 Paul Smolen
Paul Smolen Pramod K. Dash
Pramod K. Dash John B. Redell
John B. Redell
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Neurosci. , 19 September 2023
Sec. Neurogenomics
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1259405
This article is part of the Research Topic Epigenetic Modification in Neurological Diseases View all 5 articles
Epidemiological studies have shown that traumatic brain injury (TBI) increases the risk for developing neurodegenerative diseases (NDs). However, molecular mechanisms that underlie this risk are largely unidentified. TBI triggers widespread epigenetic modifications. Similarly, NDs such as Alzheimer’s or Parkinson’s are associated with numerous epigenetic changes. Although epigenetic changes can persist after TBI, it is unresolved if these modifications increase the risk of later ND development and/or dementia. We briefly review TBI-related epigenetic changes, and point out putative feedback loops that might contribute to long-term persistence of some modifications. We then focus on evidence suggesting persistent TBI-associated epigenetic changes may contribute to pathological processes (e.g., neuroinflammation) which may facilitate the development of specific NDs – Alzheimer’s disease, Parkinson’s disease, or chronic traumatic encephalopathy. Finally, we discuss possible directions for TBI therapies that may help prevent or delay development of NDs.
Epigenetic information includes reversible modifications to DNA, or to DNA-associated histone proteins involved in regulating gene expression (Fitz-James and Cavalli, 2022). Long-lasting epigenetic changes occur in many disease states (Lardenoije et al., 2018; Cavalli and Heard, 2019; Bertogliat et al., 2020) and play important roles in aging and long-term memory (Kim and Kaang, 2017; Bellver-Sanchis et al., 2021). Epigenetic modifications and their putative impact on the pathology of neurodegenerative diseases (NDs), including Alzheimer’s disease (AD), and Parkinson’s disease (PD), have been extensively studied (Pavlou and Outeiro, 2017; Berson et al., 2018; Stoccoro and Coppedè, 2018; Bennett et al., 2019; Nikolac Perkovic et al., 2021; Lee et al., 2023). Fewer studies, however, have examined the nature and persistence of epigenetic changes arising from traumatic brain injury (TBI). For some NDs and dementias; such as AD, PD, and chronic traumatic encephalopathy (CTE); evidence suggests TBI (including repeated mild TBI) is a risk factor (Goldman et al., 2006; Gardner et al., 2015; Gardner and Yaffe, 2015; Delic et al., 2020; Schneider et al., 2021; Brett et al., 2022; Graham et al., 2022; Mielke et al., 2022). Epigenetic modifications observed after TBI have also been observed in these NDs. Some changes correlate with persistent neuroinflammation, which commonly follows TBI and may predispose for NDs up to years later (Faden and Loane, 2015; Dams-O'Connor et al., 2016; Brett et al., 2022). These modifications can potentially be pharmacologically targeted to prevent or delay NDs, by targeting the enzymes that add (epigenetic writers), remove (erasers), or decode (readers), epigenetic modifications (Kelly et al., 2010; Cheng et al., 2019; Majchrzak-Celińska et al., 2021).
For brevity, we focus on two common modification types, DNA methylation and histone acetylation. These modifications associate with NDs and TBI and could serve as therapeutic targets (Bruno et al., 2022).
DNA methyltransferases (DNMTs) are writer enzymes that add a methyl (-CH3) group to a cytosine base (Okano et al., 1999; Jin and Robertson, 2013; Coppede, 2022), commonly at 5′-cytosine-guanine-3′ (CpG) dinucleotides in a promoter region. Cytosine methylation primarily represses transcription (Cedar and Bergman, 2009), although there are activation examples (Harris et al., 2018; Rauluseviciute et al., 2020). Methylated DNA recruits readers (Fitz-James and Cavalli, 2022) that facilitate local chromatin compaction and can recruit other writers (Bennett and Licht, 2018; Schmidt et al., 2020). Methylation is reversed by erasers of the ten-eleven translocation (Tet) family (Yamaguchi et al., 2013). Most intracellular methylation reactions utilize the universal methyl donor S-adenosyl methionine (SAM; Landgraf et al., 2016), which can be derived from dietary methionine and from folate (vitamin B9). Vitamins B2 and B12, and choline, also help maintain SAM levels and methylation homeostasis (Abbasi et al., 2017; Froese et al., 2019; McNulty et al., 2019).
Each nucleosome of eukaryotic chromatin consists of a histone octamer wound by ~2 turns of DNA (Kornberg, 1974). Attraction between DNA and positively charged histones tends to compactify chromatin, hindering transcription complex access to the promoter (Bartke and Kouzarides, 2011). Acetylation of histone lysine (Gräff and Tsai, 2013) or arginine (Cura and Cavarelli, 2021) residues by acetyltransferase writers neutralizes positive charge, facilitating gene expression. Specificity is mediated by readers that identify patterns of acetylated residues and modulate transcription (Xu et al., 2017; Poulard et al., 2021; Chen et al., 2022). Other histone modifications include methylation, which can be long-lasting (Fitz-James and Cavalli, 2022); phosphorylation, and ubiquitination. Roles of these modifications following TBI have not been well characterized, thus we mainly discuss acetylation.
We focus on three NDs for which there is evidence that TBI is a risk factor, AD, PD, and CTE. For AD, a meta-analysis of 15 case–control studies (Fleminger et al., 2003) and a veteran cohort study (Plassman et al., 2000) identified TBI as a risk factor. For PD, TBI is repeatedly reported as a risk factor (Bower et al., 2003; Lee et al., 2012; Jafari et al., 2013; Delic et al., 2020; although see Rugbjerg et al., 2008; Marras et al., 2014). CTE is a tauopathy with progressive accumulation of Tau protein (McKee et al., 2009; Stein et al., 2014) and has been delineated as a consequence of repeated mild TBI (Gardner and Yaffe, 2015; Smith et al., 2019; McKee et al., 2023). A relationship was found in American football players (McKee, 2020), with CTE-related pathology reported in most players’ post-mortem brains (Mez et al., 2017). Other contact sports, and military injuries, are a risk factor for CTE and likely other ND-related dementias (Chauhan, 2014; Johnson et al., 2017; Smith et al., 2019; Delic et al., 2020).
TBI commonly engenders life-long consequences (Masel and DeWitt, 2010) including cognitive impairments. Experimental TBI, and human studies, have begun exploring post-TBI epigenetic changes (Wong and Langley, 2016; Mateen et al., 2017; Nagalakshmi et al., 2018; Bertogliat et al., 2020). In a rat blast injury model, hippocampal DNA methylation was increased 2 weeks post-injury, as was expression of the writers DNMT1 and DNMT3b (Bailey et al., 2015). Other murine TBI models exhibited hypomethylated as well as hypermethylated loci in cortex (Haghighi et al., 2015; Meng et al., 2017). Serotonin N-acetyltransferase (Aanat) was hypermethylated and downregulated (Haghighi et al., 2015), reducing conversion of serotonin to melatonin, possibly contributing to insomnia (Cruz-Sanabria et al., 2023). The superoxide dismutase 2 (Sod2) promoter was hypermethylated and its expression downregulated, which could increase oxidative stress (Balasubramanian et al., 2021a). In amygdala, enhanced DNMT activity correlated with brain-derived neurotrophic factor (Bdnf) methylation and reduced expression (Sagarkar et al., 2017). After repeated TBI, mitofusin 2 (Mfn2) hypermethylation and repression triggers mitochondrial dysfunction (Kulkarni et al., 2023). Hypomethylation in perilesional microglia was associated with inflammation (Zhang et al., 2007). DNA methylation changes, most commonly hypomethylation, were found in blood cells following human TBI (Bahado-Singh et al., 2020).
Histone hypoacetylation has been observed in murine cortex post-TBI (Gao et al., 2006) and in hippocampus (Kumari et al., 2023). Increased hippocampal histone deacetylase (HDAC) activity has also been observed (Sagarkar et al., 2019) as was neuropeptide Y promoter hypoacetylation (Balasubramanian et al., 2021b). TBI was reported to upregulate HDAC2-5 and HDAC11 (Sagarkar et al., 2019) and downregulate HDAC4-5 (Kamal et al., 2022). HDAC2 upregulation is of interest because its activity has been implicated in AD (see below). It is not yet known if altered histone acetylation persists chronically post-TBI.
Overall, substantial epigenetic changes are induced by TBI. However, only a few changes correlate with data suggesting long-term persistence of these changes or links to development of NDs years later. We discuss links that, although hypothetical, are plausible from data. Thus, below, we do not comprehensively discuss epigenetic changes associated with NDs, but focus on a subset of changes that could be linked to a preceding TBI.
Inflammatory biomarkers correlate with cognitive impairment in AD patients (de Oliveira et al., 2021), and persistent neuroinflammation is one hypothesized driver of AD development (see below). Genetic polymorphisms associated with immune system regulation, such as in TREM2, CR1, and APOE, enhance AD risk and correlate with increased neuroinflammation (Karch and Goate, 2015; Lee et al., 2018; de Oliveira et al., 2021). Neuroinflammation and reduced microglia phagocytic function increase Aβ deposition and Tau hyperphosphorylation (Kitazawa et al., 2005; Lee et al., 2008; Li et al., 2022). In turn, Tau hyperphosphorylation promotes Tau aggregation (Limorenko and Lashuel, 2021), plausibly contributing to CTE. In mouse tauopathy, microglia activation preceded deposition of Tau neurofibrillary tangles (Yoshiyama et al., 2007). The cytokine interleukin-1β enhances Tau phosphorylation (Ghosh et al., 2013; Collins-Praino and Corrigan, 2017). Indeed, some data may support a hypothetical positive feedback loop with reciprocal activation of inflammation and Tau hyperphosphorylation/aggregation. Secretion of hyperphosphorylated Tau enhances glia overactivation and cytokine production, and neuroinflammation, in turn, enhances Tau phosphorylation (Lee et al., 2010; Laurent et al., 2018; Fesharaki-Zadeh, 2019; Al-Ghraiybah et al., 2022). Chronic neuroinflammation may also predispose to PD (Rasheed et al., 2021). Inflammation upregulates caspase-1 which in turn increases α-synuclein aggregation (Wang et al., 2016).
Following TBI, neuroinflammation, characterized in part by microglia overactivation, can persist for many years (Ramlackhansingh et al., 2011; Johnson et al., 2013; Coughlin et al., 2015; Makinde et al., 2017; Risbrough et al., 2022) and DNA hypomethylation has been identified in microglia that can enhance neuroinflammation (Zhang et al., 2007). However, does persistent neuroinflammation post-TBI result, at least in part, from persistence of epigenetic changes? A recent study may suggest a link. In mouse cortex, up to 2 years post-TBI, genes in the complement activation and effector pathways were upregulated (Toutonji et al., 2021). Complement inhibition ameliorated this upregulation. These data suggest a hypothetical positive feedback loop between complement activity, persistent inflammation, and transcription. DNA hypomethylation after TBI in microglia suggests such feedback may involve epigenetic modifications. Hippocampal DNA hypomethylation is also reported in AD patients (Chouliaras et al., 2013).
Currently ~6 million Americans live with AD, expected to increase to ~13 million by 2050 (Alzheimer’s Association, www.alz.org). AD correlates with neuronal accumulation of toxic amyloid β (Aβ), with some evidence for causation (Selkoe and Hardy, 2016; Abu Hamdeh et al., 2018; Hampel et al., 2021). Alternative hypotheses for AD causation include accumulation of toxic Tau protein as a primary driver (Guo et al., 2017; Nasb et al., 2023), cumulative oxidative stress (Roy et al., 2023), or persistent inflammation and glial senescence (Lau et al., 2023).
Following human TBI, diffuse Aβ plaques have been observed to accumulate in brain, up to decades post-TBI (Roberts et al., 1991; Johnson et al., 2012; Scott et al., 2016). Aβ accumulation is observed in only a minority of post-TBI human brains, and may correlate with genetic susceptibility (Roberts et al., 1994; Nicoll et al., 1995; DeKosky et al., 2007; Johnson et al., 2009; Smith and Stewart, 2018). Following human TBI, methylation changes in or near the amyloid precursor protein (APP), MAPT (encoding Tau protein isoforms), and neurofilament genes (NEFH, NEFM, and NEFL) have been reported in brain (Abu Hamdeh et al., 2021). Thus, epigenetic upregulation of APP expression, or MAPT, may play a role in any post-TBI Aβ or Tau accumulation. However, these changes have not yet been shown to be long-lasting or to upregulate expression. Thus, more research is necessary to investigate links between epigenetic changes post-TBI and subsequent AD, and investigate whether pathology is driven primarily by Aβ or Tau accumulation, or oxidative stress, and/or inflammation.
Hypomethylation of presenilin 1 (PSEN1) correlates with AD, and with increased PSEN1 expression, which may indirectly enhance production of toxic Aβ (Monti et al., 2020). It would be of interest to examine PSEN1 epigenetic modifications at late time points post-TBI. In contrast, repeated TBI hypermethylates and decreases expression of Sod2 in murine hippocampus (Balasubramanian et al., 2021a). This decrease could lead to oxidative damage, increasing risk for NDs including AD. DNMT inhibition normalized Sod2 methylation and expression and ameliorated neurodegeneration and learning deficits.
Neuronal and glial epigenetic changes characterize PD (Lee et al., 2023). Hypomethylation in/near the α-synuclein gene (SNCA) occurs in PD patients and enhances SNCA expression (Matsumoto et al., 2010). Consequent α-synuclein accumulation may accelerate PD (Kontopoulos et al., 2006). In turn, α-synuclein was observed to interact with, and mislocalize, DNMT1 (Desplats et al., 2011) which may contribute to hypomethylation and further activation of SNCA. These interactions constitute a putative positive feedback loop that could contribute to late α-synuclein accumulation, and consequent PD, following an initial increase due to TBI. α-synuclein has occasionally been observed to increase post-TBI. In rat models, α-synuclein was increased in rat substantia nigra (SN) 60 days post-TBI (Acosta et al., 2015), and α-synuclein aggregation was enhanced in SN and striatum (Acosta et al., 2019). In two studies, the majority of postmortem human brains with a history of recent severe TBI showed α-synuclein accumulation (Ikonomovic et al., 2004; Uryu et al., 2007). However, another study did not find α-synuclein accumulation in brain samples from 53 individuals with a history of remote TBI (Postupna et al., 2021).
Human TBI can induce accumulation of Tau (Collins-Praino and Corrigan, 2017; Clark et al., 2021). In the Sydney Brain Bank, observed prevalence of CTE pathology was relatively low (0.79%), suggesting single TBI may be unlikely to cause long-lasting Tau accumulation or CTE, with repeated TBI plausibly required (McCann et al., 2022). In rats, TBI can induce a phosphorylated Tau species that correlates with neuropathology (Hintermayer et al., 2020). Following a TBI-induced Tau increase, positive feedback favoring further Tau phosphorylation could lead to CTE tauopathy. A recent study (Wang et al., 2021) suggests epigenetics could drive such a positive feedback loop. In P301S Tau mutant mouse cortex, Tau is hyperphosphorylated and aggregates, and activity of euchromatic histone-lysine N-methyltransferase (EHMT), which methylates histones, is elevated. EHMT inhibition reduced hyperphosphorylated Tau, supporting the existence of positive feedback in which EHMT activity elevation indirectly drives further Tau phosphorylation. Alternatively, EHMT could mediate such feedback via a non-histone substrate.
Table 1 lists representative genes found to be epigenetically modified either after TBI, or in an ND (either in humans or in rodent TBI or ND models), putative links to other pathologies, and whether the modification is persistent, if known.
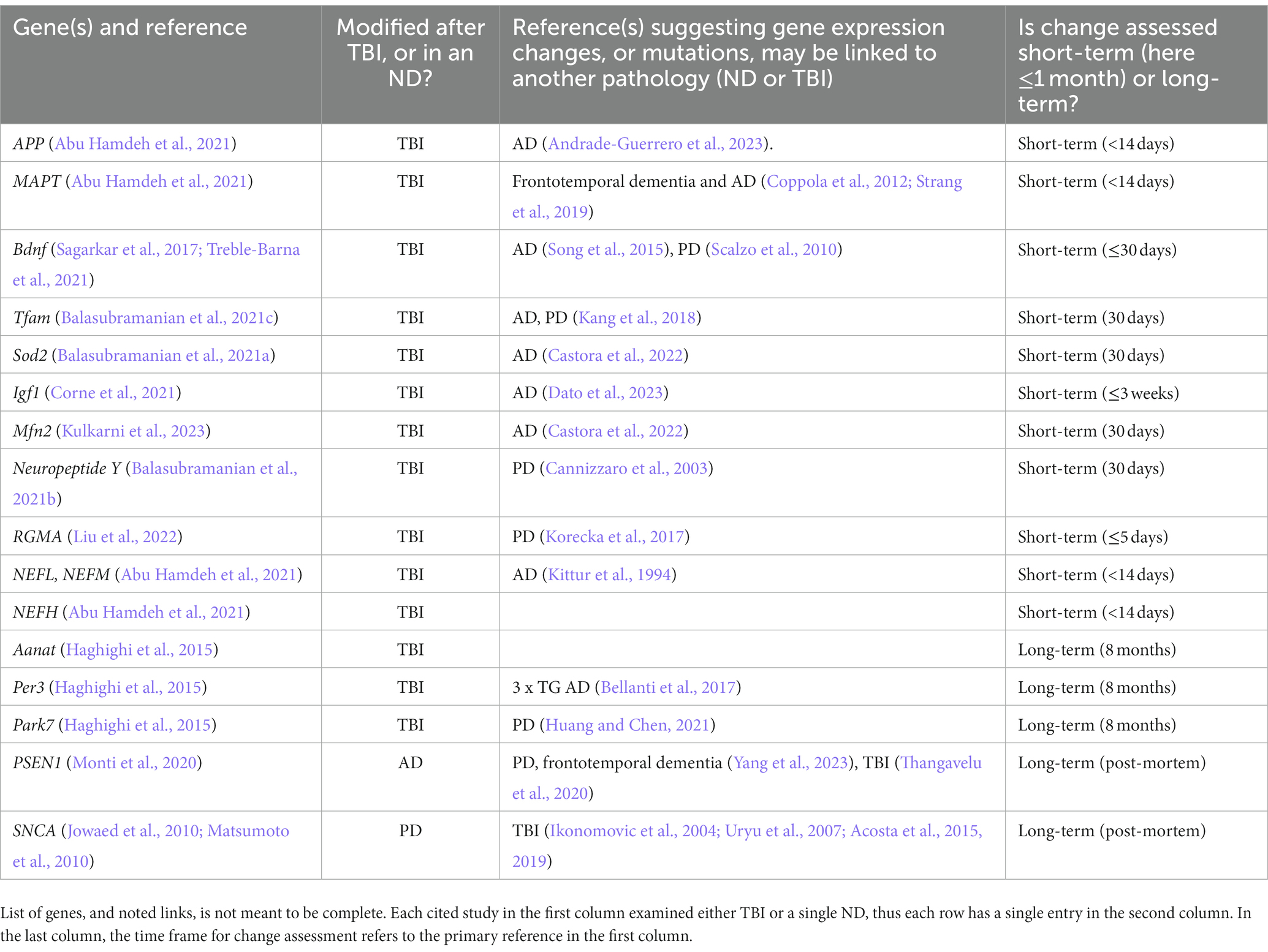
Table 1. Representative genes epigenetically altered due to TBI or in AD or PD.
Considering the above putative epigenetic links between TBI and NDs suggests strategies that might prevent or delay NDs.
Blood levels of SAM and methionine are decreased in TBI patients (Dash et al., 2016). S-adenosyl-L-methionine decarboxylase, which synthesizes SAM, is decreased in rat motor cortex post-TBI, which may contribute to reduced epigenetic methylation (Henley et al., 1997). SAM administration alters DNA methylation in macrophages and reduces inflammation (Pfalzer et al., 2014). These data suggest SAM might ameliorate some adverse TBI sequelae (Schieffler and Matta, 2022; Zima et al., 2022). In one clinical trial, administration of SAM post-concussion correlated with a 77% reduction in adverse clinical scores, compared to a 49% reduction with placebo (Bacci Ballerini et al., 1983). Global DNA hypomethylation appears to correlate with long-lasting neuroinflammation (Gonzalez-Jaramillo et al., 2019) which might be alleviated by SAM. In a murine AD model, SAM reduced Aβ deposition and rescued cognitive deficits (Do Carmo et al., 2016).
Thus, treatment or dietary supplementation by SAM or B vitamins might ameliorate short-term TBI sequelae and provide some ND protection. However, there are caveats. Hypermethylation of SOD2 occurs post-TBI and suppresses its expression as noted above. Methyl donors might further suppress SOD2 expression. Supplementing mouse fibroblast cultures with SAM was reported to overproduce adenine and methylthioadenosine, leading to toxicity (Fukumoto et al., 2022). It will be important to examine whether different doses of SAM or alternative methyl donors, such as B vitamins, show fewer potential adverse effects. Epigenetics may not underlie all therapeutic effects of methyl donors. SAM is a cofactor for enzymes that do not catalyze methylation but associate with human diseases (Landgraf et al., 2016). The potential therapeutic usefulness of methyl donors is further discussed in Coppede (2022). It is important to note that for methyl donors as well as other potential therapies discussed here, our discussion is relatively speculative, insofar as no clinical results have been published demonstrating the efficacy, for any ND, of these potential treatments. As of August 2023, clinical trials are examining the efficacy of B vitamin supplementation, exercise, and complement inhibition as therapies post-TBI (ClinicalTrials.gov).
In case of adverse effects, more targeted modifications might be preferred. Specific isoforms of demethylating (or methylating) enzymes involved might be targeted. Or, methylation might be altered at specific gene loci. This could be useful if, for example, a treatment could break positive feedback in which aberrant methylation of a locus, induced by TBI, becomes self-sustaining. This direction would require development of vectors crossing the blood–brain barrier, plausibly using CRISPR/Cas-based gene editors (Urbano et al., 2019; Zou et al., 2022).
As discussed above, positive feedback may occur between neuroinflammation and Tau aggregation/propagation (Laurent et al., 2018; Fesharaki-Zadeh, 2019; Al-Ghraiybah et al., 2022). Post-TBI anti-inflammatory treatment might break such a feedback loop (Selvaraj et al., 2021; Kip and Parr-Brownlie, 2022; McGovern et al., 2022; Smith et al., 2022). Gene expression in the complement activation and effector pathways was upregulated up to 2 years post-TBI in mouse (Toutonji et al., 2021). Thus, hypothetically, complement inhibitor therapy may ameliorate neuroinflammation and ND development.
Although late persistence of post-TBI histone hypoacetylation has not been established, it would be interesting to explore possible links with ND development. Following rodent TBI, spatial and fear learning and other cognitive measures are improved by HDAC inhibitors (HDACIs; Dash et al., 2009; Shein et al., 2009; Yu et al., 2013; Tai et al., 2014; Sagarkar et al., 2019) and microglia inflammatory responses reduced (Zhang et al., 2008). The HDACI Scriptaid improves neuronal survival and other aspects of pathology (Wang et al., 2013; Meng et al., 2020). HDAC inhibition may also decrease expression of pro-inflammatory factors in neurons and immune cells (Dai et al., 2021; Ghiboub et al., 2021). These data suggest HDAC inhibition may inhibit the process – neuroinflammation – that currently appears to provide the strongest long-term link between TBI and subsequent NDs. HDAC2 activity is increased in AD patient brains, and HDAC2 knockdown in a mouse neurodegeneration model rescued learning and memory impairments and related gene expression (Gräff et al., 2012). Thus, HDACIs specific to HDAC2 may be of particular interest.
Vorinostat, a relatively broad-spectrum HDAC inhibitor, is in a clinical trial as a potential AD treatment (Bondarev et al., 2021). However, there is concern regarding potential toxicity of long-term HDAC inhibition in humans, with more study of targeted inhibitors needed (Coppede, 2022). One study suggests HDAC1 protects against neurodegeneration (Patnaik et al., 2021) so that an HDAC1 activator might also be therapeutic.
As discussed above positive feedback may sustain Tau hyperphosphorylation and histone methylation, and inhibiting histone n-lysine methyl transferase (EHMT) reduced hyperphosphorylated Tau (Wang et al., 2021). It would be of interest to investigate whether post-TBI Tau aggregation could be reduced in this way. This might help prevent CTE, or other tauopathies.
Evidence suggests habitual exercise can prevent or delay NDs and reduce chronic neuroinflammation (Coppede, 2022; Ribarič, 2022; Sujkowski et al., 2022). Numerous epigenetic changes in rodent brain tissue have been observed following endurance exercise (Fernandes et al., 2017). Prominent are altered histone acetylation, enhanced expression of DNA demethylases, and DNA methylation changes at/near genes including Bdnf (Sellami et al., 2021; Xu et al., 2021). Levels of BDNF and another growth factor, IGF-1, are enhanced by exercise (Vaynman et al., 2004; Ding et al., 2006), which would help to counter an age-associated BDNF decrease (Xu et al., 2021). Enhanced growth factor activity likely helps maintain neural circuits, improving TBI recovery and reducing ND susceptibility. Thus an exercise program for TBI survivors might delay or prevent NDs.
Finally, studies have found a “Mediterranean-style” diet, rich in fruits and vegetables, and in flavonoids and antioxidants, to associate with a reduced risk of AD and other NDs (Holland et al., 2020; Migliore and Coppede, 2022), suggesting recommendation of these diets post-TBI.
Studies of epigenetic changes post-TBI, and their possible relationships with subsequent ND development, are beginning to provide suggestions for therapeutic directions that may prevent or reduce the risk for human NDs. But many gaps in knowledge need to be addressed. Prominently: (1) For most cases, the possible persistence over months or years of specific epigenetic changes that correlate with TBI has not been examined, thus their potential relevance to subsequent ND development cannot be assessed. (2) If changes are found to be persistent, what are the mechanisms underlying persistence, e.g., positive feedback? Mechanistic understanding may prove essential to develop therapies. (3) There is a profound lack of clinical studies examining the long-term effects of potential therapies.
Overall, we are at very early stages in understanding the epigenetic effects of TBI and their relationship with NDs. This active field is primed to deliver numerous insights into epigenetic events, and treatments, in years to come.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
PS: Conceptualization, Investigation, Writing – original draft, Writing – review and editing. PD: Conceptualization, Supervision, Writing – review and editing, Funding acquisition. JR: Conceptualization, Project administration, Supervision, Writing – review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported in part by an NIH grant (NS121261; PD) and by the Nina and Michael Zilkha Distinguished Chair in Neurodegenerative Research (PD).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aβ, Amyloid β; AD, Alzheimer’s disease; APP, Amyloid precursor protein; CTE, Chronic traumatic encephalopathy; DNMT:, DNA methyl transferase; EHMT, Euchromatic histone-lysine N-methyltransferase; HDAC, Histone deacetylase; HDACI, HDAC inhibitor; LTP, Long-term potentiation; ND, Neurodegenerative disease; PD, Parkinson’s disease; PSEN1, Presenilin 1; SNCA, α-Synuclein; SAM, S-Adenosyl methionine; Sod2, Superoxide dismutase 2; TBI, Traumatic brain injury.
Abbasi, I. H. R., Abbasi, F., Soomro, R. N., Abd El-Hack, M. E., Abdel-Latif, M. A., Li, W., et al. (2017). Considering choline as methionine precursor, lipoproteins transporter, hepatic promoter and antioxidant agent in dairy cows. AMB Express 7:214. doi: 10.1186/s13568-017-0513-z
Abu Hamdeh, S., Ciuculete, D. M., Sarkisyan, D., Bakalkin, G., Ingelsson, M., Schiöth, H. B., et al. (2021). Differential DNA methylation of the genes for amyloid precursor protein, tau, and neurofilaments in human traumatic brain injury. J. Neurotrauma 38, 1679–1688. doi: 10.1089/neu.2020.7283
Abu Hamdeh, S., Waara, E. R., Möller, C., Söderberg, L., Basun, H., Alafuzoff, I., et al. (2018). Rapid amyloid-β oligomer and protofibril accumulation in traumatic brain injury. Brain Pathol. 28, 451–462. doi: 10.1111/bpa.12532
Acosta, G., Race, N., Herr, S., Fernandez, J., Tang, J., Rogers, E., et al. (2019). Acrolein-mediated α-synuclein pathology involvement in the early post-injury pathogenesis of mild blast-induced parkinsonian neurodegeneration. Mol. Cell. Neurosci. 98, 140–154. doi: 10.1016/j.mcn.2019.06.004
Acosta, S. A., Tajiri, N., de la Pena, I., Bastawrous, M., Sanberg, P. R., Kaneko, Y., et al. (2015). α-Synuclein as a pathological link between chronic traumatic brain injury and Parkinson's disease. J. Cell. Physiol. 230, 1024–1032. doi: 10.1002/jcp.24830
Al-Ghraiybah, N. F., Wang, J., Alkhalifa, A. E., Roberts, A. B., Raj, R., Yang, E., et al. (2022). Glial cell-mediated neuroinflammation in Alzheimer's disease. Int. J. Mol. Sci. 23:10572. doi: 10.3390/ijms231810572
Andrade-Guerrero, J., Santiago-Balmaseda, A., Jeronimo-Aguilar, P., Vargas-Rodriguez, I., Cadena-Suarez, A. R., Sanchez-Garibay, C., et al. (2023). Alzheimer's disease: an updated overview of its genetics. Int. J. Mol. Sci. 24:3754. doi: 10.3390/ijms24043754
Bacci Ballerini, F., López Anguera, A., Alcaraz, P., and Hernández Reyes, N. (1983). Treatment of postconcussion syndrome with S-adenosylmethionine. Med. Clin. (Barc.) 80, 161–164.
Bahado-Singh, R. O., Vishweswaraiah, S., Er, A., Aydas, B., Turkoglu, O., Taskin, B. D., et al. (2020). Artificial intelligence and the detection of pediatric concussion using epigenomic analysis. Brain Res. 1726:146510. doi: 10.1016/j.brainres.2019.146510
Bailey, Z. S., Grinter, M. B., De La Torre Campos, D., and VandeVord, P. J. (2015). Blast induced neurotrauma causes overpressure dependent changes to the DNA methylation equilibrium. Neurosci. Lett. 604, 119–123. doi: 10.1016/j.neulet.2015.07.035
Balasubramanian, N., Jadhav, G., and Sakharkar, A. J. (2021c). Repeated mild traumatic brain injuries perturb the mitochondrial biogenesis via DNA methylation in the hippocampus of rat. Mitochondrion 61, 11–24.
Balasubramanian, N., Sagarkar, S., Choudhary, A. G., Kokare, D. M., and Sakharkar, A. J. (2021a). Epigenetic blockade of hippocampal SOD2 via DNMT3b-mediated DNA methylation: implications in mild traumatic brain injury-induced persistent oxidative damage. Mol. Neurobiol. 58, 1162–1184. doi: 10.1007/s12035-020-02166-z
Balasubramanian, N., Sagarkar, S., Jadhav, M., Shahi, N., Sirmaur, R., and Sakharkar, A. J. (2021b). Role for histone deacetylation in traumatic brain injury-induced deficits in neuropeptide Y in arcuate nucleus: possible implications in feeding behavior. Neuroendocrinology 111, 1187–1200. doi: 10.1159/000513638
Bartke, T., and Kouzarides, T. (2011). Decoding the chromatin modification landscape. Cell Cycle 10:182. doi: 10.4161/cc.10.2.14477
Bellanti, F., Iannelli, G., Blonda, M., Tamborra, R., Villani, R., Romano, A., et al. (2017). Alterations of clock gene RNA expression in brain regions of a triple transgenic model of Alzheimer's disease. J. Alzheimers Dis. 59, 615–631. doi: 10.3233/JAD-160942
Bellver-Sanchis, A., Pallàs, M., and Griñán-Ferré, C. (2021). The contribution of epigenetic inheritance processes on age-related cognitive decline and Alzheimer's disease. Epigenomes 5:15. doi: 10.3390/epigenomes5020015
Bennett, R. L., and Licht, J. D. (2018). Targeting epigenetics in cancer. Annu. Rev. Pharmacol. Toxicol. 58, 187–207. doi: 10.1146/annurev-pharmtox-010716-105106
Bennett, S. A., Tanaz, R., Cobos, S. N., and Torrente, M. P. (2019). Epigenetics in amyotrophic lateral sclerosis: a role for histone post-translational modifications in neurodegenerative disease. Transl. Res. 204, 19–30. doi: 10.1016/j.trsl.2018.10.002
Berson, A., Nativio, R., Berger, S. L., and Bonini, N. M. (2018). Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 41, 587–598. doi: 10.1016/j.tins.2018.05.005
Bertogliat, M. J., Morris-Blanco, K. C., and Vemuganti, R. (2020). Epigenetic mechanisms of neurodegenerative diseases and acute brain injury. Neurochem. Int. 133:104642. doi: 10.1016/j.neuint.2019.104642
Bondarev, A. D., Attwood, M. M., Jonsson, J., Chubarev, V. N., Tarasov, V. V., and Schiöth, H. B. (2021). Recent developments of HDAC inhibitors: emerging indications and novel molecules. Br. J. Clin. Pharmacol. 87, 4577–4597. doi: 10.1111/bcp.14889
Bower, J. H., Maraganore, D. M., Peterson, B. J., McDonnell, S. K., Ahlskog, J. E., and Rocca, W. A. (2003). Head trauma preceding PD: a case-control study. Neurology 60, 1610–1615. doi: 10.1212/01.WNL.0000068008.78394.2C
Brett, B. L., Gardner, R. C., Godbout, J., Dams-O'Connor, K., and Keene, C. D. (2022). Traumatic brain injury and risk of neurodegenerative disorder. Biol. Psychiatry 91, 498–507. doi: 10.1016/j.biopsych.2021.05.025
Bruno, S., Williams, R. J., and Del Vecchio, D. (2022). Epigenetic cell memory: the gene's inner chromatin modification circuit. PLoS Comput. Biol. 18:e1009961. doi: 10.1371/journal.pcbi.1009961
Cannizzaro, C., Tel, B. C., Rose, S., Zeng, B. Y., and Jenner, P. (2003). Increased neuropeptide Y mRNA expression in striatum in Parkinson's disease. Brain Res. Mol. Brain Res. 110, 169–176. doi: 10.1016/S0169-328X(02)00555-7
Castora, F. J., Kerns, K. A., Pflanzer, H. K., Hitefield, N. L., Gershon, B., Shugoll, J., et al. (2022). Expression changes in mitochondrial genes affecting mitochondrial morphology, transmembrane potential, fragmentation, amyloidosis, and neuronal cell death found in brains of Alzheimer's disease patients. J. Alzheimers Dis. 90, 119–137. doi: 10.3233/JAD-220161
Cavalli, G., and Heard, E. (2019). Advances in epigenetics link genetics to the environment and disease. Nature 571, 489–499. doi: 10.1038/s41586-019-1411-0
Cedar, H., and Bergman, Y. (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 10, 295–304. doi: 10.1038/nrg2540
Chauhan, N. B. (2014). Chronic neurodegenerative consequences of traumatic brain injury. Restor. Neurol. Neurosci. 32, 337–365. doi: 10.3233/RNN-130354
Chen, Y. C., Koutelou, E., and Dent, S. Y. R. (2022). Now open: evolving insights to the roles of lysine acetylation in chromatin organization and function. Mol. Cell 82, 716–727. doi: 10.1016/j.molcel.2021.12.004
Cheng, Y., He, C., Wang, M., Ma, X., Mo, F., Yang, S., et al. (2019). Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 4:62. doi: 10.1038/s41392-019-0095-0
Chouliaras, L., Mastroeni, D., Delvaux, E., Grover, A., Kenis, G., Hof, P. R., et al. (2013). Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 34, 2091–2099. doi: 10.1016/j.neurobiolaging.2013.02.021
Clark, A. L., Weigand, A. J., Bangen, K. J., Thomas, K. R., Eglit, G. M. L., Bondi, M. W., et al. (2021). Higher cerebrospinal fluid tau is associated with history of traumatic brain injury and reduced processing speed in Vietnam-era veterans: a Department of Defense Alzheimer's disease neuroimaging initiative (DOD-ADNI) study. Alzheimers Dement. 13:e12239. doi: 10.1002/dad2.12239
Collins-Praino, L. E., and Corrigan, F. (2017). Does neuroinflammation drive the relationship between tau hyperphosphorylation and dementia development following traumatic brain injury? Brain Behav. Immun. 60, 369–382. doi: 10.1016/j.bbi.2016.09.027
Coppede, F. (2022). Targeting the epigenome to treat neurodegenerative diseases or delay their onset: a perspective. Neural Regen. Res. 17, 1745–1747. doi: 10.4103/1673-5374.332145
Coppola, G., Chinnathambi, S., Lee, J. J., Dombroski, B. A., Barker, M. C., Soto-Ortolaza, A. I., et al. (2012). Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer's disease. Hum. Mol. Genet. 21, 3500–3512. doi: 10.1093/hmg/dds161
Corne, R., Besson, V., Slimane, S. A., Coutan, M., Palhas, M. L. C., Shen, F. X., et al. (2021). Insulin-like growth factors may be markers of both traumatic brain injury and fear-related stress. Neuroscience 466, 205–221. doi: 10.1016/j.neuroscience.2021.04.013
Coughlin, J. M., Wang, Y., Munro, C. A., Ma, S., Yue, C., Chen, S., et al. (2015). Neuroinflammation and brain atrophy in former NFL players: An in vivo multimodal imaging pilot study. Neurobiol. Dis. 74, 58–65. doi: 10.1016/j.nbd.2014.10.019
Cruz-Sanabria, F., Carmassi, C., Bruno, S., Bazzani, A., Carli, M., Scarselli, M., et al. (2023). Melatonin as a chronobiotic with sleep-promoting properties. Curr. Neuropharmacol. 21, 951–987. doi: 10.2174/1570159X20666220217152617
Cura, V., and Cavarelli, J. (2021). Structure, activity and function of the PRMT2 protein arginine methyltransferase. Life 11:1263. doi: 10.3390/life11111263
Dai, Y., Wei, T., Shen, Z., Bei, Y., Lin, H., and Dai, H. (2021). Classical HDACs in the regulation of neuroinflammation. Neurochem. Int. 150:105182. doi: 10.1016/j.neuint.2021.105182
Dams-O'Connor, K., Guetta, G., Hahn-Ketter, A. E., and Fedor, A. (2016). Traumatic brain injury as a risk factor for Alzheimer's disease: current knowledge and future directions. Neurodegener. Dis. Manag. 6, 417–429. doi: 10.2217/nmt-2016-0017
Dash, P. K., Hergenroeder, G. W., Jeter, C. B., Choi, H. A., Kobori, N., and Moore, A. N. (2016). Traumatic brain injury alters methionine metabolism: implications for pathophysiology. Front. Syst. Neurosci. 10:36. doi: 10.3389/fnsys.2016.00036
Dash, P. K., Orsi, S. A., and Moore, A. N. (2009). Histone deactylase inhibition combined with behavioral therapy enhances learning and memory following traumatic brain injury. Neuroscience 163, 1–8. doi: 10.1016/j.neuroscience.2009.06.028
Dato, S., De Rango, F., Crocco, P., Pallotti, S., Belloy, M. E., Le Guen, Y., et al. (2023). Sex- and APOE-specific genetic risk factors for late-onset Alzheimer's disease: evidence from gene-gene interaction of longevity-related loci. Aging Cell, doi: 10.1111/acel.13938 :e13938. Epub ahead of print.
de Oliveira, J., Kucharska, E., Garcez, M. L., Rodrigues, M. S., Quevedo, J., Moreno-Gonzalez, I., et al. (2021). Inflammatory cascade in Alzheimer's disease pathogenesis: a review of experimental findings. Cells 10:2581. doi: 10.3390/cells10102581
DeKosky, S. T., Abrahamson, E. E., Ciallela, J. R., Paljug, W. R., Wisniewski, S. R., Clark, R. S. B., et al. (2007). Association of increased cortical soluble Aβ42 levels with diffuse plaques after severe brain injury in humans. Arch. Neurol. 64, 541–544. doi: 10.1001/archneur.64.4.541
Delic, V., Beck, K. D., Pang, K. C. H., and Citron, B. A. (2020). Biological links between traumatic brain injury and Parkinson's disease. Acta Neuropathol. Commun. 8:45. doi: 10.1186/s40478-020-00924-7
Desplats, P., Spencer, B., Coffee, E., Patel, P., Michael, S., Patrick, C., et al. (2011). α-Synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 286, 9031–9037. doi: 10.1074/jbc.C110.212589
Ding, Q., Vaynman, S., Akhavan, M., Ying, Z., and Gomez-Pinilla, F. (2006). Insulin-like growth factor I interfaces with brain-derived neurotrophic factor-mediated synaptic plasticity to modulate aspects of exercise-induced cognitive function. Neuroscience 140, 823–833. doi: 10.1016/j.neuroscience.2006.02.084
Do Carmo, S., Hanzel, C. E., Jacobs, M. L., Machnes, Z., Iulita, M. F., Yang, J., et al. (2016). Rescue of early bace-1 and global DNA demethylation by S-adenosylmethionine reduces amyloid pathology and improves cognition in an Alzheimer's model. Sci. Rep. 6:34051. doi: 10.1038/srep34051
Faden, A. I., and Loane, D. J. (2015). Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics 12, 143–150. doi: 10.1007/s13311-014-0319-5
Fernandes, J., Arida, R. M., and Gomez-Pinilla, F. (2017). Physical exercise as an epigenetic modulator of brain plasticity and cognition. Neurosci. Biobehav. Rev. 80, 443–456. doi: 10.1016/j.neubiorev.2017.06.012
Fesharaki-Zadeh, A. (2019). Chronic traumatic encephalopathy: a brief overview. Front. Neurol. 10:713. doi: 10.3389/fneur.2019.00713
Fitz-James, M. H., and Cavalli, G. (2022). Molecular mechanisms of transgenerational epigenetic inheritance. Nat. Rev. Genet. 23, 325–341. doi: 10.1038/s41576-021-00438-5
Fleminger, S., Oliver, D. L., Lovestone, S., Rabe-Hesketh, S., and Giora, A. (2003). Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication. J. Neurol. Neurosurg. Psychiatry 74, 857–862. doi: 10.1136/jnnp.74.7.857
Froese, D. S., Fowler, B., and Baumgartner, M. R. (2019). Vitamin B(12), folate, and the methionine remethylation cycle-biochemistry, pathways, and regulation. J. Inherit. Metab. Dis. 42, 673–685. doi: 10.1002/jimd.12009
Fukumoto, K., Ito, K., Saer, B., Taylor, G., Ye, S., Yamano, M., et al. (2022). Excess S-adenosylmethionine inhibits methylation via catabolism to adenine. Commun. Biol. 5:313. doi: 10.1038/s42003-022-03280-5
Gao, W. M., Chadha, M. S., Kline, A. E., Clark, R. S., Kochanek, P. M., Dixon, C. E., et al. (2006). Immunohistochemical analysis of histone H3 acetylation and methylation--evidence for altered epigenetic signaling following traumatic brain injury in immature rats. Brain Res. 1070, 31–34. doi: 10.1016/j.brainres.2005.11.038
Gardner, R. C., Burke, J. F., Nettiksimmons, J., Goldman, S., Tanner, C. M., and Yaffe, K. (2015). Traumatic brain injury in later life increases risk for Parkinson disease. Ann. Neurol. 77, 987–995. doi: 10.1002/ana.24396
Gardner, R. C., and Yaffe, K. (2015). Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol. Cell. Neurosci. 66, 75–80. doi: 10.1016/j.mcn.2015.03.001
Ghiboub, M., Elfiky, A. M. I., de Winther, M. P. J., Harker, N. R., Tough, D. F., and de Jonge, W. J. (2021). Selective targeting of epigenetic readers and histone deacetylases in autoimmune and inflammatory diseases: recent advances and future perspectives. J. Pers. Med. 11:36. doi: 10.3390/jpm11050336
Ghosh, S., Wu, M. D., Shaftel, S. S., Kyrkanides, S., LaFeria, F. M., Olschowka, J. A., et al. (2013). Sustained interleukin-1b overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer's mouse model. J. Neurosci. 33, 5053–5064. doi: 10.1523/JNEUROSCI.4361-12.2013
Goldman, S. M., Tanner, C. M., Oakes, D., Bhudhikanok, G. S., Gupta, A., and Langston, J. W. (2006). Head injury and Parkinson's disease risk in twins. Ann. Neurol. 60, 65–72. doi: 10.1002/ana.20882
Gonzalez-Jaramillo, V., Portilla-Fernandez, E., Glisic, M., Voortman, T., Ghanbari, M., Bramer, W., et al. (2019). Epigenetics and inflammatory markers: a systematic review of the current evidence. Int. J. Inflamm. 2019:6273680. doi: 10.1155/2019/6273680
Gräff, J., Rei, D., Guan, J. S., Wang, W. Y., Seo, J., Hennig, K. M., et al. (2012). An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–226. doi: 10.1038/nature10849
Gräff, J., and Tsai, L. H. (2013). Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 14, 97–111. doi: 10.1038/nrn3427
Graham, A., Livingston, G., Purnell, L., and Huntley, J. (2022). Mild traumatic brain injuries and future risk of developing Alzheimer's disease: systematic review and meta-analysis. J. Alzheimers Dis. 87, 969–979. doi: 10.3233/JAD-220069
Guo, T., Noble, W., and Hanger, D. P. (2017). Roles of tau protein in health and disease. Acta Neuropathol. 133, 665–704. doi: 10.1007/s00401-017-1707-9
Haghighi, F., Ge, Y., Chen, S., Xin, Y., Umali, M. U., De Gasperi, R., et al. (2015). Neuronal DNA methylation profiling of blast-related traumatic brain injury. J. Neurotrauma 32, 1200–1209. doi: 10.1089/neu.2014.3640
Hampel, H., Hardy, J., Blennow, K., Chen, C., Perry, G., Kim, S. H., et al. (2021). The amyloid-β pathway in Alzheimer's disease. Mol. Psychiatry 26, 5481–5503. doi: 10.1038/s41380-021-01249-0
Harris, C. J., Scheibe, M., Wongpalee, S. P., Liu, W., Cornett, E. M., Vaughan, R. M., et al. (2018). A DNA methylation reader complex that enhances gene transcription. Science 362, 1182–1186. doi: 10.1126/science.aar7854
Henley, C. M., Wey, K., Takashima, A., Mills, C., Granmayeh, E., Krishnappa, I., et al. (1997). S-adenosylmethionine decarboxylase activity is decreased in the rat cortex after traumatic brain injury. J. Neurochem. 69, 259–265. doi: 10.1046/j.1471-4159.1997.69010259.x
Hintermayer, M. A., Volkening, K., Moszczynski, A. J., Donison, N., and Strong, M. J. (2020). Tau protein phosphorylation at Thr(175) initiates fibril formation via accessibility of the N-terminal phosphatase-activating domain. J. Neurochem. 155, 313–326. doi: 10.1111/jnc.14942
Holland, T. M., Agarwal, P., Wang, Y., Leurgans, S. E., Bennett, D. A., Booth, S. L., et al. (2020). Dietary flavonols and risk of Alzheimer dementia. Neurology 94, e1749–e1756. doi: 10.1212/WNL.0000000000008981
Huang, M., and Chen, S. (2021). DJ-1 in neurodegenerative diseases: pathogenesis and clinical application. Prog. Neurobiol. 204:102114. doi: 10.1016/j.pneurobio.2021.102114
Ikonomovic, M. D., Uryu, K., Abrahamson, E. E., Ciallella, J. R., Trojanowski, J. Q., Lee, V. M., et al. (2004). Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp. Neurol. 190, 192–203. doi: 10.1016/j.expneurol.2004.06.011
Jafari, S., Etminan, M., Aminzadeh, F., and Samii, A. (2013). Head injury and risk of Parkinson disease: a systematic review and meta-analysis. Mov. Disord. 28, 1222–1229. doi: 10.1002/mds.25458
Jin, B., and Robertson, K. D. (2013). DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 754, 3–29. doi: 10.1007/978-1-4419-9967-2_1
Johnson, V. E., Stewart, W., Arena, J. D., and Smith, D. H. (2017). Traumatic brain injury as a trigger of neurodegeneration. Adv. Neurobiol. 15, 383–400. doi: 10.1007/978-3-319-57193-5_15
Johnson, V. E., Stewart, J. E., Begbie, F. D., Trojanowski, J. Q., Smith, D. H., and Stewart, W. (2013). Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136, 28–42. doi: 10.1093/brain/aws322
Johnson, V. E., Stewart, W., Graham, D. I., Stewart, J. E., Praestgaard, A. H., and Smith, D. H. (2009). A neprilysin polymorphism and amyloid-β plaques after traumatic brain injury. J. Neurotrauma 26, 1197–1202. doi: 10.1089/neu.2008.0843
Johnson, V. E., Stewart, W., and Smith, D. H. (2012). Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 22, 142–149. doi: 10.1111/j.1750-3639.2011.00513.x
Jowaed, A., Schmitt, I., Kaut, O., and Wullner, U. (2010). Methylation regulates a-synuclein expression and is decreased in Parkinson's disease patients' brains. J. Neurosci. 30, 6355–6359. doi: 10.1523/JNEUROSCI.6119-09.2010
Kamal, S. R., Potukutchi, S., Gelovani, D. J., Bonomi, R. E., Kallakuri, S., Cavanaugh, J. M., et al. (2022). Spatial and temporal dynamics of HDACs class IIa following mild traumatic brain injury in adult rats. Mol. Psychiatry 27, 1683–1693. doi: 10.1038/s41380-021-01369-7
Kang, I., Chu, C. T., and Kaufman, B. A. (2018). The mitochondrial transcription factor TFAM in neurodegeneration: emerging evidence and mechanisms. FEBS Lett. 592, 793–811. doi: 10.1002/1873-3468.12989
Karch, C. M., and Goate, A. M. (2015). Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. doi: 10.1016/j.biopsych.2014.05.006
Kelly, T. K., De Carvalho, D. D., and Jones, P. A. (2010). Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 28, 1069–1078. doi: 10.1038/nbt.1678
Kim, S., and Kaang, B. K. (2017). Epigenetic regulation and chromatin remodeling in learning and memory. Exp. Mol. Med. 49:e281. doi: 10.1038/emm.2017.152
Kip, E., and Parr-Brownlie, L. C. (2022). Reducing neuroinflammation via therapeutic compounds and lifestyle to prevent or delay progression of Parkinson's disease. Ageing Res. Rev. 78:101618. doi: 10.1016/j.arr.2022.101618
Kitazawa, M., Oddo, S., Yamasaki, T. R., Green, K. N., and LaFerla, F. M. (2005). Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer's disease. J. Neurosci. 25, 8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005
Kittur, S., Hoh, J., Endo, H., Tourtellote, W., Weeks, B. S., Markesbery, W., et al. (1994). Cytoskeletal neurofilament gene expression in brain tissue from Alzheimer's disease patients. I. Decrease in NF-L and NF-M message. J. Geriatr. Psychiatry Neurol. 7, 153–158. doi: 10.1177/089198879400700305
Kontopoulos, E., Parvin, J. D., and Feany, M. B. (2006). α-Synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 15, 3012–3023. doi: 10.1093/hmg/ddl243
Korecka, J. A., Moloney, E. B., Eggers, R., Hobo, B., Scheffer, S., Ras-Verloop, N., et al. (2017). Repulsive guidance molecule a (RGMa) induces neuropathological and behavioral changes that closely resemble Parkinson's disease. J. Neurosci. 37, 9361–9379. doi: 10.1523/JNEUROSCI.0084-17.2017
Kornberg, R. D. (1974). Chromatin structure: a repeating unit of histones and DNA. Science 184, 868–871. doi: 10.1126/science.184.4139.868
Kulkarni, P. G., Balasubramanian, N., Manjrekar, R., Banerjee, T., and Sakharkar, A. (2023). DNA methylation-mediated Mfn2 gene regulation in the brain: a role in brain trauma-induced mitochondrial dysfunction and memory deficits. Cell. Mol. Neurobiol. doi: 10.1007/s10571-023-01358-0 [Epub ahead of print].
Kumari, M., Arora, P., Sharma, P., Hasija, Y., Rana, P., D’souza, M. M., et al. (2023). Acute metabolic alterations in the hippocampus are associated with decreased acetylation after blast induced TBI. Metabolomics 19:5. doi: 10.1007/s11306-022-01970-z
Landgraf, B. J., McCarthy, E. L., and Booker, S. J. (2016). Radical S-adenosylmethionine enzymes in human health and disease. Annu. Rev. Biochem. 85, 485–514. doi: 10.1146/annurev-biochem-060713-035504
Lardenoije, R., Pishva, E., Lunnon, K., and van den Hove, D. L. (2018). Neuroepigenetics of aging and age-related neurodegenerative disorders. Prog. Mol. Biol. Transl. Sci. 158, 49–82. doi: 10.1016/bs.pmbts.2018.04.008
Lau, V., Ramer, L., and Tremblay, M. E. (2023). An aging, pathology burden, and glial senescence build-up hypothesis for late onset Alzheimer's disease. Nat. Commun. 14:1670. doi: 10.1038/s41467-023-37304-3
Laurent, C., Buée, L., and Blum, D. (2018). Tau and neuroinflammation: what impact for Alzheimer's disease and tauopathies? Biom. J. 41, 21–33. doi: 10.1016/j.bj.2018.01.003
Lee, P. C., Bordelon, Y., Bronstein, J., and Ritz, B. (2012). Traumatic brain injury, paraquat exposure, and their relationship to Parkinson disease. Neurology 79, 2061–2066. doi: 10.1212/WNL.0b013e3182749f28
Lee, C. Y. D., Daggett, A., Gu, X., Jiang, L. L., Langfelder, P., Li, X., et al. (2018). Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer's disease models. Neuron 97, 1032–1048.e5. doi: 10.1016/j.neuron.2018.02.002
Lee, A. J., Kim, C., Park, S., Joo, J., Choi, B., Yang, D., et al. (2023). Characterization of altered molecular mechanisms in Parkinson's disease through cell-type resolved multiomics analysis. Sci. Adv. 9:eabo2467. doi: 10.1126/sciadv.abo2467
Lee, J. W., Lee, Y. K., Yuk, D. Y., Choi, D. Y., Ban, S. B., Oh, K. W., et al. (2008). Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of β-amyloid generation. J. Neuroinflammation 5:37. doi: 10.1186/1742-2094-5-37
Lee, D. C., Rizer, J., Selenica, M. L., Reid, P., Kraft, C., Johnson, A., et al. (2010). LPS-induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J. Neuroinflammation 7:56. doi: 10.1186/1742-2094-7-56
Li, N., Deng, M., Hu, G., Li, N., Yuan, H., and Zhou, Y. (2022). New insights into microglial mechanisms of memory impairment in Alzheimer's disease. Biomol. Ther. 12:1722. doi: 10.3390/biom12111722
Limorenko, G., and Lashuel, H. A. (2021). To target Tau pathologies, we must embrace and reconstruct their complexities. Neurobiol. Dis. 161:105536. doi: 10.1016/j.nbd.2021.105536
Liu, D., Zusman, B. E., Shaffer, J. R., Li, Y., Arockiaraj, A. I., Liu, S., et al. (2022). Decreased DNA methylation of RGMA is associated with intracranial hypertension after severe traumatic brain injury: an exploratory epigenome-wide association study. Neurocrit. Care. 37, 26–37. doi: 10.1007/s12028-021-01424-9
Majchrzak-Celińska, A., Warych, A., and Szoszkiewicz, M. (2021). Novel approaches to epigenetic therapies: from drug combinations to epigenetic editing. Genes (Basel) 12:208. doi: 10.3390/genes12020208
Makinde, H. M., Just, T. B., Cuda, C. M., Perlman, H., and Schwulst, S. J. (2017). The role of microglia in the etiology and evolution of chronic traumatic encephalopathy. Shock 48, 276–283. doi: 10.1097/SHK.0000000000000859
Marras, C., Hincapié, C. A., Kristman, V. L., Cancelliere, C., Soklaridis, S., Li, A., et al. (2014). Systematic review of the risk of Parkinson's disease after mild traumatic brain injury: results of the international collaboration on mild traumatic brain injury prognosis. Arch. Phys. Med. Rehabil. 95, S238–S244. doi: 10.1016/j.apmr.2013.08.298
Masel, B. E., and DeWitt, D. S. (2010). Traumatic brain injury: a disease process, not an event. J. Neurotrauma 27, 1529–1540. doi: 10.1089/neu.2010.1358
Mateen, B. A., Hill, C. S., Biddie, S. C., and Menon, D. K. (2017). DNA methylation: basic biology and application to traumatic brain injury. J. Neurotrauma 34, 2379–2388. doi: 10.1089/neu.2017.5007
Matsumoto, L., Takuma, H., Tamaoka, A., Kurisaki, H., Date, H., Tsuji, S., et al. (2010). CpG demethylation enhances α-synuclein expression and affects the pathogenesis of Parkinson's disease. PLoS One 5:e15522. doi: 10.1371/journal.pone.0015522
McCann, H., Bahar, A. Y., Burkhardt, K., Gardner, A. J., Halliday, G. M., Iverson, G. L., et al. (2022). Prevalence of chronic traumatic encephalopathy in the Sydney brain bank. Brain Commun. 4:fcac189. doi: 10.1093/braincomms/fcac189
McGovern, A. J., González, J., Ramírez, D., and Barreto, G. E. (2022). Identification of HMGCR, PPGARG and prohibitin as potential druggable targets of dihydrotestosterone for treatment against traumatic brain injury using system pharmacology. Int. Immunopharmacol. 108:108721. doi: 10.1016/j.intimp.2022.108721
McKee, A. C. (2020). The neuropathology of chronic traumatic encephalopathy: the status of the literature. Semin. Neurol. 40, 359–369. doi: 10.1055/s-0040-1713632
McKee, A. C., Cantu, R. C., Nowinski, C. J., Hedley-Whyte, E. T., Gavett, B. E., Budson, A. E., et al. (2009). Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 68, 709–735. doi: 10.1097/NEN.0b013e3181a9d503
McKee, A. C., Mez, J., Abdolmohammadi, B., Butler, M., Huber, B. R., Uretsky, M., et al. (2023). Neuropathologic and clinical findings in young contact sport athletes exposed to repetitive head injuries. JAMA Neurol. doi: 10.1001/jamaneurol.2023.2907 [Epub ahead of print].
McNulty, H., Ward, M., Hoey, L., Hughes, C. F., and Pentieva, K. (2019). Addressing optimal folate and related B-vitamin status through the lifecycle: health impacts and challenges. Proc. Nutr. Soc. 78, 449–462. doi: 10.1017/S0029665119000661
Meng, Q., Yang, G., Yang, Y., Ding, F., and Hu, F. (2020). Protective effects of histone deacetylase inhibition by Scriptaid on brain injury in neonatal rat models of cerebral ischemia and hypoxia. Int. J. Clin. Exp. Pathol. 13, 179–191.
Meng, Q., Zhuang, Y., Ying, Z., Agrawal, R., Yang, X., and Gomez-Pinilla, F. (2017). Traumatic brain injury induces genome-wide transcriptomic, methylomic, and network perturbations in brain and blood predicting neurological disorders. EBioMedicine 16, 184–194. doi: 10.1016/j.ebiom.2017.01.046
Mez, J., Daneshvar, D. H., Kiernan, P. T., Abdolmohammadi, B., Alvarez, V. E., Huber, B. R., et al. (2017). Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA 318, 360–370. doi: 10.1001/jama.2017.8334
Mielke, M. M., Ransom, J. E., Mandrekar, J., Turcano, P., Savica, R., and Brown, A. W. (2022). Traumatic brain injury and risk of Alzheimer's disease and related dementias in the population. J. Alzheimers Dis. 88, 1049–1059. doi: 10.3233/JAD-220159
Migliore, L., and Coppede, F. (2022). Gene-environment interactions in Alzheimer disease: the emerging role of epigenetics. Nat. Rev. Neurol. 18, 643–660. doi: 10.1038/s41582-022-00714-w
Monti, N., Cavallaro, R. A., Stoccoro, A., Nicolia, V., Scarpa, S., Kovacs, G. G., et al. (2020). CpG and non-CpG Presenilin1 methylation pattern in course of neurodevelopment and neurodegeneration is associated with gene expression in human and murine brain. Epigenetics 15, 781–799. doi: 10.1080/15592294.2020.1722917
Nagalakshmi, B., Sagarkar, S., and Sakharkar, A. J. (2018). Epigenetic mechanisms of traumatic brain injuries. Prog. Mol. Biol. Transl. Sci. 157, 263–298. doi: 10.1016/bs.pmbts.2017.12.013
Nasb, M., Tao, W., and Chen, N. (2023). Alzheimer's disease puzzle: delving into pathogenesis hypotheses. Aging Dis. doi: 10.14336/AD.2023.0608 [Epub ahead of print].
Nicoll, J. A., Roberts, G. W., and Graham, D. I. (1995). Apolipoprotein E epsilon 4 allele is associated with deposition of amyloid β-protein following head injury. Nat. Med. 1, 135–137. doi: 10.1038/nm0295-135
Nikolac Perkovic, M., Videtic Paska, A., Konjevod, M., Kouter, K., Svob Strac, D., Nedic Erjavec, G., et al. (2021). Epigenetics of Alzheimer's disease. Biomolecules 11:195. doi: 10.3390/biom11020195
Okano, M., Bell, D. W., Haber, D. A., and Li, E. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cells 99, 247–257. doi: 10.1016/S0092-8674(00)81656-6
Patnaik, D., Pao, P. C., Zhao, W. N., Silva, M. C., Hylton, N. K., Chindavong, P. S., et al. (2021). Exifone is a potent HDAC1 activator with neuroprotective activity in human neuronal models of neurodegeneration. ACS Chem. Neurosci. 12, 271–284. doi: 10.1021/acschemneuro.0c00308
Pavlou, M. A. S., and Outeiro, T. F. (2017). Epigenetics in Parkinson's disease. Adv. Exp. Med. Biol. 978, 363–390. doi: 10.1007/978-3-319-53889-1_19
Pfalzer, A. C., Choi, S. W., Tammen, S. A., Park, L. K., Bottiglieri, T., Parnell, L. D., et al. (2014). S-adenosylmethionine mediates inhibition of inflammatory response and changes in DNA methylation in human macrophages. Physiol. Genomics 46, 617–623. doi: 10.1152/physiolgenomics.00056.2014
Plassman, B. L., Havlik, R. J., Steffens, D. C., Helms, M. J., Newman, T. N., Drosdick, D., et al. (2000). Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology 55, 1158–1166. doi: 10.1212/WNL.55.8.1158
Postupna, N., Rose, S. E., Gibbons, L. E., Coleman, N. M., Hellstern, L. L., Ritchie, K., et al. (2021). The delayed neuropathological consequences of traumatic brain injury in a community-based sample. Front. Neurol. 12:624696. doi: 10.3389/fneur.2021.624696
Poulard, C., Noureddine, L. M., Pruvost, L., and Le Romancer, M. (2021). Structure, activity, and function of the protein lysine methyltransferase G9a. Life 11:1082. doi: 10.3390/life11101082
Ramlackhansingh, A. F., Brooks, D. J., Greenwood, R. J., Bose, S. K., Turkheimer, F. E., Kinnunen, M. K., et al. (2011). Inflammation after trauma: microglial activation and traumatic brain injury. Ann. Neurol. 70, 374–383. doi: 10.1002/ana.22455
Rasheed, M., Liang, J., Wang, C., Deng, Y., and Chen, Z. (2021). Epigenetic regulation of neuroinflammation in Parkinson's disease. Int. J. Mol. Sci. 22:4956. doi: 10.3390/ijms22094956
Rauluseviciute, I., Drabløs, F., and Rye, M. B. (2020). DNA hypermethylation associated with upregulated gene expression in prostate cancer demonstrates the diversity of epigenetic regulation. BMC Med. Genet. 13:6. doi: 10.1186/s12920-020-0657-6
Ribarič, S. (2022). Physical exercise, a potential non-pharmacological intervention for attenuating neuroinflammation and cognitive decline in Alzheimer's disease patients. Int. J. Mol. Sci. 23:3245. doi: 10.3390/ijms23063245
Risbrough, V. B., Vaughn, M. N., and Friend, S. F. (2022). Role of inflammation in traumatic brain injury-associated risk for neuropsychiatric disorders: state of the evidence and where do we go from here. Biol. Psychiatry 91, 438–448. doi: 10.1016/j.biopsych.2021.11.012
Roberts, G. W., Gentleman, S. M., Lynch, A., and Graham, D. I. (1991). β A4 amyloid protein deposition in brain after head trauma. Lancet 338, 1422–1423. doi: 10.1016/0140-6736(91)92724-G
Roberts, G. W., Gentleman, S. M., Lynch, A., Murray, L., Landon, M., and Graham, D. I. (1994). β amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 57, 419–425. doi: 10.1136/jnnp.57.4.419
Roy, R. G., Mandal, P. K., and Maroon, J. C. (2023). Oxidative stress occurs prior to amyloid Aβ plaque formation and tau phosphorylation in Alzheimer's disease: role of glutathione and metal ions. ACS Chem. Neurosci., doi: 10.1021/acschemneuro.3c00486, Epub ahead of print.
Rugbjerg, K., Ritz, B., Korbo, L., Martinussen, N., and Olsen, J. H. (2008). Risk of Parkinson's disease after hospital contact for head injury: population based case-control study. BMJ 337:a2494. doi: 10.1136/bmj.a2494
Sagarkar, S., Balasubramanian, N., Mishra, S., Choudhary, A. G., Kokare, D. M., and Sakharkar, A. J. (2019). Repeated mild traumatic brain injury causes persistent changes in histone deacetylase function in hippocampus: implications in learning and memory deficits in rats. Brain Res. 1711, 183–192. doi: 10.1016/j.brainres.2019.01.022
Sagarkar, S., Bhamburkar, T., Shelkar, G., Choudhary, A. G., Kokare, D. M., and Sakharkar, A. J. (2017). Minimal traumatic brain injury causes persistent changes in DNA methylation at BDNF gene promoters in rat amygdala: a possible role in anxiety-like behaviors. Neurobiol. Dis. 106, 101–109. doi: 10.1016/j.nbd.2017.06.016
Scalzo, P., Kummer, A., Bretas, T. L., Cardoso, F., and Teixeira, A. L. (2010). Serum levels of brain-derived neurotrophic factor correlate with motor impairment in Parkinson's disease. J. Neurol. 257, 540–545. doi: 10.1007/s00415-009-5357-2
Schieffler, D. A., and Matta, S. E. (2022). Evidence to support the use of S-adenosylmethionine for treatment of post-concussive sequelae in the military. Mil. Med. 187, e1182–e1192. doi: 10.1093/milmed/usab130
Schmidt, A., Zhang, H., and Cardoso, M. C. (2020). MeCP2 and chromatin compartmentalization. Cells 9:878. doi: 10.3390/cells9040878
Schneider, A. L. C., Selvin, E., Latour, L., Turtzo, L. C., Coresh, J., Mosley, T., et al. (2021). Head injury and 25-year risk of dementia. Alzheimers Dement. 17, 1432–1441. doi: 10.1002/alz.12315
Scott, G., Ramlackhansingh, A. F., Edison, P., Hellyer, P., Cole, J., Veronese, M., et al. (2016). Amyloid pathology and axonal injury after brain trauma. Neurology 86, 821–828. doi: 10.1212/WNL.0000000000002413
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Sellami, M., Bragazzi, N., Prince, M. S., Denham, J., and Elrayess, M. (2021). Regular, intense exercise training as a healthy aging lifestyle strategy: preventing DNA damage, telomere shortening and adverse DNA methylation changes over a lifetime. Front. Genet. 12:652497. doi: 10.3389/fgene.2021.652497
Selvaraj, P., Tanaka, M., Wen, J., and Zhang, Y. (2021). The novel monoacylglycerol lipase inhibitor MJN110 suppresses neuroinflammation, normalizes synaptic composition and improves behavioral performance in the repetitive traumatic brain injury mouse model. Cells 10:3454. doi: 10.3390/cells10123454
Shein, N. A., Grigoriadis, N., Alexandrovich, A. G., Simeonidou, C., Lourbopoulos, A., Polyzoidou, E., et al. (2009). Histone deacetylase inhibitor ITF2357 is neuroprotective, improves functional recovery, and induces glial apoptosis following experimental traumatic brain injury. FASEB J. 23, 4266–4275. doi: 10.1096/fj.09-134700
Smith, D. H., Johnson, V. E., Trojanowski, J. Q., and Stewart, W. (2019). Chronic traumatic encephalopathy - confusion and controversies. Nat. Rev. Neurol. 15, 179–183. doi: 10.1038/s41582-018-0114-8
Smith, A. N., Shaughness, M., Collier, S., Hopkins, D., and Byrnes, K. R. (2022). Therapeutic targeting of microglia mediated oxidative stress after neurotrauma. Front. Med. 9:1034692. doi: 10.3389/fmed.2022.1034692
Smith, D. H., and Stewart, W. (2018). Traumatic brain injury: a platform for studies in Aβ processing: commentary on: "rapid Aβ oligomer and protofibril accumulation in traumatic brain injury". Brain Pathol. 28, 463–465. doi: 10.1111/bpa.12534
Song, J. H., Yu, J. T., and Tan, L. (2015). Brain-derived neurotrophic factor in Alzheimer's disease: risk, mechanisms, and therapy. Mol. Neurobiol. 52, 1477–1493. doi: 10.1007/s12035-014-8958-4
Stein, T. D., Alvarez, V. E., and McKee, A. C. (2014). Chronic traumatic encephalopathy: a spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimers Res. Ther. 6:4. doi: 10.1186/alzrt234
Stoccoro, A., and Coppedè, F. (2018). Role of epigenetics in Alzheimer's disease pathogenesis. Neurodegener. Dis. Manag. 8, 181–193. doi: 10.2217/nmt-2018-0004
Strang, K. H., Golde, T. E., and Giasson, B. I. (2019). MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Investig. 99, 912–928. doi: 10.1038/s41374-019-0197-x
Sujkowski, A., Hong, L., Wessells, R. J., and Todi, S. V. (2022). The protective role of exercise against age-related neurodegeneration. Ageing Res. Rev. 74:101543. doi: 10.1016/j.arr.2021.101543
Tai, Y. T., Lee, W. Y., Lee, F. P., Lin, T. J., Shih, C. L., Wang, J. Y., et al. (2014). Low dose of valproate improves motor function after traumatic brain injury. Biomed. Res. Int. 2014:980657. doi: 10.1155/2014/980657
Thangavelu, B., Wilfred, B. S., Johnson, D., Gilsdorf, J. S., Shear, D. A., and Boutte, A. M. (2020). Penetrating ballistic-like brain injury leads to micro-RNA dysregulation, BACE1 upregulation, and amyloid precursor protein loss in lesioned rat brain tissues. Front. Neurosci. 14:915. doi: 10.3389/fnins.2020.00915
Toutonji, A., Mandava, M., Guglietta, S., and Tomlinson, S. (2021). Chronic complement dysregulation drives neuroinflammation after traumatic brain injury: a transcriptomic study. Acta Neuropathol. Commun. 9:126. doi: 10.1186/s40478-021-01226-2
Treble-Barna, A., Heinsberg, L. W., Puccio, A. M., Shaffer, J. R., Okonkwo, D. O., Beers, S. R., et al. (2021). Acute brain-derived neurotrophic factor DNA methylation trajectories in cerebrospinal fluid and associations with outcomes following severe traumatic brain injury in adults. Neurorehabil. Neural Repair 35, 790–800. doi: 10.1177/15459683211028245
Urbano, A., Smith, J., Weeks, R. J., and Chatterjee, A. (2019). Gene-specific targeting of DNA methylation in the mammalian genome. Cancers 11:1515. doi: 10.3390/cancers11101515
Uryu, K., Chen, X. H., Martinez, D., Browne, K. D., Johnson, V. E., Graham, D. I., et al. (2007). Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 208, 185–192. doi: 10.1016/j.expneurol.2007.06.018
Vaynman, S., Ying, Z., and Gomez-Pinilla, F. (2004). Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur. J. Neurosci. 20, 2580–2590. doi: 10.1111/j.1460-9568.2004.03720.x
Wang, W., Cao, Q., Tan, T., Yang, F., Williams, J. B., and Yan, Z. (2021). Epigenetic treatment of behavioral and physiological deficits in a tauopathy mouse model. Aging Cell 20:e13456. doi: 10.1111/acel.13456
Wang, G., Jiang, X., Pu, H., Zhang, W., An, C., Hu, X., et al. (2013). Scriptaid, a novel histone deacetylase inhibitor, protects against traumatic brain injury via modulation of PTEN and AKT pathway: Scriptaid protects against TBI via AKT. Neurotherapeutics 10, 124–142. doi: 10.1007/s13311-012-0157-2
Wang, W., Nguyen, L. T., Burlak, C., Chegini, F., Guo, F., Chataway, T., et al. (2016). Caspase-1 causes truncation and aggregation of the Parkinson's disease-associated protein α-synuclein. Proc. Natl. Acad. Sci. U. S. A. 113, 9587–9592. doi: 10.1073/pnas.1610099113
Wong, V. S., and Langley, B. (2016). Epigenetic changes following traumatic brain injury and their implications for outcome, recovery and therapy. Neurosci. Lett. 625, 26–33. doi: 10.1016/j.neulet.2016.04.009
Xu, Y., Zhang, S., Lin, S., Guo, Y., Deng, W., Zhang, Y., et al. (2017). WERAM: a database of writers, erasers and readers of histone acetylation and methylation in eukaryotes. Nucleic Acids Res. 45, D264–d270. doi: 10.1093/nar/gkw1011
Xu, M., Zhu, J., Liu, X. D., Luo, M. Y., and Xu, N. J. (2021). Roles of physical exercise in neurodegeneration: reversal of epigenetic clock. Transl. Neurodegener. 10:30. doi: 10.1186/s40035-021-00254-1
Yamaguchi, S., Shen, L., Liu, Y., Sendler, D., and Zhang, Y. (2013). Role of Tet1 in erasure of genomic imprinting. Nature 504, 460–464. doi: 10.1038/nature12805
Yang, Y., Bagyinszky, E., and An, S. S. A. (2023). Presenilin-1 (PSEN1) mutations: clinical phenotypes beyond Alzheimer's disease. Int. J. Mol. Sci. 24:8417. doi: 10.3390/ijms24098417
Yoshiyama, Y., Higuchi, M., Zhang, B., Huang, S. M., Iwata, N., Saido, T. C., et al. (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. doi: 10.1016/j.neuron.2007.01.010
Yu, F., Wang, Z., Tanaka, M., Chiu, C. T., Leeds, P., Zhang, Y., et al. (2013). Posttrauma cotreatment with lithium and valproate: reduction of lesion volume, attenuation of blood-brain barrier disruption, and improvement in motor coordination in mice with traumatic brain injury. J. Neurosurg. 119, 766–773. doi: 10.3171/2013.6.JNS13135
Zhang, B., West, E. J., Van, K. C., Gurkoff, G. G., Zhou, J., Zhang, X. M., et al. (2008). HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res. 1226, 181–191. doi: 10.1016/j.brainres.2008.05.085
Zhang, Z. Y., Zhang, Z., Fauser, U., and Schluesener, H. J. (2007). Global hypomethylation defines a sub-population of reactive microglia/macrophages in experimental traumatic brain injury. Neurosci. Lett. 429, 1–6. doi: 10.1016/j.neulet.2007.09.061
Zima, L., West, R., Smolen, P., Kobori, N., Hergenroeder, G., Choi, H. A., et al. (2022). Epigenetic modifications and their potential contribution to traumatic brain injury pathobiology and outcome. J. Neurotrauma 39, 1279–1288. doi: 10.1089/neu.2022.0128
Keywords: acetylation, Alzheimer’s disease, dementia, feedback loop, epigenetics, methylation, Parkinson’s disease, encephalopathy
Citation: Smolen P, Dash PK and Redell JB (2023) Traumatic brain injury-associated epigenetic changes and the risk for neurodegenerative diseases. Front. Neurosci. 17:1259405. doi: 10.3389/fnins.2023.1259405
Edited by:
Mojgan Rastegar, University of Manitoba, CanadaReviewed by:
Angels Almenar, University of California, San Diego, United StatesCopyright © 2023 Smolen, Dash and Redell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul Smolen, cGF1bC5kLnNtb2xlbkB1dGgudG1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.