 Youngshin Lim1,2*
Youngshin Lim1,2*- 1Department of Pathology and Laboratory Medicine, Cedars-Sinai Medical Center, Los Angeles, CA, United States
- 2Department of Biomedical Science Education, Charles R. Drew University of Medicine and Science, Los Angeles, CA, United States
Higher cognition in humans, compared to other primates, is often attributed to an increased brain size, especially forebrain cortical surface area. Brain size is determined through highly orchestrated developmental processes, including neural stem cell proliferation, differentiation, migration, lamination, arborization, and apoptosis. Disruption in these processes often results in either a small (microcephaly) or large (megalencephaly) brain. One of the key mechanisms controlling these developmental processes is the spatial and temporal transcriptional regulation of critical genes. In humans, microcephaly is defined as a condition with a significantly smaller head circumference compared to the average head size of a given age and sex group. A growing number of genes are identified as associated with microcephaly, and among them are those involved in transcriptional regulation. In this review, a subset of genes encoding transcription factors (e.g., homeobox-, basic helix-loop-helix-, forkhead box-, high mobility group box-, and zinc finger domain-containing transcription factors), whose functions are important for cortical development and implicated in microcephaly, are discussed.
1 Introduction
Microcephaly is a clinical condition where the occipito-frontal head circumference (OCF) of an individual is significantly smaller than the average head size of the population for a given sex and age (Lim and Golden, 2020). Typically, a cutoff of more than 2 standard deviations (SDs) below the mean or less than the 3rd percentile is used for microcephaly diagnosis; however, other cutoff values such as more than 3 SD and less than the 5th or 10th percentile can be used as well (Opitz and Holt, 1990; Raymond and Holmes, 1994; Ashwal et al., 2009). The prevalence of microcephaly is between 1.5 and 8.7 per 10,000 births in Europe and the US, respectively (Cragan et al., 2016; Morris et al., 2016), and ~15–20% of children with developmental delay are reported to have microcephaly (Sassaman and Zartler, 1982; Watemberg et al., 2002; Aggarwal et al., 2013). Microcephaly can manifest as the only phenotype without other obvious morphologic or functional abnormalities (e.g., cortical- or extra-cortical malformations, and intellectual disability), as in the case of “isolated microcephaly”; or it can be accompanied by neurological or psychiatric conditions, but without cortical or extra-cortical malformations, as in the case of “non-syndromic microcephaly”. In contrast, “syndromic microcephaly” often presents together with other cortical malformations (e.g., polymicrogyria, lissencephaly, periventricular nodular heterotopia, and agenesis of corpus callosum) or can be part of a broader syndrome involving other organ systems (more than 700 genetic syndromes are reported to have microcephaly) (Pirozzi et al., 2018).
Depending on the time of presentation, microcephaly can be classified as primary (congenital) or secondary (postnatal) microcephaly. Congenital microcephaly is usually caused by deficiencies in the number of neurons (either reduced production or increased loss), while postnatal microcephaly is more often attributed to defective brain growth and/or neurodegeneration causing brain atrophy (Gilmore and Walsh, 2013). However, this classification is by no means strict. For example, a defect in neuronal generation can still result in postnatal microcephaly if the proliferation defect is not severe enough to manifest at birth.
The recent explosion in the use of next-generation sequencing has enabled the discovery of many novel disease-causing genetic variants in microcephaly. For example, 32 primary microcephaly (MCPH, microcephaly primary hereditary) genes have been identified (Asif et al., 2023), and the list is rapidly growing. Most of these genes encode centrosome-specific proteins, spindle-associated proteins, microtubule-associated proteins, and cell cycle checkpoint proteins (e.g., ASPM, CDK6, CDK5RAP2, CENPJ, CEP63/135/152, NIN, PLK4, STIL, TUBGCP6, and WDR62, among others) that cause cell cycle-related defects when mutated (Lim and Golden, 2020). Another group of the prominent MCPH genes encode proteins in various DNA damage responses and repair pathways (e.g., PNKP, ERCC6, ERCC8, CHEK2, NHEJ1, XRCC4, XRCC5, and XRCC6, among others) (Lim and Golden, 2020). In addition, the list also includes genes encoding proteins involved in transcriptional regulation (e.g., transcription factors and chromatin remodeling proteins), although only a small number of them are identified in MCPH (Jayaraman et al., 2018). If syndromic microcephalies are also considered, many additional transcription-related proteins associated with microcephaly can be included (see below).
In this review, transcription factors associated with microcephaly will be discussed, focusing on their reported functions and pathogenic mechanisms in model systems, which can be linked to clinical features in humans. Selected members of five different transcription factor families will be highlighted, including (1) homeobox genes such as ARX, LHX2, MEIS, NKX2-1, OTX1, OTX2, and PAX6; (2) bHLH (basic helix-loop-helix) genes such as MYCN and TCF4; (3) FOX (forkhead box) genes, FOXG1 and FOXR1; (4) SOX (sex-determining regions Y-related HMG box) genes belonging to SOXB1 (SOX2 and SOX3) and SOXC (SOX4 and SOX11) subfamily; and finally (5) zinc finger genes, ZNF238 and ZNF355 (Table 1). While not covered in this review, chromatin remodeling proteins are also critical factors in transcriptional regulation that can result in microcephaly when mutated. For instance, the SMARCB1, SMARCA4, SMARCE1, ARID1A, and ARID1B genes encode subunits of the BAF complex (also known as the SWI/SNF complex in yeast), and they are responsible for 55–70% of Coffin–Siris syndrome cases, where microcephaly can manifest as one of the phenotypes (Kosho et al., 2014; Vergano et al., 2021).
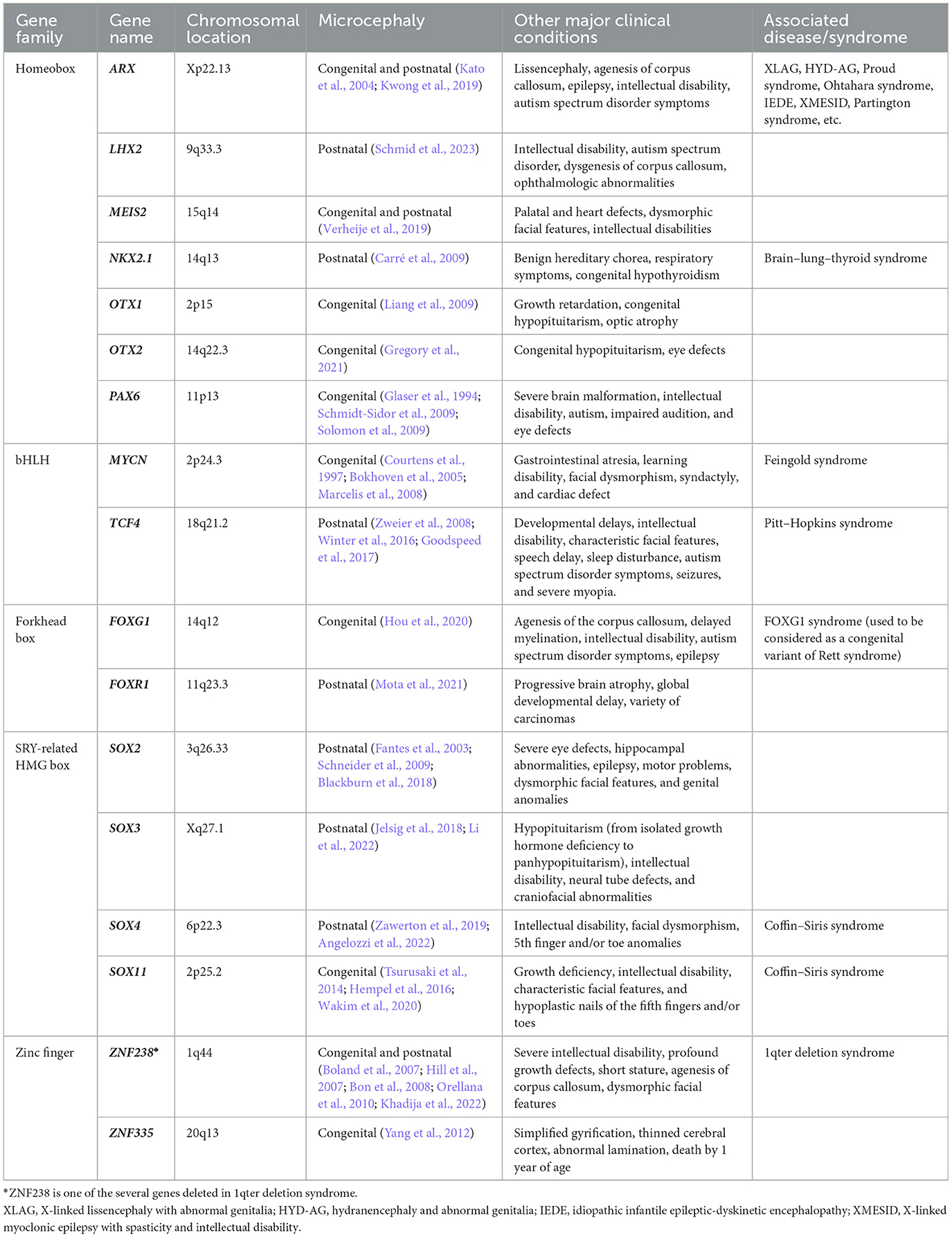
Table 1. Transcription factors associated with microcephaly.
2 Transcription factors associated with microcephaly
2.1 Homeobox genes: ARX, LHX2, Meis, NKX2-1, OTX1, OTX2, and PAX6
The homeobox gene family members are characterized by 180-bp DNA sequences known as the “homeobox” encoding a 60-amino acid protein domain called the “homeodomain” (Figure 1). Most homeodomains contain a helix-loop-helix-turn-helix structure responsible for DNA binding (Gehring et al., 1994; Bürglin and Affolter, 2016). The DNA-binding motif is typically located at the second or third helix and recognizes and binds to the major groove of DNA at specific consensus sites (Kappen, 2000). The proteins encoded by homeobox genes are called homeodomain proteins, or simply homeoproteins. In addition to the homeodomain, homeoproteins often contain other domains or motifs (e.g., paired domain and “PRD”) that can contribute to DNA and/or co-factor binding (Leung et al., 2022). Often, these additional DNA-binding domains and co-factor binding sites confer additional DNA specificity to these proteins, apart from the consensus binding sequence (Holland et al., 2007). Homeoproteins can be divided into various different classes, such as the Antennapedia (ANTP), Paired (PRD), and LIM classes (Holland et al., 2007), based on the variations in the homeodomain and additional domains or motifs. Well-known examples of homeoproteins belonging to these three classes include HOX (ANTP class), PAX (PRD class), and LHX (LIM class) (Holland et al., 2007).
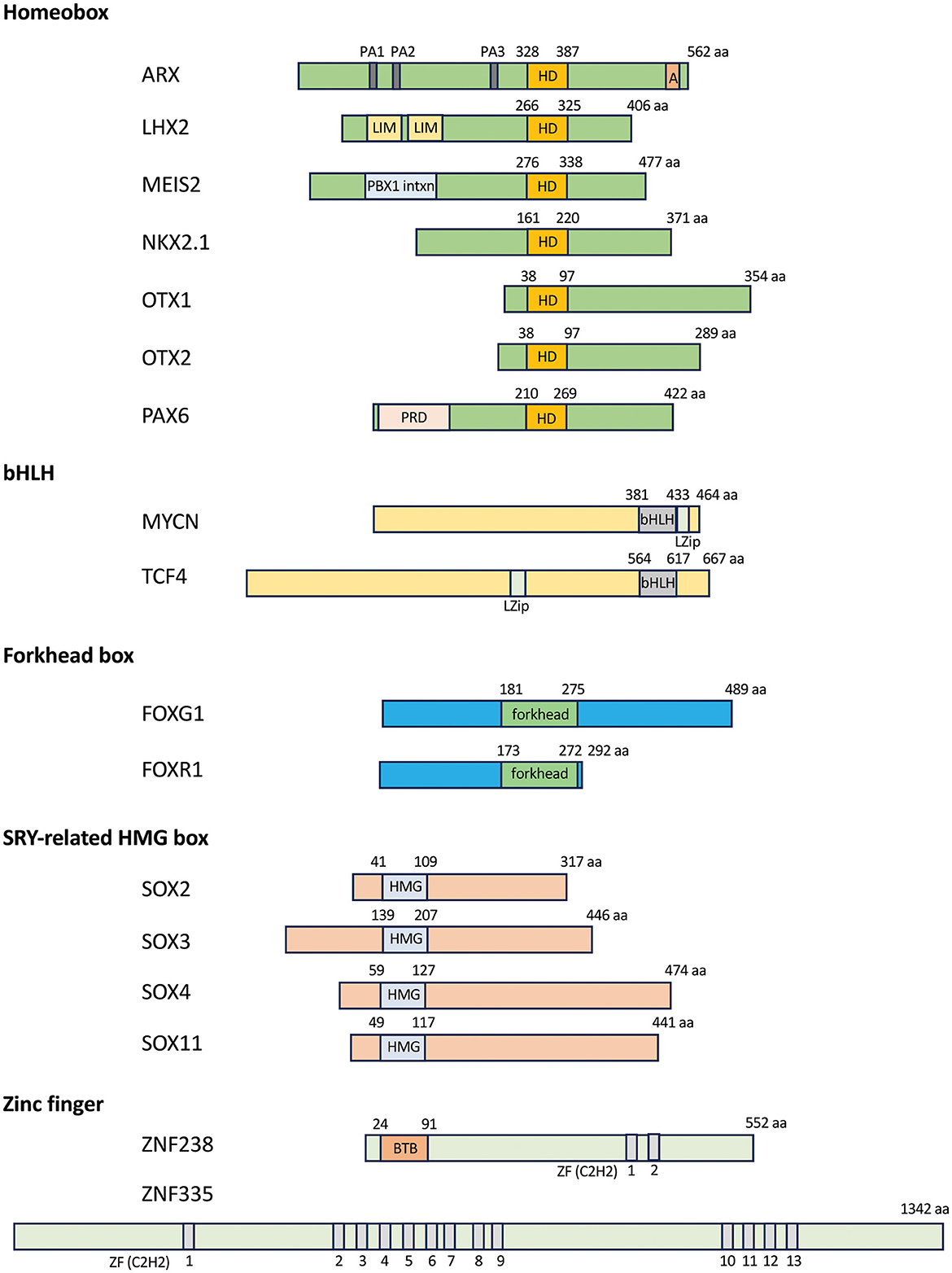
Figure 1. Transcription factors associated with microcephaly. Schematics of the domain structure of the homeobox, bHLH, forkhead box, HMG box, and zinc finger transcription factors. Numbers over the DNA-binding domain of each selected transcription factor indicate amino acid residues, while numbers below the gray domains in the zinc finger genes indicate each C2H2 zinc finger domain. A, aristaless domain; BTB, BTB domain [Broad-Complex, Tram track, and Bric-a-brac, also known as POZ (poxvirus zinc finger) domain; bHLH, basic helix-loop-helix domain; C2H2, cysteine–cysteine–histidine–histidine; forkhead, forkhead box domain; HD, homeobox domain; HMG, high mobility group; LIM, LIM domain (Lin-11, Isl1, and Mec-3); LZip, leucine zipper domain; PA1-3, poly alanine track 1–3; PBX1 intxn, PBX1 interaction domain; PRD, paired domain; ZF, zinc finger domain.
Mutations in homeobox genes were first described in Drosophila, which resulted in homeotic transformation (normal structure developing at an abnormal body position, e.g., leg growing from the location expected to be antennae) (Gehring et al., 1994). It is now known that homeoproteins control many cellular processes crucial for embryonic development, including proliferation, differentiation, apoptosis, cell shape, cell adhesion, and migration (Pearson et al., 2005). Furthermore, homeobox genes are known to be associated with many developmental brain disorders and cancers in humans (Leung et al., 2022). In this study, I focus on several homeobox genes (ARX, LHX2, MEIS2, NKX2-1, OTX1, OTX2, and PAX6) implicated in the pathogenesis of microcephaly (Table 1). The expression patterns of these homeobox genes in the embryonic mouse brains (Figure 2), their functional roles during brain development, and their human genetic variants and clinical features associated with microcephaly will be highlighted.
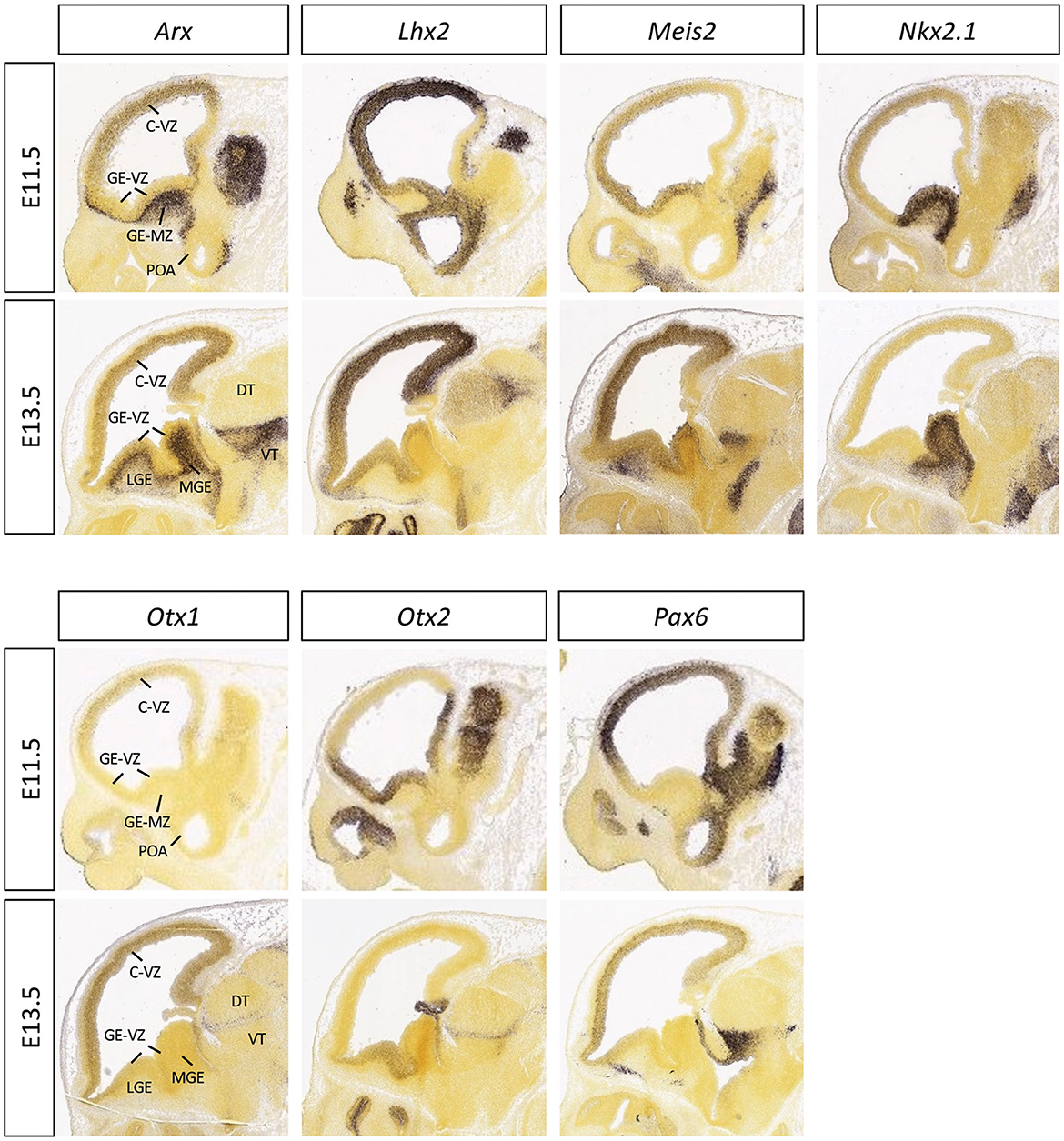
Figure 2. Expression patterns of the selected homeobox transcription factors in embryonic mouse brain. The mid-sagittal sections of E11.5 or E13.5 embryonic brains showing RNA in situ hybridization results of Arx, Lhx2, Meis2, Nkx2.1, Otx1, Otx2, and Pax6, adapted from Allen Brain Atlas (https://developingmouse.brain-map.org).
2.1.1 ARX (Aristaless-related homeobox)
Aristaless-related homeobox gene (ARX) belongs to the PRD class, one of the three largest classes of homeobox genes. It is the vertebrate homolog of Drosophila aristaless (al), essential for the formation of the head segment and the axis specification of appendages (Miura et al., 1997). It is located on the X chromosome (Xp22.13 in humans), and mutations in ARX are associated with a spectrum of neurodevelopmental conditions that can be classified into two groups—those with and those without structural abnormalities. The malformation group can include lissencephaly, microcephaly, and/or agenesis of corpus callosum and is associated with several syndromes: X-linked lissencephaly with abnormal genitalia (XLAG), hydranencephaly and abnormal genitalia (HYD-AG), and Proud syndrome (agenesis of corpus callosum with abnormal genitalia) (Dobyns et al., 1999; Ogata et al., 2000; Kitamura et al., 2002; Kato et al., 2004). The second group of ARX-associated conditions, typically without severe malformations, frequently includes a variety of infantile and childhood seizure phenotypes and non-syndromic X-linked intellectual disability (Bienvenu et al., 2002; Strømme et al., 2002; Kato et al., 2003). In rare cases, these relatively less severe conditions can also present with brain malformations (e.g., microcephaly in a case with Ohtahara syndrome) (Absoud et al., 2010). More severe phenotypes harboring brain malformations seem to arise from premature termination mutations (large deletions, frameshifts, and nonsense mutations) or missense mutations in the homeodomain or nuclear localization sequences, while missense mutations outside of the homeodomain or mutations in polyalanine tracts (expansion or deletion) are associated with less severe phenotypes (Friocourt and Parnavelas, 2010).
Interestingly, individuals with ARX variants can have either congenital or postnatal microcephaly (Friocourt and Parnavelas, 2010). For example, XLAG patients with nonsense mutation (c.1117C<T (Q373X)) or with missense mutation (c.1561G<A (A521T)) in the aristaless domain tend to have congenital microcephaly (Kato et al., 2004), while XLAG patients with missense mutation (c.989G > A; p.Arg330) located in the nuclear localization sequence (NLS2) or with a deletion mutation (790delC) predicted to cause premature truncation were reported to have postnatal (progressive) microcephaly (Kwong et al., 2019). Of note, in some cases of postnatal microcephaly, the underlying pathogenic mechanism is distinct from that of the congenital microcephaly—proliferation defects in congenital vs. growth defects or neurodegeneration in postnatal microcephaly (Absoud et al., 2010). However, in other cases, it is possible that the embryonic proliferation defects, which are not severe enough to be detected at birth, can manifest as postnatal microcephaly.
The function of ARX in the brain is well reflected by its expression pattern. In the dorsal telencephalon (developing cortex), Arx is expressed in the proliferating cells located in the ventricular zone (VZ) and subventricular zone (SVZ) (Figure 2). However, its expression is turned off once cells exit the cell cycle and begin to migrate out of the VZ and SVZ (Lim et al., 2022). In contrast, in the ventral forebrain, Arx is not expressed in the VZ; instead, it is strongly expressed in the SVZ and mantle zone of the lateral ganglionic eminence (LGE) and medial ganglionic eminence (MGE) (Figure 2) and remains expressed in non-radially migrating neurons emanating from the MGE to the cerebral cortex (Lim et al., 2022). Interestingly, when Arx is genetically ablated in mice from the dorsal telencephalon, neural progenitor expansion is disrupted, mainly due to premature cell cycle exit (Colasante et al., 2015). The brains of these mice are smaller, resembling the microcephaly observed in humans with an ARX mutation; however, these mice do not develop seizures (Friocourt and Parnavelas, 2010; Colasante et al., 2015). In contrast, when Arx is abrogated from the ventral telencephalon, the brains are of normal size, but all of the male mice exhibit a range of seizures (Marsh et al., 2009).
What is the pathogenic mechanism for ARX mutations to result in microcephaly? It appears that ARX regulates the expansion of cortical progenitors by regulating the level of Cdkn1c expression (Colasante et al., 2015). CDKN1C (also known as p57/KIP2) regulates the G1-S transition of cortical progenitors (Sherr and Roberts, 1999). When overexpressed in mice, it promotes progenitor cell cycle exit and a transition from proliferation to differentiation, while its loss leads to increased proliferation and macrocephaly (Mairet-Coello et al., 2012). Targeted ablation of Arx in cortical progenitors leads to an abnormal increase in Cdkn1c expression (ARX normally represses Cdkn1c expression), resulting in premature cell cycle exit and depleting progenitor pools, leading to a reduced brain size. Furthermore, the loss of Arx disrupts PAX6 and TBR2 expression in progenitor cells (Lim et al., 2019). PAX6 and TBR2 are also transcription factors expressed in progenitor cells, where they also play a role in proliferation (PAX6 for radial glial progenitors; TBR2 for intermediate progenitors). Their decreased expression in Arx mutant mice appears to be a second contributing mechanism leading to the microcephaly (Lim et al., 2019).
2.1.2 LHX2 (LIM homeobox 2)
LHX2 is a member of the LIM (Lin-11, Isl1, and Mec-3) class of homeobox gene family. The protein contains a homeodomain (for DNA binding) and two cysteine-rich LIM zinc finger domains (required for zinc binding and functioning as a modular protein-binding interface to mediate protein–protein interactions) (Feuerstein et al., 1994; Schmeichel and Beckerle, 1994). LHX2 can form multimeric complexes with other co-factors such as LIM-domain-binding-1 (LDB1) via the LIM domains, allowing homeodomain-mediated DNA binding to activate its target genes (Schmeichel and Beckerle, 1994; Agulnick et al., 1996; Breen et al., 1998; Kadrmas and Beckerle, 2004).
LHX2/Lhx2 is a vertebrate ortholog of the Drosophila “selector” gene apterous (Ap) which is essential for wing development (Cohen et al., 1992). Selector genes are known to function cell-autonomously to specify cell identity while suppressing alternative fates (Lawrence and Struhl, 1996; Irvine and Rauskolb, 2001). Lhx2, which is expressed in cortical precursor cells but not adjacent cells (choroid plexus epithelium and cortical hem), has been shown to act as a selector gene in the developing mouse cerebral cortex, specifying cortical identity in a cell-autonomous fashion while suppressing hippocampal organizer fate (Mangale et al., 2008). Lhx2 homozygous knockout mice are embryonic lethal, likely due to severe anemia. They also show forebrain hypoplasia along with defects in eye development (anophthalmia) (Porter et al., 1997). Furthermore, dorsal forebrain conditional Lhx2 mutant mice have dramatically smaller cerebral cortices when compared to controls (Chou et al., 2009), again resembling with human microcephaly phenotype observed in patients with LHX2 variants (Schmid et al., 2023).
Until recently, LHX2 defects had not been linked to neurodevelopmental disorders, with the exception of a few variants described in a large study on developmental disorders (Kaplanis et al., 2020) and in an autism cohort (Zhou et al., 2022). However, LHX2 haploinsufficiency has now been linked to variable neurodevelopmental disorders (Schmid et al., 2023); the authors identified de novo deletions (likely gene-disrupting) and missense variants in LHX2 from 19 individuals (18 families) presenting with a variable neurodevelopmental phenotype, including microcephaly, intellectual disability, autism spectrum disorder, and other behavioral anomalies. Among 10 patients carrying likely pathogenic variants with occipito-frontal head circumference (OFC) records available, 7 patients showed microcephaly at the time of investigation (age ranging from 2 to 8 years; OFC at birth not available); 4 of them had deletions or nonsense variants (likely gene-disrupting), and 3 had missense variants in the LIM domain or homeodomain (Schmid et al., 2023). These data implicate LHX2 as a causative gene for microcephaly. Functional analysis of these variants revealed altered subcellular localization (nucleolar accumulation) for two missense variants located in the homeodomain, impaired interaction with co-factor LDB1 for another variant located in the LIM domain, and impaired transcriptional activation for four missense variants. These data suggest a loss-of-function effect of LHX2. Importantly, the identification of more genetic cases in future will strengthen the argument for the role of LHX2 in microcephaly.
2.1.3 MEIS2 (Myeloid ecotropic viral integration site 2)
MEIS2 belongs to the three-amino acid-loop extension (TALE) superfamily of the HOX class of homeobox genes, a homolog of the Drosophila homothorax gene (Leung et al., 2022). MEIS2 appears to function as a HOX co-factor, which binds to HOX proteins or pre-B cell leukemia homeobox (PBX) transcription factor to form dimeric or trimeric complexes to enhance the specificity and affinity of DNA binding (Chang et al., 1997). There are three mammalian MEIS transcription factors, MEIS 1, MEIS2, and MEIS3, and they are characterized by a three amino acid residue loop insertion between helix 1 and helix 2 of the homeodomain, which is an important feature for protein–protein interactions (Bürglin, 1997).
The expression of Meis2 in many embryonic tissues has been described in mice, including the forebrain, midbrain, hindbrain, spinal cord, and heart (Machon et al., 2015). In the developing mouse forebrain, it is expressed in both the dorsal and ventral telencephalon—dorsally it is mainly expressed in the VZ, while ventral expression predominates in the SVZ of the LGE and in the striatum, excluding the MGE and globus pallidus (Figure 2) (Su et al., 2022). It has been shown that MEIS2 function is important for proliferation and neuron differentiation in the striatum as well as in the olfactory bulb (Toresson et al., 2000; Agoston et al., 2012). In addition, zebrafish studies have demonstrated a role in the development of the mesencephalon, craniofacial skeleton, and heart (Glickman and Yelon, 2002).
In humans, MEIS2 is expressed in the proliferative zones of the fetal forebrain as well as in the adult brain (Larsen et al., 2010). Interestingly, microcephaly has been observed (not fully penetrant) in patients with chromosome 15q14 microdeletions, which encompasses MEIS2 and is a well-known chromosomal cause of palatal defects co-occurring with congenital heart defects and intellectual disability. In a recent study, 8 of 17 patients (47%) with a 15q14 deletion presented with microcephaly compared to 2 of 11 patients (18%) with de novo MEIS2 variants (Verheije et al., 2019), suggesting MEIS2 as a responsible gene for microcephaly. Given that microcephaly is more prevalent in 15q14 deletion than in de novo variants or intragenic deletions in MEIS2 (Verheije et al., 2019), it is possible that unidentified variants in a nearby region have a synergistic influence on the phenotype when combined with MEIS2 loss or variants.
Due to the embryonic lethality of mice with zygotic inactivation of the Meis2 allele, it is difficult to assess the brain size defect after birth, although the embryonic brain size (as well as the whole body size) is smaller in null mice when compared to controls (Machon et al., 2015). Conditional depletion of Meis2 in the forebrain would be a valuable experiment to further elucidate the role of Meis2 in microcephaly.
2.1.4 NKX2-1 (NK2 homeobox 1)
NKX2-1, also known as thyroid transcription factor 1 (TTF1) or thyroid-specific enhancer binding protein (T/EBP), belongs to the ANTP class of homeobox genes and is the mammalian homolog of the Drosophila scarecrow (scro) (Guazzi et al., 1990; Mizuno et al., 1991). The protein contains a homeodomain and an NK2 box domain of 18 amino acid sequences at the C-terminus of the NK2-type proteins, which is an important region for the regulation of transcriptional activity (Uhler et al., 2007). NKX2-1 was first identified as a nuclear protein that binds to the thyroglobulin gene promoter (Civitareale et al., 1989; Carré et al., 2009) and is known to play a role in telencephalon and diencephalon development as well as in thyroid, lung, and pituitary development (Guazzi et al., 1990; Mizuno et al., 1991; Kimura et al., 1996; Sussel et al., 1999).
In the embryonic mouse forebrain, Nkx2.1 is expressed in the progenitor and post-mitotic cells of the MGE and preoptic area (Figure 2). NKX2-1-positive progenitors give rise to GABAergic neurons that migrate tangentially from the MGE to the neocortex, as well as cholinergic neurons located in the striatum (Anderson et al., 2001; Magno et al., 2017). Nkx2.1 homozygous mutant mice die at birth with lung and thyroid defects, along with complex malformations of the ventral telencephalon structures, including the basal ganglia, hypothalamus, and pituitary (Kimura et al., 1996; Takuma et al., 1998; Sussel et al., 1999). In these mutant mice, the MGE is respecified into LGE (thus devoid of the mainly MGE-derived globus pallidus, but with the expansion of the LGE-derived striatum) and they have a reduced number of GABAergic neurons in the neocortex and cholinergic neurons in the striatum (Sussel et al., 1999). Studies with conditional knockout mice further demonstrated a role for Nkx2.1 in interneuron specification and migration (Butt et al., 2008; Nóbrega-Pereira et al., 2008).
In humans, the NKX2-1 gene is localized on chromosome 14q13. During early development, it is expressed in the prosencephalon (which gives rise to the telencephalon and diencephalon) and thyroid bud, and later in the lung epithelium (Carré et al., 2009). NKX2-1 mutations have been associated with brain–thyroid–lung syndrome, characterized by benign hereditary chorea (movement disorder), congenital hypothyroidism, and infant respiratory distress symptoms (Willemsen et al., 2005). Chromosome 14q13 deletions (that include NKX2-1) as well as nonsense mutations in NKX2-1 have also been linked to microcephaly that occasionally co-occurs with brain–thyroid–lung syndrome (Carré et al., 2009). For example, three of the six families with 14q13 deletion (0.9–17.9Mb) presented with microcephaly, as well as one of the two cases with a nonsense mutation in NKX2-1 (c.338G>A, p.Trp113*) (Carré et al., 2009). In another report on two siblings with hypothyroidism and respiratory failure due to the 14q12-13.3 deletion, both were reported to have postnatal microcephaly (Iwatani et al., 2000), further implicating the potential role of NKX2-1 in microcephaly accompanying brain–thyroid–lung syndrome. Microcephaly manifested in individuals with NKX2-1 variants suggests two possibilities: either a direct contribution of the loss of NKX2-1 function in the brain or an indirect result from its loss in other places such as the thyroid, hypothalamus, and pituitary, manifesting as an overall systematic growth defect.
2.1.5 OTX1 (Orthodenticle homeobox 1) and OTX2
OTX1 and OTX2 belong to the PRD class of homeobox genes and are the human orthologs of the Drosophila orthodenticle (otd) gene, which regulates anterior patterning as well as brain and eye development (Leung et al., 2022). Studies in mice have shown that Otx2 is essential for the early specification of the rostral neuroectoderm, Otx1 for corticogenesis, and both for sensory organ development (Simeone, 1998). During early mouse embryogenesis (gastrulation), Otx2 is expressed in the anterior visceral endoderm and prechordal mesendoderm, which emit signals for early specification and patterning of the neural plate, as well as in the epiblast and anterior neuroectoderm, which respond to these instructing signals (Simeone, 1998; Acampora et al., 1999). Otx2-/- null mutant mice are early embryonic lethal and display major abnormalities in their body plan and the absence of the rostral neuroectoderm that normally gives rise to forebrain, midbrain, and rostral hindbrain (Acampora et al., 1995; Matsuo et al., 1995; Ang et al., 1996; Simeone, 1998). On the other hand, Otx1 expression is first detected at the 1–3 somite stage in the forebrain and midbrain neuroepithelium (Simeone, 1998; Acampora et al., 1999). Its expression in the dorsal telencephalon is restricted to the VZ (where the proliferative, self-renewing, and multipotent neuroepithelial precursors reside) in the earlier stage (Figure 3) but at the end of gestation, it becomes prominent in the cortical plate consisting of layer 5 and 6 neurons, while the VZ expression becomes weaker. Otx1-/- null mutant mice mostly die at birth, although 30% survive to weaning age and even into adulthood when on specific genetic backgrounds (Acampora et al., 1996; Suda et al., 1996). The adult null mice that survive have severe morphological anomalies in the eyes, inner ears, pituitary gland, and brain. The weight and size of the brain are reduced; the dorsal telencephalic cortex shows a significant reduction in thickness and cell number, particularly in the temporal and perirhinal cortex, and the cortical lamination is disorganized. These mice also develop spontaneous seizures (Acampora et al., 1996).
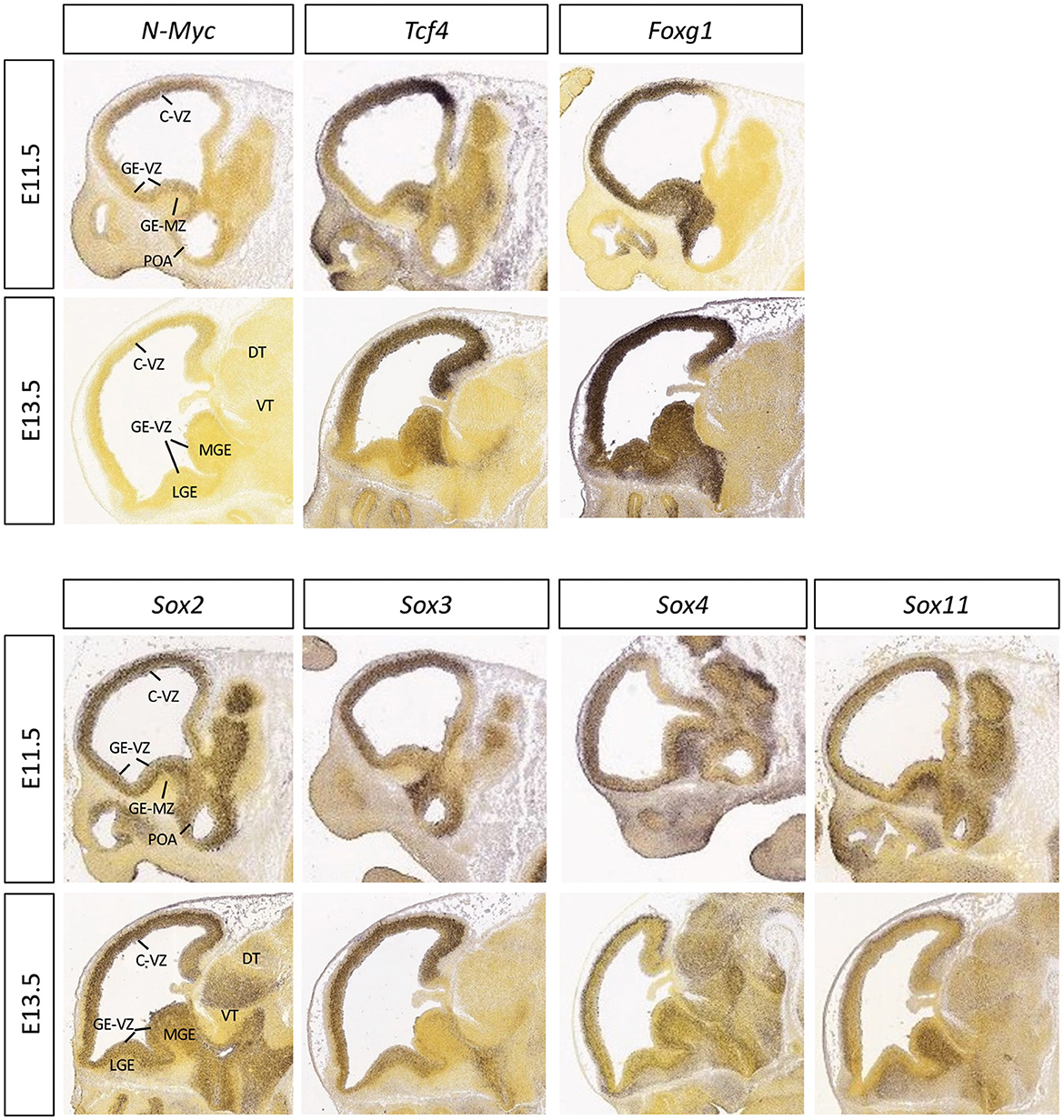
Figure 3. Expression patterns of the selected transcription factors in embryonic mouse brain. The mid-sagittal sections of E11.5 or E13.5 embryonic brains showing RNA in situ hybridization results of N-Myc, Tcf4, Foxg1, Sox2, Sox3, Sox4, and Sox11, adapted from Allen Brain Atlas (https://developingmouse.brain-map.org).
These phenotypes in murine models are consistent with those in human patients carrying OTX1 or OTX2 variants. In humans, OTX1 is localized on chromosome 2p15 and OTX2 on 14q22.3. Mutations in OTX1, including chromosomal deletions encompassing OTX1, cause developmental delay, short stature, autistic behavior, dysmorphic features, and microcephaly (Liang et al., 2009). For example, one female with the 2p15-16.1 deletion was reported to have all the above-mentioned features, including microcephaly (Liang et al., 2009). In another study, six out of seven patients with variable chromosomal deletion (OTX1 being the only gene deleted in every case) showed genitourinary defects, and two of these six had microcephaly (Jorgez et al., 2014). Similarly, mutations in OTX2 have been described in patients with eye defects, variable congenital hypopituitarism, and one individual with microcephaly (Gregory et al., 2021). Despite the low prevalence of microcephaly in patients with OTX1 or OTX2 variants, it is worth focusing on these OTX genes as potential causal genes for microcephaly, given the severity of the brain phenotypes observed in mice (Acampora et al., 1995; Matsuo et al., 1995; Ang et al., 1996; Simeone, 1998).
2.1.6 PAX6 (Paired box 6)
PAX6 is a member of the PRD class of the homeobox gene family and encodes one of the nine PAX transcription factors identified in mammals (Leung et al., 2022). Its Drosophila homolog eyeless is essential for segmentation, eye and brain development (Noveen et al., 2000; Callaerts et al., 2001; Clements et al., 2009), and in mammals, PAX6 is well known for its important role in the development of the brain, spinal cord, eye, pancreas, and pituitary (Walther and Gruss, 1991; Gehring, 1996). During mouse embryonic development, Pax6 is expressed in the forebrain, hindbrain, cerebellum, and spinal cord, as well as in the developing eye, pituitary gland, and nasal epithelium (Walther and Gruss, 1991). In the forebrain, Pax6 is expressed in the proliferating cells in the VZ of the dorsal telencephalon, as well as in the pallial-subpallial boundary, but it is not present in the ventral telencephalon (Figure 2) (Bishop et al., 2000; Hirata et al., 2002).
Pax6–/– null mice die at birth with the malformation in the cerebral cortex, and Pax6+/– mice have thinner cortices with small/reduced eyes (Hill et al., 1991; Tyas et al., 2003; Haubst et al., 2004; Quinn et al., 2007; Mi et al., 2013). Conditional Pax6 knockout mice with a specific deletion in cortical cells (with Emx1-Cre) have reduced cortical size (Piñon et al., 2008). Explaining these abnormal cortical phenotypes, PAX6 is thought to control the cell cycle length, the transition from symmetrical division to asymmetrical division, and the onset of expression of neural-specific markers, thus regulating the balance between neural progenitor cell proliferation and differentiation. During early neurogenesis, the length of the cell cycle becomes shorter, and the number of S-phase cells (undergoing proliferation) is increased in Pax6 mutant mice than in the wild type (Estivill-Torrus et al., 2002). Furthermore, the transition from symmetrical (expanding progenitor pool) to asymmetrical (generating post-mitotic cells) division is more rapid in the mutant. As cortical development progresses (E15.5), the cell cycle length becomes longer in the mutant mice. Therefore, the loss of Pax6 in mice seems to cause depletion of progenitor cell populations in early cortical development as a result of shortened cell length and a faster transition to asymmetrical divisions. However, it should be noted that the cell cycle length change was not detected in another Pax6 mutant mouse study, although this study found that cell cycle re-entry was reduced (cell cycle exit increased) and the proportion of differentiating neurons was increased (Quinn et al., 2007).
In humans, PAX6 is located on chromosome 11p13, and its mutations can result in neurological disorders including intellectual disability, autism, impaired audition, and eye defects, as mostly seen in individuals with heterozygous mutations (Malandrini et al., 2001; Davis et al., 2008). Cases with mutations in both alleles of PAX6 (compound heterozygosity) showed severe brain malformations with obvious microcephaly (Glaser et al., 1994; Schmidt-Sidor et al., 2009; Solomon et al., 2009). In these patients, increased germinal proliferation has been described, which is consistent with Pax6 mutant mouse phenotype that shows increased proliferation in early corticogenesis, which causes depletion of the progenitor pools (Piñon et al., 2008), and other cortical malformations such as polymicrogyria, heterotopia, agenesis of the corpus callosum, holoprosencephaly, etc., as well as defects in eye development (e.g., microphthalmia and aniridia) (Schmidt-Sidor et al., 2009), In addition, 11p13 chromosomal deletions, as well as duplications, encompassing PAX6 also lead to microcephaly, suggesting that the right level of PAX6, not just the presence of PAX6, is important for proper brain development, although the potential contribution by other genes in the affected chromosome cannot be ruled out.
2.2 Basic helix-loop-helix genes: MYCN and TCF4
Basic helix-loop-helix (bHLH) superfamily genes contain two highly conserved and functionally distinct domains: the “basic domain” at the N-terminus and the “HLH domain” at the C-terminus (Jones, 2004; Murre, 2019). The basic domain binds to DNA at a six-nucleotide consensus sequence (CANNTG) called an E-box; different families of bHLH proteins recognize different E-box consensus sequences (Murre et al., 1989). The HLH domain facilitates protein–protein interactions, forming homo- and hetero-dimeric complexes; many different combinations of dimeric structures are possible with different binding affinities (Fairman et al., 1993). These features of bHLH transcription factors make it possible for them to regulate diverse developmental functions through transcriptional regulation (Murre et al., 1989; Fairman et al., 1993; Jones, 2004), including lineage specification, commitment, self-renewal, proliferation, differentiation, and so on (Murre, 2019). bHLH transcription factors can be grouped into several classes based on expression patterns, DNA-binding specificities, and dimerization selectivity (Murre, 2019). Among them, two members, MYCN and TCF4, will be highlighted here for their association with microcephaly.
2.2.1 MYCN (myelocytomatosis oncogene neuroblastoma derived)
MYCN is a member of the MYC family of proto-oncogenes and encodes one of the basic helix-loop-helix-zipper (bHLHZ) classes of transcription factors, MYC-N (or N-MYC). The protein forms sequence-specific DNA-binding heterodimers with the bHLHZ protein MAX (Facchini and Penn, 1998; Grandori et al., 2000). The MYC family of proteins is known to be involved in fundamental cellular processes, including proliferation, differentiation, apoptosis, cell growth, and metabolism through regulating target gene transcription. They regulate cell cycle progression-related genes (e.g., CDK4, Cdc25A, cyclin D2, id2, gas1, gadd45, p15Ink4b, and p21Cip1) (Galaktionov et al., 1996; Lee et al., 1997; Marhin et al., 1997; Bouchard et al., 1999; Perez-Roger et al., 1999; Hermeking et al., 2000; Lasorella et al., 2000; Gartel et al., 2001; Staller et al., 2001), as well as genes controlling cell size and growth, including ribosomal proteins, translation factors, and metabolic enzymes (Rosenwald et al., 1993; Coller et al., 2000; Guo et al., 2000; Boon et al., 2001; Schuhmacher et al., 2001).
N-Myc knockout mice are embryonically lethal at mid-gestation (Charron et al., 1992; Stanton et al., 1992; Sawai et al., 1993), and hypomorphic mutations show delayed lethality (Moens et al., 1992, 1993). Conditional knockout mice targeting neuronal progenitor cells display profound microcephaly with small eyes (brain mass reduced 2-folds while body mass reduced 25%) (Knoepfler et al., 2002). In the cKO mouse brains, the VZ thickness and the BrdU incorporation rate were notably decreased, while the proportion of βTubulin III-positive cells (a marker of differentiating and differentiated neurons) increased, suggesting decreased proliferation and increased differentiation (precocious) as the underlying cause of microcephaly (Knoepfler et al., 2002).
In humans, MYCN is located on chromosome 2p24.3, and its mutations (loss-of-function) are known to cause type I Feingold syndrome (FS1), which is characterized by variable combinations of microcephaly, limb malformation/digit anomalies, intestinal atresia (blockage/obstruction), and mild to moderate intellectual disability (Courtens et al., 1997; Celli et al., 2003; Bokhoven et al., 2005). Pathogenic variants of MYCN are found in ~70% of the patients with FS1; 60% are point mutations, and 10% are chromosomal deletions encompassing the entire MYCN locus (Celli et al., 2000; Blaumeiser et al., 2008; Chen et al., 2012; Atik et al., 2016; Tedesco et al., 2021). Microcephaly has been reported in almost 90% of the cases (Bokhoven et al., 2005; Marcelis et al., 2008), which is in line with the small brain phenotype observed in N-Myc mutant mice (Knoepfler et al., 2002).
2.2.2 TCF4 (transcription factor 4)
Another bHLH transcription factor gene, TCF4, is highly expressed during brain development (Figure 3) (Jung et al., 2018). This gene (TCF4, Gene ID: 6925) is often confused with the transcription factor 7-like 2 gene (Gene ID: 6934, official gene symbol: TCF7L2), which is downstream of the WNT pathway and referred to as T-cell factor 4 and often mistakenly abbreviated as TCF4. Haploinsufficiency of TCF4 is known to cause Pitt–Hopkins syndrome, which is characterized by developmental delays with severe intellectual disability, dysmorphic facial features, and episodic hyperventilation and/or breath-holding while awake, among other features. Microcephaly has been reported in up to 60% of Pitt–Hopkins syndrome (Zweier et al., 2008; Goodspeed et al., 2017) and postnatal head growth defect in 26% (Winter et al., 2016). TCF4 mutations in patients with Pitt–Hopkins syndrome may be deletions, translocations, frameshift, nonsense, or missense mutations (Sepp et al., 2012; Forrest et al., 2014).
Some Tcf4 mouse models show Pitt–Hopkins syndrome-like phenotypes, including deficits in social interaction and memory as well as abnormal cortical development, neuronal migration, and oligodendrocyte differentiation (Flora et al., 2007; Chen et al., 2016; Kennedy et al., 2016; Thaxton et al., 2018; Li et al., 2019; Wang et al., 2019; Mesman et al., 2020; Wedel et al., 2020). However, mouse lines carrying heterozygous Tcf4 mutations, which is a clinically more relevant state than homozygous mutations, exhibit mild phenotypes only, without the severe symptoms observed in patients (Papes et al., 2022). A recent study using brain organoids derived from individuals with Pitt–Hopkins syndrome carrying TCF4 mutations (Papes et al., 2022) demonstrated that neural progenitors bearing these mutations have reduced proliferation and impaired capacity to differentiate into neurons (Papes et al., 2022), providing clear evidence for the role of TCF4 in neural proliferation and differentiation and ultimately microcephaly.
2.3 Forkhead box genes: FOXG1 and FOXR1
The forkhead box (FOX) family of transcription factors is named after the ectopic head structures of the Drosophila mutants harboring mutations in the forkhead (fkh) gene. Head involution is blocked in mutant embryos, causing an alteration of the head exoskeleton (called “forkhead”) (Jürgens and Weigel, 1988; Weigel et al., 1989). Since the discovery of fkh in Drosophila (Hannenhalli and Kaestner, 2009), hundreds of forkhead box genes have been identified from yeasts to humans, and their roles in cell proliferation and cell fate specification have been extensively studied (Carlsson and Mahlapuu, 2002; Lehmann et al., 2003). These transcription factors contain a core forkhead DNA-binding domain (monomeric), which consists of three α-helices connected to a pair of loops resembling butterfly wings or a “winged-helix” (which is why forkhead genes are also called winged helix genes) (Gajiwala and Burley, 2000). The forkhead domain contains ~100 amino acids, sharing no similarity with the previously identified DNA-binding motif (Gajiwala and Burley, 2000). Among several FOX genes implicated in human brain development (FOXG1, FOXC2, FOXL2, FOXP1, and FOXP2) (Lehmann et al., 2003; Golson and Kaestner, 2016), FOXG1 and FOXR1 will be highlighted here for their roles in the pathogenesis of microcephaly.
2.3.1 FOXG1 (Forkhead box G1)
FOXG1 was originally named brain factor 1 (BF-1) when it was identified as the Hepatocyte Nuclear Factor 3 (HNF-3) homolog expressed in the developing rat forebrain (Tao and Lai, 1992). Its alterations are known to significantly affect the formation and function of the cerebral cortex (Hannenhalli and Kaestner, 2009; Golson and Kaestner, 2016). In addition to its highly conserved forkhead DNA-binding domain, FOXG1 contains a mammalian-unique N-terminal domain (truncated in non-mammalian vertebrate) and a C-terminal domain responsible for antagonizing the transforming growth factor β (TGF-β) pathway (Hou et al., 2020).
It has been well established that FOXG1 is a master regulator of brain development, controlling cell proliferation (Hanashima et al., 2002), regional patterning (Hanashima et al., 2007; Miyoshi and Fishell, 2012), cell migration (Miyoshi and Fishell, 2012), and circuit assembly (Hanashima et al., 2002; Cargnin et al., 2018). It is strongly expressed in the embryonic forebrain (Figure 3). Its dysfunction leads to multiple congenital brain disorders, including variants of Rett syndrome (FOXG1 syndrome), microcephaly, infantile spasms, autism spectrum disorder, and schizophrenia, and it is associated with various types of cancer (Wang et al., 2018; Hou et al., 2020).
FOXG1's role in brain size control has been revealed through a Foxg1 knockout mouse model. Constitutive Foxg1 knockout mice die at birth and present with a severe reduction in the size of the cerebral hemispheres (agenesis of ventral and dorsal telencephalon) (Xuan et al., 1995). Loss of FOXG1 function leads to lengthening of the cell cycle (i.e., reduction in the rate of proliferation) and an increase in cell cycle exit events (i.e., reduction in the fraction of cells that can continue to divide) (Xuan et al., 1995; Hanashima et al., 2002). Premature onset of neuronal differentiation, shown with MAP2 staining, was also detected in Foxg1 KO mice (Xuan et al., 1995). These results indicate that during normal development, FOXG1 promotes neural stem cell proliferation and suppresses premature neuronal differentiation (Xuan et al., 1995; Martynoga et al., 2005). Studies in cancer models have demonstrated that overexpression of FOXG1 i) inhibited the FOXO/SMAD pathway (which facilitates cortical neuron differentiation), resulting in a reduction in both CDKN1A (cyclin-dependent kinase inhibitor 1A) and cyclin B1 expression, and ii) decreased the proportion of cells in the G2 phase (Wang et al., 2018). Consequently, FOXG1 prevents the cell cycle exit of neural stem cells and promotes stem cell pool expansion. In contrast, FOXG1 knockdown has the opposite effect (Wang et al., 2018).
In humans, haploinsufficiency of FOXG1 is associated with microcephaly, complete agenesis of the corpus callosum, and cognitive disability (Hou et al., 2020). Patients with FOXG1 mutations also exhibit features of Rett syndrome—a genetic disorder primarily caused by MECP2 mutations—including microcephaly, epilepsy, hyperkinetic movement, impaired sleep patterns, and intellectual disability. However, due to the significant differences in neurological phenotypes of the underlying FOXG1 mutations (e.g., agenesis of the corpus callosum, blunted gyrification, and reduction in white matter volume in some cases) compared to MECP2-mediated Rett syndrome, “FOXG1 syndrome” is now considered a distinct disorder (Kortüm et al., 2011). Microcephaly is one of the three core features of FOXG1 syndrome, along with agenesis of the corpus callosum and delayed myelination. Patients presenting with these core features have heterozygous variants of FOXG1 ranging from truncation, frameshift, missense, and nonsense mutations to duplications in the 14q12 FOXG1 locus (Yeung et al., 2009; Brunetti-Pierri et al., 2011; Seltzer et al., 2014), suggesting the homozygous mutation state is likely lethal, as observed in mice.
2.3.2 FOXR1 (Forkhead box R1)
FOXR1, also known as FOXN5 or DLNB13, is a highly conserved FOX gene containing a forkhead DNA-binding domain. It is expressed in all brain regions during embryonic and postnatal development and also in the reproductive organs based on the human brain transcriptome analysis (Mota et al., 2021). Consistent with the human expression data, mouse Foxr1 mRNA is also detected in the brain and other tissues (Mota et al., 2021). Although little is known about its function, a study in zebrafish showed that Foxr1 is an essential maternal effect gene required for proper cell division and survival (Cheung et al., 2018). In agreement with zebrafish findings, Foxr1 knockout mice showed a severe survival deficit with embryonic lethality in some mice and progressive death in surviving ones (~34% of knockout mice survived to P0 and 23.5% to weaning age). The analysis of newborn mutant mice found cortical thinning with enlarged ventricles (Mota et al., 2021). Together, these results suggest that Foxr1 is required for survival and normal cortical development.
Recently, a single de novo missense variant in FOXR1 (M280L) in an individual with severe neurological symptoms, including postnatal microcephaly, progressive brain atrophy, and global developmental delay, has been reported (Mota et al., 2021). The phenotypes described in this individual are consistent with those detected in Foxr1 knockout mice, suggesting that FOXR1 would be a responsible gene for the observed phenotypes, including microcephaly; however, validation in additional cases is warranted before drawing definitive conclusions.
It is well established that the FOX family of transcription factors induces heat shock protein (HSP, chaperone proteins that prevent protein misfolding) expression, thus providing cells with protective machinery against environmental stressors. FOXR1 appears to play this role as well, given that its most responsive target genes are two members of the HSP70 family (HSPA1A and HSPA6) and a mitochondrial reductase enzyme, DHRS2, each of which plays a role in protective stress response (Mota et al., 2021). The aforementioned M280L variant compromises FOXR1's ability to respond to cellular stressors. Interestingly, some of the upregulated genes by FOXR1 overexpression are involved in ribosome biogenesis (e.g., ribosome biogenesis regulator 1 and RRS1), an essential driver in neurodevelopment, whose dysregulation is associated with microcephaly and other neurodevelopmental syndromes (Hetman and Slomnicki, 2019). It is possible that during normal development, FOXR1 plays a role in protecting against proteotoxic stress during ribosome assembly—an energy-demanding process that, if disrupted, can lead to proteotoxic stress in cells (Albert et al., 2019), but further investigations are warranted to unravel the mechanisms underlying FOXR1-associated microcephaly.
2.4 SRY-related high mobility group box genes: SOX2, SOX3, SOX4, and SOX11
The SOX (sex-determining region Y-related high mobility group box) gene family members encode SOX transcription factors, which belong to the HMG (high mobility group) box superfamily of DNA-binding proteins (Stevanovic et al., 2021). They were first identified based on their high acidic and basic amino acid content and high mobility during polyacrylamide gel electrophoresis (Goodwin et al., 1973). HMG box is a conserved domain in HMG proteins and is responsible for DNA-binding activity. While all HMG family members share a similar HMG box that recognizes DNA structure without apparent sequence specificity, only SOX (and TCF) proteins carry HMG boxes with sequence-specific DNA-binding ability, and they are considered non-canonical HMG proteins (Bustin, 2001).
Based on the structure, expression profiles, and similarity between the proteins they encode, SOX gene family members can be divided into eight groups (A to H), with group B further divided into B1 and B2 (Stevanovic et al., 2021). Within the same group, SOX proteins have an overall high degree of homology, both within and outside the HMG domain, and they have functional redundancy. In contrast, SOX proteins from different groups show poor amino acid sequence homology, especially outside of the HMG domain, and do not show functional redundancy (Wegner, 1999; Bowles et al., 2000). In this study, I will focus on two members of the SOXB1 group (SOX2 and SOX3) and two members of the SOXC group (SOX4 and SOX11) for their associations with microcephaly.
2.4.1 SOX2 and SOX3
SOX2 and SOX3 belong to the SOXB1 group (together with SOX1) and were first identified in the screen for homologous genes to the sex-determining gene Sry, which contains an HMG box domain (Gubbay et al., 1990). At the neural induction stage, Sox2 and Sox3 are expressed prominently in the neuroectodermal cells, and when neurogenesis begins, it becomes restricted to the VZ, where the proliferative neuroepithelial precursors reside (Figure 3) (Collignon et al., 1996; Guth and Wegner, 2008). At later stages of brain development, their expression is restricted to distinct subsets of mature neurons (e.g., GABAergic neurons in the cortex, striatum, and thalamus) (Cavallaro et al., 2008). Overlapping expression patterns between Sox1, Sox2, and Sox3 appear to suggest a redundant role during CNS development (Uwanogho et al., 1995).
Studies with targeted null mice have demonstrated that Sox2 functions in the maintenance of the early pluripotent stem cells of the epiblast (Avilion et al., 2003) as well as neural stem cells and their differentiation into neurons in the brain and eye (Pevny and Placzek, 2005). It is also well known for its ability to re-establish pluripotency in terminally differentiated cells by reprogramming them into induced pluripotent stem cells (Takahashi and Yamanaka, 2006). For Sox3, when mouse ES cells targeted with Sox3 null mutations were injected into blastocysts, the chimeras showed early lethality due to a gastrulation defect (Rizzoti et al., 2004). Although one-third of the Sox3 conditional knockouts were normal, some exhibited lethality around weaning age with craniofacial defects, reduced size, and decreased fertility (Rizzoti et al., 2004). The analyses of these mice demonstrated that Sox3 is required for normal pituitary function and the formation of the hypothalamic-pituitary axis. In ovo electroporation of the fusion protein between Sox3 HMG domain and VP16 (transcription activation domain of the viral protein VP16) (HMG-VP16) or EnR (transcription repression domain of the D. melanogaster Engrailed protein) (HMG-EnR) into chick embryos revealed that SOX3 is necessary for the formation of neuroectoderm, maintenance of the neural progenitor state, and suppression of neuronal differentiation (Bylund et al., 2003; Schneider et al., 2009). Furthermore, it has been shown that the downregulation of Sox1, Sox2, and Sox3 gene expression by the proneural gene (Neurogenin 2) is essential for neuronal differentiation (Bylund et al., 2003).
In humans, individuals carrying heterozygous loss-of-function mutations in SOX2 mainly have ocular phenotypes (anophthalmia/microphthalmia), but some patients also present with microcephaly and other variable phenotypes, including hippocampal abnormalities, epilepsy, and motor problems, as well as dysmorphic facial features and genital anomalies (Fantes et al., 2003; Sisodiya et al., 2006; Schneider et al., 2009; Blackburn et al., 2018). The variability in phenotypes points to the complex interactions of SOX2 with other genetic factors, which affect the outcome of SOX2 deficiency in different ways. Similarly, SOX3 genetic variants have been reported to result in microcephaly with variable penetrance (Jelsig et al., 2018), in addition to hypopituitarism (ranging from isolated growth hormone deficiency to panhypopituitarism), intellectual disability, neural tube defects, and craniofacial abnormalities (Rizzoti et al., 2004; Arya et al., 2020). Notably, the most common mutations identified are duplications or deletions of the whole or part of SOX3, with very few examples of point mutations (Jelsig et al., 2018; Li et al., 2022). The microcephaly in some patients with SOX3 mutations may be the result of growth hormone deficiency due to hypopituitarism as well as neurogenesis defects.
2.4.2 SOX4 and SOX11
SOX4 and SOX11 belong to the SOXC transcription factor subfamily of genes that are necessary for the survival of neural precursor cells (Bhattaram et al., 2010) and the establishment of their neuronal properties (Bhattaram et al., 2010). In contrast to SOXB1s (SOX1-3), which are expressed in neural precursor cells, SOXC transcripts and proteins (SOX4, SOX11, and SOX12) are mostly expressed in neural cells committed to neuronal differentiation as well as uncommitted precursors (Figure 3) (Hargrave et al., 1997; Kuhlbrodt et al., 1998; Dy et al., 2008). The loss of either Sox4 or Sox11 leads to embryonic or postnatal lethality and many other developmental disturbances, although the nervous system defects are not as severe, likely due to their functional redundancy (Schilham et al., 1996; Cheung et al., 2000; Sock et al., 2004; Hoser et al., 2008). Sox11 conditional knockout mice have reduced body size and small brains with reduced cortical thickness noted as early as E11.5 and persisting to birth (Sock et al., 2004; Wang et al., 2013). Forced expression of SOX11 results in premature induction of neuronal markers, while its deficiency induces apoptosis in the developing nervous system (Bergsland et al., 2006; Bhattaram et al., 2010; Thein et al., 2010). Mice engineered to be conditionally mutant for both Sox4 and Sox11 die at birth with microcephaly and an ear phenotype (Gnedeva and Hudspeth, 2015).
In humans, SOX4 and SOX11 heterozygous variants have been described in patients with neurodevelopmental syndromes, mild dysmorphisms, and other variable anomalies (Tsurusaki et al., 2014; Hempel et al., 2016; Zawerton et al., 2019; Wakim et al., 2020). The disease phenotypes caused by these two genes are similar, which is consistent with findings in animal models, demonstrating SOX4 and SOX11 are co-expressed in various progenitor cell types and have additive or redundant roles in many developing organs including the brain, skeleton, heart, and eye (Angelozzi et al., 2022). SOX4- and SOX11-related syndromes often share some common features with Coffin–Siris syndrome, which is characterized by abnormal head size (microcephaly or macrocephaly) with characteristic facial features, digits, and eye abnormalities (Angelozzi et al., 2022). Coffin–Siris syndrome is also called “BAFopathy” because its causal genes encode chromatin modeling BAF complex components (e.g., ARID1B and SMARCB1) (Vasko et al., 2021). SOX4 and SOX11 are BAF-complex targets (Feng et al., 2013), which accounts for why SOX4- and SOX11-related syndromes are similar to Coffin–Siris syndrome (Angelozzi et al., 2022). As in other SOX gene-associated brain size defects, the microcephaly phenotype observed in individuals with SOX4 or SOX11 variants has low penetrance, suggesting a complex interplay between SOX genes and other unidentified genes that variably affect the consequences of the SOX gene deficiency.
2.5 Zinc finger genes: ZNF238 and ZNF 335
Zinc finger proteins (ZNFs) contain a zinc finger domain that can interact with DNA, RNA, PAR (poly-ADP-ribose), and other proteins and are involved in a wide range of cellular processes such as transcriptional regulation, ubiquitin-mediated protein degradation, signal transduction, actin targeting, DNA repair, and cell migration, among others (Cassandri et al., 2017). In this review, only those with transcriptional activity will be focused on, especially the ones associated with microcephaly.
Zinc finger transcription factors constitute the largest family of transcription factors in the human genome. The zinc finger structure is maintained by the zinc ion, which coordinates cysteine (C) and histidine (H) in different combinations (e.g., classical zinc finger has C2H2; non-classical zinc fingers have C2-HC, C2-CH, and C2-C2) (Cassandri et al., 2017). Furthermore, zinc finger motifs can be classified into several different types based on their main-chain conformation and secondary structure around their zinc-binding sites (Krishna et al., 2003; Jen and Wang, 2016). In addition to these zinc motifs, zinc finger transcription factors contain several domains that play different roles in cellular processes, including BTB (Broad-Complex, Tram tracks, and Bric-a-brac), also known as the POZ (poxvirus zinc finger) domain, KRAB (Kruppel-Associated Box) domain, SET domain and SCAN (SRE-ZBP, CTfin51, AW-1, and Number 18 cDNA) domain (Jen and Wang, 2016). Due to the diversity of zinc finger motifs and these additional domains, zinc finger transcription factors can play dynamic roles in gene regulation under various cellular environments and extracellular stimuli. In this study, two zinc finger proteins, ZNF238 and ZNF338, will be highlighted.
2.5.1 ZNF238 (Zinc finger protein, also known as RP58 or ZBTB18)
ZNF238, also known as RP58 encodes a highly conserved (95% homology in the amino acid sequences between humans and mice) transcription factor containing four zinc finger domains (responsible for DNA binding) and a BTB/POZ domain (multifaceted protein–protein interaction motif) (Aoki et al., 1998; Tatard et al., 2010). Deletion of the distal arm of human chromosome-1q, termed “1qter deletion,” “1q4 deletion,” or “terminal 1q deletion”, is linked to microcephaly with agenesis of the corpus callosum (Boland et al., 2007; Hill et al., 2007; Bon et al., 2008), and the patients carrying this deletion have severe intellectual disability and short stature with profound growth defects (Khadija et al., 2022). A critical region contains a handful of genes, including ZNF238 (Boland et al., 2007; Hill et al., 2007; Bon et al., 2008; Orellana et al., 2010).
During early mouse cortical development (E12.5), Znf238 expression is detected in a subset of cells in the VZ as well as in the developing neurons in the preplate. At E16.5, while its expression persists in some cells in the VZ, it is also detected in the cortical plate (CP), intermediate zone (IZ), and subventricular zone (SVZ), but not in the marginal zone (MZ). Double-labeling studies with PAX6 and TBR2 showed that the onset of ZNF238 expression coincides with the transition from PAX6-positive cells (radial glial progenitor cells) to TBR2-positive cells (intermediate progenitor cells), the initial stage of intermediate progenitor cells (Okado et al., 2009). Constitutive Znf238 knockout mice die at birth and show dysplasia of the neocortex and hippocampus, a reduction in the number of cortical neurons, and abnormal laminar organization (Okado et al., 2009). The increased cell death in post-mitotic zones and the expansion of VZ/SVZ in the knockout mice support that ZNF238 is required for the survival and maturation of neurons (excitatory neurons in particular) in the cortex (Okado et al., 2009). Conditional knockout mice with CNS-specific loss exhibit profound postnatal microencephaly, agenesis of the corpus callosum, and cerebellar hypoplasia, which resembles the human phenotype of 1qter deletion syndrome (Xiang et al., 2012), supporting ZNF238 as a critical, responsible gene for 1qter deletion syndrome whose main features include microcephaly. However, other genes in the deleted region may also contribute to the microcephaly phenotype in 1qter deletion syndrome (Boland et al., 2007; Hill et al., 2007; Bon et al., 2008; Orellana et al., 2010).
2.5.2 ZNF335 (Zinc finger protein 335, also known as NIF1)
ZNF335 (NIF1) encodes a nuclear zinc finger protein known as a coregulator of nuclear hormone signaling and part of the H3K4 methyltransferase complex (Garapaty et al., 2009). Autosomal recessive primary microcephaly 10 (MCPH10) is caused by a homozygous or compound heterozygous mutation in this gene on chromosome 20q13 (Jayaraman et al., 2018; Lim and Golden, 2020). It is essential for methylation and expression of brain-specific genes, and one of the critical downstream genes of ZNF335 is the master progenitor regulator REST/NRSF [repressor element 1 (RE1)-silencing transcription factor (REST)/neuron-restrictive silencer factor (NRSF)] (Yang et al., 2012). ZNF335 mutations were first identified in a large consanguineous Arab Israeli family where seven individuals presented with one of the most severe microcephalies reported (9 SD below mean) and death in all except one (Yang et al., 2012). Extremely small brain size with severely simplified gyrification was revealed by MRI, and histopathological analyses demonstrated a thinned cerebral cortex and neuronal disorganization, with only ~20% of the cortex showing the normal six cortical layers (Yang et al., 2012).
In agreement with findings in humans, Znf335 null mutant mice also show embryonic lethality as early as E7.5, and conditional KO leads to severely reduced cortical size and abnormal cortical layers (Yang et al., 2012). Knockdown of ZNF335 disrupts progenitor cell proliferation (premature cell cycle exit causing precocious depletion of the progenitor pool), cell fate determination, and neuronal differentiation, indicating that ZNF335 is essential for these processes (Yang et al., 2012).
3 Discussion/conclusion
In this review, selected transcription factors that are associated with microcephaly have been discussed and summarized. These transcription factors contain unique amino acid domains or motifs responsible for DNA binding (recognizing specific sequences or structures) and protein–protein interactions. Genetic variants of these transcription factors cause a spectrum of neurodevelopmental conditions ranging from mild learning disabilities without obvious structural abnormalities of the brain to severe brain malformations (e.g., microcephaly, lissencephaly, and polymicrogyria) resulting in lethality during early life.
In understanding the pathophysiologic mechanisms underlying microcephaly, knockout mouse models have been useful. Accumulating data suggest that transcription factors can control brain growth and size by directly regulating the expression of the target genes that act on cycle progression, cell cycle exit, neurogenesis, or cell survival (e.g., Galaktionov et al., 1996; Colasante et al., 2015). When target gene expression is compromised due to mutations in transcription factors (e.g., ARX and PAX6), the most common consequences appear to be either an abnormal increase or decrease in neural precursor cell proliferation, and premature cell cycle exit (Estivill-Torrus et al., 2002; Colasante et al., 2015). These defects can lead to the depletion of the self-renewable stem cell populations or a reduction in the number of neurons generated, eventually resulting in microcephaly. In other cases, microcephaly seems to occur as a secondary consequence of overall growth defects (due to growth hormone deficiency, etc.), as observed in hypopituitarism or hypothyroidism caused by mutations in some transcription factors (e.g., NK2.1, OTX2, and SOX3). Furthermore, some transcription factors (e.g., MYCN) regulate the genes important for cell size and growth, such as ribosomal proteins, translation initiation or elongation factors, and metabolic enzymes (Rosenwald et al., 1993; Coller et al., 2000; Guo et al., 2000; Boon et al., 2001; Schuhmacher et al., 2001).
Patients with microcephaly associated with the genetic variants in these transcription factors often exhibit variable penetrance in the severity of the phenotype and in other features co-occurring, which seem to arise from specific types of genetic variants. For instance, mutations causing premature termination (e.g., deletions, nonsense mutations, and frameshift mutations) or missense mutations in critical domains such as homeodomains tend to give rise to a more severe microcephaly with higher prevalence. Alternatively, it is also possible that these variabilities could arise from the involvement of additional genetic interactors that could affect the severity or prevalence of a transcription factor-associated microcephaly (i.e., an unidentified mutation in another gene in addition to the mutation in a given transcription factor).
The knowledge we have built from the studies of individual transcription factors will further our effort to understand brain development in a more systematic way, focusing on the orchestration among all the transcription factors in transcriptional networks. Given that most transcription factors (~75%) work as heterodimers (Walhout, 2006), it will be important to map out interactions between transcription factors involved in microcephaly and to delineate the consequences of their interactions (e.g., changes in DNA-binding preference, changes in DNA-binding affinity, and changes in activity mode between transcriptional activation and repression). Furthermore, systematic analysis and comparison of their target genes using transcriptome analysis (by RNA-seq) and chromatin immunoprecipitation followed by sequencing (ChIP-seq) will provide significant tools to elucidate the role of the transcriptional network in the pathogenesis of microcephaly.
Author contributions
YL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The author was partially supported by an NIH Grant (R01NS100007).
Acknowledgments
The author thanks Dr. Jeffrey Golden and Norma Hylton for their critical reading and feedback on this manuscript. Some primary publications could not be cited due to space limitations.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Absoud, M., Parr, J. R., Halliday, D., Pretorius, P., Zaiwalla, Z., and, Jayawant, S. (2010). A novel ARX phenotype: rapid neurodegeneration with Ohtahara syndrome and a dyskinetic movement disorder. Dev. Med. Child Neurol. 52, 305–307. doi: 10.1111/j.1469-8749.2009.03470.x
Acampora, D., Barone, P., and Simeone, A. (1999). Otx genes in corticogenesis and brain development. Cereb. Cortex 9, 533–542. doi: 10.1093/cercor/9.6.533
Acampora, D., Mazan, S., Avantaggiato, V., Barone, P., Tuorto, F., Lallemand, Y., et al. (1996). Epilepsy and brain abnormalities in mice lacking the Otx1 gene. Nat. Genet. 14, 218–222. doi: 10.1038/ng1096-218
Acampora, D., Mazan, S., Lallemand, Y., Avantaggiato, V., Maury, M., Simeone, A., et al. (1995). Forebrain and midbrain regions are deleted in Otx2–/– mutants due to a defective anterior neuroectoderm specification during gastrulation. Development 121, 3279–3290. doi: 10.1242/dev.121.10.3279
Aggarwal, A., Mittal, H., Patil, R., Debnath, S., and Rai, A. (2013). Clinical profile of children with developmental delay and microcephaly. J. Neurosci. Rural Pr. 4, 288–291. doi: 10.4103/0976-3147.118781
Agoston, Z., Li, N., Haslinger, A., Wizenmann, A., and Schulte, D. (2012). Genetic and physical interaction of Meis2, Pax3 and Pax7 during dorsal midbrain development. BMC Dev. Biol. 12, 10. doi: 10.1186/1471-213X-12-10
Agulnick, A. D., Taira, M., Breen, J. J., Tanaka, T., Dawid, I. B., and Westphal, H. (1996). Interactions of the LIM-domain-binding factor Ldbl with LIM homeodomain proteins. Nature 384, 270–272. doi: 10.1038/384270a0
Albert, B., Kos-Braun, I. C., Henras, A. K., Dez, C., Rueda, M. P., Zhang, X., et al. (2019). A ribosome assembly stress response regulates transcription to maintain proteome homeostasis. eLife 8, e45002. doi: 10.7554/eLife.45002.029
Anderson, S. A., Marin, O., Horn, C., Jennings, K., and Rubenstein, J. L. (2001). Distinct cortical migrations from the medial and lateral ganglionic eminences. Development 128, 353–363. doi: 10.1242/dev.128.3.353
Ang, S.-L., Jin, O., Rhinn, M., Daigle, N., Stevenson, L., and Rossant, J. (1996). A targeted mouse Otx2 mutation leads to severe defects in gastrulation and formation of axial mesoderm and to deletion of rostral brain. Development 122, 243–252. doi: 10.1242/dev.122.1.243
Angelozzi, M., Karvande, A., Molin, A. N., Ritter, A. L., Leonard, J. M. M., Savatt, J. M., et al. (2022). Consolidation of the clinical and genetic definition of a SOX4-related neurodevelopmental syndrome. J. Méd. Genet. 59, 1058–1068. doi: 10.1136/jmedgenet-2021-108375
Aoki, K., Meng, G., Suzuki, K., Takashi, T., Kameoka, Y., Nakahara, K., et al. (1998). RP58 Associates with condensed chromatin and mediates a sequence-specific transcriptional repression. J. Biol. Chem. 273, 26698–26704. doi: 10.1074/jbc.273.41.26698
Arya, V. B., Chawla, G., Nambisan, A. K. R., Muhi-Iddin, N., Vamvakiti, E., Ajzensztejn, M., et al. (2020). Xq27.1 duplication encompassing SOX3: variable phenotype and smallest duplication associated with hypopituitarism to date – a large case series of unrelated patients and a literature review. Horm. Res. Paediatr. 92, 382–389. doi: 10.1159/000503784
Ashwal, S., Michelson, D., Plawner, L., Dobyns, W. B., and Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. (2009). Practice parameterandcolon; evaluation of the child with microcephaly (an evidence-based review). Neurology 73, 887–897. doi: 10.1212/WNL.0b013e3181b783f7
Asif, M., Abdullah, U., Nürnberg, P., Tinschert, S., and Hussain, M. S. (2023). Congenital microcephaly: a debate on diagnostic challenges and etiological paradigm of the shift from isolated/non-syndromic to syndromic microcephaly. Cells 12, 642. doi: 10.3390/cells12040642
Atik, T., Güvenç, M. S., and Onay, H., Özkinay, F., and Çogulu, Ö. (2016). Genetic counselling in feingold syndrome and a novel mutation. Genet. Couns. 27, 381–384.
Avilion, A. A., Nicolis, S. K., Pevny, L. H., Perez, L., Vivian, N., and Lovell-Badge, R. (2003). Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 17, 126–140. doi: 10.1101/gad.224503
Bergsland, M., Werme, M., Malewicz, M., Perlmann, T., and Muhr, J. (2006). The establishment of neuronal properties is controlled by Sox4 and Sox11. Genes Dev. 20, 3475–3486. doi: 10.1101/gad.403406
Bhattaram, P., Penzo-Méndez, A., Sock, E., Colmenares, C., Kaneko, K. J., Vassilev, A., et al. (2010). Organogenesis relies on SoxC transcription factors for the survival of neural and mesenchymal progenitors. Nat. Commun. 1, 9. doi: 10.1038/ncomms1008
Bienvenu, T., Poirier, K., Friocourt, G., Bahi, N., Beaumont, D., Fauchereau, F., et al. (2002). ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet 11, 981–991. doi: 10.1093/hmg/11.8.981
Bishop, K. M., Goudreau, G., and O'Leary, D. D. (2000). Regulation of area identity in the mammalian neocortex by Emx2 and Pax6. Science 288, 344–349. doi: 10.1126/science.288.5464.344
Blackburn, P. R., Chacon-Camacho, O. F., Ortiz-González, X. R., Reyes, M., Lopez-Uriarte, G. A., Zarei, S., et al. (2018). Extension of the mutational and clinical spectrum of SOX2 related disorders: Description of six new cases and a novel association with suprasellar teratoma. Am. J. Méd. Genet. Part A 176, 2710–2719. doi: 10.1002/ajmg.a.40644
Blaumeiser, B., Oehl-Jaschkowitz, B., Borozdin, W., and Kohlhase, J. (2008). Feingold syndrome associated with two novel MYCN mutations in sporadic and familial cases including monozygotic twins. Am. J. Méd. Genet. Part A 146A, 2304–2307. doi: 10.1002/ajmg.a.32444
Bokhoven, H., van Celli, J., Reeuwijk, J., van Rinne, T., Glaudemans, B., Beusekom, E., et al. (2005). MYCN haploinsufficiency is associated with reduced brain size and intestinal atresias in Feingold syndrome. Nat. Genet. 37, 465–467. doi: 10.1038/ng1546
Boland, E., Clayton-Smith, J., Woo, V. G., McKee, S., Manson, F. D. C., Medne, L., et al. (2007). Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am. J. Hum. Genet. 81, 292–303. doi: 10.1086/519999
Bon, B. W. M., van Koolen, D. A., Borgatti, R., Magee, A., Garcia-Minaur, S., Rooms, L., et al. (2008). Clinical and molecular characteristics of 1qter microdeletion syndrome: delineating a critical region for corpus callosum agenesis/hypogenesis. J. Méd. Genet. 45, 346. doi: 10.1136/jmg.2007.055830
Boon, K., Caron, H. N., Asperen, R., van Valentijn, L., Hermus, M., Sluis, P., et al. (2001). N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 20, 1383–1393. doi: 10.1093/emboj/20.6.1383
Bouchard, C., Thieke, K., Maier, A., Saffrich, R., Hanley-Hyde, J., Ansorge, W., et al. (1999). Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 18, 5321–5333. doi: 10.1093/emboj/18.19.5321
Bowles, J., Schepers, G., and Koopman, P. (2000). Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev. Biol. 227, 239–255. doi: 10.1006/dbio.2000.9883
Breen, J. J., Agulnick, A. D., Westphal, H., and Dawid, I. B. (1998). Interactions between LIM domains and the LIM domain-binding protein Ldb1*. J. Biol. Chem. 273, 4712–4717. doi: 10.1074/jbc.273.8.4712
Brunetti-Pierri, N., Paciorkowski, A. R., Ciccone, R., Mina, E. D., Bonaglia, M. C., Borgatti, R., et al. (2011). Duplications of FOXG1 in 14q12 are associated with developmental epilepsy, mental retardation, and severe speech impairment. Eur. J. Hum. Genet. 19, 102–107. doi: 10.1038/ejhg.2010.142
Bürglin, T. R. (1997). Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucleic Acids Res. 25, 4173–4180. doi: 10.1093/nar/25.21.4173
Bürglin, T. R., and Affolter, M. (2016). Homeodomain proteins: an update. Chromosoma 125, 497–521. doi: 10.1007/s00412-015-0543-8
Bustin, M. (2001). Revised nomenclature for high mobility group (HMG) chromosomal proteins. Trends Biochem. Sci. 26, 152–153. doi: 10.1016/S0968-0004(00)01777-1
Butt, S. J. B., Sousa, V. H., Fuccillo, M. V., Hjerling-Leffler, J., Miyoshi, G., Kimura, S., et al. (2008). The requirement of Nkx2-1 in the temporal specification of cortical interneuron subtypes. Neuron 59, 722–732. doi: 10.1016/j.neuron.2008.07.031
Bylund, M., Andersson, E., Novitch, B. G., and Muhr, J. (2003). Vertebrate neurogenesis is counteracted by Sox1–3 activity. Nat. Neurosci. 6, 1162–1168. doi: 10.1038/nn1131
Callaerts, P., Leng, S., Clements, J., Benassayag, C., Cribbs, D., Kang, Y. Y., et al. (2001). Drosophila Pax-6/eyeless is essential for normal adult brain structure and function. J. Neurobiol. 46, 73–88. doi: 10.1002/1097-4695(20010205)46:2<73::AID-NEU10>3.0.CO;2-N
Cargnin, F., Kwon, J.-S., Katzman, S., Chen, B., Lee, J. W., and Lee, S.-K. (2018). FOXG1 orchestrates neocortical organization and cortico-cortical connections. Neuron, 100, 1083–1096.e5. doi: 10.1016/j.neuron.2018.10.016
Carlsson, P., and Mahlapuu, M. (2002). Forkhead transcription factors: key players in development and metabolism. Dev. Biol. 250, 1–23. doi: 10.1006/dbio.2002.0780
Carré, A., Szinnai, G., Castanet, M., Sura-Trueba, S., Tron, E., Broutin-L'Hermite, I., et al. (2009). Five new TTF1/NKX2.1 mutations in brain–lung–thyroid syndrome: rescue by PAX8 synergism in one case. Hum. Mol. Genet. 18, 2266–2276. doi: 10.1093/hmg/ddp162
Cassandri, M., Smirnov, A., Novelli, F., Pitolli, C., Agostini, M., Malewicz, M., et al. (2017). Zinc-finger proteins in health and disease. Cell Death Discov. 3, 17071. doi: 10.1038/cddiscovery.2017.71
Cavallaro, M., Mariani, J., Lancini, C., Latorre, E., Caccia, R., Gullo, F., et al. (2008). Impaired generation of mature neurons by neural stem cells from hypomorphic Sox2 mutants. Development 135, 541–557. doi: 10.1242/dev.010801
Celli, J., Beusekom, E., van Hennekam, R. C. M., Gallardo, M. E., Smeets, D. F. C. M., Córdoba, S. R., et al. (2000). Familial syndromic esophageal atresia maps to 2p23-p24. Am. J. Hum. Genet. 66, 436–444. doi: 10.1086/302779
Celli, J., Bokhoven, H., and van Brunner, H. G. (2003). Feingold syndrome: clinical review and genetic mapping. Am. J. Méd. Genet. Part A 122A, 294–300. doi: 10.1002/ajmg.a.20471
Chang, C.-P., Jacobs, Y., Nakamura, T., Jenkins, N. A., Copeland, N. G., and Cleary, M. L. (1997). Meis proteins are major in vivo DNA binding partners for wild-type but not chimeric Pbx proteins. Mol. Cell. Biol. 17, 5679–5687. doi: 10.1128/MCB.17.10.5679
Charron, J., Malynn, B. A., Fisher, P., Stewart, V., Jeannotte, L., Goff, S. P., et al. (1992). Embryonic lethality in mice homozygous for a targeted disruption of the N-myc gene. Genes Dev. 6, 2248–2257. doi: 10.1101/gad.6.12a.2248
Chen, C.-P., Lin, S.-P., Chern, S.-R., Wu, P.-S., Chang, S.-D., Ng, S.-H., et al. (2012). A de novo 4.4-Mb microdeletion in 2p24.3 → p24.2 in a girl with bilateral hearing impairment, microcephaly, digit abnormalities and Feingold syndrome. Eur. J. Méd. Genet. 55, 666–669. doi: 10.1016/j.ejmg.2012.07.003
Chen, T., Wu, Q., Zhang, Y., Lu, T., Yue, W., and Zhang, D. (2016). Tcf4 controls neuronal migration of the cerebral cortex through regulation of Bmp7. Front. Mol. Neurosci. 9, 94. doi: 10.3389/fnmol.2016.00094
Cheung, C. T., Patinote, A., Guiguen, Y., and Bobe, J. (2018). Foxr1 is a novel maternal-effect gene in fish that is required for early embryonic success. PeerJ 6, e5534. doi: 10.7717/peerj.5534
Cheung, M., Abu-Elmagd, M., Clevers, H., and Scotting, P. J. (2000). Roles of Sox4 in central nervous system development. Mol. Brain Res. 79, 180–191. doi: 10.1016/s0169-328x(00)00109-1
Chou, S.-J., Perez-Garcia, C. G., Kroll, T. T., and O'Leary, D. D. M. (2009). Lhx2 specifies regional fate in Emx1 lineage of telencephalic progenitors generating cerebral cortex. Nat. Neurosci. 12, 1381–1389. doi: 10.1038/nn.2427
Civitareale, D., Lonigro, R., Sinclair, A. J., and Lauro, R. D. (1989). A thyroid-specific nuclear protein essential for tissue-specific expression of the thyroglobulin promoter. EMBO J. 8, 2537–2542. doi: 10.1002/j.1460-2075.1989.tb08391.x
Clements, J., Hens, K., Merugu, S., Dichtl, B., Couet, H. G., and de Callaerts, P. (2009). Mutational analysis of the eyeless gene and phenotypic rescue reveal that an intact Eyeless protein is necessary for normal eye and brain development in Drosophila. Dev. Biol. 334, 503–512. doi: 10.1016/j.ydbio.2009.08.003
Cohen, B., McGuffin, M. E., Pfeifle, C., Segal, D., and Cohen, S. M. (1992). apterous, a gene required for imaginal disc development in Drosophila encodes a member of the LIM family of developmental regulatory proteins. Genes Dev. 6, 715–729. doi: 10.1101/gad.6.5.715
Colasante, G., Simonet, J. C., Calogero, R., Crispi, S., Sessa, A., Cho, G., et al. (2015). ARX regulates cortical intermediate progenitor cell expansion and upper layer neuron formation through repression of Cdkn1c. Cereb Cortex 25, 322–335. doi: 10.1093/cercor/bht222
Coller, H. A., Grandori, C., Tamayo, P., Colbert, T., Lander, E. S., Eisenman, R. N., et al. (2000). Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc. Natl. Acad. Sci. 97, 3260–3265. doi: 10.1073/pnas.97.7.3260
Collignon, J., Sockanathan, S., Hacker, A., Cohen-Tannoudji, M., Norris, D., Rastan, S., et al. (1996). A comparison of the properties of Sox-3 with Sry and two related genes, Sox-1 and Sox-2. Development 122, 509–520. doi: 10.1242/dev.122.2.509
Courtens, W., Levi, S., Verbelen, F., Verloes, A., and Vamos, E. (1997). Feingold syndrome: report of a new family and review. Am. J. Méd. Genet. 73, 55–60. doi: 10.1002/(SICI)1096-8628(19971128)73:1<55::AID-AJMG11>3.0.CO;2-Q
Cragan, J. D., Isenburg, J. L., Parker, S. E., Alverson, C. J., Meyer, R. E., Stallings, E. B., et al. (2016). Population-based microcephaly surveillance in the United States, 2009 to 2013: an analysis of potential sources of variation. Birth Defects Res. Part A: Clin. Mol. Teratol. 106, 972–982. doi: 10.1002/bdra.23587
Davis, L. K., Meyer, K. J., Rudd, D. S., Librant, A. L., Epping, E. A., Sheffield, V. C., et al. (2008). Pax6 3′ deletion results in aniridia, autism and mental retardation. Hum. Genet. 123, 371–378. doi: 10.1007/s00439-008-0484-x
Dobyns, W. B., Berry-Kravis, E., Havernick, N. J., Holden, K. R., and Viskochil, D. (1999). X-linked lissencephaly with absent corpus callosum and ambiguous genitalia. Am. J. Med. Genet. 86, 331–337. doi: 10.1002/(SICI)1096-8628(19991008)86:4<331::AID-AJMG7>3.0.CO;2-P
Dy, P., Penzo-Méndez, A., Wang, H., Pedraza, C. E., Macklin, W. B., and Lefebvre, V. (2008). The three SoxC proteins—Sox4, Sox11 and Sox12—exhibit overlapping expression patterns and molecular properties. Nucleic Acids Res. 36, 3101–3117. doi: 10.1093/nar/gkn162
Estivill-Torrus, G., Pearson, H., Heyningen, V., van Price, D. J., and Rashbass, P. (2002). Pax6 is required to regulate the cell cycle and the rate of progression from symmetrical to asymmetrical division in mammalian cortical progenitors. Development 129, 455–466. doi: 10.1242/dev.129.2.455
Facchini, L. M., and Penn, L. Z. (1998). The molecular role of Myc in growth and transformation: recent discoveries lead to new insights. FASEB J. 12, 633–651. doi: 10.1096/fasebj.12.9.633
Fairman, R., Beran-Steed, R. K., Anthony-Cahill, S. J., Lear, J. D., Stafford, W. F., DeGrado, W. F., et al. (1993). Multiple oligomeric states regulate the DNA binding of helix-loop-helix peptides. Proc. Natl. Acad. Sci. 90, 10429–10433. doi: 10.1073/pnas.90.22.10429
Fantes, J., Ragge, N. K., Lynch, S.-A., McGill, N. I., Collin, J. R. O., Howard-Peebles, P. N., et al. (2003). Mutations in SOX2 cause anophthalmia. Nat. Genet. 33, 462–463. doi: 10.1038/ng1120
Feng, W., Khan, M. A., Bellvis, P., Zhu, Z., Bernhardt, O., Herold-Mende, C., et al. (2013). The chromatin remodeler CHD7 regulates adult neurogenesis via activation of SoxC transcription factors. Cell Stem Cell 13, 62–72. doi: 10.1016/j.stem.2013.05.002
Feuerstein, R., Wang, X., Song, D., Cooke, N. E., and Liebhaber, S. A. (1994). The LIM/double zinc-finger motif functions as a protein dimerization domain. Proc. Natl. Acad. Sci. 91, 10655–10659. doi: 10.1073/pnas.91.22.10655
Flora, A., Garcia, J. J., Thaller, C., and Zoghbi, H. Y. (2007). The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc. Natl. Acad. Sci. 104, 15382–15387. doi: 10.1073/pnas.0707456104
Forrest, M. P., Hill, M. J., Quantock, A. J., Martin-Rendon, E., and Blake, D. J. (2014). The emerging roles of TCF4 in disease and development. Trends Mol. Med. 20, 322–331. doi: 10.1016/j.molmed.2014.01.010
Friocourt, G., and Parnavelas, J. G. (2010). Mutations in ARX result in several defects involving GABAergic neurons. Front Cell Neurosci 4, 4. doi: 10.3389/fncel.2010.00004
Gajiwala, K. S., and Burley, S. K. (2000). Winged helix proteins. Curr. Opin. Struct. Biol. 10, 110–116. doi: 10.1016/S0959-440X(99)00057-3
Galaktionov, K., Chen, X., and Beach, D. (1996). Cdc25 cell-cycle phosphatase as a target of c-myc. Nature 382, 511–517. doi: 10.1038/382511a0
Garapaty, S., Xu, C.-F., Trojer, P., Mahajan, M. A., Neubert, T. A., and Samuels, H. H. (2009). Identification and Characterization of a Novel Nuclear Protein Complex Involved in Nuclear Hormone Receptor-mediated Gene Regulation. J. Biol. Chem. 284, 7542–7552. doi: 10.1074/jbc.M805872200
Gartel, A. L., Ye, X., Goufman, E., Shianov, P., Hay, N., Najmabadi, F., et al. (2001). Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. 98, 4510–4515. doi: 10.1073/pnas.081074898
Gehring, W. J. (1996). The master control gene for morphogenesis and evolution of the eye. Genes Cells 1, 11–15. doi: 10.1046/j.1365-2443.1996.11011.x
Gehring, W. J., Affolter, M., and Burglin, T. (1994). Homeodomain proteins. Annu. Rev. Biochem. 63, 487–526. doi: 10.1146/annurev.bi.63.070194.002415
Gilmore, E. C., and Walsh, C. A. (2013). Genetic causes of microcephaly and lessons for neuronal development. WIREs Dev. Biol. 2, 461–478. doi: 10.1002/wdev.89
Glaser, T., Jepeal, L., Edwards, J. G., Young, S. R., Favor, J., and Maas, R. L. (1994). PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat. Genet. 7, 463–471. doi: 10.1038/ng0894-463
Glickman, N. S., and Yelon, D. (2002). Cardiac development in zebrafish: coordination of form and function. Semin. Cell Dev. Biol. 13, 507–513. doi: 10.1016/S1084952102001040
Gnedeva, K., and Hudspeth, A. J. (2015). SoxC transcription factors are essential for the development of the inner ear. Proc. Natl. Acad. Sci. 112, 14066–14071. doi: 10.1073/pnas.1517371112
Golson, M. L., and Kaestner, K. H. (2016). Fox transcription factors: from development to disease. Development 143, 4558–4570. doi: 10.1242/dev.112672
Goodspeed, K., Newsom, C., Morris, M. A., Powell, C., Evans, P., and Golla, S. (2017). Pitt-Hopkins syndrome: a review of current literature, clinical approach, and 23-patient case series. J. Child Neurol. 33, 233–244. doi: 10.1177/0883073817750490
Goodwin, G. H., Sanders, C., and Johns, E. W. (1973). A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur. J. Biochem. 38, 14–19. doi: 10.1111/j.1432-1033.1973.tb03026.x
Grandori, C., Cowley, S. M., James, L. P., and Eisenman, R. N. (2000). The MYC/MAX/MAD network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 16, 653–699. doi: 10.1146/annurev.cellbio.16.1.653
Gregory, L. C., Gergics, P., Nakaguma, M., Bando, H., Patti, G., McCabe, M. J., et al. (2021). The phenotypic spectrum associated with OTX2 mutations in humans. Eur. J. Endocrinol. 185, 121–135. doi: 10.1530/EJE-20-1453
Guazzi, S., Price, M., Felice, M. D., Damante, G., Mattei, M. G., and Lauro, R. D. (1990). Thyroid nuclear factor 1 (TTF-1) contains a homeodomain and displays a novel DNA binding specificity. EMBO J. 9, 3631–3639. doi: 10.1002/j.1460-2075.1990.tb07574.x
Gubbay, J., Collignon, J., Koopman, P., Capel, B., Economou, A., Münsterberg, A., et al. (1990). A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature 346, 245–250. doi: 10.1038/346245a0
Guo, Q. M., Malek, R. L., Kim, S., Chiao, C., He, M., Ruffy, M., et al. (2000). Identification of c-myc responsive genes using rat cDNA microarray. Cancer Res. 60, 5922–5928.
Guth, S. I. E., and Wegner, M. (2008). Having it both ways: sox protein function between conservation and innovation. Cell. Mol. Life Sci. 65, 3000–3018. doi: 10.1007/s00018-008-8138-7
Hanashima, C., Fernandes, M., Hebert, J. M., and Fishell, G. (2007). The role of Foxg1 and dorsal midline signaling in the generation of cajal-retzius subtypes. J. Neurosci. 27, 11103–11111. doi: 10.1523/JNEUROSCI.1066-07.2007
Hanashima, C., Shen, L., Li, S. C., and Lai, E. (2002). Brain factor-1 controls the proliferation and differentiation of neocortical progenitor cells through independent mechanisms. J. Neurosci. 22, 6526–6536. doi: 10.1523/JNEUROSCI.22-15-06526.2002
Hannenhalli, S., and Kaestner, K. H. (2009). The evolution of Fox genes and their role in development and disease. Nat. Rev. Genet. 10, 233–240. doi: 10.1038/nrg2523
Hargrave, M., Wright, E., Kun, J., Emery, J., Cooper, L., and Koopman, P. (1997). Expression of the Sox11 gene in mouse embryos suggests roles in neuronal maturation and epithelio-mesenchymal induction. Dev. Dyn. 210, 79–86. doi: 10.1002/(SICI)1097-0177(199710)210:2<79::AID-AJA1>3.0.CO;2-6
Haubst, N., Berger, J., Radjendirane, V., Graw, J., Favor, J., Saunders, G. F., et al. (2004). Molecular dissection of Pax6 function: the specific roles of the paired domain and homeodomain in brain development. Development 131, 6131–6140. doi: 10.1242/dev.01524
Hempel, A., Pagnamenta, A. T., Blyth, M., Mansour, S., McConnell, V., Kou, I., et al. (2016). Deletions and de novo mutations of SOX11 are associated with a neurodevelopmental disorder with features of Coffin–Siris syndrome. J. Méd. Genet. 53, 152. doi: 10.1136/jmedgenet-2015-103393
Hermeking, H., Rago, C., Schuhmacher, M., Li, Q., Barrett, J. F., Obaya, A. J., et al. (2000). Identification of CDK4 as a target of c-MYC. Proc. Natl. Acad. Sci. 97, 2229–2234. doi: 10.1073/pnas.050586197
Hetman, M., and Slomnicki, L. P. (2019). Ribosomal biogenesis as an emerging target of neurodevelopmental pathologies. J. Neurochem. 148, 325–347. doi: 10.1111/jnc.14576
Hill, A. D., Chang, B. S., Hill, R. S., Garraway, L. A., Bodell, A., Sellers, W. R., et al. (2007). A 2-Mb critical region implicated in the microcephaly associated with terminal 1q deletion syndrome. Am. J. Méd. Genet. Part A 143A, 1692–1698. doi: 10.1002/ajmg.a.31776
Hill, R. E., Favor, J., Hogan, B. L. M., Ton, C. C. T., Saunders, G. F., Hanson, I. M., et al. (1991). Mouse Small eye results from mutations in a paired-like homeobox-containing gene. Nature 354, 522–525. doi: 10.1038/354522a0
Hirata, T., Nomura, T., Takagi, Y., Sato, Y., Tomioka, N., Fujisawa, H., et al. (2002). Mosaic development of the olfactory cortex with Pax6-dependent and -independent components. Dev. Brain Res. 136, 17–26. doi: 10.1016/S0165-3806(02)00304-8
Holland, P. W., Booth, H. A. F., and Bruford, E. A. (2007). Classification and nomenclature of all human homeobox genes. BMC Biol. 5, 47. doi: 10.1186/1741-7007-5-47
Hoser, M., Potzner, M. R., Koch, J. M. C., Bösl, M. R., Wegner, M., and Sock, E. (2008). Sox12 deletion in the mouse reveals nonreciprocal redundancy with the related Sox4 and Sox11 transcription factors. Mol. Cell. Biol. 28, 4675–4687. doi: 10.1128/MCB.00338-08
Hou, P.-S., hAilín, D. Ó., Vogel, T., and Hanashima, C. (2020). Transcription and beyond: delineating FOXG1 function in cortical development and disorders. Front. Cell. Neurosci. 14, 35. doi: 10.3389/fncel.2020.00035
Irvine, K. D., and Rauskolb, C. (2001). boundaries in development: formation and function. Annu. Rev. Cell Dev. Biol. 17, 189–214. doi: 10.1146/annurev.cellbio.17.1.189
Iwatani, N., Mabe, H., Devriendt, K., Kodama, M., and Miike, T. (2000). Deletion of NKX2.1 gene encoding thyroid transcription factor-1 in two siblings with hypothyroidism and respiratory failure. J. Pediatr. 137, 272–276. doi: 10.1067/mpd.2000.107111
Jayaraman, D., Bae, B.-I., and Walsh, C. A. (2018). The genetics of primary microcephaly. Annu. Rev. Genom. Hum. Genet. 19, annurev-genom-083117-021441-24. doi: 10.1146/annurev-genom-083117-021441
Jelsig, A. M., Diness, B. R., Kreiborg, S., Main, K. M., Larsen, V. A., and Hove, H. (2018). A complex phenotype in a family with a pathogenic SOX3 missense variant. Eur. J. Méd. Genet. 61, 168–172. doi: 10.1016/j.ejmg.2017.11.012
Jen, J., and Wang, Y.-C. (2016). Zinc finger proteins in cancer progression. J. Biomed. Sci. 23, 53. doi: 10.1186/s12929-016-0269-9
Jones, S. (2004). An overview of the basic helix-loop-helix proteins. Genome Biol. 5, 226. doi: 10.1186/gb-2004-5-6-226
Jorgez, C. J., Rosenfeld, J. A., Wilken, N. R., Vangapandu, H. V., Sahin, A., Pham, D., et al. (2014). Genitourinary defects associated with genomic deletions in 2p15 encompassing OTX1. PLoS ONE 9, e107028. doi: 10.1371/journal.pone.0107028
Jung, M., Häberle, B. M., Tschaikowsky, T., Wittmann, M.-T., Balta, E.-A., Stadler, V.-C., et al. (2018). Analysis of the expression pattern of the schizophrenia-risk and intellectual disability gene TCF4 in the developing and adult brain suggests a role in development and plasticity of cortical and hippocampal neurons. Mol. Autism 9, 20. doi: 10.1186/s13229-018-0200-1
Jürgens, G., and Weigel, D. (1988). Terminal versus segmental development in the Drosophila embryo: the role of the homeotic gene fork head. Roux's Arch. Dev. Biol. 197, 345–354. doi: 10.1007/BF00375954
Kadrmas, J. L., and Beckerle, M. C. (2004). The LIM domain: from the cytoskeleton to the nucleus. Nat. Rev. Mol. Cell Biol. 5, 920–931. doi: 10.1038/nrm1499
Kaplanis, J., Samocha, K. E., Wiel, L., Zhang, Z., Arvai, K. J., Eberhardt, R. Y., et al. (2020). Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 586, 757–762. doi: 10.1038/s41586-020-2832-5
Kappen, C. (2000). The homeodomain: an ancient evolutionary motif in animals and plants. Comput. Chem. 24, 95–103. doi: 10.1016/S0097-8485(00)80009-1
Kato, M., Das, S., Petras, K., Kitamura, K., Morohashi, K., Abuelo, D. N., et al. (2004). Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum. Mutat. 23, 147–159. doi: 10.1002/humu.10310
Kato, M., Das, S., Petras, K., Sawaishi, Y., and Dobyns, W. B. (2003). Polyalanine expansion of ARX associated with cryptogenic West syndrome. Neurology. 61, 267–276. doi: 10.1212/01.WNL.0000068012.69928.92
Kennedy, A. J., Rahn, E. J., Paulukaitis, B. S., Savell, K. E., Kordasiewicz, H. B., Wang, J., et al. (2016). Tcf4 regulates synaptic plasticity, DNA methylation, and memory function. Cell Rep. 16, 2666–2685. doi: 10.1016/j.celrep.2016.08.004
Khadija, B., Rjiba, K., Dimassi, S., Dahleb, W., Kammoun, M., Hannechi, H., et al. (2022). Clinical and molecular characterization of 1q43q44 deletion and corpus callosum malformations: 2 new cases and literature review. Mol. Cytogenet. 15, 42. doi: 10.1186/s13039-022-00620-2
Kimura, S., Hara, Y., Pineau, T., Fernandez-Salguero, P., Fox, C. H., Ward, J. M., et al. (1996). The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 10, 60–69. doi: 10.1101/gad.10.1.60
Kitamura, K., Yanazawa, M., Sugiyama, N., Miura, H., Iizuka-Kogo, A., Kusaka, M., et al. (2002). Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat. Genet. 32, 359–369. doi: 10.1038/ng1009
Knoepfler, P. S., Cheng, P. F., and Eisenman, R. N. (2002). N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 16, 2699–2712. doi: 10.1101/gad.1021202
Kortüm, F., Das, S., Flindt, M., Morris-Rosendahl, D. J., Stefanova, I., Goldstein, A., et al. (2011). The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis. J. Méd. Genet. 48, 396. doi: 10.1136/jmg.2010.087528
Kosho, T., Miyake, N., and Carey, J. C. (2014). Coffin–Siris syndrome and related disorders involving components of the BAF (mSWI/SNF) complex: Historical review and recent advances using next generation sequencing. Am. J. Méd. Genet. Part C Semin. Méd. Genet. 166, 241–251. doi: 10.1002/ajmg.c.31415
Krishna, S. S., Majumdar, I., and Grishin, N. V. (2003). Structural classification of zinc fingers survey and summary. Nucleic Acids Res. 31, 532–550. doi: 10.1093/nar/gkg161
Kuhlbrodt, K., Herbarth, B., Sock, E., Enderich, J., Hermans-Borgmeyer, I., and Wegner, M. (1998). Cooperative function of POU proteins and SOX proteins in glial cells. J. Biol. Chem. 273, 16050–16057. doi: 10.1074/jbc.273.26.16050
Kwong, A. K.-Y., Chu, V. L.-Y., Rodenburg, R. J. T., Smeitink, J., and Fung, C.-W. (2019). ARX-associated infantile epileptic-dyskinetic encephalopathy with responsiveness to valproate for controlling seizures and reduced activity of muscle mitochondrial complex IV. Brain Dev. 41, 883–887. doi: 10.1016/j.braindev.2019.07.003
Larsen, K. B., Lutterodt, M. C., Laursen, H., Græm, N., Pakkenberg, B., Møllgård, K., et al. (2010). Spatiotemporal distribution of PAX6 and MEIS2 expression and total cell numbers in the ganglionic eminence in the early developing human forebrain. Dev. Neurosci. 32, 149–162. doi: 10.1159/000297602
Lasorella, A., Noseda, M., Beyna, M., Yokota, Y., and Iavarone, A. (2000). Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature 407, 592–598. doi: 10.1038/35036504
Lawrence, P. A., and Struhl, G. (1996). Morphogens, compartments, and pattern: lessons from Drosophila? Cell 85, 951–961. doi: 10.1016/S0092-8674(00)81297-0
Lee, T. C., Li, L., Philipson, L., and Ziff, E. B. (1997). Myc represses transcription of the growth arrest gene gas1. Proc. Natl. Acad. Sci. 94, 12886–12891. doi: 10.1073/pnas.94.24.12886
Lehmann, O. J., Sowden, J. C., Carlsson, P., Jordan, T., and Bhattacharya, S. S. (2003). Fox's in development and disease. Trends Genet. 19, 339–344. doi: 10.1016/S0168-9525(03)00111-2
Leung, R. F., George, A. M., Roussel, E. M., Faux, M. C., Wigle, J. T., and Eisenstat, D. D. (2022). Genetic regulation of vertebrate forebrain development by homeobox genes. Front. Neurosci. 16, 843794. doi: 10.3389/fnins.2022.843794
Li, H., Zhu, Y., Morozov, Y. M., Chen, X., Page, S. C., Rannals, M. D., et al. (2019). Disruption of TCF4 regulatory networks leads to abnormal cortical development and mental disabilities. Mol. Psychiatry 24, 1235–1246. doi: 10.1038/s41380-019-0353-0
Li, J., Zhong, Y., Guo, T., Yu, Y., and Li, J. (2022). Case report: a novel point mutation of SOX3 in a subject with growth hormone deficiency, hypogonadotrophic hypogonadism, and borderline intellectual disability. Front. Endocrinol. 13, 810375. doi: 10.3389/fendo.2022.810375
Liang, J.-S., Shimojima, K., Ohno, K., Sugiura, C., Une, Y., Ohno, K., et al. (2009). A newly recognised microdeletion syndrome of 2p15-16.1 manifesting moderate developmental delay, autistic behaviour, short stature, microcephaly, and dysmorphic features: a new patient with 3.2 Mb deletion. J. Méd. Genet. 46, 645. doi: 10.1136/jmg.2008.059220
Lim, Y., Cho, I., Golden, J. A., and Cho, G. (2022). Generation of FLAG-tagged Arx knock-in mouse model. Genesis 60, e23479. doi: 10.1002/dvg.23479
Lim, Y., Cho, I.-T., Shi, X., Grinspan, J. B., Cho, G., and Golden, J. A. (2019). Arx expression suppresses ventralization of the developing dorsal forebrain. Sci. Rep-UK 9, 226. doi: 10.1038/s41598-018-36194-6
Lim, Y., and Golden, J. A. (2020). “Chapter 16: Congenital and postnatal microcephalies,” in Neurodevelopmental Disorders: Comprehensive Developmental Neuroscience, eds J. Rubenstein, B. Chen, P. Rakic, and K. Y. Kwan (London: Academic Press), 377–408.
Machon, O., Masek, J., Machonova, O., Krauss, S., and Kozmik, Z. (2015). Meis2 is essential for cranial and cardiac neural crest development. BMC Dev. Biol. 15, 40. doi: 10.1186/s12861-015-0093-6
Magno, L., Barry, C., Schmidt-Hieber, C., Theodotou, P., Häusser, M., and Kessaris, N. (2017). NKX2-1 is required in the embryonic septum for cholinergic system development, learning, and memory. Cell Rep. 20, 1572–1584. doi: 10.1016/j.celrep.2017.07.053
Mairet-Coello, G., Tury, A., Buskirk, E. V., Robinson, K., Genestine, M., and DiCicco-Bloom, E. (2012). p57(KIP2) regulates radial glia and intermediate precursor cell cycle dynamics and lower layer neurogenesis in developing cerebral cortex. Development 139, 475–487. doi: 10.1242/dev.067314
Malandrini, A., Mari, F., Palmeri, S., Gambelli, S., Berti, G., Bruttini, M., et al. (2001). PAX6 mutation in a family with aniridia, congenital ptosis, and mental retardation. Clin. Genet. 60, 151–154. doi: 10.1034/j.1399-0004.2001.600210.x
Mangale, V. S., Hirokawa, K. E., Satyaki, P. R. V., Gokulchandran, N., Chikbire, S., Subramanian, L., et al. (2008). Lhx2 selector activity specifies cortical identity and suppresses hippocampal organizer fate. Science 319, 304–309. doi: 10.1126/science.1151695
Marcelis, C. L. M., Hol, F. A., Graham, G. E., Rieu, P. N. M. A., Kellermayer, R., Meijer, R. P. P., et al. (2008). Genotype–phenotype correlations in MYCN-related Feingold syndrome. Hum. Mutat. 29, 1125–1132. doi: 10.1002/humu.20750
Marhin, W. W., Chen, S., Facchini, L. M. Jr, and Penn, L. Z. (1997). Myc represses the growth arrest gene gadd45. Oncogene 14, 2825–2834. doi: 10.1038/sj.onc.1201138
Marsh, E., Fulp, C., Gomez, E., Nasrallah, I., Minarcik, J., Sudi, J., et al. (2009). Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain 132, 1563–1576. doi: 10.1093/brain/awp107
Martynoga, B., Morrison, H., Price, D. J., and Mason, J. O. (2005). Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev. Biol. 283, 113–127. doi: 10.1016/j.ydbio.2005.04.005
Matsuo, I., Kuratani, S., Kimura, C., Takeda, N., and Aizawa, S. (1995). Mouse Otx2 functions in the formation and patterning of rostral head. Genes Dev. 9, 2646–2658. doi: 10.1101/gad.9.21.2646
Mesman, S., Bakker, R., and Smidt, M. P. (2020). Tcf4 is required for correct brain development during embryogenesis. Mol. Cell. Neurosci. 106, 103502. doi: 10.1016/j.mcn.2020.103502
Mi, D., Carr, C. B., Georgala, P. A., Huang, Y.-T., Manuel, M. N., Jeanes, E., et al. (2013). Pax6 exerts regional control of cortical progenitor proliferation via direct repression of Cdk6 and hypophosphorylation of pRb. Neuron 78, 269–284. doi: 10.1016/j.neuron.2013.02.012
Miura, H., Yanazawa, M., Kato, K., and Kitamura, K. (1997). Expression of a novel aristaless related homeobox gene “Arx” in the vertebrate telencephalon, diencephalon and floor plate. Mech. Dev. 65, 99–109. doi: 10.1016/S0925-4773(97)00062-2
Miyoshi, G., and Fishell, G. (2012). Dynamic FoxG1 expression coordinates the integration of multipolar pyramidal neuron precursors into the cortical plate. Neuron 74, 1045–1058. doi: 10.1016/j.neuron.2012.04.025
Mizuno, K., Gonzalez, F. J., and Kimura, S. (1991). Thyroid-specific enhancer-binding protein (T/EBP): cDNA cloning, functional characterization, and structural identity with thyroid transcription factor TTF-1. Mol. Cell. Biol. 11, 4927–4933. doi: 10.1128/MCB.11.10.4927
Moens, C. B., Auerbach, A. B., Conlon, R. A., Joyner, A. L., and Rossant, J. (1992). A targeted mutation reveals a role for N-myc in branching morphogenesis in the embryonic mouse lung. Genes Dev. 6, 691–704. doi: 10.1101/gad.6.5.691
Moens, C. B., Stanton, B. R., Parada, L. F., and Rossant, J. (1993). Defects in heart and lung development in compound heterozygotes for two different targeted mutations at the N-myc locus. Development 119, 485–499. doi: 10.1242/dev.119.2.485
Morris, J. K., Rankin, J., Garne, E., Loane, M., Greenlees, R., Addor, M.-C., et al. (2016). Prevalence of microcephaly in Europe: population based study. BMJ 354, i4721. doi: 10.1136/bmj.i4721
Mota, A., Waxman, H. K., Hong, R., Lagani, G. D., Niu, S.-Y., Bertherat, F. L., et al. (2021). FOXR1 regulates stress response pathways and is necessary for proper brain development. PLoS Genet. 17, e1009854. doi: 10.1371/journal.pgen.1009854
Murre, C. (2019). Helix–loop–helix proteins and the advent of cellular diversity: 30 years of discovery. Genes Dev. 33, 6–25. doi: 10.1101/gad.320663.118
Murre, C., McCaw, P. S., Vaessin, H., Caudy, M., Jan, L. Y., Jan, Y. N., et al. (1989). Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell 58, 537–544. doi: 10.1016/0092-8674(89)90434-0
Nóbrega-Pereira, S., Kessaris, N., Du, T., Kimura, S., Anderson, S. A., and Marín, O. (2008). Postmitotic Nkx2-1 controls the migration of telencephalic interneurons by direct repression of guidance receptors. Neuron 59, 733–745. doi: 10.1016/j.neuron.2008.07.024
Noveen, A., Daniel, A., and Hartenstein, V. (2000). Early development of the Drosophila mushroom body: the roles of eyeless and dachshund. Development 127, 3475–3488. doi: 10.1242/dev.127.16.3475
Ogata, T., Matsuo, N., Hiraoka, N., and Hata, J. I. (2000). X-linked lissencephaly with ambiguous genitalia: delineation of further case. Am. J. Med. Genet. 94, 174–176. doi: 10.1002/1096-8628(20000911)94:2<174::AID-AJMG11>3.0.CO;2-O
Okado, H., Ohtaka-Maruyama, C., Sugitani, Y., Fukuda, Y., Ishida, R., Hirai, S., et al. (2009). The transcriptional repressor RP58 is crucial for cell-division patterning and neuronal survival in the developing cortex. Dev. Biol. 331, 140–151. doi: 10.1016/j.ydbio.2009.04.030
Opitz, J. M., and Holt, M. C. (1990). Microcephaly: general considerations and aids to nosology. J. Craniofacial Genet. Dev. Biol. 10, 175–204.
Orellana, C., Roselló, M., Monfort, S., Oltra, S., Quiroga, R., Ferrer, I., et al. (2010). Corpus callosum abnormalities and the controversy about the candidate genes located in 1q44. Cytogenet. Genome Res. 127, 5–8. doi: 10.1159/000279261
Papes, F., Camargo, A. P., Souza, J. S., de Carvalho, V. M. A., Szeto, R. A., LaMontagne, E., et al. (2022). Transcription Factor 4 loss-of-function is associated with deficits in progenitor proliferation and cortical neuron content. Nat. Commun. 13, 2387. doi: 10.1038/s41467-022-29942-w
Pearson, J. C., Lemons, D., and McGinnis, W. (2005). Modulating Hox gene functions during animal body patterning. Nat. Rev. Genet. 6, 893–904. doi: 10.1038/nrg1726
Perez-Roger, I., Kim, S., Griffiths, B., Sewing, A., and Land, H. (1999). Cyclins D1 and D2 mediate Myc-induced proliferation via sequestration of p27Kip1 and p21Cip1. EMBO J. 18, 5310–5320. doi: 10.1093/emboj/18.19.5310
Pevny, L., and Placzek, M. (2005). SOX genes and neural progenitor identity. Curr. Opin. Neurobiol. 15, 7–13. doi: 10.1016/j.conb.2005.01.016
Piñon, M. C., Tuoc, T. C., Ashery-Padan, R., Molnár, Z., and Stoykova, A. (2008). Altered molecular regionalization and normal thalamocortical connections in cortex-specific Pax6 knock-out mice. J. Neurosci. 28, 8724–8734. doi: 10.1523/JNEUROSCI.2565-08.2008
Pirozzi, F., Nelson, B., and Mirzaa, G. (2018). From microcephaly to megalencephaly: determinants of brain size. Dial. Clin. Neurosci. 20, 267–282. doi: 10.31887/DCNS.2018.20.4/gmirzaa
Porter, F. D., Drago, J., Xu, Y., Cheema, S. S., Wassif, C., Huang, S.-P., et al. (1997). Lhx2, a LIM homeobox gene, is required for eye, forebrain, and definitive erythrocyte development. Development 124, 2935–2944. doi: 10.1242/dev.124.15.2935
Quinn, J. C., Molinek, M., Martynoga, B. S., Zaki, P. A., Faedo, A., Bulfone, A., et al. (2007). Pax6 controls cerebral cortical cell number by regulating exit from the cell cycle and specifies cortical cell identity by a cell autonomous mechanism. Dev. Biol. 302, 50–65. doi: 10.1016/j.ydbio.2006.08.035
Raymond, G. V., and Holmes, L. B. (1994). Head circumference standards in neonates. J. Child Neurol. 9, 63–66. doi: 10.1177/088307389400900116
Rizzoti, K., Brunelli, S., Carmignac, D., Thomas, P. Q., Robinson, I. C., and Lovell-Badge, R. (2004). SOX3 is required during the formation of the hypothalamo-pituitary axis. Nat. Genet. 36, 247–255. doi: 10.1038/ng1309
Rosenwald, I. B., Rhoads, D. B., Callanan, L. D., Isselbacher, K. J., and Schmidt, E. V. (1993). Increased expression of eukaryotic translation initiation factors eIF-4E and eIF-2 alpha in response to growth induction by c-myc. Proc. Natl. Acad. Sci. 90, 6175–6178. doi: 10.1073/pnas.90.13.6175
Sassaman, E. A., and Zartler, A. S. (1982). Mental retardation and head growth abnormalities. J. Pediatr. Psychol. 7, 149–156. doi: 10.1093/jpepsy/7.2.149
Sawai, S., Shimono, A., Wakamatsu, Y., Palmes, C., Hanaoka, K., and Kondoh, H. (1993). Defects of embryonic organogenesis resulting from targeted disruption of the N-myc gene in the mouse. Development 117, 1445–1455. doi: 10.1242/dev.117.4.1445
Schilham, M. W., Oosterwegel, M. A., Moerer, P., Ya, J., de Boer, P. A. J., de Wetering, M., et al. (1996). Defects in cardiac outflow tract formation and pro-B-lymphocyte expansion in mice lacking Sox-4. Nature 380, 711–714.
Schmeichel, K. L., and Beckerle, M. C. (1994). The LIM domain is a modular protein-binding interface. Cell 79, 211–219. doi: 10.1016/0092-8674(94)90191-0
Schmid, C. M., Gregor, A., Costain, G., Morel, C. F., Massingham, L., Schwab, J., et al. (2023). LHX2 haploinsufficiency causes a variable neurodevelopmental disorder. Genet. Med. 25, 100839. doi: 10.1016/j.gim.2023.100839
Schmidt-Sidor, B., Szymańska, K., Williamson, K., Heyningen, V., van Roszkowski, T., Wierzba-Bobrowicz, T., et al. (2009). Malformations of the brain in two fetuses with a compound heterozygosity for two PAX6 mutations. Folia Neuropathol. 47, 372–382.
Schneider, A., Bardakjian, T., Reis, L. M., Tyler, R. C., and Semina, E. V. (2009). Novel SOX2 mutations and genotype–phenotype correlation in anophthalmia and microphthalmia. Am. J. Méd. Genet. Part A 149A, 2706–2715. doi: 10.1002/ajmg.a.33098
Schuhmacher, M., Kohlhuber, F., Hölzel, M., Kaiser, C., Burtscher, H., Jarsch, M., et al. (2001). The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res. 29, 397–406. doi: 10.1093/nar/29.2.397
Seltzer, L. E., Ma, M., Ahmed, S., Bertrand, M., Dobyns, W. B., Wheless, J., et al. (2014). Epilepsy and outcome in FOXG1-related disorders. Epilepsia 55, 1292–1300. doi: 10.1111/epi.12648
Sepp, M., Pruunsild, P., and Timmusk, T. (2012). Pitt–Hopkins syndrome-associated mutations in TCF4 lead to variable impairment of the transcription factor function ranging from hypomorphic to dominant-negative effects. Hum. Mol. Genet. 21, 2873–2888. doi: 10.1093/hmg/dds112
Sherr, C. J., and Roberts, J. M. (1999). CDK inhibitors: positive and negative regulators of G1-phase progression. Gene Dev. 13, 1501–1512. doi: 10.1101/gad.13.12.1501
Simeone, A. (1998). Otx1 and Otx2 in the development and evolution of the mammalian brain. EMBO J. 17, 6790–6798. doi: 10.1093/emboj/17.23.6790
Sisodiya, S. M., Ragge, N. K., Cavalleri, G. L., Hever, A., Lorenz, B., Schneider, A., et al. (2006). Role of SOX2 mutations in human hippocampal malformations and epilepsy. Epilepsia 47, 534–542. doi: 10.1111/j.1528-1167.2006.00464.x
Sock, E., Rettig, S. D., Enderich, J., Bösl, M. R., Tamm, E. R., and Wegner, M. (2004). Gene targeting reveals a widespread role for the high-mobility-group transcription factor Sox11 in tissue remodeling. Mol. Cell. Biol. 24, 6635–6644. doi: 10.1128/MCB.24.15.6635-6644.2004
Solomon, B. D., Pineda-Alvarez, D. E., Balog, J. Z., Hadley, D., Gropman, A. L., Nandagopal, R., et al. (2009). Compound heterozygosity for mutations in PAX6 in a patient with complex brain anomaly, neonatal diabetes mellitus, and microophthalmia. Am. J. Méd. Genet. Part A 149A, 2543–2546. doi: 10.1002/ajmg.a.33081
Staller, P., Peukert, K., Kiermaier, A., Seoane, J., Lukas, J., Karsunky, H., et al. (2001). Repression of p15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 3, 392–399. doi: 10.1038/35070076
Stanton, B. R., Perkins, A. S., Tessarollo, L., Sassoon, D. A., and Parada, L. F. (1992). Loss of N-myc function results in embryonic lethality and failure of the epithelial component of the embryo to develop. Genes Dev. 6, 2235–2247. doi: 10.1101/gad.6.12a.2235
Stevanovic, M., Drakulic, D., Lazic, A., Ninkovic, D. S., Schwirtlich, M., and Mojsin, M. (2021). SOX transcription factors as important regulators of neuronal and glial differentiation during nervous system development and adult neurogenesis. Front. Mol. Neurosci. 14, 654031. doi: 10.3389/fnmol.2021.654031
Strømme, P., Mangelsdorf, M. E., Shaw, M. A., Lower, K. M., Lewis, S. M. E., Bruyere, H., et al. (2002). Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat. Genet. 30, 441–445. doi: 10.1038/ng862
Su, Z., Wang, Z., Lindtner, S., Yang, L., Shang, Z., Tian, Y., et al. (2022). Dlx1/2-dependent expression of Meis2 promotes neuronal fate determination in the mammalian striatum. Development 149, 35. doi: 10.1242/dev.200035
Suda, Y., Matsuo, I., Kuratani, S., and Aizawa, S. (1996). Otx1 function overlaps with Otx2 in development of mouse forebrain and midbrain. Genes Cells 1, 1031–1044. doi: 10.1046/j.1365-2443.1996.900288.x
Sussel, L., Marin, O., Kimura, S., and Rubenstein, J. L. R. (1999). Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development 126, 3359–3370. doi: 10.1242/dev.126.15.3359
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Takuma, N., Sheng, H. Z., Furuta, Y., Ward, J. M., Sharma, K., Hogan, B. L. M., et al. (1998). Formation of Rathke's pouch requires dual induction from the diencephalon. Development 125, 4835–4840. doi: 10.1242/dev.125.23.4835
Tao, W., and Lai, E. (1992). Telencephalon-restricted expression of BF-1, a new member of the HNF-3/fork head gene family, in the developing rat brain. Neuron 8, 957–966. doi: 10.1016/0896-6273(92)90210-5
Tatard, V. M., Xiang, C., Biegel, J. A., and Dahmane, N. (2010). ZNF238 is expressed in postmitotic brain cells and inhibits brain tumor growth. Cancer Res. 70, 1236–1246. doi: 10.1158/0008-5472.CAN-09-2249
Tedesco, M. G., Lonardo, F., Ceccarini, C., Cesarano, C., Digilio, M. C., Magliozzi, M., et al. (2021). Clinical and molecular characterizations of 11 new patients with type 1 Feingold syndrome: Proposal for selecting diagnostic criteria and further genetic testing in patients with severe phenotype. Am. J. Méd. Genet. Part A 185, 1204–1210. doi: 10.1002/ajmg.a.62068
Thaxton, C., Kloth, A. D., Clark, E. P., Moy, S. S., Chitwood, R. A., and Philpot, B. D. (2018). Common pathophysiology in multiple mouse models of Pitt–Hopkins syndrome. J. Neurosci. 38, 918–936. doi: 10.1523/JNEUROSCI.1305-17.2017
Thein, D. C., Thalhammer, J. M., Hartwig, A. C., Bryan Crenshaw, E., III, and Lefebvre, V., Wegner, M., et al. (2010). The closely related transcription factors Sox4 and Sox11 function as survival factors during spinal cord development. J. Neurochem. 115, 131–141. doi: 10.1111/j.1471-4159.2010.06910.x
Toresson, H., Parmar, M., and Campbell, K. (2000). Expression of Meis and Pbx genes and their protein products in the developing telencephalon: implications for regional differentiation. Mech. Dev. 94, 183–187. doi: 10.1016/S0925-4773(00)00324-5
Tsurusaki, Y., Koshimizu, E., Ohashi, H., Phadke, S., Kou, I., Shiina, M., et al. (2014). De novo SOX11 mutations cause Coffin–Siris syndrome. Nat. Commun. 5, 4011. doi: 10.1038/ncomms5011
Tyas, D. A., Pearson, H., Rashbass, P., and Price, D. J. (2003). Pax6 regulates cell adhesion during cortical development. Cereb. Cortex 13, 612–619. doi: 10.1093/cercor/13.6.612
Uhler, J., Zhang, H., Syu, L.-J., and Mellerick, D. M. (2007). The Nk-2 box of the Drosophila homeodomain protein, Vnd, contributes to its repression activity in a Groucho-dependent manner. Mech. Dev. 124, 1–10. doi: 10.1016/j.mod.2006.07.009
Uwanogho, D., Rex, M., Cartwright, E. J., Pearl, G., Healy, C., Scotting, P. J., et al. (1995). Embryonic expression of the chicken Sox2, Sox3 and Sox11 genes suggests an interactive role in neuronal development. Mech. Dev. 49, 23–36. doi: 10.1016/0925-4773(94)00299-3
Vasko, A., Drivas, T. G., and Vergano, S. A. S. (2021). Genotype-phenotype correlations in 208 individuals with Coffin-Siris syndrome. Genes 12, 937. doi: 10.3390/genes12060937
Vergano, S. S., Santen, G., Wieczorek, D., Wollnik, B., Matsumoto, N., and Deardorff, M. A. (2021). “Coffin-Siris Syndrome,” in GeneReviews®, eds M. P. Adam, J. Feldman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Gripp, and A. Amemiya (Seattle, WA: University of Washington).
Verheije, R., Kupchik, G. S., Isidor, B., Kroes, H. Y., Lynch, S. A., Hawkes, L., et al. (2019). Heterozygous loss-of-function variants of MEIS2 cause a triad of palatal defects, congenital heart defects, and intellectual disability. Eur. J. Hum. Genet. 27, 278–290. doi: 10.1038/s41431-018-0281-5
Wakim, V., Nair, P., Delague, V., Bizzari, S., Al-Ali, M. T., Castro, C., et al. (2020). SOX11-related syndrome: report on a new case and review. Clin. Dysmorphol. 30, 44–49. doi: 10.1097/MCD.0000000000000348
Walhout, A. J. M. (2006). Unraveling transcription regulatory networks by protein–DNA and protein–protein interaction mapping. Genome Res. 16, 1445–1454. doi: 10.1101/gr.5321506
Walther, C., and Gruss, P. (1991). Pax-6, a murine paired box gene, is expressed in the developing CNS. Development 113, 1435–1449. doi: 10.1242/dev.113.4.1435
Wang, L., Wang, J., Jin, T., Zhou, Y., and Chen, Q. (2018). FoxG1 facilitates proliferation and inhibits differentiation by downregulating FoxO/Smad signaling in glioblastoma. Biochem. Biophys. Res. Commun. 504, 46–53. doi: 10.1016/j.bbrc.2018.08.118
Wang, Y., Lin, L., Lai, H., Parada, L. F., and Lei, L. (2013). Transcription factor Sox11 is essential for both embryonic and adult neurogenesis. Dev. Dyn. 242, 638–653. doi: 10.1002/dvdy.23962
Wang, Y., Lu, Z., Zhang, Y., Cai, Y., Yun, D., Tang, T., et al. (2019). Transcription factor 4 safeguards hippocampal dentate gyrus development by regulating neural progenitor migration. Cereb. Cortex 30, 3102–3115. doi: 10.1093/cercor/bhz297
Watemberg, N., Silver, S., Harel, S., and Lerman-Sagie, T. (2002). Significance of microcephaly among children with developmental disabilities. J. Child Neurol. 17, 117–122. doi: 10.1177/088307380201700205
Wedel, M., Fröb, F., Elsesser, O., Wittmann, M.-T., Lie, D. C., Reis, A., et al. (2020). Transcription factor Tcf4 is the preferred heterodimerization partner for Olig2 in oligodendrocytes and required for differentiation. Nucleic Acids Res. 48, 4839–4857. doi: 10.1093/nar/gkaa218
Wegner, M. (1999). From head to toes: the multiple facets of Sox proteins. Nucleic Acids Res. 27, 1409–1420. doi: 10.1093/nar/27.6.1409
Weigel, D., Jürgens, G., Küttner, F., Seifert, E., and Jäckle, H. (1989). The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell 57, 645–658. doi: 10.1016/0092-8674(89)90133-5
Willemsen, M. A. A. P., Breedveld, G. J., Wouda, S., Otten, B. J., Yntema, J. L., Lammens, M., et al. (2005). Brain-Thyroid-Lung syndrome: a patient with a severe multi-system disorder due to a de novo mutation in the thyroid transcription factor 1 gene. Eur. J. Pediatr. 164, 28–30. doi: 10.1007/s00431-004-1559-x
Winter, C. F., de Baas, M., Bijlsma, E. K., Heukelingen, J., van Routledge, S., and Hennekam, R. C. M. (2016). Phenotype and natural history in 101 individuals with Pitt-Hopkins syndrome through an internet questionnaire system. Orphanet J. Rare Dis. 11, 37. doi: 10.1186/s13023-016-0422-2
Xiang, C., Baubet, V., Pal, S., Holderbaum, L., Tatard, V., Jiang, P., et al. (2012). RP58/ZNF238 directly modulates proneurogenic gene levels and is required for neuronal differentiation and brain expansion. Cell Death Differ. 19, 692–702. doi: 10.1038/cdd.2011.144
Xuan, S., Baptista, C. A., Balas, G., Tao, W., Soares, V. C., and Lai, E. (1995). Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron 14, 1141–1152. doi: 10.1016/0896-6273(95)90262-7
Yang, Y. J., Baltus, A. E., Mathew, R. S., Murphy, E. A., Evrony, G. D., Gonzalez, D. M., et al. (2012). Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 151, 1097–1112. doi: 10.1016/j.cell.2012.10.043
Yeung, A., Bruno, D., Scheffer, I. E., Carranza, D., Burgess, T., Slater, H. R., et al. (2009). 4.45 Mb microduplication in chromosome band 14q12 including FOXG1 in a girl with refractory epilepsy and intellectual impairment. Eur. J. Méd. Genet. 52, 440–442. doi: 10.1016/j.ejmg.2009.09.004
Zawerton, A., Yao, B., Yeager, J. P., Pippucci, T., Haseeb, A., Smith, J. D., et al. (2019). De novo SOX4 variants cause a neurodevelopmental disease associated with mild dysmorphism. Am. J. Hum. Genet. 104, 246–259. doi: 10.1016/j.ajhg.2018.12.014
Zhou, X., Feliciano, P., Shu, C., Wang, T., Astrovskaya, I., Hall, J. B., et al. (2022). Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat. Genet. 54, 1305–1319. doi: 10.1038/s41588-022-01148-2
Keywords: brain size, cortical development, microcephaly, proliferation, neurogenesis, transcription factors
Citation: Lim Y (2023) Transcription factors in microcephaly. Front. Neurosci. 17:1302033. doi: 10.3389/fnins.2023.1302033
Received: 25 September 2023; Accepted: 06 November 2023;
Published: 29 November 2023.
Edited by:
Zhong-Wei Zhou, Sun Yat-sen University, ChinaReviewed by:
Ferdinando Di Cunto, University of Turin, ItalyTodd Schoborg, University of Wyoming, United States
Copyright © 2023 Lim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youngshin Lim, eW91bmcubGltQGNzaHMub3Jn