 Randall J. Eck
Randall J. Eck Jade G. Stair3†
Jade G. Stair3† Nicole F. Liachko
Nicole F. Liachko- 1Graduate Program in Neuroscience, University of Washington, Seattle, WA, United States
- 2Division of Gerontology and Geriatric Medicine, Department of Medicine, University of Washington, Seattle, WA, United States
- 3Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA, United States
- 4Department of Psychiatry and Behavioral Sciences, University of Washington, Seattle, WA, United States
- 5Department of Laboratory Medicine and Pathology, University of Washington, Seattle, WA, United States
The nematode Caenorhabditis elegans are a powerful model system to study human disease, with numerous experimental advantages including significant genetic and cellular homology to vertebrate animals, a short lifespan, and tractable behavioral, molecular biology and imaging assays. Beginning with the identification of SOD1 as a genetic cause of amyotrophic lateral sclerosis (ALS), C. elegans have contributed to a deeper understanding of the mechanistic underpinnings of this devastating neurodegenerative disease. More recently this work has expanded to encompass models of other types of ALS and the related disease frontotemporal lobar degeneration (FTLD-TDP), including those characterized by mutation or accumulation of the proteins TDP-43, C9orf72, FUS, HnRNPA2B1, ALS2, DCTN1, CHCHD10, ELP3, TUBA4A, CAV1, UBQLN2, ATXN3, TIA1, KIF5A, VAPB, GRN, and RAB38. In this review we summarize these models and the progress and insights from the last ten years of using C. elegans to study the neurodegenerative diseases ALS and FTLD-TDP.
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease characterized by progressive muscle denervation and motor neuron loss in the brain and spinal cord. ALS affects one in 350 individuals, with higher rates of ALS in some populations including military veterans (Tai et al., 2017; van Es et al., 2017). Although the majority of cases of ALS are sporadic, with no known genetic cause, approximately 5%–10% of cases have a familial-inherited causative mutation. To date there are more than 45 human genes implicated as genetic drivers of ALS (Smukowski et al., 2022). ALS-causing gene mutations provide insight into cellular mechanisms that initiate disease and can be a starting point to model ALS in the laboratory. In human disease, most patients with sporadic ALS (sALS) and familial-inherited ALS (fALS) exhibit inclusions of the transactive response DNA binding protein (TDP-43) in disease affected neurons. However, patients with fALS mutations in the SOD1 gene accumulate aggregates of the protein SOD1, while patients with FUS mutations accumulate aggregates of the protein FUS. Approximately half of all patients with frontotemporal lobar degeneration (FTLD), another neurodegenerative disease, also exhibit TDP-43 pathology (FTLD-TDP). A subset of FTLD-TDP patients exhibit motor symptoms, while some ALS patients exhibit FTLD-like cognitive changes. Some genetic causes of ALS can lead to ALS, FTLD-TDP, or mixed ALS/FTLD presentations within the same family, leading to the recognition that ALS and FTLD-TDP represent a clinical spectrum of related diseases (Strong et al., 2017).
The nematode Caenorhabditis elegans was established in the 1960s as a tractable model organism for scientific research (Brenner, 1973). C. elegans are optically transparent, have a relatively short lifespan averaging 21 days, and can self-fertilize resulting in genetically identical progeny (Johnson, 2003). Adult hermaphrodite C. elegans contain ~300 neurons with a defined and consistent connectome controlling sensory, motor, and interneuron signaling with relevant human neurotransmitters such as glutamate, gamma-aminobutyric acid (GABA), acetylcholine, dopamine, and serotonin (Cook et al., 2019). At least 40% of the C. elegans protein coding genome, or 7,943 genes, are orthologues or paralogs of human genes, including a significant number of genes related to human genetic disease (Shaye and Greenwald, 2011; Kim et al., 2018). Disease relevant biological pathways are also conserved in C. elegans (Shaye and Greenwald, 2011; Kim et al., 2018). These experimental advantages have fueled the use of C. elegans to model neurodegenerative diseases (Silverman et al., 2009; Apfeld and Alper, 2018; Caldwell et al., 2020).
Approaches to modeling ALS/FTLD in C. elegans either manipulate the endogenous C. elegans homolog of a known disease gene or utilize transgenes to express a human disease associated gene (Figure 1). When an ALS/FTLD-associated gene is conserved in C. elegans, researchers employ a variety of strategies. These include deletion or partial reduction of the C. elegans gene, overexpression, introduction of human ALS/FTLD-associated mutations into the endogenous C. elegans homolog at conserved sites, or generation of a chimera of the C. elegans protein with key domains from the wild-type or mutant human protein. To directly examine the consequences of human ALS/FTLD genes, researchers can transgenically express wild-type or mutant human disease-associated genes in muscles, neurons, or throughout the C. elegans body, express individual protein domains, or replace the C. elegans homolog with a single-copy knock-in of the wild-type or mutant human gene. More recent efforts to model neurodegenerative diseases in C. elegans have included the development of a photoconvertible fluorescent protein tag to track protein dynamics in vivo (Pigazzini and Kirstein, 2020), the conditional expression or inducible aggregation of neurotoxic proteins in aging (Lim et al., 2020), the use of natural genetic variation to study resistance and resilience to protein aggregation in disease (Alexander-Floyd et al., 2020), the study of synergies between distinct pathological proteinopathies (Benbow et al., 2020; Latimer et al., 2022), the exploration of glia–neuron communication in protein quality control (Bar-Ziv et al., 2023), and the development of models to study prion-like seeding or spread of disease-causing proteins in neurons (Gallrein et al., 2021; Zanier et al., 2021). These approaches may inspire future ALS/FTLD models in C. elegans.
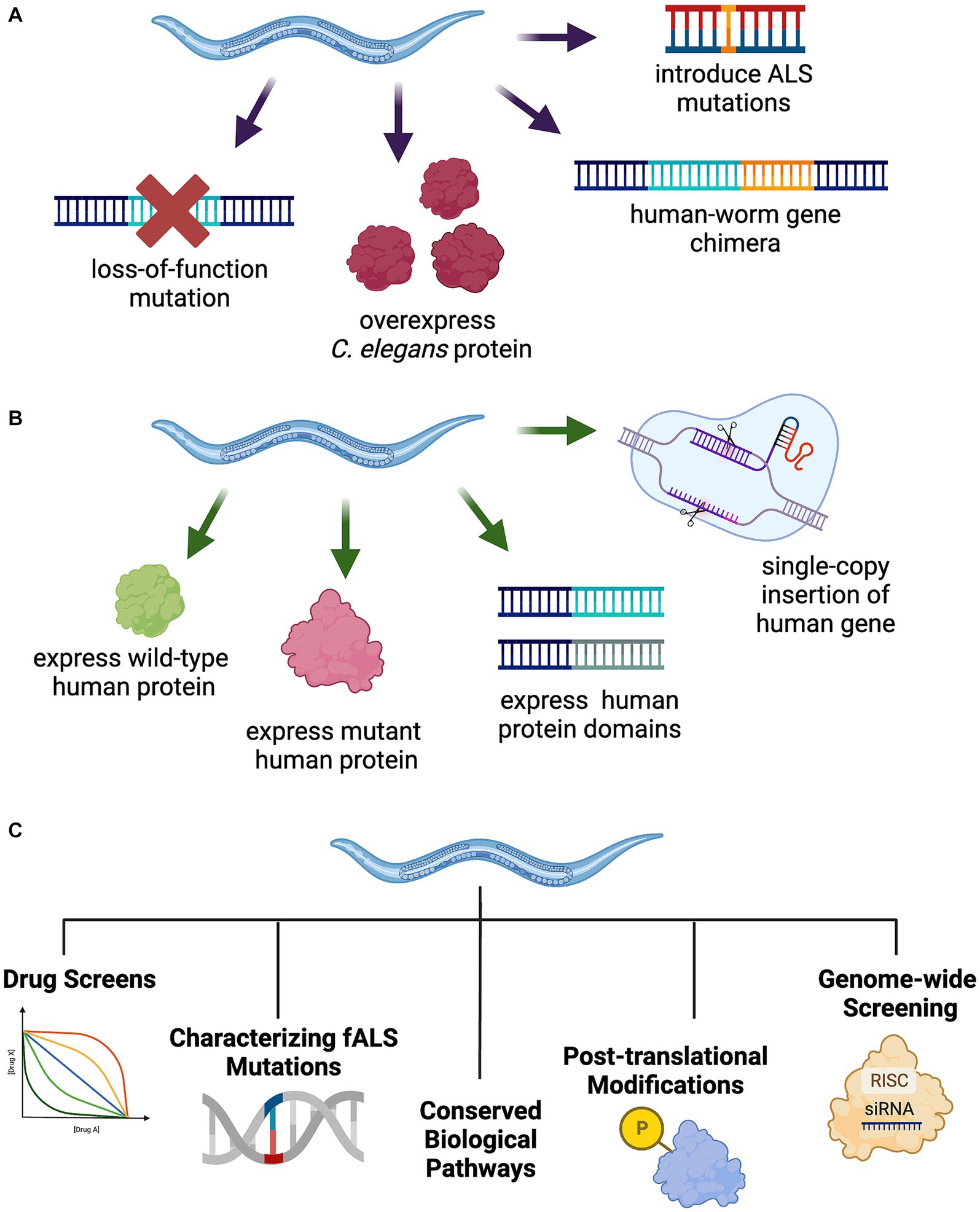
Figure 1. Strategies to model TDP-43-driven ALS and FTLD-TDP in C. elegans. (A) Manipulation of the endogenous C. elegans ALS gene homolog through introduction of fALS mutations at conserved loci, protein overexpression, human-worm gene chimeras, or loss-of-function mutations. (B) Generation of transgenic C. elegans expressing full-length wild-type or mutant human protein, select protein domains, or a single-copy insertion of the human gene into the genome, potentially replacing the endogenous worm homolog. (C) C. elegans model utility for a broad range of unbiased and hypothesis driven research including drug screening, fALS mutation characterization, cellular and molecular pathway exploration, protein post-translational modifications, and genome-wide applications including forward and reverse genetics approaches.
A comprehensive review of C. elegans ALS models was published in 2014 (Therrien and Parker, 2014); however, significant progress has been made in the last 10 years studying genetic and sporadic forms of ALS using C. elegans, refining our understanding of ALS and its relationship with FTLD, and identifying new pathways and targets for therapeutic development. This review will focus on these advances from 2014 to 2023, identifying progress and highlighting areas for further investigation.
Caenorhabditis elegans models of ALS/FTLD by proteinopathy
TDP-43
Cytoplasmic aggregates of TDP-43 are found in the motor neurons of approximately 97% of ALS patients (Arai et al., 2006; Neumann et al., 2006; Tan et al., 2017). Encoded by the TARDBP gene, TDP-43 regulates transcription, pre-mRNA and alternative splicing, mRNA stability and transport, and microRNA biogenesis (reviewed in Nilaver and Urbanski, 2023). TDP-43 contains two RNA-binding domains (RRM1 and RRM2), a nuclear localization signal (NLS), and a C-terminal glycine-rich, low-complexity, intrinsically disordered region. More than 30 different mutations have been identified in TARDBP that cause fALS (Smukowski et al., 2022). The majority of fALS mutations are located in the C-terminus and potentiate a variety of changes in protein function, including altering TDP-43 liquid–liquid phase separation (reviewed in Hurtle et al., 2023). Both loss of normal TDP-43 functions and gains of toxic function may contribute to disease (as reviewed in Gao et al., 2018; Prasad et al., 2019). For example, loss of TDP-43 nuclear function alters the splicing of thousands of mRNA transcripts, while cytoplasmic aggregates of TDP-43 in disease sequester RNA binding proteins and RNA, which may also contribute to cellular dysfunction (Barmada, 2015; Mehta et al., 2023).
Loss of function mutations in the C. elegans TDP-43 homolog tdp-1 (tdp-1(ok803) or tdp-1(ok781)) do not cause motor deficits or neurodegeneration (Table 1) (Ash et al., 2010; Saldi et al., 2018; Mitra et al., 2019). However, loss of tdp-1 increases C. elegans sensitivity to DNA damage and oxidative stress, enhances the efficacy of nuclear RNA interference, produces double stranded RNA foci, and alters exon inclusion in mRNA splicing (Saldi et al., 2014, 2018; Melnick et al., 2019; Mitra et al., 2019; Lins et al., 2023; Taylor et al., 2023). Loss of tdp-1 also causes temperature dependent lifespan extension (Zhang et al., 2012; Vaccaro et al., 2012c) by modifying DAF-16/FOXO signaling (Table 1) (Zhang et al., 2014). On the other hand, overexpression of TDP-1 in neurons, under the snb-1 promoter, is sufficient to trigger motor deficits (Ash et al., 2010). TDP-1 overexpression under its endogenous promoter also decreases lifespan of C. elegans grown at both 20°C and 25°C (Table 1) (Vaccaro et al., 2012c). Like in human neurons, TDP-1 localizes to cytoplasmic granules during osmotic stress, as visualized by a P snb-1 ::TDP-1::YFP fusion (Zhang et al., 2014). A CRISPR-Cas9 generated tdp-1 true null allele, tdp-1(tgx58), exhibits increased sensitivity to moderate oxidative stress, as evidenced by increased loss of glutamatergic sensory neurons. This phenotype was rescued by insertion of wild-type human TDP-43 sequences at the endogenous tdp-1 locus, demonstrating a conservation of function (Lins et al., 2023).
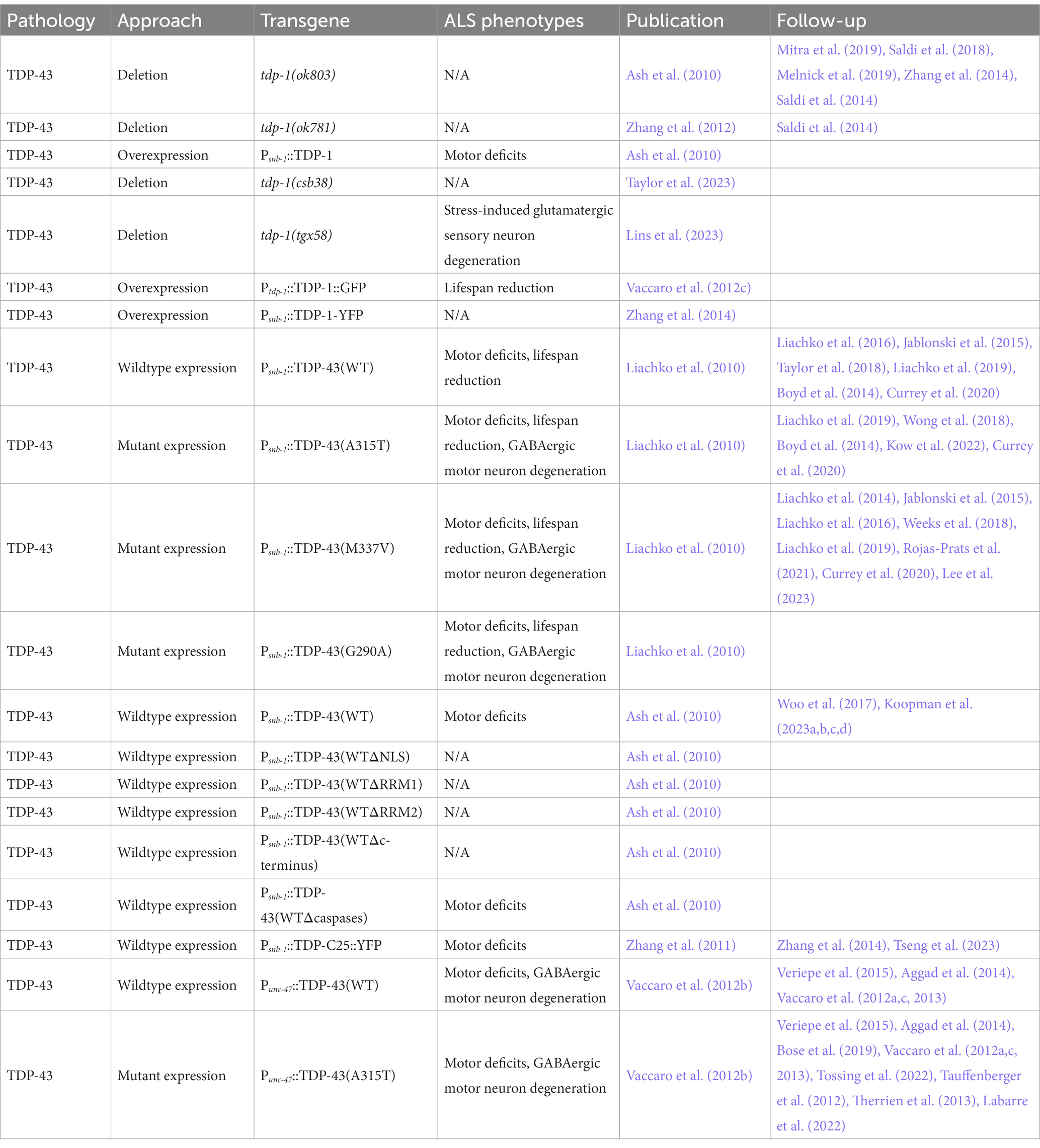
Table 1. C. elegans models of TDP-43-driven ALS and FTLD-TDP.
Transgenic expression of wild-type human TDP-43 or fALS mutant TDP-43(A315T), TDP-43(M337V), and TDP-43(G290A) in C. elegans neurons under the snb-1 promoter results in several ALS associated phenotypes including progressive motor deficits, GABAergic motor neuron degeneration, a reduction in lifespan, disease-associated TDP-43 phosphorylation at epitopes S409/410, and the formation of insoluble TDP-43 aggregates in the nucleus, but not the cytoplasm (Table 1) (Liachko et al., 2010). The snb-1-driven expression of human wild-type TDP-43 also results in decreased fecundity and disrupted chemotaxis (Ash et al., 2010; Koopman et al., 2023b,d), in addition to motor deficits (Koopman et al., 2023c). TDP-43 exhibits temperature-sensitive increases in cytoplasmic mislocalization, accompanied by exacerbated aggregation and pathological phosphorylation at S409/S410, highlighting the importance of cytoplasmic mislocalization in disease pathogenesis (Koopman et al., 2023a). In this model, TDP-43’s NLS, both RNA binding domains, and the C-terminus, but not caspase cleavage sites, are required for TDP-43 neurotoxicity (Ash et al., 2010). The TDP-43 C-terminus alone, when expressed in neurons under the snb-1 promoter, is sufficient to trigger robust aggregation and motor deficits (Zhang et al., 2011). Expressing TDP-43 or TDP-43(A315T) exclusively in GABAergic motor neurons using the unc-47 promoter also produces progressive motor deficits, axonal GABAergic neuron degeneration, lowered fecundity, reduced chemotaxis, and accumulation of TDP-43 aggregates in both the nucleus and the cytoplasm, but not a reduction in lifespan (Table 1) (Vaccaro et al., 2012b,c). These transgenic overexpression models suggest fALS mutations G290A, A315T, and M337V in the C-terminus of TDP-43 are gain-of-function mutations, since mutant TDP-43 results in more severe motor deficits, neurodegeneration, lifespan reduction, aggregation, and pathological phosphorylation compared to wild-type expression, and these phenotypes do not align with the effects of loss of tdp-1 function.
TDP-43 phosphorylation serves as a robust and consistent clinical marker of pathological TDP-43 inclusions in ALS patient motor neurons (Arai et al., 2006; Neumann et al., 2006; Hasegawa et al., 2008). In C. elegans, the kinases PRKD2/3, CDC7 and TTBK1/2 can phosphorylate TDP-43 in vivo (Liachko et al., 2014). rgef-1 promoter mediated neuronal expression of TTBK1, but not TTBK2, exacerbates motor deficits and increases accumulation and S409/S410 phosphorylation of wild-type TDP-43 (Taylor et al., 2018). These results suggest dysregulation of TDP-43 phosphorylation contributes to the progression of ALS and is a possible therapeutic target (Eck et al., 2021).
In C. elegans, there is a long history of using forward genetic and whole genome screens to identify novel gain of function mutations and genes that modify neurodegenerative phenotypes (reviewed in Kutscher and Shaham, 2014, Sin et al., 2014). These screens reveal critical biological pathways and new therapeutic targets in disease. A genome wide RNAi screen identified several modifiers of motor deficits in C. elegans expressing TDP-43 and mutant TDP-43(M337V) in neurons. Of these, loss of function mutations in hse-5(tm472)/GLCE, zig-3(tm924)/HMCN1, paqr-1(tm3262)/ADIPOR1, gly-8(tm1156)/GALNT11, and sax-2(ky216)/FRYL reduce accumulation and pathological TDP-43 phosphorylation, indicating they act in pathways critical to the development of TDP-43 toxicity (Table 2). In addition, hse-5 restores synaptic transmission in GABAergic motor neurons (Liachko et al., 2019). Another modifier of mutant TDP-43(M337V) is rad-23. RAD-23, and its human homologs RAD23A and RAD23B, are part of the endoplasmic-reticulum (ER) associated protein degradation pathway and function in substrate clearance and DNA damage repair. Loss of function mutations rad-23(tm3690) and rad-23(tm2595) rescue motor deficits, GABAergic motor neuron degeneration, and TDP-43(M337V) aggregation (Table 2) (Jablonski et al., 2015).
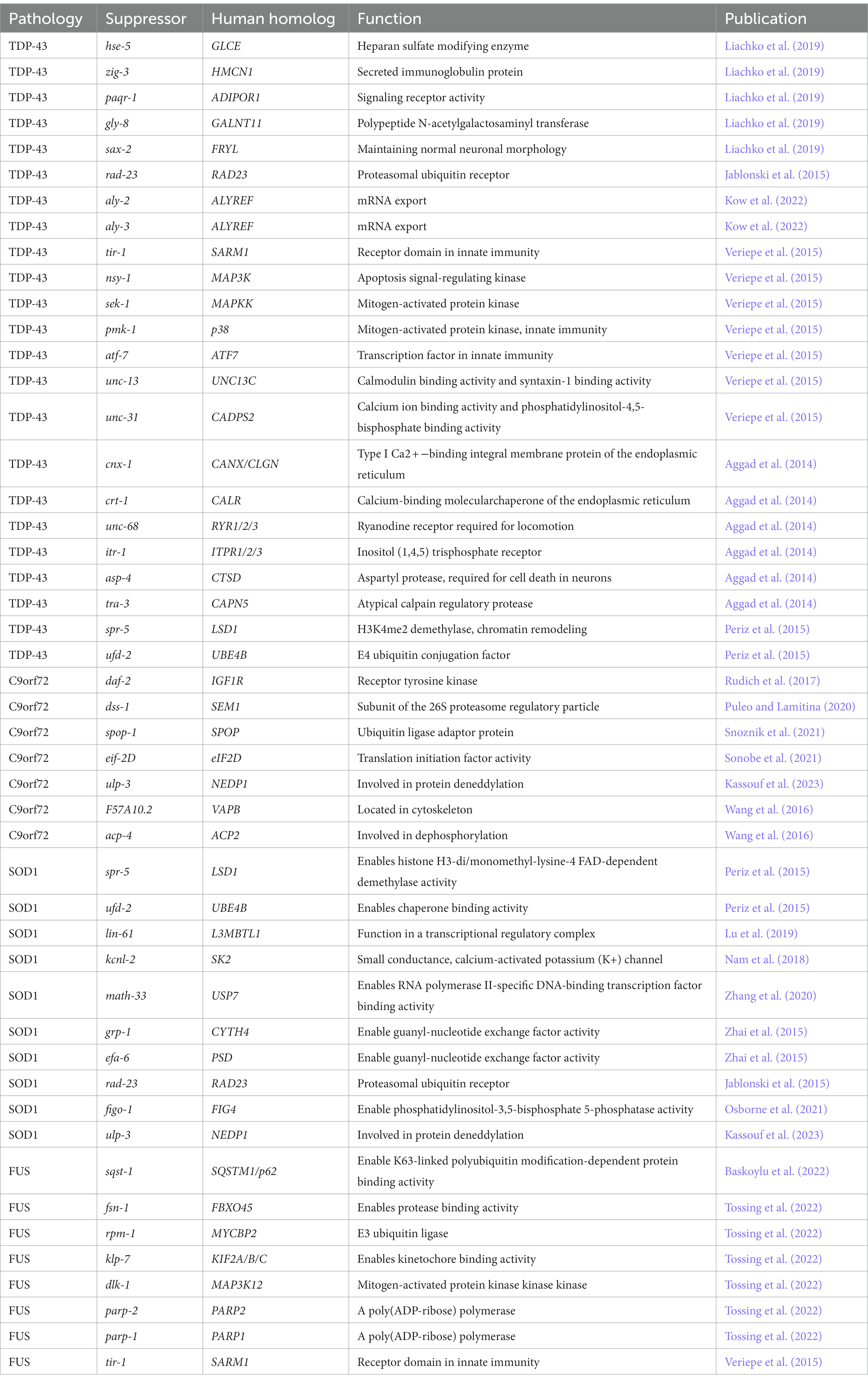
Table 2. A list of genes that modify ALS/FTLD-TDP phenotypes in C. elegans models of disease.
In C. elegans expressing TDP-43(A315T) in neurons, loss of function mutations in the RNA export factor ALYREF homologs aly-2(bk3079) and aly-3(bk3069) together rescue motor deficits (Table 2) (Kow et al., 2022). When expressed in GABAergic motor neurons specifically, TDP-43(A315T) triggers an innate immune response in motor neurons and surrounding tissue. Suppressing this innate immune response by a tir-1(qd4) deletion allele rescues motor deficits and neurodegeneration without altering TDP-43(A315T) levels. The TIR-1 receptor is critical to the innate immune response and is homologous to human Sarm1, which also regulates axon degeneration. Loss of function mutations in TIR-1 pathway genes nsy-1(ok593)/MAP3K, sek-1(km4)/MAPKK, pmk-1(km25)/p38, and transcription factor atf-7(qd22)/ATF7 also reduce motor deficits and neurodegeneration. Loss of function mutations in the neurosecretory genes unc-13(e540)/UNC13C and unc-31(e928)/CADPS2 rescue neurodegeneration as well, suggesting neurosecretion is critical to innate immune induction in this model (Table 2) (Veriepe et al., 2015). Another pathway required for TDP-43(A315T) toxicity in GABAergic motor neurons is ER calcium-regulated calpain and aspartyl protease activity. Null mutations cnx-1(nr2009)/CANX, crt-1(bz30)/CALR, unc-68(e540)/RYR1/2/3, itr-1(sa73)/ITPR2/3, asp-4(ok2693)/CTSD, and tra-3(ok2207)/CAPN5 disrupt this pathway and rescue TDP-43(A315T) driven progressive paralysis as well as reduce neurodegeneration without altering TDP-43(A315T) levels (Table 2). None of these mutants improve C. elegans expressing wild-type TDP-43 (Aggad et al., 2014).
Cold temperature, which also extends lifespan, reduces the accumulation of TDP-43 protein in TDP-43(M337V) transgenics grown at 15°C. Knockdown of psme-3/PSME3, a proteasome regulator, reverses this reduction (Lee et al., 2023). In addition to aging, protein quality control genes also modify TDP-43 aggregation. Loss of function mutations in spr-5(by134)/LSD1 and ufd-2(tm1380)/UBE4B together dramatically improve motor deficits and reduce the aggregation of C-terminally truncated TDP-43 (TDP-43-C25) by upregulating proteasomal and autophagic degradation (Table 2) (Periz et al., 2015).
Taken together, these modifiers of TDP-43 identified through genetic screens in several C. elegans models of ALS represent a group of compelling therapeutic targets and implicate the activity or disruption of ER-associated protein homeostasis, RNA metabolism, protease activity, the innate immune response, proteasomal and autophagic degradation, and other biological pathways in ALS pathogenesis.
In addition to genetic screens, C. elegans models enable cost-effective high-throughput screens of drug and novel chemical libraries for basic efficacy and toxicity, shaping the drug discovery pipeline (O’Reilly et al., 2014; Giunti et al., 2021). For ALS, several screens in C. elegans models have identified translatable neuroprotective compounds. One screen of 3,768 small molecules in C. elegans expressing mutant TDP-43(A315T) in GABAergic motor neurons identified 11 compounds that improve motor deficits when delivered at a concentration of 20 μM for 6 hour in a liquid culture. Of these, the most potent, TRVA242, also decreases neurodegeneration by an unknown mechanism (Bose et al., 2019). C. elegans can also screen derivatives of chemical compounds aimed at lowering their necessary dosage for action. C. elegans expressing mutant TDP-43(A315T) in neurons pinpointed more effective inhibitors of CDC7 kinase activity, which reduce TDP-43 phosphorylation in vivo (Rojas-Prats et al., 2021). In the same model, a screen of ethosuximide-based compounds found α-methyl-α-phenylsuccinimide (MPS) significantly improves motor deficits, rescues shortened lifespan, and reduces GABAergic motor neuron degeneration through a pathway that requires the FOXO transcription factor DAF-16 (Wong et al., 2018).
Outside of large library or derivative screens, C. elegans can also validate results from cell-based screens or test known neuroprotective compounds. For example, compound LDN-0130436, identified in a screen of 75,000 compounds in cells, rescues motor deficits and decreases GABAergic motor neuron degeneration in C. elegans expressing TDP-43 and mutant TDP-43(A315T) in all neurons (Boyd et al., 2014). Recently, a series of proteolysis targeting chimeras (PROTACs), an alternative to classical chemical agonists, were also screened in a C. elegans model expressing the C-terminus of TDP-43 in neurons. PROTACS are designed to scaffold an interaction between a protein of interest and an E3 ligase, which target the protein for degradation by the proteasome. In C. elegans, PROTAC2 decreases TDP-43-C25 aggregation and partially rescues motor deficits (Tseng et al., 2023).
Overall, the expression of both wild-type and mutant TDP-43 in C. elegans neurons provides a platform for the study of loss and gain of function mechanisms in both sALS and fALS. These models have facilitated the identification of dozens of novel genetic modifiers of TDP-43 proteinopathy in ALS and many neuroprotective chemical compounds.
C9orf72
The most common genetic cause of ALS is an expansion of the non-coding hexanucleotide repeat G4C2 in the first intron of the C9ORF72 gene (DeJesus-Hernandez et al., 2011; Renton et al., 2011). This expansion is relatively rare in Asia and the Middle East; the majority of families affected are of European descent (Majounie et al., 2012). Normally, C9ORF72’s first intron contains less than 12 hexanucleotide repeats, but in fALS, this region is oftentimes hundreds or even thousands of hexanucleotide repeats long (Balendra and Isaacs, 2018). There remains disagreement on the threshold of repeat length necessary for fALS, but one meta-analysis suggested repeats as short as 24 may be sufficient (Van Mossevelde et al., 2017; Iacoangeli et al., 2019). These hexanucleotide expansions could contribute to neurodegeneration by altering C9orf72 expression, through the aggregation of repeat RNAs, or through aggregation of dipeptide repeat proteins (DPRs) (Tang et al., 2020). C9orf72 regulates autophagy, endosomal trafficking, lysosomal biogenesis, and inflammation by interacting with Rab-GTPases and other partners (Farg et al., 2014; Sellier et al., 2016; Webster et al., 2016; Burberry et al., 2020; Tang et al., 2020). In human motor neurons and mice, haploinsufficiency of C9orf72 leads to neurodegeneration (Shi et al., 2018; Shao et al., 2019). The G4C2 expansion can be bidirectionally transcribed into RNA that form foci or hexanucleotide repeat RNA can be translated as five DPRs (poly-GA, poly-GP poly-GR, poly-PA, and poly-PR) following repeat-associated non-AUG-dependent (RAN) translation (Ash et al., 2013; Gendron et al., 2013; Zu et al., 2013; Mori et al., 2013a,b). RNA foci can adopt stable conformations and sequester critical RNA binding proteins, resulting in nucleolar stress and other disruptions in cell culture, potentially contributing to neurodegeneration (Haeusler et al., 2014; Balendra and Isaacs, 2018). On the other hand, DPR expression in zebrafish, mice, Drosophila, and cell culture is neurotoxic, especially the expression of poly-GR, poly-PR, and poly-GA, dysregulating translation, phase-separated condensates, RNA binding protein function, and other biological pathways (as reviewed in Balendra and Isaacs, 2018, Tang et al., 2020).
In C. elegans, deletion of the C9ORF72 orthologue, alfa-1(ok3062), accelerates age-related paralysis, increases the rate of GABAergic motor neuron degeneration in aging, and reduces resistance to osmotic stress (Table 3). The motor deficits of alfa-1 mutants add to the toxicity of mutant TDP-43(A315T) in GABAergic motor neurons, suggesting several pathways may lead to neurodegeneration in ALS (Therrien et al., 2013). More recent work has shown that loss of alfa-1 also causes defects in lysosomal homeostasis which can be partially rescued by the expression of human C9orf72, suggesting both a pathogenic mechanism and conserved protein function (Corrionero and Horvitz, 2018; Ji et al., 2020). alfa-1 lacks the G4C2 repeats responsible for C9orf72 RNA and DPR toxicity.
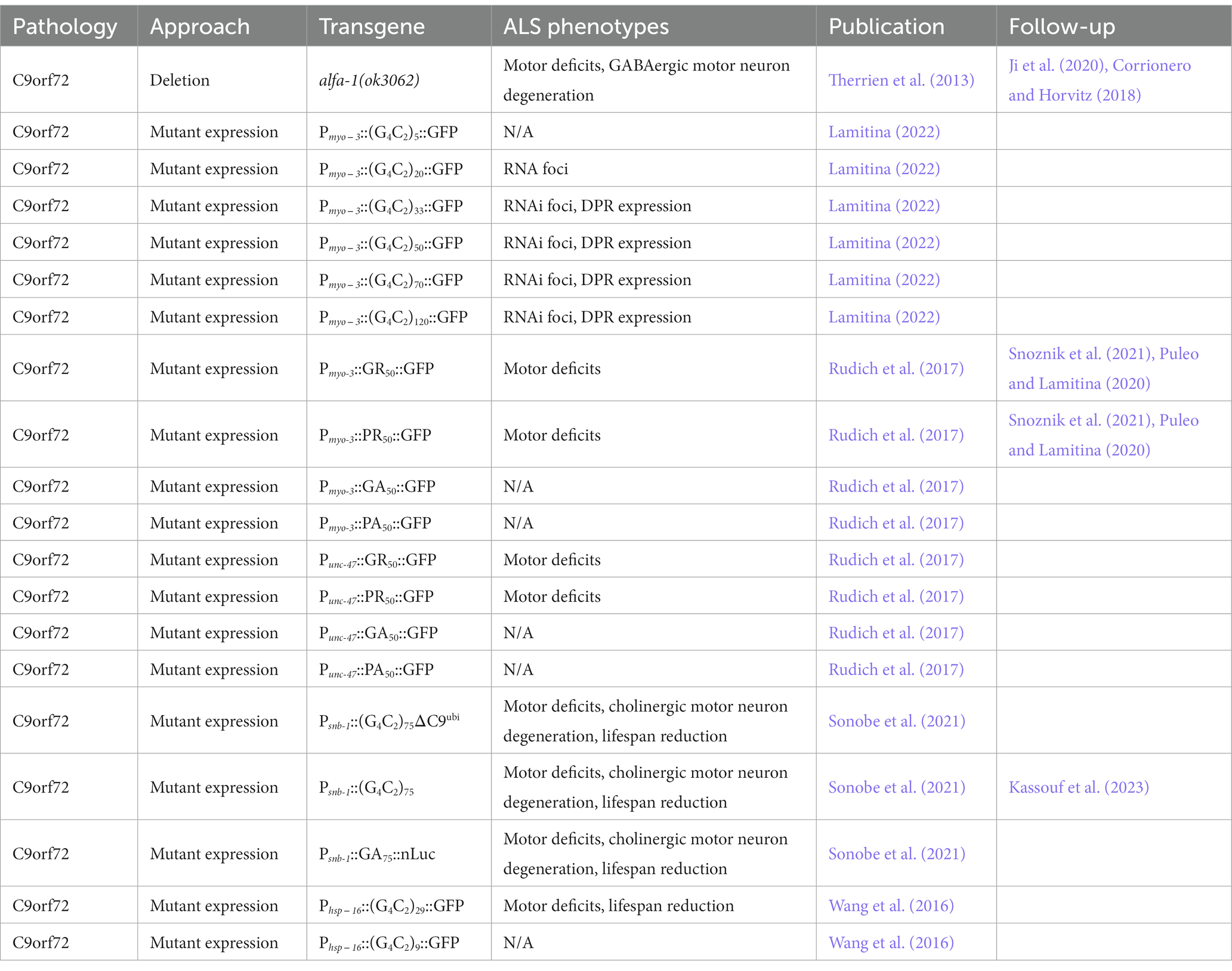
Table 3. C. elegans models of C9orf72-driven ALS and FTLD.
The expression of these pure G4C2 repeats in C. elegans muscles under the myo-3 promoter is sufficient to trigger the formation of RNA foci and RAN translation of DPRs, even without any accompanying human C9orf72 intronic regions (Table 3). By examining transgenics expressing 5, 20, 33, 50, 70, and 120 G4C2 repeats, the number of G4C2 repeats required to initiate RNAi foci formation in C. elegans was determined to be greater than 5, but less than 20. For RAN translation of DPRs, the repeat threshold falls between 20 and 33, which aligns with the proposed threshold in humans (Lamitina, 2022).
The RAN translation of G4C2 repeats can result in five DPR proteins depending on the open-reading frame, including (PA)n, (GA)n, (PR)n, and (GR)n. In C. elegans, the individual expression of DPRs (GR)50 and (PR)50, but not (PA)50 and (GA)50, in muscles under the myo-3 promoter significantly accelerates age-related paralysis (Table 3) (Rudich et al., 2017; Puleo and Lamitina, 2020; Snoznik et al., 2021). (PR)50 DPRs are soluble and localize to the nucleolus. At least 25 repeats and continued expression of poly-PR DPRs are required for paralysis. The forced nuclear localization of (PR)50 DPRs accelerates paralysis, while mutations decreasing the rate of physiological aging delay DPR toxicity (Rudich et al., 2017).
A pilot and then genome-wide RNAi screen in C. elegans expressing (PR)50 DPRs in muscles identified several novel modifiers of C9orf72 toxicity. Two were further characterized: dss-1 and spop-1 (Table 2) (Puleo and Lamitina, 2020; Snoznik et al., 2021). Knockdown of dss-1, whose human homolog Sem1 functions in mRNA export and nuclear pore function, partially rescues a progressive paralysis phenotype in both (PR)50 and (GR)50 expressing transgenic animals but does not alter poly-PR nuclear localization (Puleo and Lamitina, 2020). Loss of function mutations in spop-1, whose human homolog SPOP is a nuclear ubiquitin ligase adaptor protein, similarly rescues a progressive paralysis phenotype in (PR)50 and (GR)50 models without changing DPR levels or localization. The increased activity of transcription factor BET-1, BRD2/3/4 in humans, which is normally degraded by SPOP, is responsible for spop-1 suppression (Snoznik et al., 2021).
The expression of pure DPRs as well as G4C2 repeats in cell types other than muscles also results in pronounced motor deficits in C. elegans. For example, the expression of DPRs (GR)50 and (PR)50, but not (PA)50 and (GA)50, in GABAergic motor neurons under the unc-47 promoter results in motor deficits and GABAergic motor neuron degeneration (Rudich et al., 2017). In addition, the pan-neuronal expression of 75 G4C2 repeats flanked by the human C9orf72 intronic regions under the snb-1 promoter results in the expression of several DPRs, progressive motor deficits, cholinergic motor neuron degeneration, and a reduced lifespan. Lifespan is also reduced when this construct is expressed under a fragment of the unc-11 promoter, known to be more exclusively active in neurons. The inclusion of the C9orf72 intronic regions allows for the robust translation of poly-GA, which generally requires an initiation codon. The neurodegenerative phenotypes of transgenics lacking poly-GA are less severe (Table 3) (Sonobe et al., 2021).
In this model, loss of function mutations in C. elegans eif-2D, or deletion of its SUI1 initiation codon recognition domain, dramatically reduce the expression of poly-GA, suggesting eif-2D human homolog eIF2D is critical for DPR translation (Table 2) (Sonobe et al., 2021). Loss of ulp-3(tm1287), whose human homolog NEDP1 regulates stress granule dynamics, also rescues motor deficits in this model and reduces the number of stress granule formed during oxidative stress (Table 2) (Kassouf et al., 2023).
The expression of both nine and 29 G4C2 repeats under the global hsp-16 promoter results in motor deficits and a reduction in lifespan as well, with 29 G4C2 repeats resulting in significantly worse phenotypes, suggesting neurotoxicity is repeat length-dependent (Table 3) (Wang et al., 2016). In this model, loss of function mutations in F57A10.2/VAPB and acp-4(gk833833)/ACP2 rescue progressive motor deficits (Table 2). Initially identified in a forward-genetic mutagenesis screen, F57A10.2 is a homolog of human VAPB, which regulates endosomal trafficking, and ACP-4 is the homolog of ACP2, a lysosomal acid phosphatase (Wang et al., 2016).
Taken together, the expression of G4C2 repeats in C. elegans is a valuable model for studying C9orf72 fALS. The expression of C9orf72 DPRs in C. elegans produce a variety of neurodegenerative phenotypes that have allowed for the identification of several novel modifiers of DPR toxicity. These modifiers suggest autophagy, the ubiquitin proteasome system, RAN translation, and stress granules as possible therapeutic targets in C9orf72 fALS.
SOD1
Mutations in the gene Cu/Zn superoxide dismutase 1 (SOD1) were the first identified genetic cause of fALS and account for approximately 8–23% of familial and 1–4% of sporadic cases without a family history of ALS (Rosen et al., 1993; Muller et al., 2022; Smukowski et al., 2022). To date more than 150 mutations throughout the SOD1 coding region have been identified, although the most commonly observed in ALS are SOD1 D90A, A4V, or G93A (Pansarasa et al., 2018). For these individuals, neuronal inclusions of the protein SOD1 are the major neuropathological marker. SOD1 localizes to mitochondria and protects these organelles from oxidative stress damage by converting harmful superoxide anions into hydrogen peroxide and oxygen. SOD1 has four cysteine residues that contribute to its stability and catalytic activity, and changes in the redox states of these residues have been found in ALS-causing mutant SOD1 (Peggion et al., 2022). Numerous mechanisms have been proposed by which changes in SOD1 function cause or contribute to ALS, including altered SOD1 protein maturation or localization, increased cellular oxidative stress, or impaired mitochondrial function.
Human SOD1 and C. elegans SOD-1 are highly homologous, sharing 71% protein similarity (Baskoylu et al., 2018). In addition to SOD1, there are two other SOD isoforms: MnSOD (manganese superoxide dismutase) and ECSOD (extracellular superoxide dismutase). MnSOD is encoded by the gene SOD2 and ECSOD is encoded by the gene SOD3 (Miao and St. Clair, 2009). However, there are five superoxide dismutase (sod) genes in C. elegans: sod-1 and sod-5 encode Cu/Zn SOD, sod-2 and sod-3 encode MnSOD, and sod-4 encodes ECSOD. These sod genes all have some degree of overlapping or compensatory functions, and coordinated gene expression regulation. For example, sod-5 expression is increased in a sod-1 mutant background (Yanase et al., 2009), while sod-1, sod-2, sod-3, and sod-4 expression are all increased in a sod-5 deletion mutant background (Table 4) (Yanase et al., 2020). In contrast, expression of sod-4 and sod-5 are decreased in a sod-2(gk257);sod-3(tm760) loss of function background (Back et al., 2010). An earlier study found no effect of sod loss of function mutations sod-1(tm776), sod-2(gk257), sod-3(tm760), sod-4(gk101), or sod-5(tm1146) on lifespan, although they had differential effects on development, fertility, and oxidative stress resistance (Doonan et al., 2008). A subsequent study found sod-1(tm776) or sod-5(tm1146) decrease lifespan compared to wild-type controls, and the double mutant sod-1(tm776); sod-5(tm1146) has a further shortened lifespan. Both insulin/ insulin-like growth factor and p38 MAPK signaling pathways coordinate sod gene regulation (Yanase et al., 2009, 2020). Under conditions of oxidative stress, loss of function sod-1(tm776) animals exhibit loss of glutamatergic but not cholinergic neurons (Table 4) (Baskoylu et al., 2018).
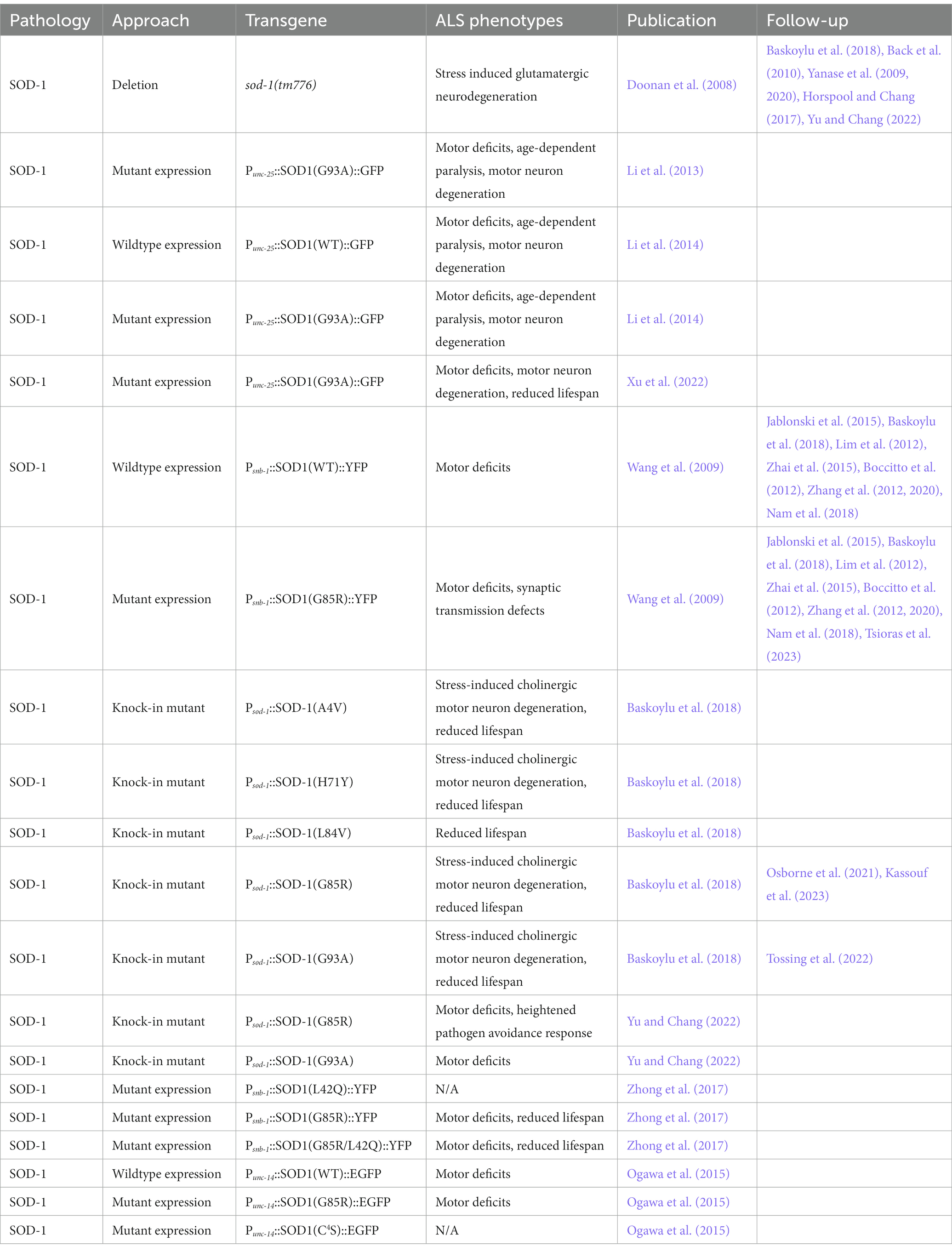
Table 4. C. elegans models of SOD1-driven ALS.
To create a model of human SOD1-driven ALS in C. elegans, fALS mutant SOD1(G85R) tagged with YFP was expressed pan-neuronally under the snb-1 promoter. This model induces motor deficits and motor neuron dysfunction, which are exacerbated by knockdown of protein turnover, chaperone, and protein modification genes (Table 4) (Wang et al., 2009). Another pan-neuronal SOD1 model expresses wild-type or mutant SOD1(G93A) tagged with GFP in GABAergic motor neurons under the unc-25 promoter (Li et al., 2013, 2014). These models exhibit several fALS phenotypes including SOD1 aggregation, motor deficits, age-dependent paralysis, axon guidance defects, and motor neuron degeneration, but no reduction in lifespan. Mutant SOD1(G93A) forms larger and more insoluble aggregates in the axons of motor neurons, correlating with more severe neurodegeneration compared to wild-type SOD1 (Table 4) (Li et al., 2014). Another model expressing SOD1(G93A) under the unc-25 promoter did have a reduced lifespan in addition to motor deficits and neurodegeneration (Table 4) (Xu et al., 2022).
C. elegans models overexpressing human mutant SOD1 preclude the evaluation of loss of function mechanisms of fALS variants. To overcome this, a series of human fALS SOD1 mutations, A4V, H71Y, L84V, G85R, or G93A, were introduced into conserved sites in the endogenous C. elegans sod-1 gene (Baskoylu et al., 2018). These strains exhibit glutamatergic and cholinergic neuron degeneration when exposed to oxidative stress, while GABAergic, dopaminergic and serotonergic neurons are relatively spared. When crossed with transgenic C. elegans expressing human wild-type SOD1::YFP in motor neurons, sod-1 A4V, H71Y, G85R, or G93A, but not L84V, increase inclusion formation of human SOD1::YFP. Loss of function sod-1(tm776) has no effect on SOD1::YFP aggregation, suggesting these mutations have a gain of function mechanism. sod-1 mutants H71Y, L84V, and G85R show similar stress-induced glutamatergic neuron dysfunction as sod-1 loss of function, indicating these mutations may have a loss of function component (Table 4) (Baskoylu et al., 2018). Additional single-copy knock-in models of SOD-1(G85R) and SOD-1(G93A) support G85R but not G93A as a loss of function fALS mutation (Yu and Chang, 2022). C. elegans lacking sod-1(tm776) or expressing SOD-1(G85R), but not SOD-1(G93A), exhibit a heightened pathogen avoidance response to the bacteria P. aeruginosa, by regulating the synaptic density of AMPA-type glutamate receptor GLR-1 in the ventral nerve cord (Table 4) (Horspool and Chang, 2017; Yu and Chang, 2022).
In humans, SOD1 shuttles between the nucleus and cytoplasm. In C. elegans, the fALS SOD1(G85R) or SOD1(L42Q/G85R) disrupt SOD1 nuclear localization. Expression of these mutations in neurons under the snb-1 promoter results in cytoplasmic SOD1 aggregates, motor deficits, and a reduced lifespan compared to wild-type SOD1 or SOD1(L42Q) (Table 4) (Zhong et al., 2017), supporting a role for SOD1 mislocalization in disease pathogenesis. In addition, SOD1’s cysteine residues are also necessary for SOD1 toxicity. The expression of human EGFP tagged SOD1 with four cystine residues mutated to serines (SOD1(C4S)) in neurons under the unc-14 promoter results in no motor deficits compared to wild-type C. elegans. This is in contrast to the expression of wild-type SOD1 or mutant SOD1(G85R) which display significant motor deficits (Table 4) (Ogawa et al., 2015), indicating the thiol/thiolate state of SOD1’s cystine residues modify fALS SOD1 neurotoxicity.
Forward genetic mutagenesis screens are a powerful method to identify novel mutations that modify a phenotype of interest. A mutagenesis screen in C. elegans expressing SOD1(G85R) under the pan-neuronal promoter snb-1 identified a combination of loss of function mutations in spr-5/LSD1 and ufd-2/UBE4B that suppress SOD1-induced neurotoxicity. spr-5(by134);ufd-2(tm1380) improves motor deficits and reduces SOD1(G85R) aggregation by upregulating proteasomal and autophagic degradation, partially through the FOXO transcription factor DAF-16 (Table 2) (Periz et al., 2015). A similar screen in the same model found mutations in lin-61, whose human homolog L3MBTL1 regulates p53 protein quality control pathways, rescues SOD1(G85R) motor deficits and aggregation (Table 2) (Lu et al., 2019). A single gain of function mutation in the C. elegans gene kcnl-2(rt462), which confers calcium sensitivity in an SK2 channel responsible for neuron excitability, also rescues motor deficits in mutant SOD1(G85R), but not wild-type SOD1, models (Nam et al., 2018). In addition to mutagenesis, a genome-wide RNAi screen identified knockdown of math-33 rescues motor deficits and SOD1(G85R)::YFP aggregation. The human homolog of MATH-33 is USP7, a ubiquitin protease (Table 2) (Zhang et al., 2020).
The disruption of proteins such as cytohesins and ADP-ribosylation factor (ARF) GTPases have also been implicated in SOD1-linked ALS. In the same model of mutant SOD1(G85R) neuron aggregation, knockdown of C. elegans cytohesin homologs grp-1 and efa-6 rescues motor defects (Table 2). Loss of function mutations in C. elegans arf-6(tm1447) and arf-1.2(ok796) do not alter SOD1(G85R) motor deficits, indicating cytohesins but not ARF GTPases are possible therapeutic targets critical for SOD1 neurotoxicity (Zhai et al., 2015). Another modifier of mutant SOD1(G85R) in this model is rad-23. RAD-23 and its human homolog are part of the endoplasmic-reticulum (ER) associated protein degradation pathway and function in substrate clearance and DNA damage repair. Loss of function mutations rad-23(tm3690) and rad-23(tm2595) rescue motor deficits and increase SOD1(G85R) solubility (Table 2) (Jablonski et al., 2015). More recently, a deletion allele of the C. elegans homolog of VCP, cdc-48.1(tm544), a fALS gene that regulates protein quality control and intracellular signaling, was found to exacerbate SOD1(G85R)-dependent motor deficits. Overexpression of cdc-48.1 under its endogenous promoter significantly rescues these motor deficits and SOD1(G85R) aggregation in C. elegans neurons, linking two fALS genes and further implicating disruptions to protein quality control in ALS (Tsioras et al., 2023).
To determine if fALS SOD1 mutations interact with other ALS-risk factor genes or fALS genes in disease, the C. elegans homologs of ALS/FTLD related genes figo-1(tm5202)/Fig 4, sqst-1(ok2892)/SQSTM1, ubql-1(tm1574)/UBQLN2, ptl-1(tm543)/MAPT, or daao-1(tm3673)/DAO, were deleted in a single-copy knock-in model of SOD-1(G85R) and wild-type control. Of these, only loss of figo-1(tm5202) reduces glutamatergic neuron degeneration by SOD-1(G85R). Therefore, loss of figo-1 partially compensates for the loss of sod-1 function, possibly through its indirect function in endosomal signaling and trafficking (Table 2) (Osborne et al., 2021). The expression SOD-1(G85R) also disrupts another conserved ALS-related pathway: stress granules. During oxidative stress, C. elegans with the SOD-1(G85R) mutation form larger stress granules as visualized by the G3BP1 homolog g3bp-1::GFP, which constitute the stress granule core. Loss of ulp-3(tm1287), whose human homolog NEDP1 regulates stress granule dynamics, reduces the formation of stress granules and rescues SOD-1(G85R) motor deficits, indicating stress granule formation may facilitate ALS-related neuron dysfunction (Table 2) (Kassouf et al., 2023).
Neuroprotective compounds have also been identified in SOD1 fALS models. For example, the diabetes drug metformin rescues motor deficits, reduces neurodegeneration, and extends lifespan in C. elegans expressing mutant SOD1(G93A) in GABAergic motor neurons. The beneficial effect of metformin is eliminated when C. elegans genes daf-16 and lgg-1 are knocked down, indicating metformin likely improves ALS phenotypes by upregulating autophagy (Xu et al., 2022).
Modeling ALS-linked SOD1 in C. elegans has provided valuable insights as to its function in disease as well as potential therapeutic methods. Future studies will continue to reveal mechanisms underlying SOD1’s toxicity and pathogenesis.
FUS
The DNA/RNA binding protein fused in sarcoma (FUS) has been identified as another ALS causative gene, with FUS mutations accounting for 1%–2% of sporadic ALS cases and 1%–5% of fALS. fALS FUS mutations are predominantly in the C-terminal low complexity, RGG-binding, or nuclear localization signal (NLS) domains. These mutations cause FUS mislocalization to the cytoplasm and aggregation, and are frequently associated with a younger onset of disease (Ishigaki and Sobue, 2018; Birsa et al., 2020). FUS functions as a DNA and RNA binding protein, and regulates DNA damage repair, mRNA splicing and transport, and stress granule formation. Evidence from disease models supports both FUS toxic loss- and gain-of-function mechanisms in disease, potentially through impaired DNA damage repair or RNA splicing defects (Shang and Huang, 2016).
The ortholog for FUS in C. elegans is FUST-1. Deletion of fust-1(tm4439) recapitulates several fALS phenotypes including progressive motor deficits and axon degeneration, as well as disrupts miRNA-mediated gene silencing and alters exon inclusion in mRNA splicing, suggesting a possible pathogenic loss of function mechanism (Table 5) (Therrien et al., 2016; Zhang et al., 2018; Taylor et al., 2023). In cholinergic motor neurons, FUST-1 localizes to the nucleus, as visualized by an N-terminal GFP tagged construct expressed under the unc-17 promoter. fALS mutations FUS(R524S) and FUS(P525L) may disrupt the nuclear localization FUS. When these fALS mutations are introduced at conserved sites in the C. elegans gene and expressed in cholinergic motor neurons under the unc-17 promoter with a fluorescent tag, GFP::FUST-1(R446S) and GFP::FUST-1(P447L), but not wild-type GFP::FUST-1, mislocalize to the cytoplasm, where they form aggregates following ER-stress induction (Baskoylu et al., 2022). These fust-1 mutants exhibit decreased survival when exposed to ER or oxidative stress, impaired autophagy, and neuromuscular junction (NMJ) dysfunction, but not lifespan reduction or neurodegeneration (Table 5). Human SQSTM1/p62 selects cargo for autophagic degradation and accumulates in FUS fALS patient neurons. A loss of function mutation in the C. elegans SQSTM1 homolog sqst-1(ok2892) rescues or partially rescues fust-1 mutation driven NMJ and motor deficits (Table 2) (Baskoylu et al., 2022).

Table 5. C. elegans models of FUS-driven ALS.
The expression of mutant human FUS(R521G, R522G, P525L, and C-terminal truncated FUS513 and FUS501), but not wild-type or FUS(R514G) and FUS(R521G), under the neuron-specific rgef-1 promoter also causes several fALS phenotypes including motor deficits, a reduced lifespan, cytoplasmic FUS mislocalization, and FUS aggregation (Murakami et al., 2012). In contrast, the expression of human wild-type FUS, mutant FUS(P525L), or FUS(FUS501) without the FUS N-terminal low complexity (LC) domain show reduced FUS aggregation, motor deficits, and lifespan defects. rgef-1-driven neuronal expression of the FUS LC domain alone or mutant FUS(S96del) is sufficient to increase FUS aggregation compared to wild-type FUS. Mutant FUS aggregates sequester ribonucleoprotein granule components, preventing new protein synthesis in axon terminals (Table 5) (Murakami et al., 2015). Compared to wild-type FUS, the cholinergic and GABAergic NMJs of C. elegans expressing neuronal truncated FUS501 are enriched for endosome-like organelles correlating with increased synaptic and vesicle dysfunction, highlighting another potential gain of function mechanism of mutant FUS (Markert et al., 2020). In the pan-neuronal mutant FUS(P525L) model, cooler growth temperatures, incubating C. elegans at 15°C rather than 20°C or 25°C, reduce the accumulation of FUS aggregates. Knockdown of psme-3/PSME3, a proteasome regulator, reverses this reduction (Table 2) (Lee et al., 2023).
Expression of human wild-type or mutant FUS(S57Δ) exclusively in GABAergic motor neurons under the unc-47 promoter also induces motor deficits, neurodegeneration, and the formation of FUS aggregates (Table 5) (Vaccaro et al., 2012b). Overexpression of mutant FUS(S57Δ) in GABAergic motor neurons triggers an innate immune response in these neurons and surrounding tissue. Deletion of innate immunity receptor TIR-1, by tir-1(qd4), rescues motor deficits and neurodegeneration without altering FUS levels, suggesting the innate immune response contributions to FUS neurotoxicity (Table 2) (Veriepe et al., 2015). The expression of a single-copy mutant FUS(S57Δ) under the unc-47 promoter is sufficient to induce age-dependent motor deficits and neurodegeneration. A single-copy wild-type FUS does not exhibit fALS-associated phenotypes (Table 5) (Labarre et al., 2021). The axonal degeneration and motor deficits in this single-copy FUS(S57Δ) model are dependent on several genes in the DLK-1/MAP3K12 axonal regeneration pathway (Tossing et al., 2022). Knockdown of fsn-1/FBXO45, rpm-1/MYCBP2, klp-7/KIF2C, or dlk-1/MAP3K12 improve a progressive paralysis phenotype, while knockdown of fsn-1/FBXO45, rpm-1/MYCBP2, or parp-2/PARP2 decrease axon degeneration. A loss of function mutation in parp-1(ok988)/PARP1 similarly rescues paralysis and degeneration (Table 2). In fact, a screen of PARP inhibitors identified several compounds that reduce FUS-driven axon degeneration. The PARP inhibitors Olaparib, Veliparib, and 3-AB also partially reduce axon degeneration in mutant SOD1(G93A) and TDP-43(A315T) models (Tossing et al., 2022). In addition to screening drugs, C. elegans can also model dietary interventions for ALS. For example, fatty acids derived from the bacteria Lacticaseibacillus rhamnosus HA-114 reduce aberrant lipid accumulation, motor deficits, and neurodegeneration in C. elegans expressing mutant FUS(S57Δ) or TDP-43(A315T) in GABAergic motor neurons. This rescue acts through genes involved in lipid homeostasis and mitochondrial β-oxidation, suggesting both pathways are critical to ALS (Labarre et al., 2022).
C. elegans models of FUS have identified mechanisms contributing to FUS-driven ALS including FUS mislocalization, autophagic disruption, and protein synthesis dependent synaptic dysfunction. In addition, work using C. elegans models have identified a number of protective genes and compounds.
Other ALS/FTLD-TDP-associated genes and risk factors
While mutations in TARDBP, C9orf72, SOD1, and FUS are the most common causes of fALS, there are more than 45 fALS or disease modifying risk factor genes (Smukowski et al., 2022). So far, 14 have been explored in C. elegans models. Briefly, evidence from C. elegans suggest mutations in CHCHD10, ALS2, DCTN1, ELP3, TUBA4A, and CAV1 are loss of function mutations, which could be rescued by supplementation with the wild-type protein (Parker et al., 2007; Pan et al., 2011; Nawa et al., 2012; Woo et al., 2017; Kawamura and Maruyama, 2019; Soh et al., 2020; Ryan et al., 2021; Ryan and Hart, 2021). On the other hand, mutations in UBQLN2, ATXN3, and TIA1 indicate a gain of function mechanism in C. elegans models of ALS (Fardghassemi et al., 2017; Andrusiak et al., 2019; Saxton and Kraemer, 2021). Evidence exists in C. elegans that both gain and loss of function mechanisms could be at play in neurodegeneration by fALS gene HnRNPA2B1, KIF5A, and VABP (Han et al., 2013; Zhang et al., 2017). Finally, the role of GRN and RAB38 mutations in FTLD have been studied in C. elegans models (Grill et al., 2007; Salazar et al., 2015; Doyle et al., 2021).
Mutations in the CHCHD10 gene cause fALS in several families. CHCHD10 encodes a mitochondrial intermembrane protein involved in mitochondria organization (Bannwarth et al., 2014; Johnson et al., 2014; Zhang et al., 2015). Loss of C. elegans CHCHD10 homolog, har-1(gk3124), leads to motor deficits, a reduction in lifespan, and reduced mitochondrial health. The general expression of human CHCHD10, but not mutant CHCHD10(R15L) or CHCHD10(S59L), under the eef-1A.1 promoter rescues these defects, suggesting CHCHD10 fALS mutations are loss of function mutations (Table 6) (Woo et al., 2017).

Table 6. C. elegans models of other ALS/FTLD-TDP-associated genes and risk factors.
The ALS2 gene encodes the Rho guanine nucleotide exchange factor Alsin, which regulates the GTPase Rab5, endosome trafficking, and neurite outgrowth. Mutations in ALS2 cause a juvenile onset form of fALS (Yang et al., 2001; Mintchev et al., 2009; Sheerin et al., 2014). While C. elegans do not have a homolog of the ALS2 gene, they do have a homolog of BTBD10, an Akt kinase activator. Overexpression of BTBD10, which is reduced in ALS motor neurons, can overcome ALS2 fALS mutations by preventing Akt3 dephosphorylation (Table 6) (Nawa et al., 2008, 2012; Kanekura et al., 2004, 2005). In C. elegans, loss of function mutations in BTBD10 homolog btbd-10(tm3335) and btbd-10(tm3607) result in motor deficits and degeneration of touch-receptor and GABAergic motor neurons, creating a possible unique model of juvenile ALS (Table 6) (Nawa et al., 2012).
Mutations in the gene DCTN1 can cause fALS. The DCTN1 protein, dynactin-1, activates the microtubule-binding motor protein dynein (Gill et al., 1991; Puls et al., 2003; Munch et al., 2004). In C. elegans, the knock-down of DCTN1 homolog dnc-1 in motor neurons by the expression of an RNAi construct under the acr-2 promoter results in motor deficits, axonal degeneration, defects in axonal transport, and the abnormal accumulation of autophagosomes (Table 6) (Ikenaka et al., 2013). A drug screen in this model identified 12 neuroprotective compounds, two of which (ALS-approved drugs riluzole and nifedipine) also rescue motor deficits in a C. elegans model of TDP-43 toxicity (Ikenaka et al., 2019).
Mutations in the elongator acetyl transferase complex subunit 3 (ELP3) are associated with sALS in several genome wide associations screens and decrease survival in cases of C9orf72 fALS (Simpson et al., 2009; Kwee et al., 2012). Part of the elongation complex for RNA polymerase II, ELP3 is known to acetylate alpha-tubulin and modify tRNA uridines, regulating mTORC2 activation and other biological pathways (Creppe et al., 2009; Chen et al., 2022). Deletion of the C. elegans homolog of ELP3, elpc-3(ok2452), results in progressive motor deficits (Kawamura and Maruyama, 2019). A second deletion allele, elpc-3(tm3120), also disrupts experience-dependent learning, neuropeptide signaling, and tRNA modification, suggesting ELP3 loss of function contributes to ALS (Chen et al., 2009). In a cell model of SOD1 ALS, ELP3 depletion increased SOD1 aggregation (Bento-Abreu et al., 2018).
Mutations in the tubulin gene tubulin alpha 4a (TUBA4A) have been identified as likely pathogenic in several rare cases of fALS and FTLD-TDP (Smith et al., 2014; Mol et al., 2021). TUBA4A is a tubulin alpha chain, one of the basic building blocks of microtubules and the cytoskeleton (Ganne et al., 2023). A mutation in the C. elegans TUBA4A homolog, mec-12(e1605), leads to progressive neuronal dysfunction in touch receptor neurons but does not alter lifespan (Pan et al., 2011). Structural and biological studies suggest TUBA4A mutations have both a loss of function component, disrupting microtubule polymerization, as well as a gain of function component, where mutant TUBA4A aggregates and disrupts microtubule dynamics in fALS (Smith et al., 2014; Ganne et al., 2023).
Mutations in the enhancer region of the CAV1 gene are a risk factor for the development of ALS. These mutations decrease the expression of CAV1 in patient-derived neurons, which encodes for a critical plasma membrane structural protein named caveolin-1 (Cooper-Knock et al., 2020). In C. elegans, the deletion of CAV1 homolog cav-1(ok2089) results in increased sensitivity to levamisole, suggesting defects in neuromuscular junction or muscle function (Table 6). Depletion of cav-1 by RNAi also worsens a temperature-sensitive motor deficit in dyn-1(ky51) mutants. Furthermore, mutations in cav-1 alter the distribution of the dyn-1 protein dynamin, a regulator of vesicle trafficking (Parker et al., 2007). Together these results suggest CAV1 acts in ALS by disrupting motor neuron function at the synapse.
Mutations in the Ubiquilin-2 (UBQLN2) gene are responsible for fALS cases in several families (Deng et al., 2011; Williams et al., 2012; Gellera et al., 2013; Fahed et al., 2014). UBQLN2 shuttles ubiquitinated proteins to the proteasome for degradation. Evidence suggests fALS mutations in UBQLN2 impair autophagy, mitochondrial function, and the delivery of UBQLN2 substrates to the proteasome (Lin et al., 2022). In C. elegans, the neuronal expression of wild-type human UBQLN2 and mutant UBQLN2(P506T) and UBQLN2(P497H) under the rgef-1 promoter results in motor deficits and degeneration of GABAergic motor neurons. C. elegans tdp-1 is not required for these phenotypes (Table 6). Co-expression of wild-type and mutant UBQLN2 with wild-type human TDP-43 exacerbates motor deficits, neurodegeneration, and increases the accumulation of TDP-43 and UBQLN2 (Saxton and Kraemer, 2021). A loss of function mutation in the UBQLN2 homolog ubql-1(tm1574) has no effect on the motor performance of a C. elegans model expressing mutant TDP-43 or on neurodegeneration in a C. elegans model expressing SOD-1(G85R) (Jablonski et al., 2015; Osborne et al., 2021).
Several genome-wide association studies in ALS patients have identified mutations in ATXN3 that increase the risk of developing ALS. ATXN3 encodes the protein Ataxin-3, an important player in protein quality control during stress. Expansions in a CAG repeat region in ATXN3 cause Machado-Joseph disease, another progressive motor disorder. ALS-associated risk mutations in ATXN3 are also thought to impact this region (Nakamura et al., 2020; Li et al., 2021; Humphrey et al., 2023). The expression of human ATXN3 with 10 CAG repeats, below the threshold to cause Machado-Joseph disease, in C. elegans GABAergic neurons under the unc-47 promoter results in motor deficits and reduced lifespan, but not neurodegeneration (Table 6) (Fardghassemi et al., 2017). The C. elegans homolog atx-3 regulates autophagy, and when deleted, enhances the stress response to heat and extends lifespan of cdc-48.1/VCP mutants (Kuhlbrodt et al., 2011; Rodrigues et al., 2011; Herzog et al., 2020).
Mutations in TIA1 have been identified in several cases of ALS (Hirsch-Reinshagen et al., 2017; Mackenzie et al., 2017). TIA1 encodes the protein TIA1 which regulates various aspects of RNA metabolism and stress granules dynamics (Rayman and Kandel, 2017), and ALS-associated mutations may enhance TIA1 phase separation and aggregation in disease (Mackenzie et al., 2017; Sekiyama et al., 2022). In C. elegans, overexpression of the TIA1 homolog, tiar-2, in mechanosensory neurons under the mec-4 promoter inhibits axon regeneration following injury. TIAR-2 phase separates into dynamic granules in C. elegans neurons (Table 6). Granule formation is required for TIAR-2’s suppression of axon regeneration and is regulated by phosphorylation of TIAR-2’s prion-like domain (Andrusiak et al., 2019).
Mutations in the HnRNPA2B1 and HnRNPA1 genes are responsible for fALS in several families (Kim et al., 2013). The HnRNPA2B1 and HnRNPA1 genes express several protein isoforms, including HNRNPA2, HNRNPA1, and HNRNPA3, which control mRNA splicing, trafficking, stability, and translation (Hutchison et al., 2002; Fahling et al., 2006; Kosturko et al., 2006; Clower et al., 2010). A loss of function mutation in the C. elegans HnRNPA2B1 and HnRNPA1 homolog gene hrpa-1(tm781) results in glutamatergic neuron degeneration (Ryan et al., 2021; Ryan and Hart, 2021). The expression of a chimeric protein of the C. elegans HRPA-1 and the human low complexity domain of mutant hnRNPA2(D290V) in glutamatergic neurons under the mec-4 promoter results in stress-induced neurodegeneration of glutamatergic neurons (Table 6). Neurodegeneration is rescued by loss of tdp-1. The expression of a chimera of HRPA-1 and the wild-type human low complexity domain does not result in neurodegeneration. Post translational modification of hnRNPA2 can influence its phase separation and aggregation, impacting ALS progression (reviewed in Farina et al., 2021). In C. elegans, the co-expression of a constitutively active Fyn kinase, which phosphorylates hnRNPA2, and the chimera HRPA-1 hnRNPA2(D290V) protein reduces stress-induced glutamatergic neuron neurodegeneration (Ryan et al., 2021). This suggests hnRNPA2 phosphorylation may be a therapeutic target in fALS.
Mutations in the C-terminal cargo-binding domain of the protein KIF5A cause fALS in several families (Brenner et al., 2018; Nicolas et al., 2018; Saez-Atienzar et al., 2020). KIF5A, or kinesin-1, is a microtubule motor protein involved in intracellular trafficking and is necessary for neuronal development and function (Wang and Brown, 2010; Nakajima et al., 2012). In C. elegans, loss of KIF5A homolog unc-116(e2310) results in progressive motor deficits and morphological defects in cholinergic motor neurons, further suggesting defects in axonal transport are key in ALS (Table 6) (Soh et al., 2020). The expression of a human fALS KIF5A splicing variant, KIF5A(∆exon27), in C. elegans touch-receptor neurons under the mec-7 promoter results in morphological defects and degeneration of touch receptor neurons. C. elegans expressing wildtype KIF5A do not show any defects, indicating KIF5A(∆exon27) has a toxic gain of function (Table 6) (Nakano et al., 2022).
A point mutation in the VAPB gene causes a late-onset form of fALS in several families (Nishimura et al., 2004; Millecamps et al., 2010). VAPB is found in the ER-membrane and regulates Golgi transport, neurotransmitter release, and calcium homeostasis (Skehel et al., 1995; Soussan et al., 1999; Amarilio et al., 2005; De Vos et al., 2012; Morotz et al., 2012). RNAi knock-down of the C. elegans homolog vpr-1 results in stress-inducible motor deficits and stress-inducible cholinergic motor neuron degeneration, suggesting VAPB loss of function may drive fALS (Table 6). PIK-93, a synthetic PI4K inhibitor, partially prevents these stress-induced motor deficits and motor neuron degeneration when delivered at a concentration of 250 nM via liquid culture. The expression of both human wild-type VAPB and mutant VAPB(P56S) in C. elegans cholinergic motor neurons under the unc-4 promoter also results in age-dependent motor deficits and progressive degeneration of cholinergic motor neurons (Table 6) (Zhang et al., 2017).
Mutations in the progranulin gene, whose protein regulates a diverse array of biological functions from cell growth and survival to repair and inflammation, cause frontotemporal lobar degeneration (FTLD) in several families (Baker et al., 2006; Cruts et al., 2006; Moore et al., 2020). Progranulin is cleaved into individual granulin peptides, a process that is disrupted in FTLD. A reduction in progranulin protein levels overall is also thought to drive disease by disrupting endolysosomes, lysosomal homeostasis, inflammation, and other pathways (Ward and Miller, 2011; Mohan et al., 2021; Amin et al., 2022). Deletion of the C. elegans progranulin homolog, pgrn-1, results in progressive motor deficits, a reduction in lifespan, and lysosomal and autophagic defects, indicating progranulin loss of function may contribute to disease (Table 6). A deletion of sphk-1(ok1097), whose human homolog SPHK1 regulates sphingolipid metabolism, restores autophagosome and autolysosome numbers in pgrn-1 mutants. RNAi knockdown of cgt-3/UGCG and asah-1/ASAH1, also involved in sphingolipid metabolism, rescue prgn-1 mutant motor deficits. A screen of molecular compounds in this model uncovered two drugs, Rivastigmine and Rottlerin, which rescue pgrn-1 mutant motor and autophagy deficits and had a prolonged positive effect in a cell culture model (Doyle et al., 2021). In another C. elegans model expressing human TDP-43, heterozygous deletion of pgrn-1(tm985) worsens motor deficits. Additionally, the selective expression of progranulin cleavage products, the granulin peptides 2 and 3, but not 1, under the pgrn-1 promoter, exacerbate motor deficits, reduce lifespan, and increase TDP-43 accumulation modeling possible interactions in FTLD-TDP (Table 6) (Salazar et al., 2015). Further studies of granulin peptides found they localize to endolysosomes, disrupt lysosomal morphology and lysosomal protease activity, and their expression inhibits ER stress resistance and impairs organismal fitness. Interestingly age and organismal stress drives increased accumulation of granulins (Butler et al., 2019). Granulins 1, 2, and 3 may have distinct phenotypic outcomes, with granulin 3 in particular impairing organismal fitness and preventing clearance of TDP-43 (Butler et al., 2019; Wang et al., 2023a).
In genome-wide association studies, mutations associated with RAB38 are linked with FTLD, specifically in patients with personality and behavioral changes (Yang et al., 2017; Reus et al., 2021). These mutations are thought to increase the risk of FTLD by altering the expression of RAB38, which encodes the transmembrane protein RAB38 that regulates lysosomal trafficking (Bultema et al., 2012; Ferrari et al., 2014). In C. elegans, loss of function mutations in the RAB38 homolog, glo-1, disrupt axon termination, likely resulting in neuron dysfunction (Table 6) (Grill et al., 2007).
Conclusions and outlook
During the last 10 years of progress, C. elegans models of ALS and FTLD-TDP have been used to identify conserved biological pathways in disease, characterize the impact of disease-causing mutations, determine the role of post-translational modifications in disease progression, uncover novel suppressors of neurodegeneration, and screen drug libraries and novel chemical compounds in the search for new therapeutic treatments. These models use diverse genetic and transgenic strategies, but recapitulate aspects of human disease, including protein or RNA aggregation, motor deficits and neurodegeneration. These characteristics have allowed for the identification of conserved, translationally relevant, and potentially targetable biological pathways in disease.
Overall, C. elegans models of ALS and FTLD-TDP have fundamentally advanced our understanding of the biology driving neurodegeneration and are important tools for future studies. They will continue to be a valuable asset in characterizing the molecular consequences of novel disease-associated mutations in known and unknown genes. Compared to cell culture and mice, C. elegans have robust behavioral, neurodegenerative, and lifespan phenotypes and are cost-effective for high throughput screens. Their short generation time enables aging studies not feasible in other systems. Recognition of limitations of the model system are also critical for interpreting results. While C. elegans lack a conserved neuroinflammatory pathway and canonical glia, both important in ALS pathology, previous work in C. elegans has been able to identify a key role for the innate immune response in ALS models. Additionally, glia-like cells in C. elegans have been shown to regulate proteostasis and stress responses in neurons and could be studied in ALS models (Veriepe et al., 2015; Bar-Ziv et al., 2023; Wang et al., 2023b). For abnormal protein conformations and inclusions that slowly develop over time, the short lifespan of C. elegans may not be long enough to observe mature pathological species. However, C. elegans models of ALS and FTLD-TDP will continue to be leveraged to screen novel neuroprotective compounds, identify novel genetic targets for therapeutic intervention, and identify key biological pathways necessary for neurodegeneration which can be further explored in human cells, vertebrate animal models, and patient tissue. C. elegans models enable evaluation of the impacts of stress, environment, and chemical insults on disease progression. Robust tools for genetic manipulation will continue to drive the development of new and refined C. elegans systems that will continue to contribute to understanding the biology of ALS.
Author contributions
RE: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. JS: Writing – original draft, Writing – review & editing. BK: Writing – review & editing, Funding acquisition, Project administration, Resources, Supervision, Validation. NL: Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing, Conceptualization, Visualization, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the Department of Veterans Affairs [I01BX002619 to BK and I01BX004044 to NL] and National Institutes of Health [F31AG082391 and T32GM136534 to RE, RF1AG055474 and R01NS064131 to BK, and R01AG066729 to NL].
Acknowledgments
We thank WormBase for essential C. elegans model organism information. Some strains discussed in this review are available for order from the Caenorhabditis Genetics Center, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440), the international C. elegans Gene Knockout Consortium, and the National Bioresource Project (Japan). Figure 1 was created with BioRender.com. This material is the result of work supported with resources and the use of facilities at the VA Puget Sound Health Care System.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aggad, D., Veriepe, J., Tauffenberger, A., and Parker, J. A. (2014). TDP-43 toxicity proceeds via calcium dysregulation and necrosis in aging Caenorhabditis elegans motor neurons. J. Neurosci. 34, 12093–12103. doi: 10.1523/JNEUROSCI.2495-13.2014
Alexander-Floyd, J., Haroon, S., Ying, M., Entezari, A. A., Jaeger, C., Vermulst, M., et al. (2020). Unexpected cell type-dependent effects of autophagy on polyglutamine aggregation revealed by natural genetic variation in C. elegans. BMC Biol. 18:18. doi: 10.1186/s12915-020-0750-5
Alzheimer’s Disease Neuroimaging InitiativeIacoangeli, A., al Khleifat, A., Jones, A. R., Sproviero, W., Shatunov, A., et al. (2019). C9orf72 intermediate expansions of 24-30 repeats are associated with ALS. Acta Neuropathol. Commun. 7:115. doi: 10.1186/s40478-019-0724-4
Amarilio, R., Ramachandran, S., Sabanay, H., and Lev, S. (2005). Differential regulation of endoplasmic reticulum structure through VAP-Nir protein interaction. J. Biol. Chem. 280, 5934–5944. doi: 10.1074/jbc.M409566200
Amin, S., Carling, G., and Gan, L. (2022). New insights and therapeutic opportunities for progranulin-deficient frontotemporal dementia. Curr. Opin. Neurobiol. 72, 131–139. doi: 10.1016/j.conb.2021.10.001
Andrusiak, M. G., Sharifnia, P., Lyu, X., Wang, Z., Dickey, A. M., Wu, Z., et al. (2019). Inhibition of axon regeneration by liquid-like TIAR-2 granules. Neuron 104, 290–304.e8. doi: 10.1016/j.neuron.2019.07.004
Apfeld, J., and Alper, S. (2018). What can we learn about human disease from the Nematode C. elegans? Methods Mol. Biol. 1706, 53–75. doi: 10.1007/978-1-4939-7471-9_4
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi: 10.1016/j.bbrc.2006.10.093
Ash, P. E., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., Dejesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Ash, P. E., Zhang, Y. J., Roberts, C. M., Saldi, T., Hutter, H., Buratti, E., et al. (2010). Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 19, 3206–3218. doi: 10.1093/hmg/ddq230
Back, P., Matthijssens, F., Vlaeminck, C., Braeckman, B. P., and Vanfleteren, J. R. (2010). Effects of sod gene overexpression and deletion mutation on the expression profiles of reporter genes of major detoxification pathways in Caenorhabditis elegans. Exp. Gerontol. 45, 603–610. doi: 10.1016/j.exger.2010.01.014
Baker, M., Mackenzie, I. R., Pickering-Brown, S. M., Gass, J., Rademakers, R., Lindholm, C., et al. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. doi: 10.1038/nature05016
Balendra, R., and Isaacs, A. M. (2018). C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558. doi: 10.1038/s41582-018-0047-2
Bannwarth, S., Ait-el-Mkadem, S., Chaussenot, A., Genin, E. C., Lacas-Gervais, S., Fragaki, K., et al. (2014). A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137, 2329–2345. doi: 10.1093/brain/awu138
Barmada, S. J. (2015). Linking RNA dysfunction and neurodegeneration in amyotrophic lateral sclerosis. Neurotherapeutics 12, 340–351. doi: 10.1007/s13311-015-0340-3
Bar-Ziv, R., Dutta, N., Hruby, A., Sukarto, E., Averbukh, M., Alcala, A., et al. (2023). Glial-derived mitochondrial signals affect neuronal proteostasis and aging. Sci. Adv. 9:eadi1411. doi: 10.1126/sciadv.adi1411
Baskoylu, S. N., Chapkis, N., Unsal, B., Lins, J., Schuch, K., Simon, J., et al. (2022). Disrupted autophagy and neuronal dysfunction in C. elegans knockin models of FUS amyotrophic lateral sclerosis. Cell Rep. 38:110195. doi: 10.1016/j.celrep.2021.110195
Baskoylu, S. N., Yersak, J., O’Hern, P., Grosser, S., Simon, J., Kim, S., et al. (2018). Single copy/knock-in models of ALS SOD1 in C. elegans suggest loss and gain of function have different contributions to cholinergic and glutamatergic neurodegeneration. PLoS Genet. 14:e1007682. doi: 10.1371/journal.pgen.1007682
Benbow, S. J., Strovas, T. J., Darvas, M., Saxton, A., and Kraemer, B. C. (2020). Synergistic toxicity between tau and amyloid drives neuronal dysfunction and neurodegeneration in transgenic C. elegans. Hum. Mol. Genet. 29, 495–505. doi: 10.1093/hmg/ddz319
Bento-Abreu, A., Jager, G., Swinnen, B., Rue, L., Hendrickx, S., Jones, A., et al. (2018). Elongator subunit 3 (ELP3) modifies ALS through tRNA modification. Hum. Mol. Genet. 27, 1276–1289. doi: 10.1093/hmg/ddy043
Birsa, N., Bentham, M. P., and Fratta, P. (2020). Cytoplasmic functions of TDP-43 and FUS and their role in ALS. Semin. Cell Dev. Biol. 99, 193–201. doi: 10.1016/j.semcdb.2019.05.023
Boccitto, M., Lamitina, T., and Kalb, R. G. (2012). Daf-2 signaling modifies mutant SOD1 toxicity in C. elegans. PLoS One 7:e33494. doi: 10.1371/journal.pone.0033494
Bose, P., Tremblay, E., Maios, C., Narasimhan, V., Armstrong, G. A. B., Liao, M., et al. (2019). The novel small molecule TRVA242 stabilizes neuromuscular junction defects in multiple animal models of amyotrophic lateral sclerosis. Neurotherapeutics 16, 1149–1166. doi: 10.1007/s13311-019-00765-w
Boyd, J. D., Lee, P., Feiler, M. S., Zauur, N., Liu, M., Concannon, J., et al. (2014). A high-content screen identifies novel compounds that inhibit stress-induced TDP-43 cellular aggregation and associated cytotoxicity. J. Biomol. Screen. 19, 44–56. doi: 10.1177/1087057113501553
Brenner, S. (1973). The genetics of behaviour. Br. Med. Bull. 29, 269–271. doi: 10.1093/oxfordjournals.bmb.a071019
Brenner, D., Yilmaz, R., Muller, K., Grehl, T., Petri, S., Meyer, T., et al. (2018). Hot-spot Kif5A mutations cause familial ALS. Brain 141, 688–697. doi: 10.1093/brain/awx370
Bultema, J. J., Ambrosio, A. L., Burek, C. L., and Di Pietro, S. M. (2012). BLOC-2, AP-3, and AP-1 proteins function in concert with Rab38 and Rab32 proteins to mediate protein trafficking to lysosome-related organelles. J. Biol. Chem. 287, 19550–19563. doi: 10.1074/jbc.M112.351908
Burberry, A., Wells, M. F., Limone, F., Couto, A., Smith, K. S., Keaney, J., et al. (2020). C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature 582, 89–94. doi: 10.1038/s41586-020-2288-7
Butler, V. J., Gao, F., Corrales, C. I., Cortopassi, W. A., Caballero, B., Vohra, M., et al. (2019). Age- and stress-associated C. elegans granulins impair lysosomal function and induce a compensatory HLH-30/TFEB transcriptional response. PLoS Genet. 15:e1008295. doi: 10.1371/journal.pgen.1008295
Caldwell, K. A., Willicott, C. W., and Caldwell, G. A. (2020). Modeling neurodegeneration in Caenorhabditiselegans. Dis. Model. Mech. 13:dmm046110. doi: 10.1242/dmm.046110
Chen, D., Nemazanyy, I., Peulen, O., Shostak, K., Xu, X., Tang, S. C., et al. (2022). Elp3-mediated codon-dependent translation promotes mTORC2 activation and regulates macrophage polarization. EMBO J. 41:e109353. doi: 10.15252/embj.2021109353
Chen, C., Tuck, S., and Bystrom, A. S. (2009). Defects in tRNA modification associated with neurological and developmental dysfunctions in Caenorhabditis elegans elongator mutants. PLoS Genet. 5:e1000561. doi: 10.1371/journal.pgen.1000561
Clower, C. V., Chatterjee, D., Wang, Z., Cantley, L. C., Vander Heiden, M. G., and Krainer, A. R. (2010). The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc. Natl. Acad. Sci. U. S. A. 107, 1894–1899. doi: 10.1073/pnas.0914845107
Cook, S. J., Jarrell, T. A., Brittin, C. A., Wang, Y., Bloniarz, A. E., Yakovlev, M. A., et al. (2019). Whole-animal connectomes of both Caenorhabditis elegans sexes. Nature 571, 63–71. doi: 10.1038/s41586-019-1352-7
Cooper-Knock, J., Zhang, S., Kenna, K. P., Moll, T., Franklin, J. P., Allen, S., et al. (2020). Rare variant burden analysis within enhancers identifies CAV1 as an ALS risk gene. Cell Rep. 33:108456. doi: 10.1016/j.celrep.2020.108456
Corrionero, A., and Horvitz, H. R. (2018). A C9orf72 ALS/FTD Ortholog acts in Endolysosomal degradation and lysosomal homeostasis. Curr. Biol. 28, 1522–1535.e5. doi: 10.1016/j.cub.2018.03.063
Creppe, C., Malinouskaya, L., Volvert, M. L., Gillard, M., Close, P., Malaise, O., et al. (2009). Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cells 136, 551–564. doi: 10.1016/j.cell.2008.11.043
Cruts, M., Gijselinck, I., Van Der Zee, J., Engelborghs, S., Wils, H., Pirici, D., et al. (2006). Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924. doi: 10.1038/nature05017
Currey, H. N., Malinkevich, A., Melquist, P., and Liachko, N. F. (2020). Arena-based activity profiling of tau and TDP-43 transgenic C. elegans. MicroPubl. Biol. 2020. doi: 10.17912/micropub.biology.000278
De Vos, K. J., Morotz, G. M., Stoica, R., Tudor, E. L., Lau, K. F., Ackerley, S., et al. (2012). VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311. doi: 10.1093/hmg/ddr559
Dejesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Deng, H. X., Chen, W., Hong, S. T., Boycott, K. M., Gorrie, G. H., Siddique, N., et al. (2011). Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211–215. doi: 10.1038/nature10353
Doonan, R., Mcelwee, J. J., Matthijssens, F., Walker, G. A., Houthoofd, K., Back, P., et al. (2008). Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 22, 3236–3241. doi: 10.1101/gad.504808
Doyle, J. J., Maios, C., Vrancx, C., Duhaime, S., Chitramuthu, B., Bennett, H. P. J., et al. (2021). Chemical and genetic rescue of in vivo progranulin-deficient lysosomal and autophagic defects. Proc. Natl. Acad. Sci. U. S. A. 118:e2022115118. doi: 10.1073/pnas.2022115118
Eck, R. J., Kraemer, B. C., and Liachko, N. F. (2021). Regulation of TDP-43 phosphorylation in aging and disease. Geroscience 43, 1605–1614. doi: 10.1007/s11357-021-00383-5
Fahed, A. C., Mcdonough, B., Gouvion, C. M., Newell, K. L., Dure, L. S., Bebin, M., et al. (2014). UBQLN2 mutation causing heterogeneous X-linked dominant neurodegeneration. Ann. Neurol. 75, 793–798. doi: 10.1002/ana.24164
Fahling, M., Mrowka, R., Steege, A., Martinka, P., Persson, P. B., and Thiele, B. J. (2006). Heterogeneous nuclear ribonucleoprotein-A2/B1 modulate collagen prolyl 4-hydroxylase, alpha (I) mRNA stability. J. Biol. Chem. 281, 9279–9286. doi: 10.1074/jbc.M510925200
Fardghassemi, Y., Tauffenberger, A., Gosselin, S., and Parker, J. A. (2017). Rescue of ATXN3 neuronal toxicity in Caenorhabditiselegans by chemical modification of endoplasmic reticulum stress. Dis. Model. Mech. 10, 1465–1480. doi: 10.1242/dmm.029736
Farg, M. A., Sundaramoorthy, V., Sultana, J. M., Yang, S., Atkinson, R. A., Levina, V., et al. (2014). C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 23, 3579–3595. doi: 10.1093/hmg/ddu068
Farina, S., Esposito, F., Battistoni, M., Biamonti, G., and Francia, S. (2021). Post-translational modifications modulate Proteinopathies of TDP-43, FUS and hnRNP-A/B in amyotrophic lateral sclerosis. Front. Mol. Biosci. 8:693325. doi: 10.3389/fmolb.2021.693325
Ferrari, R., Hernandez, D. G., Nalls, M. A., Rohrer, J. D., Ramasamy, A., Kwok, J. B., et al. (2014). Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 13, 686–699. doi: 10.1016/S1474-4422(14)70065-1
Gallrein, C., Iburg, M., Michelberger, T., Kocak, A., Puchkov, D., Liu, F., et al. (2021). Novel amyloid-beta pathology C. elegans model reveals distinct neurons as seeds of pathogenicity. Prog. Neurobiol. 198:101907. doi: 10.1016/j.pneurobio.2020.101907
Ganne, A., Balasubramaniam, M., Ayyadevara, H., Kiaei, L., Shmookler Reis, R. J., Varughese, K. I., et al. (2023). In silico analysis of TUBA4A mutations in amyotrophic lateral sclerosis to define mechanisms of microtubule disintegration. Sci. Rep. 13:2096. doi: 10.1038/s41598-023-28381-x
Gao, J., Wang, L., Huntley, M. L., Perry, G., and Wang, X. (2018). Pathomechanisms of TDP-43 in neurodegeneration. J. Neurochem. 146, 7–20. doi: 10.1111/jnc.14327
Gellera, C., Tiloca, C., Del Bo, R., Corrado, L., Pensato, V., Agostini, J., et al. (2013). Ubiquilin 2 mutations in Italian patients with amyotrophic lateral sclerosis and frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 84, 183–187. doi: 10.1136/jnnp-2012-303433
Gendron, T. F., Bieniek, K. F., Zhang, Y. J., Jansen-West, K., Ash, P. E., Caulfield, T., et al. (2013). Antisense transcripts of the expanded C9orf72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126, 829–844. doi: 10.1007/s00401-013-1192-8
Gill, S. R., Schroer, T. A., Szilak, I., Steuer, E. R., Sheetz, M. P., and Cleveland, D. W. (1991). Dynactin, a conserved, ubiquitously expressed component of an activator of vesicle motility mediated by cytoplasmic dynein. J. Cell Biol. 115, 1639–1650. doi: 10.1083/jcb.115.6.1639
Giunti, S., Andersen, N., Rayes, D., and De Rosa, M. J. (2021). Drug discovery: insights from the invertebrate Caenorhabditis elegans. Pharmacol. Res. Perspect. 9:e00721. doi: 10.1002/prp2.721
Grill, B., Bienvenut, W. V., Brown, H. M., Ackley, B. D., Quadroni, M., and Jin, Y. (2007). C. elegans RPM-1 regulates axon termination and synaptogenesis through the Rab GEF GLO-4 and the Rab GTPase GLO-1. Neuron 55, 587–601. doi: 10.1016/j.neuron.2007.07.009
Haeusler, A. R., Donnelly, C. J., Periz, G., Simko, E. A., Shaw, P. G., Kim, M. S., et al. (2014). C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200. doi: 10.1038/nature13124
Han, S. M., El Oussini, H., Scekic-Zahirovic, J., Vibbert, J., Cottee, P., Prasain, J. K., et al. (2013). VAPB/ALS8 MSP ligands regulate striated muscle energy metabolism critical for adult survival in caenorhabditis elegans. PLoS Genet. 9:e1003738. doi: 10.1371/journal.pgen.1003738
Hasegawa, M., Arai, T., Nonaka, T., Kametani, F., Yoshida, M., Hashizume, Y., et al. (2008). Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64, 60–70. doi: 10.1002/ana.21425
Herzog, L. K., Kevei, E., Marchante, R., Bottcher, C., Bindesboll, C., Lystad, A. H., et al. (2020). The Machado-Joseph disease deubiquitylase ataxin-3 interacts with Lc3C/Gabarap and promotes autophagy. Aging Cell 19:e13051. doi: 10.1111/acel.13051
Hirsch-Reinshagen, V., Pottier, C., Nicholson, A. M., Baker, M., Hsiung, G. R., Krieger, C., et al. (2017). Clinical and neuropathological features of ALS/FTD with TIA1 mutations. Acta Neuropathol. Commun. 5:96. doi: 10.1186/s40478-017-0493-x
Horspool, A. M., and Chang, H. C. (2017). Superoxide dismutase SOD-1 modulates C. elegans pathogen avoidance behavior. Sci. Rep. 7:45128. doi: 10.1038/srep45128
Humphrey, J., Venkatesh, S., Hasan, R., Herb, J. T., De Paiva Lopes, K., Kucukali, F., et al. (2023). Integrative transcriptomic analysis of the amyotrophic lateral sclerosis spinal cord implicates glial activation and suggests new risk genes. Nat. Neurosci. 26, 150–162. doi: 10.1038/s41593-022-01205-3
Hurtle, B. T., Xie, L., and Donnelly, C. J. (2023). Disrupting pathologic phase transitions in neurodegeneration. J. Clin. Invest. 133:e168549. doi: 10.1172/JCI168549
Hutchison, S., Lebel, C., Blanchette, M., and Chabot, B. (2002). Distinct sets of adjacent heterogeneous nuclear ribonucleoprotein (hnRNP) A1/A2 binding sites control 5′ splice site selection in the hnRNP A1 mRNA precursor. J. Biol. Chem. 277, 29745–29752. doi: 10.1074/jbc.M203633200
Ikenaka, K., Kawai, K., Katsuno, M., Huang, Z., Jiang, Y. M., Iguchi, Y., et al. (2013). Dnc-1/dynactin 1 knockdown disrupts transport of autophagosomes and induces motor neuron degeneration. PLoS One 8:e54511. doi: 10.1371/journal.pone.0054511
Ikenaka, K., Tsukada, Y., Giles, A. C., Arai, T., Nakadera, Y., Nakano, S., et al. (2019). A behavior-based drug screening system using a Caenorhabditis elegans model of motor neuron disease. Sci. Rep. 9:10104. doi: 10.1038/s41598-019-46642-6
Ishigaki, S., and Sobue, G. (2018). Importance of functional loss of FUS in FTLD/ALS. Front. Mol. Biosci. 5:44. doi: 10.3389/fmolb.2018.00044
Jablonski, A. M., Lamitina, T., Liachko, N. F., Sabatella, M., Lu, J., Zhang, L., et al. (2015). Loss of RAD-23 protects against models of motor neuron disease by enhancing mutant protein clearance. J. Neurosci. 35, 14286–14306. doi: 10.1523/JNEUROSCI.0642-15.2015
Ji, Y. J., Ugolino, J., Zhang, T., Lu, J., Kim, D., and Wang, J. (2020). C9orf72/ALFA-1 controls TFEB/HLH-30-dependent metabolism through dynamic regulation of rag GTPases. PLoS Genet. 16:e1008738. doi: 10.1371/journal.pgen.1008738
Johnson, T. E. (2003). Advantages and disadvantages of Caenorhabditis elegans for aging research. Exp. Gerontol. 38, 1329–1332. doi: 10.1016/j.exger.2003.10.020
Johnson, J. O., Glynn, S. M., Gibbs, J. R., Nalls, M. A., Sabatelli, M., Restagno, G., et al. (2014). Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain 137:e311. doi: 10.1093/brain/awu265
Kanekura, K., Hashimoto, Y., Kita, Y., Sasabe, J., Aiso, S., Nishimoto, I., et al. (2005). A Rac1/phosphatidylinositol 3-kinase/Akt3 anti-apoptotic pathway, triggered by AlsinLF, the product of the ALS2 gene, antagonizes Cu/Zn-superoxide dismutase (SOD1) mutant-induced motoneuronal cell death. J. Biol. Chem. 280, 4532–4543. doi: 10.1074/jbc.M410508200
Kanekura, K., Hashimoto, Y., Niikura, T., Aiso, S., Matsuoka, M., and Nishimoto, I. (2004). Alsin, the product of ALS2 gene, suppresses SOD1 mutant neurotoxicity through RhoGEF domain by interacting with Sod1 mutants. J. Biol. Chem. 279, 19247–19256. doi: 10.1074/jbc.M313236200
Kassouf, T., Shrivastava, R., Meszka, I., Bailly, A., Polanowska, J., Trauchessec, H., et al. (2023). Targeting the NEDP1 enzyme to ameliorate ALS phenotypes through stress granule disassembly. Sci. Adv. 9:eabq7585. doi: 10.1126/sciadv.abq7585
Kawamura, K., and Maruyama, I. N. (2019). Forward genetic screen for Caenorhabditis elegans mutants with a shortened locomotor healthspan. G3 (Bethesda) 9, 2415–2423. doi: 10.1534/g3.119.400241
Kim, H. J., Kim, N. C., Wang, Y. D., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. doi: 10.1038/nature11922
Kim, W., Underwood, R. S., Greenwald, I., and Shaye, D. D. (2018). OrthoList 2: a new comparative genomic analysis of human and Caenorhabditis elegans genes. Genetics 210, 445–461. doi: 10.1534/genetics.118.301307
Koopman, M., Gungordu, L., Seinstra, R. I., Hogewerf, W., and Nollen, E. A. A. (2023a). Neuronal overexpression of hTDP-43 in Caenorhabditis elegans mimics the cellular pathology commonly observed in TDP-43 proteinopathies. MicroPubl. Biol. 2023. doi: 10.17912/micropub.biology.000767
Koopman, M., Gungordu, L., Seinstra, R. I., and Nollen, E. A. A. (2023b). Neuronal overexpression of hTDP-43 in Caenorhabditis elegans impairs different neuronally controlled behaviors and decreases fecundity. MicroPubl. Biol. 2023. doi: 10.17912/micropub.biology.000769
Koopman, M., Gungordu, L., Seinstra, R. I., and Nollen, E. A. A. (2023c). Neuronal overexpression of hTDP-43 in Caenorhabditis elegans impairs motor function. MicroPubl. Biol. 2023. doi: 10.17912/micropub.biology.000768
Koopman, M., Gungordu, L., Seinstra, R. I., and Nollen, E. A. A. (2023d). Neuronal overexpression of human TDP-43 in Caenorhabditis elegans causes a range of sensorimotor phenotypes. MicroPubl. Biol. 2023. doi: 10.17912/micropub.biology.000766
Kosturko, L. D., Maggipinto, M. J., Korza, G., Lee, J. W., Carson, J. H., and Barbarese, E. (2006). Heterogeneous nuclear ribonucleoprotein (hnRNP) E1 binds to hnRNP A2 and inhibits translation of A2 response element mRNAs. Mol. Biol. Cell 17, 3521–3533. doi: 10.1091/mbc.e05-10-0946
Kow, R. L., Black, A. H., Saxton, A. D., Liachko, N. F., and Kraemer, B. C. (2022). Loss of aly/ALYREF suppresses toxicity in both tau and TDP-43 models of neurodegeneration. Geroscience 44, 747–761. doi: 10.1007/s11357-022-00526-2
Kuhlbrodt, K., Janiesch, P. C., Kevei, E., Segref, A., Barikbin, R., and Hoppe, T. (2011). The Machado-Joseph disease deubiquitylase Atx-3 couples longevity and proteostasis. Nat. Cell Biol. 13, 273–281. doi: 10.1038/ncb2200
Kutscher, L. M., and Shaham, S. (2014). Forward and reverse mutagenesis in C. elegans. WormBook, 1–26.
Kwee, L. C., Liu, Y., Haynes, C., Gibson, J. R., Stone, A., Schichman, S. A., et al. (2012). A high-density genome-wide association screen of sporadic ALS in US veterans. PLoS One 7:e32768. doi: 10.1371/journal.pone.0032768
Labarre, A., Tossing, G., Maios, C., Doyle, J. J., and Parker, J. A. (2021). A single copy transgenic mutant FUS strain reproduces age-dependent ALS phenotypes in C. elegans. MicroPubl. Biol. 2021. doi: 10.17912/micropub.biology.000473
Labarre, A., Guitard, E., Tossing, G., Forest, A., Bareke, E., Labrecque, M., et al. (2022). Fatty acids derived from the probiotic Lacticaseibacillus rhamnosus HA-114 suppress age-dependent neurodegeneration. Commun Biol. 5:1340. doi: 10.1038/s42003-022-04295-8
Lamitina, T. (2022). Length-dependent RNA foci formation and repeat associated non-Aug dependent translation in a C. elegans G (4) C (2) model. MicroPubl. Biol. 2022. doi: 10.17912/micropub.biology.000600
Latimer, C. S., Stair, J. G., Hincks, J. C., Currey, H. N., Bird, T. D., Keene, C. D., et al. (2022). TDP-43 promotes tau accumulation and selective neurotoxicity in bigenic Caenorhabditis elegans. Dis. Model. Mech. 15:dmm049323. doi: 10.1242/dmm.049323
Lee, H. J., Alirzayeva, H., Koyuncu, S., Rueber, A., Noormohammadi, A., and Vilchez, D. (2023). Cold temperature extends longevity and prevents disease-related protein aggregation through Pa28gamma-induced proteasomes. Nat Aging 3, 546–566. doi: 10.1038/s43587-023-00383-4
Li, J., Huang, K. X., and Le, W. D. (2013). Establishing a novel C. elegans model to investigate the role of autophagy in amyotrophic lateral sclerosis. Acta Pharmacol. Sin. 34, 644–650. doi: 10.1038/aps.2012.190
Li, J., Li, T., Zhang, X., Tang, Y., Yang, J., and Le, W. (2014). Human superoxide dismutase 1 overexpression in motor neurons of Caenorhabditis elegans causes axon guidance defect and neurodegeneration. Neurobiol. Aging 35, 837–846. doi: 10.1016/j.neurobiolaging.2013.09.003
Li, C. Y., Yang, T. M., Ou, R. W., Wei, Q. Q., and Shang, H. F. (2021). Genome-wide genetic links between amyotrophic lateral sclerosis and autoimmune diseases. BMC Med. 19:27. doi: 10.1186/s12916-021-01903-y
Liachko, N. F., Guthrie, C. R., and Kraemer, B. C. (2010). Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J. Neurosci. 30, 16208–16219. doi: 10.1523/JNEUROSCI.2911-10.2010
Liachko, N. F., Mcmillan, P. J., Strovas, T. J., Loomis, E., Greenup, L., Murrell, J. R., et al. (2014). The tau tubulin kinases TTBK1/2 promote accumulation of pathological TDP-43. PLoS Genet. 10:e1004803. doi: 10.1371/journal.pgen.1004803
Liachko, N. F., Saxton, A. D., Mcmillan, P. J., Strovas, T. J., Currey, H. N., Taylor, L. M., et al. (2016). The phosphatase calcineurin regulates pathological TDP-43 phosphorylation. Acta Neuropathol 132, 545–561. doi: 10.1007/s00401-016-1600-y
Liachko, N. F., Saxton, A. D., Mcmillan, P. J., Strovas, T. J., Keene, C. D., Bird, T. D., et al. (2019). Genome wide analysis reveals heparan sulfate epimerase modulates TDP-43 proteinopathy. PLoS Genet. 15:e1008526. doi: 10.1371/journal.pgen.1008526
Lim, C. H., Kaur, P., Teo, E., Lam, V. Y. M., Zhu, F., Kibat, C., et al. (2020). Application of optogenetic amyloid-beta distinguishes between metabolic and physical damages in neurodegeneration. elife 9:e52589. doi: 10.7554/eLife.52589
Lim, M. A., Selak, M. A., Xiang, Z., Krainc, D., Neve, R. L., Kraemer, B. C., et al. (2012). Reduced activity of amp-activated protein kinase protects against genetic models of motor neuron disease. J. Neurosci. 32, 1123–1141. doi: 10.1523/JNEUROSCI.6554-10.2012
Lin, B. C., Higgins, N. R., Phung, T. H., and Monteiro, M. J. (2022). UBQLN proteins in health and disease with a focus on UBQLN2 in ALS/FTD. FEBS J. 289, 6132–6153. doi: 10.1111/febs.16129
Lins, J., Brock, T. J., Hopkins, C. E., and Hart, A. C. (2023). Generation of a C. elegans tdp-1 null allele and humanized TARDBP containing human disease-variants. MicroPubl. Biol. 2023. doi: 10.17912/micropub.biology.000693
Lu, J., Periz, G., Lu, Y. N., Tang, Q., Liu, Y., Zhang, T., et al. (2019). L3MBTL1 regulates ALS/FTD-associated proteotoxicity and quality control. Nat. Neurosci. 22, 875–886. doi: 10.1038/s41593-019-0384-5
Mackenzie, I. R., Nicholson, A. M., Sarkar, M., Messing, J., Purice, M. D., Pottier, C., et al. (2017). TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95, 808–816.e9. doi: 10.1016/j.neuron.2017.07.025
Majounie, E., Renton, A. E., Mok, K., Dopper, E. G., Waite, A., Rollinson, S., et al. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330. doi: 10.1016/S1474-4422(12)70043-1
Markert, S. M., Skoruppa, M., Yu, B., Mulcahy, B., Zhen, M., Gao, S., et al. (2020). Overexpression of an ALS-associated FUS mutation in C. elegans disrupts NMJ morphology and leads to defective neuromuscular transmission. Biol Open 9:bio055129. doi: 10.1242/bio.055129
Mehta, P. R., Brown, A. L., Ward, M. E., and Fratta, P. (2023). The era of cryptic exons: implications for ALS-FTD. Mol. Neurodegener. 18:16. doi: 10.1186/s13024-023-00608-5
Melnick, M., Gonzales, P., Cabral, J., Allen, M. A., Dowell, R. D., and Link, C. D. (2019). Heat shock in C. elegans induces downstream of gene transcription and accumulation of double-stranded RNA. PLoS One 14:e0206715. doi: 10.1371/journal.pone.0206715
Miao, L., and St. Clair, D. (2009). Regulation of superoxide dismutase genes: Implications in disease. Free Radic. Biol. Med. 47, 344–356. doi: 10.1016/j.freeradbiomed.2009.05.018
Millecamps, S., Salachas, F., Cazeneuve, C., Gordon, P., Bricka, B., Camuzat, A., et al. (2010). SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J. Med. Genet. 47, 554–560. doi: 10.1136/jmg.2010.077180
Mintchev, N., Zamba-Papanicolaou, E., Kleopa, K. A., and Christodoulou, K. (2009). A novel ALS2 splice-site mutation in a Cypriot juvenile-onset primary lateral sclerosis family. Neurology 72, 28–32. doi: 10.1212/01.wnl.0000338530.77394.60
Mitra, J., Guerrero, E. N., Hegde, P. M., Liachko, N. F., Wang, H., Vasquez, V., et al. (2019). Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. U. S. A. 116, 4696–4705. doi: 10.1073/pnas.1818415116
Mohan, S., Sampognaro, P. J., Argouarch, A. R., Maynard, J. C., Welch, M., Patwardhan, A., et al. (2021). Processing of progranulin into granulins involves multiple lysosomal proteases and is affected in frontotemporal lobar degeneration. Mol. Neurodegener. 16:51. doi: 10.1186/s13024-021-00472-1
Mol, M. O., Wong, T. H., Melhem, S., Basu, S., Viscusi, R., Galjart, N., et al. (2021). Novel TUBA4A variant associated with familial frontotemporal dementia. Neurol Genet 7:e596. doi: 10.1212/NXG.0000000000000596
Moore, K. M., Nicholas, J., Grossman, M., Mcmillan, C. T., Irwin, D. J., Massimo, L., et al. (2020). Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol. 19, 145–156. doi: 10.1016/S1474-4422(19)30394-1
Mori, K., Arzberger, T., Grasser, F. A., Gijselinck, I., May, S., Rentzsch, K., et al. (2013a). Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126, 881–893. doi: 10.1007/s00401-013-1189-3
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013b). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Morotz, G. M., De Vos, K. J., Vagnoni, A., Ackerley, S., Shaw, C. E., and Miller, C. C. (2012). Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum. Mol. Genet. 21, 1979–1988. doi: 10.1093/hmg/dds011
Muller, K., Oh, K. W., Nordin, A., Panthi, S., Kim, S. H., Nordin, F., et al. (2022). De novo mutations in SOD1 are a cause of ALS. J. Neurol. Neurosurg. Psychiatry 93, 201–206. doi: 10.1136/jnnp-2021-327520
Munch, C., Sedlmeier, R., Meyer, T., Homberg, V., Sperfeld, A. D., Kurt, A., et al. (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63, 724–726. doi: 10.1212/01.WNL.0000134608.83927.B1
Murakami, T., Qamar, S., Lin, J. Q., Schierle, G. S., Rees, E., Miyashita, A., et al. (2015). ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88, 678–690. doi: 10.1016/j.neuron.2015.10.030
Murakami, T., Yang, S. P., Xie, L., Kawano, T., Fu, D., Mukai, A., et al. (2012). ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum. Mol. Genet. 21, 1–9. doi: 10.1093/hmg/ddr417
Nakajima, K., Yin, X., Takei, Y., Seog, D. H., Homma, N., and Hirokawa, N. (2012). Molecular motor KIF5A is essential for GABA(A) receptor transport, and KIF5A deletion causes epilepsy. Neuron 76, 945–961. doi: 10.1016/j.neuron.2012.10.012
Nakamura, R., Misawa, K., Tohnai, G., Nakatochi, M., Furuhashi, S., Atsuta, N., et al. (2020). A multi-ethnic meta-analysis identifies novel genes, including ACSL5, associated with amyotrophic lateral sclerosis. Commun Biol 3:526. doi: 10.1038/s42003-020-01251-2
Nakano, J., Chiba, K., and Niwa, S. (2022). An ALS-associated KIF5A mutant forms oligomers and aggregates and induces neuronal toxicity. Genes Cells 27, 421–435. doi: 10.1111/gtc.12936
Nam, Y. W., Baskoylu, S. N., Gazgalis, D., Orfali, R., Cui, M., Hart, A. C., et al. (2018). A V-to-F substitution in SK2 channels causes Ca(2+) hypersensitivity and improves locomotion in a C. elegans ALS model. Sci. Rep. 8:10749. doi: 10.1038/s41598-018-28783-2
Nawa, M., Kage-Nakadai, E., Aiso, S., Okamoto, K., Mitani, S., and Matsuoka, M. (2012). Reduced expression of BTBD10, an Akt activator, leads to motor neuron death. Cell Death Differ. 19, 1398–1407. doi: 10.1038/cdd.2012.19
Nawa, M., Kanekura, K., Hashimoto, Y., Aiso, S., and Matsuoka, M. (2008). A novel Akt/PKB-interacting protein promotes cell adhesion and inhibits familial amyotrophic lateral sclerosis-linked mutant SOD1-induced neuronal death via inhibition of Pp2A-mediated dephosphorylation of Akt/PKB. Cell. Signal. 20, 493–505. doi: 10.1016/j.cellsig.2007.11.004
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nicolas, A., Kenna, K. P., Renton, A. E., Ticozzi, N., Faghri, F., Chia, R., et al. (2018). Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268–1283.e6. doi: 10.1016/j.neuron.2018.02.027
Nilaver, B. I., and Urbanski, H. F. (2023). Mechanisms underlying TDP-43 pathology and neurodegeneration: an updated mini-review. Front. Aging Neurosci. 15:1142617. doi: 10.3389/fnagi.2023.1142617
Nishimura, A. L., Mitne-Neto, M., Silva, H. C., Richieri-Costa, A., Middleton, S., Cascio, D., et al. (2004). A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 822–831. doi: 10.1086/425287
Ogawa, M., Shidara, H., Oka, K., Kurosawa, M., Nukina, N., and Furukawa, Y. (2015). Cysteine residues in Cu,Zn-superoxide dismutase are essential to toxicity in Caenorhabditis elegans model of amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 463, 1196–1202. doi: 10.1016/j.bbrc.2015.06.084
O’Reilly, L. P., Luke, C. J., Perlmutter, D. H., Silverman, G. A., and Pak, S. C. (2014). C. elegans in high-throughput drug discovery. Adv. Drug Deliv. Rev. 69-70, 247–253. doi: 10.1016/j.addr.2013.12.001
Osborne, J. F., Yanagi, K. S., and Hart, A. C. (2021). Genetic interactions in a C. elegans sod-1 ALS model: glutamatergic neuron degeneration. MicroPubl. Biol. 2021. doi: 10.17912/micropub.biology.000338
Pan, C. L., Peng, C. Y., Chen, C. H., and Mcintire, S. (2011). Genetic analysis of age-dependent defects of the Caenorhabditis elegans touch receptor neurons. Proc. Natl. Acad. Sci. U. S. A. 108, 9274–9279. doi: 10.1073/pnas.1011711108
Pansarasa, O., Bordoni, M., Diamanti, L., Sproviero, D., Gagliardi, S., and Cereda, C. (2018). Sod1 in amyotrophic lateral sclerosis: "ambivalent" behavior connected to the disease. Int. J. Mol. Sci. 19:1345. doi: 10.3390/ijms19051345
Parker, S., Peterkin, H. S., and Baylis, H. A. (2007). Muscular dystrophy associated mutations in caveolin-1 induce neurotransmission and locomotion defects in Caenorhabditis elegans. Invertebr. Neurosci. 7, 157–164. doi: 10.1007/s10158-007-0051-5
Peggion, C., Scalcon, V., Massimino, M. L., Nies, K., Lopreiato, R., Rigobello, M. P., et al. (2022). Sod1 in ALS: taking stock in pathogenic mechanisms and the role of glial and muscle cells. Antioxidants (Basel) 11:614. doi: 10.3390/antiox11040614
Periz, G., Lu, J., Zhang, T., Kankel, M. W., Jablonski, A. M., Kalb, R., et al. (2015). Regulation of protein quality control by UBE4B and LSD1 through p53-mediated transcription. PLoS Biol. 13:e1002114. doi: 10.1371/journal.pbio.1002114
Pigazzini, M. L., and Kirstein, J. (2020). In vivo quantification of protein turnover in aging C. elegans using photoconvertible Dendra2. JoVE 160:e61196. doi: 10.3791/61196
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A., and Patel, B. K. (2019). Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 12:25. doi: 10.3389/fnmol.2019.00025
Puleo, N., and Lamitina, T. (2020). The conserved multi-functional nuclear protein dss-1/sem1 is required for C9orf72-associated ALS/FTD dipeptide toxicity. MicroPubl. Biol. 2020. doi: 10.17912/micropub.biology.000262
Puls, I., Jonnakuty, C., Lamonte, B. H., Holzbaur, E. L., Tokito, M., Mann, E., et al. (2003). Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455–456. doi: 10.1038/ng1123
Rayman, J. B., and Kandel, E. R. (2017). TIA-1 is a functional prion-like protein. Cold Spring Harb. Perspect. Biol. 9:a030718. doi: 10.1101/cshperspect.a030718
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9orf72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Reus, L. M., Pasaniuc, B., Posthuma, D., Boltz, T., Pijnenburg, Y. A. L., Ophoff, R. A., et al. (2021). Gene expression imputation across multiple tissue types provides insight into the genetic architecture of frontotemporal dementia and its clinical subtypes. Biol. Psychiatry 89, 825–835. doi: 10.1016/j.biopsych.2020.12.023
Rodrigues, A. J., Neves-Carvalho, A., Teixeira-Castro, A., Rokka, A., Corthals, G., Logarinho, E., et al. (2011). Absence of ataxin-3 leads to enhanced stress response in C. elegans. PLoS One 6:e18512. doi: 10.1371/journal.pone.0018512
Rojas-Prats, E., Martinez-Gonzalez, L., Gonzalo-Consuegra, C., Liachko, N. F., Perez, C., Ramirez, D., et al. (2021). Targeting nuclear protein TDP-43 by cell division cycle kinase 7 inhibitors: a new therapeutic approach for amyotrophic lateral sclerosis. Eur. J. Med. Chem. 210:112968. doi: 10.1016/j.ejmech.2020.112968
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rudich, P., Snoznik, C., Watkins, S. C., Monaghan, J., Pandey, U. B., and Lamitina, S. T. (2017). Nuclear localized C9orf72-associated arginine-containing dipeptides exhibit age-dependent toxicity in C. elegans. Hum. Mol. Genet. 26, 4916–4928. doi: 10.1093/hmg/ddx372
Ryan, V. H., and Hart, A. C. (2021). Sym-2 loss-of-function causes glutamatergic neurodegeneration after oxidative stress. MicroPubl. Biol. 2021. doi: 10.17912/micropub.biology.000363
Ryan, V. H., Perdikari, T. M., Naik, M. T., Saueressig, C. F., Lins, J., Dignon, G. L., et al. (2021). Tyrosine phosphorylation regulates hnRNPA2 granule protein partitioning and reduces neurodegeneration. EMBO J. 40:e105001. doi: 10.15252/embj.2020105001
Saez-Atienzar, S., Dalgard, C. L., Ding, J., Chio, A., Alba, C., Hupalo, D. N., et al. (2020). Identification of a pathogenic intronic KIF5A mutation in an ALS-FTD kindred. Neurology 95, 1015–1018. doi: 10.1212/WNL.0000000000011064
Salazar, D. A., Butler, V. J., Argouarch, A. R., Hsu, T. Y., Mason, A., Nakamura, A., et al. (2015). The progranulin cleavage products, granulins, exacerbate TDP-43 toxicity and increase TDP-43 levels. J. Neurosci. 35, 9315–9328. doi: 10.1523/JNEUROSCI.4808-14.2015
Saldi, T. K., Ash, P. E., Wilson, G., Gonzales, P., Garrido-Lecca, A., Roberts, C. M., et al. (2014). TDP-1, the Caenorhabditis elegans ortholog of TDP-43, limits the accumulation of double-stranded RNA. EMBO J. 33, 2947–2966. doi: 10.15252/embj.201488740
Saldi, T. K., Gonzales, P., Garrido-Lecca, A., Dostal, V., Roberts, C. M., Petrucelli, L., et al. (2018). The Caenorhabditis elegans Ortholog of TDP-43 regulates the chromatin localization of the heterochromatin protein 1 homolog HPL-2. Mol. Cell. Biol. 38:e00668-17. doi: 10.1128/MCB.00668-17
Saxton, A. D., and Kraemer, B. C. (2021). Human Ubiquilin 2 and TDP-43 copathology drives neurodegeneration in transgenic Caenorhabditis elegans. G3 (Bethesda) 11:jkab158. doi: 10.1093/g3journal/jkab158
Sekiyama, N., Takaba, K., Maki-Yonekura, S., Akagi, K. I., Ohtani, Y., Imamura, K., et al. (2022). ALS mutations in the Tia-1 prion-like domain trigger highly condensed pathogenic structures. Proc. Natl. Acad. Sci. U. S. A. 119:e2122523119. doi: 10.1073/pnas.2122523119
Sellier, C., Campanari, M. L., Julie Corbier, C., Gaucherot, A., Kolb-Cheynel, I., Oulad-Abdelghani, M., et al. (2016). Loss of C9orf72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 35, 1276–1297. doi: 10.15252/embj.201593350
Shang, Y., and Huang, E. J. (2016). Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 1647, 65–78. doi: 10.1016/j.brainres.2016.03.036
Shao, Q., Liang, C., Chang, Q., Zhang, W., Yang, M., and Chen, J. F. (2019). C9orf72 deficiency promotes motor deficits of a C9als/FTD mouse model in a dose-dependent manner. Acta Neuropathol. Commun. 7:32. doi: 10.1186/s40478-019-0685-7
Shaye, D. D., and Greenwald, I. (2011). OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One 6:e20085. doi: 10.1371/journal.pone.0020085
Sheerin, U. M., Schneider, S. A., Carr, L., Deuschl, G., Hopfner, F., Stamelou, M., et al. (2014). ALS2 mutations: juvenile amyotrophic lateral sclerosis and generalized dystonia. Neurology 82, 1065–1067. doi: 10.1212/WNL.0000000000000254
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W. H., Hung, S. T., et al. (2018). Haploinsufficiency leads to neurodegeneration in C9orf72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313–325. doi: 10.1038/nm.4490
Silverman, G. A., Luke, C. J., Bhatia, S. R., Long, O. S., Vetica, A. C., Perlmutter, D. H., et al. (2009). Modeling molecular and cellular aspects of human disease using the nematode Caenorhabditis elegans. Pediatr. Res. 65, 10–18. doi: 10.1203/PDR.0b013e31819009b0
Simpson, C. L., Lemmens, R., Miskiewicz, K., Broom, W. J., Hansen, V. K., Van Vught, P. W., et al. (2009). Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum. Mol. Genet. 18, 472–481. doi: 10.1093/hmg/ddn375
Sin, O., Michels, H., and Nollen, E. A. (2014). Genetic screens in Caenorhabditis elegans models for neurodegenerative diseases. Biochim. Biophys. Acta 1842, 1951–1959. doi: 10.1016/j.bbadis.2014.01.015
Skehel, P. A., Martin, K. C., Kandel, E. R., and Bartsch, D. (1995). A VAMP-binding protein from Aplysia required for neurotransmitter release. Science 269, 1580–1583. doi: 10.1126/science.7667638
Smith, B. N., Ticozzi, N., Fallini, C., Gkazi, A. S., Topp, S., Kenna, K. P., et al. (2014). Exome-wide rare variant analysis identifies Tuba4A mutations associated with familial ALS. Neuron 84, 324–331. doi: 10.1016/j.neuron.2014.09.027
Smukowski, S. N., Maioli, H., Latimer, C. S., Bird, T. D., Jayadev, S., and Valdmanis, P. N. (2022). Progress in amyotrophic lateral sclerosis gene discovery: reflecting on classic approaches and leveraging emerging technologies. Neurol Genet 8:e669. doi: 10.1212/NXG.0000000000000669
Snoznik, C., Medvedeva, V., Mojsilovic-Petrovic, J., Rudich, P., Oosten, J., Kalb, R. G., et al. (2021). The nuclear ubiquitin ligase adaptor SPOP is a conserved regulator of C9orf72 dipeptide toxicity. Proc. Natl. Acad. Sci. U. S. A. 118:e2104664118. doi: 10.1073/pnas.2104664118
Soh, M. S., Cheng, X., Vijayaraghavan, T., Vernon, A., Liu, J., and Neumann, B. (2020). Disruption of genes associated with Charcot-Marie-tooth type 2 lead to common behavioural, cellular and molecular defects in Caenorhabditis elegans. PLoS One 15:e0231600. doi: 10.1371/journal.pone.0231600
Sonobe, Y., Aburas, J., Krishnan, G., Fleming, A. C., Ghadge, G., Islam, P., et al. (2021). A C. elegans model of C9orf72-associated ALS/FTD uncovers a conserved role for eIF2D in RAN translation. Nat. Commun. 12:6025. doi: 10.1038/s41467-021-26303-x
Soussan, L., Burakov, D., Daniels, M. P., Toister-Achituv, M., Porat, A., Yarden, Y., et al. (1999). ERG30, a VAP-33-related protein, functions in protein transport mediated by COPI vesicles. J. Cell Biol. 146, 301–312. doi: 10.1083/jcb.146.2.301
Strong, M. J., Abrahams, S., Goldstein, L. H., Woolley, S., Mclaughlin, P., Snowden, J., et al. (2017). Amyotrophic lateral sclerosis – frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph. Lateral Scler. Frontotemporal Degener. 18, 153–174. doi: 10.1080/21678421.2016.1267768
Tai, H., Cui, L., Shen, D., Li, D., Cui, B., and Fang, J. (2017). Military service and the risk of amyotrophic lateral sclerosis: a meta-analysis. J. Clin. Neurosci. 45, 337–342. doi: 10.1016/j.jocn.2017.08.035
Tan, R. H., Ke, Y. D., Ittner, L. M., and Halliday, G. M. (2017). ALS/FTLD: experimental models and reality. Acta Neuropathol. 133, 177–196. doi: 10.1007/s00401-016-1666-6
Tang, X., Toro, A., Sahana, T. G., Gao, J., Chalk, J., Oskarsson, B. E., et al. (2020). Divergence, convergence, and therapeutic implications: a cell biology perspective of C9orf72-ALS/FTD. Mol. Neurodegener. 15:34. doi: 10.1186/s13024-020-00383-7
Tauffenberger, A., Vaccaro, A., Aulas, A., Vande Velde, C., and Parker, J. A. (2012). Glucose delays age-dependent proteotoxicity. Aging Cell 11, 856–866. doi: 10.1111/j.1474-9726.2012.00855.x
Taylor, M., Marx, O., and Norris, A. (2023). TDP-1 and FUST-1 co-inhibit exon inclusion and control fertility together with transcriptional regulation. Nucleic Acids Res. 51, 9610–9628. doi: 10.1093/nar/gkad665
Taylor, L. M., Mcmillan, P. J., Liachko, N. F., Strovas, T. J., Ghetti, B., Bird, T. D., et al. (2018). Pathological phosphorylation of tau and TDP-43 by TTBK1 and TTBK2 drives neurodegeneration. Mol. Neurodegener. 13:7. doi: 10.1186/s13024-018-0237-9
Therrien, M., and Parker, J. A. (2014). Worming forward: amyotrophic lateral sclerosis toxicity mechanisms and genetic interactions in Caenorhabditis elegans. Front. Genet. 5:85. doi: 10.3389/fgene.2014.00085
Therrien, M., Rouleau, G. A., Dion, P. A., and Parker, J. A. (2013). Deletion of C9orf72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One 8:e83450. doi: 10.1371/journal.pone.0083450
Therrien, M., Rouleau, G. A., Dion, P. A., and Parker, J. A. (2016). FET proteins regulate lifespan and neuronal integrity. Sci. Rep. 6:25159. doi: 10.1038/srep25159
Tossing, G., Livernoche, R., Maios, C., Bretonneau, C., Labarre, A., and Parker, J. A. (2022). Genetic and pharmacological PARP inhibition reduces axonal degeneration in C. elegans models of ALS. Hum. Mol. Genet. 31, 3313–3324. doi: 10.1093/hmg/ddac116
Tseng, Y. L., Lu, P. C., Lee, C. C., He, R. Y., Huang, Y. A., Tseng, Y. C., et al. (2023). Degradation of neurodegenerative disease-associated TDP-43 aggregates and oligomers via a proteolysis-targeting chimera. J. Biomed. Sci. 30:27. doi: 10.1186/s12929-023-00921-7
Tsioras, K., Smith, K. C., Edassery, S. L., Garjani, M., Li, Y., Williams, C., et al. (2023). Analysis of proteome-wide degradation dynamics in ALS SOD1 iPSC-derived patient neurons reveals disrupted VCP homeostasis. Cell Rep. 42:113160. doi: 10.1016/j.celrep.2023.113160
Vaccaro, A., Patten, S. A., Aggad, D., Julien, C., Maios, C., Kabashi, E., et al. (2013). Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol. Dis. 55, 64–75. doi: 10.1016/j.nbd.2013.03.015
Vaccaro, A., Patten, S. A., Ciura, S., Maios, C., Therrien, M., Drapeau, P., et al. (2012a). Methylene blue protects against TDP-43 and FUS neuronal toxicity in C. Elegans and D. rerio. PLoS One 7:e42117. doi: 10.1371/journal.pone.0042117
Vaccaro, A., Tauffenberger, A., Aggad, D., Rouleau, G., Drapeau, P., and Parker, J. A. (2012b). Mutant TDP-43 and FUS cause age-dependent paralysis and neurodegeneration in C. elegans. PLoS One 7:e31321. doi: 10.1371/journal.pone.0031321
Vaccaro, A., Tauffenberger, A., Ash, P. E., Carlomagno, Y., Petrucelli, L., and Parker, J. A. (2012c). TDP-1/TDP-43 regulates stress signaling and age-dependent proteotoxicity in Caenorhabditis elegans. PLoS Genet. 8:e1002806. doi: 10.1371/journal.pgen.1002806
van Es, M. A., Hardiman, O., Chio, A., Al-Chalabi, A., Pasterkamp, R. J., Veldink, J. H., et al. (2017). Amyotrophic lateral sclerosis. Lancet 390, 2084–2098. doi: 10.1016/S0140-6736(17)31287-4
Van Mossevelde, S., Van Der Zee, J., Cruts, M., and Van Broeckhoven, C. (2017). Relationship between C9orf72 repeat size and clinical phenotype. Curr. Opin. Genet. Dev. 44, 117–124. doi: 10.1016/j.gde.2017.02.008
Veriepe, J., Fossouo, L., and Parker, J. A. (2015). Neurodegeneration in C. elegans models of ALS requires TIR-1/Sarm1 immune pathway activation in neurons. Nat. Commun. 6:7319. doi: 10.1038/ncomms8319
Wang, Y., Arnold, M. L., Smart, A. J., Wang, G., Androwski, R. J., Morera, A., et al. (2023b). Large vesicle extrusions from C. elegans neurons are consumed and stimulated by glial-like phagocytosis activity of the neighboring cell. elife 12:e82227. doi: 10.7554/eLife.82227
Wang, L., and Brown, A. (2010). A hereditary spastic paraplegia mutation in kinesin-1A/KIF5A disrupts neurofilament transport. Mol. Neurodegener. 5:52. doi: 10.1186/1750-1326-5-52
Wang, J., Farr, G. W., Hall, D. H., Li, F., Furtak, K., Dreier, L., et al. (2009). An ALS-linked mutant Sod1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 5:e1000350. doi: 10.1371/journal.pgen.1000350
Wang, X., Hao, L., Saur, T., Joyal, K., Zhao, Y., Zhai, D., et al. (2016). Forward genetic screen in Caenorhabditis elegans suggests F57A10.2 and acp-4 as suppressors of C9orf72 related phenotypes. Front. Mol. Neurosci. 9:113. doi: 10.3389/fnmol.2016.00113
Wang, A. L., Mambou, E. A., and Kao, A. W. (2023a). The progranulin cleavage product granulin 3 exerts a dominant negative effect on animal fitness. Hum. Mol. Genet. :ddad184. doi: 10.1093/hmg/ddad184
Ward, M. E., and Miller, B. L. (2011). Potential mechanisms of progranulin-deficient FTLD. J. Mol. Neurosci. 45, 574–582. doi: 10.1007/s12031-011-9622-3
Webster, C. P., Smith, E. F., Bauer, C. S., Moller, A., Hautbergue, G. M., Ferraiuolo, L., et al. (2016). The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 35, 1656–1676. doi: 10.15252/embj.201694401
Weeks, J. C., Roberts, W. M., Leasure, C., Suzuki, B. M., Robinson, K. J., Currey, H., et al. (2018). Sertraline, paroxetine, and chlorpromazine are rapidly acting anthelmintic drugs capable of clinical repurposing. Sci. Rep. 8:975. doi: 10.1038/s41598-017-18457-w
Williams, K. L., Warraich, S. T., Yang, S., Solski, J. A., Fernando, R., Rouleau, G. A., et al. (2012). UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol. Aging 33:2527.e3–e10. doi: 10.1016/j.neurobiolaging.2012.05.008
Wong, S. Q., Pontifex, M. G., Phelan, M. M., Pidathala, C., Kraemer, B. C., Barclay, J. W., et al. (2018). Alpha-methyl-alpha-phenylsuccinimide ameliorates neurodegeneration in a C. elegans model of TDP-43 proteinopathy. Neurobiol. Dis. 118, 40–54. doi: 10.1016/j.nbd.2018.06.013
Woo, J. A., Liu, T., Trotter, C., Fang, C. C., De Narvaez, E., Lepochat, P., et al. (2017). Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun. 8:15558. doi: 10.1038/ncomms15558
Xu, H., Jia, C., Cheng, C., Wu, H., Cai, H., and Le, W. (2022). Activation of autophagy attenuates motor deficits and extends lifespan in a C. elegans model of ALS. Free Radic. Biol. Med. 181, 52–61. doi: 10.1016/j.freeradbiomed.2022.01.030
Yanase, S., Onodera, A., Tedesco, P., Johnson, T. E., and Ishii, N. (2009). Sod-1 deletions in Caenorhabditis elegans alter the localization of intracellular reactive oxygen species and show molecular compensation. J. Gerontol. A Biol. Sci. Med. Sci. 64, 530–539. doi: 10.1093/gerona/glp020
Yanase, S., Yasuda, K., and Ishii, N. (2020). Interaction between the ins/IGF-1 and p38 MAPK signaling pathways in molecular compensation of sod genes and modulation related to intracellular ROS levels in C. elegans. Biochem Biophys Rep 23:100796. doi: 10.1016/j.bbrep.2020.100796
Yang, Y., Hentati, A., Deng, H. X., Dabbagh, O., Sasaki, T., Hirano, M., et al. (2001). The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 29, 160–165. doi: 10.1038/ng1001-160
Yang, X., Zheng, J., Tian, S., Chen, Y., An, R., Zhao, Q., et al. (2017). HLA-DRA/HLA-DRB5 polymorphism affects risk of sporadic ALS and survival in a southwest Chinese cohort. J. Neurol. Sci. 373, 124–128. doi: 10.1016/j.jns.2016.12.055
Yu, C. Y., and Chang, H. C. (2022). Glutamate signaling mediates C. elegans behavioral plasticity to pathogens. iScience 25:103919. doi: 10.1016/j.isci.2022.103919
Zanier, E. R., Barzago, M. M., Vegliante, G., Romeo, M., Restelli, E., Bertani, I., et al. (2021). C. elegans detects toxicity of traumatic brain injury generated tau. Neurobiol. Dis. 153:105330. doi: 10.1016/j.nbd.2021.105330
Zhai, J., Zhang, L., Mojsilovic-Petrovic, J., Jian, X., Thomas, J., Homma, K., et al. (2015). Inhibition of Cytohesins protects against genetic models of motor neuron disease. J. Neurosci. 35, 9088–9105. doi: 10.1523/JNEUROSCI.5032-13.2015
Zhang, T., Baldie, G., Periz, G., and Wang, J. (2014). RNA-processing protein TDP-43 regulates FOXO-dependent protein quality control in stress response. PLoS Genet. 10:e1004693. doi: 10.1371/journal.pgen.1004693
Zhang, W., Colavita, A., and Ngsee, J. K. (2017). Mitigating motor neuronal loss in C. elegans model of ALS8. Sci. Rep. 7:11582. doi: 10.1038/s41598-017-11798-6
Zhang, T., Hwang, H. Y., Hao, H., Talbot, C. Jr., and Wang, J. (2012). Caenorhabditis elegans RNA-processing protein TDP-1 regulates protein homeostasis and life span. J. Biol. Chem. 287, 8371–8382. doi: 10.1074/jbc.M111.311977
Zhang, T., Mullane, P. C., Periz, G., and Wang, J. (2011). TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/Igf-1 signaling. Hum. Mol. Genet. 20, 1952–1965. doi: 10.1093/hmg/ddr076
Zhang, T., Periz, G., Lu, Y. N., and Wang, J. (2020). USP7 regulates ALS-associated proteotoxicity and quality control through the NEDD4L-SMAD pathway. Proc. Natl. Acad. Sci. U. S. A. 117, 28114–28125. doi: 10.1073/pnas.2014349117
Zhang, T., Wu, Y. C., Mullane, P., Ji, Y. J., Liu, H., He, L., et al. (2018). FUS regulates activity of MicroRNA-mediated gene silencing. Mol. Cell 69, 787–801.e8. doi: 10.1016/j.molcel.2018.02.001
Zhang, M., Xi, Z., Zinman, L., Bruni, A. C., Maletta, R. G., Curcio, S. A., et al. (2015). Mutation analysis of CHCHD10 in different neurodegenerative diseases. Brain 138:e380. doi: 10.1093/brain/awv082
Zhong, Y., Wang, J., Henderson, M. J., Yang, P., Hagen, B. M., Siddique, T., et al. (2017). Nuclear export of misfolded SOD1 mediated by a normally buried NES-like sequence reduces proteotoxicity in the nucleus. elife 6:e23759. doi: 10.7554/eLife.23759
Keywords: C. elegans, neurodegeneration, amyotrophic lateral sclerosis, frontotemporal lobar degeneration, TDP-43, C9orf72, SOD1, FUS
Citation: Eck RJ, Stair JG, Kraemer BC and Liachko NF (2024) Simple models to understand complex disease: 10 years of progress from Caenorhabditis elegans models of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Front. Neurosci. 17:1300705. doi: 10.3389/fnins.2023.1300705
Edited by:
Zainuddin Quadri, University of Kentucky, United StatesReviewed by:
Haripriya Vittal Rao, University of Rhode Island, United StatesLalitha Venkataraman, Nationwide Children's Hospital, United States
Copyright © 2024 Eck, Stair, Kraemer and Liachko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicole F. Liachko, bmxpYWNoa29AdXcuZWR1
†These authors have contributed equally to this work