
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Neurosci., 24 October 2023
Sec. Neurodegeneration
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1283742
This article is part of the Research TopicTissue Culture to Clinical Relevance: Brain Organoids to Investigate Brain Disorders Rooted in Aging and DevelopmentView all 4 articles
 Jonas Cerneckis1,2
Jonas Cerneckis1,2 Yanhong Shi1,2*
Yanhong Shi1,2*Alzheimer's disease (AD) remains a leading cause of dementia and poses a heavy burden on aging populations worldwide (Long and Holtzman, 2019). Amyloid-targeting antibodies have recently gained accelerated (aducanumab) and full approval (lecanemab) by the Food and Drug Administration, ushering in a new era of AD therapeutics targeting neuronal pathology (Shi et al., 2022). Dysfunction of glial cells has also emerged as a major factor in AD progression, contributing to neuroinflammation, impaired lipid metabolism, myelin breakdown, and other disease-associated phenotypes (De Strooper and Karran, 2016; Long and Holtzman, 2019). Given the species-specific differences between rodent models and humans, the study of glial cell dysregulation in AD has been greatly facilitated by the induced pluripotent stem cell (iPSC) technology that enables differentiation of human glial cells in vitro (Shi et al., 2017; Li and Shi, 2020; Chen et al., 2021b). Moreover, brain organoids—three-dimensional (3D) iPSC-derived human brain-like tissues—recapitulate intricate cell-cell interactions of the human brain that deteriorate in AD (Di Lullo and Kriegstein, 2017; Cerneckis et al., 2023). Although astrocyte- and microglia-containing brain organoids have been widely applied to study glial cell type-specific responses to AD pathology, oligodendrocyte-containing myelin organoids (myelinoids) are yet to be used to model myelin breakdown in AD (Cerneckis et al., 2023). In this opinion, we discuss recent findings on the importance of myelin dysfunction in AD pathogenesis and highlight potential applications of myelinoids to test specific hypotheses in vitro.
The myelin membrane is produced by oligodendrocytes and ensheaths axons to provide electrical insulation, facilitate saltatory conduction of action potentials, and support neuronal metabolism (Lee et al., 2012; Stadelmann et al., 2019). Extensive myelination and enlargement of prefrontal white matter distinguish the human brain from other species but may also confer unique vulnerability to neurological disorders associated with myelin dysfunction (Schoenemann et al., 2005; Bartzokis, 2011, 2012). Myelin volume decreases in aging, which may contribute to the onset and progression of age-related neurodegeneration (Braak and Braak, 1996; Bartzokis, 2011). Indeed, magnetic resonance imaging (MRI) has revealed white matter deficits and inflammation in asymptomatic individuals who later progress to AD (Brickman et al., 2015; Caballero et al., 2018; Wang et al., 2019) and an association between white matter damage and classical amyloid-β (Aβ) and phosphorylated tau (p-tau) pathology (Dean et al., 2017; Wang et al., 2019). Myelin loss (Chen J. F. et al., 2021) and changes in oligodendrocyte gene expression programs (Kenigsbuch et al., 2022; Sadick et al., 2022; Murdock and Tsai, 2023) are also evident in postmortem brain tissue of AD patients and AD mouse models. Single-cell profiling of oligodendrocyte gene expression has revealed aberrant expression of genes required for myelin synthesis, impaired lipid metabolism, and a population of disease-associated oligodendrocytes (DOLs) in AD (Kenigsbuch et al., 2022; Sadick et al., 2022; Murdock and Tsai, 2023). Elucidating whether myelin dysfunction can trigger Aβ and p-tau pathology as an upstream event in AD pathogenesis would provide strong rationale for therapeutic development targeting myelin pathology at early stages of the disease (Bartzokis, 2011). Although the idea of myelin breakdown as an event upstream of Aβ and p-tau pathology was proposed by a neuroscientist George Bartzokis (1956–2014) two decades ago (Bartzokis, 2004, 2011), only recently has experimental evidence been acquired to support this hypothesis (Depp et al., 2023). Depp et al. (2023) have demonstrated that myelin-defective neurons form axonal swellings that accumulate the amyloid precursor protein (APP) processing machinery, leading to increased production of Aβ. These findings suggest that breakdown of myelin can act as an upstream cause of classical AD pathology, whereas limiting myelin dysfunction might be a promising therapeutic strategy to treat cognitive decline and AD (Depp et al., 2023). Indeed, pharmacologically enhancing remyelination with clemastine improves cognitive functions of aging mice (Wang et al., 2020).
Genetic risk factors for AD identified in genome-wide association studies (GWAS) (Wightman et al., 2021), such as the E4 variant of apolipoprotein E (APOE4), also contribute to myelin dysfunction. The detrimental effects of APOE4, the strongest genetic risk factor for AD, have been characterized in different brain cell types, including oligodendrocytes (Lin et al., 2018; Blanchard et al., 2022; Martens et al., 2022). In APOE4 carriers, dysregulation of lipid metabolism and cholesterol accumulation in oligodendrocytes lead to impaired myelination, including formation of thinner myelin sheaths, as compared to APOE3 carriers (Blanchard et al., 2022). Interestingly, 2-hydroxypropyl-β-cyclodextrin, a compound that promotes cholesterol transport, ameliorates cholesterol accumulation in iPSC-derived APOE4 oligodendrocytes, promotes myelination in vitro and in vivo, and improves cognition of APOE4 targeted replacement mice (Blanchard et al., 2022). Conditional removal of APOE4 in neurons is associated with decreased expression of DOL cluster genes and improved myelination, indicating that ApoE4 secreted by other cell types also influences oligodendrocyte phenotypes (Koutsodendris et al., 2023). We have recently shown that the clusterin (CLU, also known as apolipoprotein J, APOJ) isoform encoded by the “C” allele of the rs11136000 polymorphism impairs oligodendrocyte progenitor cell (OPC) proliferation and myelination through paracrine signaling from astrocytes (Liu et al., 2023). In particular, iPSC-derived astrocytes carrying the “C” allele exhibit increased activity of the interferon response pathway and higher secretion of the C-X-C motif ligand 10 (CXCL10) as compared to astrocytes carrying the “T” allele (Liu et al., 2023). These findings highlight the involvement of paracrine signaling and cell-cell interactions in myelin breakdown that can be investigated further using myelinoids.
As professional phagocytes, microglia play a major role in myelin homeostasis and clearance of myelin debris (Safaiyan et al., 2016; McNamara et al., 2023). A unique profile of white matter-associated microglia (WAM), including their increased phagocytic activity and altered lipid metabolism, suggests that the myelin-rich environment shapes microglial identity and gene expression (Safaiyan et al., 2021). A partial overlap between gene expression profiles of WAM and disease-associated microglia (DAM), such as upregulation of APOE expression, may indicate a common response to disease-associated debris (Safaiyan et al., 2021). Although clearance of myelin debris and recycling of lipid species, such as cholesterol, promote myelin repair, excessive myelin debris overwhelms microglia (Safaiyan et al., 2016; Cantuti-Castelvetri et al., 2018; Depp et al., 2023). Accumulation of high-molecular weight myelin fragments in microglia leads to formation of insoluble lipofuscin, which likely further burdens the lysosomal compartment required for efficient breakdown of AD-associated debris (Depp et al., 2023). Interestingly, microglia engage myelin debris at the expense of amyloid plaque clearance, which may worsen classical AD pathology (Depp et al., 2023).
Overall, defining how Aβ and p-tau pathology, genetic and other risk factors for AD, and deterioration of cell-cell interactions interplay to drive myelin dysfunction may reveal novel targets for therapeutic development for AD. In the remainder of this opinion, we discuss the potential applications of myelinoids to study these phenotypes.
Human iPSCs can be differentiated into oligodendrocytes by mimicking the in vivo developmental trajectory of oligodendroglia (Douvaras and Fossati, 2015; Li et al., 2018; Liu et al., 2023). Similarly, exposure of differentiating brain organoids to factors that promote oligodendrocyte fate, including thyroid hormone (T3), insulin-like growth factor 1 (IGF-1), and platelet-derived growth factor AA (PDGF-AA), yields oligodendrocyte-containing myelinoids (Madhavan et al., 2018; Kim et al., 2019; Marton et al., 2019; James et al., 2021; Feng et al., 2023). Accelerated differentiation of oligodendrocytes in neural spheroids can also be achieved by overexpression of master transcription factors involved in oligodendrogenesis, SOX10 and OLIG2 (Ma et al., 2022). Several groups have developed cortical myelinoids patterned toward the forebrain identity by dual SMAD inhibition (Madhavan et al., 2018; Kim et al., 2019; Marton et al., 2019); these organoids contain OPCs and myelinating oligodendrocytes that ensheath axons in the 3D space. Furthermore, James et al. (2021) have shown that myelinoids patterned toward ventral spinal cord identity, where oligodendrogenesis begins during development, exhibit advanced myelination patterns, including myelin internodes, paranodal junctions, and nodes of Ranvier. Although cortical myelinoids might be more suitable for modeling AD, improved myelination of spinal cord myelinoids makes them an attractive alternative to investigate the specificities of myelin sheath maintenance and dysfunction. An important advantage of modeling AD with iPSC-derived myelinoids is their human origin that enables the study of human-specific phenotypes that may not be recapitulated in animal models (Fang et al., 2022; Gargareta et al., 2022). Moreover, varying observations of myelin dysfunction in AD mouse models across different studies (Zhang et al., 2020; Chen J. F. et al., 2021; Depp et al., 2023) further support the use of myelinoids to clarify disease-relevant phenotypes. Feng et al. (2023), James et al. (2021), and Madhavan et al. (2018) have shown that myelinoids recapitulate key aspects of genetic myelin disorders, including Canavan disease (CD) and Pelizaeus–Merzbacher disease. For example, exposure of aspartoacylase (ASPA)-deficient myelinoids derived from iPSCs of CD patients to N-acetyl-aspartate (NAA) induces extensive damage to myelin sheaths, mimicking spongy degeneration of the white matter in CD patients (Feng et al., 2023). Furthermore, myelinoids are responsive to pharmacological intervention; for example, pro-myelinating drugs ketoconazole and clemastine promote oligodendrogenesis in myelinoids (Madhavan et al., 2018). These findings indicate that myelinoids can be applied to study myelin dysfunction in human diseases, including AD, and develop novel therapeutics for modulating myelination.
Myelinoids should be first employed to assess the effects of classical AD pathology—Aβ and p-tau aggregates—on oligodendrocyte and myelin homeostasis (Figure 1A). Aβ and p-tau pathology robustly develops in brain organoids derived from iPSCs of patients carrying disease-causing mutations, such as those in presenilin 1 (PSEN1) and microtubule-associated protein tau (MAPT) genes (Cerneckis et al., 2023). Aβ and p-tau pathology can also be induced acutely by treating brain organoids with a small-molecule compound Aftin-5 (Pavoni et al., 2018) and by delivering a mutant MAPT expression cassette using adeno-associated virus (Shimada et al., 2022), respectively. Whether acute demyelination can also induce Aβ and p-tau formation can be assessed by treating myelinoids with demyelinating compounds, such as lysolecithin (Marton et al., 2019). These models of myelin dysfunction may clarify the interplay between classical AD pathology and myelination. It will also be important to characterize the OPC pool, such as their proliferation and differentiation capacity in the presence of toxic Aβ and p-tau aggregates, and define oligodendrocyte gene expression profiles that may reveal distinct cellular states upon Aβ and p-tau challenge in myelinoids. The impact of myelin breakdown on neuronal network connectivity in myelinoids can be assessed using multi-electrode array (MEA) analysis with ever improving spatiotemporal resolution of MEA devices (Floch et al., 2022; Huang et al., 2022a).
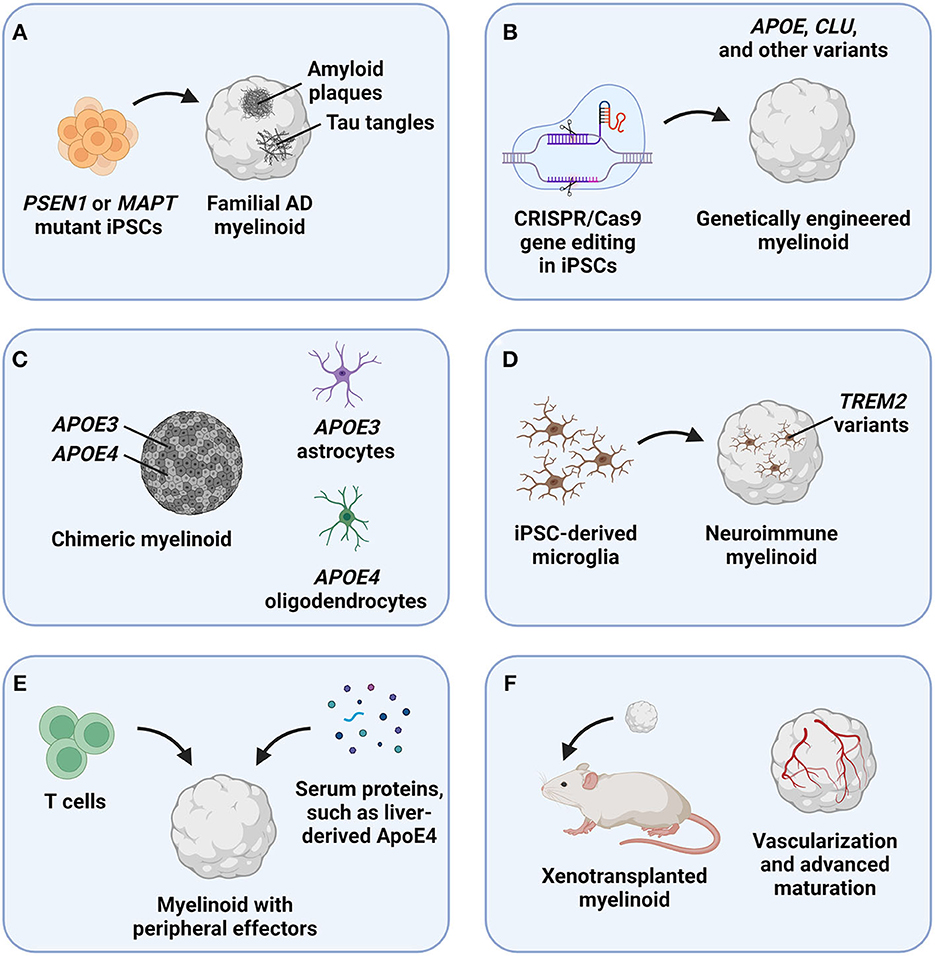
Figure 1. Potential applications of myelinoids to study myelin dysfunction in Alzheimer's disease (AD). (A) The effects of classical AD pathology, including amyloid plaques and tau tangles, on oligodendroglia and myelin health can be studied by deriving myelinoids from induced pluripotent stem cells (iPSCs) carrying disease-causing mutations, such as those in the presenilin 1 (PSEN1) and microtubule-associated protein tau (MAPT) genes. (B) The effects of genetic risk factors for late-onset AD, such as variants of the apolipoprotein E (APOE) and clusterin (CLU) genes, can be studied in myelinoids derived from CRISPR/Cas9-edited iPSCs. (C) To discern between cell-autonomous and non-autonomous effects of genetic risk factors expressed in multiple cell types, such as APOE, chimeric myelinoids with cell type-specific genotypes can be derived. (D) The roles of microglia in clearing myelin debris and recycling myelin lipids can be studied in neuroimmune myelinoids that contain iPSC-derived microglia. (E) The effects of peripheral factors, such as immune cells and serum proteins, can be modeled by exposing myelinoids to these effectors in vitro. (F) To achieve advanced myelinoid maturation, these organoids can be xenotransplanted into rodent models.
The roles of genetic risk factors for AD in myelin homeostasis can be defined by deriving genetically engineered myelinoids that express genetic risk variants, such as APOE4 (Figure 1B). As a transporter of cholesterol, ApoE plays an important role in the recycling of myelin lipids and in myelin repair; notably, neurons and astrocytes exhibit dysregulated cholesterol biosynthesis and accumulate cholesterol in APOE4 brain organoids (Zhao et al., 2023). Therefore, myelinoids with different ApoE isoforms can be used to characterize how each isoform affects lipid metabolism and myelination that require coordinated interactions and metabolite exchange between different brain cell types. Because APOE is expressed in multiple brain cell types, cell autonomous and non-autonomous effects of ApoE could be assessed using chimeric myelinoids, where only a specific cell type expresses APOE4 (Figure 1C) (Huang et al., 2022b). Another effector involved in lipid metabolism and implicated in AD pathogenesis, triggering receptor expressed on myeloid cells 2 (TREM2), is expressed in microglia and mediates extracellular lipid sensing (Deczkowska et al., 2020). However, the lipid sensing ability of the R47H isoform of TREM2 is impaired (Wang et al., 2015). To study the effects of TREM2 and other genetic risk variants specific to microglia, neuroimmune myelinoids can be generated by seeding iPSC-derived microglia onto the myelinoid to allow microglia infiltration and interaction with other cell types in the organoid (Figure 1D) (Cerneckis and Shi, 2023a; Cerneckis et al., 2023). Given the major roles that microglia play in clearing myelin debris, recycling lipids, and mediating neuroinflammation in AD, neuroimmune myelinoids will serve as a robust platform to define the interplay between microglia and myelin dysfunction in AD. It will also be important to clarify whether disruption of lipid and myelin homeostasis can drive neuroinflammation by activating microglia or astrocytes and which lipid species released with myelin debris are inflammatory. For example, astrocyte activation is evident in myelin-rich regions in a mouse model of sulfatide deficiency, contributing to neuroinflammation (Qiu et al., 2021).
Peripheral effectors, such as immune cells and serum proteins have been recently implicated in AD pathogenesis as well (Gate et al., 2020; Chen et al., 2021a; Kaya et al., 2022; Liu et al., 2022). For example, liver-derived ApoE4 impairs cognition in a mouse model (Liu et al., 2022), whereas exposure of brain organoids to human serum precipitates Aβ and p-tau pathology (Chen et al., 2021a). Moreover, oligodendrocytes co-localize with CD8+ T cells in the aging mouse brain, leading to oligodendrocyte activation and death (Kaya et al., 2022). To gain a deeper understanding on how peripheral effectors impact myelin homeostasis in AD, myelinoids can be co-cultured with primary or iPSC-derived immune cells (Sommer et al., 2018), exposed to other peripheral factors (Chen et al., 2021a), or assembled into state-of-the-art microfluidic organoids-on-a-chip with a separated peripheral compartment (Figure 1E) (Cho et al., 2021; Ronaldson-Bouchard et al., 2022).
Finally, various myelinoids discussed earlier can be transplanted in vivo to promote advanced myelinoid maturation and vascularization (Figure 1F). Indeed, brain organoid transplantation paradigm has recently emerged as a powerful approach to achieve improved organoid maturation, revealing subtle disease phenotypes that are not recapitulated in vitro (Mansour et al., 2018; Revah et al., 2022; Cerneckis and Shi, 2023b; Schafer et al., 2023). It can be expected that xenotransplanted myelinoids will contain a robust pool of OPCs and mature oligodendrocytes as well as exhibit improved myelination patterns, making them especially suitable for modeling myelin dysfunction in AD. Transplantation of myelinoids carrying causal mutations or genetic risk factors for AD into wild-type mice can be performed to improve myelinoid maturation and study organoid-specific phenotypes. Alternatively, myelinoid transplantation into transgenic AD mouse models will enable the study of complex disease phenotypes, such as Aβ propagation into the myelinoid, dysfunction of organoid-invading murine vasculature, and the effects of blood-derived factors or systemic perturbations on AD progression.
It is also important to consider various limitations of the brain organoid technology for modeling AD, so that appropriate experimental controls can be utilized (Cerneckis et al., 2023). Most notably, iPSC-derived cellular models, including brain organoids, exhibit fetal-like gene expression signatures and represent early human brain development, whereas AD is an age-related neurodegenerative disease (Cornacchia and Studer, 2017). Furthermore, brain organoids are a simplified model of the human brain and often lack important cellular components, such as endothelial cells, that can also contribute to AD progression (Cerneckis et al., 2023). Therefore, myelinoids that lack aging-associated phenotypes and complete cellular diversity may not recapitulate all disease-relevant phenotypes. Nonetheless, the effects of causal disease mutations, genetic and non-genetic risk factors, and specific cell types on AD progression have been successfully modeled using brain organoids (Cerneckis et al., 2023). Limited yet sufficient complexity of brain organoids may even be advantageous to pinpoint key dysregulated pathways that drive AD pathogenesis. If necessary, neurodegenerative phenotypes can be amplified by exposing myelinoids to a combination of genetic and non-genetic risk factors for AD, whereas additional cell types can be introduced by established techniques (Di Lullo and Kriegstein, 2017). Importantly, novel phenotypes obtained from myelinoid studies should be validated by a comparative analysis of primary human brain tissue whenever possible.
Although AD has historically been considered a gray matter disease, emerging evidence indicates concurrent white matter dysfunction during AD progression. The versatile myelinoid platform offers unprecedented access to human brain myelination models that can be tailored to address specific hypotheses pertaining to myelin dysfunction in AD. For example, microglia can be incorporated into myelinoids described in Figures 1A–C, E, F to study the interplay between microglia, various genetic and non-genetic risk factors that contribute to AD progression, and myelin dysfunction. Myelinoids can also be combined with other technological advancements, such as high-throughput drug screening and high-content imaging assays, to systematically assess disease-relevant pathology and drug efficacy (James et al., 2021; Park et al., 2021). Overall, we anticipate that myelinoids will facilitate the elucidation of human-specific molecular mechanisms and cellular phenotypes governing oligodendrocyte function and advance the development of novel therapeutics targeting impaired myelination in AD.
JC: Writing—original draft, Writing—review and editing. YS: Writing—review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Institute of Aging of the National Institutes of Health RF1 AG061794, R01 AG072291, and RF1 AG079307 to YS. JC is a predoctoral scholar in the Stem Cell Biology and Regenerative Medicine Research Training Program of the California Institute for Regenerative Medicine (CIRM).
The authors would like to thank Louise and Herbert Horvitz, the Christopher Family, the Judy and Bernard Briskin Fund, and the Sidell Kagan Foundation for their generosity and forethought. Figure 1 was created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bartzokis, G. (2004). Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer's disease. Neurobiol. Aging 25, 5–18. doi: 10.1016/j.neurobiolaging.2003.03.001
Bartzokis, G. (2011). Alzheimer's disease as homeostatic responses to age-related myelin breakdown. Neurobiol. Aging 32, 1341–1371. doi: 10.1016/j.neurobiolaging.2009.08.007
Bartzokis, G. (2012). Neuroglialpharmacology: myelination as a shared mechanism of action of psychotropic treatments. Neuropharmacology 62, 2137–2153. doi: 10.1016/j.neuropharm.2012.01.015
Blanchard, J. W., Akay, L. A., Davila-Velderrain, J., von Maydell, D., Mathys, H., Davidson, S. M., et al. (2022). APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 611, 769–779. doi: 10.1038/s41586-022-05439-w
Braak, H., and Braak, E. (1996). Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol. 92, 197–201. doi: 10.1007/s004010050508
Brickman, A. M., Zahodne, L. B., Guzman, V. A., Narkhede, A., Meier, I. B., Griffith, E. Y., et al. (2015). Reconsidering harbingers of dementia: progression of parietal lobe white matter hyperintensities predicts Alzheimer's disease incidence. Neurobiol. Aging 36, 27–32. doi: 10.1016/j.neurobiolaging.2014.07.019
Caballero, M. Á. A., Suárez-Calvet, M., Duering, M., Franzmeier, N., Benzinger, T., Fagan, A. M., et al. (2018). White matter diffusion alterations precede symptom onset in autosomal dominant Alzheimer's disease. Brain 141, 3065–3080. doi: 10.1093/brain/awy229
Cantuti-Castelvetri, L., Fitzner, D., Bosch-Queralt, M., Weil, M.-T., Su, M., Sen, P., et al. (2018). Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 359, 684–688. doi: 10.1126/science.aan4183
Cerneckis, J., Bu, G., and Shi, Y. (2023). Pushing the boundaries of brain organoids to study Alzheimer's disease. Trends Mol. Med. 29, 659–672. doi: 10.1016/j.molmed.2023.05.007
Cerneckis, J., and Shi, Y. (2023a). Modeling brain macrophage biology and neurodegenerative diseases using human iPSC-derived neuroimmune organoids. Front. Cell. Neurosci. 17, 1198715. doi: 10.3389/fncel.2023.1198715
Cerneckis, J., and Shi, Y. (2023b). Context matters: hPSC-derived microglia thrive in a humanized brain environment in vivo. Cell Stem Cell 30, 909–910. doi: 10.1016/j.stem.2023.05.013
Chen, J.-F., Liu, K., Hu, B., Li, R.-R., Xin, W., Chen, H., et al. (2021). Enhancing myelin renewal reverses cognitive dysfunction in a murine model of Alzheimer's disease. Neuron 109, 2292–2307. e2295. doi: 10.1016/j.neuron.2021.05.012
Chen, X., Sun, G., Tian, E., Zhang, M., Davtyan, H., Beach, T. G., et al. (2021a). Modeling sporadic Alzheimer's disease in human brain organoids under serum exposure. Adv. Sci. 8, e2101462. doi: 10.1002/advs.202101462
Chen, X., Yang, Q., Liu, Z., and Shi, Y. (2021b). “Human induced pluripotent stem cell-based modeling of Alzheimer's disease, a glial perspective,” in iPSCs for Modeling Central Nervous System Disorders (Academic Press), 21–35. doi: 10.1016/B978-0-323-85764-2.00006-5
Cho, A.-N., Jin, Y., An, Y., Kim, J., Choi, Y. S., Lee, J. S., et al. (2021). Microfluidic device with brain extracellular matrix promotes structural and functional maturation of human brain organoids. Nat. Commun. 12, 4730. doi: 10.1038/s41467-021-24775-5
Cornacchia, D., and Studer, L. (2017). Back and forth in time: directing age in iPSC-derived lineages. Brain Res. 1656, 14–26. doi: 10.1016/j.brainres.2015.11.013
De Strooper, B., and Karran, E. (2016). The cellular phase of Alzheimer's disease. Cell 164, 603–615. doi: 10.1016/j.cell.2015.12.056
Dean, D. C., Hurley, S. A., Kecskemeti, S. R., O'Grady, J. P., Canda, C., Davenport-Sis, N. J., et al. (2017). Association of amyloid pathology with myelin alteration in preclinical Alzheimer disease. JAMA Neurol. 74, 41–49. doi: 10.1001/jamaneurol.2016.3232
Deczkowska, A., Weiner, A., and Amit, I. (2020). The physiology, pathology, and potential therapeutic applications of the TREM2 signaling pathway. Cell 181, 1207–1217. doi: 10.1016/j.cell.2020.05.003
Depp, C., Sun, T., Sasmita, A. O., Spieth, L., Berghoff, S. A., Nazarenko, T., et al. (2023). Myelin dysfunction drives amyloid-beta deposition in models of Alzheimer's disease. Nature 618, 349–357. doi: 10.1038/s41586-023-06120-6
Di Lullo, E., and Kriegstein, A. R. (2017). The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 18, 573–584. doi: 10.1038/nrn.2017.107
Douvaras, P., and Fossati, V. (2015). Generation and isolation of oligodendrocyte progenitor cells from human pluripotent stem cells. Nat. Protoc. 10, 1143–1154. doi: 10.1038/nprot.2015.075
Fang, R., Xia, C., Close, J. L., Zhang, M., He, J., Huang, Z., et al. (2022). Conservation and divergence of cortical cell organization in human and mouse revealed by MERFISH. Science 377, 56–62. doi: 10.1126/science.abm1741
Feng, L., Chao, J., Zhang, M., Pacquing, E., Hu, W., and Shi, Y. (2023). Developing a human iPSC-derived three dimensional myelin spheroid platform for modeling myelin diseases iScience. 26:108037. doi: 10.1016/j.isci.2023.108037
Floch, P. L., Li, Q., Lin, Z., Zhao, S., Liu, R., Tasnim, K., et al. (2022). Stretchable mesh nanoelectronics for 3D single-cell chronic electrophysiology from developing brain organoids. Adv. Mater. Weinheim. 34, e2106829. doi: 10.1002/adma.202106829
Gargareta, V.-I., Reuschenbach, J., Siems, S. B., Sun, T., Piepkorn, L., Mangana, C., et al. (2022). Conservation and divergence of myelin proteome and oligodendrocyte transcriptome profiles between humans and mice. Elife 11, e77019. doi: 10.7554/eLife.77019.sa2
Gate, D., Saligrama, N., Leventhal, O., Yang, A. C., Unger, M. S., Middeldorp, J., et al. (2020). Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer's disease. Nature 577, 399–404. doi: 10.1038/s41586-019-1895-7
Huang, Q., Tang, B., Romero, J. C., Yang, Y., Elsayed, S. K., Pahapale, G., et al. (2022a). Shell microelectrode arrays (MEAs) for brain organoids. Sci. Adv. 8, eabq5031. doi: 10.1126/sciadv.abq5031
Huang, S., Zhang, Z., Cao, J., Yu, Y., and Pei, G. (2022b). Chimeric cerebral organoids reveal the essentials of neuronal and astrocytic APOE4 for Alzheimer's tau pathology. Signal. Transduct. Target Ther. 7, 176. doi: 10.1038/s41392-022-01006-x
James, O. G., Selvaraj, B. T., Magnani, D., Burr, K., Connick, P., Barton, S. K., et al. (2021). iPSC-derived myelinoids to study myelin biology of humans. Dev. Cell 56, 1346–1358. doi: 10.1016/j.devcel.2021.04.006
Kaya, T., Mattugini, N., Liu, L., Ji, H., Cantuti-Castelvetri, L., Wu, J., et al. (2022). CD8(+) T cells induce interferon-responsive oligodendrocytes and microglia in white matter aging. Nat. Neurosci. 25, 1446–1457. doi: 10.1038/s41593-022-01183-6
Kenigsbuch, M., Bost, P., Halevi, S., Chang, Y., Chen, S., Ma, Q., et al. (2022). A shared disease-associated oligodendrocyte signature among multiple CNS pathologies. Nat. Neurosci. 25, 876–886. doi: 10.1038/s41593-022-01104-7
Kim, H., Xu, R., Padmashri, R., Dunaevsky, A., Liu, Y., Dreyfus, C. F., et al. (2019). Pluripotent stem cell-derived cerebral organoids reveal human oligodendrogenesis with dorsal and ventral origins. Stem Cell Rep. 12, 890–905. doi: 10.1016/j.stemcr.2019.04.011
Koutsodendris, N., Blumenfeld, J., Agrawal, A., Traglia, M., Grone, B., Zilberter, M., et al. (2023). Neuronal APOE4 removal protects against tau-mediated gliosis, neurodegeneration and myelin deficits. Nat Aging 3, 275–296. doi: 10.1038/s43587-023-00368-3
Lee, Y., Morrison, B. M., Li, Y., Lengacher, S., Farah, M. H., Hoffman, P. N., et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. doi: 10.1038/nature11314
Li, L., and Shi, Y. (2020). When glia meet induced pluripotent stem cells (iPSCs). Mol. Cell. Neurosci. 109, 103565. doi: 10.1016/j.mcn.2020.103565
Li, L., Tian, E., Chen, X., Chao, J., Klein, J., Qu, Q., et al. (2018). GFAP mutations in astrocytes impair oligodendrocyte progenitor proliferation and myelination in an hiPSC model of Alexander disease. Cell Stem Cell 23, 239–251. e236. doi: 10.1016/j.stem.2018.07.009
Lin, Y. T., Seo, J., Gao, F., Feldman, H. M., Wen, H. L., Penney, J., et al. (2018). APOE4 causes widespread molecular and cellular alterations associated with Alzheimer's disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1141–1154. doi: 10.1016/j.neuron.2018.05.008
Liu, C.-C., Zhao, J., Fu, Y., Inoue, Y., Ren, Y., Chen, Y., et al. (2022). Peripheral apoE4 enhances Alzheimer's pathology and impairs cognition by compromising cerebrovascular function. Nat. Neurosci. 25, 1020–1033. doi: 10.1038/s41593-022-01127-0
Liu, Z., Chao, J., Wang, C., Sun, G., Roeth, D., Liu, W., et al. (2023). Astrocytic response mediated by the CLU risk allele inhibits OPC proliferation and myelination in a human iPSC model. Cell Rep. 42, 112841. doi: 10.1016/j.celrep.2023.112841
Long, J. M., and Holtzman, D. M. (2019). Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179, 312–339. doi: 10.1016/j.cell.2019.09.001
Ma, L., Mei, Y., Xu, P., Cheng, Y., You, Z., Ji, X., et al. (2022). Fast generation of forebrain oligodendrocyte spheroids from human embryonic stem cells by transcription factors. iScience 25, 105172. doi: 10.1016/j.isci.2022.105172
Madhavan, M., Nevin, Z. S., Shick, H. E., Garrison, E., Clarkson-Paredes, C., Karl, M., et al. (2018). Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 15, 700–706. doi: 10.1038/s41592-018-0081-4
Mansour, A. A., Gonçalves, J. T., Bloyd, C. W., Li, H., Fernandes, S., Quang, D., et al. (2018). An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 36, 432–441. doi: 10.1038/nbt.4127
Martens, Y. A., Zhao, N., Liu, C. C., Kanekiyo, T., Yang, A. J., Goate, A. M., et al. (2022). ApoE cascade hypothesis in the pathogenesis of Alzheimer's disease and related dementias. Neuron. doi: 10.1016/j.neuron.2022.03.004
Marton, R. M., Miura, Y., Sloan, S. A., Li, Q., Revah, O., Levy, R. J., et al. (2019). Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 22, 484–491. doi: 10.1038/s41593-018-0316-9
McNamara, N. B., Munro, D. A., Bestard-Cuche, N., Uyeda, A., Bogie, J. F., Hoffmann, A., et al. (2023). Microglia regulate central nervous system myelin growth and integrity. Nature 613, 120–129. doi: 10.1038/s41586-022-05534-y
Murdock, M. H., and Tsai, L. H. (2023). Insights into Alzheimer's disease from single-cell genomic approaches. Nat. Neurosci. 26, 181–195. doi: 10.1038/s41593-022-01222-2
Park, J.-C., Jang, S.-Y., Lee, D., Lee, J., Kang, U., Chang, H., et al. (2021). A logical network-based drug-screening platform for Alzheimer's disease representing pathological features of human brain organoids. Nat. Commun. 12, 280. doi: 10.1038/s41467-020-20440-5
Pavoni, S., Jarray, R., Nassor, F., Guyot, A.-C., Cottin, S., Rontard, J., et al. (2018). Small-molecule induction of Abeta-42 peptide production in human cerebral organoids to model Alzheimer's disease associated phenotypes. PLoS ONE 13, e0209150. doi: 10.1371/journal.pone.0209150
Qiu, S., Palavicini, J. P., Wang, J., Gonzalez, N. S., He, S., Dustin, E., et al. (2021). Adult-onset CNS myelin sulfatide deficiency is sufficient to cause Alzheimer's disease-like neuroinflammation and cognitive impairment. Mol. Neurodegener. 16, 1–24. doi: 10.1186/s13024-021-00488-7
Revah, O., Gore, F., Kelley, K. W., Andersen, J., Sakai, N., Chen, X., et al. (2022). Maturation and circuit integration of transplanted human cortical organoids. Nature 610, 319–26. doi: 10.1038/s41586-022-05277-w
Ronaldson-Bouchard, K., Teles, D., Yeager, K., Tavakol, D. N., Zhao, Y., Chramiec, A., et al. (2022). A multi-organ chip with matured tissue niches linked by vascular flow. Nat. Biomed. Eng. 6, 351–371. doi: 10.1038/s41551-022-00882-6
Sadick, J. S., O'Dea, M. R., Hasel, P., Dykstra, T., Faustin, A., and Liddelow, S. A. (2022). Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer's disease. Neuron 110, 1788–1805. e1710. doi: 10.1016/j.neuron.2022.03.008
Safaiyan, S., Besson-Girard, S., Kaya, T., Cantuti-Castelvetri, L., Liu, L., Ji, H., et al. (2021). White matter aging drives microglial diversity. Neuron 109, 1100–1117. doi: 10.1016/j.neuron.2021.01.027
Safaiyan, S., Kannaiyan, N., Snaidero, N., Brioschi, S., Biber, K., Yona, S., et al. (2016). Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 19, 995–998. doi: 10.1038/nn.4325
Schafer, S. T., Mansour, A. A., Schlachetzki, J. C. M., Pena, M., Ghassemzadeh, S., Mitchell, L., et al. (2023). An in vivo neuroimmune organoid model to study human microglia phenotypes. Cell 186, 2111–2126. e2120. doi: 10.1016/j.cell.2023.04.022
Schoenemann, P. T., Sheehan, M. J., and Glotzer, L. D. (2005). Prefrontal white matter volume is disproportionately larger in humans than in other primates. Nat. Neurosci. 8, 242–252. doi: 10.1038/nn1394
Shi, M., Chu, F., Zhu, F., and Zhu, J. (2022). Impact of anti-amyloid-beta monoclonal antibodies on the pathology and clinical profile of alzheimer's disease: a focus on aducanumab and lecanemab. Front. Aging Neurosci. 14, 870517. doi: 10.3389/fnagi.2022.870517
Shi, Y., Inoue, H., Wu, J. C., and Yamanaka, S. (2017). Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov. 16, 115–130. doi: 10.1038/nrd.2016.245
Shimada, H., Sato, Y., Sasaki, T., Shimozawa, A., Imaizumi, K., Shindo, T., et al. (2022). A next-generation iPSC-derived forebrain organoid model of tauopathy with tau fibrils by AAV-mediated gene transfer. Cell Rep. Methods 2, 100289. doi: 10.1016/j.crmeth.2022.100289
Sommer, A., Marxreiter, F., Krach, F., Fadler, T., Grosch, J., Maroni, M., et al. (2018). Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson's disease. Cell Stem Cell 23, 123–131. doi: 10.1016/j.stem.2018.06.015
Stadelmann, C., Timmler, S., Barrantes-Freer, A., and Simons, M. (2019). Myelin in the central nervous system: structure, function, and pathology. Physiol. Rev. 99, 1381–1431. doi: 10.1152/physrev.00031.2018
Wang, F., Ren, S.-Y., Chen, J.-F., Liu, K., Li, R.-X., Li, Z.-F., et al. (2020). Myelin degeneration and diminished myelin renewal contribute to age-related deficits in memory. Nat. Neurosci. 23, 481–486. doi: 10.1038/s41593-020-0588-8
Wang, Q., Wang, Y., Liu, J., Sutphen, C. L., Cruchaga, C., Blazey, T., et al. (2019). Quantification of white matter cellularity and damage in preclinical and early symptomatic Alzheimer's disease. Neuroimage Clin. 22, 101767. doi: 10.1016/j.nicl.2019.101767
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Wightman, D. P., Jansen, I. E., Savage, J. E., Shadrin, A. A., Bahrami, S., Holland, D., et al. (2021). A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer's disease. Nat. Genet. 53, 1276–1282. doi: 10.1038/s41588-021-00921-z
Zhang, X., Wang, R., Hu, D., Sun, X., Fujioka, H., Lundberg, K., et al. (2020). Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer's disease. Sci. Adv. 6:eabb8680. doi: 10.1126/sciadv.abb8680
Keywords: Alzheimer's disease, myelin, myelinoids, oligodendrocytes, microglia
Citation: Cerneckis J and Shi Y (2023) Myelin organoids for the study of Alzheimer's disease. Front. Neurosci. 17:1283742. doi: 10.3389/fnins.2023.1283742
Received: 27 August 2023; Accepted: 09 October 2023;
Published: 24 October 2023.
Edited by:
Jeffrey Jones, Salk Institute for Biological Studies, United StatesCopyright © 2023 Cerneckis and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanhong Shi, eXNoaUBjb2gub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.