
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Neurosci. , 02 October 2023
Sec. Neurodegeneration
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1251228
 Minggang Fang1*
Minggang Fang1* Sara K. Deibler1Alissa L. Nana2
Sara K. Deibler1Alissa L. Nana2 Sarat C. Vatsavayai2
Sarat C. Vatsavayai2 Shahid Banday1You Zhou3,4
Shahid Banday1You Zhou3,4 Sandra Almeida3Alexandra Weiss3Robert H. Brown3
Sandra Almeida3Alexandra Weiss3Robert H. Brown3 William W. Seeley2
William W. Seeley2 Fen-Biao Gao3,4Michael R. Green1
Fen-Biao Gao3,4Michael R. Green1A common pathological hallmark of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) is the cytoplasmic mislocalization and aggregation of the DNA/RNA-binding protein TDP-43, but how loss of nuclear TDP-43 function contributes to ALS and FTD pathogenesis remains largely unknown. Here, using large-scale RNAi screening, we identify TARDBP, which encodes TDP-43, as a gene whose loss-of-function results in elevated DNA mutation rate and genomic instability. Consistent with this finding, we observe increased DNA damage in induced pluripotent stem cells (iPSCs) and iPSC-derived post-mitotic neurons generated from ALS patients harboring TARDBP mutations. We find that the increase in DNA damage in ALS iPSC-derived neurons is due to defects in two major pathways for DNA double-strand break repair: non-homologous end joining and homologous recombination. Cells with defects in DNA repair are sensitive to DNA damaging agents and, accordingly, we find that ALS iPSC-derived neurons show a marked reduction in survival following treatment with a DNA damaging agent. Importantly, we find that increased DNA damage is also observed in neurons with nuclear TDP-43 depletion from ALS/FTD patient brain tissues. Collectively, our results demonstrate that ALS neurons with loss of nuclear TDP-43 function have elevated levels of DNA damage and contribute to the idea that genomic instability is a defining pathological feature of ALS/FTD patients with TDP-43 pathology.
Amyotrophic lateral sclerosis (ALS) is one of the major adult-onset neurodegenerative disorders, with a prevalence in the United States of 5 in 100,000 people (Mehta et al., 2022). The disease is characterized by the degeneration of motor neurons of the motor cortex, brainstem, and spinal cord, resulting in progressive muscle weakness, paralysis and eventual respiratory failure (Masrori and Van Damme, 2020). Approximately 10–15% of patients with ALS also develop frontotemporal dementia (FTD), marked by degeneration in the frontal, insular, and temporal cortex, resulting in a range of cognitive, behavioral and language deficits (Yang et al., 2020). The etiology of ALS is complex, with a combination of both genetic and environmental factors playing a role in the neurodegenerative process (Mejzini et al., 2019). Most cases of ALS are sporadic, whereas approximately 5–10% are caused by genetic mutations, most commonly a hexanucleotide repeat expansion in the C9ORF72 gene or mutation in SOD1, FUS or TARDBP (Alsultan et al., 2016). In total, more than 50 genes associated with ALS risk and pathogenesis have been identified, and are involved in diverse biological functions (Mejzini et al., 2019). The large number of genes and cellular processes implicated in ALS has led to the notion that the pathophysiology of the disease is multifactorial, with several mechanisms contributing to neurodegeneration.
A major determinant of ALS pathogenesis is the TARDBP gene, which encodes transactive response DNA-binding protein 43 (TDP-43), a ubiquitously expressed DNA/RNA-binding protein that predominantly resides in the nucleus, but is capable of shuttling to the cytoplasm (Suk and Rousseaux, 2020). In the nucleus, TDP-43 plays diverse roles in gene regulation and RNA processing, with reported roles in transcription, RNA stability, microRNA biogenesis, and pre-mRNA splicing (Lagier-Tourenne et al., 2010; Liao et al., 2022). TDP-43 is a key component of RNA transport granules in neurons (Alami et al., 2014; Liu-Yesucevitz et al., 2014), and plays an important role in neuronal plasticity by regulating local protein synthesis in dendrites (Diaper et al., 2013). Mutations in TARDBP are rare in ALS and have been found in 1–5% of sporadic and familial ALS cases (Mackenzie et al., 2010b; Ravanidis et al., 2018). However, TDP-43 is a major component of cytoplasmic ubiquitin-positive protein inclusions that are the pathological hallmark of the disease and are found in ~97% of ALS patients regardless of genetic etiology (Neumann et al., 2006; Ling et al., 2013). The formation of TDP-43-positive cytoplasmic aggregates is almost always associated with clearing of TDP-43 from the nucleus and the presumptive loss of nuclear TDP-43 function (Neumann et al., 2006). Similar inclusions are seen in ~50% of patients with FTD (Neumann et al., 2006). A thorough understanding of the nuclear functions of TDP-43 is crucial to determining whether loss of these functions may play a role in disease pathogenesis.
Similar to other age-related neurodegenerative disorders, ALS/FTD is associated with an impaired DNA damage response and resultant increase in genomic instability, which is thought to contribute to motor neuron degeneration and disease pathogenesis (Konopka and Atkin, 2022). Indeed, many ALS-associated genes have roles in the DNA damage response, and defects in these genes lead to deficiencies in DNA repair (Sun et al., 2020). For example, mutations in C9ORF72 are associated with increased DNA double-strand breaks and defects in ATM-mediated repair in cells and spinal cord tissues from C9ORF72-ALS patients (Lopez-Gonzalez et al., 2016; Farg et al., 2017; Walker et al., 2017) and induce neurodegeneration in mice (Walker et al., 2017). Moreover, ALS-associated mutations in the FUS gene lead to defects in DNA repair (Wang et al., 2013) and increase DNA damage in mice (Qiu et al., 2014). Several studies have implicated a role for TDP-43 in maintaining genomic stability through diverse mechanisms including promoting non-homologous end-joining DNA repair (Mitra et al., 2019; Konopka et al., 2020) and preventing accumulation of genotoxic R-loops (Hill et al., 2016; Giannini et al., 2020; Wood et al., 2020). Defective DNA repair and genomic instability represent a potential unifying etiology that underlies the diverse pathophysiological mechanisms of ALS/FTD (Sun et al., 2020).
Here, using an unbiased, large-scale loss-of-function screening approach, we identify TARDBP as a gene required for genomic stability. We find that post-mitotic neurons derived from ALS patient induced pluripotent stem cells (iPSCs) harboring TARDBP mutations are defective in DNA repair and display increased DNA damage, which we also observe in neurons lacking nuclear TDP-43 in ALS/FTD patient brain tissues. We show that the increase in DNA damage in ALS iPSC-derived neurons is due to defects in two major pathways for DNA double-strand break repair. Our results provide support for the model that loss of nuclear TDP-43 function underlies genomic instability and molecular pathogenesis in ALS/FTD.
The RNAi Consortium (TRC) lentiviral mouse shRNA library (Dharmacon/Horizon Discovery), divided into 24 pools (5,000 shRNAs per pool), was obtained through the UMass Chan Medical School RNAi Core Facility. The lentiviral pools were generated with titers of ~2–3 × 107 cfu/mL, as previously described (Gazin et al., 2007). Because ethyl methanesulfonate (EMS) treatment alone will result in a small number of Aprt−/− cells, a pilot experiment was performed to determine the minimal number of Aprt−/− 3C4 cells required to form colonies in the presence of 2,6-diaminopurine. Toward this end, 1 × 104, 1 × 105, or 1 × 106 3C4 cells expressing a non-silencing (NS) negative control shRNA, or as a positive control an shRNA targeting the DNA mismatch repair gene Mlh1, whose inactivation is known to increase mutation rate (Hsieh and Yamane, 2008), were treated with EMS and then cultured in the presence of 2,6-diaminopurine for 10 days to select for Aprt−/− cells. In the presence of EMS, 1 × 104 Mlh1 knockdown 3C4 cells were sufficient to form colonies, whereas 3C4 cells expressing a control NS shRNA did not form any colonies (see Supplementary Figure S1A). Thus, for an shRNA to be represented in 1 × 104 cells to form a colony, a minimum of 5 × 107 cells would need to be transduced. For the primary screen, 5 × 107 mouse embryonic stem cells (clone 3C4; provided by Jay Tischfield, Rutgers University), maintained in embryonic stem cell culture as previously (Cervantes et al., 2002), were transduced with each pool of the shRNA library at an MOI of 0.2 and selected with 1.5 μg/mL puromycin for 4 days. Cells were then treated with EMS (Sigma, 300 μg/mL) for 5 h, after which the cells were washed with media. Cells were plated on 100-mm dishes, incubated with media containing 2,6 di-aminopurine (Sigma, 5 μg/mL). After 10 days, DAP-resistant colonies were harvested for genomic DNA isolation, and shRNAs were identified by sequence analysis as previously described (Gazin et al., 2007).
For validation assays, individual knockdown cell lines were generated by stable transduction of mouse 3C4 cells (1 × 106) or human A549 cells (1 × 105, obtained from the ATCC) with a single shRNA (Supplementary Table S1) followed by puromycin selection and incubation with 5 μg/mL 2,6 di-aminopurine (for 3C4 cells) or 0.75 μg/mL 6-thioguanine (Sigma) (for A549 cells) for 10 days. Surviving colonies were fixed with 50% ethanol, stained with crystal violet, and counted. Mutation rate was calculated by the P0 method as previously described (Cervantes et al., 2002) and corrected for colony forming efficiency. Rates are presented per cell per generation.
Total RNA was isolated, and reverse transcription was performed as described (Gazin et al., 2007), followed by qRT-PCR using Power SYBR Green PCR Master Mix (Applied Biosystems) and TDP-43 gene-specific primers (forward, 5′-ATTCAAAGGGGTTTGGCTTT-3′, and reverse, 5′-CAGTCACACCATCGTCCATC-3′). GAPDH was used as an internal reference gene for normalization.
Human control iPSCs [clones 2#20 (WT-1), 37#20 (WT-2) and 35#11 (WT-3)] (Almeida et al., 2012; Zhang et al., 2013; Freibaum et al., 2015) and ALS patient-derived iPSCs (clones HPS0292 ((M337V)-1), HPS0293 ((M337V)-2), HPS0290 ((Q343R)-1) and HPS0291 ((Q343R)-1)) (Egawa et al., 2012), provided by the RIKEN BRC through the National BioResource Project of the MEXT/AMED, Japan, were cultured in mTeSR1 media (STEMCELL Technologies) on matrigel-coated plates. To generate iPSC-derived neurons, iPSCs were differentiated by lentiviral gene transfer and genomic integration of doxycycline-inducible Neurogenin1/2 and rTA3 vectors as previously described (Busskamp et al., 2014). To confirm neuronal differentiation, neurons were fixed with 4% paraformaldehyde in PBS for 10 min, blocked with 10% normal goat serum (Vector Laboratories), and then stained with a TUJ1 (Covance) antibody for 1 h at room temperature. Cells were then rinsed several times with PBS, incubated with an Alexa 488-conjugated donkey anti-mouse secondary antibody (Molecular Probes) and DAPI (Molecular Probes) in the appropriate buffer for 1 h at room temperature. After several more rinses, cells were mounted with Vectashield (Vector Laboratories) and imaged with a Zeiss Axiovert microscope and 10X objective equipped with a Zeiss Axiocam digital camera.
Control or mutant TDP-43 ALS iPSC or iPSC-derived neurons at day 20 were harvested, counted for comet assays. Single-cell gel electrophoresis under alkaline conditions was performed using a Comet assay kit (Trevigen, 4250-050-K). Samples were stained with SYBR-green and observed using a Zeiss AXIO Imager Z2 microscope. Images were analyzed using ImageJ software.
The small-plasmid-based NHEJ assay was performed as previously described (Wilson et al., 1999). Briefly, iPSC-derived neurons (at day 4 of the differentiation process) were co-transfected with a linearized small ampicillin plasmid (pGL-4.14) and a circularized kanamycin plasmid (pDSRed-N1). Forty-eight hours post-transfection, plasmids were isolated and transformed into E. coli strain DH10B. The NHEJ frequency was determined by calculating the ratio of ampicillin-resistant colonies to kanamycin-resistant colonies, and the result was normalized to that observed in control neurons, which was set to 1. The chromosomal NHEJ assay was performed with HEK293/pPHW1 cells as previously described (Zhuang et al., 2009). The NHEJ frequency was calculated from the ratio of the number of XHATM-resistant colonies to the total number of cells seeded and normalized for transfection and plating efficiencies.
The small-plasmid-based HR assay was performed as previously described (Thyagarajan et al., 1996; Fang et al., 2013). Briefly, plasmids pSV2neoDL, pSV2neoDR, and pRSVed1884 were transfected into iPSC-derived neurons (at day 4 of the differentiation process) using an Amaxa Cell Line Optimization Nucleofector Kit (Lonza). Forty-eight hours post-transfection, plasmids were isolated, digested with DpnI, and transformed into E. coli strain DH10B. The HR frequency was determined by calculating the ratio of kanamycin-resistant colonies to ampicillin-resistant colonies, and the result was normalized to that observed in control neurons, which was set to 1. The chromosomal HR assay was performed with HCT116/HN5 cells as previously described (Mohindra et al., 2004; Wen et al., 2008). The HR frequency was calculated from the ratio of the number of G418-resistant colonies to the total number of cells seeded and normalized for transfection and plating efficiencies.
Immunocytochemical analysis was performed as previously described (Sorensen et al., 2005). Briefly, cells were treated in the presence or absence of 0.2 mM hydroxyurea (Sigma) for 24 h. Cells were stained with anti-RAD51 (Santa Cruz) and 4,6-diamidino-2-phenylindole (DAPI) and analyzed on a Zeiss AXIO Imager Z2 microscope. Nuclei with >5 foci were considered positive for hydroxyurea-induced RAD51 foci. The percentages of cells with positive RAD51 were determined in at least two separate experiments by counting at least 300 nuclei on each slide.
Control or mutant TDP-43 ALS iPSCs (5 × 105) were seeded in 60-mm dishes, and 24 h later incubated with a range of concentrations of etoposide (Sigma) or 5-fluorouracil (Sigma) for 14 days. Colonies were fixed, stained with crystal violet and counted. Control or mutant TDP-43 ALS iPSC-derived neurons (1 × 105) at day 20 were seeded in one well of 6-well plates, 24 hours later were incubated with a series of concentration of etoside or 5-fluorouracil for 3 days. Neuron viability was measured with alamarBlue assay (Thermo Fisher Scientific). The results were normalized to 0 uM (DMSO solvent), which was set to 1.0.
Post-mortem human brain tissue samples were obtained from the UCSF Neurodegenerative Disease Brain Bank. Consent for brain donation was obtained from all subjects or their surrogates in accordance with the Declaration of Helsinki, and the research was approved by the University of California, San Francisco Committee on Human Research. Clinical diagnoses of bvFTD were made according to prevailing international consensus criteria at the time of assessment (Neary et al., 1998; Rascovsky et al., 2011). Brains were cut fresh into 8–10 mm thick coronal slabs and immersion fixed on a platform in 10% neutral buffered formalin (Fisher Scientific, United States). Neuropathological diagnoses were made following consensus diagnostic criteria (Mackenzie et al., 2010a, 2011; Montine et al., 2012) using previously described immunohistochemical and histological methods (Tartaglia et al., 2010). Four patients with FTLD-TDP were selected per brain region (2 sporadic and 2 carrying the C9ORF72 expansion) based on clinical and neuropathological diagnoses (Supplementary Table S2).
Eight-micrometer thick sections of precentral gyrus were cut from formalin fixed paraffin-embedded tissue blocks taken for neuropathological diagnosis (Vatsavayai et al., 2016). Blocks of the frontoinsula were dissected from fixed slabs, cryoprotected, and sectioned at 50 μm as previously described (Kim et al., 2012; Nana et al., 2019). DNA damage was assessed using an antibody against phosphorylated-Serine139 to Histone H2AX (H2AX, mouse, 1:5 k, Millipore Sigma 05-636). An antibody against TDP-43 (rabbit, 1:4,000, Proteintech 10782-2-AP) was used to identify neurons lacking normal nuclear TDP-43 and bearing a TDP-43 cytoplasmic inclusion, and layer 5 neurons were identified with MAP2 (chicken, 1:3,000, EnCor Biotechnology Inc. CPCA-MAP2) using previously described immunofluorescence methods (Nana et al., 2019). All sections were counterstained with NeuroTrace 435/455 Blue Fluorescent Nissl Stain (Invitrogen).
For the precentral gyrus, non-overlapping Z-stack images at x60 magnification were captured in layer 5 based on Nissl and MAP2 channels and blind to TDP-43 and γH2AX status on a Zeiss LSM 880 confocal laser scanning microscope. For the FI, to ensure image capture of at least one TDP-inclusion bearing neuron per image, 20 TDP-inclusion-bearing MAP2-positive neurons from layer 5 were marked at random at x10 magnification using only the Nissl, MAP2, and TDP-43 channels, then z- stacks were captured at each marker location at x40 magnification from all four channels. Neurons from both regions were counted manually using Zeiss Zen software. For each field of view, investigators first marked all neurons using the MAP2 channel only. Next, each neuron was classified as TDP-43 inclusion-bearing or normal, blinded to γH2AX status. The TDP-43 channel and markers were then hidden, and each neuron was assessed for the presence of γH2AX foci. γH2AX staining showed two patterns of immunoreactivity: (1) 1–3 large punctate γH2AX positive foci in the nucleus (Figure 1A) or (2) a diffuse pan-nuclear γH2AX staining. Given that in brain tissue focal but not pan-nuclear γH2AX staining is associated with DNA DSBs (Shanbhag et al., 2019), we only counted neurons with nuclear foci. A total of 285 neurons from precentral gyrus and 979 neurons from frontoinsular cortex were counted.
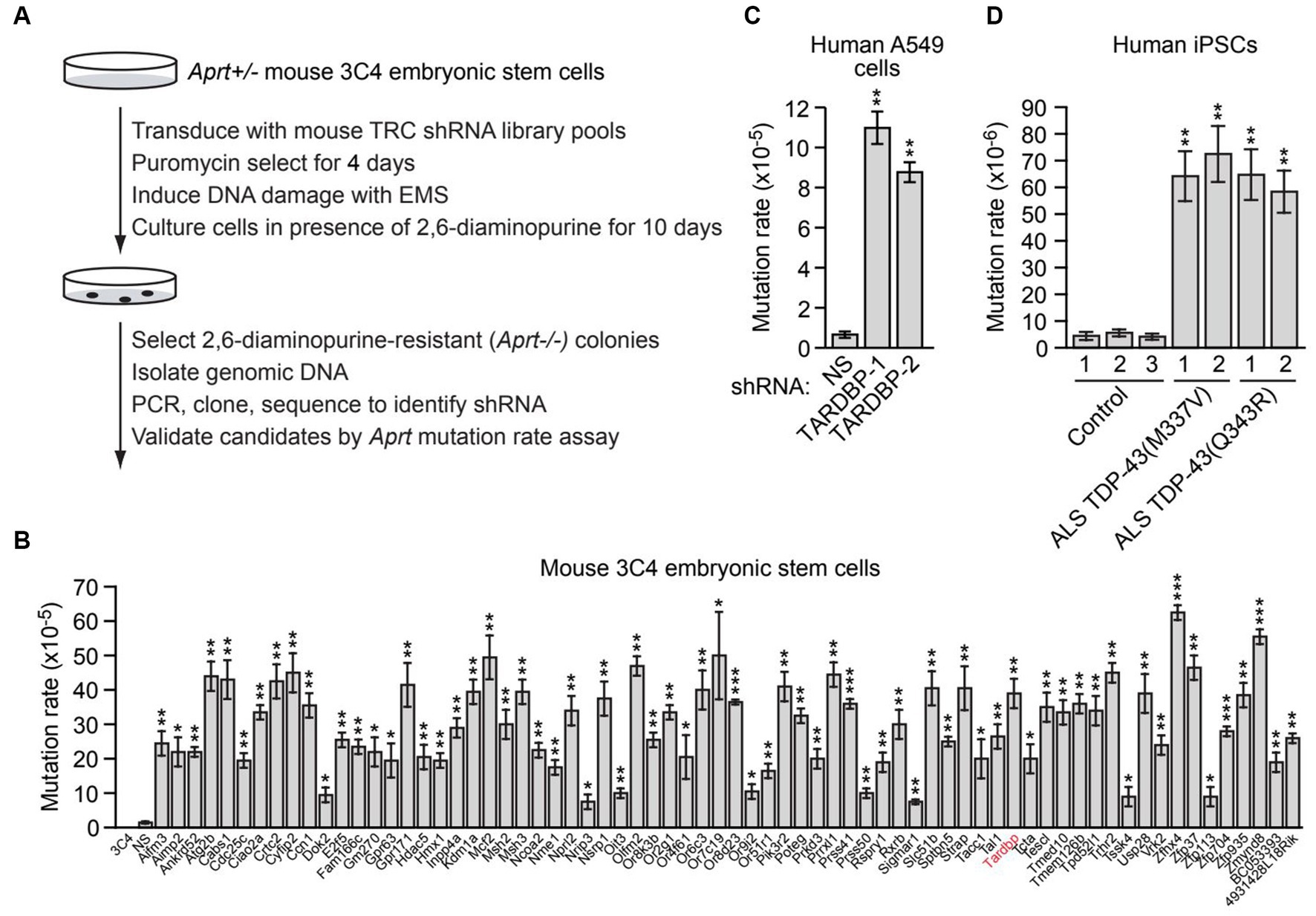
Figure 1. A large-scale RNAi screen identifies genes required for genomic stability, including TARDBP. (A) Schematic summary of the large-scale RNAi screening strategy. (B) Aprt mutation rate in mouse 3C4 embryonic stem cells expressing an shRNA targeting one of 70 genes identified in the initial RNAi screen. Shown as controls are parental 3C4 cells, and 3C4 cells expressing a non-silencing (NS) shRNA. Not shown are the results with an Aprt shRNA, which, as expected, was isolated in the primary screen. (C) Spontaneous HPRT mutation frequency in A549 cells expressing a NS shRNA or one of two unrelated TARDBP shRNAs, TARDBP-1 and TARDBP-2. (D) Spontaneous HPRT mutation frequency in iPSC lines generated from two familial ALS patients harboring a TDP-43(M337V) or TDP-43(Q343R) mutation, or in three control iPSC lines. Data are represented as mean ± SD; Statistical analysis was done with Students’ t test, *p < 0.05, **p < 0.01, ***p < 0.001.
Statistical analysis was performed using R version 4.0.5 in RStudio 1.1.1093 using logistic regression (binomial glm) in the lme4 package (http://www.r-project.org). The presence or absence of γH2AX foci was used as the outcome, and TDP-43 inclusion status as the primary categorical predictor, with C9ORF72 status and subject as covariates. A separate model was run for each region. A p < 0.05 (two-tailed) was considered statistically significant.
To identify new genes required for genomic stability, we performed a large-scale loss-of-function RNAi screen for genes that, when knocked down, result in an increased mutation rate. For the screen readout, we used a well-established genotoxic assay based on loss of heterozygosity of the adenine phosphoribosyl transferase (Aprt) gene, which confers resistance to the drug 2,6-diaminopurine. The screen was performed in mouse 3C4 embryonic stem cells, which are heterozygous for the Aprt gene (Aprt+/−) (Cervantes et al., 2002). Previous studies have shown that the spontaneous mutation rate at Aprt in these cells is relatively low (8.3 × 10−8) (Cervantes et al., 2002), therefore the primary screen was performed in the presence of the chemical mutagen ethyl methanesulfonate (EMS) to increase the mutation rate and reduce the number of cells required for large-scale screening. For the primary screen, a genome-wide mouse short hairpin RNA (shRNA) library was divided into 24 pools (5,000 shRNAs per pool), which were then packaged into lentivirus particles to stably transduce 5 × 107 3C4 cells so that on average, each shRNA was represented in a sufficient number of cells that could enable colony formation (Supplementary Figure S1A). Cells were treated with EMS and then cultured in the presence of 2,6-diaminopurine for 10 days to select for Aprt−/− cells, which were then harvested and subjected to sequence analysis to identify the shRNAs (Figure 1A). Individual positive candidate shRNAs were validated in a second Aprt mutation rate assay performed in the absence of EMS, in which the number of 2,3-diaminopurine-resistant colonies were counted and used to calculate the spontaneous mutation rate. Using this approach, we identified 70 genes whose knockdown significantly increased the spontaneous mutation rate ≥2-fold compared to a control non-silencing (NS) shRNA (Figure 1B). The 70 genes encode proteins that are involved in a variety of biological processes, including apoptosis and autophagy, cell cycle regulation, signal transduction, and transcription (Supplementary Table S3). Notably, the screen identified the mismatch repair genes Msh2 and Msh3, whose inactivation is known to increase mutation rate (Hsieh and Yamane, 2008). The screen also identified several other factors known to play a role in genomic stability, such as Aimp2 (Han et al., 2008), Atg2b (Kang et al., 2009), Cdc25c (Thanasoula et al., 2012), Crtc2 (Fang et al., 2015), Kdm1a (Mosammaparast et al., 2013), Nme1 (Kaetzel et al., 2015), Nprl2 (Ma et al., 2017), Tardbp (Hill et al., 2016; Mitra et al., 2019; Giannini et al., 2020; Konopka et al., 2020), Usp28 (Zhang et al., 2006), and Zmynd8 (Gong et al., 2015), demonstrating the validity of our experimental approach. Validation of a representative subset of candidates using a second, unrelated shRNA confirmed that target gene knockdown increased spontaneous Aprt mutation rate (Supplementary Figure S1B).
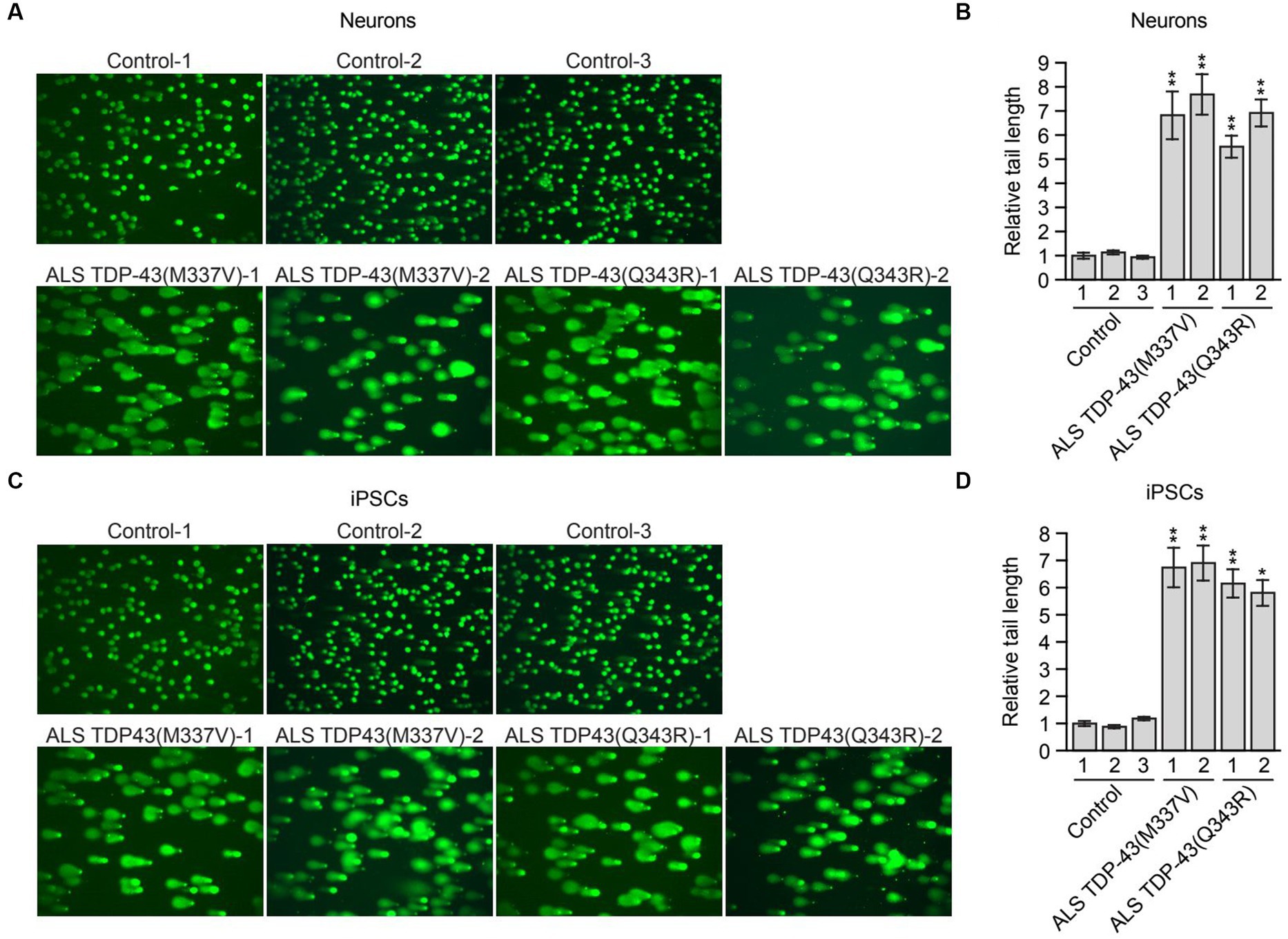
Figure 2. Mutant TDP-43 ALS iPSC-derived neurons have elevated levels of DNA damage. (A) Comet assay in control, TDP-43(M337V) or TDP-43(Q343R) ALS iPSC-derived neurons. Representative images are shown. (B) Quantification of the comet assays shown in (A). (C) Comet assay in control, TDP-43(M337V) and TDP-43(Q343R) ALS iPSCs. Representative images are shown. (D) Quantification of the comet assays shown in (C). Data are represented as mean ± SD; Statistical analysis was done with Students’ t test, *p < 0.05, **p < 0.01.
We elected to focus our further investigations on TARDBP. To confirm the elevated mutation rate in TARDBP-knockdown cells, we performed a second mutation rate assay based on hypoxanthine-guanine phosphoribosyltransferase (HPRT) deficiency. In this assay, human lung adenocarcinoma A549 cells harboring mutations in the essential HPRT gene are detected by positive selection using 6-thioguanine (6-TG). We found that knockdown of TARDBP using one of two unrelated shRNAs (Supplementary Figure S1C) significantly increased the spontaneous HPRT mutation rate (Figure 1C).
Because of the importance of TDP-43 in ALS/FTD pathophysiology, we sought to confirm and extend our results in ALS patient cells. We therefore measured the spontaneous mutation rate in four induced pluripotent stem cell (iPSC) lines generated from two familial ALS patients harboring a heterozygous TDP-43(M337V) or TDP-43(Q343R) mutation (Egawa et al., 2012), and in three control iPSC lines (Zhang et al., 2013). Figure 1D shows that the HPRT mutation rate in iPSCs containing TDP-43(M337V) or TDP-43(Q343R) (hereafter called mutant TDP-43 ALS iPSCs) was substantially higher than that observed in control iPSCs, indicating increased genomic instability in ALS patient cells harboring TDP-43 mutations.
Previous studies have shown that loss of nuclear TDP-43 function results in reduced DNA repair and elevated levels of DNA damage (Mitra et al., 2019; Konopka et al., 2020). These prior studies were performed in TDP-43-depleted neuronal cell lines, normal iPSCs and iPSC-derived neural progenitor cells (Mitra et al., 2019) or in neuron-like cells transfected with plasmids expressing TDP-43 mutants (Konopka et al., 2020). To determine whether increased DNA damage is also observed in ALS patient neurons harboring TARDBP mutations, we performed a comet assay, a sensitive method to evaluate DNA lesions at a single-cell level (Olive and Banath, 2006). For these experiments, we differentiated control and mutant TDP-43 ALS iPSCs into neurons as previously described (Busskamp et al., 2014), and confirmed neuronal differentiation by staining for the neuronal marker TUJ1 (Supplementary Figure S2A). As expected, very few comet tails were observed in neurons differentiated from multiple control iPSC lines, indicating a very low level of endogenous DNA lesions (Figure 2A). By contrast, mutant TDP-43 ALS neurons differentiated from multiple patient iPSC lines displayed a significant increase in comet tail length (Figures 2A,B). To examine if the increased DNA damage upon TDP43 mutation is specific to neuron or common to other cell types, TDP-43 mutant iPSC cells were also examined. Similarly, mutant TDP-43 ALS iPSCs also showed increased comet tail length compared to control iPSCs (Figures 2C,D). As expected, an increase in comet tail length was also observed in control neurons following shRNA-mediated knockdown of TARDBP (Supplementary Figure S2B).
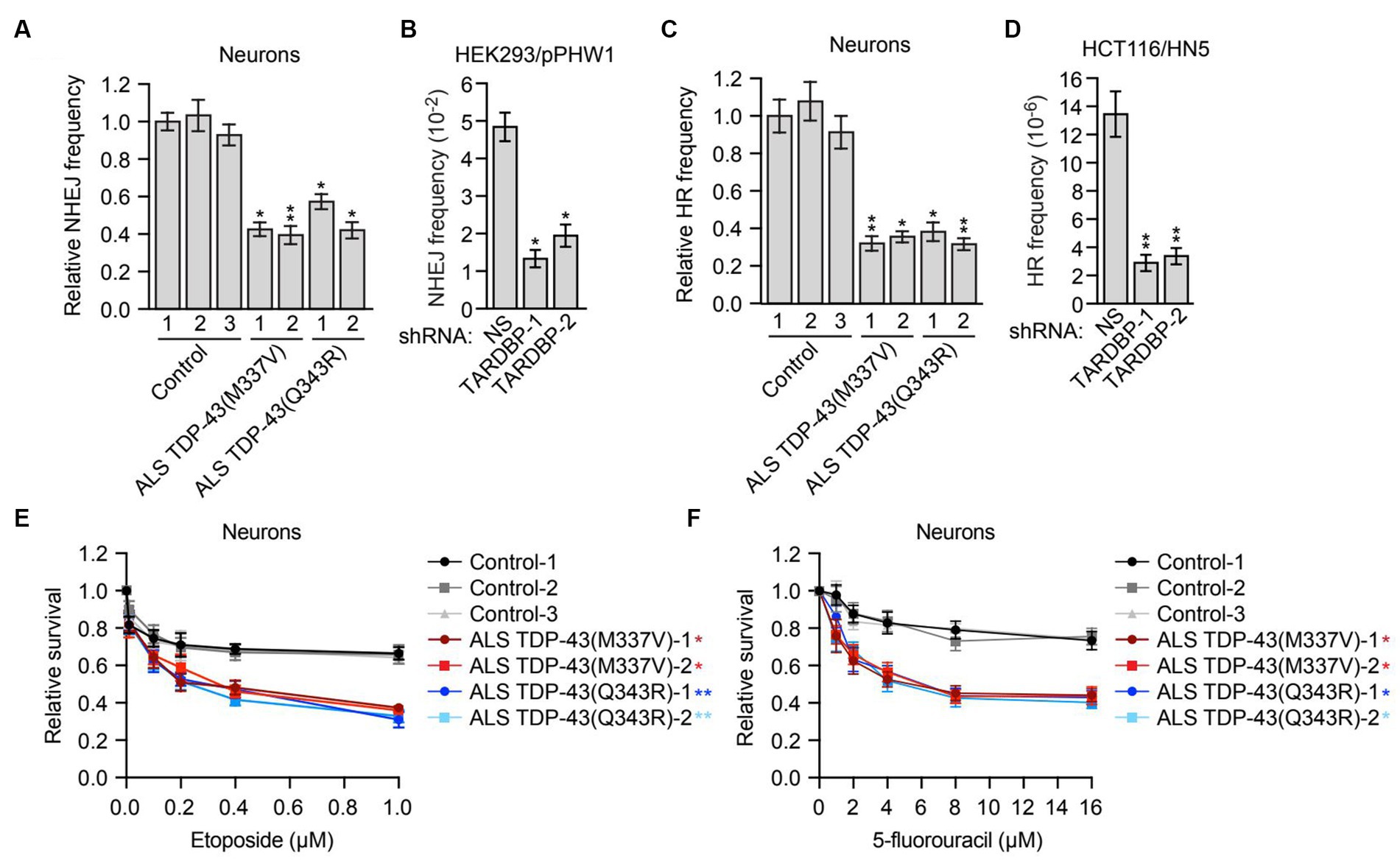
Figure 3. Mutant TDP-43 ALS iPSC-derived neurons are deficient in NHEJ and HR DNA repair pathways and are sensitive to DNA damaging agents. (A) Plasmid-based NHEJ assay in ALS iPSC-derived neurons containing TDP-43(M337V) or TDP-43(Q343R). The results were normalized to that obtained in control neurons, which was set to 1. (B) Chromosomal-based NHEJ assay in HEK293/pPHW1 cells expressing a NS or TARDBP shRNA. (C) Plasmid-based HR assay in control, TDP-43(M337V) or TDP-43(Q343R) ALS iPSC-derived neurons. (D) Chromosomal-based HR assay in HCT116/HN5 cells expressing a NS or TARDBP shRNA. (E,F) Survival curves for control, TDP-43(M337V) or TDP-43(Q343R) ALS iPSC-derived neurons treated with increasing doses of etoposide (E) or 5-fluorouracil (F). The results were normalized to those obtained in the absence of drug, which was set to 1. Data are represented as mean ± SD; Statistical analysis was done with Students’ t test, *p < 0.05, **p < 0.01. In (E,F), p value comparisons are shown for treatment with 1 μM etoposide and 16 μM 5-fluorouracil, respectively.
In mammalian cells, there are two major pathways for DNA double-strand break repair: non-homologous end joining (NHEJ) and homologous recombination (HR) (Chapman et al., 2012). As mentioned above, previous studies have shown that depletion of TDP-43 results in reduced NHEJ repair (Mitra et al., 2019). To investigate whether, as expected, NHEJ is impaired in mutant TDP-43 ALS neurons, we first used a well-established assay that is based upon NHEJ-directed repair of a plasmid-borne ampicillin-resistance reporter gene (Wilson et al., 1999). Briefly, control or mutant TDP-43 ALS neurons were co-transfected with a linearized plasmid encoding an ampicillin-resistance gene and a circularized plasmid encoding a kanamycin-resistance gene. Generation of a functional ampicillin-resistance gene is dependent on NHEJ. Following isolation of the plasmids and transformation into bacteria, NHEJ frequency was calculated as the ratio of ampicillin-resistant (indicative of repair) to kanamycin-resistant (indicative of total plasmid recovery) colonies. We found that NHEJ activity in mutant TDP-43 ALS neurons was ~2-fold lower than that in control neurons (Figure 3A). We also performed a chromosomal-based NHEJ assay in which TARDBP was knocked down in HEK293/pPHW1 cells, which contain a non-functional integrated GPT reporter gene that can be repaired by NHEJ (Zhuang et al., 2009). We found that shRNA-mediated knockdown of TARDBP in HEK293/pPHW1 cells (Supplementary Figure S3A) resulted in a 2.5–4-fold decrease in NHEJ activity (Figure 3B).
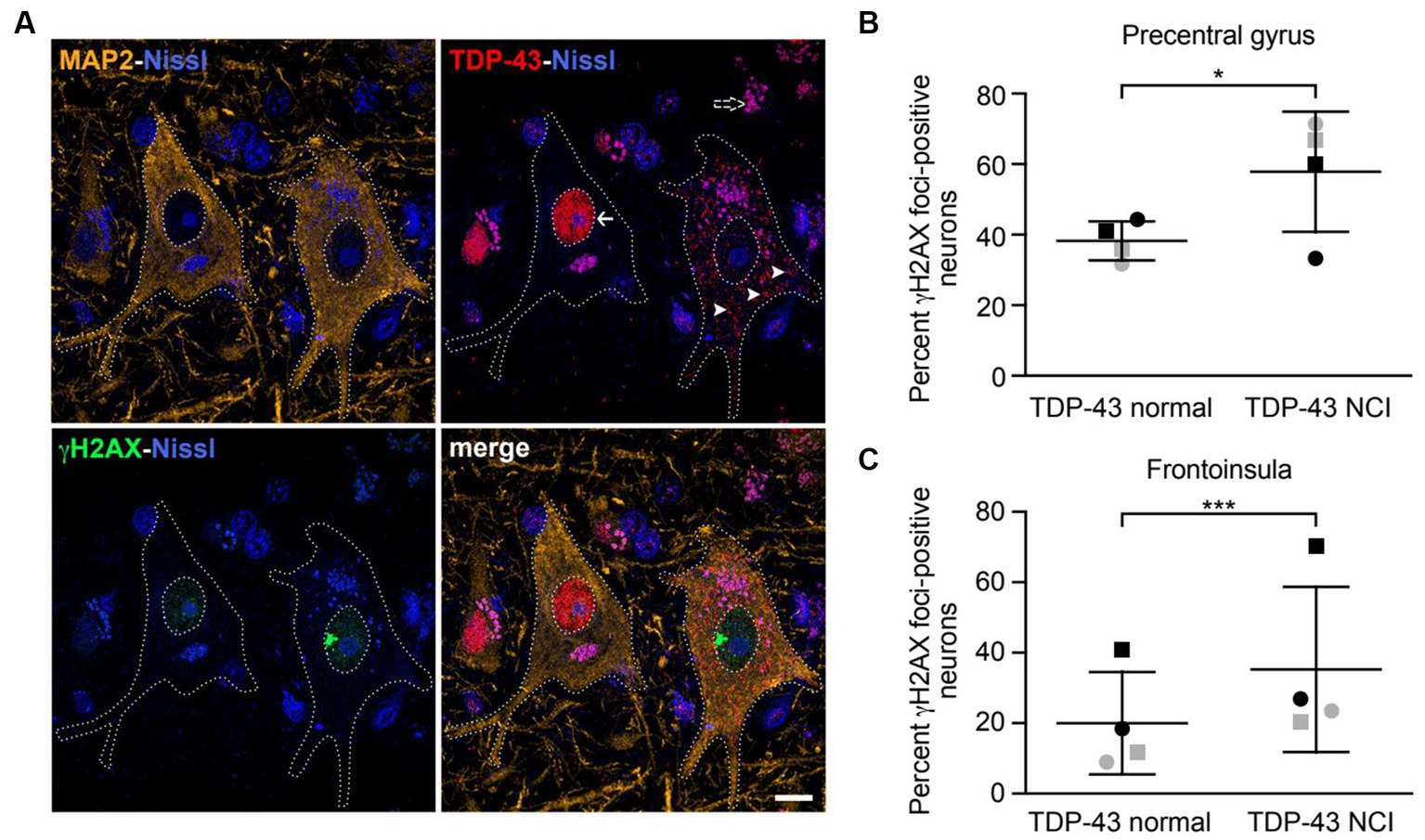
Figure 4. Neurons with nuclear TDP-43 depletion are associated with increased DNA damage in ALS/FTD patient brain tissues. (A) Confocal images showing two representative upper motor neurons from the precentral gyrus with either normal nuclear TDP-43 (arrow) or loss of nuclear TDP-43 and accompanying cytoplasmic inclusions (arrowheads) with a large γH2AX-positive focus in the nucleus, consistent with DNA double-strand breakage. Dotted lines represent cell and nuclear boundaries derived from the MAP2 immunostaining. All sections were counterstained with fluorescent Nissl stain (blue). Purple granular staining (dashed arrow) is lipofuscin autofluorescence background seen across channels. Images are maximum intensity projections of a confocal z-stack. Scale bar equals 10 μm. (B,C) Percentage of neurons showing γH2AX foci in cells with normal nuclear TDP-43 or TDP-43 neuronal cytoplasmic inclusions (NCIs) in the precentral gyrus (B) and frontoinsula (C) of patients with either C9ORF72-associated (gray) or sporadic (black) FTD-ALS. Data are represented as mean ± SD; Statistical analysis was performed using R version 4.0.5 in RStudio 1.1.1093, *p < 0.05, ***p < 0.001.
Although in post-mitotic neurons the prevalent DNA double-strand break repair pathway is NHEJ (Merlo et al., 2016), several reports have shown that HR also occurs in neurons (Cox et al., 2015; Welty et al., 2018; Yu et al., 2018). We therefore tested whether HR is also affected in mutant TDP-43 ALS neurons. First we used a plasmid-based assay similar to that described above in which two plasmids carrying non-overlapping deletions within the coding region of the kanamycin-resistance gene are co-transfected into cells, and generation of a functional kanamycin gene is dependent on inter-plasmid HR (Thyagarajan et al., 1996). We found that the HR frequency was 2–3-fold lower in mutant TDP-43 ALS neurons compared to control neurons (Figure 3C). To confirm this result, we also carried out a chromosomal-based HR assay. We knocked down TARDBP in HCT116/HN5 cells (Supplementary Figure S3B), a reporter cell line that contains two chromosomally-integrated non-functional portions of a neomycin-resistance gene (Mohindra et al., 2004; Wen et al., 2008). HR can generate an intact neomycin-resistance gene, enabling colony formation in the presence of neomycin. We found that shRNA-mediated knockdown of TARDBP resulted in a 4–5-fold decrease in HR efficiency (Figure 3D).
To further investigate the possibility that loss of TDP-43 impairs HR activity, we analyzed RAD51 foci formation, a characteristic marker of HR-directed DNA repair (Moynahan and Jasin, 2010). Human A549 cells stably expressing a TARDBP shRNA were subjected to DNA replication stress by the addition of hydroxyurea, a DNA replication inhibitor, and 24 h later, RAD51 foci were detected by immunofluorescence. We found that following TARDBP knockdown, there was a substantial decrease in the percentage of RAD51 foci-positive cells (Supplementary Figure S3C).
Cells that are defective for DNA repair are sensitive to DNA damaging agents (Bouwman and Jonkers, 2012). We therefore measured the relative sensitivity of control and mutant TDP-43 ALS neurons to DNA damage. Using increasing doses of the DNA damaging agent etoposide (Figure 3E) or 5-fluorouracil (Figure 3F), we generated survival curves for control and mutant TDP-43 ALS neurons and found that compared to control neurons, mutant TDP-43 ALS neurons displayed a reduction in survival following treatment with either DNA damaging agent. Similar results were obtained in iPSCs (Supplementary Figure S4). Collectively, these results indicate that mutant TDP-43 ALS neurons in culture are deficient in both NHEJ and HR repair pathways, leading to elevated levels of DNA damage.
A previous report found increased DNA double-strand breaks in spinal cord tissues from ALS patients with TDP-43 pathology (i.e., formation of TDP-43-positive cytoplasmic inclusions and nuclear TDP-43 depletion) compared to matched controls (Mitra et al., 2019). However, whether increased DNA damage is observed in the same neurons that have TDP-43 pathology had not been carefully examined. We therefore obtained brain tissues from patients with either C9ORF72 mutations or sporadic FTD-ALS, both of which were accompanied by typical TDP-43 pathology. ALS is characterized by loss of upper motor neurons of the motor cortex, the majority of which are located in layer 5 (Ragagnin et al., 2019). FTD is associated with degeneration targeting a similar population of large, layer 5 projection neurons within the frontoinsular and anterior cingulate cortices (Nana et al., 2019). We therefore investigated layer 5 neurons within the precentral gyrus (targeted in ALS) and frontoinsular cortex (targeted in FTD), in patients with a blended FTD-ALS presentation. We combined immunofluorescence staining for γH2AX, a widely used marker for DNA double-strand breaks (Sedelnikova and Bonner, 2006), and TDP-43 (Figure 4A). Cells were also co-stained with the neuronal marker MAP2. In precentral gyrus, γH2AX foci were observed in 62% of TDP-43 inclusion-positive neurons (n = 26) compared to 38% of neurons with normal nuclear TDP-43 (n = 259) (Figure 4B). Similarly, in the frontoinsula, γH2AX foci were observed in 35% of TDP-43 inclusion-positive neurons (n = 139) compared to 21% in neurons without TDP-43 pathology (n = 782) (Figure 4C). In both regions, we found a significant increase in γH2AX foci in neurons with depleted nuclear TDP-43 and cytoplasmic aggregation (precentral gyrus z = 2.275, p = 0.0229; frontoinsula z = 3.991, p < 0.001). Collectively, these results indicate that neurons with loss of nuclear TDP-43 function display increased DNA double-strand breaks in patient brain tissues.
In this study, we identified TARDBP in an unbiased large-scale RNAi screen for genes involved in maintaining genomic stability, as assessed by increased mutation frequency. In addition to TARDBP, the screen identified several other genes with known roles in genomic stability, as well as dozens of candidate genes with no previously reported role in genomic stability. Thus, the results of our study will provide a foundation for further studies of the molecular mechanisms underlying genomic stability.
Consistent with previous studies reporting increased DNA damage following TDP-43 depletion (Mitra et al., 2019; Konopka et al., 2020), we found elevated DNA damage in iPSCs generated from ALS patients with specific genetic mutations in TARDBP and in post-mitotic neurons differentiated from these iPSC lines, suggesting that these defects caused by mutant TDP-43 are not cell-type specific, indicating a molecular consequence of partial loss of TDP-43 function. Although isogenic mutant TDP-43 iPSC lines were not available to us for this study, we observed increased DNA damage selectively in multiple ALS patient iPSC lines but not in multiple control iPSC lines, as well as in TARDBP knockdown cells. A potential pitfall of control cell line from healthy individuals is a large heterogeneity due to cell line and genetic differences. Isogenic cell lines from well characterized pre-existing iPSC cell line provides a more accurate interpretation of the data. Most importantly, we observed a strong correlation between TDP-43 nuclear depletion and increased DNA damage in the same neurons from ALS/FTD patient brain tissues. Our findings contribute to the idea that DNA damage is a defining pathological feature of ALS/FTD patients with TDP-43 pathology.
TDP-43 is a key component of the cytoplasmic ubiquitin-positive protein inclusions that are the pathological hallmark of the disease and are found in approximately 97% of ALS patients regardless of genetic etiology (Neumann et al., 2006; Ling et al., 2013). Mutations in TARDBP are uncommon in ALS and have been found in 1-5% of sporadic and familial ALS cases (Mackenzie et al., 2010b; Ravanidis et al., 2018). TDP-43 clearance from the nucleus is almost always linked to the creation of TDP-43-positive cytoplasmic aggregates. We observed a considerable increase in H2AX foci in neurons with decreased nuclear TDP-43 and cytoplasmic aggregation from the ALS patient’s neuron in Figure 4. Our findings suggest TARDBP mutation is not the only cause of higher mutation rate in ALS with TDP-43 pathology, the increased mutation rate is common to ALS/FTD with TDP-43 pathology. Previous studies showed that loss of nuclear TDP-43 function is associated with increased DNA double-strand breaks (Mitra et al., 2019; Giannini et al., 2020; Konopka et al., 2020), however, these studies focused only on the NHEJ DNA repair pathway (Mitra et al., 2019; Konopka et al., 2020). Here we showed that in addition to NHEJ, the HR repair pathway is also misregulated in ALS iPSC-derived neurons. Cells with defects in DNA repair are sensitive to DNA damaging agents and, accordingly, we find that ALS iPSCs and iPSC-derived neurons show a marked reduction in survival following treatment with the DNA damaging agent etoposide or 5-fluorouracil. Our alamar blue cell viability assay shows that cells with TDP43 shRNA grew a little slower than NS shRNA at basal level without DNA damage agents, but not statistical significance (data not shown). Although the HR pathway has been studied mainly in dividing cells, previous reports have shown that HR also occurs in neurons (Cox et al., 2015; Welty et al., 2018; Yu et al., 2018). Our findings expand the role the TDP-43 in maintaining genomic stability beyond the NHEJ pathway and underscore the multi-faceted role of TDP-43 in DNA repair.
Cells frequently sustain DNA lesions that, if not repaired, can have serious effects, including cell death. Since mutations in the DDR and DNA repair genes have been discovered to cause a number of neurodegenerative illnesses, DNA damage and DNA repair deficiencies have long been linked to neurodegeneration. Post-mitotic cells have different DNA repair pathways than mitotic cells. Therefore, DNA repair may be less effective in neuronal cells, increasing the susceptibility of neurons to DNA damage. It has been demonstrated that long-term TDP-43 depletion causes a long-term increase in pATM, persistent DDR activation and eventually cell death (Mitra et al., 2019). Similar to other age-related neurodegenerative diseases, ALS/FTD is linked to an increase in genomic instability. This association is assumed to have a role in the etiology of the disease and the degeneration of motor neurons. Our data are consistent with these findings and support that DNA damage is a primary or secondary factor in the pathogenesis of ALS, not a symptom or consequence.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by University of California, San Francisco Committee on Human Research. The patients/participants provided their written informed consent to participate in this study.
MF and MG conceived and designed the experiments. MF designed and conducted the RNAi screen and performed the majority of the experiments. SB and YZ validated some key results. AN and SV performed the histological experiments in FTD-ALS patient brain tissues. SA and F-BG provided iPSC lines and related technical assistance. AW and RB provided key reagents during the early stages of the project. MF, SD, WS, F-BG, and MG analyzed and interpreted the data and wrote the manuscript. All authors reviewed the paper and provided comments.
This work was supported by NIH grants R01GM035490 to MG, R37NS057553 and R01NS101986 to F-BG, R21NS112766 and R21NS119952 to SA, and R01NS104437 to WS. F-BG and WS were also funded by the Muscular Dystrophy Association. Human tissue samples were provided by the Neurodegenerative Disease Brain Bank at the University of California, San Francisco, which receives support from NIH grants P30AG062422, P01AG019724, U01AG057195, and U19AG063911, as well as the Rainwater Charitable Foundation and the Bluefield Project to Cure FTD.
This paper is dedicated to MG who passed away unexpectedly on February 10, 2023. We thank Jay Tischfield for reagents, the UMass Chan Medical School RNAi Core Facility for providing shRNA clones and libraries, and the UMass Chan Medical School Morphology Core Facility for assistance with immunofluorescence experiments.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2023.1251228/full#supplementary-material
Alami, N. H., Smith, R. B., Carrasco, M. A., Williams, L. A., Winborn, C. S., Han, S. S., et al. (2014). Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543. doi: 10.1016/j.neuron.2013.12.018
Almeida, S., Zhang, Z., Coppola, G., Mao, W., Futai, K., Karydas, A., et al. (2012). Induced pluripotent stem cell models of progranulin-deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Rep. 2, 789–798. doi: 10.1016/j.celrep.2012.09.007
Alsultan, A. A., Waller, R., Heath, P. R., and Kirby, J. (2016). The genetics of amyotrophic lateral sclerosis: current insights. Degener. Neurol. Neuromuscul. Dis. 6, 49–64. doi: 10.2147/DNND.S84956
Bouwman, P., and Jonkers, J. (2012). The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 12, 587–598. doi: 10.1038/nrc3342
Busskamp, V., Lewis, N. E., Guye, P., Ng, A. H., Shipman, S. L., Byrne, S. M., et al. (2014). Rapid neurogenesis through transcriptional activation in human stem cells. Mol. Syst. Biol. 10:760. doi: 10.15252/msb.20145508
Cervantes, R. B., Stringer, J. R., Shao, C., Tischfield, J. A., and Stambrook, P. J. (2002). Embryonic stem cells and somatic cells differ in mutation frequency and type. Proc. Natl. Acad. Sci. U. S. A. 99, 3586–3590. doi: 10.1073/pnas.062527199
Chapman, J. R., Taylor, M. R., and Boulton, S. J. (2012). Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 47, 497–510. doi: 10.1016/j.molcel.2012.07.029
Cox, D. B., Platt, R. J., and Zhang, F. (2015). Therapeutic genome editing: prospects and challenges. Nat. Med. 21, 121–131. doi: 10.1038/nm.3793
Diaper, D. C., Adachi, Y., Sutcliffe, B., Humphrey, D. M., Elliott, C. J., Stepto, A., et al. (2013). Loss and gain of Drosophila TDP-43 impair synaptic efficacy and motor control leading to age-related neurodegeneration by loss-of-function phenotypes. Hum. Mol. Genet. 22, 1539–1557. doi: 10.1093/hmg/ddt005
Egawa, N., Kitaoka, S., Tsukita, K., Naitoh, M., Takahashi, K., Yamamoto, T., et al. (2012). Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 4:145ra104. doi: 10.1126/scitranslmed.3004052
Fang, M., Pak, M. L., Chamberlain, L., Xing, W., Yu, H., and Green, M. R. (2015). The CREB coactivator CRTC2 is a lymphoma tumor suppressor that preserves genome integrity through transcription of DNA mismatch repair genes. Cell Rep. 11, 1350–1357. doi: 10.1016/j.celrep.2015.04.052
Fang, M., Xia, F., Mahalingam, M., Virbasius, C. M., Wajapeyee, N., and Green, M. R. (2013). MEN1 is a melanoma tumor suppressor that preserves genomic integrity by stimulating transcription of genes that promote homologous recombination-directed DNA repair. Mol. Cell. Biol. 33, 2635–2647. doi: 10.1128/MCB.00167-13
Farg, M. A., Konopka, A., Soo, K. Y., Ito, D., and Atkin, J. D. (2017). The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet. 26, 2882–2896. doi: 10.1093/hmg/ddx170
Freibaum, B. D., Lu, Y., Lopez-Gonzalez, R., Kim, N. C., Almeida, S., Lee, K. H., et al. (2015). GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133. doi: 10.1038/nature14974
Gazin, C., Wajapeyee, N., Gobeil, S., Virbasius, C. M., and Green, M. R. (2007). An elaborate pathway required for Ras-mediated epigenetic silencing. Nature 449, 1073–1077. doi: 10.1038/nature06251
Giannini, M., Bayona-Feliu, A., Sproviero, D., Barroso, S. I., Cereda, C., and Aguilera, A. (2020). TDP-43 mutations link amyotrophic lateral sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLoS Genet. 16:e1009260. doi: 10.1371/journal.pgen.1009260
Gong, F., Chiu, L. Y., Cox, B., Aymard, F., Clouaire, T., Leung, J. W., et al. (2015). Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev. 29, 197–211. doi: 10.1101/gad.252189.114
Han, J. M., Park, B. J., Park, S. G., Oh, Y. S., Choi, S. J., Lee, S. W., et al. (2008). AIMP2/p38, the scaffold for the multi-tRNA synthetase complex, responds to genotoxic stresses via p53. Proc. Natl. Acad. Sci. U. S. A. 105, 11206–11211. doi: 10.1073/pnas.0800297105
Hill, S. J., Mordes, D. A., Cameron, L. A., Neuberg, D. S., Landini, S., Eggan, K., et al. (2016). Two familial ALS proteins function in prevention/repair of transcription-associated DNA damage. Proc. Natl. Acad. Sci. U. S. A. 113, E7701–E7709. doi: 10.1073/pnas.1611673113
Hsieh, P., and Yamane, K. (2008). DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech. Ageing Dev. 129, 391–407. doi: 10.1016/j.mad.2008.02.012
Kaetzel, D. M., Leonard, M. K., Cook, G. S., Novak, M., Jarrett, S. G., Yang, X., et al. (2015). Dual functions of NME1 in suppression of cell motility and enhancement of genomic stability in melanoma. Naunyn Schmiedeberg's Arch. Pharmacol. 388, 199–206. doi: 10.1007/s00210-014-1010-4
Kang, M. R., Kim, M. S., Oh, J. E., Kim, Y. R., Song, S. Y., Kim, S. S., et al. (2009). Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J. Pathol. 217, 702–706. doi: 10.1002/path.2509
Kim, E. J., Sidhu, M., Gaus, S. E., Huang, E. J., Hof, P. R., Miller, B. L., et al. (2012). Selective frontoinsular von Economo neuron and fork cell loss in early behavioral variant frontotemporal dementia. Cereb. Cortex 22, 251–259. doi: 10.1093/cercor/bhr004
Konopka, A., and Atkin, J. D. (2022). DNA damage, defective DNA repair, and neurodegeneration in amyotrophic lateral sclerosis. Front. Aging Neurosci. 14:786420. doi: 10.3389/fnagi.2022.786420
Konopka, A., Whelan, D. R., Jamali, M. S., Perri, E., Shahheydari, H., Toth, R. P., et al. (2020). Impaired NHEJ repair in amyotrophic lateral sclerosis is associated with TDP-43 mutations. Mol. Neurodegener. 15:51. doi: 10.1186/s13024-020-00386-4
Lagier-Tourenne, C., Polymenidou, M., and Cleveland, D. W. (2010). TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 19, R46–R64. doi: 10.1093/hmg/ddq137
Liao, Y. Z., Ma, J., and Dou, J. Z. (2022). The role of TDP-43 in neurodegenerative disease. Mol. Neurobiol. 59, 4223–4241. doi: 10.1007/s12035-022-02847-x
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Liu-Yesucevitz, L., Lin, A. Y., Ebata, A., Boon, J. Y., Reid, W., Xu, Y. F., et al. (2014). ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. J. Neurosci. 34, 4167–4174. doi: 10.1523/JNEUROSCI.2350-13.2014
Lopez-Gonzalez, R., Lu, Y., Gendron, T. F., Karydas, A., Tran, H., Yang, D., et al. (2016). Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 92, 383–391. doi: 10.1016/j.neuron.2016.09.015
Ma, Y., Silveri, L., LaCava, J., and Dokudovskaya, S. (2017). Tumor suppressor NPRL2 induces ROS production and DNA damage response. Sci. Rep. 7:15311. doi: 10.1038/s41598-017-15497-0
Mackenzie, I. R., Munoz, D. G., Kusaka, H., Yokota, O., Ishihara, K., Roeber, S., et al. (2011). Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol. 121, 207–218. doi: 10.1007/s00401-010-0764-0
Mackenzie, I. R., Neumann, M., Bigio, E. H., Cairns, N. J., Alafuzoff, I., Kril, J., et al. (2010a). Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 119, 1–4. doi: 10.1007/s00401-009-0612-2
Mackenzie, I. R., Rademakers, R., and Neumann, M. (2010b). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9, 995–1007. doi: 10.1016/S1474-4422(10)70195-2
Masrori, P., and Van Damme, P. (2020). Amyotrophic lateral sclerosis: a clinical review. Eur. J. Neurol. 27, 1918–1929. doi: 10.1111/ene.14393
Mehta, P., Raymond, J., Punjani, R., Han, M., Larson, T., Kaye, W., et al. (2022). Prevalence of amyotrophic lateral sclerosis in the United States using established and novel methodologies, 2017. Amyotroph. Lateral Scler Frontotemporal Degener. 24, 108–116. doi: 10.1080/21678421.2022.2059380
Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., and Akkari, P. A. (2019). ALS genetics, mechanisms, and therapeutics: where are we now? Front. Neurosci. 13:1310. doi: 10.3389/fnins.2019.01310
Merlo, D., Mollinari, C., Racaniello, M., Garaci, E., and Cardinale, A. (2016). DNA double Strand breaks: a common theme in neurodegenerative diseases. Curr. Alzheimer Res. 13, 1208–1218. doi: 10.2174/1567205013666160401114915
Mitra, J., Guerrero, E. N., Hegde, P. M., Liachko, N. F., Wang, H., Vasquez, V., et al. (2019). Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. U. S. A. 116, 4696–4705. doi: 10.1073/pnas.1818415116
Mohindra, A., Bolderson, E., Stone, J., Wells, M., Helleday, T., and Meuth, M. (2004). A tumour-derived mutant allele of XRCC2 preferentially suppresses homologous recombination at DNA replication forks. Hum. Mol. Genet. 13, 203–212. doi: 10.1093/hmg/ddh022
Montine, T. J., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Dickson, D. W., et al. (2012). National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 123, 1–11. doi: 10.1007/s00401-011-0910-3
Mosammaparast, N., Kim, H., Laurent, B., Zhao, Y., Lim, H. J., Majid, M. C., et al. (2013). The histone demethylase LSD1/KDM1A promotes the DNA damage response. J. Cell Biol. 203, 457–470. doi: 10.1083/jcb.201302092
Moynahan, M. E., and Jasin, M. (2010). Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 11, 196–207. doi: 10.1038/nrm2851
Nana, A. L., Sidhu, M., Gaus, S. E., Hwang, J. L., Li, L., Park, Y., et al. (2019). Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol. 137, 27–46. doi: 10.1007/s00401-018-1942-8
Neary, D., Snowden, J. S., Gustafson, L., Passant, U., Stuss, D., Black, S., et al. (1998). Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546–1554. doi: 10.1212/wnl.51.6.1546
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Olive, P. L., and Banath, J. P. (2006). The comet assay: a method to measure DNA damage in individual cells. Nat. Protoc. 1, 23–29. doi: 10.1038/nprot.2006.5
Qiu, H., Lee, S., Shang, Y., Wang, W. Y., Au, K. F., Kamiya, S., et al. (2014). ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Invest. 124, 981–999. doi: 10.1172/JCI72723
Ragagnin, A. M. G., Shadfar, S., Vidal, M., Jamali, M. S., and Atkin, J. D. (2019). Motor neuron susceptibility in ALS/FTD. Front. Neurosci. 13:532. doi: 10.3389/fnins.2019.00532
Rascovsky, K., Hodges, J. R., Knopman, D., Mendez, M. F., Kramer, J. H., Neuhaus, J., et al. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477. doi: 10.1093/brain/awr179
Ravanidis, S., Kattan, F. G., and Doxakis, E. (2018). Unraveling the pathways to neuronal homeostasis and disease: mechanistic insights into the role of RNA-binding proteins and associated factors. Int. J. Mol. Sci. 19:2280. doi: 10.3390/ijms19082280
Sedelnikova, O. A., and Bonner, W. M. (2006). GammaH2AX in cancer cells: a potential biomarker for cancer diagnostics, prediction and recurrence. Cell Cycle 5, 2909–2913. doi: 10.4161/cc.5.24.3569
Shanbhag, N. M., Evans, M. D., Mao, W., Nana, A. L., Seeley, W. W., Adame, A., et al. (2019). Early neuronal accumulation of DNA double strand breaks in Alzheimer's disease. Acta Neuropathol. Commun. 7:77. doi: 10.1186/s40478-019-0723-5
Sorensen, C. S., Hansen, L. T., Dziegielewski, J., Syljuasen, R. G., Lundin, C., Bartek, J., et al. (2005). The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 7, 195–201. doi: 10.1038/ncb1212
Suk, T. R., and Rousseaux, M. W. C. (2020). The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 15:45. doi: 10.1186/s13024-020-00397-1
Sun, Y., Curle, A. J., Haider, A. M., and Balmus, G. (2020). The role of DNA damage response in amyotrophic lateral sclerosis. Essays Biochem. 64, 847–861. doi: 10.1042/EBC20200002
Tartaglia, M. C., Sidhu, M., Laluz, V., Racine, C., Rabinovici, G. D., Creighton, K., et al. (2010). Sporadic corticobasal syndrome due to FTLD-TDP. Acta Neuropathol. 119, 365–374. doi: 10.1007/s00401-009-0605-1
Thanasoula, M., Escandell, J. M., Suwaki, N., and Tarsounas, M. (2012). ATM/ATR checkpoint activation downregulates CDC25C to prevent mitotic entry with uncapped telomeres. EMBO J. 31, 3398–3410. doi: 10.1038/emboj.2012.191
Thyagarajan, B., McCormick-Graham, M., Romero, D. P., and Campbell, C. (1996). Characterization of homologous DNA recombination activity in normal and immortal mammalian cells. Nucleic Acids Res. 24, 4084–4091. doi: 10.1093/nar/24.20.4084
Vatsavayai, S. C., Yoon, S. J., Gardner, R. C., Gendron, T. F., Vargas, J. N., Trujillo, A., et al. (2016). Timing and significance of pathological features in C9orf72 expansion-associated frontotemporal dementia. Brain 139, 3202–3216. doi: 10.1093/brain/aww250
Walker, C., Herranz-Martin, S., Karyka, E., Liao, C., Lewis, K., Elsayed, W., et al. (2017). C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat. Neurosci. 20, 1225–1235. doi: 10.1038/nn.4604
Wang, W. Y., Pan, L., Su, S. C., Quinn, E. J., Sasaki, M., Jimenez, J. C., et al. (2013). Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 16, 1383–1391. doi: 10.1038/nn.3514
Welty, S., Teng, Y., Liang, Z., Zhao, W., Sanders, L. H., Greenamyre, J. T., et al. (2018). RAD52 is required for RNA-templated recombination repair in post-mitotic neurons. J. Biol. Chem. 293, 1353–1362. doi: 10.1074/jbc.M117.808402
Wen, Q., Scorah, J., Phear, G., Rodgers, G., Rodgers, S., and Meuth, M. (2008). A mutant allele of MRE11 found in mismatch repair-deficient tumor cells suppresses the cellular response to DNA replication fork stress in a dominant negative manner. Mol. Biol. Cell 19, 1693–1705. doi: 10.1091/mbc.E07-09-0975
Wilson, S., Warr, N., Taylor, D. L., and Watts, F. Z. (1999). The role of Schizosaccharomyces pombe Rad32, the Mre11 homologue, and other DNA damage response proteins in non-homologous end joining and telomere length maintenance. Nucleic Acids Res. 27, 2655–2661. doi: 10.1093/nar/27.13.2655
Wood, M., Quinet, A., Lin, Y. L., Davis, A. A., Pasero, P., Ayala, Y. M., et al. (2020). TDP-43 dysfunction results in R-loop accumulation and DNA replication defects. J. Cell Sci. 133:jcs244129. doi: 10.1242/jcs.244129
Yang, Q., Jiao, B., and Shen, L. (2020). The development of C9orf72-related amyotrophic lateral sclerosis and frontotemporal dementia disorders. Front. Genet. 11:562758. doi: 10.3389/fgene.2020.562758
Yu, H., Harrison, F. E., and Xia, F. (2018). Altered DNA repair; an early pathogenic pathway in Alzheimer's disease and obesity. Sci. Rep. 8:5600. doi: 10.1038/s41598-018-23644-4
Zhang, Z., Almeida, S., Lu, Y., Nishimura, A. L., Peng, L., Sun, D., et al. (2013). Downregulation of microRNA-9 in iPSC-derived neurons of FTD/ALS patients with TDP-43 mutations. PLoS One 8:e76055. doi: 10.1371/journal.pone.0076055
Zhang, D., Zaugg, K., Mak, T. W., and Elledge, S. J. (2006). A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cells 126, 529–542. doi: 10.1016/j.cell.2006.06.039
Keywords: amyotrophic lateral sclerosis, frontotemporal dementia, TDP-43, DNA repair, genomic instability, homologous recombination
Citation: Fang M, Deibler SK, Nana AL, Vatsavayai SC, Banday S, Zhou Y, Almeida S, Weiss A, Brown RH, Seeley WW, Gao F-B and Green MR (2023) Loss of TDP-43 function contributes to genomic instability in amyotrophic lateral sclerosis. Front. Neurosci. 17:1251228. doi: 10.3389/fnins.2023.1251228
Edited by:
Renshi Xu, Jiangxi Provincial People's Hospital, ChinaReviewed by:
Michael Kurnellas, Independent Researcher, South San Francisco, CA, United StatesCopyright © 2023 Fang, Deibler, Nana, Vatsavayai, Banday, Zhou, Almeida, Weiss, Brown, Seeley, Gao and Green. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Minggang Fang, bWluZ2dhbmcuZmFuZ0B1bWFzc21lZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.