 Jie Wu
Jie Wu Jiyue Chen†
Jiyue Chen† Zhiwei Ding
Zhiwei Ding Pu Dai
Pu Dai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurosci., 13 November 2023
Sec. Auditory Cognitive Neuroscience
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1281884
This article is part of the Research TopicThe Effects of Auditory Neural Disorders on Speech Production and PerceptionView all 8 articles
Background: Cochlear implantation (CI) outcomes in patients with auditory neuropathy (AN) are variable, which hampers patients’ decisions on CI.
Objective: This study aims to assess the outcomes of CI in individuals diagnosed with AN and to examine the various factors that may influence the effectiveness of this intervention.
Methods: A total of 75 patients diagnosed with AN were included in the study. The hearing threshold, the score of categories of auditory performance (CAP), speech intelligibility rating (SIR), and speech audiometry were tested. Genetic testing was conducted by medical exome sequencing in 46 patients.
Results: After CI, the average aided hearing threshold for patients with prelingual and post-lingual onset was 38.25 ± 6.63 dB and 32.58 ± 9.26 dB, respectively; CAP score improved to 5.52 ± 1.64 (p < 0.001) and 6.00 ± 0.96 (p < 0.001), respectively; SIR score increased to 3.57 ± 1.22 (p < 0.001) and 4.15 ± 0.95 (p < 0.001), respectively. Maximum speech recognition ranged from 58 to 93% for prelingual onset patients and 43 to 98% for those with post-lingual onset. Speech outcomes of CI in cases with cochlear nerve (CN) deficiency were significantly poorer (p = 0.008). Molecular etiologies, including TWIST1, ACTG1, m.A7445G, and a copy-number variant (CNV) carrying ACTB, were related to AN here.
Conclusion: CI is a viable therapy option for patients with AN; CN deficiency might impact outcomes of CI.
Auditory neuropathy (AN) or auditory neuropathy spectrum disorder (ANSD) is a specific type of sensorineural hearing loss characterized by the presence of otoacoustic emissions (OAE) and/or cochlear microphonics with the absent or abnormal auditory brainstem responses (ABR) (Moser and Starr, 2016). Nearly 9.85% of 1,025 children with sensorineural hearing loss were identified to have ANSD (Almishaal et al., 2022).
In patients with AN, the degree of hearing loss (HL) varies from mild to profound, and it can occur prelingually or post-lingually. In contrast, the hearing may be affected bilaterally or unilaterally. AN may occur simply or as part of some syndromes. Simultaneously, patients of AN may also have inner-ear malformations. Risk factors, including hyperbilirubinemia, infection, premature, ototoxic drug exposure, cochlear nerve (CN) deficiency, and congenital brain anomalies (Rajput et al., 2019; Natale et al., 2020; Liddle et al., 2022), are highly related to AN.
Many genes have been found as the molecular causes of AN (Moser and Starr, 2016; Shearer and Hansen, 2019), with hereditary patterns involving autosomal recessive, autosomal dominant, X-linked, and mitochondrial. More novel genes associated with AN are expected to be discovered, which would promote our insights into the pathogenesis of AN and guide the treatment and prevention of this disorder. Considering the treatment, cochlear implantation (CI) can benefit patients with AN (Sarankumar et al., 2018; Alzhrani et al., 2019). Studies have demonstrated that factors including the age of implantation, duration of cochlear, and genetic lesion sites could impact outcomes of CI (Daneshi et al., 2018; Shearer and Hansen, 2019). Speech performances in patients with AN receiving CI were variable (Harrison et al., 2015; Chaudhry et al., 2020), which traps the patients’ decision on CI.
This study enrolled 75 unrelated patients with AN who received cochlear implantation. Outcomes of CI in patients were evaluated through auditory assessment, speech audiometry, and questionnaires, including categories of auditory performance (CAP) and speech intelligibility rate (SIR). Meanwhile, for 46 patients, medical exome sequencing was performed for genetic testing. The molecular etiology of this specific patient subgroup was thoroughly elucidated. Furthermore, examining candidate factors, including hereditary influences that may affect the IC outcomes of individuals with AN, was also assessed to provide more reliable information for the decision of the rehabilitation approach for AN.
In this study, 84 patients diagnosed as AN and received CI in Chinese PLA General Hospital from 2010–08 to 2020–11 were enrolled. Exclusion criteria were: (1) having a tumor (e.g., acoustic neuroma), (2) severe cognitive abnormality hampering speech development, (3) otitis media, and (4) loss to follow-up. Finally, 75 subjects were included. The Ethics Committee of Chinese PLA General Hospital approved this study. It was conducted as per the Declaration of Helsinki. Written informed consent was obtained from the participants or the parents of the minors.
Auditory brainstem responses (ABR), distortion-product otoacoustic emissions (DPOAE), cochlear microphonics, and tympanometry were conducted to diagnose AN. Pure tone audiometry (PTA) was used for auditory threshold determination in subjects >5 years old who could complete this testing; behavior audiometry was performed in subjects ≤5 years old, including strength vision audiometer (subjects >1 and ≤ 2.5 years old) and play audiometer (subjects >2.5 and ≤ 5 years old). ABR and auditory steady-state response (ASSR) were measured for patients who could not undergo the aforementioned auditory tests. The degree of hearing loss was determined by the average threshold at frequencies of 0.5, 1, 2, and 4 kHz or response thresholds in ABR. The level of HL was graded as mild (26–40 dB), moderate (41–55 dB), moderately severe (56–70 dB), severe (71–90 dB), and profound (>90 dB). To evaluate the hearing sensitivity pre-operation and post-operation, categories of auditory performance (CAP) were also used.
Hierarchic speech audiometry was estimated based on patients’ age and their capability of hearing and speech. The Mandarin Early Speech Perception (MESP) test was performed for patients aged 2 to 5 years to evaluate the children’s recognition of words. The Mandarin Pediatric Speech Intelligibility (MPSI) test was chosen for patients aged 3 to 6 years to assess the children’s perception of sentences (Zheng et al., 2009a,b; Ji et al., 2015). Speech audiometry was performed to test Mandarin monosyllables, disyllables, and sentences in quiet for subjects who could complete this test (Ji et al., 2014). Additionally, speech intelligibility rating (SIR) was used to evaluate the communication ability before and after IC at least six months post-operation (Ji et al., 2015).
In order to assess the condition of the inner ear and auditory nerve, a comprehensive diagnostic approach was employed, which included high-resolution computed tomography (CT) scanning of the temporal bone, magnetic resonance imaging (MRI) of the brain, and magnetic resonance hydrography (MRH) of the inner ear.
The genetic cause of patients was identified through medical exome sequencing (Trio exome sequencing was performed for probands whose parents’ DNA samples were available). The peripheral blood was obtained, and DNA was extracted according to the standard protocol. DNA sequencing (targeting 52.9 Mbp of the genome, covering the exons of 19,608 genes), bioinformatics analysis, and variant interpretation were performed following an earlier described protocol (Wu et al., 2022). Copy number variation was detected as described earlier (Baux et al., 2017), and the method of in-run correction and the Z-test analysis was applied in CNV calling. Candidate variants were confirmed by Sanger sequencing, while CNV was confirmed by real-time quantitative PCR. Variants were classified based on the American College of Medical Genetics and Genomics guidelines. Novel variants identified here were submitted to the ClinVar database with the accession number from SCV002568090 to SCV002568102.
Mann–Whitney U test in SPSS Statistics 25 was applied for significance analysis. The statistical significance was defined as p < 0.05.
Of the 75 patients enrolled here, 61 had prelingual HL with the onset or awareness age ≤ 3 years old, and the remaining 14 subjects had post-lingual HL (>3 years old). Among this subgroup, the onset or awareness age of HL in 13 patients ranged from 3 to 16 years of age. Unilateral AN was identified in three cases, with sensorineural HL determined in the contralateral ear. The average implantation age was 2.67 ± 2.17 years for subjects with prelingual HL and 20.29 ± 8.44 years for patients with post-lingual HL. Bilateral implantation was conducted in eight cases with the same type of device (Table 1).
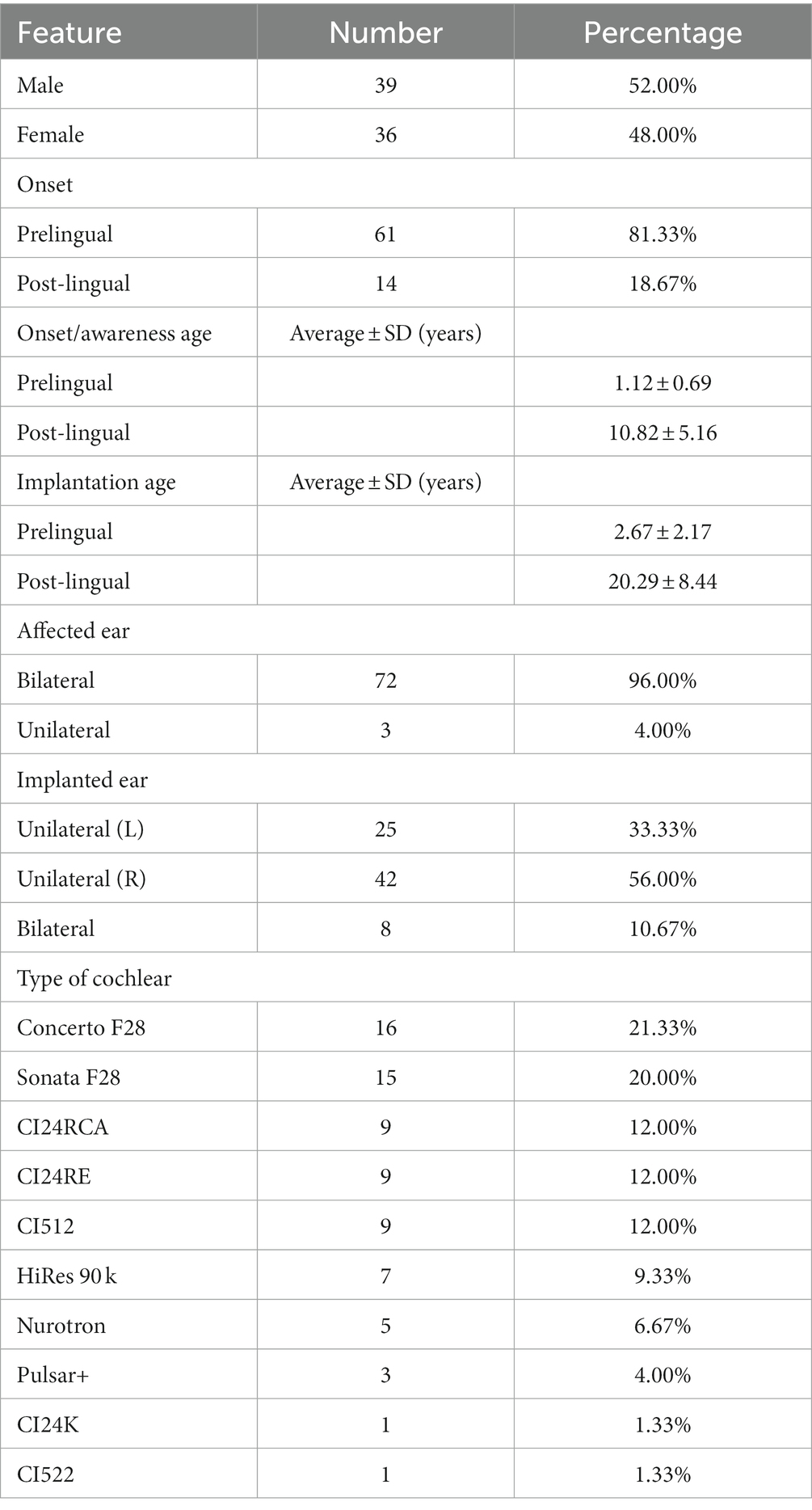
Table 1. Clinical details of 75 patients with AN enrolled in this study.
After a careful review of the disease history of the patients, risk factors related to AN were identified in 14 patients, including neonatal hyperbilirubinemia (n = 3), neonatal hyperbilirubinemia combined with Kawasaki syndrome (n = 1), neonatal hyperbilirubinemia combined with favism (n = 1), neonatal RH hemolysis combined with acute bilirubin encephalopathy (n = 1), meningitis (n = 1), neonatal septicemia combined with intrauterine distress and mixed acidosis (n = 1), hypoxic–ischemic encephalopathy (n = 1), premature (n = 1), trauma (n = 1), expose to ototoxic medicine (n = 1), hydrocephalus (n = 1), and development delay (n = 1) (Supplementary Table S1).
Based on imaging results, 10 patients had bilateral cochlear nerve (CN) deficiency, including hypoplastic CN and absent CN; two cases had a bilateral cochlear malformation, and another case had enlarged vestibular aqueduct (EVA) (Supplementary Table S1; Sennaroğlu and Bajin, 2017).
Other relevant comorbidities included epilepsy (n = 1) and unilateral renal agenesis (n = 1).
Tympanograms of all 147 ears, including 74 left and 73 right ears (three cases with unilateral AN), were normal as A type. For the ABR test, 140 ears had no response to stimulants (69 left ears, 71 right ears), while the response threshold of the remaining seven ears ranged from 75 dB to 100 dB. Of 74 left ears, 9, 9, 9, 26, 33, 28, 40, 37, 39, 38 ears passed DPOAE at frequencies 0.125, 0.25, 0.5, 1, 2, 1.5, 3, 4, 6, 8 kHz, respectively, while 7, 7, 8, 26, 34, 33, 39, 38, 38, 35 of 73 right ears passed DPOAE at frequencies as mentioned above, respectively. In 58 left ears and 55 right ears, cochlear microphonics waveforms were detected.
Of the patients with prelingual HL (61 subjects), behavior audiometry was completed in 20 cases, with the average hearing threshold being 98.38 ± 16.28 dB for left ears and 102.56 ± 17.14 dB for right ears (Figures 1A,B). ASSR results of this patient subgroup (n = 56) are shown in Figures 1C,D, with the average hearing threshold being 81.48 ± 15.23 dB for left ears and 83.34 ± 15.40 dB for right ears.
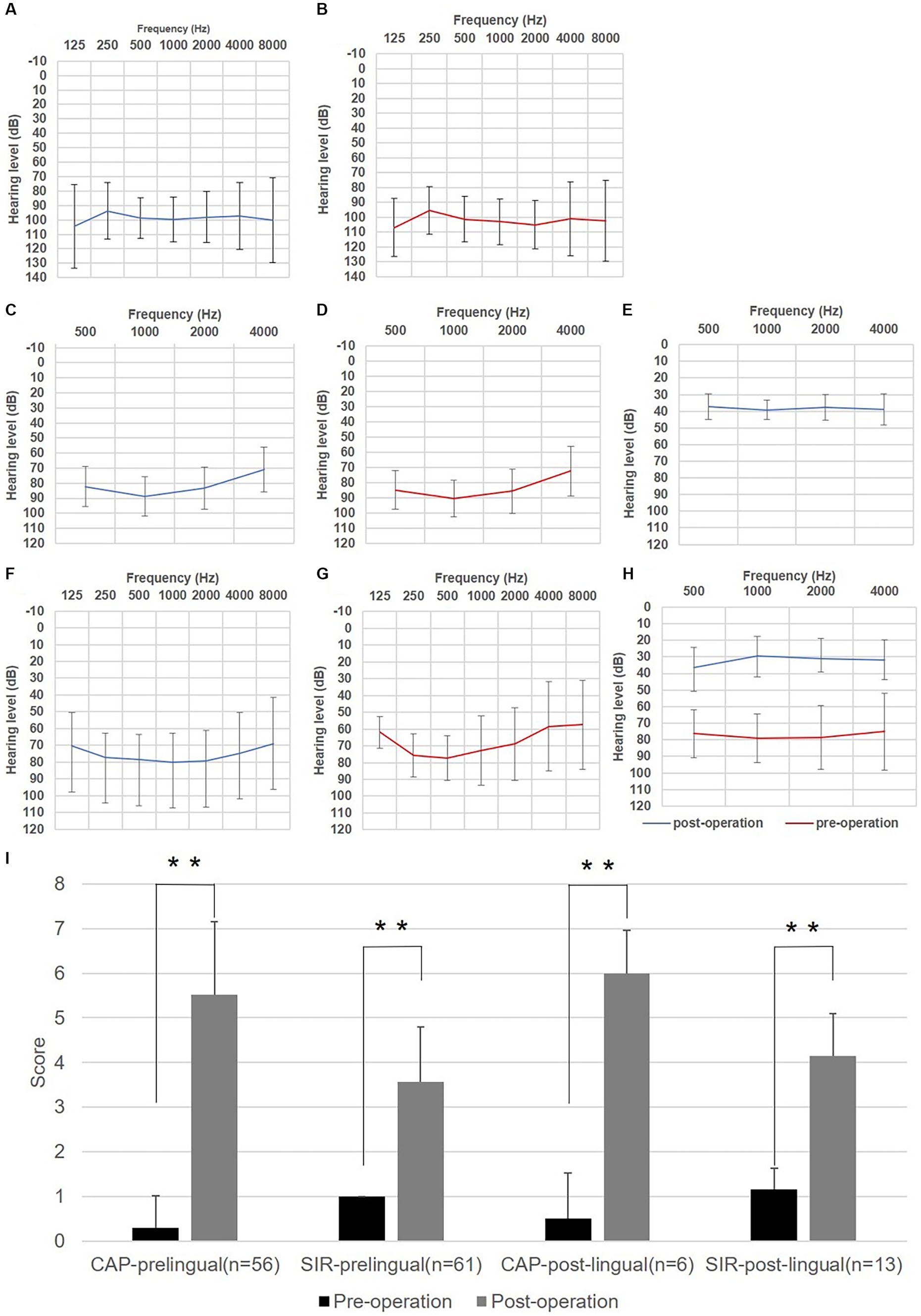
Figure 1. Auditory and speech outcomes before and after cochlear implantation (CI) in enrolled auditory neuropathy patients. (A) Hearing thresholds of left ears for subjects with prelingual onset determined by behavior audiometry (n = 20). (B) Hearing thresholds of right ears for subjects with prelingual onset determined by behavior audiometry. (C) Hearing thresholds of left ears for subjects with prelingual onset determined by the auditory steady-state response (ASSR) (n = 56). (D) Hearing thresholds of right ears for subjects with prelingual onset determined by ASSR. (E) Average aided hearing threshold after CI for subjects with prelingual onset determined by behavior audiometry (n = 30). (F) Hearing thresholds of left ears for subjects with post-lingual onset determined by pure tone audiometry (n = 11). (G) Hearing threshold of right ears for subjects with post-lingual onset determined by Pure tone audiometry (PTA). (H) Average aided hearing threshold after CI for subjects with post-lingual onset determined by PTA (n = 11). (I) Categories of auditory performance (CAP) and speech intelligibility rating (SIR) scores of enrolled subjects before and after CI. Whitney U test was applied for significance analysis, and statistical significance was defined as **p < 0.01.
For the remaining 14 cases with post-lingual onset, the average hearing threshold through PTA was 79.08 ± 15.11 dB for left ears and 74.83 ± 22.58 dB for right ears (Figures 1F,G). At the same time, maximum speech recognition scores ranged from 0 to 36% (n = 5) for this subgroup by speech audiometer, indicating profound speech disabilities.
After operation, behavior audiometry was conducted in thirty cases with prelingual HL, and the average aided hearing threshold was 38.25 ± 6.63 dB, with the average hearing threshold at a frequency of 0.5, 1, 2, 4 kHz being 37.17 ± 7.62, 39.17 ± 5.88, 37.67 ± 7.85, and 39.00 ± 9.32 dB, respectively (Figure 1E), implying that subjects obtained good practical hearing due to CI. After implantation, the CAP score of the patients with prelingual HL (n = 56) improved from 0.29 ± 0.73 to 5.52 ± 1.64 (p < 0.001), and the SIR score (n = 61) increased from 1.00 ± 0.00 (before implantation, connected speech was unintelligible for all patients of this subgroup, with the primary mode of communication being manual) to 3.57 ± 1.22 (p < 0.001) (Figure 1I). Five patients of this subgroup completed speech audiometry post-operation, and four patients’ max speech recognition scores were > 60% (Table 2).
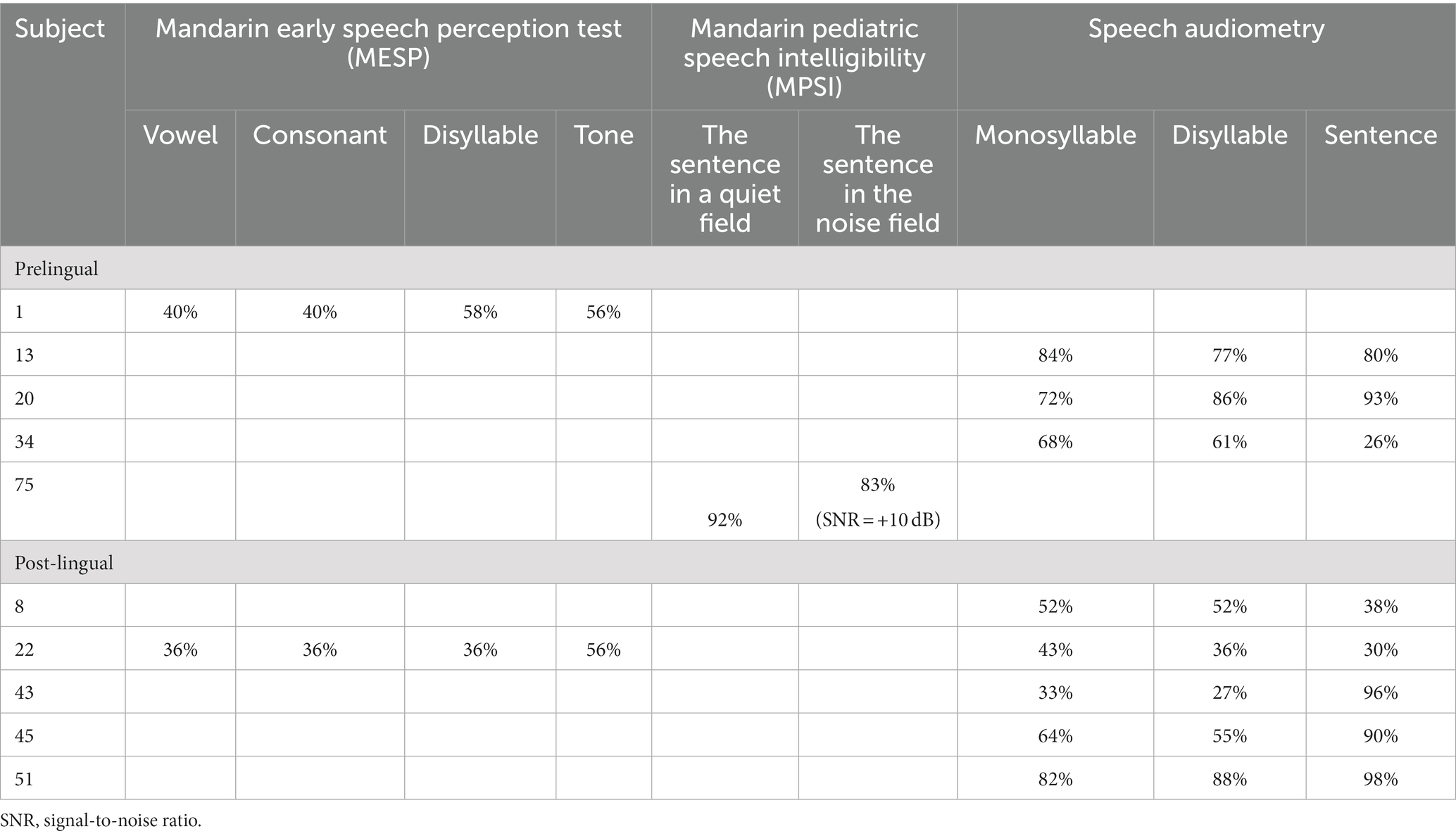
Table 2. Speech recognition rate of subjects after cochlear implantation.
Among post-lingual hearing loss patients (n = 11), the average hearing threshold after CI through pure tone audiometry improved from 78.33 ± 17.34 dB to 32.58 ± 9.26 dB (p < 0.001) (Figure 1H). CAP score (n = 6) was enhanced from 0.50 ± 1.03 to 6.00 ± 0.96 (p < 0.001), and SIR score (n = 13) increased from 1.15 ± 0.48 to 4.15 ± 0.95 (p < 0.001) (Figure 1I). In this subgroup, the maximum speech recognition scores ranged from 52 to 98% for five subjects who underwent speech audiometry after CI (Table 2).
In this study, 46 patients (39 with the prelingual onset and seven with post-lingual onset) participated in genetic testing using high throughput sequencing. When only pathogenic or likely pathogenic variants were considered, molecular etiologies were identified in 25 patients (20 patients of prelingual onset and five cases of post-lingual onset), with two leading responsive genes being OTOF in 13 prelingual onset patients and AIFM1 in three cases of post-lingual hearing loss. Genetic testing results are presented in Table 3.

Table 3. The molecular causes of 25 diagnosed subjects.
Of 13 patients caused by mutations in OTOF attributed to CI, CAP score (n = 10) improved dramatically from 0.10 ± 0.32 to 6.20 ± 1.32 (p < 0.001), and SIR score (n = 13) increased significantly from 1.00 ± 0.00 to 3.83 ± 1.10 (p < 0.001), while in proband 71, SIR score increased to 2 after the operation, which indicated the poor effect of CI.
In three subjects (cases 46, 51, 68), simple hearing loss was identified to be caused by mutations in AIFM1. In case 46, the average hearing threshold improved from 78.25 dB to 40 dB, the CAP score increased from 0 to 3, and the SIR score improved from 1 to 5 after three years of operation. In case 51, the speech recognition score of monosyllables, disyllables, and sentences in quiet was 0, 0, and 0% before implantation. In comparison, the score was 82, 88, and 98%, respectively, after 1.5 years of implantation in the left ear. In patient 68, the CAP score increased from 2 to 5, and the SIR score increased from 1 to 3 after one year of implantation.
Two mutations in TIMM8A were detected in patients 61 and 70, respectively. Case 61 was found to have progressive hearing impairment at two years old, and he had no other obvious abnormalities until this report. After six months of implantation in the right ear, his average hearing threshold improved from 116.25 dB to 40 dB, his CAP score increased from 0 to 5, and his SIR score increased from 1 to 4. Case 70 was noticed to have hearing loss at two months old, with the molecular diagnosis being established at the age of one year. The maternal aunt of Proband 70 also had hearing loss with the same pathogenic variant as Proband, which was revealed when he was 30. Case 70 and his maternal aunt had no other obvious abnormalities. After one year of implantation in the left ear, the average hearing threshold of case 70 improved from 91.25 dB to 36.25 dB, the CAP score increased from 0 to 6, and the SIR score rose from 1 to 3.
For case 15, progressive hearing loss combined with tinnitus was noticed at nine years of age, and no other abnormality was reported. The mother of Proband was also affected by hearing loss combined with nystagmus and ataxia at three years old. A known pathogenic variant in ATP1A3 (p.Glu818Lys) (Han et al., 2017) was found in Proband and confirmed to be inherited from his mother. The average hearing threshold for this patient improved from 71.25 dB to 35 dB. The speech recognition score of vowel, consonant, disyllable, and tone was 72, 80, 70, and 72%, respectively by the MESP test; the SIR score increased from 1 to 3 after unilateral cochlear implantation of half a year.
In case 22, compound heterozygote variants in the TWNK gene were detected. He walked unsteadily and fell quickly after three years old; both eyes had strabismus and astigmia when he was four years old and progressive hearing loss at half past five years old. He was later diagnosed as AN with profound hearing impairment. His speech, understanding, emotion management, and body development were delayed. The sibling of Proband had similar clinical manifestations and had the same compound heterozygote variants in TWNK. Accordingly, these two siblings were diagnosed with Perrault syndrome. For the Proband, after one year of implantation, the average hearing threshold improved from 98.75 dB to 38.25 dB, and vowel, consonant, disyllable, and tone recognition scores in the quiet field were 36, 36, 36, and 56%, respectively. In contrast, the speech recognition rate was 0% for all subtests before CI by the MESP test. The CAP score increased from 0 to 6, and the SIR score improved from 1 to 3 after two years of implantation.
Case 30 had congenital hearing loss combined with ovarian dysfunction, manifested as primary amenorrhea and undeveloped mammary gland. Compound heterozygote variants in the TWNK gene were also identified in this patient. The CAP score increased from 1 to 4, and the SIR score rose from 1 to 3 after two years of implantation.
Case 11 had congenital hearing loss and was later diagnosed as AN with profound hearing impairment. This patient had ptosis on the right eye, hypertelorism, skull shape asymmetry, and bilateral semicircular canal abnormalities. She had a pathogenic variant in the TWIST1 gene (rs104894054). Accordingly, the patient was diagnosed as having Saethre-Chotzen syndrome. The average hearing threshold improved from 102.5 dB to 35 dB, the CAP score increased from 0 to 5, and the SIR score was raised from 1 to 2 after six months of implantation (Figure 2).

Figure 2. Case 11, 39, 74 caused by molecular etiologies related to AN. (A) Pedigree of case 11 caused by c.309C>A in TWIST. (B) Hearing thresholds of case 11 before and after CI. (C) Pedigree of case 39 caused by m.A7445G in MT-CO1. (D) Palmar keratoderma on the left hand of maternal grandmother of Proband. (E) A de novo variant identified in ACTG1 in case 74 and the high conservation of the base in ACTG1 among species. (F) The variant of p.T126I would impact the secondary bone between T126 and I122, E83, leading to the reduction of affinity between subdomains or amino acid chains (ΔΔG affinity = −0.136 kcal/mol).
For case 39, profound hearing loss was detected at half years old and later diagnosed as AN. Genetic testing revealed m.A7445G in homoplasmic status, with no other variant related to hearing loss detected in this case. This variant was inherited from the mother, whose hearing was normal. The maternal grandmother of Proband had normal hearing but palmar keratoderma on the left hand (Figure 2). The m.A7445G variant was identified as the molecular cause of hearing loss of the Proband. After four years of implantation, the CAP score was raised from 0 to 7, and the SIR score increased from 1 to 5.
Severe hearing loss was found at one year old in case 74, and no other obvious abnormalities were found till this report when this case was 13 years old. A likely pathogenic de novo variant in ACTG1 was identified by Trio exome sequencing as the cause of this disease, and no other variant leading to hearing loss was revealed in this patient. After seven years of implantation, the average hearing threshold improved from 70.25 dB to 43.75 dB, the CAP score increased from 2 to 5, and the SIR score was raised from 1 to 2.
Trio exome sequencing found a pathogenic de novo CNV to be a possible cause of AN in case 56 (hearing levels of this patient’s parents are normal). In addition, three variants with uncertain significance were also detected in this patient, including c.3041C>T in MYO7A (NM_000260.3), c.1007G>T in USH1C (NM_001297764.1), and c.1461G>T in PDZD7 (NM_001195263.1), which was inherited from father, mother, mother of this patient, respectively. Profound hearing loss was observed when case 56 was one year old, and the parents of this case reported no other abnormalities until this report. After four years of implantation, the average hearing threshold improved from 97.75 dB to 30 dB after implantation of half years. The CAP score was increased from 0 to 7, and the SIR score was raised from 1 to 5.
In this study, based on SIR score (n = 74) before and after CI, candidate impact factors on speech performances attributed to CI were analyzed, including the age of onset, age of implantation, with or without risk factors, with or without CN deficiency, bilateral or unilateral implantation, etc. (Figure 3). As we observed, no significant difference in the speech capabilities was observed between subjects with implantation age younger or older than 3 years of age (p = 0.671), subjects with an interval time between onset age and implantation age less or more than three years (p = 0.502), subjects with or without risk factors (p = 0.925), and subjects with bilateral or unilateral implantation (p = 0.394); while speech outcomes of patients with no CN deficiency were better than those of subjects with CN deficiency (p = 0.008). As for subjects with prelingual and post-lingual hearing loss, there was no significant difference in speech outcomes after CI (p = 0.260), but for speech performances before IC, patients with post-lingual onset were better than that of subjects with prelingual onset (p = 0.002).
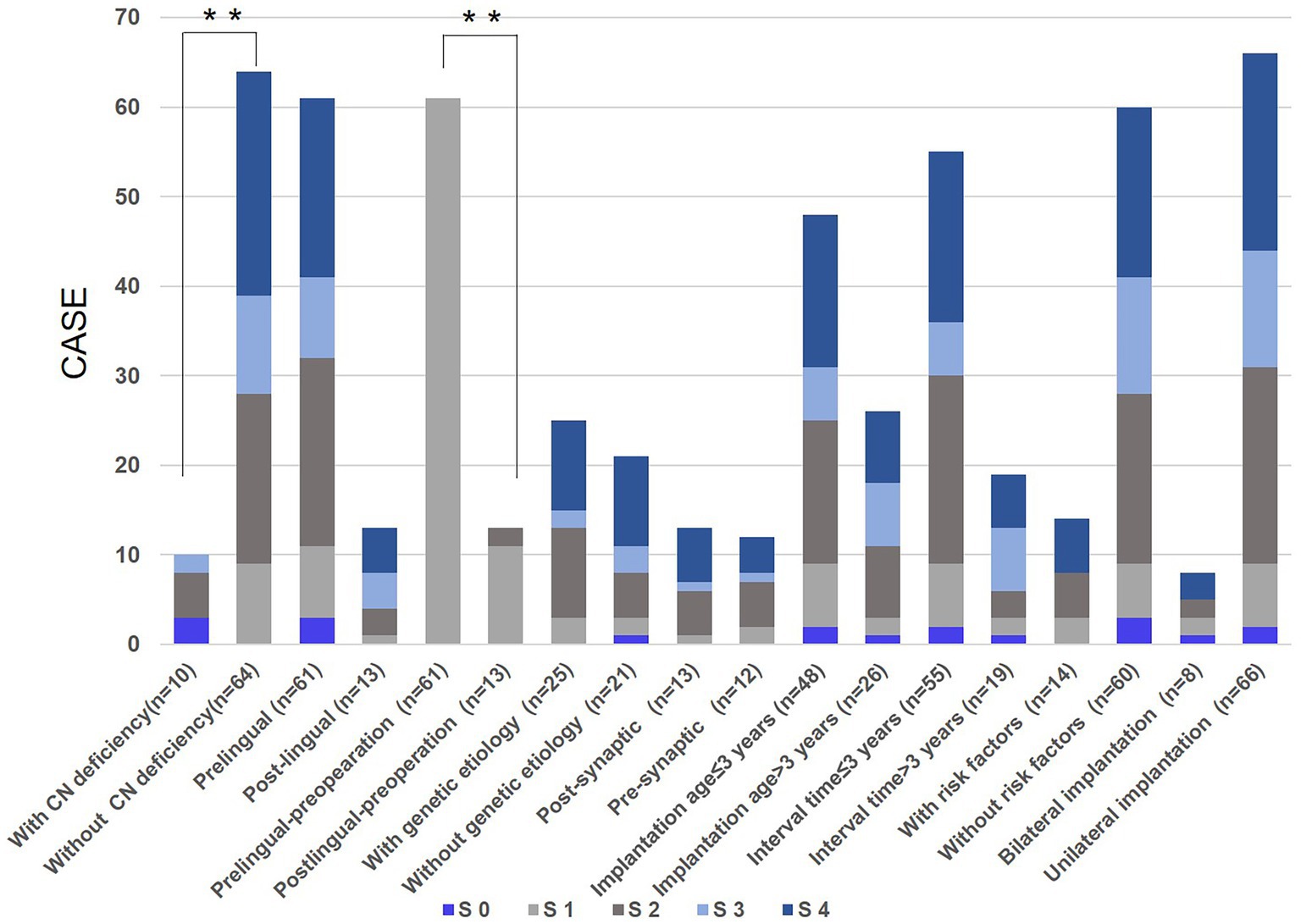
Figure 3. Candidate impact factors on speech outcomes due to cochlear implantation (CI). S 0: number of cases whose SIR score after CI did not improve compared with that before CI; S 1, S 2, S 3, S 4: number of cases in whom speech intelligibility rating (SIR) score after CI was improved by one, two, three, four compared with that before CI, respectively; Whitney U test was applied for significance analysis and statistical significance was defined as **p < 0.01.
Similarly, 46 patients participating in the genetic testing had no significant difference in speech outcomes between subjects with or without positive genetic etiology (p = 0.597). Furthermore, there was no significant difference in speech performance for subjects with OTOF variants and subjects with remaining genetic lesions (p = 0.503).
Auditory and speech outcomes of CI in subjects with AN are always the primary issues to be addressed. Though many studies have focused on this topic (Alzhrani et al., 2019; Shearer and Hansen, 2019), it is still difficult for patients or their parents to decide on CI. In this retrospective study, 75 unrelated patients with AN (61 subjects with the prelingual onset and 14 with post-lingual onset) were enrolled to evaluate auditory and speech performances after CI. For both prelingual and post-lingual recipients, there was a significant improvement in hearing and speech performances compared with those at pre-operation, according to the results of hearing threshold, CAP score, and speech audiometry, SIR score. Generally, cochlear implantation is efficient for patients with AN with severe to profound hearing impairment or those who could not benefit sufficiently from hearing aids.
In the present study, CN deficiency was detected in 10 patients. Second, neonatal hyperbilirubinemia was detected in five patients, including two with other disorders. In another patient, bilirubin encephalopathy combined with neonatal RH hemolysis was also detected. This finding confirms the association between hyperbilirubinemia and AN occurrence (Almishaal et al., 2022). Of 10 patients with CN deficiency and 14 patients identified with other risk factors, nine underwent genetic testing, while molecular etiology was identified in none of these patients. In addition, none of the 75 patients underwent CMV screening. As a result, the risk factor of CMV infection was not analyzed here.
Speech performances of the patient subgroup with CN deficiency were significantly poorer than those of the subgroup with no CN deficiency after CI. At the same time, other candidate impact factors evaluated here had no impacts on CI outcomes, indicating that we should be cautious with the decision of CI in patients with CN deficiency. For 25 patients with a positive molecular diagnosis, there was no significant difference in speech outcomes of patients with mutations in OTOF, which was believed to be related to presynaptic pathology, and mutations from the remaining eight genes or CNV, that were thought to be associated with postsynaptic pathology or affecting spiral ganglion and auditory nerve (Shearer and Hansen, 2019; Chaudhry et al., 2020).
Saethre-Chotzen syndrome is a rare, dominant condition characterized by craniosynostosis and syndactyly (Pelc and Mikulewicz, 2018). Diagnosis of this syndrome is based on typical clinical manifestations and identification of pathogenic variants in the TWIST1 gene. Sensorineural, conductive, and mixed hearing loss might be identified in this condition (Rosen et al., 2011). However, to our knowledge, hearing loss type of AN had never been reported in this syndrome before, which would now broaden the phenotype spectrum of this disorder.
A variant of m.A7445G in mitochondrial DNA was implicated in sensorineural hearing loss and nonepidermolytic palmoplantar keratoderma, with incomplete penetrance and variable clinical findings (Maász et al., 2008). The severity of hearing loss caused by m.A7445G varied from mild to profound, while onset age ranged from infancy to adulthood (Maász et al., 2008; Matsushima et al., 2018), although m.A7445G was first reported to be related to AN in this study.
ACTG1 gene, encoding γ-actins, is the responsive gene of DFNA20/26 and Baraitser–Winter cerebrofrontofacial syndrome (Verloes et al., 2015; Sorrentino et al., 2021). The characteristic clinical phenotype of DFNA20/26 is progressive post-lingual hearing loss. However, congenital severe isolated hearing loss caused by a variant in ACTG1 was also reported earlier (Lee et al., 2018). Here, case 74 had a likely pathogenic de novo variant in ACTG1 (T126I) was identified. The wild type of this variant was highly conserved among species (Figure 2; Liu et al., 2008). Moreover, the secondary bond between T126 and I122 (present in subdomain 4) and E83 (present in subdomain 1) amino acid residues would change in mutation type, which would reduce the affinity between amino acid chains or subdomains of protein1 (Figure 2; Liu et al., 2008).
A pathogenic CNV encompassing 1.08 Mb, a recurrent microdeletion in 7p22.1 (Shimojima et al., 2016), was detected and identified as the possible genetic cause in case 56. This CNV covered 12 genes, including SDK1, FOXK1, AP5Z1, RADIL, MMD2, RBAK, WIPI2, SLC29A4, TNRC18, FBXL18, ACTB, and RNF216. Among these genes, like ACTG1, ACTB encoding β-actins (β- and γ-actin differ at their conserved N-terminal ends by only 4 amino acids) is also associated with Baraitser–Winter cerebrofrontofacial syndrome. Clinical presentations of this syndrome identified sensorineural hearing loss (Verloes et al., 2015). Additionally, haploinsufficiency of ACTB has been reported to be the possible responsive gene of 7p22.1 microdeletion (Palumbo et al., 2018). Therefore, we inferred that the heterozygous deletion of the ACTB gene might be the main cause of AN in this case.
Perrault syndrome characterizes sensorineural hearing loss with or without progressive neurological deficit in both sexes, combined with ovarian dysfunction only in females (Fiumara et al., 2004). TWNK is one of the responsive genes of this syndrome. Meanwhile, the molecular etiology of this syndrome was not identified in approximately 60% of the patients (Lerat et al., 2016; Demain et al., 2017). Herein, we present the first report of a Chinese patient with AN associated with Perrault syndrome, further confirming AN as an audiological feature of this syndrome (Ołdak et al., 2017; Forli et al., 2021). In case 58, hearing loss occurred at eight years of age, combined with primary amenorrhea and hypoplasia of the uterus, and the genetic cause could not be revealed by gene testing. This patient was also diagnosed with Perrault syndrome.
The presynaptic processes of AN encompass both dysfunction and/or loss of inner hair cells, as well as abnormalities in the ribbon synapses in these cells. The postsynaptic mechanisms of AN encompass various pathological conditions, including abnormalities in dendritic nerve terminals, axonal neuropathies, disorders affecting auditory ganglion cells, myelin disorders, hypoplasia of the auditory nerve, and auditory nerve conduction disorders (Rance and Starr, 2015). CI technology is specifically engineered to directly stimulate the auditory nerve, circumventing the signal transmission process between ribbon synapses in inner cells and terminal dendrites of the auditory nerve. The CI outcomes are influenced by the specific locations of malfunction (Shearer and Hansen, 2019). Oxygen deprivation-induced auditory system impairment was linked to inner hair cell dysfunction, and the auditory system’s sensitivity to hyperbilirubinemia involved presynaptic terminal deficits and damages in neuronal Ca2+ homeostasis, while congenital auditory nerve malformation was found in the postsynaptic site (Rance and Starr, 2015; Moser and Starr, 2016). The findings of this study indicate that the outcomes of CI in patients with CN deficiency were poorer than other patients.
Regarding genetic causes, the products of OTOF have been identified as playing a role in the exocytosis process of glutamatergic ribbon synapses. It has been reported that mutations in OTOF can lead to presynaptic synaptopathy (Moser and Starr, 2016). The study findings indicate that patients with OTOF mutations have demonstrated favorable outcomes of CI (Zheng and Liu, 2020). Mutations occurring in the ATP1A3 gene have probably contributed to the development of postsynaptic synaptopathy (Han et al., 2017). The present study has demonstrated that individuals who have AN resulting from ATP1A3 mutations may have potential advantages from CI (the SIR score ≥ 3, or the maximum speech recognition score > 60% after CI). Similarly, it has been reported that two of four CAPOS patients caused by the same mutation as we have identified in this study have markedly benefitted from CI (Tranebjærg et al., 2018). The study indicated a correlation between mutations in TIMM8A and the degeneration of cochlear, vestibular, and optic neurons (Bahmad et al., 2007). Additionally, patients with mutations in AIFM1 experienced delayed development of cochlear nerve hypoplasia (Zong et al., 2015). TIMM8A and AIFM1 mutation has been identified as potential factors contributing to the development of postsynaptic neuropathy (Shearer and Hansen, 2019). Nevertheless, as previously documented, three individuals exhibiting mutations in AIFM1 and two patients with TIMM8A mutations all experienced positive outcomes from CI. The precise pathological mechanisms and specific locations of lesions associated with mutations in TWNK, TWIST1, m.A7445G, ACTG1, and the pathogenic CNV in 7p22.1, remain uncertain. CI outcomes are few and documented in individuals who possess mutations in the TWNK and TWIST1 genes, as well as pathogenic CNVs in the 7p22.1 region. Positive outcomes have been obtained in patients with sensory hearing loss attributed to m.A7445G following CI (Love and Bird, 2013), same results have been observed in individual with AN (case 39) in this study as well. Previous reports have indicated that patients with mutations in ACTG1 gene exhibited satisfactory speech performances after CI (Liu et al., 2019). However, in the present case (case 74), the outcomes of CI did not meet the anticipated level of speech recognition. Understanding the specific locations of lesions can provide valuable insights for guiding the therapeutic therapy of AN.
A few limitations of this study include the relatively small subject size, especially leading to the small size of some subgroups in the analysis of impact factors on speech performances. The auditory and speech examinations administered to the recruited patients varied due to factors such as their age and hearing and speech abilities and experiences. In the interim, the length of CI among patients varied from six months to nine years, a factor that was considered significant in the context of speech rehabilitation.
In conclusion, 82.67% (62/75) of enrolled patients in the present study could benefit from CI (the SIR score of 3 to 5, or the maximum speech recognition score > 60% after CI). Ten patients were CN deficient whose speech performances after CI were poorer than those who were not CN deficient (p = 0.008). Other risk factors were identified in 14 patients, and the leading factor was neonatal hyperbilirubinemia. Molecular diagnosis was established in 25 (54.35%) of 46 patients participating in the comprehensive genetic testing.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
The studies involving humans were approved by the Ethics Committee of Chinese PLA General Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
JW: Formal analysis, Investigation, Writing – original draft. JC: Formal analysis, Investigation, Writing – original draft. ZD: Data curation, Writing – original draft. JF: Data curation, Writing – original draft. QW: Formal analysis, Writing – original draft. PD: Conceptualization, Writing – review & editing. DH: Conceptualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. National Natural Science Foundation of China (No. 81770991) to DH; Nature Science Foundation of Beijing (No. 7191011) to PD; Postdoctoral Research Foundation of China (No. 2018M633731) to JW.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2023.1281884/full#supplementary-material
Almishaal, A. A., Saleh, S., Alferaih, H., and Alhelo, O. (2022). Prevalence, risk factors, and audiological characteristics of auditory neuropathy. Int. J. Audiol. 61, 1018–1026. doi: 10.1080/14992027.2021.2014074
Alzhrani, F., Yousef, M., Almuhawas, F., and Almutawa, H. (2019). Auditory and speech performance in cochlear implanted ANSD children. Acta Otolaryngol. 139, 279–283. doi: 10.1080/00016489.2019.1571283
Bahmad, F. Jr., Merchant, S. N., Nadol, J. B. Jr., and Tranebjaerg, L. (2007). Otopathology in Mohr-Tranebjaerg syndrome. Laryngoscope 117, 1202–1208. doi: 10.1097/MLG.0b013e3180581944
Baux, D., Vaché, C., Blanchet, C., Willems, M., Baudoin, C., Moclyn, M., et al. (2017). Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci. Rep. 7:16783. doi: 10.1038/s41598-017-16846-9
Chaudhry, D., Chaudhry, A., Muzaffar, J., Monksfield, P., and Bance, M. (2020). Cochlear implantation outcomes in post synaptic auditory neuropathies: a systematic review and narrative synthesis. J. Int. Adv. Otol. 16, 411–431. doi: 10.5152/iao.2020.9035
Daneshi, A., Mirsalehi, M., Hashemi, S. B., Ajalloueyan, M., Rajati, M., Ghasemi, M. M., et al. (2018). Cochlear implantation in children with auditory neuropathy spectrum disorder: a multicenter study on auditory performance and speech production outcomes. Int. J. Pediatr. Otorhinolaryngol. 108, 12–16. doi: 10.1016/j.ijporl.2018.02.004
Demain, L. A. M., Urquhart, J. E., O'Sullivan, J., Williams, S. G., Bhaskar, S. S., Jenkinson, E. M., et al. (2017). Expanding the genotypic spectrum of Perrault syndrome. Clin. Genet. 91, 302–312. doi: 10.1111/cge.12776
Fiumara, A., Sorge, G., Toscano, A., Parano, E., Pavone, L., and Opitz, J. M. (2004). Perrault syndrome: evidence for progressive nervous system involvement. Am. J. Med. Genet. 128A, 246–249. doi: 10.1002/ajmg.a.20616
Forli, F., Bruschini, L., Franciosi, B., Battini, R., Marinella, G., Berrettini, S., et al. (2021). A rare case of Perrault syndrome with auditory neuropathy Spectrum disorder: Cochlear implantation treatment and literature review. Audiol. Res. 11, 609–617. doi: 10.3390/audiolres11040055
Han, K.-H., Oh, D.-Y., Lee, S., Lee, C., Han, J. H., Kim, M. Y., et al. (2017). ATP1A3 mutations can cause progressive auditory neuropathy: a new gene of auditory synaptopathy. Sci. Rep. 7:16504. doi: 10.1038/s41598-017-16676-9
Harrison, R. V., Gordon, K. A., Papsin, B. C., Negandhi, J., and James, A. L. (2015). Auditory neuropathy spectrum disorder (ANSD) and cochlear implantation. Int. J. Pediatr. Otorhinolaryngol. 79, 1980–1987. doi: 10.1016/j.ijporl.2015.10.006
Ji, F., Li, J., Hong, M., Chen, A., Jiao, Q., Sun, L., et al. (2015). Determination of benefits of Cochlear implantation in children with auditory neuropathy. PLoS One 10:e0127566. doi: 10.1371/journal.pone.0127566
Ji, F., Li, J.-N., Hong, M.-D., Wang, Q., and Yang, S.-M. (2014). Preliminary performance of cochlear implants in post-lingual patients with auditory neuropathy. Acta Otolaryngol. 134, 280–285. doi: 10.3109/00016489.2013.852689
Lee, C. G., Jang, J., and Jin, H. S. (2018). A novel missense mutation in the ACTG1 gene in a family with congenital autosomal dominant deafness: a case report. Mol. Med. Rep. 17, 7611–7617. doi: 10.3892/mmr.2018.8837
Lerat, J., Jonard, L., Loundon, N., Christin-Maitre, S., Lacombe, D., Goizet, C., et al. (2016). An application of NGS for molecular investigations in Perrault syndrome: study of 14 families and review of the literature. Hum. Mutat. 37, 1354–1362. doi: 10.1002/humu.23120
Liddle, K., Fitzgibbons, E. J., Beswick, R., and Driscoll, C. (2022). Cochlear nerve deficiency is an important cause of auditory neuropathy spectrum disorder at a population level in children. Int. J. Pediatr. Otorhinolaryngol. 158:111171. doi: 10.1016/j.ijporl.2022.111171
Liu, W.-H., Chang, P.-Y., Chang, S.-C., Lu, J.-J., and Wu, C.-M. (2019). Mutation screening in non-syndromic hearing loss patients with cochlear implantation by massive parallel sequencing in Taiwan. PLoS One 14:1261. doi: 10.1371/journal.pone.0211261
Liu, P., Li, H., Ren, X., Mao, H., Zhu, Q., Zhu, Z., et al. (2008). Novel ACTG1 mutation causing autosomal dominant non-syndromic hearing impairment in a Chinese family. J. Genet. Genomics 35, 553–558. doi: 10.1016/S1673-8527(08)60075-2
Love, R. L., and Bird, P. (2013). Cochlear implantation in mitochondrial deafness due to A7445G mutation. Cochlear Implants Int. 14, 28–31. doi: 10.1179/1754762811Y.0000000030
Maász, A., Komlósi, K., Hadzsiev, K., Szabó, Z., Willems, P. J., Gerlinger, I., et al. (2008). Phenotypic variants of the deafness-associated mitochondrial DNA A7445G mutation. Curr. Med. Chem. 15, 1257–1262. doi: 10.2174/092986708784534910
Matsushima, K., Nakano, A., Arimoto, Y., Mutai, H., Yamazawa, K., Murayama, K., et al. (2018). High-level heteroplasmy for the m.7445A>G mitochondrial DNA mutation can cause progressive sensorineural hearing loss in infancy. Int. J. Pediatr. Otorhinolaryngol. 108, 125–131. doi: 10.1016/j.ijporl.2018.02.037
Moser, T., and Starr, A. (2016). Auditory neuropathy — neural and synaptic mechanisms. Nat. Rev. Neurol. 12, 135–149. doi: 10.1038/nrneurol.2016.10
Natale, F., de Curtis, M., Bizzarri, B., Orlando, M. P., Ralli, M., Liuzzi, G., et al. (2020). Isolated auditory neuropathy at birth in congenital cytomegalovirus infection. Ital. J. Pediatr. 46:3. doi: 10.1186/s13052-019-0767-y
Ołdak, M., Oziębło, D., Pollak, A., Stępniak, I., Lazniewski, M., Lechowicz, U., et al. (2017). Novel neuro-audiological findings and further evidence for TWNK involvement in Perrault syndrome. J. Transl. Med. 15:25. doi: 10.1186/s12967-017-1129-4
Palumbo, O., Accadia, M., Palumbo, P., Leone, M. P., Scorrano, A., Palladino, T., et al. (2018). Refinement of the critical 7p22.1 deletion region: Haploinsufficiency of ACTB is the cause of the 7p22.1 microdeletion-related developmental disorders. Eur. J. Med. Genet. 61, 248–252. doi: 10.1016/j.ejmg.2017.12.008
Pelc, A., and Mikulewicz, M. (2018). Saethre-Chotzen syndrome: case report and literature review. Dent Med Probl. 55, 217–225. doi: 10.17219/dmp/91050
Rajput, K., Saeed, M., Ahmed, J., Chung, M., Munro, C., Patel, S., et al. (2019). Findings from aetiological investigation of auditory neuropathy Spectrum disorder in children referred to cochlear implant programs. Int. J. Pediatr. Otorhinolaryngol. 116, 79–83. doi: 10.1016/j.ijporl.2018.10.010
Rance, G., and Starr, A. (2015). Pathophysiological mechanisms and functional hearing consequences of auditory neuropathy. Brain 138, 3141–3158. doi: 10.1093/brain/awv270
Rosen, H., Andrews, B. T., Meara, J. G., Stoler, J. M., Mulliken, J. B., and Rogers, G. F. (2011). Audiologic findings in Saethre-Chotzen syndrome. Plast. Reconstr. Surg. 127, 2014–2020. doi: 10.1097/PRS.0b013e31820cf16a
Sarankumar, T., Arumugam, S. V., Goyal, S., Chauhan, N., Kumari, A., and Kameswaran, M. (2018). Outcomes of Cochlear implantation in auditory neuropathy Spectrum disorder and the role of cortical auditory evoked potentials in benefit evaluation. Turk Otolarengoloji Arsivi/Turkish. Arch. Otolaryngol. 1, 15–20. doi: 10.5152/tao.2017.2537
Sennaroğlu, L., and Bajin, M. D. (2017). Classification and current Management of Inner ear Malformations. Balkan Med. J. 34, 397–411. doi: 10.4274/balkanmedj.2017.0367
Shearer, A. E., and Hansen, M. R. (2019). Auditory synaptopathy, auditory neuropathy, and cochlear implantation. Laryngoscope Invest. Otolaryngol. 4, 429–440. doi: 10.1002/lio2.288
Shimojima, K., Narai, S., Togawa, M., Doumoto, T., Sangu, N., Vanakker, O. M., et al. (2016). 7p22.1 microdeletions involving ACTB associated with developmental delay, short stature, and microcephaly. Eur. J. Med. Genet. 59, 502–506. doi: 10.1016/j.ejmg.2016.09.008
Sorrentino, U., Piccolo, C., Rigon, C., Brasson, V., Trevisson, E., Boaretto, F., et al. (2021). DFNA20/26 and other ACTG1-associated phenotypes: a case report and review of the literature. Audiol. Res. 11, 582–593. doi: 10.3390/audiolres11040052
Tranebjærg, L., Strenzke, N., Lindholm, S., Rendtorff, N. D., Poulsen, H., Khandelia, H., et al. (2018). The CAPOS mutation in ATP1A3 alters Na/K-ATPase function and results in auditory neuropathy which has implications for management. Hum. Genet. 137, 111–127. doi: 10.1007/s00439-017-1862-z
Verloes, A., di Donato, N., Masliah-Planchon, J., Jongmans, M., Abdul-Raman, O. A., Albrecht, B., et al. (2015). Baraitser–winter cerebrofrontofacial syndrome: delineation of the spectrum in 42 cases. Eur. J. Hum. Genet. 23, 292–301. doi: 10.1038/ejhg.2014.95
Wu, J., Cao, Z., Su, Y., Wang, Y., Cai, R., Chen, J., et al. (2022). Molecular diagnosis of a large hearing loss population from China by targeted genome sequencing. J. Hum. Genet. 67, 643–649. doi: 10.1038/s10038-022-01066-5
Zheng, D., and Liu, X. (2020). Cochlear implantation outcomes in patients with OTOF mutations. Front. Neurosci. 14:447. doi: 10.3389/fnins.2020.00447
Zheng, Y., Meng, Z.-L., Wang, K., Tao, Y., Xu, K., and Soli, S. D. (2009a). Development of the mandarin early speech perception test: children with normal hearing and the effects of dialect exposure. Ear Hear. 30, 600–612. doi: 10.1097/AUD.0b013e3181b4aba8
Zheng, Y., Soli, S. D., Wang, K., Meng, J., Meng, Z., Xu, K., et al. (2009b). Development of the mandarin pediatric speech intelligibility (MPSI) test. Int. J. Audiol. 48, 718–728. doi: 10.1080/14992020902902658
Keywords: auditory neuropathy, cochlear implantation, impact factor, cochlear nerve, genetic testing
Citation: Wu J, Chen J, Ding Z, Fan J, Wang Q, Dai P and Han D (2023) Outcomes of cochlear implantation in 75 patients with auditory neuropathy. Front. Neurosci. 17:1281884. doi: 10.3389/fnins.2023.1281884
Edited by:
Meisam Arjmandi, University of South Carolina, United StatesReviewed by:
Lisbeth Tranebjærg, University of Copenhagen, DenmarkCopyright © 2023 Wu, Chen, Ding, Fan, Wang, Dai and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pu Dai, ZGFpcHUzMDFAdmlwLnNpbmEuY29t; Dongyi Han, aGR5MzAxQDI2My5uZXQ=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.