and equipment offer enhanced support for researching, diagnosing, and treating ACHM, including the implementation of optical coherence tomography (OCT) and adaptive optics scanning laser ophthalmoscopy (AOSLO). OCT has the capability to assess the retinal structure in vivo through the outer hyperreflective bands that correspond to various aspects of photoreceptor cells. Despite this, traditional OCT devices lack the lateral resolution necessary to differentiate individual photoreceptors (specifically, the structure of rods and cones). Therefore, OCT only provides a general assessment of photoreceptor structure in ACHM. However, with AOSLO, we can capture non-invasive images of rods and cones with cellular level resolution.
As a result of advancements in technology and equipment, additional symptoms linked to pathogenic mutations have been uncovered in addition to the typical ones. Certain studies propose that GNAT2 mutations can permit some ACHM patients to retain portions of their color vision (Michaelides et al., 2003; Rosenberg et al., 2004). A study suggested that the retention of color vision may be related to variants that result in some functional protein products (Rosenberg et al., 2004). Although ACHM is traditionally deemed a disease that remains stable throughout one’s lifetime, recent research indicates that patients with CNGA3 (Thomas et al., 2012), CNGB3 (Thiadens et al., 2010; Thomas et al., 2012), and PDE6C (Thiadens et al., 2010; Georgiou et al., 2019) variants have evidence of progressive cone cell involvement, that even affect rod cells. Subsequent studies have also demonstrated that, in addition to ACHM, mutations in PDE6C can cause cone dystrophy (CD) and cone-rod dystrophy (CRD; Thiadens et al., 2009; Grau et al., 2011; Weisschuh et al., 2018). Full field ERG showed severe cone system dysfunction, but shortwave sensitivity was relatively preserved, similar to another rare form of ACHM (GNAT2-ACHM; Michaelides et al., 2003) and similar to blue cone monochromatic lesions (Gardner et al., 2014). Myopia and slowly progressive maculopathy are common features among patients with PDE6C mutations (Georgiou et al., 2019). In ACHM patients caused by CNGA3 or CNGB3 mutations, the ellipsoid zone (EZ) band (also known as the inner segment/outer segment [IS/OS] band) was disrupted or deleted in approximately 70% of cases (Sundaram et al., 2014; Langlo et al., 2016). Furthermore, foveal hypoplasia with minimal foveal pit formation has been observed in all subjects with ATF6-ACHM to date (Kohl et al., 2015; Xu et al., 2015). And relevant research data show that ACHM caused by ATF6 gene mutation has almost no vertebral structure and fewer targets for cone gene therapy (Mastey et al., 2019).
Currently, six genes that cause achromatopsia have been discovered, including CNGA3, CNGB3, GNAT2, PDE6C, PDE6H and ATF6 (Felden et al., 2019). Among them, CNGA3, CNGB3, GNAT2, PDE6C and PDE6H all encode cone cell-specific proteins. Over 70% of ACHM cases are caused by mutations in CNGA3 and CNGB3 genes with the proportion of ACHM cases caused by CNGB3 mutation reached 50% (Kohl et al., 2005; Mayer et al., 2017; Georgiou et al., 2020; Brunetti-Pierri et al., 2021). Mutations in the other four genes account for less than 6%–8% of cases (Felden et al., 2019; Georgiou et al., 2021). The remaining 24% of unexplained cases may be due to undetected pathogenic genes or unidentified mutations (Weisschuh et al., 2018, 2020a; Brunetti-Pierri et al., 2021; Figure 2).
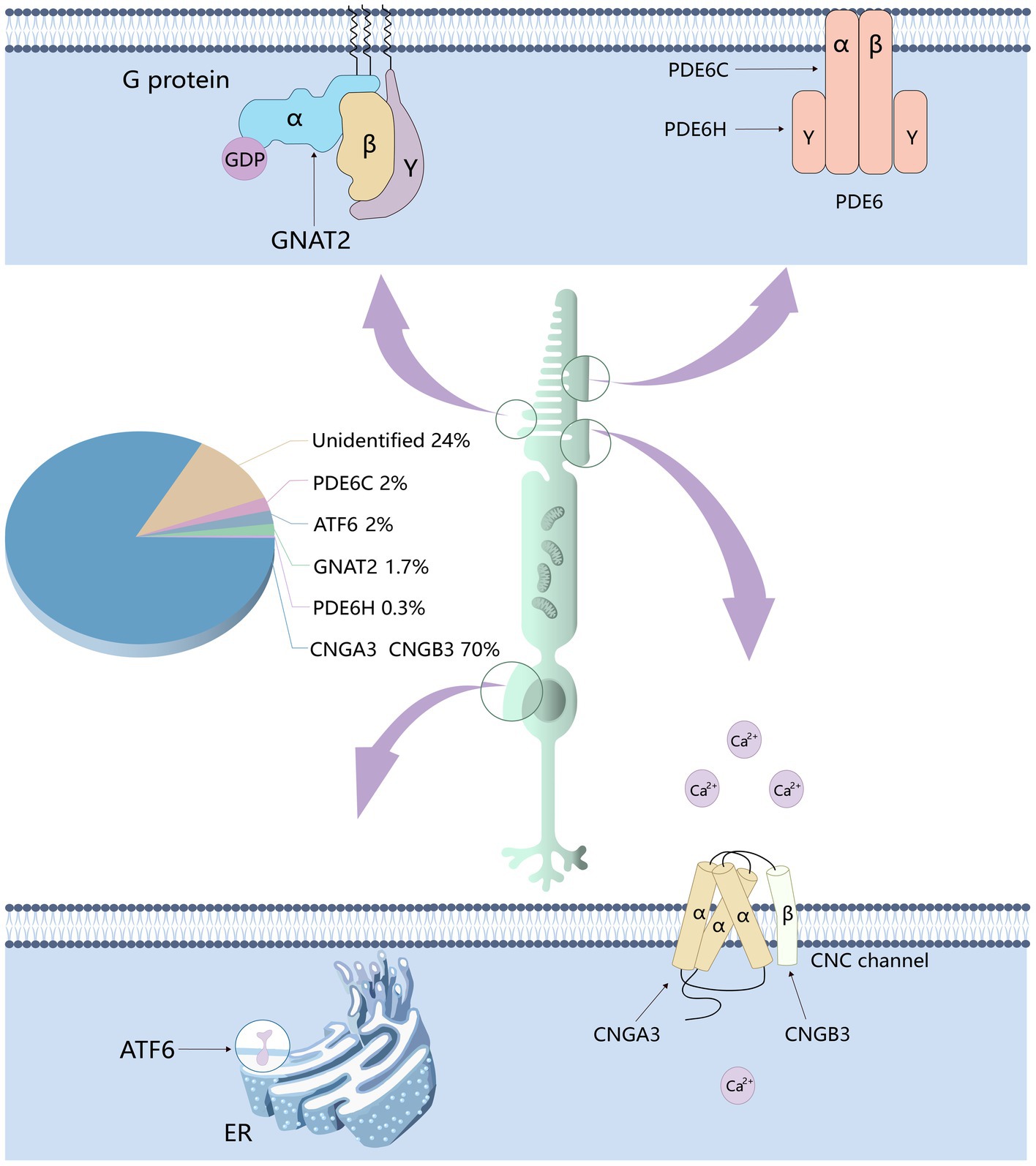
Figure 2. The genes responsible for achromatopsia and the proportion of cases thereof. Six genes related to achromatopsia have been identified: CNGA3, CNGB3, PDE6C, PDE6H, GNAT2, and ATF6. CNGA3 and CNGB3 encode the α and β subunits of CNG channel respectively, which affect the flow of calcium ions. PDE6C and PDE6H encode the α and γ subunits of PDE respectively, which affect the concentration of cGMP. GNAT2 encodes cone cell specificity G protein α subunit, which affects the activity of PDE.
Rod cells and cone cells consist of four primary structural and functional regions: outer segment, inner segment, cell body and synaptic terminal (Figure 1B). The outer segment is densely packed with a dense stack of membrane disks that are spaced at intervals of approximately 28 nm intervals. These disks contain the visual pigments (rhodopsin in rod cells and cone pigment in cone cells) and other transduction components, either as transmembrane or peripheral membrane proteins. Visual pigment is the most abundant protein in the outer segment. An intriguing variation between rods and cones is that the rod disks (excluding nascent disks at the base of the outer segment) are entirely internalized and thus physically separated from the plasma membrane, whereas the cone disks remain as the folding of the plasma membrane. The open cone-shaped discs provide a greater surface area, enabling speedy transfer of substances between the exterior and interior of cells. This facilitates the efficient transfer of chromophores for pigment regeneration and swift calcium dynamics during light adjustment (Fu and Yau, 2007).
Rod cells and cone cells undergo continuous renewal of their outer segments (Young, 1967; Young and Droz, 1968). The newly formed disk at the base of the outer segment gradually pushes the previously formed disk toward the top. The intersegmental disk, situated at the apex of the lateral segment, sheds daily and is phagocytized by adjacent retinal pigment epithelial cells (RPE; Young and Bok, 1969). The rate of disc formation and shedding is roughly equivalent, thereby ensuring a relatively constant outer segment length in the adult retina.
The inner segment contains a substantial amount of endoplasmic reticulum (ER) and Golgi apparatus. To satisfy the elevated metabolic energy requirements related to phototransduction, a significant number of mitochondria are present. All proteins that travel to the outer segment must traverse the narrow channel between the outer segment and the inner segment.
The synaptic end converts light signals into electrical signals and transmits them to subsequent neurons in the retina: bipolar cells and ganglion cells. In the dark, a stable inward current (“dark current”) is generated by the influx of cations on the outer segment membrane, which depolarizes the rod or cone and maintains the stable release of glutamate from the synapse. The light signal stops the influx of cations (“photosensitive” conductance, consisting of cGMP-gated channels) to stop the dark current and produce membrane hyperpolarization. This hyperpolarization reduces or terminates the release of glutamate. The signal is then further processed by other neurons in the retina before being transmitted to the higher center of the brain.
Human vision of sunlight and color depends on three types of retinal cone photoreceptors with complete functions. However, cone cells are not only responsible for providing color vision, they are also responsible for mediating high acuity spatial vision, achromatic black-white vision (Neitz and Neitz, 2017) and central visual acuity (Kohl et al., 2002). Cone cells sensitive to short wavelengths (blue), medium wavelength (green), and long wavelength (red), namely S-cones, M-cones and L-cones, express visual pigments (cone proteins) that are sensitive to specific wavelength of light, respectively, with peak spectral sensitivity of 419, 531, and 560 nm (Dartnall et al., 1983; Figure 1C). But different classes of cones respond to a wide range of wavelengths of light, so they have overlapping sensitivity curves (Simunovic, 2010). The cone pigment is composed of 11-cis retinal and opsin. All eutherian mammalian cone pigments share the same 11-cis retinal chromophore (Wald, 1968). Binding of opsins to chromophores red-shifts the absorption spectrum of the chromophore, and differences in the amino acid sequence between opsins are responsible for the different spectral characteristics of each cone pigment (Wald, 1968; Kosower, 1988; Chen et al., 1989). The cone cells respond very quickly to changes in light. Even in dim light (when the response is slowest), only 20 ms is needed to reach the peak response of the cone to the superimposed flash (van Hateren and Lamb, 2006). As the background intensity increases, the response of the cones becomes faster, and in very bright light, the photopic vision system can detect flickers of more than 100 Hz in the surrounding retina (Tyler and Hamer, 1990). In the human retina, L-cones and M-cones constitute approximately 95% of the total cone population. They are densely packed in a hexagonal pattern in the central fovea, the foveola. The S-cone is located in the periphery of the retina and is not present in the fovea in humans (Curcio et al., 1991; Mustafi et al., 2009). Color recognition depends on the differential excitation of cone pigments by light stimulation of specific wavelengths and the correct processing of receptors after stimulation. Color vision impairment occurs when the function of one or more cone photoreceptors is lost or altered (caused by mutations, deletions, or rearrangements of opsin genes).
Total color blindness is a rare autosomal recessive retinal disease caused by the complete loss of the function of the three cone pigments, also referred to as rod monochromatism. In addition to ATF6 (activating transcription factor 6), CNGA3, CNGB3, GNAT2, PDE6C, and PDE6H genes all encode the phototransduction cascade components of cone cells, which are responsible for converting light signals into electrical and calcium signals (Michalakis et al., 2022). Mutation of any one of these genes will result in the paralysis of the entire phototransduction cascade system.
The CNGA3 (ACHM2, OMIM600053) gene is located on chromosome 2 and encodes the alpha subunit of CNG channel (Cyclic nucleotide gated ion channel) in cones (Table 1). The CNGB3 (ACHM3, OMIM605080) gene is located on chromosome 8 and encodes the beta subunit of the CNG channel (Michalakis et al., 2017) in cones. CNGB3 is the major ACHM pathogenic gene in Europe and North America (Weisschuh et al., 2020b). CNG channels are heterotetramers composed of alpha subunit and beta subunit. They are the key component of the cone cells phototransduction cascade assembly. Although recent studies have shown that ACHM due to CNGA3 or CNGB3 mutations cannot be clinically distinguished, they are quite different (Kohl et al., 2002). Previous studies have shown that CNGA3 is an ion conduction subunit and the CNGA3 homologous channel is fully functional in heterologous expression systems (Gerstner et al., 2000; Faillace et al., 2004). However, when the CNGB3 is expressed alone, it cannot form functional homologous channels in the heterologous expression system (Chen et al., 1993; Gerstner et al., 2000). Therefore, the beta subunit is only considered to confer specific biophysical properties of the CNG channel complex and to regulate the function of the cone CNG channel, while the alpha subunit is considered to be the main subunit (Kaupp and Seifert, 2002; Schön et al., 2013). Related researches also point out that most mutations in CNGA3 are missense mutations (Kohl et al., 1993; Biel and Michalakis, 2009; Sun and Zhang, 2019), causes the CNG channel to lose its function. The working mechanism of CNG channels is that the high concentration of cGMP outside the photoreceptor keeps the CNG channel open to cation influx in the dark, while light stimulation leads to a decrease in cGMP level, and subsequently the CNG channel closes. This closure terminates the stable inward current, thereby generating a membrane hyperpolarizing signal that reduces glutamate release from photoreceptor synapses (Müller and Kaupp, 1998). Some studies have pointed out that the degeneration of cone cells caused by spontaneous CNG channel activity may be the pathogenic factor of color blindness (Zheng et al., 2022).
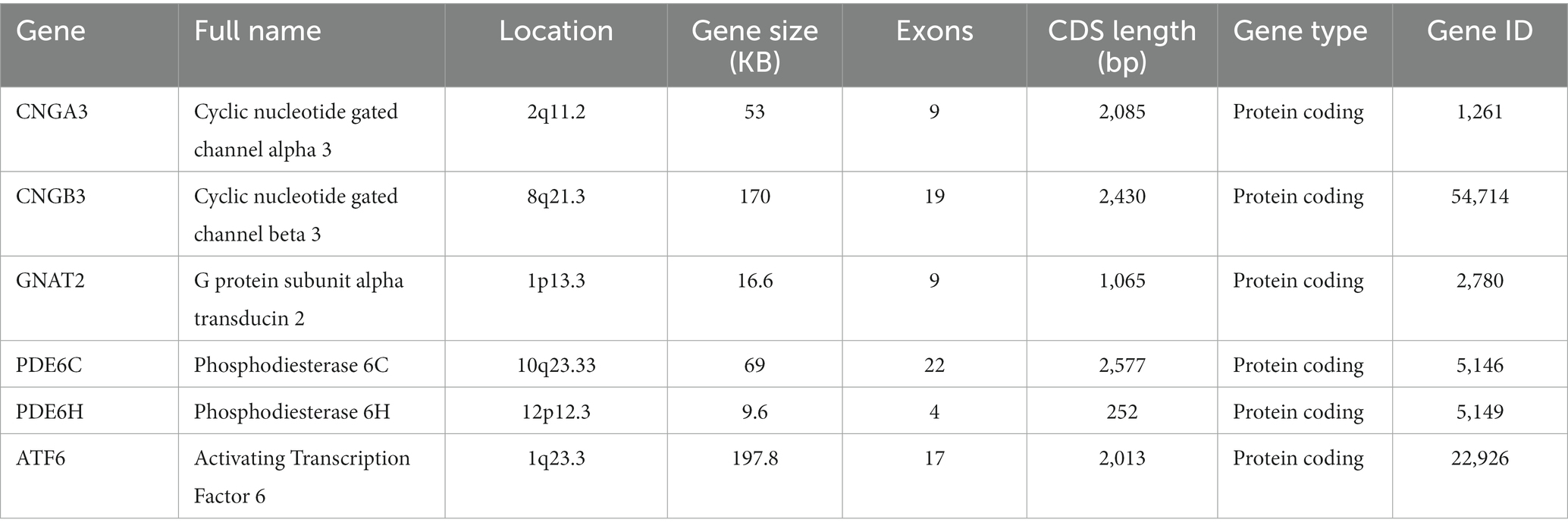
Table 1. ACHM pathogenic genes.
The GNAT2 (ACHM4, OMIM139340) gene is located on chromosome 1 and encodes cone cell specificity alpha subunit, a heterotrimeric G protein coupled to cone cytochrome in the outer segment of the cone photoreceptor. The beta and gamma subunits interact with photoactivated photopigments to exchange GTP with GDP, thereby releasing the α-subunit. The activated GTP • transduction complex binds and activates phosphodiesterase, which subsequently hydrolyzes cGMP, thereby effectively reducing its intracellular concentration. This leads to the closure of cGMP-gated channels, followed by membrane hyperpolarization (Stryer, 1991). All GNAT2 mutations identified to date result in premature translation termination and protein truncation at the carboxyl terminus (Aligianis et al., 2002; Kohl et al., 2002). A recent study by Felden et al. pointed out that the prevalence of ACHM caused by GNAT2 mutations was estimated to be 1.7% in a cohort of 1,116 independent ACHM families (Felden et al., 2019; Figure 3).
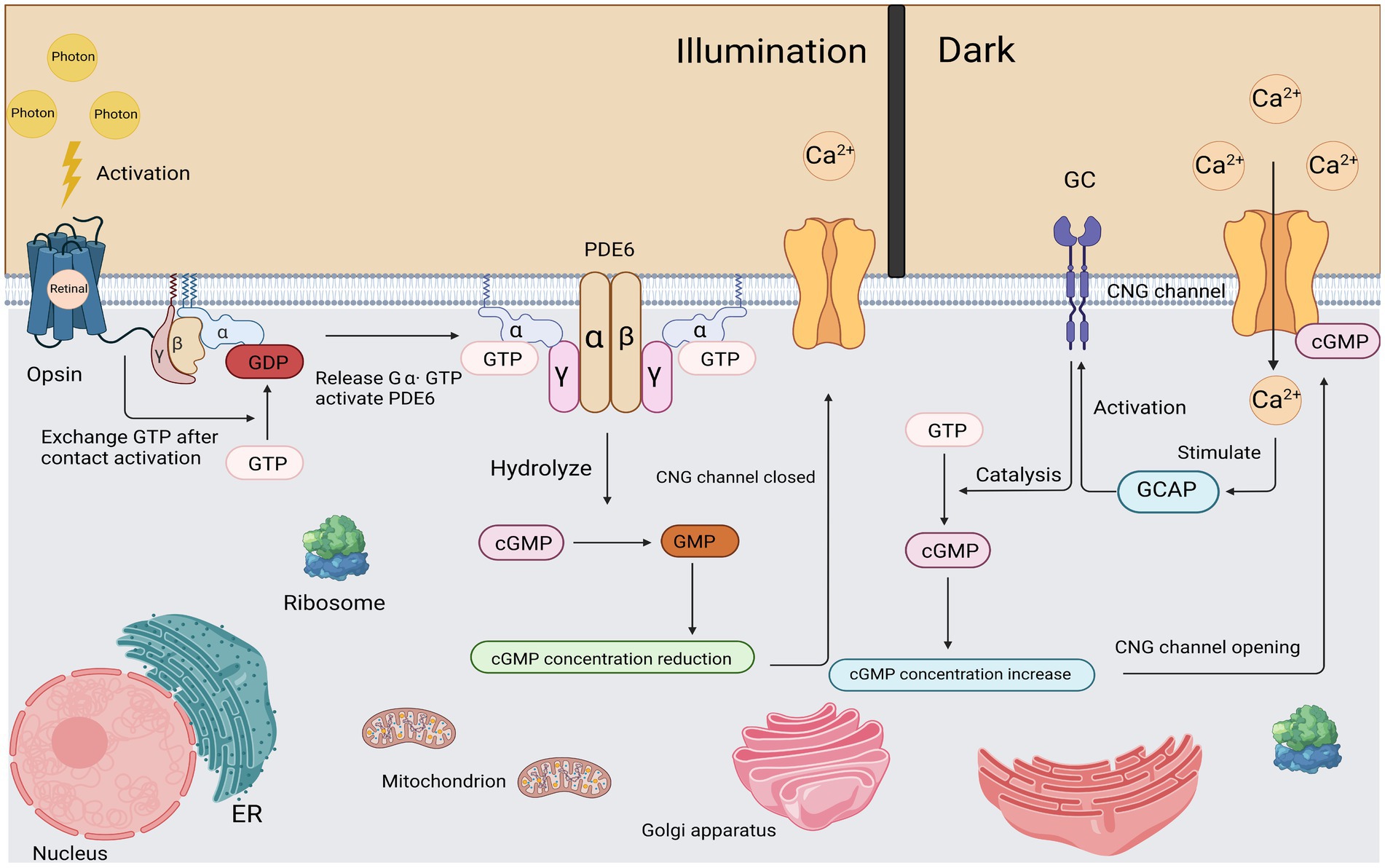
Figure 3. Light activation and recovery of cone cells. After corresponding stimulation, Gα subunit was released from Gβγ subunit and the exchange between GDP and GTP was completed. The subsequently activated Gα subunit binds to the γ subunit of PDE and causes it to contract to complete the activation of PDE. Activated PDE begins to hydrolyze cGMP efficiently. As the cGMP concentration decreases, the CNG channel closed. In a dark environment, a certain concentration of cGMP maintains the opening of CNG channels.
PDE6C (ACHM5, OMIM600827) is located on chromosome 10 and encodes the catalytic alpha subunit of pyramidal phosphodiesterase (PDE; Uzunov and Weiss, 1972). PDE is the third component of the vertebrate phototransduction system, and is a tetrameric protein consisting of two equally active catalytic subunits α and β and two identical inhibitory subunits γ (Baehr et al., 1979; Hurley and Stryer, 1982). In the dark, two γ subunits act as inhibition subunits by binding to two catalytic subunits and preventing the hydrolysis of cGMP. PDE is anchored to the disk membrane by isoprenylation of the C termini of the two catalytic subunits (Li et al., 1990; Catty et al., 1992; Qin et al., 1992). It was reported that PDE* hydrolyzes cGMP at a rate close to the limit set by aqueous diffusion, with a Km of ~10 μM and a Kcat of 2,200 s−1 (Leskov et al., 2000). After the initiation of phototransduction, transducing proteins shrink their inhibitory γ subunit activates PDE6C. Followed by the last step of the cascade, the hydrolysis of cGMP by active PDE6C leads to the closure of the extrapyramidal cation channel. Then hyperpolarization of the photoreceptor membrane was performed (Zhang and Cote, 2005). The PDE6C gene mutation leads to its loss of function, which reduces the hydrolysis level of cGMP in cells, thereby causing the cGMP level to rise. The increase of cGMP level mainly leads to the excessive opening of cGMP-gated channels in the extrapyramidal segment, resulting in an unrestricted inflow of Ca2+. CGMP gated channel belongs to the family of cyclic nucleotide gated (CNG) channels and is a non-selective cationic channel (Kaupp and Seifert, 2002). This channel is located on the plasma membrane of photoreceptor cells and is the last component in the activation stage of phototransduction. In the dark, the basal concentration of one to several micromoles of cGMP keeps a small number of CNG channels open (Yau, 1994). The drop in cGMP concentration upon exposure to light causes the channel to close rapidly with a sub-millisecond delay (Karpen et al., 1988). Related studies have shown that uncontrolled increased concentrations of cytoplasmic cGMP and Ca2+ ions can lead to the death of cones (Iribarne and Masai, 2017). A recent study by Weisscheh et al. estimated that the prevalence of PDE6C mutations was 2.4% in a cohort of 1,074 independent ACHM families (Weisschuh et al., 2018). Studies of ACHM caused by PDE6C mutations have shown conflicting results, and Thiadens et al. concluded in a cohort of 5 PDE6C patients (20%) that ACHM is usually a progressive disease (Thiadens et al., 2010). However, Hirji et al. concluded in a cohort of one PDE6C subject (2%) that ACHM is mainly a quiescent disease (Hirji et al., 2018). This issue requires further research.
PDE6H (ACHM6, OMIM601190) is located on chromosome 12 and encodes the pyramidal cell phosphodiesterase inhibitory γ subunit (Kohl et al., 2012). The PDE6H mutation functionally means that PDE activity is continuously increased, cGMP levels in the outer cone segment are decreased, and cGMP gated channels are permanently closed, which is similar to permanent light stimulation (Lisman and Fain, 1995). One study estimated a prevalence of 0.3% in 680 independent ACHM cohorts (Kohl et al., 2012).
ATF6 (ACHM7, OMIM605537) is a widely expressed gene located on chromosome 1 that encodes cyclic AMP-dependent activating transcription factor-6 alpha that plays a key role in unfolded protein response (UPR) and endoplasmic reticulum homeostasis (Walter and Ron, 2011). ATF6 is located at the autosomal recessive cone-rod dystrophy (CORD8) locus, which was previously mapped to the 11.53 cM region on chromosomes 1q12-q24 in a Pakistani family (Khaliq et al., 2000; Ismail et al., 2006). As one of the three transmembrane proteins that regulate the UPR, ATF6 is activated upon ER stress, transcriptionally upregulates ER chaperones and ER protein folding enzymes, helping to alleviate ER stress and restore cellular homeostasis (Walter and Ron, 2011; Chan et al., 2016; Wang and Kaufman, 2016). When cones trapped in ER stress due to excessive light damage, ATF6 mediates early UPR to relieve ER stress and restore homeostasis (Nakanishi et al., 2013). Additionally, ATF6 also activates IRE1 (Inositol requiring enzyme-1), which mediates the late UPR, during which the activation of ATF6 decreases, and severe or sustained ER stress can lead to apoptosis (Lin et al., 2007; Nakanishi et al., 2013). Apoptotic cell death caused by ER stress has been recognized as a common pathway in retinal degenerative diseases, including ACHM (Thapa et al., 2012). Pathogenic sequence variants in the ATF6 gene cause dysfunction of this critical signaling pathway. As one of the most active cells in human metabolism, this is particularly harmful to photoreceptor cells (Sung and Chuang, 2010; Wong-Riley, 2010). A recent study showed that the ATF6 pathway is essential for color vision not only within photoreceptors, but also in the inner layer of the retina and is a potential target for the treatment of inherited or acquired retinal diseases (Ansar et al., 2015). Research shows that the proportion of ACHM cases caused by ATF6 mutation is less than 2%.
Due to the scarcity of cone cells and the fragility of COS (cone outer segment), as well as a number of other reasons, the phototransduction mechanism of cone cells cannot be accurately described (Fu and Yau, 2007). However, its mechanism is generally similar to the phototransduction mechanism of rod cells, and there is nothing inherently different between the processes of activation of the cone and rod phototransduction (Astakhova et al., 2015). Here, we take the phototransduction mechanism of rod cells as an example:
First, activation of the Rhodopsin, a photon of light is absorbed by the rhodopsin molecules in the outer segments of the rods, isomerizing the 11-cis retinal (the chromophore of cone and rod cells) to all-trans retinal, thereby inducing a conformational change in the structure of the rhodopsin molecule, becoming the activated form R*.
Second, activation of the G-protein. All proteins move within (or on the surface) of the disc membrane, so they diffuse laterally; thus, activated R* and inactivated G contact and bind instantaneously. In this R*-G state, the GDP molecule bound to the G protein alpha subunit (Gα) is released, allowing the GTP molecule in the cytoplasm to bind to its place. This process, called nucleotide exchange, activates the G-protein (forming G-GTP), which then dissociates from the R*. Most importantly, the activated rhodopsin (R*) is not altered in any way by this interaction, so it can come into contact with another G protein molecule by diffusion, triggering nucleotide exchange again. While R* remains active, this process can be repeated indefinitely, so R* can be viewed as an enzyme that catalyzes the activation of numerous molecules of G proteins. Activated G protein is denoted as G*.
Third, activation of the PDE. As a result of lateral diffusion, G* contacts the PDE and binds to one of its two regulated γ subunits, thereby partially activating the PDE in the form of PDE* that we denote. Subsequently, the second G* can bind to the second γ-subunit to fully activate the PDE. Unlike step 2, this activation does not have an amplification effect: a single G* can only activate at most one PDE catalytic subunit.
Fourth, hydrolysis of Cyclic GMP (cGMP). In the dark resting state, there is a stable balance between cyclic GMP synthesis by guanylate cyclase (GC) and the slow hydrolysis of cyclic GMP by normal PDE, so there is a stable, moderate and appropriate cytoplasmic concentration of cyclic GMP (a few micromoles). Upon exposure to light stimulation, disc proteins are activated (Steps 1–3), and the resulting PDE* activation increases the rate of cyclic GMP hydrolysis, thereby decreasing cyclic GMP concentrations.
Finally, closure of Ion Channels. At its resting concentration in the dark, cyclic GMP binds to, and holds open, a proportion of the ion channels in the cell’s plasma membrane. Although this proportion is quite small (typically just a few percent), the number of open channels is sufficient to conduct the substantial cation current (tens of pA) that flows into the outer segment in darkness. When the concentration of cyclic GMP drops in the light, cyclic GMP unbinds from the channels, causing the channels to close, and thereby generating the cell’s electrical response—a reduction in the circulating current and a consequent hyperpolarization (Lamb and Pugh, 2006; Figure 3).
While the underlying mechanisms exhibit a broad similarity, notable distinctions exist. Firstly, in cones, it is the cone visual pigment rather than rhodopsin that receive incoming light signals. Secondly, in vitro biochemical analyses have consistently demonstrated that the activation rate of cone visual pigment is 2–5 times slower compared to rhodopsin. It has been confirmed in humans (Vissers et al., 1998), mice (Kojima et al., 2014) and chickens (Imai et al., 1997; Kojima et al., 2014). Thirdly, the fast quenching of cone R* is ensured by phosphorylation via cone-specific rhodopsin-kinase (G protein coupled receptor kinase 7, GRK7), whose level of expression and specific activity are substantially higher than those of GRK1 in rods (Rinner et al., 2005; Tachibanaki et al., 2005; Wada et al., 2006; Arinobu et al., 2010; Vogalis et al., 2011). Fourth, rod cells can respond to a single photon, whereas cone cells need to be stimulated by more than a hundred photons. Fifth, in the rod outer segment (ROS), the disc membrane is separated from and surrounded by the plasma membrane. Instead, the cone outer segment (COS) is formed by a stacked invagination of the plasma membrane (Mustafi et al., 2009). As a result, the rod discs are isolated from the extracellular space by the plasma membrane, while the cone discs are open to the extracellular matrix. The structure of the outer segment of the cone greatly increases the surface area of its disc membrane and the surface-to-volume ratio of the cone. The continuous and open structure of the outer segments of the cones facilitates the rapid response of light transmission and metabolism in the cones, and ensures the rapid transport of substances between the cones and the interphotoreceptor matrix (IPM; Yau, 1994). Sixth, the chromophore quenching and regeneration rate of cone cells are faster than rod cells (Wang and Kefalov, 2011).
To put it succinctly, the process of light transduction in cone cells can be described as follows: Upon exposure to light, the cone visual pigment molecules within the cells are stimulated. This stimulation activates the transducing protein α subunit (GNAT2 = Gα). At the guanosine binding site, guanosine diphosphate (GDP) is exchanged for guanosine triphosphate (GTP), resulting in the release of the inhibitory βγ subunit and the formation of the activated form of the G protein (G α-GTP). Activated Gα-GTP leads to a significant increase in phosphodiesterase (PDE) activity. PDE is responsible for hydrolyzing cyclic guanosine monophosphate (cGMP) and effectively reducing its concentration in the outer segment of the cone cell. The decrease in cGMP concentration causes the closure of heterotetrameric cGMP-gated cation channels (CNGA3/CNGB3), resulting in membrane hyperpolarization of the cone cell. The membrane hyperpolarization leads to a decreased release of the excitatory neurotransmitter, glutamate. Finally, the cone photoreceptor cells recover from the light response through a series of quenching/terminating reactions of the activated phototransduction proteins, which ultimately restore the cells to a dark-adapted state. These intricate steps ensure efficient light detection and signal processing in cone photoreceptor cells.
The genes responsible for coding the red cone protein and green cone protein are located on the X chromosome at Xq28, encode the protein components of the light-sensitive photopigments that are specifically expressed in the L cones and M cones (Neitz and Neitz, 2021), with mutations leading to red-green color blindness. Patients with red-green color blindness commonly experience have symptoms such as decreased vision, photophobia, nystagmus and myopia (Gardner et al., 2009). Despite female carriers being asymptomatic, confocal AOSLO has demonstrated variably reduced cone density, increased spacing, and disrupted organization, with phenotypic variability likely relating to random X-chromosome inactivation (Carroll et al., 2010).
The red-green color blindness is an X-linked recessive genetic disorder caused by mutations in two genes, OPN1MW (Opsin 1 medium wavelength gene, OMIM300821) and OPN1LW (Opsin 1 long wavelength gene, OMIM300822), resulting in a lack of long-wavelength photosensitive opsin protein and medium-wavelength photosensitive opsin protein (Table 2). In humans, OPN1MW and OPN1LW genes are linked together on the X chromosome in a head-to-tail arrangement (Vollrath et al., 1988). Both OPN1LW and OPN1MW genes have 6 exons each (Nathans et al., 1986). OPN1LW and OPN1MW are almost identical to each other, with more than 98% nucleotide sequence homology. Because of their similarity, L and M opsin genes are prone to unequal homologous recombination, which has profound implications for visual function (Neitz and Neitz, 2011). Exons 1 and 6 are highly conserved with almost no changes (Winderickx et al., 1993). Exon 5 encodes amino acid differences that functionally separates the L and M photopigments (Neitz et al., 1991). Exons 2, 3 and 4 differ between L-opsin (red) and M-opsin genes (green). The selective expression of OPN1LW and OPN1MW is regulated by specific proximal promoters and a single upstream locus control region (LCR), ensuring that only one opsin gene is expressed in each cone photoreceptor (Wang et al., 1992; Yamaguchi et al., 1997; Smallwood et al., 2002). At present, there are four pathological mechanisms of red-green color blindness: the first is the partial or complete deletion of the locus control region, resulting in transcriptional termination of an opsin gene array (Nathans et al., 1989). The second is non-homologous recombination between long-wave opsin and medium-wave opsin gene arrays followed by inactivating mutations. The third is the deletion of a complete exon in the single opsin gene array (Ladekjaer-Mikkelsen et al., 1996). The fourth is the gene conversion with mutation transfer between OPN1LW and OPN1MW (Reyniers et al., 1995). In addition, in some cases, it is also associated with cone dystrophy (Gardner et al., 2010; McClements et al., 2013; Figure 4).
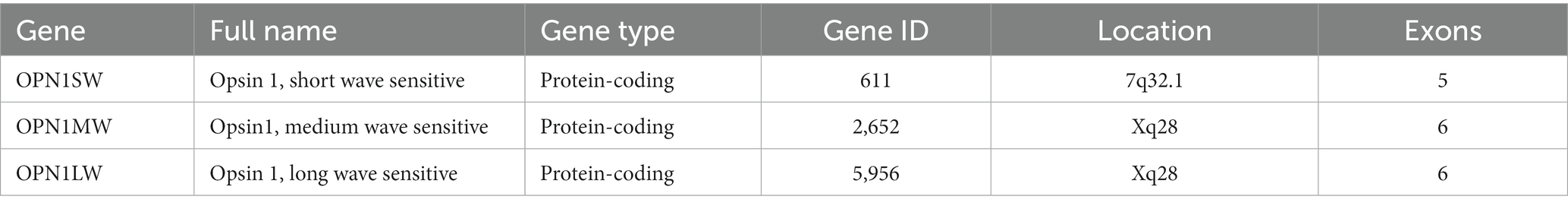
Table 2. Opsin genes.
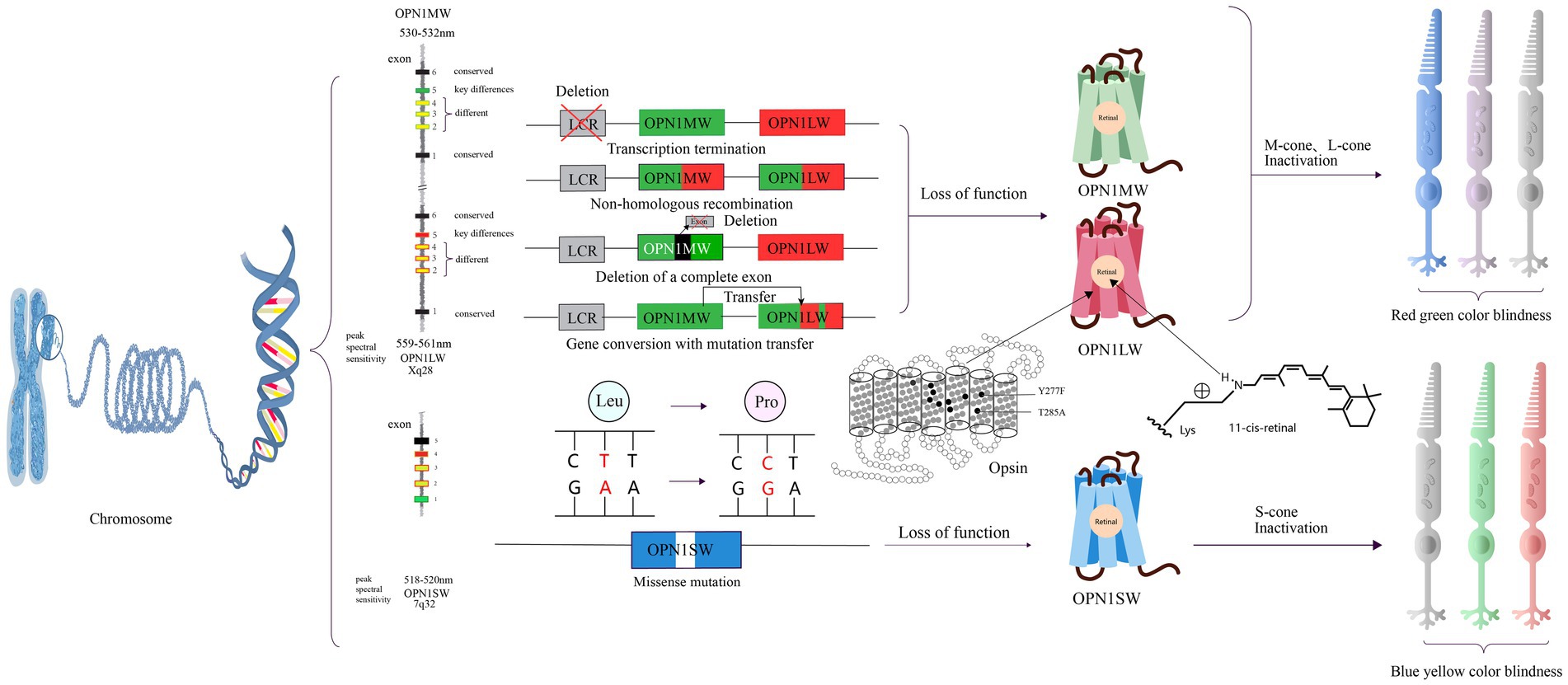
Figure 4. The pathogenic mechanisms of red-green color blindness and blue-yellow color blindness and opsin structures. There are four mutation patterns for OPNIMW and OPNILW, resulting in the loss of function of the corresponding visual pigment and causing red-green color blindness. For OPNI S W, there is only one missense mutation pattern, leading to blue-yellow color blindness. Y277F and T285A are amino acids that determine key differences between retinoids. The gray cone represents loss of function.
The gene responsible for encoding the blue cone protein is located on chromosome 7 at position 7q32. This gene, referred to as OPN1SW (Opsin 1 short wavelength gene, OMIM190900), has a nucleotide sequence similarity of only about 40% with the other two opsin genes. Mutations in OPN1SW lead to abnormal functionality of the blue cone protein, resulting in a condition known as blue-yellow color blindness. Affected individuals experience difficulty in effectively perceiving colors in the short wavelength range, but they can still distinguish colors in the medium and long wavelength regions.
Blue-yellow color blindness is a dominant genetic disorder with a simple pathology. Missense mutations in the gene encoding the blue cone opsin protein result in amino acid substitutions within the blue cone opsin sequence. As a consequence, individuals with these mutations experience blue-yellow color blindness (Weitz et al., 1992a, b). Other studies have suggest that blue-yellow color blindness could be linked to defects in OPN1SW splicing defects and haploinsufficiency (Neitz et al., 2020; Figure 4).
Currently, there are no pharmaceutical treatments or therapeutic methods exist for achromatopsia (Simunovic, 2010). The management of this condition primarily involves specialized genetic counseling to provide guidance and support to affected individuals and their families. Low vision devices may also be used as visual aids to assist with visual tasks. To alleviate symptoms of photophobia, individuals with achromatopsia may also benefit from wearing colored contact lenses or glasses (Kohl and Hamel, 2013). A study has reported that the use of red glasses or red contact lenses may provide relief for patients experiencing photophobia (Park and Sunness, 2004). Clinical trials have shown that these visual aids such as colored contact lenses or glasses are not fully effective in addressing the challenges related to color vision disorders (Martínez-Domingo et al., 2019).
However, it is noteworthy that the coding sequences of all genes associated with total color blindness are relatively small, typically less than 2,600 base pairs (BP). This compact size allows for their encapsulation within adeno-associated virus (AAV) gene therapy vectors, which holds promise for potential gene therapy interventions (Kotterman and Schaffer, 2014). Although AAV has inherent limitations as a gene delivery system, advancements in vector engineering offer the potential to develop improved recombinant AAV (rAAV) variants with enhanced delivery capabilities. These engineered rAAV vectors aim to overcome the size constraints and improve the efficiency of gene transfer, thereby expanding their therapeutic potential in various gene therapy applications (Michalakis et al., 2022). Currently, several studies have provided compelling evidence demonstrating the efficacy of gene-based or alternative therapies in restoring pyramidal function in animal models of achromatopsia (ACHM) with various genotypes (Alexander et al., 2007; Komáromy et al., 2010; Michalakis et al., 2010; Carvalho et al., 2011; Figure 5). Studies have reported that in cases of achromatopsia (ACHM) caused by GNAT2 mutations, cone cells exhibit a relatively well-preserved state. This finding suggests a potential broad therapeutic window for interventions targeted specifically at cone cells (Georgiou et al., 2020). In a study conducted on Gnatcpfl3 (Cone photoreceptor function loss-3) mice, the delivery of GNAT2 cDNA was shown to result in the restoration of cone function, as evidenced by improvements observed in photo-adapted electroretinogram (ERG) and optomotor response (OMR) tests (Alexander et al., 2007). Another study used AAV5 vector expressing human CNGB3 to treat two color blind dog models and successfully improved their response to maze tasks and functional vision (Komáromy et al., 2010). Among mammals, only primates have trichromatic vision. Therefore, these studies based on non-primate color blindness models still need to be further developed. And it is worth noting that the cone residues of ACHM caused by different pathogenic gene mutations are different, which cannot be generalized.
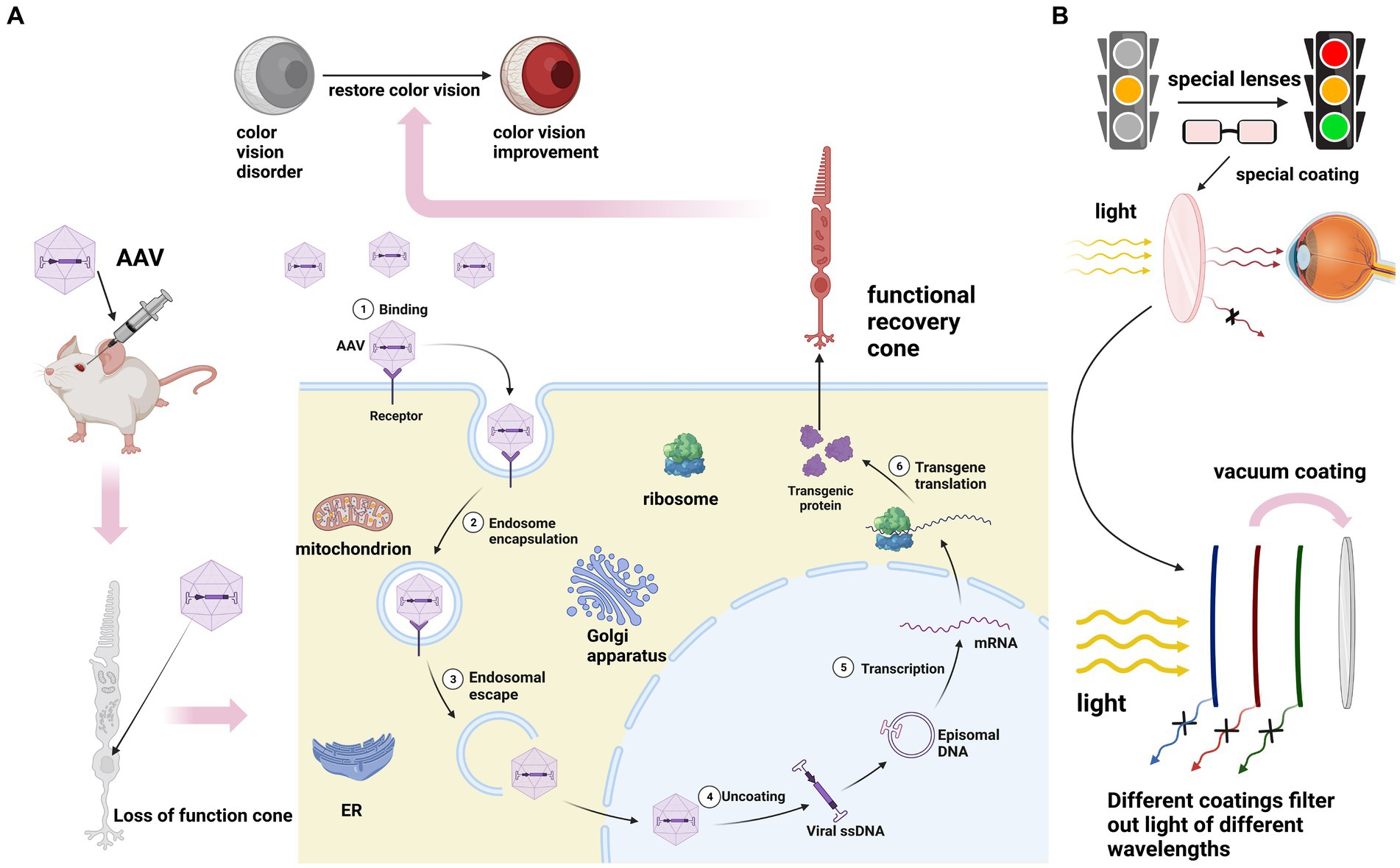
Figure 5. (A) The principle of gene therapy for ACHM. After intraocular injection of AAV, the exogenous normal genes carried by AAV replace the mutated genes to perform their functions. Transcription translation produces proteins with normal functions to save color vision. (B) The principles of physical therapy for red-green color blindness and blue-yellow color blindness involve applying special coatings on lenses to filter out specific wavelengths of light. It’s important to note that currently, all treatment methods for these three types of color blindness are still immature.
Interestingly, previous research has produced conflicting results. Specifically, several studies have demonstrated that the therapeutic window for gene therapy decreases with age (Komáromy et al., 2010; Carvalho et al., 2011). However, studies have indicated that a clear relationship between the degradation of retinal structure or function in individuals with ACHM and age has not yet been established. This implies that gene therapy may have a wider potential window for intervention in specific cases than previously assumed (Sundaram et al., 2014).
It is important to emphasize that in current research, there is a shift toward utilizing subretinal (SR) injection as a delivery method instead of intravitreal injection. This change is driven by the limitations of intravitreal injections in effectively targeting the outer retina and the increased risk of missing the desired location in the inner retina (MacLaren et al., 2016). Although gratifying progress has been made in gene therapy, safety studies on sheep and non-human primate models have shown that injection can lead to inflammation. However, it is reassuring to note that several gene therapies have demonstrated a generally acceptable safety profile, indicating the feasibility of their clinical application in the future (Seitz et al., 2017; Ofri et al., 2018; Reichel et al., 2018; Tobias et al., 2019).
Another research study has reported therapeutic effects of CNTF (ciliary neurotrophic factor) on both rod and cone photoreceptors in degenerative diseases. This finding highlights the potential of CNTF as a treatment option for preserving and promoting the function of photoreceptor cells affected by degenerative conditions (Lipinski et al., 2015). And it has been proved that CNTF has a neuroprotective effect on rod photoreceptor cells in several animal models (Wen et al., 2012). Recent studies have shown that CNTF can also produce protective effects on cone photoreceptors (Li et al., 2010; Talcott et al., 2011), and promote regeneration of the outer segments of degenerated cones (Li et al., 2010). These research findings have garnered significant attention from the scientific community. Currently, a phase I/II clinical trial (National Clinical Trial number: NCT01648452) is underway to evaluate the efficacy of CNTF treatment in patients with CNGB3 mutations. However, the trial results have indicated that CNTF did not lead to a substantial improvement in cone function. This suggests that there may be species-specific differences in the response of cone CNGB3 to CNTF, which highlights the need for further investigation and understanding of the underlying mechanisms in humans (Zein et al., 2014). Interestingly, the combined administration of CNTF has been shown to overcome a major limitation of gene therapy alone, which is the inability to achieve therapeutic effects in the majority of dogs over 1 year old (Komáromy et al., 2013). This promising finding highlights the potential of CNTF as an adjunct therapy in combination with gene therapy, suggesting a new avenue for therapeutic intervention in these cases.
ACHM caused by CNGA3 and CNGB3 mutations accounts for 70% of all cases, making them the most extensively studied genes. In fact, relevant progress has been made in North America, Europe and the Middle East, and the first phase of clinical trials for the treatment of color blindness with AAV carriers has been started, which are Germany (CNGA3, NCT02610582), the United States and Israel (CNGA3, NCT02935517), the United States (CNGB3, NCT0259922) and the United Kingdom (CNGB3, NCT03001310; Table 3). Several clinical trials have already yielded results:
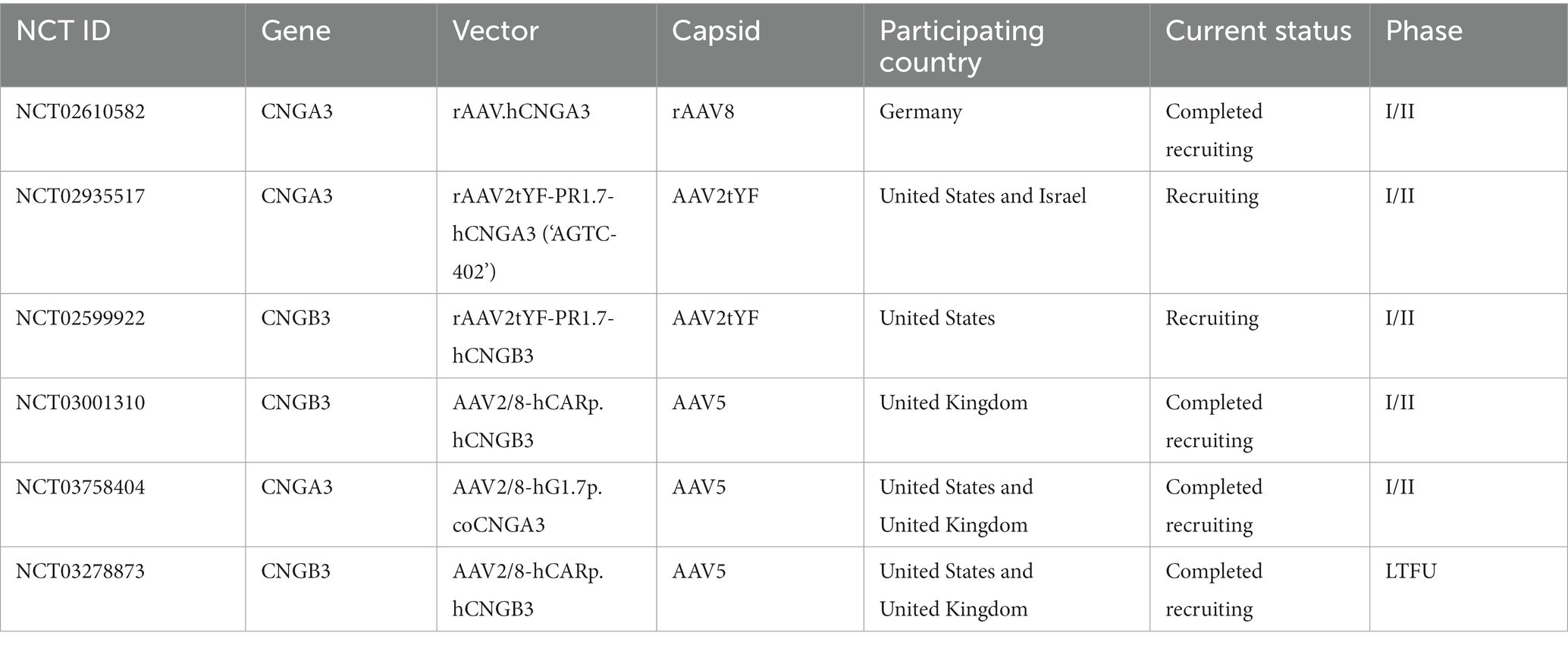
Table 3. AAV treatment project.
Following successful gene augmentation therapy in a naturally occurring ovine model (Banin et al., 2015; Gootwine et al., 2017), a phase I/II a human trial was initiated in some research centers in the United States (ClinicalTrials.gov, ID NCT02935517). In this trial, patients are treated with a single subretinal injection of an AAV2tYF capsid variant carrying the CNGA3 transgene under control of the engineered PR1.7 cone-specific opsin promoter in their worse eye. Although there is some improvement in the patient’s vision after 1 year of treatment, there is no evidence of improvement in color vision (McKyton et al., 2021).
In another study (ClinicalTrials.gov, ID NCT02610582), researchers obtained even more exciting results. This gene therapy study for achromatopsia found an excellent safety profile associated with subretinally delivered AAV8.CNGA3 and functional improvement in patients 1 year after treatment, although the absence of randomized concurrent control individuals precludes determining a cause-and-effect relationship. Improvement in functional variables, such as visual acuity, contrast sensitivity, and color vision, were noted despite the limited cohort size. In addition, patient-reported outcome measures provided additional support for a possible beneficial treatment effect. For example, the ability to differentiate colors improved after treatment with AAV8.CNGA3, as did measures for vision and identification of letters and numbers—key tasks in everyday life. As such, we found that targeting cone photoreceptors for gene supplementation with recombinant AAV8 can be applied safely and successfully. Of more importance, these data provide evidence that cone photoreceptor function can be gained in adult patients with complete achromatopsia (Fischer et al., 2020).
In a study to treat CNGB3-associated ACHM (ClinicalTrials.gov, ID NCT03001310), researchers achieved promising results. This is a phase 1/2, open-label, nonrandomized, dose-escalation/expansion study. In this trial, researchers treated 23 participants (11 adults, 12 children) with AAV8-hCARp.hCNGB3. It resulted in an adverse event (AE) profile that was expected and manageable. Overall, AAV8-hCARp.hCNGB3 demonstrated an acceptable safety and tolerability profile. There was no systematic pattern of change from baseline to week 24 for any specific efficacy assessment between the treated and untreated eye across the entire study population, within either cohort (adult or child), or with respect to dose. However, favorable changes were observed for individual participants across several assessments, participants’ performance improved after treatment on several efficacy assessments including PA (light sensitivity) and VRQoL (vision-related quality of life). Improvements in several efficacy parameters indicate that AAV8-hCARp.hCNGB3 gene therapy may provide benefit (Michaelides et al., 2023). The research results of other projects have not been announced yet.
Due to the low prevalence of ACHM caused by GNAT2 mutations, a limited number of studies have focused on phenotypic analysis despite advances in the field of gene therapy (Georgiou et al., 2020). The proportion of ACHM cases caused by PDE6C, PDE6H, and ATF6 mutations is relatively low, and therefore, the research focus on these genes is not as extensive as that of CNGA3 and CNGB3. Currently, there are no ongoing clinical trials specifically targeting these genes.
To date, there is still no effective treatment for red-green color blindness. Treatment approaches currently being explored can be broadly categorized into physical therapy and gene therapy.
In the realm of physical therapy, significant efforts are being directed toward the research and development of wearable devices. At present, there are special lenses that are covered with a variety of coatings to filter out specific wavelengths of light (Randhawa et al., 2010). These innovative lenses aim to enhance color perception and improve the overall visual experience for individuals with red-green color blindness. In addition, a kind of dyed contact lenses has been developed to improve patients’ color perception, which has the advantages of non-toxicity, low cost and customizability (Badawy et al., 2018). Another study showed that contact lenses made of gold nanoparticles (GNPs) and hydrogel matrix also have the function of improving color vision (Salih et al., 2021). At present, the leader in the color blindness glasses market is Enchroma, which first released its products in 2012. The glasses use a multi-notch filter to remove the overlap of red and green. The lens material used in this product is Trivex, which is lighter, thinner, and stronger than commonly used glass materials (CR-39 and polycarbonate), by coating the lens surface with a dye with a narrow absorption band to avoid affecting the patient’s normal ability to distinguish colors. However, one study tested its products using color assessment and diagnosis (CAD), and the results showed that the products did not significantly improve the symptoms of patients with color vision disorders. In addition to Enchroma, there are VINO, Colorlite and other companies engaged in the production of related products. Nevertheless, the current corrective eyeglasses available for red-green color blindness present several limitations. They tend to be expensive, bulky, and incompatible with other vision correction lenses. Furthermore, their effectiveness and stability in improving color perception are often unsatisfactory.
In the field of gene therapy, there is currently no mature and reliable drug or treatment available for red-green color blindness. However, certain research findings have shown promising indications for researchers in this field. For instance, a study demonstrated that adult primates with red-green color blindness, when supplemented with a third opsin, were able to achieve trichromatic vision. This suggests that trichromatic vision can be attained by introducing a single additional type of cone, without the need for early developmental processes. These findings offer a promising outlook for gene therapy in treating color vision disorders in adult humans (Mancuso et al., 2009). However, it should be noted that other studies have indicated that none of the conducted studies so far have definitively shown that the experimental animals have gained the ability to perceive new colors. Additionally, it should be noted that there is currently no evidence indicating that these experimental animals can differentiate between the colors red and green, despite their ability to detect both colors against a gray background (Cornelissen and Brenner, 2015). The findings of this study raise doubts regarding the efficacy of gene therapy in treating red-green color blindness, suggesting that it is premature to conclude that gene therapy can effectively address this condition in humans.
However, there is also encouraging news. Reports indicate that gene therapy has shown potential in rescuing the remaining M-cones in aged OPN1MW−/− mice (M-opsin knockout), offering hope for the treatment of color vision deficiency (Deng et al., 2019). In addition, a study provided encouraging evidence by demonstrating that the introduction of exogenous human opsin resulted in the regeneration of the outer cone segment and restoration of M cone function in OPN1MW−/− mice (Deng et al., 2018). The findings from these studies hold promise for the potential development of a cure for individuals with color vision disorders, particularly middle-aged and elderly patients.
Another study showed that the electroretinogram (ERG) function of M-cone was successfully restored in congenital S-cone monochromatic rat model by using a vector containing rat M-cone-opsin (Zhang et al., 2011). This result provides a direction for human treatment of color blindness. Recent studies have shown that there are still residual L-cone and M-cone at 1.5 mm in the center of the retina of red-green color blindness patients (Luo et al., 2015). This retinal structural evidence suggests that red-green color blindness deserves consideration for human clinical trials of L-cone and M-cone gene therapy (Cideciyan et al., 2013).
In addition to these scientific studies, certain technology companies, such as Google and Apple, have also taken initiatives to address the challenges faced by individuals with red-green color blindness. For instance, Google is developing an interface and application specifically designed to mitigate the negative impact of color vision impairment on individuals with red-green color blindness. Apple is committed to manufacturing end products for patients with red-green color blindness, improving the impact of red-green color blindness on patients’ daily life from a mechanical level.
There is no effective treatment for blue-yellow blindness. Clinical outcomes of assistive devices like glasses or contact lenses have generally been underwhelming. The relatively low prevalence of blue-yellow color blindness and its limited impact on patients, as S-cones only make up around 5% of total cone cells, have resulted in a scarcity of related research. Existing studies mainly focus on investigating the underlying pathological mechanisms rather than developing viable treatments. Therefore, there is an urgent need for researchers to address and find solutions for the treatment of blue-yellow color blindness.
Advances in various technologies have greatly enhanced our comprehension of the clinical symptoms and underlying pathological mechanisms of congenital color vision disorders. Nonetheless, there are still instances where the etiology of the disease remains inexplicable, and our grasp of the underlying cellular and molecular mechanisms is not yet complete. Further in-depth research by dedicated scientists and researchers is necessary to fill the knowledge gaps and expand our understanding of congenital color vision disorders.
Achieving cell-level resolution imaging holds paramount importance for future diagnosis and research in the field of congenital color vision disorders. Split-detection adaptive optics scanning laser ophthalmoscopy (AOSLO) imaging is a promising tool for objectively visualizing and assessing the structure of living cells. The high-resolution capabilities of split-detection AOSLO imaging enable detailed examination of cellular-level changes and abnormalities, providing valuable insights into the pathophysiology of these disorders. Its potential as a tool for diagnosis and research tool makes it a compelling avenue for further investigation within the field.
Currently, there is a shortage of effective physical treatment methods for congenital color vision disorders. Likewise, biological-level interventions, including gene therapy, are in the early stages of development and necessitate further research prior to their clinical implementation. Although gene therapy approaches are primarily being studied in animal models or clinical trials, there have been promising advancements and encouraging results have been observed in some studies, indicating potential for future treatment of these disorders in humans. The ongoing research and progress in this field offer hope for the development of effective therapies in the future.
While studies have shown therapy assisted by gene therapy can provide therapeutic effects of gene therapy in middle-aged and elderly models of color blindness, current research indicates that treatment interventions for color blindness are most effective when implemented during childhood, especially before the age of 6. It is believed that early intervention during the developmental stages of visual processing provides the best opportunity for optimal therapeutic outcomes. Although performing injection surgery on young patients presents additional technical challenges, the potential benefits of comprehensive treatment make it a worthwhile consideration.
In summary, while there is still a long journey ahead to fully understand the pathological mechanisms of color vision disorders and develop safe and effective treatments, the prospect of finding a cure is promising and warrants our unwavering commitment and perseverance.
ZY: Writing – original draft. LY: Writing – original draft. WZ: Writing – original draft, Writing – review & editing. JQ: Writing – original draft, Writing – review & editing. WA: Visualization, Writing – original draft. KY: Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (no. 31970930), Hubei Natural Science Foundation (nos. 2020CFA069 and 2018CFB434), and Neuroscience Team Development Project of Wuhan University of Science and Technology (no. 1180002).
The authors thank the members of Yao Laboratory for their kind suggestion and technical assistance. Picture drawn using biorender.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aboshiha, J., Dubis, A. M., Carroll, J., Hardcastle, A. J., and Michaelides, M. (2016). The cone dysfunction syndromes. Br. J. Ophthalmol. 100, 115–121. doi: 10.1136/bjophthalmol-2014-306505
Alexander, J. J., Umino, Y., Everhart, D., Chang, B., Min, S. H., Li, Q., et al. (2007). Restoration of cone vision in a mouse model of achromatopsia. Nat. Med. 13, 685–687. doi: 10.1038/nm1596
Aligianis, I. A., Forshew, T., Johnson, S., Michaelides, M., Johnson, C. A., Trembath, R. C., et al. (2002). Mapping of a novel locus for achromatopsia (ACHM4) to 1p and identification of a germline mutation in the alpha subunit of cone transducin (GNAT2). J. Med. Genet. 39, 656–660. doi: 10.1136/jmg.39.9.656
Ansar, M., Santos-Cortez, R. L., Saqib, M. A., Zulfiqar, F., Lee, K., Ashraf, N. M., et al. (2015). Mutation of ATF6 causes autosomal recessive achromatopsia. Hum. Genet. 134, 941–950. doi: 10.1007/s00439-015-1571-4
Arinobu, D., Tachibanaki, S., and Kawamura, S. (2010). Larger inhibition of visual pigment kinase in cones than in rods. J. Neurochem. 115, 259–268. doi: 10.1111/j.1471-4159.2010.06925.x
Astakhova, L., Firsov, M., and Govardovskii, V. (2015). Activation and quenching of the phototransduction cascade in retinal cones as inferred from electrophysiology and mathematical modeling. Mol. Vis. 21, 244–263.
Badawy, A. R., Hassan, M. U., Elsherif, M., Ahmed, Z., Yetisen, A. K., and Butt, H. (2018). Contact lenses for color blindness. Adv. Healthc. Mater. 7:e1800152. doi: 10.1002/adhm.201800152
Baehr, W., Devlin, M. J., and Applebury, M. L. (1979). Isolation and characterization of cGMP phosphodiesterase from bovine rod outer segments. J. Biol. Chem. 254, 11669–11677. doi: 10.1016/S0021-9258(19)86536-5
Banin, E., Gootwine, E., Obolensky, A., Ezra-Elia, R., Ejzenberg, A., Zelinger, L., et al. (2015). Gene augmentation therapy restores retinal function and visual behavior in a sheep model of CNGA3 Achromatopsia. Mol. Ther. 23, 1423–1433. doi: 10.1038/mt.2015.114
Biel, M., and Michalakis, S. (2009). Cyclic nucleotide-gated channels. Handb. Exp. Pharmacol. 2009, 111–136. doi: 10.1007/978-3-540-68964-5_7
Brasil, A., Castro, A. J., Martins, I. C., Lacerda, E. M., Souza, G. S., Herculano, A. M., et al. (2015). Colour vision impairment in Young alcohol consumers. PloS One 10:e0140169. doi: 10.1371/journal.pone.0140169
Brunetti-Pierri, R., Karali, M., Melillo, P., Di Iorio, V., De Benedictis, A., Iaccarino, G., et al. (2021). Clinical and molecular characterization of Achromatopsia patients: a longitudinal study. Int. J. Mol. Sci. 22:1681. doi: 10.3390/ijms22041681
Carroll, J., Rossi, E. A., Porter, J., Neitz, J., Roorda, A., Williams, D. R., et al. (2010). Deletion of the X-linked opsin gene array locus control region (LCR) results in disruption of the cone mosaic. Vision Res. 50, 1989–1999. doi: 10.1016/j.visres.2010.07.009
Carvalho, L. S., Xu, J., Pearson, R. A., Smith, A. J., Bainbridge, J. W., Morris, L. M., et al. (2011). Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum. Mol. Genet. 20, 3161–3175. doi: 10.1093/hmg/ddr218
Catty, P., Pfister, C., Bruckert, F., and Deterre, P. (1992). The cGMP phosphodiesterase-transducin complex of retinal rods. Membrane binding and subunits interactions. J. Biol. Chem. 267, 19489–19493.
Chan, P., Stolz, J., Kohl, S., Chiang, W. C., and Lin, J. H. (2016). Endoplasmic reticulum stress in human photoreceptor diseases. Brain Res. 1648, 538–541. doi: 10.1016/j.brainres.2016.04.021
Chapanis, A. (1947). The dark adaptation of the color anomalous measured with lights of different hues. J. Gen. Physiol. 30, 423–437. doi: 10.1085/jgp.30.5.423
Chen, J. G., Nakamura, T., Ebrey, T. G., Ok, H., Konno, K., Derguini, F., et al. (1989). Wavelength regulation in iodopsin, a cone pigment. Biophys. J. 55, 725–729. doi: 10.1016/S0006-3495(89)82871-1
Chen, T. Y., Peng, Y. W., Dhallan, R. S., Ahamed, B., Reed, R. R., and Yau, K. W. (1993). A new subunit of the cyclic nucleotide-gated cation channel in retinal rods. Nature 362, 764–767. doi: 10.1038/362764a0
Cideciyan, A. V., Hufnagel, R. B., Carroll, J., Sumaroka, A., Luo, X., Schwartz, S. B., et al. (2013). Human cone visual pigment deletions spare sufficient photoreceptors to warrant gene therapy. Hum. Gene Ther. 24, 993–1006. doi: 10.1089/hum.2013.153
Cornelissen, F. W., and Brenner, E. (2015). Is adding a new class of cones to the retina sufficient to cure color-blindness? J. Vis. 15:22. doi: 10.1167/15.13.22
Curcio, C. A., Allen, K. A., Sloan, K. R., Lerea, C. L., Hurley, J. B., Klock, I. B., et al. (1991). Distribution and morphology of human cone photoreceptors stained with anti-blue opsin. J. Comp. Neurol. 312, 610–624. doi: 10.1002/cne.903120411
Dartnall, H. J., Bowmaker, J. K., and Mollon, J. D. (1983). Human visual pigments: microspectrophotometric results from the eyes of seven persons. Proc. R. Soc. Lond. B Biol. Sci. 220, 115–130.
Deng, W. T., Li, J., Zhu, P., Chiodo, V. A., Smith, W. C., Freedman, B., et al. (2018). Human L- and M-opsins restore M-cone function in a mouse model for human blue cone monochromacy. Mol. Vis. 24, 17–28.
Deng, W. T., Li, J., Zhu, P., Freedman, B., Smith, W. C., Baehr, W., et al. (2019). Rescue of M-cone function in aged Opn1mw−/− mice, a model for late-stage blue cone Monochromacy. Invest. Ophthalmol. Vis. Sci. 60, 3644–3651. doi: 10.1167/iovs.19-27079
Faillace, M. P., Bernabeu, R. O., and Korenbrot, J. I. (2004). Cellular processing of cone photoreceptor cyclic GMP-gated ion channels: a role for the S4 structural motif. J. Biol. Chem. 279, 22643–22653. doi: 10.1074/jbc.M400035200
Felden, J., Baumann, B., Ali, M., Audo, I., Ayuso, C., Bocquet, B., et al. (2019). Mutation spectrum and clinical investigation of achromatopsia patients with mutations in the GNAT2 gene. Hum. Mutat. 40, 1145–1155. doi: 10.1002/humu.23768
Fischer, M. D., Michalakis, S., Wilhelm, B., Zobor, D., Muehlfriedel, R., Kohl, S., et al. (2020). Safety and vision outcomes of subretinal gene therapy targeting cone photoreceptors in Achromatopsia: a nonrandomized controlled trial. JAMA Ophthalmol 138, 643–651. doi: 10.1001/jamaophthalmol.2020.1032
Fu, Y., and Yau, K. W. (2007). Phototransduction in mouse rods and cones. Pflugers Arch. 454, 805–819. doi: 10.1007/s00424-006-0194-y
Gardner, J. C., Liew, G., Quan, Y. H., Ermetal, B., Ueyama, H., Davidson, A. E., et al. (2014). Three different cone opsin gene array mutational mechanisms with genotype-phenotype correlation and functional investigation of cone opsin variants. Hum. Mutat. 35, 1354–1362. doi: 10.1002/humu.22679
Gardner, J. C., Michaelides, M., Holder, G. E., Kanuga, N., Webb, T. R., Mollon, J. D., et al. (2009). Blue cone monochromacy: causative mutations and associated phenotypes. Mol. Vis. 15, 876–884.
Gardner, J. C., Webb, T. R., Kanuga, N., Robson, A. G., Holder, G. E., Stockman, A., et al. (2010). X-linked cone dystrophy caused by mutation of the red and green cone opsins. Am. J. Hum. Genet. 87, 26–39. doi: 10.1016/j.ajhg.2010.05.019
Georgiou, M., Fujinami, K., and Michaelides, M. (2021). Inherited retinal diseases: therapeutics, clinical trials and end points-a review. Clin. Exp. Ophthalmol. 49, 270–288. doi: 10.1111/ceo.13917
Georgiou, M., Robson, A. G., Singh, N., Pontikos, N., Kane, T., Hirji, N., et al. (2019). Deep phenotyping of PDE6C-associated Achromatopsia. Invest. Ophthalmol. Vis. Sci. 60, 5112–5123. doi: 10.1167/iovs.19-27761
Georgiou, M., Singh, N., Kane, T., Robson, A. G., Kalitzeos, A., Hirji, N., et al. (2020). Photoreceptor structure in GNAT2-associated Achromatopsia. Invest. Ophthalmol. Vis. Sci. 61:40. doi: 10.1167/iovs.61.3.40
Gerstner, A., Zong, X., Hofmann, F., and Biel, M. (2000). Molecular cloning and functional characterization of a new modulatory cyclic nucleotide-gated channel subunit from mouse retina. J. Neurosci. 20, 1324–1332. doi: 10.1523/JNEUROSCI.20-04-01324.2000
Goodman, G., Ripps, H., and Siegel, I. M. (1963). Cone dysfunction syndromes. Arch. Ophthalmol. 70, 214–231. doi: 10.1001/archopht.1963.00960050216013
Gootwine, E., Ofri, R., Banin, E., Obolensky, A., Averbukh, E., Ezra-Elia, R., et al. (2017). Safety and efficacy evaluation of rAAV2tYF-PR1.7-hCNGA3 vector delivered by subretinal injection in CNGA3 mutant Achromatopsia sheep. Hum. Gene. Ther. Clin. Dev. 28, 96–107. doi: 10.1089/humc.2017.028
Grau, T., Artemyev, N. O., Rosenberg, T., Dollfus, H., Haugen, O. H., Cumhur Sener, E., et al. (2011). Decreased catalytic activity and altered activation properties of PDE6C mutants associated with autosomal recessive achromatopsia. Hum. Mol. Genet. 20, 719–730. doi: 10.1093/hmg/ddq517
Haegerstrom-Portnoy, G., Schneck, M. E., Verdon, W. A., and Hewlett, S. E. (1996). Clinical vision characteristics of the congenital achromatopsias. I. Visual acuity, refractive error, and binocular status. Optom. Vis. Sci. 73, 446–456. doi: 10.1097/00006324-199607000-00001
Hathibelagal, A. R. (2022). Implications of inherited color vision deficiency on occupations: a neglected entity! Indian J. Ophthalmol. 70, 256–260. doi: 10.4103/ijo.IJO_1100_21
Hirji, N., Georgiou, M., Kalitzeos, A., Bainbridge, J. W., Kumaran, N., Aboshiha, J., et al. (2018). Longitudinal assessment of retinal structure in Achromatopsia patients with long-term follow-up. Invest. Ophthalmol. Vis. Sci. 59, 5735–5744. doi: 10.1167/iovs.18-25452
Holroyd, E., and Hall, D. M. (1997). A re-appraisal of screening for colour vision impairments. Child Care Health Dev. 23, 391–398. doi: 10.1111/j.1365-2214.1997.tb00906.x
Hurley, J. B., and Stryer, L. (1982). Purification and characterization of the gamma regulatory subunit of the cyclic GMP phosphodiesterase from retinal rod outer segments. J. Biol. Chem. 257, 11094–11099. doi: 10.1016/S0021-9258(18)33937-1
Imai, H., Kojima, D., Oura, T., Tachibanaki, S., Terakita, A., and Shichida, Y. (1997). Single amino acid residue as a functional determinant of rod and cone visual pigments. Proc. Natl. Acad. Sci. U. S. A. 94, 2322–2326. doi: 10.1073/pnas.94.6.2322
Iribarne, M., and Masai, I. (2017). Neurotoxicity of cGMP in the vertebrate retina: from the initial research on rd mutant mice to zebrafish genetic approaches. J. Neurogenet. 31, 88–101. doi: 10.1080/01677063.2017.1358268
Ismail, M., Abid, A., Anwar, K., Qasim Mehdi, S., and Khaliq, S. (2006). Refinement of the locus for autosomal recessive cone-rod dystrophy (CORD8) linked to chromosome 1q23-q24 in a Pakistani family and exclusion of candidate genes. J. Hum. Genet. 51, 827–831. doi: 10.1007/s10038-006-0028-y
Karpen, J. W., Zimmerman, A. L., Stryer, L., and Baylor, D. A. (1988). Gating kinetics of the cyclic-GMP-activated channel of retinal rods: flash photolysis and voltage-jump studies. Proc. Natl. Acad. Sci. U. S. A. 85, 1287–1291. doi: 10.1073/pnas.85.4.1287
Kaupp, U. B., and Seifert, R. (2002). Cyclic nucleotide-gated ion channels. Physiol. Rev. 82, 769–824. doi: 10.1152/physrev.00008.2002
Kelly, J. P., Crognale, M. A., and Weiss, A. H. (2003). ERGs, cone-isolating VEPs and analytical techniques in children with cone dysfunction syndromes. Doc. Ophthalmol. 106, 289–304. doi: 10.1023/A:1022909328103
Khaliq, S., Hameed, A., Ismail, M., Anwar, K., Leroy, B. P., Mehdi, S. Q., et al. (2000). Novel locus for autosomal recessive cone-rod dystrophy CORD8 mapping to chromosome 1q12-Q24. Invest. Ophthalmol. Vis. Sci. 41, 3709–3712.
Khan, N. W., Wissinger, B., Kohl, S., and Sieving, P. A. (2007). CNGB3 achromatopsia with progressive loss of residual cone function and impaired rod-mediated function. Invest. Ophthalmol. Vis. Sci. 48, 3864–3871. doi: 10.1167/iovs.06-1521
Kohl, S., Baumann, B., Rosenberg, T., Kellner, U., Lorenz, B., Vadalà, M., et al. (2002). Mutations in the cone photoreceptor G-protein alpha-subunit gene GNAT2 in patients with achromatopsia. Am. J. Hum. Genet. 71, 422–425. doi: 10.1086/341835
Kohl, S., Coppieters, F., Meire, F., Schaich, S., Roosing, S., Brennenstuhl, C., et al. (2012). A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia. Am. J. Hum. Genet. 91, 527–532. doi: 10.1016/j.ajhg.2012.07.006
Kohl, S., and Hamel, C. (2013). Clinical utility gene card for: Achromatopsia—update 2013. Eur. J. Hum. Genet. 21, 1–3. doi: 10.1038/ejhg.2013.44
Kohl, S., Jägle, H., Wissinger, B., and Zobor, D. (1993). “Achromatopsia” in GeneReviews. eds. M. P. Adam, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Bean, and K. W. Gripp, et al. (Seattle (WA): University of Washington), 1993–2022.
Kohl, S., Varsanyi, B., Antunes, G. A., Baumann, B., Hoyng, C. B., Jagle, H., et al. (2005). CNGB3 mutations account for 50% of all cases with autosomal recessive achromatopsia. Eur. J. Hum. Genet. 13, 302–308. doi: 10.1038/sj.ejhg.5201269
Kohl, S., Zobor, D., Chiang, W. C., Weisschuh, N., Staller, J., Gonzalez Menendez, I., et al. (2015). Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat. Genet. 47, 757–765. doi: 10.1038/ng.3319
Kojima, K., Imamoto, Y., Maeda, R., Yamashita, T., and Shichida, Y. (2014). Rod visual pigment optimizes active state to achieve efficient G protein activation as compared with cone visual pigments. J. Biol. Chem. 289, 5061–5073. doi: 10.1074/jbc.M113.508507
Komáromy, A. M., Alexander, J. J., Rowlan, J. S., Garcia, M. M., Chiodo, V. A., Kaya, A., et al. (2010). Gene therapy rescues cone function in congenital achromatopsia. Hum. Mol. Genet. 19, 2581–2593. doi: 10.1093/hmg/ddq136
Komáromy, A. M., Rowlan, J. S., Corr, A. T., Reinstein, S. L., Boye, S. L., Cooper, A. E., et al. (2013). Transient photoreceptor deconstruction by CNTF enhances rAAV-mediated cone functional rescue in late stage CNGB3-achromatopsia. Mol. Ther. 21, 1131–1141. doi: 10.1038/mt.2013.50
Kosower, E. M. (1988). Assignment of groups responsible for the "opsin shift" and light absorptions of rhodopsin and red, green, and blue iodopsins (cone pigments). Proc. Natl. Acad. Sci. U. S. A. 85, 1076–1080. doi: 10.1073/pnas.85.4.1076
Kotterman, M. A., and Schaffer, D. V. (2014). Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 15, 445–451. doi: 10.1038/nrg3742
Ladekjaer-Mikkelsen, A. S., Rosenberg, T., and Jørgensen, A. L. (1996). A new mechanism in blue cone monochromatism. Hum. Genet. 98, 403–408. doi: 10.1007/s004390050229
Lamb, T. D., and Pugh, E. N. (2006). Phototransduction, dark adaptation, and rhodopsin regeneration the proctor lecture. Invest. Ophthalmol. Vis. Sci. 47, 5137–5152. doi: 10.1167/iovs.06-0849
Langlo, C. S., Patterson, E. J., Higgins, B. P., Summerfelt, P., Razeen, M. M., Erker, L. R., et al. (2016). Residual foveal cone structure in CNGB3-associated Achromatopsia. Invest. Ophthalmol. Vis. Sci. 57, 3984–3995. doi: 10.1167/iovs.16-19313
Leskov, I. B., Klenchin, V. A., Handy, J. W., Whitlock, G. G., Govardovskii, V. I., Bownds, M. D., et al. (2000). The gain of rod phototransduction: reconciliation of biochemical and electrophysiological measurements. Neuron 27, 525–537. doi: 10.1016/S0896-6273(00)00063-5
Li, Y., Tao, W., Luo, L., Huang, D., Kauper, K., Stabila, P., et al. (2010). CNTF induces regeneration of cone outer segments in a rat model of retinal degeneration. PloS One 5:e9495. doi: 10.1371/journal.pone.0009495
Li, T. S., Volpp, K., and Applebury, M. L. (1990). Bovine cone photoreceptor cGMP phosphodiesterase structure deduced from a cDNA clone. Proc. Natl. Acad. Sci. U. S. A. 87, 293–297. doi: 10.1073/pnas.87.1.293
Lin, J. H., Li, H., Yasumura, D., Cohen, H. R., Zhang, C., Panning, B., et al. (2007). IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944–949. doi: 10.1126/science.1146361
Lipinski, D. M., Barnard, A. R., Singh, M. S., Martin, C., Lee, E. J., Davies, W. I. L., et al. (2015). CNTF gene therapy confers lifelong neuroprotection in a mouse model of human retinitis Pigmentosa. Mol. Ther. 23, 1308–1319. doi: 10.1038/mt.2015.68
Lisman, J., and Fain, G. (1995). Support for the equivalent light hypothesis for RP. Nat. Med. 1, 1254–1255. doi: 10.1038/nm1295-1254
Luo, X., Cideciyan, A. V., Iannaccone, A., Roman, A. J., Ditta, L. C., Jennings, B. J., et al. (2015). Blue cone monochromacy: visual function and efficacy outcome measures for clinical trials. PloS One 10:e0125700. doi: 10.1371/journal.pone.0125700
MacLaren, R. E., Bennett, J., and Schwartz, S. D. (2016). Gene therapy and stem cell transplantation in retinal Disease: the new frontier. Ophthalmology 123, S98–s106. doi: 10.1016/j.ophtha.2016.06.041
Mancuso, K., Hauswirth, W. W., Li, Q., Connor, T. B., Kuchenbecker, J. A., Mauck, M. C., et al. (2009). Gene therapy for red-green colour blindness in adult primates. Nature 461, 784–787. doi: 10.1038/nature08401
Martínez-Domingo, M. A., Gómez-Robledo, L., Valero, E. M., Huertas, R., Hernández-Andrés, J., Ezpeleta, S., et al. (2019). Assessment of VINO filters for correcting red-green color vision deficiency. Opt. Express 27, 17954–17967. doi: 10.1364/OE.27.017954
Mastey, R. R., Georgiou, M., Langlo, C. S., Kalitzeos, A., Patterson, E. J., Kane, T., et al. (2019). Characterization of retinal structure in ATF6-associated Achromatopsia. Invest. Ophthalmol. Vis. Sci. 60, 2631–2640. doi: 10.1167/iovs.19-27047
Mayer, A. K., Van Cauwenbergh, C., Rother, C., Baumann, B., Reuter, P., De Baere, E., et al. (2017). CNGB3 mutation spectrum including copy number variations in 552 achromatopsia patients. Hum. Mutat. 38, 1579–1591. doi: 10.1002/humu.23311
McClements, M., Davies, W. I., Michaelides, M., Carroll, J., Rha, J., Mollon, J. D., et al. (2013). X-linked cone dystrophy and colour vision deficiency arising from a missense mutation in a hybrid L/M cone opsin gene. Vision Res. 80, 41–50. doi: 10.1016/j.visres.2012.12.012
McKyton, A., Averbukh, E., Marks Ohana, D., Levin, N., and Banin, E. (2021). Cortical visual mapping following ocular gene augmentation therapy for Achromatopsia. J. Neurosci. 41, 7363–7371. doi: 10.1523/JNEUROSCI.3222-20.2021
Michaelides, M., Aligianis, I. A., Holder, G. E., Simunovic, M., Mollon, J. D., Maher, E. R., et al. (2003). Cone dystrophy phenotype associated with a frameshift mutation (M280fsX291) in the alpha-subunit of cone specific transducin (GNAT2). Br. J. Ophthalmol. 87, 1317–1320. doi: 10.1136/bjo.87.11.1317
Michaelides, M., Hirji, N., Wong, S. C., Besirli, C. G., Zaman, S., Kumaran, N., et al. (2023). First-in-human gene therapy trial of AAV8-hCARp.hCNGB3 in adults and children with CNGB3-associated Achromatopsia. Am. J. Ophthalmol. 253, 243–251. doi: 10.1016/j.ajo.2023.05.009
Michalakis, S., Gerhardt, M., Rudolph, G., Priglinger, S., and Priglinger, C. (2022). Achromatopsia: genetics and gene therapy. Mol. Diagn. Ther. 26, 51–59. doi: 10.1007/s40291-021-00565-z
Michalakis, S., Mühlfriedel, R., Tanimoto, N., Krishnamoorthy, V., Koch, S., Fischer, M. D., et al. (2010). Restoration of cone vision in the CNGA3−/− mouse model of congenital complete lack of cone photoreceptor function. Mol. Ther. 18, 2057–2063. doi: 10.1038/mt.2010.149
Michalakis, S., Schon, C., Becirovic, E., and Biel, M. (2017). Gene therapy for achromatopsia. J. Gene Med. 19:2944. doi: 10.1002/jgm.2944
Müller, F., and Kaupp, U. B. (1998). Signal transduction in photoreceptor cells. Naturwissenschaften 85, 49–61. doi: 10.1007/s001140050453
Mustafi, D., Engel, A. H., and Palczewski, K. (2009). Structure of cone photoreceptors. Prog. Retin. Eye Res. 28, 289–302. doi: 10.1016/j.preteyeres.2009.05.003
Nakanishi, T., Shimazawa, M., Sugitani, S., Kudo, T., Imai, S., Inokuchi, Y., et al. (2013). Role of endoplasmic reticulum stress in light-induced photoreceptor degeneration in mice. J. Neurochem. 125, 111–124. doi: 10.1111/jnc.12116
Nathans, J., Davenport, C. M., Maumenee, I. H., Lewis, R. A., Hejtmancik, J. F., Litt, M., et al. (1989). Molecular genetics of human blue cone monochromacy. Science 245, 831–838. doi: 10.1126/science.2788922
Nathans, J., Thomas, D., and Hogness, D. S. (1986). Molecular genetics of human color vision: the genes encoding blue, green, and red pigments. Science 232, 193–202. doi: 10.1126/science.2937147
Neitz, M., Krekling, E. D., Hagen, L. A., Pedersen, H. R., Rowlan, J., Barborek, R., et al. (2020). Tritan color vision deficiency may be associated with an OPN1SW splicing defect and haploinsufficiency. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 37, A26–a34. doi: 10.1364/JOSAA.381919
Neitz, J., and Neitz, M. (2011). The genetics of normal and defective color vision. Vision Res. 51, 633–651. doi: 10.1016/j.visres.2010.12.002
Neitz, J., and Neitz, M. (2017). Evolution of the circuitry for conscious color vision in primates. Eye (Lond.) 31, 286–300. doi: 10.1038/eye.2016.257
Neitz, M., and Neitz, J. (2021). Intermixing the OPN1LW and OPN1MW genes disrupts the Exonic splicing code causing an Array of vision disorders. Genes 12:1180. doi: 10.3390/genes12081180
Neitz, M., Neitz, J., and Jacobs, G. H. (1991). Spectral tuning of pigments underlying red-green color vision. Science 252, 971–974. doi: 10.1126/science.1903559
Ofri, R., Averbukh, E., Ezra-Elia, R., Ross, M., Honig, H., Obolensky, A., et al. (2018). Six years and counting: restoration of Photopic retinal function and visual behavior following gene augmentation therapy in a sheep model of CNGA3 Achromatopsia. Hum. Gene Ther. 29, 1376–1386. doi: 10.1089/hum.2018.076
Park, W. L., and Sunness, J. S. (2004). Red contact lenses for alleviation of photophobia in patients with cone disorders. Am. J. Ophthalmol. 137, 774–775. doi: 10.1016/j.ajo.2003.09.061
Qin, N., Pittler, S. J., and Baehr, W. (1992). In vitro isoprenylation and membrane association of mouse rod photoreceptor cGMP phosphodiesterase alpha and beta subunits expressed in bacteria. J. Biol. Chem. 267, 8458–8463. doi: 10.1016/S0021-9258(18)42466-0
Randhawa, S., González, M. U., Renger, J., Enoch, S., and Quidant, R. (2010). Design and properties of dielectric surface plasmon Bragg mirrors. Opt. Express 18, 14496–14510. doi: 10.1364/OE.18.014496
Reichel, F. F., Peters, T., Wilhelm, B., Biel, M., Ueffing, M., Wissinger, B., et al. (2018). Humoral immune response after intravitreal but not after subretinal AAV8 in Primates and patients. Invest. Ophthalmol. Vis. Sci. 59, 1910–1915. doi: 10.1167/iovs.17-22494
Reyniers, E., Van Thienen, M. N., Meire, F., De Boulle, K., Devries, K., Kestelijn, P., et al. (1995). Gene conversion between red and defective green opsin gene in blue cone monochromacy. Genomics 29, 323–328. doi: 10.1006/geno.1995.9998
Rinner, O., Makhankov, Y. V., Biehlmaier, O., and Neuhauss, S. C. (2005). Knockdown of cone-specific kinase GRK7 in larval zebrafish leads to impaired cone response recovery and delayed dark adaptation. Neuron 47, 231–242. doi: 10.1016/j.neuron.2005.06.010
Rosenberg, T., Baumann, B., Kohl, S., Zrenner, E., Jorgensen, A. L., and Wissinger, B. (2004). Variant phenotypes of incomplete achromatopsia in two cousins with GNAT2 gene mutations. Invest. Ophthalmol. Vis. Sci. 45, 4256–4262. doi: 10.1167/iovs.04-0317
Salih, A. E., Elsherif, M., Alam, F., Yetisen, A. K., and Butt, H. (2021). Gold nanocomposite contact lenses for color blindness management. ACS Nano 15, 4870–4880. doi: 10.1021/acsnano.0c09657
Salih, A. E., Elsherif, M., Ali, M., Vahdati, N., Yetisen, A. K., and Butt, H. (2020). Ophthalmic wearable devices for color blindness management. Adv Material Technol 5:1901134. doi: 10.1002/admt.201901134
Schön, C., Biel, M., and Michalakis, S. (2013). Gene replacement therapy for retinal CNG channelopathies. Mol. Genet. Genomics 288, 459–467. doi: 10.1007/s00438-013-0766-4
Seitz, I. P., Michalakis, S., Wilhelm, B., Reichel, F. F., Ochakovski, G. A., Zrenner, E., et al. (2017). Superior retinal gene transfer and biodistribution profile of subretinal versus intravitreal delivery of AAV8 in nonhuman Primates. Invest. Ophthalmol. Vis. Sci. 58, 5792–5801. doi: 10.1167/iovs.17-22473
Simunovic, M. P., and Moore, A. T. (1998). The cone dystrophies. Eye 12, 553–565. doi: 10.1038/eye.1998.145
Simunovic, M. P., Regan, B. C., and Mollon, J. D. (2001). Is color vision deficiency an advantage under scotopic conditions? Invest. Ophthalmol. Vis. Sci. 42, 3357–3364.
Smallwood, P. M., Wang, Y., and Nathans, J. (2002). Role of a locus control region in the mutually exclusive expression of human red and green cone pigment genes. Proc. Natl. Acad. Sci. U. S. A. 99, 1008–1011. doi: 10.1073/pnas.022629799
Stryer, L. (1991). Visual excitation and recovery. J. Biol. Chem. 266, 10711–10714. doi: 10.1016/S0021-9258(18)99072-1
Sun, W., and Zhang, Q. (2019). Diseases associated with mutations in CNGA3: genotype-phenotype correlation and diagnostic guideline. Prog. Mol. Biol. Transl. Sci. 161, 1–27. doi: 10.1016/bs.pmbts.2018.10.002
Sundaram, V., Wilde, C., Aboshiha, J., Cowing, J., Han, C., Langlo, C. S., et al. (2014). Retinal structure and function in achromatopsia: implications for gene therapy. Ophthalmology 121, 234–245. doi: 10.1016/j.ophtha.2013.08.017
Sung, C. H., and Chuang, J. Z. (2010). The cell biology of vision. J. Cell Biol. 190, 953–963. doi: 10.1083/jcb.201006020
Swanson, W. H., and Cohen, J. M. (2003). Color vision. Ophthalmol. Clin. North Am. 16, 179–203. doi: 10.1016/S0896-1549(03)00004-X
Tachibanaki, S., Arinobu, D., Shimauchi-Matsukawa, Y., Tsushima, S., and Kawamura, S. (2005). Highly effective phosphorylation by G protein-coupled receptor kinase 7 of light-activated visual pigment in cones. Proc. Natl. Acad. Sci. U. S. A. 102, 9329–9334. doi: 10.1073/pnas.0501875102
Talcott, K. E., Ratnam, K., Sundquist, S. M., Lucero, A. S., Lujan, B. J., Tao, W., et al. (2011). Longitudinal study of cone photoreceptors during retinal degeneration and in response to ciliary neurotrophic factor treatment. Invest. Ophthalmol. Vis. Sci. 52, 2219–2226. doi: 10.1167/iovs.10-6479
Thapa, A., Morris, L., Xu, J., Ma, H., Michalakis, S., Biel, M., et al. (2012). Endoplasmic reticulum stress-associated cone photoreceptor degeneration in cyclic nucleotide-gated channel deficiency. J. Biol. Chem. 287, 18018–18029. doi: 10.1074/jbc.M112.342220
Thiadens, A. A., den Hollander, A. I., Roosing, S., Nabuurs, S. B., Zekveld-Vroon, R. C., Collin, R. W., et al. (2009). Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am. J. Hum. Genet. 85, 240–247. doi: 10.1016/j.ajhg.2009.06.016
Thiadens, A. A., Somervuo, V., van den Born, L. I., Roosing, S., van Schooneveld, M. J., Kuijpers, R. W., et al. (2010). Progressive loss of cones in achromatopsia: an imaging study using spectral-domain optical coherence tomography. Invest. Ophthalmol. Vis. Sci. 51, 5952–5957. doi: 10.1167/iovs.10-5680
Thomas, M. G., McLean, R. J., Kohl, S., Sheth, V., and Gottlob, I. (2012). Early signs of longitudinal progressive cone photoreceptor degeneration in achromatopsia. Br. J. Ophthalmol. 96, 1232–1236. doi: 10.1136/bjophthalmol-2012-301737
Tobias, P., Philipp, S. I., Stylianos, M., Martin, B., Barbara, W., Felix, R., et al. (2019). Safety and toxicology of ocular gene therapy with recombinant AAV vector rAAV.hCNGA3 in nonhuman Primates. Hum. Gene. Ther. Clin. Dev. 30, 50–56. doi: 10.1089/humc.2018.188
Tyler, C. W., and Hamer, R. D. (1990). Analysis of visual modulation sensitivity. IV. Validity of the ferry-Porter law. J. Opt. Soc. Am. A 7, 743–758. doi: 10.1364/JOSAA.7.000743
Uzunov, P., and Weiss, B. (1972). Separation of multiple molecular forms of cyclic adenosine-3′,5′-monophosphate phosphodiesterase in rat cerebellum by polyacrylamide gel electrophoresis. Biochim. Biophys. Acta 284, 220–226. doi: 10.1016/0005-2744(72)90060-5
van Hateren, J. H., and Lamb, T. D. (2006). The photocurrent response of human cones is fast and monophasic. BMC Neurosci. 7:34. doi: 10.1186/1471-2202-7-34
Vissers, P. M., Bovee-Geurts, P. H., Portier, M. D., Klaassen, C. H., and Degrip, W. J. (1998). Large-scale production and purification of the human green cone pigment: characterization of late photo-intermediates. Biochem. J. 330, 1201–1208. doi: 10.1042/bj3301201
Vogalis, F., Shiraki, T., Kojima, D., Wada, Y., Nishiwaki, Y., Jarvinen, J. L., et al. (2011). Ectopic expression of cone-specific G-protein-coupled receptor kinase GRK7 in zebrafish rods leads to lower photosensitivity and altered responses. J. Physiol. 589, 2321–2348. doi: 10.1113/jphysiol.2010.204156
Vollrath, D., Nathans, J., and Davis, R. W. (1988). Tandem array of human visual pigment genes at Xq28. Science 240, 1669–1672. doi: 10.1126/science.2837827
Wada, Y., Sugiyama, J., Okano, T., and Fukada, Y. (2006). GRK1 and GRK7: unique cellular distribution and widely different activities of opsin phosphorylation in the zebrafish rods and cones. J. Neurochem. 98, 824–837. doi: 10.1111/j.1471-4159.2006.03920.x
Wald, G. (1968). The molecular basis of visual excitation. Nature 219, 800–807. doi: 10.1038/219800a0
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Wang, M., and Kaufman, R. J. (2016). Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326–335. doi: 10.1038/nature17041
Wang, J. S., and Kefalov, V. J. (2011). The cone-specific visual cycle. Prog. Retin. Eye Res. 30, 115–128. doi: 10.1016/j.preteyeres.2010.11.001
Wang, Y., Macke, J. P., Merbs, S. L., Zack, D. J., Klaunberg, B., Bennett, J., et al. (1992). A locus control region adjacent to the human red and green visual pigment genes. Neuron 9, 429–440. doi: 10.1016/0896-6273(92)90181-C
Weisschuh, N., Obermaier, C. D., Battke, F., Bernd, A., Kuehlewein, L., Nasser, F., et al. (2020a). Genetic architecture of inherited retinal degeneration in Germany: a large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat. 41, 1514–1527. doi: 10.1002/humu.24064
Weisschuh, N., Stingl, K., Audo, I., Biskup, S., Bocquet, B., Branham, K., et al. (2018). Mutations in the gene PDE6C encoding the catalytic subunit of the cone photoreceptor phosphodiesterase in patients with achromatopsia. Hum. Mutat. 39, 1366–1371. doi: 10.1002/humu.23606
Weisschuh, N., Sturm, M., Baumann, B., Audo, I., Ayuso, C., Bocquet, B., et al. (2020b). Deep-intronic variants in CNGB3 cause achromatopsia by pseudoexon activation. Hum. Mutat. 41, 255–264. doi: 10.1002/humu.23920
Weitz, C. J., Miyake, Y., Shinzato, K., Montag, E., Zrenner, E., Went, L. N., et al. (1992a). Human tritanopia associated with two amino acid substitutions in the blue-sensitive opsin. Am. J. Hum. Genet. 50, 498–507.
Weitz, C. J., Went, L. N., and Nathans, J. (1992b). Human tritanopia associated with a third amino acid substitution in the blue-sensitive visual pigment. Am. J. Hum. Genet. 51, 444–446.
Wen, R., Tao, W., Li, Y., and Sieving, P. A. (2012). CNTF and retina. Prog. Retin. Eye Res. 31, 136–151. doi: 10.1016/j.preteyeres.2011.11.005
Winderickx, J., Battisti, L., Hibiya, Y., Motulsky, A. G., and Deeb, S. S. (1993). Haplotype diversity in the human red and green opsin genes: evidence for frequent sequence exchange in exon 3. Hum. Mol. Genet. 2, 1413–1421. doi: 10.1093/hmg/2.9.1413
Wissinger, B., Gamer, D., Jägle, H., Giorda, R., Marx, T., Mayer, S., et al. (2001). CNGA3 mutations in hereditary cone photoreceptor disorders. Am. J. Hum. Genet. 69, 722–737. doi: 10.1086/323613
Wong-Riley, M. T. (2010). Energy metabolism of the visual system. Eye Brain 2, 99–116. doi: 10.2147/EB.S9078
Xu, M., Gelowani, V., Eblimit, A., Wang, F., Young, M. P., Sawyer, B. L., et al. (2015). ATF6 is mutated in early onset photoreceptor degeneration with macular involvement. Invest. Ophthalmol. Vis. Sci. 56, 3889–3895. doi: 10.1167/iovs.15-16778
Yamaguchi, T., Motulsky, A. G., and Deeb, S. S. (1997). Visual pigment gene structure and expression in human retinae. Hum. Mol. Genet. 6, 981–990. doi: 10.1093/hmg/6.7.981
Yau, K. W. (1994). Phototransduction mechanism in retinal rods and cones. The Friedenwald Lecture. Invest. Ophthalmol. Vis. Sci. 35, 9–32.
Young, R. W. (1967). The renewal of photoreceptor cell outer segments. J. Cell Biol. 33, 61–72. doi: 10.1083/jcb.33.1.61
Young, R. W., and Bok, D. (1969). Participation of the retinal pigment epithelium in the rod outer segment renewal process. J. Cell Biol. 42, 392–403. doi: 10.1083/jcb.42.2.392
Young, R. W., and Droz, B. (1968). The renewal of protein in retinal rods and cones. J. Cell Biol. 39, 169–184. doi: 10.1083/jcb.39.1.169
Zein, W. M., Jeffrey, B. G., Wiley, H. E., Turriff, A. E., Tumminia, S. J., Tao, W., et al. (2014). CNGB3-achromatopsia clinical trial with CNTF: diminished rod pathway responses with no evidence of improvement in cone function. Invest. Ophthalmol. Vis. Sci. 55, 6301–6308. doi: 10.1167/iovs.14-14860
Zhang, X., and Cote, R. H. (2005). cGMP signaling in vertebrate retinal photoreceptor cells. Front. Biosci. 10, 1191–1204. doi: 10.2741/1612
Zhang, Z., Pang, J., Feng, X., Guo, Q., Li, L., Jing, A., et al. (2011). AAV-mediated gene therapy restores cone function in a rat with an M-cone opsin deficiency, a model for blue cone Monochromacy. Invest. Ophthalmol. Vis. Sci. 52
Keywords: color blindness, achromatopsia, cone, mechanism, treatment
Citation: Yang Z, Yan L, Zhang W, Qi J, An W and Yao K (2024) Dyschromatopsia: a comprehensive analysis of mechanisms and cutting-edge treatments for color vision deficiency. Front. Neurosci. 18:1265630. doi: 10.3389/fnins.2024.1265630
Edited by:
Ione Fine, University of Washington, United StatesReviewed by:
Mugen Liu, Huazhong University of Science and Technology, ChinaCopyright © 2024 Yang, Yan, Zhang, Qi, An and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Yao, a3lhbzIxQG91dGxvb2suY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.