 Foster D. Ritchie
Foster D. Ritchie Sofia B. Lizarraga
Sofia B. Lizarraga- Department of Biological Sciences and Center for Childhood Neurotherapeutics, University of South Carolina, Columbia, SC, United States
Brain size is controlled by several factors during neuronal development, including neural progenitor proliferation, neuronal arborization, gliogenesis, cell death, and synaptogenesis. Multiple neurodevelopmental disorders have co-morbid brain size abnormalities, such as microcephaly and macrocephaly. Mutations in histone methyltransferases that modify histone H3 on Lysine 36 and Lysine 4 (H3K36 and H3K4) have been identified in neurodevelopmental disorders involving both microcephaly and macrocephaly. H3K36 and H3K4 methylation are both associated with transcriptional activation and are proposed to sterically hinder the repressive activity of the Polycomb Repressor Complex 2 (PRC2). During neuronal development, tri-methylation of H3K27 (H3K27me3) by PRC2 leads to genome wide transcriptional repression of genes that regulate cell fate transitions and neuronal arborization. Here we provide a review of neurodevelopmental processes and disorders associated with H3K36 and H3K4 histone methyltransferases, with emphasis on processes that contribute to brain size abnormalities. Additionally, we discuss how the counteracting activities of H3K36 and H3K4 modifying enzymes vs. PRC2 could contribute to brain size abnormalities which is an underexplored mechanism in relation to brain size control.
1. Introduction
Neurodevelopment is a complex process that depends on the precise regulation of gene transcription. In particular, the development of the cerebral cortex, the structure in the brain that gives us our highest cognitive functions, is reliant on the careful orchestration of multiple cellular processes including the proliferation of neuronal progenitors, as well as the migration, and differentiation of neuronal cells. Early in development a thin neuroepithelium constituted by neuroepithelial stem cells (NES) lines up the ventricles of the developing telencephalon. As development proceeds, NES give rise to Radial Glial Cells (RGCs) which are neuronal progenitor cells (NPCs) located in the ventricular zone (VZ) (Namba and Huttner, 2017). RGCs have an apical and a basal process that sense signals from the environment. In addition, the RGC basal processes also provide structural support for the migration of the newly born neurons (Noctor et al., 2002). In lyssencephalic mammals (e.g., mice), a second proliferative region is located away from the ventricle in the subventricular zone (SVZ). The NPC population at the SVZ is composed of both outer (or basal) Radial Glia Cells (oRGCs) and intermediate progenitor cells (IPCs) which lack an apical process (Namba and Huttner, 2017). In gyrated mammals (e.g., ferrets and primates) there is an expansion of the SVZ forming an inner (iSVZ) and an outer (oSVZ) SVZ (Lui et al., 2011). In particular, the oSVZ composed of both IPCs and oRGCs is proposed to have driven the rapid evolutionary expansion of the superficial layers of the cerebral cortex contributing to a larger brain to body size ratio in humans compared to other non-human primates (Lui et al., 2011; Nowakowski et al., 2016).
Changes in the mode of NPC cell division are proposed to contribute to the diversity of the daughter cell population. Before the onset of neurogenesis, NES and RGC cells divide symmetrically to generate two identical daughter cells (Brand and Rakic, 1979). This mode of cell division occurs with the division plane perpendicular to the neuroepithelial ventricular surface. After the start of neurogenesis at the VZ, RGCs undergo asymmetric or symmetric cell division. In RGCs, a parallel or oblique plane of division with respect to the neuroepithelium ventricular surface is proposed to result in the asymmetric inheritance of the basal process and cell fate determining factors giving rise to two distinct daughter cells–an NPC and a neuron for example (Chenn and McConnell, 1995; Huttner and Brand, 1997; Haubensak et al., 2004). In contrast, oRGCs undergo proliferative symmetric cell divisions to expand the NPC population, or non-proliferative symmetric terminal divisions to give rise to two post-mitotic neurons (Shitamukai et al., 2011).
After birth newly born neurons migrate away from the VZ to their final location into the nascent cortical plate along the RGC basal processes. The earliest born neurons will form the pre-plate that later on will be split into the marginal zone and sub-plate by the first wave of migrating neurons. Subsequent waves of newborn neurons will form the transcriptionally and functionally distinct layers in the neocortex (Molnar et al., 2019a,b). Early in neurogenesis the earlier born projection neurons populate the deeper layers of the cerebral cortex, while during mid-neurogenesis the later born neurons start to populate the more superficial layers of the developing cortex (Rakic, 1972, 2009; Clowry et al., 2010). As neurogenesis ends, there is a switch from a neurogenic to a gliogenic fate, for which RGC competence changes to generate glial cells (astrocytes and oligodendrocytes) at late embryonic or perinatal stages (Ohtsuka and Kageyama, 2019).
Improper regulation of NPC division during cortical development has been proposed as a major mechanism underlying defects in brain size. Primary microcephaly has been associated with mutations in genes that modulate centrosome function and mitotic spindle formation, which are essential for proper cell division (Cox et al., 2006; Jean et al., 2020). Alternatively, postnatal microcephaly, or the failure of the brain to grow after birth (Seltzer and Paciorkowski, 2014), has been associated (albeit not exclusively) with mutations in transcriptional and chromatin regulators that modulate the expression of genes important to the development of the forebrain and hindbrain (Seltzer and Paciorkowski, 2014). In contrast, an increased head size, or macrocephaly, has also been associated with mutations in developmentally important genes that regulate neurogenesis and gliogenesis (Chenn and Walsh, 2002; Geisert et al., 2002; McCaffery and Deutsch, 2005). Alterations in brain size could ultimately alter the cytoarchitecture of the developing brain and impair the development of the neuronal circuitry. Not surprisingly, microcephaly has been associated with disorders of neuronal connectivity including autism spectrum disorders (ASD), and intellectual disability (ID), while macrocephaly has been primarily correlated with ASD (Fombonne et al., 1999; Miles et al., 2000).
Proper development of the brain architecture and wiring requires the careful regulation of multiple gene programs that may need to seamlessly transition between transcriptional activation or repression states in a cell type specific manner at distinct developmental timepoints. Epigenetic mechanisms such as DNA methylation, histone post-translational modifications, and chromatin remodeling are essential for these transitions. Histone methylation is a type of post-translational histone modification that is highly dynamic and essential to the regulation of transcriptional activation and repression. Work from multiple experimental systems has demonstrated that during development histone methylation is essential for the establishment of different cell lineages (Jambhekar et al., 2019). For instance, the evolutionarily conserved Polycomb Group of Proteins Complex (PcG) is a major regulator of histone methylation that is essential for the switching between neurogenic to gliogenic fates in the developing cerebral cortex (Pereira et al., 2010). Therefore, the precise regulation of a wide array of distinct histone methylation marks modulates transcriptional programs in a spatial and temporal manner to ensure normal brain development (Jambhekar et al., 2019).
In particular, histone methyltransferases that modify Histone H3 on Lysines 4, 36, and 27 are emerging as major regulators of neuronal structure and function (Figures 1A–C). Tri-methylation of Histone H3 on Lysine 4 (H3K4me3) and di or tri-methylation of Histone H3 on Lysine 36 (H3K36me2 and H3K36me3) have been associated with transcriptional activation (Bannister et al., 2005; Barski et al., 2007; Wagner and Carpenter, 2012; Zhu et al., 2013; Tables 1–4). In contrast to the activating effect on transcription of H3K4me3 and H3K36me2/3, tri-methylation of Histone H3 on Lysine 27 (H3K27me3) (Yu J. et al., 2019) by the Polycomb repressor complex 2 (PRC2) has been linked to transcriptional silencing. Histone modifications can either facilitate or hinder the recruitment or activation of other chromatin factors (Kim and Kim, 2012; Gates et al., 2017). Both H3K36me2/3 and H3K4me3 have been proposed to either directly or indirectly sterically hinder the deposition of H3K27me3 by the PcG which acts as a transcriptional repressor during development (Tie et al., 2014; Schuettengruber et al., 2017). While these mechanisms have been widely studied in cancer and general development, we focused on the evolutionary conserved counteracting activities of H3K36 (SETD2, SETD5, NSD1, NSD2, and ASH1L), and H3K4 (MLL1/KMT2A, MLL2/KMT2B, MLL3/KMT2C, MLL4/KMT2D, SETD1A, and SETD1B) modifiers vs. PcG (PRC2), because their antagonizing activities are understudied in the regulation of brain architecture, wiring and brain size control (Figures 1, 2 and Tables 1–4).
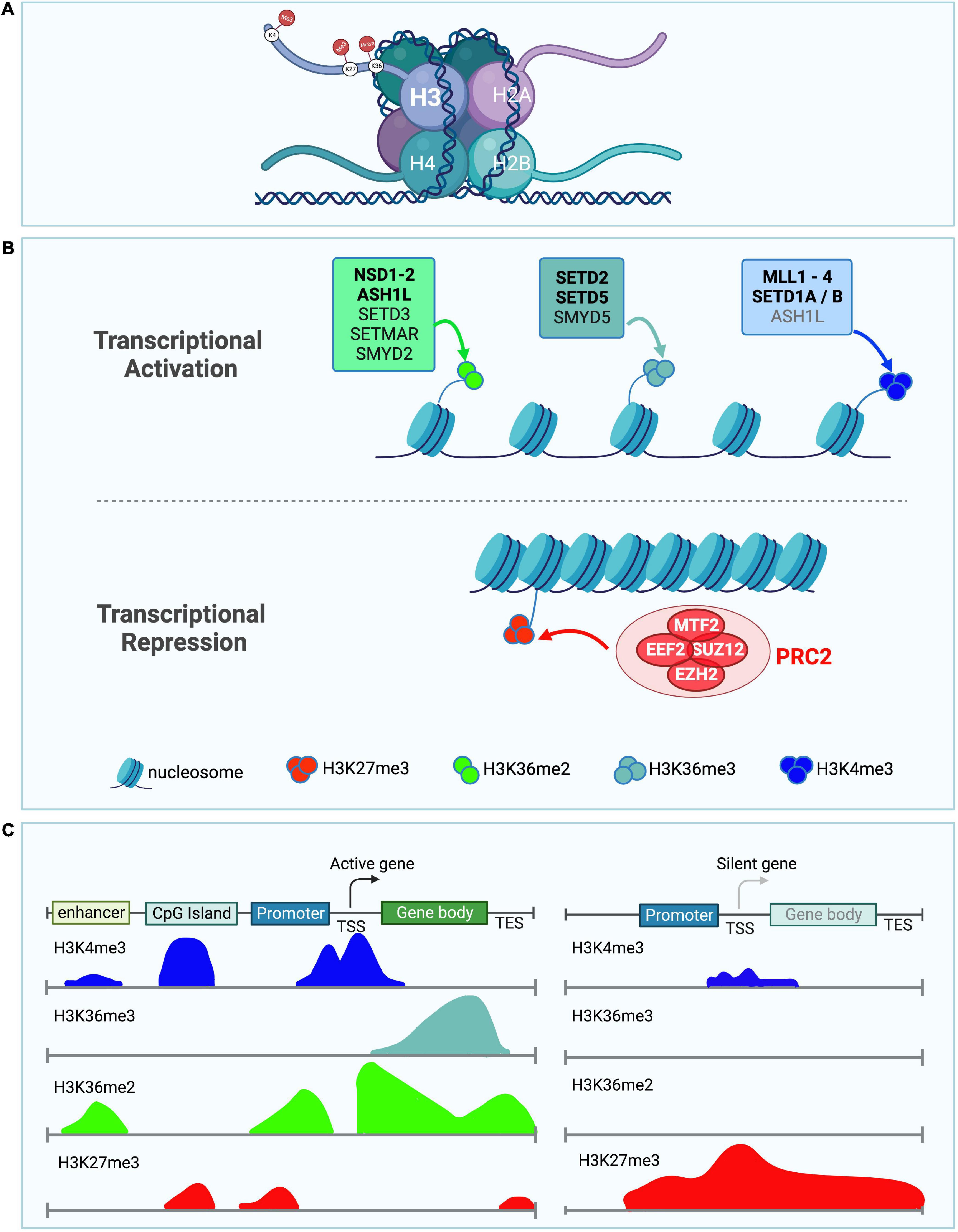
Figure 1. Role of H3K36me2/3, H3K4me3, and H3K27me3 in transcriptional activation and repression. (A) Depiction of a nucleosome showing the location of the different lysine histone modifications in the tail of histone H3. (B) Schematic showing histone methyltransferases and their corresponding histone marks associated with transcriptional activation (top panel) and repression (bottom panel). H3K36me2 (light green), H3K36me3 (light blue), and H3K4me3 (dark blue) histone marks are associated with transcriptional activation. Histone methyltransferases associated with deposition of either H3K36me2, H3K36me3, or H3K4me3 are shown. ASH1L is in gray for H3K4me3 as it might indirectly affect the levels of this histone mark. Bold enzyme names are associated with neurodevelopmental disorders or have neuronal phenotypes. H3K27me3 (red) histone mark is associated with transcriptional repression. The PRC2 complex catalyzes H3K27 methylation, its individual core subunits are shown in red. (C) Diagram illustrates the distribution of the different histone marks in different genomic locations including the enhancer, CpG island, promoter, transcription start site (TSS), gene body, and transcription end site (TES). Left panel depicts the histone modification distribution in a transcriptionally active gene. Right panel shows histone modifications in a transcriptionally silent gene (Lim et al., 2010).
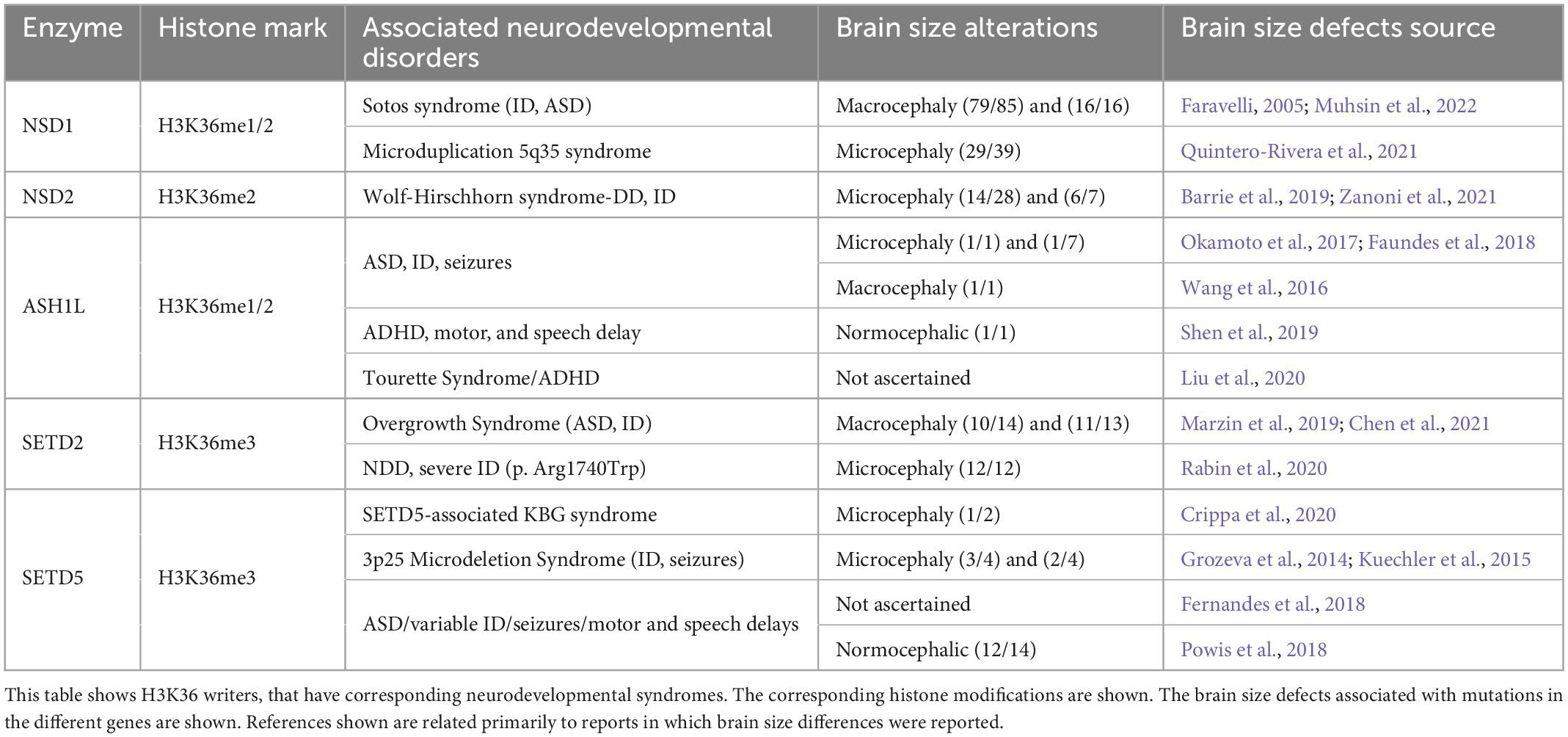
Table 1. Neurodevelopmental disorders associated with H3K36 histone methyltransferases.
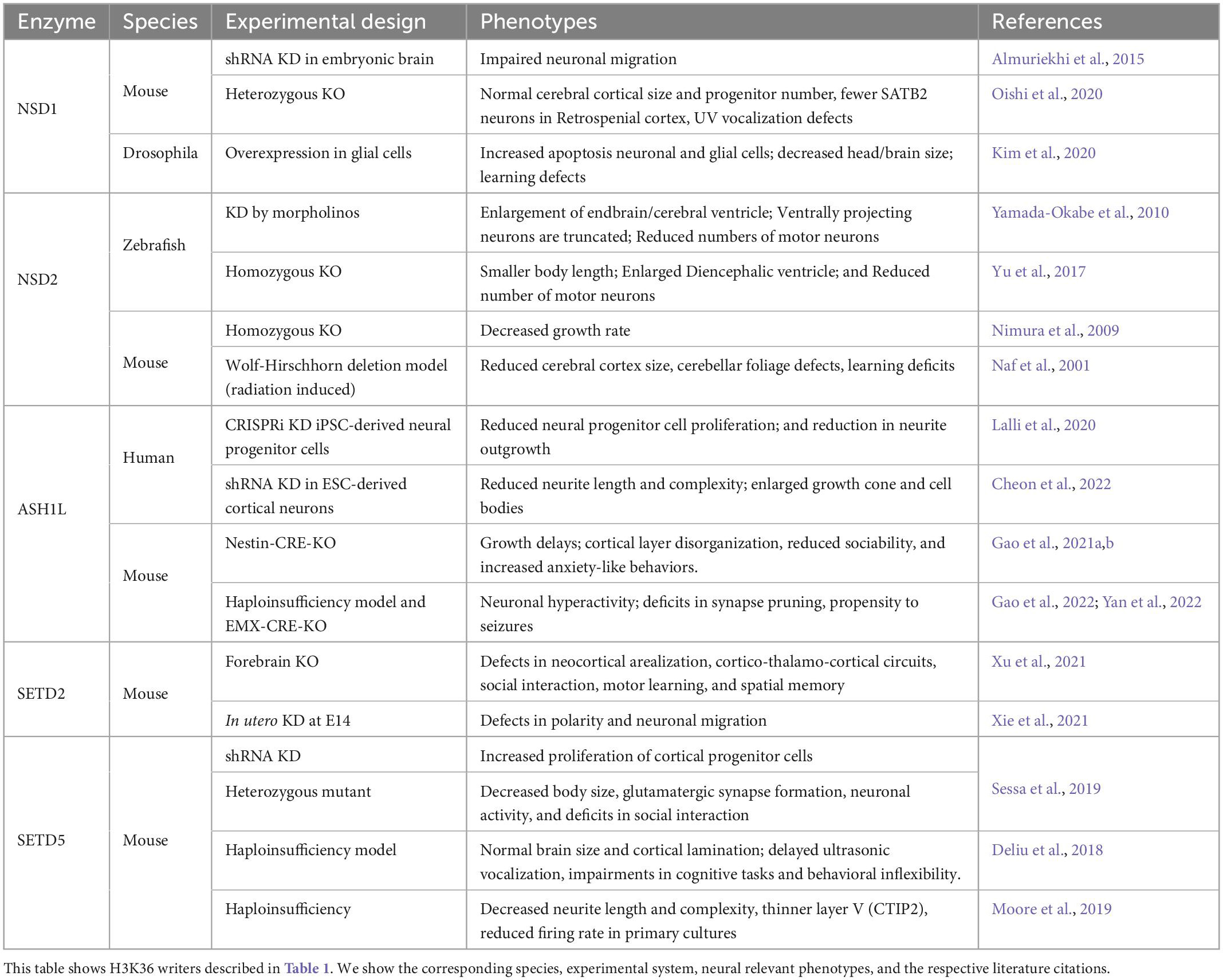
Table 2. Neuronal phenotypes associated with mutations in H3K36 histone methyltransferases.
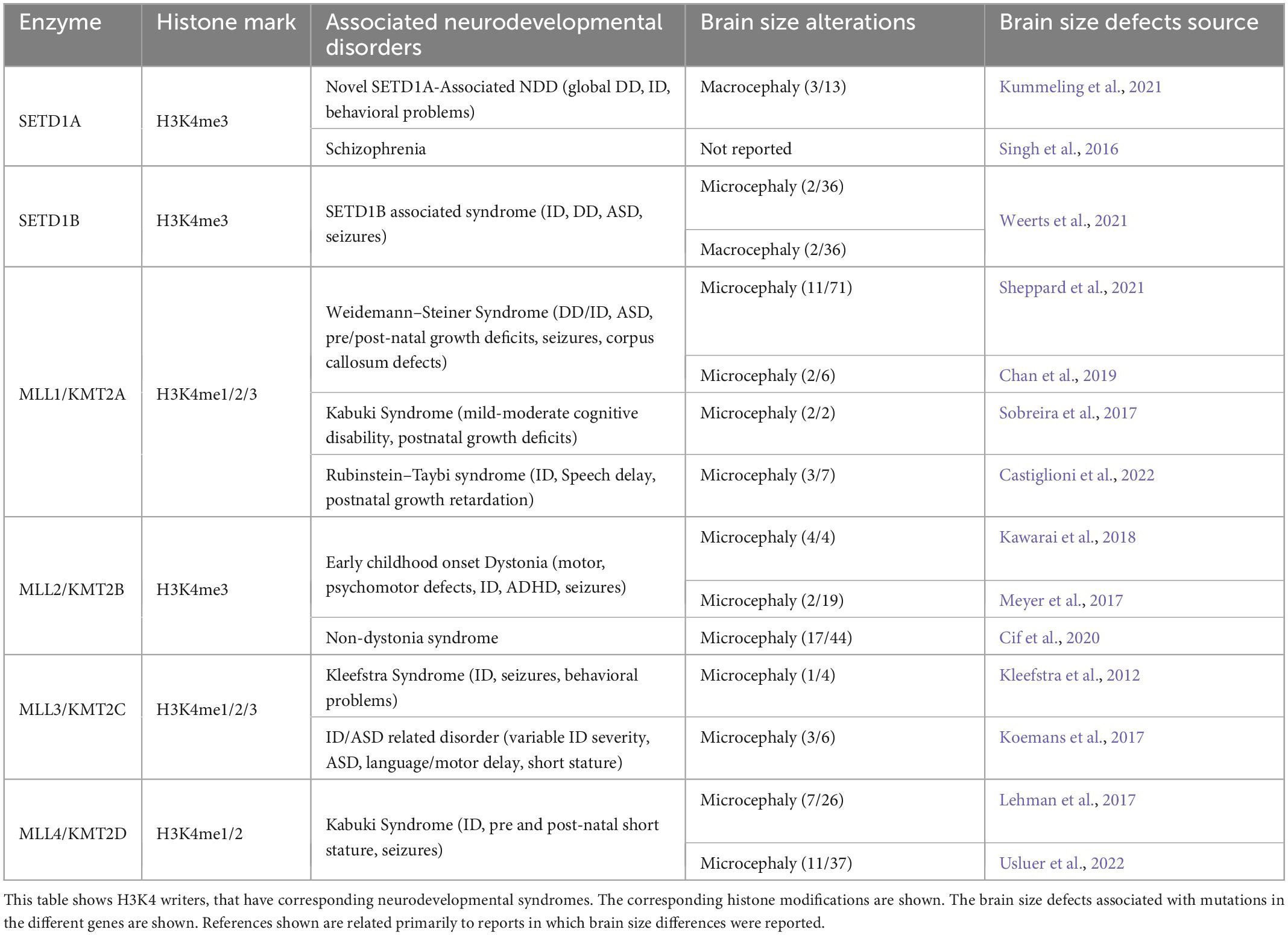
Table 3. Neurodevelopmental disorders associated with H3K4 histone methyltransferases.
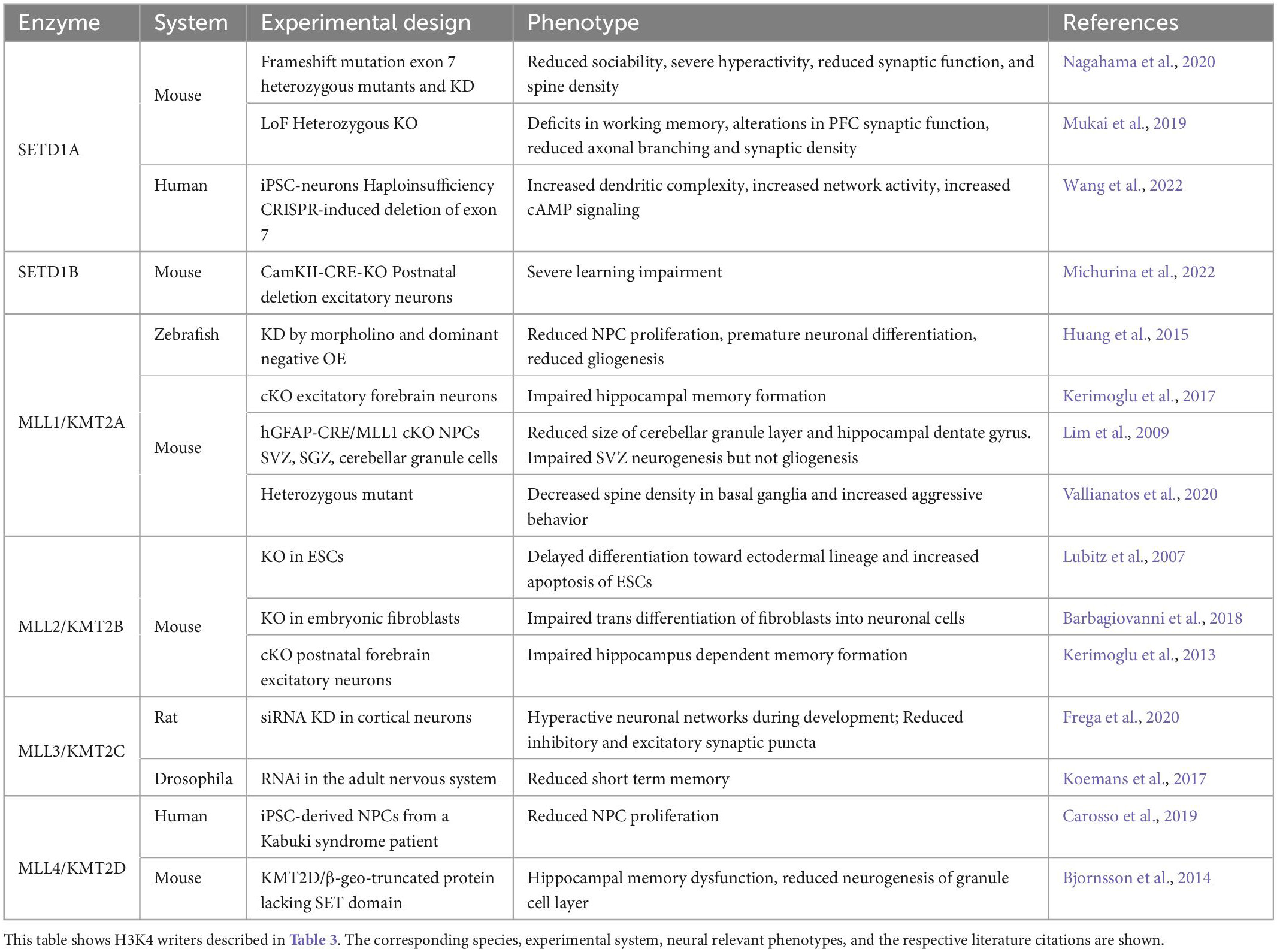
Table 4. Neuronal phenotypes associated with mutations in H3K4 histone methyltransferases.
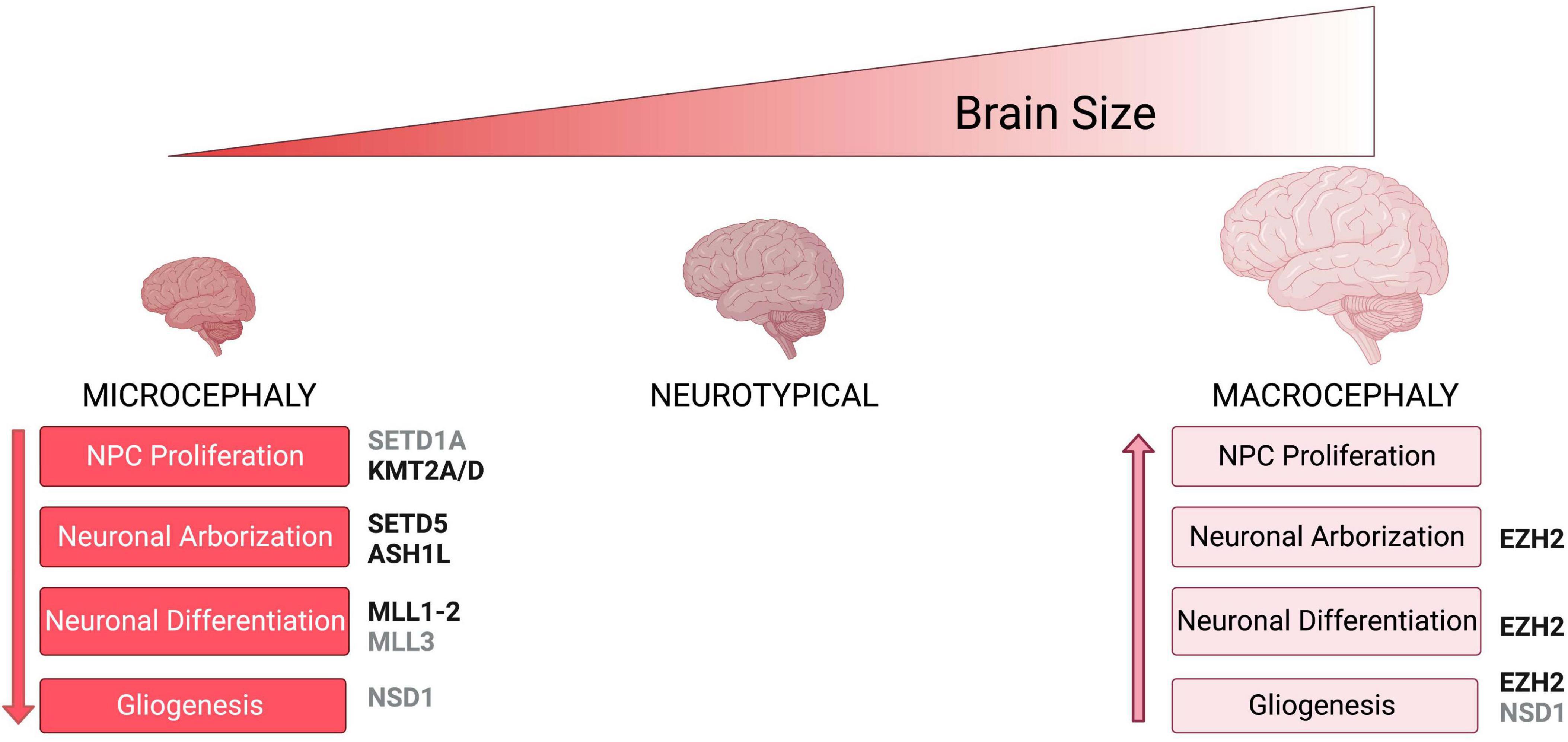
Figure 2. Neuronal phenotypes associated with brain size abnormalities. Schematic shows brain size abnormalities and potential cellular defects associated with differences in brain size. Microcephaly mechanism are shown in red while macrocephaly mechanisms are shown in light pink. Histone methyltransferases and their corresponding neuronal phenotypes that have been associated with either microcephaly or macrocephaly are shown in black, while enzymes for which there is no conclusive evidence on specific phenotypes are shown in gray.
2. H3K36 methylation and neurodevelopment
Methylation of Lysine 36 on Histone H3 (H3K36) has important biological implications due to its involvement in the regulation of transcriptional activation, splicing, and the control of spurious transcription (Wang et al., 2007; Pradeepa et al., 2012; Huang and Zhu, 2018). H3K36 methylation occurs in three different forms: mono-methylation (H3K36me), di-methylation (H3K36me2), and tri-methylation (H3K36me3). H3K36me is widely considered an intermediate step, but a function for H3K36me in the repair of double-strand breaks has been reported (Jha and Strahl, 2014). Both H3K36me2 and H3K36me3 are mainly contained within lightly packed regions of chromatin, indicating their role in transcriptional activation (Bannister et al., 2005; Wagner and Carpenter, 2012; Figure 1). H3K36me2 is mostly found in intergenic regions (Weinberg et al., 2019; Xu et al., 2020), while H3K36me3 localizes along gene bodies (Weinberg et al., 2019), and is proposed to serve as a hub to recruit proteins involved in RNA splicing (Pradeepa et al., 2012; Iwamori et al., 2016). Control of H3K36 methylation is modulated by H3K36 demethylases and methyltransferases. Mutations in these proteins have been associated with several neurodevelopmental disorders. Next, we highlight the cellular functions and neurological clinical findings associated with histone methyltransferases that deposit the H3K36me1/2/3 histone marks (NSD1, NSD2, ASH1L, SETD2, and SETD5) (Figure 1 and Tables 1, 2).
2.1. NSD1 gene dosage and the balancing act of large vs. small brains
Nuclear receptor SET domain-containing protein 1 (NSD1) catalyzes both the mono-methylation and di-methylation of H3K36 (Li et al., 2009). The fact that H3K36me1/2 could serve as a substrate for the tri-methylation of H3K36 (H3K36me3), suggests that NSD1 function might also contribute to the total levels of H3K36me3 (Lucio-Eterovic et al., 2010). Haploinsufficiency of NSD1 has been associated with syndromic (Kurotaki et al., 2002; Buxbaum et al., 2007; Koshimizu et al., 2013; Lane et al., 2017) and idiopathic ASD (De Rubeis et al., 2014; Iossifov et al., 2015; Yuen et al., 2017; Satterstrom et al., 2020). Loss of function mutations in NSD1 are a major cause of Sotos Syndrome (Kurotaki et al., 2002, 2005) which presents with macrocephaly, pre- and postnatal overgrowth, facial dysmorphism, ID, autistic features, developmental delay, and in some cases seizures (Sotos et al., 1964; Fortin et al., 2021). The macrocephaly phenotype associated with NSD1 mutations in Sotos syndrome has variable penetrance as 72–95% of patients present with larger head circumference (Cecconi et al., 2005; Faravelli, 2005; Saugier-Veber et al., 2007; Fortin et al., 2021; Muhsin et al., 2022). In contrast, duplication of NSD1 in the microduplication 5q35 syndrome leads to microcephaly in 74% of cases, with undergrowth, and developmental delay in close to 90% of the cases (Chen et al., 2006; Franco et al., 2010; Dikow et al., 2013; Rosenfeld et al., 2013; Quintero-Rivera et al., 2021). Therefore, differences in NSD1 gene dosage that elicit gain or loss of function phenotypes result in either microcephaly or macrocephaly, respectively. However, the precise molecular and cellular mechanisms regulated by NSD1 that influence the control of brain size are underexplored.
At the molecular level, NSD1 is proposed to regulate the recruitment of RNA Polymerase II to bone morphogenetic protein 4 (BMP4) nascent mRNA, indicating a role for NSD1 in transcriptional activation during development (Lucio-Eterovic et al., 2010). In embryonic stem cells (ESCs), NSD1 counteracts PRC2 activity by preventing non-specific deposition of H3K27me3 along the genome, which will prevent random transcriptional gene repression by PRC2 (Streubel et al., 2018). Therefore, NSD1 promotes a chromatin environment that is permissive of transcriptional activation to ensure the normal progression of organismal development. In the developing human cortex, NSD1 is highly expressed in RGCs, oRGCs, IPCs, and subsets of excitatory and inhibitory neurons suggesting a major role for NSD1 in the regulation of these neural cell types (Speir et al., 2021). In fact, acute knockdown of Nsd1 in mouse cortical progenitors led to migration defects in cortical neurons and correlated with differential expression of gene programs that regulate neuronal migration and cell fate decisions (Almuriekhi et al., 2015). In contrast, studies in Nsd1 heterozygous mutant mice showed that while they did not present defects in overall brain architecture, they did show reduced social novelty which is a hallmark of an autism-like behavior (Oishi et al., 2020). There are currently no duplication mouse models for NSD1 function. However, overexpression studies in Drosophila showed that increased dosage of Nsd1 in glial cells lead to cell death of glial and neuronal cells resulting in smaller head sizes (Kim et al., 2020). Taken together these studies suggest that while NSD1 contributes to the development of the brain architecture and circuitry there might be cell type and species-specific differences associated with its function.
2.2. NSD2 influencing brain size through NPC proliferation and survival dynamics
Nuclear receptor SET domain-containing protein 2 (NSD2) is a catalyst for the di-methylation of H3K36. In humans, NSD2 is located in a critical region of microdeletion 4p16.3 which is associated with Wolf-Hirschhorn syndrome (Bergemann et al., 2005), which presents with microcephaly at birth (Bernardini et al., 2018; Zanoni et al., 2021), global developmental delay, postnatal growth deficiency, ID, distinct craniofacial features, and seizures (Kagitani-Shimono et al., 2005; Battaglia et al., 2021; Wiel et al., 2022). Loss of function mutations in NSD2 have been associated with ASD across multiple genetics studies (De Rubeis et al., 2014; Wang et al., 2016; Yuen et al., 2017), and replicate several of the clinical presentations of Wolf–Hirschhorn syndrome including microcephaly, global developmental delay, and ID, but do not present seizures (Boczek et al., 2018; Barrie et al., 2019; Wiel et al., 2022). Therefore, the clinical evidence suggests an essential role for NSD2 in the development of human neuronal circuitry.
In non-neuronal cells, NSD2 is proposed to regulate cellular proliferation and survival (Park et al., 2018). Therefore, it is possible that the microcephaly phenotypes in humans with mutations in NSD2 could arise from defects in NPC proliferation. Mouse models with different deletions that affect Nsd2 present with a reduced cerebral cortex (Naf et al., 2001), while Zebrafish null for nsd2 showed reduced numbers of motor neurons with delayed axon outgrowth (Yamada-Okabe et al., 2010; Yu et al., 2017). The reduction in cerebral cortex size in mice or the reduction in the numbers of motor neurons could be a consequence of reduced NPC proliferation, altered cell fate or increase neuronal cell death. However, additional mechanistic studies using human systems are needed to address the role of NSD2 in the control of brain size and neuronal function at large.
2.3. ASH1L a potential regulator of prenatal and postnatal brain growth
Similar, to the NSD proteins, the Absent, small, or homeotic-1 like (ASH1L) methyltransferase catalyzes both the mono- and di-methylation of H3K36 (H3K36me1, H3K36me2) (Tanaka et al., 2007, 2011). ASH1L might also be indirectly associated with tri-methylation of H3K4 (H3K4me3) (Gregory et al., 2007). De novo loss of function and missense mutations in ASH1L reached genome wide significance across large genetics studies in ASD (Willsey et al., 2013; Iossifov et al., 2014, 2015; Tammimies et al., 2015; Wang et al., 2016; Stessman et al., 2017). Mutations in ASH1L have also been reported in severe forms of ASD, presenting with ID, seizures, speech difficulties, and in a small number of cases microcephaly and macrocephaly (Homsy et al., 2015; Wang et al., 2016; Faundes et al., 2017, 2018; Okamoto et al., 2017; Stessman et al., 2017). Additional pathogenic variants in ASH1L have been associated with Attention-deficit/hyperactivity disorder (ADHD) (Satterstrom et al., 2019), Tourette syndrome (Liu et al., 2020; Zhang et al., 2021), and epilepsy (Tang et al., 2020). The association of ASH1L deficits with multiple neurological disorders suggests that common downstream mechanisms could underlie the diverse neuropathology in ASH1L-related disorders.
ASH1L is emerging as a major modulator of neurodevelopment and insight into its function has come from multiple studies that integrate mouse and human experimental systems. CRISPR-inactivation of ASH1L in LUHMES neural-like progenitor cells, which are of human mesencephalic origin and give rise to dopaminergic-like neurons, resulted in delayed neuronal differentiation (Lalli et al., 2020). Similarly, introduction of truncating frameshift mutations in ASH1L by CRISPR/CAS9 genome editing led to reduced neuronal output that correlated with increased neural stem cell production in a human induced pluripotent stem cell (iPSC)-model of prefrontal cortex neurogenesis (Cederquist et al., 2020). Together these studies suggest a role for ASH1L in the regulation of neuronal numbers. However, additional evidence suggests that ASH1L functions in a cell-type dependent manner affecting additional processes important for neuronal morphogenesis and synaptic function. For example, knockdown of ASH1L in human ESC-derived neurons led to severe reduction in neurite outgrowth (Lalli et al., 2020; Cheon et al., 2022). In vivo, knockout of Ash1l in mice lead to an overall growth retardation at later postnatal stages and revealed cortical malformations (Gao et al., 2021b). Specifically, knockout of Ash1l led to a disorganized cortical architecture, with SATB Homeobox 2 (Satb2), a marker specific for cortical layer 2/4 being observed in deeper layers (Gao et al., 2021b). These results suggested that Ash1l knockout led to the delayed lamination of neuronal cells. Recently, conditional knockout of Ash1l in mice demonstrated a role for ASH1L in synaptic function and synapse remodeling. Ash1l -haploinsufficiency resulted in neuronal hyperactivity (Gao et al., 2022) and increased synapse numbers due to deficits in synapse elimination (Yan et al., 2022). Together the clinical and mechanistic studies discussed above, suggest that ASH1L regulates gene programs relevant to the proper development of the brain architecture, wiring, and possibly the control of brain size.
2.4. SETD2 gain or loss of function differentially impact brain size
In contrast to the NSDs and ASH1L proteins, the Set Domain Containing 2 (SETD2) catalyzes exclusively the tri-methylation of H3K36 (Edmunds et al., 2008), which is a histone mark mostly found in the gene body of transcriptionally active genes. Several clinical studies suggest that loss of SETD2 could impact neuronal connectivity and brain size. De novo loss of function mutations in SETD2 have been associated with idiopathic ASD (O’Roak et al., 2012a,b; Wang et al., 2020), and a syndrome that presents with variable degrees of ID, autistic behaviors, overgrowth, seizures, speech delays, facial dysmorphisms, and macrocephaly (Lumish et al., 2015; van Rij et al., 2018; Marzin et al., 2019; Chen et al., 2021). In contrast, a recent report of patients with a gain of function mutation (p. Arg1740Trp) in SETD2, presented microcephaly and epilepsy in 12 out of 12 cases (Rabin et al., 2020). Therefore, SETD2 like the NSDs and ASH1L, appears to be important for the proper development of neuronal connectivity and for the control of brain size.
SETD2 is a multifaceted protein that regulates transcriptional activation, modulates transcriptional elongation through its interaction with RNA Polymerase II (Kizer et al., 2005), is essential in the control of co-transcriptional splicing (Leung et al., 2019), and contributes to DNA repair through promotion of homologous recombination (Pfister et al., 2014). In the human developing cortex SETD2 is widely expressed in oRGCs, RGCs, IPCs, astrocytes, excitatory, and inhibitory neurons (Speir et al., 2021), which suggests widespread functions in multiple neural cell types. Hence cell specific knockout studies will be important to define cell-type specific mechanisms governed by SETD2 during the development of neuronal circuitry. In fact, knockout of Setd2 in the developing forebrain revealed abnormal cortical arealization and aberrant thalamo-cortico-thalamic circuits in mice that also had defects in social interaction, motor learning and spatial memory (Xu et al., 2021). The observed neural phenotypes associated with SETD2 dysfunction could be related in part to changes in the chromatin environment. For instance, SETD2 interacts with histone H2 variant H2A.Z to regulate the H3K36me3 modification of the NK2 Homeobox 4 (Nkx2.4) gene (Shen et al., 2018; Zhang et al., 2020). Deficits in both Nkx2.4 and H2A.Z promote NPC proliferation and inhibit neuronal differentiation (Shen et al., 2018; Zhang et al., 2020). However, only deficits in H2A.Z lead to dendritic arborization defects (Shen et al., 2018). Together deficits in NPC proliferation, neuronal differentiation, and arborization could contribute to the defects in arealization and the aberrant thalamo-cortical circuits associated with loss of Setd2 (Xu et al., 2021). Therefore, we posit that the modulation of the chromatin environment by SETD2 plays an important role in the regulation of cell fate transitions during neurogenesis, and the structural development of neuronal circuits.
2.5. SETD5 dysfunction in microcephaly related syndrome
Like SETD2, the SET Domain Containing 5 (SETD5) histone methyltransferase appears to exclusively catalyze the tri-methylation of H3K36 (Sessa et al., 2019). SETD5 is one of 3 genes located in a critical region of the 3p25 microdeletion syndrome. Large deletions in 3p25 present with varying degrees of ID, and facial dysmorphisms and could include seizures, cardiac defects, and microcephaly, yet smaller deletions containing the critical interval only present with ID and facial dysmorphisms (Kellogg et al., 2013; Grozeva et al., 2014; Kuechler et al., 2015). Similarly, haploinsufficiency of SETD5 has been identified as a frequent cause of ID based on whole exome sequencing studies (Kuechler et al., 2015) and has been associated with variable dysmorphic features, ASD, and ADHD (De Rubeis et al., 2014; Grozeva et al., 2014; Szczałuba et al., 2016; Rawlins et al., 2017; Fernandes et al., 2018; Powis et al., 2018; Satterstrom et al., 2020). Finally, loss of function mutation in SETD5 was identified in a patient with KBG syndrome that also presented with microcephaly (Crippa et al., 2020).
Loss of SETD5 causes abnormal transcription within neural stem cells (Nakagawa et al., 2020). In neonatal mouse brains, knockdown of Setd5 led to an increase in the proliferation of cortical progenitor cells, and a significant decrease in glutamatergic synaptic formation (Sessa et al., 2019). In addition, Setd5 haploinsufficiency in mice was associated with decreased neurite length, reduced arbor complexity and autism-like behaviors (Moore et al., 2019). Furthermore, a different study of mice haploinsufficient for SETD5 showed that despite an overall normal brain architecture and cortical lamination, the animals presented defects in ultrasonic vocalization and cognitive tasks (Deliu et al., 2018). Taken together these studies demonstrate an essential role for H3K36me3 methylation mediated by SETD5 during neurogenesis, neuronal differentiation, maturation, and synaptic function.
In summary, mounting evidence suggests a major role for H3K36 methyltransferases in the regulation of early brain development and the proper wiring of neural circuits. H3K36 methyltransferases have been implicated in multiple processes that can contribute to the control of brain size including cell fate transitions, NPC proliferation and neuronal arborization. Next, we present an overview of another group of histone Lysine methyltransferases with major roles in neuronal development.
3. H3K4 methylation and neurodevelopment
H3K4 methylation occurs in three states: mono-, di-, and tri-methylation (H3K4me, H3K4me2, and H3k4me3) and has previously been associated with transcriptional activation (Barski et al., 2007; Zhu et al., 2013; Figure 1). The three different H3K4 methylation states are differentially distributed across the genome, with H3K4me1 being present primarily at enhancer regions, while H3K4me2/3 are enriched at transcriptional active promoters (Collins et al., 2019). Histone H3K4 methyltransferases and demethylases control the transition between these different methylation states (Shilatifard, 2008). H3K4 histone methyltransferases have been widely studied in cancer biology (Deb et al., 2014; Li et al., 2018; Yang et al., 2021), and have been strongly implicated in various neurodevelopmental disorders, including ASD and ID (Wynder et al., 2010; Vallianatos and Iwase, 2015; Kim et al., 2017; Tables 3, 4).
3.1. MLL1/KMT2A a central contributor to the pathogenesis of multiple microcephaly associated syndromes
Mixed lineage-leukemia 1 (MLL1), also known as Lysine methyltransferase 2A (KMT2A), is a member of the Trithorax Group (TrxG) of proteins. MLL1/KMT2A catalyzes the mono, di and tri-methylation of H3K4 (H3K4me1/2/3). Dominant de novo mutations in MLL1/KMT2A are associated with Weidemann–Steiner syndrome. Patients with Weidemann–Steiner syndrome exhibit ID, microcephaly, short stature, and a subset present with ASD (Jones et al., 2012; Strom et al., 2014; Chan et al., 2019; Sheppard et al., 2021). Large scale exome sequencing studies identified mutations in MLL1/KMT2A among patients presenting with developmental delay and ID (Deciphering Developmental Disorders Study, 2015; Lelieveld et al., 2016; Trujillano et al., 2017). Clinical genetics studies identified MLL1/KMT2A pathogenic variants in patients diagnosed with either Kabuki syndrome or Rubinstein–Taiby syndrome which are characterized by ID, postnatal growth defects and microcephaly (Sobreira et al., 2017; Castiglioni et al., 2022).
MLL1/KMT2A-mediated H3K4 methylation has been associated with the regulation of transcriptional initiation through recruitment of RNA Polymerase II (Wang et al., 2009). Morpholino knockdown of the MLL1/KMT2A homologue in Zebrafish embryos showed reduced NPC proliferation, premature neuronal differentiation, and reduced numbers of glial cells (Huang et al., 2015). Similarly, in human iPSC-derived neural stem cells loss of function mutations in MLL1/KMT2A introduced by CRISPR/CAS9 genome editing led to neurogenesis defects that resulted in the depletion of the neural stem cell population and premature neuronal differentiation (Cederquist et al., 2020). In contrast, Mll1/Kmt2a—deficient SVZ adult neural stem cells showed severe impairment of neuronal differentiation but normal gliogenesis (Lim et al., 2009). In addition to its role in neurogenesis, MLL1/KMT2A also modulates synaptic function. Mice heterozygous for Mll1/Kmt2a showed reduced numbers of dendritic spines (Vallianatos et al., 2020), and the specific ablation of Mll1/Kmt2a in mice prefrontal cortical neurons lead to defects in short-term synaptic plasticity (Jakovcevski et al., 2015). Furthermore, the synaptic defects identified in Mll1/Kmt2a mutant mice lead to increased aggressive behaviors and social dominance (Vallianatos et al., 2020), as well as deficits in working memory (Jakovcevski et al., 2015). In summary, MLL1/KMT2A control of H3K4 methylation might be species and cell type specific but is overall essential for the proper development of brain architecture and wiring.
3.2. MLL2/KMT2B in dystonia and brain size control
Mixed lineage-leukemia 2 (MLL2), also known as Lysine methyltransferase 2B (KMT2B), tri-methylates H3K4 (Denissov et al., 2014). Mutations in MLL2/KMT2B have been commonly associated with early onset generalized dystonia, which is a disorder characterized by sustained muscle contractions causing abnormal, repetitive movements, and in a small number of cases has been associated with ID, facial dysmorphisms and microcephaly (Albanese et al., 2013; Zech et al., 2016; Meyer et al., 2017; Kawarai et al., 2018). In contrast, a subset of non-dystonia patients with mutations in MLL2/KMT2B presented with neurodevelopmental delay, ID, microcephaly, short stature, and facial dysmorphic features (Cif et al., 2020).
Knockout of Mll2/Kmt2b in mouse ESCs resulted in a severe delay in differentiation toward the ectodermal lineage which gives rise to the neuroectoderm (Lubitz et al., 2007). Further, inactivation of Mll2/Kmt2b resulted in impaired trans differentiation of mouse fibroblasts into neuronal cells, and disruption of a network of genes responsible for neuronal maturation, suggesting that MLL2/KMT2B is required for the activation of the neuronal maturation programs during this process (Barbagiovanni et al., 2018). In vivo, post-natal deletion of Mll2/Kmt2b from excitatory forebrain neurons led to impaired memory consolidation (Kerimoglu et al., 2013). Therefore, similar to MLL1/KMT2A, MLL2/KMT2B is also essential for the regulation of gene networks that modulate memory processes. However, the gene programs modulated by both proteins are almost non-overlapping (Kerimoglu et al., 2017).
3.3. MLL3/KMT2C
Mixed lineage-leukemia 3 (MLL3), also known as Lysine methyltransferase 2C (KMT2C), catalyzes the mono- and di-methylation of H3K4 (Hu et al., 2013; Lee et al., 2013). H3K4me and H3K4me2 have been identified at enhancer regions leading to transcriptional activation (Hu et al., 2013). Pathogenic mutations in MLL3 are associated with Kleefstra syndrome, ID, behavioral problems, and epileptic seizures as its core features, while in some cases microcephaly is present (Kleefstra et al., 2012; Frega et al., 2020). Pathogenic variants in MLL3 were identified in large whole exon sequencing studies in patients with ASD (De Rubeis et al., 2014; Iossifov et al., 2014), and have been associated with an ID/ASD related syndrome that presents microcephaly in 50% of the cases reported (Koemans et al., 2017). Therefore, suggesting the importance of this member of the MLL family of proteins in the development of neuronal circuitry.
MLL3/KMT2C is widely expressed in the developing human fetal brain (Johnson et al., 2009). Introduction of frameshift mutations in MLL3/KMT2C by CRISPR/CAS9 genome editing in iPSC-derived neural stem cells render them unable to generate neurons (Cederquist et al., 2020). Work in other species suggests that MLL3/KMT2C might have additional roles outside of the control of neurogenesis, as knockdown of MLL3/KMT2C in rat primary neurons led to a hyperactive state of mature neuronal networks compared to control neurons (Frega et al., 2020). In Drosophila, knockdown of the MLL3/KMT2C homologue in the adult nervous system led to reduced short-term memory, but no effect on cell fate decisions or increase in cell death was observed (Koemans et al., 2017). Taken together, these studies across multiple species suggest that MLL3/KMT2C drives temporally distinct programs as development proceeds.
3.4. MLL4/KMT2D in Kabuki syndrome associated with microcephaly
Like MLL3/KMT2C the paralogous MLL4/KMT2D can also catalyze the mono- and di-methylation of H3K4 (Hu et al., 2013; Lee et al., 2013). Mutations in MLL4/KMT2D have been identified as the most common cause of Kabuki syndrome, which is characterized by distinctive facial features, microcephaly, growth delay, cardiac anomalies, and varying degrees of ID (Lehman et al., 2017; Di Candia et al., 2022; Usluer et al., 2022; Table 3).
Work in human iPSC-derived NPCs from a Kabuki syndrome patient with a nonsense mutation in MLL4/KMT2D showed a reduction in NPC proliferation that resulted in precocious neuronal differentiation (Carosso et al., 2019). Similarly, a study of a mouse with a truncation mutation in the catalytic domain of Mll4/Kmt2d led to reduced neurogenesis of the granule cell layer. Taken together, these studies suggest a major role for MLL4/KMT2D regulating the numbers of NPCs during embryonic and adult neurogenesis.
3.5. SETD1A is a major factor in neuropsychiatric disorders
SET Domain Containing 1A (SETD1A) catalyzes the tri-methylation of H3K4 (Lee and Skalnik, 2008), and has been shown to be important for the maintenance of genome stability during DNA replication (Higgs et al., 2018). Haploinsufficiency of SETD1A was identified in patients with a novel neurodevelopmental disorder, characterized by global developmental delay, ID, facial dysmorphisms, psychosis, behavioral and psychiatric abnormalities, and in some cases autistic-like behaviors (Kummeling et al., 2021). In contrast, a de novo mutation in SETD1A was associated with macrocephaly but was not co-diagnosed with ASD (Zhang et al., 2022). Recently, a large human genetics study of schizophrenia identified rare coding variants in SETD1A raising above genome-wide significance (Singh et al., 2022). Clearly, SETD1A has a major role in the proper development of neuronal circuits, however, additional evidence is needed to determine how common is the macrocephaly phenotype in patients with SETD1A mutations.
SETD1A has been proposed to regulate neuronal progenitor proliferation through its interaction with Histone Cell Cycle Regulator (HIRA) (Li and Jiao, 2017) could contribute to the regulation of neuronal differentiation and excitability by transcriptionally activating β-catenin (Li and Jiao, 2017), a key component of the Wnt/β-catenin pathway which is essential for neurogenesis (Hirabayashi et al., 2004; Zhang et al., 2011; Rubio et al., 2020). In mice, a missense mutation in SETD1A resulted in faster migration of neurons within the cortex (Yu X. et al., 2019), suggesting that neuronal migration could also be regulated by SETD1A. Mice heterozygous for Setd1a showed decreased axonal branching, reduced dendritic spines, and altered cortical synaptic dynamics that are associated with defects in working memory and social interaction (Mukai et al., 2019; Nagahama et al., 2020). In contrast, iPSC-derived human glutamatergic and GABAergic neurons haploinsufficient for SETD1A had enlarged neuronal arbors, increased network activity, and synaptic connectivity (Wang et al., 2022). Taken together, mounting evidence suggests an essential role for SETD1A in the regulation of neurogenesis and neuronal connectivity, that might differ in a species-specific manner.
3.6. SETD1B and the balancing act between a large vs. a small brain
SET Domain Containing 1B (SETD1B) is the catalytic subunit of the Complex Proteins Associated with SET1 (COMPASS) that catalyzes the tri-methylation of H3K4 (Lee et al., 2007). Human pathogenic variants in coding regions of SETD1B have been associated with ASD, ID, and epilepsy (Den et al., 2019; Hiraide et al., 2019; Roston et al., 2021). Additionally, SETD1B has been identified as a candidate gene for 12q24.31 microdeletion syndrome, which presents with ID, autism-like features, facial dysmorphisms, and epilepsy (Baple et al., 2010; Palumbo et al., 2015; Labonne et al., 2016). Both microcephaly and macrocephaly have been associated with SETD1B mutations. However, this is a variable phenotype as in each only 2 out of 36 patients were reported to have significant changes in head circumference (Weerts et al., 2021). In mice, deletion of Setd1b at early embryonic stages did not appear to be required for neural stem cell (NSC) survival or proliferation (Bledau et al., 2014). However, postnatal deletion of Setd1b in excitatory forebrain cortical neurons led to severe learning impairment and altered the expression of neural genes involved in learning and memory (Michurina et al., 2022). Therefore, SETD1B appears to control primarily neuronal function.
In summary, the different H3K4 methyltransferases control various stages of neuronal development that can influence brain size and cognition by controlling different H3 methylation states that influence distinct gene programs.
4. The role of H3K27 methylation by PRC2 in neurodevelopment and brain size control
Polycomb group complex genes are a family of chromatin regulators closely associated with gene silencing. In particular, PRC2 plays a vital role in the epigenetic regulation of gene expression during normal development (Deevy and Bracken, 2019). PRC2 catalyzes H3K27me3 (Yu J. et al., 2019), which is a repressive histone modification associated with gene silencing (Figure 1). PRC2 is composed of four core subunits: EZH1/2, SUZ12, EED, and RbAp46/48. Enhancer of Zeste Homolog 1 or 2 (EZH1/2) is the main catalytic subunit of PRC2 and it catalyzes the mono-, di-, or tri-methylation of H3K27 (Di Croce and Helin, 2013). Embryonic ectoderm development (EED) subunit of PRC2 plays an important role in maintaining H3K27me3 through the binding of H3K27me3 to its C-terminal domain. Suppressor of zeste 12 (SUZ12) helps to maintain the stability and catalytic activity of EZH2. Retinoblastoma-associated protein 46/48 (RbAP46/48) is required for the association of PRC2 to histone tails.
Different core subunits of PRC2 have been linked to multiple neurodevelopmental disorders. Mutations in EZH2 (catalytic subunit of PRC2) have been associated with Weaver Syndrome, which is characterized by overgrowth and ID (Tatton-Brown et al., 2013). Further, mutations in EZH2 have also been suggested to contribute to the genetic ethology of ASD in the Chinese Han population (Li et al., 2016). Previous studies suggest that a subset of ASD cases might be associated with larger brain size (Sacco et al., 2015). However, the extent to which ASD patients with EZH2 pathogenic variants also present macrocephaly is unknown. Similar to EZH2, several mutations in EED have also been identified in patients with Weaver syndrome (Cooney et al., 2017; Cyrus et al., 2019; Griffiths et al., 2019), while mutations in SUZ12 have been associated with a Weaver-like syndrome presenting with macrocephaly, postnatal overgrowth, and skeletal abnormalities (Imagawa et al., 2017, 2018). Similarly, mutations in non-core subunits of PRC2 have also been shown to have an important role in neuronal development. Jumonji and AT-Rich Interaction Domain Containing 2 (JARID2) is a non-core subunit of PRC2 that acts as an activator of Polycomb (Kasinath et al., 2021). Mutations in JARID2 have been associated with developmental delay, variable degrees of ID, facial dysmorphisms (Verberne et al., 2021), and ASD (Weiss et al., 2009; Ramos et al., 2012). However, to date no brain size alterations correlate with pathogenic variants in JARID2.
Based on the clinical studies, PRC2 has an important role in neuronal development and can impact brain size. We posit that PRC2 modulates brain growth through the control of gene programs that impact neural cell fate decisions and neuronal morphogenesis (Pereira et al., 2010; Liu et al., 2018). In particular the tight regulation of the switch from a neurogenic to an astrocytic cell fate could alter brain size. Knockdown of EED in mice NPCs showed a lengthening of the neurogenic phase while there was a shortening of the astrocytic phase (Hirabayashi et al., 2009). Similarly, deletion of EZH2 in cortical progenitors led to an increase in the number of neurons (Pereira et al., 2010) which correlated with a lengthening of the neurogenic phase (Hirabayashi et al., 2009). Loss of EZH2 in primary neurons led to increased neuronal arborization and advanced migration of cells to the upper layers (Pereira et al., 2010). Alterations in neuronal arborization is a mechanism that could contribute to changes in brain size postnatally. The defects in neuronal arborization associated with EZH2 dysfunction might be mediated in part by alterations in the Brain-derived Neurotrophic Factor (BDNF)/TrkB signaling pathway which is a major regulator of neuronal arborization, synaptic function and survival (Gonzalez et al., 2016). EZH2 regulates BDNF expression through its interaction with Chromodomain Y Like (CDYL). CDYL recognizes H3K27me3 and recruits additional PRC2, leading to a restriction of dendritic morphogenesis (Qi et al., 2014), which could result from increased repression of gene expression by elevated levels of H3K27me2 repressive marks. Similarly, deletion of Lysine demethylase 6A (UTX), an H3K27 demethylase, resulted in abnormal dendritic development and reduced synaptic formation coinciding with increased levels of H3K27me3 (Tang et al., 2017). Taken together, these results implicate the restriction of PRC2 activity as essential for proper neuronal differentiation, migration, and morphogenesis.
5. Counteracting activities between H3K36 and H3K4 methylation and PRC2-mediated H3K27 methylation
The interplay between PRC2 methylation of H3K27 and Trithorax methylation of H3K36 in the regulation of gene expression is a known mechanism essential during development. However, the implications of PRC2/Trithorax counteracting activities in the formation of proper neuronal architecture and circuitry are largely understudied (Schuettengruber et al., 2017). Deposition of H3K36 methylation marks provides a steric hindrance to PRC2 activity by preventing it from methylating H3K27me3. The majority of our current knowledge on how these counteracting epigenetic mechanisms work is based on studies of non-neuronal systems. For example, knockdown of Nsd1 in mouse ESCs resulted in a genome-wide reduction of H3K36me2 levels coupled with increased levels in H3K27me3. Additionally, NSD1 is required in ESCs for the demarcation of H3K27me3 and H3K27me2, further corroborating the importance of the counteracting activity between H3K36 histone methyltransferase activity and PRC2 activity (Streubel et al., 2018). However, the mechanism by which NSD1 regulates the demarcation of H3K27me3 and H3K27me2 in neurons is not currently known. RNA-sequencing of a Nsd1 knockout mouse revealed counteracting regulation of PRC2-associated genes (Shirane et al., 2020). Knockdown of both Nsd1 and Nsd2 in mice brown preadipocytes resulted in decreased H3K36me2 and increased H3K27me3, with a more substantial change seen when Nsd2 was knocked down compared to Nsd1 (Zhuang et al., 2018). Similarly, in Drosophila, ASH1L antagonizes PRC2 activity by preventing the methylation of H3K27me3 (Yuan et al., 2011). ASH1L knockdown in bovine cumulus cells resulted in increased mRNA expression of EZH2 and SUZ12 which in turn could lead to higher levels of H3K27me3 methylation. These studies demonstrate that the opposing activities of H3K36 modifying enzymes and PRC2-mediated methylation of H3K27 is a conserved epigenetic mechanism. ASH1L has also been shown to form a functional interaction with CREB-binding protein (CBP) in Drosophila (Bantignies et al., 2000). CBP is a histone acetyltransferase that acetylates H3K27, preventing the methylation of H3K27 (Tie et al., 2014). Therefore, ASH1L by directly methylating H3K36me2 and indirectly through its interaction with CBP could oppose the catalytic activity of PRC2.
However, despite the antagonizing relationship between H3K36 methyltransferase activity and PRC2 activity, there is some evidence that PRC2 directly binds to regions containing H3K36 methylation. The introduction of PRC2 to H3K36 methylated regions is driven by PHD Finger Protein 1 (PHF1) and PHD Finger Protein 19 (PHF19) (Cai et al., 2013), which contain Tudor domains that recognize H3K36me3. Recognition of H3K36 methylation marks by PHF1 or PHF19 promotes the introduction of PRC2, leading to gene silencing in these regions. PHF1 is also important for the stabilization of PRC2 on chromatin (Cai et al., 2013). These results suggest a more direct interaction between these two histone methylation marks.
Finally, besides the opposing activities of H3K36 vs. H3K27 methylation states, H3K4 methylation also provides a counterbalancing mechanism to H3K27 modifiers. Mechanistically, H3K4 histone marks are proposed to indirectly prevent the methylation of H3K27 (Tie et al., 2014). Specifically, methylated H3K4 binds CBP, which by acetylating H3K27 prevents its methylation (Tie et al., 2014). Comparably, H3K27 tri-methylation has been shown to inhibit the binding of the SET1-like H3K4 methyltransferase complex to histone H3 (Kim et al., 2013). In addition, the MLL3/4 COMPASS-like complex contains the histone demethylase KDM6/UTX, which is able to remove the H3K27me3 repressive mark produced by PRC2 (Agger et al., 2007; Kim et al., 2014; Piunti and Shilatifard, 2016). Therefore, there is an interplay of opposing activities between H3K4 and H3K27 modifying enzymes that modulate the transition between transcriptional activation and repression (Piunti and Shilatifard, 2016; Schuettengruber et al., 2017).
6. Impact of histone methyltransferases on brain size and circuitry development–Integrating the clinical findings with the underlying biological mechanisms
6.1. Microcephaly
Different cellular mechanisms can contribute to a reduced brain size. Here we discuss two major forms of microcephaly (primary and postnatal) associated with genetic causes and the cellular mechanisms that could underlie the development of a microcephalic brain. Primary microcephaly is characterized by a small brain size in the absence of major brain malformations at birth. Some of the major cellular mechanisms that underlie primary microcephaly pathogenesis are reduced NPC proliferation, increased cell death of NPC, and changes in cell fate determination (associated with premature cell cycle exit) (Wollnik, 2010). This is not surprising as several of the key primary microcephaly genes (CDK5RAP2, ASPM, WDR62, and MCPH1) (Jackson et al., 2002; Bond et al., 2005; Shen et al., 2005; Yu et al., 2010) are essential for the control of mitosis and cell cycle progression (Cox et al., 2006; Jean et al., 2020).
Alternately, postnatal microcephaly has been associated with defects in axon and dendritic arborization (Kwon et al., 2006; Dindot et al., 2008; Belichenko et al., 2009; Zikopoulos and Barbas, 2010), synaptogenesis (Giedd et al., 1999), gliogenesis (Zuchero and Barres, 2015) and potentially increased postnatal neural cell death. In particular, defects in the BDNF/TrkB signaling pathway might constitute a common molecular underpinning of neurodevelopmental disorders associated with postnatal microcephaly. Variants in NTRK2, the gene encoding the BDNF receptor TrkB, have been identified in patients presenting with microcephaly (Yoganathan et al., 2021). During neuronal development the BDNF/TrkB signaling pathway is a major regulator of axonal and dendritic arborization (Gonzalez et al., 2016), as well as synaptogenesis (Yoshii and Constantine-Paton, 2010). Christianson syndrome (Ouyang et al., 2013) and Angelman syndrome (Judson et al., 2017) are two neurodevelopmental disorders associated with postnatal microcephaly that share defects in neurotrophin signaling. Mouse models of both syndromes showed reductions in neuronal arborization that correlate with deficits in neurotrophin signaling. However, the extent to which the different histone methyltransferases that have been associated with postnatal microcephaly could modulate brain size by modulating the BDNF/TrkB signaling pathway is underexplored. Recently, mice with loss of Ash1l revealed growth delays and ASD/ID-like social behaviors (Gao et al., 2021b). Knockdown of ASH1L in human ESC-derived cortical neurons lead to decreased neurite length and arbor complexity (Cheon et al., 2022). Similarly, depletion of ASH1L in human iPSC-derived NPCs resulted in delayed neural differentiation and maturation (Lalli et al., 2020). The neuronal arborization defects associated with loss of ASH1L might correlate with defects in the BDNF/TrkB signaling pathway (Cheon et al., 2022). Taken together, these data are suggestive of a mechanism through which deficits in ASH1L could underlie postnatal microcephaly through its regulation of the neurotrophin signaling pathway.
In addition to the potential role of ASH1L in the control of brain size postnatally, clinical studies suggest that mutations in either NSD1, NSD2, SETD2, or SETD5 have also been implicated in microcephaly. Several clinical studies on NSD1 suggest that the microcephaly associated with NSD1 increased dosage appears to be postnatal (Dikow et al., 2013). Based on overexpression studies in Drosophila, elevating the levels of the NSD1 fly homologue in glial cells but not in neuronal cells led to reduced brain size that was associated with increased cell death of both glial and neuronal cells (Kim et al., 2020). However, further work in animal models and human iPSC-derived neurons with duplication of NSD1 will be necessary to elucidate the role of this histone methyltransferase in the control of brain size in vertebrates. In contrast to patients with NSD1 duplications, patients with small 4p.16.3 deletions that implicate NSD2 have been reported to be microcephalic at birth (Bernardini et al., 2018) which suggests that NSD2 controls brain size by mechanisms independent of NSD1. Mouse models with different deletions that affect Nsd2 present with reduced cerebral cortex, and reduction of Nsd2 in zebrafish led to reduced numbers of motor neurons; however, to date there is a lack of mechanistic studies in these systems (Naf et al., 2001). In hematopoietic B cells, loss of NSD2 reduced B cell proliferation and led to alterations in cell fate commitment (Campos-Sanchez et al., 2017). Therefore, it is possible that during brain development, NSD2 could control the proliferation and cell fate decisions of NPCs which if impaired could be a mechanism contributing to the congenital microcephaly associated with deficits in NSD2.
In contrast, multiple mechanisms could be implicated in microcephaly associated with deficits in SETD5. For example, decreased formation of glutamatergic synapses was observed in mice with knockdown of Setd5 (Sessa et al., 2019), which suggests defects in synaptogenesis that could lead to a reduced brain size postnatally. Similarly, defects in neuronal arborization outgrowth and complexity due to Setd5 downregulation could also be a potential mechanism contributing to SETD5-associated microcephaly (Moore et al., 2019). However, based on the current clinical literature it is not clear whether the microcephaly associated with SETD5 is congenital or postnatal.
Additional insights into the mechanistic underpinnings of microcephaly associated with histone methyltransferases will be gained by the study of the MLL family of H3K4 methyltransferases. Deficits in both MLL1 and MLL2 have been associated with delays or impairments of neuronal differentiation observed in mice models targeting these two genes (Lubitz et al., 2007; Lim et al., 2009). Similar to MLL1, loss of MLL4 leads to reduced NPC proliferation (Lim et al., 2009; Huang et al., 2015; Carosso et al., 2019). Therefore, convergent downstream mechanisms modulated by MLL1, MLL2, and MLL4 could lead to a reduced brain size. In contrast, knockdown of MLL3 in rat cortical neurons showed increased hyperactive neuronal networks which correlated with decreased inhibitory and excitatory synaptic puncta (Frega et al., 2020). However, MLL3 is widely expressed in developing human fetal brain which suggests that it could have additional important functions during human neuronal development (Nagase et al., 2000; Johnson et al., 2009). To gain further understanding of the chromatin regulatory mechanisms that control brain size, studies that consider the counterbalance between MLLs’ driven H3K4 methylation and PRC2 driven H3K27 methylation will be needed. PRC2 is known to repress genes implicated in neural differentiation (Dietrich et al., 2012). Therefore, genome wide scale loss of H3K4 methylation could result in increased levels of H3K27me3 which could reduce neuronal differentiation for example.
6.2. Macrocephaly
Macrocephaly or enlarged brain in the absence of hydrocephalus has been linked to defects in axon/dendritic arborization, synaptogenesis and hyperproliferation of neuronal progenitors. A classic molecular mechanism previously implicated in macrocephaly is the deletion of Phosphatase and Tensin Homolog (PTEN). Loss of PTEN in neuronal cell populations of the mouse cerebral cortex resulted in progressive macrocephaly, axonal hypertrophy, and increased synapse formation (Kwon et al., 2006). PTEN is involved with the PI3K/AKT pathway, which regulates neurite growth and dendritic arborization (Crowder and Freeman, 1998; Markus et al., 2002; Jaworski et al., 2005). Mutations in Mammalian target of rapamycin (MTOR), which is downstream of the PI3K/AKT pathway, have been identified in patients with macrocephaly (Baynam et al., 2015). The MTOR pathway has been identified as important for proper synaptogenesis, suggesting a mechanistic role for abnormal synaptogenesis in the etiology of macrocephaly (Takei et al., 2004).
The mechanism through which loss of function mutations in NSD1 contribute to macrocephaly in patients with Sotos Syndrome and Weaver Syndrome is largely understudied. Mice heterozygous for Nsd1 presented deficits in social behaviors but did not show enlarged brain size (Oishi et al., 2020). Analysis of homozygous Nsd1 mutant embryos at embryonic day 9.5 showed they had a smaller prosencephalon which was unexpected based on the human clinical findings on Sotos syndrome (Oishi et al., 2020). Therefore, there could be species specific differences, and it will be important to model Sotos syndrome using human organoids for example. Macrocephaly has been shown to be co-morbid with cortical malformations that could result from impaired neuronal migration (Andrews and Hulette, 1993; Robertson et al., 2000; Tenney et al., 2011; Mirzaa and Poduri, 2014; Winden et al., 2015). In humans, mutations in Adenomatous Polyposis coli 2 (APC2), a protein with a crucial role in the control of neuronal migration and axon guidance (Mimori-Kiyosue et al., 2000; Shintani et al., 2009, 2012), have been identified by whole exome sequencing as an additional cause of Sotos syndrome (Almuriekhi et al., 2015). APC2 is a downstream target of NSD1 as overexpression of Apc2 in mice with Nsd1 knockdown rescued the neuronal migration phenotype associated with loss of Nsd1 (Almuriekhi et al., 2015). In contrast to the Nsd1 mutant mice that does not show an enlarged head circumference, loss of function mutation in Apc2 resulted in a Sotos Syndrome-like phenotype in mice, with increased head circumference and dilated brain ventricles (Almuriekhi et al., 2015). Similar to NSD1, both SETD2 and SETD1A have also been implicated in the control of neuronal migration (Yu X. et al., 2019; Xu et al., 2021). However, divergent mechanisms could underlie a larger head size associated with loss of function mutations in SETD2 and SETD1A. For instance, deficits in SETD2 could increase NPC proliferation and lead to an enlarged brain (Li and Jiao, 2017; Shen et al., 2018; Xu et al., 2021). In contrast, loss of SETD1A might result in a macrocephalic brain due to increased neuronal arborization (Wang et al., 2022).
Impairing the function of EZH2, the catalytic subunit of PRC2, leads to increased neuronal differentiation which correlates with decreased NPC proliferation and accelerated gliogenesis (Liu et al., 2017). While it is unclear whether these cellular mechanisms contribute to PRC2-associated macrocephaly, as brain size was not investigated in this EZH2-knockout mouse model, an additional mechanism that could contribute to the increase in brain size associated with PRC2 defects is increased neuronal arborization (Di Meglio et al., 2013; Qi et al., 2014). Studies on the effects of PRC2 dysregulation and its interaction with CDYL provide support to the idea of increased neuronal arborization as a mechanism underlying PRC2-associated macrocephaly. The interaction between CDYL and EZH2 leads to inhibition of BDNF (Qi et al., 2014). If CDYL is unable to recruit EZH2 to inhibit BDNF, this could potentially lead to increased neuronal arborization and possibly synaptogenesis through upregulation of the BDNF/TrkB pathway. In fact, loss of EZH2 has been associated with increases in neuronal arborization and astrogliogenesis (Hirabayashi et al., 2009; Corley and Kroll, 2015), providing additional mechanisms that could contribute to PRC2-associated macrocephaly.
7. Final remarks
Brain size abnormalities, including microcephaly and macrocephaly, have previously been identified in a variety of neurodevelopmental disorders. Gaining insight into the mechanisms that produce brain size abnormalities will increase our understanding of the pathogenesis resulting in these neurodevelopmental disorders. Mutations in H3K36 and H3K4 histone methyltransferases have been associated with both microcephaly and macrocephaly related disorders (Figure 2 and Tables 1, 3). Methylation of H3K36 is vital to a variety of cellular processes, including transcriptional activation, DNA repair, and RNA splicing (Wang et al., 2007; Pradeepa et al., 2012; Huang and Zhu, 2018). Similarly, H3K4 methylation is also important to many cellular processes, including transcriptional activation and DNA repair (Barski et al., 2007; Zhu et al., 2013). Studies have shown a major role for H3K36 and H3K4 methylation during neuronal development. Histone methyltransferases responsible for H3K36 and H3K4 methylation have been implicated in neural stem cell proliferation (Zhang et al., 2011; Sessa et al., 2019), neuronal migration (Almuriekhi et al., 2015; Yu X. et al., 2019), neuronal arborization (Moore et al., 2019; Lalli et al., 2020; Gao et al., 2021b), and the control of gliogenesis which constitute essential mechanisms for proper brain development.
Additionally, gene silencing via PRC2-mediated H3K27 methylation is implicated in many different neurodevelopmental processes, including neuronal migration (Pereira et al., 2010), neural stem cell proliferation, and neuronal differentiation (Pereira et al., 2010; Dietrich et al., 2012). Mutations within the subunits of PRC2 are associated with several neurodevelopmental disorders, some of which also have reported brain size abnormalities (Cooney et al., 2017; Imagawa et al., 2017, 2018; Cyrus et al., 2019; Griffiths et al., 2019; Verberne et al., 2021). Both H3K36 and H3K4 methylation can directly or indirectly counteract PRC2 function by preventing the methylation of H3K27me3 (Agger et al., 2007; Yuan et al., 2011; Kim et al., 2014; Piunti and Shilatifard, 2016; Schuettengruber et al., 2017; Streubel et al., 2018; Zhuang et al., 2018; Shirane et al., 2020). While the balance between H3K36 and H3K4 methyltransferases and PRC2-mediated H3K27 tri-methylation have been well-studied in cancer and general development, the potential role of this counteracting activity to the etiology of different neurodevelopmental disorders is understudied. Potential increases in H3K27me3 caused by mutations in H3K36 or H3K4 methyltransferases could provide a convergent epigenetic mechanism leading to abnormal neurodevelopment.
It is important to note that the activity of proteins associated with H3K36 and H3K4 methylation is not necessarily restricted to only histone methylation. There is increasing evidence that several H3K36 methyltransferases also function as methylators of cytoskeletal components. Specifically, recent studies have shown that SETD2 is responsible for the trimethylation of Lysine 40 on α-tubulin (Park et al., 2016; Koenning et al., 2021). This cytoskeletal methylation has been identified in the mouse brain, with high concentrations of this mark found in post-mitotic neurites and growth cones (Koenning et al., 2021). Mutations to the Set2-Rpb1 interaction (SRI) domain of SETD2, which is responsible for substrate recognition, resulted in increased anxiety-like behavior and reduced dendritic arborization and axon length in mice (Koenning et al., 2021). Both α- and β-tubulin have been associated with maintenance of brain size, as mutations in both of these genes have been associated with microcephaly (Keays et al., 2007; Breuss et al., 2012). Additionally, SETD2 has been shown to tri-methylate actin on Lysine 68 through its interaction with the Huntington protein and the actin-binding adapter HIP1R (Seervai et al., 2020). This interaction was shown to be important for proper cell migration in human embryonic kidney 293T cells (Seervai et al., 2020). However, it is unknown if this interaction contributes to neuronal cell migration. SMYD2 is another H3K36 methyltransferase that has been linked to methylation of α-tubulin at Lysine 394 through cross-talk with cyclin-dependent kinases 4 and 6 (CDK4/6) (Li et al., 2020). Taken together, these studies suggest that direct methylation of cytoskeletal components is another mechanism that should be considered when attempting to uncover epigenetic mechanisms contributing to brain size abnormalities.
Epigenetic mechanisms play an important role in the regulation of gene programs responsible for brain development (Keverne, 2014). Here we have discussed how mutations across different histone methyltransferases lead to dysregulation of neurodevelopmental process, including NPC proliferation, neuronal migration, and neuronal arborization. Additionally, we connect these neurodevelopmental processes to brain size abnormalities identified in disorders associated with multiple histone methyltransferases. The counteracting mechanisms associated with H3K27me3 vs. H3K36me2 or H3k4me3 modifications bring forward the exciting opportunity to test rescue strategies of the different neural phenotypes. For example, inhibition of PRC2 catalytic activity using a pre-clinical inhibitor of EZH2 rescued ASH1L neuronal arborization defects in human ESC-derived neurons (Cheon et al., 2022). Similarly, acetylation of H3K27, which can inhibit the methylation of the same site by PRC2, has been used as a rescue strategy for H3K4- and H3K36-related neuronal phenotypes. For instance, inhibition of histone deacetylation which would maintain chromatin in a hyperacetylated state improved autistic-like behaviors of an Ash1l mutant mice (Gao et al., 2021a). Therefore, gaining an in depth understanding of the molecular signatures and cellular phenotypes associated with the pathogenic mutations in H3K36 and H3K4 histone methyltransferases in the etiology of various neurodevelopmental disorders will be instrumental in the potential development of mechanistic based therapeutic strategies.
Finally, a major remaining question moving forward in the field of neurodevelopmental epigenetics will be how to explain the phenotypic divergency associated with mutations in the different H3K4-H3K36- and H3K27 methyltransferases. Are the differences associated with cell type specific expression? Based on single cell data from developing human cortex ASH1L, KMT2A, KMT2C, SETD2, and SETD5 are widely expressed in multiple subtypes of NPCs, inhibitory and excitatory neurons as well as glial cells (Nowakowski et al., 2017; Speir et al., 2021). In contrast, NSD1, KMT2B, KMT2D, SETD1A, and SETD1B seem to have higher expression in different NPC subtypes but are not as highly expressed in excitatory and inhibitory neurons in the developing cortex (Nowakowski et al., 2017; Speir et al., 2021). Therefore, the differences in spatial and temporal expression could contribute to the divergence of phenotypes associated with mutations in the different histone methyltransferases. In addition, despite having similar cellular functions the differences in the gene programs regulated by the different histone methyltransferases could also contribute to the phenotypic diversity associated with disruptions in the different enzymes. For example, the paralogous enzymes MLL1/KMT2A and MLL2/KMT2B contribute to memory formation through the regulation of distinct gene programs (Kerimoglu et al., 2017). The differences in substrate specificity and fine control of the chromatin environment, could be further contributed by the different complexes these enzymes form part of, which can give them their target specificity (Piunti and Shilatifard, 2016). Therefore, future studies will need to address the dynamic nature of these interactions during cortical development.
Author contributions
Both authors designed, illustrated, wrote, and edited the manuscript and approved the submitted version.
Funding
SBL was supported by an NIH/NIMH R01 MH127081-01A1, NIH/NIGMS COBRE 4P20GM103641-06, SC EPSCOR stimulus award 18-SR04, and ASPIRE awards Track I and Track II from the VPR office at University of South Carolina.
Acknowledgments
We thank Dr. Shannon Davis and Dr. Calvin Leung for providing critical feedback on this manuscript. Illustrations were made with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agger, K., Cloos, P., Christensen, J., Pasini, D., Rose, S., Rappsilber, J., et al. (2007). UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449, 731–734.
Albanese, A., Bhatia, K., Bressman, S., Delong, M., Fahn, S., Fung, V., et al. (2013). Phenomenology and classification of dystonia: A consensus update. Mov. Disord. 28, 863–873.
Almuriekhi, M., Shintani, T., Fahiminiya, S., Fujikawa, A., Kuboyama, K., Takeuchi, Y., et al. (2015). Loss-of-Function mutation in APC2 causes sotos syndrome features. Cell Rep. 10, 1585–1598. doi: 10.1016/j.celrep.2015.02.011
Andrews, P., and Hulette, C. (1993). An infant with macrocephaly, abnormal neuronal migration and persistent olfactory ventricles. Clin. Neuropathol. 12, 13–18.
Bannister, A., Schneider, R., Myers, F., Thorne, A., Crane-Robinson, C., and Kouzarides, T. (2005). Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J. Biol. Chem. 280, 17732–17736. doi: 10.1074/jbc.M500796200
Bantignies, F., Goodman, R., and Smolik, S. (2000). Functional interaction between the coactivator Drosophila CREB-binding protein and ASH1, a member of the trithorax group of chromatin modifiers. Mol. Cell. Biol. 20, 9317–9330. doi: 10.1128/MCB.20.24.9317-9330.2000
Baple, E., Palmer, R., and Hennekam, R. C. A. (2010). microdeletion at 12q24.31 can mimic beckwith-wiedemann syndrome neonatally. Mol. Syndromol. 1, 42–45. doi: 10.1159/000275671
Barbagiovanni, G., Germain, P., Zech, M., Atashpaz, S., Lo Riso, P., D’Antonio-Chronowska, A., et al. (2018). KMT2B is selectively required for neuronal transdifferentiation, and its loss exposes dystonia candidate genes. Cell Rep. 25, 988–1001.
Barrie, E., Alfaro, M., Pfau, R., Goff, M., McBride, K., Manickam, K., et al. (2019). De novo loss-of-function variants in NSD2 (WHSC1) associate with a subset of Wolf-Hirschhorn syndrome. Cold Spring Harb. Mol. Case Stud. 5:a004044. doi: 10.1101/mcs.a004044
Barski, A., Cuddapah, S., Cui, K., Roh, T., Schones, D., Wang, Z., et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837.
Battaglia, A., Lortz, A., and Carey, J. (2021). Natural history study of adults with Wolf-Hirschhorn syndrome 1: Case series of personally observed 35 individuals. Am. J. Med. Genet. A 185, 1794–1802. doi: 10.1002/ajmg.a.62176
Baynam, G., Overkov, A., Davis, M., Mina, K., Schofield, L., Allcock, R., et al. (2015). A germline MTOR mutation in Aboriginal Australian siblings with intellectual disability, dysmorphism, macrocephaly, and small thoraces. Am. J. Med. Genet. A 167, 1659–1667. doi: 10.1002/ajmg.a.37070
Belichenko, P., Wright, E., Belichenko, N., Masliah, E., Li, H., Mobley, W., et al. (2009). Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: Evidence for disruption of neuronal networks. J. Comp. Neurol. 514, 240–258. doi: 10.1002/cne.22009
Bergemann, A., Cole, F., and Hirschhorn, K. (2005). The etiology of Wolf-Hirschhorn syndrome. Trends Genet. 21, 188–195.
Bernardini, L., Radio, F., Acquaviva, F., Gorgone, C., Postorivo, D., Torres, B., et al. (2018). Small 4p16.3 deletions: Three additional patients and review of the literature. Am. J. Med. Genet. A 176, 2501–2508.
Bjornsson, H., Benjamin, J., Zhang, L., Weissman, J., Gerber, E., Chen, Y., et al. (2014). Histone deacetylase inhibition rescues structural and functional brain deficits in a mouse model of Kabuki syndrome. Sci. Transl. Med. 6:256ra135. doi: 10.1126/scitranslmed.3009278
Bledau, A., Schmidt, K., Neumann, K., Hill, U., Ciotta, G., Gupta, A., et al. (2014). The H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development 141, 1022–1035. doi: 10.1242/dev.098152
Boczek, N., Lahner, C., Nguyen, T., Ferber, M., Hasadsri, L., Thorland, E., et al. (2018). Developmental delay and failure to thrive associated with a loss-of-function variant in WHSC1 (n.d.). Am. J. Med. Genet. A 176, 2798–2802. doi: 10.1002/ajmg.a.40498
Bond, J., Roberts, E., Springell, K., Lizarraga, S., Scott, S., Higgins, J., et al. (2005). A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat. Genet. 37, 353–355.
Brand, S., and Rakic, P. (1979). Genesis of the primate neostriatum: [3H]thymidine autoradiographic analysis of the time of neuron origin in the rhesus monkey. Neuroscience 4, 767–778. doi: 10.1016/0306-4522(79)90005-8
Breuss, M., Heng, J., Poirier, K., Tian, G., Jaglin, X., Qu, Z., et al. (2012). Mutations in the β-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell Rep. 2, 1554–1562. doi: 10.1016/j.celrep.2012.11.017
Buxbaum, J., Cai, G., Nygren, G., Chaste, P., Delorme, R., Goldsmith, J., et al. (2007). Mutation analysis of the NSD1 gene in patients with autism spectrum disorders and macrocephaly. BMC Med. Genet. 8:68. doi: 10.1186/1471-2350-8-68
Cai, L., Rothbart, S., Lu, R., Xu, B., Chen, W., Tripathy, A., et al. (2013). An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol. Cell 49, 571–582. doi: 10.1016/j.molcel.2012.11.026
Campos-Sanchez, E., Deleyto-Seldas, N., Dominguez, V., Carrillo-de-Santa-Pau, E., Ura, K., Rocha, P., et al. (2017). Wolf-hirschhorn syndrome candidate 1 is necessary for correct hematopoietic and B cell development. Cell Rep. 19, 1586–1601. doi: 10.1016/j.celrep.2017.04.069
Carosso, G., Boukas, L., Augustin, J., Nguyen, H., Winer, B., Cannon, G., et al. (2019). Precocious neuronal differentiation and disrupted oxygen responses in Kabuki syndrome. JCI Insight 4:e129375. doi: 10.1172/jci.insight.129375
Castiglioni, S., Di Fede, E., Bernardelli, C., Lettieri, A., Parodi, C., Grazioli, P., et al. (2022). KMT2A: Umbrella gene for multiple diseases. Genes 13:514.
Cecconi, M., Forzano, F., Milani, D., Cavani, S., Baldo, C., Selicorni, A., et al. (2005). Mutation analysis of the NSD1 gene in a group of 59 patients with congenital overgrowth. Am. J. Med. Genet. A 134, 247–253. doi: 10.1002/ajmg.a.30492
Cederquist, G., Tchieu, J., Callahan, S., Ramnarine, K., Ryan, S., Zhang, C., et al. (2020). A multiplex human pluripotent stem cell platform defines molecular and functional subclasses of autism-related genes. Cell Stem Cell 27, 35–49.e6. doi: 10.1016/j.stem.2020.06.004
Chan, A., Cytrynbaum, C., Hoang, N., Ambrozewicz, P., Weksberg, R., Drmic, I., et al. (2019). Expanding the neurodevelopmental phenotypes of individuals with de novo KMT2A variants. NPJ Genom. Med. 4:9. doi: 10.1038/s41525-019-0083-x
Chen, C., Lin, S., Lin, C., Chen, Y., Chern, S., Li, Y., et al. (2006). Molecular cytogenetic analysis of de novo dup(5)(q35.2q35.3) and review of the literature of pure partial trisomy 5q. Am. J. Med. Genet. A 140, 1594–1600. doi: 10.1002/ajmg.a.31329
Chen, M., Quan, Y., Duan, G., Wu, H., Bai, T., Wang, Y., et al. (2021). Mutation pattern and genotype-phenotype correlations of SETD2 in neurodevelopmental disorders. Eur. J. Med. Genet. 64:104200. doi: 10.1016/j.ejmg.2021.104200
Chenn, A., and McConnell, S. (1995). Cleavage orientation and the asymmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell 82, 631–641. doi: 10.1016/0092-8674(95)90035-7
Chenn, A., and Walsh, C. (2002). Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297, 365–369.
Cheon, S., Culver, A., Bagnell, A., Ritchie, F., Vacharasin, J., McCord, M., et al. (2022). Counteracting epigenetic mechanisms regulate the structural development of neuronal circuitry in human neurons. Mol. Psychiatry 27, 2291–2303. doi: 10.1038/s41380-022-01474-1
Cif, L., Demailly, D., Lin, J., Barwick, K., Sa, M., Abela, L., et al. (2020). KMT2B-related disorders: Expansion of the phenotypic spectrum and long-term efficacy of deep brain stimulation. Brain 143, 3242–3261. doi: 10.1093/brain/awaa304
Clowry, G., Molnar, Z., and Rakic, P. (2010). Renewed focus on the developing human neocortex. J. Anat. 217, 276–288.
Collins, B., Greer, C., Coleman, B., and Sweatt, J. (2019). Histone H3 lysine K4 methylation and its role in learning and memory. Epigenetics Chromatin 12:7.
Cooney, E., Bi, W., Schlesinger, A., Vinson, S., and Potocki, L. (2017). Novel EED mutation in patient with Weaver syndrome. Am. J. Med. Genet. A 173, 541–545.
Corley, M., and Kroll, K. (2015). The roles and regulation of Polycomb complexes in neural development. Cell Tissue Res. 359, 65–85.
Cox, J., Jackson, A., Bond, J., and Woods, C. (2006). What primary microcephaly can tell us about brain growth. Trends Mol. Med. 12, 358–366. doi: 10.1016/j.molmed.2006.06.006
Crippa, M., Bestetti, I., Maitz, S., Weiss, K., Spano, A., Masciadri, M., et al. (2020). SETD5 Gene haploinsufficiency in three patients with suspected KBG syndrome. Front. Neurol. 11:631. doi: 10.3389/fneur.2020.00631
Crowder, R., and Freeman, R. (1998). Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J. Neurosci. 18, 2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998
Cyrus, S., Burkardt, D., Weaver, D., and Gibson, W. (2019). PRC2-complex related dysfunction in overgrowth syndromes: A review of EZH2, EED, and SUZ12 and their syndromic phenotypes. Am. J. Med. Genet. C Semin. Med. Genet. 181, 519–531. doi: 10.1002/ajmg.c.31754
De Rubeis, S., He, X., Goldberg, A., Poultney, C., Samocha, K., Cicek, A., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215.
Deb, M., Kar, S., Sengupta, D., Shilpi, A., Parbin, S., Rath, S., et al. (2014). Chromatin dynamics: H3K4 methylation and H3 variant replacement during development and in cancer. Cell. Mol. Life Sci. 71, 3439–3463.
Deciphering Developmental Disorders Study (2015). Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228.
Deevy, O., and Bracken, A. (2019). PRC2 functions in development and congenital disorders. Development 146:dev181354.
Deliu, E., Arecco, N., Morandell, J., Dotter, C., Contreras, X., Girardot, C., et al. (2018). Haploinsufficiency of the intellectual disability gene SETD5 disturbs developmental gene expression and cognition. Nat. Neurosci. 21, 1717–1727. doi: 10.1038/s41593-018-0266-2
Den, K., Kato, M., Yamaguchi, T., Miyatake, S., Takata, A., Mizuguchi, T., et al. (2019). A novel de novo frameshift variant in SETD1B causes epilepsy. J. Hum. Genet. 64, 821–827. doi: 10.1038/s10038-019-0617-1
Denissov, S., Hofemeister, H., Marks, H., Kranz, A., Ciotta, G., Singh, S., et al. (2014). Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development 141, 526–537. doi: 10.1242/dev.102681
Di Candia, F., Fontana, P., Paglia, P., Falco, M., Rosano, C., Piscopo, C., et al. (2022). Clinical heterogeneity of Kabuki syndrome in a cohort of Italian patients and review of the literature. Eur. J. Pediatr. 181, 171–187. doi: 10.1007/s00431-021-04108-w
Di Croce, L., and Helin, K. (2013). Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 20, 1147–1155.
Di Meglio, T., Kratochwil, C., Vilain, N., Loche, A., Vitobello, A., Yonehara, K., et al. (2013). Ezh2 orchestrates topographic migration and connectivity of mouse precerebellar neurons. Science 339, 204–207. doi: 10.1126/science.1229326
Dietrich, N., Lerdrup, M., Landt, E., Agrawal-Singh, S., Bak, M., Tommerup, N., et al. (2012). REST-mediated recruitment of polycomb repressor complexes in mammalian cells. PLoS Genet. 8:e1002494. doi: 10.1371/journal.pgen.1002494
Dikow, N., Maas, B., Gaspar, H., Kreiss-Nachtsheim, M., Engels, H., Kuechler, A., et al. (2013). The phenotypic spectrum of duplication 5q35.2-q35.3 encompassing NSD1: Is it really a reversed Sotos syndrome? Am. J. Med. Genet. A 161A, 2158–2166. doi: 10.1002/ajmg.a.36046
Dindot, S., Antalffy, B., Bhattacharjee, M., and Beaudet, A. (2008). The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 17, 111–118. doi: 10.1093/hmg/ddm288
Edmunds, J., Mahadevan, L., and Clayton, A. (2008). Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 27, 406–420. doi: 10.1038/sj.emboj.7601967
Faravelli, F. (2005). NSD1 mutations in Sotos syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 137C, 24–31.
Faundes, V., Newman, W., Bernardini, L., Canham, N., Clayton-Smith, J., Dallapiccola, B., et al. (2018). Histone lysine methylases and demethylases in the landscape of human developmental disorders. Am. J. Hum. Genet. 102, 175–187. doi: 10.1016/j.ajhg.2017.11.013
Faundes, V., Santa Maria, L., Morales, P., Curotto, B., and Alliende, M. (2017). [Microarrays in 236 patients with neurodevelopmental disorders and congenital abnormalities]. Rev. Med. Chil. 145, 854–861.
Fernandes, I., Cruz, A., Ferrasa, A., Phan, D., Herai, R., and Muotri, A. (2018). Genetic variations on SETD5 underlying autistic conditions. Dev. Neurobiol. 78, 500–518. doi: 10.1002/dneu.22584
Fombonne, E., Rogé, B., Claverie, J., Courty, S., and Frémolle, J. (1999). Microcephaly and macrocephaly in autism. J. Autism Dev. Disord. 29, 113–119.
Fortin, O., Vincelette, C., Khan, A., Berrahmoune, S., Dassi, C., Karimi, M., et al. (2021). Seizures in Sotos syndrome: Phenotyping in 49 patients. Epilepsia Open 6, 425–430. doi: 10.1002/epi4.12484
Franco, L., de Ravel, T., Graham, B., Frenkel, S., Van Driessche, J., Stankiewicz, P., et al. (2010). A syndrome of short stature, microcephaly and speech delay is associated with duplications reciprocal to the common Sotos syndrome deletion. Eur. J. Hum. Genet. 18, 258–261. doi: 10.1038/ejhg.2009.164
Frega, M., Selten, M., Mossink, B., Keller, J., Linda, K., Moerschen, R., et al. (2020). Distinct pathogenic genes causing intellectual disability and autism exhibit a common neuronal network hyperactivity phenotype. Cell Rep. 30, 173–186.e6. doi: 10.1016/j.celrep.2019.12.002
Gao, Y., Aljazi, M., and He, J. (2022). Neural hyperactivity is a core pathophysiological change induced by deletion of a high autism risk gene Ash1L in the mouse brain. Front. Behav. Neurosci. 16:873466. doi: 10.3389/fnbeh.2022.873466
Gao, Y., Duque-Wilckens, N., Aljazi, M., Wu, Y., Moeser, A., Mias, G., et al. (2021b). Loss of histone methyltransferase ASH1L in the developing mouse brain causes autistic-like behaviors. Commun. Biol. 4:756.
Gao, Y., Aljazi, M., Wu, Y., and He, J. (2021a). Vorinostat, a histone deacetylase inhibitor, ameliorates the sociability and cognitive memory in an Ash1L-deletion-induced ASD/ID mouse model. Neurosci. Lett. 764:136241. doi: 10.1016/j.neulet.2021.136241
Gates, L., Foulds, C., and O’Malley, B. (2017). Histone marks in the ‘Driver’s Seat’: Functional roles in steering the transcription cycle. Trends Biochem. Sci. 42, 977–989. doi: 10.1016/j.tibs.2017.10.004
Geisert, E. Jr., Williams, R., Geisert, G., Fan, L., Asbury, A., Maecker, H., et al. (2002). Increased brain size and glial cell number in CD81-null mice. J. Comp. Neurol. 453, 22–32. doi: 10.1002/cne.10364
Giedd, J., Blumenthal, J., Jeffries, N., Castellanos, F., Liu, H., Zijdenbos, A., et al. (1999). Brain development during childhood and adolescence: A longitudinal MRI study. Nat. Neurosci. 2, 861–863.
Gonzalez, A., Moya-Alvarado, G., Gonzalez-Billaut, C., and Bronfman, F. (2016). Cellular and molecular mechanisms regulating neuronal growth by brain-derived neurotrophic factor. Cytoskeleton 73, 612–628.
Gregory, G., Vakoc, C., Rozovskaia, T., Zheng, X., Patel, S., Nakamura, T., et al. (2007). Mammalian ASH1L is a histone methyltransferase that occupies the transcribed region of active genes. Mol. Cell. Biol. 27, 8466–8479. doi: 10.1128/MCB.00993-07
Griffiths, S., Loveday, C., Zachariou, A., Behan, L., Chandler, K., Cole, T., et al. (2019). EED and EZH2 constitutive variants: A study to expand the Cohen-Gibson syndrome phenotype and contrast it with Weaver syndrome. Am. J. Med. Genet. A 179, 588–594. doi: 10.1002/ajmg.a.61066
Grozeva, D., Carss, K., Spasic-Boskovic, O., Parker, M., Archer, H., Firth, H., et al. (2014). De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. Am. J. Hum. Genet. 94, 618–624. doi: 10.1016/j.ajhg.2014.03.006
Haubensak, W., Attardo, A., Denk, W., and Huttner, W. (2004). Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: A major site of neurogenesis. Proc. Natl. Acad. Sci. U.S.A. 101, 3196–3201. doi: 10.1073/pnas.0308600100
Higgs, M., Sato, K., Reynolds, J., Begum, S., Bayley, R., Goula, A., et al. (2018). Histone Methylation by SETD1A Protects Nascent DNA through the Nucleosome Chaperone Activity of FANCD2. Mol. Cell 71, 25–41.e6. doi: 10.1016/j.molcel.2018.05.018
Hirabayashi, Y., Itoh, Y., Tabata, H., Nakajima, K., Akiyama, T., Masuyama, N., et al. (2004). The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 131, 2791–2801. doi: 10.1242/dev.01165
Hirabayashi, Y., Suzki, N., Tsuboi, M., Endo, T., Toyoda, T., Shinga, J., et al. (2009). Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 63, 600–613. doi: 10.1016/j.neuron.2009.08.021
Hiraide, T., Hattori, A., Ieda, D., Hori, I., Saitoh, S., Nakashima, M., et al. (2019). De novo variants in SETD1B cause intellectual disability, autism spectrum disorder, and epilepsy with myoclonic absences. Epilepsia Open 4, 476–481. doi: 10.1002/epi4.12339
Homsy, J., Zaidi, S., Shen, Y., Ware, J., Samocha, K., Karczewski, K., et al. (2015). De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350, 1262–1266.
Hu, D., Gao, X., Morgan, M., Herz, H., Smith, E., and Shilatifard, A. (2013). The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell. Biol. 33, 4745–4754. doi: 10.1128/MCB.01181-13
Huang, C., and Zhu, B. (2018). Roles of H3K36-specific histone methyltransferases in transcription: Antagonizing silencing and safeguarding transcription fidelity. Biophys. Rep. 4, 170–177. doi: 10.1007/s41048-018-0063-1
Huang, Y., Shih, H., Lin, S., Chiu, C., Ma, T., Yeh, T., et al. (2015). The epigenetic factor Kmt2a/Mll1 regulates neural progenitor proliferation and neuronal and glial differentiation. Dev. Neurobiol. 75, 452–462. doi: 10.1002/dneu.22235
Huttner, W., and Brand, M. (1997). Asymmetric division and polarity of neuroepithelial cells. Curr. Opin. Neurobiol. 7, 29–39.
Imagawa, E., Albuquerque, E., Isidor, B., Mitsuhashi, S., Mizuguchi, T., Miyatake, S., et al. (2018). Novel SUZ12 mutations in Weaver-like syndrome. Clin. Genet. 94, 461–466. doi: 10.1111/cge.13415
Imagawa, E., Higashimoto, K., Sakai, Y., Numakura, C., Okamoto, N., Matsunaga, S., et al. (2017). Mutations in genes encoding polycomb repressive complex 2 subunits cause Weaver syndrome. Hum. Mutat. 38, 637–648.
Iossifov, I., Levy, D., Allen, J., Ye, K., Ronemus, M., Lee, Y., et al. (2015). Low load for disruptive mutations in autism genes and their biased transmission. Proc. Natl. Acad. Sci. U.S.A. 112, E5600–E5607. doi: 10.1073/pnas.1516376112
Iossifov, I., O’Roak, B., Sanders, S., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221.
Iwamori, N., Tominaga, K., Sato, T., Riehle, K., Iwamori, T., Ohkawa, Y., et al. (2016). MRG15 is required for pre-mRNA splicing and spermatogenesis. Proc. Natl. Acad. Sci. U.S.A. 113, E5408–E5415. doi: 10.1073/pnas.1611995113
Jackson, A., Eastwood, H., Bell, S., Adu, J., Toomes, C., Carr, I., et al. (2002). Identification of microcephalin, a protein implicated in determining the size of the human brain. Am. J. Hum. Genet. 71, 136–142. doi: 10.1086/341283
Jakovcevski, M., Ruan, H., Shen, E., Dincer, A., Javidfar, B., Ma, Q., et al. (2015). Neuronal Kmt2a/Mll1 histone methyltransferase is essential for prefrontal synaptic plasticity and working memory. J. Neurosci. 35, 5097–5108. doi: 10.1523/JNEUROSCI.3004-14.2015
Jambhekar, A., Dhall, A., and Shi, Y. (2019). Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 20, 625–641.
Jaworski, J., Spangler, S., Seeburg, D., Hoogenraad, C., and Sheng, M. (2005). Control of dendritic arborization by the phosphoinositide-3’-kinase-Akt-mammalian target of rapamycin pathway. J. Neurosci. 25, 11300–11312.
Jean, F., Stuart, A., and Tarailo-Graovac, M. (2020). Dissecting the genetic and etiological causes of primary microcephaly. Front. Neurol. 11:570830. doi: 10.3389/fneur.2020.570830
Jha, D., and Strahl, B. (2014). An RNA polymerase II-coupled function for histone H3K36 methylation in checkpoint activation and DSB repair. Nat. Commun. 5:3965.
Johnson, M., Kawasawa, Y., Mason, C., Krsnik, Z., Coppola, G., Bogdanoviæ, D., et al. (2009). Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62, 494–509.
Jones, W., Dafou, D., McEntagart, M., Woollard, W., Elmslie, F., Holder-Espinasse, M., et al. (2012). De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am. J. Hum. Genet. 91, 358–364. doi: 10.1016/j.ajhg.2012.06.008
Judson, M., Burette, A., Thaxton, C., Pribisko, A., Shen, M., Rumple, A., et al. (2017). Decreased axon caliber underlies loss of fiber tract integrity, disproportional reductions in white matter volume, and microcephaly in angelman syndrome model mice. J. Neurosci. 37, 7347–7361. doi: 10.1523/JNEUROSCI.0037-17.2017
Kagitani-Shimono, K., Imai, K., Otani, K., Kamio, N., Okinaga, T., Toribe, Y., et al. (2005). Epilepsy in Wolf-Hirschhorn syndrome (4p-). Epilepsia 46, 150–155.
Kasinath, V., Beck, C., Sauer, P., Poepsel, S., Kosmatka, J., Faini, M., et al. (2021). JARID2 and AEBP2 regulate PRC2 in the presence of H2AK119ub1 and other histone modifications. Science 371. doi: 10.1126/science.abc3393
Kawarai, T., Miyamoto, R., Nakagawa, E., Koichihara, R., Sakamoto, T., Mure, H., et al. (2018). Phenotype variability and allelic heterogeneity in KMT2B-Associated disease. Parkinsonism Relat. Disord. 52, 55–61. doi: 10.1016/j.parkreldis.2018.03.022
Keays, D., Tian, G., Poirier, K., Huang, G., Siebold, C., Cleak, J., et al. (2007). Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 128, 45–57.
Kellogg, G., Sum, J., and Wallerstein, R. (2013). Deletion of 3p25.3 in a patient with intellectual disability and dysmorphic features with further definition of a critical region. Am. J. Med. Genet. A 161A, 1405–1408. doi: 10.1002/ajmg.a.35876
Kerimoglu, C., Agis-Balboa, R., Kranz, A., Stilling, R., Bahari-Javan, S., Benito-Garagorri, E., et al. (2013). Histone-methyltransferase MLL2 (KMT2B) is required for memory formation in mice. J. Neurosci. 33, 3452–3464.
Kerimoglu, C., Sakib, M., Jain, G., Benito, E., Burkhardt, S., Capece, V., et al. (2017). KMT2A and KMT2B mediate memory function by affecting distinct genomic regions. Cell Rep. 20, 538–548. doi: 10.1016/j.celrep.2017.06.072
Keverne, E. (2014). Significance of epigenetics for understanding brain development, brain evolution and behaviour. Neuroscience 264, 207–217.
Kim, D., Tang, Z., Shimada, M., Fierz, B., Houck-Loomis, B., Bar-Dagen, M., et al. (2013). Histone H3K27 trimethylation inhibits H3 binding and function of SET1-like H3K4 methyltransferase complexes. Mol. Cell. Biol. 33, 4936–4946. doi: 10.1128/MCB.00601-13
Kim, J., and Kim, H. (2012). Recruitment and biological consequences of histone modification of H3K27me3 and H3K9me3. ILAR J. 53, 232–239.
Kim, J., Lee, J., Lee, I., Lee, S., and Cho, K. (2017). Histone lysine methylation and neurodevelopmental disorders. Int. J. Mol. Sci. 18:1404.
Kim, J., Sharma, A., Dhar, S., Lee, S., Gu, B., Chan, C., et al. (2014). UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 74, 1705–1717.
Kim, T., Shin, H., Song, B., Won, C., Yoshida, H., Yamaguchi, M., et al. (2020). Overexpression of H3K36 methyltransferase NSD in glial cells affects brain development in Drosophila. Glia 68, 2503–2516. doi: 10.1002/glia.23867
Kizer, K., Phatnani, H., Shibata, Y., Hall, H., Greenleaf, A., and Strahl, B. D. (2005). A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol. Cell. Biol. 25, 3305–3316. doi: 10.1128/MCB.25.8.3305-3316.2005
Kleefstra, T., Kramer, J., Neveling, K., Willemsen, M., Koemans, T., Vissers, L., et al. (2012). Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am. J. Hum. Genet. 91, 73–82. doi: 10.1016/j.ajhg.2012.05.003
Koemans, T., Kleefstra, T., Chubak, M., Stone, M., Reijnders, M., de Munnik, S., et al. (2017). Functional convergence of histone methyltransferases EHMT1 and KMT2C involved in intellectual disability and autism spectrum disorder. PLoS Genet. 13:e1006864. doi: 10.1371/journal.pgen.1006864
Koenning, M., Wang, X., Karki, M., Jangid, R., Kearns, S., Tripathi, D., et al. (2021). Neuronal SETD2 activity links microtubule methylation to an anxiety-like phenotype in mice. Brain 144, 2527–2540. doi: 10.1093/brain/awab200
Koshimizu, E., Miyatake, S., Okamoto, N., Nakashima, M., Tsurusaki, Y., Miyake, N., et al. (2013). Performance comparison of bench-top next generation sequencers using microdroplet PCR-based enrichment for targeted sequencing in patients with autism spectrum disorder. PLoS One 8:e74167. doi: 10.1371/journal.pone.0074167
Kuechler, A., Zink, A., Wieland, T., Ludecke, H., Cremer, K., Salviati, L., et al. (2015). Loss-of-function variants of SETD5 cause intellectual disability and the core phenotype of microdeletion 3p25.3 syndrome. Eur. J. Hum. Genet. 23, 753–760. doi: 10.1038/ejhg.2014.165
Kummeling, J., Stremmelaar, D., Raun, N., Reijnders, M., Willemsen, M., Ruiterkamp-Versteeg, M., et al. (2021). Characterization of SETD1A haploinsufficiency in humans and Drosophila defines a novel neurodevelopmental syndrome. Mol. Psychiatry 26, 2013–2024. doi: 10.1038/s41380-020-0725-5
Kurotaki, N., Imaizumi, K., Harada, N., Masuno, M., Kondoh, T., Nagai, T., et al. (2002). Haploinsufficiency of NSD1 causes Sotos syndrome. Nat. Genet. 30, 365–366.
Kurotaki, N., Stankiewicz, P., Wakui, K., Niikawa, N., and Lupski, J. (2005). Sotos syndrome common deletion is mediated by directly oriented subunits within inverted Sos-REP low-copy repeats. Hum. Mol. Genet. 14, 535–542. doi: 10.1093/hmg/ddi050
Kwon, C., Luikart, B., Powell, C., Zhou, J., Matheny, S., Zhang, W., et al. (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388. doi: 10.1016/j.neuron.2006.03.023
Labonne, J., Lee, K., Iwase, S., Kong, I., Diamond, M., Layman, L., et al. (2016). An atypical 12q24.31 microdeletion implicates six genes including a histone demethylase KDM2B and a histone methyltransferase SETD1B in syndromic intellectual disability. Hum. Genet. 135, 757–771. doi: 10.1007/s00439-016-1668-4
Lalli, M., Avey, D., Dougherty, J., Milbrandt, J., and Mitra, R. (2020). High-throughput single-cell functional elucidation of neurodevelopmental disease-associated genes reveals convergent mechanisms altering neuronal differentiation. Genome Res. 30, 1317–1331. doi: 10.1101/gr.262295.120
Lane, C., Milne, E., and Freeth, M. (2017). Characteristics of autism spectrum disorder in sotos syndrome. J. Autism Dev. Disord. 47, 135–143.
Lee, J., and Skalnik, D. (2008). Wdr82 is a C-terminal domain-binding protein that recruits the Setd1A Histone H3-Lys4 methyltransferase complex to transcription start sites of transcribed human genes. Mol. Cell. Biol. 28, 609–618. doi: 10.1128/MCB.01356-07
Lee, J., Tate, C., You, J., and Skalnik, D. (2007). Identification and characterization of the human Set1B histone H3-Lys4 methyltransferase complex. J. Biol. Chem. 282, 13419–13428. doi: 10.1074/jbc.M609809200
Lee, J., Wang, C., Xu, S., Cho, Y., Wang, L., Feng, X., et al. (2013). H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife 2:e01503. doi: 10.7554/eLife.01503
Lehman, N., Mazery, A., Visier, A., Baumann, C., Lachesnais, D., Capri, Y., et al. (2017). Molecular, clinical and neuropsychological study in 31 patients with Kabuki syndrome and KMT2D mutations. Clin. Genet. 92, 298–305. doi: 10.1111/cge.13010
Lelieveld, S., Reijnders, M., Pfundt, R., Yntema, H., Kamsteeg, E., de Vries, P., et al. (2016). Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 19, 1194–1196. doi: 10.1038/nn.4352
Leung, C., Douglass, S., Morselli, M., Obusan, M., Pavlyukov, M., Pellegrini, M., et al. (2019). H3K36 methylation and the chromodomain protein Eaf3 are required for proper cotranscriptional spliceosome assembly. Cell Rep. 27, 3760–3769.e4. doi: 10.1016/j.celrep.2019.05.100
Li, J., You, Y., Yue, W., Yu, H., Lu, T., Wu, Z., et al. (2016). Chromatin remodeling gene EZH2 involved in the genetic etiology of autism in Chinese Han population. Neurosci. Lett. 610, 182–186. doi: 10.1016/j.neulet.2015.10.074
Li, L., Zhou, J., Wang, X., Zhang, H., Harris, P., Calvet, J., et al. (2020). Cross-talk between CDK4/6 and SMYD2 regulates gene transcription, tubulin methylation, and ciliogenesis. Sci. Adv. 6:eabb3154. doi: 10.1126/sciadv.abb3154
Li, S., Shen, L., and Chen, K. (2018). Association between H3K4 methylation and cancer prognosis: A meta-analysis. Thorac. Cancer 9, 794–799. doi: 10.1111/1759-7714.12647
Li, Y., and Jiao, J. (2017). Histone chaperone HIRA regulates neural progenitor cell proliferation and neurogenesis via β-catenin. J. Cell Biol. 216, 1975–1992. doi: 10.1083/jcb.201610014
Li, Y., Trojer, P., Xu, C., Cheung, P., Kuo, A., Drury, W. III, et al. (2009). The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J. Biol. Chem. 284, 34283–34295. doi: 10.1074/jbc.M109.034462
Lim, D., Huang, Y., Swigut, T., Mirick, A., Garcia-Verdugo, J., Wysocka, J., et al. (2009). Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 458, 529–533. doi: 10.1038/nature07726
Lim, P. S., Shannon, M. F., and Hardy, K. (2010). Epigenetic control of inducible gene expression in the immune system. Epigenomics 2, 775–795. doi: 10.2217/epi.10.55
Liu, P., Tang, G., Xu, Y., Zeng, Y., Zhang, S., Du, H., et al. (2017). MiR-203 Interplays with polycomb repressive complexes to regulate the proliferation of neural stem/progenitor cells. Stem Cell Rep. 9, 190–202. doi: 10.1016/j.stemcr.2017.05.007
Liu, P., Xu, Y., Teng, Z., and Liu, C. (2018). Polycomb repressive complex 2: Emerging roles in the central nervous system. Neuroscientist 24, 208–220.
Liu, S., Tian, M., He, F., Li, J., Xie, H., Liu, W., et al. (2020). Mutations in ASH1L confer susceptibility to Tourette syndrome. Mol. Psychiatry 25, 476–490. doi: 10.1038/s41380-019-0560-8
Lubitz, S., Glaser, S., Schaft, J., Stewart, A., and Anastassiadis, K. (2007). Increased apoptosis and skewed differentiation in mouse embryonic stem cells lacking the histone methyltransferase Mll2. Mol. Biol. Cell 18, 2356–2366. doi: 10.1091/mbc.e06-11-1060
Lucio-Eterovic, A., Singh, M., Gardner, J., Veerappan, C., Rice, J., and Carpenter, P. (2010). Role for the nuclear receptor-binding SET domain protein 1 (n.d.) methyltransferase in coordinating lysine 36 methylation at histone 3 with RNA polymerase II function. Proc. Natl. Acad. Sci. U.S.A. 107, 16952–16957. doi: 10.1073/pnas.1002653107
Lui, J., Hansen, D., and Kriegstein, A. (2011). Development and evolution of the human neocortex. Cell 146, 18–36.
Lumish, H., Wynn, J., Devinsky, O., and Chung, W. (2015). Brief Report: SETD2 mutation in a child with autism, intellectual disabilities and epilepsy. J. Autism Dev. Disord. 45, 3764–3770. doi: 10.1007/s10803-015-2484-8
Markus, A., Zhong, J., and Snider, W. (2002). Raf and akt mediate distinct aspects of sensory axon growth. Neuron 35, 65–76. doi: 10.1016/s0896-6273(02)00752-3
Marzin, P., Rondeau, S., Aldinger, K., Alessandri, J., Isidor, B., Heron, D., et al. (2019). SETD2 related overgrowth syndrome: Presentation of four new patients and review of the literature. Am. J. Med. Genet. C Semin. Med. Genet. 181, 509–518. doi: 10.1002/ajmg.c.31746
McCaffery, P., and Deutsch, C. (2005). Macrocephaly and the control of brain growth in autistic disorders. Prog. Neurobiol. 77, 38–56.
Meyer, E., Carss, K., Rankin, J., Nichols, J., Grozeva, D., Joseph, A., et al. (2017). Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat. Genet. 49, 223–237.
Michurina, A., Sakib, M., Kerimoglu, C., Kruger, D., Kaurani, L., Islam, M., et al. (2022). Postnatal expression of the lysine methyltransferase SETD1B is essential for learning and the regulation of neuron-enriched genes. EMBO J. 41:e106459. doi: 10.15252/embj.2020106459
Miles, J., Hadden, L., Takahashi, T., and Hillman, R. (2000). Head circumference is an independent clinical finding associated with autism. Am. J. Med. Genet. 95, 339–350.
Mimori-Kiyosue, Y., Shiina, N., and Tsukita, S. (2000). Adenomatous polyposis coli (APC) protein moves along microtubules and concentrates at their growing ends in epithelial cells. J. Cell Biol. 148, 505–518.
Mirzaa, G., and Poduri, A. (2014). Megalencephaly and hemimegalencephaly: Breakthroughs in molecular etiology. Am. J. Med. Genet. C Semin. Med. Genet. 166c, 156–172. doi: 10.1002/ajmg.c.31401
Molnar, Z., Clowry, G., and Co-Chairs of the Summer Meeting (2019a). Human cerebral cortex development. J. Anat. 235:431.
Molnar, Z., Clowry, G., Sestan, N., Alzu’bi, A., Bakken, T., Hevner, R., et al. (2019b). New insights into the development of the human cerebral cortex. J. Anat. 235, 432–451.
Moore, S., Seidman, J., Ellegood, J., Gao, R., Savchenko, A., Troutman, T., et al. (2019). Setd5 haploinsufficiency alters neuronal network connectivity and leads to autistic-like behaviors in mice. Transl. Psychiatry 9:24. doi: 10.1038/s41398-018-0344-y
Muhsin, E., Basak, G., Banu, D., Alper, G., and Mustafa, S. (2022). Neurodevelopment and genetic evaluation of sotos syndrome cases with a novel mutation: A single-center experience. J. Mol. Neurosci. 72, 149–157. doi: 10.1007/s12031-021-01897-5
Mukai, J., Cannavò, E., Crabtree, G., Sun, Z., Diamantopoulou, A., Thakur, P., et al. (2019). Recapitulation and reversal of schizophrenia-related phenotypes in setd1a-deficient mice. Neuron 104, 471–487.e12. doi: 10.1016/j.neuron.2019.09.014
Naf, D., Wilson, L., Bergstrom, R., Smith, R., Goodwin, N., Verkerk, A., et al. (2001). Mouse models for the Wolf-Hirschhorn deletion syndrome. Hum. Mol. Genet. 10, 91–98.
Nagahama, K., Sakoori, K., Watanabe, T., Kishi, Y., Kawaji, K., Koebis, M., et al. (2020). Setd1a insufficiency in mice attenuates excitatory synaptic function and recapitulates schizophrenia-related behavioral abnormalities. Cell Rep. 32:108126. doi: 10.1016/j.celrep.2020.108126
Nagase, T., Kikuno, R., Ishikawa, K., Hirosawa, M., and Ohara, O. (2000). Prediction of the coding sequences of unidentified human genes. XVII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 7, 143–150. doi: 10.1093/dnares/7.2.143
Nakagawa, T., Hattori, S., Nobuta, R., Kimura, R., Nakagawa, M., Matsumoto, M., et al. (2020). The autism-related protein SETD5 controls neural cell proliferation through epigenetic regulation of rDNA Expression. iScience 23:101030. doi: 10.1016/j.isci.2020.101030
Namba, T., and Huttner, W. (2017). Neural progenitor cells and their role in the development and evolutionary expansion of the neocortex. Wiley Interdiscip. Rev. Dev. Biol. 6:e256.
Nimura, K., Ura, K., Shiratori, H., Ikawa, M., Okabe, M., Schwartz, R., et al. (2009). A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature 460, 287–291. doi: 10.1038/nature08086
Noctor, S., Flint, A., Weissman, T., Wong, W., Clinton, B., and Kriegstein, A. (2002). Dividing precursor cells of the embryonic cortical ventricular zone have morphological and molecular characteristics of radial glia. J. Neurosci. 22, 3161–3173. doi: 10.1523/JNEUROSCI.22-08-03161.2002
Nowakowski, T., Bhaduri, A., Pollen, A., Alvarado, B., Mostajo-Radji, M., Di Lullo, E., et al. (2017). Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323. doi: 10.1126/science.aap8809
Nowakowski, T., Pollen, A., Sandoval-Espinosa, C., and Kriegstein, A. (2016). Transformation of the radial glia scaffold demarcates two stages of human cerebral cortex development. Neuron 91, 1219–1227. doi: 10.1016/j.neuron.2016.09.005
Ohtsuka, T., and Kageyama, R. (2019). Regulation of temporal properties of neural stem cells and transition timing of neurogenesis and gliogenesis during mammalian neocortical development. Semin Cell Dev. Biol. 95, 4–11. doi: 10.1016/j.semcdb.2019.01.007
Oishi, S., Zalucki, O., Vega, M., Harkins, D., Harvey, T., Kasherman, M., et al. (2020). Investigating cortical features of Sotos syndrome using mice heterozygous for Nsd1. Genes Brain Behav. 19:e12637. doi: 10.1111/gbb.12637
Okamoto, N., Miya, F., Tsunoda, T., Kato, M., Saitoh, S., Yamasaki, M., et al. (2017). Novel MCA/ID syndrome with ASH1L mutation. Am. J. Med. Genet. A 173, 1644–1648. doi: 10.1002/ajmg.a.38193
O’Roak, B., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B., et al. (2012b). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
O’Roak, B., Vives, L., Fu, W., Egertson, J., Stanaway, I., Phelps, I., et al. (2012a). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622.
Ouyang, Q., Lizarraga, S., Schmidt, M., Yang, U., Gong, J., Ellisor, D., et al. (2013). Christianson syndrome protein NHE6 modulates TrkB endosomal signaling required for neuronal circuit development. Neuron 80, 97–112. doi: 10.1016/j.neuron.2013.07.043
Palumbo, O., Palumbo, P., Delvecchio, M., Palladino, T., Stallone, R., Crisetti, M., et al. (2015). Microdeletion of 12q24.31: Report of a girl with intellectual disability, stereotypies, seizures and facial dysmorphisms. Am. J. Med. Genet. A 167a, 438–444. doi: 10.1002/ajmg.a.36872
Park, I., Powell, R., Tripathi, D., Dere, R., Ho, T., Blasius, T., et al. (2016). Dual chromatin and cytoskeletal remodeling by SETD2. Cell 166, 950–962.
Park, J., Chae, Y., Kim, J., Oh, H., and Seo, S. (2018). Methylation of Aurora kinase A by MMSET reduces p53 stability and regulates cell proliferation and apoptosis. Oncogene 37, 6212–6224. doi: 10.1038/s41388-018-0393-y
Pereira, J., Sansom, S., Smith, J., Dobenecker, M., Tarakhovsky, A., and Livesey, F. (2010). Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc. Natl. Acad. Sci. U.S.A. 107, 15957–15962. doi: 10.1073/pnas.1002530107
Pfister, S., Ahrabi, S., Zalmas, L., Sarkar, S., Aymard, F., Bachrati, C., et al. (2014). SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 7, 2006–2018. doi: 10.1016/j.celrep.2014.05.026
Piunti, A., and Shilatifard, A. (2016). Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 352:aad9780. doi: 10.1126/science.aad9780
Powis, Z., Farwell Hagman, K., Mroske, C., McWalter, K., Cohen, J., Colombo, R., et al. (2018). Expansion and further delineation of the SETD5 phenotype leading to global developmental delay, variable dysmorphic features, and reduced penetrance. Clin. Genet. 93, 752–761. doi: 10.1111/cge.13132
Pradeepa, M., Sutherland, H., Ule, J., Grimes, G., and Bickmore, W. (2012). Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 8:e1002717. doi: 10.1371/journal.pgen.1002717
Qi, C., Liu, S., Qin, R., Zhang, Y., Wang, G., Shang, Y., et al. (2014). Coordinated regulation of dendrite arborization by epigenetic factors CDYL and EZH2. J. Neurosci. 34, 4494–4508. doi: 10.1523/JNEUROSCI.3647-13.2014
Quintero-Rivera, F., Eno, C., Sutanto, C., Jones, K., Nowaczyk, M., Wong, D., et al. (2021). 5q35 duplication presents with psychiatric and undergrowth phenotypes mediated by NSD1 overexpression and mTOR signaling downregulation. Hum. Genet. 140, 681–690. doi: 10.1007/s00439-020-02240-5
Rabin, R., Radmanesh, A., Glass, I., Dobyns, W., Aldinger, K., Shieh, J., et al. (2020). Genotype-phenotype correlation at codon 1740 of SETD2. Am. J. Med. Genet. A 182, 2037–2048. doi: 10.1002/ajmg.a.61724
Rakic, P. (1972). Mode of cell migration to the superficial layers of fetal monkey neocortex. J. Comp. Neurol. 145, 61–83. doi: 10.1002/cne.901450105
Rakic, P. (2009). Evolution of the neocortex: A perspective from developmental biology. Nat. Rev. Neurosci. 10, 724–735.
Ramos, P., Sajuthi, S., Langefeld, C., and Walker, S. (2012). Immune function genes CD99L2, JARID2 and TPO show association with autism spectrum disorder. Mol. Autism 3:4. doi: 10.1186/2040-2392-3-4
Rawlins, L., Stals, K., Eason, J., and Turnpenny, P. (2017). De novo SETD5 nonsense mutation associated with diaphragmatic hernia and severe cerebral cortical dysplasia. Clin. Dysmorphol. 26, 95–97. doi: 10.1097/MCD.0000000000000144
Robertson, S., Gattas, M., Rogers, M., and Adès, L. (2000). Macrocephaly–cutis marmorata telangiectatica congenita: Report of five patients and a review of the literature. Clin. Dysmorphol. 9, 1–9.
Rosenfeld, J., Kim, K., Angle, B., Troxell, R., Gorski, J., Westemeyer, M., et al. (2013). Further evidence of contrasting phenotypes caused by reciprocal deletions and duplications: Duplication of NSD1 causes growth retardation and microcephaly. Mol. Syndromol. 3, 247–254. doi: 10.1159/000345578
Roston, A., Evans, D., Gill, H., McKinnon, M., Isidor, B., Cogné, B., et al. (2021). SETD1B-associated neurodevelopmental disorder. J. Med. Genet. 58, 196–204.
Rubio, C., Taddei, E., Acosta, J., Custodio, V., and Paz, C. (2020). Neuronal excitability in epileptogenic zones regulated by the Wnt/B-Catenin Pathway. CNS Neurol. Disord. Drug Targets 19, 2–11. doi: 10.2174/1871527319666200120143133
Sacco, R., Gabriele, S., and Persico, A. (2015). Head circumference and brain size in autism spectrum disorder: A systematic review and meta-analysis. Psychiatry Res. 234, 239–251. doi: 10.1016/j.pscychresns.2015.08.016
Satterstrom, F., Kosmicki, J., Wang, J., Breen, M., De Rubeis, S., An, J., et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23. doi: 10.1016/j.cell.2019.12.036
Satterstrom, F., Walters, R., Singh, T., Wigdor, E., Lescai, F., Demontis, D., et al. (2019). Autism spectrum disorder and attention deficit hyperactivity disorder have a similar burden of rare protein-truncating variants. Nat. Neurosci. 22, 1961–1965. doi: 10.1038/s41593-019-0527-8
Saugier-Veber, P., Bonnet, C., Afenjar, A., Drouin-Garraud, V., Coubes, C., Fehrenbach, S., et al. (2007). Heterogeneity of NSD1 alterations in 116 patients with Sotos syndrome. Hum. Mutat. 28, 1098–1107. doi: 10.1002/humu.20568
Schuettengruber, B., Bourbon, H., Di Croce, L., and Cavalli, G. (2017). Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 171, 34–57. doi: 10.1016/j.cell.2017.08.002
Seervai, R., Jangid, R., Karki, M., Tripathi, D., Jung, S., Kearns, S., et al. (2020). The Huntingtin-interacting protein SETD2/HYPB is an actin lysine methyltransferase. Sci. Adv. 6:eabb7854. doi: 10.1126/sciadv.abb7854
Seltzer, L., and Paciorkowski, A. (2014). Genetic disorders associated with postnatal microcephaly. Am. J. Med. Genet. C Semin. Med. Genet. 166C, 140–155.
Sessa, A., Fagnocchi, L., Mastrototaro, G., Massimino, L., Zaghi, M., Indrigo, M., et al. (2019). SETD5 regulates chromatin methylation state and preserves global transcriptional fidelity during brain development and neuronal wiring. Neuron 104, 271–289.e13. doi: 10.1016/j.neuron.2019.07.013
Shen, J., Eyaid, W., Mochida, G., Al-Moayyad, F., Bodell, A., Woods, C., et al. (2005). ASPM mutations identified in patients with primary microcephaly and seizures. J. Med. Genet. 42, 725–729.
Shen, T., Ji, F., Wang, Y., Lei, X., Zhang, D., and Jiao, J. (2018). Brain-specific deletion of histone variant H2A.z results in cortical neurogenesis defects and neurodevelopmental disorder. Nucleic Acids Res. 46, 2290–2307. doi: 10.1093/nar/gkx1295
Shen, W., Krautscheid, P., Rutz, A., Bayrak-Toydemir, P., and Dugan, S. (2019). De novo loss-of-function variants of ASH1L are associated with an emergent neurodevelopmental disorder. Eur. J. Med. Genet. 62, 55–60. doi: 10.1016/j.ejmg.2018.05.003
Sheppard, S., Campbell, I., Harr, M., Gold, N., Li, D., Bjornsson, H., et al. (2021). Expanding the genotypic and phenotypic spectrum in a diverse cohort of 104 individuals with Wiedemann-Steiner syndrome. Am. J. Med. Genet. A 185, 1649–1665.
Shilatifard, A. (2008). Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr. Opin. Cell Biol. 20, 341–348. doi: 10.1016/j.ceb.2008.03.019
Shintani, T., Ihara, M., Tani, S., Sakuraba, J., Sakuta, H., and Noda, M. (2009). APC2 plays an essential role in axonal projections through the regulation of microtubule stability. J. Neurosci. 29, 11628–11640.
Shintani, T., Takeuchi, Y., Fujikawa, A., and Noda, M. (2012). Directional neuronal migration is impaired in mice lacking adenomatous polyposis coli 2. J. Neurosci. 32, 6468–6484. doi: 10.1523/JNEUROSCI.0590-12.2012
Shirane, K., Miura, F., Ito, T., and Lorincz, M. (2020). NSD1-deposited H3K36me2 directs de novo methylation in the mouse male germline and counteracts Polycomb-associated silencing. Nat. Genet. 52, 1088–1098. doi: 10.1038/s41588-020-0689-z
Shitamukai, A., Konno, D., and Matsuzaki, F. (2011). Oblique radial glial divisions in the developing mouse neocortex induce self-renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. J. Neurosci. 31, 3683–3695. doi: 10.1523/JNEUROSCI.4773-10.2011
Singh, T., Kurki, M., Curtis, D., Purcell, S., Crooks, L., McRae, J., et al. (2016). Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci. 19, 571–577.
Singh, T., Poterba, T., Curtis, D., Akil, H., Al Eissa, M., Barchas, J., et al. (2022). Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 604, 509–516. doi: 10.1038/s41586-022-04556-w
Sobreira, N., Brucato, M., Zhang, L., Ladd-Acosta, C., Ongaco, C., Romm, J., et al. (2017). Patients with a Kabuki syndrome phenotype demonstrate DNA methylation abnormalities. Eur. J. Hum. Genet. 25, 1335–1344.
Sotos, J., Dodge, P., Muirhead, D., Crawford, J., and Talbot, N. (1964). Cerebral gigantism in childhood. A syndrome of excessively rapid growth and acromegalic features and a nonprogressive neurologic disorder. N. Engl. J. Med. 271, 109–116. doi: 10.1056/NEJM196407162710301
Speir, M., Bhaduri, A., Markov, N., Moreno, P., Nowakowski, T., Papatheodorou, I., et al. (2021). UCSC cell browser: Visualize your single-cell data. Bioinformatics 37, 4578–4580. doi: 10.1093/bioinformatics/btab503
Stessman, H., Xiong, B., Coe, B., Wang, T., Hoekzema, K., Fenckova, M., et al. (2017). Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 49, 515–526. doi: 10.1038/ng.3792
Streubel, G., Watson, A., Jammula, S., Scelfo, A., Fitzpatrick, D., Oliviero, G., et al. (2018). The H3K36me2 Methyltransferase Nsd1 Demarcates PRC2-Mediated H3K27me2 and H3K27me3 Domains in Embryonic Stem Cells. Mol. Cell 70, 371–379.e5. doi: 10.1016/j.molcel.2018.02.027
Strom, S., Lozano, R., Lee, H., Dorrani, N., Mann, J., O’Lague, P., et al. (2014). De novo variants in the KMT2A (MLL) gene causing atypical Wiedemann-Steiner syndrome in two unrelated individuals identified by clinical exome sequencing. BMC Med. Genet. 15:49. doi: 10.1186/1471-2350-15-49
Szczałuba, K., Brzezinska, M., Kot, J., Rydzanicz, M., Walczak, A., Stawiński, P., et al. (2016). SETD5 loss-of-function mutation as a likely cause of a familial syndromic intellectual disability with variable phenotypic expression. Am. J. Med. Genet. A 170, 2322–2327. doi: 10.1002/ajmg.a.37832
Takei, N., Inamura, N., Kawamura, M., Namba, H., Hara, K., Yonezawa, K., et al. (2004). Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J. Neurosci. 24, 9760–9769. doi: 10.1523/JNEUROSCI.1427-04.2004
Tammimies, K., Marshall, C., Walker, S., Kaur, G., Thiruvahindrapuram, B., Lionel, A., et al. (2015). Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA 314, 895–903.
Tanaka, Y., Katagiri, Z., Kawahashi, K., Kioussis, D., and Kitajima, S. (2007). Trithorax-group protein ASH1 methylates histone H3 lysine 36. Gene 397, 161–168.
Tanaka, Y., Kawahashi, K., Katagiri, Z., Nakayama, Y., Mahajan, M., and Kioussis, D. (2011). Dual function of histone H3 lysine 36 methyltransferase ASH1 in regulation of Hox gene expression. PLoS One 6:e28171. doi: 10.1371/journal.pone.0028171
Tang, G., Zeng, Y., Liu, P., Mi, T., Zhang, S., Dai, S., et al. (2017). The Histone H3K27 Demethylase UTX regulates synaptic plasticity and cognitive behaviors in mice. Front. Mol. Neurosci. 10:267. doi: 10.3389/fnmol.2017.00267
Tang, S., Addis, L., Smith, A., Topp, S., Pendziwiat, M., Mei, D., et al. (2020). Phenotypic and genetic spectrum of epilepsy with myoclonic atonic seizures. Epilepsia 61, 995–1007.
Tatton-Brown, K., Murray, A., Hanks, S., Douglas, J., Armstrong, R., Banka, S., et al. (2013). Weaver syndrome and EZH2 mutations: Clarifying the clinical phenotype. Am. J. Med. Genet. A 161A, 2972–2980. doi: 10.1002/ajmg.a.36229
Tenney, J., Hopkin, R., and Schapiro, M. (2011). Deletion of 14-3-3{varepsilon} and CRK: A clinical syndrome with macrocephaly, developmental delay, and generalized epilepsy. J. Child Neurol. 26, 223–227. doi: 10.1177/0883073810379638
Tie, F., Banerjee, R., Saiakhova, A., Howard, B., Monteith, K., Scacheri, P., et al. (2014). Trithorax monomethylates histone H3K4 and interacts directly with CBP to promote H3K27 acetylation and antagonize Polycomb silencing. Development 141, 1129–1139. doi: 10.1242/dev.102392
Trujillano, D., Bertoli-Avella, A., Kumar Kandaswamy, K., Weiss, M., Köster, J., Marais, A., et al. (2017). Clinical exome sequencing: Results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 25, 176–182. doi: 10.1038/ejhg.2016.146
Usluer, E., Sayin, G., Gunes, N., Kasap, B., and Tuysuz, B. (2022). Investigation of genetic and phenotypic heterogeneity in 37 Turkish patients with Kabuki and Kabuki-like phenotype. Am. J. Med. Genet. A 188, 2976–2987. doi: 10.1002/ajmg.a.62944
Vallianatos, C., and Iwase, S. (2015). Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics 7, 503–519. doi: 10.2217/epi.15.1
Vallianatos, C., Raines, B., Porter, R., Bonefas, K., Wu, M., Garay, P., et al. (2020). Mutually suppressive roles of KMT2A and KDM5C in behaviour, neuronal structure, and histone H3K4 methylation. Commun. Biol. 3:278.
van Rij, M., Hollink, I., Terhal, P., Kant, S., Ruivenkamp, C., van Haeringen, A., et al. (2018). Two novel cases expanding the phenotype of SETD2-related overgrowth syndrome. Am. J. Med. Genet. A 176, 1212–1215. doi: 10.1002/ajmg.a.38666
Verberne, E., Goh, S., England, J., van Ginkel, M., Rafael-Croes, L., Maas, S., et al. (2021). JARID2 haploinsufficiency is associated with a clinically distinct neurodevelopmental syndrome. Genet. Med. 23, 374–383.
Wagner, E., and Carpenter, P. (2012). Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 13, 115–126. doi: 10.1038/nrm3274
Wang, G., Cai, L., Pasillas, M., and Kamps, M. (2007). NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell. Biol. 9, 804–812. doi: 10.1038/ncb1608
Wang, P., Lin, C., Smith, E., Guo, H., Sanderson, B., Wu, M., et al. (2009). Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell. Biol. 29, 6074–6085. doi: 10.1128/MCB.00924-09
Wang, S., Rhijn, J., Akkouh, I., Kogo, N., Maas, N., Bleeck, A., et al. (2022). Loss-of-function variants in the schizophrenia risk gene SETD1A alter neuronal network activity in human neurons through the cAMP/PKA pathway. Cell Rep. 39:110790. doi: 10.1016/j.celrep.2022.110790
Wang, T., Guo, H., Xiong, B., Stessman, H., Wu, H., Coe, B., et al. (2016). De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 7:13316. doi: 10.1038/ncomms13316
Wang, T., Hoekzema, K., Vecchio, D., Wu, H., Sulovari, A., Coe, B., et al. (2020). Large-scale targeted sequencing identifies risk genes for neurodevelopmental disorders. Nat. Commun. 11:4932.
Weerts, M., Lanko, K., Guzman-Vega, F., Jackson, A., Ramakrishnan, R., Cardona-Londono, K., et al. (2021). Delineating the molecular and phenotypic spectrum of the SETD1B-related syndrome. Genet. Med. 23, 2122–2137. doi: 10.1038/s41436-021-01246-2
Weinberg, D., Papillon-Cavanagh, S., Chen, H., Yue, Y., Chen, X., Rajagopalan, K., et al. (2019). The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281–286.
Weiss, L., Arking, D., Daly, M., and Chakravarti, A. (2009). A genome-wide linkage and association scan reveals novel loci for autism. Nature 461, 802–808.
Wiel, L., Bruno, I., Barbi, E., and Sirchia, F. (2022). From Wolf-Hirschhorn syndrome to NSD2 haploinsufficiency: A shifting paradigm through the description of a new case and a review of the literature. Ital. J. Pediatr. 48, 72. doi: 10.1186/s13052-022-01267-w
Willsey, A., Sanders, S., Li, M., Dong, S., Tebbenkamp, A., Muhle, R., et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. doi: 10.1016/j.cell.2013.10.020
Winden, K., Yuskaitis, C., and Poduri, A. (2015). Megalencephaly and macrocephaly. Semin. Neurol. 35, 277–287.
Wynder, C., Stalker, L., and Doughty, M. (2010). Role of H3K4 demethylases in complex neurodevelopmental diseases. Epigenomics 2, 407–418.
Xie, X., Wang, S., Li, M., Diao, L., Pan, X., Chen, J., et al. (2021). alpha-TubK40me3 is required for neuronal polarization and migration by promoting microtubule formation. Nat. Commun. 12:4113. doi: 10.1038/s41467-021-24376-2
Xu, L., Zheng, Y., Li, X., Wang, A., Huo, D., Li, Q., et al. (2021). Abnormal neocortex arealization and Sotos-like syndrome-associated behavior in Setd2 mutant mice. Sci. Adv. 7:eaba1180. doi: 10.1126/sciadv.aba1180
Xu, W., Li, J., Rong, B., Zhao, B., Wang, M., Dai, R., et al. (2020). DNMT3A reads and connects histone H3K36me2 to DNA methylation. Protein Cell 11, 150–154.
Yamada-Okabe, T., Imamura, K., Kawaguchi, N., Sakai, H., Yamashita, M., and Matsumoto, N. (2010). Functional characterization of the zebrafish WHSC1-related gene, a homolog of human NSD2. Biochem. Biophys. Res. Commun. 402, 335–339. doi: 10.1016/j.bbrc.2010.10.027
Yan, Y., Tian, M., Li, M., Zhou, G., Chen, Q., Xu, M., et al. (2022). ASH1L haploinsufficiency results in autistic-like phenotypes in mice and links Eph receptor gene to autism spectrum disorder. Neuron 110, 1156–1172.e9. doi: 10.1016/j.neuron.2021.12.035
Yang, L., Jin, M., and Jeong, K. (2021). Histone H3K4 methyltransferases as targets for drug-resistant cancers. Biology 10:581.
Yoganathan, S., Arunachal, G., Gowda, V., Vinayan, K., Thomas, M., Whitney, R., et al. (2021). NTRK2-related developmental and epileptic encephalopathy: Report of 5 new cases. Seizure 92, 52–55. doi: 10.1016/j.seizure.2021.08.008
Yoshii, A., and Constantine-Paton, M. (2010). Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev. Neurobiol. 70, 304–322.
Yu, C., Yao, X., Zhao, L., Wang, P., Zhang, Q., Zhao, C., et al. (2017). Wolf-Hirschhorn Syndrome Candidate 1 (whsc1) Functions as a Tumor Suppressor by Governing Cell Differentiation. Neoplasia 19, 606–616. doi: 10.1016/j.neo.2017.05.001
Yu, J., Lee, C., Oksuz, O., Stafford, J., and Reinberg, D. (2019). PRC2 is high maintenance. Genes Dev. 33, 903–935.
Yu, X., Yang, L., Li, J., Li, W., Li, D., Wang, R., et al. (2019). De novo and inherited SETD1A variants in early-onset epilepsy. Neurosci. Bull. 35, 1045–1057. doi: 10.1007/s12264-019-00400-w
Yu, T., Mochida, G., Tischfield, D., Sgaier, S., Flores-Sarnat, L., Sergi, C., et al. (2010). Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat. Genet. 42, 1015–1020. doi: 10.1038/ng.683
Yuan, W., Xu, M., Huang, C., Liu, N., Chen, S., and Zhu, B. (2011). H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 286, 7983– 7989.
Yuen, R. C., Merico, D., Bookman, M., Howe, J. L., Thiruvahindrapuram, B., Patel, R., et al. (2017).. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 20, 602–611.
Zanoni, P., Steindl, K., Sengupta, D., Joset, P., Bahr, A., Sticht, H., et al. (2021). Loss-of-function and missense variants in NSD2 cause decreased methylation activity and are associated with a distinct developmental phenotype. Genet. Med. 23, 1474– 1483.
Zech, M., Boesch, S., Maier, E., Borggraefe, I., Vill, K., Laccone, F., et al. (2016). Haploinsufficiency of KMT2B, encoding the lysine-specific histone methyltransferase 2B, results in early-onset generalized dystonia. Am. J. Hum. Genet. 99, 1377–1387. doi: 10.1016/j.ajhg.2016.10.010
Zhang, C., Xu, L., Zheng, X., Liu, S., and Che, F. (2021). Role of Ash1l in Tourette syndrome and other neurodevelopmental disorders. Dev. Neurobiol. 81, 79–91. doi: 10.1002/dneu.22795
Zhang, J., Tao, Q., Yang, Z., Li, Y., and Gan, J. (2022). De novo variant of SETD1A causes neurodevelopmental disorder with dysmorphic facies: A case report. Psychiatry Clin. Neurosci. 76, 58–59. doi: 10.1111/pcn.13310
Zhang, L., Yang, X., Yang, S., and Zhang, J. (2011). The Wnt?/β-catenin signaling pathway in the adult neurogenesis. Eur. J. Neurosci. 33, 1–8.
Zhang, M., Zhao, J., Lv, Y., Wang, W., Feng, C., Zou, W., et al. (2020). Histone Variants and Histone Modifications in Neurogenesis. Trends Cell Biol. 30, 869–880.
Zhu, J., Adli, M., Zou, J., Verstappen, G., Coyne, M., Zhang, X., et al. (2013). Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 152, 642–654.
Zhuang, L., Jang, Y., Park, Y., Lee, J., Jain, S., Froimchuk, E., et al. (2018). Depletion of Nsd2-mediated histone H3K36 methylation impairs adipose tissue development and function. Nat. Commun. 9, 1796. doi: 10.1038/s41467-018-04127-6
Zikopoulos, B., and Barbas, H. (2010). Changes in prefrontal axons may disrupt the network in autism. J. Neurosci. 30, 14595–14609. doi: 10.1523/JNEUROSCI.2257-10.2010
Keywords: autism, histone methyltransferase, brain size, neurodevelopment, chromatin, microcephaly, macrocephaly
Citation: Ritchie FD and Lizarraga SB (2023) The role of histone methyltransferases in neurocognitive disorders associated with brain size abnormalities. Front. Neurosci. 17:989109. doi: 10.3389/fnins.2023.989109
Received: 08 July 2022; Accepted: 17 January 2023;
Published: 10 February 2023.
Edited by:
Debra Silver, Duke University, United StatesReviewed by:
Jessica L. MacDonald, Syracuse University, United StatesShan Wang, Radboud University Medical Centre, Netherlands
Copyright © 2023 Ritchie and Lizarraga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sofia B. Lizarraga,  c29maWFfbGl6YXJyYWdhQGJyb3duLmVkdQ==
c29maWFfbGl6YXJyYWdhQGJyb3duLmVkdQ==
†Present address: Sofia B. Lizarraga, Department of Molecular Biology, Cell Biology and Biochemistry, Brown University, Providence, RI, United States Center for Translational Neuroscience, Carney Institute for Brain Science and Brown Institute for Translational Science, Brown University, Providence, RI, United States