
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 16 November 2023
Sec. Neurodegeneration
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1297984
 Matin Ramezani1
Matin Ramezani1 Malika Fernando1
Malika Fernando1 Shaun Eslick1
Shaun Eslick1 Prita R. Asih1,2Sina Shadfar3
Prita R. Asih1,2Sina Shadfar3 Ekanayaka M. S. Bandara4
Ekanayaka M. S. Bandara4 Heidi Hillebrandt1Silochna Meghwar1Maryam Shahriari1
Heidi Hillebrandt1Silochna Meghwar1Maryam Shahriari1 Pratishtha Chatterjee1
Pratishtha Chatterjee1 Rohith Thota1Cintia B. Dias1
Rohith Thota1Cintia B. Dias1 Manohar L. Garg1
Manohar L. Garg1 Ralph N. Martins1,4*
Ralph N. Martins1,4*Alzheimer’s disease (AD) is the most common form of dementia. AD is a progressive neurodegenerative disorder characterized by cognitive dysfunction, including learning and memory deficits, and behavioral changes. Neuropathology hallmarks of AD such as amyloid beta (Aβ) plaques and neurofibrillary tangles containing the neuron-specific protein tau is associated with changes in fluid biomarkers including Aβ, phosphorylated tau (p-tau)-181, p-tau 231, p-tau 217, glial fibrillary acidic protein (GFAP), and neurofilament light (NFL). Another pathological feature of AD is neural damage and hyperactivation of astrocytes, that can cause increased pro-inflammatory mediators and oxidative stress. In addition, reduced brain glucose metabolism and mitochondrial dysfunction appears up to 15 years before the onset of clinical AD symptoms. As glucose utilization is compromised in the brain of patients with AD, ketone bodies (KBs) may serve as an alternative source of energy. KBs are generated from the β-oxidation of fatty acids, which are enhanced following consumption of ketogenic diets with high fat, moderate protein, and low carbohydrate. KBs have been shown to cross the blood brain barrier to improve brain energy metabolism. This review comprehensively summarizes the current literature on how increasing KBs support brain energy metabolism. In addition, for the first time, this review discusses the effects of ketogenic diet on the putative AD biomarkers such as Aβ, tau (mainly p-tau 181), GFAP, and NFL, and discusses the role of KBs on neuroinflammation, oxidative stress, and mitochondrial metabolism.
Alzheimer’s disease (AD) has emerged as one of the most severe health-threatening conditions in the 21st century (Rolandi et al., 2020). The incidence rate of AD is expected to rise with a disproportionate increase in low- and middle-income societies (Zhang et al., 2021). From 1990 to 2019, the prevalence and mortality rates of disease have doubled (Javaid et al., 2021). Currently, more than 50 million people worldwide are living with dementia, particularly AD accounting for an estimated 50–70% of all dementia cases (Zhang et al., 2021). This number is estimated to increase to 150 million by the end of 2050 (Patterson, 2018). As AD prevalence and mortality rates are increasing worldwide, it is crucial to advance our understanding of the disease (Patterson, 2018), which will considerably support the development of therapies (Cunnane et al., 2011).
Aging is the most prominent risk factor for developing AD with predominantly diagnosed as late-onset Alzheimer’s disease in people above 65 years old (Hoogmartens et al., 2021). More than 90% of AD is sporadic predominantly with a late onset, due to a combination of genetic variants (70%) such as APOE4 and environmental factors (30%), such as hormonal and molecular changes, diet and toxicological exposure (Dorszewska et al., 2016). However, early onset of AD can predominantly be diagnosed in patients with familial AD, due to mutations in the Amyloid Precursor Protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2) genes (Piaceri et al., 2013; Hoogmartens et al., 2021).
Neuropathology of AD starts approximately 15–20 years prior to its clinical symptoms (Ashton et al., 2019). This preclinical phase in AD is a “symptom free” stage, and individuals are cognitively unimpaired with amyloid aggregation (Vermunt et al., 2022). The more progressive Aβ aggregation would be found in prodromal phase of AD referring to the mild cognitive impairment (MCI) with an impairment in at least one cognitive domain (Douglas and Scharre, 2019; Vermunt et al., 2022), and severe AD dementia or symptomatic AD is the late-stage AD characterized by a progressive functional impairments, and more definitive clinical symptoms (Douglas and Scharre, 2019).
Classical neuropathological hallmarks of AD, such as amyloid beta (Aβ) plaques and neurofibrillary tangles containing the neuron-specific protein tau has been reported in the preclinical phase of AD (Gunes et al., 2022). The most advanced AD biomarkers with a greater prognostic and diagnostic value that are changed in cerebrospinal fluid (CSF), and plasma include amyloid beta (Aβ), phosphorylated tau (p-tau), neurofilament light (NFL) and glial fibrillary acidic protein (GFAP) (Benedet et al., 2021; Chatterjee et al., 2023). Any neuropathological changes such as reactive astrogliosis (Salvadó et al., 2022) and disruption of the neural axonal cytoskeletal structure (Poff et al., 2021; Alberti et al., 2022) can change the level of these circulating biomarkers (Poff et al., 2021; Alberti et al., 2022), thereby making these biomarkers as the promising indicators for the early diagnosis of AD (Khoury and Ghossoub, 2019; Ausó et al., 2020; Klyucherev et al., 2022). Understanding these changes will proceed early diagnosis and therefore more effective disease-modifying approaches (Arvanitakis et al., 2019).
Furthermore, AD-affected brain shows glucose hypometabolism due to alleviated glucose uptake and utilization through different types of glucose transporters (GLUTs) (Szablewski, 2021). Predominantly, glucose uptake into the brain occurs using transporters GLUT1 and GLUT3 (Kyrtata et al., 2021). In AD brain, a reduced expression of GLUT1 carriers localized in brain microvasculature and astrocytes (Jurcovicova, 2014) as well as a decline in GLUT3 expressed in neurons have been indicated (Szablewski, 2021). In contrast, GLUT2 as an insulin-sensitive glucose transporter is highly expressed in AD pathology assumed to be due to astrogliosis (Kyrtata et al., 2021). These subsequently result in reduced ATP production from glucose metabolism by 50% that continues to decrease further with disease progression (Szablewski, 2021). Brain energy deficits arising from the aforementioned processes are further attributed to AD-related neuropathological changes (Poff et al., 2021; Alberti et al., 2022; Salvadó et al., 2022).
In the brain neurons produce the majority of ATP through the oxidative phosphorylation of ADP. However, glia cells are also responsible for ATP synthesis (Beard et al., 2022). Preferentially, astrocytes undergo glycolysis to synthesize lactate and pyruvate from glucose (Chamberlain and Sheng, 2019; Beard et al., 2022). Astrocytes due to having glycolytic enzymes are capable of using 80% of the glucose via glycolysis, while glycolytic enzymes are inhibited in the neurons (Chamberlain and Sheng, 2019). Albeit decreased glycolysis is associated with early cognitive impairment, contributing to AD progression (Goyal et al., 2023).
These abnormal glucose homeostasis in the brain (An et al., 2018) such as reduced uptake and utilization of brain glucose, perturbed glucose metabolism, reduced glycolysis and insulin and insulin-like growth factor-1 (IGF-1) resistance can cause a deficit in brain energy metabolism reported in AD brain (De La Monte, 2012; Szablewski, 2021). Glucose deficiency can subsequently result in reduced ATP production by 50% that continues to decrease further with disease progression (Hoyer, 1992; Szablewski, 2021). Brain energy deficits arising from the aforementioned processes are further attributed to AD-related neuropathological changes (Poff et al., 2021; Alberti et al., 2022; Salvadó et al., 2022). Glucose deficiency not only cause an energy crisis which affect ATP and the NAD+/NADH ratio, but also it can detrimentally affect the biosynthesis of different components such as neurotransmitters (Dienel, 2019) and hepatic sialic acid (Peng et al., 2023). Moreover, 2 NADH shuttles (pentose phosphate shunt, malate–aspartate) contributing to glycolysis and glycogen turnover would be affected by glucose deficiency (Dienel, 2019).
Impaired brain energy metabolism can be compensated by selected dietary approaches which result in an increase in plasma ketone bodies (KBs) from the catabolism of fatty acids (Phillips et al., 2021). Ketogenic diets, which have been effective in treating pediatric epilepsy (Barañano and Hartman, 2008), can facilitate brain energy function by inducing nutritional ketosis (Ota et al., 2016; Jensen et al., 2020; Ashton et al., 2021). Several studies have shown a positive association between various ketogenic diets and a better cognitive performance (Krikorian et al., 2012; Xu et al., 2020; Ferraris et al., 2021; Roy et al., 2021). For instance, it has been reported that 3 months medium chain triglycerides (MCT) intervention (17.3 g/day) can improve cognitive performance in mild to moderate AD patients (Xu et al., 2020). In another study, an average dosage of 25.2 g of MCT containing 99.3% caprylic acids, 0.6% capric acids, and 0.1% lauric acid for 4 months significantly improved the cognitive performance in AD patients (Juby et al., 2022). However, relatively few studies have reported the effects of this diet on AD-putative CSF and blood biomarkers such as Aβ, p-tau, GFAP, and NFL (Table 1). Moreover, there has been no human studies to date that have been conducted on the effects of ketogenic diet on these blood biomarkers. While recent studies indicate that ketogenic diet may impact AD biomarkers, it has not yet been determined whether increased KBs per se causes such changes or that KBs acts indirectly by increasing brain energy metabolism. Therefore, we aim to discuss how a ketogenic diet could provide an alternative energy source when glucose is not accessible. Furthermore, the influence of this diet on AD-associated biomarkers and other related risk factors is discussed. To the best of our knowledge, this is the first review article discussing recent studies on how different ketogenic diets or supplementations can affect these putative AD biomarkers and other AD related risk factors.
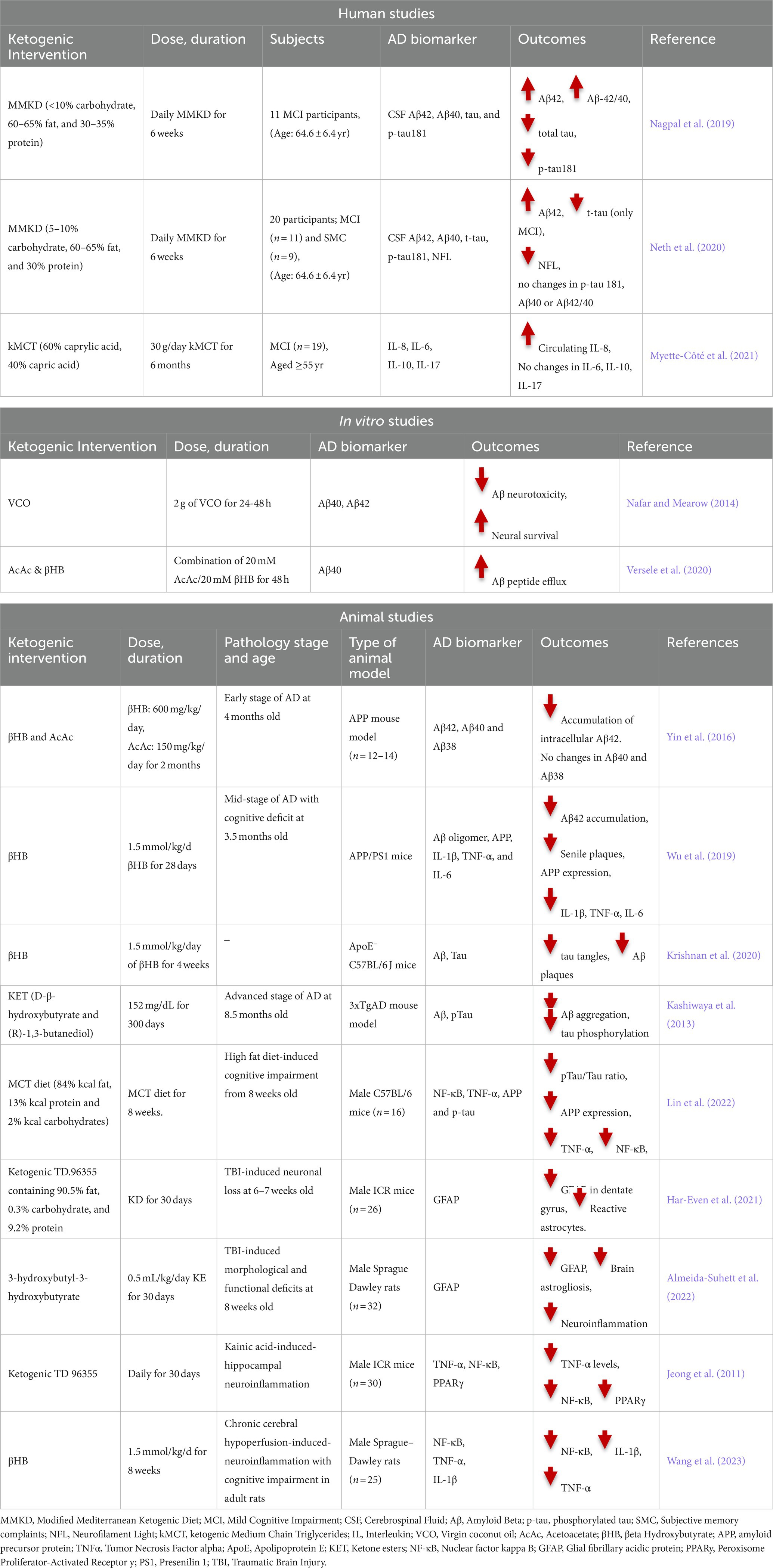
Table 1. Human, in vitro and animal studies on the association between ketogenic intervention and AD biomarkers.
Dietary approaches, with their holistic properties, have gained much attention over the last 4 decades (Mazzucca et al., 2021). Evidence has shown that dietary approaches can prevent or treat chronic diseases such as cardiovascular diseases, cancer, chronic respiratory diseases, and diabetes (Ojo, 2019). A ketogenic diet is a high-fat, moderate protein and low-carbohydrate diet primarily used for the treatment of drug-resistant epilepsy (Jiang et al., 2022). In this diet, the total calories are largely obtained from fat with protein and carbohydrates making a relatively lower contribution (D’Andrea Meira et al., 2019).
Based on the percentage of fat, carbohydrate and protein, ketogenic diets are categorized into different groups which include the following: Classic Ketogenic Diet (CKD), traditional MCT diet, modified MCT diet (Schwartz et al., 1989) and Modified Mediterranean Ketogenic Diet (MMKD) (Nagpal et al., 2019). CKD is typically comprised of 90% fat, 7% protein and 3% carbohydrate and is the most stringent ketogenic diet. Due to its anticonvulsant properties, CKD was used for treating epilepsy (Bough and Rho, 2007; Ferraris et al., 2021; Pietrzak et al., 2022). Two alternative forms of CKD are traditional MCT (60% MCT oil, 21% proteins and 19% carbohydrates) and modified MCT (30% MCT oils, 40% long chain saturated fat, 11% proteins and 19% carbohydrates) diets which consist of ketogenic kMCT (Schwartz et al., 1989; Neal et al., 2009). MMKD encompasses 60–65% fat, less than 10% carbohydrate, and 30–35% protein (Nagpal et al., 2019).
kMCT was first recognized by Huttenlocher in 1971 as a more tolerant and palatable form of CKD (Huttenlocher et al., 1971). MCT is a 6- to 12-chain-length carbon atom found abundant in coconut, palm kernel, and mammalian milk (Taylor et al., 2019; Mett and Müller, 2021). Based on their chain length, MCT is classified into hexanoic acid (caproic acid; C6), octanoic acid (caprylic acid; C8), decanoic acid (capric acid; C10), and dodecanoic acid (lauric acid; C12) (Nimbkar et al., 2022). Due to the higher capric/caprylic content with a higher potential to produce KBs, MMKD and kMCT have been suggested to have greater ketogenic properties than CKD (Huttenlocher, 1976; St-Pierre et al., 2019). Unlike longer chain fatty acids which require specific transporters such as CD36, fatty acid transport proteins (FATPs) and carnitine shuttle (Heidt et al., 2023), medium chain fatty acid (MCFA) arising from MCT do not rely on specific transporters to pass through the mitochondrial membrane (Miyagawa et al., 2018; Heidt et al., 2023). Through passive diffusion, MCFA can be easily and directly transported into the mitochondrial matrix (Heidt et al., 2023), and enhances beta-oxidation (β-oxidation) rate in hepatocytes, which ultimately increases the serum KB concentration (Ameen et al., 2022). In response to the increased levels of KBs, brain energy metabolism and cognitive functions are improved significantly (Ameen et al., 2022). Increased levels of KB and brain energy might be important especially for the treatment of patients with AD, impaired brain energy metabolism and cognitive dysfunction (Krikorian et al., 2012).
Although glucose is a primary energy source for the brain, KBs provide up to 60% of brain energy during glucose restrictions (Pietrzak et al., 2022). MCT ketogenic diet mimics fasting-associated metabolisms, during which glucose is replaced with fatty acids (Augustin et al., 2018; Włodarek, 2019). Similar to fasting, MCT- and any kind of ketone-rich diet induce nutritional ketosis (Włodarek, 2019). This is generally characterized by elevated concentration of KBs from a normal range (~0.5 mM) to higher levels (3 mM) which considers an optimal range of KBs in serum (Harvey et al., 2019). This increased level of serum KBs is achieved via shortage of carbohydrates (Harvey et al., 2019). The metabolic pathway of ketone bodies from synthesis in liver and astrocytes to energy generation in the brain is shown in Figure 1. Shortly after the consumption of a ketogenic meal, MCT is hydrolyzed into MCFA. Via portal circulation, MCFAs are transported into the liver and in the hepatocytes, they undergo β-oxidation, which are converted to acetyl-CoA, thereby initiating ketogenesis to yield KBs. Three types of KBs, including beta-hydroxybutyrate (βHB), acetoacetate (AcAc), and acetone, are generated from the hydrolyzation of fatty acids (Watanabe et al., 2020). These KBs are produced in the liver as the primary site of ketone synthesis, albeit they cannot be utilized by hepatocytes as the liver does not have ketolysis-related enzymes (enzyme Oxct1/SCOT1) to metabolize Acetoacetyl-CoA (AcAc-CoA) (Bendridi et al., 2022). The newly synthesized KBs flow to the extra-hepatic tissues, including brain (Bendridi et al., 2022). As their levels increase in the bloodstream, KBs enter the brain via monocarboxylate transporters located in Blood Brain Barriers (BBB) (Jensen et al., 2020). KBs are delivered to neurons via different types of monocarboxylate transporters localized in astrocytes and neurons. Monocarboxylate transporters 1, 3 and 4 expressed in astrocytes and monocarboxylate transporter 2 expressed in neurons are responsible for KB transportations (Ardanaz et al., 2022). However, the expression of these fundamental transporters, as the brain bioenergetic carriers, is reduced in AD pathology (Ding et al., 2020). In the healthy individuals, after KBs were transported into the neurons, inside the mitochondria, they induce ketolysis. KBs are then converted to Acetyl-CoA, which enters the tricarboxylic acid cycle to produce energy for the neurons (Jensen et al., 2020).
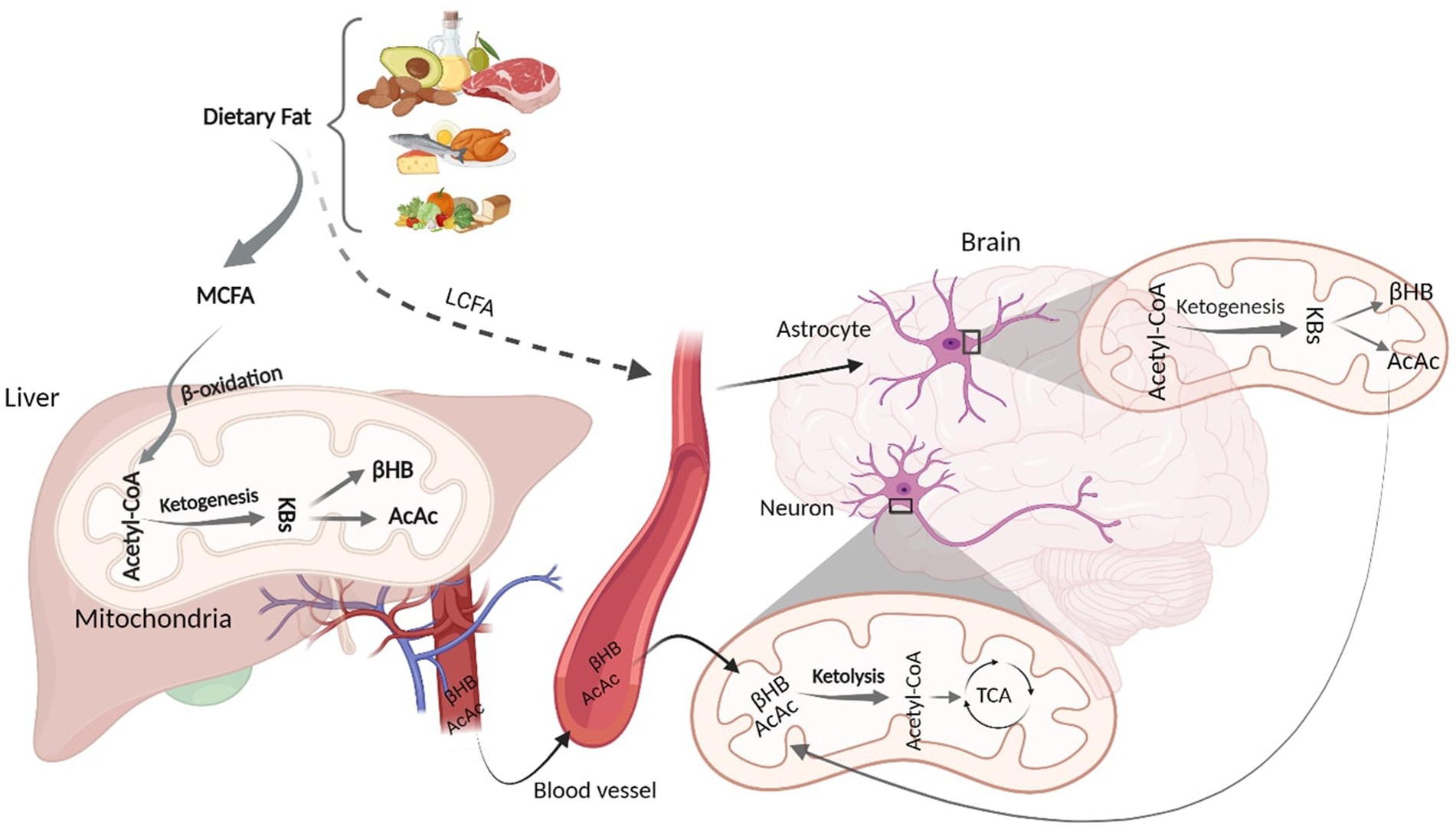
Figure 1. Schematic figure of Ketone Body Synthesis and Metabolism. Dietary MCT is hydrolysed into MCFA, which undergoes β-oxidation to produce acetyl-CoA. In the liver mitochondria, excessive acetyl-CoA induces ketogenesis and produces KBs (βHB and AcAc). Via circulation, KBs enter the brain and inside the neural mitochondria, they induce ketolysis to produce acetyl-CoA. LCFAs that bypass liver metabolism goes to the astrocytes and initiate β-oxidation, generating acetyl-CoA that undergoes ketogenesis to provide surplus KBs, which go to the neurons to furnish further energy. KD, Ketogenic Diet; MCFA, Medium Chain Fatty Acid; KBs, Ketone Bodies; βHB, βeta Hydroxybutyrate; AcAc, Acetoacetate; TCA, Tricarboxylic Acid. Adapted from Biorender.com.
Although hepatocytes are the primary site of KBs production, to a lesser extent, other extrahepatic tissues, such as astrocytes, have the capacity to produce KBs from some fatty acids (long-chain; >12 carbons) (Takahashi, 2020). Both in vitro and animal studies have demonstrated that astrocytes can synthesize KBs due to their ability to oxidize fatty acids (Yudkoff et al., 1997; Yoon and Jo, 2012). It has been found that some fatty acids can bypass liver metabolism and undergo ketogenesis in astrocytes (Yudkoff et al., 1997; Yoon and Jo, 2012; Nonaka et al., 2016). Astrocytes are the main site of mitochondrial β-oxidation and are the only source of KB generation in the brain (Yang et al., 2022). Fatty acids, transported to the astrocytes, can initiate β-oxidation to produce acetyl-CoA. The produced acetyl-CoA undergoes ketogenesis to provide surplus KBs (Nonaka et al., 2016; Silva et al., 2022), which then moves to the neighboring neurons via monocarboxylate transporters. In the neurons, KBs undergo ketolysis to produce Acetyl-CoA to make further energy fuels for the brain (Nonaka et al., 2016; Jensen et al., 2020; Takahashi, 2020). This can ameliorate the energy crisis in the brain when glucose is not accessible (Poff et al., 2021). Therefore, an adequate and continuous brain energy supply provided by KBs can repair the brain metabolism (Jensen et al., 2020). In addition to improved brain energy metabolism, ketogenic diets are associated with alteration in AD CSF biomarkers (Neth et al., 2020). However, the mechanism of action of ketogenic diets in altering AD biomarker levels remains to be elucidated.
The beneficial effects of ketogenic diets on cognitive performance have been reported widely in healthy individuals as well as those with mild, moderate, and severe AD (Fortier et al., 2019, 2021; Xu et al., 2020; Yomogida et al., 2021; Juby et al., 2022). In two human studies, 30 g/day of kMCT containing 12% Captex 355 (60% caprylic acid, 40% capric acid) mixed with lactose-free skim milk improved executive function (Fortier et al., 2019, 2021). An improvement in episodic memory, processing speed and language has been reported in participants with MCI after a 6-month treatment with kMCT (Fortier et al., 2019, 2021). An average dosage of 25.2 g of MCT containing 99.3% caprylic acids, 0.6% capric acids, and 0.1% lauric acid for 4 months significantly improved the cognitive performance in AD patients (Juby et al., 2022).
Daily consumption of a jelly preparation containing 17.3 g MCT within 3 months showed improved cognitive ability in mild to moderate AD patients (Xu et al., 2020). Meiji817-B is a MCT meal containing ketogenic milk with 30.3 g caprylic acid and 9.8 g capric acid per 100 g total fat (Yomogida et al., 2021). Meiji817-B exhibited improved executive function, including working memory or inhibitory control in healthy elderly subjects (Yomogida et al., 2021). These cognitive benefits were positively associated across various ketogenic diets including MCT oil, MCT powder and MCT jelly (Fortier et al., 2019, 2021; Xu et al., 2020; Yomogida et al., 2021; Juby et al., 2022). However, there is a paucity of information on the effects of this diet on AD CSF or plasma biomarkers (Table 1). Therefore, herein, we aim to review the novel studies on the effects of ketogenic diet on several AD fluid biomarkers such as Aβ, tau, GFAP, and NFL as well as to explore their involvement in other AD related risk factors.
Amyloid plaque, which is the extracellular abnormal deposition of Aβ, is a major neuropathological hallmark of AD (Murpy and LeVine III, 2010; Qiu et al., 2015; Moore et al., 2018; Tarawneh, 2020). Increased brain Aβ load along with reduced concentration of Aβ in the CSF and plasma have been widely reported (Hansson et al., 2019; Zaretsky et al., 2022; Chatterjee et al., 2023).
BBB is essential in maintaining Aβ metabolism, and any abnormalities in BBB might impair Aβ normal transport, thereby causing Aβ accumulation and deposition (Wang et al., 2021). It has been demonstrated that the expression of different Aβ transporters in BBB is decreased in the AD brain (Wang et al., 2021). Three major proteins, including low-density lipoprotein receptor-related protein 1 (LRP1), AΒCB1 as P-glycoprotein (P-gp), and phosphatidylinositol-binding clathrin assembly protein (PICALM) have a pivotal role in the efflux of Aβ peptides across BBB (Storck et al., 2018). Reduced LRP1 and P-gp, as the major Aβ transporters across the BBB, further contributes to the poor clearance of brain Aβ (Figure 2). In contrast, increased KBs can facilitate the efflux of Aβ peptides across a human in vitro BBB model by enhancing LRP1, PICALM, and p-gp (Figure 3). This improves the Aβ transportation and clearance resulting in less Aβ plaque deposition and slower release of soluble Aβ (Versele et al., 2020).

Figure 2. The Schematic Figure of Various Pathological Status of AD Biomarkers in the Fluid. Increased Aβ plaques inside the brain suppress the Aβ clearance and reduce Aβ efflux. This mitigates 3 Aβ transporters including LRP1, RAGE and P-gp within the BBB. Aβ deposition in the brain leads to reduced CSF and plasma Aβ. Injured neurons increase the levels of CSF and blood p-tau. Reactive astrogliosis increases GFAP levels in both CSF and blood. Axonal neural damage also releases higher NFL. Although disruption of these biomarkers can be found in both CSF and blood, their concentrations are significantly higher in the CSF ( ) rather than on blood (
) rather than on blood ( ). In addition, Mitochondrial deficits, neuroinflammation and apoptosis are all closely linked to the AD pathology. Aβ, Amyloid Beta; GFAP, Glial fibrillary acidic protein; NFL, Neurofilament Light; ROS, Reactive Oxygen Species; NF-κB, Nuclear factor kappa B; TNFα, Tumor Necrosis Factor alpha; IL, Interleukin; TGF, Transforming Growth Factor; BAX, Bcl-2-associated X; PPAR, Peroxisome Proliferator-Activated Receptor; P-gp, P-glycoprotein; LRP1, lipoprotein receptor-related protein-1; PICALM, Phosphatidylinositol-binding clathrin assembly protein; BBB, Blood Brain Barrier. Adapted from Biorender.com.
). In addition, Mitochondrial deficits, neuroinflammation and apoptosis are all closely linked to the AD pathology. Aβ, Amyloid Beta; GFAP, Glial fibrillary acidic protein; NFL, Neurofilament Light; ROS, Reactive Oxygen Species; NF-κB, Nuclear factor kappa B; TNFα, Tumor Necrosis Factor alpha; IL, Interleukin; TGF, Transforming Growth Factor; BAX, Bcl-2-associated X; PPAR, Peroxisome Proliferator-Activated Receptor; P-gp, P-glycoprotein; LRP1, lipoprotein receptor-related protein-1; PICALM, Phosphatidylinositol-binding clathrin assembly protein; BBB, Blood Brain Barrier. Adapted from Biorender.com.
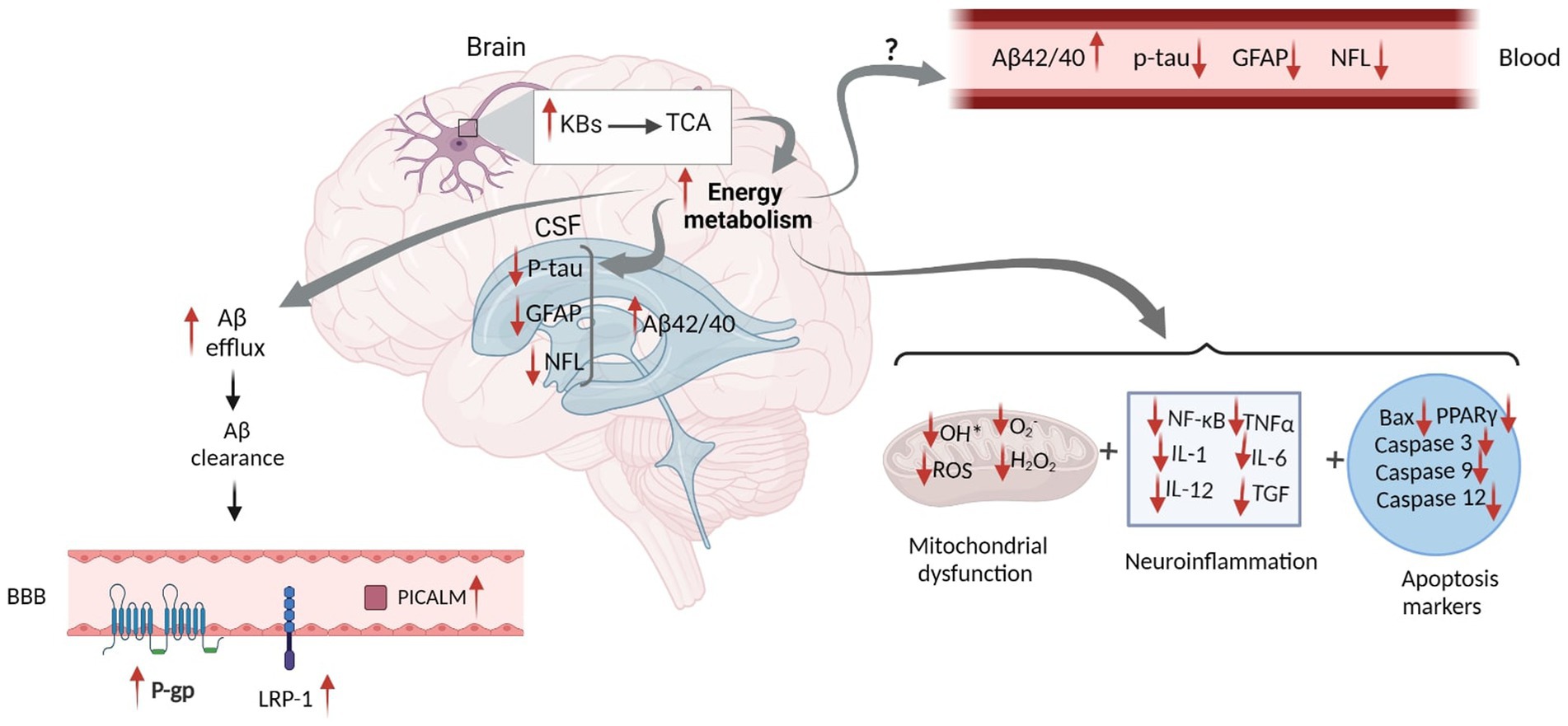
Figure 3. Proposed Schematic Figure for the Effects of Ketogenic Intervention on Different AD Related Markers in the CSF and Blood. KBs in the neural mitochondrial go to the TCA cycle, which increases energy metabolism. This increases Aβ efflux and facilitates Aβ clearance in the brain. Higher Aβ clearance across the BBB leads to the elevation of LRP-1, P-gp, and PICALM proteins, which altogether result in the increased concentration of Aβ in the CSF. Increased energy metabolism might reduce tau hyperphosphorylation, GFAP expression, and NFL release in the brain. However, the mechanism by which KBs can eliminate their levels in the CSF and blood has not yet been discovered. In addition, increased energy metabolism might actively impact mitochondrial function and reduce neuroinflammation and apoptosis markers. Aβ, Amyloid Beta; GFAP, Glial fibrillary acidic protein; NFL, Neurofilament Light; ROS, Reactive Oxygen Species; NF-κB, Nuclear factor kappa B; TNFα, Tumor Necrosis Factor alpha; IL, Interleukin; TGF, Transforming Growth Factor; BAX, Bcl-2-associated X; PPAR, Peroxisome Proliferator-Activated Receptor; P-gp, P-glycoprotein; LRP1, lipoprotein receptor-related protein-1; PICALM, Phosphatidylinositol-binding clathrin assembly protein; BBB, Blood Brain Barrier. Adapted from Biorender.com.
Increased Aβ efflux reduces Aβ plaque deposition and mitigates soluble Aβ (Wang et al., 2021). Reduced levels of soluble oligomer Aβ42 was reported followed by the combined treatment of AcAc and βHB (Yin et al., 2016). An in vitro study showed a significant decline in neurotoxicity due to lowered Aβ plaque deposition and reduced soluble Aβ after consuming coconut oil (CoOil) (Nafar and Mearow, 2014). βHB therapy (1.5 mmol/kg/d) in AD model of mice for 28 days suppressed APP expression, enhanced the expression levels of neprilysin, as a degradation enzyme for Aβ, reduced number of senile amyloid plaques, and mitigated soluble and insoluble Aβ42 and Aβ40 (Wu et al., 2019).
Increased oxidative stress and changes in brain energy availability are associated with impaired ATP-sensitive potassium (KATP) channels, found in both glia and neurons. KATP channels known as metabolic sensors are changed across the AD continuum (Grizzanti et al., 2022). Activation of Kir6.2, one of the main subunits of KATP channels, is increased as Aβ pathology is elevated (Grizzanti et al., 2022). In an animal study, APP/PSE1 mice knocking out Kir6.2, showed no significant increase in Aβ pathology, while activation of KATP channel showed Aβ deposition (Grizzanti et al., 2023). On the other hand, in an in vitro study, a ketone cocktail (BHB and AcAc; each 1 mM) treatment through their interactions with KATP channels guaranteed neural survival, increase ATP production (Kim et al., 2015; Pietrzak et al., 2022) and decrease Aβ aggregations (Pietrzak et al., 2022).
In a human trial, a 6-week MMKD intervention containing 60–65% fat, 30% protein and 5–10% carbohydrate potentially increased CSF Aβ42 and Aβ42/tau ratio in the at-risk adults compared to control groups who consumed 55–65% carbohydrate, 15–20% fat, and 20–30% protein (Neth et al., 2020). Likewise, in another human study, compared to cognitively unimpaired participants, MCT-treated participants showed a significantly increase CSF Aβ40 and Aβ42 over a six-week MMKD therapy containing >10% carbohydrate, 60–65% fat, and 30–35% (Nagpal et al., 2019). Accordingly, it is suggested that KBs arising from various types of ketogenic interventions can decrease Aβ neurotoxicity, reduce Aβ aggregation and modulate Aβ peptides in the circulation. Ketone molecules are also able to increase Aβ efflux and mitigate soluble Aβ.
Tau protein, a core hallmark of AD, is the main constituent of the paired helical filaments (PHF), which forms neurofibrillary tangles (NFTs) in the AD brain (Serrano-Pozo et al., 2011). Increased CSF and plasma phosphorylated tau, commences decades before the clinical presentation of the disease (Rajmohan and Reddy, 2017; Chatterjee et al., 2022). However, the neuroprotective antioxidant properties of compounds present in some ketogenic diets can ameliorate abnormal tau aggregation and induce neural survival (Guo et al., 2013; Baek et al., 2020; Krishnan et al., 2020).
There are few evidence supporting neuroprotective features induced by ketogenic diet on tau biomarkers, though some animal studies and only 2 human studies were conducted to show their association. For example, 4 weeks βHB therapy, in the C57BL/6 mice models of ApoE-deficient AD, which causes progressive p-tau Ser202/Thr205 accumulation, significantly ameliorated tau tangles colocalized in the hippocampal region of ApoE4 transgenic mice (Krishnan et al., 2020). This significantly reduced the risk of AD progression in these mice (Krishnan et al., 2020). Increased number of intracellular p-tau in the amygdala, subiculum, CA1 and CA3 of the hippocampus in AD male 3xTgAD mice models has been modified followed by taking a ketone ester diet comprising of D-β-hydroxybutyrate and (R)-1,3-butanediol (Kashiwaya et al., 2013). Prolonged consumption (16 weeks) of a high-fat-high cholesterol diet in C57BL/6 mice model of AD showed hyperphosphorylation of p-tau S396 and increased neuroinflammation in cortex and hippocampus (Lin et al., 2022). Albeit, an eight-week treatment of MCT diet composed of 84% fat, 2% carbohydrates and 13% protein significantly reduced the ratio of p-tau S396 /total-tau (t-tau) in these regions (Lin et al., 2022). MCT mitigated the hyperphosphorylation of p-tau S396 and reduced neuroinflammation (Lin et al., 2022).
A decline in CSF t-tau concentration was found in the MCI subjects after a 6-week consumption of MMKD (Neth et al., 2020). However, the same study showed no significant changes in the levels of p-tau181 in the subjective memory complainer (SMC) group (Neth et al., 2020). Compared to American Heart Association Diet (AHAD), a low-fat and higher-carbohydrate diet, MMKD over a 6-week therapy significantly impeded CSF t-tau in MCI or SMC patients (Nagpal et al., 2019). Therefore, it can be suggested that the levels of tau in some isoforms is reduced followed by increased levels of KBs in the circulation.
AD pathogenesis is not exclusively limited to the formation of Aβ plaque and tau phosphorylation. It includes some other factors that contribute to neuropathological processes (Pereira et al., 2021). Astroglial-dependent toxicity plays a leading role in the development of AD pathology. Astrogliopathy refers to hyperactivation, astroglial atrophy and loss of function due to the destruction of adjacent neurons (Verkhratsky et al., 2019). In AD, reactive astrocytes acquire neurotoxicity arising from astrocyte hypertrophy (Perez-Nievas and Serrano-Pozo, 2018), which can provide an anatomical substrate for the aberrant growth of newborn dentate granule cells (Robinson et al., 2016). Intermediate filament (IF) cytoskeleton changes lead to the overexpression of IF proteins such as GFAP, an index for astroglia activation, which gradually increases followed by any neurodegenerative injuries (Smit et al., 2021). Astrogliopathy can affect the level of biomarkers and can be found in the early stages of AD (Verkhratsky et al., 2019; Pereira et al., 2021). Astrocytic phagocytosis also mediates the Aβ clearance in the brain through the influx and degeneration of soluble form of Aβ (Frost and Li, 2017). It has been reported that even with a slight deficiency or decline in Aβ clearance, neurotoxicity occurs, which is correlated to a higher risk of AD (Yoon and Jo, 2012). In astrogliosis, the levels of some biomarkers such as GFAP (Chatterjee et al., 2021) and vimentin (Dai et al., 2023) are elevated significantly.
Higher levels of GFAP in the hippocampus and cortex have been reported caused by neural loss, cognitive and memory deficits in a TBI mouse model (Har-Even et al., 2021). However, a 30-day treatment of ketogenic diet (90.5% fat, 9.2% protein, and 0.3% carbohydrate) attenuated neural loss, and improve memory function through mitigating reactive astrocytes. GFAP concentration in the dentate gyrus but not in the cortex was significantly reduced, followed by an increase in blood KBs levels (Har-Even et al., 2021).
Increased levels of βHB significantly reduced the hyperactivation of astrocytes and three other hyperactivated microglial markers, including ionized calcium-binding adaptor molecule 1 (Iba-1), a M1 microglial marker (CD16/32), and a marker of macroglia (CD68) (Zhang et al., 2020). Higher levels of GFAP in response to regional astrogliosis in the mouse hypothalamus and the hippocampal network is associated with memory decline and further neural damage (Bondan et al., 2019). However, by producing KBs, ketone monoester and 3-hydroxy butyl-3-hydroxybutyrate provides an alternative brain fuel, which compensates for the reduction in glucose utilization, and combats astrogliosis, and microgliosis in the prefrontal cortex, cortex, amygdala, and hippocampus (Le Foll and Levin, 2016; Morris et al., 2020; Almeida-Suhett et al., 2022). Thirty days treatment with 3-hydroxybutyl-3-hydroxybutyrate (0.5 mL/kg/day) were able to reduce prominent GFAP positive reactive astrocytes in the TBI-induced behavioral and neuropathological alterations (Almeida-Suhett et al., 2022). The neuroprotective mechanism of KBs is a possible mechanism that ultimately changes the levels of GFAP (Figure 3).
It has been shown that 8 weeks intervention therapy with MCT composed of 84% fat, 2% carbohydrates, and 13% protein could significantly decrease GFAP expression in the cortical and hippocampal regions of the C57BL/6 mice brain (Lin et al., 2022). This effect was ascribed to the presence of caprylic acid and capric acid in the MCT diet, which significantly elevated βHB in the circulation (Lin et al., 2022). Therefore, it is suggested that βHB or combined KBs arising from different types of KDs mitigate reactive astrogliosis and reduce the levels of GFAP in circulation.
NFL, a neuronal cytoplasmic biomarker, has emerged as one of the most promising candidates for the diagnosis and progression of neurodegenerative disorder (Dhiman et al., 2020). In neurological disorders including inflammatory, neurodegenerative, traumatic, and vascular diseases, the release of this biomarker is highly increased in response to severe axonal damage (Gaetani et al., 2019). Elevated NFL levels correlate with poorer cognitive performance, brain hypometabolism, and atrophy, making this biomarker a promising candidate for detecting neurodegeneration and AD (Mattsson et al., 2017).
Little is known about the association between ketogenic intervention and changes in NFL concentrations. The modified ketogenic diet may impact axonal neural injuries (Neth et al., 2020). Six-week treatment with MMKD could significantly reduce CSF NFL biomarkers in the older populations with MCI, suggesting ketogenic intervention by inducing ketosis can constructively impact neurodegeneration-related injuries. While only one human study has been conducted on the impact of KDs on NFL levels (Neth et al., 2020), it is proposed that KBs can mitigate NFL levels in the circulation.
In addition to aforementioned biomarkers with prognostic and diagnostic value, AD is characterized by increased multiple metabolic interactions and comorbidities that promote its progression (Karikari et al., 2020; Verberk et al., 2022). As the disease progresses, AD patients exhibit further abnormalities, such as down regulation of neurotrophic factors including brain-derived neurotrophic factor (BDNF) through dysregulation of the glutamatergic N-methyl-D-aspartate receptor (NMDAR) which can cause Aβ-induced neuronal loss and dendritic atrophy (Meng et al., 2013). Downregulation of BDNF as a potential diagnostic biomarker is manifested during prodromal stage to severe AD (Bessi et al., 2020). On the contrary, 14 days injection of 100 mg/kg AcAc showed a higher expression of hippocampal BDNF in the mice model of familial AD (Wu et al., 2022), which is assumed to be due to neuroprotective properties induced by AcAc (Murugan and Boison, 2020; Zhang et al., 2023). Expression of BDNF increased followed by 2 weeks intermittent fasting in the mouse models of Parkinson disease (Ojha et al., 2023). In addition, in a human study on 15 healthy subjects, combined caprylic acid (20 g) and coconut oil (30 g) after 4 h significantly increased serum levels of precursor BDNF, but not mature BDNF (Norgren et al., 2021).
In 2004, the mitochondrial cascade hypothesis was reported, describing it as a prerequisite that leads to disease progression in AD (Swerdlow and Khan, 2004). The percentage of mitochondrial dysfunction and depolarization is increased with age which ultimately increases the level of free radicals and insoluble Aβ from APP (Swerdlow and Khan, 2004). Increased levels of free radicals, such as Reactive Oxygen Species (ROS) and hydrogen peroxide (H2O2) are associated with cellular oxidative damage and disruption of cellular integrity (Agrawal and Jha, 2020). One of the main events after increased oxidative stress is DNA damage and consequently cell death which is considered to be one of the main events in neurodegeneration (Shadfar et al., 2022, 2023). However, the effects of ketogenic diet on oxidative stress and DNA damage in AD models have not yet been investigated.
It has been revealed that through reducing excessive levels of H2O2-induced neural injuries, MCFA capric acid can significantly suppress intracellular oxidative stress in the neuroblastoma cell line (Mett and Müller, 2021). Capric acid significantly inhibits the natural release of cellular H2O2 and reduces ROS levels (Mett and Müller, 2021).
In addition to mitochondrial dysfunction and surplus ROS production, several apoptosis-associated mediators such as p38, p21, mitogen-activated protein kinase (MAPK), and caspase 2, 3 and 9 can worsen the pathology of disease (Obulesu and Lakshmi, 2014; Misrani et al., 2021). Inversely, βHB can modulate neural apoptosis induced by low glucose accessibility due to mitochondrial dysfunction (Lin et al., 2022). βHB not only provides an alternative energy fuel for the brain, but also modulates cellular signaling transduction related to apoptotic pathogenesis (Lin et al., 2022). The antiapoptotic feature of βHB decreased apoptosis-related proteins, including p38 and caspase 3 and increase p-ERK (Lin et al., 2022). Exogenous βHB can suppress overexpression of p53, caspase-3, caspase-9, and caspase-12 in Aβ-induced cell apoptosis in the hippocampal network (Xie et al., 2015).
Pro-inflammatory mediators are other major markers for AD progression (Rajesh and Kanneganti, 2022), tightly coupled with oxidative stress (Agrawal and Jha, 2020). Increased nuclear factor kappa-B (NF-κB), interleukin-1 (IL-1), IL-6, IL-12, transforming growth factor beta (TGFβ), and tumor necrosis factor alpha (TNFα) increase the risk of AD (Su et al., 2016). However, treatment with ketogenic Harlan Teklad TD 96355 diet containing 90.5% fat, 0.3% carbohydrate, and 9.2% protein reduced hippocampal TNF-α and PPARγ activation and down regulate the expression of hippocampal COX-2. Reduced level of these markers results in the reduction of neurotoxicity and neuroinflammation (Jeong et al., 2011). Due to their anti-inflammatory properties, KBs can reduce the expression of pro-inflammatory factors IL-6 (Platero et al., 2020), TNF-α and IL-1β (Wu et al., 2019; Wang et al., 2023). In a human study, 6 months of kMCT drink in MCI patients could significantly increase circulating IL-8 levels with minor side effects (Myette-Côté et al., 2021). The nod-like receptor pyrin domain expression levels containing 3 (NLRP3) inflammasome and pro-inflammatory cytokines such as IL- 1β and TNF-α were significantly reduced after ketone therapy (Zhang et al., 2020). Reduced levels of NLRP3 could inhibit caspase-1 activation and pro-inflammatory pathways and suppress NF-κB which lead to neural survival (Gough et al., 2021). According to these studies, KBs can modulate inflammatory cytokines, reduce free radicals and apoptosis.
Ketone bodies are known to be the main energy source for the brain when glucose is restricted. To date, many studies have examined the effects of ketogenic diet on cognitive function and glucose metabolism from the early preclinical stages to severe AD. However, there are major gaps in the existing literature that need to be addressed. Since a wide variety of intervention dose and duration has been reported, it is crucial to find out the most optimal dose and duration for ketogenic intervention with no/few side effects and maximum tolerability. To improve cognitive outcomes in AD, long-term adherence to ketogenic diet is imperative, therefore identifying a tolerable ketogenic diet that causes limited or few side effects will reduce dropouts and allow for a longer-term adherence.
Secondly, differing fatty acids stimulate differing levels of ketone body production. For instance, compared to LCFAs, MCFAs stimulate greater levels of ketone body production. It is hypothesized that brain energy resulting from increased ketone body response may delay AD progression and may subsequently result in a better outcome for AD biomarkers. So, as different ketogenic diets can provide differing ratios of fatty acids, it is important to consider the composition of fatty acids in the ketogenic diet in future studies.
While studies have shown improvements in cognition and brain energy metabolism following consumption of ketogenic intervention, few studies have reported the impact of ketogenic diets on AD biomarkers. Recent evidence has shown that AD biomarkers are capable of diagnosing disease 15 to 20 years prior to clinical onset. Therefore, more robust clinical studies are needed to investigate the effect of ketogenic diet on AD biomarkers to evaluate its effectiveness as a therapeutic approach to delay AD progression.
Lastly, the exact molecular mechanism on how KBs can make these changes has not yet been determined. As such, future studies are required to investigate whether KBs themselves directly alter the levels of biomarkers or whether it is mediated by its action to alter brain energy metabolism. Therefore, investigation of the mechanisms behind such possible changes should be a research priority for future clinical trials.
The current review of the clinical trials undertaken to date indicates that the majority of ketogenic dietary interventions induce ketosis to produce KBs which ultimately lead to the improvements in cognition. The KBs generated as a result of these interventions provide essential energy for the brain and thereby help to retard neurodegeneration, though more convincing evidence is needed. In addition to changes in brain energy metabolism, KBs can modulate fluid biomarkers associated with AD pathology. Animal, in vitro and some human studies have demonstrated that ketogenic intervention not only modulates AD putative biomarkers, such as Aβ 42/40, p-tau, GFAP and NFL, but also plays an effective role in the improvement of oxidative stress, inflammation, and mitochondrial mechanism. However, blood biomarkers while having the potential as both diagnostic and prognostic markers need considerably more investigation before their significance and contribution to brain health can be determined.
MR: Writing – original draft, Investigation, writing review and editing. MF: Writing – review & editing, Investigation. SE: Writing – review & editing, Investigation. PA: Writing – review & editing. SS: Writing – review & editing. EB: Writing – review & editing, Investigation. HH: Writing – review & editing. SM: Writing – review & editing. MS: Writing – review & editing. PC: Conceptualization, Writing – original draft, Investigation, and Visualization. RT: Writing – review & editing, Investigation and Visualization. CD: Writing – review & editing. MG: Writing – review & editing, Investigation, Conceptualization and Visualization. RM: Writing – review & editing, Conceptualization, Project administration, Supervision.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agrawal, I., and Jha, S. (2020). Mitochondrial dysfunction and Alzheimer’s disease: role of microglia. Front. Aging Neurosci. 12:252. doi: 10.3389/fnagi.2020.00252
Alberti, P., Semperboni, S., Cavaletti, G., and Scuteri, A. (2022). Neurons: the interplay between cytoskeleton, ion channels/transporters and mitochondria. Cells 11:2499. doi: 10.3390/cells11162499
Almeida-Suhett, C., Namboodiri, A. M., Clarke, K., and Deuster, P. A. (2022). The ketone ester, 3-hydroxybutyl-3-hydroxybutyrate, attenuates neurobehavioral deficits and improves neuropathology following controlled cortical impact in male rats. Nutr. Neurosci. 25, 1287–1299. doi: 10.1080/1028415X.2020.1853414
Ameen, A. O., Freude, K., and Aldana, B. I. (2022). Fats, friends or foes: investigating the role of short-and medium-chain fatty acids in Alzheimer’s disease. Biomedicine 10:2778. doi: 10.3390/biomedicines10112778
An, Y., Varma, V. R., Varma, S., Casanova, R., Dammer, E., Pletnikova, O., et al. (2018). Evidence for brain glucose dysregulation in Alzheimer's disease. Alzheimers Dement. 14, 318–329. doi: 10.1016/j.jalz.2017.09.011
Ardanaz, C. G., Ramírez, M. J., and Solas, M. (2022). Brain metabolic alterations in Alzheimer’s disease. Int. J. Mol. Sci. 23:3785. doi: 10.3390/ijms23073785
Arvanitakis, Z., Shah, R. C., and Bennett, D. A. (2019). Diagnosis and Management of Dementia: review. JAMA 322, 1589–1599. doi: 10.1001/jama.2019.4782
Ashton, J. S., Roberts, J. W., Wakefield, C. J., Page, R. M., Maclaren, D. P., Marwood, S., et al. (2021). The effects of medium chain triglyceride (MCT) supplementation using a C8: C10 ratio of 30: 70 on cognitive performance in healthy young adults. Physiol. Behav. 229:113252. doi: 10.1016/j.physbeh.2020.113252
Ashton, N. J., Nevado-Holgado, A. J., Barber, I. S., Lynham, S., Gupta, V., Chatterjee, P., et al. (2019). A plasma protein classifier for predicting amyloid burden for preclinical Alzheimer’s disease. Sci. Adv. 5:eaau7220. doi: 10.1126/sciadv.aau7220
Augustin, K., Khabbush, A., Williams, S., Eaton, S., Orford, M., Cross, J. H., et al. (2018). Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 17, 84–93. doi: 10.1016/S1474-4422(17)30408-8
Ausó, E., Gómez-Vicente, V., and Esquiva, G. (2020). Biomarkers for Alzheimer’s disease early diagnosis. J. Pers. Med. 10:114. doi: 10.3390/jpm10030114
Baek, M. S., Cho, H., Lee, H. S., Lee, J. H., Ryu, Y. H., and Lyoo, C. H. (2020). Effect of APOE ε4 genotype on amyloid-β and tau accumulation in Alzheimer’s disease. Alzheimers Res. Ther. 12, 1–12. doi: 10.1186/s13195-020-00710-6
Barañano, K. W., and Hartman, A. L. (2008). The ketogenic diet: uses in epilepsy and other neurologic illnesses. Curr. Treat. Options Neurol. 10, 410–419. doi: 10.1007/s11940-008-0043-8
Beard, E., Lengacher, S., Dias, S., Magistretti, P. J., and Finsterwald, C. (2022). Astrocytes as key regulators of brain energy metabolism: new therapeutic perspectives. Front. Physiol. 12:825816. doi: 10.3389/fphys.2021.825816
Bendridi, N., Selmi, A., Balcerczyk, A., and Pirola, L. (2022). Ketone bodies as metabolites and Signalling molecules at the crossroad between inflammation and epigenetic control of Cardiometabolic disorders. Int. J. Mol. Sci. 23:14564. doi: 10.3390/ijms232314564
Benedet, A. L., Milà-Alomà, M., Vrillon, A., Ashton, N. J., Pascoal, T. A., Lussier, F., et al. (2021). Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 78, 1471–1483. doi: 10.1001/jamaneurol.2021.3671
Bessi, V., Mazzeo, S., Bagnoli, S., Padiglioni, S., Carraro, M., Piaceri, I., et al. (2020). The implication of BDNF Val66Met polymorphism in progression from subjective cognitive decline to mild cognitive impairment and Alzheimer’s disease: a 9-year follow-up study. Eur. Arch. Psychiatry Clin. Neurosci. 270, 471–482. doi: 10.1007/s00406-019-01069-y
Bondan, E. F., Cardoso, C. V., Martins, M. D. F. M., and Otton, R. (2019). Memory impairments and increased GFAP expression in hippocampal astrocytes following hypercaloric diet in rats. Arq. Neuropsiquiatr. 77, 601–608. doi: 10.1590/0004-282x20190091
Bough, K. J., and Rho, J. M. (2007). Anticonvulsant mechanisms of the ketogenic diet. Epilepsia 48, 43–58. doi: 10.1111/j.1528-1167.2007.00915.x
Chamberlain, K. A., and Sheng, Z. H. (2019). Mechanisms for the maintenance and regulation of axonal energy supply. J. Neurosci. Res. 97, 897–913. doi: 10.1002/jnr.24411
Chatterjee, P., Pedrini, S., Ashton, N. J., Tegg, M., Goozee, K., Singh, A. K., et al. (2022). Diagnostic and prognostic plasma biomarkers for preclinical Alzheimer's disease. Alzheimers Dement. 18, 1141–1154. doi: 10.1002/alz.12447
Chatterjee, P., Pedrini, S., Doecke, J. D., Thota, R., Villemagne, V. L., Doré, V., et al. (2023). Plasma Aβ42/40 ratio, p-tau181, GFAP, and NfL across the Alzheimer's disease continuum: a cross-sectional and longitudinal study in the AIBL cohort. Alzheimers Dement. 19, 1117–1134. doi: 10.1002/alz.12724
Chatterjee, P., Pedrini, S., Stoops, E., Goozee, K., Villemagne, V. L., Asih, P. R., et al. (2021). Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer’s disease. Transl. Psychiatry 11:27. doi: 10.1038/s41398-020-01137-1
Cunnane, S., Nugent, S., Roy, M., Courchesne-Loyer, A., Croteau, E., Tremblay, S., et al. (2011). Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 27, 3–20. doi: 10.1016/j.nut.2010.07.021
D’andrea Meira, I., Romão, T. T., Pires Do Prado, H. J., Krüger, L. T., Pires, M. E. P., and Da Conceição, P. O. (2019). Ketogenic diet and epilepsy: what we know so far. Front. Neurosci. 13:5. doi: 10.3389/fnins.2019.00005
Dai, D. L., Li, M., and Lee, E. B. (2023). Human Alzheimer’s disease reactive astrocytes exhibit a loss of homeostastic gene expression. Acta Neuropathol. Commun. 11:127. doi: 10.1186/s40478-023-01624-8
De La Monte, S. M. (2012). Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs 72, 49–66. doi: 10.2165/11597760-000000000-00000
Dhiman, K., Gupta, V. B., Villemagne, V. L., Eratne, D., Graham, P. L., Fowler, C., et al. (2020). Cerebrospinal fluid neurofilament light concentration predicts brain atrophy and cognition in Alzheimer's disease. Alzheimers Dement 12:e12005. doi: 10.1002/dad2.12005
Dienel, G. A. (2019). Brain glucose metabolism: integration of energetics with function. Physiol. Rev. 99, 949–1045. doi: 10.1152/physrev.00062.2017
Ding, R., Tan, Y., Du, A., Wen, G., Ren, X., Yao, H., et al. (2020). Redistribution of monocarboxylate 1 and 4 in hippocampus and spatial memory impairment induced by long-term ketamine administration. Front. Behav. Neurosci. 14:60. doi: 10.3389/fnbeh.2020.00060
Dorszewska, J., Prendecki, M., Oczkowska, A., Dezor, M., and Kozubski, W. (2016). Molecular basis of familial and sporadic Alzheimer's disease. Curr. Alzheimer Res. 13, 952–963. doi: 10.2174/1567205013666160314150501
Douglas, W., and Scharre, M. (2019). Preclinical, prodromal, and dementia stages of Alzheimer’s disease. Pract. Neurol. 2019, 36–42.
Ferraris, C., Meroni, E., Casiraghi, M. C., Tagliabue, A., De Giorgis, V., and Erba, D. (2021). One month of classic therapeutic ketogenic diet decreases short chain fatty acids production in epileptic patients. Front. Nutr. 8:613100. doi: 10.3389/fnut.2021.613100
Fortier, M., Castellano, C.-A., Croteau, E., Langlois, F., Bocti, C., St-Pierre, V., et al. (2019). A ketogenic drink improves brain energy and some measures of cognition in mild cognitive impairment. Alzheimers Dement. 15, 625–634. doi: 10.1016/j.jalz.2018.12.017
Fortier, M., Castellano, C. A., St-Pierre, V., Myette-Côté, É., Langlois, F., Roy, M., et al. (2021). A ketogenic drink improves cognition in mild cognitive impairment: results of a 6-month RCT. Alzheimers Dement. 17, 543–552. doi: 10.1002/alz.12206
Frost, G. R., and Li, Y.-M. (2017). The role of astrocytes in amyloid production and Alzheimer's disease. Open Biol. 7:170228. doi: 10.1098/rsob.170228
Gaetani, L., Blennow, K., Calabresi, P., Di Filippo, M., Parnetti, L., and Zetterberg, H. (2019). Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry 90, 870–881. doi: 10.1136/jnnp-2018-320106
Gough, S. M., Casella, A., Ortega, K. J., and Hackam, A. S. (2021). Neuroprotection by the ketogenic diet: evidence and controversies. Front. Nutr. 8:782657. doi: 10.3389/fnut.2021.782657
Goyal, M. S., Blazey, T., Metcalf, N. V., Mcavoy, M. P., Strain, J. F., Rahmani, M., et al. (2023). Brain aerobic glycolysis and resilience in Alzheimer disease. Proc. Natl. Acad. Sci. 120:e2212256120. doi: 10.1073/pnas.2212256120
Grizzanti, J., Moritz, W. R., Pait, M. C., Stanley, M., Kaye, S. D., Carroll, C. M., et al. (2022). Kir6. 2-containing KATP channels are necessary for glucose dependent increases in amyloid-beta and Alzheimer’s-related pathology. bioRxiv 2002:481215. doi: 10.1101/2022.02.20.481215
Grizzanti, J., Moritz, W. R., Pait, M. C., Stanley, M., Kaye, S. D., Carroll, C. M., et al. (2023). KATP channels are necessary for glucose-dependent increases in amyloid-β and Alzheimer’s disease–related pathology. JCI insight 8:8. doi: 10.1172/jci.insight.162454
Gunes, S., Aizawa, Y., Sugashi, T., Sugimoto, M., and Rodrigues, P. P. (2022). Biomarkers for Alzheimer’s disease in the current state: a narrative review. Int. J. Mol. Sci. 23:4962. doi: 10.3390/ijms23094962
Guo, C., Sun, L., Chen, X., and Zhang, D. (2013). Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 8, 2003–2014. doi: 10.3969/j.issn.1673-5374.2013.21.009
Hansson, O., Lehmann, S., Otto, M., Zetterberg, H., and Lewczuk, P. (2019). Advantages and disadvantages of the use of the CSF amyloid β (Aβ) 42/40 ratio in the diagnosis of Alzheimer’s disease. Alzheimers Res. Ther. 11, 1–15. doi: 10.1186/s13195-019-0485-0
Har-Even, M., Rubovitch, V., Ratliff, W. A., Richmond-Hacham, B., Citron, B. A., and Pick, C. G. (2021). Ketogenic diet as a potential treatment for traumatic brain injury in mice. Sci. Rep. 11:23559. doi: 10.1038/s41598-021-02849-0
Harvey, K. L., Holcomb, L. E., and Kolwicz, S. C. Jr. (2019). Ketogenic diets and exercise performance. Nutrients 11:2296. doi: 10.3390/nu11102296
Heidt, C., Fobker, M., Newport, M., Feldmann, R., Fischer, T., and Marquardt, T. (2023). Beta-Hydroxybutyrate (BHB), glucose, insulin, Octanoate (C8), and Decanoate (C10) responses to a medium-chain triglyceride (MCT) oil with and without glucose: a single-center study in healthy adults. Nutrients 15:1148. doi: 10.3390/nu15051148
Hoogmartens, J., Cacace, R., and Van Broeckhoven, C. (2021). Insight into the genetic etiology of Alzheimer's disease: a comprehensive review of the role of rare variants. Alzheimers Dement. 13:e12155. doi: 10.1002/dad2.12155
Hoyer, S. (1992). Oxidative energy metabolism in Alzheimer brain: studies in early-onset and late-onset cases. Mol. Chem. Neuropathol. 16, 207–224. doi: 10.1007/BF03159971
Huttenlocher, P., Wilbourn, A., and Signore, J. (1971). Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology 21, 1097–1103. doi: 10.1212/WNL.21.11.1097
Huttenlocher, P. R. (1976). Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatr. Res. 10, 536–540. doi: 10.1203/00006450-197605000-00006
Javaid, S. F., Giebel, C., Khan, M. A., and Hashim, M. J. (2021). Epidemiology of Alzheimer’s disease and other dementias: rising global burden and forecasted trends. F1000Research 10:425. doi: 10.12688/f1000research.50786.1
Jensen, N. J., Wodschow, H. Z., Nilsson, M., and Rungby, J. (2020). Effects of ketone bodies on brain metabolism and function in neurodegenerative diseases. Int. J. Mol. Sci. 21:8767. doi: 10.3390/ijms21228767
Jeong, E. A., Jeon, B. T., Shin, H. J., Kim, N., Lee, D. H., Kim, H. J., et al. (2011). Ketogenic diet-induced peroxisome proliferator-activated receptor-γ activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp. Neurol. 232, 195–202. doi: 10.1016/j.expneurol.2011.09.001
Jiang, Z., Yin, X., Wang, M., Chen, T., Wang, Y., Gao, Z., et al. (2022). Effects of ketogenic diet on neuroinflammation in neurodegenerative diseases. Aging Dis. 13, 1146–1165. doi: 10.14336/AD.2021.1217
Juby, A. G., Blackburn, T. E., and Mager, D. R. (2022). Use of medium chain triglyceride (MCT) oil in subjects with Alzheimer's disease: a randomized, double-blind, placebo-controlled, crossover study, with an open-label extension. Alzheimers Dement. 8:e12259. doi: 10.1002/trc2.12259
Jurcovicova, J. (2014). Glucose transport in brain-effect of inflammation. Endocr. Regul. 48, 35–48. doi: 10.4149/endo_2014_01_35
Karikari, T. K., Pascoal, T. A., Ashton, N. J., Janelidze, S., Benedet, A. L., Rodriguez, J. L., et al. (2020). Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 19, 422–433. doi: 10.1016/S1474-4422(20)30071-5
Kashiwaya, Y., Bergman, C., Lee, J.-H., Wan, R., King, M. T., Mughal, M. R., et al. (2013). A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer's disease. Neurobiol. Aging 34, 1530–1539. doi: 10.1016/j.neurobiolaging.2012.11.023
Khoury, R., and Ghossoub, E. (2019). Diagnostic biomarkers of Alzheimer’s disease: a state-of-the-art review. Biomark. Neuropsychiatry 1:100005. doi: 10.1016/j.bionps.2019.100005
Kim, D. Y., Abdelwahab, M. G., Lee, S. H., O’neill, D., Thompson, R. J., Duff, H. J., et al. (2015). Ketones prevent oxidative impairment of hippocampal synaptic integrity through KATP channels. PLoS One 10:e0119316. doi: 10.1371/journal.pone.0119316
Klyucherev, T. O., Olszewski, P., Shalimova, A. A., Chubarev, V. N., Tarasov, V. V., Attwood, M. M., et al. (2022). Advances in the development of new biomarkers for Alzheimer’s disease. Transl. Neurodegen. 11, 1–24. doi: 10.1186/s40035-022-00296-z
Krikorian, R., Shidler, M. D., Dangelo, K., Couch, S. C., Benoit, S. C., and Clegg, D. J. (2012). Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 33:425.e19. doi: 10.1016/j.neurobiolaging.2010.10.006
Krishnan, M., Hwang, J. S., Kim, M., Kim, Y. J., Seo, J. H., Jung, J., et al. (2020). β-Hydroxybutyrate impedes the progression of Alzheimer’s disease and atherosclerosis in ApoE-deficient mice. Nutrients 12:471. doi: 10.3390/nu12020471
Kyrtata, N., Emsley, H. C., Sparasci, O., Parkes, L. M., and Dickie, B. R. (2021). A systematic review of glucose transport alterations in Alzheimer's disease. Front. Neurosci. 15:626636. doi: 10.3389/fnins.2021.626636
Le Foll, C., and Levin, B. E. (2016). Fatty acid-induced astrocyte ketone production and the control of food intake. Am. J. Phys. Regul. Integr. Comp. Phys. 310, R1186–R1192. doi: 10.1152/ajpregu.00113.2016
Lin, D.-T., Kao, N.-J., Cross, T.-W. L., Lee, W.-J., and Lin, S.-H. (2022). Effects of ketogenic diet on cognitive functions of mice fed high-fat-high-cholesterol diet. J. Nutr. Biochem. 104:108974. doi: 10.1016/j.jnutbio.2022.108974
Mattsson, N., Andreasson, U., Zetterberg, H., Blennow, K., and Initiative, A. S. D. N. (2017). Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 74, 557–566. doi: 10.1001/jamaneurol.2016.6117
Mazzucca, S., Arredondo, E. M., Hoelscher, D. M., Haire-Joshu, D., Tabak, R. G., Kumanyika, S. K., et al. (2021). Expanding implementation research to prevent chronic diseases in community settings. Annu. Rev. Public Health 42, 135–158. doi: 10.1146/annurev-publhealth-090419-102547
Meng, C., He, Z., and Xing, D. (2013). Low-level laser therapy rescues dendrite atrophy via upregulating BDNF expression: implications for Alzheimer's disease. J. Neurosci. 33, 13505–13517. doi: 10.1523/JNEUROSCI.0918-13.2013
Mett, J., and Müller, U. (2021). The medium-chain fatty acid decanoic acid reduces oxidative stress levels in neuroblastoma cells. Sci. Rep. 11:6135. doi: 10.1038/s41598-021-85523-9
Misrani, A., Tabassum, S., and Yang, L. (2021). Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease. Front. Aging Neurosci. 13:57. doi: 10.3389/fnagi.2021.617588
Miyagawa, Y., Mori, T., Goto, K., Kawahara, I., Fujiwara-Tani, R., Kishi, S., et al. (2018). Intake of medium-chain fatty acids induces myocardial oxidative stress and atrophy. Lipids Health Dis. 17, 1–7. doi: 10.1186/s12944-018-0908-0
Moore, B. D., Martin, J., De Mena, L., Sanchez, J., Cruz, P. E., Ceballos-Diaz, C., et al. (2018). Short Aβ peptides attenuate Aβ42 toxicity in vivo. J. Exp. Med. 215, 283–301. doi: 10.1084/jem.20170600
Morris, G., Maes, M., Berk, M., Carvalho, A. F., and Puri, B. K. (2020). Nutritional ketosis as an intervention to relieve astrogliosis: possible therapeutic applications in the treatment of neurodegenerative and neuroprogressive disorders. Eur. Psychiatry 63:e8. doi: 10.1192/j.eurpsy.2019.13
Murpy, M., and Levine Iii, H. (2010). Alzheimer’s disease and the β-amyloid peptide. J. Alzheimers Dis. 19, 311–323. doi: 10.3233/JAD-2010-1221
Murugan, M., and Boison, D. (2020). Ketogenic diet, neuroprotection, and antiepileptogenesis. Epilepsy Res. 167:106444. doi: 10.1016/j.eplepsyres.2020.106444
Myette-Côté, É., St-Pierre, V., Beaulieu, S., Castellano, C.-A., Fortier, M., Plourde, M., et al. (2021). The effect of a 6-month ketogenic medium-chain triglyceride supplement on plasma cardiometabolic and inflammatory markers in mild cognitive impairment. Prostaglandins Leukot. Essent. Fat. Acids 169:102236. doi: 10.1016/j.plefa.2020.102236
Nafar, F., and Mearow, K. M. (2014). Coconut oil attenuates the effects of amyloid-β on cortical neurons in vitro. J. Alzheimers Dis. 39, 233–237. doi: 10.3233/JAD-131436
Nagpal, R., Neth, B. J., Wang, S., Craft, S., and Yadav, H. (2019). Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer's disease markers in subjects with mild cognitive impairment. EBioMedicine 47, 529–542. doi: 10.1016/j.ebiom.2019.08.032
Neal, E. G., Chaffe, H., Schwartz, R. H., Lawson, M. S., Edwards, N., Fitzsimmons, G., et al. (2009). A randomized trial of classical and medium-chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia 50, 1109–1117. doi: 10.1111/j.1528-1167.2008.01870.x
Neth, B. J., Mintz, A., Whitlow, C., Jung, Y., Sai, K. S., Register, T. C., et al. (2020). Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: a pilot study. Neurobiol. Aging 86, 54–63. doi: 10.1016/j.neurobiolaging.2019.09.015
Nimbkar, S., Leena, M. M., Moses, J., and Anandharamakrishnan, C. (2022). Medium chain triglycerides (MCT): state-of-the-art on chemistry, synthesis, health benefits and applications in food industry. Compr. Rev. Food Sci. Food Saf. 21, 843–867. doi: 10.1111/1541-4337.12926
Nonaka, Y., Takagi, T., Inai, M., Nishimura, S., Urashima, S., Honda, K., et al. (2016). Lauric acid stimulates ketone body production in the KT-5 astrocyte cell line. J. Oleo Sci. 65, 693–699. doi: 10.5650/jos.ess16069
Norgren, J., Daniilidou, M., Kåreholt, I., Sindi, S., Akenine, U., Nordin, K., et al. (2021). Serum proBDNF is associated with changes in the ketone body β-hydroxybutyrate and shows superior repeatability over mature BDNF: secondary outcomes from a cross-over trial in healthy older adults. Front. Aging Neurosci. 13:716594. doi: 10.3389/fnagi.2021.716594
Obulesu, M., and Lakshmi, M. J. (2014). Apoptosis in Alzheimer’s disease: an understanding of the physiology, pathology and therapeutic avenues. Neurochem. Res. 39, 2301–2312. doi: 10.1007/s11064-014-1454-4
Ojha, U., Khanal, S., Park, P.-H., Hong, J. T., and Choi, D.-Y. (2023). Intermittent fasting protects the nigral dopaminergic neurons from MPTP-mediated dopaminergic neuronal injury in mice. J. Nutr. Biochem. 112:109212. doi: 10.1016/j.jnutbio.2022.109212
Ota, M., Matsuo, J., Ishida, I., Hattori, K., Teraishi, T., Tonouchi, H., et al. (2016). Effect of a ketogenic meal on cognitive function in elderly adults: potential for cognitive enhancement. Psychopharmacology 233, 3797–3802. doi: 10.1007/s00213-016-4414-7
Peng, J., Yu, L., Huang, L., Paschoal, V. A., Chu, H., De Souza, C. O., et al. (2023). Hepatic sialic acid synthesis modulates glucose homeostasis in both liver and skeletal muscle. Mol. Metab. 78:101812. doi: 10.1016/j.molmet.2023.101812
Pereira, J. B., Janelidze, S., Smith, R., Mattsson-Carlgren, N., Palmqvist, S., Teunissen, C. E., et al. (2021). Plasma GFAP is an early marker of amyloid-β but not tau pathology in Alzheimer’s disease. Brain 144, 3505–3516. doi: 10.1093/brain/awab223
Perez-Nievas, B. G., and Serrano-Pozo, A. (2018). Deciphering the astrocyte reaction in Alzheimer’s disease. Front. Aging Neurosci. 10:114. doi: 10.3389/fnagi.2018.00114
Phillips, M. C., Deprez, L. M., Mortimer, G., Murtagh, D. K., Mccoy, S., Mylchreest, R., et al. (2021). Randomized crossover trial of a modified ketogenic diet in Alzheimer’s disease. Alzheimers Res. Ther. 13, 1–12. doi: 10.1186/s13195-021-00783-x
Piaceri, I., Nacmias, B., and Sorbi, S. (2013). Genetics of familial and sporadic Alzheimer’s disease. Front. Biosci. Elite E5, 167–177. doi: 10.2741/E605
Pietrzak, D., Kasperek, K., Rękawek, P., and Piątkowska-Chmiel, I. (2022). The therapeutic role of ketogenic diet in neurological disorders. Nutrients 14:1952. doi: 10.3390/nu14091952
Platero, J. L., Cuerda-Ballester, M., Ibáñez, V., Sancho, D., Lopez-Rodríguez, M. M., Drehmer, E., et al. (2020). The impact of coconut oil and epigallocatechin gallate on the levels of IL-6, anxiety and disability in multiple sclerosis patients. Nutrients 12:305. doi: 10.3390/nu12020305
Poff, A. M., Moss, S., Soliven, M., and D'agostino, D. P. (2021). Ketone supplementation: meeting the needs of the brain in an energy crisis. Front. Nutr. 8:659. doi: 10.3389/fnut.2021.783659
Qiu, T., Liu, Q., Chen, Y. X., Zhao, Y. F., and Li, Y. M. (2015). Aβ42 and Aβ40: similarities and differences. J. Pept. Sci. 21, 522–529. doi: 10.1002/psc.2789
Rajesh, Y., and Kanneganti, T.-D. (2022). Innate immune cell death in neuroinflammation and Alzheimer’s disease. Cells 11:1885. doi: 10.3390/cells11121885
Rajmohan, R., and Reddy, P. H. (2017). Amyloid-beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer’s disease neurons. J. Alzheimers Dis. 57, 975–999. doi: 10.3233/JAD-160612
Robinson, C., Apgar, C., and Shapiro, L. A. (2016). Astrocyte hypertrophy contributes to aberrant neurogenesis after traumatic brain injury. Neural Plast. 2016, 1–10. doi: 10.1155/2016/1347987
Rolandi, E., Zaccaria, D., Vaccaro, R., Abbondanza, S., Pettinato, L., Davin, A., et al. (2020). Estimating the potential for dementia prevention through modifiable risk factors elimination in the real-world setting: a population-based study. Alzheimers Res. Ther. 12, 1–9. doi: 10.1186/s13195-020-00661-y
Roy, M., Fortier, M., Rheault, F., Edde, M., Croteau, E., Castellano, C. A., et al. (2021). A ketogenic supplement improves white matter energy supply and processing speed in mild cognitive impairment. Alzheimers Dement. 7:e12217. doi: 10.1002/trc2.12217
Salvadó, G., Milà-Alomà, M., Shekari, M., Ashton, N. J., Operto, G., Falcon, C., et al. (2022). Reactive astrogliosis is associated with higher cerebral glucose consumption in the early Alzheimer’s continuum. Eur. J. Nucl. Med. Mol. Imaging 49, 4567–4579. doi: 10.1007/s00259-022-05897-4
Schwartz, R. H., Eaton, J., Bower, B., and Aynsley-Green, A. (1989). Ketogenic diets in the treatment of epilepsy: short-term clinical effects. Dev. Med. Child Neurol. 31, 145–151. doi: 10.1111/j.1469-8749.1989.tb03972.x
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Shadfar, S., Brocardo, M., and Atkin, J. D. (2022). The complex mechanisms by which neurons die following DNA damage in neurodegenerative diseases. Int. J. Mol. Sci. 23:2484. doi: 10.3390/ijms23052484
Shadfar, S., Parakh, S., Jamali, M. S., and Atkin, J. D. (2023). Redox dysregulation as a driver for DNA damage and its relationship to neurodegenerative diseases. Transl. Neurodegen. 12:18. doi: 10.1186/s40035-023-00350-4
Silva, B., Mantha, O. L., Schor, J., Pascual, A., Plaçais, P.-Y., Pavlowsky, A., et al. (2022). Glia fuel neurons with locally synthesized ketone bodies to sustain memory under starvation. Nat. Metab. 4, 213–224. doi: 10.1038/s42255-022-00528-6
Smit, T., Deshayes, N. A., Borchelt, D. R., Kamphuis, W., Middeldorp, J., and Hol, E. M. (2021). Reactive astrocytes as treatment targets in Alzheimer's disease—systematic review of studies using the APPswePS1dE9 mouse model. Glia 69, 1852–1881. doi: 10.1002/glia.23981
St-Pierre, V., Vandenberghe, C., Lowry, C.-M., Fortier, M., Castellano, C.-A., Wagner, R., et al. (2019). Plasma ketone and medium chain fatty acid response in humans consuming different medium chain triglycerides during a metabolic study day. Front. Nutr. 6:46. doi: 10.3389/fnut.2019.00046
Storck, S. E., Hartz, A. M., Bernard, J., Wolf, A., Kachlmeier, A., Mahringer, A., et al. (2018). The concerted amyloid-beta clearance of LRP1 and ABCB1/P-gp across the blood-brain barrier is linked by PICALM. Brain Behav. Immun. 73, 21–33. doi: 10.1016/j.bbi.2018.07.017
Su, F., Bai, F., and Zhang, Z. (2016). Inflammatory cytokines and Alzheimer’s disease: a review from the perspective of genetic polymorphisms. Neurosci. Bull. 32, 469–480. doi: 10.1007/s12264-016-0055-4
Swerdlow, R. H., and Khan, S. M. (2004). A “mitochondrial cascade hypothesis” for sporadic Alzheimer's disease. Med. Hypotheses 63, 8–20. doi: 10.1016/j.mehy.2003.12.045
Szablewski, L. (2021). Brain glucose transporters: role in pathogenesis and potential targets for the treatment of alzheimer’s disease. Int. J. Mol. Sci. 22:8142. doi: 10.3390/ijms22158142
Takahashi, S. (2020). Metabolic compartmentalization between astroglia and neurons in physiological and pathophysiological conditions of the neurovascular unit. Neuropathology 40, 121–137. doi: 10.1111/neup.12639
Tarawneh, R. (2020). Biomarkers: our path towards a cure for Alzheimer disease. Biomark. Insights 15:117727192097636. doi: 10.1177/1177271920976367
Taylor, M. K., Swerdlow, R. H., and Sullivan, D. K. (2019). Dietary neuroketotherapeutics for Alzheimer’s disease: an evidence update and the potential role for diet quality. Nutrients 11:1910. doi: 10.3390/nu11081910
Verberk, I. M., Misdorp, E. O., Koelewijn, J., Ball, A. J., Blennow, K., Dage, J. L., et al. (2022). Characterization of pre-analytical sample handling effects on a panel of Alzheimer's disease–related blood-based biomarkers: results from the standardization of Alzheimer's blood biomarkers (SABB) working group. Alzheimers Dement. 18, 1484–1497. doi: 10.1002/alz.12510
Verkhratsky, A., Parpura, V., Rodriguez-Arellano, J. J., and Zorec, R. (2019). Astroglia in Alzheimer’s disease. Neuroglia Neurodegen. Dis. 2019, 273–324. doi: 10.1007/978-981-13-9913-8_11
Vermunt, L., Sikkes, S., and Van Den Hout, A. (2022). Duration of preclinical, prodromal and dementia Alzheimer disease stages in relation to age, sex, and APOE genotype. Alzheimers Dement. 15, 888–898. doi: 10.1016/j.jalz.2019.04.001
Versele, R., Corsi, M., Fuso, A., Sevin, E., Businaro, R., Gosselet, F., et al. (2020). Ketone bodies promote amyloid-β1–40 clearance in a human in vitro blood–brain barrier model. Int. J. Mol. Sci. 21:934. doi: 10.3390/ijms21030934
Wang, D., Chen, F., Han, Z., Yin, Z., Ge, X., and Lei, P. (2021). Relationship between amyloid-β deposition and blood–brain barrier dysfunction in Alzheimer’s disease. Front. Cell. Neurosci. 15:695479. doi: 10.3389/fncel.2021.695479
Wang, Z., Li, T., Du, M., Zhang, L., Xu, L., Song, H., et al. (2023). β-Hydroxybutyrate improves cognitive impairment caused by chronic cerebral hypoperfusion via amelioration of neuroinflammation and blood-brain barrier damage. Brain Res. Bull. 193, 117–130. doi: 10.1016/j.brainresbull.2022.12.011
Watanabe, M., Tozzi, R., Risi, R., Tuccinardi, D., Mariani, S., Basciani, S., et al. (2020). Beneficial effects of the ketogenic diet on nonalcoholic fatty liver disease: a comprehensive review of the literature. Obes. Rev. 21:e13024. doi: 10.1111/obr.13024
Włodarek, D. (2019). Role of ketogenic diets in neurodegenerative diseases (Alzheimer’s disease and Parkinson’s disease). Nutrients 11:169. doi: 10.3390/nu11010169
Wu, X.-J., Shu, Q.-Q., Wang, B., Dong, L., and Hao, B. (2022). Acetoacetate improves memory in alzheimer’s mice via promoting brain-derived neurotrophic factor and inhibiting inflammation. Am. J. Alzheimers Dis. Other Dement. 37:153331752211249. doi: 10.1177/15333175221124949
Wu, Y., Gong, Y., Luan, Y., Li, Y., Liu, J., Yue, Z., et al. (2019). BHBA treatment improves cognitive function by targeting pleiotropic mechanisms in a transgenic mouse model of Alzheimer's disease. FASEB J 34, 1412–1429. doi: 10.2139/ssrn.3421582
Xie, G., Tian, W., Wei, T., and Liu, F. (2015). The neuroprotective effects of β-hydroxybutyrate on Aβ-injected rat hippocampus in vivo and in Aβ-treated PC-12 cells in vitro. Free Radic. Res. 49, 139–150. doi: 10.3109/10715762.2014.987274
Xu, Q., Zhang, Y., Zhang, X., Liu, L., Zhou, B., Mo, R., et al. (2020). Medium-chain triglycerides improved cognition and lipid metabolomics in mild to moderate Alzheimer's disease patients with APOE4−/−: a double-blind, randomized, placebo-controlled crossover trial. Clin. Nutr. 39, 2092–2105. doi: 10.1016/j.clnu.2019.10.017
Yang, D., Wang, X., Zhang, L., Fang, Y., Zheng, Q., Liu, X., et al. (2022). Lipid metabolism and storage in neuroglia: role in brain development and neurodegenerative diseases. Cell Biosci. 12, 1–16. doi: 10.1186/s13578-022-00828-0
Yin, J. X., Maalouf, M., Han, P., Zhao, M., Gao, M., Dharshaun, T., et al. (2016). Ketones block amyloid entry and improve cognition in an Alzheimer's model. Neurobiol. Aging 39, 25–37. doi: 10.1016/j.neurobiolaging.2015.11.018
Yomogida, Y., Matsuo, J., Ishida, I., Ota, M., Nakamura, K., Ashida, K., et al. (2021). An fMRI investigation into the effects of ketogenic medium-chain triglycerides on cognitive function in elderly adults: a pilot study. Nutrients 13:2134. doi: 10.3390/nu13072134
Yoon, S.-S., and Jo, S. A. (2012). Mechanisms of amyloid-β peptide clearance: potential therapeutic targets for Alzheimer’s disease. Biomol. Ther. 20, 245–255. doi: 10.4062/biomolther.2012.20.3.245
Yudkoff, M., Daikhin, Y., Nissim, I., Grunstein, R., and Nissim, I. (1997). Effects of ketone bodies on astrocyte amino acid metabolism. J. Neurochem. 69, 682–692. doi: 10.1046/j.1471-4159.1997.69020682.x
Zaretsky, D. V., Zaretskaia, M. V., Molkov, Y. I., and Initiative, A. S. D. N. (2022). Patients with Alzheimer’s disease have increased cellular amyloid uptake. medRxiv 2022:22269196. doi: 10.1101/2022.01.12.22269196
Zhang, N., Liu, C., Zhang, R., Jin, L., Yin, X., Zheng, X., et al. (2020). Amelioration of clinical course and demyelination in the cuprizone mouse model in relation to ketogenic diet. Food Funct. 11, 5647–5663. doi: 10.1039/C9FO02944C
Zhang, X.-X., Tian, Y., Wang, Z.-T., Ma, Y.-H., Tan, L., and Yu, J.-T. (2021). The epidemiology of Alzheimer’s disease modifiable risk factors and prevention. J. Prev Alzheimers Dis. 8, 313–321. doi: 10.14283/jpad.2021.15
Keywords: ketogenic intervention, disease-modifying therapy, Alzheimer’s disease, circulating biomarkers, metabolic interaction, ketogenesis, brain energy fuel
Citation: Ramezani M, Fernando M, Eslick S, Asih PR, Shadfar S, Bandara EMS, Hillebrandt H, Meghwar S, Shahriari M, Chatterjee P, Thota R, Dias CB, Garg ML and Martins RN (2023) Ketone bodies mediate alterations in brain energy metabolism and biomarkers of Alzheimer’s disease. Front. Neurosci. 17:1297984. doi: 10.3389/fnins.2023.1297984
Edited by:
Pradip Kumar Kamat, Augusta University, United StatesReviewed by:
Ankit Seth, Augusta University, United StatesCopyright © 2023 Ramezani, Fernando, Eslick, Asih, Shadfar, Bandara, Hillebrandt, Meghwar, Shahriari, Chatterjee, Thota, Dias, Garg and Martins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralph N. Martins, cmFscGgubWFydGluc0BtcS5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.